ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к способам лечения межбровных морщин (англ. glabellar lines, далее сокращенно GL) и боковых периорбитальных морщин (морщин у наружных уголков глаз или морщин кантальной области, так называемых "гусиных лапок", англ. lateral canthal lines, далее сокращенно LCL), имеющих выраженность от умеренной до очень сильной, посредством применения композиций, содержащих ботулинический нейротоксин. Изобретение также относится к жидким композициям, содержащим ботулинический нейротоксин.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Приведенная ниже справочная информация об уровне техники предоставлена исключительно для лучшего понимания существующего уровня техники и не составляет и не описывает предшествующий уровень техники.
В настоящее время описаны семь различающихся в общем иммунологическом отношении ботулинических нейротоксинов - серотипы ботулинического нейротоксина А, В, С, D, Е, F и G - каждый из которых может быть отличен от других по нейтрализации типоспецифических антител. В качестве примера можно привести товарный знак BOTOX®, который относится к ботулотоксину типа A (BoNT-A), очищенному комплексу нейротоксина, коммерчески поставляемому Компанией Allergan, Inc. (Irvine, Калифорния). Инъекции BOTOX® являются популярной косметической терапией, которая временно уменьшает выраженность мелких и более глубоких морщин.
В настоящее время для лечения (терапии) GL имеются четыре одобренных в США продукта BoNT-A (BOTOX COSMETIC®, DYSPORT®, XEOMIN® и JEUVEAU®), из которых для введения взрослым пациентам применяют (BOTOX COSMETIC® и XEOMIN®) или для введения взрослым пациентам в возрасте до 65 лет применяют (DYSPORT®). Для обеспечения стабильности все эти продукты хранят в лиофилизованном виде или в высушенном сублимацией виде. Перед введением пациенту врач должен восстанавливать такие композиции добавлением стерильного солевого раствора. Такой этап восстановления связан с потерей времени врачом, с риском ошибки в разбавлении и риском загрязнения. Кроме того, поставщик ботулотоксина должен проинструктировать врачей для того, чтобы этап восстановления был проведен адекватным образом.
Таким образом, в настоящее время имеется необходимость в создании композиции, содержащей ботулинический нейротоксин, предпочтительно в жидком виде, подходящем для хранения и применения в лечении.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
В целом, настоящее изобретение относится к композициям и способам, которые обеспечивают потребность в стабильных жидких композициях, содержащих ботулотоксин, которые могут быть легко введены без их восстановления или смешивания врачом.
Один из аспектов настоящего изобретения относится к жидким композициям, включающим ботулинический нейротоксин и от 1 до 5 буферных агентов, по меньшей мере один стабилизатор и по меньшей мере одно поверхностно-активное вещество.
Другой аспект настоящего изобретения относится к способам лечения межбровных морщин и/или боковых периорбитальных морщин, имеющих выраженность от умеренной до сильной, у субъекта-человека, где способы включают введение субъекту терапевтически эффективного количества жидкой композиции, включающей ботулинический нейротоксин, что приводит к снижению выраженности межбровных морщин и/или боковых периорбитальных морщин, имеющих выраженность от умеренной до сильной, причем жидкая композиция дополнительно включает от 1 до 5 буферных агентов, по меньшей мере один стабилизатор и по меньшей мере одно поверхностно-активное вещество.
В некоторых примерах осуществления жидкая композиция включает по меньшей мере 2 буферных агента, по меньшей мере 3 буферных агента или по меньшей мере 4 буферных агента.
В некоторых примерах осуществления жидкая композиция включает первый буферный агент, и в некоторых примерах осуществления первый буферный агент присутствует в концентрации, составляющей от приблизительно 100 до приблизительно 300 мМ, или в концентрации, составляющей приблизительно от 0,1 до 10 мг/мл.
В некоторых примерах осуществления жидкая композиция включает второй буферный агент, и в некоторых примерах осуществления второй буферный агент присутствует в концентрации, составляющей от приблизительно 1 до приблизительно 25 мМ, или в концентрации, составляющей приблизительно от 0,1 до 1,0 мг/мл.
В некоторых примерах осуществления жидкая композиция включает третий буферный агент, и в некоторых примерах осуществления третий буферный агент присутствует в концентрации, составляющей от приблизительно 1 до приблизительно 25 мМ, или в концентрации, составляющей приблизительно от 0,1 до 1,0 мг/мл.
В некоторых примерах осуществления жидкая композиция включает четвертый буферный агент, и в некоторых примерах осуществления четвертый буферный агент присутствует в концентрации, составляющей от приблизительно 1 до приблизительно 25 мМ, или в концентрации, составляющей приблизительно от 0,1 до 1,0 мг/мл.
В некоторых примерах осуществления жидкая композиция включает пятый буферный агент, и в некоторых примерах осуществления пятый буферный агент присутствует в концентрации, составляющей от приблизительно 1 до приблизительно 25 мМ, или в концентрации, составляющей приблизительно от 0,1 до 1,0 мг/мл.
В некоторых примерах осуществления буферные агенты выбраны из группы, состоящей из хлорида натрия, хлорида калия, фосфата натрия, фосфата калия, безводного гидрофосфата динатрия и безводного дигидрофосфата натрия.
В некоторых примерах осуществления по меньшей мере один стабилизатор представляет собой аминокислоту. Например, аминокислоту выбирают из группы, состоящей из аланина, валина, лейцина, изолейцина, метионина, фенилаланина, тирозина и триптофана. Кроме того, аминокислота может находиться в D изоформе или L изоформе. В некоторых примерах осуществления аминокислота присутствует в концентрации, составляющей от приблизительно 0,1 до приблизительно 3,0 мг/мл.
В некоторых примерах осуществления по меньшей мере одно поверхностно-активное вещество представляет собой неионное поверхностно-активное вещество (например, полисорбат 20 или полисорбат 80), и в некоторых примерах осуществления неионное поверхностно-активное вещество присутствует в концентрации, составляющей от приблизительно 0,01% (об./об.) до приблизительно 5,0% (об./об.), или в концентрации, составляющей от приблизительно 0,1 до приблизительно 3,0 мг/мл.
В некоторых примерах осуществления ботулинический нейротоксин выбран из группы, состоящей из ботулинического нейротоксина типов А, В, С, D, Е, F и G; например, ботулинический нейротоксин может представлять собой ботулинический нейротоксин типа А (т.е. BoNT-A). В некоторых примерах осуществления молекулярная масса ботулинического нейротоксина составляет приблизительно 150 кДа.
В некоторых примерах осуществления рН жидкой композиции составляет от 6,6 до 6,9. В некоторых примерах осуществления осмотическая концентрация (осмолярность) жидкой композиции составляет от 270 мосмоль/кг до 310 мосмоль/кг.
В некоторых примерах осуществления жидкая композиция включает от 10 до 200 единиц ботулинического нейротоксина на 1 мл, например, в некоторых примерах осуществления жидкая композиция может включать 100 единиц ботулинического нейротоксина на 1 мл.
В некоторых примерах осуществления способа согласно изобретению субъекту вводят от 1 до 100 единиц ботулотоксина. В некоторых примерах осуществления способа согласно изобретению субъекту вводят от 10 до 75 единиц ботулотоксина. В некоторых примерах осуществления способа согласно изобретению субъекту вводят от 25 до 75 единиц ботулотоксина. В некоторых примерах осуществления способа согласно изобретению субъекту вводят 10, 25, 30, 45, 50, 60, 75 или 90 единиц ботулотоксина.
В некоторых примерах осуществления способа согласно изобретению жидкую композицию вводят посредством инъекции, например, подкожной, чрескожной (трансдермальной), внутрикожной (интрадермальной) или внутримышечной инъекции. В некоторых примерах осуществления способа согласно изобретению субъекту вводят совокупность инъекций в межбровную область. В некоторых примерах осуществления способа согласно изобретению соседние инъекции вводят в межбровную область на расстоянии друг от друга, составляющем от приблизительно 0,5 до приблизительно 10 см, и в некоторых примерах осуществления соседние инъекции вводят на расстоянии друг от друга, составляющем от приблизительно 1,5 до приблизительно 3 см. В некоторых примерах осуществления способа согласно изобретению инъекции вводят в мышцу гордецов и мышцы, сморщивающие бровь, находящиеся на каждой стороне лица, и в некоторых примерах осуществления инъекции сначала делают в мышцу гордецов, а затем в мышцы, сморщивающие бровь, находящиеся на каждой стороне лица, перемещаясь от середины к периферии. В некоторых примерах осуществления способа согласно изобретению все инъекции производят в область, лежащую приблизительно на 1 см выше верхнего глазничного валика, и внутри области, ограниченной линиями, проходящими через середину зрачков. В некоторых примерах осуществления способа согласно изобретению все инъекции производят в область, лежащую по меньшей мере на 1 см выше центральной части надбровной дуги или остеоидной надбровной дуги. В некоторых примерах осуществления субъекту вводят совокупность инъекций в область, расположенную ниже латеральной спайки век (ниже наружного угла глаза), в наружную часть круговой мышцы глаза и/или на расстоянии от 1 до 2 см от глазничного валика. В некоторых примерах осуществления субъекту вводят совокупность инъекций в межбровную область и в область ниже латеральной спайки век, в наружную часть круговой мышцы глаза и/или на расстоянии от 1 до 2 см от глазничного валика.
Некоторые примеры осуществления способа согласно изобретению дополнительно включают приложение к верхнему глазничному валику давления во время инъекции с целью минимизации рисков возникновения локального влияния ботулинического нейротоксина.
В некоторых примерах осуществления способа согласно изобретению способ повторяют через определенные промежутки времени, составляющие от приблизительно 1 месяца до приблизительно 6 месяцев, для подавления повторного проявления; например, для подавления повторного проявления способ может быть повторен через промежутки времени, составляющие от приблизительно 3 месяцев до приблизительно 6 месяцев, или для подавления повторного проявления способ может быть повторен через промежутки времени, составляющие приблизительно 4 месяца.
Изобретение также относится к жидким композициям ботулинического нейротоксина согласно любому из предшествующих примеров для применения в лечении межбровных морщин и/или боковых периорбитальных морщин.
Изобретение также относится к применению согласно любому из предшествующих примеров осуществления жидких композиций ботулинического нейротоксина для лечения межбровных морщин и/или боковых периорбитальных морщин у субъекта, где применение включает введение субъекту композиции согласно изобретению.
Приведенное ниже подробное описание является иллюстративным и предлагаемым для лучшего разъяснения изобретения, но не является ограничивающим.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
На Фиг. 1 представлена демонстрация выраженности межбровных морщин (GL) согласно шкале, созданной Компанией Merz Aesthetic (англ. шкала Merz Aesthetic Scale, сокращенно "MAS"), в состоянии покоя и согласно шкале MAS для GL в динамике.
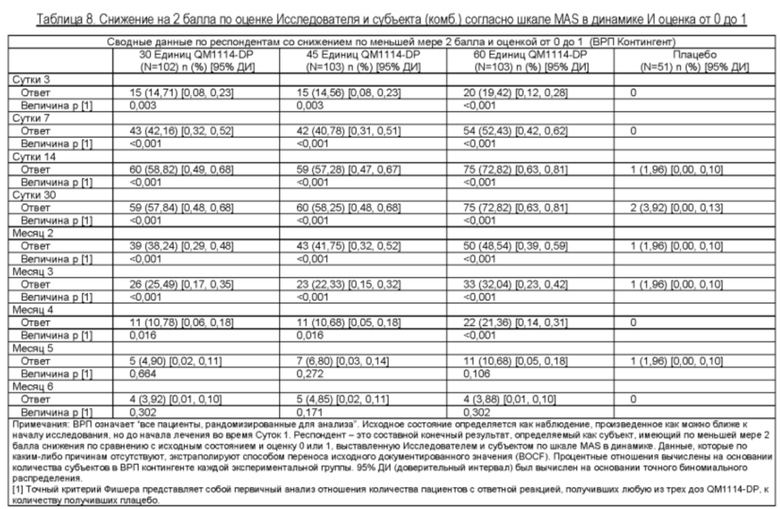
На Фиг. 2 показано, что процент пациентов с ответной реакцией на лечение, включающее введение BoNT (композиции QM1114-DP) в дозировке 30, 45 и 60 единиц, была значительно выше, чем в группе плацебо.
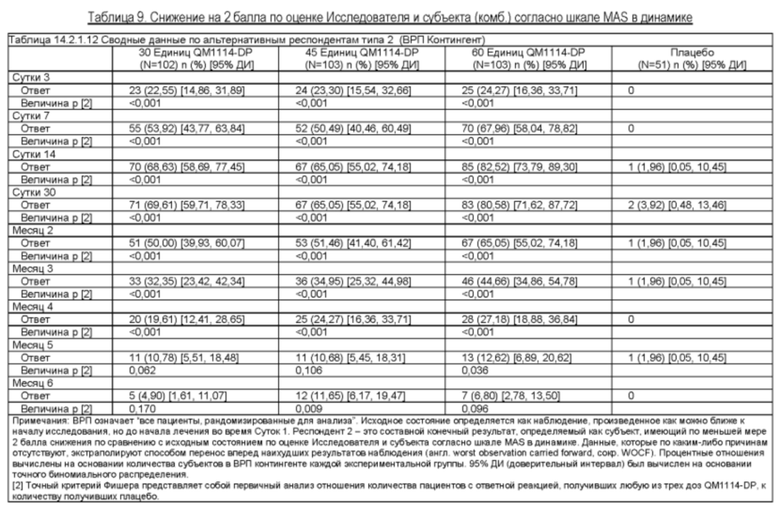
На Фиг. 3 представлен процент пациентов с ответной реакцией на лечение композицией QM1114-DP, включающей BoNT, за весь период исследования, составляющий шесть месяцев.
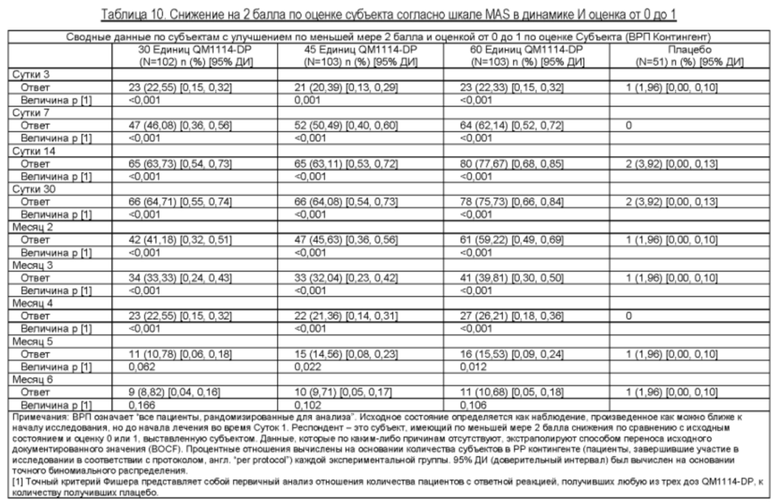
На Фиг. 4 представлен процент пациентов с ответной реакцией на лечение композицией QM1114-DP BoNT, т.е. с улучшением на 2 балла и при этом с оценкой 0 или 1, как по мнению Исследователя, так и по мнению и субъекта.
На Фиг. 5 представлено сравнение лечения ботулинический нейротоксином (композицией QM1114-DP) с лечением, в котором применяют ботулотоксины, коммерчески доступные в настоящее время в США.
На Фиг. 6 представлена длительность ответной реакции на лечение ботулинический нейротоксином (композицией QM1114-DP). Длительность ответной реакции определяли с точки зрения исследователя и с точки зрения субъекта по оценке выраженности GL согласно шкале MAS в динамике как промежуток времени до первого появления снижения по меньшей мере на 2 балла и до возврата к исходному состоянию.
На Фиг. 7 представлена удовлетворенность пациента результатами лечения ботулинический нейротоксином (композицией QM1114-DP).
На Фиг. 8А - 8В представлены нежелательные явления и побочные эффекты лечения ботулинический нейротоксином. На Фиг. 8А представлена частота возникновения боли в месте инъекции, а на Фиг. 8В представлено количество субъектов, которые сообщали о птозе (опущении верхнего века).
На Фиг. 9А - 9В представлены различные оценки глубины межбровных морщин в различных условиях. На Фиг. 9А представлена оценка вида межбровных морщин, образующихся при максимальной сознательной мышечной активности (т.е. межбровных морщин при максимальном нахмуривании), и на Фиг. 9В представлены межбровные морщины в состоянии покоя.
На Фиг. 10 представлена блок-схема клинического исследования.
На Фиг. 11 представлены точки ввода инъекций для лечения межбровных морщин.
На Фиг. 12 представлены точки ввода инъекций для лечения боковых периорбитальных морщин (LCL). В зависимости от расположения LCL у субъекта, точки ввода инъекций могут быть расположены так, как показано на изображении (А) или (В).
На Фиг. 13 представлены средние оценки состояния морщин в группе, получавшей лечение, в течение периода времени (Исследователь): при самой широкой улыбке (А) и в состоянии покоя (В).
На Фиг. 14 представлены средние оценки состояния морщин в группе, получавшей лечение, в течение периода времени (Субъект): при самой широкой улыбке (А) и в состоянии покоя (В).
На Фиг. 15 представлены средние оценки состояния морщин в группе, получавшей лечение, в течение периода времени (Исследователь): при максимальном нахмуривании/самой широкой улыбке (А) и в состоянии покоя (В).
На Фиг. 16 представлены средние оценки состояния морщин в группе, получавшей лечение, в течение периода времени (Субъект): при максимальном нахмуривании/самой широкой улыбке (А) и в состоянии покоя (В).
СВЕДЕНИЯ, ПОДТВЕРЖДАЮЩИЕ ВОЗМОЖНОСТЬ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
Далее более подробно описаны примеры осуществления настоящего изобретения. Однако аспекты изобретения могут быть воплощены в различных формах и не должны рассматриваться лишь как аспекты, ограниченные описанными в настоящей работе примерами осуществления. Напротив, приведенные примеры осуществления даны для более полного и понятного описания изобретения и полностью раскрывают объем изобретения для специалистов в данной области техники. Терминология, применяемая в настоящем описании, использована лишь с целью рассмотрения конкретных примеров осуществления и не должна рассматриваться как ограничивающая.
Если не указано иное, все термины (включая технические и научные термины), употребляемые в настоящей работе, имеют значения, обычно приписываемые им специалистами в той области техники, к которой относится изобретение. Также следует понимать, что значения таких терминов, как термины, рассмотренные в обычно используемых словарях, должны интерпретироваться как соответствующие значениям этих терминов в контексте настоящей заявки и соответствующей области техники и не должны интерпретироваться в идеализированном или чрезмерно формальном смысле, если в тексте ясно не указано иное. Несмотря на то, что это не указано ясно ниже, такие термины должны интерпретироваться согласно их обычному значению.
Терминология, употребляемая в настоящем описании, предназначена лишь для описания конкретных примеров осуществления и не ограничивает настоящее изобретение. Все публикации, патентные заявки, патенты и другие упоминаемые здесь источники полностью включены в настоящую работу посредством ссылки.
Если из контекста ясно не следует иное, то предполагается, что различные признаки изобретения, описанные в настоящей работе, могут быть применены в любой комбинации. Кроме того, также согласно изобретению, в некоторых примерах осуществления любой рассмотренный признак или комбинация рассмотренных признаков может быть исключена или пропущена. Так, если в описании указано, что комплекс включает компоненты А, В и С, то предполагается, что любой из компонентов А, В или С или их комбинация могут быть исключены и пропущены, как по отдельности, так и в любой комбинации.
Если из контекста ясно не следует иное, то все рассмотренные примеры осуществления, признаки и термины включают как приведенный пример осуществления, признак или термин, так и его биологические эквиваленты.
I. Определения
Употребляемые в настоящей работе формы единственного числа и указательные формы относятся как к единственному, так и к множественному числу, если явно не указано, что они обозначают только формы единственного числа.
Несмотря на то, что это не всегда явно указано, следует понимать, что всем числовым обозначениям предшествует термин "приблизительно". Термин "приблизительно" означает, что рассматриваемое число не ограничено точной указанной цифрой и включает числа, по существу находящиеся в области приближения к указанному числу, но при этом не выходит за пределы объема изобретения. Употребляемый в настоящей работе термин "приблизительно" должен быть понятен специалистам в данной области техники и может до некоторой степени изменяться в том контексте, в котором его употребляют. Если имеются области применения, в которых этот термин оказывается неясным для специалистов в данной области техники, учитывая контекст, в котором его употребляют, термин "приблизительно" означает плюс или минус 15%, 10%, 5%, 1%, или 0,1% от конкретной величины.
Также употребляемое в настоящей работе выражение "и/или" относится к и включает любые и все возможные комбинации одного или более связанных с ним перечисленных пунктов, а также отсутствие комбинаций, если выбрана альтернатива ("или").
Употребляемые в настоящей работе термины "вводить", "введение" или "введенный" относятся к (1) предоставлению, выдаче, дозированию и/или назначению, например, медицинским работником или его или ее уполномоченным представителем или под его/ее руководством, и (2) внесению внутрь, приему или потреблению, например, с помощью медицинского работника или самим субъектом. Введение включает, без ограничений, пероральное, парентеральное введение (например, внутримышечное, внутрибрюшинное, внутривенное, интрацеребральное введение, интрацистернальную инъекцию или инфузию, подкожную инъекцию или через имплантат), введение через ингаляционный спрей, назальное, вагинальное, ректальное, подъязычное, уретральное введение (например, через уретральный суппозиторий) или топические виды введения (например, в виде геля, мази, крема, аэрозоля и т.д.), и препарат может быть получен, как таковой или совместно, в виде подходящих стандартных лекарственных форм, содержащих традиционные нетоксичные фармацевтически приемлемые носители, адъюванты, наполнители и несущие среды, подходящие для каждого из путей введения. Изобретение не ограничено типом пути введения, композиции или режима дозирования. Фармацевтические композиции, описанные в настоящей работе, "вводят локально" (локальное введение), то есть их вводят в или вблизи участка, в котором требуется достичь терапевтического результата или эффекта.
Употребляемый в настоящей работе термин "лечение" или "терапия" включает снижение или смягчение выраженности межбровных морщин (GL) и/или боковых периорбитальных морщин (LCL) или одного или более их симптомов, независимо от того, считаются ли GL или LCL "вылеченными" или "устраненными", и независимо от того, считаются ли все симптомы исчезнувшими. Этот термин также включает уменьшение или предотвращение прогрессирования GL, LCL и/или одного или более их симптомов и достижение любого терапевтического и/или профилактического полезного эффекта.
Термин "ботулотоксин" означает ботулинический нейротоксин типа А, В, С, D, Е, F или G либо в виде чистого токсина (т.е. нейротоксичный компонент с молекулярной массой приблизительно 150 килоДальтон), либо в виде комплекса ботулотоксина (с молекулярной массой от приблизительно 300 до приблизительно 900 килоДальтон), включающего рекомбинантные, химерные, гибридные, перепрограммированные ботулинические нейротоксины и ботулинические нейротоксины с модифицированной последовательностью аминокислот, но исключая ботулотоксины, которые не являются нейротоксинами, такие как цитотоксичные ботулотоксины С2 и С3.
"Локальное введение" означает введение (т.е. посредством подкожного, внутримышечного, внутрикожного введения, введения под кожу/шкуру, введения внутрь органа, например, инъекцией в стенку мочевого пузыря или в тело простаты, или чрескожного введения) фармацевтического агента в или вблизи целевой ткани, мышцы или подкожного участка несистемным путем. Таким образом, локальное введение исключает системные пути введения (т.е. в кровеносную систему), такие как внутривенное или пероральное введение. Периферическое введение означает введение в периферию (т.е. в участок на или в лице, конечности, туловище или голове пациента), в отличие от введения во внутренний орган или кишечник (т.е. во внутренности).
"Фармацевтическая композиция" означает композицию, в которой активным ингредиентом (активным агентом) может быть ботулинический нейротоксин. Термин "композиция" означает, что, кроме активного агента, фармацевтическая композиция содержит по меньшей мере один дополнительный ингредиент. Таким образом, фармацевтическая композиция - это композиция, подходящая для диагностического или терапевтического введения (т.е. посредством подкожной или внутримышечной инъекции) субъекту, такому как пациент-человек.
"Эффективным количеством" является количество, достаточное для достижения благоприятных или требуемых результатов, таких как уменьшение, смягчение или разглаживание межбровных морщин и/или боковых периорбитальных морщин. Эффективное количество согласно изобретению также включает количество, достаточное для предотвращения усиления образования межбровных морщин или боковых периорбитальных морщин или для уменьшения уже образовавшихся межбровных морщин. Таким образом, точное "эффективное количество" не может быть установлено. Эффективное количество может быть введено за одно или более введений, нанесений или дозировок. Доставка агента зависит от ряда переменных величин, которые включают период времени, в течение которого применяют индивидуальную стандартную лекарственную форму, участок введения агента, выраженность межбровных морщин, путь введения и т.д. Однако следует понимать, что конкретные дозировки терапевтических агентов согласно настоящему изобретению для любого конкретного субъекта зависят от ряда различных факторов, которые включают активность конкретного применяемого соединения, возраст, массу тела, общее состояние здоровья, пол и диету субъекта, момент введения, скорость выведения из организма, комбинацию лекарственных средств и тяжесть конкретного подвергаемого лечению нарушения и форму введения. Для оптимизации безопасности и эффективности, терапевтические дозировки могут быть оттитрованы. Дозировка может быть определена врачом и отрегулирована, в зависимости от необходимости, для соответствия наблюдаемого эффекта от лечения.
II. Жидкая композиция
Изобретение относится к жидким композициям, включающим ботулинический нейротоксин и буфер, которые подходят для хранения в жидком виде и для лечения межбровных морщин без дополнительного восстановления или смешивания.
Ботулинический нейротоксин (BoNT) представляет собой белковый димер массой 150 килоДальтон (кДа), состоящий из тяжелой цепи с массой 100 кДа и легкой цепи с массой 50 кДа. Эти две цепи соединены дисульфидной связью, формируемой двумя цистеиновыми остатками. Легкая цепь представляет собой фермент, который разрезает синаптосомально-ассоциированный белок массой 25 кДа (SNAP-25). Тяжелая цепь опосредует связывание и интернализацию белка токсина. В отличие от других коммерчески доступных BoNT, ботулинический нейротоксин в жидкой композиции согласно изобретению (далее обозначаемой QM1114) стабилен в жидком виде и не требует восстановления или смешивания перед использованием. В некоторых примерах осуществления BoNT в составе жидкой композиции представляет собой ботулинический нейротоксин типа A (BoNT-А1).
Жидкая композиция согласно изобретению включает буфер, который включает ионы натрия, хлорид-ионы и/или фосфат-ионы. Добавление таких ионов обычно производят добавлением буферных солей.
Например, жидкая композиция может включать по меньшей мере один источник хлорид-ионов, такой как хлорид натрия, хлорид калия или другой источник хлорид-ионов, в концентрации, составляющей от приблизительно 100 до приблизительно 300 мМ, такой как 100, 110, 120, 130, 140, 150, 160, 170, 180, 190, 200, 210, 220, 230, 240, 250, 260, 270, 280, 290 или 300 мМ, или в концентрации, составляющей от приблизительно 1 до приблизительно 25 мМ, такой как 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25 мМ. В некоторых примерах осуществления жидкая композиция может включать более одного источника хлорид-ионов в одинаковых или в различных концентрациях, например, хлорид натрия или другой источник ионов натрия или хлорид-ионов в концентрации, составляющей от приблизительно 100 до приблизительно 300 мМ, такой как 100, 110, 120, 130, 140, 150, 160, 170, 180, 190, 200, 210, 220, 230, 240, 250, 260, 270, 280, 290 или 300 мМ, и хлорид калия или другой источник хлорид-ионов в концентрации, составляющей от приблизительно 1 до приблизительно 25 мМ, такой как 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25 мМ. В некоторых примерах осуществления один или более источников ионов натрия/хлорид-ионов могут присутствовать в одинаковых или в различных концентрациях, составляющих приблизительно от 0,1 до 10 мг/мл, таких как 0,1, 0,2, 0,3, 0,4, 0,5, 0,6, 0,7, 0,8, 0,9, 1,0, 1,2, 1,3, 1,4, 1,5, 1,6, 1,7, 1,8, 1,9, 2,0, 2,1, 2,2, 2,3, 2,4, 2,5, 2,6, 2,7, 2,8, 2,9, 3,0, 3,1, 3,2, 3,3, 3,4, 3,5, 3,6, 3,7, 3,8, 3,9, 4,0, 4,1, 4,2, 4,3, 4,4, 4,5, 4,6, 4,7, 4,8, 4,9, 5,0, 5,1, 5,2, 5,3, 5,4, 5,5, 5,6, 5,7, 5,8, 5,9, 6,0, 6,1, 6,2, 6,3, 6,4, 6,5, 6,6, 6,7, 6,8, 6,9, 7,0, 7,1, 7,2, 7,3, 7,4, 7,5, 7,6, 7,7, 7,8, 7,9, 8,0, 8,1, 8,2, 8,3, 8,4, 8,5, 8,6, 8,7, 8,8, 8,9, 9,0, 9,1, 9,2, 9,3, 9,4, 9,5, 9,6, 9,7, 9,8, 9,9 или 10 мг/мл или составляющих любое значение в промежутке между указанными величинами.
Аналогично, жидкая композиция может включать по меньшей мере один источник фосфат-ионов, такой как фосфат натрия, фосфат калия, безводный гидрофосфат динатрия, безводный дигидрофосфат натрия или другой источник фосфат-ионов, в концентрации, составляющей от приблизительно 1 до приблизительно 50 мМ или от приблизительно 5 до приблизительно 15 мМ, такой как 1, 5, 10, 15, 20, 25, 30, 35, 40, 45, или 50 мМ. В некоторых примерах осуществления жидкая композиция может включать более одного источника фосфат-ионов в одинаковых или в различных концентрациях, например, фосфат натрия, фосфат калия, безводный гидрофосфат динатрия, безводный дигидрофосфат натрия или другой источник фосфат-ионов, в концентрации, составляющей от приблизительно 1 до приблизительно 50 мМ или от приблизительно 5 до приблизительно 15 мМ, такой как 1, 5, 10, 15, 20, 25, 30, 35, 40, 45 или 50 мМ, и другой источник фосфат-ионов, выбранный из фосфата натрия, фосфата калия, безводного гидрофосфата динатрия, безводного дигидрофосфата натрия или другого источника фосфат-ионов в концентрации, составляющей от приблизительно 1 до приблизительно 50 мМ или приблизительно от 5 до приблизительно 15 мМ, такой как 1, 5, 10, 15, 20, 25, 30, 35, 40, 45 или 50 мМ. В некоторых примерах осуществления один или более источников фосфат-ионов могут присутствовать в одинаковых или в различных концентрациях, составляющих приблизительно от 0,1 до 1,0 мг/мл, таких как 0,1, 0,2, 0,3, 0,4, 0,5, 0,6, 0,7, 0,8, 0,9 или 1,0 мг/мл или составляющих любое значение в промежутке между указанными величинами.
В некоторых примерах осуществления жидкая композиция может включать от 1 до 5 или более буферных агентов. Таким образом, жидкая композиция может включать 1, 2, 3, 4 или 5 или более буферных агентов, примеры которых включают, без ограничений, хлорид натрия, хлорид калия, фосфат натрия, фосфат калия, безводный гидрофосфат динатрия или безводный дигидрофосфат натрия. Буферные агенты в количестве 1, 2, 3, 4 или 5 или более могут присутствовать в одинаковых или в различных концентрациях. Например, в некоторых примерах осуществления первый буферный агент (например, хлорид натрия, хлорид калия, фосфат натрия, фосфат калия, безводный гидрофосфат динатрия или безводный дигидрофосфат натрия) может присутствовать в концентрации, составляющей от приблизительно 100 до приблизительно 300 мМ, такой как 100, 110, 120, 130, 140, 150, 160, 170, 180, 190, 200, 210, 220, 230, 240, 250, 260, 270, 280, 290 или 300 мМ, или в концентрации, составляющей приблизительно от 0,1 до 10 мг/мл, такой как 0,1, 0,2, 0,3, 0,4, 0,5, 0,6, 0,7, 0,8, 0,9, 1,0, 1,2, 1,3, 1,4, 1,5, 1,6, 1,7, 1,8, 1,9, 2,0, 2,1, 2,2, 2,3, 2,4, 2,5, 2,6, 2,7, 2,8, 2,9, 3,0, 3,1, 3,2, 3,3, 3,4, 3,5, 3,6, 3,7, 3,8, 3,9, 4,0, 4,1, 4,2, 4,3, 4,4, 4,5, 4,6, 4,7, 4,8, 4,9, 5,0, 5,1, 5,2, 5,3, 5,4, 5,5, 5,6, 5,7, 5,8, 5,9, 6,0, 6,1, 6,2, 6,3, 6,4, 6,5, 6,6, 6,7, 6,8, 6,9, 7,0, 7,1, 7,2, 7,3, 7,4, 7,5, 7,6, 7,7, 7,8, 7,9, 8,0, 8,1, 8,2, 8,3, 8,4, 8,5, 8,6, 8,7, 8,8, 8,9, 9,0, 9,1, 9,2, 9,3, 9,4, 9,5, 9,6, 9,7, 9,8, 9,9 или 10 мг/мл или составляющей любое значение в промежутке между указанными величинами. В некоторых примерах осуществления второй буферный агент (например, хлорид натрия, хлорид калия, фосфат натрия, фосфат калия, безводный гидрофосфат динатрия или безводный дигидрофосфат натрия) может присутствовать в концентрации, составляющей от приблизительно 1 до приблизительно 25 мМ, такой как 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25 мМ, или в концентрации, составляющей приблизительно от 0,1 до 1,0 мг/мл, такой как 0,1, 0,2, 0,3, 0,4, 0,5, 0,6, 0,7, 0,8, 0,9 или 1,0 мг/мл или составляющей любое значение в промежутке между указанными величинами. В некоторых примерах осуществления третий буферный агент (например, хлорид натрия, хлорид калия, фосфат натрия, фосфат калия, безводный гидрофосфат динатрия или безводный дигидрофосфат натрия) может присутствовать в концентрации, составляющей от приблизительно 1 до приблизительно 25 мМ, такой как 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25 мМ, или в концентрации, составляющей приблизительно от 0,1 до 1,0 мг/мл, такой как 0,1, 0,2, 0,3, 0,4, 0,5, 0,6, 0,7, 0,8, 0,9 или 1,0 мг/мл или составляющей любое значение в промежутке между указанными величинами. В некоторых примерах осуществления четвертый буферный агент (например, хлорид натрия, хлорид калия, фосфат натрия, фосфат калия, безводный гидрофосфат динатрия или безводный дигидрофосфат натрия) может присутствовать в концентрации, составляющей от приблизительно 1 до приблизительно 25 мМ, такой как 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25 мМ, или в концентрации, составляющей приблизительно от 0,1 до 1,0 мг/мл, такой как 0,1, 0,2, 0,3, 0,4, 0,5, 0,6, 0,7, 0,8, 0,9 или 1,0 мг/мл или составляющей любое значение в промежутке между указанными величинами. В некоторых примерах осуществления пятый буферный агент (например, хлорид натрия, хлорид калия, фосфат натрия, фосфат калия, безводный гидрофосфат динатрия или безводный дигидрофосфат натрия) может присутствовать в концентрации, составляющей от приблизительно 1 до приблизительно 25 мМ, такой как 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25 мМ, или в концентрации, составляющей приблизительно от 0,1 до 1,0 мг/мл, такой как 0,1, 0,2, 0,3, 0,4, 0,5, 0,6, 0,7, 0,8, 0,9 или 1,0 мг/мл или составляющей любое значение в промежутке между указанными величинами.
Для повышения стабильности или улучшения других свойств композиции, в жидкую композицию также могут быть включены другие компоненты. Например, подходящие для применения стабилизаторы могут включать, без ограничений, аминокислоты (например, аланин, валин, лейцин, серии, треонин, лизин, гистидин, триптофан, аспарагиновую кислоту или глутаминовую кислоту), гидросульфит натрия, цитрат натрия или другие цитраты и т.д. В некоторых примерах осуществления аминокислота может представлять собой аминокислоту с гидрофобной боковой цепью (например, аланин, валин, лейцин, изолейцин, метионин, фенилаланин, тирозин и триптофан). В некоторых примерах осуществления аминокислота может быть в D изоформе, и в некоторых примерах осуществления аминокислота может быть в L изоформе. Таким образом, в некоторых примерах осуществления жидкая композиция может включать по меньшей мере одну D- или L-аминокислоту (например, аланин, валин, лейцин, серии, треонин, лизин гистидин, триптофан, аспарагиновую кислоту или глутаминовую кислоту) в концентрации, составляющей от приблизительно 0,1 до приблизительно 3,0 мг/мл, от приблизительно 0,5 до приблизительно 2,5 мг/мл или от приблизительно 0,75 до приблизительно 2,25 мг/мл. В некоторых примерах осуществления жидкая композиция может включать по меньшей мере одну D- или L-аминокислоту (например, аланин, валин, лейцин, серии, треонин, лизин гистидин, триптофан, аспарагиновую кислоту или глутаминовую кислоту) в концентрации, составляющей 0,1, 0,2, 0,3, 0,4, 0,5, 0,6, 0,7, 0,8, 0,9, 1,0, 1,1, 1,2, 1,3, 1,4, 1,5, 1,6, 1,7, 1,8, 1,9, 2,0, 2,1, 2,2, 2,3, 2,4, 2,5, 2,6, 2,7, 2,8, 2,9 или 3,0 мг/мл или составляющей любое значение в промежутке между указанными величинами. В некоторых примерах осуществления жидкая композиция может включать по меньшей мере одну D- или L-аминокислоту (например, аланин, валин, лейцин, серии, треонин, лизин гистидин, триптофан, аспарагиновую кислоту или глутаминовую кислоту) в концентрации, составляющей приблизительно 0,1, приблизительно 0,2, приблизительно 0,3, приблизительно 0,4, приблизительно 0,5, приблизительно 0,6, приблизительно 0,7, приблизительно 0,8, приблизительно 0,9, приблизительно 1,0, приблизительно 1,1, приблизительно 1,2, приблизительно 1,3, приблизительно 1,4, приблизительно 1,5, приблизительно 1,6, приблизительно 1,7, приблизительно 1,8, приблизительно 1,9, приблизительно 2,0, приблизительно 2,1, приблизительно 2,2, приблизительно 2,3, приблизительно 2,4, приблизительно 2,5, приблизительно 2,6, приблизительно 2,7, приблизительно 2,8, приблизительно 2,9 или приблизительно 3,0 мг/мл или составляющей любое значение в промежутке между указанными величинами.
В некоторых примерах осуществления жидкая композиция может дополнительно включать одно или более поверхностно-активных веществ (например, неионных поверхностно-активных веществ, таких как полисорбат (например, полисорбат 80 или полисорбат 20) или ноноксинолы; анионных поверхностно-активных веществ, таких как докузат; или катионных поверхностно-активных веществ, таких как четвертичные аммонийные соли). Таким образом, в некоторых примерах осуществления жидкая композиция может включать неионное поверхностно-активное вещество, примеры которого включают, без ограничений, полисорбат (например, полисорбат 80 или полисорбат 20) или ноноксинол. В некоторых примерах осуществления жидкая композиция может включать анионное поверхностно-активное вещество, примеры которого включают, без ограничений, докузат. В некоторых примерах осуществления жидкая композиция может включать катионное поверхностно-активное вещество, примеры которого включают, без ограничений, четвертичную аммонийную соль. В некоторых примерах осуществления поверхностно-активное вещество может присутствовать в концентрации, составляющей от приблизительно 0,01% (об./об.) до приблизительно 5,0% (об./об.), от приблизительно 0,05% (об./об.) до приблизительно 2,5% (об./об.) или от приблизительно 0,1% (об./об.) до приблизительно 1,5% (об./об.). В некоторых примерах осуществления по меньшей мере одно поверхностно-активное вещество может присутствовать в концентрации, составляющей 0,01, 0,02, 0,03, 0,04, 0,05, 0,06, 0,07, 0,08, 0,09, 0,1, 0,2, 0,3, 0,4, 0,5, 0,6, 0,7, 0,8, 0,9, 1,0, 1,1, 1,2, 1,3, 1,4, 1,5, 1,6, 1,7, 1,8, 1,9, 2,0, 2,1, 2,2, 2,3, 2,4, 2,5, 2,6, 2,7, 2,8, 2,9, 3,0, 3,1, 3,2, 3,3, 3,4, 3,5, 3,6, 3,7, 3,8, 3,9, 4,0, 4,1, 4,2, 4,3, 4,4, 4,5, 4,6, 4,7, 4,8, 4,9 или 5,0% (об./об.) или составляющей любое значение в промежутке между указанными величинами. В некоторых примерах осуществления по меньшей мере одно поверхностно-активное вещество может присутствовать в концентрации, составляющей приблизительно 0,01, приблизительно 0,02, приблизительно 0,03, приблизительно 0,04, приблизительно 0,05, приблизительно 0,06, приблизительно 0,07, приблизительно 0,08, приблизительно 0,09, приблизительно 0,1, приблизительно 0,2, приблизительно 0,3, приблизительно 0,4, приблизительно 0,5, приблизительно 0,6, приблизительно 0,7, приблизительно 0,8, приблизительно 0,9, приблизительно 1,0, приблизительно 1,1, приблизительно 1,2, приблизительно 1,3, приблизительно 1,4, приблизительно 1,5, приблизительно 1,6, приблизительно 1,7, приблизительно 1,8, приблизительно 1,9, приблизительно 2,0, приблизительно 2,1, приблизительно 2,2, приблизительно 2,3, приблизительно 2,4, приблизительно 2,5, приблизительно 2,6, приблизительно 2,7, приблизительно 2,8, приблизительно 2,9, приблизительно 3,0, приблизительно 3,1, приблизительно 3,2, приблизительно 3,3, приблизительно 3,4, приблизительно 3,5, приблизительно 3,6, приблизительно 3,7, приблизительно 3,8, приблизительно 3,9, приблизительно 4,0, приблизительно 4,1, приблизительно 4,2, приблизительно 4,3, приблизительно 4,4, приблизительно 4,5, приблизительно 4,6, приблизительно 4,7, приблизительно 4,8, приблизительно 4,9 или приблизительно 5,0% (об./об.) или составляющей любое значение в промежутке между указанными величинами. В некоторых примерах осуществления жидкая композиция может включать по меньшей мере одно поверхностно-активное вещество в концентрации, составляющей 0,1, 0,2, 0,3, 0,4, 0,5, 0,6, 0,7, 0,8, 0,9, 1,0, 1,1, 1,2, 1,3, 1,4, 1,5, 1,6, 1,7, 1,8, 1,9, 2,0, 2,1, 2,2, 2,3, 2,4, 2,5, 2,6, 2,7, 2,8, 2,9 или 3,0 мг/мл или составляющей любое значение в промежутке между указанными величинами. В некоторых примерах осуществления жидкая композиция может включать по меньшей мере одно поверхностно-активное вещество в концентрации, составляющей приблизительно 0,1, приблизительно 0,2, приблизительно 0,3, приблизительно 0,4, приблизительно 0,5, приблизительно 0,6, приблизительно 0,7, приблизительно 0,8, приблизительно 0,9, приблизительно 1,0, приблизительно 1,1, приблизительно 1,2, приблизительно 1,3, приблизительно 1,4, приблизительно 1,5, приблизительно 1,6, приблизительно 1,7, приблизительно 1,8, приблизительно 1,9, приблизительно 2,0, приблизительно 2,1, приблизительно 2,2, приблизительно 2,3, приблизительно 2,4, приблизительно 2,5, приблизительно 2,6, приблизительно 2,7, приблизительно 2,8, приблизительно 2,9 или приблизительно 3,0 мг/мл или составляющей любое значение в промежутке между указанными величинами.
В некоторых примерах осуществления жидкая композиция может дополнительно включать один или более эмульгаторов (например, соевый лецитин), смачивающих агентов, наполнителей (лактозу, маннит, глюкозу, микрокристаллическую целлюлозу, коллоидный оксид кремния, крахмал и т.д.), связующие вещества (гидроксипропилцеллюлозу, поливинилпирролидон, мета силикат-алюминат магния и т.д.), дезинтегрирующий агент (крахмал, L-гидроксипропилцеллюлозу, карбоксиметилцеллюлозу, натрий-кроскармеллозу, целлюлозу-гликолят кальция и т.д.), скользящие вещества (стеарат магния и т.д.), агенты, вызывающие набухание (гидроксипропилцеллюлозу, гидроксипропилметилцеллюлозу, карбопол, карбоксиметилцеллюлозу, поливиниловый спирт, ксантановую камедь и гуаровую камедь и т.д.), вспомогательные средства, вызывающие набухание (глюкозу, фрукутозу, маннит, ксилит, эритрит, мальтозу, трегалозу, соли фосфаты, цитраты, силикаты, глицин, глутамат, аргинин и т.д.) и/или солюбилизирующие агенты (полиэтиленгликоль, пропиленгликоль и т.д.).
В некоторых примерах осуществления жидкая композиция может включать от 1 до 5 или более буферных агентов (например, хлорид натрия, хлорид калия, фосфат натрия, фосфат калия, безводный гидрофосфат динатрия или безводный дигидрофосфат натрия); один или более стабилизаторов (например, аланин, валин, лейцин, серии, треонин, лизин, гистидин, триптофан, аспарагиновую кислоту или глутаминовую кислоту); и одно или более поверхностно-активных веществ (например, неионные поверхностно-активные вещества, такие как полисорбат (например, полисорбат 80 или полисорбат 20) или ноноксинолы; анионные поверхностно-активные вещества, такие как докузат; или катионные поверхностно-активные вещества, такие как четвертичные аммонийные соли). В некоторых примерах осуществления жидкая композиция может включать: (i) первый буферный агент (например, хлорид натрия, хлорид калия, фосфат натрия, фосфат калия, безводный гидрофосфат динатрия или безводный дигидрофосфат натрия) в концентрации, составляющей от приблизительно 100 до приблизительно 300 мМ, или в концентрации, составляющей приблизительно от 0,1 до 10 мг/мл; (ii) второй буферный агент (например, хлорид натрия, хлорид калия, фосфат натрия, фосфат калия, безводный гидрофосфат динатрия или безводный дигидрофосфат натрия) в концентрации, составляющей от приблизительно 1 до приблизительно 25 мМ, или в концентрации, составляющей приблизительно от 0,1 до 1,0 мг/мл; (iii) третий буферный агент (например, хлорид натрия, хлорид калия, фосфат натрия, фосфат калия, безводный гидрофосфат динатрия или безводный дигидрофосфат натрия) в концентрации, составляющей от приблизительно 1 до приблизительно 25 мМ, или в концентрации, составляющей приблизительно от 0,1 до 1,0 мг/мл; (iv) четвертый буферный агент (например, хлорид натрия, хлорид калия, фосфат натрия, фосфат калия, безводный гидрофосфат динатрия или безводный дигидрофосфат натрия) в концентрации, составляющей от приблизительно 1 до приблизительно 25 мМ, или в концентрации, составляющей приблизительно от 0,1 до 1,0 мг/мл; (v) один или более стабилизаторов (например, аланин, валин, лейцин, серии, треонин, лизин, гистидин, триптофан, аспарагиновую кислоту или глутаминовую кислоту) в концентрации, составляющей от приблизительно 0,1 до приблизительно 3,0 мг/мл; и (vi) одно или более поверхностно-активных веществ (например, неионных поверхностно-активных веществ, таких как полисорбат (например, полисорбат 80 или полисорбат 20) или ноноксинолы; анионных поверхностно-активных веществ, таких как докузат; или катионных поверхностно-активных веществ, таких как четвертичные аммонийные соли) в концентрации, составляющей от приблизительно 0,05% (об./об.) до приблизительно 2,5% (об./об.), или в концентрации, составляющей от приблизительно 0,1 до приблизительно 3,0 мг/мл. В некоторых примерах осуществления стабилизатор может представлять собой аминокислоту, и в некоторых примерах осуществления поверхностно-активное вещество может быть неионным поверхностно-активным веществом, таким как полисорбат. При осуществлении настоящего изобретения следует понимать, что примеры QM1114-DP могут включать все описанные выше примеры осуществления.
Величина рН жидкой композиции согласно изобретению составляет от 5,5 до 8. Согласно предпочтительному примеру осуществления, рН составляет от 6,0 до 7,5, например, приблизительно 6,3, 6,35, 6,4, 6,45, 6,5, 6,55, 6,6, 6,65, 6,7, 6,75, 6,8, 6,85, 6,9, 6,95, 7,0, 7,05, 7,1, 7,15, 7,2, 7,25, 7,3, 7,35, 7,4, 7,45 или 7,5. Предпочтительно рН составляет от 6,6 до 6,9. Жидкая композиция предпочтительно включает водный разбавитель, более предпочтительно воду, например, стерильную воду, воду для инъекций, очищенную воду и стерильную воду для инъекций.
Предпочтительно жидкая композиция подходит для инъекций пациенту, в частности, пациенту-человеку. Количество ботулинического нейротоксина обычно выражают в единицах LD50 (сокр. от англ. "lethal dose 50", то есть "летальная доза, приводящая к гибели 50% испытуемых организмов") для мышей, определяемой как средняя летальная доза для мышей при интраперитонеальном введении. Единица LD50 для мышей (англ. mouse lethal dose 50, сокращенно MLD50) для ботулотоксина не является стандартизованной единицей. Действительно, исследования, проводимые разными изготовителями поставляемых на рынок токсинов, различаются, в частности, выбором буфера разбавления. Например, в испытаниях DYSPORT® применяли желатино-фосфатный буфер, в то время как в исследовании BOTOX® в качестве разбавителя применяли солевой раствор. Полагают, что желатиновые буферы защищают токсин при высоких степенях разбавления, используемых при определении LD50. Напротив, полагают, что применение в качестве разбавителя солевого раствора приводит к некоторой потере активности. Этим можно объяснить тот факт, что в испытаниях с DYSPORT® одна единица BOTOX® эквивалентна приблизительно трем единицам DYSPORT® (Straughan, D. W., 2006, AT LA 34(3), 305-313; Hambleton, Pickett, Hambleton, P., A. M. Pickett., 1994, Journal of the Royal Society of Medicine 87.11: 719).
Предпочтительно буфер разбавления, применяемый для определения LD50 для мышей, представляет собой желатино-фосфатный буфер. Например, LD50 для мышей может быть определена, как описано в публикации Hambleton, Р. с соавт. "Получение, очистка и получение обезвреженного токсина Clostridium Botulinum типа A (Production, purification and toxoiding of Clostridium botulinum type A toxin)", изд. G. E. Jr Lewis, P. S. Angel. Academic Press, Inc., New York, USA, 1981, стр. 248. Вкратце, образцы ботулотоксина последовательно разбавляют в 0,2% (масс./об.) желатинового буфера, содержащего 0.07М Na2HPO4, с рН 6,5. Группам мышей (например, содержащим от 4 до 8 мышей в группе) с массой тела приблизительно 20 г интраперитонеально вводят образец разбавленного токсина (например, 0,5 мл одному животному). Для определения дозы 50% летальности выбирают группы разбавления, например, 5 групп разбавления. За мышами наблюдают в течение периода, составляющего до 96 часов, и оценивают летальную дозу, приводящую к гибели 50% испытуемых мышей (MLD50).
Жидкая композиция согласно изобретению предпочтительно включает от 4 до 10000 единиц LD50 ботулинического нейротоксина на 1 мл, более предпочтительно от 10 до 200 единиц LD50 ботулинического нейротоксина на 1 мл, например, 10, 15, 20, 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100, 105, 110, 115, 120, 125, 130, 135, 140, 145, 150, 155, 160, 165, 170, 175, 180, 185, 190, 195, 200 единиц LD50 ботулинического нейротоксина на 1 мл. Количество ботулинического нейротоксина также может быть выражено в нг (нанограммах).
Жидкая композиция согласно настоящему изобретению имеет осмотическую концентрацию, составляющую от 200 до 400 мосмоль/кг, предпочтительно от 270 до 310 мосмоль/кг, например, 270, 275, 280, 285, 290, 295, 300, 305 или 310 мосмоль/кг или составляющую любое значение в промежутке между указанными величинами.
III. Лечение
Изобретение также относится к способам лечения у субъекта-человека межбровных морщин и/или боковых периорбитальных морщин, имеющих выраженность от умеренной до сильной, где способы включают введение субъекту терапевтически эффективного количества жидкой композиции, включающей ботулинический нейротоксин, что приводит к снижению выраженности межбровных морщин, имеющих выраженность от умеренной до сильной. Жидкая композиция может соответствовать любому из описанных выше в разделе II примеров осуществления.
В некоторых примерах осуществления субъекту вводят от 1 до 100 единиц ботулотоксина. В некоторых примерах осуществления субъекту вводят от 10 до 75 единиц ботулотоксина. В некоторых примерах осуществления субъекту вводят от 25 до 75 единиц ботулотоксина. В некоторых примерах осуществления субъекту вводят 10, 15, 20, 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95 или 100 единиц ботулотоксина. В некоторых примерах осуществления субъекту вводят от 50 до 250 единиц ботулотоксина. В некоторых примерах осуществления субъекту вводят от 75 до 200 единиц ботулотоксина. В некоторых примерах осуществления концентрация жидкой композиции, вводимой субъекту, составляет от 1 до 300 единиц ботулотоксина/мл, например, 1, 5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100, 110, 115, 120, 125, 130, 135, 140, 145, 150, 155, 160, 165, 170, 175, 180, 185, 190, 195, 200, 210, 215, 220, 225, 230, 235, 240, 245, 250, 255, 260, 265, 270, 275, 280, 285, 290, 295 или 300 единиц/мл.
В некоторых примерах осуществления композицию вводят посредством инъекции. В некоторых примерах осуществления инъекцию производят подкожно, чрескожно, внутрикожно или внутримышечно. В некоторых примерах осуществления способ включает многократное введение инъекций в межбровную область. В некоторых примерах осуществления соседние участки, в которые вводят инъекции, расположены на расстоянии друг от друга, составляющем приблизительно от 0,5 до 10 см. В некоторых примерах осуществления соседние участки, в которые вводят инъекции, расположены на расстоянии друг от друга, составляющем приблизительно от 1,5 до 3 см.
В некоторых примерах осуществления инъекции производят в мышцу гордецов и мышцы, сморщивающие бровь, на каждой стороне лица, и в некоторых примерах осуществления инъекции производят в определенном порядке, например, начиная с мышцы гордецов, и затем в мышцы, сморщивающие бровь, на каждой стороне лица, перемещаясь от середины к периферии. В некоторых примерах осуществления все инъекции производят в область, лежащую приблизительно на 1 см выше верхнего глазничного валика, и внутри области, ограниченной линиями, проходящими через середину зрачков.
В некоторых примерах осуществления способ дополнительно включает приложение к верхнему глазничному валику давления во время инъекции с целью минимизации рисков от локального влияния нейротоксина.
В некоторых примерах осуществления все инъекции производят в область, лежащую по меньшей мере на 1 см выше центральной части надбровной дуги или остеоидной надбровной дуги.
В некоторых примерах осуществления композиция может быть введена для лечения, предотвращения ухудшения состояния или для улучшения состояния боковых периорбитальных морщин (LCL). В некоторых примерах осуществления лечение может включать приблизительно три инъекции (одна инъекция на одну точку ввода инъекции). Например, лечение может включать 1, 2, 3, 4 или 5 инъекций. Точки введения инъекций могут быть уточнены в соответствии с рисунком (расположением) LCL морщин конкретного субъекта. В зависимости от расположения морщин конкретных субъектов, если морщины в LCL области находятся выше и ниже латеральной спайки век, то инъекции вводят, как показано, например, на Фиг. 12А. В альтернативном варианте, если морщины в LCL области конкретного субъекта в основном расположены ниже латеральной спайки век, то инъекции вводят, как показано, например, на Фиг. 12В. В некоторых примерах осуществления точки инъекции могут располагаться в наружной части круговой мышцы глаза и, если это возможно, на расстоянии приблизительно 1-2 см от глазничного валика. Некоторые примеры осуществления могут включать введение трех инъекций равного объема (100 мкл) в каждую из сторон лица (т.е. всего шесть инъекций).
В некоторых примерах осуществления композиция может быть введена для лечения, предотвращения ухудшения состояния или для улучшения состояния одновременно и GL, и LCL. В некоторых примерах осуществления лечение может включать, например, 11 инъекций (одну инъекцию на одну точку ввода инъекции) равного объема (100 мкл), вводимых в область GL (пять инъекций) и область LCL (три инъекции в каждую из сторон лица). В некоторых примерах осуществления одновременное лечение GL и LCL может включать 5, 6, 7, 8, 9, 10, 11, 12, 13 или 14 или более инъекций. Например, при лечении GL пять точек ввода инъекций могут включать две инъекции в каждую мышцу, сморщивающую бровь, и одну инъекцию в мышцу гордецов, а при лечении LCL расположение инъекций может быть отрегулировано в соответствии с расположением LCL морщин конкретного субъекта. В зависимости от расположения морщин конкретных субъектов, если морщины в LCL области находятся выше и ниже латеральной спайки век, то инъекции могут быть введены, как показано, на Фиг. 12А. В альтернативном варианте, если морщины в LCL области конкретного субъекта в основном расположены ниже латеральной спайки век, то инъекции могут быть введены, как показано на Фиг. 12В. В некоторых примерах осуществления точки инъекций для лечения LCL могут располагаться в наружной части круговой мышцы глаза и, если это возможно, на расстоянии приблизительно от 1 до 2 см от глазничного валика.
В некоторых примерах осуществления для подавления повторного проявления способ повторяют через определенные промежутки времени, составляющие от приблизительно 3 месяцев до приблизительно 6 месяцев. В некоторых примерах осуществления для подавления повторного проявления способ повторяют через определенные промежутки времени, составляющие приблизительно 4 месяца.
Осуществление способа согласно изобретению приводит к временному снижению выраженности межбровных морщин и/или боковых периорбитальных морщин у субъекта. Эффективность лечения может быть оценена способами, известными специалистам в данной области техники. Иллюстративные способы оценки приведены ниже и в Примерах.
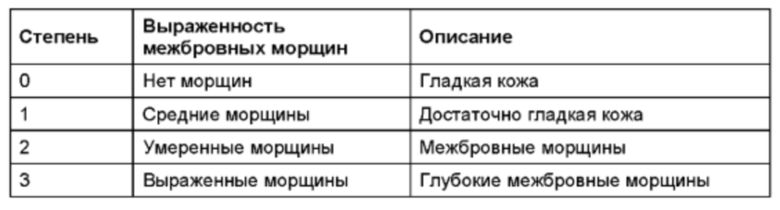
а. Оценка выраженности межброеных морщин на фотографическом изображении по 4-балльной шкале: Оценка Исследователем в реальных условиях (GL ОИР)
Обоснованная оценка выраженности межбровных морщин на фотографическом изображении по 4-балльной шкале включает две системы оценивания: одну - оценку Исследователем в реальных условиях при максимальном нахмуривании, и одну - оценку Исследователем в реальных условиях в состоянии покоя. Согласно шкале, межбровные морщины имеют следующую выраженность: отсутствие (степень 0), средние (степень 1), умеренные (степень 2) и выраженные межбровные морщины (степень 3). Каждая степень также запечатлена на индивидуальной фотографии и в описательном тексте. Исследователи обучаются оцениванию фотографического изображения по 4-балльной шкале. Исследователи используют оценку фотографического изображения по 4-балльной шкале для прямого сравнения с живым лицом субъекта во время отборочных визитов, при установлении исходного состояния (до начала лечения) и во время всех визитов по завершении лечения. Субъекты производят свое оценивание выраженности своих межбровных морщин независимо оценки исследователя. Субъектов просят оценить состояние собственных межбровных морщин при максимальном нахмуривании во время отборочных визитов, при установлении исходного состояния (до начала лечения) и во время всех визитов по завершении лечения по статичной категориальной 4-балльной шкале.
b. Шкала общего эстетического улучшения (англ. Global Aesthetic Improvement Scale, сокращенно GAIS))
Субъекты оценивают общее эстетическое улучшение своих межбровных морщин при максимальном нахмуривании в сравнении с их выраженностью до начала лечения, используя следующую категориальную шкалу во время всех визитов по окончании лечения:

Субъектов спрашивают: "Как бы Вы оценили изменения в выраженности Ваших межбровных морщин (морщин между бровями) при максимальном нахмуривании по сравнению с состоянием непосредственно до инъекции?"
Субъектов просят выбрать одну категорию, которая наилучшим образом описывает изменение выраженности их межбровных морщин при максимальном нахмуривании по сравнению с исходным состоянием. Для помощи в оценивании, субъект может посмотреть фотографию исходного состояния.
c. Дневник пациента
Субъектов просят записывать их оценку ответной реакции на исследуемое лечение в дневник пациента, начиная с суток после получения лечения (Сутки 1) до Суток 7 (визит 3 в рамках исследования). Их просят ответить "да" или "нет" на следующий вопрос: "Заметили ли Вы с момента введения инъекции улучшение внешнего вида Ваших межбровных морщин (морщин между бровями)?" Субъекты должны были ежесуточно заполнять дневник пациента и вернуть дневник в исследовательский центр во время визита в Сутки 7.
d. FACE-Q
FACE-Q - это инструмент для сбора информации от пациентов путем анкетирования, предназначенный для оценки опыта и результатов процедур по эстетической коррекции лица с точки зрения субъекта. FACE-Q состоит из более чем 40 шкал (анкет), относящихся к четырем областям (Удовлетворенность внешним видом лица, Качество жизни, обусловленное состоянием здоровья, Неблагоприятные явления и Процесс ухода). В каждой области имеется одна или более независимо функционирующих шкал. Для целей настоящего исследования и с учетом состояния, подвергаемого лечению, была выбрана шкала Физиологических Функций, которую субъекты заполняли в моменты времени, указанные в Графике оценивания.
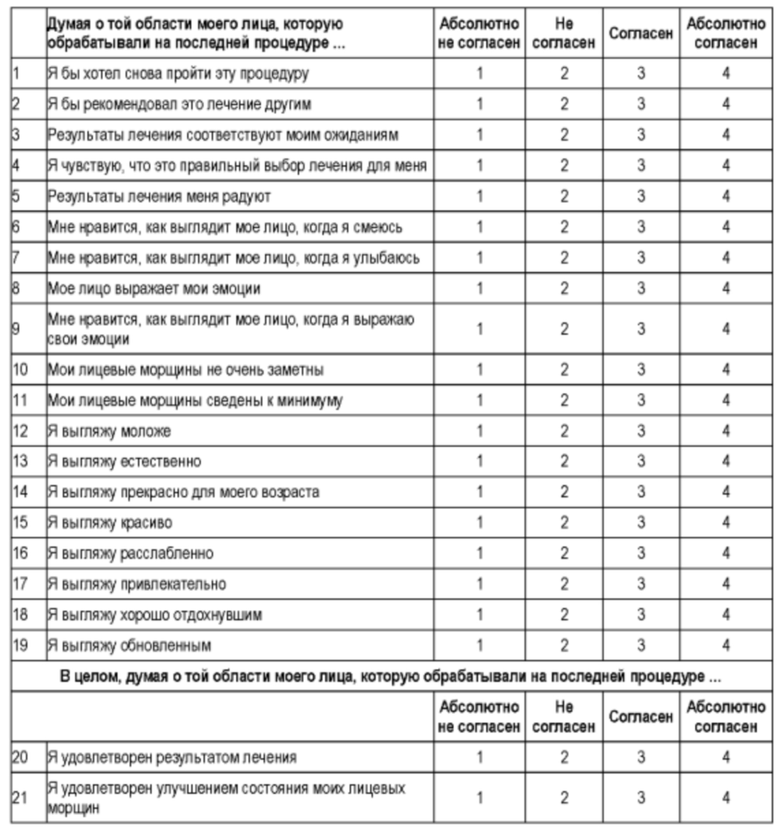
е. Анкета удовлетворенности лечением лицевых морщин
Указанные вопросы относятся к той области лица субъекта, которую обрабатывали в последней перед опросом процедуре. Субъекты выбирают ответ, который наилучшим образом отражает их согласие с приведенным утверждением. Пример анкеты представлен ниже. На каждый вопрос может быть дан только один ответ.
f. Анкета удовлетворенности состоянием морщин (УСМ)
В исходном состоянии (до начала лечения) и при всех визитах по окончании лечения, субъектов просят заполнить обоснованную анкету УСМ, составленную Galderma.
g. Независимая оценка по фотографии (НОФ)
В трех НОФ, ослепленных в отношении рандомизированного лечения субъектов, производят оценивание по фотографии GL каждого субъекта при максимальном нахмуривании в соответствии с обоснованной оценкой выраженности межбровных морщин по 4-балльной шкале на фотографическом изображении. Оценку НОФ всех субъектов производят в конце исследования. Для НОФ применяют шкалу для сравнения фотографий GL каждого субъекта в исходном состоянии при максимальном нахмуривании с аналогичными фотографиями во время каждого визита после проведенного лечения. Количество баллов НОФ определяют в виде среднего всех оценок, выставленных тремя экспертами. НОФ не включены в другие аспекты исследования.
ОПИСАНИЕ ПРИМЕРОВ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
Пример 1
Лечение межбровных морщин (GL) от умеренных до сильно выраженных
Для оценки применения различных доз ботулинического нейротоксина для лечения морщин в верхней части лица, имеющих выраженность от умеренной до сильной, включающих межбровные морщины (GL), боковые периорбитальные морщины (LCL) и комбинацию GL/LCL, проводили многоцентровые рандомизированные двойные слепые плацебо-контролируемые исследования. Пациенты с GL получали 4 дозировки (10, 25, 50 и 75 единиц). Пациенты с LCL получали 3 дозировки (30, 60 и 90 единиц). Пациенты с комбинацией GL/LCL получали 2 дозировки (50/60 и 50/90 единиц).
Фазу I исследования проводили для изучения безопасности и переносимости лечения. В этом исследовании не наблюдали серьезных нежелательных явлений (СНЯ), и ни один субъект не покинул исследование из-за НЯ (нежелательных явлений) или по любой другой причине. Большинство НЯ имели слабую интенсивность. Все дозы были безопасными и хорошо переносимыми.
При всех уровнях доз введение ботулинического нейротоксина эффективно снижало выраженность GL при максимальном нахмуривании и в состоянии покоя в течение периода, составляющего до 28 суток. При всех уровнях доз введение ботулинического нейротоксина эффективно снижало выраженность LCL при максимальном нахмуривании и в состоянии покоя в течение периода, составляющего до 28 суток. При всех уровнях доз введение ботулинического нейротоксина эффективно снижало выраженность GL и LCL, которые обрабатывали в комбинации, в течение периода, составляющего до 28 суток при максимальном нахмуривании. Эффект был менее очевиден в состоянии покоя.
В Фазе II исследования Исследователем и субъектом по отдельности был оценен эффект после получения 3 доз (30, 45 и 60 единиц) BoNT в Сутки 14 проведения лечения GL. Как показано на Фиг. 2, по сравнению с плацебо, основная задача была выполнена при всех уровнях доз ботулинического нейротоксина. При каждом уровне дозы в группе, получавшей лечение, количество пациентов с ответной реакцией было значительно больше, чем в группе плацебо.
Наиболее часто встречающиеся НЯВЛ (нежелательные явления, возникшие при лечении) в группах, получавших ботулинический нейротоксин, включают боль в месте инъекции (15,9%), головную боль (10,4%), опущение века (3,9%), и опухание места ввода инъекции (1,3%).
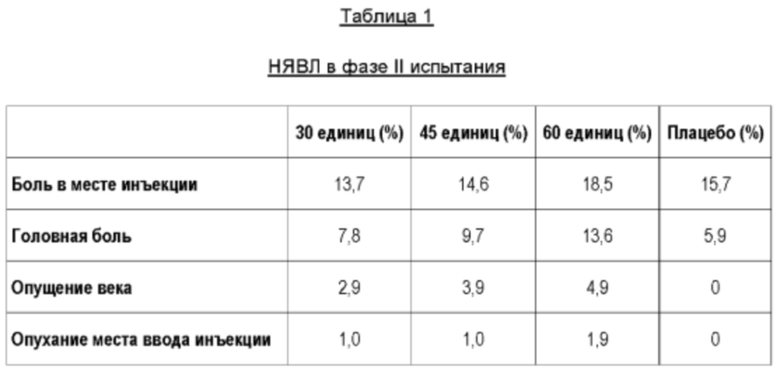
Из представленных данных фазы II исследования можно заключить, что ботулинический нейротоксин безопасен, и его введение во всех исследованных дозировках эффективно уменьшает GL. Средняя длительность ответной реакции составляет приблизительно 6 месяцев, как показано на Фиг. 3. Удовлетворенность пациентов высокая.
Фаза III исследования
Состояние межбровных морщин оценивали в соответствии со шкалой, созданной Компанией Merz Aesthetic (англ. шкала Merz Aesthetic Severity, сокращенно "MAS"), в состоянии покоя и при максимальном нахмуривании (в динамике).
Основные квалификационные критерии включали:
1. Субъекты в возрасте 18 лет или старше, имеющие GL при максимальном нахмуривании с выраженностью от умеренной до сильной по оценке субъекта и Исследователя в соответствии со шкалой MAS (в динамике), и имеющие по меньшей мере слабо выраженные GL в состоянии покоя в соответствии со шкалой MAS (в состоянии покоя).
2. Отсутствие проводимого ранее лечения любым ботулиническим нейротоксином (BoNT), отсутствие морщин в межбровной области, которые нельзя было бы разгладить вручную, растягивая кожу.
3. Отсутствие любого нерассасывающегося или частично нерассасывающегося материала, гилауроновой кислоты или коллагеновых филлеров, введенных ранее в межбровную область, и отсутствие пластических операций лица в области выше нижнего глазничного валика; отсутствие запланированных пластических операций лица или эстетических процедур в течение периода исследования или абляционной шлифовки кожи или химического пилинга в области выше нижнего глазничного валика в течение предыдущих 12 месяцев или в течение периода исследования.
4. Отсутствие в анамнезе опущения века или брови или амблиопии. Исследование контингента приведено ниже в Таблице 2.

Исходные демографические данные
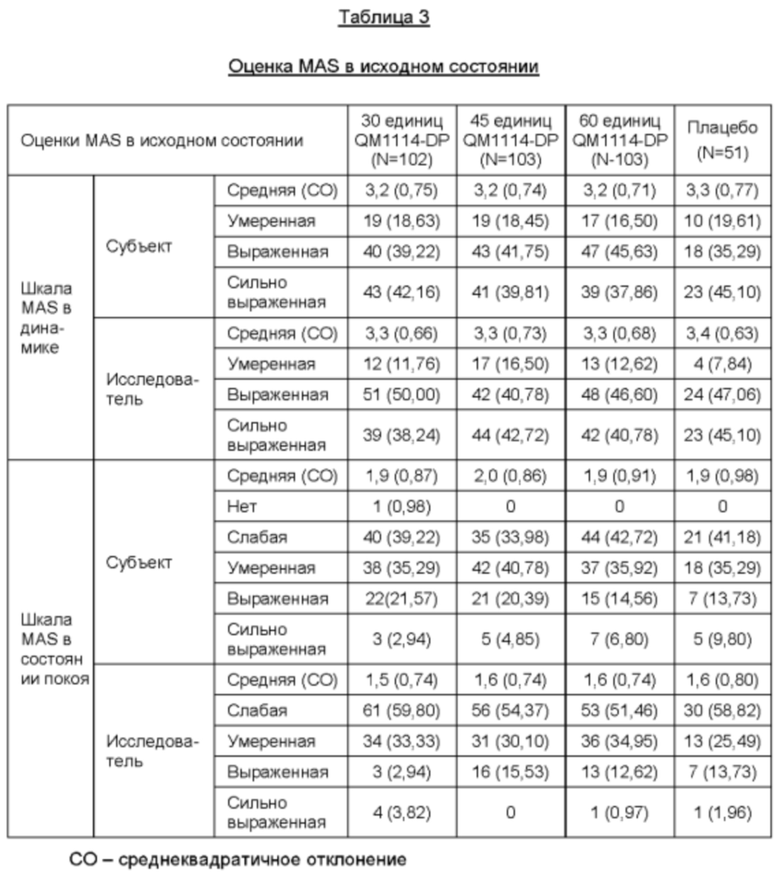
В исходном состоянии средние оценки субъектом и Исследователем по шкале MAS в динамике были близки, но средние оценки субъектом по шкале MAS в состоянии покоя были более высокими, чем оценки Исследователем.
Первичная оценка конечных результатов
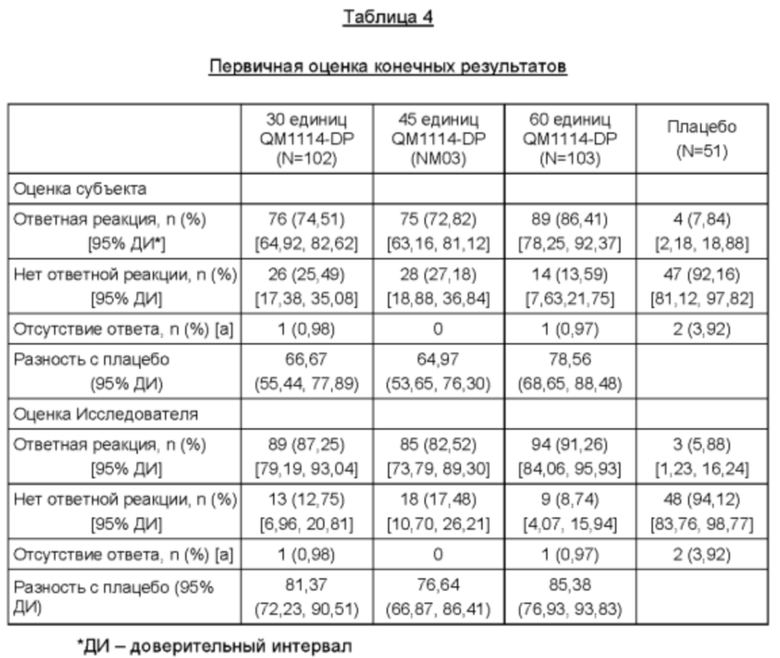
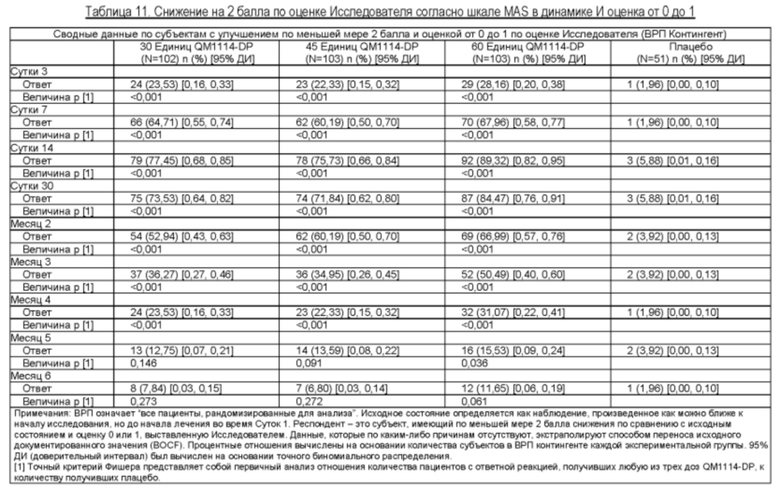
Как показано выше в Таблице 4, разность количеств пациентов с ответной реакцией по шкале MAS в динамике на Сутки 14 при вводе 60 единиц, 45 единиц и 30 единиц ботулинического нейротоксина по сравнению с плацебо была статистически значимой для обеих сопервичных переменных (р<0,001).
Вторичные конечные результаты
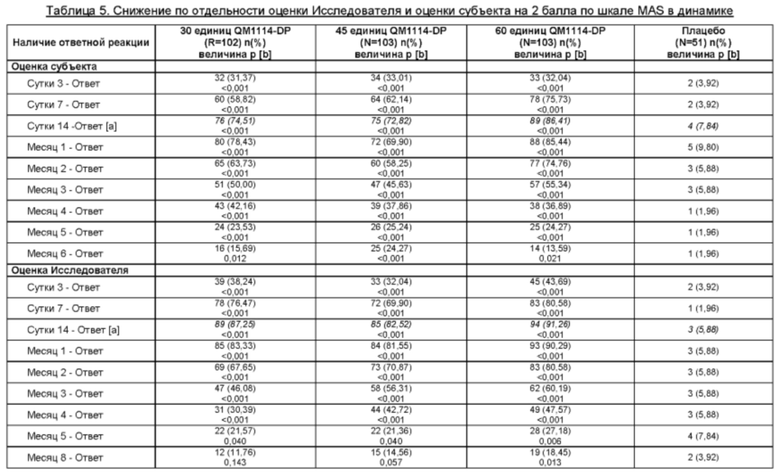
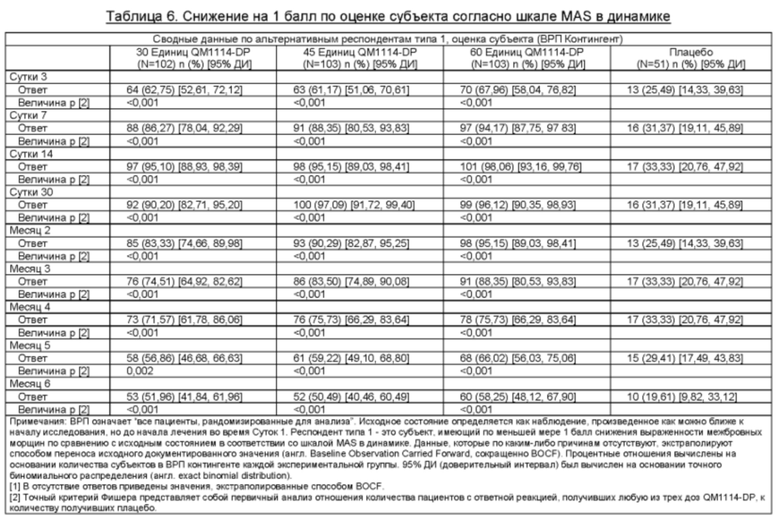
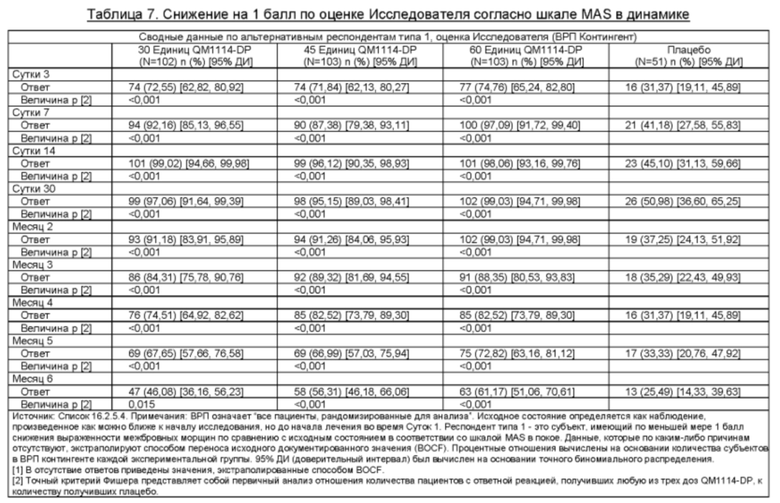
Оценка по шкале MAS в динамике: При вторичном анализе эффективности, респондента определяли как субъекта, демонстрирующего снижение по меньшей мере на 2 балла по сравнению с исходным состоянием выраженности GL по шкале MAS в динамике, отдельно по оценке субъекта и отдельно по оценке Исследователя. Оценивание субъектом проводили в течение периода, составляющего до 6 месяцев, во всех группах, получивших ботулинический нейротоксин. Оценивание Исследователем проводили в течение периода, составляющего до 5 месяцев, в группах, получивших 30 единиц и 45 единиц, и в течение периода, составляющего до 6 месяцев, в группе, получившей 60 единиц.
Оценка по шкале MAS в состоянии покоя: При вторичном анализе эффективности, респондента определяли как субъекта, демонстрирующего снижение по меньшей мере на 1 балл по сравнению с исходным состоянием выраженности GL по шкале MAS в состоянии покоя. Оценивание субъектом проводили в течение периода, составляющего до 6 месяцев, во всех группах, получивших ботулинический нейротоксин. Оценивание Исследователем проводили в течение периода, составляющего до 4 месяцев, в группах, получивших 30 единиц и 45 единиц, и в течение периода, составляющего до 5 месяцев, в группе, получившей 60 единиц.
Результаты: Для этого исследования вводили специальное определение длительности ответной реакции. Длительность ответной реакции (согласно исследованию) Исследователь и субъект определяли по отдельности в соответствии со шкалой MAS выраженности GL в динамике как время, прошедшее от момента снижения оценки по меньшей мере на 2 балла до возврата к исходному состоянию.
По оценке Исследователя, средняя длительность ответной реакции (определенная выше) была близка к 6 месяцам в группах, получивших 30 единиц и 45 единиц, и при этом в группе, получившей 60 единиц, 53% субъектов, имеющих ответную реакцию, не возвратились к исходному состоянию по истечении 6 месяца. В группе плацебо средняя длительность ответной реакции составила приблизительно 3,5 месяца.
По оценке субъекта, разница в состояниях групп, получавших ботулинический нейротоксин и плацебо, была менее выражена, и средняя длительность ответной реакции составила приблизительно 6 месяцев в группах, получавших ботулинический нейротоксин, и 4,5 месяца в группе, получившей плацебо.
Период до начала действия (согласно настоящему исследованию) Исследователь и субъект определяли по отдельности по шкале MAS выраженности GL в динамике как время, прошедшее до первого снижения оценки на 1 балл по сравнению с исходным состоянием, и время, прошедшее до первого снижения оценки на 2 балла по сравнению с исходным состоянием.
Результаты показывают, что в группах, получавших ботулинический нейротоксин, период времени до начала снижения по меньшей мере на 1 балл по оценкам Исследователя и субъектов был приблизительно одинаковым и в среднем составлял 8 суток. В группах, получавших ботулинический нейротоксин, период времени до начала снижения по меньшей мере на 2 балла по оценкам Исследователя и субъектов был приблизительно одинаковым и в среднем составлял 8 или 9 суток.
Кроме того, доля субъектов, которые были «очень довольны» или «довольны» была выше в группах, получавших ботулинический нейротоксин, чем в группе плацебо в течение всего времени. Также верно было обратное, т.е. доля субъектов, которые были «недовольны» или «очень недовольны» была меньше в группах, получавших ботулинический нейротоксин, чем в группе плацебо. Самый высокий уровень удовлетворенности лечением наблюдали на протяжении Месяца 1. Явной взаимосвязи между величиной дозы и уровнем удовлетворенности лечением не наблюдали.
Безопасность
В этом исследовании ботулинический нейротоксин был безопасен, и все исследуемые дозировки переносились хорошо. Отмеченные связанные с лечением и выявляемые при лечении нежелательные явления (НЯВЛ) были аналогичны явлениям, наблюдаемым при введении других токсинов для лечения GL. Наиболее частыми (по меньшей мере 1% субъектов в любой группе, получавшей ботулинический нейротоксин) НЯВЛ, связанными с лечением, были боль в месте инъекции, головная боль, опущение века, опухание места ввода инъекции, зуд в точке ввода инъекции и нарушение зрения. Большинство связанных с лечением НЯВЛ демонстрировали зависимость от дозы. Большинство связанных с лечением НЯВЛ были слабыми или умеренными.
Связанное с лечением опущение века было обнаружено: у 3 субъектов (2,9%) группы, получающей 30 единиц токсина; у 4 субъектов (3,9%) группы, получающей 45 единиц токсина; и у 5 субъектов (4,9%) группы, получающей 60 единиц токсина. Связанных с лечением НЯВЛ, предполагающих большее распространение действия токсина, не наблюдали.
Отмечали отсутствие связанных с лечением серьезных нежелательных явлений (СНЯ). В исследовании отмечали четыре не связанных с лечением СНЯ, которые включали положительный тест на синдром Дауна у беременной пациентки (30 единиц), большую депрессию (45 единиц), рождение живого ребенка с расщеплением неба (45 единиц) и панкреатит (60 единиц). Во время исследования не было зарегистрировано смертельных исходов или НЯ, приводящих к выходу из исследования.
В заключение следует отметить, что первичная оценка конечных результатов эффективности, проведенная в Сутки 14, подтвердила эффективность действия всех доз ботулинического нейротоксина (30, 45 и 60 единиц) по сравнению с плацебо. На Сутки 14 доля пациентов с ответной реакцией по шкале MAS в динамике была высокой во всех группах, получавших ботулинический нейротоксин (от 73% до 91%), по сравнению с плацебо (от 6% до 8%). Результаты также показывают, что обработка ботулинический нейротоксином эффективно снижает выраженность GL. В частности, в сравнении с плацебо эффект сохраняется при максимальном нахмуривании в любой момент течение периода времени, составляющего до Месяца 6 по оценке субъекта, и по оценке Исследователя до Месяца 5 в группах, получавших 30 единиц и 45 единиц токсина, и до Месяца 6 в группе, получавшей 60 единиц токсина. В сравнении с плацебо эффект сохраняется в состоянии покоя при всех величинах доз ботулинического нейротоксина в течение всего времени до Месяца 6 по оценке субъекта, и по оценкам Исследователя до Месяца 4 в группах, получавших 30 единиц и 45 единиц токсина, и до Месяца 5 в группе, получавшей 60 единиц токсина. Начало снижения выраженности GL по шкале MAS также показывало аналогичную тенденцию удовлетворенности субъекта результатами лечения в этом исследовании.
Наиболее часто встречающимися НЯВЛ, связанными с лечением, были: боль в месте инъекции, головная боль, опущение века, опухание места ввода инъекции, зуд в точке ввода инъекции и нарушение зрения. Частот возникновения связанных с лечением НЯВЛ была самой большой в группах, получавших 45 единиц (33%) и 60 единиц (36%) ботулинического нейротоксина. Частота возникновения опущения века в этом исследовании составила от 3% до 5%.
Не было отмечено связанных с лечением СНЯ (в исследовании отмечали 4 не связанных с лечением СНЯ, которые были внесены в базу данных безопасности), и не было смертельных исходов. Во время исследования не отмечали НЯ, приводящих к выходу из исследования. Во время исследования стало известно о трех беременностях, одной беременности в группе получавших 30 единиц и о двух беременностях в группе получавших 45 единиц.
Результаты приведены в таблицах ниже.
Пример 2
Рандомизированное двойное слепое плацебо-контролируемое исследование с увеличением дозы с однократной обработкой для оценки безопасности и эффективности QM1114-DP при лечении здоровых мужчин и женщин, имеющих морщины в верхней части лица с выраженностью от умеренной до сильной
Доклинические исследования
Программа доклинических фармако-токсикологических исследований для обоснования фазы I клинических испытаний включала доклинические фармакологические исследования, обеспечивающие экспериментальную проверку продукта, применяемого по показаниям, впервые проводимую с участием людей (англ. first-in-man, сокращенно FIM). Доклиническая программа также включала подходящие токсикологические исследования для обеспечения безопасности впервые испытываемой на людях композиции QM1114-DP.
Фармакологические исследования для экспериментальной проверки эффекта QM1114-DP производили в опытах, в которых вызывали паралич задних конечностей у мышей; было показано, что внутримышечное (ВМ) введение продукта вызывает частичный паралич мышц вблизи участков ввода инъекций аналогично действию коммерчески доступных в настоящее время продуктов BoNT-A. Также проводили доклиническое фармакологическое исследование, включающее более раннюю версию исследования LD50 у мышей (стандартное исследование для оценки активности BoNT-A) и опыты, в которых вызывали паралич задних конечностей у мышей.
Фармакологические или фармакокинетические исследования безопасности QM1114-DP не проводили, поскольку предполагаемое использование продукта не подразумевает системного воздействия при введении продукта в виде единичной ВМ инъекции в определенные мышцы лица в дозировках, подходящих для клинического исследования. Кроме того, BoNT-A связывается с синапсами нейронов в локализованном участке инъекции с высоким сродством (Montecucco с соавт., 2004). Таким образом, метаболизм и удаление продукта происходят в локализованном участке инъекции.
Проводили общие токсикологические исследования QM1114-DP, которые включали пилотные исследования токсичности посредством ВМ введения однократной дозы крысам породы Вистар и собакам породы Бигль. Исследования проводили не в соответствии с НЛП (не в соответствии с Надлежащей лабораторной практикой); целью исследований было определение максимальной переносимой дозы QM1114-DP, позволяющей установить подходящие величины доз для опорного токсикологического исследования в соответствии с НЛП. Результаты пилотных токсикологических исследований ясно показали, что крысы породы Вистар были более чувствительны к токсичному действию QM1114-DP, чем собаки, и, таким образом, опорное токсикологическое исследование в соответствии с НЛП проводили только с крысами породы Вистар.
Отдельных исследований местной переносимости QM1114-DP не проводили. Вместо этого местную переносимость внимательно оценивали при клинических наблюдениях и гистопатологическом анализе участков ввода инъекций, который составлял часть опорного токсикологического исследования в соответствии с НЛП на крысах породы Вистар. При этих дополнительных наблюдениях не было получено никаких неожиданных результатов, которые могли бы указывать на проблемы с переносимостью продукта.
Клинические исследования
В Примере 1 (только GL) настоящего клинического исследования (43QM1302 [n=30]) оценивали безопасность и переносимость доз 10, 25, 50 и 75 единиц QM1114-DP. Всего было заявлено о 48 нежелательных явлениях (НЯ) у 23 субъектов, ни одно из которых не было серьезным, и большинство из них были слабо выраженными. Исследователи выявили у 16 субъектов тридцать два (32) НЯ, связанные с лечением. Из 32 связанных с лечением НЯ, большая часть (17 эпизодов) было выявлено у пациентов, которым вводили 75 единиц QM1114-DP, и из них наиболее часто возникающими НЯ были общие нарушения и нарушения в месте введения, и следующим по частоте были нарушения состояния кожи и подкожной ткани. Оказалось, что частота возникновения нарушений состояния глаз, общих нарушений и нарушений в месте введения, а также нарушений состояния кожи и подкожной ткани связана с величиной дозы. При дозах 10 единиц и 25 единиц QM1114-DP ни у одного из субъектов не возникали нарушения состояния глаз, такие как ослабление зрения; однако дозы 50 единиц и 75 единиц QM1114-DP вызывали с умеренное повышение частоты возникновения нарушений состояния глаз. Аналогично, эту тенденцию также наблюдали для общих нарушений и нарушений в месте введения. Кроме указанных тенденций, ни одна из обработок не привела к обнаружению других клинически значимых результатов, влияющих на безопасность, которые включали бы результаты клинических лабораторных анализов, показатели жизнедеятельности, параметры данные физического обследования и электрокардиограммы (ЭКГ).
Эффективность
В Части 1 настоящего клинического исследования 43QM1302 (n=30) Исследователь и субъект оценивали эффективность воздействия 10, 25, 50 и 75 единиц QM1114-DP на выраженность морщин в межбровной области в состоянии покоя и при максимальном нахмуривании в соответствии с 5-балльной фотошкалой Merz Aesthetic ("MAS"). В шкале применяют следующие степени выраженности GL: 0 (нет), 1 (слабо), 2 (умеренно), 3 (выражено) и 4 (сильно выражено). Анализ эффективности показал, что при всех уровнях доз обработка QM1114-DP эффективно снижала выраженность GL при максимальном нахмуривании в течение периода, составляющего до 28 суток, по сравнению с субъектами группы плацебо. По оценке Исследователя и субъекта выраженность GL в состоянии покоя снижалась в группах, получавших действующее лечение, в сравнении с группой плацебо. Этот эффект сохранялся в течение периода, составляющего до 28 суток. Оценка субъектом удовлетворенности состоянием GL показывает, что за исключением одного субъекта, все субъекты в группах, получавших действующее лечение, были либо очень довольны, либо довольны лечением в течение времени, составляющего до 28 суток. Кроме того, при всех уровнях доз уменьшение выраженности морщин было очевидно в течение времени, составляющего до 28 суток. В заключение следует отметить, что результаты исследования эффективности показывают, что введение всех исследуемых доз QM1114-DP эффективно снижало выраженность GL при максимальном нахмуривании по сравнению с GL субъектов группы плацебо.
Перед исследованием, описанным в этом примере, не проводили анализ эффективности снижения выраженности боковых периорбитальных морщин под действием QM1114-DP.
Задачи исследования
Первичная задача: оценка безопасности и переносимости каждой из исследуемых дозировок препарата QM1114-DP.
Вторичные задачи: оценка эффективности действия каждой из дозировок препарата QM1114-DP, где действие состоит во временном улучшении состояния межбровных морщин (GL) и боковых периорбитальных морщин (LCL); и оценка эффективности действия каждой из дозировок препарата QM1114-DP, где действие состоит во временном улучшении состояния GL и LCL, которые обрабатывают в комбинации.
Результаты исследования
Безопасность: Частота возникновения и выраженность НЯ
Эффективность:
• Оценка выраженности LCL Исследователем при самой широкой улыбке в течение периода до Суток 28 (неделя 4) в реальных условиях по шкале Merz Aesthetic Scale (MAS) LCL - в динамике;
• Оценка выраженности LCL Исследователем в состоянии покоя в течение периода до Суток 28 (неделя 4) в реальных условиях по шкале MAS LCL - в состоянии покоя (в статике);
• Оценка выраженности LCL субъектом при самой широкой улыбке в течение периода до Суток 28 (неделя 4) в реальных условиях по шкале MAS LCL - в динамике;
• Оценка выраженности LCL субъектом в состоянии покоя в течение периода до Суток 28 (неделя 4) в реальных условиях по шкале MAS LCL - в состоянии покоя (в статике); и
• Оценка удовлетворенности субъекта в течение периода до Суток 28 (неделя 4) по 4-балльной шкале.
Экспериментальная часть:
• Оценка выраженности LCL Исследователем при самой широкой улыбке до того срока, когда состояние морщин сровняется с исходным состоянием, или в течение срока, не превышающего 6 месяцев, в реальных условиях по шкале MAS LCL - в динамике;
• Оценка выраженности LCL Исследователем в состоянии покоя до того срока, когда состояние морщин сровняется с исходным состоянием, или в течение срока, не превышающего 6 месяцев, в реальных условиях по шкале MAS LCL - в состоянии покоя;
• Оценка выраженности LCL субъектом при самой широкой улыбке до того срока, когда состояние морщин сровняется с исходным состоянием, или в течение срока, не превышающего 6 месяцев, в реальных условиях по шкале MAS LCL - в динамике;
• Оценка выраженности LCL субъектом в состоянии покоя до того срока, когда состояние морщин сровняется с исходным состоянием, или в течение срока, не превышающего 6 месяцев, в реальных условиях по шкале MAS LCL - в состоянии покоя; и
• Оценка удовлетворенности субъекта по 4-балльной шкале.
Субъекты, которые участвовали в экспериментальной фазе этого исследования, посещали отделение клинической фармакологии (сокращенно "ОКФ") каждый месяц. Конечные результаты этого эксперимента не были включены в данный пример.
План исследования
Общая схема и план исследования
В этом примере Фазы I клинического исследования рассматривали только LCL. Фаза I исследования также включала в виде отдельной части (когорты 1-4) обработку GL увеличивающимися дозами, как указано выше. Отдельная фаза (когорты 8 и 9), в которых оценивали лечение GL в комбинации с LCL, индивидуально рассмотрена в Примере 3.
Исследование представляло собой рандомизированное двойное слепое плацебо-контролируемое исследование для оценки безопасности и эффективности увеличивающихся доз ботулотоксина типа A (QM1114-DP; активный компонент), вводимых здоровым мужчинам и женщинам в возрасте от 18 до 65 лет, имеющих LCL от умеренных до выраженных. Всего в исследовании участвовали 24 субъекта, которых разделяли на 3 когорты (когорты 5, 6 и 7), и в каждой когорте находилось 8 субъектов (6 получали активный компонент: 2 - плацебо).
Перед подписанием ФИС (формы информированного согласия), каждый здоровый субъект получал вербальную (от врача-исследователя) и письменную информацию. Субъектов проверяли на соответствие требованиям в течение 28 суток перед Сутками 1, в которые начинали исследуемое лечение, и субъекты совершали визиты в рамках исследования для оценки безопасности и эффективности до последующего визита в Сутки 28 (неделя 4). Всех субъектов помещали в ОКФ, где их оставляли в течение Суток -1 и затем в течение ночи, предшествующей суткам, в которые производили введение дозировок, т.е. перед Сутками 1. Для минимизации рисков, которым подвергались субъекты, для каждой из исследуемых доз были созданы сигнальные группы. Соответственно, в когортах 5, 6 и 7 первым 2 субъектам потребовалась более длительная госпитализация в ОКФ (1 получал активный ингредиент и 1 плацебо). Этих субъектов помещали в ОКФ в Сутки -1, подвергали лечению в Сутки 1 и выписывали из ОКФ в Сутки 2, после чего они совершали визиты в рамках исследования до последующего визита в Сутки 28 (неделя 4). Исследование состояло из следующих визитов: Отбор, Сутки -1, Сутки 1, 2, 3, 7, 14, 21 и 28.
Исследователи и субъекты оценивали выраженность LCL по шкале Merz в динамике и в состоянии покоя при отборе, во время визитов перед лечением и во время всех визитов по окончании лечения LCL. Субъекты также оценивали свою удовлетворенность состоянием своих LCL по 4-балльной шкале во время всех визитов по окончании лечения (Ascher с соавт., 2009).
Субъектов рандомизировали для одной обработки исследуемым лекарственным средством (QM1114-DP или плацебо). Каждая обработка включала три инъекции равного объема (100 мкл) в каждую из сторон лица. Места ввода инъекции уточняли в соответствии с расположением LCL морщин у конкретного субъекта. Во всех случаях инъекции вводили в наружную часть круговой мышцы глаза и, если это было возможно, на расстоянии приблизительно 1-2 см от глазничного валика.
Вводили следующие дозировки QM1114-DP:
• Когорта 5, Доза 1: 5 единиц QM1114-DP на участок ввода инъекции (всего 30 единиц за одну обработку);
• Когорта 6, Доза 2: 10 единиц QM1114-DP на участок ввода инъекции (всего 60 единиц за одну обработку); и
• Когорта 7, Доза 3: 15 единиц QM1114-DP на участок ввода инъекции (всего 90 единиц за одну обработку).
Проводимые в этом исследовании процедуры и соответствующее оценивание приведено в Таблице 12 в виде данных по визитам в рамках исследования. Клинические исследования включали отбор и визиты до недели 4 (последующие). Если оценка, выставленная Исследователем (при максимальной улыбке), возвращалась к оценке, соответствующей исходному состоянию, то последний визит субъектов в ОКФ приходился на Сутки 28 (неделя 4). В этот момент, согласно первичной оценке конечных результатов, для таких субъектов исследование считалось законченным. Субъекты, у которых оценка выраженности морщин, выставленная Исследователем (при максимальной улыбке), не возвращалась к оценке, соответствующей исходному состоянию, на Сутки 28 (неделя 4), участвовали в экспериментальной фазе настоящего исследования и посещали ОКФ каждый месяц до тех пор, пока их морщины по оценкам не возвращались к исходному состоянию. Данные по последующей эффективности описанной экспериментальной обработки будут опубликованы отдельно и не в этом отчете о клиническом исследовании.
Это исследование представляло собой рандомизированное плацебо-контролируемое исследование оценки безопасности и переносимости каждой из доз QM1114-DP и оценки эффективности введения каждой из доз QM1114-DP для достижения временного улучшения состояния морщин на лице в левой и правой боковой периорбитальной области. Критерии отбора были определены таким образом, чтобы субъекты, отобранные для участия в исследовании, не имели никаких заболеваний.
Отбор контингента для исследования
В течение 28 суток до начала исследования, субъекты проходили полную процедуру отбора, как описано в Таблице 12.
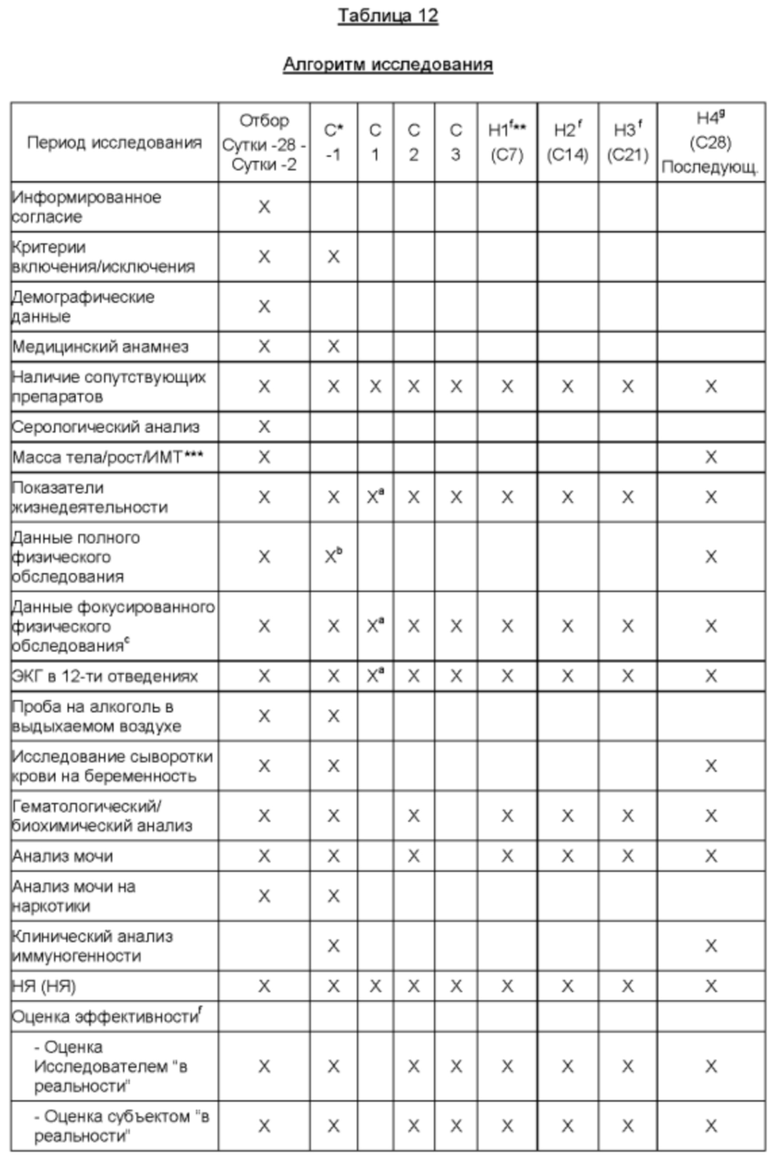

Критерии включения
Для включения в Часть 2 исследования каждый субъект должен был соответствовать каждому из следующих критериев:
• Здоровый мужчина или здоровая женщина в возрасте от 18 и 65 лет (включительно) на момент отбора.
• Ранее не подвергались лечению BoNT-A или В.
• По оценке Исследователя на момент отбора и визитов для оценки исходного состояния в соответствии с обоснованной фотошкалой (MAS, LCL в динамике) имеет LCL с обеих сторон лица от умеренных до выраженных (Степень 2 или 3) при максимальной улыбке.
• По оценке Исследователя на момент отбора и визитов для оценки исходного состояния в соответствии с обоснованной фотошкалой (MAS, LCL в состоянии покоя) имеет LCL с обеих сторон лица в состоянии покоя от слабых до выраженных (Степень 1, 2 или 3).
• Субъекты согласны применять один из нескольких приемлемых способов контрацепции.
• Субъекты были способны понять и согласиться с требованиями протокола и подписали ФИС перед проведением любых процедур, связанных с исследованием.
• Не имеет клинически значимого заболевания или аномальных результатов лабораторного анализа, ЭКГ или показателей жизнедеятельности, указанных в медицинском анамнезе, данных физического обследования или других анализах, проводимых на момент отбора или при приеме в ОКФ.
Критерии исключения
Субъект считается неподходящим для включения в это исследование, если он отвечает любому из следующих критериев:
• Ранее получал лечение BoNT-А или В.
• Имеет морщины в области LCL, которые нельзя было бы по существу разгладить вручную, растягивая кожу.
• Субъекту ранее вводили любой нерассасывающийся или частично нерассасывающийся материал или ранее вводили кожный наполнитель в область LCL морщин.
• Субъекту была сделана инъекция в область LCL в течение 14 суток перед визитом для оценки исходного состояния.
• Присутствие в анамнезе опущения века или брови.
• Имеет сухость глаз, ярко выраженные мешки под глазами или отечность век по утрам.
• Имеет раковые или предраковые поражения, острые или хронические кожные заболевания, воспаления или связанные с ними состояния, включающие шрамы вблизи или в межбровной области.
• Перенесенные ранее пластические операции лица (например, блефаропластику, операцию вокруг глаз, подтяжку лица, подтяжку бровей, подтяжку век или операцию на бровях).
• Перенес лазерную или световую обработку лица, микрошлифовку кожи или поверхностный пилинг в течение 6 месяцев до начала отбора для участия в исследовании.
• Имеет в анамнезе паралич лицевого нерва.
• Применял препараты для топического нанесения, которые заявлены как обладающие способностью разглаживать морщины, в течение 7 суток перед введением исследуемого препарата или планирует применять такие препараты во время проведения исследования.
• Субъект планировал делать на лице пластическую операцию или косметические процедуры в течение периода исследования.
• В течение 14 суток перед сутками, на которые назначены инъекции, наблюдаются симптомы, подобные симптомам ОРВИ.
• Принимает в настоящее время или принимал аминогликозидные антибиотики, курареподобные лекарственные средства, хинидин, сукцинилхолин, полимиксины, антихолинестеразы, сульфат магния или линкозамиды. Прием любых других назначаемых или не назначаемых препаратов допускается под ответственность Исследователя, при условии, что они не оказывают негативного влияния на безопасность субъекта или корректность данных исследования.
• Беременность или грудное вскармливание (женщины детородного возраста, участвующие в исследовании, перед рандомизацией должны иметь отрицательный результат теста на беременность [по сыворотке крови]).
• Имеет подтвержденный положительный анализ мочи на наличие лекарственных средств в организме, указывающий на наркотическую зависимость, включающий наличие: опиатов, барбитуратов, метаболитов кокаина, метадона, бензодиазепина, каннабиноида или амфетамина.
• Имеет в анамнезе алкоголизм или наркотическую зависимость или проявляет клинические признаки алкоголизма или наркотической зависимости. Алкогольную зависимость определяют как регулярное еженедельное употребление более 14 алкогольных единиц для женщин и 21 алкогольных единиц для мужчин (согласно alcohol tracker nhs.uk/Tools/Pages/NHSAIcoholtracker.aspx); наркотическую зависимость определяют как связанное с непреодолимым влечением, многократное и/или хроническое употребление наркотиков или других веществ, вызывающих или не вызывающих проблем, связанные с их употреблением, и/или если прекращение употребления или снижение дозировки приводит к синдрому отмены.
• Имеет положительный результат анализа на гепатит В, включающий обнаружение HBsAG (поверхностного антигена гепатита В), положительный результат анализа на анти-HCV (антитела к вирусу гепатита С) и положительный результат анализа вирус иммунодефицита человека (ВИЧ).
• Имеет в анамнезе эпизоды затрудненного глотания, пневмопатии вдоха или выдоха.
• Имеет в анамнезе нарушения свертываемости крови.
• Имеет в анамнезе аутоиммунное заболевание, которое потенциально может влиять на результаты исследования.
• Имеет сочетанное заболевание в стадии обострения, которое потенциально может влиять на безопасность или оценку результатов исследования, примеры которого включают, без ограничений, клинически значимое сердечнососудистое, респираторное нарушение, нарушение печени/желчного пузыря, почечное, желудочно-кишечное, эндокринное, психическое или неврологическое нарушение.
• Имеет в анамнезе тяжелую миастению или другое нарушение нейротрансмиссии.
• Имеет нейромышечные нарушения, которые потенциально могут влиять на результаты субъекта.
• Уже получал любой из исследуемых продуктов в течение 90 суток перед отбором для участия в данном исследовании.
• Выкуривает более 10 сигарет или эквивалентное количество табака в сутки и не может прекратить курение во время пребывания в ОКФ.
• Имеет любое состояние (состояния), которое, по мнению Главного Исследователя (ГИ), нарушает безопасность субъекта или предотвращает участие субъекта в исследовании до полного его завершения.
• Не может установить надежное взаимодействие с исследователем.
• Субъект является вегетарианцем, веганом или придерживается ограничений питания по медицинским показаниям, которые не позволяют ему придерживаться стандартизованного меню, принятого в исследовании.
Отстранение субъектов от лечения или оценивания
Для определения тяжести НЯ применяли стандартную оценку токсичности в соответствии с общими терминологическими критериями нежелательных явлений, установленными Национальным институтом онкологии США (англ. National Cancer Institute Common Terminology Criteria, сокращенно NCI CTC, версия 4.0). Эти критерии применяют, поскольку они являются единственным доступным набором стандартных универсальных критериев, и было доказано, что они подходят для Фазы I исследований с участием здоровых добровольцев. Использовали нормальные показатели, получаемые в местной лаборатории. Для обеспечения достоверности и для исключения технических ошибок, аномальные лабораторные показатели и данные других испытаний всегда повторно проверяли перед вынесением оценки. При определении того, вызваны ли обнаруженные аномалии токсичным действием лекарственного средства, и, если это возможно, при оценивании всегда принимали во внимание суточные колебания лабораторных показателей и других измерений, а также исходный статус и состояние (например, синдром Жильбера).
Критерии СТС для НЯ представлены Степенями I-V с подробным клиническим описанием выраженности каждого НЯ в соответствии с общим руководством. Определения степеней приведены ниже:
Степень I: Слабая; отсутствуют или слабо выражены симптомы; только клинические или диагностические наблюдения; вмешательство не предполагается.
Степень II: Умеренная; показано минимальное, местное или неинвазивное вмешательство; ограничение подходящих по возрасту инструментальных ADL*.
* инструментальные ADL (англ. Activities of Daily Living) - инструментальные действия по самообслуживанию в повседневной жизни, к которым относится приготовление пищи, покупка продуктов или одежды, использование телефона, распоряжение денежными средствами и т.д.
Степень III: Выраженная или клинически значимая, но не несущая немедленной угрозы жизни; показана госпитализация или удлинение срока госпитализации; потеря трудоспособности; ограничение ADL самообслуживания**.
** ADL самообслуживания относятся к мытью, одеванию и раздеванию, самостоятельному приему пищи, пользованию туалетом, приему медицинских препаратов, но не к лежачим больным.
Степень IV: Последствия, угрожающие жизни; показано срочное вмешательство.
Степень V: Смертельный исход, вызванный НЯ.
Критерии СТС для НЯ и их интерпретация согласуются со стандартными оценками интенсивности НЯ при клинических испытаниях: Степень I: слабая, Степень II: умеренная, Степень III: выраженная или клинически значимая, но не несущая немедленной угрозы жизни, могут наблюдаться серьезные нежелательные явления (СНЯ)/предполагаемая непредвиденная серьезная нежелательная реакция (сокращенно "ПНСНР"). Степени IV и V связаны с проявлением СНЯ/ПНСНР.
Исследование необходимо прекратить, если возникают некоторые нежелательные явления, которые с большой долей вероятности вызваны ИМП (исследуемым медицинским продуктом, англ. investigational medicinal product, сокр. IMP).
Выход субъектов из исследования
В соответствии с Хельсинкской декларацией, субъекты могут выйти из исследования в любе время, но, если обработка или дозирование уже произведены, то должны быть предприняты все меры для продолжения оценивания состояния с целью обеспечения безопасности субъекта. Конкретные причины для выхода субъекта из настоящего исследования включали:
• Добровольное решение субъекта прекратить участие в исследовании, поскольку субъект имеет право в любое время прекратить свое участие в исследовании.
• Выраженное несогласие или принципиальное отклонение от протокола по мнению Исследователя и/или Спонсора.
• Некорректное включение, т.е. при последующем рассмотрении оказалось, что на момент включения субъект не соответствовал квалификационным критериям, требуемым для исследования.
• Другие причины, выдвигаемые ГИ.
Субъекты могли в любое время без ущерба выйти из исследования (выход из исследования по соглашению). Таких субъектов всегда спрашивали о причине (причинах) и наличии каких-либо НЯ. Если это было возможно, то субъекты, вышедшие из исследования после дозирования и до выполнения визита на неделе 4, должны были посетить прием ГИ или уполномоченного лица и пройти оценивание и процедуры, запланированные для следующего визита. Во всех случаях, где это было возможно, НЯ следовало отслеживать до их полного исчезновения, возвращения к исходному состоянию или стабилизации, если была установлена причина, не связанная с исследованием.
Замена субъектов
Для оценки безопасности и переносимости заданной дозы QM1114-DP или плацебо (7 суток после обработки) в каждой когорте должно быть запланированное количество субъектов. Если количество заполняющих группу субъектов было меньше, чем минимум, требующийся для данной когорты, для принятия решения об увеличении дозы спонсор и ГИ могли постановить, что этих субъектов нужно заменить, и перед следующим увеличением дозы этих субъектов соответственно подготавливали и оценивали.
Лечение
Получаемое лечение
Субъекты получали одну терапевтическую процедуру введения 30 единиц, 60 единиц или 90 единиц QM1114-DP или плацебо равного объема (100 мкл) в область LCL. Каждая обработка включала три инъекции (одну инъекцию на одну точку ввода инъекции). Места ввода инъекций уточняли в соответствии с расположением морщин LCL конкретного субъекта. В зависимости от расположения морщин у конкретных субъектов, если морщины располагались в области LCL выше и ниже латеральной спайки век, то инъекции вводили, как показано на Фиг. 12А. В альтернативном варианте, если морщины в области LCL конкретного субъекта в основном находились ниже латеральной спайки век, то инъекции вводили, как показано на Фиг. 12В. Во всех случаях инъекции вводили в наружную часть круговой мышцы глаза и, если это было возможно, на расстоянии приблизительно 1-2 см от глазничного валика.
Распределение субъектов по терапевтическим группам
Субъектов, которые подписали информированное согласие на исследование и затем прошли отборочные процедуры, в течение соответствующего периода идентифицировали с помощью уникального номера RPL ID (идентификационного номера из базы добровольцев RPL) и скринингового номера.
Номера субъектов присваивали субъектам, отвечающим квалификационным критериям, перед введением доз утром Суток 1. Субъектам присваивали номера от 201 до 208 (Когорта 5), от 221 до 228 (Когорта 6) и от 241 до 248 (Когорта 7). Номера присваивали субъектам последовательно, и эти номера соответствовали номерам в созданном компьютером рандомизованном списке, который определял последовательность получения лечения. Если субъект прекращал свое участие в исследовании, то номер субъекта больше не использовали, и субъекту не разрешали вновь вступать в ряды участников исследования.
Выбор доз
Для определения подходов для впервые проводимого с участием людей клинического исследования любого нового продукта BoNT-A необходимо учитывать клинические параметры уже существующих продуктов того же серотипа и происхождения.
Начальная доза для лечения LCL составляла 5 единиц в одной инъекции (всего 30 единиц), после чего дозу увеличивали (после принятия решений SRC, англ. Safety Review Committee - Комитет по рассмотрению безопасности) до 10 единиц в одной инъекции (всего 60 единиц) и затем до 15 единиц в одной инъекции (всего 90 единиц). Максимальную дозу, принятую в исследовании лечения LCL, выбирали на основании проведенных ранее исследований, в которых для лечения LCL в каждый участок вводили 15, 30 и 45 единиц BoNT (всего вводили 30, 60 и 90 единиц). Частота возникновения связанных с лечением НЯ была сходной при всех исследованных дозировках.
Выбор времени введения доз
В этом исследовании субъектам однократно вводили 30, 60 или 90 единиц QM1114-DP или плацебо утром Суток 1 Исследования.
Процедуры исследования
Процедуры отбора, сутки, входящие в исследование, и последующие процедуры указаны в Таблице 12.
Все измерения, проводимые в исследовании, описаны ниже.
Если на основании просмотра данных безопасности по конкретному субъекту возникали какие-либо опасения за его безопасность, то это могло быть показанием для дополнительного определения показателей жизнедеятельности, записи ЭКГ и/или лабораторного анализа образцов для подтверждения безопасности. Общий объем крови, отбираемой для исследования у одного субъекта, не должен превышать 300 мл.
Показатели жизнедеятельности
Кровяное давление, частота сердечных сокращений, температура в наружном слуховом проходе и частота дыхательных движений
Артериальное давление в положении лежа на спине (КД - сокр. от "кровяное давление") и частоту сердечных сокращений (ЧСС) измеряли с помощью полуавтоматического устройства, записывающего КД (мониторы Critikon Dinamap®) с подходящим размером манжеты. Для измерения частоты дыхательных движений, перед измерением субъекты должны были находиться в покое в положении лежа на спине в течение по меньшей мере 10 минут. Расписание индивидуальных измерений указано в алгоритме исследования (Таблица 12). Время проведения всех измерений, выполняемых в процессе исследования, могло быть изменено в зависимости от текущего состояния данных безопасности и переносимости. Температуру тела (в наружном слуховом проходе) определяли (одно измерение) в градусах Цельсия с помощью автоматического термометра согласно расписанию, указанному в алгоритме исследования (Таблица 12). Для обеспечения безопасности по инициативе ГИ или уполномоченного лица могут быть проведены дополнительные измерения температуры.
Снятие ЭКГ
ЭКГ регистрировали в 12-ти отведениях трижды с помощью устройства GE Marquette MAC1200®/MAC1200ST®, соединенного фиксированным сетевым соединением с кардиологической информационной системой MUSE® (MUSE). Все зарегистрированные во время исследования ЭКГ хранили в электронном виде в информационной системе MUSE. Для любых целей, не являющихся оценкой безопасности, имеющими силу считались только ЭКГ, записанные в электронном виде. Распечатки ЭКГ вкладывали в ИРК (индивидуальную регистрационную карту) субъекта для проверки медицинской безопасности.
В тех случаях, когда это возможно, ЭКГ всех субъектов регистрировали одним и тем же регистрирующим устройством. Каждое устройство для регистрации ЭКГ настраивали в соответствии с требуемыми техническими условиями, и каждое устройство содержало информацию, необходимую для идентификации записей. Каждая запись ЭКГ могла быть легко идентифицирована (ID субъекта, запланированное время ввода доз препарата и реальное время регистрации ЭКГ).
Запись ЭКГ в 12-ти отведениях проводили в соответствии с расписанием, указанным в алгоритме исследования (Таблица 12), после того, как субъекты находились в состоянии покоя в положении лежа на спине в течение по меньшей мере 10 минут. Субъектов просили не изменять позу во время записи ЭКГ, и персонал клиники следил за тем, чтобы во время записи ЭКГ субъекты находились в бодрствующем состоянии.
Все записанные ЭКГ оперативно просматривал врач-исследователь, и результаты анализа записывались в ИРК. Если на каком-либо этапе у субъекта наблюдали аномальную ЭКГ, то проводили дополнительные исследования для безопасности (включающие применение оборудования Holter с 5 или 12 отведениями), и, при необходимости, аномалию прослеживали до ее полного устранения.
Время проведения всех измерений, выполняемых в процессе исследования, может быть изменено в соответствии с оперативными просмотрами данных безопасности и переносимости.
Клинические лабораторные определения безопасности
Клинические лабораторные параметры безопасности представлены ниже в Таблице 13.
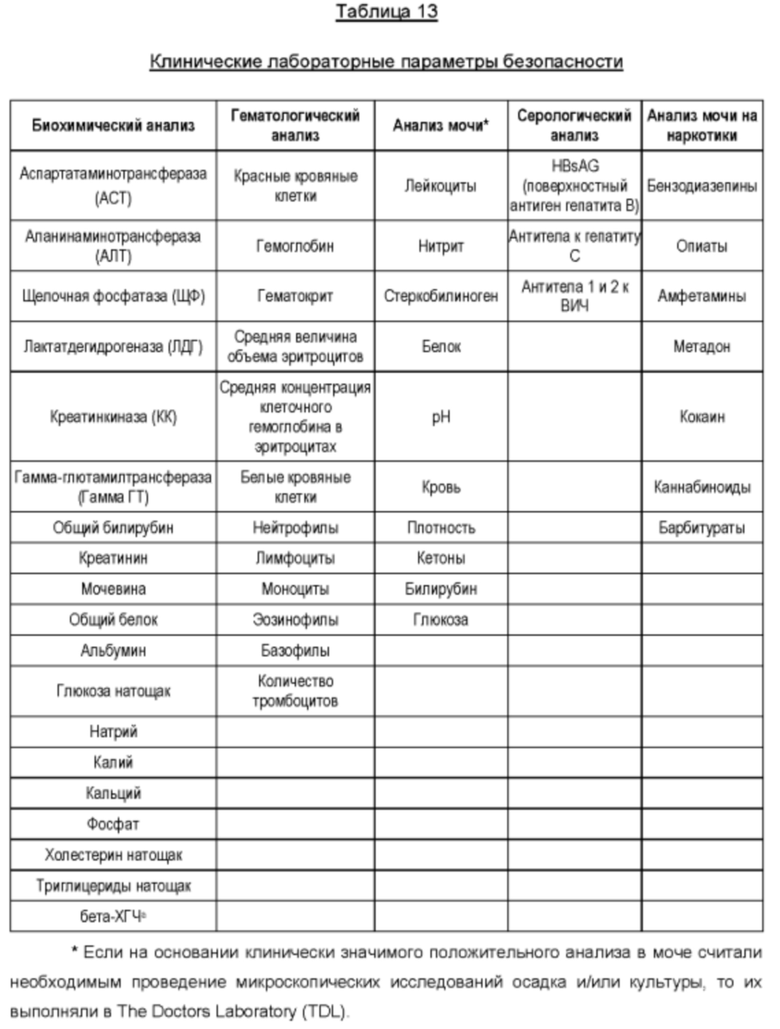

Гематологический и биохимический анализ
Образцы крови для определения гематологических и биохимических параметров отбирали в соответствии с расписанием, указанным в алгоритме исследования (Таблица 12). Дата и время отбора образцов записывали в соответствующую ИРК. Анализы проводили в лаборатории TDL обычными способами. Образцы крови для определения стандартных гематологических параметров отбирали в пробирки емкостью 4 мл, содержащие КЗ ЭДТА, и образцы крови для определения стандартных биохимических параметров отбирали в пробирки для отделения сыворотки (англ. Serum Separator Tubes, сокращенно SST) емкостью 5 мл.
Если результаты лабораторных анализов, которые, как предполагалось, имели клиническую значимость, выходили за пределы нормального диапазона значений, то такие анализы проводили повторно. Субъекты, результаты которых, имеющие предполагаемую клиническую значимость, подтверждались при повторном анализе, не включались в исследование, или, если они уже были включены, могли быть освобождены от последующего участия в исследовании, и/или их могли наблюдать до нормализации значений или до тех пор, пока Исследователь или уполномоченное лицо считали, что это необходимо.
Серологический анализ
Серологический анализ выполняли, как указано в алгоритме исследования (Таблица 12). На момент отборочного визита у всех субъектов проверяли все параметры, перечисленные в Таблице 13. Это было сделано для безопасности исследовательского персонала, и результаты проверки не включали в базу данных исследования. Если в каком-либо из указанных испытаний было обнаружено, что субъект (субъекты) имеет (имеют) положительные результаты, то такого субъекта подвергали дополнительным исследованиям и процедурам и не включали в исследование. Серологические анализы проводили на тех же образцах крови, которые использовали для клинического химического анализа (собранные в SST пробирки объемом 5 мл). Образцы анализировали в лаборатории TDL.
бета-ХГЧ
Для исключения возможной беременности в тех случаях, когда ее можно было бы ожидать, в образцах сыворотки крови анализировали бета-ХГЧ, как описано в Таблице 12. Любого субъекта с положительным результатом теста на беременность исключали или выводили из исследования.
Анализ мочи
Образцы мочи для анализа параметров мочи отбирали в соответствии с расписанием, указанным в алгоритме исследования (Таблица 12). В анализе мочи с помощью тест-полосок определяли параметры, указанные в Таблице 13 RPL. При необходимости, в случае клинически значимого положительного результата в лаборатории TDL проводили микроскопическое исследование осадка и/или культуры.
Анализ на наркотики
Анализ мочи на наркотики выполняли в RPL в соответствии с расписанием, указанным в Таблице 12. Если субъект не проходил испытание на наркотическую зависимость, то его/ее исключали из исследования. Повторную проверку на наркотики проводили только в тех случаях, когда были методологические основания полагать, что результат является ложноположительным. В тех случаях, если получаемые положительные результаты, близкие к граничным, не могли быть объяснены предшествующим состоянием, такие результаты считались положительными, и субъекта исключали из исследования. Если обнаруживали, что субъект имеет положительный результат из-за приема медикамента, например, противопростудных/противогриппозных препаратов, проводили повторный тест на наркотики, если субъект все еще находился в пределах скринингового окна. Результаты этих анализов не размещали в базе данных.
Проба на алкоголь в выдыхаемом воздухе
Пробу на алкоголь в выдыхаемом воздухе получали с помощью устройства для измерения уровня алкоголя (расписание анализов указано в алгоритме исследования в Таблице 12). Результаты этого анализа не вносили в базу данных клинического исследования. Если субъект имел положительный результат пробы на алкоголь в выдыхаемом воздухе, то его исключали из исследования.
Данные физического обследования, рост и масса тела
Расписание индивидуальных обследований указано в алгоритме исследования (Таблица 12). Данные физического обследования на момент отбора и в последующие периоды (сутки 28) включали следующие данные полного физического обследования: общее состояние, состояние кожи, головы, шеи, лимфатических узлов, щитовидной железы, органов брюшной полости, опорно-двигательной системы, сердечно-сосудистой системы, дыхательной системы и нервной системы.
Физическое обследование, производимое в Сутки -1, представляло собой поверхностное обследование, нацеленное на обнаружение любых изменений, которые могли произойти с момента отбора.
Рост определяли в сантиметрах, а массу - в килограммах. Во время измерений субъекты были одеты легко, без обуви, и для всех измерений применяли калиброванные устройства. Индекс массы тела (ИМТ) вычисляли, исходя из роста и массы тела.
Данные фокусированного физического обследования
Фокусированное физическое обследование глаз и лица (расписание - см. Таблицу 12) выполняли с использованием стандартизованной системы оценок, в которой оценивали местную температуру, зуд, боль, отек/уплотнение, покраснение и гематомы. Была принята следующая система оценок:
• Слабые изменения или
• Умеренные изменения. Оценка эффективности
Оценку эффективности проводили в соответствии с расписанием, указанным в алгоритме исследования (Таблица 12).
Оценка выраженности морщин Исследователем и субъектом
В течение каждого визита Исследователь и субъект независимо оценивали выраженность морщин субъекта "в состоянии покоя" и "при самой широкой улыбке". Исследователь оценивал морщины согласно шкале MAS LCL в динамике и в состоянии покоя в соответствии со следующими критериями (используя для оценивания фотографии с высоким разрешением):
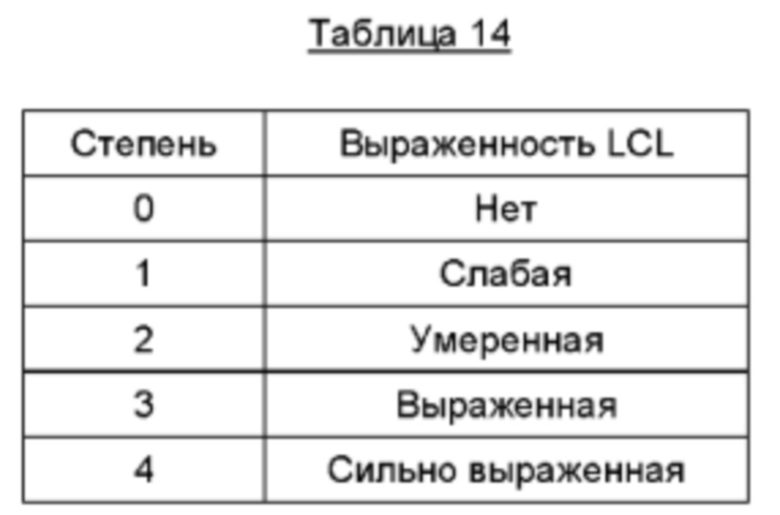
Оценка удовлетворенности субъекта
Во время каждого визита после лечения субъект оценивал степень своей удовлетворенности лечением по следующей 4-балльной шкале:

Фотография
В соответствии с расписанием, указанным в алгоритме исследования (Таблица 12), были сделаны стандартные цифровые фотографии. Фотографии использовали только для иллюстрации эффекта применения ИМП (исследуемого медицинского продукта) и не использовали для оценки эффективности. Субъектов просили удалять декоративную косметику перед приходом в клинику или в самой клинике перед фотографированием.
Делали следующие фотографии:
• Анфас в состоянии покоя (без улыбки); и
• Фотографии области LCL (одна при самой широкой улыбке и одна в состоянии покоя).
Отбор образцов
Клинические исследования иммуногенности: Анализ для определения нейтрализующих антител к QM1114-DP будет разработан позже. Образцы крови отбирали согласно расписанию, указанному в алгоритме исследования (Таблица 12), для определения количества антител в будущем, после создания соответствующего анализа.
Нежелательные явления
Ниже приведены определения НЯ, нежелательных реакций на препарат (НРП), СНЯ и предполагаемых непредвиденных серьезных нежелательных реакций (ПНСНР). Очень важно, чтобы весь персонал, участвующий в проведении клинического исследования, был ознакомлен с данным разделом.
Определения
Нежелательное явление (НЯ) - НЯ состоит в развитии нежелательного медицинского нарушения или ухудшения уже существующего медицинского состояния после или во время воздействия фармацевтического продукта, независимо от того, связно ли это явление с продуктом. Нежелательными медицинскими нарушениями могут быть симптомы (например, тошнота, боль в груди), признаки (например, тахикардия, увеличенная печень) или аномальные результаты исследования (например, результаты лабораторных анализов, ЭКГ). В клинических исследованиях НЯ может включать нежелательное медицинское нарушение, возникающее в любое время, начиная с момента подписания информированного согласия до окончания участия в исследовании, т.е. до вывода субъекта из исследования или в альтернативном варианте до завершения исследования (Сутки 28).
Обусловленность НЯ (т.е. причинно-следственную связь между исследуемой лечением и НЯ) оценивал Исследователь (Исследователи), который при заполнении соответствующей индивидуальной регистрационной формы должен отвечать "да" или "нет" на вопрос "Считаете ли Вы, что имеется обоснованная возможность того, что этот эпизод мог быть вызван одним из следующих: исследуемым препаратом; другим препаратом?".
Следует отметить, что также необходимо сообщать о СНЯ, которые могут быть связаны с любыми процедурами исследования.
Нежелательная реакция на препарат (НРП) - НРП представляет собой любое НЯ, при котором причинная взаимосвязь с ИМП имеет по меньшей мере обоснованную возможность.
Серьезное нежелательное явление (СНЯ) - СНЯ представляет собой НЯ, возникающее в любой фазе исследования (т.е. во время вводного периода, лечения, периода выведения и/или позднее) и при любой дозе ИМП или плацебо, которое соответствует одному или более из следующих критериев:
• Приводит к смертельному исходу;
• Угрожает жизни;
• Требует пребывания пациента в стационаре или продолжения текущей госпитализации;
• Приводит к трудноизлечимому или значительному нарушению функций или нетрудоспособности;
• Является врожденной аномалией или врожденным дефектом;
• Является важным медицинским эпизодом, который может угрожать субъекту или может требовать медицинского вмешательства для предотвращения одного из исходов, перечисленных выше.
Обусловленность СНЯ (т.е. причинно-следственную связь между исследуемым лечением и СНЯ) оценивали также, как и несерьезные НЯ. Следует отметить, что также необходимо сообщать о СНЯ, которые могут быть связаны с любыми процедурами исследования.
Предполагаемая непредвиденная серьезная нежелательная реакция (ПНСНР) - ПНСНР представляет собой любое СНЯ, при котором причинная взаимосвязь с ИМП имеет по меньшей мере обоснованную возможность, но это явление не указано в Брошюре Исследователя и/или в Сводных данных о Характеристиках Продукта.
Соответствие измерений
Все проводимые в этом исследовании определения эффективности и безопасности были стандартными, широко применяемыми и имеющими подтвержденную надежность и точность.
Анализ результатов эффективности
Эффективность оценивали по обоснованной 5-балльной шкале MAS и шкале оценивания удовлетворенности субъектов. Данные по эффективности обобщали с применением методик описательной статистики (n, среднее, СО (среднеквадратичное отклонение), минимальное, среднее и максимальное) в соответствии с этапами исследования и типам получающих лечение групп в состоянии покоя и при самой широкой улыбке по отдельности.
Изменения выраженности морщин в состоянии покоя и при максимальном нахмуривании/самой широкой улыбке по сравнению с исходным состоянием согласно оценкам Исследователя и субъектов по отдельности оценивали с применением модели повторных измерений (смешанной модели). Анализ выполняли с помощью SAS PROC MIXED. В качестве фиксированных эффектов смешанная модель включала лечение, время и зависимость эффекта лечения от времени, и в качестве коварианта - параметры исходного состояния. В этой модели в качестве повторного эффекта выбирали время и субъектно идентифицируемые группы коррелированных данных. Исходное состояние определяли как состояние в момент времени Сутки -1.
С помощью смешанной модели были рассчитаны вычисляемые способом наименьших квадратов средние значения для каждой из групп с соответствующей дозировкой в моменты времени, соответствующие различным суткам, и разности между группами, получавшими различные дозы, в моменты времени, соответствующие различным суткам, а также 95% доверительный интервал.
Размер выборки n=8 субъектов в одной когорте посчитали достаточным для исследования безопасности и переносимости однократных доз QM1114-DP, вводимых для временного улучшения лицевых морщин в области LCL у здоровых мужчин и женщин.
Исследуемые субъекты
Для этого исследования были отобраны 24 здоровых субъекта, 6 мужчин и 18 женщин, которых рандомизировали и которым вводили дозы препарата в соответствии с ПКИ (протоколом клинического исследования). Все участники полностью завершили исследование и прошли анализы для оценки безопасности и эффективности.
Описательная статистика демографических параметров представлена ниже в Таблице 16. Двадцать три (23) субъекта имели европейскую внешность, и один субъект был чернокожим; средний возраст в когорте составлял от 42,2 до 51,0 года (включительно). Восемнадцать (18) субъектов были женщинами, и 6 субъектов были мужчинами. Средняя масса тела составляла от 66,8 до 71,7 кг, и ИМТ всех субъектов в среднем составлял от 24,0 до 26,4 кг/м2. При включении в исследование в анамнезе субъектов не было никаких клинически значимых событий медицинского или хирургического вмешательства. Каких либо существенных отклонений в данных физического обследования конкретных субъектов во время исследования также не было обнаружено.
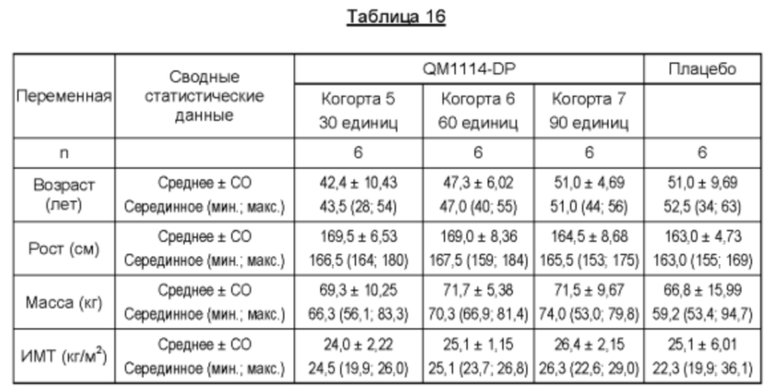
Оценка эффективности
Выборка для анализа эффективности состояла из всех субъектов (24 субъектов), которые получали однократную дозу QM1114-DP или Плацебо и для которых имелись данные эффективности после введения дозы.
Одной из задач настоящего исследования была оценка эффективности действия каждой из доз QM1114-DP, направленного на временное улучшение состояния лицевых морщин в боковой периорбитальной области. На Фиг. 13 и Фиг. 14 представлены выставляемые в течение определенного периода времени Исследователем и субъектом средние оценки состояния морщин в левой и правой боковой периорбитальной области в состоянии покоя и при самой широкой улыбке у лиц, состоящих в группе, получавшей лечение. Смоделированная статистика по оценкам выраженности морщин в состоянии покоя и при самой широкой улыбке, выставляемых Исследователем и субъектом в группах, получавших разные дозы, представлена в Таблицах 17-20. Эффективность оценивали по обоснованной шкале MAS, которую Исследователь и субъект использовали для оценивания выраженности морщин. Применяли следующие степени выраженности морщин: 0 (нет), 1 (слабая), 2 (умеренная), 3 (выраженная) и 4 (сильно выраженная).
Выраженность морщин в состоянии покоя
Данные по эффективности показывают, что после введения QM1114-DP выраженность LCL в состоянии покоя в группах, получавших действующую лечение, снижалась по оценке Исследователя и субъекта в сравнении с группой плацебо (Фиг. 13 и Фиг. 14). Изменения в выраженности морщин LCL становились очевидными при дозировках QM1114-DP 30 и 60 единиц, начиная с Суток 2.
При дозировке QM1114-DP 90 единиц уменьшение выраженности морщин начиналось в период времени между Сутками 3 и Сутками 7. Оценка выраженности морщин Исследователем указывала на то, что максимальный эффект в состоянии покоя достигался в период времени между Сутками 21 и Сутками 28 при введении любой из исследуемых доз. Если максимальный эффект был достигнут на Сутки 21, то эффект сохранялся до Суток 28. По оценке выраженности морщин субъектом максимальный эффект был достигнут быстрее. Это было заметно по выраженности морщин в области LCL слева. Оценка субъектом выраженности морщин в области слева показывает, что при дозировках QM1114-DP 30 и 90 единиц максимальный эффект был достигнут на Сутки 14 (средняя оценка 0,7 [30 единиц QM1114-DP] и 1,0 [90 единиц QM1114-DP] по шкале MAS). Максимальный эффект при дозировке QM1114-DP 60 единиц был достигнут на Сутки 7 (средняя оценка 0,5 по шкале MAS), и этот эффект сохранялся до Суток 28. На Сутки 28 субъекты в группе, получавшей 30 единиц QM1114-DP, имели среднюю оценку 0,7 по шкале MAS, и субъекты в группе, получавшей 60 единиц QM1114-DP, имели среднюю оценку, составляющую от 0,2 до 0,8 по шкале MAS. На Сутки 28 субъекты в группе, получавшей 90 единиц QM1114-DP, имели среднюю оценку шкале MAS, составляющую от 0,8 до 1,0.
Выраженность морщин при самой широкой улыбке
Аналогичные тенденции к снижению также наблюдали в оценках выраженности LCL Исследователем и субъектом при самой широкой улыбке (Фиг. 13 и Фиг. 14). При самой широкой улыбке выраженность LCL по оценке Исследователя в группах, получавших действующее лечение, снижалась, начиная с Суток 2 (Фиг. 13). Аналогичную тенденцию также наблюдали в оценках выраженности морщин субъектом (Фиг. 14). При дозировке QM1114-DP 30 единиц максимальный эффект (средняя оценка 1,0 по шкале MAS) по оценке Исследователя был достигнут на Сутки 14. Максимальный эффект при дозировке QM1114-DP 60 единиц был достигнут на Сутки 7 (средняя оценка 0,7 [LCL слева] и 0,8 [LCL справа] по шкале MAS), и некоторый эффект сохранялся до Суток 28 (средняя оценка 1,0 [LCL слева] и 0,8 [LCL справа] по шкале MAS). Максимальный эффект при дозировке QM1114-DP 90 единиц был достигнут на Сутки 7 (средняя оценка 1,3 [LCL слева] по шкале MAS) и Сутки 21 (средняя оценка 1,2 [LCL справа] по шкале MAS). Эту тенденцию также наблюдали при оценке субъектом выраженности морщин, за исключением того, что для всех субъектов максимальный эффект (средняя оценка от 0,7 до 1,5 по шкале MAS) был достигнут на Сутки 28. На Сутки 28 субъекты в группе, получавшей 30 единиц QM1114-DP, имели среднюю оценку 1,0 по шкале MAS, и субъекты в группе, получавшей 60 единиц QM1114-DP, имели среднюю оценку, составляющую от 0,7 до 1,0 по шкале MAS. На Сутки 28 субъекты в группе, получавшей 90 единиц QM1114-DP, имели среднюю оценку MAS, составляющую от 1,2 до 1,5.
Оценка удовлетворенности субъекта
В каждый визит после проведения лечения субъект оценивал свою степень удовлетворенности лечением по четырехбалльной шкале. Шкала включала следующие степени удовлетворенности: 0 (очень доволен), 1 (доволен), 2 (недоволен) и 3 (очень недоволен).
Оценка удовлетворенности субъекта показывает, что за исключением одного субъекта (субъект 208, Сутки 2) в группе, получавшей 30 единиц QM1114-DP, который был недоволен, и одного субъекта (субъект 246, Сутки 7 и 14) в группе, получавшей 90 единиц, который был очень недоволен лечением, все субъекты в группах, получавших 30, 60 и 90 единиц QM1114-DP, были либо очень довольны, либо довольны лечением, начиная с Суток 2 и до Суток 28. В группе плацебо от 2 до 3 субъектов были либо очень недовольны, либо недовольны лечением при каждом визите в рамках исследования, начиная с Суток 2 и до Суток 28. Самую большую часть субъектов (50%), которые были либо очень недовольны, либо недовольны при введении плацебо, наблюдали на Сутки 2, Сутки 3 и Сутки 21.
Анализ е рамках смешанной модели
Изменения по сравнению с исходным состоянием по оценке выраженности морщин Исследователем и субъектам по отдельности в состоянии покоя и при самой широкой улыбке оценивали в рамках модели повторных измерений (смешанной модели). Оценки выраженности морщин Исследователем (различия между результатами лечения различными дозами в разные дни) представлены в Таблицах 17-18 (в состоянии покоя и при самой широкой улыбке). Оценки выраженности морщин субъектом (различия между результатами лечения различными дозами в разные дни) представлены в Таблицах 19-20 (в состоянии покоя и при самой широкой улыбке). Анализируемой и представленной в этом разделе переменной является изменение выраженности морщин в состоянии покоя и при самой широкой улыбке по сравнению с исходным состоянием по оценке Исследователем и субъектами по отдельности.
Оценка Исследователем морщин в левой и правой боковых периорбитальных областях при самой широкой улыбке показывает, что, начиная с Суток 3 и до Суток 28, изменение средних оценок по шкале MAS по сравнению с исходным состоянием в группах, получавших 30, 60 и 90 единиц QM1114-DP, имеет статистически значимое отличие (Р<0,05) от оценок в группе плацебо (Таблица 17). При оценивании субъектом морщин на левой и правой сторонах лица при самой широкой улыбке (Таблица 19), статистически значимые отличия (Р<0,05) от плацебо были выявлены, начиная с Суток 3 или с недели 1 до Суток 28, в группах, получавших 30 и 60 единиц QM1114-DP. Напротив, при сравнении результатов в области LCL на левой стороне лица у группы, получавшей 90 единиц QM1114-DP, не было выявлено статистически значимых отличий от результатов группы плацебо. Однако о статистически значимом результате было сообщено при рассмотрении LCL на правой стороне лица на Сутки 28 (Р=0,012) при сравнении группы, получавшей 90 единиц QM1114-DP, с группой плацебо.
Оценка Исследователем морщин боковой периорбитальной области в состоянии покоя показывает, что оценки LCL (на левой и на правой сторонах) в период с недели 1 до недели 4 (Сутки 28) в группе, получавшей 30 единиц QM1114-DP, имеют статистически значимые отличия (Р<0,05) от оценок группы плацебо. За исключением Суток 28 (LCL слева), аналогичные результаты в период с недели 1 до недели 4 также были получены в группе, получавшей 60 единиц QM1114-DP. Для максимальной дозировки (в группе, получавшей 90 единиц QM1114-DP), оценки, начиная с Суток 3 (LCL справа) или недели 1 (LCL слева), до недели 4, также статистически отличаются (Р<0,05) от оценок группы плацебо (Таблица 18).
Оценки морщин LCL на правой стороне лица в состоянии покоя субъектами начинали статистически значимо отличаться (Р<0,05) от оценок в группе плацебо, начиная с Суток 3, и отличались до Суток 28 в группах, получавших 30 единиц и 60 единиц QM1114-DP (Таблица 20В). За исключением Суток 3 (в группе, получавшей 60 единиц QM1114-DP) и недели 1 (в группе, получавшей 30 единиц QM1114-DP), начиная с Суток 3 и до Суток 28, также регистрировали статистически значимое отличие оценок LCL на левой стороне лица в группах, получавших 30 единиц и 60 единиц QM1114-DP, от оценок группы плацебо (Таблица 20А). В группе, получавшей 90 единиц QM1114-DP, результаты различались. В оценках LCL на правой стороне лица статистически значимые отличия в выраженности морщин наблюдали, начиная с Суток 14 и до Суток 28 (Таблица 20В). Напротив, в оценках LCL на левой стороне лица, статистически значимые отличия от плацебо наблюдали только на Сутки 14 и Сутки 28.
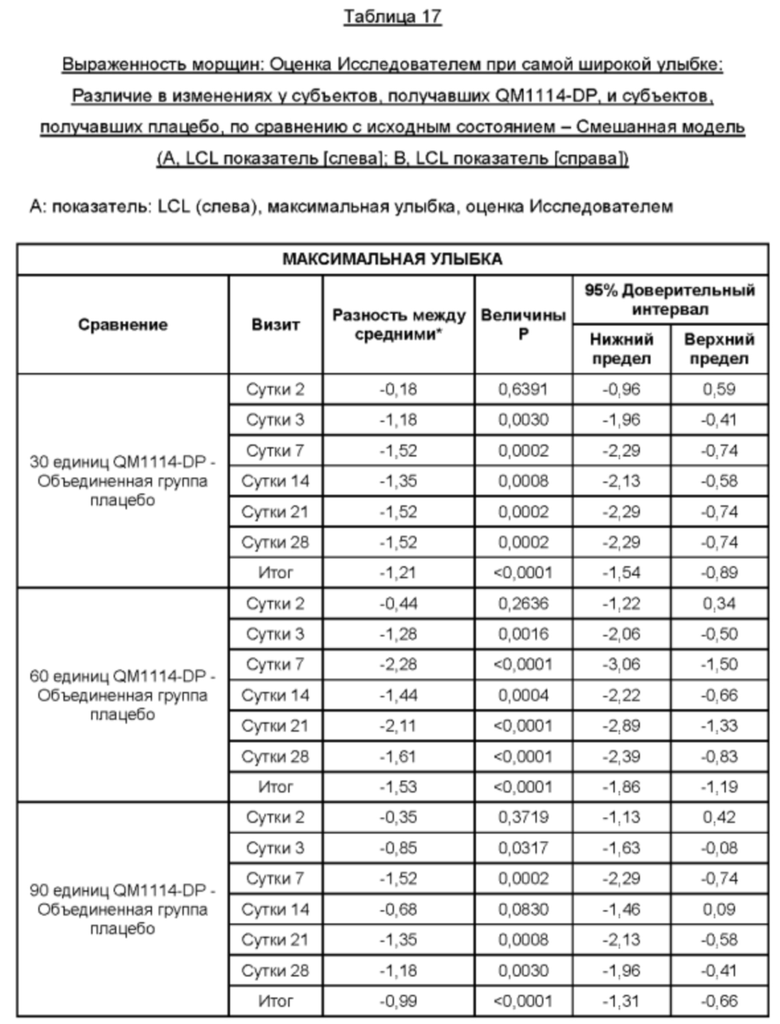
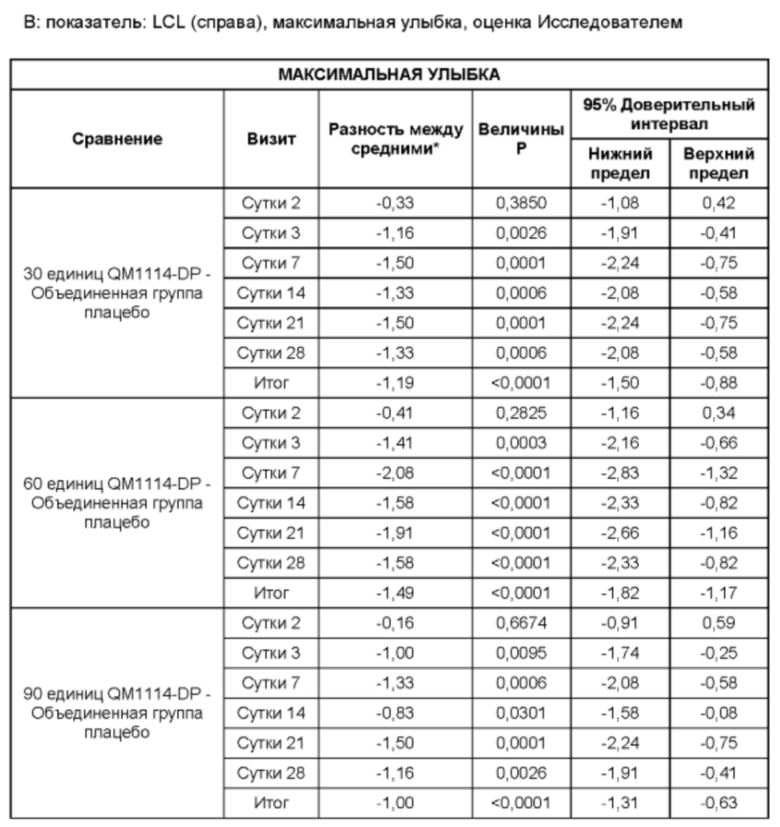
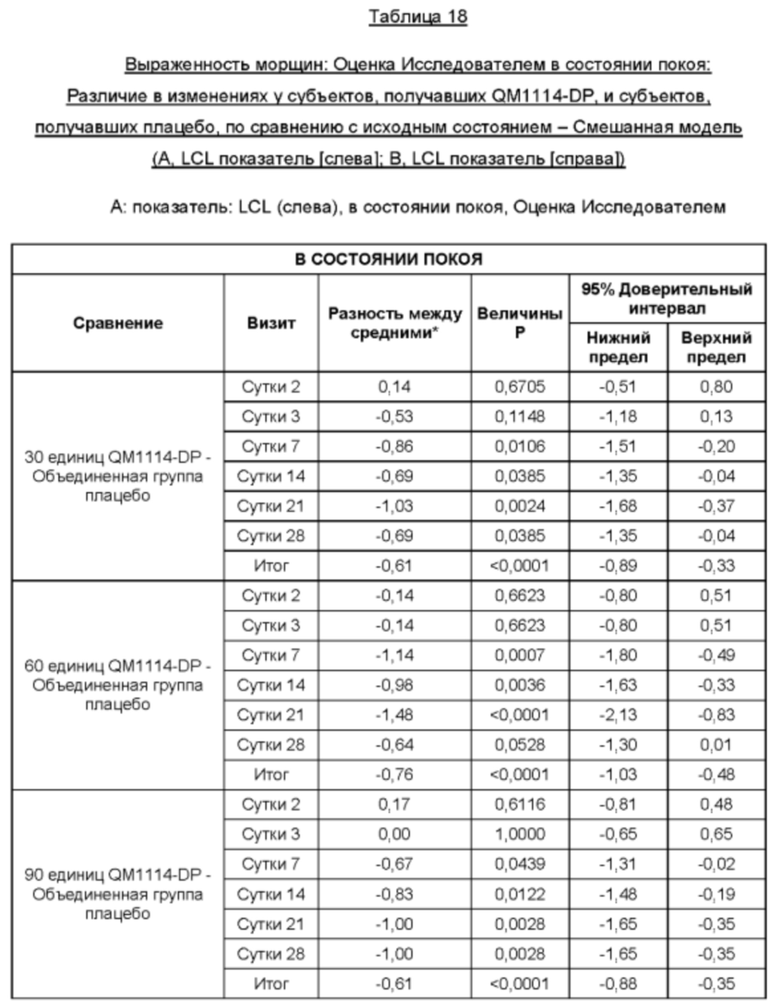
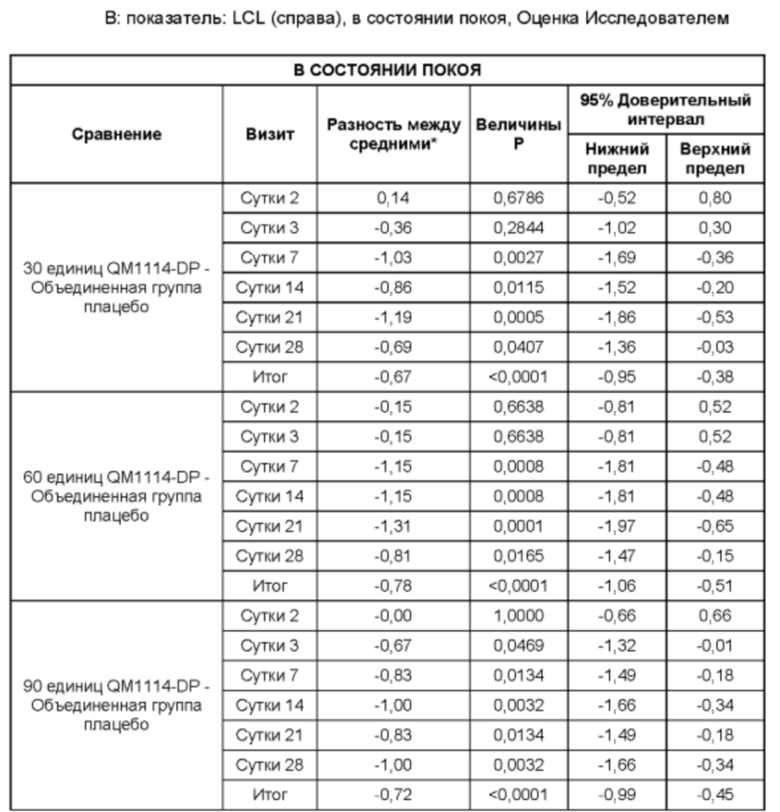
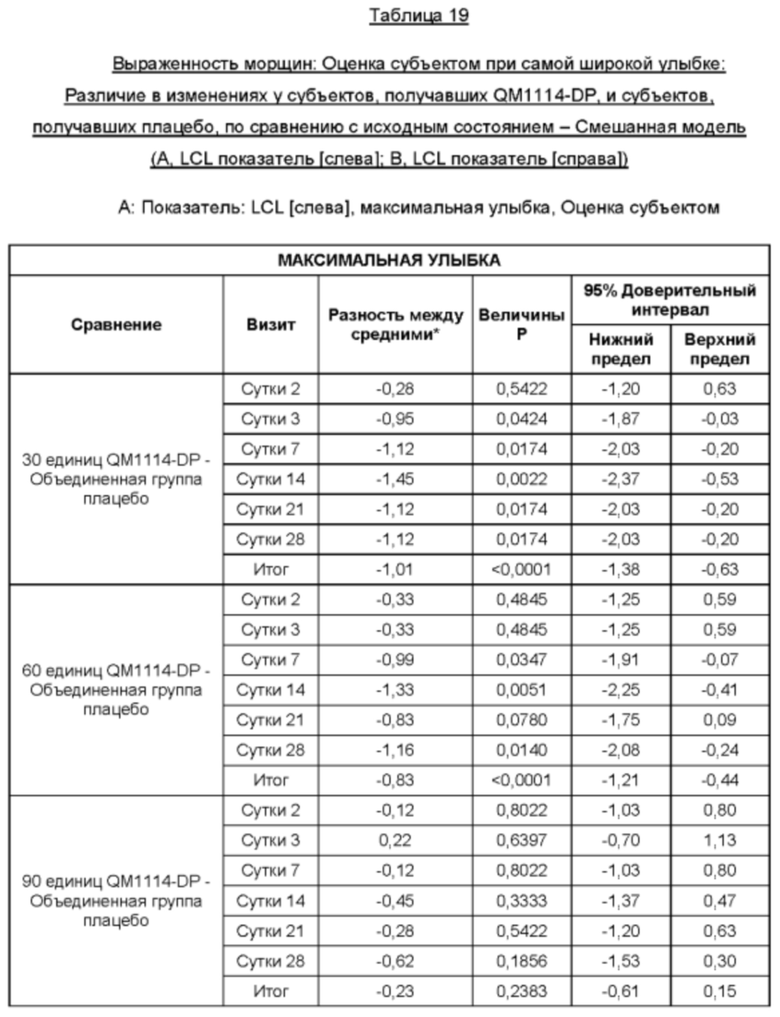
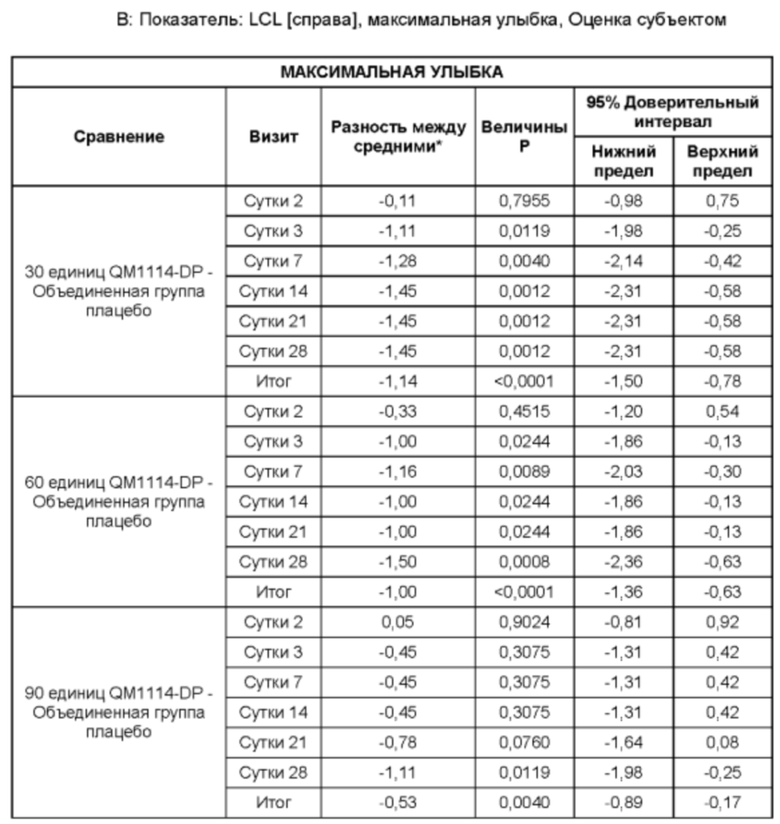
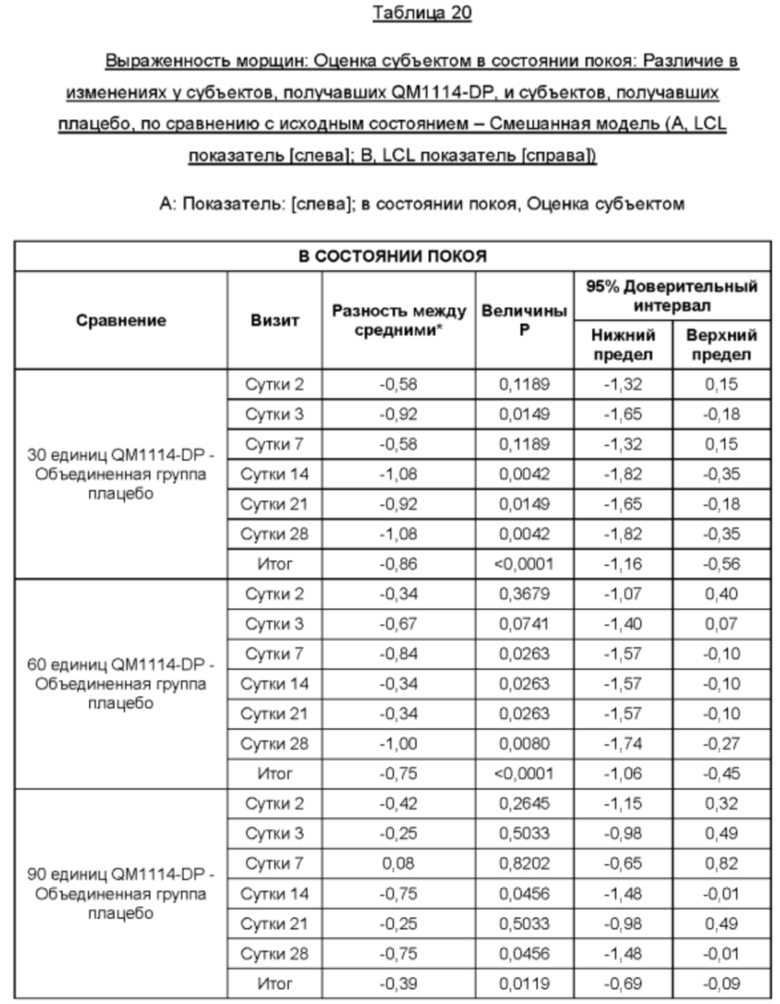
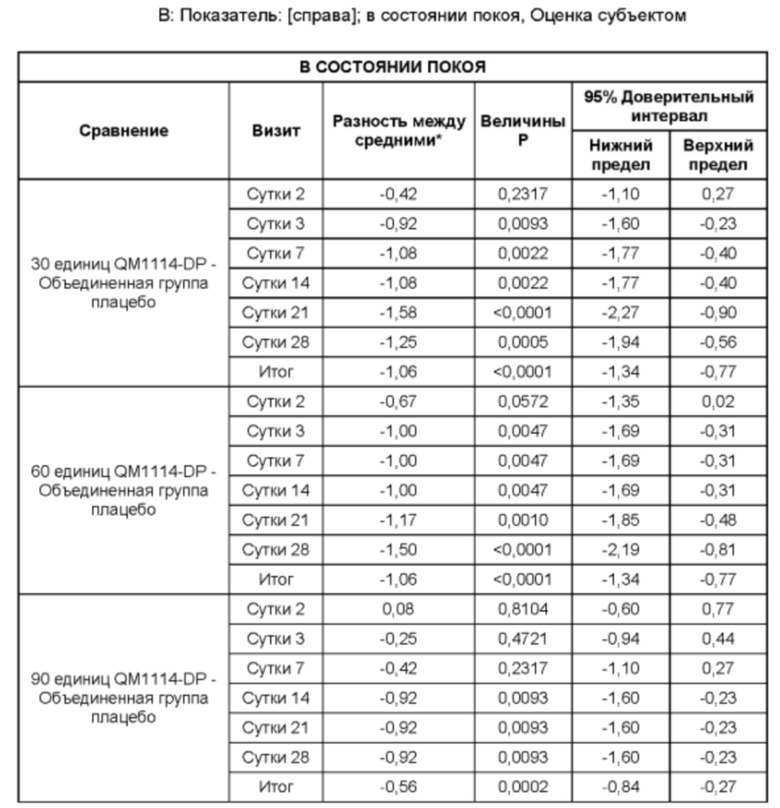
Заключение по эффективности
За исключением оценки субъектом LCL (в группе, получавшей 90 единиц QM1114-DP), результаты эффективности по оценкам исследователя и субъекта LCL при самой широкой улыбке ясно показывают, что препарат QM1114-DP эффективно снижал выраженность морщин LCL при самой широкой улыбке по сравнению с их выраженностью у субъектов группы плацебо. Уменьшение выраженности морщин сохранялось в дальнейшем (Сутки 28). Аналогично, и по оценке Исследователя, и по оценке субъекта, снижение выраженности LCL в состоянии покоя было более заметным, за некоторыми исключениями, в группах, получавших действующее лечение, чем в группе плацебо. Оценка субъектом удовлетворенности состоянием LCL показывает, что, за исключением двух субъектов, все субъекты в группах, получавших действующее лечение, были либо очень довольны, либо довольны лечением до Суток 28. В группе плацебо до 50% субъектов были недовольны или очень недовольны лечением.
Оценка безопасности
В анализе безопасности участвовали все 24 субъекта, которые получили однократную дозу препарата, изучаемого в рандомизированном исследовании.
Шесть субъектов получили 30 единиц QM1114-DP, шесть субъектов получили 60 единиц QM1114-DP, и шесть субъектов получили максимальную дозу, составляющую 90 единиц. Шесть субъектов получили плацебо.
В приведенном в данном разделе анализе НЯ в основном рассмотрены НЯ, возникающие в процессе лечения, т.е. возникающие после введения дозы ИМП (исследуемого медицинского продукта), и НРП (с нежелательные реакции на препарат), т.е. те случаи, в которых взаимосвязь с ИМП имеет по меньшей мере обоснованную возможность. Во время исследования не сообщалось ни о СНЯ, ни о ПНСНР, и ни один из субъектов не вышел из исследования по соображениям безопасности.
Обзор НЯ, появившихся в группе, получавшей лечение, представлен в Таблице 21. НЯ были выявлены при каждой дозировке. Сорок четыре (44) НЯ было выявлено у 18 из 24 (75,0%) субъектов, и из них всего 31 НЯ у 15 из 24 (62,5%) субъектов, по мнению Исследователя, могли быть или вероятно были связаны с лечением. Было выявлено четырнадцать (14) НЯ у 5 из 6 субъектов, получавших 30 единиц QM1114-DP, девять НЯ у 5 из 6 субъектов, получавших 60 единиц QM1114-DP, и четырнадцать (14) у 3 из 6 (50,0%) субъектов, получавших максимальную дозировку (90 единиц QM1114-DP). В группе плацебо было выявлено семь НЯ у 5 из 6 субъектов. В Таблице 22 представлен обзор НЯ по классам органов и систем органов (сокр. КОС) (англ. System Organ Class, сокращенно SOC) и терминам предпочтительного употребления (сокр. ТПУ) (англ. preferred term, сокращенно РТ). Связанные с лечением НЯ по классам по поражению органов и систем органов и терминам предпочтительного употребления представлены в Таблице 23.
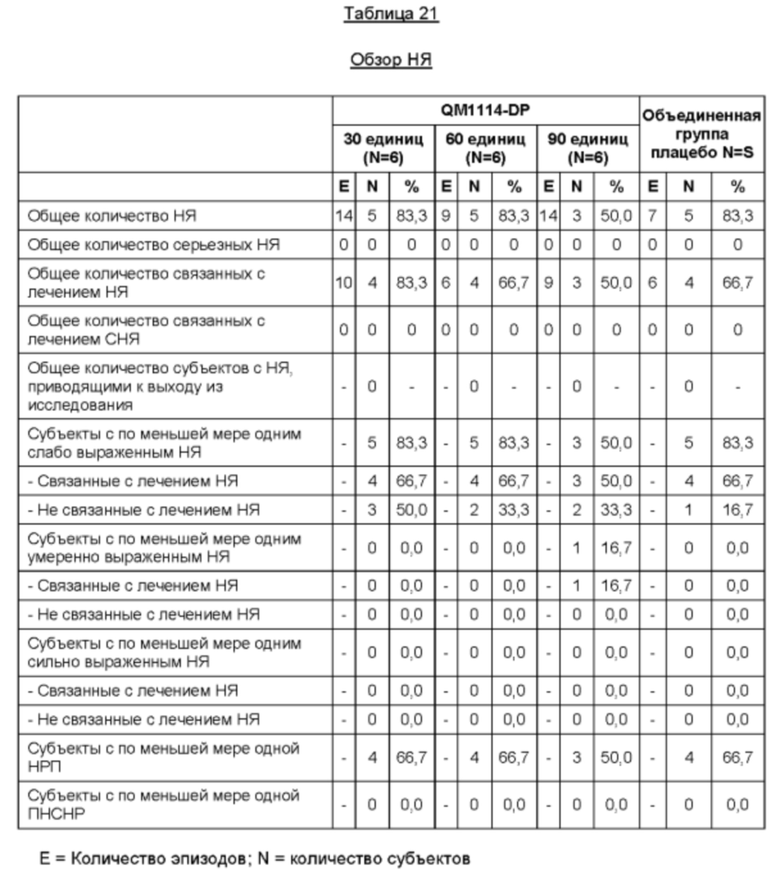
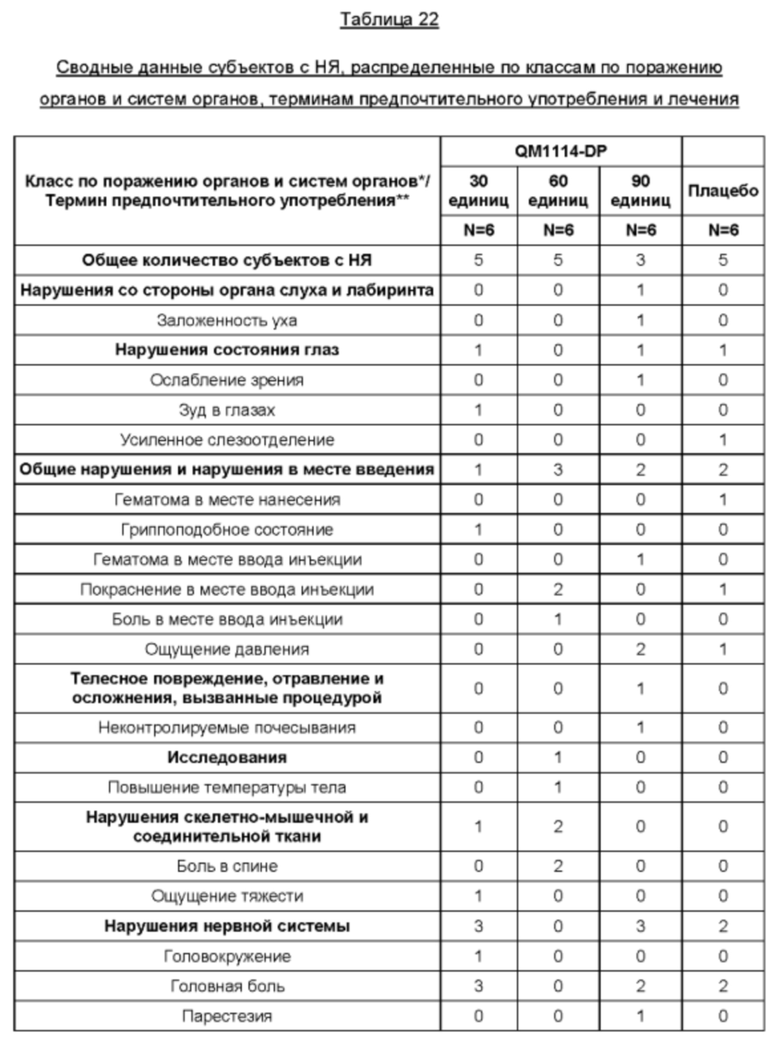
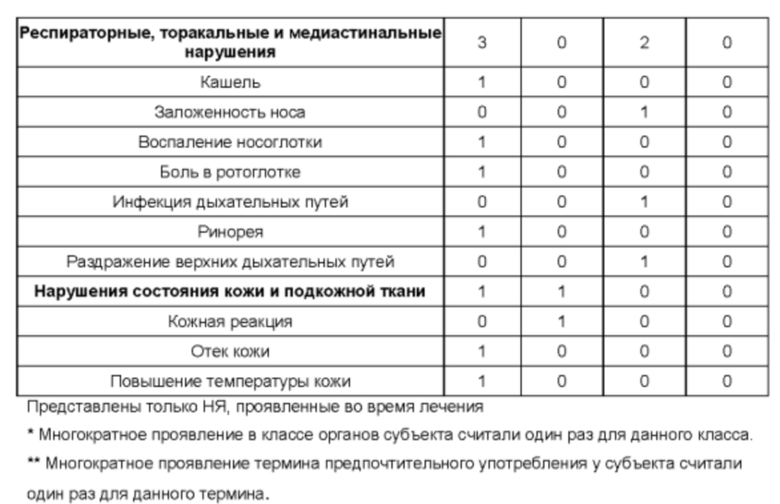
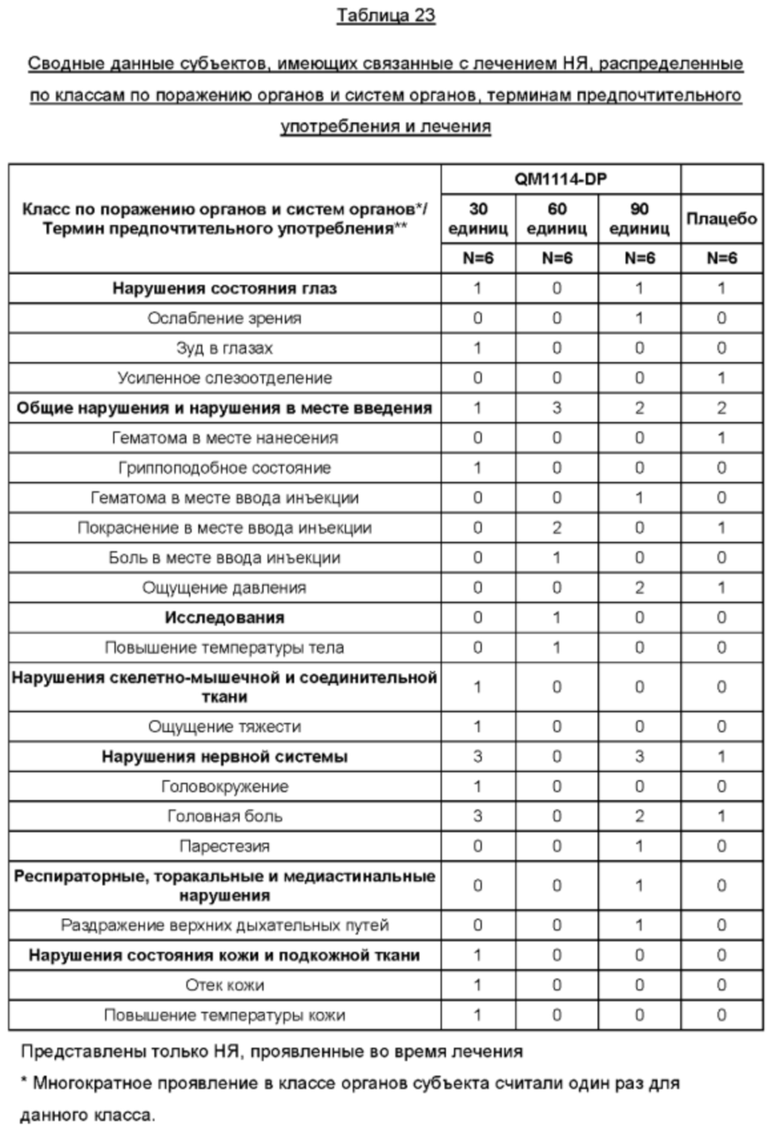

Заключение о безопасности
В целом, наиболее часто возникающие НЯ у участников групп, получавших лечение, представляли собой нарушения нервной системы, общие нарушения и состояние участка введения. Это верно и для связанных с лечением НЯ. Большинство нарушений нервной системы составляла головная боль, о которой сообщали пять субъектов, и большинство общих нарушений и неприятных состояний участка введения у субъектов состояло в покраснении места ввода инъекции и ощущении давления.
Первичной задачей этой Фазы I исследования было определение безопасности и переносимости каждой из исследуемых доз QM1114-DP здоровыми мужчинами и женщинами при лечении морщин в межбровной и боковой периорбитальной областях. В этой части исследования, включающей обработку LCL, СНЯ не были выявлены, и не было выходов из исследования из-за возникновения НЯ или по любой другой причине. Сорок четыре (44) НЯ было выявлено у 18 из 24 (75,0%) субъектов, и из них всего 31 НЯ у 15 (62,5%) субъектов, по мнению Исследователя, могли быть или вероятно были связаны с лечением. По мнению Исследователя, все другие НЯ не были связаны с лечением. За исключением одного НЯ умеренной интенсивности (головная боль, в группе, получавшей 90 единиц QM1114-DP), все другие НЯ были слабо выраженными. НЯ было выявлено при каждом уровне дозы, причем самое большое количество было выявлено при максимальной (90 единиц) дозировке и наименьшее - при минимальной (30 единиц) дозировке. Наименьшее количество НЯ (7 НЯ) было выявлено в группе плацебо.
КОС, наиболее часто упоминаемые при выявлении НЯ среди групп, получавших лечение, относились к нарушениям нервной системы, общим нарушениям и состояниям участка введения. Это также верно для связанных с лечением НЯ. Большинство нарушений нервной системы составляла головная боль, о которой сообщали семь субъектов, и большинство общих нарушений и неприятных состояний участка введения у субъектов состояло в покраснении места ввода инъекции и ощущении давления. В группе, получавшей 60 единиц QM1114-DP, нарушений нервной системы выявлено не было.
Во время исследования не наблюдали клинически значимых изменений гематологических и биохимических параметров и анализа мочи, и при физическом обследовании не было обнаружено клинически значимых отклонений. Отдельные отклонения от нормальных диапазонов показателей жизнедеятельности и данных ЭКГ, по мнению Исследователя, не были клинически значимыми и не были связаны с получением исследуемого лекарственного средства.
Обсуждение и итоговые заключения
Первичной задачей этой Фазы I исследования была оценка безопасности и переносимости QM1114-DP при каждом уровне дозы, и вторичная задача состояла в оценке эффективности действия каждой из дозировок QM1114-DP, которые вводили с целью временного улучшения состояния лицевых морщин в межбровной и боковых периорбитальных областях здоровым мужчинам и женщинам.
Наиболее часто возникающими НЯ у конкретных субъектов были нарушения нервной системы. Это также верно для связанных с лечением НЯ. Наиболее часто встречающимся индивидуальным НЯ, которое, по мнению Исследователя, связанно с лечением, была головная боль (восемь связанных с лечением НЯ), о которой сообщали всего семь субъектов. Этот результат сравним с результатом, полученным Части 1 (только GL), в которой всего у шести субъектов было выявлено шесть случаев связанной с лечением головной боли. За исключением одного НЯ умеренной интенсивности (головная боль), все НЯ были слабо выраженными, и при этом не наблюдалось СНЯ или выходов из исследования из-за проблем с НЯ. Во время исследования не происходило клинически значимых изменений гематологических и биохимических параметров и результатов анализа мочи. Кроме того, не наблюдали клинически значимых физических отклонений. Во время исследования субъекты имели показатели жизнедеятельности в пределах диапазона нормальных значений, и клинически значимых отклонений в данных ЭКГ не наблюдали.
Суммируя можно утверждать, что большая часть связанных с лечением НЯ относились к нарушениям нервной системы, общим нарушениям и к состоянию участка введения. Учитывая имеющуюся информацию по безопасности, эти НЯ, связанные с лечением, были ожидаемы и соответствовали НЯ, наблюдаемыми в предыдущих клинических исследованиях других продуктов класса BoNT-A.
Полученные в настоящем исследовании данные по эффективности показали, что лечение QM1114-DP эффективно снижала выраженность LCL при самой широкой улыбке при всех уровнях доз по сравнению с субъектами группы плацебо. Анализ согласно смешанной модели оценки Исследователем морщин в левой и правой боковых периорбитальных областях при самой широкой улыбке показывает, что, начиная с Суток 3 и до Суток 28, изменения по сравнению с исходным состоянием средних оценок по шкале MAS в группах, получавших 30, 60 и 90 единиц QM1114-DP, показывают статистически значимое отличие (Р<0,05) от оценок группы плацебо. В группах, получавших 30 и 60 единиц QM1114-DP, по оценкам субъектами морщин на левой и правой сторонах лица при самой широкой улыбке статистически значимые отличия (Р<0,05) от плацебо были выявлены в период от Суток 3 или недели 1 до Суток 28. Напротив, при оценке LCL на левой стороне лица в группе, получавшей 90 единиц QM1114-DP, не было выявлено статистически значимых отличий по сравнению с оценками в группе плацебо. Оценка субъектами удовлетворенности состоянием LCL показывает, что, за исключением двух субъектов, все субъекты в группах, получавших действующее лечение, были либо очень довольны, либо довольны лечением в период до Суток 28.
В группе плацебо не менее 50% субъектов были недовольны или очень недовольны лечением.
В заключение следует отметить, что, согласно представленным данным, введение субъектам QM1114-DP эффективно снижало выраженность LCL при самой широкой улыбке по сравнению с субъектами группы плацебо.
Итоговые заключения:
• Все дозы были безопасны и хорошо переносились.
• В этом исследовании не было выявлено СНЯ.
• Не было выходов субъектов из исследования из-за проблем с НЯ или по другим причинам.
• За исключением одного НЯ умеренной интенсивности, все НЯ были слабо выраженными.
• В результатах клинических лабораторных исследований, данных физического обследования, показателях жизнедеятельности и параметрах ЭКГ в 12-ти отведениях не было выявлено клинически значимых изменений.
• По оценке Исследователя и субъекта, в сравнении с группой плацебо, выраженность LCL в состоянии покоя в группах, получавших действующее лечение, снижалась, начиная с Суток 7. Этот эффект сохранялся в течение периода, составляющего до 28 суток.
• За исключением оценки LCL при самой широкой улыбке субъектами (в группе, получавшей 90 единиц QM1114-DP), по оценкам Исследователя и субъекта в сравнении с группой плацебо, выраженность LCL в группах, получавших действующее лечение, снижалась. Этот эффект сохранялся в течение периода, составляющего до 28 суток.
• Оценка субъектами удовлетворенности состоянием LCL показывает, что большинство субъектов в группах, получавших действующее лечение, были либо очень довольны, либо довольны лечением в период до Суток 28.
ПРИМЕР 3
Рандомизированное двойное слепое плацебо-контролируемое исследование с
увеличением дозы с однократной обработкой для оценки безопасности и эффективности QM1114-DP при лечении здоровых мужчин и женщин, имеющих морщины в верхней части лица с выраженностью от умеренной до сильной
Доклинические исследования
Кроме доклинических исследований, описанных выше в Примере 2, проводили общие токсикологические исследования QM1114-DP, которые включали пилотные исследования токсичности посредством ВМ введения однократной дозы крысам породы Вистар и собакам породы Бигль. Исследования проводили не в соответствии с НЛП (не в соответствии с Надлежащей лабораторной практикой); исследования выполняли с целью определения максимальной переносимой дозы QM1114-DP, позволяющей установить подходящие величины доз для опорного токсикологического исследования в соответствии с НЛП. Результаты пилотных токсикологических исследований ясно показали, что крысы породы Вистар были более чувствительны к токсичному действию QM1114-DP, чем собаки, и, таким образом, опорное токсикологическое исследование в соответствии с НЛП проводили только с крысами породы Вистар.
Отдельных исследований местной переносимости QM1114-DP не проводили. Вместо этого местную переносимость внимательно оценивали при клинических наблюдениях и гистопатологическом анализе участков ввода инъекций, который составлял часть опорного токсикологического исследования в соответствии с НЛП на крысах породы Вистар. При этих дополнительных наблюдениях не было получено никаких неожиданных результатов, которые могли бы указывать на проблемы с переносимостью продукта.
Клинические исследования
В Примерах 1 и 2 для лечения морщин в межбровной или боковой периорбитальной области, имеющих выраженность от умеренной до сильной, субъектам вводили QM1114-DP.
Пример 1 - Межбровные морщины
В Примере 1 (только GL) оценивали безопасность и переносимость введения 10, 25, 50 и 75 единиц QM1114-DP здоровым мужчинам и женщинам в возрасте от 18 до 65 лет, имеющих GL от умеренных до выраженных. Четыре субъекта получили 10 единиц, шесть субъектов получили 25 единиц, шесть субъектов получили 50 единиц, и шесть субъектов получили максимальную дозу, составляющую 75 единиц QM1114-DP. Восемь (8) субъектов получали плацебо. Всего было выявлено 48 нежелательных явлений (НЯ) у 23 субъектов.
Тридцать восемь (38) НЯ было выявлено у 17 субъектов, получивших QM1114-DP, и 10 НЯ было выявлено у шести субъектов, получивших плацебо; ни одно из НЯ не было серьезным, и большинство из них были слабо выраженными. Одно НЯ было оценено как явление высокой интенсивности (рвота [субъект 144, в группе, получавшей 50 единиц QM1114-DP]), и НЯ два были оценены как явления умеренной интенсивности (вазовагальная реакция [предобморочное состояние], проявившаяся непосредственно после инъекции у субъекта 126 [в группе, получавшей 25 единиц QM1114-DP] и субъекта 146 [в группе, получавшей 50 единиц QM1114-DP]).
Большая часть (17) из 32 связанных с лечением нежелательных явлений была выявлена в группе, получавшей 75 единиц QM1114-DP, и наиболее часто встречающимися НЯ были реакции в месте инъекции, т.е. покраснение, отек, раздражение, сыпь, зуд, боль, дискомфорт, покалывание и гематома. Наиболее часто встречающимся индивидуальным НЯ, которое, по мнению Исследователя, было связано с лечением, была головная боль (шесть связанных с лечением НЯ), один из случаев которой наблюдали при максимальной дозировке.
В результатах клинических лабораторных исследований безопасности, данных физического обследования, показателях жизнедеятельности и параметрах ЭКГ в 12-ти отведениях также не было выявлено клинически значимых изменений.
Пример 2 - Боковые периорбитальные морщины
Во второй части исследования 43QM1302 (только LCL) здоровым мужчинам и женщинам в возрасте от 18 до 65 лет с выраженностью LCL от умеренной до сильной вводили 30, 60 или 90 единиц QM1114-DP. Шесть субъектов получили 30 единиц, шесть субъектов получили 60 единиц, и 6 субъектов получали максимальную дозу, составляющую 90 единиц QM1114-DP. Шесть (6) субъектов получали плацебо. В этом исследовании было выявлено всего 44 НЯ у 18 субъектов.
Тридцать семь (37) НЯ было выявлено у 13 субъектов, получавших QM1114-DP, и семь НЯ было выявлено у пяти субъектов, получавших плацебо; ни одно из этих НЯ не было серьезным, за исключением одного НЯ умеренной интенсивности (головная боль у субъекта 246, в группе, получавшей 90 единиц QM1114-DP); все НЯ были слабо выраженными.
Из 31 связанного с лечением НЯ, нежелательным явлением, наиболее часто возникающим в группах, получавших лечение, были нарушения нервной системы, общие нарушения и состояния участка введения. Наиболее часто встречающимся нарушением нервной системы была головная боль, о которой сообщали семь субъектов, и наиболее часто встречающимися общими нарушениями и состояниями участка введения у субъектов были покраснение места ввода инъекции и ощущение давления.
Кроме указанных тенденций, ни в одной из терапевтических групп не было выявлено клинически значимых отклонений, связанных с безопасностью, включая относящиеся к безопасности результаты клинического лабораторного анализа, показатели жизнедеятельности, данные физического обследования и параметры ЭКГ.
Эффективность
Эффективность QM1114-DP при лечении GL (n=22) и LCL (n=18) у здоровых мужчин и женщин оценивали в исследовании 43QM1302 (Примеры 1 и 2). Эффективность оценивали по обоснованным 5-балльным фотошкалам Merz (шкала Merz Aesthetic Scale, сокращенно MAS), по которым Исследователь и субъекты оценивали выраженность морщин в состоянии покоя и при максимальном нахмуривании/самой широкой улыбке. Полученные результаты показывают, что внутримышечное введение QM1114-DP в дозировке до 75 единиц (показатель GL) или до 90 единиц (показатель LCL) эффективно снижало выраженность GL или LCL при максимальном нахмуривании (показатель GL) или максимальной улыбке (показатель LCL) по сравнению с субъектами группы плацебо. Кроме того, оценка субъектом удовлетворенности состоянием GL или LCL показывает, что все субъекты в группах, получавших QM1114-DP, были либо очень довольны, либо довольны лечением в период до Суток 28.
В заключение следует отметить, что данные по эффективности, полученные в Примерах 1 и 2, показывают, что при всех изученных дозировках QM1114-DP эффективно снижает выраженность GL при максимальном нахмуривании и выраженность LCL при самой широкой улыбке по сравнению с субъектами группы плацебо.
Задачи исследования
Первичная задача: оценка безопасности и переносимости каждой из дозировок препарата QM1114-DP, вводимого с целью временного улучшения состояния межбровных морщин (GL) и боковых периорбитальных морщин (LCL), обрабатываемых в комбинации.
Вторичные задачи: оценка эффективности действия каждой из дозировок препарата QM1114-DP, где действие состоит во временном улучшении состояния GL и LCL, и оценка эффективности действия каждой из дозировок препарата QM1114-DP, где действие состоит во временном улучшении состояния GL и LCL, которые обрабатывают в комбинации.
Результаты исследования
Безопасность: Частота возникновения и выраженность НЯ
Эффективность:
• Оценка Исследователем выраженности GL при максимальном нахмуривании и выраженности LCL при самой широкой улыбке в течение периода до Суток 28 (неделя 4) в реальных условиях по шкале MAS, GL - в динамике и LCL -в динамике;
• Оценка Исследователем выраженности GL и LCL в состоянии покоя в течение периода до Суток 28 (неделя 4) в реальных условиях по шкале MAS, GL - в состоянии покоя и LCL- в состоянии покоя (в статике);
• Оценка субъектом выраженности GL при максимальном нахмуривании и выраженности LCL при самой широкой улыбке в течение периода до Суток 28 (неделя 4) в реальных условиях по шкале MAS, GL - в динамике и LCL - в динамике; и
• Оценка субъектом выраженности GL и LCL в состоянии покоя в течение периода до Суток 28 (неделя 4) в реальных условиях по шкале MAS, GL - в состоянии покоя и LCL - в состоянии покоя (в статике);
• Оценка удовлетворенности субъекта в течение периода до Суток 28 (неделя 4) по 4-балльной шкале.
Экспериментальная часть:
• Оценка Исследователем выраженности GL при максимальном нахмуривании и выраженности LCL при самой широкой улыбке в реальных условиях по шкале MAS, GL - в динамике и LCL - в динамике, до тех пор, пока оценка состояния морщин не вернется к исходному значению;
• Оценка Исследователем выраженности GL и LCL в состоянии покоя в реальных условиях по шкале MAS, GL - в состоянии покоя и/или LCL - в состоянии покоя, до тех пор, пока оценка состояния морщин не вернется к исходному значению;
• Оценка субъектом выраженности GL при максимальном нахмуривании и выраженности LCL при самой широкой улыбке в реальных условиях по шкале MAS, GL - в динамике и LCL - в динамике, до тех пор, пока оценка состояния морщин не вернется к исходному значению;
• Оценка субъектом выраженности GL и LCL в состоянии покоя в реальных условиях по шкале MAS, GL - в состоянии покоя и LCL - в состоянии покоя, до тех пор, пока оценка состояния морщин не вернется к исходному значению; и
• Оценка удовлетворенности субъекта по 4-балльной шкале.
Субъекты, которые участвовали в экспериментальной фазе этого исследования, посещали отделение клинической фармакологии (ОКФ) каждый месяц. Экспериментальная оценка конечных результатов не была включена в настоящий отчет о клиническом исследовании.
План исследования
В этом Примере рассмотрено исследование безопасности и эффективности применения QM1114-DP для временного улучшения состояния LCL и GL, обрабатываемых в комбинации. Отдельная часть исследования также включала лечение GL и LCL в когортах 1-4 (Пример 1 [GL]) и когортах 5-7 (Пример 2 [LCL]) с применением увеличивающихся доз, как было рассмотрено выше.
Исследование представляло собой рандомизированное двойное слепое плацебо-контролируемое исследование для оценки безопасности и эффективности увеличивающихся доз ботулотоксина типа A (QM1114-DP; активный компонент), вводимых здоровым мужчинам и женщинам в возрасте от 18 до 65 лет, имеющих GL и LCL от умеренных до выраженных. Всего в исследовании участвовали 16 субъектов, которых разделяли на две когорты (когорты 8 и 9), и в каждой когорте находилось 8 субъектов (6 получали активный компонент: 2 - плацебо).
Перед подписанием ФИС (формы информированного согласия), каждый здоровый субъект получал вербальную (от врача-исследователя) и письменную информацию. Субъектов проверяли на соответствие требованиям в течение 28 суток перед проведением лечения в момент времени Сутки 1, и они совершали визиты в рамках исследования для оценки безопасности и эффективности до последующего визита в Сутки 28 (неделя 4). Всех субъектов помещали в ОКФ, где их оставляли в течение Суток -1 и затем в течение ночи, предшествующей суткам, в которые производили введение дозировок, т.е. Суткам 1. Для минимизации рисков, которым подвергались субъекты, при каждом уровне дозы были созданы сигнальные группы. Соответственно, в когортах 8 и 9 первым 2 субъектам потребовалась более длительная госпитализация в ОКФ (1 получал активный ингредиент и 1 плацебо). Этих субъектов помещали в ОКФ в Сутки -1, подвергали лечению в Сутки 1 и выписывали из ОКФ в Сутки 2, после чего они совершали визиты в рамках исследования до последующего визита в Сутки 28 (неделя 4). Исследование состояло из следующих визитов: Отбор, Сутки -1, Сутки 1, 2, 3, 7, 14, 21 и 28.
Исследователи и субъекты оценивали выраженность по шкале MAS для GL и LCL в динамике и в состоянии покоя при отборе, во время визитов перед лечением и во время всех визитов по окончании лечения GL и LCL. Субъекты также оценивали свою удовлетворенность состоянием своих GL и LCL по 4-балльной шкале во время всех визитов по окончании лечения (Ascher с соавт., 2009).
Субъектов рандомизировали для получения одной обработки исследуемым лекарственным средством (QM1114-DP или плацебо). Каждая обработка включала 11 инъекций равного объема (100 мкл) в область GL (пять инъекций) и область LCL (три инъекции в каждую из сторон лица). Пять точек ввода инъекций в область GL включали две инъекции в каждую мышцу, сморщивающую бровь, и одну инъекцию в мышцу гордецов. В области LCL места ввода инъекции уточняли в соответствии с расположением LCL морщин у конкретного субъекта. Во всех случаях инъекции вводили в наружную часть круговой мышцы глаза и, если это было возможно, на расстоянии приблизительно 1-2 см от глазничного валика.
Вводили следующие дозировки QM1114-DP:
• Когорта 8, Доза 1: всего 110 единиц QM1114-DP за одну обработку (10 единиц на участок ввода инъекции [всего 50 единиц в область GL и всего 60 единиц в область LCL]).
• Когорта 9, Доза 2: всего QM1114-DP 140 единиц за одну обработку (10 единиц на участок ввода инъекции в области GL [всего 50 единиц в область GL] и 15 единиц на участок ввода инъекции в области LCL [всего 90 единиц в область LCL]).
Проводимые в этом исследовании процедуры и соответствующее оценивание приведено в Таблице 1 в виде данных по визитам в рамках исследования. Клинические исследования включали отбор и визиты до недели 4 (последующие). Если оценка, выставленная Исследователем (при максимальном нахмуривании [GL] и максимальной улыбке [LCL]), возвращалась к оценке, соответствующей исходному состоянию, то последний визит субъектов в ОКФ приходился на Сутки 28 (неделя 4). В этот момент исследование формально завершалось. Субъекты, у которых оценка выраженности морщин, выставленная Исследователем (при максимальном нахмуривании [GL] и максимальной улыбке [LCL]), не возвращалась к оценке, соответствующей исходному состоянию, на Сутки 28 (неделя 4), участвовали в экспериментальной фазе настоящего исследования и посещали ОКФ каждый месяц до тех пор, пока их морщины по оценкам не возвращались к исходному состоянию. Данные по последующей эффективности описанной экспериментальной обработки будут опубликованы отдельно и не в этом отчете о клиническом исследовании.
Отбор контингента для исследования
Это исследование представляло собой рандомизированное плацебо-контролируемое исследование оценки безопасности и переносимости каждой из доз QM1114-DP и оценки эффективности введения каждой из доз QM1114-DP для достижения временного улучшения состояния морщин на лице в левой и правой боковой периорбитальной области и межбровной области. Критерии отбора были определены таким образом, чтобы субъекты, отобранные для участия в исследовании, не имели никаких значимых заболеваний.
В течение 28 суток до начала исследования, субъекты проходили полную процедуру отбора, как описано в Таблице 24.
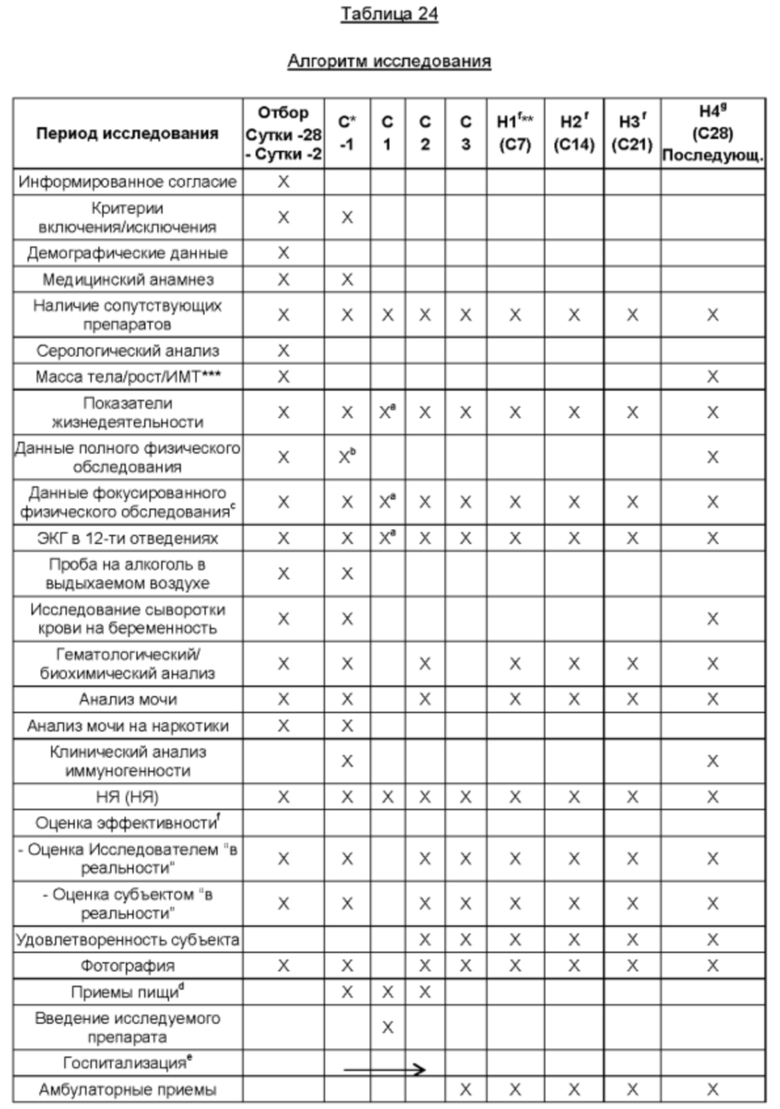

Критерии включения
Для включения в это исследование каждый субъект должен был соответствовать каждому из следующих критериев:
• Здоровый мужчина или здоровая женщина в возрасте от 18 и 65 лет (включительно) на момент отбора.
• Ранее не подвергались лечению BoNT-A или В.
• По оценке Исследователя на момент отбора и визитов для оценки исходного состояния в соответствии с обоснованной фотошкалой (MAS, GL в динамике) имеет морщины по меньшей мере умеренной выраженности (степень 2) при максимальном нахмуривании.
• По оценке Исследователя на момент отбора и визитов для оценки исходного состояния в соответствии с обоснованной фотошкалой (MAS, LCL в динамике) имеет LCL с обеих сторон лица от умеренных до выраженных (Степень 2 или 3) при максимальной улыбке.
• По оценке Исследователя на момент отбора и визитов для оценки исходного состояния в соответствии с обоснованной фотошкалой (MAS, GL в состоянии покоя) имеет по меньшей мере слабо выраженные (степень 1) морщины в состоянии покоя.
• По оценке Исследователя на момент отбора и визитов для оценки исходного состояния в соответствии с обоснованной фотошкалой (MAS, LCL в состоянии покоя) имеет LCL с обеих сторон лица в состоянии покоя от слабых до выраженных (Степень 1, 2 или 3).
• Субъекты согласны применять приемлемый способ контрацепции.
• Субъекты были способны понять и согласиться с требованиями протокола и подписали ФИС перед проведением любых процедур, связанных с исследованием.
• Не имеет клинически значимого заболевания или аномальных результатов лабораторного анализа, ЭКГ или показателей жизнедеятельности, указанных в медицинском анамнезе, данных физического обследования или других анализах, проводимых на момент отбора или при приеме в ОКФ.
Критерии исключения
Субъект считается неподходящим для включения в Часть 3 настоящего исследования, если он отвечает любому из следующих критериев:
• Ранее получал лечение BoNT-А или В.
• Имеет морщины в области GL и/или LCL, которые нельзя было бы по существу разгладить вручную, растягивая кожу.
• Субъекту ранее вводили любой нерассасывающийся или частично нерассасывающийся материал или ранее вводили кожный наполнитель в область GL и/или LCL морщин.
• Субъекту была сделана инъекция в область GL и/или LCL в течение 14 суток перед визитом для оценки исходного состояния.
• Присутствие в анамнезе опущения века или брови.
• Имеет сухость глаз, ярко выраженные мешки под глазами или отечность век по утрам.
• Имеет раковые или предраковые поражения, острые или хронические кожные заболевания, воспаления или связанные с ними состояния, включающие шрамы вблизи или в межбровной области.
• Перенесенные ранее пластические операции лица (например, блефаропластику, операцию вокруг глаз, подтяжку лица, подтяжку бровей, подтяжку век или операцию на бровях).
• Перенес лазерную или световую обработку лица, микрошлифовку кожи или поверхностный пилинг в течение 6 месяцев до начала отбора для участия в исследовании.
• Имеет в анамнезе паралич лицевого нерва.
• Применял препараты для топического нанесения, которые заявлены как обладающие способностью разглаживать морщины, в течение 7 суток перед введением исследуемого препарата или планирует применять такие препараты во время проведения исследования.
• Субъекту планирует делать на лице пластическую операцию или косметические процедуры в течение периода исследования.
• В течение 14 суток перед сутками, на которые назначены инъекции, наблюдаются симптомы, подобные симптомам ОРВИ.
• Принимает в настоящее время или принимал аминогликозидные антибиотики, курареподобные лекарственные средства, хинидин, сукцинилхолин, полимиксины, антихолинестеразы, сульфат магния или линкозамиды. Прием любых других назначаемых или не назначаемых препаратов допускается под ответственность Исследователя, при условии, что они не оказывают негативного влияния на безопасность субъекта или корректность данных исследования.
• Беременность или грудное вскармливание (женщины детородного возраста, участвующие в исследовании, перед рандомизацией должны иметь отрицательный результат теста на беременность [по сыворотке крови]).
• Имеет подтвержденный положительный анализ мочи на наличие лекарственных средств в организме, указывающий на наркотическую зависимость, включающий наличие: опиатов, барбитуратов, метаболитов кокаина, метадона, бензодиазепина, каннабиноида или амфетамина.
• Имеет в анамнезе алкоголизм или наркотическую зависимость или проявляет клинические признаки алкоголизма или наркотической зависимости.
• Имеет положительный результат анализа на гепатит В, включающий обнаружение HBsAG (поверхностного антигена гепатита В), положительный результат анализа на анти-HCV (антитела к вирусу гепатита С) и положительный результат анализа вирус иммунодефицита человека (ВИЧ).
• Имеет в анамнезе эпизоды затрудненного глотания, пневмопатии вдоха или выдоха.
• Имеет в анамнезе нарушения свертываемости крови.
• Имеет в анамнезе аутоиммунное заболевание, которое потенциально может влиять на результаты исследования.
• Имеет сочетанное заболевание в стадии обострения, которое потенциально может влиять на безопасность или оценку результатов исследования, примеры которого включают, без ограничений, клинически значимое сердечнососудистое, респираторное нарушение, нарушение печени/желчного пузыря, почечное, желудочно-кишечное, эндокринное, психическое или неврологическое нарушение.
• Имеет в анамнезе тяжелую миастению или другое нарушение нейротрансмиссии.
• Имеет нейромышечные нарушения, которые потенциально могут влиять на результаты субъекта.
• Уже получал любой из исследуемых продуктов в течение 90 суток перед отбором для участия в данном исследовании.
• Выкуривает более 10 сигарет или эквивалентное количество табака в сутки и не может прекратить курение во время пребывания в ОКФ.
• Имеет любое состояние (состояния), которое, по мнению Главного Исследователя (ГИ), нарушает безопасность субъекта или предотвращает участие субъекта в исследовании до полного его завершения.
• Не может установить надежное взаимодействие с исследователем.
• Субъект является вегетарианцем, веганом или придерживается ограничений питания по медицинским показаниям, которые не позволяют ему придерживаться стандартизованного меню, принятого в исследовании.
Отстранение субъектов от лечения или оценивания
Для определения тяжести НЯ применяли стандартную оценку токсичности в соответствии с общими терминологическими критериями нежелательных явлений, установленными Национальным институтом онкологии США (англ. National Cancer Institute Common Terminology Criteria, сокращенно NCI СТС, версия 4.0). Эти критерии применяют, поскольку они являются единственным доступным набором стандартных универсальных критериев, и было доказано, что они подходят для Фазы I исследований с участием здоровых добровольцев. Использовали нормальные показатели, получаемые в местной лаборатории. Для обеспечения достоверности и для исключения технических ошибок, аномальные лабораторные показатели и данные других испытаний всегда повторно проверяли перед вынесением оценки. При определении того, вызваны ли обнаруженные аномалии токсичным действием лекарственного средства, и при оценивании, если это возможно, всегда принимали во внимание суточные колебания лабораторных показателей и других измерений, а также исходный статус и состояние (например, синдром Жильбера).
Критерии СТС для НЯ представлены Степенями I-V с подробным клиническим описанием выраженности каждого НЯ в соответствии с общим руководством. Определения степеней приведены ниже:
Степень I: Слабая; отсутствуют или слабо выражены симптомы; только клинические или диагностические наблюдения; вмешательство не предполагается.
Степень II: Умеренная; показано минимальное, местное или неинвазивное вмешательство; ограничение подходящих по возрасту инструментальных ADL*.
* инструментальные ADL (англ. Activities of Daily Living) - инструментальные действия по самообслуживанию в повседневной жизни, к которым относится приготовление пищи, покупка продуктов или одежды, использование телефона, распоряжение денежными средствами и т.д.
Степень III: Выраженная или клинически значимая, но не несущая немедленной угрозы жизни; показана госпитализация или удлинение срока госпитализации; потеря трудоспособности; ограничение ADL самообслуживания**.
**ADL самообслуживания относятся к мытью, одеванию и раздеванию, самостоятельному приему пищи, пользованию туалетом, приему медицинских препаратов, но не к лежачим больным.
Степень IV: Последствия, угрожающие жизни; показано срочное вмешательство.
Степень V: Смертельный исход, вызванный НЯ.
Критерии СТС для НЯ и их интерпретация согласуются со стандартными оценками интенсивности НЯ при клинических испытаниях: Степень I: слабая, Степень II: умеренная, Степень III: выраженная или клинически значимая, но не несущая немедленной угрозы жизни, могут наблюдаться серьезные нежелательные явления (СНЯ)/предполагаемая непредвиденная серьезная нежелательная реакция (сокращенно "ПНСНР"). Степени IV и V связаны с проявлением СНЯ/ПНСНР.
Исследование необходимо прекратить, если возникают некоторые нежелательные явления, которые с большой долей вероятности вызваны ИМП (исследуемым медицинским продуктом, англ. investigational medicinal product, сокр. IMP).
Выход субъектов из исследования
В соответствии с Хельсинкской декларацией, субъекты могут выйти из исследования в любе время, но, если обработка или дозирование уже произведены, то должны быть предприняты все меры для продолжения оценивания состояния с целью обеспечения безопасности субъекта. Конкретные причины для выхода субъекта из настоящего исследования включали:
• Добровольное решение субъекта прекратить участие в исследовании, поскольку субъект имеет право в любое время прекратить свое участие в исследовании.
• Выраженное несогласие или принципиальное отклонение от протокола по мнению Исследователя и/или Спонсора.
• Некорректное включение, т.е. при последующем рассмотрении оказалось, что на момент включения субъект не соответствовал квалификационным критериям, требуемым для исследования.
• Другие причины, выдвигаемые ГИ.
Субъекты могли в любое время без ущерба выйти из исследования (выход из исследования по соглашению). Таких субъектов всегда спрашивали о причине (причинах) и наличии каких-либо НЯ. Если это было возможно, то субъекты, вышедшие из исследования после дозирования и до выполнения визита на неделе 4, должны были посетить прием ГИ или уполномоченного лица и пройти оценивание и процедуры, запланированные для следующего визита. Во всех случаях. Где это было возможно, НЯ следовало отслеживать до их полного исчезновения, возвращения к исходному состоянию или стабилизации, если была установлена причина, не связанная с исследованием.
Замена субъектов
Для оценки безопасности и переносимости заданной дозы QM1114-DP или плацебо (7 суток после обработки) в каждой когорте должно быть запланированное количество субъектов. Если количество заполняющих группу субъектов было меньше, чем минимум, требующийся для данной когорты, для принятия решения об увеличении дозы спонсор и ГИ могли постановить, что этих субъектов нужно заменить, и перед следующим увеличением дозы этих субъектов соответственно подготавливают и оценивают.
Лечение
Получаемое лечение
Субъектов рандомизировали для получения одной обработки исследуемым лекарственным средством (QM1114-DP или плацебо). Каждая обработка включала 11 инъекций (одну инъекцию на одну точку ввода инъекции) равного объема (100 мкл) в область GL (пять инъекций) и область LCL (три инъекции в каждую из сторон лица). В области GL пять точек ввода инъекций включали две инъекции в каждую мышцу, сморщивающую бровь, и одну инъекцию в мышцу гордецов. В области LCL места ввода инъекций уточняли в соответствии с расположением морщин LCL у конкретного субъекта. В зависимости от расположения морщин у конкретных субъектов, если морщины располагались в области LCL выше и ниже латеральной спайки век, то инъекции вводили, как показано на Фиг. 12А. В альтернативном варианте, если морщины в области LCL конкретного субъекта в основном находились ниже латеральной спайки век, то инъекции вводили, как показано на Фиг. 12В. Во всех случаях инъекции вводили в наружную часть круговой мышцы глаза и, если это было возможно, на расстоянии приблизительно 1-2 см от глазничного валика.
Распределение субъектов по терапевтическим группам
Субъектов, которые подписали информированное согласие на исследование и затем прошли отборочные процедуры, в течение соответствующего периода идентифицировали с помощью уникального номера RPL ID (идентификационного номера из базы добровольцев RPL) и скринингового номера.
Номера субъектов присваивали субъектам, отвечающим квалификационным критериям, перед введением доз утром Суток 1. Субъектам присваивали номера от 301 до 308 (Когорта 8) и от 321 до 328 (Когорта 9). Номера присваивали субъектам последовательно, и эти номера соответствовали номерам в созданном компьютером рандомизованном списке, который определял последовательность получения лечения. Если субъект прекращал свое участие в исследовании, то номер субъекта больше не использовали, и субъекту не разрешали вновь вступать в ряды участников исследования.
Выбор доз в исследовании
Уже имеющиеся в наличии клинические данные считали репрезентативными для выбора доз QM1114-DP для введения в область GL. Выбор доз для первого исследования QM1114-DP с участием человека был основан на клиническом опыте лечения тех же участков лица, но учитывал собранные к моменту начала анализа данные фармакологических и токсикологических исследований, которые позволяли предполагать эквивалентный принцип действия, общие ответные реакции и обратимый эффект. Стандартная клиническая доза при лечении GL составляет пять инъекций, содержащих 10 единиц в одной инъекции (всего 50 единиц, также называемых единицами Спейвуд, англ. Speywood Unit, сокращено "sU"). В Примере 1 (только GL), в качестве предосторожности предлагаемая начальная доза QM1114-DP составляла 2 единицы в одной инъекции (всего 10 единиц), после чего дозу увеличивали (после принятия решений SRC, англ. Safety Review Committee -Комитет по рассмотрению безопасности) в три этапа до достижения 5 единиц в одной инъекции (всего 25 единиц), затем 10 единиц в одной инъекции (всего 50 единиц) и затем 15 единиц в одной инъекции (всего 75 единиц).
Начальная доза для лечения LCL (Пример 2) составляла 5 единиц в одной инъекции (всего 30 единиц), после чего дозу увеличивали (после принятия решений SRC) до 10 единиц в одной инъекции (всего 60 единиц) и затем до 15 единиц в одной инъекции (всего 90 единиц). Максимальную дозу, принятую в исследовании лечения LCL, выбирали на основании проведенных ранее исследований. В проведенных ранее исследованиях BoNT для лечения LCL в каждый участок вводили 15, 30 и 45 единиц (всего вводили 30, 60 и 90 единиц). Частота возникновения связанных с лечением НЯ была сходной при всех исследованных дозировках.
Выбор и время введения доз
В этом исследовании субъектам однократно вводили 110 или 140 единиц QM1114-DP или плацебо утром Суток 1 Исследования.
Процедуры исследования
Процедуры отбора, сутки, входящие в исследование, и последующие процедуры указаны в Таблице 24.
Все измерения, проводимые в исследовании, описаны ниже.
Если на основании просмотра данных безопасности по конкретному субъекту возникали какие-либо опасения за его безопасность, то это могло быть показанием для дополнительного определения показателей жизнедеятельности, записи ЭКГ и/или лабораторного анализа образцов для подтверждения безопасности. Общий объем крови, отбираемой для исследования у одного субъекта, не должен превышать 300 мл.
Показатели жизнедеятельности
Кровяное давление, частота сердечных сокращений, температура в наружном слуховом проходе и частота дыхательных движений
Артериальное давление в положении лежа на спине (КД, сокр. от "кровяное давление") и частоту сердечных сокращений (ЧСС) измеряли с помощью полуавтоматического устройства, записывающего КД (мониторы Critikon Dinamap®) с подходящим размером манжеты. Для измерения частоты дыхательных движений, перед измерением субъекты должны были находиться в покое в положении лежа на спине в течение по меньшей мере 10 минут. Расписание индивидуальных измерений указано в алгоритме исследования (Таблица 24). Время проведения всех измерений, выполняемых в процессе исследования, могло быть изменено в зависимости от текущего состояния данных безопасности и переносимости. Температуру тела (в наружном слуховом проходе) определяли (одно измерение) в градусах Цельсия с помощью автоматического термометра согласно расписанию, указанному в алгоритме исследования (Таблица 24). Для обеспечения безопасности по инициативе ГИ или уполномоченного лица могут быть проведены дополнительные измерения температуры.
Снятие ЭКГ
ЭКГ регистрировали в 12-ти отведениях трижды с помощью устройства GE Marquette MAC1200®/MAC1200ST®, соединенного фиксированным сетевым соединением с кардиологической информационной системой MUSE® (MUSE). Все зарегистрированные во время исследования ЭКГ хранили в электронном виде в информационной системе MUSE. Для любых целей, не являющихся оценкой безопасности, имеющими силу считались только ЭКГ, записанные в электронном виде. Распечатки ЭКГ вкладывали в ИРК (индивидуальную регистрационную карту) субъекта для проверки медицинской безопасности.
В тех случаях, когда это возможно, ЭКГ всех субъектов регистрировали одним и тем же регистрирующим устройством. Каждое устройство для регистрации ЭКГ настраивали в соответствии с требуемыми техническими условиями, и каждое устройство содержало информацию, необходимую для идентификации записей. Каждая запись ЭКГ могла быть легко идентифицирована (ID субъекта, запланированное время ввода доз препарата и реальное время регистрации ЭКГ).
Запись ЭКГ в 12-ти отведениях проводили в соответствии с расписанием, указанным в алгоритме исследования (Таблица 24), после того, как субъекты находились в состоянии покоя в положении лежа на спине в течение по меньшей мере 10 минут. Субъектов просили не изменять позу во время записи ЭКГ, и персонал клиники следил за тем, чтобы во время записи ЭКГ субъекты находились в бодрствующем состоянии.
Все записанные ЭКГ оперативно просматривал врач-исследователь, и результаты анализа записывались в ИРК. Если на каком-либо этапе у субъекта наблюдали аномальную ЭКГ, то проводили дополнительные исследования для безопасности (включающие применение оборудования Holter с 5 или 12 отведениями), и, при необходимости, аномалию прослеживали до ее полного устранения.
Время проведения всех измерений, выполняемых в процессе исследования, может быть изменено в соответствии с оперативными просмотрами данных безопасности и переносимости.
Клинические лабораторные определения безопасности
Клинические лабораторные параметры безопасности представлены ниже в Таблице 25.
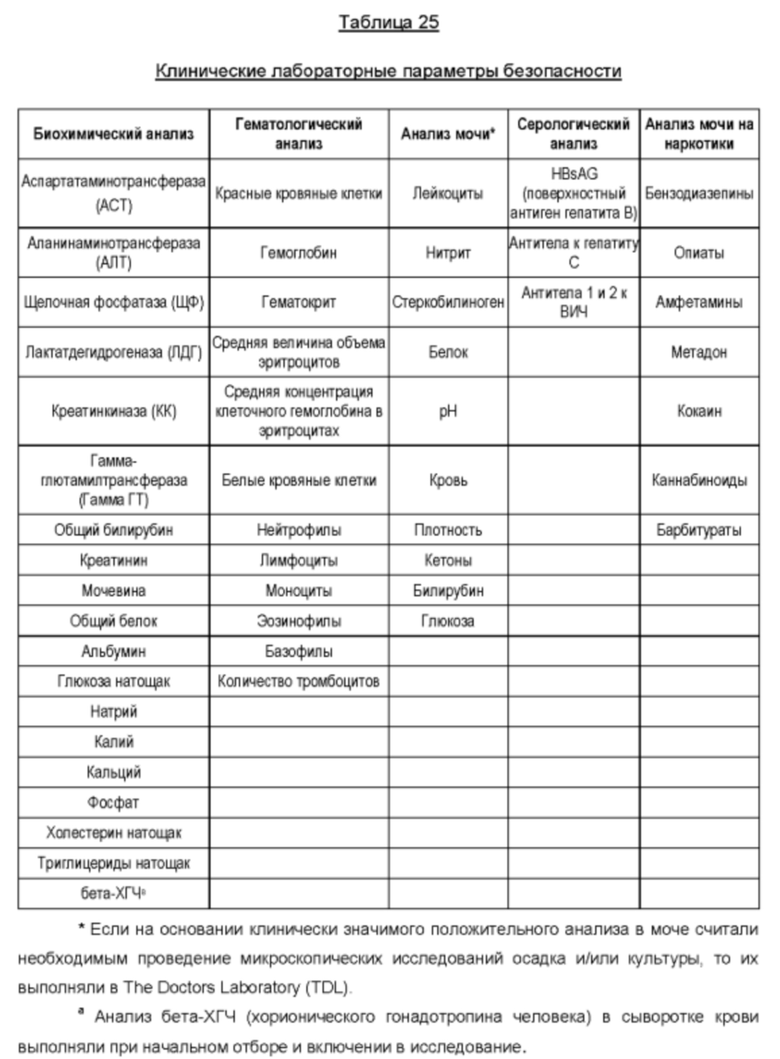
Гематологический и биохимический анализ
Образцы крови для определения гематологических и биохимических параметров отбирали в соответствии с расписанием, указанным в алгоритме исследования (Таблица 24). Дата и время отбора образцов записывали в соответствующую ИРК. Анализы проводили в лаборатории TDL обычными способами. Образцы крови для определения стандартных гематологических параметров отбирали в пробирки емкостью 4 мл, содержащие КЗ ЭДТА, и образцы крови для определения стандартных биохимических параметров отбирали в пробирки для отделения сыворотки (англ. Serum Separator Tubes, сокращенно SST) емкостью 5 мл.
Если результаты лабораторных анализов, которые, как предполагалось, имели клиническую значимость, выходили за пределы нормального диапазона значений, то такие анализы проводили повторно. Субъекты, результаты которых, имеющие предполагаемую клиническую значимость, подтверждались при повторном анализе, не включались в исследование, или, если они уже были включены, могли быть освобождены от последующего участия в исследовании, и/или их могли наблюдать до нормализации значений или до тех пор, пока Исследователь или уполномоченное лицо считали, что это необходимо.
Серологический анализ
Серологический анализ выполняли, как указано в алгоритме исследования (Таблица 24). На момент отборочного визита у всех субъектов проверяли все параметры, перечисленные в Таблице 25. Это было сделано для безопасности исследовательского персонала, и результаты проверки не включали в базу данных исследования. Если в каком-либо из указанных испытаний было обнаружено, что субъект (субъекты) имеет (имеют) положительные результаты, то такого субъекта подвергали дополнительным исследованиям и процедурам и не включали в исследование. Серологические анализы проводили на тех же образцах крови, которые использовали для клинического химического анализа (собранные в SST пробирки объемом 5 мл). Образцы анализировали в лаборатории TDL.
бета-ХГЧ
Для исключения возможной беременности в тех случаях, когда ее можно было бы ожидать, в образцах сыворотки крови анализировали бета-ХГЧ, как описано в Таблице 24. Любого субъекта с положительным результатом теста на беременность исключали или выводили из исследования.
Анализ мочи
Образцы мочи для анализа параметров мочи отбирали в соответствии с расписанием, указанным в алгоритме исследования (Таблица 24). В анализе мочи с помощью тест-полосок определяли параметры, указанные в Таблице 25 RPL. При необходимости, в случае клинически значимого положительного результата в лаборатории TDL проводили микроскопическое исследование осадка и/или культуры.
Анализ на наркотики
Анализ мочи на наркотики выполняли в RPL в соответствии с расписанием, указанным в Таблице 24. Если субъект не проходил испытание на наркотическую зависимость, то его/ее исключали из исследования. Повторную проверку на наркотики проводили только в тех случаях, когда были методологические основания полагать, что результат является ложноположительным. В тех случаях, если получаемые положительные результаты, близкие к граничным, не могли быть объяснены предшествующим состоянием, такие результаты считались положительными, и субъекта исключали из исследования. Если обнаруживали, что субъект имеет положительный результат из-за приема медикамента, например, противопростудных/противогриппозных препаратов, проводили повторный тест на наркотики, если субъект все еще находился в пределах скринингового окна. Результаты этих анализов не размещали в базе данных.
Проба на алкоголь в выдыхаемом воздухе
Пробу на алкоголь в выдыхаемом воздухе получали с помощью устройства для измерения уровня алкоголя (расписание анализов указано в алгоритме исследования в Таблице 24). Результаты этого анализа не вносили в базу данных клинического исследования. Если субъект имел положительный результат пробы на алкоголь в выдыхаемом воздухе, то его исключали из исследования.
Данные физического обследования, рост и масса тела
Расписание индивидуальных обследований указано в алгоритме исследования (Таблица 24). Данные физического обследования на момент отбора и в последующие периоды (сутки 28) включали следующие данные полного физического обследования: общее состояние, состояние кожи, головы, шеи, лимфатических узлов, щитовидной железы, органов брюшной полости, опорно-двигательной системы, сердечно-сосудистой системы, дыхательной системы и нервной системы.
Физическое обследование, производимое в Сутки -1, представляло собой поверхностное обследование, нацеленное на обнаружение любых изменений, которые могли произойти с момента отбора.
Рост определяли в сантиметрах, а массу - в килограммах. Во время измерений субъекты были одеты легко, без обуви, и для всех измерений применяли калиброванные устройства. Индекс массы тела (ИМТ) вычисляли, исходя из роста и массы тела.
Данные фокусированного физического обследования
Фокусированное физическое обследование глаз и лица (расписание - см. Таблицу 24) выполняли с использованием стандартизованной системы оценок, в которой оценивали местную температуру, зуд, боль, отек/уплотнение, покраснение и гематомы. Была принята следующая система оценок:
• Слабые изменения или
• Умеренные изменения.
Оценка эффективности
Оценку эффективности проводили в соответствии с расписанием, указанным в алгоритме исследования (Таблица 24).
Оценка выраженности морщин Исследователем и субъектом
В течение каждого визита Исследователь и субъект независимо оценивали выраженность морщин субъекта "в состоянии покоя" и "при максимальном нахмуривании" (показатель GL) и "при самой широкой улыбке" (показатель LCL). Исследователь оценивал морщины согласно шкале MAS CL в динамике и в состоянии покоя и/или MAS LCL в динамике и в состоянии покоя в соответствии со следующими критериями (используя для оценивания фотографии с высоким разрешением):
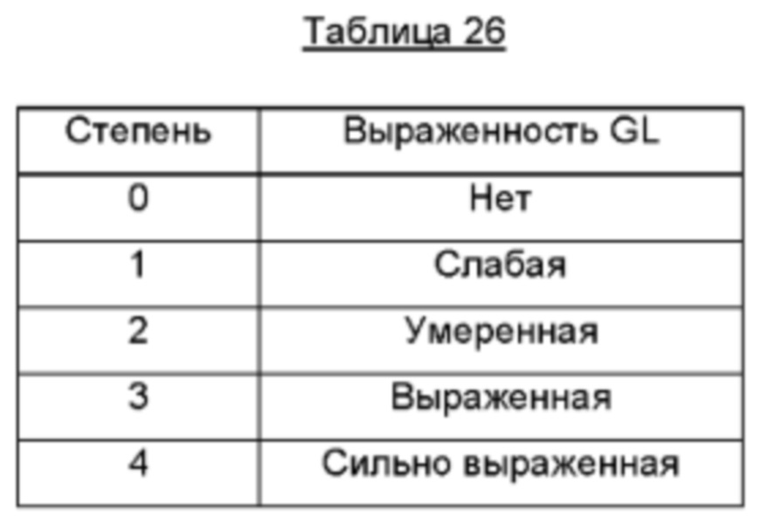
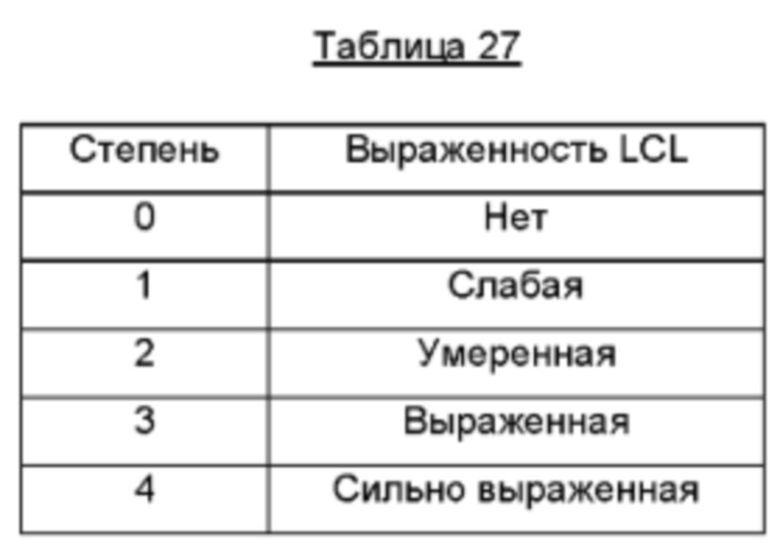
Оценка удовлетворенности субъекта
Во время каждого визита после лечения субъект оценивал степень своей удовлетворенности лечением по следующей 4-балльной шкале:
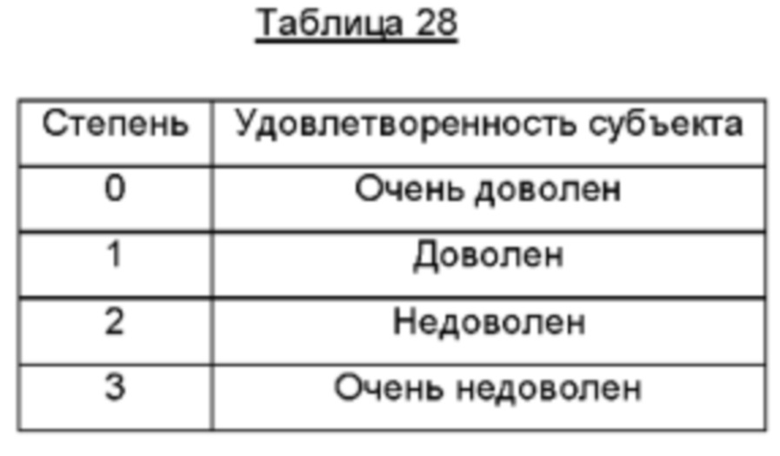
Фотография
В соответствии с расписанием, указанным в алгоритме исследования (Таблица 24), были сделаны стандартные цифровые фотографии. Фотографии использовали только для иллюстрации эффекта применения ИМП (исследуемого медицинского продукта) и не использовали для оценки эффективности. Субъектов просили удалять декоративную косметику перед приходом в клинику или в самой клинике перед фотографированием.
Делали следующие фотографии:
• Анфас в состоянии покоя (без улыбки); и
• Фотографии области GL и/или области LCL (одна при максимальном нахмуривании показатель [GL] или при максимальной улыбке [показатель LCL] и одна в состоянии покоя).
Отбор образцов
Клинические исследования иммуногенности: Анализ для определения нейтрализующих антител к QM1114-DP будет разработан позже. Образцы крови отбирали согласно расписанию, указанному в алгоритме исследования (Таблица 24), для определения количества антител в будущем, после создания соответствующего анализа.
Нежелательные явления
Ниже приведены определения НЯ, нежелательных реакций на препарат (НРП), СНЯ и предполагаемых непредвиденных серьезных нежелательных реакций (ПНСНР). Очень важно, чтобы весь персонал, участвующий в проведении клинического исследования, был ознакомлен с данным разделом.
Определения
Нежелательное явление (НЯ) - НЯ состоит в развитии нежелательного медицинского нарушения или ухудшения уже существующего медицинского состояния после или во время воздействия фармацевтического продукта, независимо от того, связно ли это явление с продуктом. Нежелательными медицинскими нарушениями могут быть симптомы (например, тошнота, боль в груди), признаки (например, тахикардия, увеличенная печень) или аномальные результаты исследования (например, результаты лабораторных анализов, ЭКГ). В клинических исследованиях НЯ может включать нежелательное медицинское нарушение, возникающее в любое время, начиная с момента подписания информированного согласия до окончания участия в исследовании, т.е. до вывода субъекта из исследования или в альтернативном варианте до завершения исследования (Сутки 28).
Обусловленность НЯ (т.е. причинно-следственную связь между исследуемым лечением и НЯ) оценивал Исследователь (Исследователи), который при заполнении соответствующей индивидуальной регистрационной формы должен отвечать "да" или "нет" на вопрос "Считаете ли Вы, что имеется обоснованная возможность того, что этот эпизод мог быть вызван одним из следующих: исследуемым препаратом; другим препаратом?".
Следует отметить, что также необходимо сообщать о СНЯ, которые могут быть связаны с любыми процедурами исследования.
Нежелательная реакция на препарат (НРП) - НРП представляет собой любое НЯ, при котором причинная взаимосвязь с ИМП имеет по меньшей мере обоснованную возможность.
Серьезное нежелательное явление (СНЯ) - СНЯ представляет собой НЯ, возникающее в любой фазе исследования (т.е. во время вводного периода, лечения, периода выведения и/или позднее) и при любой дозе ИМП или плацебо, которое соответствует одному или более из следующих критериев:
• Приводит к смертельному исходу;
• Угрожает жизни;
• Требует пребывания пациента в стационаре или продолжения текущей госпитализации;
• Приводит к трудноизлечимому или значительному нарушению функций или нетрудоспособности;
• Является врожденной аномалией или врожденным дефектом;
• Является важным медицинским эпизодом, который может угрожать субъекту или может требовать медицинского вмешательства для предотвращения одного из исходов, перечисленных выше.
Обусловленность СНЯ (т.е. причинно-следственную связь между исследуемым лечением и СНЯ) оценивали также, как и несерьезные НЯ. Следует отметить, что также необходимо сообщать о СНЯ, которые могут быть связаны с любыми процедурами исследования.
Предполагаемая непредвиденная серьезная нежелательная реакция (ПНСНР) - ПНСНР представляет собой любое СНЯ, при котором причинная взаимосвязь с ИМП имеет по меньшей мере обоснованную возможность, но это явление не указано в Брошюре Исследователя и/или в Сводных данных о Характеристиках Продукта.
Соответствие измерений
Все проводимые в этом исследовании определения эффективности и безопасности были стандартными, широко применяемыми и имеющими подтвержденную надежность и точность.
Исследуемые субъекты
Для этого исследования были отобраны 16 здоровых субъектов, 6 мужчин и 10 женщин, которых рандомизировали и которым вводили дозы препарата в соответствии с протоколом клинического исследования (ПКИ). Все участники полностью завершили исследование и прошли анализы для оценки безопасности и эффективности.
Описательная статистика демографических параметров представлена ниже в Таблице 29. Четырнадцать (14) субъектов имели европейскую внешность, один субъект имел азиатскую внешность, и один субъект относился к другой смешанной расе. Возраст субъектов составлял от 35 до 57 лет (включительно). Масса тела составляла от 55,1 до 103,7 кг, и ИМТ составлял от 20,5 до 32,7 кг/м2 для всех субъектов. При включении в исследование в анамнезе субъектов не было никаких клинически значимых событий медицинского или хирургического вмешательства. Каких-либо существенных отклонений в данных физического обследования конкретных субъектов во время исследования также не было обнаружено.

Оценка эффективности
Выборка для анализа эффективности состояла из всех субъектов (16 субъектов), которые получали однократную дозу QM1114-DP или Плацебо и для которых имелись данные эффективности после введения дозы.
Одной из задач настоящего исследования была оценка эффективности действия каждой из доз QM1114-DP, направленного на временное улучшение состояния лицевых морщин в межбровной и боковой периорбитальной области, обрабатываемых в комбинации. На Фиг. 15 и Фиг. 16 представлены средние оценки состояния морщин в межбровной и боковой периорбитальной области (на левой и на правой сторонах) выставляемые Исследователем и субъектом в течение периода времени лицам в группе, получавшей лечение, причем оценки выставлялись в состоянии покоя, при максимальном нахмуривании (GL) и при самой широкой улыбке (LCL). Анализ оценок выраженности морщин Исследователем и субъектом в состоянии покоя, при максимальном нахмуривании (GL) и при самой широкой улыбке (LCL), проведенный согласно смешанной модели, представлен в Таблице 30, Таблице 31 (Оценка Исследователем), Таблице 32 и Таблице 33 (Оценка субъектом). Эффективность оценивали по обоснованной шкале MAS, которую Исследователь и субъект использовали для оценивания выраженности морщин. Применяли следующие степени выраженности морщин: 0 (нет), 1 (слабая), 2 (умеренная), 3 (выраженная) и 4 (сильно выраженная).
Выраженность морщин в состоянии покоя Межбровные морщины (GL):
Данные по эффективности показывают, что после введения QM1114-DP, по оценке Исследователя и субъекта в сравнении с группой плацебо, выраженность морщин в межбровной области в состоянии покоя снижалась в группах, получавших действующее лечение (Фиг. 15 и Фиг. 16). Оценка выраженности морщин Исследователем указывала на то, что максимальный эффект в состоянии покоя был достигнут на Сутки 28 в группе, получавшей 50/60 единиц QM1114-DP (средняя оценка 0,5 по шкале MAS), в то время как в группе, получавшей 50/90 единиц QM1114-DP, максимальный эффект был достигнут быстрее (между Сутками 3 и Сутками 14), и средняя оценка по шкале MAS составила от 0,5 до 0,7. В Сутки 28 в группе, получавшей 50/90 единиц QM1114-DP, средняя оценка по шкале MAS составила 0,7. При оценке субъектом выраженности морщин в состоянии покоя максимальный эффект был достигнут к Суткам 21 (средняя оценка 0,2 по шкале MAS), и в группе, получавшей 50/60 единиц QM1114-DP, он сохранялся до Суток 28. В группе, получавшей 50/90 единиц QM1114-DP, максимальный эффект был достигнут на Сутки 28 (средняя оценка 0,3 по шкале MAS).
Боковые периорбитальные морщины (LCL):
После введения QM1114-DP, выраженность LCL в состоянии покоя, по оценке Исследователя и субъекта, снижалась в группах, получавших действующее лечение, в сравнении с группой плацебо (Фиг. 15 и Фиг. 16). Оценка выраженности морщин Исследователем указывала на то, что в группах, получавших лечение, максимальный эффект достигался в период времени между Сутками 14 и Сутками 28, и средние оценки составляли от 0,7 до 0,8 (LCL слева) и 0,8 (LCL справа) по шкале MAS. Аналогичные тенденции к снижению были отмечены в оценках выраженности морщин субъектом. В группах, получавших лечение, максимальный эффект достигался в период времени между Сутками 14 и Сутками 21, и средние оценки составляли от 0,5 до 0,8 (LCL слева) и 0,7 до 0,8 (LCL справа) по шкале MAS.
Выраженность морщин при максимальном нахмуривании (показатель GL)
При максимальном нахмуривании, выраженность морщин в межбровной области, по оценке Исследователя и субъекта, в группах, получавших действующую лечение, снижалась, начиная с Суток 2 (Фиг. 15 и Фиг. 16). Максимальный эффект (средняя оценка 0,7 по шкале MAS) в группе, получавшей 50/60 единиц QM1114-DP, по оценке Исследователя, был достигнут к Суткам 14, и этот эффект сохранялся до Суток 28 (средняя оценка 0,7 по шкале MAS). В группе, получавшей 50/90 единиц QM1114-DP, максимальный эффект был достигнут к Суткам 7, средняя оценка по шкале MAS GL в динамике составляла 0,5, и эффект сохранялся до Суток 28. По оценке выраженности морщин субъектом, максимальный эффект (средняя оценка 0,5 по шкале MAS) в группе, получавшей 50/60 единиц QM1114-DP, был достигнут к Суткам 14 и сохранялся до Суток 28 (средняя оценка 0,5 по шкале MAS). В группе, получавшей 50/90 единиц QM1114-DP, промежуток времени до достижения максимального эффекта был больше (Сутки 28, средняя оценка 0,3 по шкале MAS).
Выраженность морщин при самой широкой улыбке (показатель LCL)
Как и в случае GL, аналогичные тенденции к снижению также наблюдали в оценках Исследователем и субъектом выраженности LCL при самой широкой улыбке (Фиг. 15 и Фиг. 16). У субъектов в группе, получавшей 50/60 единиц QM1114-DP, максимальный эффект в разглаживании LCL слева по оценке Исследователя был достигнут к Суткам 14 (средняя оценка 0,8 по шкале MAS) и к Суткам 21 для LCL справа (средняя оценка 1,0 по шкале MAS), и некоторый эффект сохранялся до Суток 28 (средняя оценка 0,8 [LCL слева] и 1,0 [LCL справа] по шкале MAS). Для сравнения, максимальный эффект в группе, получавшей 50/90 единиц QM1114-DP, был достигнут на Сутки 7 (средняя оценка 1,0 [LCL слева] и 1,0 [LCL справа] по шкале MAS), и эффект сохранялся до Суток 28 (средняя оценка 1,0 [LCL слева] и 1,0 [LCL справа] по шкале MAS). Аналогичная тенденция к снижению также наблюдалась в оценках выраженности морщин субъектом (Фиг. 16). Максимальное воздействие на выраженность морщин LCL слева у субъектов в группе, получавшей 50/60 единиц QM1114-DP, наблюдали на Сутки 14 (средняя оценка 0,8 по шкале MAS), и некоторый эффект сохранялся до Суток 28 (средняя оценка 1,2 по шкале MAS). По оценкам морщин LCL справа, максимальный эффект был достигнут на Сутки 14 (средняя оценка 1,0 по шкале MAS), и некоторый эффект сохранялся до Суток 28 (средняя оценка 1,3 по шкале MAS). У субъектов в группе, получавшей 50/90 единиц QM1114-DP, максимальное воздействие на LCL на левой стороне лица было достигнуто на Сутки 21 (средняя оценка 1,0 по шкале MAS), и максимальное воздействие на LCL на правой стороне лица было достигнуто на Сутки 28 (средняя оценка 0,8 по шкале MAS).
Оценка удовлетворенности субъекта
В каждый визит после проведения лечения субъект оценивал свою степень удовлетворенности лечением по четырехбалльной шкале. Шкала включала следующие степени удовлетворенности: 0 (очень доволен), 1 (доволен), 2 (недоволен) и 3 (очень недоволен).
Оценка удовлетворенности субъекта показывает, что большинство субъектов в группах, получавших 50/60 и 50/90 единиц QM1114-DP, были либо очень довольны (0), либо довольны (1) лечением, начиная с Суток 2 и до Суток 28. Некоторые субъекты, такие как субъект 301 (с Суток 7 до Суток 28) и субъект 305 (на момент Суток 28) в группе, получавшей 50/60 единиц, были либо очень недовольны (3), либо недовольны (2) лечением. Кроме того, оказалось, что один субъект (субъект 324) из группы, получавшей 50/90 единиц, был очень недоволен (на Сутки 21) или недоволен (на Сутки 28) лечением. Возможным фактором, который внес свой вклад в неудовлетворенность субъектов, было то, что у субъектов 301, 305 и 324 проявились НЯ. В группе плацебо, начиная с Суток 14 и до Суток 28, недовольны лечением оказались три субъекта.
Анализ в рамках смешанной модели
Изменения по сравнению с исходным состоянием, по оценке выраженности морщин Исследователем и субъектам по отдельности в состоянии покоя и при максимальном нахмуривании/самой широкой улыбке, исследовали в рамках модели повторных измерений (смешанной модели). Оценки выраженности морщин Исследователем (различия при лечении различными дозами в разные дни) представлены в Таблицах 30-31 (в состоянии покоя и при максимальном нахмуривании/самой широкой улыбке), и оценки выраженности морщин субъектом (различия при лечении различными дозами в разные дни) представлены в Таблицах 32-33 (в состоянии покоя и при максимальном нахмуривании/самой широкой улыбке). Анализируемой и представленной в этом разделе переменной является изменение выраженности морщин в состоянии покоя и при самой широкой улыбке по сравнению с исходным состоянием по оценке Исследователем и субъектами по отдельности.
Межбровные морщины (GL):
Оценка Исследователем морщин при максимальном нахмуривании показывает, что, начиная с Суток 2 и до Суток 28, изменения средних оценок по шкале MAS по сравнению с исходным состоянием в группе, получавшей 50/60 единиц QM1114-DP, имеют статистически значимое отличие от оценок в группе плацебо. Значимый результат (Р<0,05) также получали, начиная с Суток 3 и до Суток 28, при сравнении изменений относительно средних оценок исходного состояния по шкале MAS в группе, получавшей 50/90 единиц QM1114-DP, с изменениями в группе плацебо (Таблица 30А). Аналогичная тенденция также наблюдалась в оценках субъектом морщин при максимальном нахмуривании (Таблица 32А), причем статистически значимые отличия от плацебо наблюдали, начиная с Суток 2 или Суток 3 и до Суток 28, при обеих дозировках QM1114-DP. По оценкам морщин в состоянии покоя Исследователем, изменения по сравнению с исходным состоянием средних оценок по шкале MAS в течение периода времени в группе, получавшей 50/60 единиц QM1114-DP, незначительно отличались оценок в группе плацебо (Таблица 31А). Однако изменения средних оценок по шкале MAS по сравнению с исходным состоянием в группе, получавшей 50/90 единиц QM1114-DP, в Сутки 3, Сутки 21 и Сутки 28 имели статистически значимое отличие (Р<0,05) от оценок группы плацебо (Таблица 31А). Изменение средних оценок морщин в состоянии покоя по шкале MAS, выставленных субъектом, относительно оценок исходного состояния были статистически значимыми (Р<0,05), начиная с Суток 7, и оставались такими до Суток 28 в обеих группах, получавших 50/60 единиц и 50/90 QM1114-DP, по сравнению с оценками в группе плацебо (Таблица 33А).
Боковые периорбитальные морщины (LCL):
Оценка Исследователем состояния морщин в левой боковой периорбитальной области при самой широкой улыбке показывает, что, начиная с Суток 2 и до Суток 28, изменения средних оценок по шкале MAS по сравнению с исходным состоянием в группе, получавшей 50/60 единиц QM1114-DP, показывают статистически значимое отличие от оценок в группе плацебо. Кроме того, за исключением Суток 2 и Суток 14, такую же тенденцию также наблюдали в группе, получавшей 50/90 единиц QM1114-DP (Таблица 30В). Напротив, эта тенденция не была столь же очевидной при оценке Исследователем морщин в правой боковой периорбитальной области при самой широкой улыбке. При оценке Исследователем морщин LCL справа, статистически значимые отличия от оценок плацебо были выявлены в Сутки 7 и Сутки 28 в группе, получавшей 50/60 единиц QM1114-DP. В группе, получавшей 50/90 единиц QM1114-DP, статистически значимые отличия от плацебо отмечались, начиная с Суток 3 и до Суток 7 и на Сутки 28 (Таблица 30С). При оценке морщин LCL слева при самой широкой улыбке субъектом, статистически значимые отличия (Р<0,05) от плацебо отмечались, начиная с Суток 14 и до Суток 28, в группе, получавшей 50/60 единиц QM1114-DP, и начиная с Суток 7 до Суток 28 в группе, получавшей 50/90 единиц QM1114-DP (Таблица 32В). Аналогичные различия в состоянии LCL справа при самой широкой улыбке также отмечались в группах, получавших лечение. По оценкам LCL справа, изменение по сравнению с исходным состоянием средних оценок по шкале MAS в группе, получавшей 50/60 единиц QM1114-DP, имело статистически значимое отличие от плацебо, начиная с Суток 14 до Суток 28 и начиная с Суток 3 до Суток 28, в группе, получавшей 50/90 единиц QM1114-DP (Таблица 32С).
Оценка Исследователем морщин боковой периорбитальной области на левой стороне лица в состоянии покоя показывает, что изменения средних оценок по шкале MAS по сравнению с исходным состоянием, начиная с недели 3 (Сутки 21) до недели 4 (Сутки 28), в группах, получавших 50/60 и 50/90 единиц QM1114-DP, имеют статистически значимое отличие (Р<0,05) от оценок группы плацебо (Таблица 31В). Однако оценка Исследователем морщин боковой периорбитальной области на правой стороне лица в состоянии покоя показывает, что изменения средних оценок по шкале MAS по сравнению с исходным состоянием в группах, получавших 50/60 и 50/90 единиц QM1114-DP значительно отличается от изменения оценок плацебо, только начиная с недели 4 (Сутки 28) (Таблица 31С).
При оценке морщин в состоянии покоя субъектом, за исключением Суток 7, статистически значимые результаты сравнения субъектов группы, получающей 50/90 единиц QM1114-DP, с субъектами группы плацебо (Р<0,05) были получены при оценивании LCL слева, начиная с Суток 2 и до Суток 28 (Таблица 33В). Однако, при сравнении средних оценок LCL слева по шкале MAS у субъектов группы, получающей 50/60 единиц QM1114-DP, с оценками группы плацебо статистически значимых изменений по сравнению с исходным состоянием выявлено не было (Таблица 33В). Выставленные субъектом средние оценки морщин LCL справа по шкале MAS в состоянии покоя по сравнению с исходным состоянием в группе, получавшей 50/60 единиц QM1114-DP, не имели статистически значимых отличий от оценок группы плацебо (Таблица 7С). Напротив, статистически значимые отличия по шкале MAS от группы плацебо были выявлены в Сутки 21 и Сутки 28 в группе, получавшей 50/90 единиц QM1114-DP (Таблица 33С).
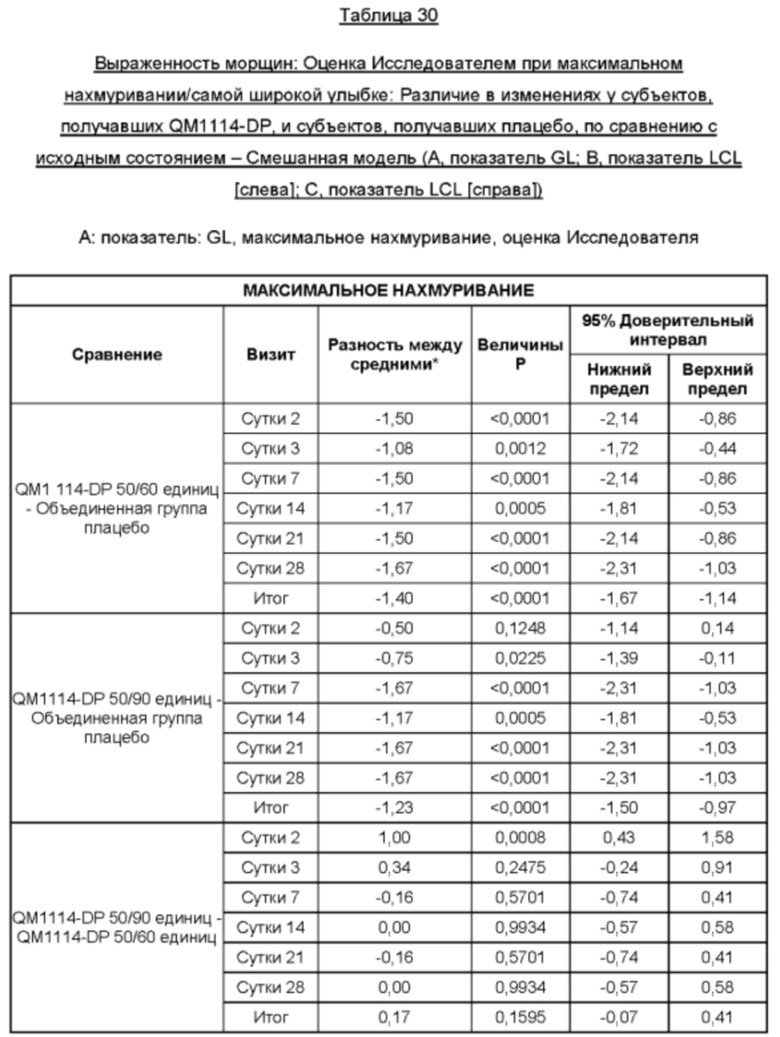
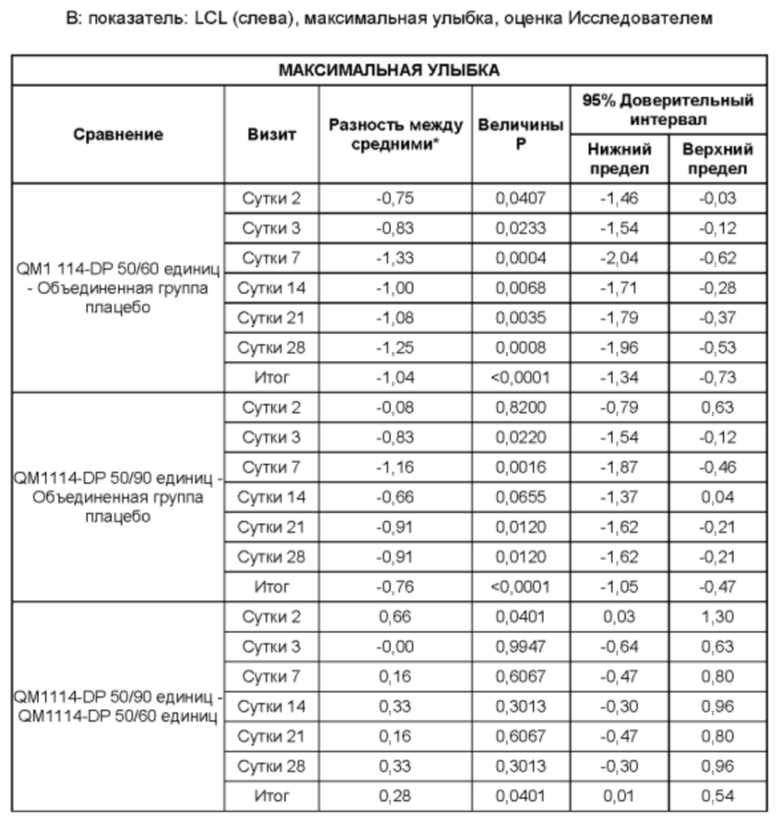
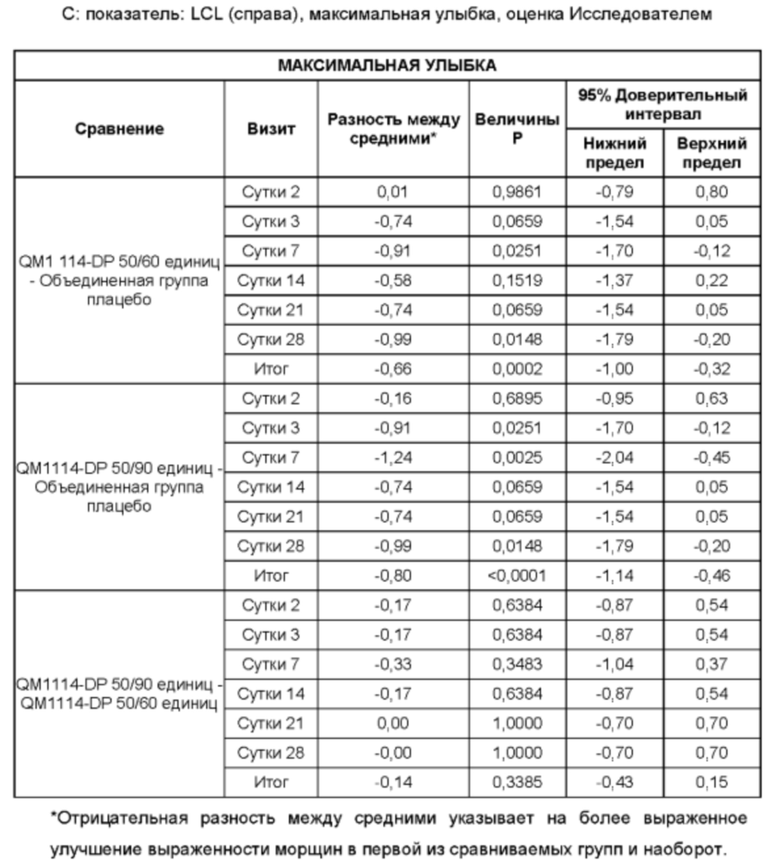
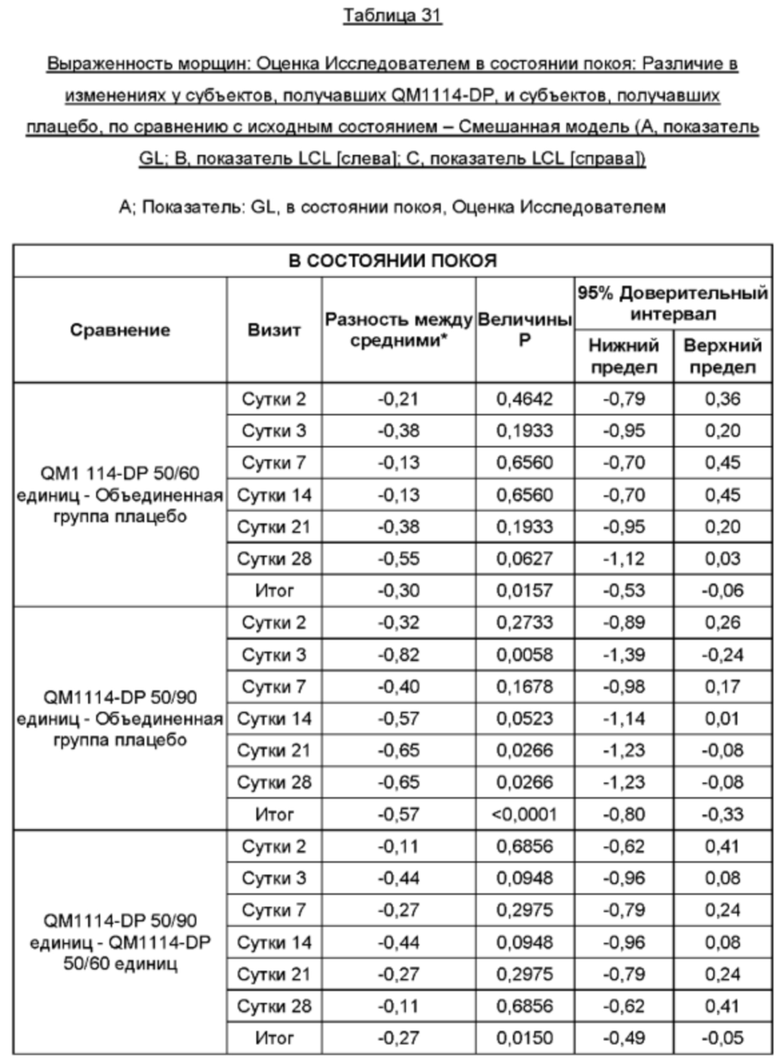
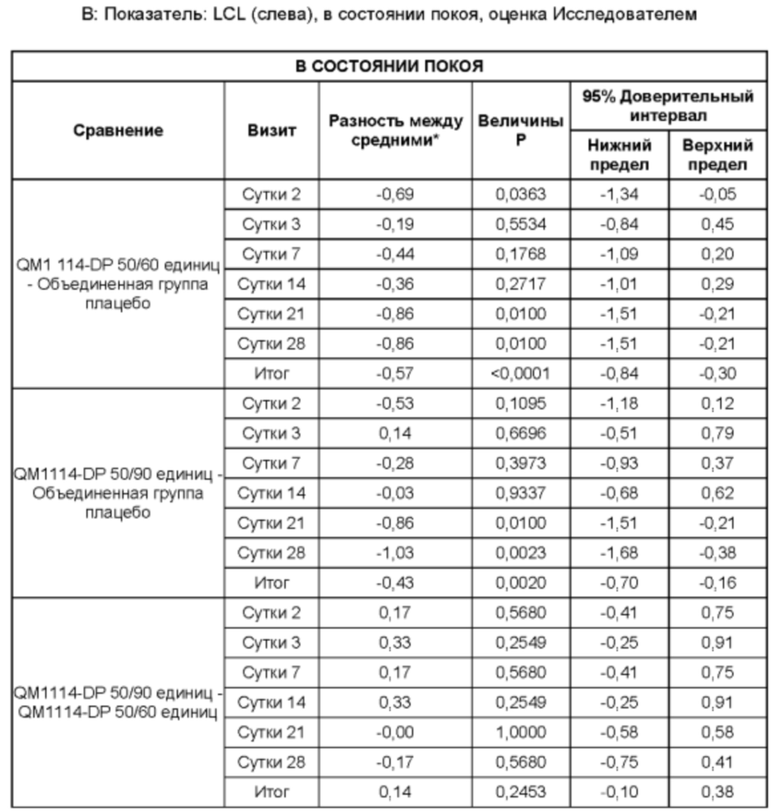
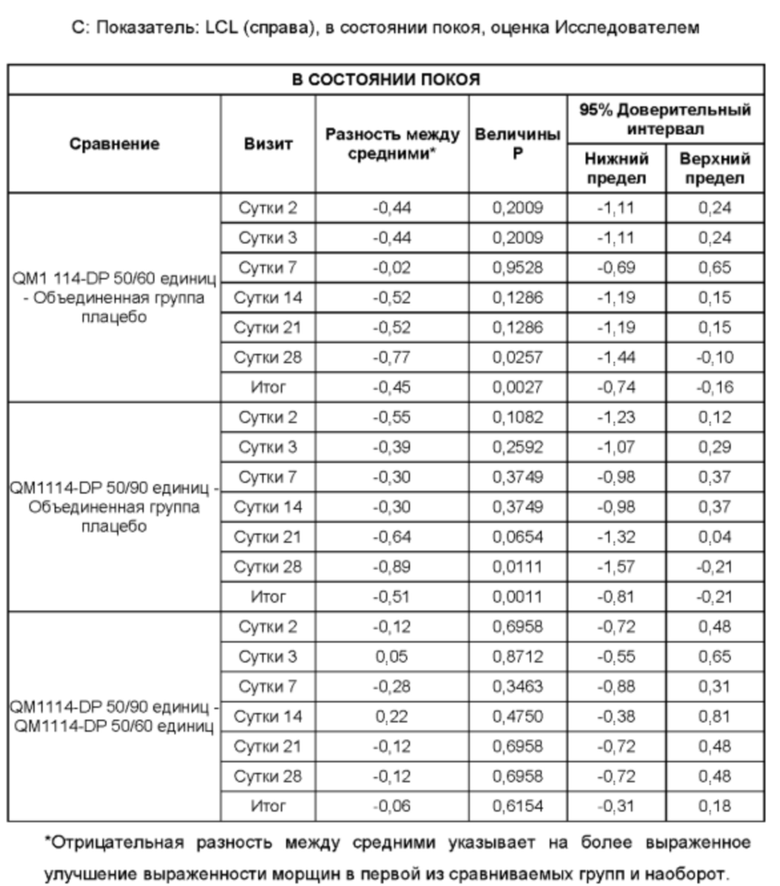
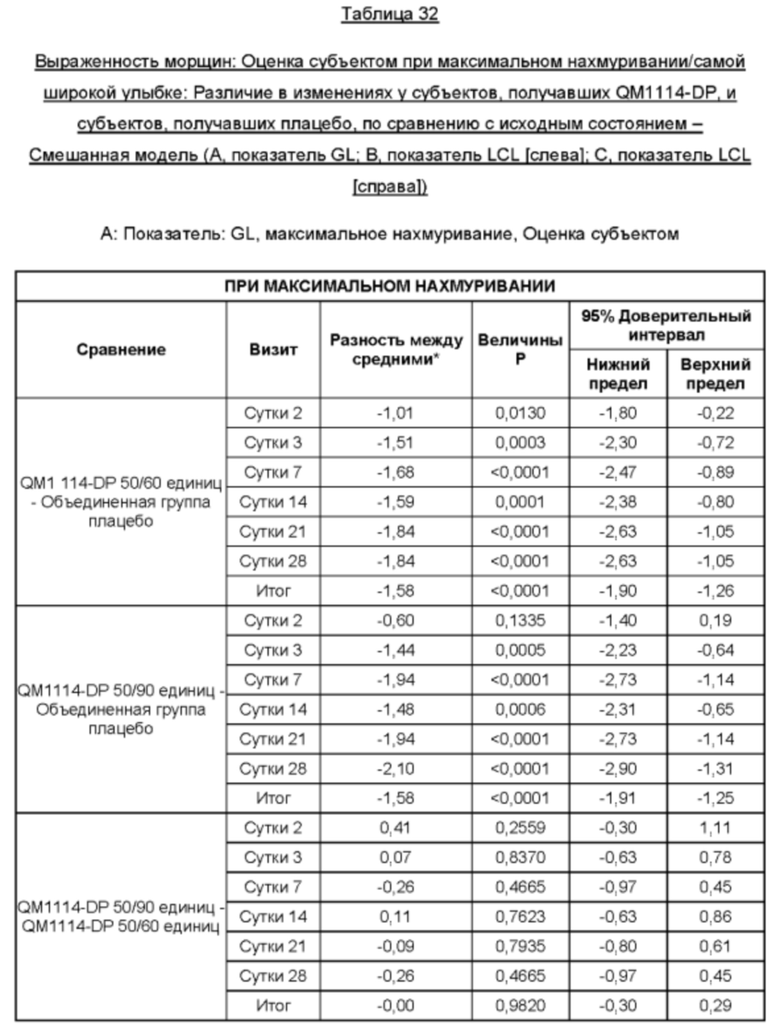
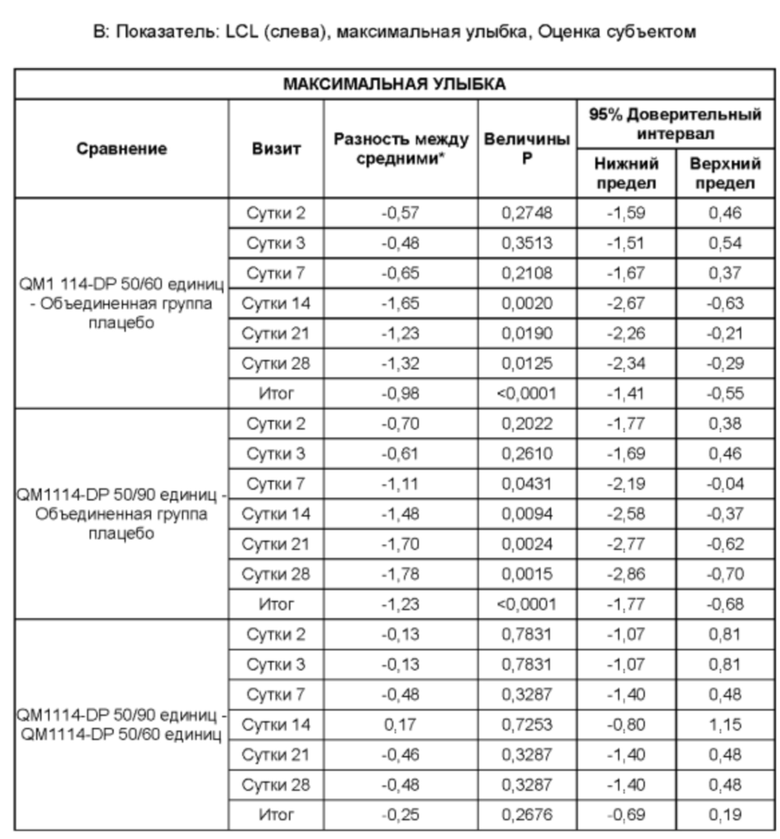
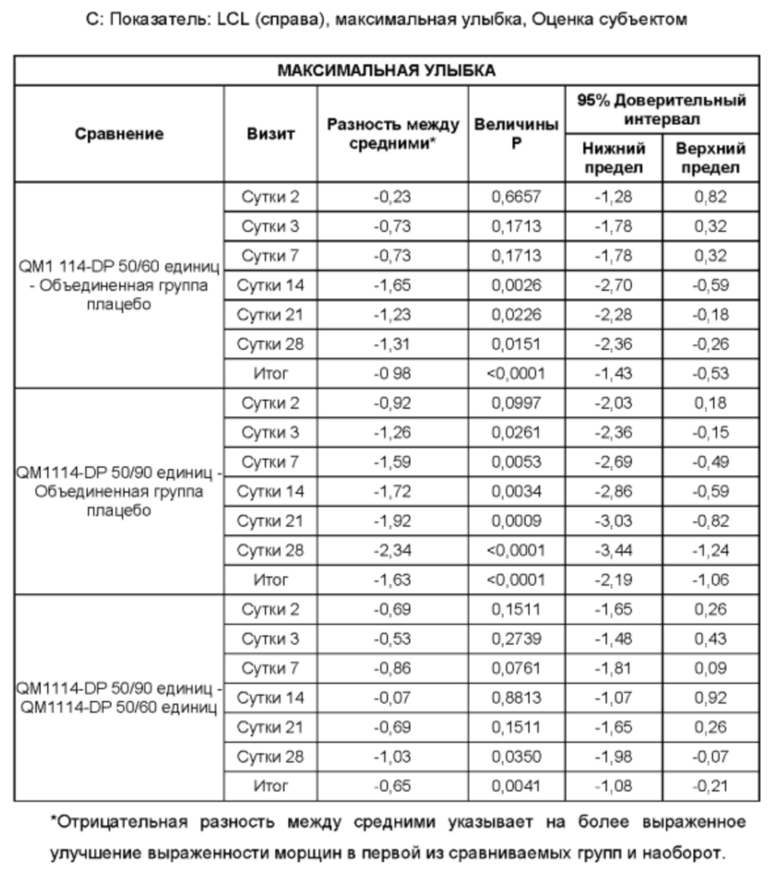
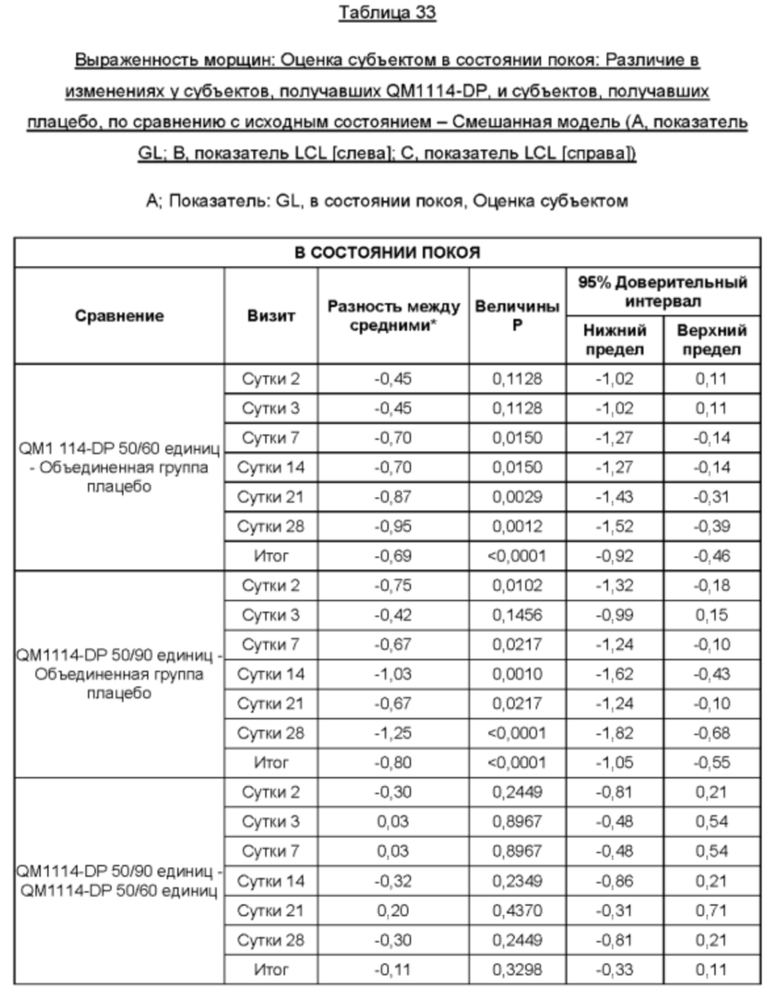
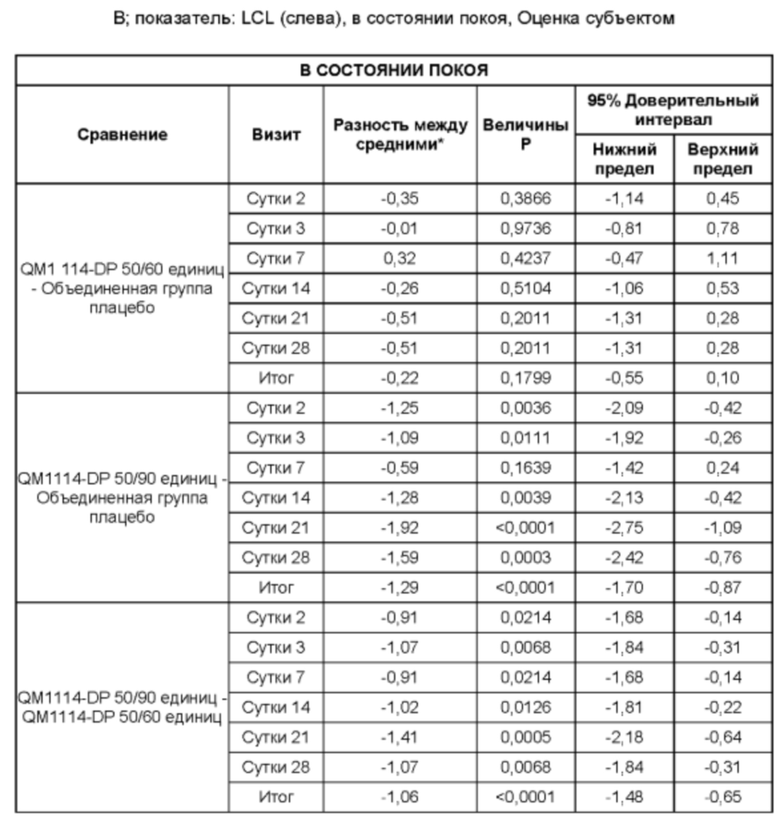
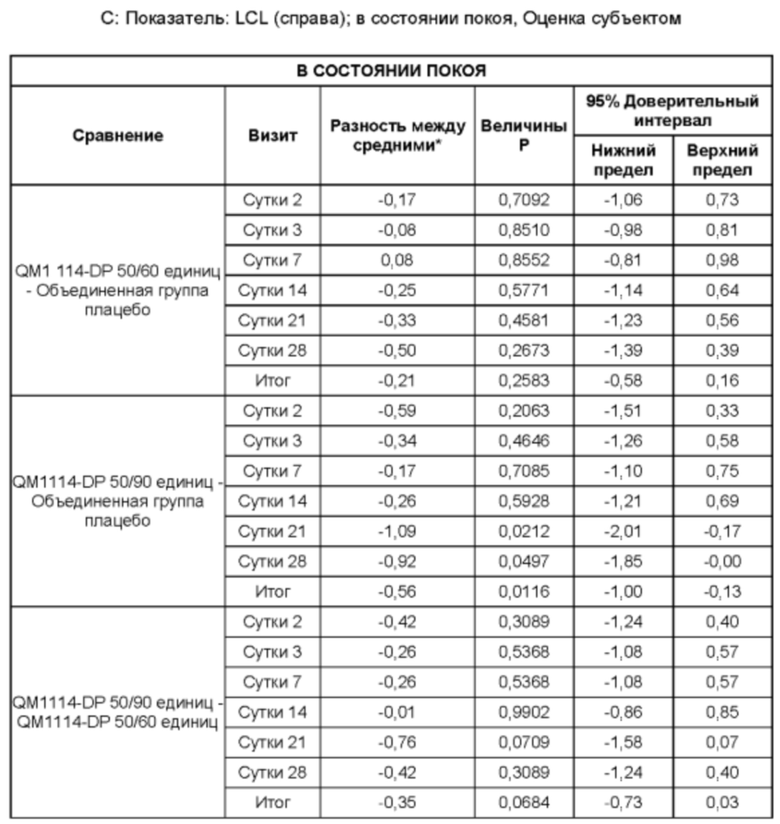
Заключение по эффективности
Результаты оценки эффективности, полученные Исследователем и субъектами по данным изменения выраженности GL при максимальном нахмуривании и LCL при самой широкой улыбке, показывают, что введение обеих дозировок QM1114-DP эффективно снижало выраженность GL и LCL (за некоторыми исключениями в последнем случае) при максимальном нахмуривании/самой широкой улыбке по сравнению с субъектами группы плацебо. Однако оценки GL и LCL в состоянии покоя показывают, что две исследуемые дозы QM1114-DP не были столь же эффективными, как в случае максимального нахмуривания и самой широкой улыбки, и оценки Исследователя и субъектов различались. Оценка субъектом удовлетворенности состоянием GL и LCL показывает, что большинство субъектов в группах, получавших действующее лечение, были либо очень довольны, либо довольны лечением в период до Суток 28. В группе плацебо три субъекта были недовольны лечением.
Оценка безопасности
В анализе безопасности участвовали все 16 субъектов, которые получили однократную дозу препарата, изучаемого в рандомизированном исследовании.
Шесть субъектов получили 110 единиц QM1114-DP (всего 50 единиц в область GL и всего 60 единиц в область LCL), и шесть субъектов получили 140 единиц QM1114-DP (всего 50 единиц в область GL и всего 90 единиц в область LCL). Четыре субъекта получали плацебо.
В приведенном в данном разделе анализе НЯ в основном рассмотрены НЯ, возникающие в процессе лечения, т.е. возникающие после введения дозы ИМП (исследуемого медицинского продукта), и НРП (с нежелательные реакции на препарат), т.е. те случаи, в которых взаимосвязь с ИМП имеет по меньшей мере обоснованную возможность. Во время исследования не сообщалось ни о СНЯ, ни о ПНСНР, и ни один из субъектов не вышел из исследования по соображениям безопасности.
Обзор НЯ, появившихся в группе, получавшей лечение, представлен в Таблице 34. НЯ были выявлены при каждой дозировке. Тридцать два (32) НЯ было выявлено у 11 из 16 (68,8%) субъектов, и из них всего 24 НЯ у 10 (62,5%) субъектов, по мнению Исследователя, могли быть или вероятно были связаны с лечением. Было выявлено четырнадцать (14) НЯ у 4 из 6 субъектов, получавших 50/60 единиц QM1114-DP и девять НЯ у 4 из 6 субъектов, получавших 50/90 единиц QM1114-DP. В группе плацебо было выявлено девять НЯ у 3 из 4 субъектов. В Таблице 35 представлен обзор НЯ по классам органов и систем органов (сокр. КОС) (англ. System Organ Class, сокращенно SOC) и терминам предпочтительного употребления (сокр. ТПУ) (англ. preferred term, сокращенно РТ). Период до развития опущения века составлял от 3 суток 22 часов (субъект 301) до 14 суток (субъект 304). Длительность опущения века составляла от 67 до 87 суток. Наиболее часто встречающимся связанным лечением нарушением нервной системы была головная боль, которая начиналась, спустя период, составляющий от 9 часов (субъект 305) до 23 часов (субъект 301). Длительность головной боли составляла от 2 суток (субъект 302) до 13 суток (субъект 305).
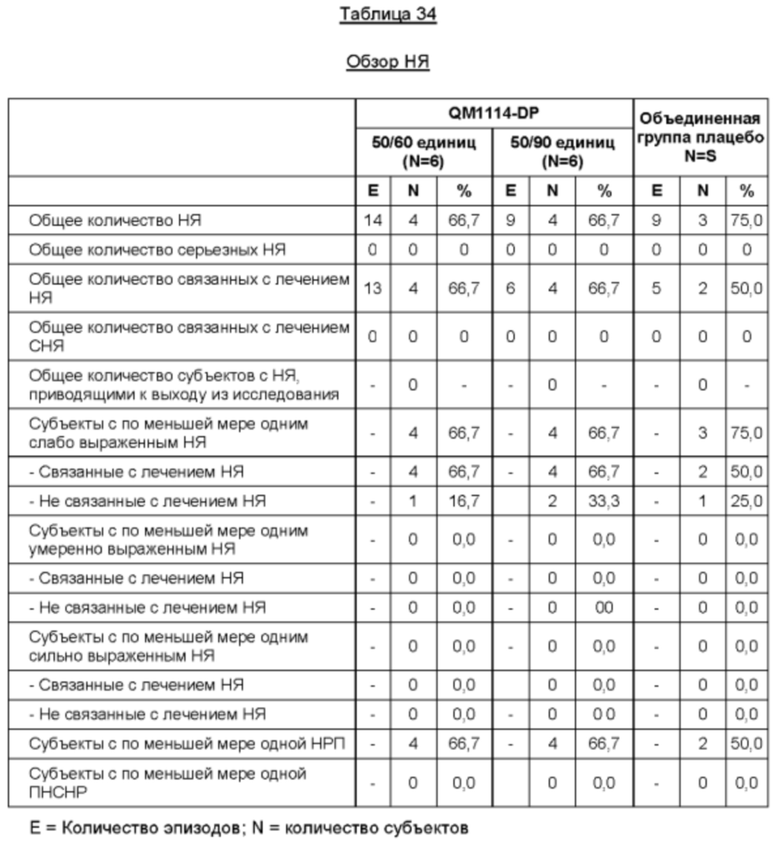
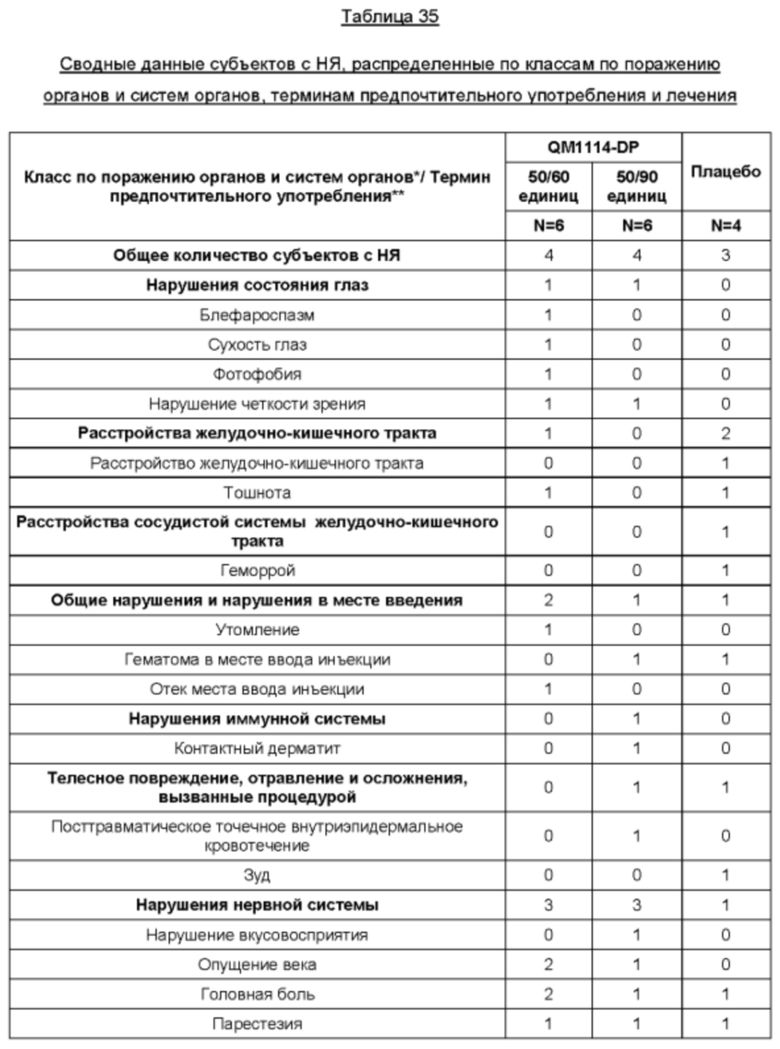
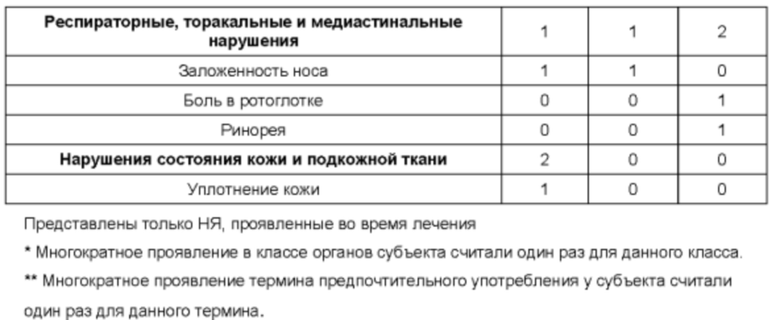
Заключение о безопасности
В целом, наиболее часто возникающие НЯ у участников групп, получавших лечение, представляли собой нарушения нервной системы, общие нарушения и состояние участка введения. Это верно и для связанных с лечением НЯ. Наиболее часто встречающимся нарушением нервной системы была головная боль, о которой сообщали четыре субъекта, и наиболее часто встречающимися общими нарушениями и неприятными состояниями участка введения у субъектов была гематома в точке ввода инъекции, о которой сообщали два субъекта. Учитывая эти результаты, в приведенных ниже разделах основное внимание будет уделено НЯ, связанным с нарушениями нервной системы, общими нарушениями и состоянием участка введения, и сопоставление рассматриваемых лечений будет проведено на примере этих НЯ.
Первичной задачей этой Фазы I исследования было определение безопасности и переносимости QM1114-DP при каждом уровне дозы здоровыми мужчинами и женщинами при лечении морщин в межбровной и боковой периорбитальной областях. В этой части исследования, включающей обработку GL и LCL в комбинации, СНЯ не были выявлены, и не было выходов из исследования из-за возникновения НЯ или по любой другой причине. Тридцать два (32) НЯ было выявлено у 11 из 16 субъектов, и из них всего 24 НЯ у 10 субъектов, по мнению Исследователя, могли быть или вероятно были связаны с лечением. По мнению Исследователя, другие НЯ (8 НЯ) не были связаны с лечением. Все НЯ были слабо выраженными. НЯ было выявлено при каждом уровне дозы, причем самое большое количество НЯ (14 НЯ) было выявлено у субъектов группы, получавшей 50/60 QM1114-DP.
При самой высокой дозировке (50/90 единиц QM1114-DP) количество НЯ (9 НЯ) было таким же, как в объединенной группе плацебо, состоящей из 4 субъектов. НЯ, наиболее часто возникающие в группах, получавших лечение, относились к нарушениям нервной системы, общим нарушениям и состояниям участка введения. Это также верно для связанных с лечением НЯ, среди которых наиболее часто встречались нарушения нервной системы, общие нарушения и нарушения состояния участка введения. Наиболее часто встречающимся нарушением нервной системы была головная боль, о которой сообщали четыре субъекта, и наиболее часто встречающимися общими нарушениями и нарушениями состояния участка введения была гематома в точке ввода инъекции. В группах, получавших действующее лечение, всего было отмечено три случая слабо выраженного опущения века у трех субъектов (субъекты 301 и 304 [в группе, получавшей 50/60 единиц QM1114-DP] и субъект 324 [в группе, получавшей 50/90 единиц QM1114-DP]).
У некоторых субъектов в некоторые дни исследования отмечалось повышение величин биохимических параметров. Однако, по мнению Исследователя, они не были связаны лечением. В заключение следует отметить, что во время исследования не наблюдали клинически значимых изменений гематологических и биохимических параметров и анализа мочи, и при физическом обследовании не было обнаружено клинически значимых отклонений. В данных фокусированного физического обследования глаз и лица с целью обнаружения локального разогрева, зуда, боли, отека/уплотнения, покраснения или гематомы во время исследования ни у одного из субъектов не было обнаружено клинически значимых результатов. За исключением одного субъекта (304), у которого обнаружилась несколько пониженная температура в наружном слуховом проходе, показатели жизнедеятельности всех субъектов были в пределах нормы. Данные ЭКГ показывают, что во время исследования у некоторых субъектов отмечалось удлинение и укорочение PR-интервала. Кроме того, во время исследования у некоторых субъектов отмечалось удлинение интервала QT с корректировкой Базетта. Однако отдельные отклонения от нормальных диапазонов ЭКГ, по мнению Исследователя, не были клинически значимыми и не были связаны с получением исследуемого лекарственного средства.
Обсуждение и итоговые заключения
Первичной задачей этой Фазы I исследования была оценка безопасности и переносимости QM1114-DP при каждом уровне дозы, и вторичная задача состояла в оценке эффективности каждой из исследуемых дозировок QM1114-DP, вводимых здоровым мужчинам и женщинам с целью временного улучшения лицевых морщин в межбровной и боковых периорбитальных областях. В клинической практике приблизительно 50% людей, получавших лечение BoNT-A с целью коррекции морщин в верхней трети лица, получали обработку GL и LCL (Rzany с соавт., 2007).
В этом исследовании всего у трех субъектов в группах, получавших действующее лечение, отмечались три случая слабо выраженного опущения века (два субъекта в группе, получавшей 50/60 единиц QM1114-DP, и один субъект в группе, получавшей 50/90 единиц QM1114-DP). Этот результат можно сравнить с результатом Примера 1 (обработка только GL), в котором слабо выраженное опущение века отмечалось только у субъектов, которым вводили максимальную дозировку (два случая у двух субъектов в группе, получавшей 75 единиц QM1114-DP).
Наиболее часто возникающими НЯ у конкретных субъектов были нарушения нервной системы. Это также верно для связанных с лечением НЯ. Наиболее часто встречающимся нарушением нервной системы была головная боль (о которой сообщали четыре субъекта), следующим по частоте проявления было опущение века (о котором сообщали три субъекта). Все НЯ были слабо выраженными, не было выявлено СНЯ и не было выходов из исследования из-за проблем с НЯ. В течение всего исследования не наблюдали клинически значимых изменений гематологических и биохимических параметров и результатов анализа мочи. Кроме того, не наблюдали клинически значимых физических отклонений. Во время исследования субъекты имели показатели жизнедеятельности в пределах диапазона нормальных значений, и клинически значимых отклонений в данных ЭКГ не наблюдали.
В заключение следует отметить, что подавляющее большинство связанных лечением НЯ относились к нарушениям нервной системы, общим нарушениям и нарушениям состояния участка введения. Эти связанные лечением НЯ были ожидаемыми, исходя из имеющейся информации по безопасности и, за исключением опущения века, согласовывались с НЯ, обнаруженными в Части 1 (GL), Части 2 (LCL) исследования и, в целом, в предшествующих клинических исследованиях других продуктов класса BoNT-A.
Полученные в настоящем исследовании данные по эффективности показали, что введение любой из рассмотренных двух доз QM1114-DP эффективно снижало выраженность комбинации GL и LCL при максимальном нахмуривании/самой широкой улыбке по сравнению с субъектами группы плацебо. Анализ согласно смешанной модели оценки Исследователем морщин в области GL показывает, что начиная с Суток 2-3 до Суток 28 изменения по сравнению с исходным состоянием средних оценок по шкале MAS в группах, получавших 50/60 и 50/90 единиц QM1114-DP, имеют статистически значимое отличие (Р<0,05) от оценок группы плацебо. Аналогичная тенденция также наблюдалась в оценке морщин при максимальном нахмуривании субъектом, причем статистически значимые отличия от плацебо наблюдали, начиная с Суток 2 или Суток 3, которые сохранялись до Суток 28, при обеих дозировках QM1114-DP.
С некоторыми исключениями, оценка Исследователем морщин в боковой периорбитальной области при самой широкой улыбке также показывает, что изменение средних оценок по шкале MAS (на левой стороне лица) по сравнению с исходным состоянием в группах, получавших 50/60 и 50/90 единиц QM1114-DP, имеют статистически значимое отличие от оценок в группе плацебо. Для оценок морщин на правой стороне лица разность между оценками состояния морщин в группах, получавших QM1114-DP, и в группе плацебо была менее выраженной. Оценка субъектом удовлетворенности состоянием GL и LCL показывает, что большинство субъектов в группах, получавших действующее лечение, были либо очень довольны, либо довольны лечением в период до Суток 28, если у субъектов не проявлялись НЯ, которые могли вносить свой вклад в неудовольствие некоторых субъектов из групп, получавших действующее лечение. Было показано, что в группе плацебо недовольными лечением оказались три субъекта.
Итоговые заключения:
• Обе дозы, примененные для временного улучшения состояния GL и LCL, которые обрабатывали в комбинации, считаются безопасными и хорошо переносимыми.
• В этом исследовании не было выявлено СНЯ.
• Не было выходов субъектов из исследования из-за проблем с НЯ или по другим причинам.
• Все НЯ были слабо выраженными.
• Оценка выраженности морщин Исследователем и субъектом при максимальном нахмуривании показывает, что введение любой из исследованных дозировок QM1114-DP эффективно снижало выраженность GL по сравнению с введением плацебо. Эффект сохранялся до Суток 28.
• Оценка выраженности морщин Исследователем и субъектом при самой широкой улыбке показывает, что, в основном, введение любой из исследованных дозировок QM1114-DP эффективно снижало выраженность LCL по сравнению с введением плацебо.
• Оценка выраженности морщин в состоянии покоя субъектом показывает, что введение любой из исследованных дозировок QM1114-DP эффективно снижало выраженность GL по сравнению с введением плацебо. Эффект сохранялся до Суток 28. Однако, согласно оценкам Исследователя, эффективность не была очевидной.
• Оценка выраженности морщин в состоянии покоя Исследователем показала, что введение любой из исследованных дозировок QM1114-DP эффективно снижает выраженность LCL по сравнению с плацебо уже к Суткам 21 и Суткам 28. Оценка Субъектом продемонстрировала эффективность более высокой дозировки (QM1114-DP 50/90 единиц) в некоторые моменты времени в рамках исследования.
• Оценка субъектом удовлетворенности состоянием GL и LCL показывает, что большинство субъектов в группах, получавших действующее лечение, были либо очень довольны, либо довольны лечением в период до Суток 28.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЛЕЧЕНИЕ МОРЩИН НА ЛИЦЕ ВЫСОКОЙ ДОЗОЙ И МАЛЫМ ОБЪЕМОМ БОТУЛИНИЧЕСКОГО ТОКСИНА | 2021 |
|
RU2835617C1 |
| ПРИМЕНЕНИЕ БОТУЛИНИЧЕСКИХ НЕЙРОТОКСИНОВ В ЛЕЧЕНИИ | 2018 |
|
RU2766147C2 |
| УЛУЧШЕННОЕ ПРИМЕНЕНИЕ БОТУЛИНИЧЕСКОГО НЕЙРОТОКСИНА В ЛЕЧЕНИИ СИАЛОРЕИ | 2018 |
|
RU2778481C2 |
| СПОСОБ УСТРАНЕНИЯ МОРЩИН | 1998 |
|
RU2123837C1 |
| СХЕМА ПРИЕМА БОТУЛОТОКСИНА ДЛЯ ПРОФИЛАКТИКИ ХРОНИЧЕСКОЙ МИГРЕНИ | 2011 |
|
RU2549981C9 |
| ЛЕЧЕНИЕ СПАСТИЧНОСТИ НИЖНИХ КОНЕЧНОСТЕЙ | 2018 |
|
RU2788039C2 |
| КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ БОТУЛИНИЧЕСКИЙ ТОКСИН | 2017 |
|
RU2743746C2 |
| КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ БОТУЛИНИЧЕСКИЙ ТОКСИН | 2017 |
|
RU2832647C1 |
| ЖИДКАЯ КОМПОЗИЦИЯ НЕЙРОТОКСИНА, СТАБИЛИЗИРОВАННАЯ ТРИПТОФАНОМ ИЛИ ТИРОЗИНОМ | 2017 |
|
RU2741497C2 |
| КОМПОЗИЦИИ И СПОСОБЫ УЛУЧШЕНИЯ МЕДИКАМЕНТОЗНОЙ ТЕРАПИИ СРЕДСТВАМИ, ЗАВИСЯЩИМИ ОТ ИОНОВ МЕТАЛЛОВ | 2010 |
|
RU2540520C2 |

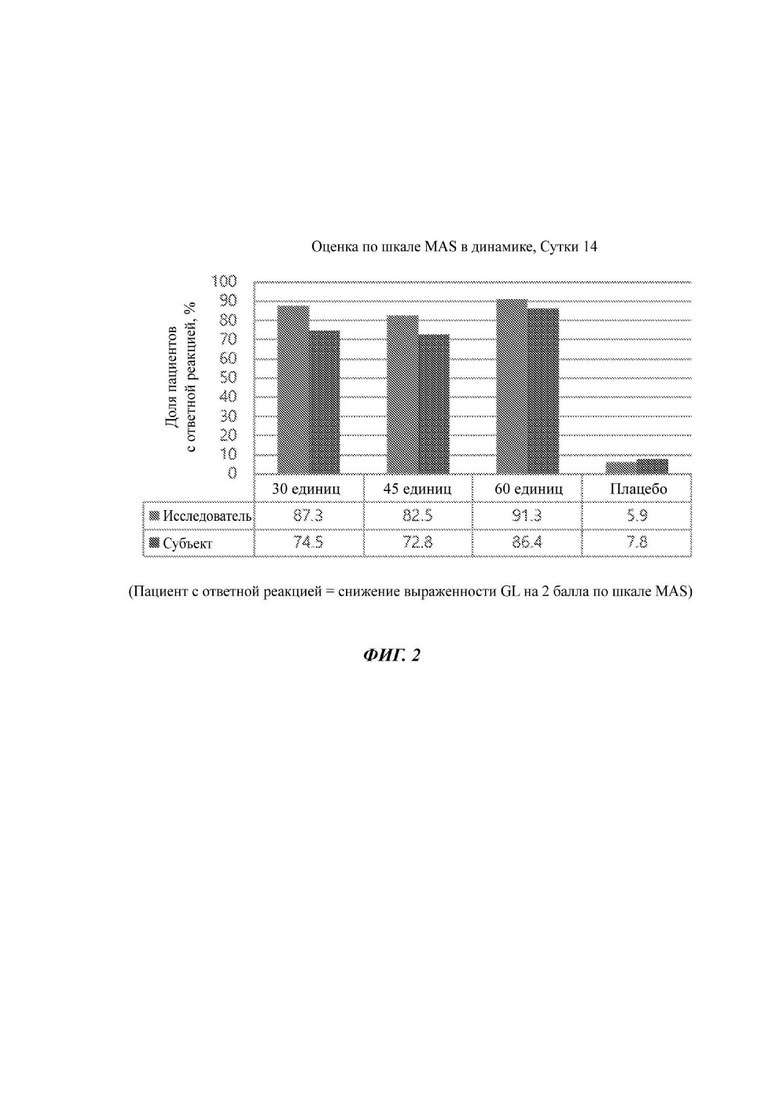

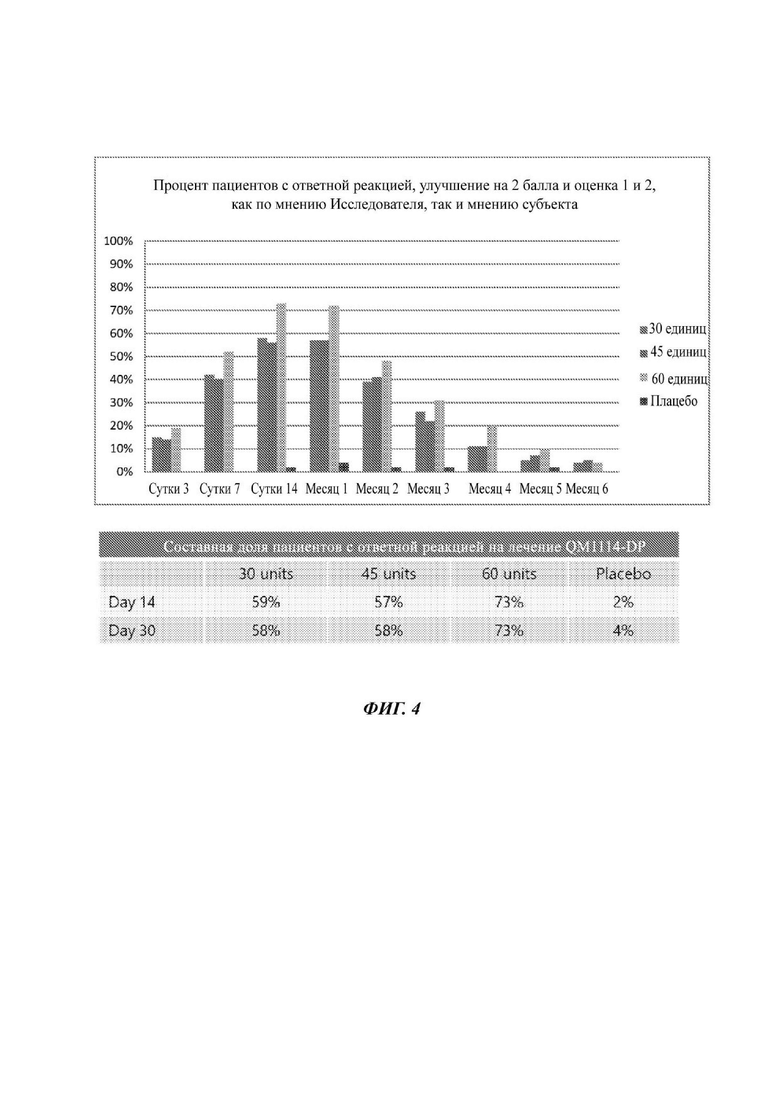
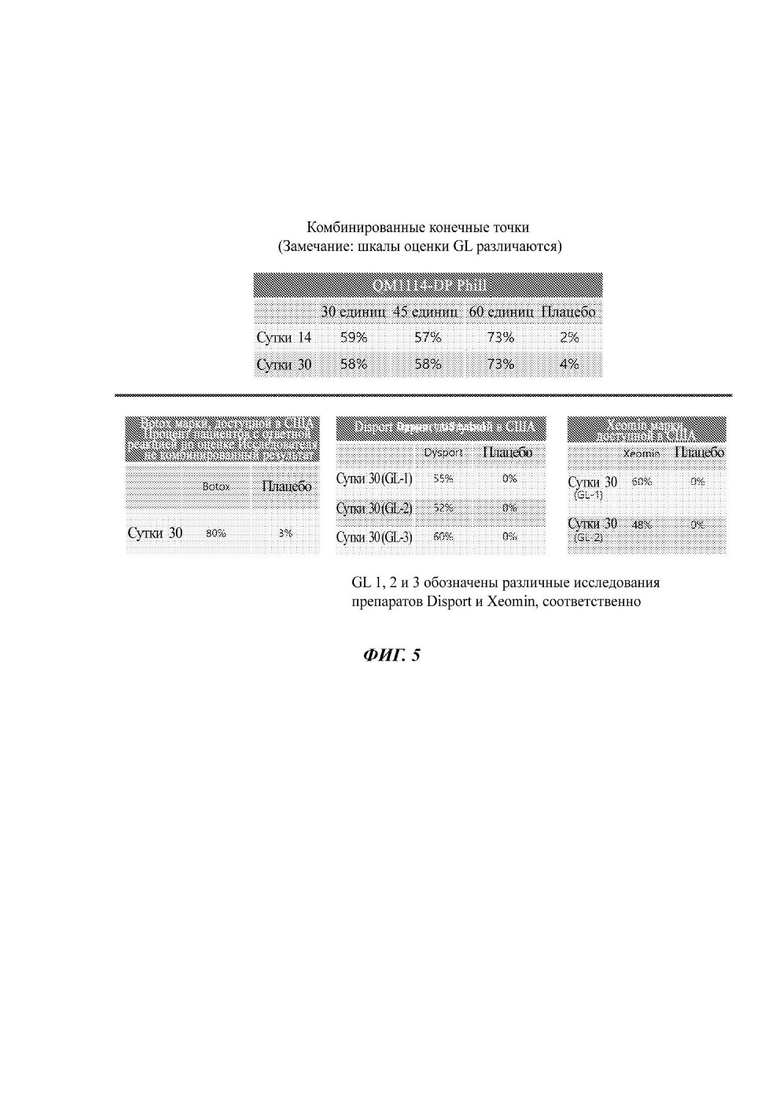
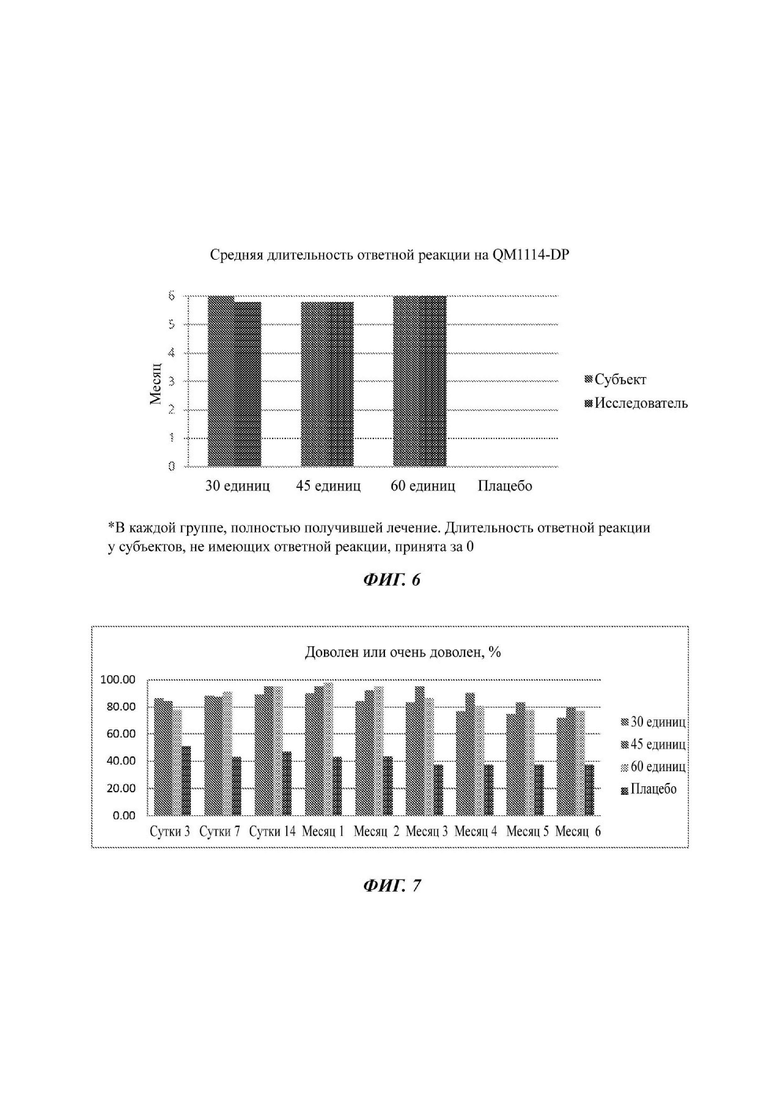
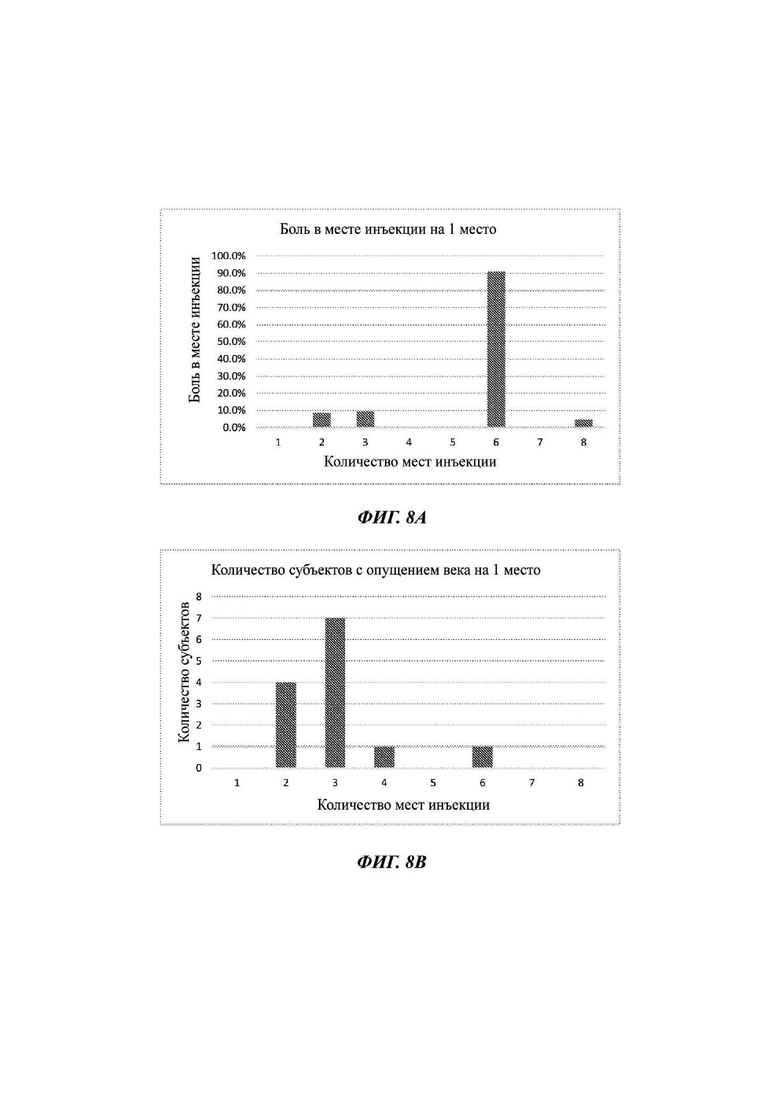
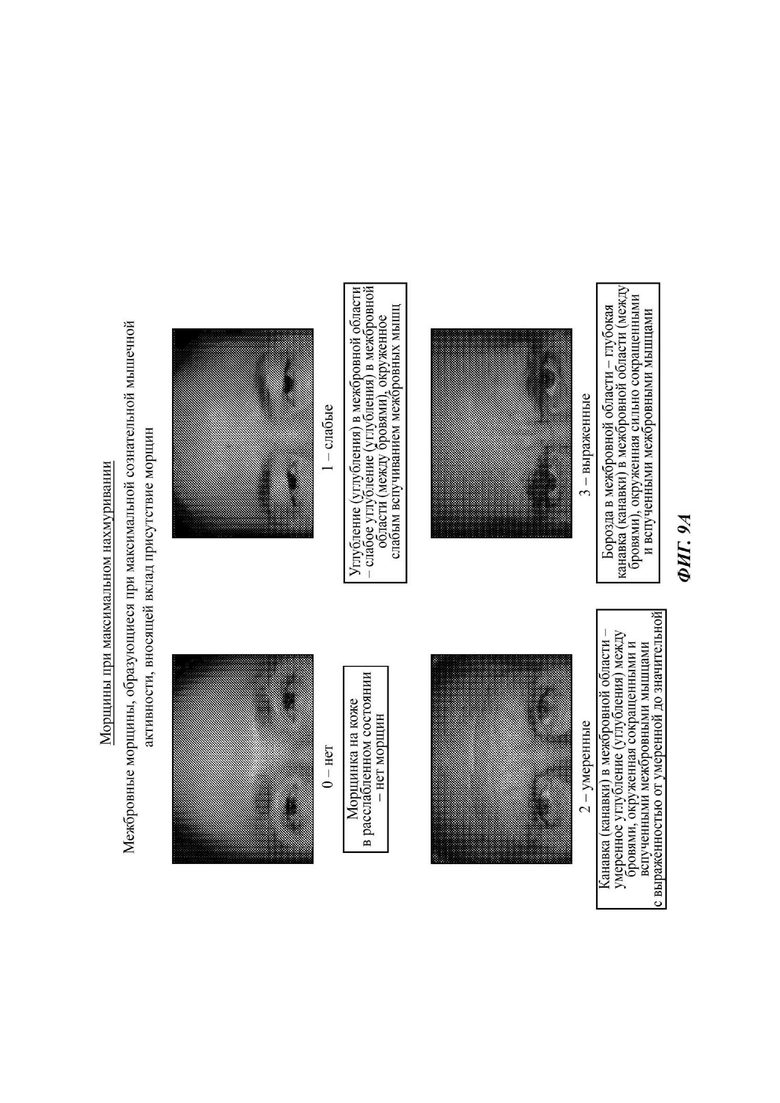
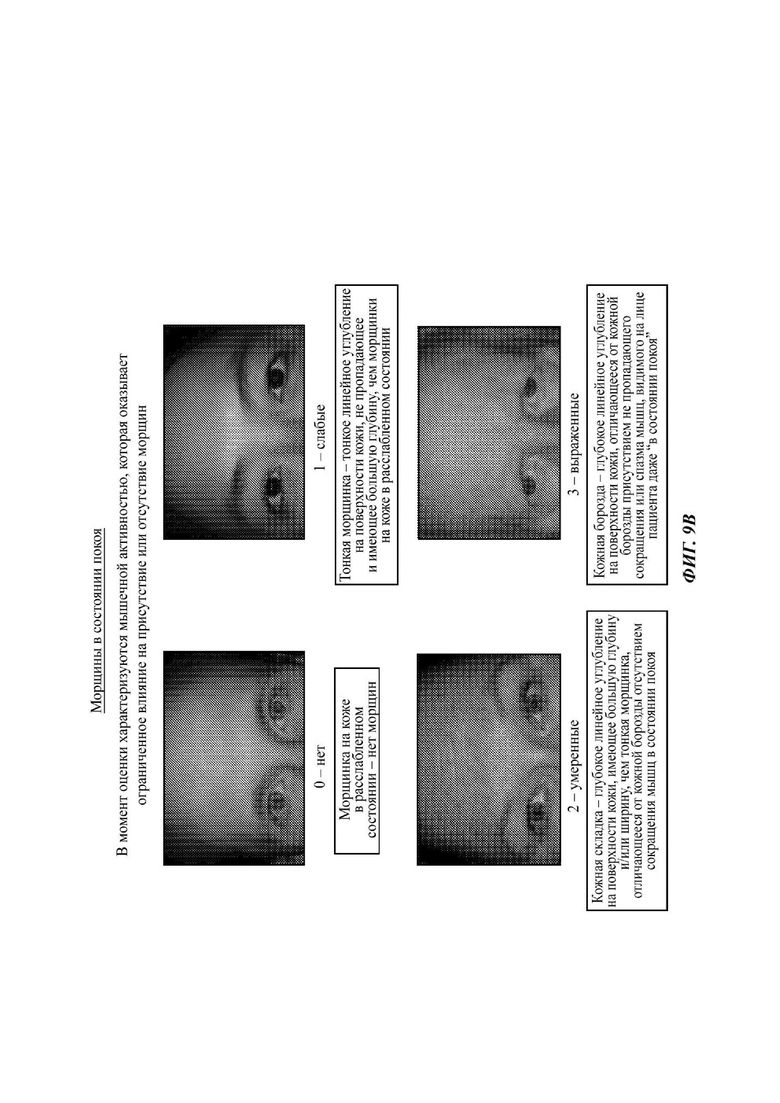
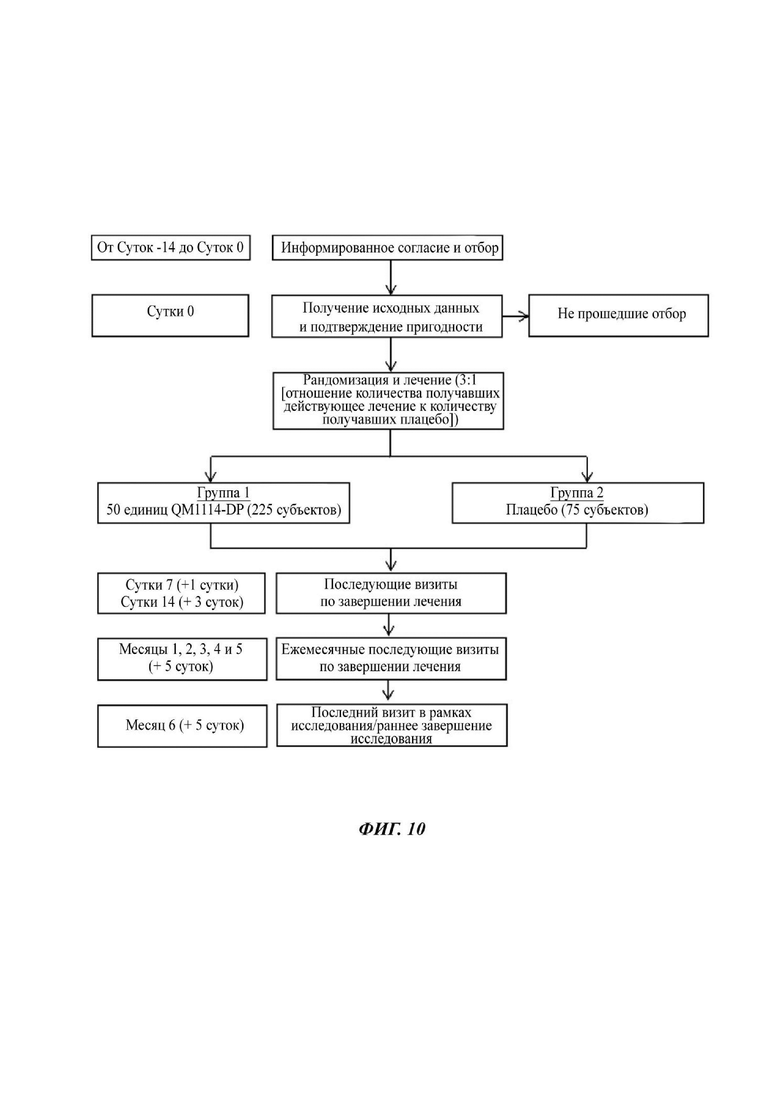
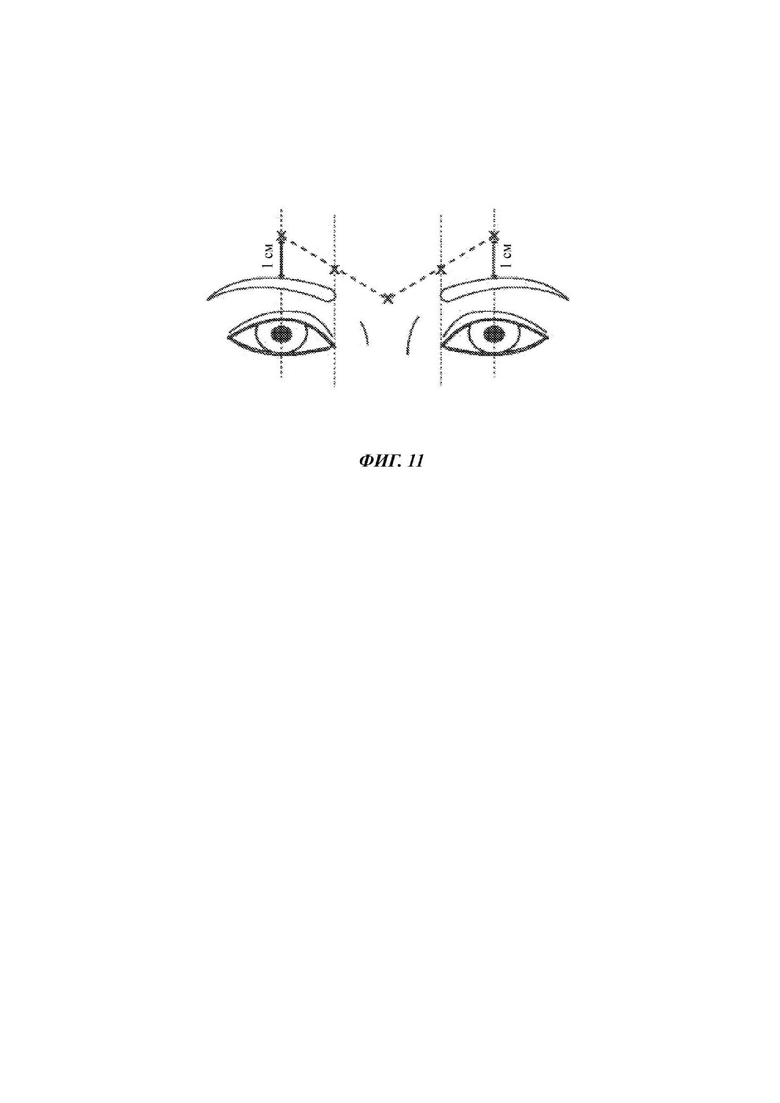
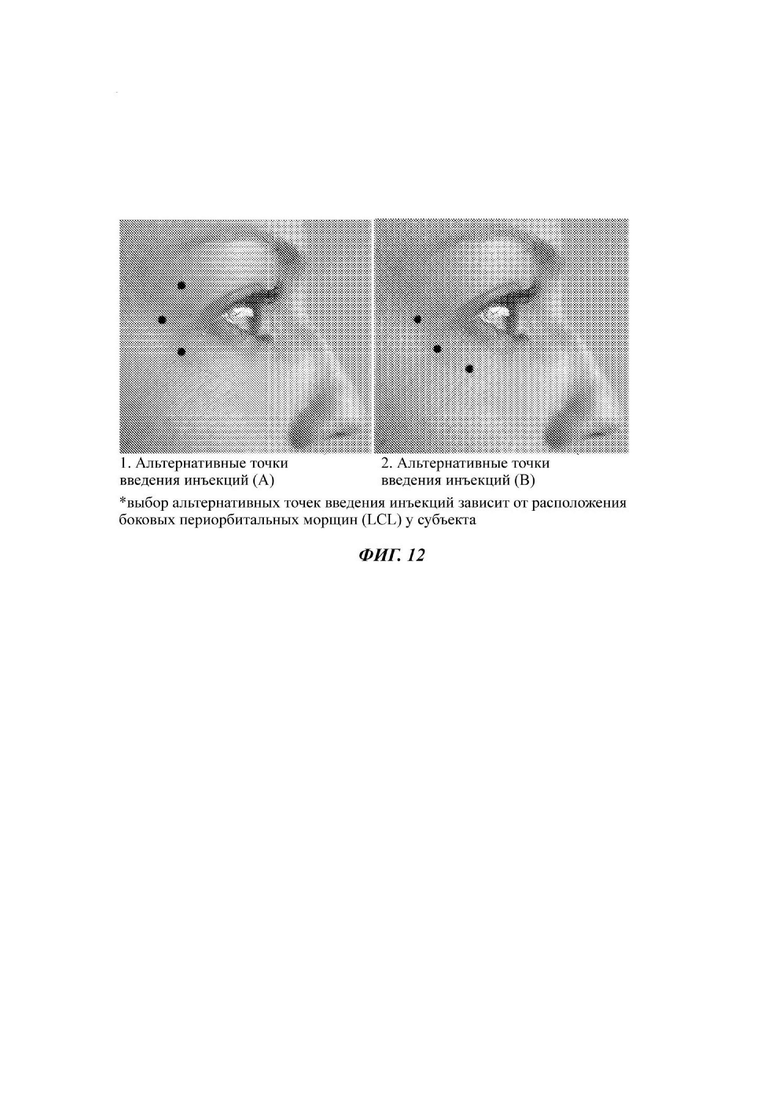
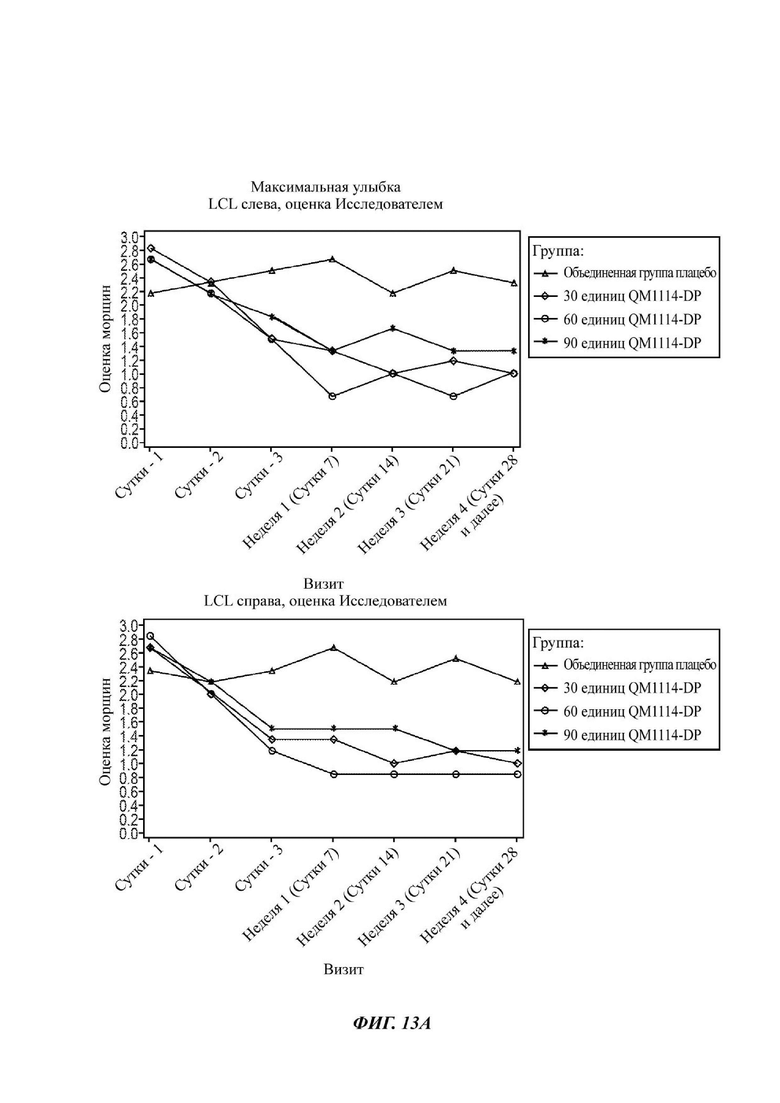
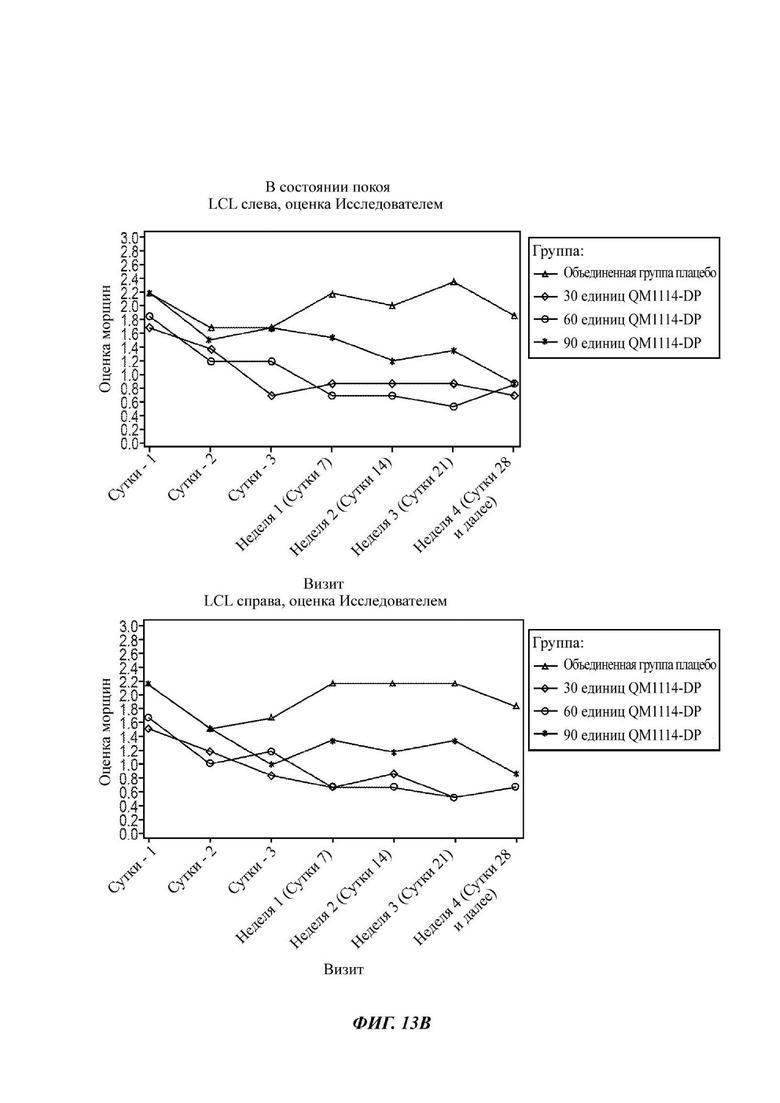
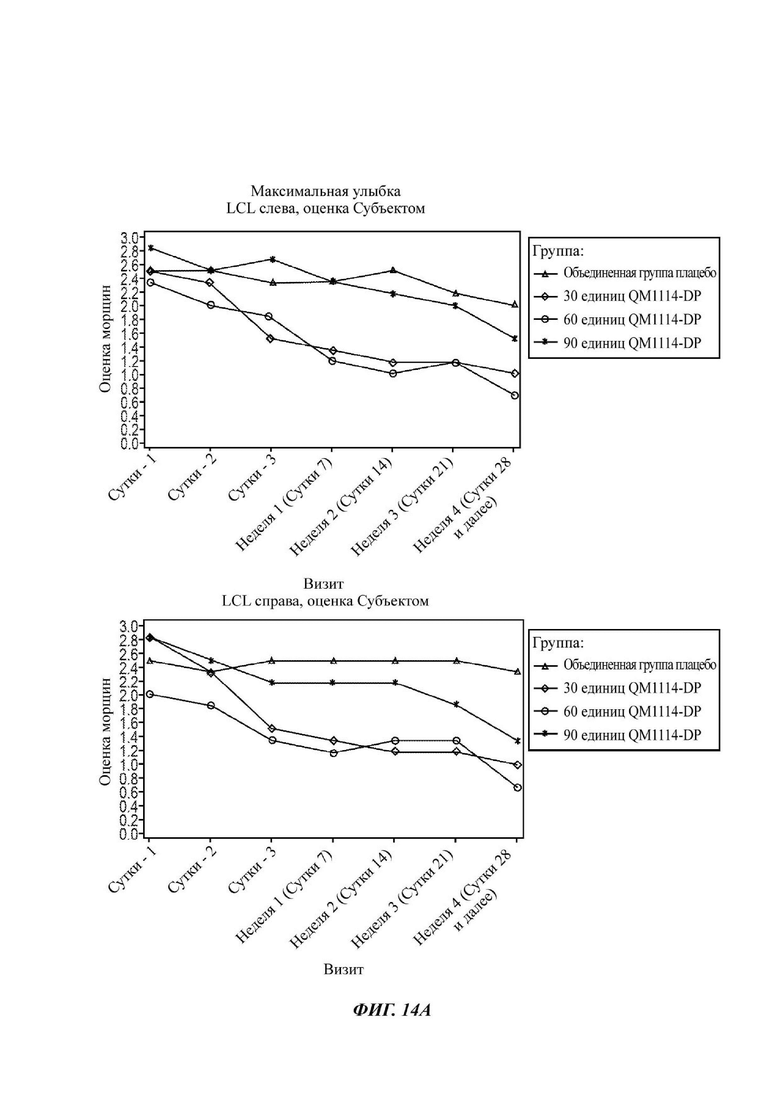
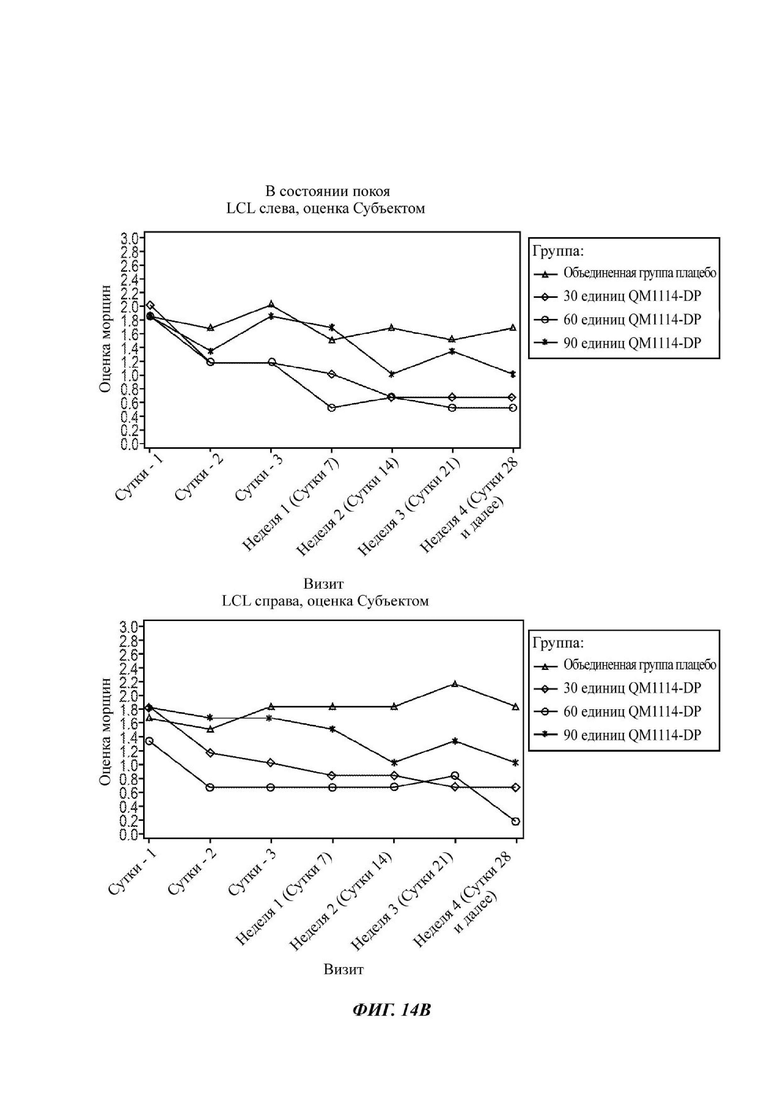
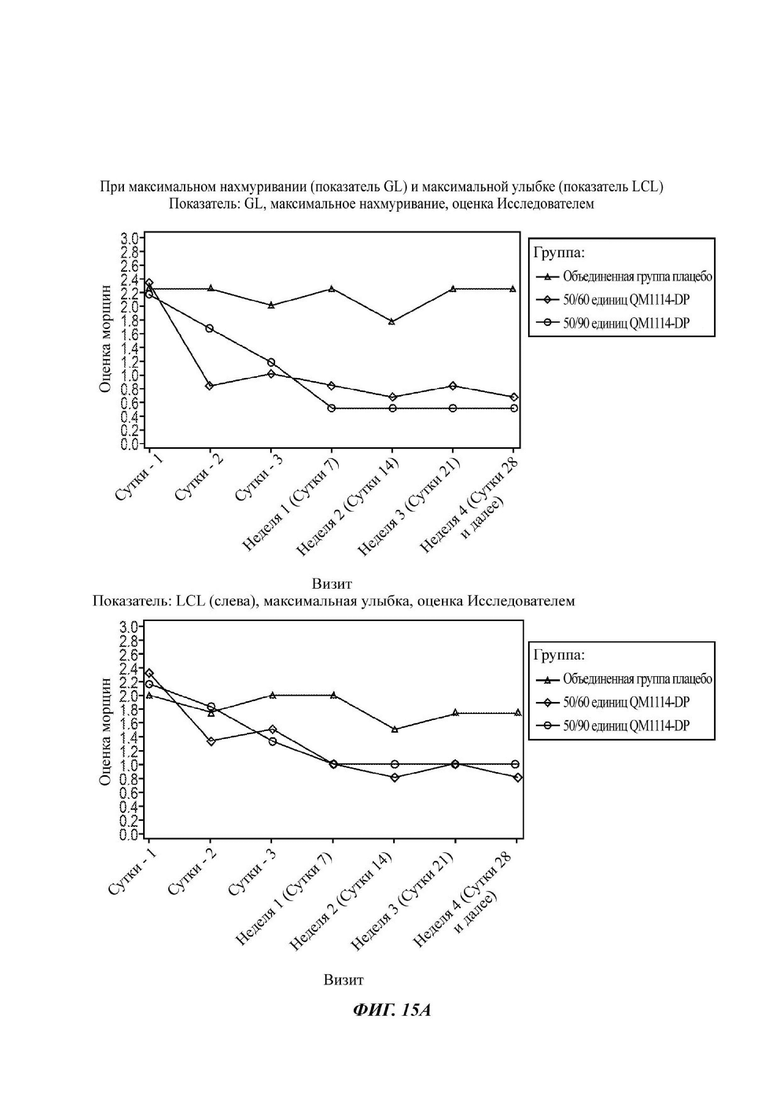
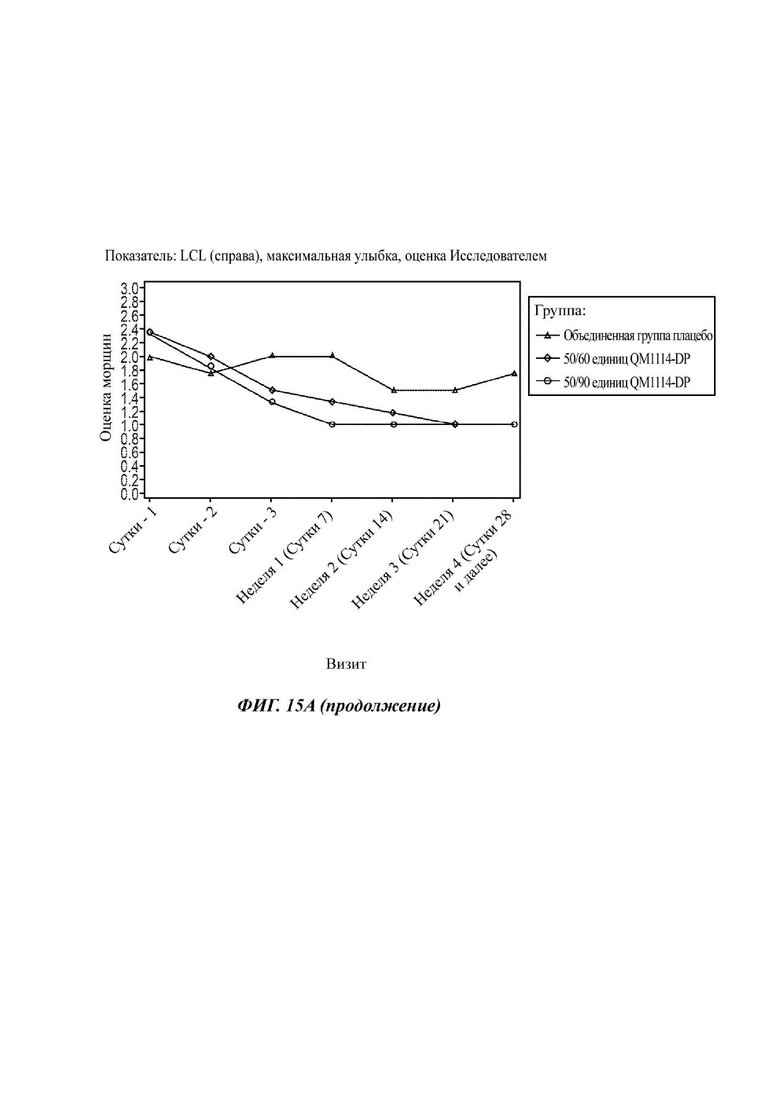
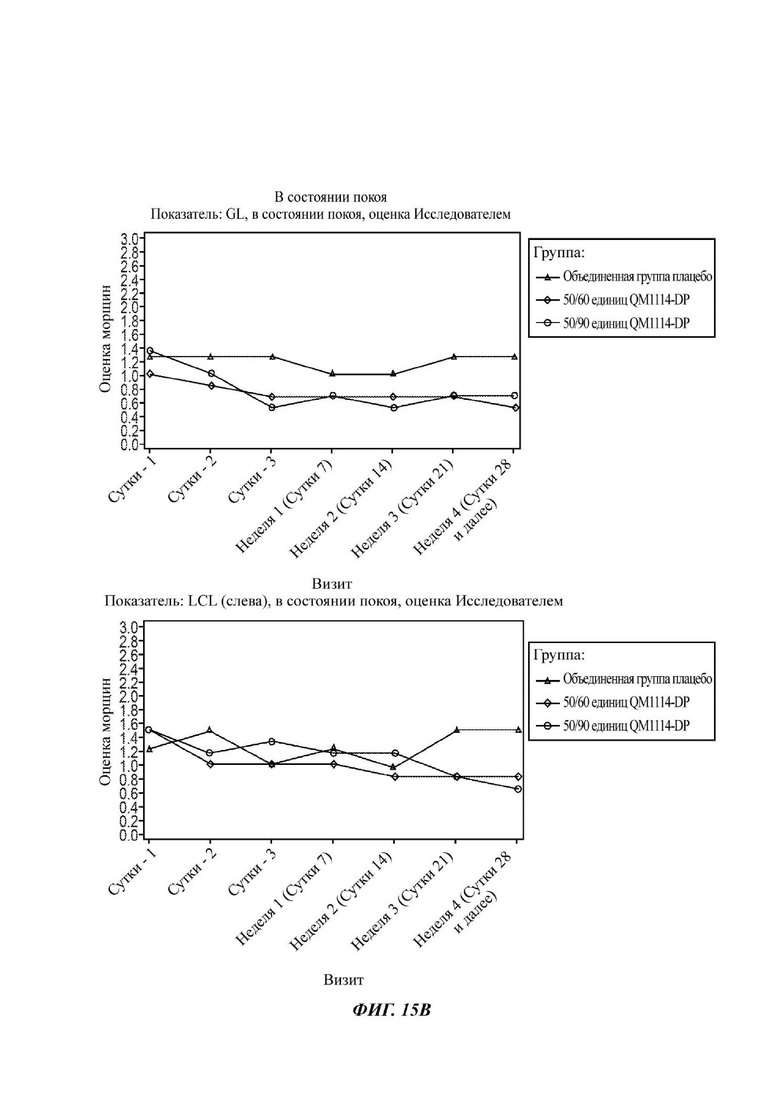
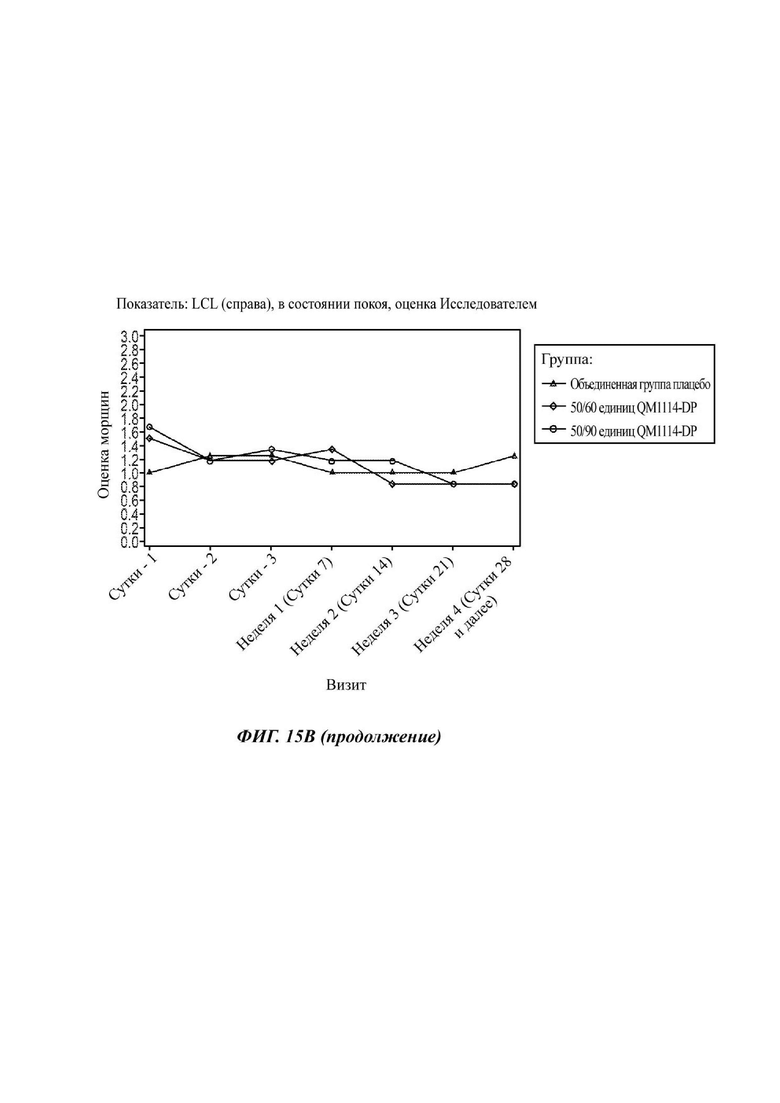
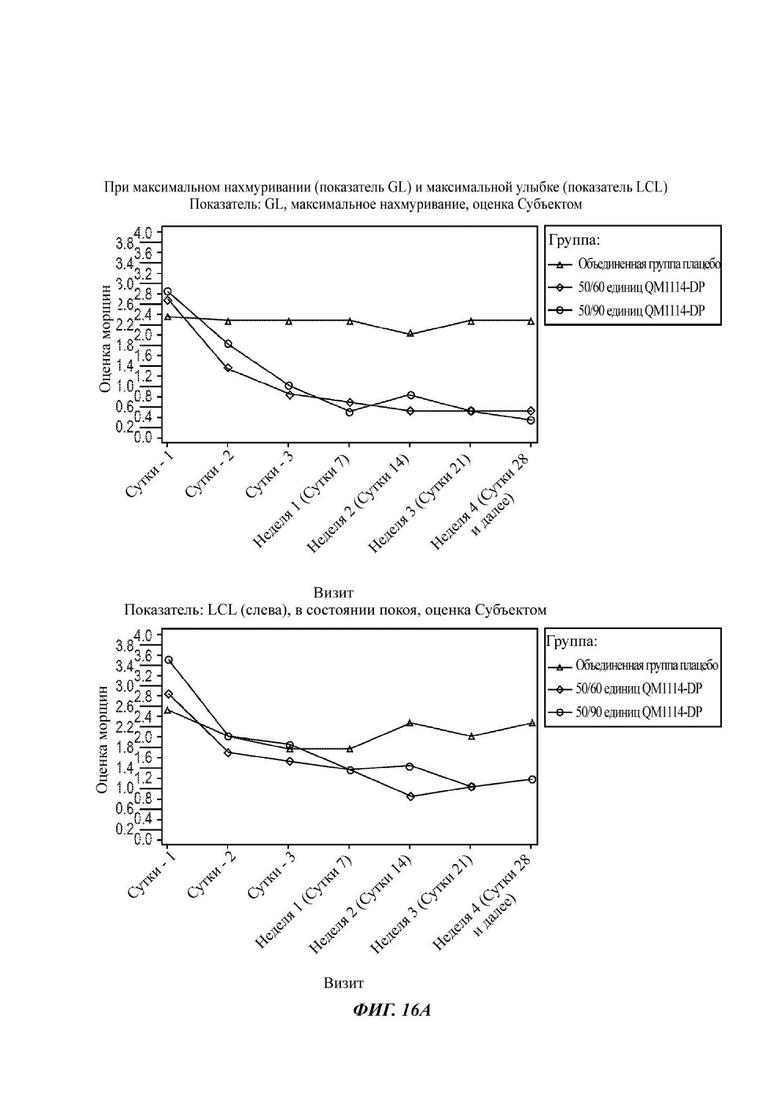
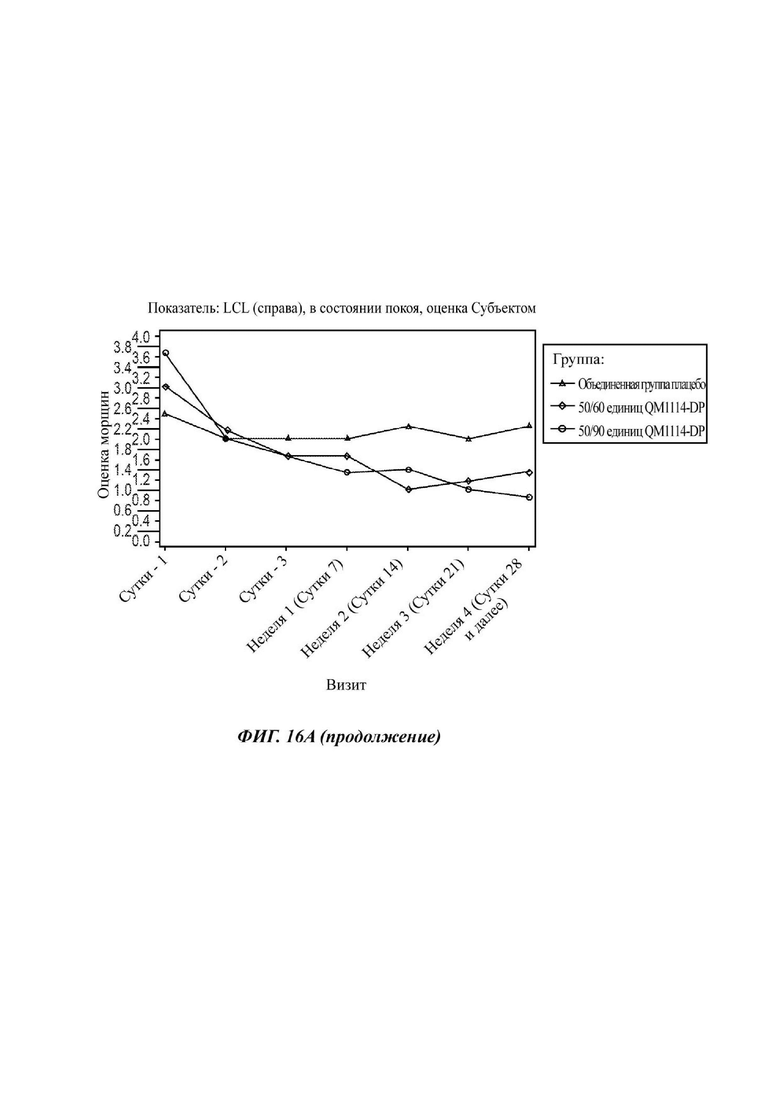
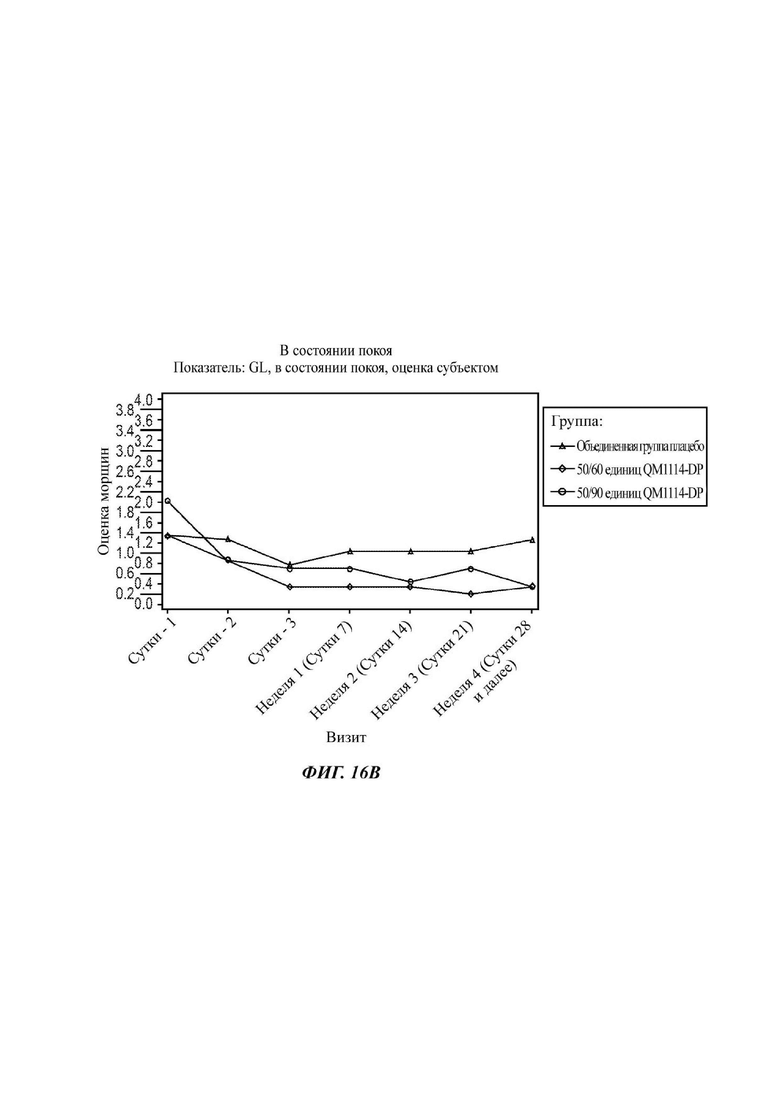
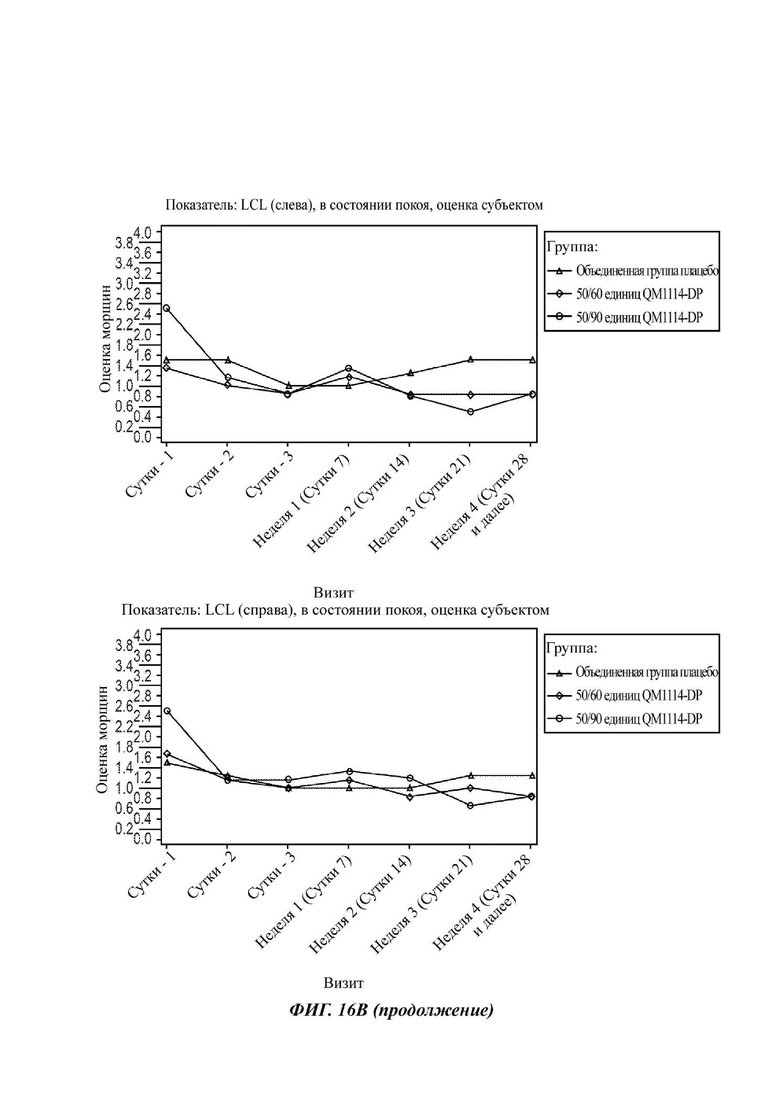
Изобретение относится к лечению межбровных морщин и боковых периорбитальных морщин. Способ лечения межбровных морщин и/или боковых периорбитальных морщин от умеренных до выраженных у субъекта-человека включает введение субъекту терапевтически эффективного количества жидкой композиции, содержащей ботулинический нейротоксин и источник хлорид-ионов, выбранный из NaCl в концентрации 100-300 мМ, KCl в концентрации 1-25 мМ; более одного источника фосфат-ионов, выбранного из фосфата натрия, фосфата калия, безводного гидрофосфата динатрия, безводного дигидрофосфата натрия, в концентрации 1-50 мМ; неионное поверхностно-активное вещество в концентрации 0,01-5,0% (об./об.); аминокислотный стабилизатор, выбранный из аланина, валина, лейцина, изолейцина, метионина, фенилаланина, тирозина и триптофана, в концентрации 0,1-3 мг/мл, где пациенту делают совокупность внутримышечных инъекций указанной композиции в межбровную область и/или ниже латеральной спайки век, в наружную часть круговой мышцы глаза. Изобретение обеспечивает стабильные жидкие композиции, содержащие ботулотоксин, которые могут быть введены без их восстановления или смешивания. 20 з.п. ф-лы, 16 ил., 35 табл., 1 пр.
1. Способ лечения межбровных морщин (GL) и/или боковых периорбитальных морщин (LCL) от умеренных до выраженных у субъекта-человека, где способ включает введение субъекту терапевтически эффективного количества жидкой композиции, содержащей ботулинический нейротоксин (BoNT) и следующие компоненты:
a. источник хлорид-ионов, выбранный из NaCl в концентрации от 100 мМ до 300 мМ и KCl в концентрации от 1 мМ до 25 мМ;
b. более одного источника фосфат-ионов, выбранного из фосфата натрия, фосфата калия, безводного гидрофосфата динатрия, безводного дигидрофосфата натрия, в концентрации от 1 мМ до 50 мМ;
c. неионное поверхностно-активное вещество в концентрации от 0,01% (об./об.) до 5,0% (об./об.); и
d. аминокислотный стабилизатор, выбранный из аланина, валина, лейцина, изолейцина, метионина, фенилаланина, тирозина и триптофана, в концентрации от 0,1 мг/мл до 3 мг/мл,
где пациенту делают совокупность внутримышечных инъекций указанной композиции в межбровную область для лечения GL; и/или
где пациенту делают совокупность внутримышечных инъекций указанной композиции ниже латеральной спайки век, в наружную часть круговой мышцы глаза для лечения LCL.
2. Способ по п. 1, где композиция содержит более одного источника хлорид-ионов.
3. Способ по п. 1 или 2, где аминокислота находится в L изоформе.
4. Способ по любому из пп. 1-3, где аминокислота присутствует в концентрации, составляющей от 0,75 до 2,25 мг/мл.
5. Способ по любому из пп. 1-4, где неионное поверхностно-активное вещество представляет собой полисорбат.
6. Способ по любому из пп. 1-5, где ботулинический нейротоксин выбран из группы, состоящей из ботулинического нейротоксина типов A, B, C, D, E, F и G, предпочтительно ботулинический нейротоксин представляет собой ботулинический нейротоксин типа A.
7. Способ по любому из пп. 1-6, где pH жидкой композиции составляет от 6,6 до 6,9.
8. Способ по любому из пп. 1-7, где осмотическая концентрация жидкой композиции составляет от 270 мосмоль/кг до 310 мосмоль/кг.
9. Способ по любому из пп. 1-8, в котором субъекту вводят от 1 до 100 единиц ботулинического нейротоксина, где 1 единица равна количеству ботулотоксина, соответствующему расчетной средней летальной дозе (LD50) для мышей; причем необязательно:
a) субъекту вводят от 10 до 75 единиц ботулинического нейротоксина;
b) субъекту вводят от 25 до 75 единиц ботулинического нейротоксина;
или
c) субъекту вводят 10, 25, 30, 45, 50, 60, 75 или 90 единиц ботулинического нейротоксина.
10. Способ по п. 1, в котором соседние инъекции в межбровную область вводят на расстоянии друг от друга, составляющем от 0,5 см до 10 см.
11. Способ по п. 1, в котором соседние инъекции в межбровную область вводят на расстоянии друг от друга, составляющем от 1,5 см до 3 см.
12. Способ по любому из пп. 1-11, в котором инъекции производят в мышцу гордецов и мышцы, сморщивающие бровь, расположенные на каждой стороне лица.
13. Способ по п. 12, в котором инъекции сначала делают в мышцу гордецов, а затем в мышцы, сморщивающие бровь, на каждой стороне лица, перемещаясь от середины к периферии.
14. Способ по любому из пп. 1-13, в котором все инъекции производят в области, расположенной на 1 см выше верхнего глазничного валика, и внутри области, ограниченной линиями, проходящими через середину зрачков.
15. Способ по любому из пп. 1-14, в котором все инъекции производят в области, расположенной по меньшей мере на 1 см выше центральной части надбровной дуги или остеоидной надбровной дуги.
16. Способ по любому из пп. 1-15, дополнительно включающий приложение к верхнему глазничному валику давления во время инъекции.
17. Способ по любому из пп. 1-9, в котором субъекту вводят совокупность инъекций в область, расположенную ниже латеральной спайки век, в наружную часть круговой мышцы глаза и/или на расстоянии от 1 до 2 см от глазничного валика.
18. Способ по любому из пп. 1-16, в котором субъекту дополнительно вводят совокупность инъекций в область, расположенную ниже латеральной спайки век, в наружную часть круговой мышцы глаза и/или на расстоянии от 1 до 2 см от глазничного валика.
19. Способ по любому из пп. 1-18, в котором для подавления повторного проявления лечение повторяют через определенные промежутки времени, составляющие от 1 месяца до 6 месяцев.
20. Способ по п. 19, в котором для подавления повторного проявления лечение повторяют через определенные промежутки времени, составляющие от 3 месяцев до 6 месяцев.
21. Способ по п. 19, в котором для подавления повторного проявления лечение повторяют через определенные промежутки времени, составляющие 4 месяца.
| WO 2017203038 A1, 30.11.2017 | |||
| Способ и устройство для гидравлической доставки ископаемых | 1932 |
|
SU30252A1 |
| US 20180071361 A1, 15.03.2018 | |||
| ЧУЕШОВ В.И | |||
| и др | |||
| Промышленная технология лекарств | |||
| Электронный учебник / Национальный фармацевтический университет, кафедра заводской технологии лекарств, Харьков, 2010, URL: https://ztl.nuph.edu.ua/medication/content.html. | |||

