Область техники
Нейродегенеративные заболевания (НДЗ) - это большая группа заболеваний преимущественно пожилого возраста, для которых характерна медленно прогрессирующая гибель нейронов, приводящая к разрыву связей между отделами центральной нервной системы (ЦНС) и, как следствие, вызывающая нарушения памяти, координации движения, мыслительных способностей человека. Наиболее известными представителями этого класса заболеваний являются болезнь Альцгеймера, болезнь Паркинсона, боковой амиотрофический склероз (БАС), хорея Гентингтона, фронто-темпоральная деменция и другие. Поскольку в мире наблюдается неуклонное старение населения, общая частота НДЗ имеет четкую тенденцию к увеличению. К сожалению, для большинства НДЗ отсутствуют радикальные методы лечения, которые позволили бы полностью остановить патологический процесс и тем более обратить его вспять. Возможности симптоматической помощи таким пациентам ограниченны, причем в поздних стадиях лечение особенно затруднено и нередко сопровождаются многочисленными осложнениями.
Болезнь Альцгеймера (БА) является самым распространенным нейродегенеративным заболеванием (50-60% всех случаев) и характеризуется прогрессирующим снижением памяти и высших корковых функций, что приводит к полной деградации умственной и интеллектуальной деятельности [Huang Y., Mucke L., Cell, 2012, 148, 1204-1222; Gandy S., DeKosky ST., Annu. Rev. Med., 2013, 64, 367-383; Tahami Monfared A. et al., Neurol. Ther., 2022, 11, 553-569]. По прогнозу экспертов число людей с деменцией в мире возрастет с 36 млн в 2010 г. до 66 млн в 2030 г. и до 115 млн к 2050 г. [Alzheimer's disease facts and figures. Alzheimers Dement., 2022, 18, 700-789]. В России насчитывается от 1,5 до 2-х миллионов больных с этим опасным нарушением, а это значит, что Россия входит в первую десятку по числу заболевших [https://demenciya.ru/altsgejmer/statistika-bolezni-altsgejmera/]. Таким образом, БА представляет серьезную медико-социальную проблему, и разработка агентов для борьбы с этим заболеванием является одним из приоритетных направлений медицинской химии [Ballard С.et al., Lancet, 2011, 377(9770), 19-25].
В настоящее время общепризнанным фактом является мультифакторная природа БА [Carreiras М.С.et al., Curr. Top.Med. Chem., 2013, 13, 1745-1770; Bachurin S.O. et al., Med. Res. Rev., 2017, 37(5), 1186-1225; Rossi M. et al., J. Med. Chem., 2021, 64, 4972-4990; Blaikie L. et al, Med. Chem. Comm., 2019, 10, 2052-2072; Blaszczyk J.W. Int. J. Mol. Sci., 2023, 24(1), 543]. К основным факторам, характеризующим ее патогенез, можно отнести нарушение работы нейромедиаторных систем, отложение аберрантных белков (бета-амилоида и тау-белка), окислительный стресс [Huang Y., Mucke L., Cell, 2012, 148, 1204-1222].
Поиск препаратов, способных компенсировать утраченные функции холинэргической системы изначально рассматривался как наиболее очевидная стратегия создания лекарств терапии БА. Ингибиторы холинэстераз (преимущественно ацетилхолинэстеразы) донепезил (Арисепт®), ривастигмин (Экселон®), галантамин, а также низкоаффинный неконкурентный антагонист NMDA-рецепторов - мемантин являются до настоящего времени основными терапевтическими средствами для лечения этого заболевания [Klafki H.W. et al., Brain., 2006, 129, 2840-2855; Mangialasche F. et al., Lancet Neurol., 2010, 9, 702-716]. Однако перечисленные препараты являются преимущественно симптоматическими, в частности, действие ингибиторов холинэстераз направлено на компенсацию дефицита нейромедиатора ацетилхолина [Martines A., Castro A., Expert Opin. Investig., 2006, 15, 1-12]. Следует отметить, что в здоровом мозге ацетилхолин преимущественно (на 80%) гидролизуется ацетилхолинэстеразой (АХЭ, КФ 3.1.1.7), тогда как бутирилхолинэстераза (БХЭ, КФ 3.1.1.8) играет вспомогательную роль. Однако по мере прогрессии БА активность АХЭ снижается, в то время как активность БХЭ постепенно увеличивается. В связи с этим возрастает роль БХЭ как терапевтической мишени для снижения наблюдаемого при БА холинергического дефицита [Furukawa-Hibi Y. et al, Behav. Brain Res., 2011, 225, 222-229; Wang L. et al., Eur. J. Med. Chem., 2014, 80, 543-561; Kamal M.A. et al., Neurochem. Res., 2008, 33, 745-753]. Полагают, что соединения, ингибирующие обе холинэстеразы - АХЭ и БХЭ, повышают эффективность лечения [Lane R.M. et al., Int. J. Neuropsychopharmacol., 2006, 9,101-124; GreigN. et al, Proc.Natl. Acad. Sci. U.S.A., 2005, 102,17213-17218].
Агрегация и отложение пептидов бета-амилоида в мозге играет ключевую роль в возникновении и прогрессии БА [Hsiao К. et al, Science, 1996, 274, 99-102; Hardy J. et al, J. Intern. Med., 2014, 275, 296-303]. Амилоидные бляшки или, более вероятно, их растворимые олигомеры, являются нейротоксичными, оказывая свое действие путем нарушения функции митохондрий, индукции апоптоза, а также стимуляции стресс-активированной протеинкиназы [Tumiatti V. et al., Curr. Med. Chem., 2010, 17, 1825-1838]. Полагают, что препараты, снижающие уровень бета-амилоида в головном мозге путем снижения скорости его образования или увеличения его клиренса, могут остановить или даже обратить вспять прогрессирование заболевания.
Известно, что АХЭ помимо классической функции гидролиза ацетилхолина обладает проагрегантными свойствами в отношении бета-амилоида [Moran М.А. et al, Acta Neuropathol., 1994, 87(3), 284-292]. АХЭ играет важную роль в процессинге бета-амилоида через участие периферического анионного сайта (PAS), который взаимодействует с растворимыми пептидами бета-амилоида, промотируя их агрегацию [Inestrosa N.C. et al., Mol. Psychiatry., 1996, 1(5), 359-361; De Ferrari G.V. et al., Biochemistry, 2001, 40(35), 10447-10457; Bartolini M. et al., Biochem. Pharmacol., 2003, 65(3), 407-416]. Эти факты привели к интенсивному исследованию соединений, проявляющих двойственную активность: как ингибиторы АХЭ и как ингибиторы агрегации бета-амилоида, т.е. такого рода соединения способны одновременно улучшать когнитивные функции и проявлять нейропротекторные свойства [Arce М.Р. et al, J. Med. Chem., 2009, 52(22), 7249-7257; de Los Ri'os C. et al., J. Med. Chem., 2010, 53(14), 5129-5143; Camps P. et al., Chem. Biol. Interact., 2010, 187(1-3), 411-415; Rouleau J. et al., Eur. J. Med. Chem., 2011, 46(6), 2193-2205].
Одним из важных факторов, отрицательно воздействующих на жизнедеятельность нейронов головного мозга, является окислительный стресс, характеризующийся дисбалансом между образованием свободных форм кислорода и их удалением с помощью различных механизмов антиоксидантной системы. Следует отметить, что по сравнению с другими тканями организма мозг более чувствителен к повреждению свободными радикалами. Эффективность работы антиоксидантной системы в мозге постепенно снижается в процессе старения, причем в мозге больных БА это снижение происходит наиболее резко. Все это обусловливает целесообразность использования антиоксидантов в терапии БА [Chakrabarti S. et al., Curr. Med. Chem., 2013, 37, 4648-4664; Ortiz G.G. et al., In Adv. Protein Chem. Struct. Biol, Academic Press: 2017; Pohanka M., Bratisl. Lek. Listy, 2018, 119, 535-543] и создание ингибиторов холинэстераз с дополнительными антиоксидантными свойствами [Rosini М. et al., Eur. J. Med. Chem., 2011, 46(11), 5435-5442; Pohanka M., Bratisl. Lek. Listy, 2018, 119, 535-543].
Принимая во внимание множественность биологических систем, вовлеченных в патогенез и прогрессию БА, крайне перспективным подходом в разработке препаратов для лечения БА представляется концепция создания мультифункциональных, мультитаргетных препаратов, оказывающих комплексное воздействие на ряд биологических мишеней, ответственных за патогенез заболевания [Rosini М. et al., Neurochem. Res., 2014, 39, 1914-1923; Agis-Torres A. et al., Curr. Neuropharmacol., 2014, 12, 2-36; Dias K.S., Viegas C, Jr. Curr. Neuropharmacol., 2014, 12, 239-255; Bolognesi M.L., Cavalli A., Chem.Med.Chem., 2016, 11, 1190-1192; Makhaeva, G.F. et al., Chem. Biol. Interact., 2019, 308, 224-234; Bachurin, S.O. et al., Russ. Chem. Bull., 2023, 72, 130-147; Makhaeva G.F. et al., Sci. Rep., 2015, 5, 13164; Bachurin S.O. et al, Sci. Rep., 2017, 7, 45627; Makhaeva G.F. et al., Int. J Mol. Sci., 2023, 24, 2285]. Создание единой химической молекулы, способной одновременно влиять на множественные патогенетические звенья заболевания, имеет дополнительные преимущества по сравнению с комбинациями лекарственных средств как с точки зрения оптимального ADMET-профиля, так и в плане снижения риска нежелательных эффектов от взаимодействий отдельных лекарственных компонентов.
Уровень техники
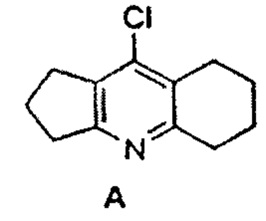
Одним из известных препаратов для лечения БА является такрин - мощный ингибитор АХЭ и БХЭ. Такрин (1,2,3,4-тетрагидроакридин-9-амин) был первым препаратом, одобренным FDA для лечения БА, но его применение было ограниченным в связи с обнаруженной гепатотоксичностью. В связи с этим, в качестве исходного фармакофора для создания мультитаргетных конъюгатов в данном изобретении был выбран схожий по структуре с такрином ингибитор холинэстераз ипидакрин (моногидрат 9-амино-2,3,5,6,7,8-гексагидро-1H-циклопента[b]хинолина моногидрохлорид, также известный как амиридин, NIK-247, нейромидин, аксамон, ипигрикс), который широко применяется в России для терапии БА и ряда других центральных и периферических неврологических расстройств. Ипидакрин был разработан в СССР в середине 1980-х годов [«Способ получения 2,3-замещенных 4-амино-6,7-дигидро-5H-1-пиридинов» SU615653A1 от 01.11.1976; Упадышева А.В. и др., Хим. гетероцикл. соед., 1983, 1, 107-111], а в 1990 г. одобрен к применению в качестве стимулятора обучения и памяти для лечения БА и других форм старческого слабоумия, а также для лечения трудностей в обучении у детей с минимальной церебральной дисфункцией [Букатина Е.Е. и др., Журн. невропатол. и псих., 1991, 91(9), 53-58]. Синтез ипидакрина и его гидрохлорида гидрата запатентован в России, США и Японии [«Способ получения производных 4-аминопиридина» RU 2069660 C1 от 27.11.1996; «Process for the preparation of Ipidacrine or Ipidacrine hydrochloride hydrate» US 6433173 B1 от 13.08.2002; «Method for producing ipidacrine hydrochloride hydrate» JP 2001106674 A от 17.04.2000; «Способ получения 9-амино-2,3,5,6,7,8-гексагидро-1H-циклопента[b]хинолина хлоргидрата, гидрата» RU 2659783 C1 от 04.07.2018]
Несмотря на структурное сходство с такрином, ипидакрин проявляет значительно меньше периферических и центральных холинергических побочных эффектов, отличается отсутствием гепатотоксичности и более широким терапевтическим окном [Yoshida S. et al., Eur. J. Pharmacol., 1993, 250(1), 117-124; Kojima J. et. al, CNS Drug Rev., 2006, 4(3), 247-259]. Ипидакрин обладает широким спектром биологической активности; в частности, это обратимый ингибитор АХЭ и БХЭ, обладающий селективностью в отношении БХЭ. Помимо этого, ипидакрин восстанавливает обмен жирных кислот и фосфолипидов, нормализует физико-химические свойства мембран, блокирует проницаемость калиевых мембран. Описана также способность ипидакрина восстанавливать нарушенную структуру тубулина микротрубочек [Шевцов П.Н., Шевцова Е.Ф., Бурбаева Г.Ш., Бюллетень экспериментальной биологии и медицины, 2008, 145(2), 178-182; Шевцов П.Н. и др., Бюллетень экспериментальной биологии и медицины, 2013, 156(12), 728-733]. Следует отметить, что ипидакрин быстро проникает в головной мозг и накапливается в основном в коре головного мозга и гиппокампе -структурах, наиболее подверженных повреждению при БА. Усиление долговременной потенциации в гиппокампе и очень слабое антагонистическое действие на центральные мускариновые рецепторы вместе могут вносить вклад в активность ипидакрина как антиамнестического средства in vivo [Kojima J et al., CNS Drug Rev., 2006, 4(3), 247-259]. Запатентованы возможности применения ипидакрина в медицине: в качестве стимулятора нервно-мышечной передачи [«Стимулятор нервно-мышечной передачи "Амиридин"» SU1528499A1 от 27.12.1979], агента для улучшения повседневной жизни пациентов с БА [«Improving agent for activity of daily living of Alzheimer's dementia» JP 2004292316 A от 21.10.2004] и терапии нейродегенеративных заболеваний [«Neurotrophic factor-like agent» JP 2002255819 A от 11.09.2002], а также в качестве агента для терапии нарушений потенции [((Pharmaceutical Compositions Containing Ipidacrine and Use Thereof to Treat Potency Disorders and Other Sexual Activity Disorders)) AU 2018200470 A1 от 19.01.2018] и лечения нарушений целостности кости [«Фармацевтические композиции на основе ипидакрина и их применение для лечения и профилактики нарушений целостности кости», RU 2512743 C1 от 10.04.2014]. Кроме того, запатентовано применение препарата Нейромидин, с действующим веществом ипидакрин, в качестве одного из компонентов для комплексной терапии различных заболеваний и травм. К таким изобретениям относятся: способ лечения травматических невропатий [«Способ лечения травматических невропатий», RU 2459642 C2 от 27.08.2012], глаукомной оптической нейропатии [«Способ лечения глаукомной оптической нейропатии», RU 2546019 C2 от 10.04.2015] и нейропатии язычного нерва [«Способ лечения нейропатии язычного нерва», RU 2665971 C1 от 05.09.2018], способ реабилитации неврологических нарушений у детей при нейроинфекциях [«Способ реабилитации неврологических нарушений у детей при нейроинфекциях», RU 2482843 C2 от 27.05.2013], способ лечения и профилактики нейросенсорной тугоухости и шумовых эффектов внутреннего уха [«Способ лечения и профилактики нейросенсорной тугоухости и шумовых эффектов внутреннего уха, связанных с воздействием производственного шума», RU 2554813 C1 от 27.06.2015] и обонятельной дисфункции при атрофическом рините [«Способ лечения пациентов с обонятельной дисфункцией при атрофическом рините», RU 2713786 C1 от 07.02.2020], способ восстановительного лечения нервно-мышечного аппарата у больных с ложным суставом шейки бедренной кости после эндопротезирования тазобедренного сустава [«Способ восстановительного лечения нервно-мышечного аппарата у больных с ложным суставом шейки бедренной кости после эндопротезирования тазобедренного сустава», RU 2528637 C1 от 20.09.2014], способ лечения острого ишемического инсульта [«Способ. лечения острого ишемического инсульта», UA95834U от 12.01.2015] и другие.
Таким образом, ипидакрин является перспективным фармакофором для создания анти-БА агентов.
В литературе имеются данные всего о нескольких производных ипидакрина, что связано с его низкой реакционной способностью. Наиболее близкими к данному изобретению можно признать описанные в трех недавних публикациях конъюгаты ипидакрина (прототипы А-С). Стратегия синтеза конъюгатов основана на использовании ипидакрина, аминогруппу которого вовлекают во взаимодействие с высокореакционноспособными хлорсодержащими агентами для введения спейсера с последующим конъюгированием со вторым фармакофором.
В работе [Makhaeva G.F. et al., Bioorg. Chem., 2021, 112, 104974] описаны конъюгаты ипидакрина с N-замещенными пиперазинами (прототип А). На первом этапе ипидакрин ацилируют хлорацетилхлоридом, а затем замещают атом хлора на производные пиперазина. Полученные соединения в зависимости от структуры заместителя в пиперазиновом фрагменте проявляют разный уровень ингибиторной активности в отношении АХЭ и БХЭ, которая, однако, не превышает антихолинэстеразную активность ипидакрина, а также обладают дополнительной биологической активностью, проявляя антиоксидантные свойства или потенциальную способность блокировать АХЭ-индуцируемую агрегацию бета-амилоида.
Реакцией ипидакрина с тиофосгеном с последующим присоединением к образовавшемуся в результате реакции изотиоцианату различных (гет)арилалкиламинов были синтезированы конъюгаты ипидакрина, содержащие тиомочевинный фрагмент (прототип Б) [Махаева Г.Ф. и др., Изв. АН. Сер. хим., 2022, 11, 2404-2415]. Полученные соединения очень слабо ингибируют АХЭ, проявляют анти-БХЭ активность, не достигая, однако, уровня ипидакрина, а также обладают антиоксидантными свойствами и антиагрегантной активностью в отношении бета-амилоида.
В статье [Makhaeva G.F. et al., Molecules, 2022, 27(3), 1060] избыток ацилированного или изотиоцианатного производных ипидакрина вводят в реакцию с различными диаминоалканами для получения бисипидакринов с бис-N-ацил- или бис-N-тиомочевино-алкиленовыми спейсерами (прототип С). Соединения проявляют ингибиторную активность в отношении АХЭ и БХЭ, антиоксидантные свойства, обладают потенциальной способностью блокировать АХЭ-индуцируемую агрегацию и самоагрегацию бета-амилоида.
Однако, существенным недостатком описанных конъюгатов (прототипы А-С) является их невысокая ингибиторная активность в отношении холинэстераз по сравнению с ипидакрином. Это связано с тем, что для функционализации ипидакрина с целью преодоления его низкой реакционной способности и присоединения спейсера использовали высокореакционные хлорсодержащие агенты (хлорангидрид хлоруксусной кислоты и тиофосген). Данное ацилирование по внешнему азоту приводит к снижению рКа ипидакринового фрагмента, его способности протонироваться и соответственно связываться с каталитическим сайтом АХЭ, что приводит к снижению анти-АХЭ активности соединения.
Объект изобретения
Задачей настоящего изобретения является расширение арсенала средств, которые могут быть использованы в качестве новых мультифункциональных соединений, обладающих способностью одновременно высокоэффективно ингибировать ацетилхолинэстеразу и бутирилхолинэстеразу, связываться с периферическим анионным сайтом ацетилхолинэстеразы, связывать свободные радикалы и ингибировать самоагрегацию бета-амилоида, и содержащим их фармацевтическим средствам, для лечения нейродегенеративных заболеваний.
Поставленная задача решается оригинальным синтезом новых конъюгатов ипидакрина с 2-замещенными фенолами общей формулы I:
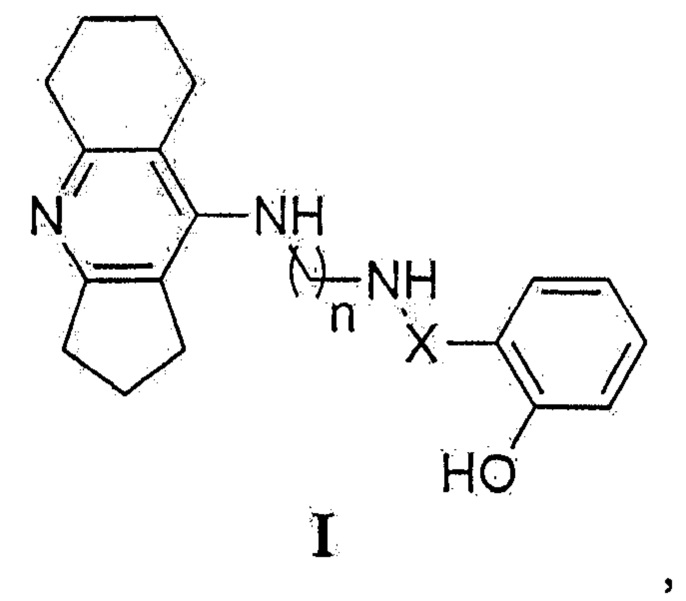
где X: -С(О)-=СН- или -СН2-; n=4, 6, 8,
обнаружением и использованием новых их свойств - способности одновременно ингибировать ацетилхолинэстеразу и бутирилхолинэстеразу, связываться с периферическим анионным сайтом ацетилхолинэстеразы, связывать свободные радикалы и ингибировать самоагрегацию бета-амилоида.
Главным аспектом настоящего изобретения является оригинальный синтез новых физиологически активных соединений, объединяющих в одной молекуле два активных фармакофора.
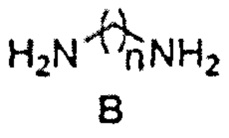
В основе предлагаемого нами синтеза лежит новый способ модификации ипидакрина путем замещения атома хлора в его хлорпроизводном А на аминогруппу диамина В, что позволяет ввести диаминоалкиленовый спейсер и соответственно получить конъюгаты с высокой ингибиторной активностью в отношении холинэстераз, которая превышает активность ипидакрина. Ранее в литературе описан метод получения хлорпроизводного ипидакрина (9-хлор-2,3,5,6,7,8-гексагидро-1H-циклопента[b]хинолина) [Жидкова A.M. и др., Хим.-фарм. журн., 1989, 23(9), 709-712]. Однако, существенным недостатком этого синтеза является то, что атом хлора в данном соединении обладает низкой реакционной способностью, а реакция замещения атома хлора на аминогруппу метиламина была проведена в жестких условиях: нагреванием хлорпроизводного ипидакрина в 25%-ом водном растворе метиламина до 210°С в автоклаве в течение 18 ч с выходом 97%.
Нами предложено проводить реакцию замещения атома хлора в соединении А на аминогруппу соединения В в пентаноле-1 в присутствии иодида щелочного металла в закрытом сосуде при 190°С в течение 12-16 ч. Применение таких условий позволило впервые ввести в ипидаркин аминополиметиленовый спейсер.
В настоящем изобретении в качестве второго фармакофора при разработке многофункциональных конъюгатов с ипидакрином выбраны замещенные фенолы, содержащие во 2-ом положении амидную, иминную или аминную группы. Значительный фармакологический потенциал таких фрагментов обусловлен широким спектром их биологической активности, включая антиоксидантные и металлохелатирующие свойства, а также способность ингибировать агрегацию бета-амилоида [Borges R.S. et al., Chem. Biol. Drag. Des., 2013, 81(3), 414-419; Borges R.S., Castle S.L., Bioorg. Med. Chem. Lett., 2015, 25(21), 4808-4811; Song Q. et al., Bioorg. Chem., 2019, 84, 137-149]. Данных о синтезе из хлорзамещенного ипидакрина аминоалкиленовых производных, а затем конъюгатов с 2-функциональнозамещенными фенолами не обнаружено.
Таким образом, авторы настоящего изобретения предлагают новые мультифункциональные соединения, которые с одной стороны, содержат оригинальный фармакофор - 2-замещенные фенолы, с другой, обладают расширенным спектром фармакологической активности. Предложенные в данном изобретении структуры проявляют способность одновременно действовать на четыре биологические мишени, ответственные за патогенез заболевания, а именно обладают способностью эффективно ингибировать АХЭ и БХЭ, связываться с периферическим анионным сайтом ацетилхолинэстеразы, ингибировать самоагрегацию бета-амилоида, а также связывать свободные радикалы.
Конъюгаты ипидакрина с 2-замещенными фенолами общей формулы I (соединения 1-9) были получены по следующей схеме:
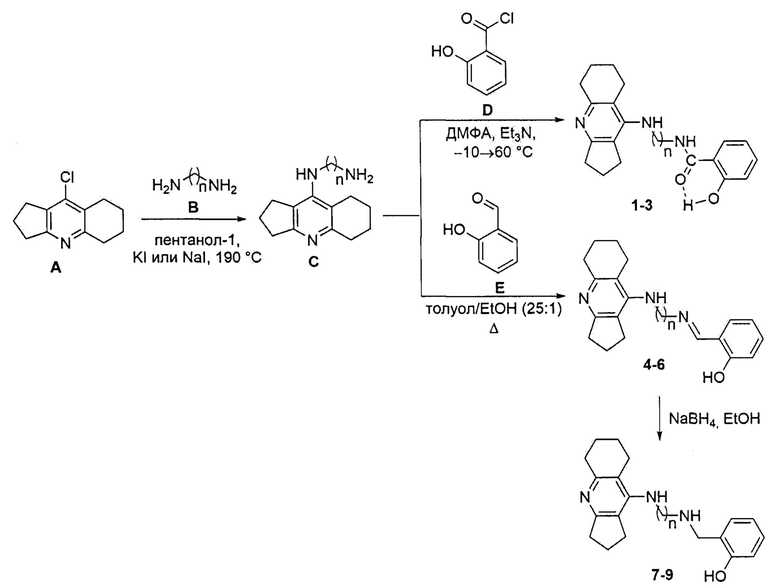
На первой стадии синтеза замещением атома хлора в производном ипидакрина А на аминогруппу диаминоалканов В вводили диаминоалкиленовый спейсер. На второй стадии взаимодействием производных ипидакрина С с хлорангидридом салициловой кислоты D в ДМФА в присутствии триэтиламина при нагревании получены конъюгаты 1-3. Конденсацией соединений С с салициловым альдегидом Е в смеси толуол/EtOH (25:1) были синтезированы производные салицилимина 4-6. Для получения аминопроизводных 7-9 проводилась третья стадия - восстановление иминов 4-6 боргидридом натрия в EtOH.
Общая методика синтеза соединений С.
К раствору хлорпроизводного ипидаркина А (0.025 моль) в пентаноле-1 (30 мл) добавляли KI или Nal (0.0001 моль) и соответствующий диамин (0.175 моль). Реакционную массу перемешивали в закрытом стальном сосуде при 190°С в течение 12-16 ч. Затем отгоняли растворитель и диамин (для С с n=8 при пониженном давлении), добавляли к реакционной смеси хлороформ (100 мл), промывали органический слой 10% раствором NaOH (3×100 мл), водой (3×100 мл), сушили над безводным сульфатом натрия, упаривали. Остаток очищали методом колоночной хроматографии (элюент CHCl3/EtOH/NH4OH 10:1:0.1→5:1:0.1). Полученное масло обрабатывали диэтиловым эфиром, образовавшийся осадок отфильтровывали, сушили.
N1-(2,3,5,6,7,8-гексагидро-1H-циклопента[b]хинолин-9-ил)бутан-1,4-диамин (А, n=4). Выход 3.950 г (61%). Бежевый порошок, т.пл. 81-83°С.
Спектр ЯМР 1H (CDCl3, δ, м.д., J/Гц): 1.31-1.38,1.42-1.49 (оба м, 4Н, 2СН2); 1.65-1.75 (м, 4Н, 2СН2); 1.91 (квинтет, J=7.5 Гц, 2Н, СН2); 2.32 (т, J=5.9 Гц, 2Н, СН2); 2.53 (т, J=6.8 Гц, 2Н, СН2); 2.57-2.64 (м, 4Н, 2СН2); 2.95 (т, J=7.2 Гц, 2Н, СН2); 3.27-3.32 (м, 2Н, СН2); 5.05 (т, J=6.2 Гц, 1Н, NHипидакрин); сигнал NH2-группы не наблюдался из-за дейтерообмена с растворителем.
Спектр ЯМР 13С (CDCl3, δ, м.д.): 22.53; 22.59; 22.71; 23.34; 28.36; 30.18; 30.62; 32.58; 33.41; 41.34; 43.76; 113.42; 114.46; 149.02; 153.22; 162.33.
Элементный анализ для C16H25N3 (259.20):
Вычислено (%): С, 74.09; Н, 9.71; N, 16.20.
Найдено (%): С, 73.91; Н, 9.64; N, 16.01.
N1'-(2,3,5,6,7,8-гексагидро-1H-циклопента[6]хинолин-9-ил)гексан-1,6-диамин (А, n=6). Выход 4.161 г (58%). Бежевый порошок, т.пл. 102-104°С.
Спектр ЯМР 1H(CDCl3, δ, м.д,, J/Гц): 1.31-1.38,1.42-1.49 (обам, 4Н, 2СН2); 1.65-1.75 (м, 4Н, 2СН2); 1.91 (квинтет, J=7.5 Гц, 2Н, СН2); 2.32 (т, J=5.9 Гц, 2Н, СН2); 2.53 (т, J=6.8 Гц, 2Н, СН2); 2.57-2.64 (м, 4Н, 2СН2); 2.95 (т, J=7.2 Гц, 2Н, СН2); 3.27-3.32 (м, 2Н, СН2); 5.05 (т, J=6.2 Гц, 1Н, NHипидакрин); сигнал NH2-группы не наблюдался из-за дейтерообмена с растворителем.
Спектр ЯМР 13С (CDCl3, δ, м.д.): 22.53; 22.59; 22.71; 23.34; 28.36; 30.18; 30.62; 32.58; 33.41; 41.34; 43.76; 113.42; 114.46; 149.02; 153.22; 162.33.
Элементный анализ для C18H29N3 (287.24):
Вычислено (%): С, 75.21; Н, 10.17; N, 14.62.
Найдено (%): С, 75.03; Н, 9.92; N, 14.45.
N1-(2,3,5,6,7,8-гексагидро-1H-циклопента[6]хинолин-9-ил)октан-1,8-диамин (А, n=8). Выход 4.332 г (55%). Бежевый порошок, т.пл. 83-85°С.
Спектр ЯМР 1H (CDCl3, δ, м.д., J/Гц): 1.22-1.26 (м, 8Н, 4СН2); 1.31-1.34,1.38-1.44 (обам, 4Н, 2СН2); 1.67-1.72 (м, 4Н, 2СН2); 1.91 (квинтет, J=7.5 Гц, 2Н, СН2); 2.31 (т, J=5.9 Гц, 2Н, СН2); 2.53 (т, J=7.0 Гц, 2Н, СН2); 2.58 (т, J=5.9 Гц, 2Н, СН2); 2.62 (т, J=7.7 Гц, 2Н, СН2); 2.93-2.96 (м, 2Н, СН2); 3.25-3.31 (м, 2Н, СН2); 5.00 (т, J=6.3 Гц, 1H, NHипидакрин); сигнал NH2-группы не наблюдался из-за дейтерообмена с растворителем. Спектр ЯМР 13С (CDCl3, δ, м.д.): 22.51; 22.58; 22.68; 23.33; 26.00; 26.15; 28.75; 28.78; 30.62; 30.79; 31.42; 32.54; 33.39; 40.78; 43.79; 113.45; 114.50; 149.05; 153.19; 162.29.
Элементный анализ для C20H33N3 (315.27):
Вычислено (%): С, 76.14; Н, 10.54; N, 13.32.
Найдено (%): С, 76.18; Н, 10.22; N, 13.57.
Общая методика синтеза соединений 1-3.
К раствору смеси хлорангидрида салициловой кислоты D (0.156 г, 0.0010 моль) и триэтиламина (0.17 мл, 0.0012 моль) в ДМФА (1 мл) при охлаждении (-10-0°С) прибавляли порциями производное ипидакрина С (0.0010 моль). Реакционную массу перемешивали при температуре 65°С в течение 3-4 ч. После охлаждения смеси до комнатной температуры добавляли хлористый метилен (30 мл), промывали органический слой водой (3x25 мл), сушили над безводным сульфатом натрия, упаривали. Образовавшийся осадок промывали этанолом (5 мл), сушили.
Ниже приведены примеры синтезов, которые иллюстрируют данное изобретение.
Пример 1. N-(4-((2,3,5,6,7,8-гексагидро-1H-циклопента[6]хинолин-9-ил)амино)бутил)-2-гидроксибензамид (1). (Соединение формулы I, где n=4, X: -С(О)-). Выход 0.233 г (62%). Белый порошок, т.пл. 192-194°С.
Спектр ЯМР 1Н (CDCl3, δ, м.д., J/Гц): 1.64-1.67, 1.70-1.75 (оба м, 4Н, 2СН2); 1.76-1.84 (м, 4Н, 2СН2); 2.01 (квинтет, J=7.5 Гц, 2Н, СН2); 2.36 (т, J=6.1 Гц, 2Н. СН2); 2.80 (т, J=6.1 Гц, 2Н, СН2); 2.84 (т, J=7.8 Гц, 2Н, СН2); 3.02 (т, J=7.3 Гц, 2Н, СН2); 3.46-3.52 (м, 4Н, 2СН2); 3.84 (т, J=6.0 Гц, 1Н, NHипидакрин); 6.63 (уш. с, 1H, NHJ, 6.82 (тд, J=7.6, 1.1 Гц, 1Н, Наром); 6.98 (дд, J=8.3, 1.0 Гц, 1H, Наром); 7.34 (дд, J=8.0, 1.5 Гц, 1Н, Наром); 7.38 (тд, J=7.8, 1.6 Гц, 1Н, Наром); сигнал ОН-группы не наблюдался из-за дейтерообмена с растворителем.
Спектр ЯМР 13С (CDCl3, δ, м.д.): 22.73; 22.98; 23.10; 23.57; 26.82; 28.58; 31.21; 32.78; 34.05; 39.28; 44.54; 113.94; 114.51; 116.03; 118.59; 118.63; 125.50; 134.15; 149.35; 154.38; 161.47; 163.52; 169.9
Элементный анализ для C23H29N3O2 (379.23):
Вычислено (%): С, 72.79; Н, 7.70; N, 11.07.
Найдено.(%): С, 72.59; Н, 7.72; N, 10.89.
Пример 2. N-(6-((2,3,5,6,7,8-гексагидро-1H-циклапенто[6]хинолин-9-ил)амино)гексил)-2-гидроксибензамид (2). (Соединение формулы I, где n=6, X: -С(О)-). Выход 0.260 г (64%). Белый порошок, т.пл. 210-212°С.
Спектр ЯМР:Н (CDCI3, 5, м.д., J/Гц): 1.41-1.45, 1.55-1.66, 1.76-1.88 (все м, 12Н, 6СН2); 2.04 (квинтет, J=7.5 Гц, 2Н, СН2); 2.35 (т, J=5.9 Гц, 2Н, СН2); 2.83 (т, J=6.0 Гц, 2Н, СН2); 2.89 (т, J=7.7 Гц, 2Н, СН2); 3.05 (т, J=7.2 Гц, 2Н, СН2); 3.41-3.49 (м, 4Н, 2СН2); 3.89 (уш. с, 1Н, NHипидакрин); 6.38 (уш. с, 1H, NHамид); 6.84 (тд, J=7.9, 0.7 Гц, 1Н, Наром); 6.99 (д, J=8.4 Гц, 1Н, Наром); 7.35 (дд, J=8.0, 1.6 Гц, 1Н, Наром); 7.39 (тд, J=8.0, 1.4 Гц, 1Н, Наром); сигнал ОН-группы не наблюдался из-за дейтерообмена с растворителем.
Спектр ЯМР 13С (CDCl3, δ, м.д.): 22.60; 22.92; 23.12; 23.47; 26.35; 26.61; 29.50; 29.69; 31.11; 31.22; 33.82; 39.45; 44.91; 113.85; 114.38; 116.04; 118.57; 118.72; 125.18; 134.17; 149.95; 153.67; 161.60; 166.39; 169.97.
Элементный анализ для C25H33N3O2 (407.26):
Вычислено (%): С, 73.68; Н, 8.16; N, 10.31.
Найдено (%): С, 73.37; Н, 8.22; N, 10.15.
Пример 3. N-(8-((2,3,5,6,7,8-гексагидро-1H-циклопента[6]хинолин-9-ил)амино)октил)-2-гидроксибензамид (3). (Соединение формулы I, где n=8, X: -С(О)-). Выход 0.261 г (60%). Белый порошок, т.пл. 148-150°С.
Спектр ЯМР 1Н (CDCl3, δ, м.д., J/Гц): 1.31-1.38 (м, 8Н, 4СН2); 1.52-1.62, 1.78-1.84 (оба м, 8Н, 4СН2); 2.03 (квинтет, J=7.5 Гц, 2Н, СН2); 2.35 (т, J=5.3 Гц, 2Н, СН2); 2.81 (т, J=5.5 Гц, 2Н, СН2); 2.85 (т, J=7.7 Гц, 2Н, СН2); 3.05 (т, J=7.1 Гц, 2Н, СН2); 3.39-3.45 (м, 4Н, 2СН2); 3.84 (неразр. т, 1H, NHипидакрин); 6.79 (уш. с, 1Н, NHамид); 6.83 (т, J=7.5 Гц, 1H, Наром); 6.97 (д, J=8.2 Гц, 1Н, Наром); 7.36 (т, J=7.4 Гц, 1Н, Наром); 7.46 (д, J=7.9 Гц, 1Н, Наром); сигнал ОН-группы не наблюдался из-за дейтерообмена с растворителем.
Спектр ЯМР 13С (CDCl3, δ, м.д.): 22.65; 22.93; 23.12; 23.44; 26.58; 26.78; 29.10; 29.14; 29.45; 31.14; 31.17; 32.46; 33.85; 39.57; 44.96; 113.76; 114.93; 115.97; 118.46; 118.48; 125.87; 133.86; 149.82; 153.86; 161.30; 163.09; 169.66.
Элементный анализ для С27Нз7Кз02 (435.29):
Вычислено (%): С, 74.45; Н, 8.56; N, 9.65.
Найдено (%): С, 74.26; Н, 8.82; N, 9.40.
Общая методика синтеза соединений 4-6
К раствору салицилового альдегида Е (0.10 мл, 0.001 моль) в толуоле (50 мл) добавляли раствор производного ипидакрина С в этаноле (2 мл). Реакционную массу кипятили с азеотропной отгонкой воды (для соединения 4) или с молекулярными ситами 4А (для соединений 5, 6) в течение 5-6 ч. В случае соединения 4 образовавшийся осадок отфильтровывали, промывали гексаном (25 мл), сушили. В случае соединений 5, 6 реакционную смесь упаривали, остаток очищали методом колоночной хроматографии (элюент CHCl3/EtOH 20:1).
Ниже приведены примеры синтезов, которые иллюстрируют данное изобретение.
Пример 4. 2-(((4-((2,3,5,6,7,8-гексагидро-1H-циклопента[6]хинолин-9-ил)амино)бутил)имино)метил)фенол (4). (Соединение формулы I, где n=4, X:=СН-). Выход 0.309 г (85%). Желтый порошок, т.пл. 152-153°С.
Спектр ЯМР 1H (CDC13, δ, м.д., J/Гц): 1.62-1.69 (м, 2Н, СН2); 1.76-1.83 (м, 6Н, 3СН2); 2.02 (квинтет, J=7.5 Гц, 2Н, СН2); 2.35 (т, J=5.8 Гц, 2Н, СН2); 2.80 (т, J=5.7 Гц, 2Н, СН2), 2.85 (т, J=7.7 Гц, 2Н, СН2), 3.04 (т, J=7.3 Гц, 2Н, СН2); 3.44-3.49 (м, 2Н, СН2); 3.63 (тд, J=6.5, 0.9 Гц, 2Н, N-CH2); 3.79 (т, J=5.8 Гц, 1Н, NHипидакрин); 6.88 (тд, J=7.5, 1.1 Гц, 1Н, Наром), 6.96 (д, J=8.3 Гц, 1Н, Наром); 7.25 (дд, J=1.1, 1.7 Гц, 1Н, Наром); 7.31 (тд, J=7.6, 1.7 Гц, 1Н, Наром); 8.35 (с, 1Н, =СН); 13.44 (с, 1Н, ОН).
Спектр ЯМР 13С (CDC13, δ, м.д., J/Гц): 22.73; 22.96; 23.09; 23.50; 28.04; 28.88; 31.17; 32.91; 34.16; 44.74; 59.16; 113.73; 115.81; 116.95; 118.56; 118.65; 131.14; 132.21; 149.26; 154.35; 161.08; 163.56; 164.99.
Элементный анализ для С23Н29K3О (363.23):
Вычислено (%): С, 76.00; Н, 8.04; N, 11.58.
Найдено (%): С, 75.88; Н, 8.19; N, 11.47.
Пример 5. 2-(((6-((2,3,5,6,7,8-гексагидро-1H-циклопента[6]хинолин-9-ил)амино)гексил)имино)метил)фенол (5). (Соединение формулы I, где n=6, X:=СН-). Выход 0.239 г (61%). Желтый порошок, т.пл. 123-125°С.
Спектр ЯМР 1Н (CDC13, δ, м.д., J/Гц): 1.42-1.43 (м, 4Н, 2СН2); 1.54-1.60, 1.69-1.74 (оба м, 4Н, 2СН2); 1.77-1.86 (м, 4Н, СН2); 2.03 (квинтет, J=7.5 Гц, 2Н, СН2); 2.35 (т, J=6.0 Гц, 2Н, СН2); 2.82 (т, J=5.9 Гц, 2Н, СН2); 2.87 (т, J=5.9 Гц, 2Н, СН2); 3.04 (т, J=7.2 Гц, 2Н, СН2); 3.40-3.44 (м, 2Н, СН2); 3.59 (тд, J=6.7, 0.6 Гц, 2Н, N-CH2); 3.83 (неразр. т, 1Н, NHипидакрин); 6.87 (тд, J=7.5, 0.9 Гц, 1Н, Наром); 6.95 (д, J=8.2 Гц, 1Н, Наром); 7.24 (дд, J=7.6, 1.6 Гц, 1Н, Наром); 7.30 (тд, J=1.1, 1.6 Гц, 1Н, Наром); 8.33 (с, 1Н,=СН); 13.61 (с, 1Н, ОН).
Спектр ЯМР 13С (CDC13, δ, м.д.): 22.67; 22.95; 23.11; 23.46; 26.48; 26.91; 30.68; 31.10; 31.18; 32.64; 34.01; 44.95; 59.35; 113.72; 115.91; 116.98; 118.46; 118.74; 131.05; 132.10; 149.65; 153.95; 161.24; 163.18; 164.60.
Элементный анализ для C25H33N30 (391.2624):
Вычислено (%): С, 76.69; Н, 8.50; N, 10.73.
Найдено (%): С, 76.48; Н, 8.38; N, 10.46.
Пример 6. 2-(((8-((2,3,5,6,7,8-гексагидро-1H-циклопента[6]хинолин-9-ил)амино)октил)имино)метил)фенол (6). (Соединение формулы I, где n=8, X:=СН-). Выход 0.249 г (59%). Желтый порошок, т.пл. 93-95°С.
Спектр ЯМР 1Н (CDCl3, δ, м.д., J/Гц): 1.34-1.39 (м, 8Н, 4СН2); 1.51-1.55,1.66-1.72 (оба м, 4Н, 2СН2); 1.77-1.86 (м, 4Н, 2СН2); 2.03 (квинтет, J=7.5 Гц, 2Н, СН2); 2.35 (т, J=6.1 Гц, 2Н, СН2); 2.81 (т, J=6.0 Гц, 2Н, СН2); 2.86 (т, J=7.7 Гц, 2Н, СН2); 3.05 (т, J=7.3 Гц, 2Н, СН2); 3.38-3.42 (м, 2Н, СН2); 3.59 (тд, J=6.7, 0.9 Гц, 2Н, N-CH2); 3.79 (неразр. т, 1Н, NHипидакрин); 6.87 (тд, J=7.5, 1.0 Гц, 1Н, Наром); 6.95 (дд, J=8.3, 0.3 Гц, 1Н, Наром); 7.24 (дд, J=7.6, 1.6 Гц, 1Н, Наром); 7.30 (тд, J=6.4, 1.7 Гц, 1Н, Наром); 8.33 (с, 1Н,=СН), 13.67 (с, 1Н, ОН).
Спектр ЯМР 13С (CDCl3, δ, м.д.): 22.75; 23.01; 23.12; 23.48; 26.67; 27.04; 29.22; 30.78; 31.16; 31.19; 32.82; 34.14; 45.05; 59.46; 113.67; 115.87; 117.00; 118.39; 118.78; 131.02; 132.03; 149.57; 154.15; 161.32; 163.38; 164.46.
Элементный анализ для С27Н37N3О (419.2937):
Вычислено (%): С, 77.28; Н, 8.89; N, 10.01.
Найдено (%): С, 76.99; Н, 8.89; N, 9.82.
Общая методика синтеза соединений 7-9
К раствору соединений 4-6 (0.0005 моль) в этаноле (20 мл) добавляли боргидрид натрия (0.076 г, 0.002 моль). Реакционную массу перемешивали при комнатной температуре в течение 1-2 ч. Затем отгоняли растворитель на ротационном испарителе, добавляли воду (30 мл), экстрагировали продукт хлористым метиленом (3×30 мл). Объединенный органический слой сушили над безводным сульфатом натрия, упаривали.
Ниже приведены примеры синтезов, которые иллюстрируют данное изобретение.
Пример 7. 2-(((4-((2,3,5,6,7,8-гексагидро-1H-циклопента[6]хинолин-9-ил)амино)бутил)амино)метил)фенол (7). (Соединение формулы I, где n=4, X: -СН2-). Выход 0.168 г (92%). Белый порошок, т.пл. 123-124°С.
Спектр ЯМР JH (CDCl3, δ, м.д., J/Гц): 1.61-1.63, 1.77-1.86 (оба м, 8Н, 4СН2); 2.04 (квинтет, J=7.5 Гц, 2Н, СН2); 2.33-2.35 (м, 2Н, СН2); 2.70-2.74 (м, 2Н, СН2); 2.83 (т, J=5.9 Гц, 2Н, СН2); 2.89 (т, J=7.7 Гц, СН2); 3.03 (т, J=7.2 Гц, СН2); 3.43-3.47 (м, 2Н, СН2); 3.90 (уш. с, 1Н, NHипидакрин); 4.00 (с, 2Н, NHCH2Ar); 6.78 (тд, J=7.4, 1.1 Гц, 1Н, Наром); 6.82 (дд, J=8.1, 0.8 Гц, 1Н, Наром); 6.98 (дд, J=7.4, 0.8 Гц, 1Н, Наром); 7.17 (тд, J=8.0, 1.6 Гц, 1Н, Наром), 11-09 (уш. с, 2Н, NH+OH).
Спектр ЯМР 13С (CDCl3, δ, м.д.): 22.49; 22.83; 23.08; 23.42; 26.81; 28.79; 31.18; 32.19; 33.68; 44.65; 48.26; 52.71; 113.90; 115.98; 116.31; 119.04; 122.36; 128.27; 128.74; 149.84; 153.47; 158.12; 162.68.
Элементный анализ для С23Н31N3O (365.25):
Вычислено (%): С, 75.40; Н, 8.78; N, 11.35.
Найдено (%): С, 75.58; Н, 8.55; N, 11.50.
Пример 8. 2-(((6-((2,3,5,6,7,8-гексагидро-1H-циклопента[6]хинолин-9-ил)амино)гексил)амино)метил)фенол (8). (Соединение формулы I, где n=6, X: -СН2-). Выход 0.175 г (89%). Бесцветное масло.
Спектр ЯМР 1Н (CDCl3, δ, м.д., J/Гц): 1.36-1.40, 1.51-1.59, 1.76-1.87 (все м, 12Н, 6СН2); 2.03 (квинтет, J=7.5 Гц, 2Н, СН2); 2.35 (т, J=5.6 Гц, 2Н, СН2); 2.68 (т, J=7.0 Гц, 2Н, СН2); 2.81 (т, J=5.8 Гц, 2Н, СН2); 2.86 (т, J=7.8 Гц, 2Н, СН2); 3.04 (т, J=7.2 Гц, 2Н, СН2); 3.38-3.43 (м, 2Н, СН2); 3.80 (т, J=4.8 Гц, 1Н, NНипидакрин); 3.99 (с, 2Н, NHCH2Ar); 6.77 (тд, J=7.4, 1.0 Гц, Наром); 6.82 (дд, J=8.1, 0.8 Гц, Наром); 6.99 (дд, J=7.3, 0.5 Гц, 1Н, Наром); 7.16 (тд, J=8.0,1.5 Гц, 1Н, Наром); 10.71 (уш. с, 2Н, NH+OH).
Спектр ЯМР 13С (CDCl3, δ, м.д.): 22.53; 22.86; 23.09; 23.40; 26.51; 26.83; 29.48; 31.10; 31.18; 32.24; 33.74; 44.88; 48.52; 52.75; 113.76; 115.93; 116.29; 118.92; 122.49; 128.19; 128.64; 149.92; 153.48; 158.26; 162.72.
Элементный анализ для C25H35N3O (393.28):
Вычислено (%): С, 76.29; Н, 8.96; N, 10.68.
Найдено (%): С, 76.01; Н, 9.15 N, 10.48.
Пример 9. 2-(((8-((2,3,5,6,7,8-гексагидро-1H-циклопента[6]хинолин-9-ил)амино)октил)амино)метил)фенол (9). (Соединение формулы I, где n=8, X: -СН2-). Выход 0.196 г (93%). Бесцветное масло.
Спектр ЯМР 1Н (CDC13, δ, м.д., J/Гц): 1.29-1.35 (м, 8Н, 4СН2); 1.50-1.57, 1.77-1.86 (оба м, 8Н, 4СН2); 2.03 (квинтет, J=7.5 Гц, 2Н, СН2); 2.35 (т, J=6.0 Гц, 2Н, СН2); 2.67 (т, J=7.1 Гц, 2Н, СН2); 2.81 (т, J=6.0 Гц, 2Н, СН2); 2.87 (т, J=7.7 Гц, 2Н, СН2); 3.05 (т, J=7.2 Гц, 2Н, СН2); 3.39-3.43 (м, 2Н, СН2); 3.80 (уш. с, 1Н, NH„ПИдaкpи„); 3.99 (с, 2Н, NHCH2Ar); 6.77 (тд, J=7.4,1.0 Гц, 1Н, Наром); 6.82 (дд, J=8.1, 0.8 Гц, 1Н, Наром); 6.98 (дд, J=7.3, 0.6 Гц, 1Н, Наром); 7.16 (тд, J=7.7, 1.5 Гц, 1Н, Наром); 11.57 (уш. с, 2Н, NH+OH).
Спектр ЯМР 13С (CDCl3, δ, м.д.): 22.68; 22.96; 23.12; 23.45; 26.65; 27.00; 29.21; 29.29; 29.55; 31.14; 31.19; 32.62; 34.00; 45.03; 48.69; 52.77; 113.69; 115.90; 116.31; 118.89; 122.57; 128.17; 128.62; 149.71; 153.93; 158.32; 163.18.
Элементный анализ для С27H39N3O (421.31):
Вычислено (%): С, 76.92; Н, 9.32; N, 9.97.
Найдено (%): С, 76.64; Н, 9.58 N, 9.88.
Другим важным аспектом изобретения является изучение биологической активности синтезированных соединений общей формулы I.
1. Определение ингибиторной активности соединений общей формулы I в отношении ацетилхолинэстеразы, бутирилхолинэстеразы и карбоксилэстеразы. Для кинетических исследований использовали коммерческие препараты («Sigma», США) ацетилхолинэстеразы эритроцитов человека (АХЭ, КФ 3.1.1.7), бутирилхолинэстеразы сыворотки лошади (БХЭ, КФ 3.1.1.8) и карбоксилэстеразы печени свиньи (КЭ, КФ 3.1.1.1). КЭ гидролизует многие терапевтически важные препараты, содержащие сложноэфирные группы [Imai Т., Drug Metab. Pharmacokinet, 2006, 21, 173-185; Laizure S.C. et al., Pharmacotherapy., 2013, 33, 210-222], в связи с чем способность антихолинэстеразных соединений, применяемых в терапии БА, ингибировать КЭ может приводить к нежелательным лекарственным взаимодействиям [Tsurkan L.G. et al., Chem. Biol. Interact., 2013, 203, 226-230].
Определение активности ферментов. Активность АХЭ и БХЭ определяли методом Эллмана (X 412 нм) [Ellman G.L. et al., Biochem. Pharmacol., 1961, 7, 88-95] с использованием в качестве субстрата ацетилтиохолина (1 мМ) и бутирилтиохолина (1 мМ), соответственно; условия определения: 0.1 М K,Na фосфатный буфер рН 7.5, 25°С. Активность КЭ определяли спектрофотометрически по выделению 4-нитрофенола (к 405 нм), субстрат - 4-нитрофенилацетат (1 мМ); условия определения: 0.1 М K,Na фосфатный буфер рН 8.0, 25°С [Sterri S.H. et al, Biochem. Pharmacol, 1985, 34(15), 2779-2785]. Измерения проводили на микропланшетном ридере FLUOStar Optima (LabTech, Germany).
Определение величин IC50 для ингибирования АХЭ, БХЭ и КЭ.
Ингибиторную активность соединений характеризовали величиной IC50 - концентрацией ингибитора, которая требуется для снижения активности фермента на 50%. Для определения IC50 ингибирования АХЭ, БХЭ и КЭ образец соответствующего фермента инкубировали с исследуемым соединением (концентрация ДМСО 2 об.%) в течение 5 минут, затем определяли остаточную активность фермента. Диапазон концентраций исследуемого соединения составлял 1×10-12-1×10_4 М. Каждый эксперимент проводили в трипликате. Измерения проводили на микропланшетном ридере FLUOStar Optima (LabTech, Germany). Вычисление IC50 проводили с использованием программы Origin 6.1.
Определение механизма ингибирования АХЭ и БХЭ соединениями общей формулы I и расчет кинетических констант ингибирования. Для выяснения механизма ингибирования определяли зависимость остаточной активности АХЭ и БХЭ от концентрации ингибитора при различных концентрациях субстрата. Каждый эксперимент проводили в трипликате. Данные анализировали в двойных обратных координатах Лайнуивера-Берка. Механизм ингибирования определяли из анализа зависимостей 1/V=f(1/S), полученных для трех возрастающих концентраций ингибитора. Вычисление кинетических констант ингибирования Ki (конкурентная составляющая) и αKi (неконкурентная составляющая) проводили с использованием программы Origin 6.1.
2. Определение степени вытеснения пропидия из периферического анионного сайта ацетилхолинэстеразы Electropfwrus electricus соединениями общей формулы I
Способность соединений общей формулы I конкурентно вытеснять пропидий - селективный лиганд периферического анионного сайта АХЭ, оценивали флуресцентным методом [Taylor P. et al., Mol. Pharmacol., 1974, 10, 703-708; Taylor P., Lappi S., Biochemistry, 1975, 14, 1989-1997]. В качестве источника фермента использовали АХЭ из Electrophorus electricus (ЕеАХЭ, фракция VI-S, Sigma-Aldrich) как высокоактивный и более дешевый фермент по сравнению с АХЭ человека, характеризующийся высокой степенью очистки. Интенсивность флуоресценции пропидия в связанном состоянии с АХЭ возрастает в несколько раз. Снижение интенсивности флуоресценции в присутствии исследуемых соединений показывает их способность связываться с периферическим анионным сайтом АХЭ, и, соответственно, блокировать агрегацию бета-амилоида.
Для определения степени вытеснения пропидия из периферического анионного сайта АХЭ проводили инкубацию.ЕеАХЭ (7 мкМ раствор) с исследуемым соединением в концентрации 20 мкМ в 1 мМ трис-HCl буфере рН 8.0, 25°С в течение 15 минут. Затем добавляли раствор пропидия иодида (конечная концентрация 8 мкМ), инкубировали 15 минут и снимали спектр флуоресценции (530 нм (возб.) и 600 нм (эмис.)). Донепезил и ипидакрин использовали как референсные соединения. Бланк содержал пропидий иодид той же концентрации в 1 мМ трис-HCl буфере рН 8.0. Измерения проводили в трипликате на микропланшетном ридере FluoSTAR Optima (BMG LabTech, Германия). Все результаты представлены как среднее ±SEM.
Степень вытеснения пропидия из периферического анионного сайта АХЭ (% вытеснения) рассчитывали по формуле:
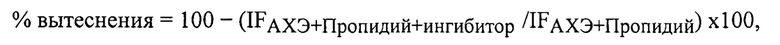
где IFАХЭ+Пропидий - интенсивность флуоресценции связанного с ЕеАХЭ пропидия в отсутствие исследуемого соединения (принимается за 100%), IFАХЭ+Пропидий+ингибитор - интенсивность флуоресценции связанного с ЕеАХЭ пропидия в присутствии исследуемого соединения.
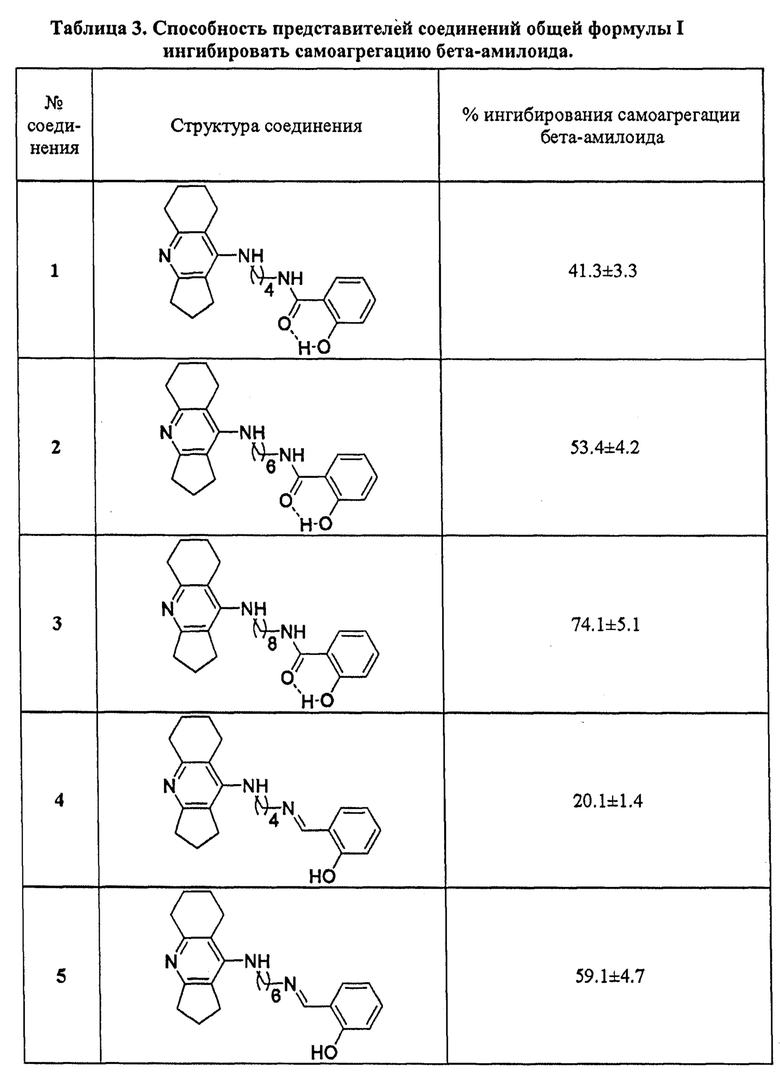
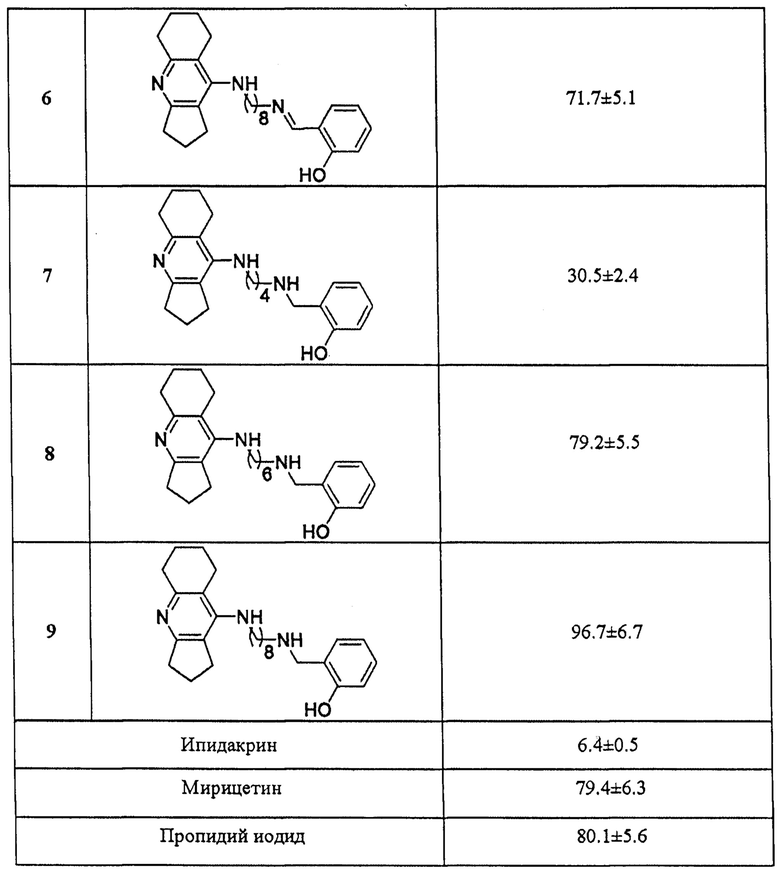
3. Определение ингибиторной активности соединений общей формулы I в отношении самоагрегации бета-амилоида
Ингибиторное действие исследуемых соединений на самоагрегацию бета-амилоида (1-42) (АР42) определяли с помощью флуоресцентного метода с использованием Тиофлавина Т (ThT) [Bartolini М. et al, Biochem. Pharmacol., 2003, 65, 407-416; Munoz-Ruiz P. et al., J. Med. Chem., 2005, 48, 7223-7233] с небольшими модификациями. Принцип метода основан на специфическом взаимодействии флуоресцентного красителя Тиофлавина Т с β-складчатыми структурами, образующимися в результате самоагрегации фибрилл бета-амилоида, которое приводит к значительному увеличению интенсивности флуоресценции связанного красителя [Biancalana М. et al., Biochim. Biophys. Acta, 2010, 1804, 1405-1412]. При этом уменьшение флуоресцентного сигнала в присутствии исследуемых соединений коррелирует с их ингибиторным эффектом в отношении образования агрегатов АР42.
1 мг лиофилизированного препарата АР42, обработанного гексафторизопропанолом (HFIP), фирмы GenicBio (Китай) растворяли в ДМСО для получения стабильного исходного раствора ([Аβ42]=500 мкМ).
Для оценки самоагрегации Аβ42 и степени ее ингибирования тестируемыми соединениями порцию приготовленного 500 мкМ раствора бета-амилоида растворяли в 215 мМ Na-фосфатном буфере рН 8.0 до конечной концентрации Аβ42 - 50 мкМ. Затем образцы инкубировали в течение 24 ч, при 37°С в термостате без перемешивания в отсутствие (базовый уровень самоагрегации Аβ42, контроль) или в присутствии тестируемых соединений. Мирицетин и пропидий иодид использовали в качестве референсных соединений. Конечная концентрация всех исследуемых соединений составляла 100 мкМ. После инкубации к образцам добавляли 5 мкМ раствор Тиофлавина Т в 50 мМ глицин-NаОН-буфере рН 8.5 и снимали спектр флуоресценции (440 нм (возб.) и 485 нм (эмис.)). Бланки состояли из 215 мМ Na-фосфатного буфера рН 8.0, содержащего ДМСО или исследуемые соединения, соответственно. Измерения проводили на мультифункциональном микропланшетном ридере FLUOstar OPTIMA (BMG Labtech, Германия). Все исследования были выполнены в трипликате, результаты представлены как среднее ±SEM.
Степень ингибирования самоагрегации Аβ42 исследуемыми соединениями (%) рассчитывали по следующей формуле:
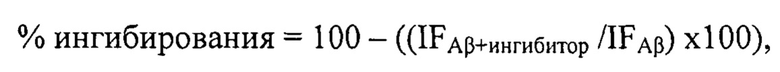
где IFAβ - интенсивность флуоресценции в отсутствие исследуемого соединения (принимается за 100%), IFAβ+ингибитор - интенсивность флуоресценции в присутствии исследуемого соединения после вычитания из них значения флуоресценции соответствующих бланков.
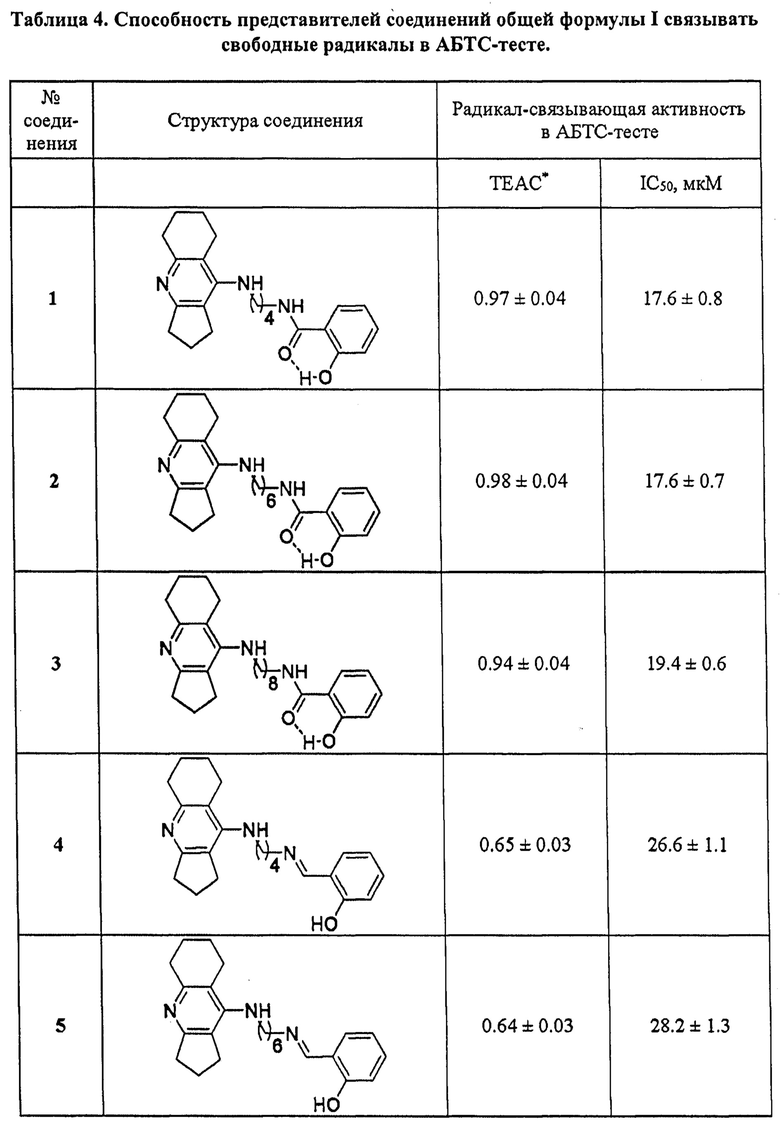
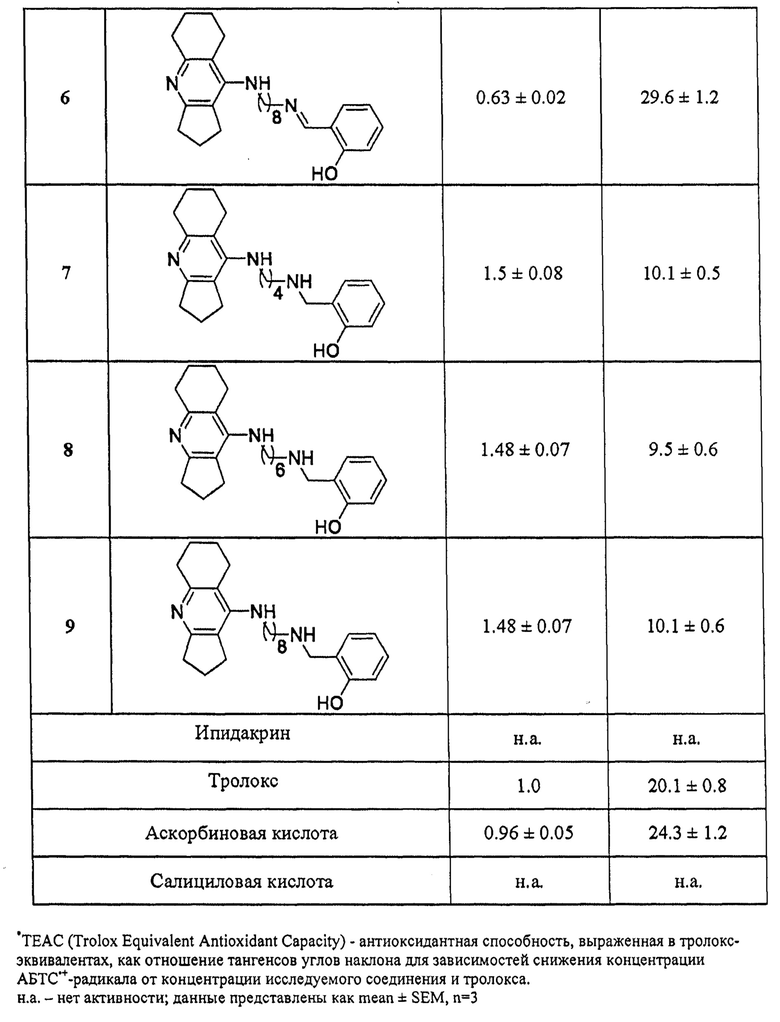
4. Определение антиоксидантной активности соединений общей формулы I
Антиоксидантную активность соединений оценивали по их способности связывать свободные радикалы в АБТС-тесте [Re R. et al., Free Radic. Biol. Med., 1999, 26, 1231-1237]. Метод основан на генерации устойчивого окрашенного катион-радикала АБТС'+темно-зеленого цвета при инкубации АБТС (2,2'-азино-бис(3-этилбензтиазолин-6-сульфоновая кислота)) с персульфатом калия. Последующее взаимодействие АБТС'+ с соединением, обладающим антиоксидантными свойствами, приводит к уменьшению светопоглощения радикала при длине волны 734 нм.
Катион-радикал АБТС'+получали в результате смешивания 7 мМ исходного водного раствора АБТС с равным объемом 2.45 мМ водного раствора персульфата калия и последующей инкубации раствора в течение 12-16 ч при комнатной температуре в темноте.
Исследуемые соединения растворяли в ДМСО, содержание которого в реакционной смеси составляло 4% (об/об), вносили в раствор АБТС'+ (конечная концентрация АБТС'+ в реакционной смеси составляла 100 мкмоль⋅л-1) и тщательно перемешивали. Реакцию проводили при 30°С в темноте, время инкубации 1 час. Степень обесцвечивания раствора АБТС'+определяли при длине волны 734 нм на микропланшетном ридере xMark BioRad (USA, Hercules). Соединения тестировали в диапазоне концентраций 1×10-6 - 2×10-4 моль⋅л-1. Все измерения проводили в пятикратном повторе для трех независимых экспериментов.
Антирадикальную активность соединений представляли в единицах ТЕАС (Trolox Equivalent Antioxidant Capacity, антиоксидантная способность, выраженная в тролокс-эквивалентах). Величины ТЕАС получали как отношение тангенсов углов наклона для зависимостей снижения концентрации АБТС'+-радикала от концентрации исследуемого соединения и Тролокса. Для всех соединений также определяли величины IC50 (концентрация соединения в мкмоль⋅л-1, при которой происходит снижение концентрации АБТС на 50%). Расчеты проводились с использованием программы Origin 6.1 для Windows (OriginLab, Northampton, MA). Все результаты представлены как среднее±SEM, рассчитанные с использованием GraphPad Prism версия 6.05 для Windows, GraphPad Software (San Diego, CA).
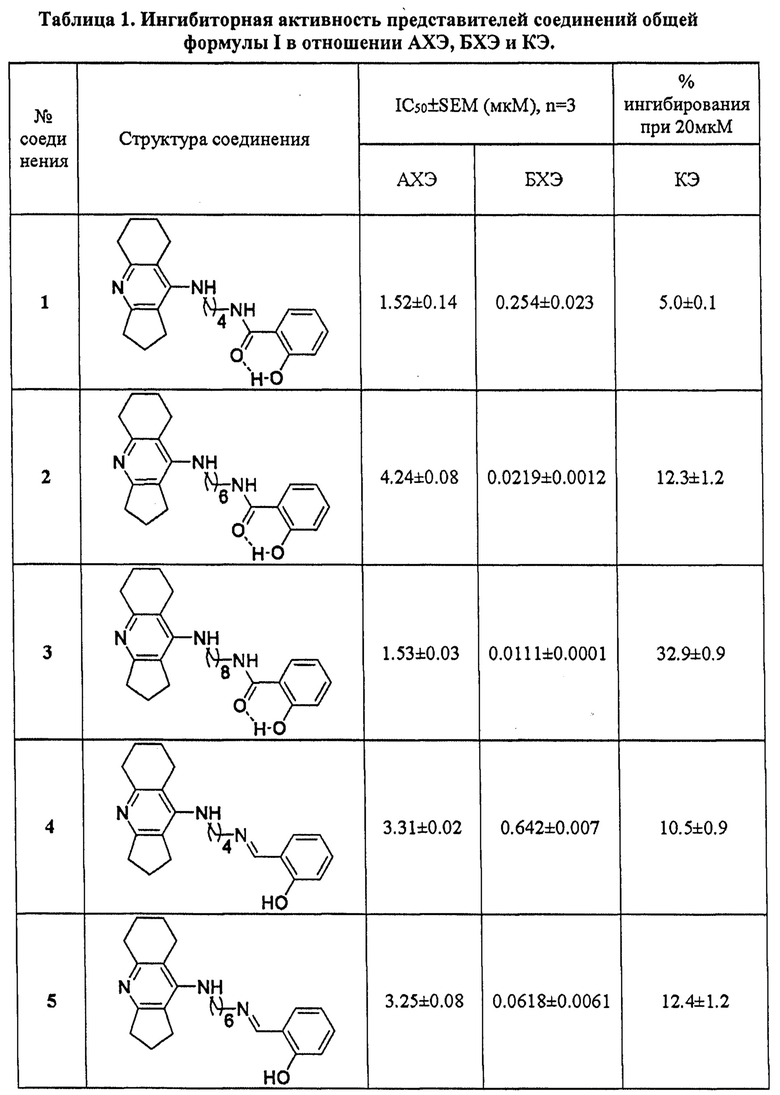
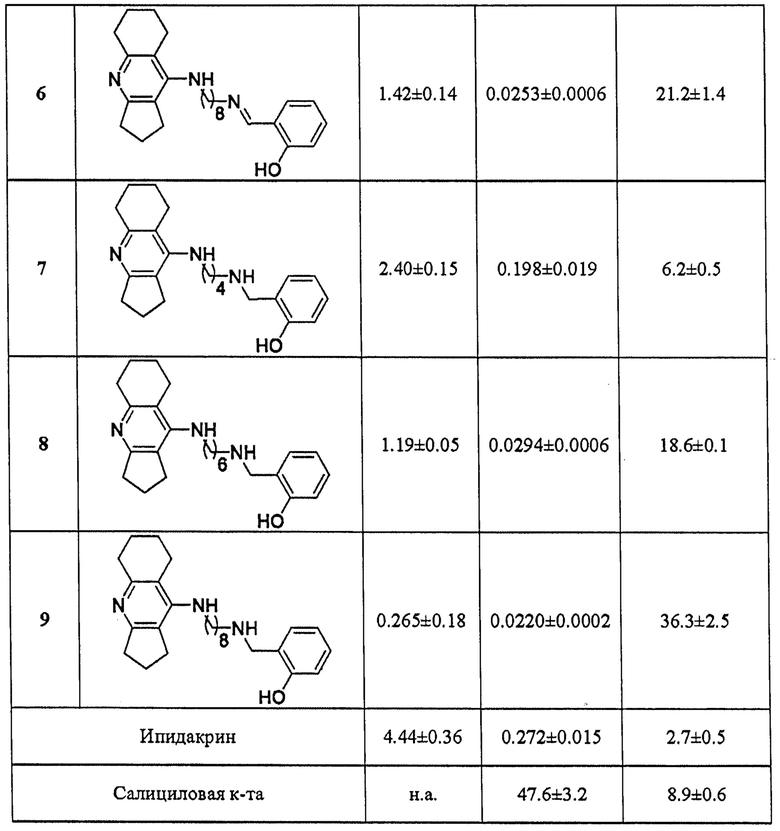
В таблицах 1-4 приведены примеры биологических испытаний соединений общей формулы I, которые иллюстрируют настоящее изобретение.
В таблице 1 приведены примеры определения ингибиторной активности представителей соединений общей формулы I в отношении АХЭ и БХЭ, а также структурно близкого холинэстеразам фермента КЭ. Результаты биологических испытаний показывают, что соединения формулы I проявляют высокую ингибиторную активность в отношении холинэстераз (АХЭ и БХЭ) с преимущественным ингибированием БХЭ и в меньшей степени ингибируют КЭ. Максимальную активность в отношении БХЭ проявляют соединения 3 и 9, которые ингибируют БХЭ в наномолярном диапазоне. Максимальную степень ингибирования в отношении АХЭ проявляет соединение 9, которое ингибируют АХЭ с величинами IC50 в субмикромолярной области. Антихолинэстеразная активность соединений формулы I сопоставима или превышает активность ипидакрина. Невысокая ингибиторная активность большинства соединений в отношении КЭ, свидетельствует об отсутствии потенциальных нежелательных лекарственных взаимодействий.
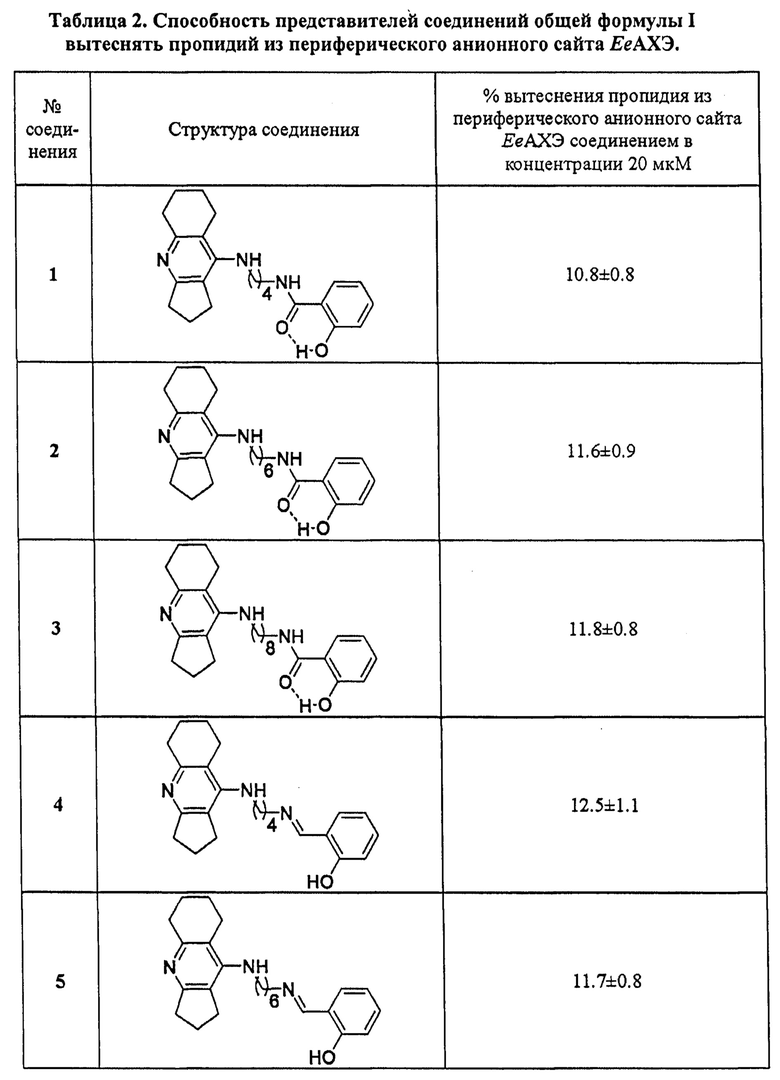
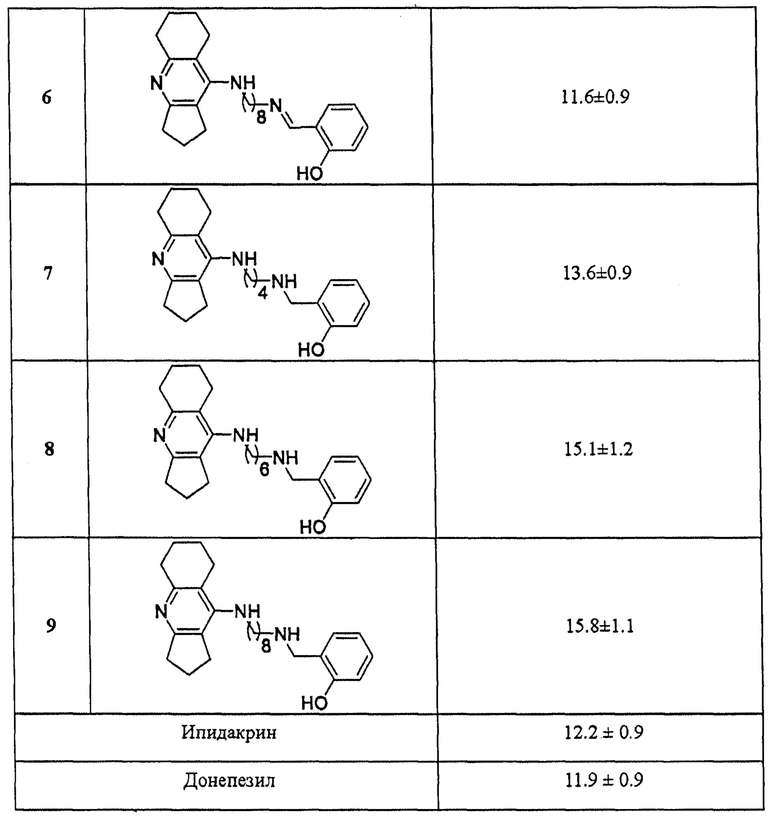
В таблице 2 приведены примеры определения степени вытеснения пропидия из периферического анионного сайта ЕеАХЭ (% вытеснения) представителями соединений общей формулы I. Примеры подтверждают, что заявляемые соединения в концентрации 20 мкМ снижают интенсивность флуоресценции на уровне (10-15%), сопоставимом или немного превышающем значения референсного соединения донепезила и исходного фармакофора ипидакрина.
Полученные результаты по вытеснению пропидия из периферического анионного сайта АХЭ наряду со смешанным механизмом ингибирования АХЭ свидетельствуют о том, что представители соединений общей формулы I связываются с периферическим анионным сайтом АХЭ и, следовательно, способны блокировать АХЭ-индуцируемую агрегацию бета-амилоида.
В таблице 3 приведены примеры ингибирования самоагрегации бета-амилоида представителями соединений общей формулы I. Примеры подтверждают, что заявляемые соединения в концентрации 100 мкМ ингибируют самоагрегацию бета-амилоида на уровне 20-96%. Соединения 3, 6, 9 со спейсером (СН2)8 проявляют максимальную ингибиторную активность в отношении бета-амилоида, что соответствует или превышает эффективность референсных соединений мирицетина (79.4±6.3%) и пропидия иодида (80.1±5.6%).
В таблице 4 приведены примеры определения антиоксидантной активности представителей соединений общей формулы I по их способности связывать свободные радикалы в АБТС-тесте. Примеры подтверждают, что заявляемые соединения проявляют достаточно высокую радикал-связывающую активность, которая превышает либо несколько ниже активности стандартных антиоксидантов тролокса и аскорбиновой кислоты. Базовые фармакофоры - ипидакрин и салициловая кислота неактивны.
Следует отметить, что синтезированные соединения общей формулы I значительно превосходят по своим фармакологическим свойствам базовые фармакофоры - ипидакрин и 2-замещенные фенолы.
Обнаружение у конъюгатов ипидакрина с 2-замещенными фенолами общей формулы I совокупности свойств, а именно способности одновременно высокоэффективно ингибировать ацетилхолинэстеразу и бутирилхолинэстеразу, связываться с периферическим анионным сайтом ацетилхолинэстеразы, связывать свободные радикалы и ингибировать самоагрегацию бета-амилоида, т.е. получены мультифункциональные конъюгаты, обладающие набором свойств, действующих однонаправленно и усиливающих терапевтический эффект конъюгата по сравнению с исходными фармкакофорами. Все это дает основания для применения этих соединений в медицинской практике как мультифункциональных препаратов для лечения нейродегенеративных заболеваний.
Применение конъюгатов ипидакрина с 2-замещенными фенолами общей формулы I возможно в качестве эффективных ингибиторов ацетилхолинэстеразы и бутирилхолинэстеразы человека.
Применение конъюгатов ипидакрина с 2-замещенными фенолами общей формулы I возможно в качестве соединений, связывающихся с периферическим анионным сайтом ацетилхолинэстеразы как потенциальных блокаторов АХЭ-индуцируемой агрегации бета-амилоида.
Применение конъюгатов ипидакрина с 2-замещенными фенолами общей формулы I возможно в качестве соединений, способных связывать свободные радикалы.
Применение конъюгатов ипидакрина с 2-замещенными фенолами общей формулы I возможно в качестве эффективных ингибиторов самоагрегации бета-амилоида.
Следующим аспектом изобретения является фармакологическое средство на основе конъюгатов ипидакрина с 2-замещенными фенолами общей формулы I, обладающее совокупностью перечисленных выше свойств, содержащее терапевтически эффективное количество соединения формулы I, применяемое в медицине для лечения нейродегенеративных заболеваний, в том числе болезни Альцгеймера. Специалисту в соответствующей области техники будет понятно, что заявляемые соединения могут входить в состав фармакологических средств для использования по указанному назначению. Заявляемое фармакологическое средство содержит активное начало и фармацевтически приемлемый носитель, новизна которого заключается в том, что в качестве активного начала используется эффективное количество соединения формулы I.
Понятие «фармакологическое средство» подразумевает использование любой лекарственной формы, содержащей соединение формулы I, которое могло бы найти профилактическое или лечебное применение в медицине в качестве средства для лечения нейродегенеративных заболеваний, в том числе болезни Альцгеймера.
Понятие «эффективное количество», используемое в данной заявке, подразумевает использование такого количества соединения формулы I, которое в сочетании с его показателями активности и токсичности, а также на основании знаний специалиста, должно быть эффективным в данной лекарственной форме.
Для получения фармакологического средства одно или несколько соединений формулы I смешивают как активный ингредиент с фармацевтически приемлемым носителем, известным в медицине, согласно принятым в фармацевтике способам. В зависимости от лекарственной формы препарата могут быть использованы различные носители.
Краткое описание чертежей
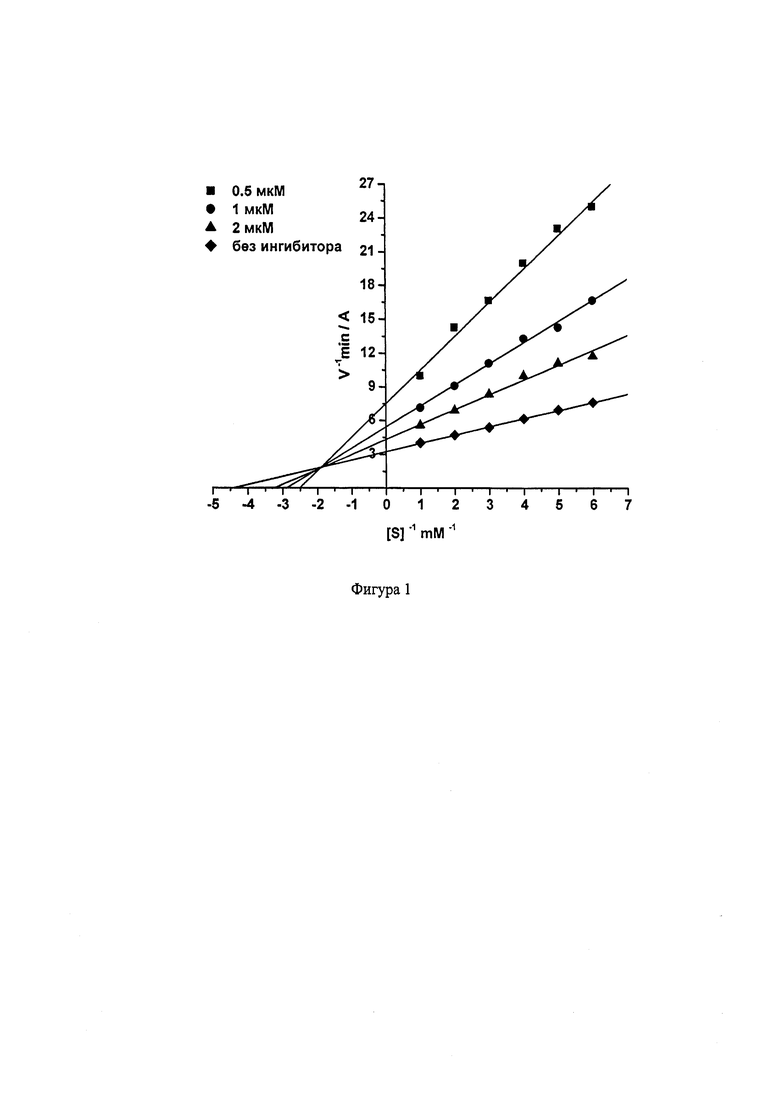
На Фиг. 1 приведена кинетика ингибирования АХЭ 2-(((6-((2,3,5,6,7,8-гексагидро-1H-циклопента[6]хинолин-9-ил)амино)гексил)амино)метил)фенолом (соединение 8).
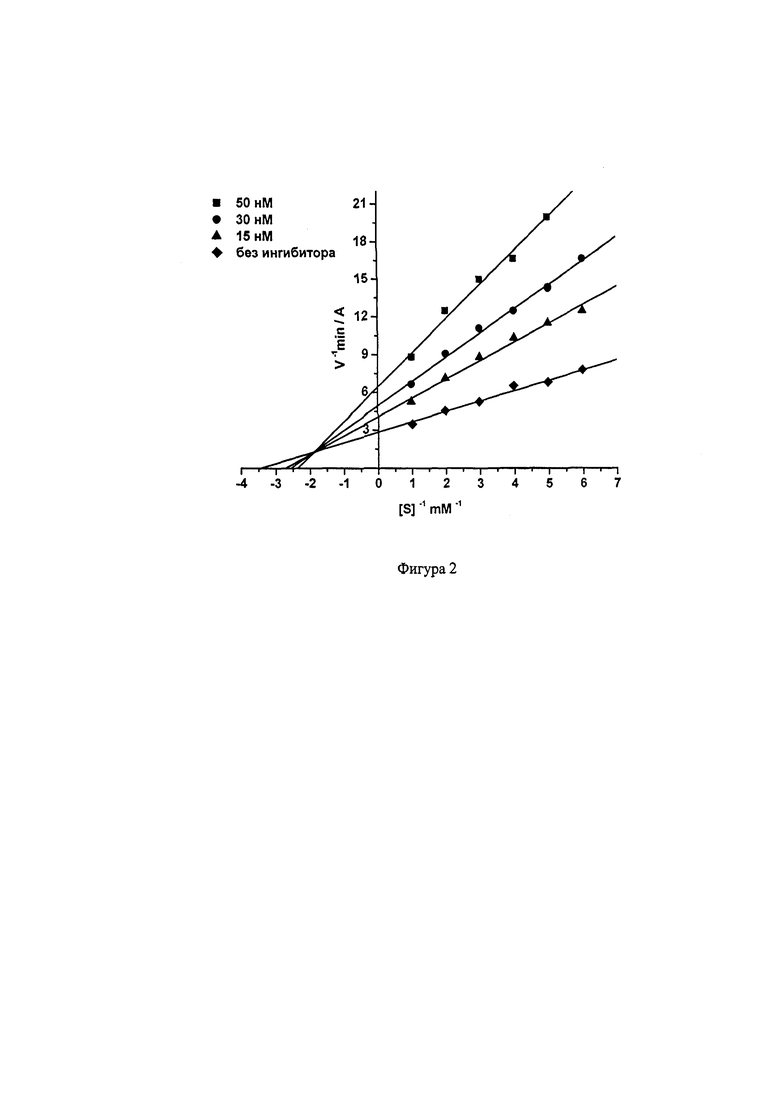
На Фиг. 2 приведена кинетика ингибирования БХЭ 2-(((6-((2,3,5,6,7,8-гексагидро-1H-циклопента[6]хинолин-9-ил)амино)гексил)амино)метил)фенолом (соединение 8).
Видно, что при связывании соединения 8 как с АХЭ, так и с БХЭ изменяются Vmax и Km, что характерно для смешанного типа ингибирования. Конкурентная (Ki) и неконкурентная (αKi) компоненты константы ингибирования АХЭ соединением 8 составляют 0.656±0.011 мкМ и 1.50±0.03 мкМ, соответственно; для БХЭ 21.03±0.61 нМ и 45.42±4.12 нМ, соответственно. Соединения 1-7, 9 ингибируют АХЭ и БХЭ по аналогичному (смешанному) механизму.
| название | год | авторы | номер документа |
|---|---|---|---|
| Мультифункциональные конъюгаты такрина и его аналогов с производными 1,2,4-тиадиазола, способ их синтеза и применение для лечения нейродегенеративных заболеваний | 2017 |
|
RU2675794C1 |
| СЕЛЕКТИВНЫЕ ИНГИБИТОРЫ КАРБОКСИЛЭСТЕРАЗЫ, ПРЕДСТАВЛЯЮЩИЕ СОБОЙ АЛКИЛ-2-АРИЛГИДРАЗИНИЛИДЕН-3-ОКСО-3-ПОЛИФТОРАЛКИЛПРОПИОНАТЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ | 2014 |
|
RU2574291C1 |
| 5-АМИНО-3-(2-АМИНОПРОПИЛ)-[1,2,4]ТИАДИАЗОЛЫ | 2011 |
|
RU2449997C1 |
| СРЕДСТВО НА ОСНОВЕ ПРОИЗВОДНОГО УРАЦИЛА ДЛЯ ТЕРАПИИ БОЛЕЗНИ АЛЬЦГЕЙМЕРА | 2014 |
|
RU2565756C1 |
| СПОСОБ ПОЛУЧЕНИЯ 2,8-БИС-(4-ГИДРОКСИФЕНИЛ, 4-КАРБОКСИФЕНИЛ)-2,3,8,9,12c,12d-ГЕКСАГИДРО-1Н,7Н-5,11-ДИОКСА-2,3a,4,6,6b,8,9a,10,12,12b-ДЕКААЗАДИЦИКЛОПЕНТА[e,l]ПИРЕНОВ | 2019 |
|
RU2736378C1 |
| ПРОИЗВОДНЫЕ 1,2,3,4-ТЕТРАГИДРОПИРИДО[4,3-b]ИНДОЛСОДЕРЖАЩИХ ФЕНОТИАЗИНОВ В КАЧЕСТВЕ ИНГИБИТОРОВ ХОЛИНЭСТЕРАЗ И БЛОКАТОРОВ СЕРОТОНИНОВЫХ РЕЦЕПТОРОВ 5-HT, СПОСОБЫ ПОЛУЧЕНИЯ ИХ ХЛОРГИДРАТОВ И ФАРМАКОЛОГИЧЕСКОЕ СРЕДСТВО НА ИХ ОСНОВЕ | 2013 |
|
RU2530881C1 |
| Адамантилсодержащие производные 1,2,4-триазола и 1,3,4-тиадиазола, имеющие монотерпеноидные фрагменты, используемые в качестве ингибиторов фермента тирозил-ДНК-фосфодиэстеразы 1 | 2020 |
|
RU2761880C1 |
| Фосфониевые соли на основе салициловой и ацетилсалициловой кислот, обладающие антибактериальной и антиоксидантной активностью" | 2019 |
|
RU2704025C1 |
| 2-ХЛОР-3-ПОЛИФТОРАЛКОКСИ-[1,4]-НАФТОХИНОНЫ, ПОВЫШАЮЩИЕ ТЕРМОСТОЙКОСТЬ ПОЛИМЕТИЛМЕТАКРИЛАТА | 2021 |
|
RU2772749C1 |
| 2,8-БИС-(5-МЕТИЛИЗОКСАЗОЛ-3-ИЛ ИЛИ 1,5-ДИМЕТИЛ-3-ОКСО-2-ФЕНИЛ-1,2-ДИГИДРО-3Н-ПИРАЗОЛ-4-ИЛ)-2,3,8,9,12c,12d-ГЕКСАГИДРО-1Н,7Н-5,11-ДИОКСА-2,3a.4,6,6b8,9a,10,12,12b-ДЕКААЗАДИЦИКЛОПЕНТА[e,l]ПИРЕНЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ В КАЧЕСТВЕ СРЕДСТВ С ЦИТОТОКСИЧЕСКОЙ АКТИВНОСТЬЮ | 2021 |
|
RU2785543C1 |
Изобретение относится к новым мультифункциональным конъюгатам ипидакрина с 2-замещенными фенолами, которые обладают способностью одновременно высокоэффективно ингибировать ацетилхолинэстеразу и бутирилхолинэстеразу, связываться с периферическим анионным сайтом ацетилхолинэстеразы, ингибировать самоагрегацию бета-амилоида и связывать свободные радикалы. Также предложен способ получения конъюгатов ипидакрина с 2-замещенными фенолами. 2 н. и 2 з.п. ф-лы, 2 ил., 4 табл., 9 пр.
1. Мультифункциональный конъюгат ипидакрина с 2-замещенным фенолом, представляющий собой соединение общей формулы I
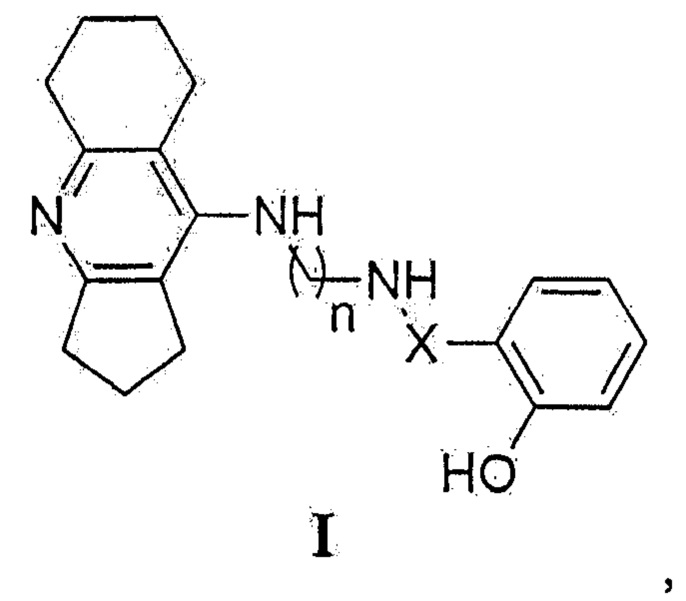
где X: -С(О)-=СН- или -СН2-; n=4, 6, 8.
2. Способ синтеза соединения общей формулы I по п. 1, включающий стадию синтеза аминоалкиленового производного ипидакрина формулы С
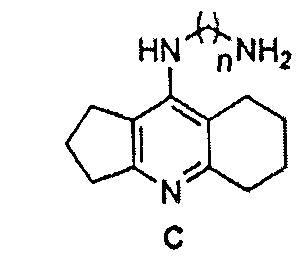 кипячением хлорпроизводного ипидакрина формулы А
кипячением хлорпроизводного ипидакрина формулы А
 и диаминоалкана формулы В
и диаминоалкана формулы В
 ,
,
где n=4, 6, 8,
в пентаноле в закрытом сосуде в присутствии иодидов щелочных металлов.
3. Способ по п. 2, где проводят конденсацию производного ипидакрина формулы С
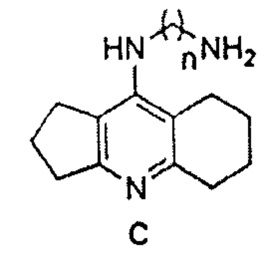 ,
,
где n=4, 6, 8,
с салициловым альдегидом с получением формулы I по п. 1, где X представляет собой =СН, при необходимости, с последующим восстановлением двойной связи енаминового фрагмента с получением соединения формулы I по п. 1, где X представляет собой -СН2.
4. Способ по п. 2, где проводят ацилирование производного ипидакрина формулы С
,
где n=4, 6, 8,
хлорангидридом салициловой кислоты с получением соединения формулы I по п. 1, где X представляет собой -С(О).
| Makhaeva, Galina F | |||
| et al | |||
| "Amiridine-piperazine hybrids as cholinesterase inhibitors and potential multitarget agents for Alzheimer's disease treatment", Bioorganic Chemistry, 2021, vol | |||
| Прялка для изготовления крученой нити | 1920 |
|
SU112A1 |
| no | |||
| Устройство для изгибания труб и профилей | 1955 |
|
SU104974A1 |
| Astakhova, Tatiana Yu | |||
| et al | |||
| "Bis-Amiridines as Acetylcholinesterase and Butyrylcholinesterase Inhibitors: N-Functionalization Determines | |||

