ТЕХНИЧЕСКАЯ ОБЛАСТЬ
Настоящее изобретение относится к областям ядерной медицины и молекулярной визуализации и, в частности, относится к соединению, фармацевтической композиции, содержащей соединение или состоящей из него, набору, содержащему соединение или фармацевтическую композицию или состоящему из них, и применению соединения или фармацевтической композиции для диагностики или лечения заболеваний, характеризующихся избыточной экспрессией белка активации фибробластов (FAP) и/или интегрина αvβ3.
УРОВЕНЬ ТЕХНИКИ
Белок активации фибробластов (FAP) представляет собой трансмембранную сериновую пептидазу, которая экспрессируется на поверхности активированных фибробластов опухолевой стромы и играет важную роль в процессах онкогенеза и прогрессирования опухолей. Предыдущие исследования показали, что FAP, как правило, не экспрессируется в нормальных тканях человека, но избирательно высоко экспрессируется на поверхностях стромальных фибробластов более чем в 90% эпителиальных злокачественных опухолей, включая рак молочной железы, рак яичников, рак легких, колоректальный рак, рак желудка, рак поджелудочной железы и тому подобное. Ввиду широкой экспрессии и важной роли в опухолях, FAP стал важной мишенью для визуализации и лечения опухолей. Меченный радионуклидами ингибитор белка активации фибробластов (FAPI), представленный производным хинолиновой кислоты, имел значительный прогресс в области точной визуализации опухолей. Например, ПЭТ/КТ-томографы, такие как FAPI-02 и FAPI-04, позволяют проводить специфическую визуализацию более 30 различных типов опухолей.
Интегрин (αvβ3) представляет собой гетеродимерный рецептор, расположенный на поверхности клеток, который редко экспрессируется в нормальных эндотелиальных и эпителиальных клетках сосудов, но экспрессируется на высоком уровне на поверхности клеток различных солидных опухолей, включая рак легких, остеосаркому, нейробластому, рак молочной железы, рак предстательной железы, рак мочевого пузыря, глиобластому, инвазивную меланому и т.п., и высоко экспрессируется в мембранах неоваскулярных эндотелиальных клеток всех опухолевых тканей, что указывает на то, что интегрин αvβ3 играет важную роль в процессах роста, инвазии и метастазирования опухолей. Полипептиды, содержащие последовательность аргинин-глицин-аспарагиновая кислота (RGD), могут специфически связываться с интегрином αvβ3. Различные RGD-пептиды, меченные радионуклидами, успешно применяются в исследованиях визуализации различных моделей животных с опухолями. В клиническом применении 18 F-Галакто-RGD стал первым неинвазивным средством для визуализации опухолей, нацеленным на интегрин αvβ3, в клинических испытаниях, успешно использовался в ПЭТ-диагностике пациентов с опухолями и имеет хорошее биологическое распределение и специфическое распознавание мишеней в клинических испытаниях глиобластомы.
Учитывая гетерогенность опухолей, для дальнейшего улучшения диагностики и эффективности лечения опухолей необходима разработка препарата, способного оказывать таргетное воздействие на две мишени, а именно FAP и интегрин αvβ3.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
С учетом вышеизложенного, основной целью настоящего изобретения является разработка нового соединения со структурой. Новое соединение позволяет синергически воздействовать на мишень FAP и мишень интегрина αvβ3 в опухолях, в результате чего количество и эффективность использования эффективных рецепторов в опухолях могут быть улучшены, тем самым улучшается эффективность поглощения опухолью и эффективность обнаружения положительной опухоли и/или эффективность лечения.
Другой целью настоящего изобретения является разработка способа получения нового соединения, позволяющего синтезировать соединение, способное синергически воздействовать на мишень FAP и мишень интегрина αvβ3 в опухолях удобным и легкодоступным способом синтеза.
Другой целью настоящего изобретения является обеспечение применения соединения в диагностике или лечении заболеваний, характеризующихся повышенной экспрессией белка активации фибробластов (FAP) и/или интегрина αvβ3.
Вышеуказанные цели настоящего изобретения достигаются путем принятия следующих технических решений.
В первом аспекте настоящее изобретение обеспечивает соединение двойного нацеливания, имеющее структуру, содержащую структуру специфического связывающего лиганда как FAP, так и интегрина αvβ3 (эта структура называется структурой «FAPI-RGD» по настоящему изобретению), соединение двойного нацеливания может одновременно нацеливаться на FAP и интегрин αvβ3, а структура соединения двойного нацеливания показана в формуле (I) или формуле (II) ниже:
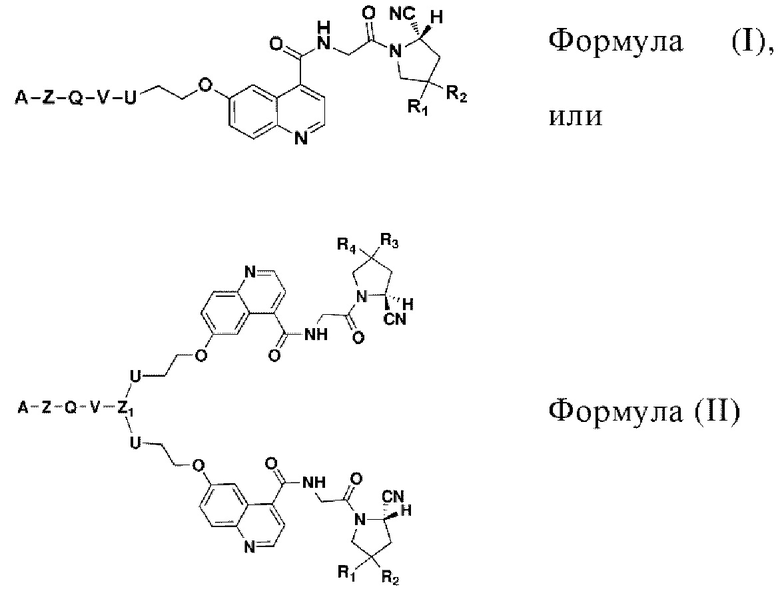
где
R1, R2, R3 и R4 могут быть независимо выбраны из Н или F, и значения R1, R2, R3 и R4 могут быть одинаковыми или отличаться.
Z, Q, V и U представляют собой одну и ту же структуру соединения или разные структуры соединения и независимо выбираются из -NH-,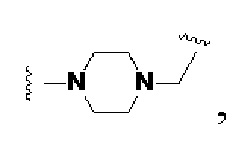

или структур замещения -(СН2)n-, соответственно.
Кроме того, когда Z, Q, V и/или U представляют собой структуры замещения на основе (СН2)n-, каждый -СН2- отдельно замещен с помощью -О-, -NH-, -(СО)-, -NH-(CO)-, -CH(NH2)- или -(CO)-NH-, или без них, причем замещение происходит, когда никакие две соседние группы -СН2- не замещены.
n - это целое число, выбранное в диапазоне от 0 до 30 (например, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29 и 30).
Z1 это 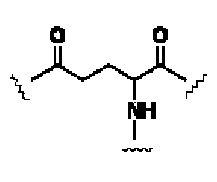 или
или 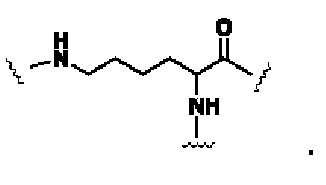
А представляет собой лигандную структуру, специфически связанную с интегрином αvβ3, причем эта структура показана в формуле (III) или формуле (IV):
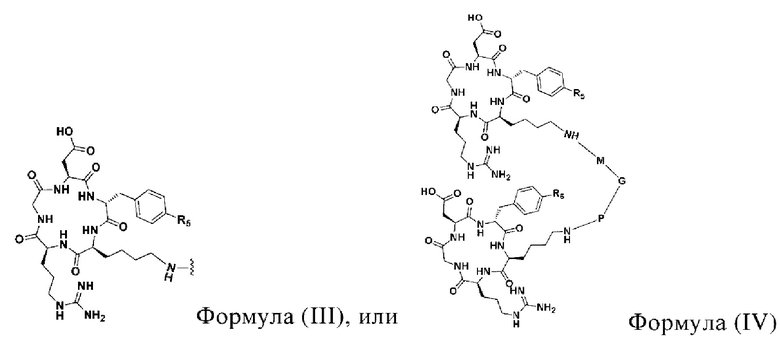
где в формуле (III) R5 выбирают из Н или ОН.
В формуле (IV) R5 и R6 являются одинаковыми или отличаются и независимо выбираются из Н или ОН.
5 В формуле (IV) М и Р являются структурами замещения, основанными
на (СН2)n-, каждый -СН2- отдельно замещен с помощью -О-, -NH-, -(СО)-, -NH-(CO)-, -CH(NH2)- или -(CO)-NH- или без них, причем замещение происходит, когда никакие две соседние группы -СН2- не замещены; a n - это целое число, выбранное в диапазоне от 0 до 30 (например, 1, 2, 3, 4, 5, 6, 7, 8, 10 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29 и 30).
В формуле (IV) G выбирают из 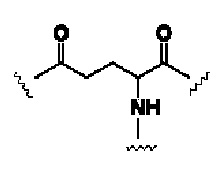 или
или 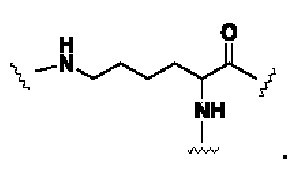
В некоторых предпочтительных вариантах осуществления, где Z, Q, V, U, М и/или Р в соответствии с любым из приведенных выше описаний являются структурами замещения, основанными на -(СН2)n-, Z, Q, V, U, М 15 и/или буква Р может быть независимо выбрана из следующих структур: -NH-СН2-(СН2-O-СН2)2-СН2-(СО)-, -NH-СН2-(СН2-O-СН2)3-СН2-(CO)-, -NH-СН2-(СН2-O-СН2)4-СН2-(CO)-, -(CO)-NH- или -(СН2)0- (а именно, «пустая» структура).
В некоторых предпочтительных вариантах осуществления R1 и R2 в структуре формулы (I) одновременно представляют собой Н (а именно, R1 представляет собой Н, a R2 представляет собой Н); в других предпочтительных вариантах осуществления R1 и R2 в структуре формулы (I) одновременно представляют собой F (а именно, R1 представляет собой F, a R2 представляет собой F); в других предпочтительных вариантах осуществления один из R1 и R2 в структуре Формулы (I) представляет собой Н, а другой представляет собой F (а именно, R1 представляет собой Н, a R2 представляет собой F; или R1 представляет собой F, и R2 представляет собой Н).
В некоторых предпочтительных вариантах осуществления R1, R2, R3 и R4 в структуре формулы (II) одновременно представляют собой Н; в других предпочтительных вариантах осуществления R1, R2, R3 и R4 в структуре формулы (II) одновременно представляют собой F; в других предпочтительных вариантах осуществления R1 и R2 в структуре формулы (II) одновременно представляют собой Н, a R3 и R4 одновременно представляют собой F; в других предпочтительных вариантах осуществления R1 и R2 в структуре формулы (II) одновременно представляют собой Н, один из R3 и R4 представляет собой Н, а другой представляет собой F (а именно, R3 представляет собой Н, a R4 представляет собой F или R3 представляет собой F, a R4 представляет собой Н); и в других предпочтительных вариантах осуществления R1 и R2 в структуре формулы (II) одновременно представляют собой F, один из R3 и R4 представляет собой Н, а другой представляет собой F (а именно, R3 представляет собой Н, a R4 представляет собой Н). представляет собой F, или R3 представляет собой F, a R4 представляет собой Н).
В некоторых предпочтительных вариантах осуществления R5 в формуле (III) равно Н; а в других предпочтительных вариантах осуществления R5 в формуле (III) равно ОН.
В некоторых предпочтительных вариантах осуществления R5 и R6 в формуле (IV) одновременно представляют собой Н (а именно, R5 представляет собой Н, a R6 представляет собой Н), в других предпочтительных вариантах осуществления R5 и R6 в формуле (IV) одновременно представляют собой F (а именно, R5 представляет собой F, a R6 представляет собой F), в других предпочтительных вариантах осуществления R5 в формуле (IV) представляет собой Н, a R6 представляет собой F; и в других предпочтительных вариантах реализации R5 в формуле (IV) представляет собой F, a R6 представляет собой Н.
В некоторых предпочтительных вариантах осуществления Z в формуле (I) или формуле (II) выбирают из -NH-СН2-(СН2-O-СН2)2-СН2-(CO)-, -NH-СН2-(СН2-O-СН2)3-СН2-(СО)-, -NH-СН2-(СН2-O-СН2)4-СН2-(CO)-, 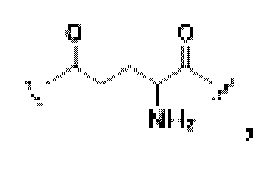
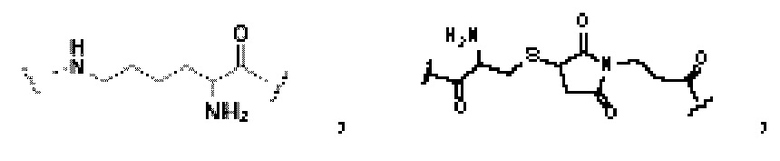 -(CO)-NH- или -(СН2)0-; и более предпочтительно Z в формуле (I) или формуле (II) выбирают из -NH-СН2-(СН2-O-СН2)2-СН2-(СО)-, -NH-СН2-(СН2-O-СН2)3-СН2-(СО)-, -NH-СН2-(СН2-O-СН2)4-СН2-(СО)-, -(СО)-NH- или -(СН2)0-.
-(CO)-NH- или -(СН2)0-; и более предпочтительно Z в формуле (I) или формуле (II) выбирают из -NH-СН2-(СН2-O-СН2)2-СН2-(СО)-, -NH-СН2-(СН2-O-СН2)3-СН2-(СО)-, -NH-СН2-(СН2-O-СН2)4-СН2-(СО)-, -(СО)-NH- или -(СН2)0-.
В некоторых предпочтительных вариантах осуществления Q в формуле (I) или формуле (II) выбирают из 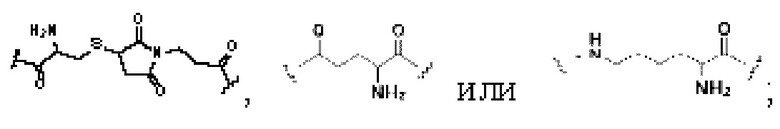 и более предпочтительно, Q в формуле (I) или формуле (II) выбирают из
и более предпочтительно, Q в формуле (I) или формуле (II) выбирают из 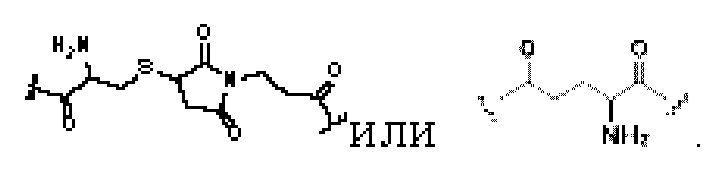
В некоторых предпочтительных вариантах осуществления, V в формуле (I) или формуле (II) выбирают из -NH-СН2-(СН2-O-СН2)2-СН2-(CO)-, -NH-СН2-(СН2-O-СН2)3-СН2-(СО)-, -NH-СН2-(СН2-O-СН2)4-СН2-(CO)-, -(СН2)0 -или -NH-(CO)-.
В некоторых предпочтительных вариантах осуществления U в формуле (I) или формуле (II) выбирают из -NH-, 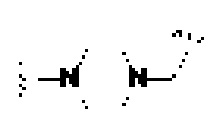 или-ИН-СН2-.
или-ИН-СН2-.
В некоторых предпочтительных вариантах осуществления Z1 в формуле (II) представляет собой 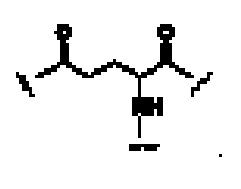
В некоторых предпочтительных вариантах осуществления М в формуле (IV) выбирают из -NH-СН2-(СН2-O-СН2)2-СН2-(CO)-, -NH-СН2-(СН2-O-СН2)3-СН2-(СО)-, -NH-СН2-(СН2-O-СН2)4-СН2-(CO)- или -(СН2)0-; и более предпочтительно, М в формуле (IV) выбирают из -NH-СН2-(СН2-O-СН2)2-СН2-ССО)- или -NH-СН2-(СН2-O-СН2)4-СН2-(CO)-.
В некоторых предпочтительных вариантах осуществления Р в формуле (IV) выбирают из -NH-СН2-(СН2-O-СН2)2-СН2-(CO)-, -NH-СН2-(СН2-O-СН2)3-СН2-(СО)-, -NH-СН2-(СН2-O-СН2)4-СН2-(CO)- или -(СН2)0-; и более предпочтительно, Р в формуле (IV) выбирают из -NH-СН2-(СН2-O-СН2)2-СН2-(СО)- или -NH-СН2-(СН2-O-СН2)4-СН2-(CO)-.
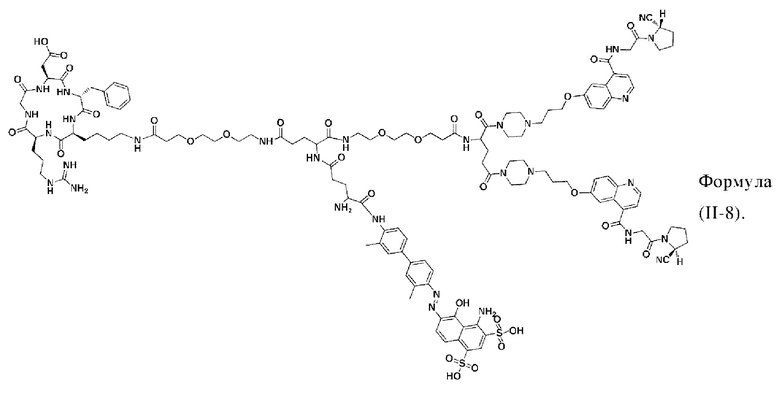
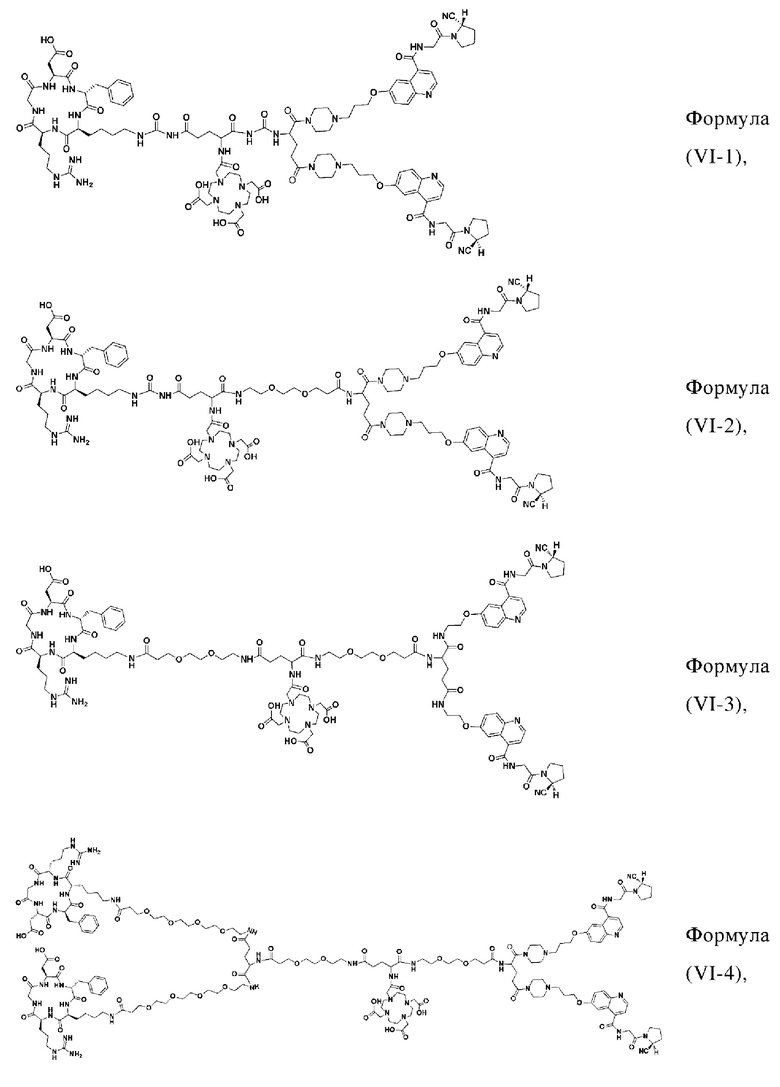
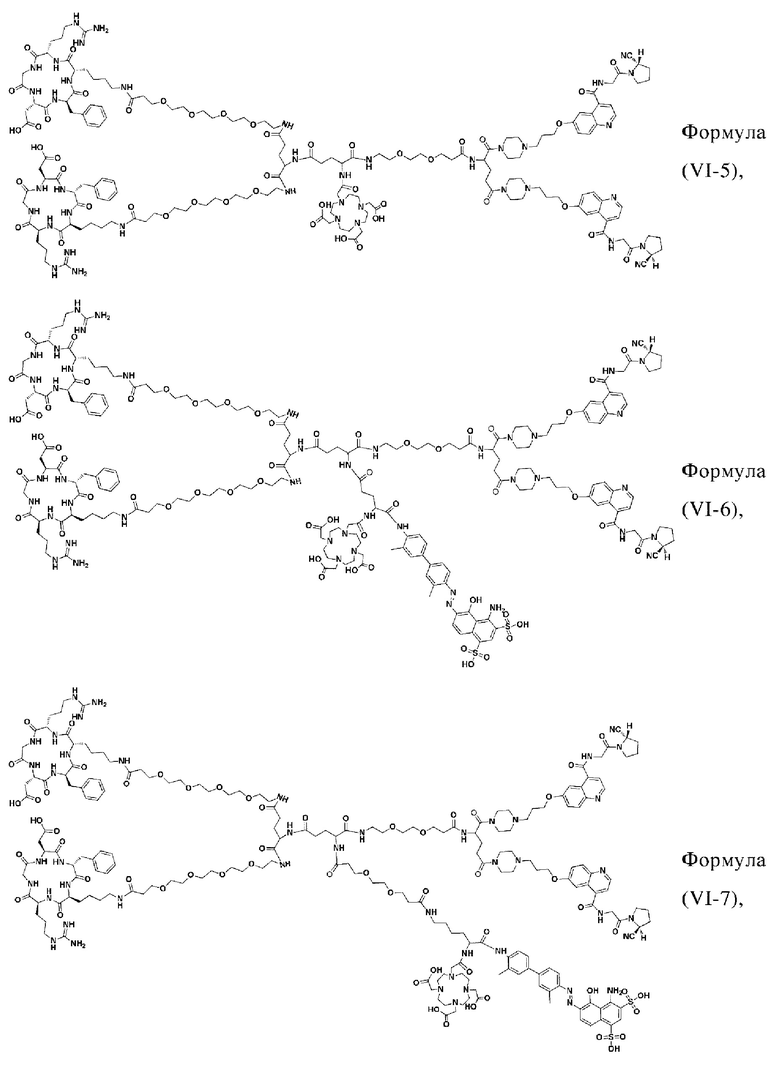
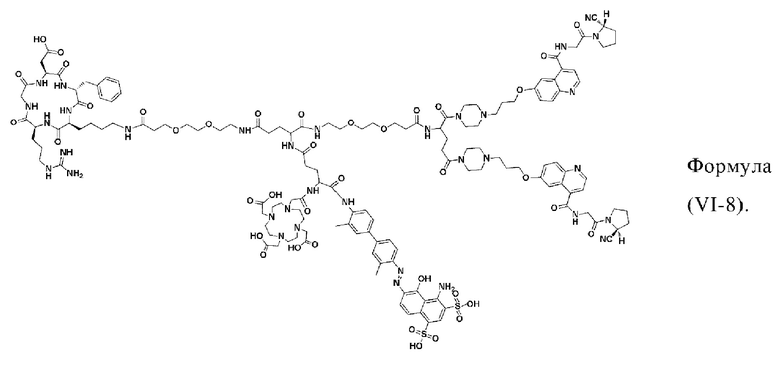
В других предпочтительных вариантах осуществления соединение двойного нацеливания выбирают из следующих структур:
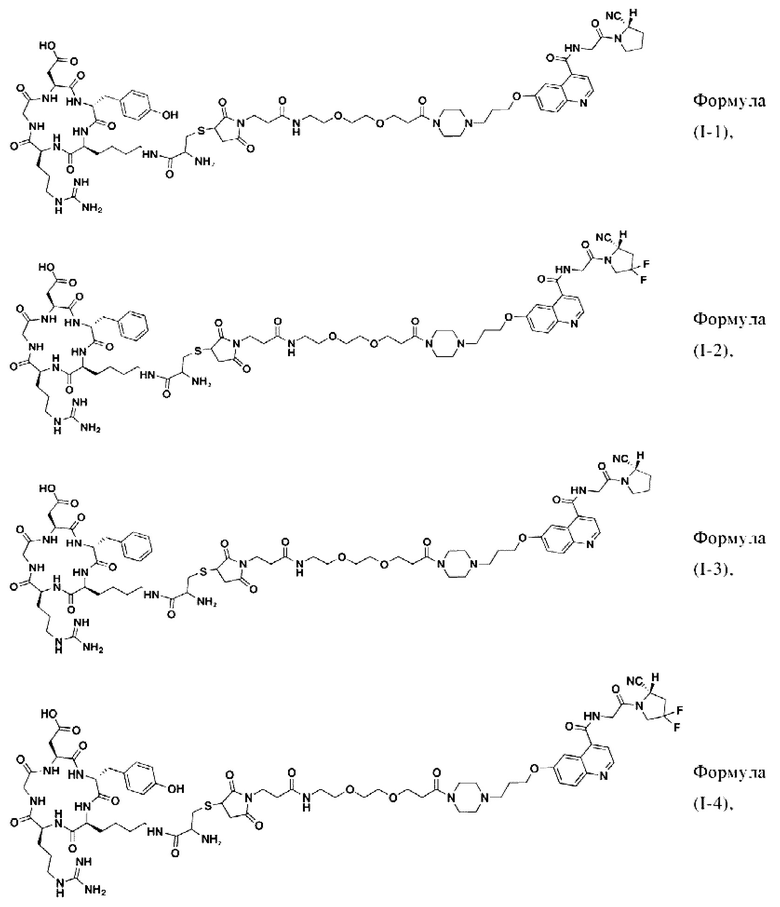
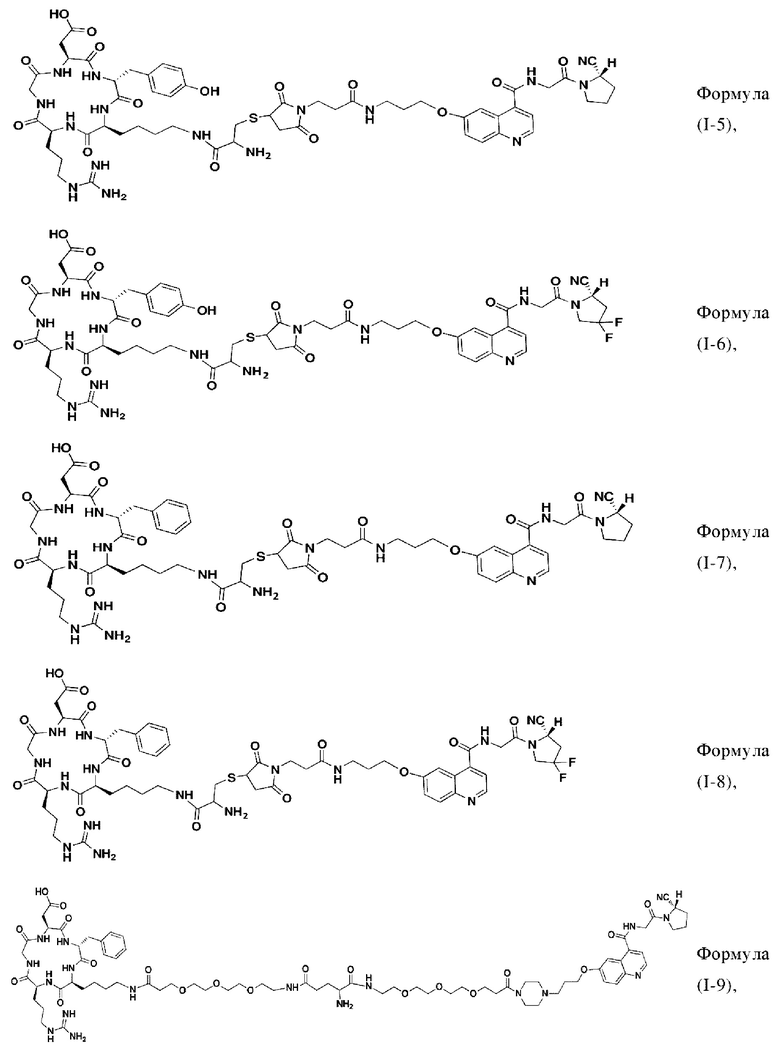
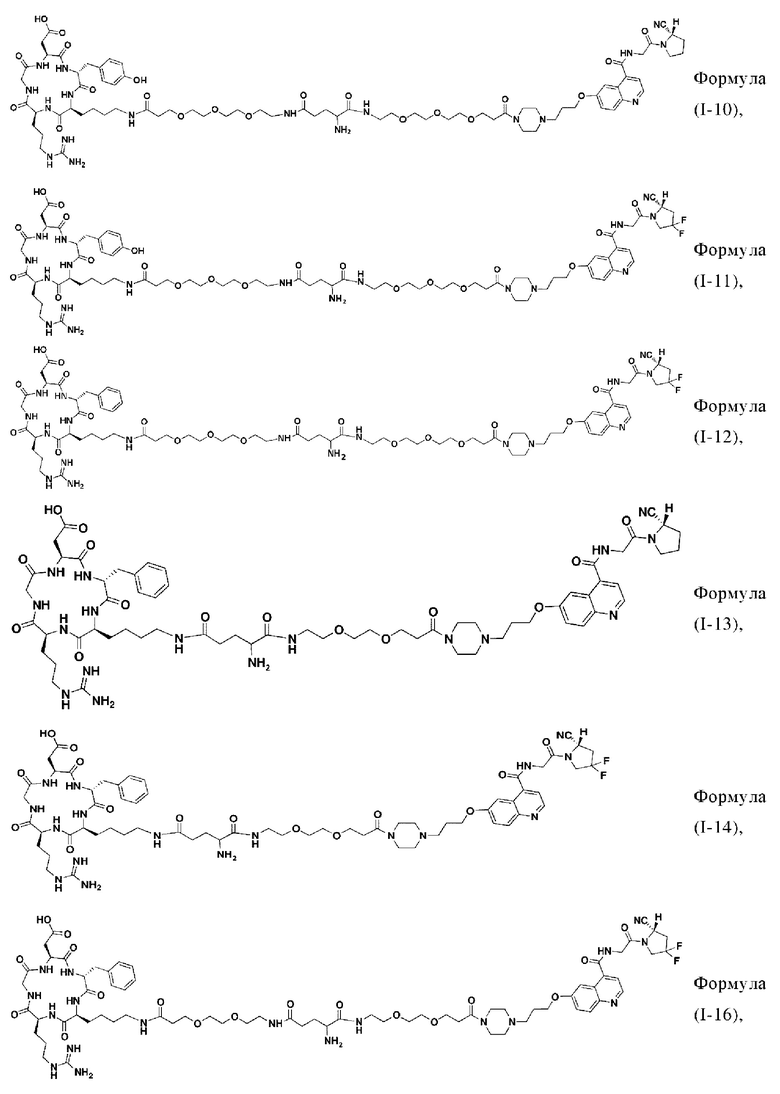
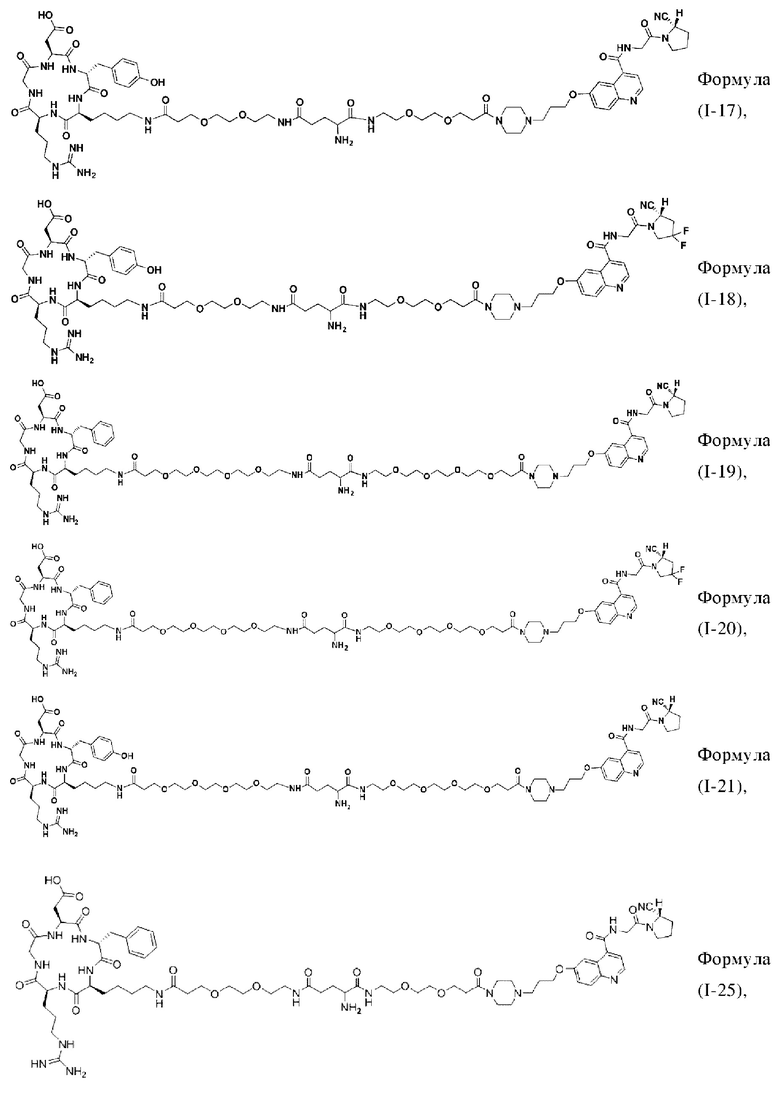
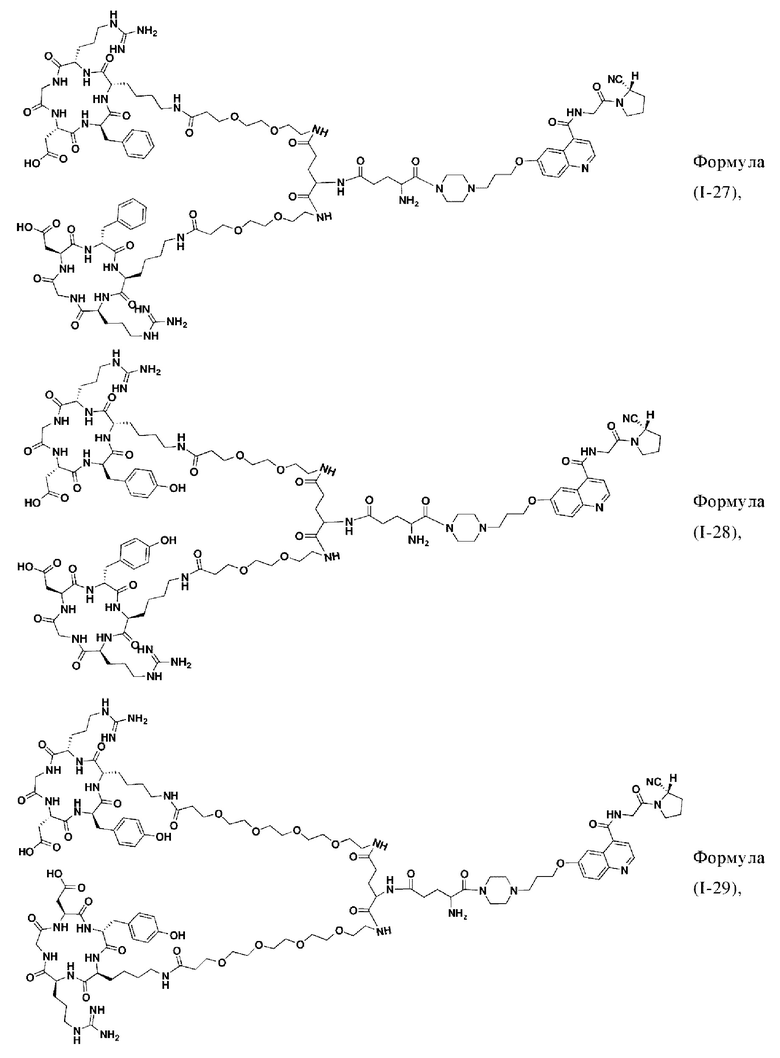
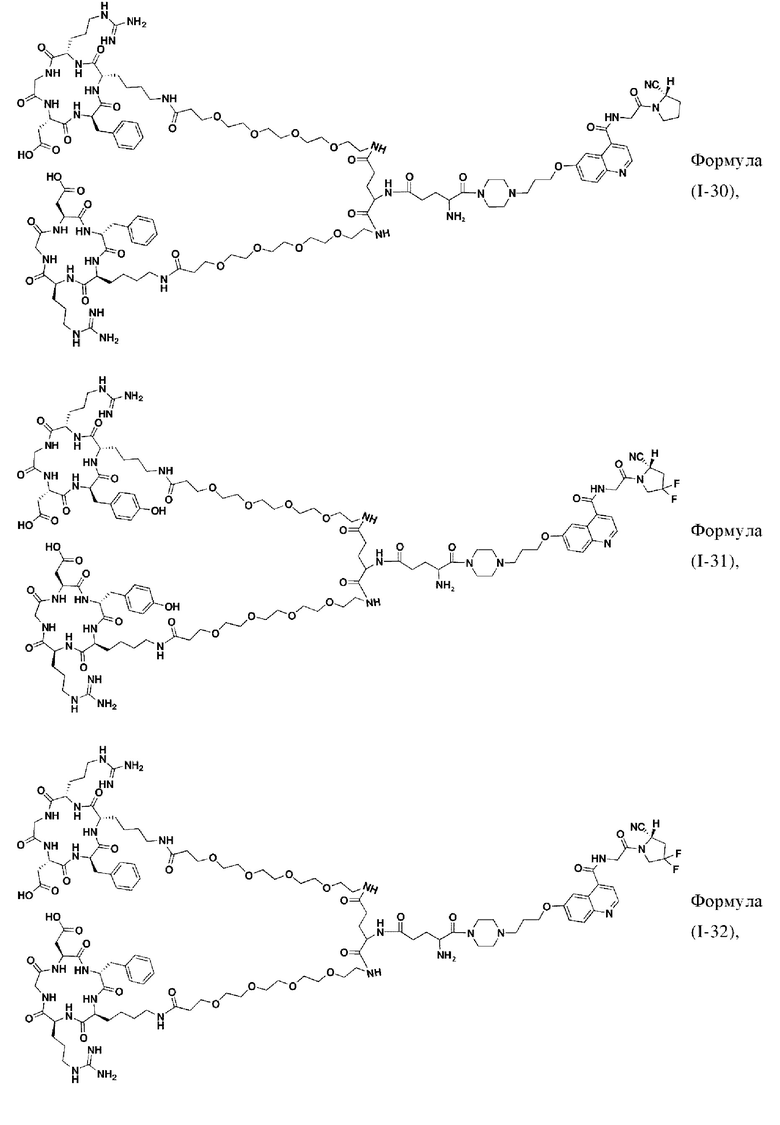
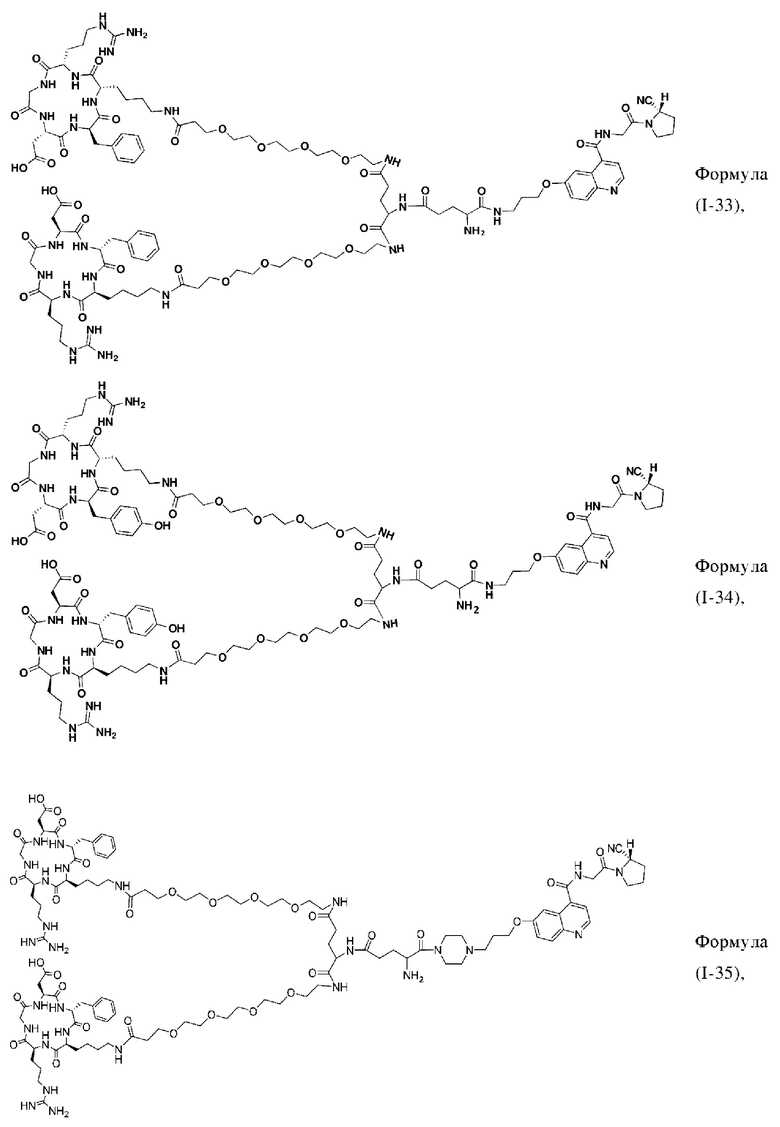
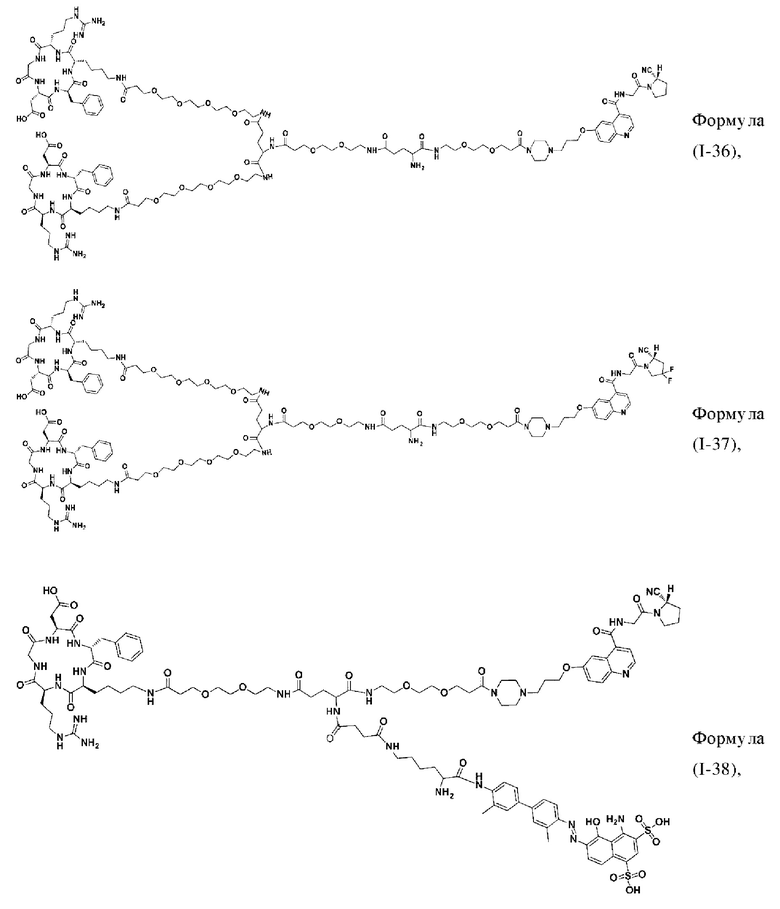
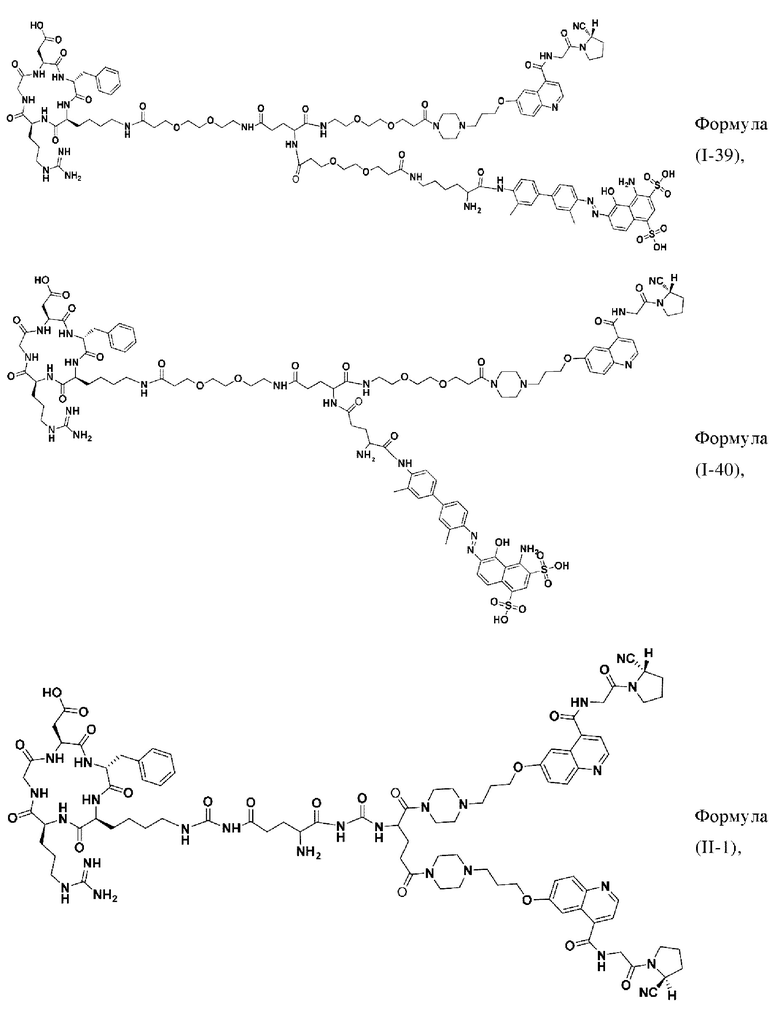
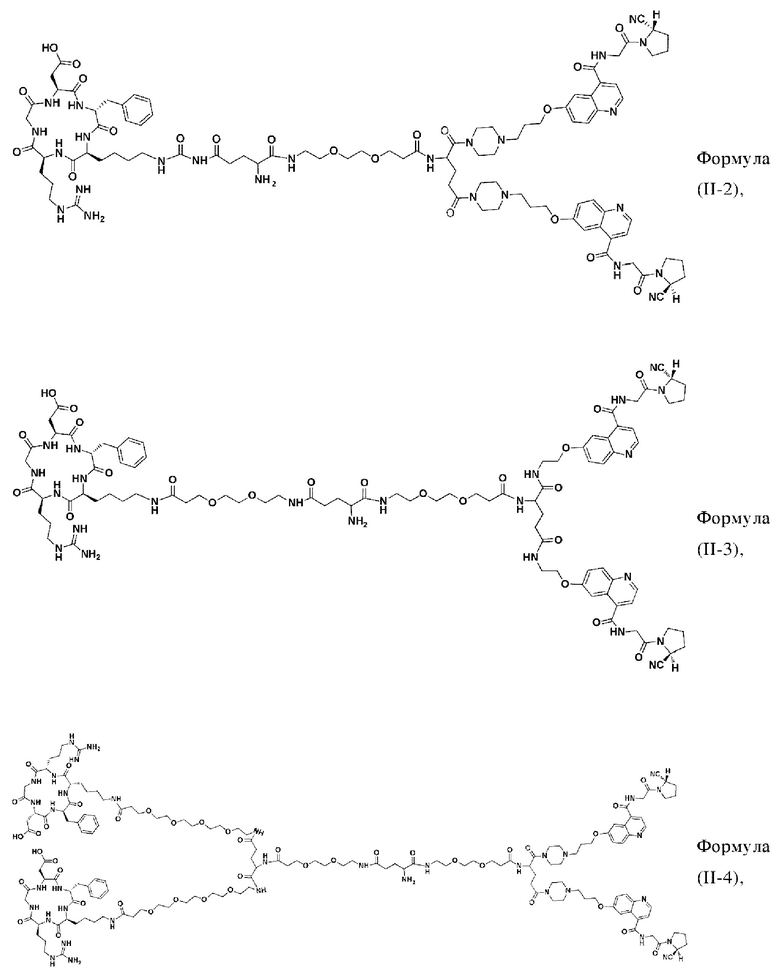
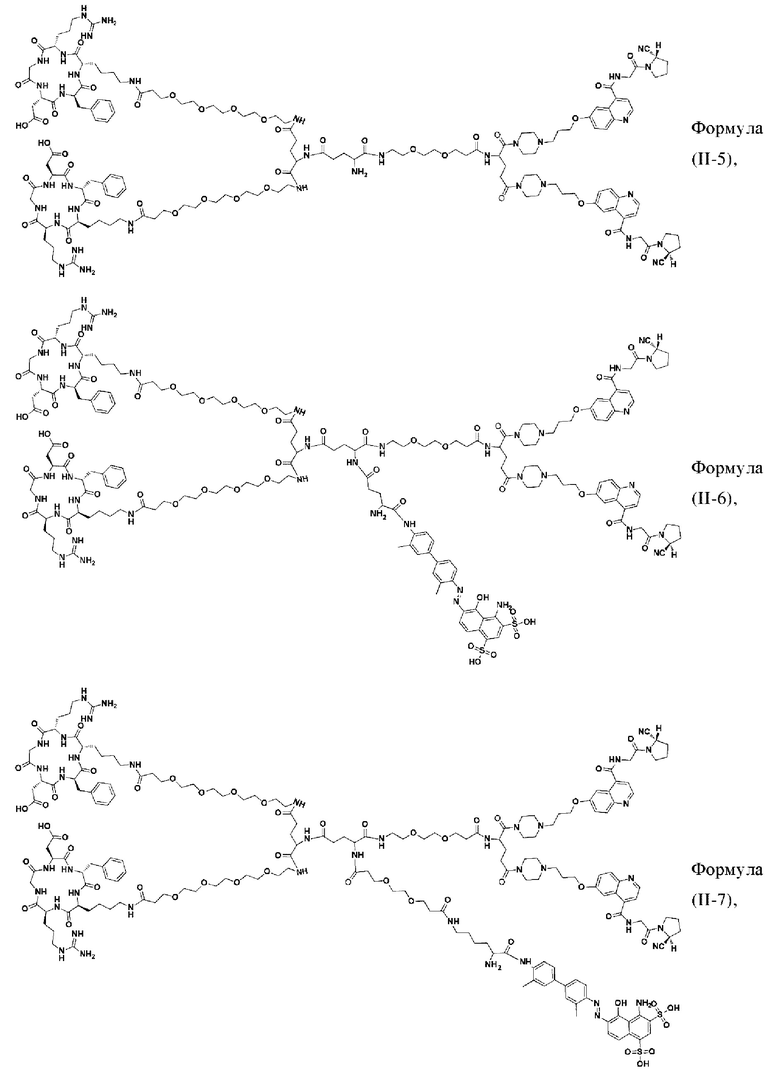
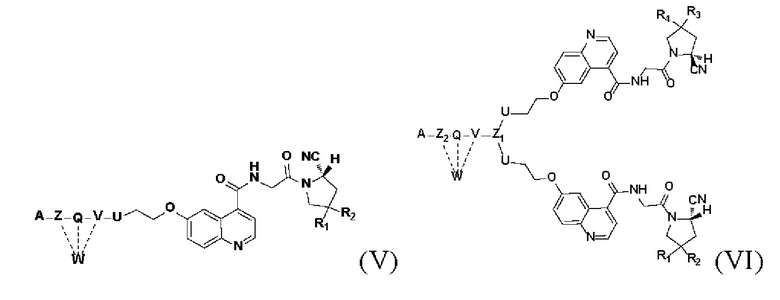
Во втором аспекте настоящее изобретение дополнительно обеспечивает соединение, которое можно метить радионуклидом на основе соединения двойного нацеливания согласно любому из приведенных выше описаний, где соединение, которое можно метить радионуклидом, образуется путем соединения аминогруппы в любой из Z-, Q- или V-структур Формулы (I) или Формулы (II) в соответствии с любым из приведенных выше описаний с хелатирующей группой нуклида, а соединение, которое можно метить радионуклидом, имеет общую формулу, показанную в формуле (V) или формуле (VI) ниже:
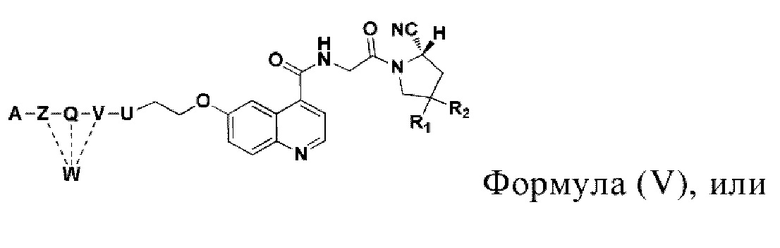
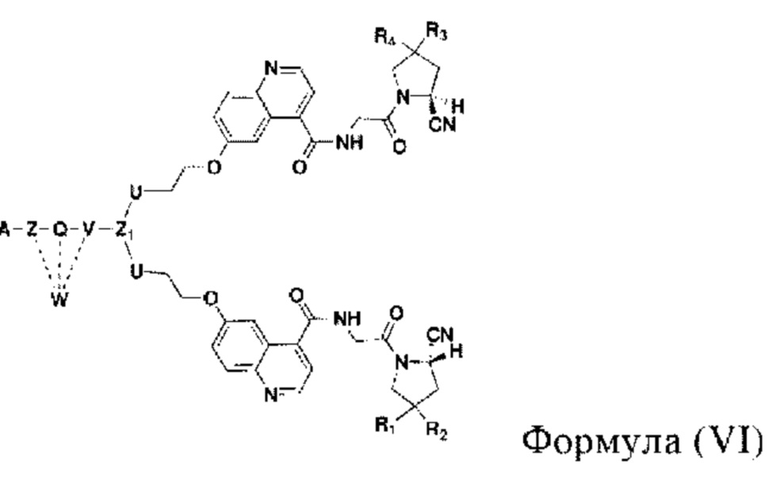
где A, Z, Q, V, U, R1 и R2 в формуле (V) определяются так же, как A, Z, Q, V, U, R1 и R2 в формуле (I); a A, Z, Q, V, U, Z1, R1, R2, R3 и R4 в формуле (VI) определяются так же, как A, Z, Q, V, U, Z1, R1, R2, R3 и R4 в формуле (II).
W представляет собой фрагмент с нуклидной хелатной группой и имеет структуру, полученную из любого из следующего: 1,4,7,10-тетраазациклододекан-N,N',N,N'-тетрауксусная кислота (DOTA), этилендиаминтетрауксусная кислота (EDTA), 1,4,7-триазациклононан-1,4,7-триуксусная кислота (NOTA), триэтилентетраамин (ТЕТА), иминодиуксусная кислота, диэтилентриамин-N,N,N',N,N''-пентауксусная кислота (DTPA), глицинат бис(карбоксиметил)имидазола или 6-гидразинопиридин-3-карбоновая кислота (HYNIC), или имеет любую из следующих структур: 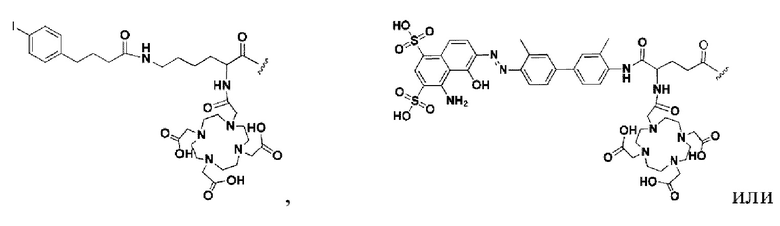
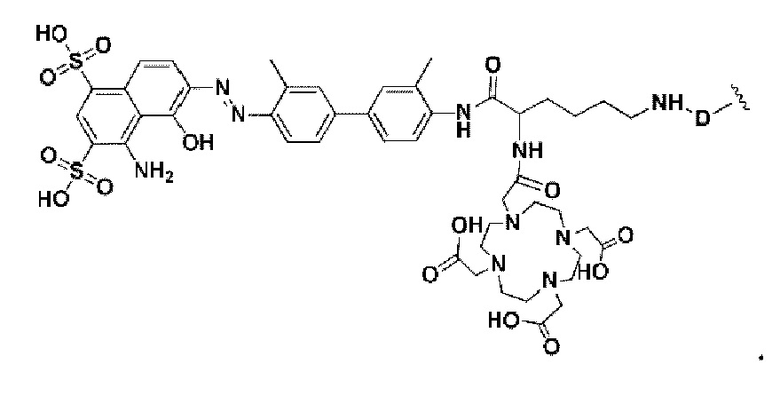
Кроме того, D согласно любому из приведенных выше описаний представляет собой структуру замещения, основанную на -(СН2)Р-, каждый -СН2- отдельно замещен с помощью -О-, -NH-, -(СО)-, -NH-(CO)-, -CH(NH2)- или -(CO)-NH- или без них, причем замещение происходит, когда никакие две соседние группы -СН2- не замещены; а р - это целое число, выбранное в диапазоне от 0 до 30 (например, 0, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29 и 30).
В некоторых предпочтительных вариантах осуществления D в соответствии с любым из приведенных выше описаний выбирают из -(СО)-СН2-СН2-(СО)-, -(СО)-СН2-(СН2-O-СН2)2-СН2-(СО)- или -(СН2)0- (применительно к структурам от формулы (V-38) до формулы (V-40)).
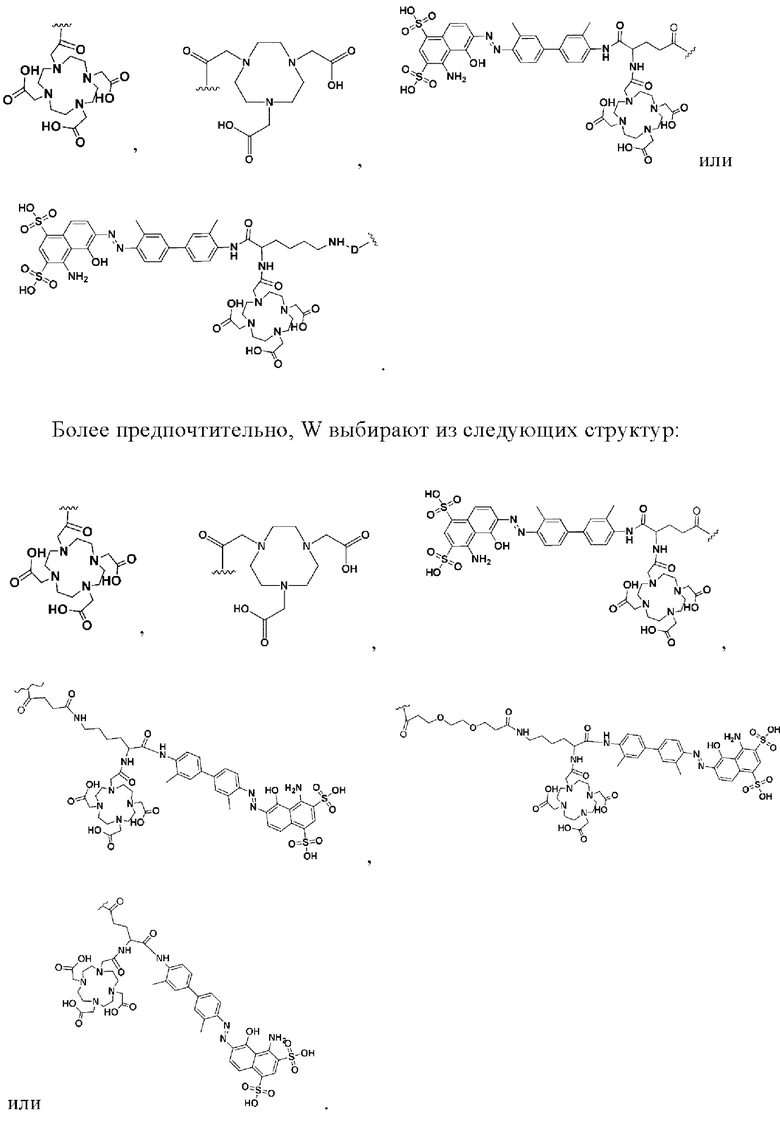
В некоторых предпочтительных вариантах осуществления W выбирают из следующих структур:
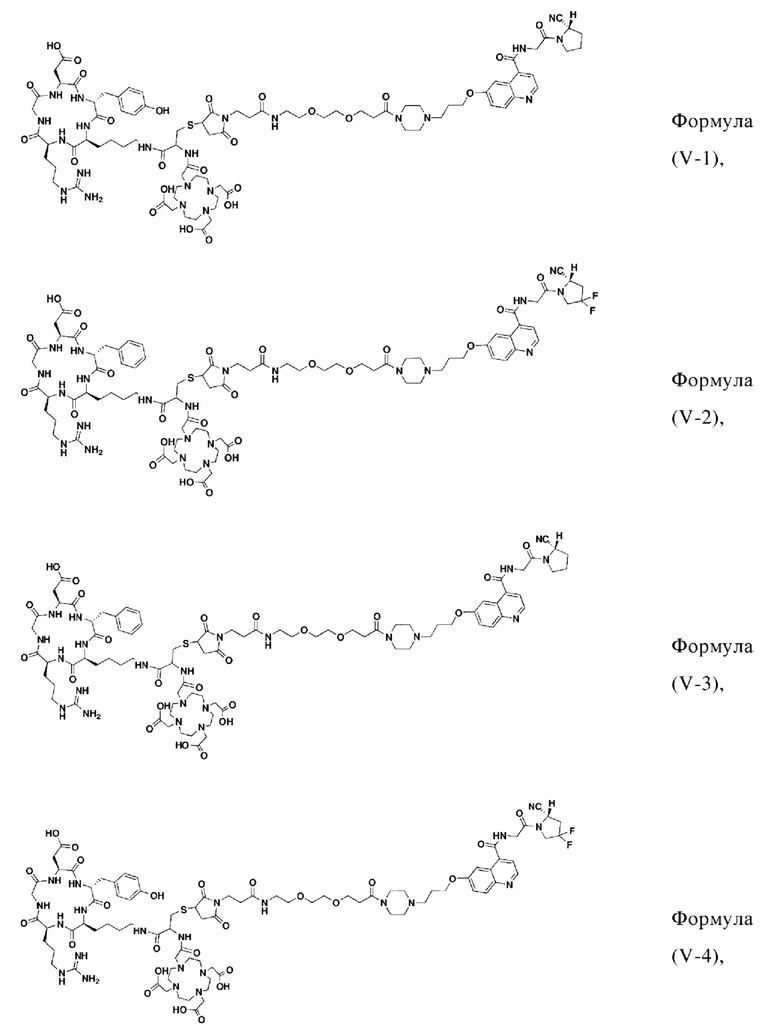
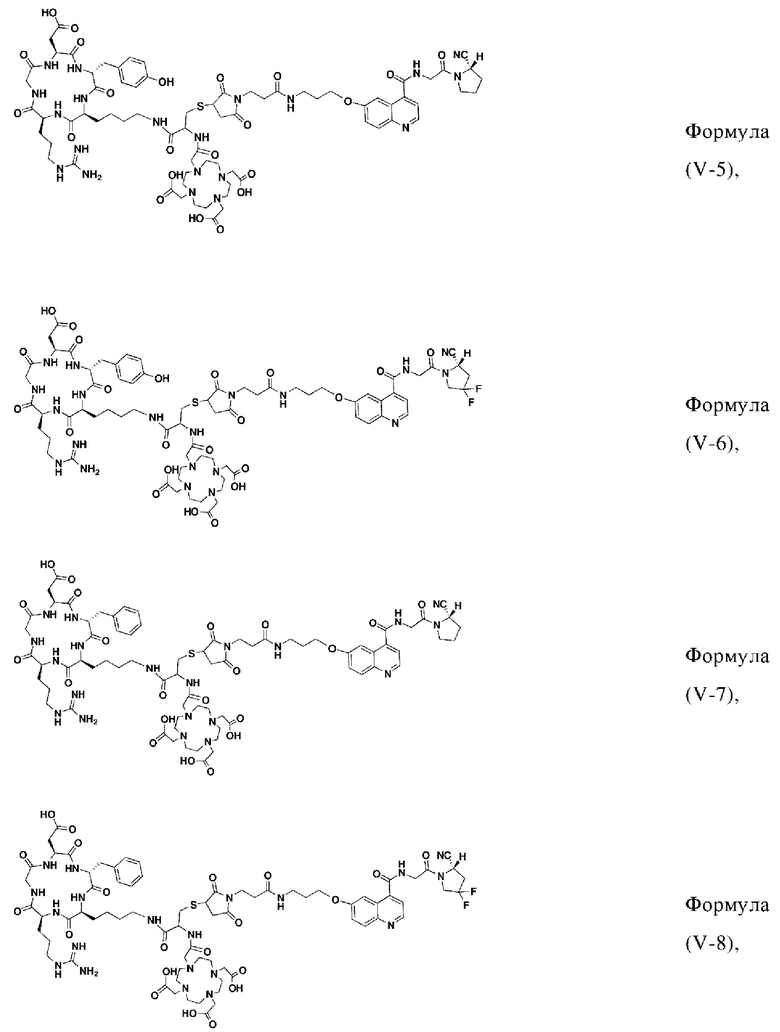
В других предпочтительных вариантах осуществления соединение, которое можно метить радионуклидом, выбирают из следующих структур:
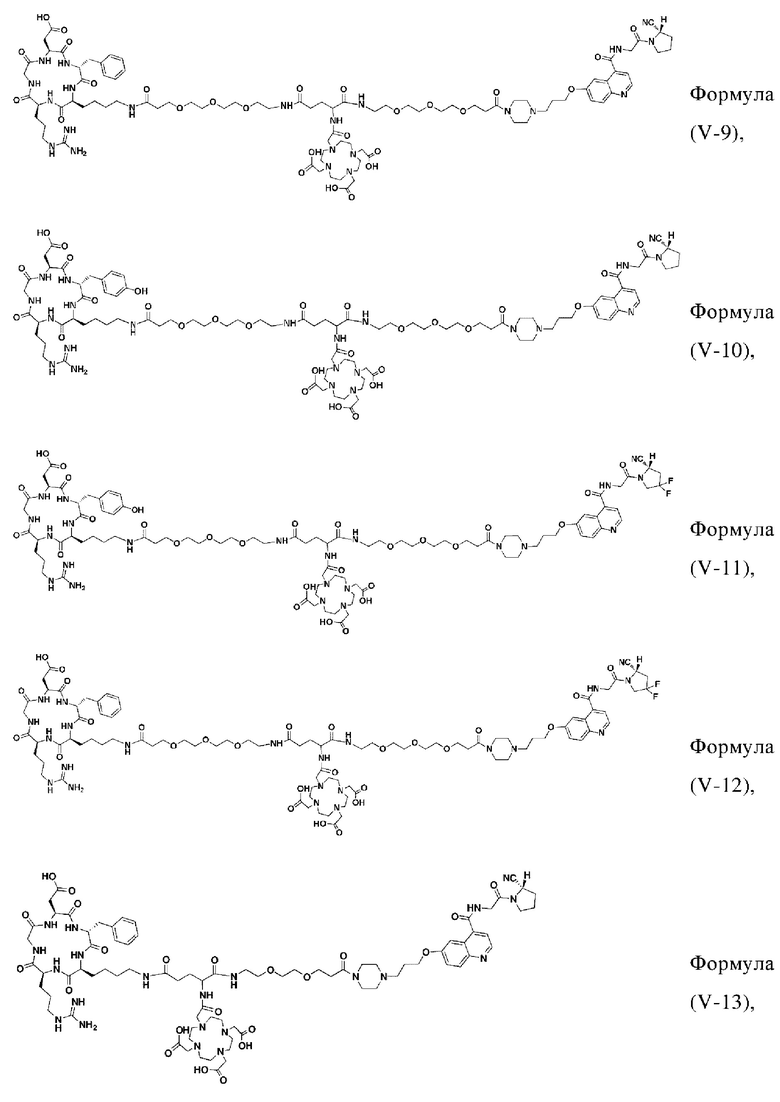
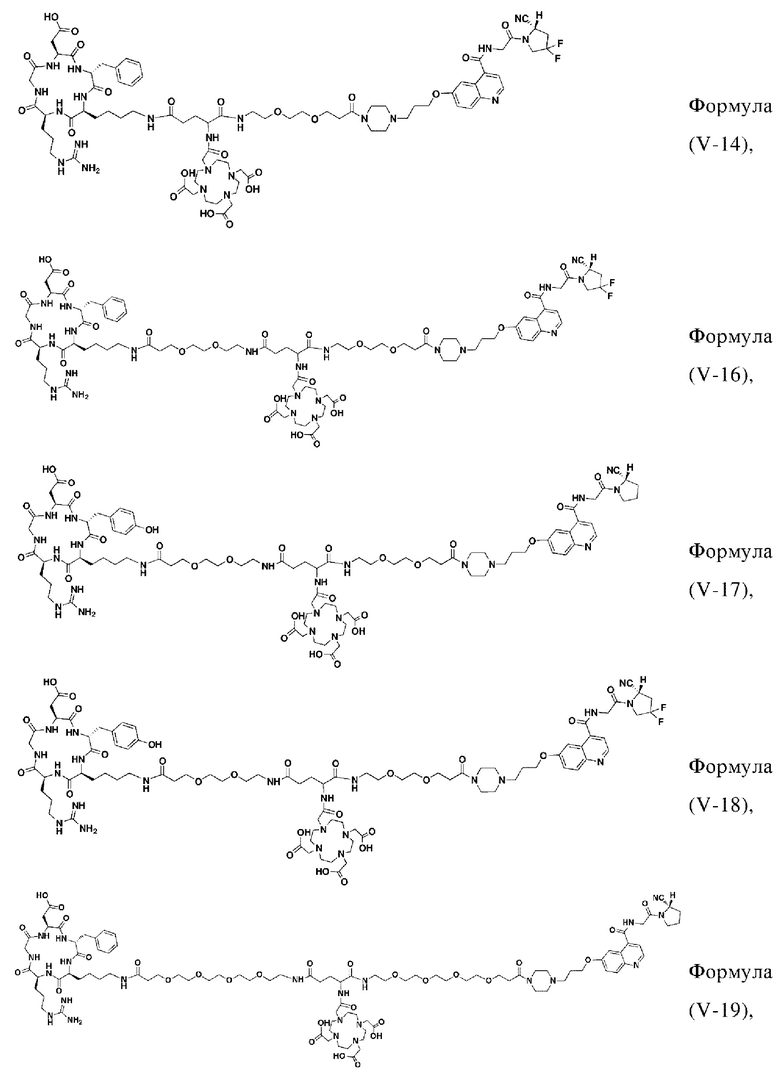
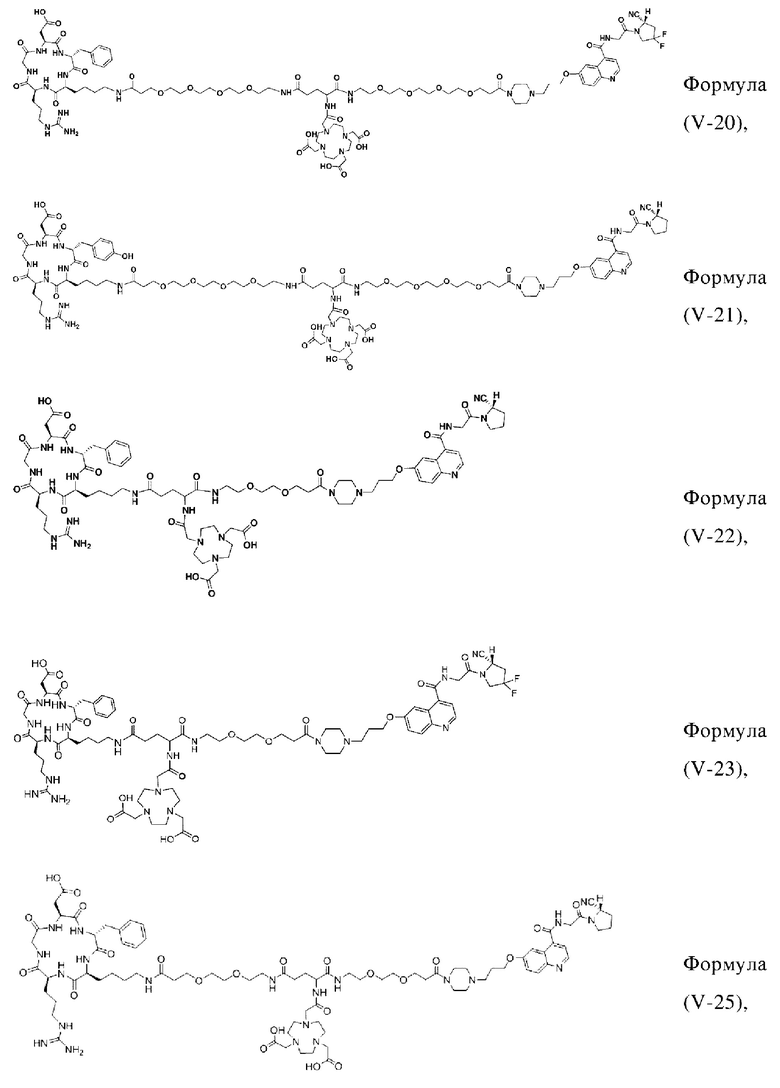
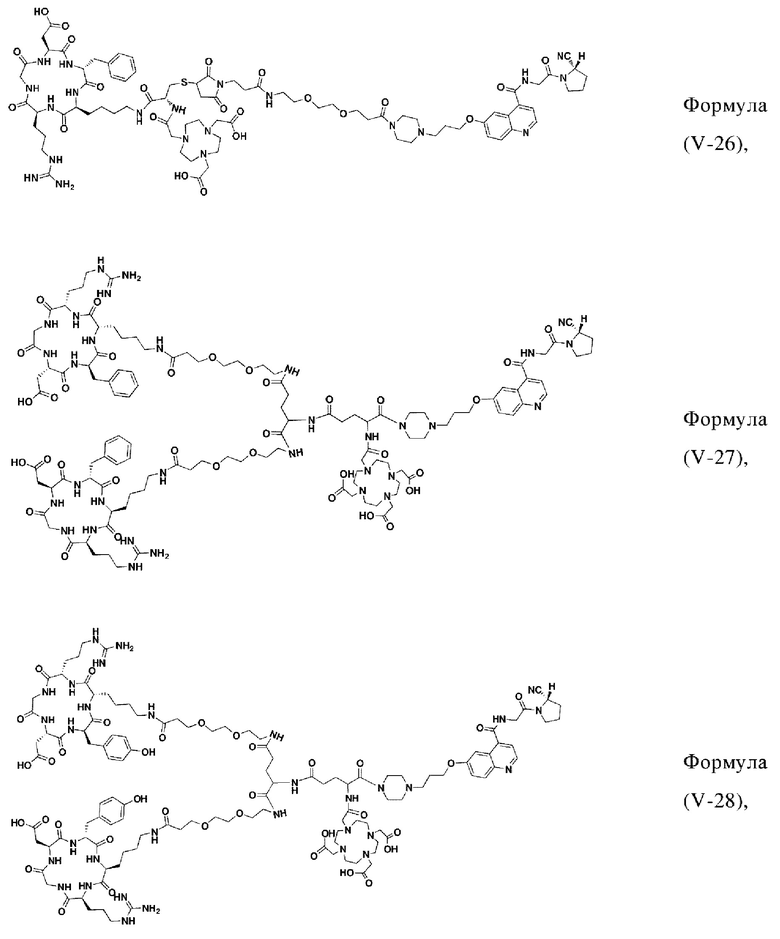
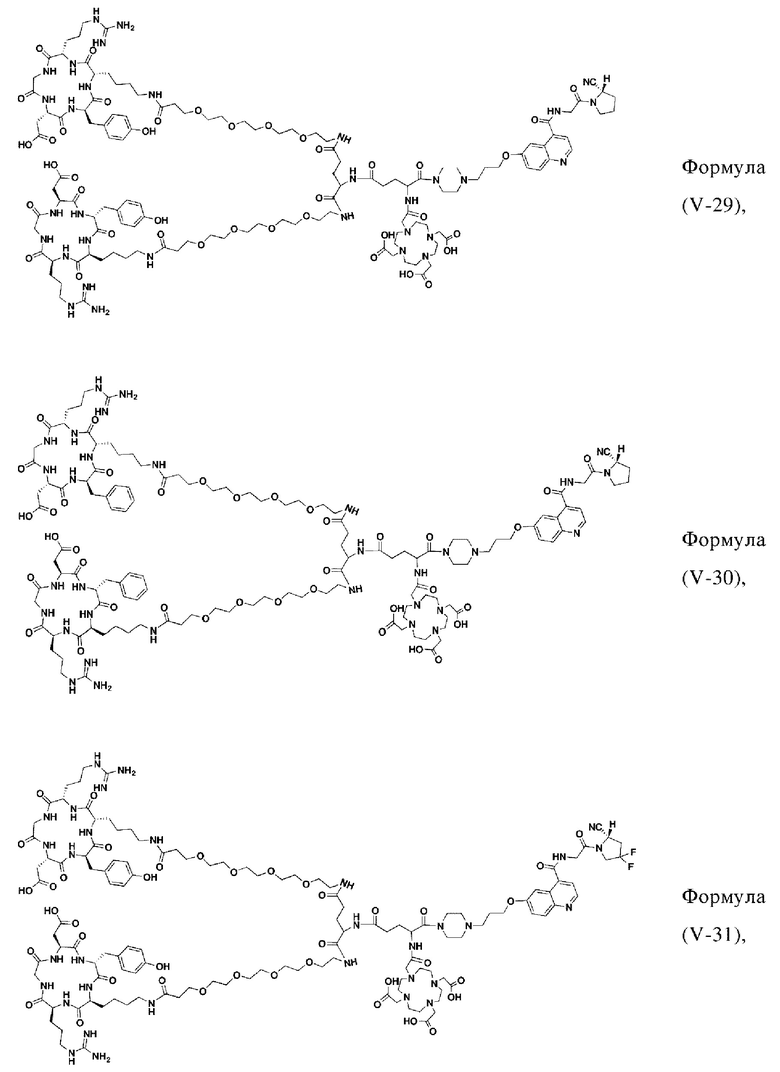
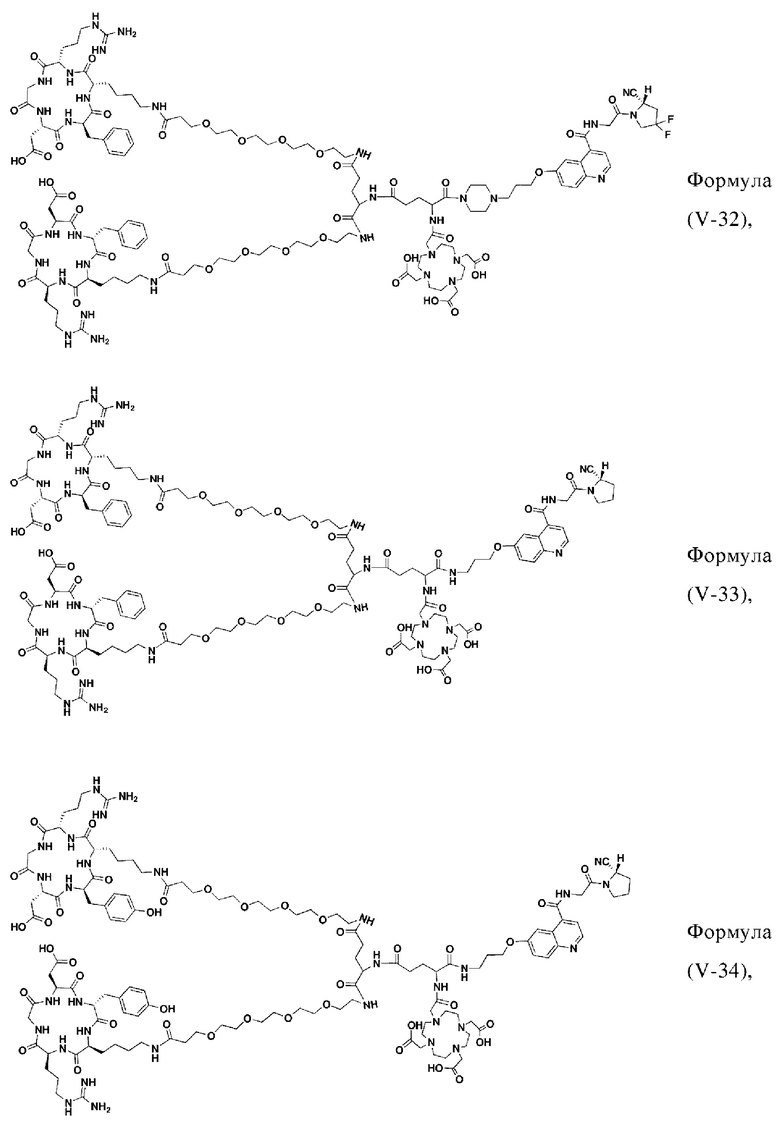
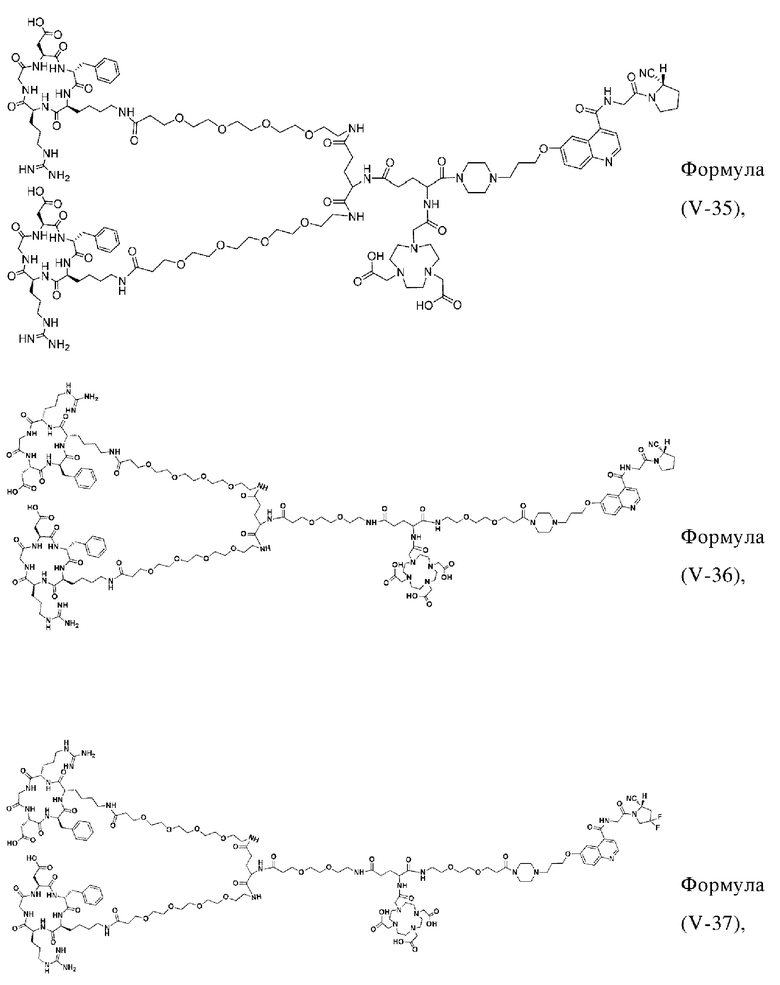
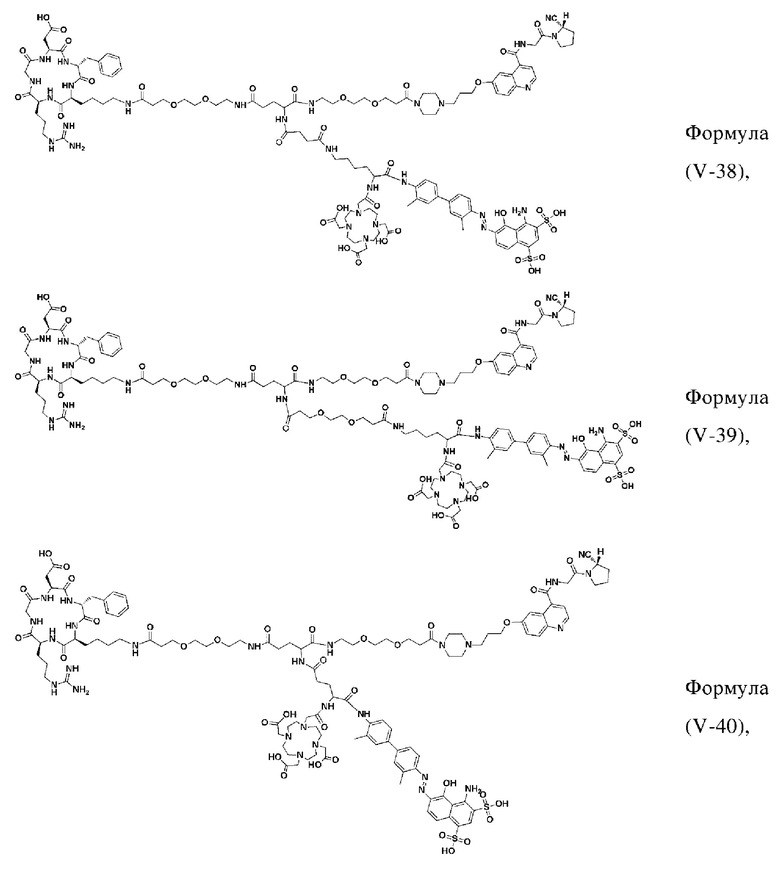
В третьем аспекте настоящее изобретение дополнительно обеспечивает меченное радионуклидами соединение двойного нацеливания на основе соединения, которое можно метить радионуклидом в соответствии с любым из приведенных выше описаний, где меченное радионуклидами соединение двойного нацеливания образуется путем хелатирования группы W в соединении формулы (V) или формула (VI) в соответствии с любым из приведенных выше описаний с радионуклидом.
Предпочтительно радионуклид может быть выбран из изотопов, испускающих α-лучи, изотопов, испускающих β-лучи, изотопов, испускающих γ-лучи, изотопов, испускающих Оже-электроны, или изотопов, испускающих рентгеновские лучи, и т.п.
Более предпочтительно, радионуклид выбирают из любого из следующего: 18F, 51Cr, 67Ga, 68Ga, 111In, 99mTc, 186Re, 188Re, 139La, 140La, 175Yb, 153Sm, 166Но, 86Y, 90Y, 149Pm, 165Dy, 169Er, 177Lu, 47Sc, 142Pr, 159Gd, 212Bi, 213Bi, 72As, 72Se, 97Ru, 109Pd, 105Rh, 101mRh, 119Sb, 128Ba, 123I, 124I, 131I,197Hg, 211At, 151Eu, 153Eu, 169Eu, 201T1, 203Pb, 212Pb, 64Cu, 67Cu, 198Au, 225Ac, 227Th, 89Zr или 199Ag.
Более предпочтительно, радионуклид представляет собой любой из следующего 18F, 64Cu, 68Ga, 89Zr, 90Y, 111In, 99mTc, 177Lu, 188Re или 225Ac. В некоторых конкретных вариантах осуществления радионуклид представляет собой 18F; в других конкретных вариантах осуществления радионуклид представляет собой 64; в других конкретных вариантах осуществления радионуклид представляет собой 68Ga; в других конкретных вариантах осуществления радионуклид представляет собой 89Zr; в других конкретных вариантах осуществления радионуклид представляет собой 90Y; в других конкретных вариантах осуществления радионуклид представляет собой 111In; в других конкретных вариантах осуществления радионуклид представляет собой 99mTc; в других конкретных вариантах осуществления радионуклид представляет собой 177Lu; в других конкретных вариантах осуществления радионуклид представляет собой 188Re; и в других конкретных вариантах осуществления радионуклид представляет собой 225Ас.
В четвертом аспекте настоящее изобретение дополнительно обеспечивает фармацевтически приемлемые таутомеры, рацематы, гидраты, сольваты или соли соединения двойного нацеливания, соединение, которое можно метить радионуклидом, и меченное радионуклидами соединение двойного нацеливания согласно любому из приведенных выше описаний.
В пятом аспекте настоящее изобретение дополнительно обеспечивает способ получения целевого соединения формулы (V) в соответствии с любым из приведенных выше описаний и его соединения, меченного радионуклидами. Способ включает в себя:
(1) сначала проводят реакцию амидной конденсации между карбоксилом 6-гидроксихинолин-4-карбоновой кислоты и амино-трет-бутиловым эфиром глицина; затем соединение Вос-защищенного пиперазинила с помощью алкильной цепи в гидроксильном положении продукта конденсации амида; удаление Вос- и трет-бутильных защитных групп в кислых условиях, а затем введение Вос-защитной группы в пиперазиновое кольцо; после чего проводят реакцию конденсации амида с гидрохлоридом (S)-пирролидин-2-карбонитрила или гидрохлоридом (S)-4,4-дифторпирролидин-2-карбонитрила; после удаления Вос-защитной группы проводят реакцию конденсации с N-Вос-3-[2-(2-аминоэтокси)этокси]пропионовой кислотой; затем проводят удаление Вос-защитной группы и последовательное проведение реакции с пропионатом малеимида и цистеином с защитной группой, либо затем проводят реакцию с глутаминовой кислотой или лизином с защитной группой; и, наконец, вводят RGD (c(RGDyK), c(RGDfK) или c(RGDyK)/c(RGDfK) с короткой цепью PEG) посредством реакции активированного сложного эфира с получением соединения двойного нацеливания;
(2) проведение реакции между соединением двойного нацеливания, полученным на стадии (1), и нуклидным хелатообразующим агентом для получения соединения, которое можно метить радионуклидом, как показано в формуле (V), согласно некоторым описаниям, где нуклидный хелатирующий агент выбирают из любого из следубщего: гидроксисукцинимид-тетраазациклододекан-N,N',N,N'-тетрауксусная кислота (DOTA-NHS), эфир НОТА-сукцинимида (HOTA-NHS), сукцинимидный активный эфир иминодиуксусной кислоты, сукцинимидный активный эфир диэтилентриамин-N,N,N',N'N'-пентауксусной кислоты (DTPA-NHS) и сукцинимидный активный эфир бис(карбоксиметил)имидазолглицината или 6-гидразинопиридин-3-карбоновой кислоты (HYNIC-NHS); а также
(3) проведение реакции между соединением, которое можно метить радионуклидом, полученным на стадии (2), и соединением, содержащим радионуклид, с помощью существующего способа мокрого мечения или способа лиофилизационной сушки для приготовления и получения меченого радионуклидами целевого соединения по настоящему изобретению.
В шестом аспекте настоящее изобретение дополнительно обеспечивает фармацевтическую композицию. Фармацевтическая композиция включает соединение двойного нацеливания, соединение, которое можно метить радионуклидом, или меченное радионуклидами соединение двойного нацеливания согласно любому из приведенных выше описаний, или любые фармацевтически приемлемые таутомеры, рацематы, гидраты, сольваты или любые фармацевтически приемлемые таутомеры, рацематы, гидраты, сольваты или их соли; или фармацевтическая композиция состоит из соединения двойного нацеливания, соединения, которое можно метить радионуклидом, или меченное радионуклидами соединение двойного нацеливания по любому из приведенных выше описаний, или любые фармацевтически приемлемые таутомеры, рацематы, гидраты, сольваты или их соли и любой фармацевтически приемлемый носитель и/или наполнитель.
В седьмом аспекте настоящее изобретение дополнительно предусматривает применение соединения двойного нацеливания, соединения, которое можно метить радионуклидом, или меченного радионуклидами соединения двойного нацеливания по любому из приведенных выше описаний при получении лекарственных средств для диагностики или лечения заболеваний, характеризующихся сверхэкспрессией белка активации фибробластов (FAP) и/или интегрина αvβ3 у животных или людей.
Настоящее изобретение дополнительно предусматривает способ диагностики или лечения заболеваний, характеризующихся сверхэкспрессией белка активации фибробластов (FAP) и/или интегрина αvβ3 у животных или людей, с использованием соединения двойного нацеливания, соединения, которое можно метить радионуклидом, или меченного радионуклидами соединения двойного нацеливания в соответствии с любым из приведенных выше описаний.
Предпочтительно заболевания, характеризующиеся сверхэкспрессией белка активации фибробластов (FAP) и/или интегрина αvβ3, включают, помимо прочего: рак, хроническое воспаление, атеросклероз, фиброз, ремоделирование тканей и рубцовые заболевания; и предпочтительно рак дополнительно выбирают из рака молочной железы, рака поджелудочной железы, рак тонкой кишки, рак толстой кишки, рак прямой кишки, рак легких, рак головы и шеи, рак яичников, гепатоцеллюлярная карцинома, рак пищевода, карцинома гортаноглотки, карцинома носоглотки, карцинома гортани, клетки миеломы, рак мочевого пузыря, холангиоцеллюлярная карцинома, светлоклеточный почечноклеточный рак, нейроэндокринное новообразование, канцерогенная остеомаляция, саркома, рак неизвестной первичной формы (CUP), карцинома тимуса, глиома, нейроглиома, астроцитома, рак шейки матки или рак простаты.
В восьмом аспекте настоящее изобретение дополнительно предоставляет набор. Набор включает в себя или состоит из целевого соединения, указанного в формуле (I) или формуле (II), соединения, указанного в формуле (V) или формуле (VI), меченного радионуклидами целевого соединения по настоящему изобретению или фармацевтической композиции по настоящему изобретению, и инструкции по диагностике заболеваний.
Соединение со структурой FAPI-RGD, предлагаемое настоящим изобретением, может синергически воздействовать на мишень FAP и мишень интегрина αvβ3 в опухолях, в результате чего количество и эффективность использования эффективных рецепторов в опухолях могут быть улучшены. Представленное далее соединение, меченное радионуклидами, на основе структуры, как ожидается, будет применяться для диагностики или лечения заболеваний, характеризующихся повышенной экспрессией белка активации фибробластов (FAP) и/или интегрина αvβ3.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Фиг. 1 представляет собой масс-спектрограмму соединения 2 в примере 1 настоящего изобретения.
Фиг.2 представляет собой ядерно-магнитный водородный спектр соединения 2 в примере 1 настоящего изобретения.
Фиг. 3 представляет собой ядерно-магнитный углеродный спектр соединения 2 в примере 1 настоящего изобретения.
Фиг. 4 представляет собой масс-спектрограмму соединения 3 в примере 1 настоящего изобретения.
Фиг. 5 представляет собой ядерно-магнитный водородный спектр соединения 3 в примере 1 настоящего изобретения.
Фиг. 6 представляет собой масс-спектрограмму соединения 4 в примере 1 настоящего изобретения.
Фиг. 7 представляет собой ядерно-магнитный водородный спектр соединения 4 в примере 1 настоящего изобретения.
Фиг. 8 представляет собой ядерно-магнитный углеродный спектр соединения 4 в примере 1 настоящего изобретения.
Фиг. 9 представляет собой масс-спектрограмму соединения 7 в примере 1 настоящего изобретения.
Фиг. 10 представляет собой ядерно-магнитный водородный спектр соединения 7 в примере 1 настоящего изобретения.
Фиг. 11 представляет собой ядерно-магнитный углеродный спектр соединения 7 в примере 1 настоящего изобретения.
Фиг.12 представляет собой масс-спектрограмму соединения 9 в примере 1 настоящего изобретения.
Фиг. 13 представляет собой масс-спектрограмму соединения 10 в примере 1 настоящего изобретения.
Фиг. 14 представляет собой масс-спектрограмму соединения 11 в примере 1 настоящего изобретения.
Фиг. 15 представляет собой масс-спектрограмму соединения формулы (V-1) в примере 1 настоящего изобретения.
Фиг. 16 представляет собой масс-спектрограмму промежуточного продукта М в примере 2 настоящего изобретения.
Фиг. 17 представляет собой масс-спектрограмму промежуточного продукта О в примере 2 настоящего изобретения.
Фиг. 18 представляет собой масс-спектрограмму промежуточного продукта В в примере 2 настоящего изобретения.
Фиг. 19 представляет собой масс-спектрограмму промежуточного продукта С в примере 2 настоящего изобретения.
Фиг. 20 представляет собой масс-спектрограмму промежуточного продукта D в примере 2 настоящего изобретения.
Фиг. 21 представляет собой масс-спектрограмму промежуточного продукта Е в примере 2 настоящего изобретения.
Фиг.22 представляет собой масс-спектрограмму промежуточного продукта F в примере 2 настоящего изобретения.
Фиг. 23 представляет собой масс-спектрограмму промежуточного продукта G в примере 2 настоящего изобретения.
Фиг. 24 представляет собой масс-спектрограмму промежуточного продукта Н в примере 2 настоящего изобретения.
Фиг. 25 представляет собой масс-спектрограмму промежуточного продукта I в примере 2 настоящего изобретения.
Фиг. 26 представляет собой масс-спектрограмму промежуточного продукта J в примере 2 настоящего изобретения.
Фиг. 27 представляет собой масс-спектрограмму промежуточного продукта Q в примере 2 настоящего изобретения.
Фиг. 28 представляет собой масс-спектрограмму соединения формулы (V-14) в примере 2 настоящего изобретения.
Фиг. 29 представляет собой масс-спектрограмму промежуточного продукта К в примере 3 настоящего изобретения.
Фиг. 30 представляет собой масс-спектрограмму соединения формулы (V-23) в примере 3 настоящего изобретения.
Фиг. 31 представляет собой масс-спектрограмму промежуточного продукта В1 в примере 4 настоящего изобретения.
Фиг.32 представляет собой масс-спектрограмму промежуточного продукта D1 в примере 4 настоящего изобретения.
Фиг. 33 представляет собой масс-спектрограмму промежуточного продукта G1 в примере 4 настоящего изобретения.
Фиг. 34 представляет собой масс-спектрограмму промежуточного продукта H1 в примере 4 настоящего изобретения.
Фиг. 35 представляет собой масс-спектрограмму промежуточного продукта I1 в примере 4 настоящего изобретения.
Фиг. 36 представляет собой масс-спектрограмму промежуточного продукта J1 в примере 4 настоящего изобретения.
Фиг. 37 представляет собой масс-спектрограмму соединения формулы (V-25) в примере 4 настоящего изобретения.
Фиг. 38 представляет собой масс-спектрограмму промежуточного продукта Н3 в примере 6 настоящего изобретения.
Фиг. 39 представляет собой масс-спектрограмму промежуточного продукта I2 в примере 6 настоящего изобретения.
Фиг. 40 представляет собой масс-спектрограмму промежуточного продукта O1 в примере 6 настоящего изобретения.
Фиг. 41 представляет собой масс-спектрограмму промежуточного продукта Р1 в примере 6 настоящего изобретения.
Фиг.42 представляет собой масс-спектрограмму соединения формулы (V-30) в примере 6 настоящего изобретения.
Фиг. 43 представляет собой масс-спектрограмму промежуточного продукта N2 в примере 7 настоящего изобретения.
Фиг. 44 представляет собой масс-спектрограмму промежуточного продукта F3 в примере 7 настоящего изобретения.
Фиг. 45 представляет собой масс-спектрограмму соединения формулы (V-35) в примере 7 настоящего изобретения.
На фиг. 46 показаны результаты контроля качества соединения 68Ga-FAPI-RGD (V-1) по настоящему изобретению с помощью высокоэффективной жидкостной хроматографии (ВЭЖХ).
Фиг. 47 показаны результаты визуализации MicroPET соединения 68Ga-FAPI-RGD (V-1) у мышей с опухолями HepG2-FAP по настоящему изобретению.
Фиг. 48 показаны результаты визуализации MicroPET соединения 68Ga-FAPI-RGD (V-1) у мышей с опухолями HepG2-FAP после совместного введения с FAPI-02 по настоящему изобретению.
Фиг. 49 показаны результаты визуализации MicroPET соединения 68Ga-FAPI-RGD (V-1) у мышей с опухолями HepG2-FAP после совместного введения с RGD по настоящему изобретению.
Фиг. 50 представляет собой статистическую диаграмму результатов поглощения соединения 68Ga-FAPI-RGD (V-1) в опухолях и жизненно важных органах после совместного введения с C(RGDfK) или FAPI-02 в течение 30 минут по настоящему изобретению (на фигуре по оси абсцисс показаны различные органы, а гистограммы для каждого органа представляют, слева направо, поглощение меченного 68Ga комплекса FAPI-RGD, поглощение 68Ga-FAPI-RGD после совместного введения с FAPI-02 для блокирования FAP и поглощение 68Ga-FAPI-RGD после совместного введения с RGD для блокирования интегрина, соответственно).
На фиг. 51 показаны результаты эксперимента по стабилизации соединения 68Ga-FAPI-RGD (V-25) в физиологическом растворе по настоящему изобретению.
На фиг. 52 показаны результаты эксперимента по поглощению клетками и связыванию с клетками соединения 68Ga-FAPI-RGD (V-25) по настоящему изобретению.
На фиг. 53 показаны результаты визуализации MicroPET соединения 68Ga-FAPI-RGD (V-25) и мономеров 68Ga-FAPI-02 и 68Ga-C(RGDfK) у мышей с опухолью HT1080-FAP по настоящему изобретению.
На фиг. 54 показаны результаты визуализации MicroPET соединения 68Ga-FAPI-RGD (V-25) после совместного введения с C(RGDfK) и/или FAPI-02 в течение 30 мин и статистическая диаграмма результатов поглощения в опухолях и жизненно важных органах по настоящему изобретению.
На фиг. 55 показаны результаты ПЭТ/КТ-визуализации соединения 68Ga-FAPI-RGD (V-25), 18 F-FDG и 68Ga-FAPI46 у пациентов с раком поджелудочной железы, немелкоклеточным раком легкого, мелкоклеточным раком легкого и карциномой носоглотки после внутривенного введения в течение 3 часов в настоящее изобретение.
Фиг. 56 представляет собой масс-спектрограмму промежуточного продукта G2 по настоящему изобретению.
Фиг. 57 представляет собой масс-спектрограмму промежуточного продукта N1 по настоящему изобретению.
Фиг. 58 представляет собой масс-спектрограмму промежуточного продукта Р по настоящему изобретению.
Фиг. 59 представляет собой масс-спектрограмму соединения формулы (V-26) по настоящему изобретению.
На фиг. 60 показана ОФЭКТ-визуализация соединения V-40, меченного радиоактивным веществом 177Lu (а именно, соединения 177Lu-FAPI-RGD (V-40)), представленного в настоящем изобретении.
ПОДРОБНОЕ ОПИСАНИЕ ВАРИАНТОВ ОСУЩЕСТВЛЕНИЯ
Технические решения настоящего изобретения далее подробно описаны без ограничений в сочетании с конкретными вариантами осуществления, приведенными ниже. Следует отметить, что приведенные ниже примеры предназначены исключительно для иллюстрации технической концепции и характеристик настоящего изобретения с целью ознакомления с содержанием настоящего изобретения и его применения специалистами в данной области техники и не предназначены для ограничения объема защиты настоящего изобретения. Любые эквивалентные варианты или модификации, выполненные в соответствии с духом и сущностью настоящего изобретения, подпадают под сферу защиты настоящего изобретения.
Пример 1: Получение соединения формулы (1-1) и соединения формулы (V-1)
Путь синтеза заключается в следующем.
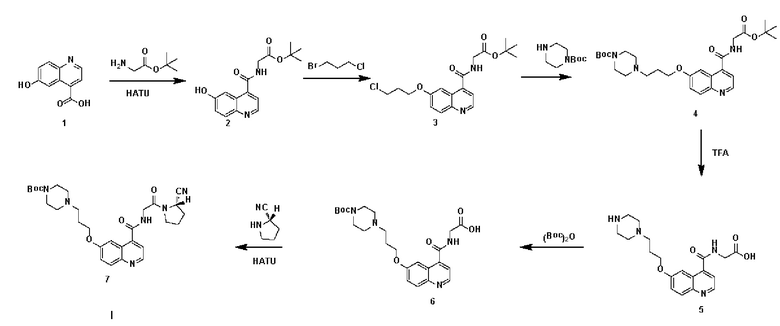
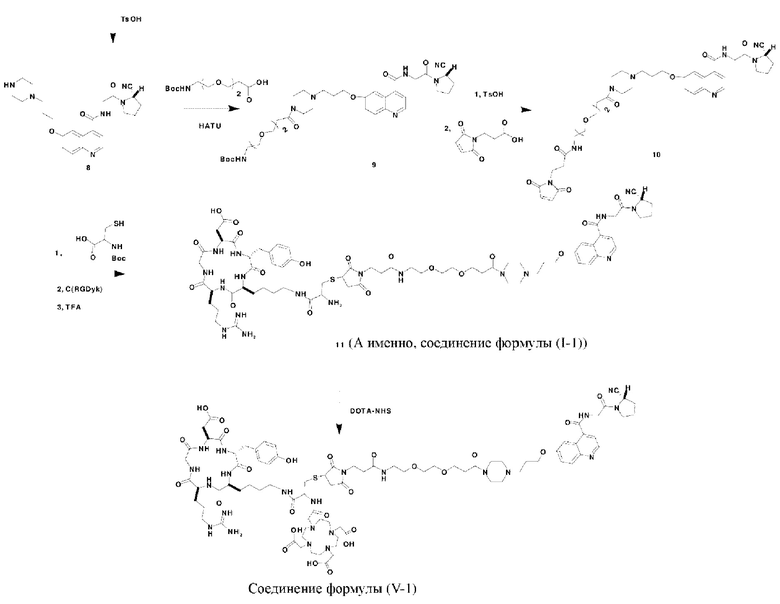
(1) Синтез соединения 2
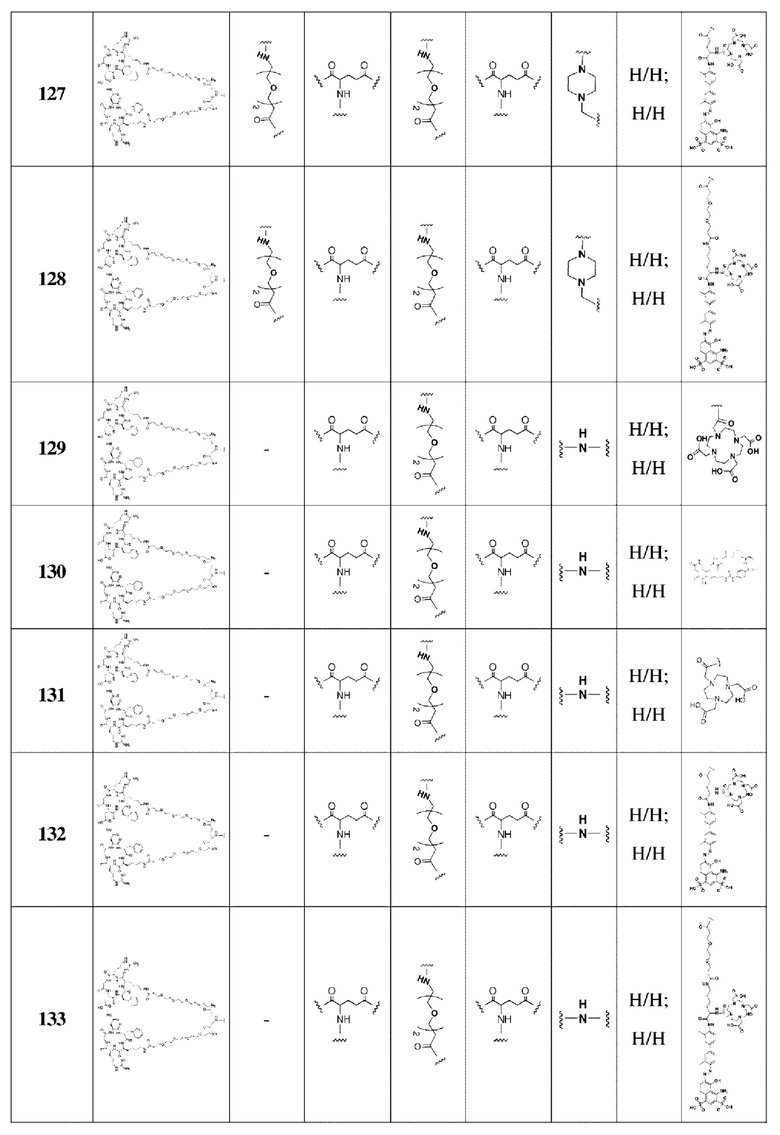
В 30 мл N,N,-диметилформамида в колбе емкостью 100 мл последовательно помещали соединение 1 (6-гидроксихинолин-4-карбоновая кислота, 1,89 г, 10,0 ммоль), трет-бутиловый эфир глицина (1,89 г, 10,0 ммоль), HATU (3,8 г, 10,0 ммоль) и N,N-диизопропилэтиламин (2,6 г, 20,0 ммоль), соответственно. Реакционную смесь перемешивали в течение ночи и подвергали дистилляции при пониженном давлении для удаления растворителя с получением неочищенного продукта. Затем проводили очистку с помощью колонки с силикагелем (с использованием дихлорметана и метанола в соотношении 30:1) для получения белого твердого соединения 2 с выходом 87%. Фиг. 1 представлена масс-спектрограмма соединения 2. На фиг. 2 показан ядерно-магнитный водородный спектр соединения 2. На фиг. 3 показан ядерно-магнитный углеродный спектр соединения 2.
(2) Синтез соединения 3
Соединение 2 (1,51 г, 5,0 ммоль), 1-бром-3-хлорпропан (1,55 г, 10,0 ммоль) и карбонат калия (1,38 г, 10,0 ммоль) последовательно вводили в 50 мл N,N-диметилформамида в колбе емкостью 100 мл, соответственно. Систему нагревали до 60°С, перемешивали в течение ночи при температуре 60°С и подвергали дистилляции под пониженным давлением для удаления растворителя с получением неочищенного продукта. Затем проводили очистку с помощью колонки с силикагелем (с использованием дихлорметана и метанола в соотношении 50:1) для получения белого твердого соединения 3 с выходом 63%. Фиг. 4 представлена масс-спектрограмма соединения 3. На фиг. 5 показан ядерно-магнитный водородный спектр соединения 3.
(3) Синтез соединения 4
Соединение 3 (0,76 г, 2,0 ммоль), трет-бутил-1-пиперазинкарбоксилат (0,55 г, 3,0 ммоль) и йодид калия (0,49 г, 3,0 ммоль) последовательно добавляли в 30 мл ацетонитрила в колбе емкостью 100 мл, соответственно. Систему нагревали до 60°С, перемешивали в течение ночи при температуре 60°С и подвергали дистилляции под пониженным давлением для удаления растворителя с получением неочищенного продукта. Затем проводили очистку с помощью колонки с силикагелем (с использованием дихлорметана и метанола в соотношении 30:1) для получения белого твердого соединения 4 с выходом 58%. MS (ESI) m/z, рассчитанное для [C28H40N4O6], составило:
528.29; найдено: 529.10 [М+Н]+. Фиг. 6 представлена масс-спектрограмма соединения 4. На фиг. 7 показан ядерно-магнитный водородный спектр соединения 4. На фиг. 8 показан ядерно-магнитный углеродный спектр соединения 4.
(4) Синтез соединения 5
Соединение 4 (0,52 г, 1,0 ммоль) растворяли в 10 мл смешанного раствора дихлорметана и трифторуксусной кислоты (в объемном соотношении 9:1) в условиях ледяной бани. Систему нагревали до комнатной температуры для проведения реакции в течение 2 ч. После завершения реакции проводили дистилляцию при пониженном давлении для удаления растворителя и растворяли систему в 10 мл N,N-диметилформамида для получения соединения 5 для последующего применения.
(5) Синтез соединения 6
Ди-трет-бутилдикарбонат (0,22 г, 1,0 ммоль) и N,N-диизопропилэтиламин (0,39 г, 3,0 ммоль) по отдельности добавляли к раствору N,N-диметилформамида соединения 5. Систему перемешивали в течение ночи при комнатной температуре, а затем подвергали дистилляции под пониженным давлением для удаления растворителя с получением неочищенного продукта. Затем проводили очистку с помощью колонки с силикагелем (с использованием дихлорметана и метанола в соотношении 10:1) для получения белого твердого соединения 6 с выходом 72%.
(6) Синтез соединения 7
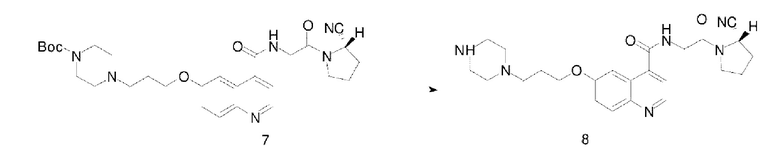
Соединение 6 (0,47 г, 1,0 ммоль), (S)-пирролидин-2-карбонитрила гидрохлорид (0,13 г, 1,0 ммоль), HATU (0,38 г, 1,0 ммоль) и N,N-диизопропилэтиламин (0,26 г, 2,0 ммоль) последовательно вводили в 10 мл N,N-диметилформамид во флаконе объемом 100 мл, соответственно. Реакционную смесь перемешивали при комнатной температуре до завершения реакции, а затем подвергали дистилляции при пониженном давлении для удаления растворителя с получением неочищенного продукта. Затем проводили очистку с помощью колонки с силикагелем (с использованием дихлорметана и метанола в соотношении 50:1) для получения белого твердого соединения 7 с выходом 85%. Фиг. 9 представлена масс-спектрограмма соединения 7. На фиг. 10 показан ядерно-магнитный водородный спектр соединения 7. На фиг. 11 показан ядерно-магнитный углеродный спектр соединения 7.
(7) Синтез соединения 8
Соединение 7 (0,55 г, 1,0 ммоль) и моногидрат п-толуолсульфоновой кислоты (0,27 г, 1,5 ммоль) последовательно добавляли в 10 мл ацетонитрила в колбе емкостью 100 мл, соответственно. Реакционную систему нагревали до 60°С, перемешивали до завершения реакции и подвергали дистилляции при пониженном давлении для удаления растворителя с получением неочищенного продукта соединения 8.
(8) Синтез соединения 9
N-Вос-3-[2-(2-аминоэтокси)этокси]пропионовую кислоту (0,27 г, 1,0 ммоль), ХАТУ (0,38 г, 1,0 ммоль), N,N-диизопропилэтиламин (0,26 г, 2,0 ммоль) и 10 мл N, N-диметилформамида помещали в реакционную колбу, содержащую соединение 8 соответственно. Реакционную смесь перемешивали в течение ночи и подвергали дистилляции при пониженном давлении для удаления растворителя с получением неочищенного продукта.Затем проводили очистку с помощью колонки с силикагелем (с использованием дихлорметана и метанола в соотношении 50:1) для получения белого твердого соединения 9 с выходом 64%. Фиг. 12 представлена масс-спектрограмма соединения 9.
(9) Синтез соединения 10
Соединение 9 (0,66 г, 1,0 ммоль) и моногидрат п-толуолсульфокислоты (0,27 г, 1,5 ммоль) последовательно помещали в 10 мл ацетонитрила в колбе емкостью 100 мл соответственно. Реакционную систему нагревали до 60°С, перемешивали до завершения реакции и подвергали перегонке при пониженном давлении для удаления растворителя с получением неочищенного продукта. Неочищенный продукт растворяли в 10 мл N,N-диметилформамида, затем добавляли HATU (0,38 г, 1,0 ммоль), N,N-диизопропилэтиламин (0,26 г, 2,0 ммоль) и пропионат малеимида (0,17 г, 1,0 ммоль) для проведения реакции в течение 3 ч, и проводили перегонку при пониженном давлении для удаления растворителя и получения неочищенного продукта. Затем проводили очистку с помощью колонки с силикагелем (с использованием дихлорметана и метанола в соотношении 50:1) для получения белого твердого соединения 10 с выходом 59%. MS (ESI) m/z, рассчитанное для [C35H51N7O8], составило: 697.38; найдено: 698.43 [М+Н]+. Фиг. 13 представлена масс-спектрограмма соединения 10.
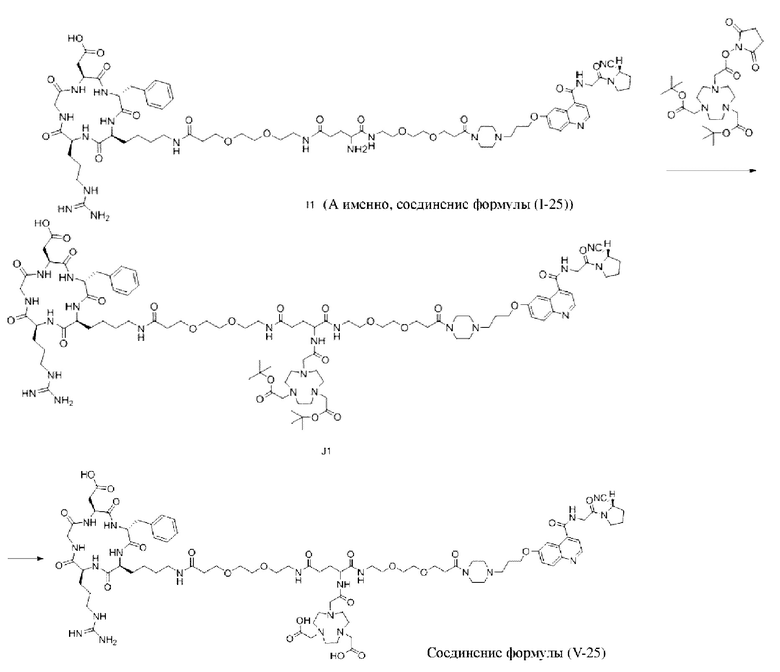
(10) Синтез соединения 11 (а именно, соединения формулы (I-1))
Соединение 10 (0,072 г, 0,1 ммоль), Вос-защищенный цистеин (0,022 г, 0,1 ммоль) и 10 мл N,N-диметилформамида помещали в колбу емкостью 25 мл соответственно и перемешивали при комнатной температуре для проведения реакции в течение 3 часов, после завершения реакции по данным мониторинга в систему добавляли 0,12 ммоль DCC и NHS, систему перемешивали для проведения реакции в течение 12 ч, затем c(RGDyK) (0,06 г, 0,1 ммоль) и N,N -диизопропилэтиламин (0,065 г, 0,05 ммоль) добавляли для проведения реакции в течение 12 часов и проводили перегонку при пониженном давлении для удаления растворителя и получения неочищенного продукта. Затем неочищенный продукт очищали с помощью колонки с обращенной фазой и подвергали сублимационной сушке для получения чистого соединения 11 (а именно соединения формулы (I-1)) с выходом 43% в две стадии. Фиг. 14 представлена масс-спектрограмма соединения 11.
(11) Синтез соединения формулы (V-1)
Соединение 11 (0,146 г, 0,1 ммоль) и тиоанизол, 1,2-этандитиол, анизол и TFA (в соотношении 5:3:2:90) помещали в колбу емкостью 25 мл соответственно для удаления трет-бутильной и Вос-защитных групп при комнатной температуре. После завершения реакции TFA удаляли потоком аргона, систему растворяли в 10 мл N,N-диметилформамида и последовательно добавляли DOTA-NHS (0,05 г, 0,1 ммоль) и N,N диизопропилэтиламин (0,04 г, 0,3 ммоль). Реакционную систему перемешивали для проведения реакции при комнатной температуре. После завершения реакции, основанной на мониторинге с помощью ВЭЖХ, была проведена дистилляция под пониженным давлением для удаления растворителя с получением неочищенного продукта. Затем неочищенный продукт очищали с помощью колонки с обращенной фазой и подвергали сублимационной сушке для получения чистого соединения формулы (V-1) с выходом 53%. На фиг. 15 представлена масс-спектрограмма соединения формулы (V-1).
Пример 2: Получение соединения формулы (1-14) и соединения формулы (V-14)
Путь синтеза заключается в следующем. 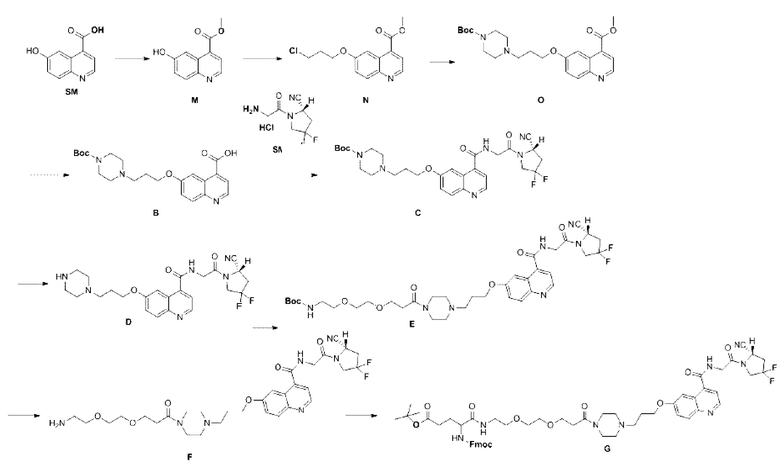
(1) Приготовление промежуточного продукта
SM (6-гидроксихинолин-4-карбоновую кислоту) растворяли в 100 мл метанола, добавляли по каплям 1 мл концентрированной серной кислоты, полученную смесь помещали во внешнюю ванну при температуре 90°С для проведения реакции в течение ночи, затем реакцию завершали на основании контроля с помощью тонкого слоя хроматография (TLC). Метанол концентрировали, и концентрированную систему добавляли по каплям в 40 мл насыщенного NaHCO3. После завершения каплеобразования систему перемешивали и кристаллизовали в течение 1 ч, а затем фильтровали и сушили для получения промежуточного продукта М с выходом 59%. Теоретическая молекулярная масса составляет 203,0582, измеренная молекулярная масса - 203,06767, а результаты масс-спектрометрии соответствуют результатам целевого продукта. Фиг. 16 представлена масс-спектрограмма промежуточного продукта М.
(2) Приготовление промежуточного продукта N
Промежуточный продукт М растворяли в 30 мл DMF и последовательно добавляли карбонат калия (2,00 г, 14,5 ммоль) и 1-бром-3-хлорпропан (2,19 г, 13,9 ммоль) для проведения реакции в течение ночи во внешней бане при температуре 25°С. После завершения реакции по данным TLC добавляли 70 мл очищенной воды, дважды экстрагировали 70 мл DCM, органические фазы объединяли, сушили и концентрировали с получением неочищенного продукта промежуточного соединения N с выходом сырья 83,8%.
(3) Приготовление промежуточного продукта О
Промежуточный продукт N, 1-Вос-пиперазин (1,67 г, 6,8 ммоль) и KI (1,11 г, 6,7 ммоль) последовательно растворяли в 10 мл DMF для проведения реакции во внешней бане при 100°С. После завершения реакции по данным TLC добавляли 60 мл воды очищенной, дважды экстрагировали по 30 мл DCM, органические фазы объединяли и промывали 30 мл очищенной воды два раза. Затем органические фазы высушивали безводным сульфатом натрия и подвергали очистке в колонке для получения неочищенного продукта с выходом 86%). Теоретическая молекулярная масса составляет 429,2264, измеренная молекулярная масса - 429,24041, а результаты масс-спектрометрии соответствуют результатам целевого продукта. Фиг. 17 представлена масс-спектрограмма промежуточного продукта О.
(4) Приготовление промежуточного продукта В
Промежуточный продукт О растворяли в 10 мл метанола и добавляли водный раствор LiOH/1V для проведения реакции во внешней ванне при температуре 25°С.После завершения реакции, основанной на мониторинге с помощью TLC, метанол в системе концентрировали, добавляли в систему 2 мл воды и медленно по каплям добавляли 1 N НС1 для доведения значения рН до 6-7. Затем систему кристаллизовали в течение 1 ч, фильтровали и сушили для получения промежуточного продукта В с выходом 85%. Теоретическая молекулярная масса составляет 415,2107, измеренная молекулярная масса 415,21775, а результаты масс-спектрометрии соответствуют результатам целевого продукта. Фиг. 18 представлена масс-спектрограмма промежуточного продукта В.
(5) Приготовление промежуточного продукта С
Промежуточный продукт В, (S)-4,4-дифтор-1-глицилпирролидин-2-карбонитрил гидрохлорид (1,11 г, 2,7 ммоль), HATU (1,06 г, 2,8 ммоль) и DIPEA (1,1 г, 8,1 ммоль) последовательно растворяли в 10 мл DMF для проведения реакции во внешней бане при 25°С. После завершения реакции по данным TLC в систему добавляли 30 мл очищенной воды, дважды экстрагировали по 3 мл DCM, органические фазы объединяли, сушили безводным сульфатом натрия, концентрировали и подвергали очистке в колонке с получением промежуточного продукта С с выходом 13,4%. Теоретическая молекулярная масса составляет 586,2715, измеренная молекулярная масса 586,28448, а результаты масс-спектрометрии соответствуют результатам целевого продукта. Фиг. 19 представлена масс-спектрограмма промежуточного продукта С.
(6) Приготовление промежуточного продукта D
Промежуточный продукт С растворяли в 10 мл ацетонитрила, добавляли моногидрат п-толуолсульфоновой кислоты (1,54 г, 8,1 ммоль) и полученную смесь помещали во внешнюю ванну при температуре 65°С для проведения реакции. После завершения реакции, основанной на мониторинге с помощью TLC, был получен неочищенный продукт промежуточного продукта D. Теоретическая молекулярная масса составляет 486,2191, измеренная молекулярная масса 486,22858, а результаты масс-спектрометрии соответствуют результатам целевого продукта. Фиг. 20 представлена масс-спектрограмма промежуточного продукта D.
(7) Приготовление промежуточного продукта Е
Промежуточный продукт D растворяли в 10 мл DMF и последовательно добавляли DIPEA (2,71 г, 21,1 ммоль) и t-Вос-N-амидо-PEG2-NHS-эфир (1,22 г, 6,3 ммоль) для проведения реакции во внешней ванне при 25°С.После завершения реакции по данным ВЭЖХ к системе добавляли 30 мл очищенной воды, дважды экстрагировали 30 мл DCM, органические фазы объединяли, сушили, концентрировали и подвергали очистке в колонке с получением промежуточного продукта Е с выходом 64,7% в две стадии. Теоретическая молекулярная масса составляет 745,3611, измеренная молекулярная масса -745,37466, а результаты масс-спектрометрии соответствуют результатам целевого продукта. Фиг. 21 представлена масс-спектрограмма промежуточного продукта Е.
(8) Приготовление промежуточного продукта F
Промежуточный продукт Е растворяли в 10 мл ацетонитрила, добавляли моногидрат п-толуолсульфоновой кислоты (2,42 г, 12,7 ммоль) и проводили реакцию во внешней ванне при 65°С. После завершения реакции на основе мониторинга с помощью ВЭЖХ систему концентрировали для получения неочищенного продукта промежуточного продукта F. Теоретическая молекулярная масса составляет 645,3086, измеренная молекулярная масса составляет 645,31807, а результаты масс-спектрометрии согласуются с результатами целевого продукта. Фиг. 22 представлена масс-спектрограмма промежуточного продукта F.
(9) Приготовление промежуточного продукта G
Промежуточный продукт F и DIPEA (1,28 г, 10,1 ммоль) последовательно растворяли в 5 мл DMF для получения системы (1). HATU (0,84 г, 2,2 ммоль) и Fmoc-Glu(OtBu)OH (0,61 г, 2,2 ммоль) последовательно растворяли в 5 мл DMF для получения системы (2). Систему (2) перемешивали при 25°С в течение 1 часа, а затем добавляли к системе (1) для проведения реакции при 25°С. После завершения реакции по данным TLC в систему добавляли 20 мл очищенной воды, дважды экстрагировали по 20 мл DCM, органические фазы объединяли, один раз промывали насыщенным раствором хлорида натрия, концентрировали и подвергали очистке в колонке с получением промежуточного продукта G с выходом 63,6% в две стадии. Теоретическая молекулярная масса составляет 1052,4819, измеренная молекулярная масса - 1052,49330, а результаты масс-спектрометрии соответствуют результатам целевого продукта. Фиг. 23 представлена масс-спектрограмма промежуточного продукта G.
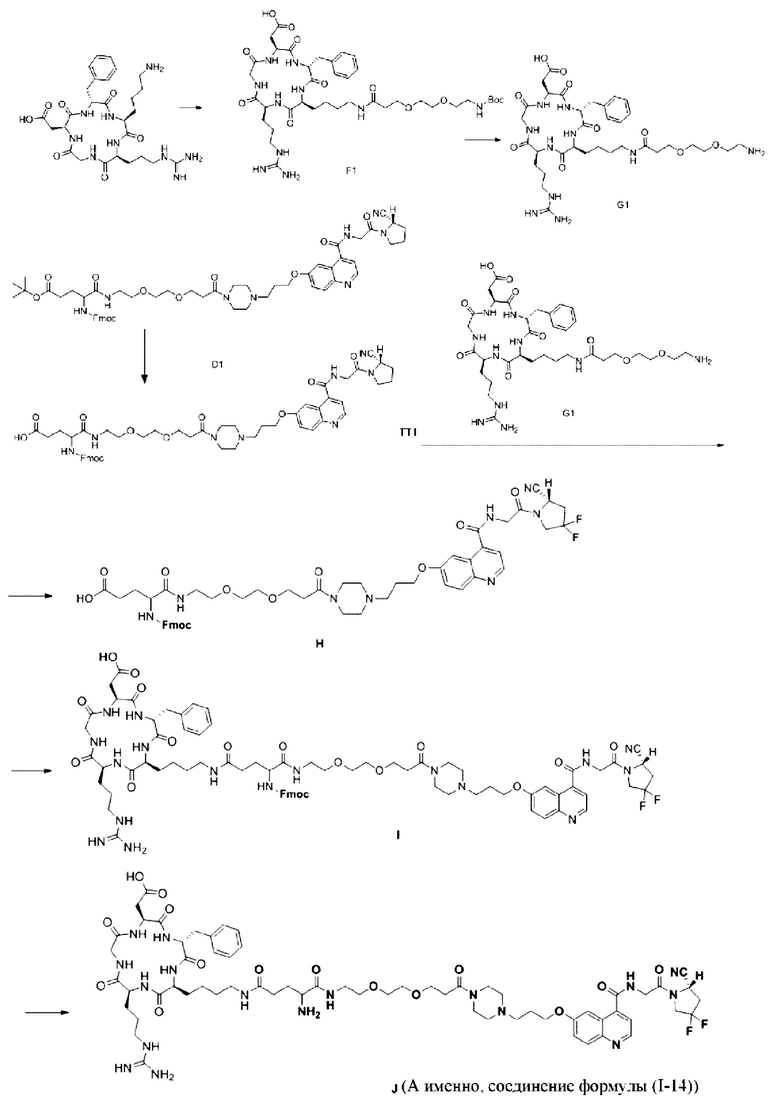
(10) Приготовление промежуточного продукта Н
Промежуточный продукт G растворяли в 10 мл ацетонитрила, добавляли моногидрат п-толуолсульфокислоты (2,87 г, 15,1 ммоль) и проводили реакцию при 65°С.После завершения реакции по данным контроля с помощью ВЭЖХ систему концентрировали и подвергали очистке в колонке с получением промежуточного продукта Н. Теоретическая молекулярная масса составляет 996,4193, измеренная молекулярная масса составляет 996,42947, а результаты масс-спектрометрии согласуются с результатами целевого продукта. Фиг. 24 представлена масс-спектрограмма промежуточного продукта Н.
(11) Приготовление промежуточного продукта I
Промежуточный продукт Н и DIPEA (1,62 г, 10,1 ммоль) растворяли в 10 мл DMF, затем последовательно добавляли HATU (1,37 г, 3,6 ммоль) и NHS (1,28 г, 5,85 ммоль) и перемешивали для проведения реакции при 30°С, чтобы получить систему (1). RGDfK (2,17 г, 3,6 ммоль) растворяли в 10 мл DMSO для получения системы (2). После завершения реакции системы (1) на основе мониторинга с помощью TLC систему (2) добавляли в систему (1) тремя порциями с интервалом в 15 мин для каждой партии. После завершения добавления проводили реакцию во внешней ванне при температуре 30°С.
После завершения реакции на основе мониторинга с помощью ВЭЖХ систему концентрировали и подвергали препаративной очистке для получения промежуточного продукта I с выходом 34,2%. Теоретическая молекулярная масса составляет 1581,7216, измеренная молекулярная масса 1581.7372, а результаты масс-спектрометрии соответствуют результатам целевого продукта. Фиг. 25 представлена масс-спектрограмма промежуточного продукта I.
(12) Получение промежуточного продукта J (а именно соединения формулы (I-14))
Промежуточный продукт I растворяли в 30 мл DMF и добавляли 2 мл пиперидина для проведения реакции при 25°С. После завершения реакции, основанной на мониторинге с помощью ВЭЖХ, в систему для кристаллизации добавляли 200 мл МТВЕ. Затем проводили отстаивание, отсасывали надосадочную жидкость и концентрировали оставшуюся часть системы для получения неочищенного промежуточного продукта J (а именно соединения формулы (I-14)), которое непосредственно применяли на следующем этапе. Теоретическая молекулярная масса составляет 1359,6536, измеренная молекулярная масса 1359,66432, а результаты масс-спектрометрии соответствуют результатам целевого продукта. Фиг. 26 представлена масс-спектрограмма промежуточного продукта J.
(13) Приготовление промежуточного продукта О
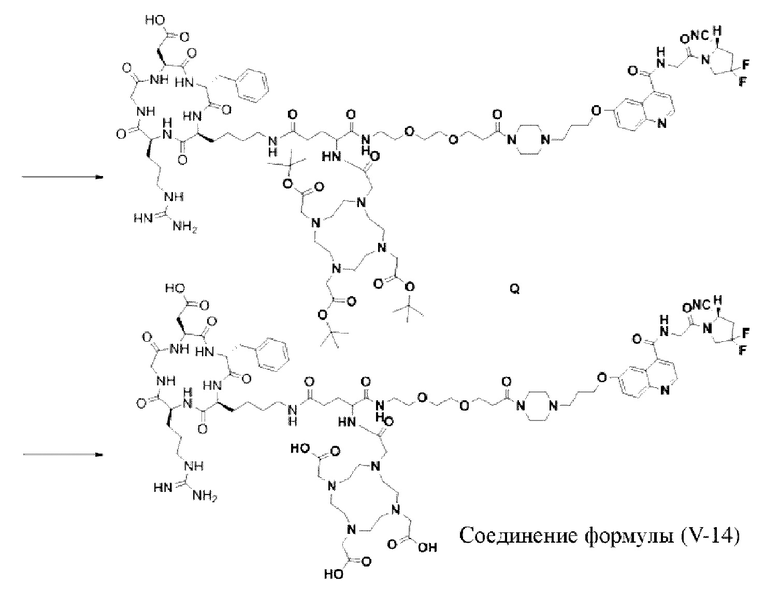
Промежуточный продукт J растворяли в 20 мл DMF и последовательно добавляли DIPEA (0,81 г, 5,0 ммоль) и сложный эфир DOTA-TRIS-TBU-NHS (0,50 г, 1,0 ммоль). После завершения реакции, основанной на мониторинге с помощью ВЭЖХ, систему концентрировали и подвергали препаративной очистке для получения промежуточного продукта Q с выходом 15,7% в две стадии. Теоретическая молекулярная масса составляет 1914,0215, измеренная молекулярная масса - 1914,03418, а результаты масс-спектрометрии соответствуют результатам целевого продукта. Фиг. 27 представлена масс-спектрограмма промежуточного продукта Q.
(14) Получение соединения формулы (V-14)
Промежуточный продукт Q растворяли в 10 мл TFA и проводили реакцию во внешней ванне при 25°С. После завершения реакции, основанной на мониторинге с помощью ВЭЖХ, в систему для кристаллизации добавляли 100 мл МТВЕ, проводили отстаивание и отсасывали надосадочную жидкость. Затем оставшуюся систему концентрировали, добавляли МТВЕ до тех пор, пока не исчезали явные остатки TFA, и подвергали препаративной очистке для получения соединения формулы (V-14). Теоретическая молекулярная масса составляет 1745,8337, измеренная молекулярная масса - 1745,84714, а результаты масс-спектрометрии соответствуют результатам целевого продукта. На фиг. 28 представлена масс-спектрограмма соединения формулы (V-14).
Пример 3: Получение соединения формулы (V-23)
Путь синтеза заключается в следующем.
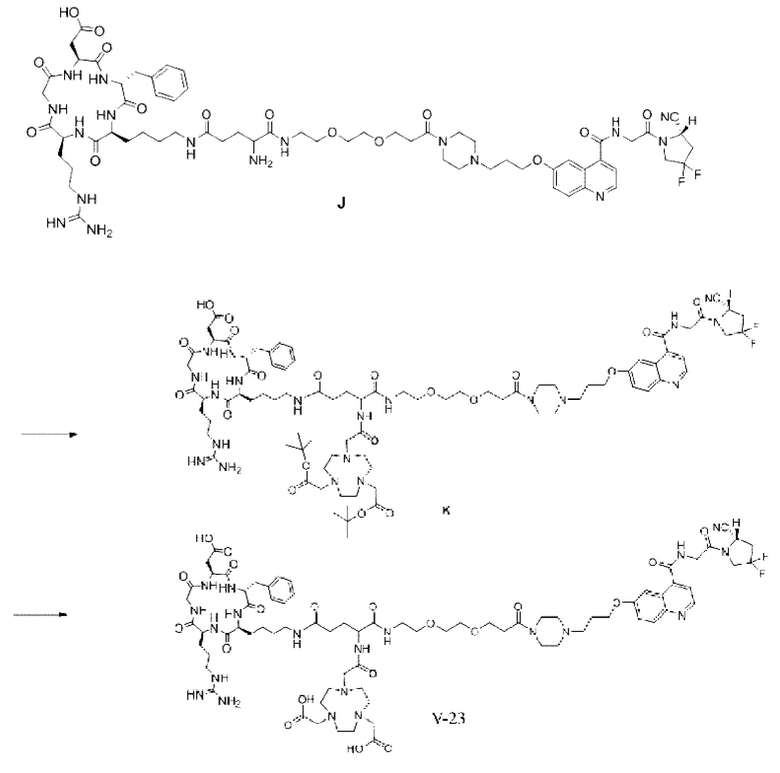
(1) Приготовление промежуточного продукта К
Промежуточный продукт J, полученное методом примера 2, растворяли в 30 мл DMF, добавляли DIPEA (0,97 г, 7,5 ммоль) и 2 экв. эфира NOTA-Bis-TBU-NHS (в расчете на основе промежуточного продукта J) и проводили реакцию во внешней бане при 25°С. После завершения реакции по данным контроля методом ВЭЖХ систему концентрировали и подвергали препаративной очистке с получением промежуточного продукта К с выходом 25,1% в две стадии. Теоретическая молекулярная масса составляет 1756,9112, измеренная молекулярная масса - 1756,92282, а результаты масс-спектрометрии соответствуют результатам целевого продукта. Фиг. 29 представлена масс-спектрограмма промежуточного продукта K.
(2) Получение соединения формулы (V-23)
Промежуточный продукт K растворяли в 30 мл TFA и проводили реакцию во внешней ванне при 25°С. После завершения реакции, основанной на мониторинге с помощью ВЭЖХ, в систему для кристаллизации добавляли 200 мл МТВЕ, проводили отстаивание и отсасывали надосадочную жидкость. Затем оставшуюся систему концентрировали, концентрировали МТВЕ до тех пор, пока не переставали оставаться очевидные остатки TFA, и подвергали препаративной очистке с получением соединения формулы (V-23) с выходом 14,2%. Теоретическая молекулярная масса составляет 1644,7860, измеренная молекулярная масса - 1644,8104, а результаты масс-спектрометрии соответствуют результатам целевого продукта. На фиг.30 представлена масс-спектрограмма соединения формулы (V-23).
Пример 4: Получение соединения формулы (I-25) и соединения формулы (V-25)
Путь синтеза заключается в следующем.
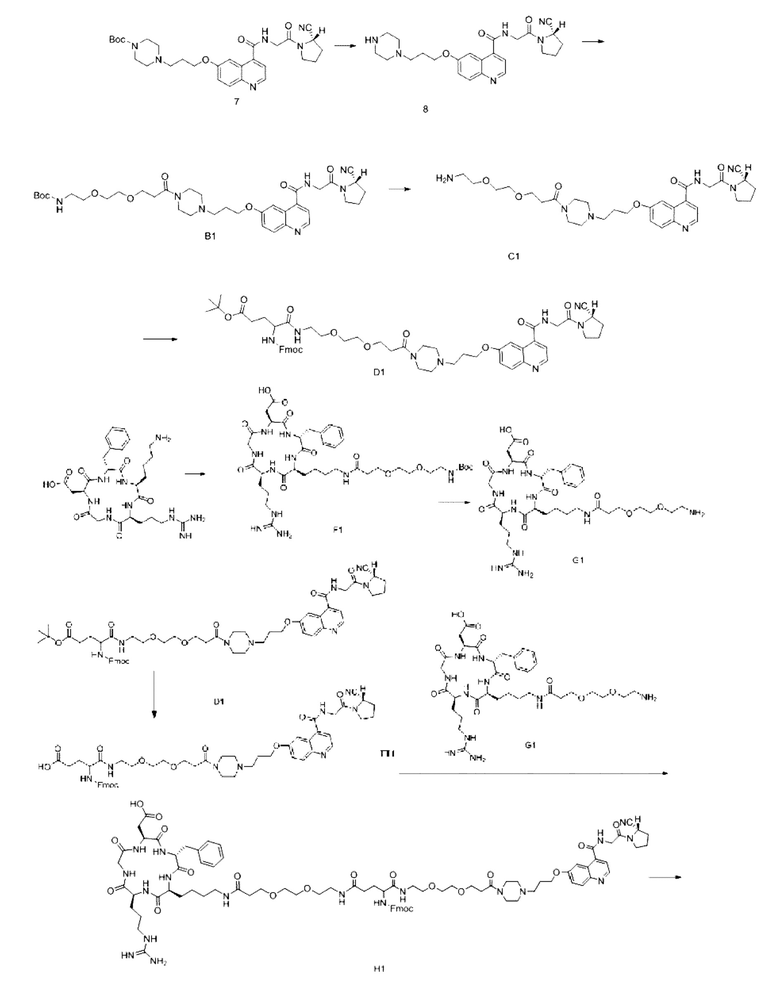
(1) Приготовление промежуточного продукта В1
В реакционную колбу добавляли соединение 7 (2,50 г, 4,5 ммоль), моногидрат n-толуолсульфокислоты (2,58 г, 13,6 ммоль) и 25 мл ацетонитрила для проведения реакции при 65°С в течение 1 часа. После завершения реакции по данным TLC (с метанолом и дихлорметаном в соотношении 5:1) проводили выпаривание при пониженном давлении и 40°С. Добавляли 14 мл DMF и DIPEA (3,05 г, 23,6 ммоль) и перемешивали при 25°С для проведения реакции, пронумерованной (1), то есть пиперазинильную защитную группу соединения 7 удаляли с получением промежуточного продукта 8. N-трет-бутилоксикарбонилдиполиэтиленгликолькарбоновую кислоту (1,62 г, 4,8 ммоль), HATU (2,60 г, 6,8 ммоль) и 10 мл DMF добавляли в другую реакционную колбу для проведения реакции, пронумерованной (2), при 25°С в течение 30 мин и реакционную систему (2) добавляли к реакционной системе (1) для проведения реакции в течение 1 часа. Выпаривание проводили при пониженном давлении и температуре 40°С. Добавляли 50 мл очищенной воды, экстрагировали DCM два раза по 50 мл каждый раз, после чего DCM объединяли. Сушку проводили безводным сульфатом натрия, а фильтрацию и выпаривание проводили для получения неочищенного продукта. Неочищенный продукт очищали методом колоночной хроматографии с получением 1,68 г целевого продукта. Теоретическая молекулярная масса составляет 709,3799, измеренная молекулярная масса - 709,38801, а результаты масс-спектрометрии соответствуют результатам целевого продукта. Фиг. 31 представлена масс-спектрограмма промежуточного продукта В1.
(2) Приготовление промежуточного продукта D1
Промежуточный продукт В1, моногидрат п-толуолсульфоновой кислоты (1,61 г, 8,5 ммоль) и 20 мл ацетонитрила добавляли в реакционную колбу для проведения реакции при 65°С в течение 1 ч и упаривали при пониженном давлении при 40°С. Добавляли 20 мл DMF и DIPEA (1,83 г, 14,2 ммоль) и перемешивали при 25°С для проведения реакции, нумерованной (1), то есть пиперазинильную защитную группу промежуточного продукта В1 удаляли с получением промежуточного продукта С1. Fmoc-O-трет-бутил-L-глутаминовую кислоту (1,43 г, 3,4 ммоль), HATU (1,29 г, 3,4 ммоль) и 20 мл DMF добавляли в другую реакционную колбу для проведения реакции, пронумерованной (2), при 25°С в течение 30 мин и реакционную систему (2) добавляли к реакционной системе (1) для проведения реакции в течение 1 часа. Выпаривание проводили при пониженном давлении и температуре 40°С с получением неочищенного продукта, причем неочищенный продукт очищали методом колоночной хроматографией с получением 1,19 г целевого продукта. Теоретическая молекулярная масса составляет 1016,5008, измеренная молекулярная масса 1016,51094, а результаты масс-спектрометрии соответствуют результатам целевого продукта. Фиг. 32 представлена масс-спектрограмма промежуточного продукта D1.
(3) Приготовление промежуточного продукта G1
В реакционную колбу добавляли c(RGDfK) (1,00 г, 1,7 ммоль), сложный эфир t-Boc-N-амидо-PEG2-NHS (0,74 г, 1,9 ммоль), DIPEA (0,44 г, 3,4 ммоль) и 20 мл DMF для проведения реакции при 30°С в течение 20 часов. Выпаривание проводили при пониженном давлении и 40°С, добавляли 10 мл метанола и по каплям добавляли 60 мл МТВЕ для осаждения твердого вещества и получения промежуточного продукта F1. Была проведена всасывающая фильтрация и вакуумная сушка при температуре 40°С в течение 2 часов. Твердый промежуточный продукт F1 добавляли в реакционную колбу, затем добавляли 30 мл TFA и 1,5 мл очищенной воды для проведения реакции при температуре 30°С в течение 1 часа и охлаждали до 0-5°С. По каплям добавляли 200 мл МТВЕ и перемешивали при 0-5°С в течение 30 минут, затем была проведена всасывающая фильтрация, промывка МТВЕ и вакуумная сушка при 40°С для получения продукта. Теоретическая молекулярная масса составляет 762,4024, измеренная молекулярная масса - 762,40768, а результаты масс-спектрометрии соответствуют результатам целевого продукта. Фиг. 33 представлена масс-спектрограмма промежуточного продукта G1.
(4) Приготовление промежуточного продукта H1
Промежуточный продукт D1, моногидрат п-толуолсуль фоновой кислоты (0,34 г, 1,8 ммоль) и 20 мл ацетонитрила добавляли в реакционную колбу для проведения реакции при 65°С в течение 4 ч и упаривали при пониженном давлении при 40°С. Добавляли 20 мл DMF, DIPEA (0,36 г, 2,8 ммоль), DCC (0,14 г, 0,7 ммоль) и NHS (0,08 г, 0,7 ммоль) для проведения реакции при 35°С в течение 15-20 ч для получения промежуточного продукта Е1. Охлаждали до 25°С, добавляли промежуточный продукт G1 для проведения реакции в течение 1 ч и проводили выпаривание при пониженном давлении при температуре 40°С для получения неочищенного продукта. Неочищенный продукт подвергали препаративной жидкостной хроматографии для получения 66,5 мг целевого продукта. Теоретическая молекулярная масса составляет 1704,8300, измеренная молекулярная масса 1704,84518, а результаты масс-спектрометрии соответствуют результатам целевого продукта. Фиг. 34 представлена масс-спектрограмма промежуточного продукта H1.
(5) Получение промежуточного продукта I1 (а именно соединения формулы (I-25))
В реакционную колбу добавляли промежуточный продукт H1, 0,5 мл пиперидина и 2 мл DMF для проведения реакции при 25°С в течение 1 ч, для кристаллизации по каплям добавляли 10 мл этилацетата, перемешивали в течение 30 мин, затем проводили фильтрацию с отсасыванием и твердое вещество подвергали вакуумной сушке при 40°С в течение 2 часов с получением 50,8 мг промежуточного продукта I1 (а именно, соединения формулы (I-25)). Теоретическая молекулярная масса составляет 1482,7619, измеренная молекулярная масса - 1482,7759, а результаты масс-спектрометрии соответствуют результатам целевого продукта. Фиг. 35 представлена масс-спектрограмма промежуточного продукта I1.
(6) Приготовление промежуточного продукта J1
Промежуточный продукт I1, эфир NOTA-Bis-TBU-NHS, DIPEA (0,010 г, 0,08 ммоль) и 2 мл DMF добавляли в реакционную колбу для проведения реакции при 25°С в течение 1 ч и упаривали при пониженное давление при 40°С. Добавляли 2 мл этилацетата, 2 мл МТВЕ для кристаллизации, перемешивали в течение 20 мин, проводили вакуум-фильтрацию и сушили в вакууме при 40°С с получением 43,2 мг продукта. Теоретическая молекулярная масса составляет 1880,0196, измеренная молекулярная масса 1880,0369, а результаты масс-спектрометрии соответствуют результатам целевого продукта. Фиг. 36 представлена масс-спектрограмма промежуточного продукта J1.
(7) Получение соединения формулы (V-25)
В реакционную колбу добавляли промежуточный продукт J1 и 2 мл трифторуксусной кислоты для проведения реакции при 25°С в течение 1 ч и проводили выпаривание при пониженном давлении при 40°С для получения неочищенного продукта. Неочищенный продукт очищали с помощью препаративной жидкостной хроматографии, а затем подвергали сублимационной сушке для получения продукта (а именно соединения формулы (V-25)). Теоретическая молекулярная масса составляет 1767,8944, измеренная молекулярная масса - 1767,91036, а результаты масс-спектрометрии соответствуют результатам целевого продукта. На фиг.37 представлена масс-спектрограмма соединения формулы (V-25).
Пример 5: Получение соединения формулы (I-3) и соединения формулы (V-26)
Путь синтеза заключается в следующем.


(1) Приготовление промежуточного продукта В1
Исходный материал 7 (5,50 г, 10 ммоль) и п-толуолсульфоновую кислоту (5,71 г, 30 ммоль) взвешивали и добавляли в реакционную колбу, добавляли 10 мл ацетонитрила, полученную смесь перемешивали и нагревали до 65°С для проведения реакции в течение 1 ч для удаления защитной группы на пиперазиновом кольце с получением промежуточного продукта 8. После завершения реакции по данным TLC (с проявителем, включающим дихлорметан и метанол в соотношении 5:1), выпаривание осуществляли при пониженном давлении и 40°С. Добавляли 4 мл DMF и DIPEA (9,05 г, 70 ммоль) и перемешивали для растворения и добавляли сложный эфир t-Boc-N-амидо-PEG2-NHS (промежуточный продукт С2, 5,62 г, 15 ммоль) для проведения реакции при 25°С в течение 3 часов. После завершения реакции по данным TLC (с проявителем, включающим дихлорметан и метанол в соотношении 5:1), выпаривание осуществляли при пониженном давлении и 40°С. Добавляли 5 мл очищенной воды, экстрагировали DCM два раза по 5 мл, органические фазы объединяли и сушили безводным сульфатом натрия (1 м/м) в течение 30 мин. Была проведена вакуумная фильтрация, после чего маточную жидкость выпаривали при пониженном давлении и температуре 40°С с получением неочищенного продукта. Очистку проводили методом колоночной хроматографии с получением промежуточного продукта В1. Продукт был непосредственно использован для подачи на следующем этапе.
(2) Приготовление промежуточного продукта G2
Промежуточный продукт В1 и п-толуолсуль фоновую кислоту (5,14 г, 27 ммоль) взвешивали и добавляли в реакционную колбу, добавляли 10 мл ацетонитрила, полученную смесь перемешивали и нагревали до 65°С для проведения реакции в течение 1 ч для удаления защитной группы с получением промежуточного продукта С1. После завершения реакции по данным TLC (с проявителем, включающим дихлорметан и метанол в соотношении 10:1), осуществляли выпаривание при пониженном давлении и 40°С. Добавляли DIPEA (5,82 г, 45 ммоль), 10 мл DMF, 3-малеимидпропионат и сложный эфир N-гидроксисукцинимида (2,88 г, 10,8 ммоль) для проведения реакции при комнатной температуре в течение 1 часа. После завершения реакции по данным TLC-пластинки (с проявителем, включающим дихлорметан и метанол в соотношении 10:1), осуществляли выпаривание при пониженном давлении и 40°С. Очистку проводили методом колоночной хроматографии с получением промежуточного продукта G2. Теоретическая молекулярная масса целевого продукта составляет 760,35443, молекулярная масса, измеренная методом жидкостной хроматографии-масс-спектрометрии, составляет 760,37090, а результаты масс-спектрометрии соответствуют результатам целевого продукта. Фиг. 56 представлена масс-спектрограмма промежуточного продукта G2.
(3) Приготовление промежуточного продукта N1
Промежуточный продукт G2 и Вос-цистеин (1,77 г, 8 ммоль) взвешивали и добавляли в реакционную колбу, затем добавляли 10 мл DMF для проведения реакции при 25°С в течение 2 ч для получения промежуточного продукта Н2. После завершения реакции по данным TLC (с проявителем, включающим дихлорметан и метанол в соотношении 5:1), добавляли DCC (1,98 г, 9,6 ммоль) и NHS (1,86 г, 9,6 ммоль) для проведения реакции при 35°С в течение 2 часов. После завершения реакции по данным TLC (с проявителем, включающим дихлорметан и метанол в соотношении 5:1), циклический пептид Cyclo(Arg-Gly-Asp-DPhe-Lys) (4,83 г, 8 ммоль) и DIPEA (3,10 г, 24 ммоль экв.) добавляли для проведения реакции при 25°С в течение 1 часа. После завершения реакции по данным TLC (с проявителем, включающим дихлорметан и метанол в соотношении 5:1), выпаривали при пониженном давлении и 40°С с получением неочищенного продукта. Неочищенный продукт очищали с помощью препаративной жидкостной хроматографии для получения промежуточного продукта N1. Теоретическая молекулярная масса целевого продукта составляет 1566,72893, молекулярная масса, измеренная методом жидкостной хроматографии-масс-спектрометрии, составляет 1566,74480, а результаты масс-спектрометрии соответствуют результатам целевого продукта. Фиг. 57 представлена масс-спектрограмма промежуточного продукта N1.
(4) Получение промежуточного продукта Р (а именно соединения формулы (1-3))
Промежуточный продукт N1 взвешивали и добавляли в реакционную колбу, а также добавляли 2 мл тиоанизола, 2 мл 1,2-этилдитиола и 20 мл трифторуксусной кислоты для проведения реакции при комнатной температуре в течение 1 ч под защитой азота. Добавляли 20 мл метил-трет-бутилового эфира до осаждения твердого вещества, проводили вакуум-фильтрацию и твердое вещество подвергали сушке в вакууме при 40°С в течение 1 часа с получением промежуточного продукта Р. Теоретическая молекулярная масса целевого продукта составляет 1466,67650, молекулярная масса, измеренная методом жидкостной хроматографии-масс-спектрометрии, составляет 1466,69746, а результаты масс-спектрометрии согласуются с таковыми целевого продукта. Фиг. 58 представлена масс-спектрограмма промежуточного продукта Р.
(5) Получение соединения формулы (V-26)
Промежуточный продукт Р и сложный эфир NOTA-Bis-TBU-NHS взвешивали и добавляли в реакционную колбу, а также добавляли 40 мл DMF для проведения реакции при комнатной температуре в течение 1 часа. Добавляли 4 мл DIPEA для проведения реакции при комнатной температуре в течение 3 часов. Выпаривание проводили при пониженном давлении и 40°С с получением промежуточного продукта S. Добавляли 30 мл трифторуксусной кислоты и перемешивали при комнатной температуре в течение 1 часа. Выпаривание осуществляли при пониженном давлении и температуре 40°С с получением неочищенного продукта, и неочищенный продукт очищали препаративной жидкостной хроматографией с получением продукта (а именно, соединения формулы (V-26)). Теоретическая молекулярная масса целевого продукта составляет 1751,80898, молекулярная масса, измеренная методом жидкостной хроматографии-масс-спектрометрии, составляет 1751,83088, а результаты масс-спектрометрии соответствуют результатам целевого продукта. На фиг.59 представлена масс-спектрограмма соединения формулы (V-26).
Пример 6: Получение соединения формулы (V-30)
Путь синтеза заключается в следующем.
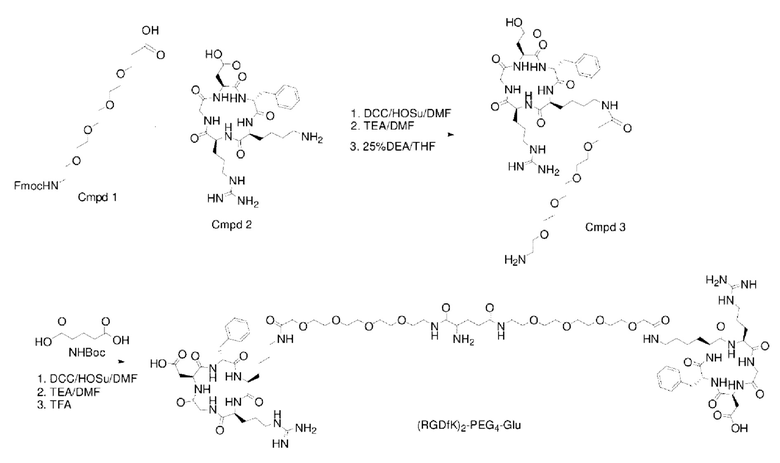
(1) Приготовление промежуточного продукта Cmpd3
Fmoc-PEG4-CH2CH2COOH (а именно соединение Cmpd1, 1,46 г, 3,0 ммоль) растворяли в DMF, а затем добавляли DCC (0,68 г, 3,3 ммоль) и HOSu (0,38 г, 3,3 ммоль) для проведения реакции при комнатной температуре в течение 6 часов, добавляли фильтрацию, к фильтрату добавляли TEA (0,90 г, 9,0 ммоль), после чего добавляли Cyclo(RGDfK) (а именно соединение Cmpd2, 2,23 г, 3,6 ммоль) для проведения реакции при комнатной температуре в течение 3 часов. Реакционный раствор сушили центрифугированием, растворяли в 25% DEA/THF для проведения реакции при комнатной температуре в течение 4 часов, концентрировали до тех пор, пока не оставалось небольшое количество раствора, а затем добавляли в 10-кратный объем этилового эфира до тех пор, пока в осадок не выпало большое количество твердого вещества. Затем проводили фильтрацию для получения неочищенного продукта Cyclo(RGDfK)-PEG4, а неочищенный продукт очищали с помощью обратнофазной препаративной жидкостной хроматографии для получения мелкодисперсного цикло-продукта (RGDfK)-PEG4 (а именно, промежуточного продукта Cmpd3), где элюент (включающий раствор А: Использовали 0,1% TFA в Н2О и раствор В: ацетонитрил).
(2) Синтез промежуточного продукта (RGDfK)2-PEG4-Glu
Boc-Glu-OH (0,4 г, 2,0 ммоль) растворяли в DMF, а затем добавляли DCC (0,45 г, 2,2 ммоль) и HOSu (0,25 г, 2,2 ммоль) для проведения реакции при комнатной температуре в течение 6 часов. Добавляли фильтрацию, к фильтрату добавляли TEA (0,60 г, 6,0 ммоль), а затем добавляли Cyclo(RGDfK)-PEG4 (а именно, промежуточный продукт Cmpd3, 2,61 г, 2,4 ммоль) для проведения реакции при комнатной температуре в течение 3 часов. Реакционный раствор сушили центрифугированием, растворяли в TFA для проведения реакции при комнатной температуре в течение 10 минут, а затем добавляли в 10-кратный объем этилового эфира до тех пор, пока в осадок не выпало большое количество твердого вещества. Затем проводили фильтрацию для получения неочищенного продукта 2(RGDfK)-PEG4-Glu, а неочищенный продукт очищали с помощью обратно фазной препаративной жидкостной хроматографии для получения очищенного (RGDfK)2-PEG4-Glu, где элюент (включающий раствор А: Использовали 0,1% TFA в H2O и раствор В: ацетонитрил). Затем рН очищенного (RGDfK)2-PEG4-Glu доводили до нейтрального с помощью TEA, снова проводили обратнофазную препаративную жидкостную хроматографию и проводили лиофилизационную сушку для получения конечного продукта (RGDfK)2-PEG4-Glu.
(3) Приготовление промежуточного продукта Н3
Промежуточный продукт 8 (0,45 г, 1 ммоль), полученный методом примера 1, Fmoc-O-трет-бутил-L-глутаминовая кислота (0,42 г, 1 ммоль), HATU (0,38 г, 1 ммоль) и DIPEA (0,58 г, 4,5 ммоль) последовательно растворяли в 10 мл DMF для проведения реакции во внешней бане при 25°С. После завершения реакции по данным ВЭЖХ к реакционной системе добавляли 20 мл очищенной воды, дважды экстрагировали 20 мл DCM, органические фазы объединяли, сушили безводным сульфатом натрия, концентрировали и подвергали очистке в колонке с получением промежуточного продукта Н3 с выходом сырья 97% в две стадии.
Теоретическая молекулярная масса составляет 735,3956, измеренная молекулярная масса - 735,40744, а результаты масс-спектрометрии соответствуют результатам целевого продукта. Фиг. 38 представлена масс-спектрограмма промежуточного продукта Н3.
(4) Приготовление промежуточного продукта I2
Промежуточный продукт Н3 растворяли в 20 мл ацетонитрила, добавляли моногидрат п-толуолсульфоновой кислоты (0,65 г, 3,4 ммоль) и проводили реакцию во внешней ванне при 70°С. После завершения реакции, основанной на мониторинге с помощью ВЭЖХ, ацетонитрил в системе концентрировался, и продукт непосредственно использовался на следующем этапе. Теоретическая молекулярная масса составляет 579,2805, измеренная молекулярная масса - 579, 28563, а результаты масс-спектрометрии соответствуют результатам целевого продукта. Фиг. 39 представлена масс-спектрограмма промежуточного продукта I2.
(5) Приготовление промежуточного продукта Q1
Промежуточный продукт 12 и DIPEA (0,64 г, 5,0 ммоль) растворяли в 10 мл DMF и добавляли сложный эфир DOTA-TRIS-TBU-NHS (1,7 г, 2,5 ммоль). После завершения реакции по данным контроля методом ВЭЖХ растворитель в системе концентрировали, а оставшуюся систему подвергали препаративной очистке для получения промежуточного продукта O1 с выходом 27,15% в две стадии. Теоретическая молекулярная масса составляет 1133,6485, измеренная молекулярная масса - 1133,65551, а результаты масс-спектрометрии соответствуют результатам целевого продукта. Фиг. 40 представлена масс-спектрограмма промежуточного продукта O1.
(6) Приготовление промежуточного продукта Р1
Промежуточный продукт O1 растворяли в 5 мл DMF и добавляли HATU (0,076 г, 0,2 ммоль) и перемешивали при комнатной температуре в течение 1 ч для получения системы (1). DIPEA (0,090 г, 0,7 ммоль) и (RGDfK)2-PEG4-Glu (0,24 г, 0,13 ммоль) растворяли в 5 мл DMSO для получения системы (2). Система (1) была добавлена к системе (2) и перемешана при температуре 28°С. После завершения реакции, основанной на контроле с помощью ВЭЖХ, DMF концентрировали, добавляли 100 мл МТВЕ для кристаллизации и проводили отстаивание. Затем сливали надосадочную жидкость, а оставшееся маслянистое соединение подвергали препаративной очистке для получения промежуточного продукта Р1 с выходом 17,09%. Теоретическая молекулярная масса составляет 2927,5797, измеренная молекулярная масса - 2927,60652, а результаты масс-спектрометрии соответствуют результатам целевого продукта. Фиг. 41 представлена масс-спектрограмма промежуточного продукта Р1.
(7) Приготовление соединения V-30
Промежуточный продукт Р1 растворяли в 5 мл TFA и проводили реакцию во внешней ванне при 25°С. После завершения реакции, основанной на мониторинге с помощью ВЭЖХ, в систему для кристаллизации добавляли 25 мл МТВЕ, проводили отстаивание и отсасывали надосадочную жидкость. Затем оставшуюся систему концентрировали МТВЕ до исчезновения видимых остатков TFA и подвергали препаративной очистке с получением V-30 с выходом 32,13%. Теоретическая молекулярная масса составляет 2759,3919, измеренная молекулярная масса - 2759,40972, а результаты масс-спектрометрии соответствуют результатам целевого продукта. На фиг.42 представлена масс-спектрограмма V-30.
Пример 7: Получение соединения формулы (V-35)
Путь синтеза заключается в следующем.
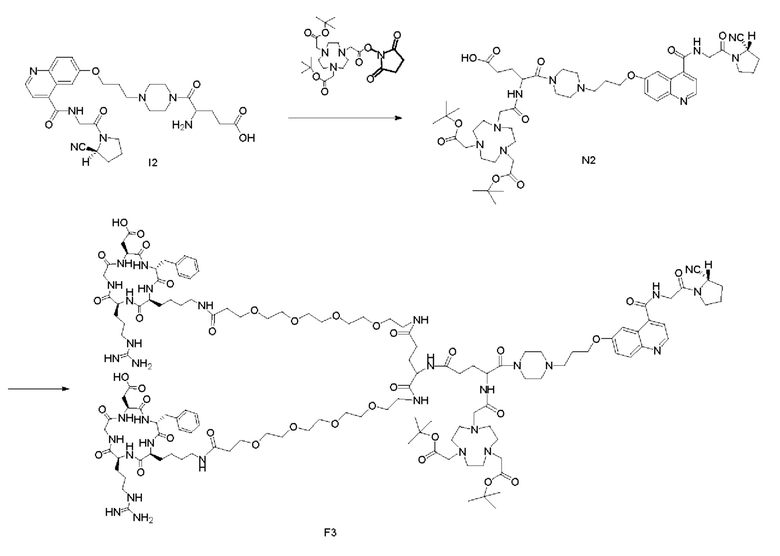
(1) Приготовление промежуточного продукта N2
Промежуточный продукт 12, полученный способом, описанным в примере 6, и DIPEA (3,90 г, 30 ммоль) растворяли в 10 мл DMF, и добавляли в систему сложный эфир NOTA-Bis-TBU-NHS (7,65 г, 15 ммоль). После завершения реакции по данным контроля методом ВЭЖХ DMF в системе концентрировали, а оставшуюся систему подвергали препаративной очистке для получения промежуточного продукта N2 с выходом 22,88% в две стадии. Теоретическая молекулярная масса составляет 976,5382, измеренная молекулярная масса - 976,56026, а результаты масс-спектрометрии соответствуют результатам целевого продукта. Фиг. 43 представлена масс-спектрограмма промежуточного продукта N2.
(2) Приготовление промежуточного продукта F3
Промежуточный продукт N2 растворяли в 10 мл DMF и добавляли HATU (0,46 г, 1,2 ммоль) для проведения реакции во внешней ванне при температуре 30°С в течение 1 ч для получения системы (1). c(RGDfK)2-PEG4-Glu (1,54 г, 0,8 ммоль) и DIPEA (0,62 г, 4,8 ммоль) растворяли в 5 мл DMF и 5 мл DMSO с получением системы (2). Система (1) была добавлена в систему (2) и перемешана во внешней ванне при температуре 30°С. После завершения реакции по данным контроля методом ВЭЖХ растворитель концентрировали, а оставшуюся систему подвергали препаративной очистке для получения промежуточного продукта F3 с выходом 15,34%. Теоретическая молекулярная масса составляет 2770,4894, измеренная молекулярная масса - 2770,49229, а результаты масс-спектрометрии соответствуют результатам целевого продукта. Фиг. 44 представлена масс-спектрограмма промежуточного продукта F3.
(3) Получение соединения формулы (V-35)
Промежуточный продукт F3 растворяли в 20 мл TFA и проводили реакцию во внешней ванне при 25°С. После завершения реакции, основанной на мониторинге с помощью ВЭЖХ, в систему для кристаллизации добавляли 50 мл МТВЕ, проводили отстаивание и сливали надосадочную жидкость. Затем оставшуюся систему концентрировали с помощью МТВЕ до тех пор, пока в системе не переставали оставаться очевидные остатки TFA, и подвергали препаративной очистке с получением соединения формулы (V-35) с выходом 2,89%. Теоретическая молекулярная масса составляет 2658,3442, измеренная молекулярная масса 2658,36508, а результаты масс-спектрометрии соответствуют результатам целевого продукта. На фиг.45 представлена масс-спектрограмма соединения формулы (V-35).
Пример 8; Получение соединения формулы (I-16), соединения формулы (I-40) и соединения формулы (V-40)
Путь синтеза заключается в следующем.

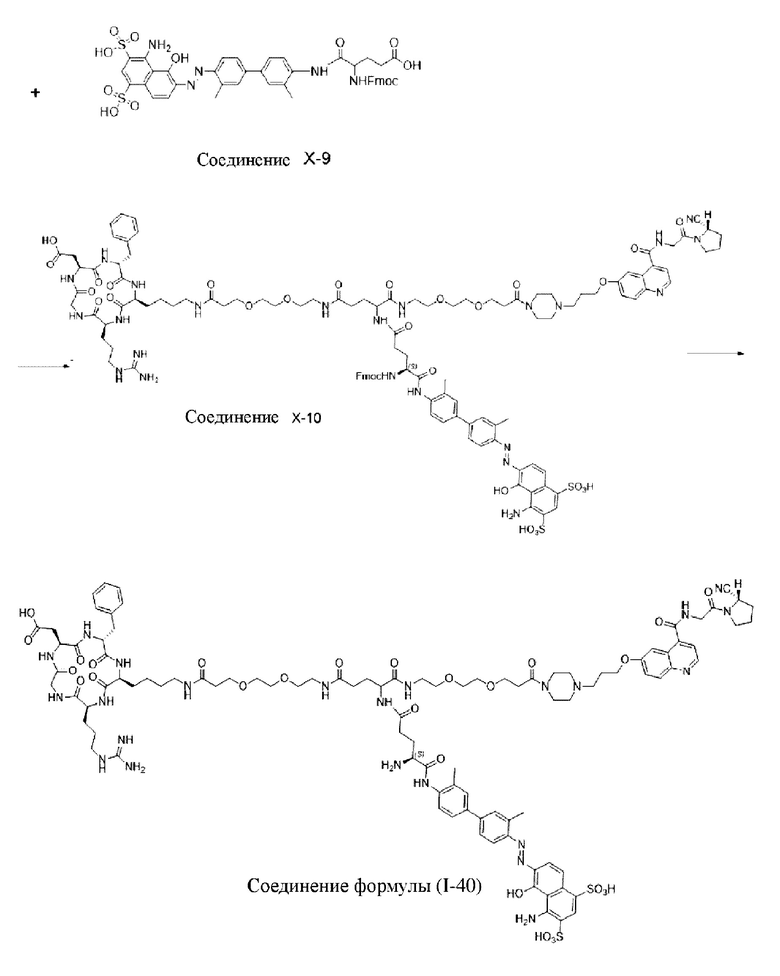
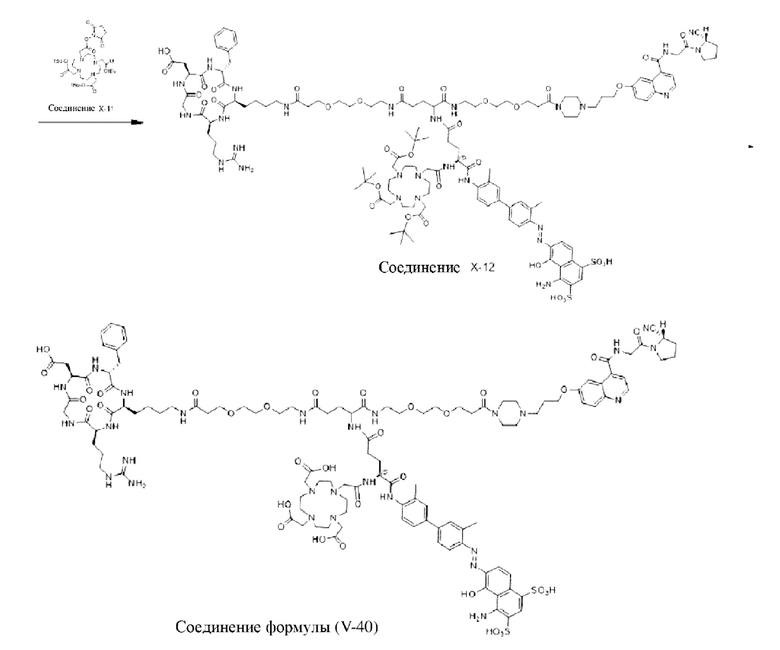
Со ссылкой на примеры получения/способы, представленные в примерах 1-7, в примере 8 были получены соединение формулы (I-16), соединение формулы (I-40) и соединение формулы (V-40). На основе примеров 1-7 специалистами в данной области могут быть произведены соответствующие замены сырья в сочетании с вышеуказанным способом приготовления, которые повторно здесь не описываются.
Примеры 9 - Примеры 45; Получение других соединений двойного нацеливания Что касается препаративных примеров/методов, представленных в примерах 1-8, соединение формулы (I-2), соединение формулы (I-4), соединение формулы (I-5), соединение формулы (I -6), соединение формулы (I-7), соединение формулы (I-8), соединение формулы (1-9), соединение формулы (I-10), соединение формулы (I-11), соединение формулы (I-12), соединение формулы (I-13), соединение формулы (I-17), соединение формулы (I-18), соединение формулы (I-19), соединение формулы (I-20), соединение формулы (I-21), соединение формулы (I-27), соединение формулы (I-28), соединение формулы (I-29), соединение формулы (I-30), соединение формулы (I-31), соединение формулы (I-32), соединение формулы (I-33), соединение формулы (I-34), соединение формулы (I-35), соединение формулы (I-36), соединение формулы (I-37), соединение формулы (I-38), соединение формулы (I-39), соединение формулы (II-1), соединение формулы (II-2), соединение формулы (II-3), соединение формулы (II-4), соединение формулы (II-5), соединение формулы (II-6), соединение формулы (II-7) и соединение формулы (II-8) были получены в примерах 9-45. На основе примеров 1-8 специалистами в данной области могут быть произведены соответствующие замены сырья, например, замена c(RGDfK) на c(RGDyK), замена c(RGDyK) на c(RGDfK) и замена гидрохлорида (S)-дифторпирролидин-2-карбонитрила на гидрохлорид (S)-4,4-дифторпирролидин-2-карбонитрила. Структуры соединений в соответствующих примерах получения описаны в приведенном выше содержании настоящего изобретения, которые повторно здесь не описываются.
Примеры 46-Примеры 83: Получение других соединений, которые могут быть помечены радионуклидом
Что касается препаративных примеров/методов, представленных в примерах 1-8, соединение формулы (V-2), соединение формулы (V-3), соединение формулы (V-4), соединение формулы (V-5), соединение формулы (V-6), соединение формулы (V-7), соединение формулы (V-8), соединение формулы (V-9), соединение формулы (V-10), соединение формулы (V-11), соединение формулы (V-12), соединение формулы (V-13), соединение формулы (V-16), соединение формулы (V-17), соединение формулы (V-18), соединение формулы (V-19), соединение формулы (V-20), соединение формулы (V-21), соединение формулы (V-22), соединение формулы (V-27), соединение формулы (V-28), соединение формулы (V-29), соединение формулы (V-31), соединение формулы (V-32), соединение формулы (V-33), соединение формулы (V-34), соединение формулы (V-36), соединение формулы (V-37), соединение формулы (V-38), соединение формулы (V-39), соединение формулы (VI-1), соединение формулы (VI-2), соединение формулы (VI-3), соединение формулы (VI-4), соединение формулы (VI-5), соединение формулы (VI-6), соединение формулы (VI-7) и соединение формулы (VI-8) были получены в примерах 46-83. На основе примеров 1-8 специалистами в данной области могут быть произведены соответствующие замены сырья, например, замена c(RGDfK) на c(RGDyK), замена c(RGDyK) на c(RGDfK) и замена гидрохлорида (S)-дифторпирролидин-2-карбонитрила на гидрохлорид (S)-4,4-дифторпирролидин-2-карбонитрила. Структуры соединений в соответствующих примерах получения описаны в приведенном выше содержании настоящего изобретения, которые повторно здесь не описываются.
Примеры 84-Примеры 133: Другие примеры получения
В соответствии со способами получения, приведенными в примерах 1-83, были получены соединения FAPI-RGD формулы (V) в таблице 1 или соединения FAPI-RGD формулы (VI) в таблице 2 ниже.
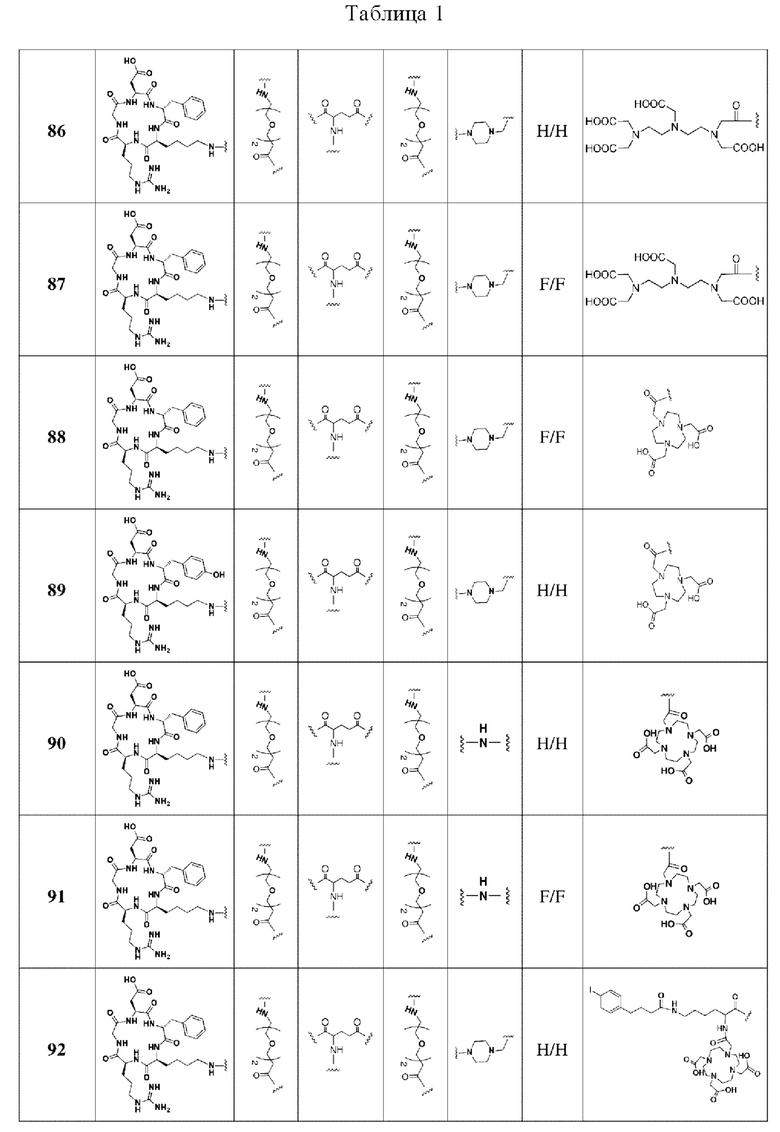
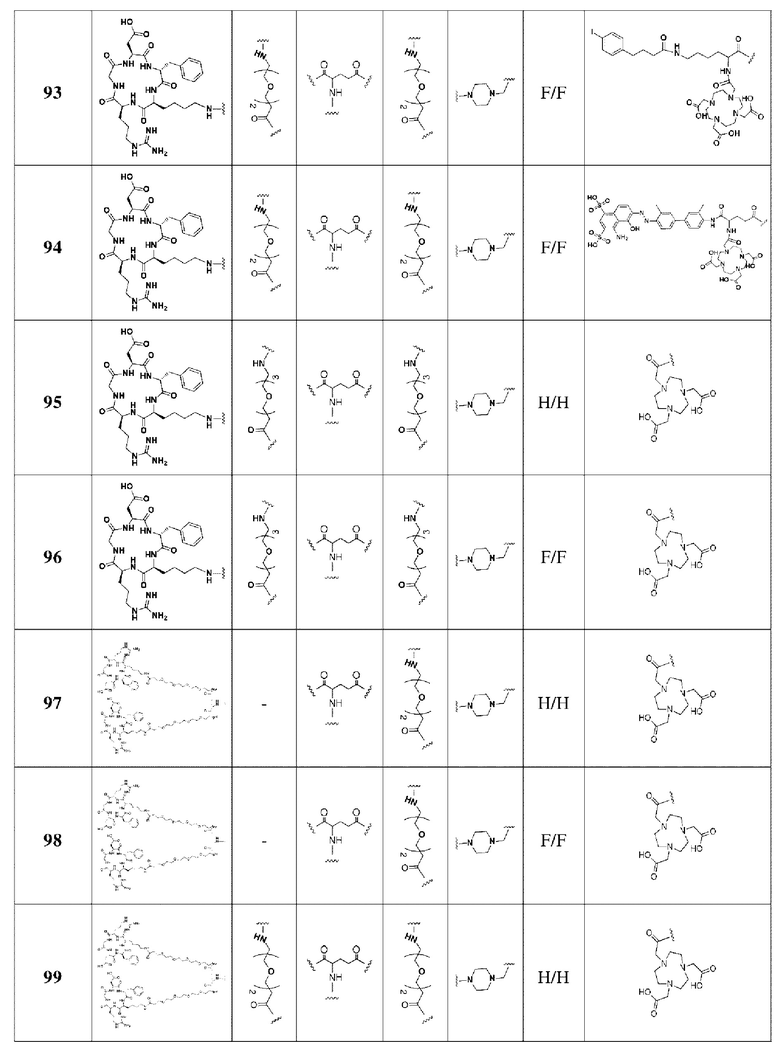

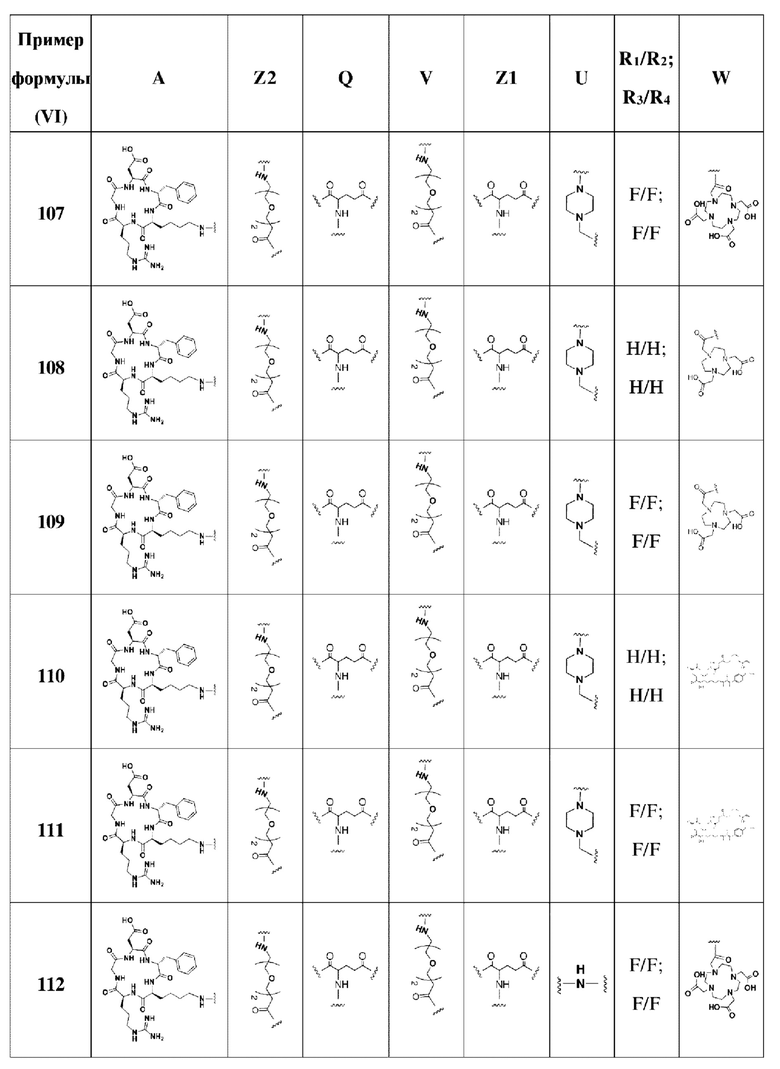
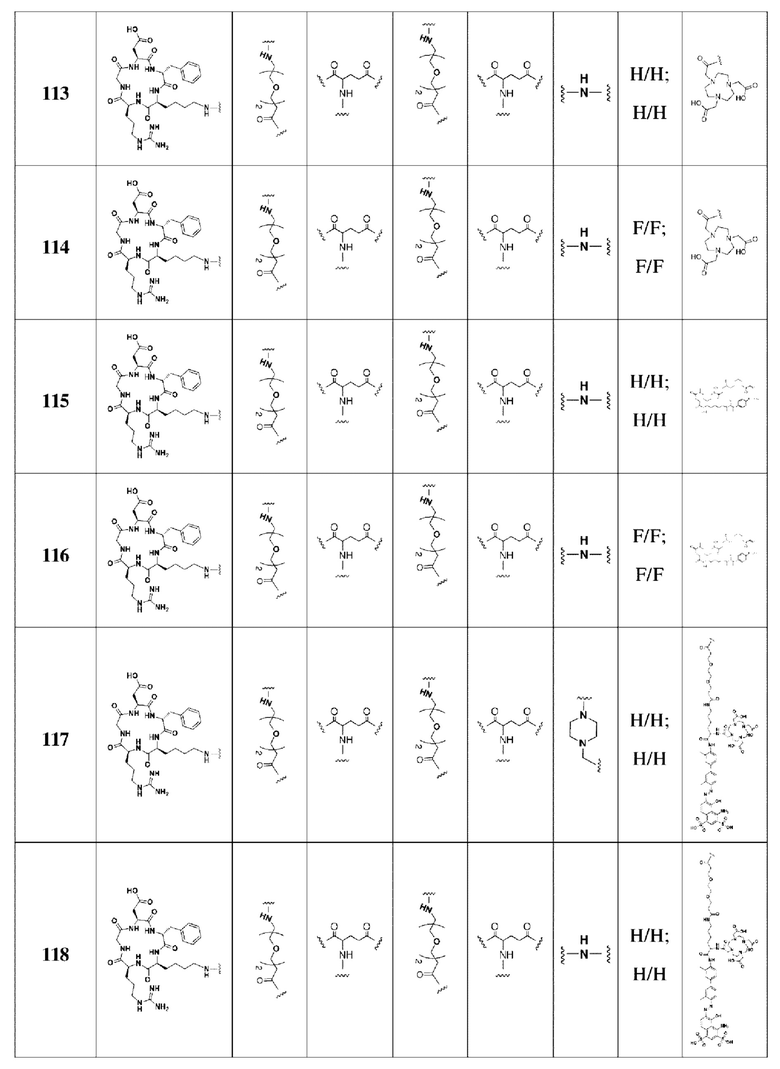
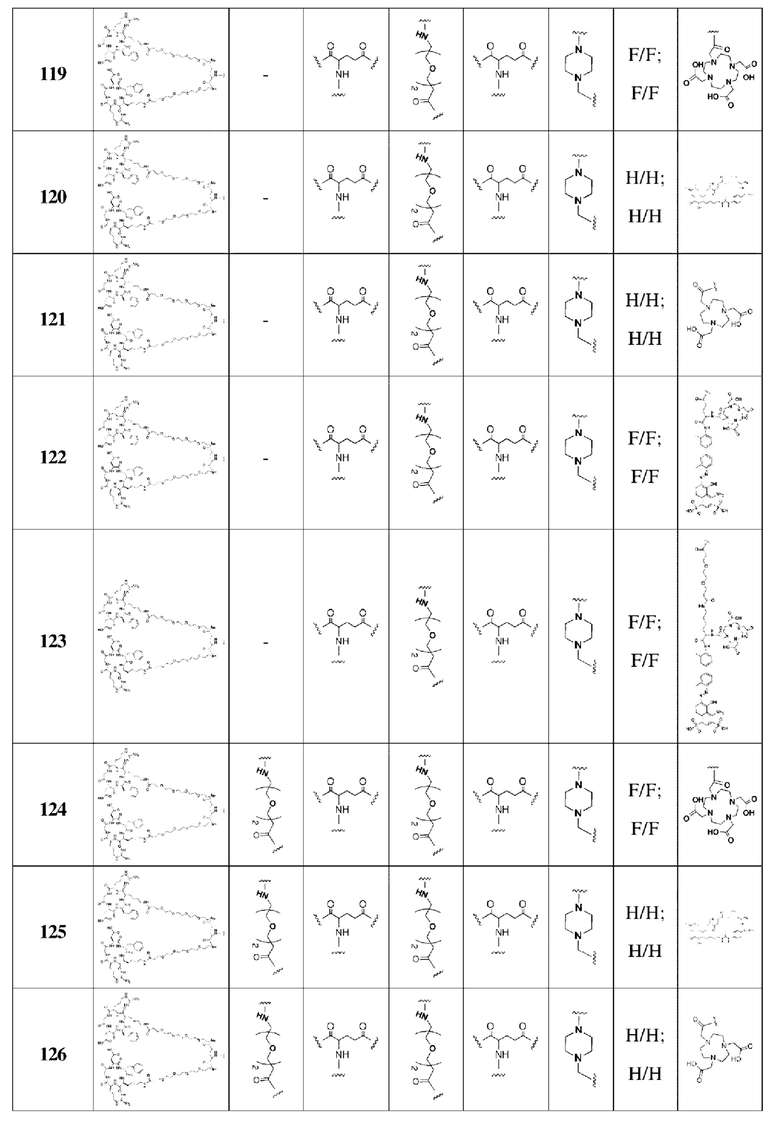
Пример 134: Общий способ получения радионуклидного маркера
(1) Мокрый способ
В этом примере был разработан общий способ получения (мокрый метод) радионуклидного маркера (с использованием Ga-68 в качестве примера) с использованием соединения формулы (V-1) в качестве примера. Раствор соляной кислоты с концентрацией примерно 18,5-1850 МБк 68GaCl3 (промытый германий-галлиевым генератором) добавляли к раствору уксусной кислоты-ацетата (1,0 г/л), содержащему 0,5 мл соединения формулы (V-1), полученного в примере 1, в центрифужной пробирке, а реакцию проводили при 37°С в течение 20 мин. Брали небольшую колонку для разделения C18, медленно промывали сначала 10 мл безводного этанола, а затем промывали 10 мл воды. После разбавления 10 мл воды меченый раствор загружали в разделительную колонку, сначала обрабатывали 10 мл воды для удаления немеченых ионов 68Ga, почле чего промывали 0,3 мл раствора этанола с концентрацией 10 мм HCl. Промытый раствор собирали, разбавляли обычным физиологическим раствором и подвергали стерильной фильтрации для получения инъекции соединения формулы (V-1), меченного 68 Ga (а именно, 68Ga-FAPI-RGD (V-1)).
(2) Способ лиофилизационной сушки
В этом примере был разработан общий метод получения (способ лиофилизационной сушки) радионуклидного маркера (с использованием Ga-68 в качестве примера) с использованием соединения формулы (V-1) в качестве примера. Раствор соляной кислоты с концентрацией около 18,5-1850 МБк 68GaCh (промытый с помощью германий-галлиевого генератора) добавляли в лиофилизированный медицинский набор, содержащий соединение формулы (V-1), и равномерно перемешивали для проведения реакции при 37°С в течение 20 мин. Брали небольшую колонку для разделения C18, медленно промывали сначала 10 мл безводного этанола, а затем промывали 10 мл воды. После разбавления 10 мл воды меченый раствор загружали в разделительную колонку, сначала обрабатывали 10 мл воды для удаления немеченых ионов 68Ga, почле чего промывали 0,3 мл раствора этанола с концентрацией 10 мм HCl для получения промытого раствора комплекса. Промытый раствор разбавляли обычным физиологическим раствором и подвергали стерильной фильтрации для получения инъекции соединения формулы (V-1), меченного 68 Ga (а именно, 68Ga-FAPI-RGD (V-1)).
Другие соединения, которые могут быть помечены радионуклидом, предусмотренным настоящим изобретением, маркируются вышеуказанными общими способами маркировки. Например, соединение формулы (V-2), соединение формулы (V-3), соединение формулы (V-4), соединение формулы (V-5), соединение формулы (V-6), соединение формулы (V-7), соединение формулы (V-8), соединение формулы (V-9), соединение формулы (V-10), соединение формулы (V-11), соединение формулы (V-12), соединение формулы (V-13), соединение формулы (V-14), соединение формулы (V-16), соединение формулы (V-17), соединение формулы (V-18), соединение формулы (V-19), соединение формулы (V-20), соединение формулы (V-21), соединение формулы (V-22), соединение формулы (V-23), соединение формулы (V-25), соединение формулы (V-26), соединение формулы (V-27), соединение формулы (V-28), соединение формулы (V-29), соединение формулы (V-30), соединение формулы (V-31), соединение формулы (V-32), соединение формулы (V-33), соединение формулы (V-34), соединение формулы (V-35), соединение формулы (V-36), соединение формулы (V-37), соединение формулы (V-38), соединение формулы (V-39), соединение формулы (V-40), соединение формулы (VI-1), соединение формулы (VI-2), соединение формулы (VI-3), соединение формулы (VI-4), соединение формулы (VI-5), соединение формулы (VI-6), соединение формулы (VI-7) и соединение формулы (VI-8), помечены 68Ga.
Кроме того, соединения, которые можно метить радионуклидом (такие как соединение формулы (V-1), соединение формулы (V-2), соединение формулы (V-3), соединение формулы (V-4), соединение формулы (V-5), соединение формулы (V-6), соединение формулы (V-7), соединение формулы (V-8), соединение формулы (V-9), соединение формулы (V-10), соединение формулы (V-11), соединение формулы (V-12), соединение формулы (V-13), соединение формулы (V-14), соединение формулы (V-16), соединение формулы (V-17), соединение формулы (V-18), соединение формулы (V-19), соединение формулы (V-20), соединение формулы (V-21), соединение формулы (V-22), соединение формулы (V-23), соединение формулы (V-25), соединение формулы (V-26), соединение формулы (V-27), соединение формулы (V-28), соединение формулы (V-29), соединение формулы (V-30), соединение формулы (V-31), соединение формулы (V-32), соединение формулы (V-33), соединение формулы (V-34), соединение формулы (V-35), соединение формулы (V-36), соединение формулы (V-37), соединение формулы (V-38), соединение формулы (V-39), соединение формулы (V-40), соединение формулы (VI-1), соединение формулы (VI-2), соединение формулы (VI-3), соединение формулы (VI-4), соединение формулы (VI-5), соединение формулы (VI-6), соединение формулы (VI-7) и соединение формулы (VI-8), то же самое ниже), предусмотренные настоящим изобретением, также могут быть помечены со ссылкой на способ мечения 18-F, предусмотренный патентами CN 102123739 В и/или CN 102066974 B. Кроме того, соединения, которые могут быть помечены радионуклидом, предусмотренным настоящим изобретением, также могут быть помечены со ссылкой на другие способы маркировки, предусмотренные в известном уровне техники (включая, но не ограничиваясь ими, способы, предусмотренные в настоящем изобретении). Радионуклид включает, но не ограничивается этим: 18F, 51Cr, 64Cu, 67Cu, 67Ga, 68Ga, 89Zr, 111In, 99mTc, 186Re, 188Re, 139La, 140La, 175Yb, 153Sm, 166Ho, 86Y, 90Y, 149Pm, 165Dy, 169Er, 177Lu, 47Sc, 142Pr, 159Gd, 212Bi, 213Bi, 72As, 72Se, 97Ru, 109Pd, 105Rh, 101mRh, 119Sb, 128Ba, 123I, 124I, 131I, 197Hg, 211At, 151Eu, 153Eu, 169Eu, 201Tl, 203Pb, 212Pb, 198Au, 225Ac, 227Th или 199Ag и тому подобное.
Экспериментальный пример 135: Анализ и эффективность применения соединения 68Ga-FAPI-RGD (V-1)
(1) Анализ и идентификация с помощью ВЭЖХ
Система ВЭЖХ была следующей: SHIMADZULC-20A; и хроматографическая колонка С18 (YMC, 3 мкм, 4,6*150 мм), используемая для анализа. Длина волны детектирования составляла 254 нм, скорость потока 1 мл/мин, градиент промывки был следующим: при 0-3 мин 10% ацетонитрила и 90% воды (50 мМ ацетата аммония) оставались неизменными; через 3-16 мин систему увеличивали до содержания 90% ацетонитрила и 10% воды (50 мМ ацетата аммония); через 16-18 мин удерживалось 90% ацетонитрила и 10% воды (50 мМ ацетата аммония); через 18-20 мин систему восстанавливали до включения 10% ацетонитрила и 90%) воды (50 мМ ацетата аммония); через 20-22 мин удерживалось 10%) ацетонитрила и 90% воды (50 мМ ацетата аммония). Результаты ВЭЖХ-контроля качества 68Ga-FAPI-RGD (V-1) приведены на фиг.46.
(2) Визуализация MicroPET у мышей с опухолями
Мышам с опухолью HepG2-FAP вводили 7,4 МБк соединения 68Ga-FAPI-RGD (V-1) путем внутривенной введения в хвост, а затем подвергали визуализации MicroPET под наркозом изофлураном после введения в течение 0-120 минут, соответственно. Результаты показаны на фиг.47. На фиг.47 показано репрезентативная корональная визуализация MicroPET мышей с опухолью HepG2-FAP (n=3) в разные моменты времени после внутривенного введения 68Ga-FAPI-RGB (V-1).B определенные моменты времени получения изображения (30 мин и 2 ч) опухоли хорошо видны. Специфическое свойство 68Ga-FAPI-RGD (V-1) связываться с интегрином и FAP in vivo доказано в эксперименте по блокированию. Кроме того, 68Ga-FAPI-RGD (V-1) вводили мышам с опухолью HepG2-FAP совместно с RGD или FAPI-02. Результаты визуализации микрочастиц и их поглощения органами показаны на фиг.48, фиг.49 и фиг.50. Как видно из фиг.48-49, поглощение 68Ga-FAPI-RGD (V-1) опухолями может быть снижено после совместного введения с RGD или FAPI-02.Как видно из фиг.50, значения поглощения (% ID/г) жизненно важными органами и опухолями получены после введения в течение 0,5 ч, а поглощение 68Ga-FAPI-RGD (V-1) опухолями частично ингибируется RGD или FAPI-02. Эксперимент по блокированию доказывает, что 68Ga-FAPI-RGD (V-1) может специфически воздействовать на опухоли in vivo путем связывания с интегрином и FAP.
Экспериментальный пример 136; Анализ и эффективность применения соединения 68Ga-FAPI-RGD (V-25)
(1) Анализ стабильности
В соответствии со способом, приведенным в примере 134, было получено соединение, меченное 68Ga, формулы (V-25) (а именно, соединение 68Ga-FAPI-RGD (V-25)). 20 мкл раствора 68Ga-FAPI-RGD (V-25) (с активностью 3,7 МБк/20 мкл) отсасывали и добавляли в центрифужную пробирку, содержащую 100 мкл физиологического раствора или PBS (со значением рН 7,4) для совместной инкубации при 37°С в течение 0,5 ч, 1 ч и 4 ч для получения раствора для совместной инкубации. Отбирали 20 мкл раствора для совместной инкубации, фильтровали с помощью игольчатой фильтрующей мембраны диаметром 0,22 мкм и анализировали методом ВЭЖХ для получения радиохимической чистоты.
Результаты тестирования показаны на фиг.51. Результаты показывают, что соединение 68Ga-FAPI-RGD (V-25) явно не разлагается после инкубации в обычном физиологическом растворе и имеет радиохимическую чистоту более 99%, что указывает на то, что 68Ga-FAPI-RGD (V-25), полученное по настоящему изобретению, обладает превосходной стабильностью.
(2) Поглощение и блокирование
Эксперимент по клеточному поглощению соединения 68Ga-FAPI-RGD (V-25) был проведен в опухолевых клетках HT1080-FAP. Результаты испытаний приведены в части А на фиг.52. Результаты показывают, что 68Ga-FAPI-RGD (V-25) быстро усваивается клетками, достигает максимального уровня усвоения после инкубации в течение 30 минут и сохраняется на аналогичном уровне усвоения в течение 2 часов.
Кроме того, в эксперименте для проведения эксперимента по блокированию также использовались «FAPI-02» и «C(RGDfK)», а также «FAPI-RGD». Результаты испытаний приведены в части А на фиг.52. Результаты показывают, что клеточное поглощение 68Ga-FAPI-RGD (V-25) может быть частично ингибировано С (RGDfK) или FAPI-02 и может быть полностью заблокировано FAPI-RGD (со ссылкой на часть А на рис. 52).
(3) Подобие
Эксперимент по связыванию клеток был проведен на опухолевых клетках HT1080-FAP и U87MG, и результаты теста показаны в частях В и С на фиг.52, соответственно. В эксперименте с клеткой HT1080-FAP соединение 68Ga-FAPI-RGD (V-25) и 68Ga-FAPI-02, как было измерено, имели значение IC50, равное 11,17 нМ и 4,14 нМ, соответственно. В эксперименте с клеткой HT1080-FAP соединение 68Ga-FAPI-RGD (V-25) и 68Ga-C(RGDfK), как было измерено, имели значение IC50, равное 18,93 нМ и 11,49 нМ соответственно. Экспериментальные результаты показывают, что по сравнению с соответствующими мономерами соединение 68Ga-FAPI-RGD (V-25) обладает сходным подобием к соответствующим рецепторам, таким как FAP и интегрин αvβ3.
(4) Визуализация MicroPET у мышей с опухолями
Мышам с опухолью HT1080-FAP, которые были случайным образом сгруппированы, вводили 7,4 МБк соединения 68Ga-FAPI-RGD (V-25), 68Ga-FAPI-02 и 68Ga-C(RGDfK) путем внутривенного введения в хвост, соответственно. Затем под анестезией изофлураном группа 68Ga-FAPI-RGD (V-25) подвергалась визуализации MicroPET после введения в течение 0-240 минут соответственно, а другие группы подвергались визуализации MicroPET после введения в течение 0-120 минут, соответственно. Результаты показаны на фиг.53. На фиг.53, часть А, часть С и часть Е показывают проекционные изображения MicroPET максимальной плотности мышей с опухолями HT1080-FAP (n=3) в разные моменты времени после внутривенного введения трем группам мышей, соответственно, а часть В, часть D и часть F показывают поглощение в различных органах или тканях (таких как кровь, печень, почки, опухоли и мышцы) трех групп мышей в разные моменты времени после введения, соответственно, где три величины поглощения в каждой группе соответствуют (слева направо) 0,5 часа, 1 часу и 2 часам после введения соответственно. На фиг.53 показано, что опухоли четко видны в моменты получения изображения, а поглощение опухолью 68Ga-FAPI-RGD (V-25) выше, чем у 68Ga-FAPI-02 и 68Ga-C(RGDfK). Специфическое свойство 68Ga-FAPI-RGD (V-25) связываться с интегрином αvβ3 и FAP in vivo доказано в эксперименте по блокированию. 68Ga-FAPI-RGD (V-25) и C(RGDfK) или FAPI-02 были совместно введены мышам с опухолью HT1080-FAP, и результаты визуализации микропетлей и поглощения их органами показаны на фиг.54. На фиг.54, четыре изображения в части А соответствуют (слева направо) изображениям, полученным путем раздельного введения 68Ga-FAPI-RGD (V-25), совместного введения 68Ga-FAPI-RGD (V-25) с C(RGDfK), совместного введения 68Ga-FAPI-RGD (V-25) с FAPI-02 и совместного введения 68Ga-FAPI-RGD (V-25) с C(RGDfK) и FAPI-02 соответственно; а в части В и части С показано поглощение 68Ga-FAPI-RGD (V-25) в различных органах или тканях (таких как кровь, печень, почки, опухоли и мышцы) мышей после введения четырьмя различными методами введения и целевое/нецелевое соотношение соответственно, где четыре гистограммы для каждого органа или ткани в части В и части С соответствуют (слева направо) четырем методам введения в части А соответственно. Как видно из фиг.54, поглощение 68Ga-FAPI-RGD (V-25) опухолями может быть снижено при совместном введении 68Ga-FAPI-RGD (V-25) с RGD или FAPI-02, а также поглощении 68Ga-FAPI-RGD (V-25) в опухолях может быть дополнительно снижено при совместном введении 68Ga-FAPI-RGD (V-25) с RGD и FAPI-02. Эксперимент по блокированию доказывает, что 68Ga-FAPI-RGD (V-25) может специфически воздействовать на опухоли путем связывания с интегрином и FAP in vivo.
(5) ПЭТ/КТ-визуализация у пациентов с опухолями
В клиническом исследовании в реальных условиях в качестве испытуемых были использованы пациент с раком поджелудочной железы (один), пациент с немелкоклеточным раком легких (один), пациент с мелкоклеточным раком легких (один) и пациент с карциномой носоглотки (один). Доза для внутривенного введения (1,8-2,2 МБк [0,05-0,06 мКи]/кг) 68Ga-FAPI-RGD (V-25) была рассчитана исходя из массы тела каждого пациента. После проведения внутривенного введения в течение 3 ч данные были получены с помощью гибридного ПЭТ/КТ-сканера (Discovery MI, GE Хэлс Кэр, Милуоки, Висконсин, США). Результаты визуализации показаны на фиг.55. Максимальное стандартное значение поглощения (SUV макс) было автоматически рассчитано с использованием исследуемой области (ROI), отображенной на трансаксиальном изображении. Значение SUVмакс для 68Ga-FAPI-RGD (V-25), нацеленного на две мишени, выше, чем у 68Ga-FAPI-46, нацеленного на одну цель, FAP, в различных типах опухолей, а значение SUVмакс увеличивается примерно на 30-50%, что доказывает, что дизайн, нацеленный на две мишени, позволяет улучшить количество и эффективность использования эффективных рецепторов в опухолях, тем самым улучшая поглощение опухолями.
Пример 137: Анализ и эффективность применения соединения V-40, меченного радиоактивным веществом 177Lu (а именно, соединения 177Lu-FAPI-RGD (V-4))
Соединение V-40, полученное в примере 8, было радиоактивно мечено 177lu с помощью обычных технических средств, известных в данной области, для получения соединения 177lu-FAPI-RGD (V-40). Был проведен эксперимент с ОФЭКТ-визуализацией для наблюдения за распределением индикатора у мышей с опухолями. Мышам с опухолью U87MG вводили 37 МБк соединения 177Lu-FAPI-RGD (V-40). После введения в течение 1 ч, 4 ч, 12 ч, 24 ч, 48 ч, 72 ч и 96 ч мышей с опухолью U87MG анестезировали и помещали на ОФЭКТ-сканер для статического ОФЭКТ-сканирования. Результаты приведены на фиг.60. Соединение 177Lu-FAPI-RGD (V-40) обладает очевидным поглощением опухолями после введения мышам с опухолью U87MG в течение 1 часа, что значительно превышает поглощение во всех органах, за исключением мочевого пузыря. При увеличении времени введения поглощение в опухолях увеличивается и остается высоким в течение 96 часов, в то время как поглощение в мочевом пузыре и других органах постепенно снижается, что доказывает превосходное поглощение и удержание зонда в опухолях и его большой потенциал для лечения глиомных опухолей.
Пример 138: Другие примеры
В настоящем изобретении анализ стабильности соединения 68Ga-FAPI-RGD (V-2) (а именно, 68Са-меченный комплекс соединения формулы (2), то же самое ниже), соединения 68Ga-FAPI-RGD (V-3), соединения 68Ga-FAPI-RGD (V-4), соединения 68Ga-FAPI-RGD (V-5), соединения 68Ga-FAPI-RGD (V-6), соединения 68Ga-FAPI-RGD (V-7), соединения 68Ga-FAPI-RGD (V-8), соединения 68Ga-FAPI-RGD (V-9), соединения 68Ga-FAPI-RGD (V-10), соединения 68Ga-FAPI-RGD (V-11), соединения 68Ga-FAPI-RGD (V-12), соединения 68Ga-FAPI-RGD (V-13), соединения 68Ga-FAPI-RGD (V-14), соединения 68Ga-FAPI-RGD (V-16), соединения 68Ga-FAPI-RGD (V-17), соединения 68Ga-FAPI-RGD (V-18), соединения 6SGa-FAPI-RGD (V-19), соединения 68Ga-FAPI-RGD (V-20), соединения 68Ga-FAPI-RGD (V-21), соединения 68Ga-FAPI-RGD (V-22), соединения 68Ga-FAPI-RGD (V-23), соединения 68Ga-FAPI-RGD (V-26), соединения 68Ga-FAPI-RGD (V-27), соединения 68Ga-FAPI-RGD (V-28), соединения 68Ga-FAPI-RGD (V-29), соединения 68Ga-FAPI-RGD (V-30), соединения 68Ga-FAPI-RGD (V-31), соединения 68Ga-FAPI-RGD (V-32), соединения 68Ga-FAPI-RGD (V-33), соединения 68Ga-FAPI-RGD (V-34), соединения 68Ga-FAPI-RGD (V-35), соединения 68Ga-FAPI-RGD (V-36), соединения 68Ga-FAPI-RGD (V-37), соединения 68Ga-FAPI-RGD (V-38), соединения 68Ga-FAPI-RGD (V-39), соединения 68Ga-FAPI-RGD (VI-1), соединения 68Ga-FAPI-RGD (VI-2), соединения 68Ga-FAPI-RGD (VI-3), соединения 68Ga-FAPI-RGD (VI-4), соединения 68Ga-FAPI-RGD (VI-5), соединения 68Ga-FAPI-RGD (VI-6), соединения 68Ga-FAPI-RGD (VI-7) и соединения 68Ga-FAPI-RGD (VI-8) (в настоящем изобретении вместе называемые соединениями, меченными радиоактивным изотопом FAPI-RGD) также проверяли с использованием соответствующего метода анализа со ссылкой на эксперимент (1) в примере 136, соответственно. Результаты показывают, что все радиоактивно меченые соединения двойного нацеливания для белка активации фибробластов (FAP) и интегрина αvβ3, представленные в настоящем изобретении, могут достигать хорошей стабильности.
В настоящем изобретении эксперименты по поглощению и блокированию и эксперимент по подобию радиоактивно меченых соединений FAPI-RGD также были дополнительно подтверждены с использованием соответствующих методов со ссылкой на эксперименты (2) и (3) в примере 136. Экспериментальные результаты показывают, что радиоактивно меченые соединения FAPI-RGD, представленные в соответствии с настоящим изобретением, могут демонстрировать быстрое клеточное поглощение в соответствующих моделях клеток, и соответствующее клеточное поглощение может блокироваться соответствующими мономерами/димерными соединениями. Кроме того, радиоактивно меченые соединения FAPI-RGD, предлагаемые настоящим изобретением, также могут проявлять сходное подобие к соответствующим рецепторам, таким как FAP и интегрины αvβ3
В настоящем изобретении визуализация MicroPET радиоактивно меченных FAPI-RGD соединений у мышей с опухолями была дополнительно проверена с использованием соответствующего метода со ссылкой на эксперимент (4) в примере 136, где экспериментальная группа подвергалась визуализации MicroPET через 0-240 минут после введения соответственно. Результаты показывают проекционные изображения MicroPET моделей животных с максимальной плотностью в разные моменты времени после внутривенного введения, опухоли четко видны в моменты времени получения изображения, а поглощение опухолью экспериментальной группы (а именно, радиоактивно меченных соединений FAPI-RGD по настоящему изобретению) выше, чем у соответствующих мономеров.
Таким образом, структура двойного нацеливания FAPI-RGD, предложенная в настоящем изобретении, обладает высоким подобием как к мишени FAP, так и к мишени интегрина αvβ3, может синергически нацеливаться на мишень FAP и мишень интегрина αvβ3 в опухолях, обладает превосходной фармакокинетикой, высоким поглощением опухолями и длительным временем удерживания в опухолях и, как ожидается, будет использоваться для диагностики или лечения заболеваний, характеризующихся сверхэкспрессией белка активации фибробластов (FAP) и/или интегрина αvβ3.
Хотя настоящее изобретение подробно описано в приведенном выше общем описании, конкретных вариантах осуществления и тестах, специалистам в данной области техники очевидно, что на основе настоящего изобретения могут быть сделаны некоторые модификации или усовершенствования. Следовательно, модификации или усовершенствования, произведенные без отступления от сути настоящего изобретения, подпадают под сферу охраны, требуемой настоящим изобретением.
| название | год | авторы | номер документа |
|---|---|---|---|
| СОЕДИНЕНИЕ ДВОЙНОГО НАЦЕЛИВАНИЯ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ | 2022 |
|
RU2838179C2 |
| ИНГИБИТОР БЕЛКА АКТИВАЦИИ ФИБРОБЛАСТОВ, МОДИФИЦИРОВАННЫЙ УСЕЧЕННЫМ СИНИМ ЭВАНСА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ | 2021 |
|
RU2841304C2 |
| ИНГИБИТОР FAP | 2019 |
|
RU2797409C2 |
| ЛИОФИЛИЗАТ ДЛЯ ПРИГОТОВЛЕНИЯ РАДИОФАРМАЦЕВТИЧЕСКОГО ПРЕПАРАТА | 2017 |
|
RU2702238C2 |
| МОДИФИЦИРОВАННЫЕ ЦИАНИНОВЫЕ КРАСИТЕЛИ И ИХ КОНЪЮГАТЫ | 2020 |
|
RU2833355C2 |
| RGD-СОДЕРЖАЩИЕ ПЕПТИДОМИМЕТИКИ И ИХ ПРИМЕНЕНИЕ | 2009 |
|
RU2519736C2 |
| АГЕНТЫ ДЛЯ ВИЗУАЛИЗАЦИИ | 2004 |
|
RU2355702C2 |
| КОНЪЮГАТЫ RGD-(БАКТЕРИО)ХЛОРОФИЛЛ ДЛЯ ФОТОДИНАМИЧЕСКОЙ ТЕРАПИИ И ВИЗУАЛИЗАЦИИ НЕКРОТИЧЕСКИХ ОПУХОЛЕЙ | 2009 |
|
RU2518296C2 |
| СОЕДИНЕНИЯ НА ОСНОВЕ ПЕПТИДОВ ДЛЯ НАПРАВЛЕННОЙ ДОСТАВКИ К РЕЦЕПТОРАМ ИНТЕГРИНОВ | 2002 |
|
RU2303042C2 |
| СОЕДИНЕНИЕ ДЛЯ ДИАГНОСТИКИ ОПУХОЛЕЙ, ЭКСПРЕССИРУЮЩИХ ПСМА, И КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ | 2019 |
|
RU2730507C1 |
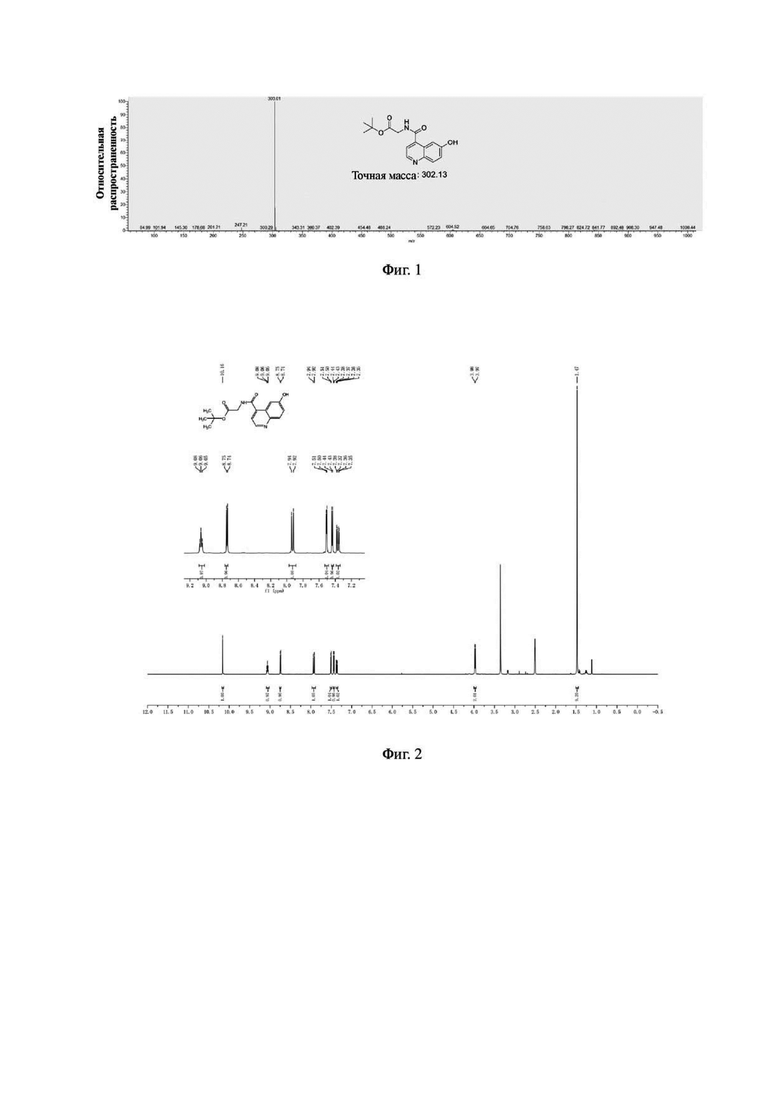
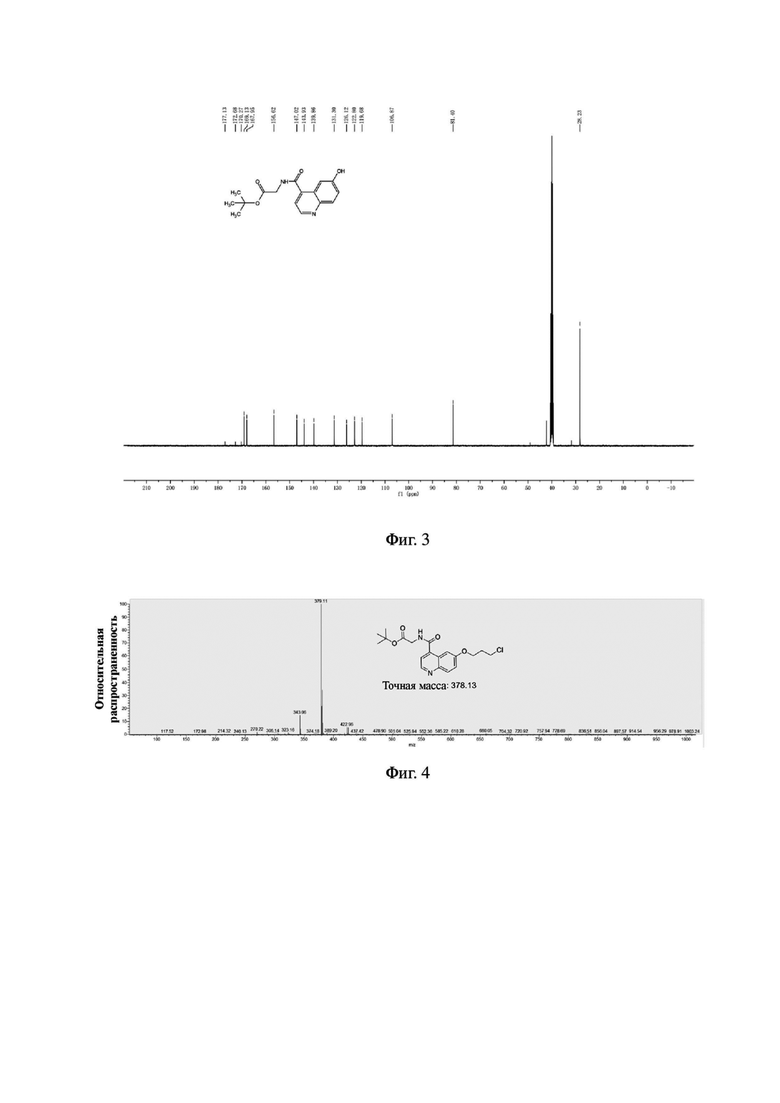
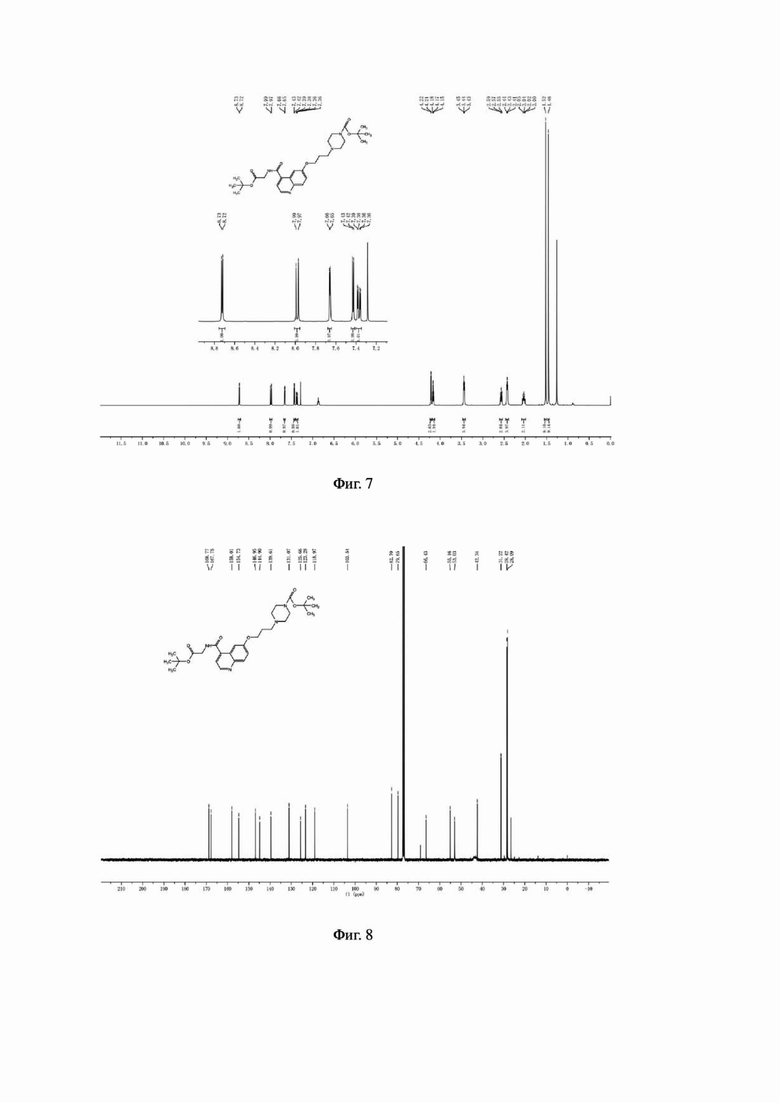
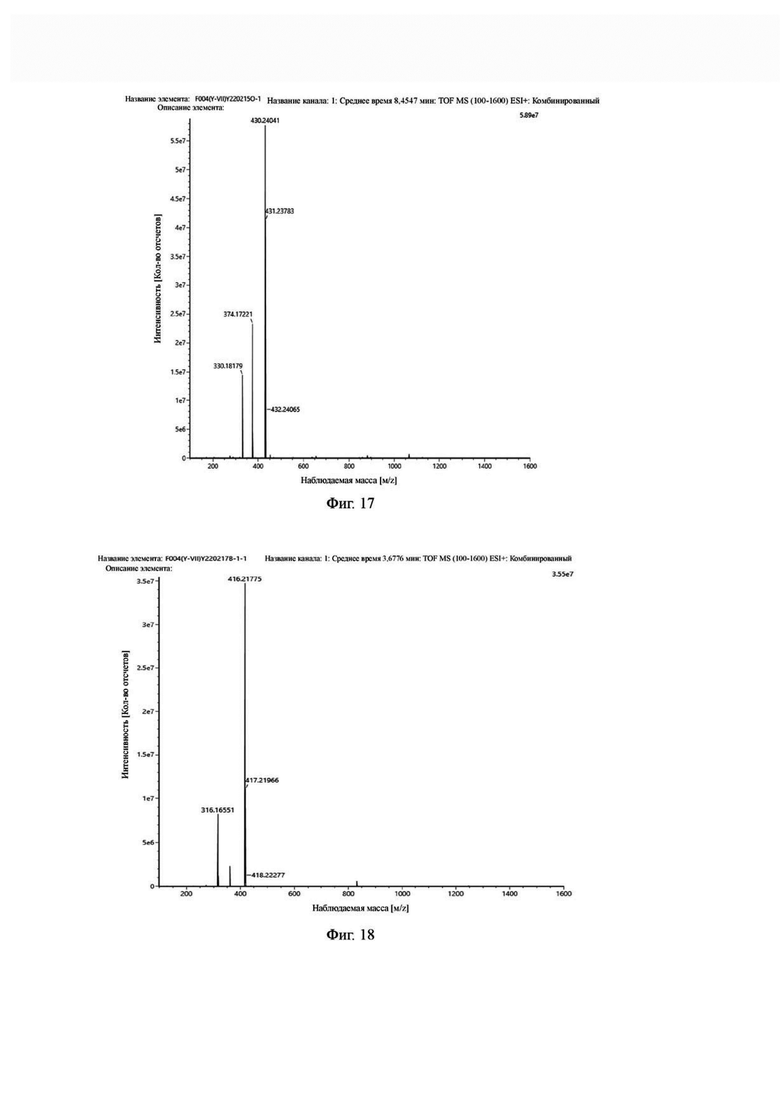
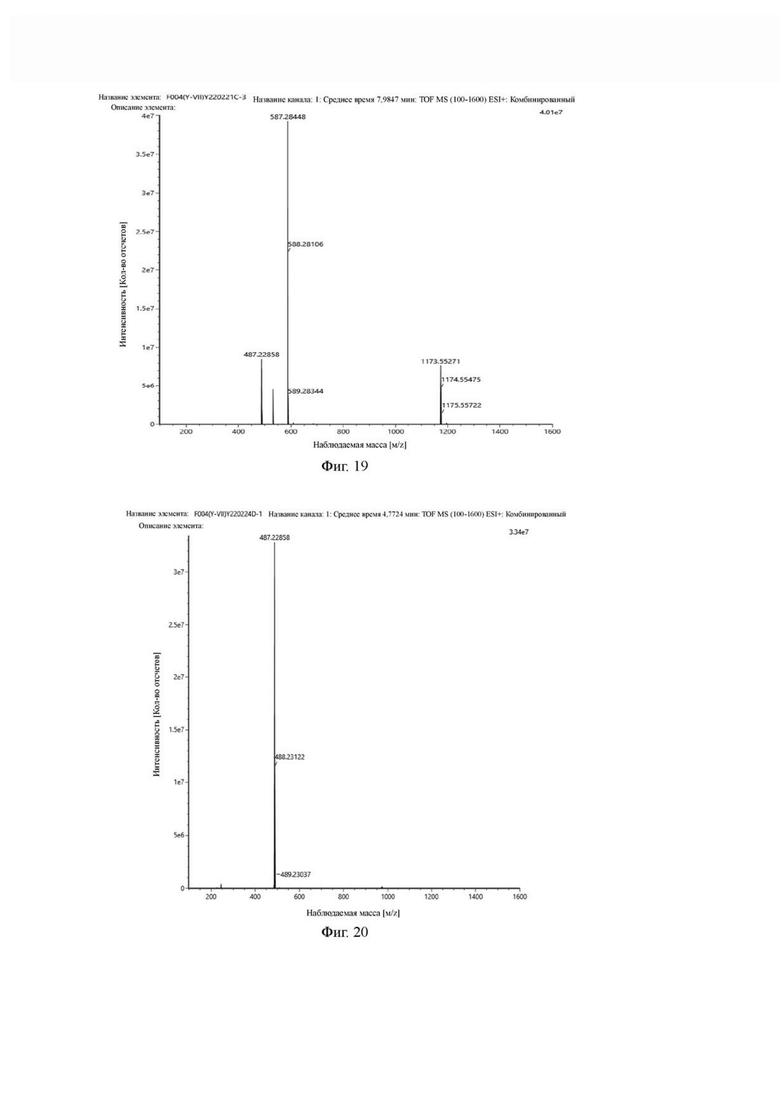
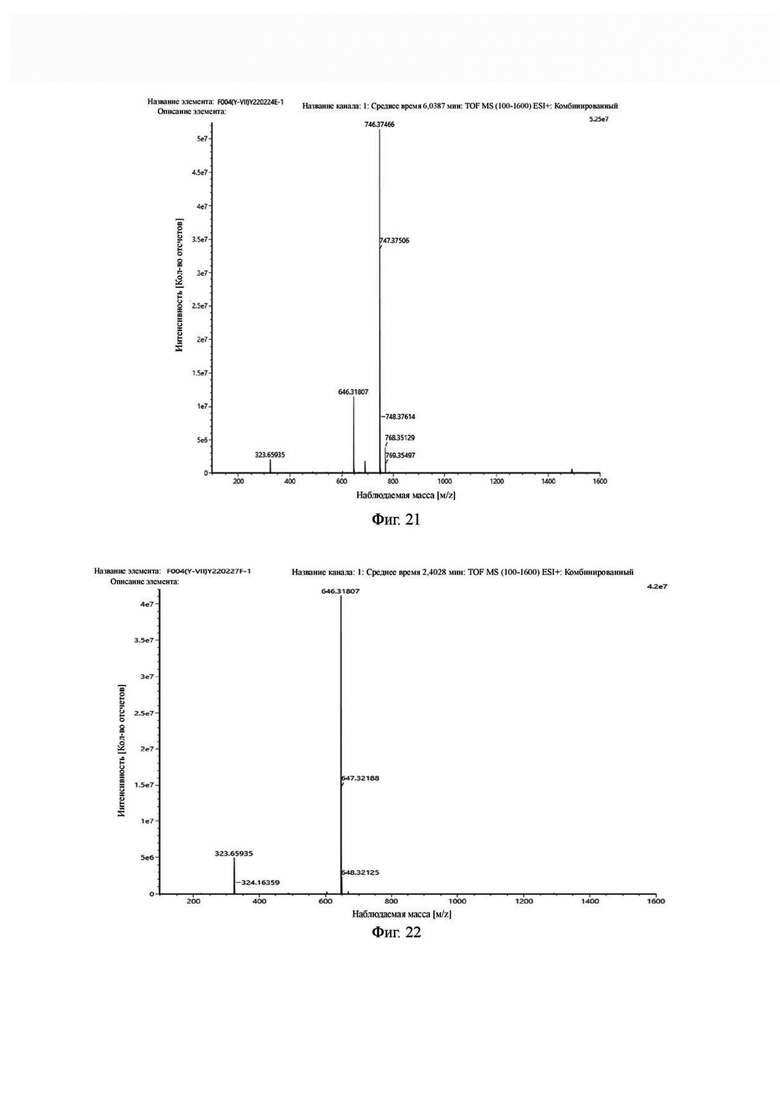
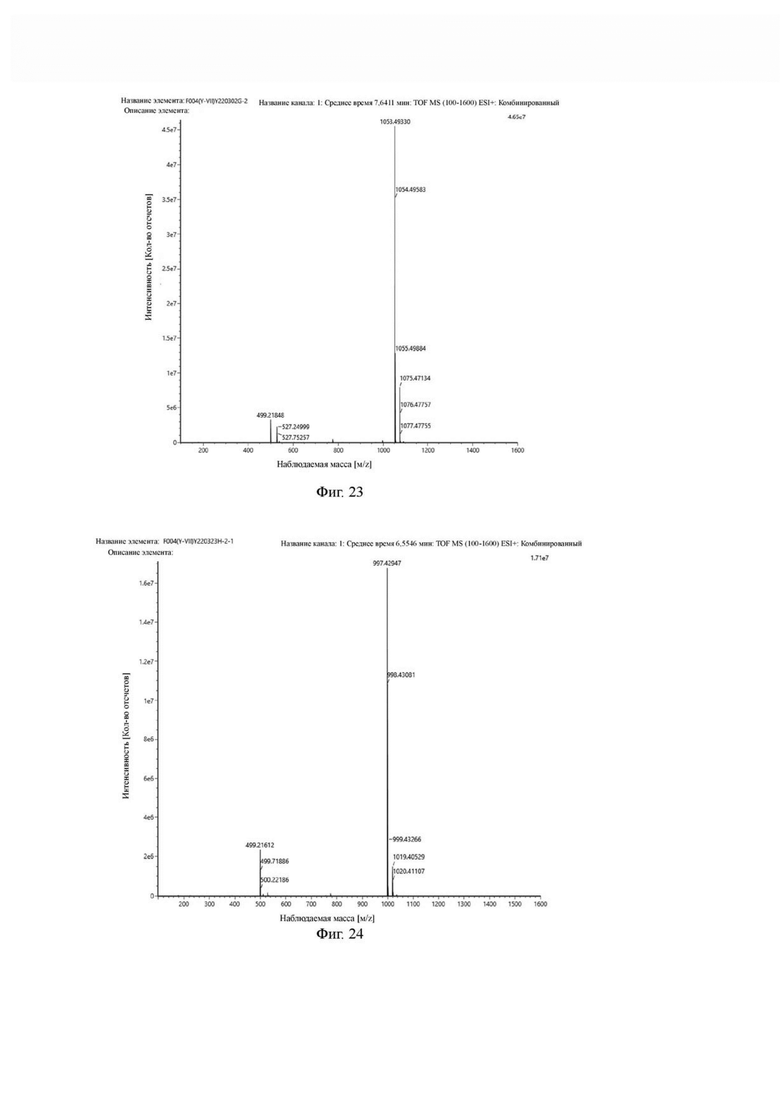
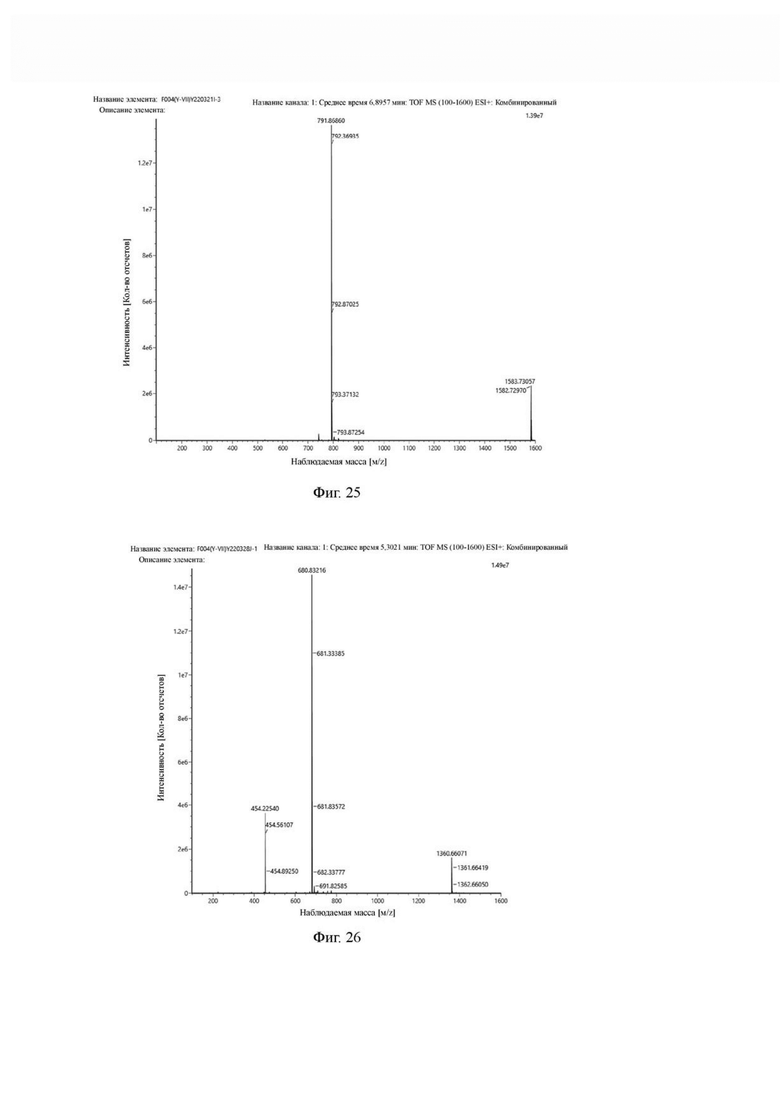
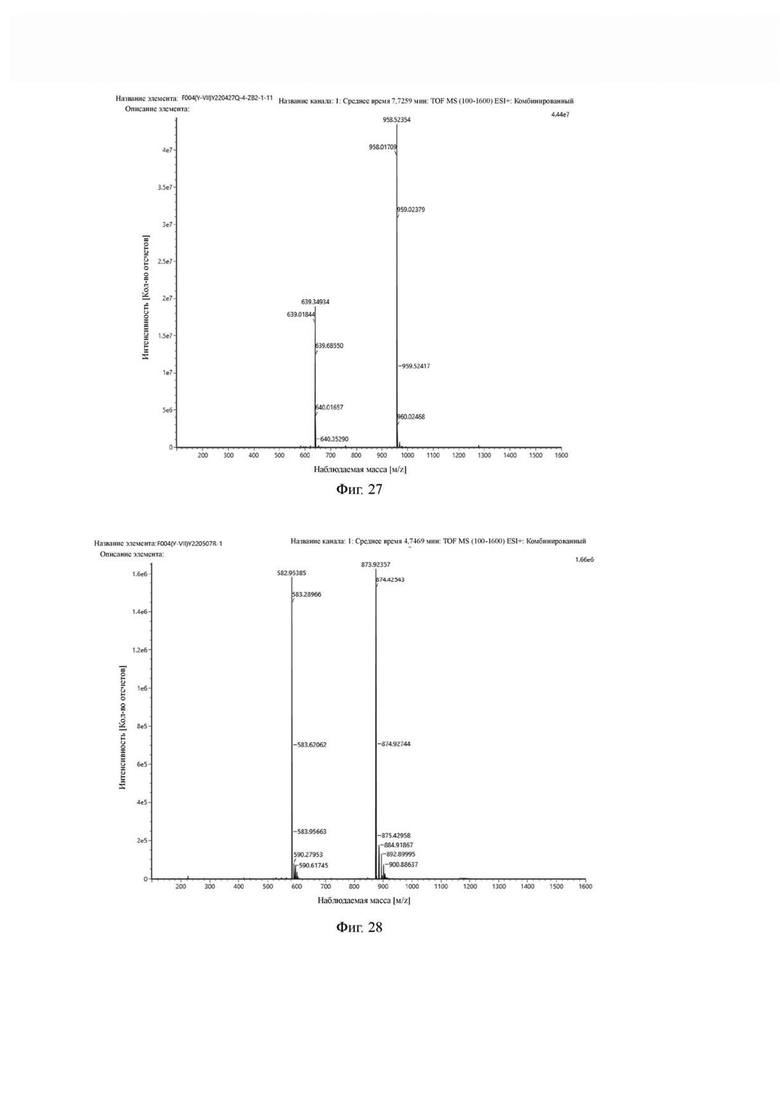
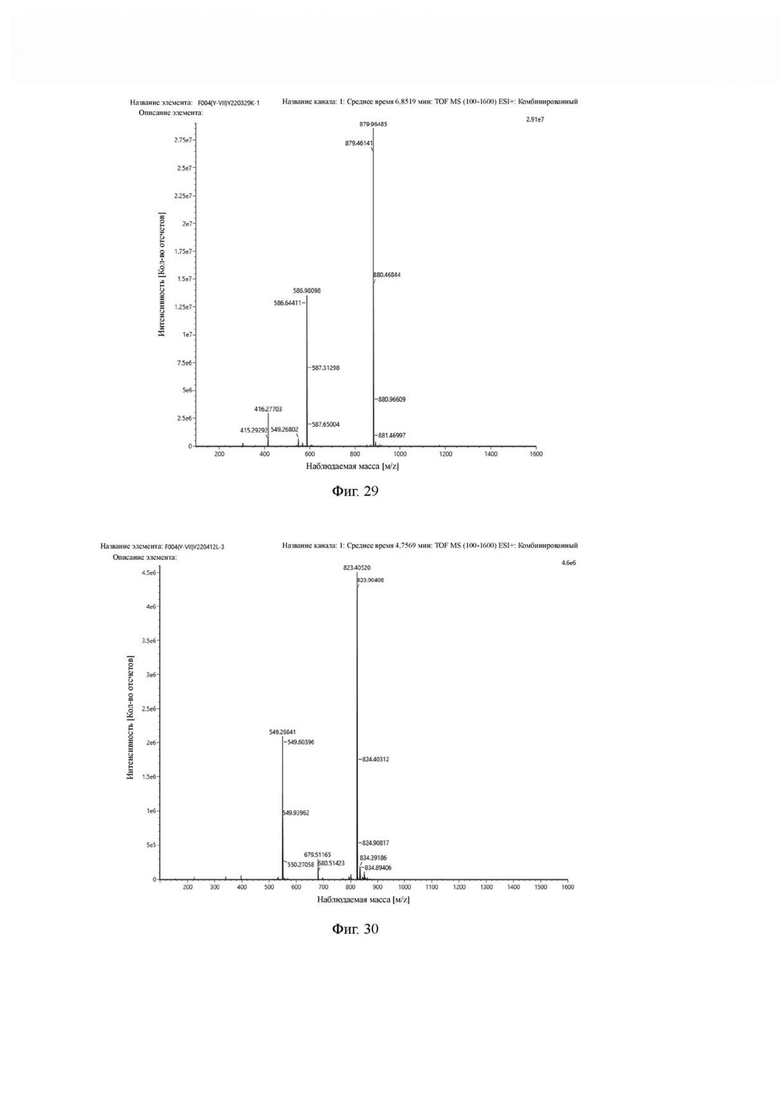
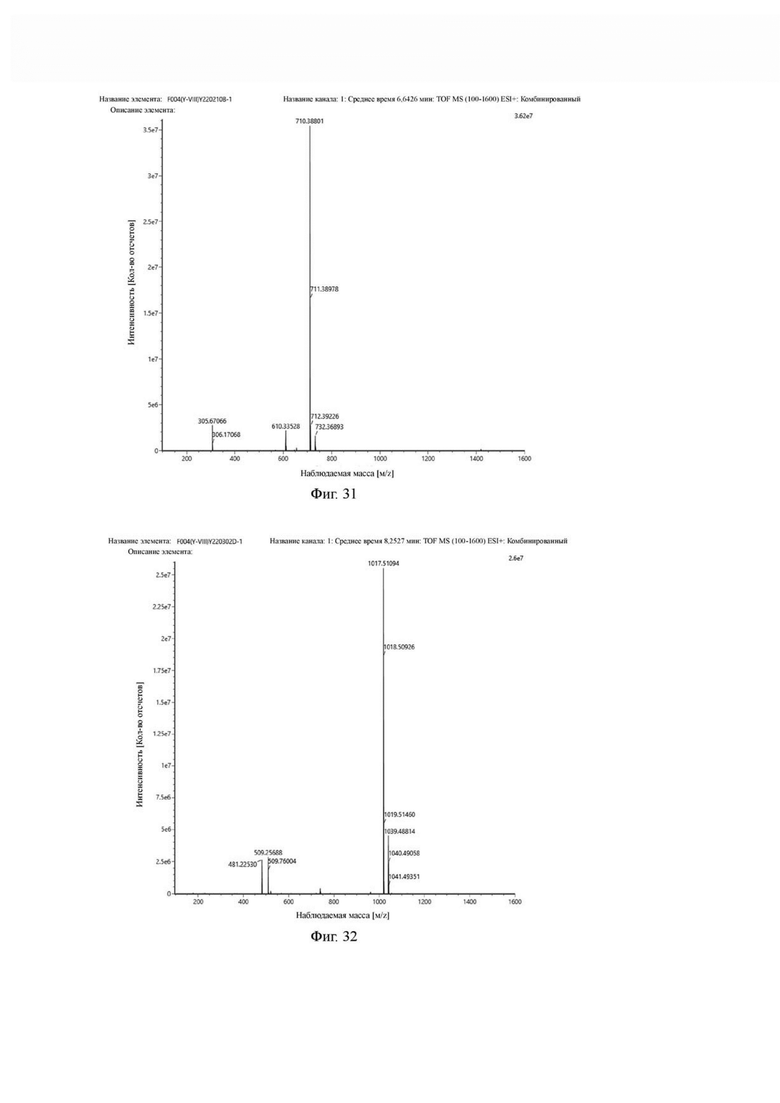
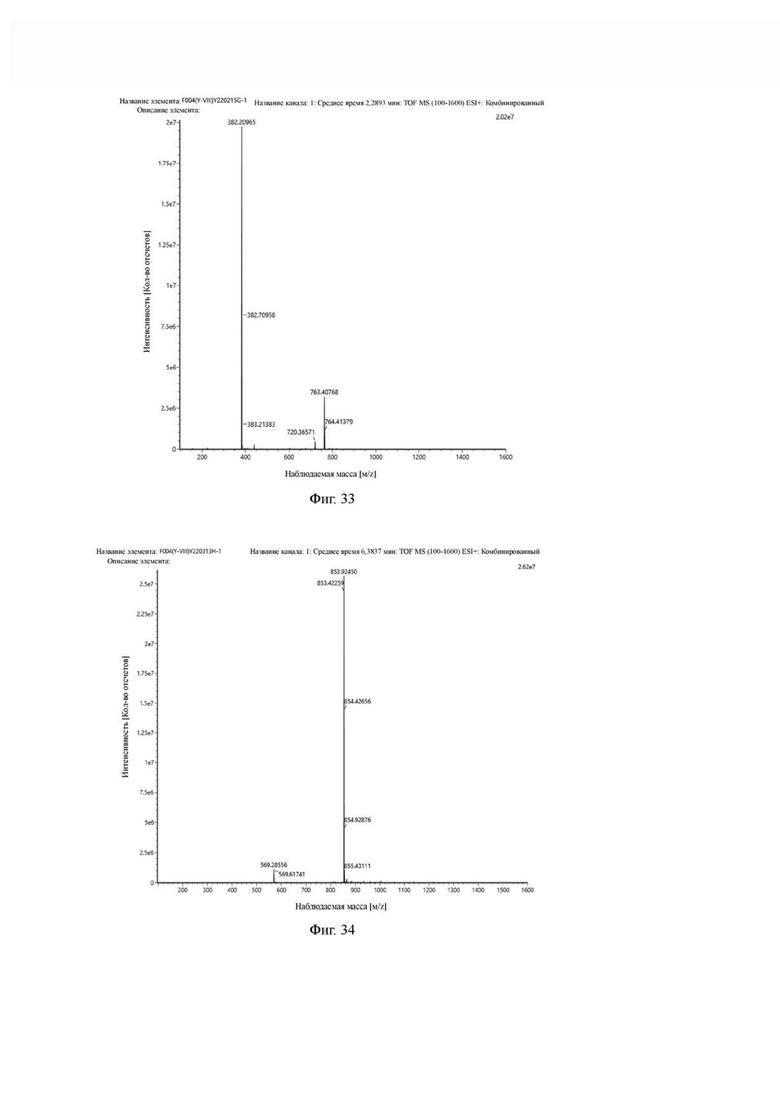
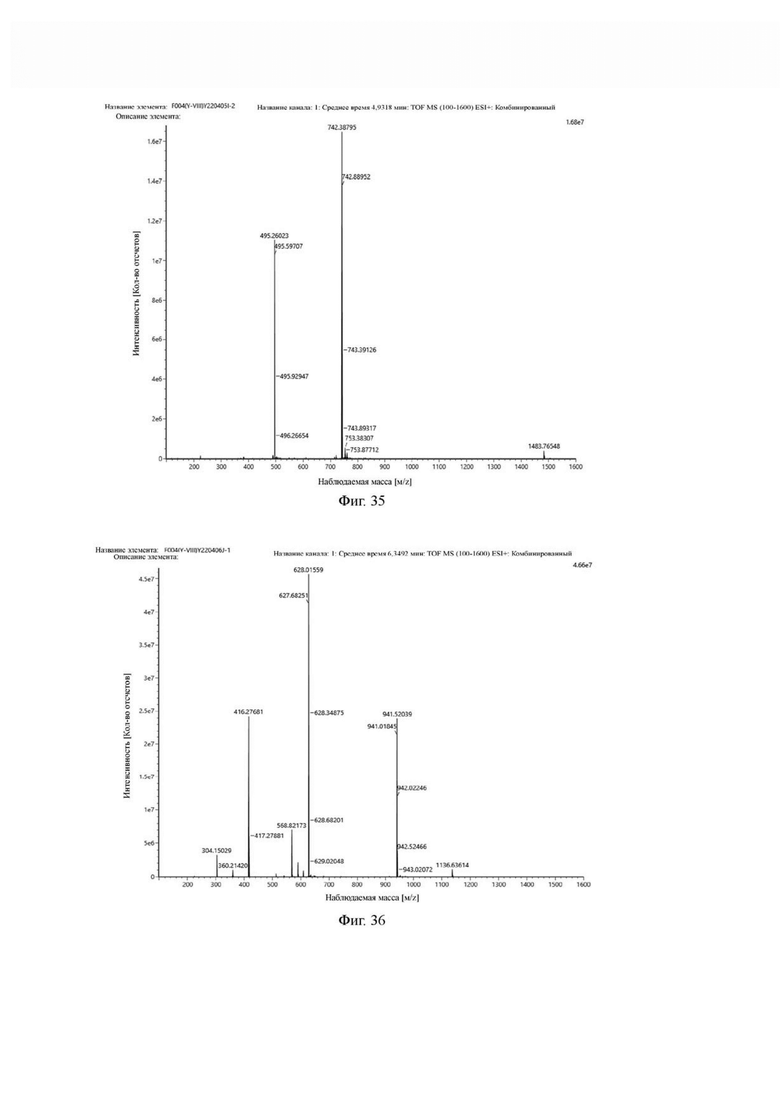
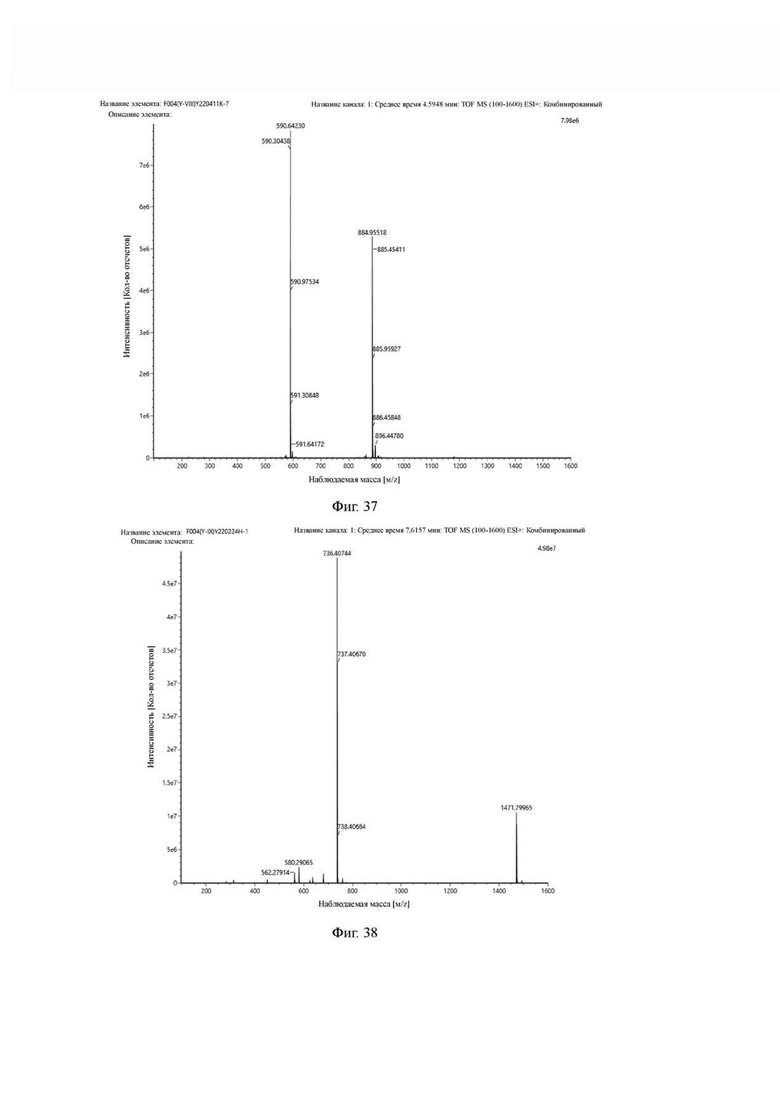
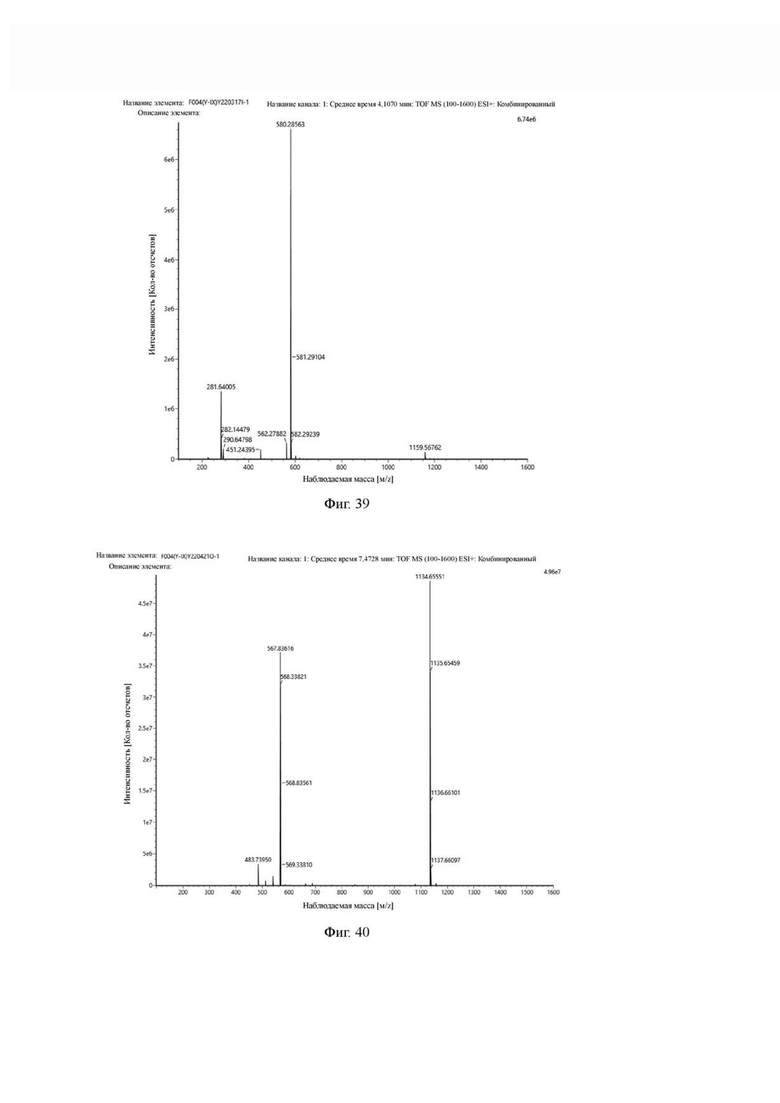
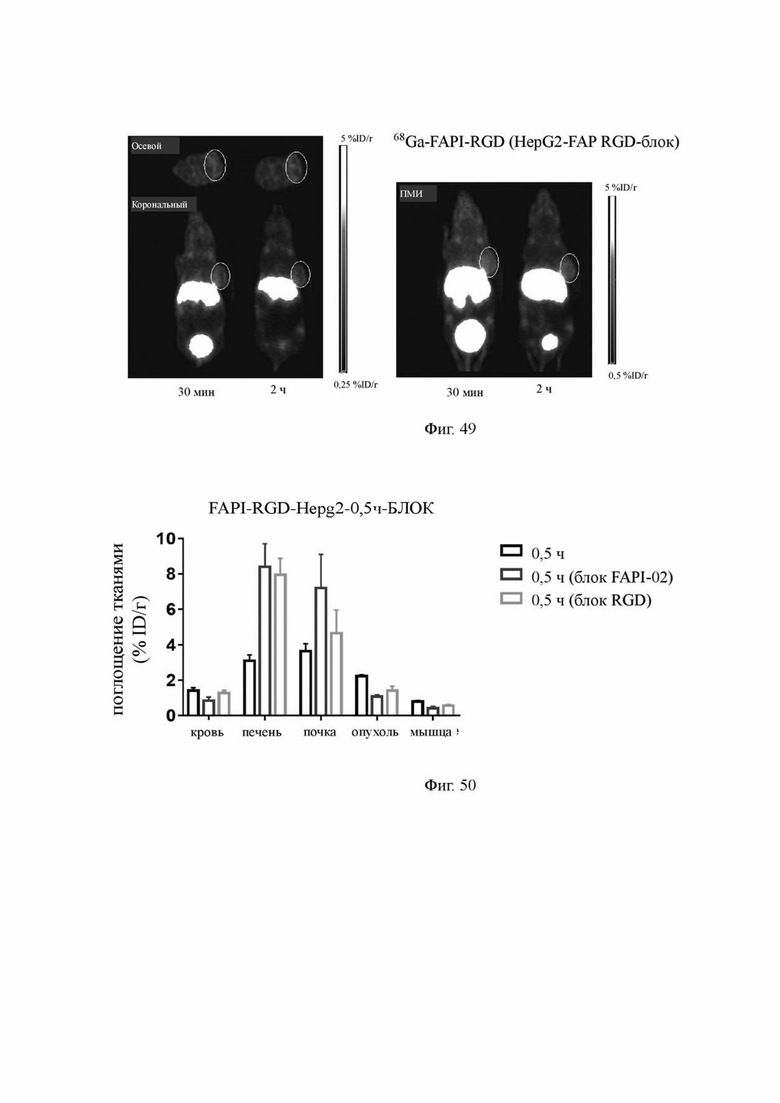
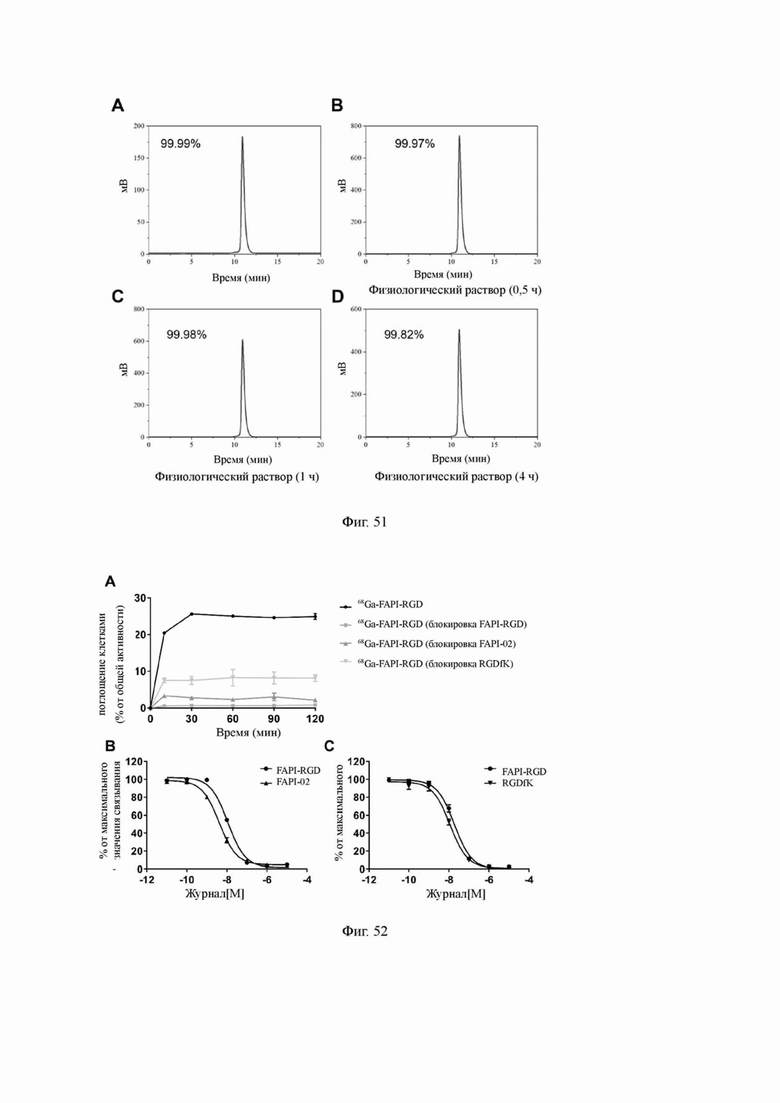
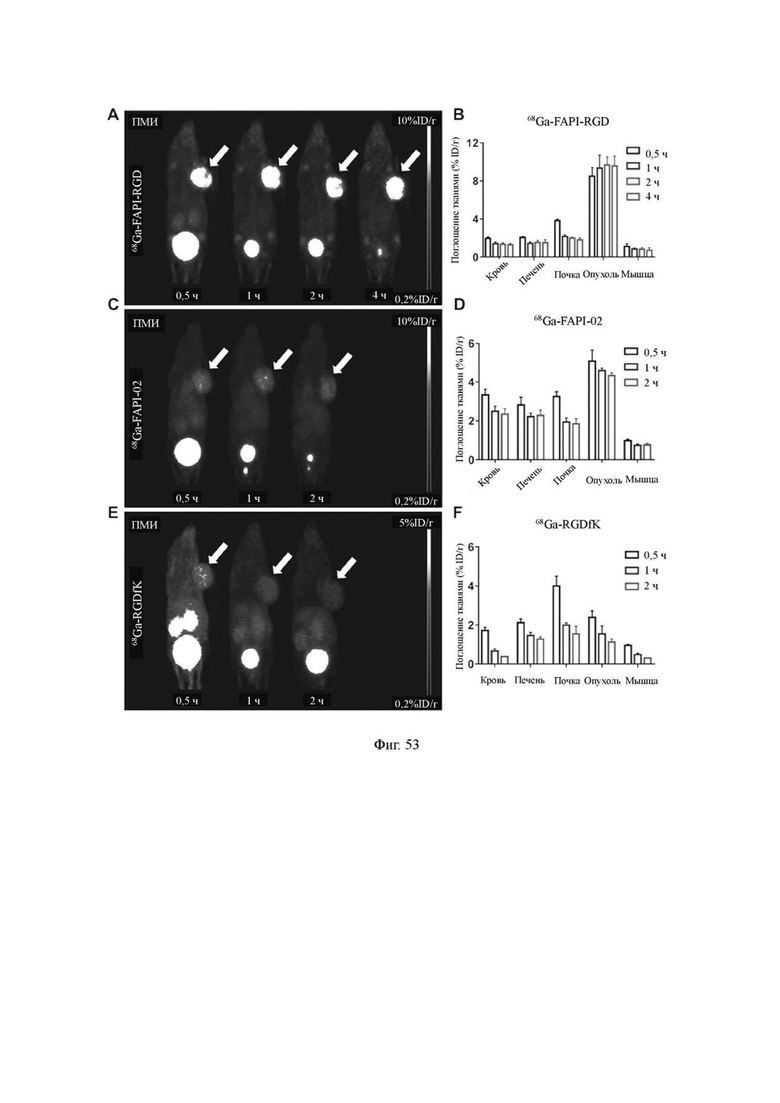

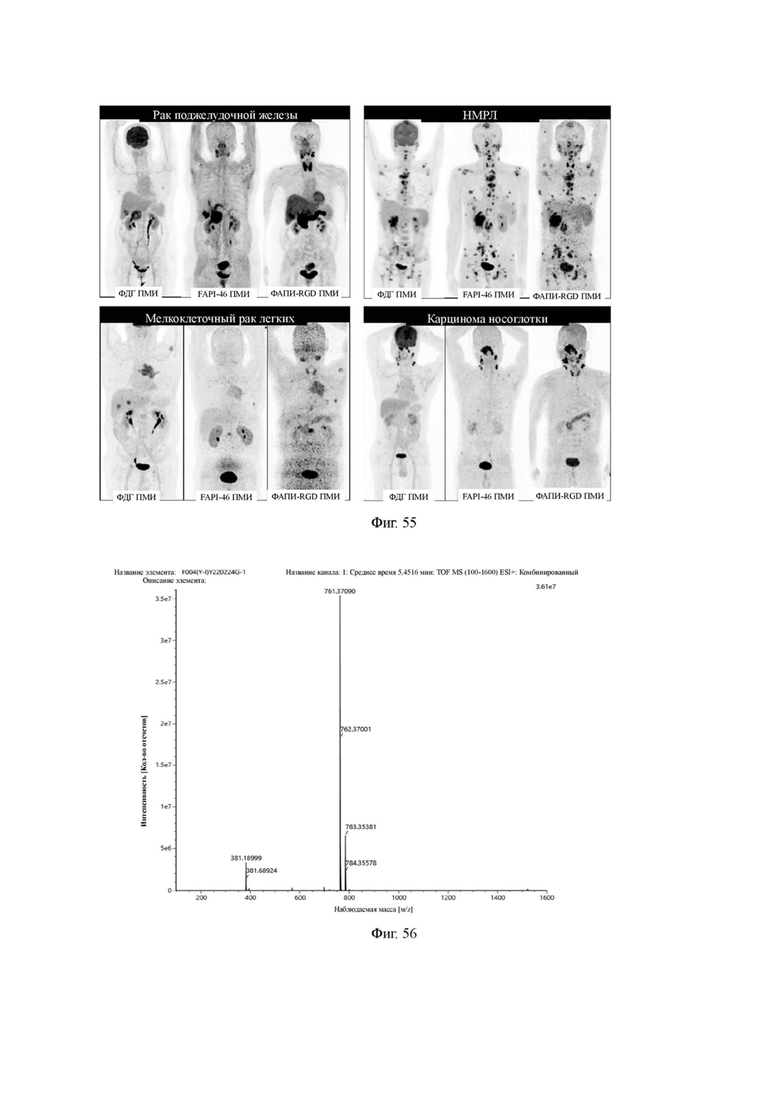
Изобретение относится к соединению двойного нацеливания, способному воздействовать на белок активации фибробластов (FAP) и интегрин αvβ3. Также предложено меченное радионуклидом соединение двойного нацеливания на основе целевого соединения, способ его получения и применение в диагностике или лечении заболеваний, характеризующихся повышенной экспрессией белка активации фибробластов (FAP) и/или интегрина αvβ3. Соединение по настоящему изобретению и его радионуклидный маркер могут синергически воздействовать на мишень FAP и мишень интегрина αvβ3 в опухолях. 7 н. и 3 з.п. ф-лы, 60 ил., 2 табл., 138 пр.
1. Соединение двойного нацеливания, в котором соединение двойного нацеливания содержит специфическую связывающую лигандную структуру белка активации фибробластов (FAP) и интегрина αvβ3, а соединение двойного нацеливания имеет структурную формулу, показанную в формуле (I) или формуле (II):
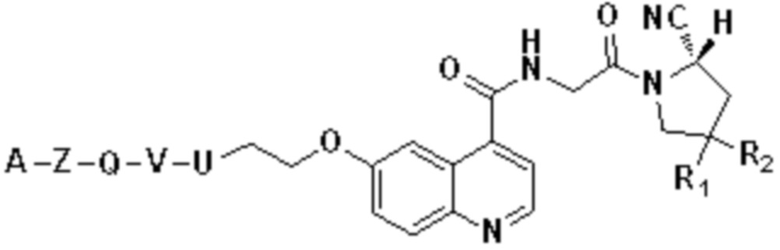 (I) или
(I) или 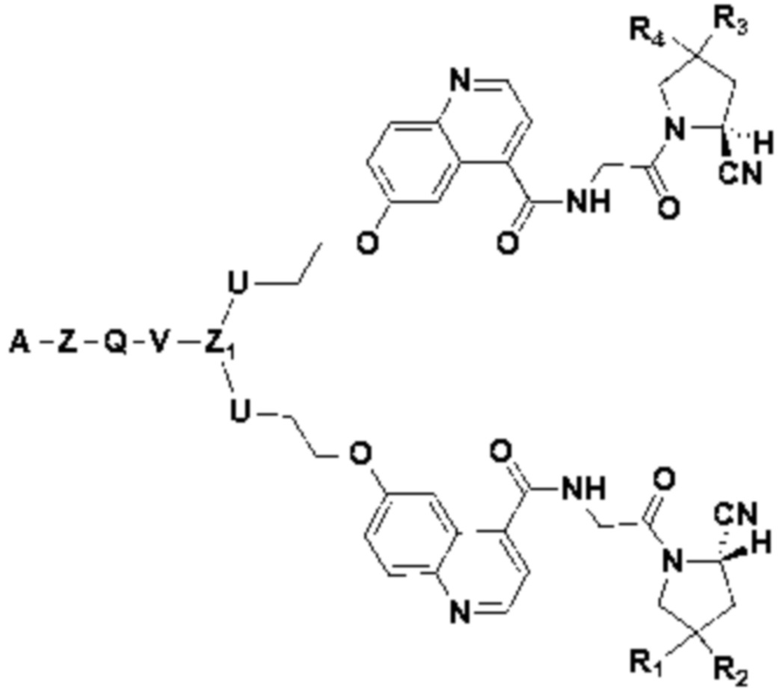 (II)
(II)
где Z в формуле (I) или формуле (II) представляет собой -NH-CH2-(CH2-O-CH2)2-CH2-(CO) или -(CH2)0-;
Q в формуле (I) или формуле (II) представляет собой 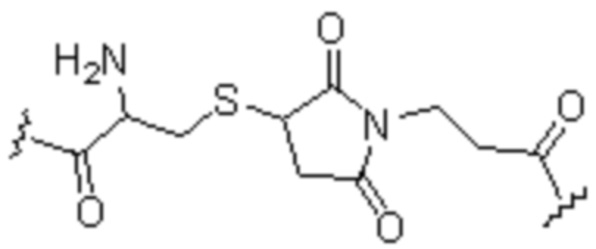 или
или  ;
;
V в формуле (I) или формуле (II) представляет собой -NH-CH2-(CH2-O-CH2)2-CH2-(CO) или -(CH2)0-;
U в формуле (I) или формуле (II) представляет собой  ;
;
Z1 в формуле (II) представляет собой  ;
;
А представляет собой лигандную структуру, специфически связанную с интегрином αvβ3, причем эта структура показана в формуле (III) или формуле (IV):
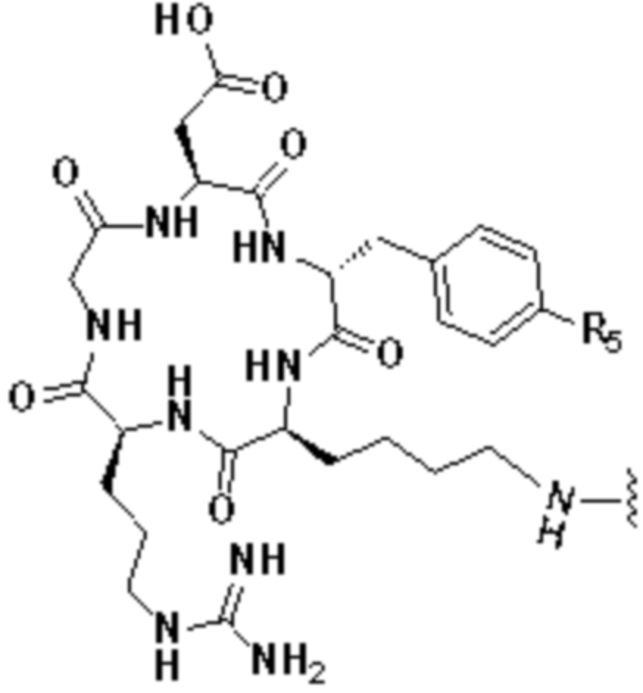 (III) или
(III) или 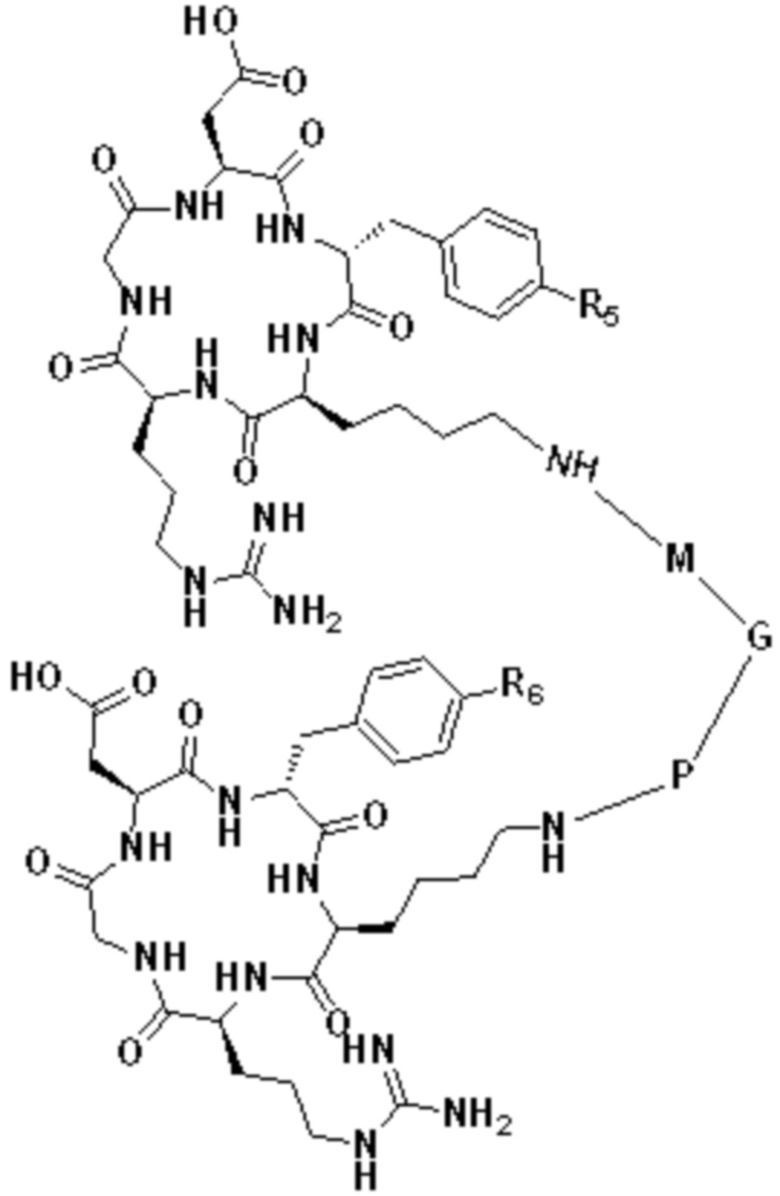 (IV),
(IV),
G в формуле (IV) представляет собой ;
M в формуле (IV) представляет собой -NH-CH2-(CH2-O-CH2)4-CH2-(CO)-; и
P в формуле (IV) представляет собой -NH-CH2-(CH2-O-CH2)4-CH2-(CO)-.
2. Соединение двойного нацеливания, которое можно метить радионуклидом, в котором соединение двойного нацеливания, которое можно метить радионуклидом, образуется путем соединения аминогруппы в любой из структур Z, Q или V формулы (I) или формулы (II) по п. 1 с нуклидной хелатирующей группой и имеет общую формулу, показанную в формуле (V) или формуле (VI) ниже:
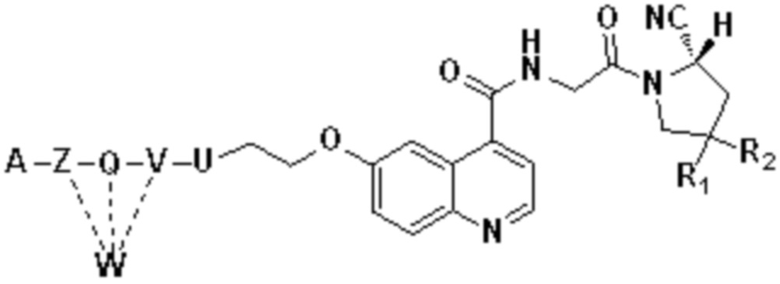 (V),
(V), 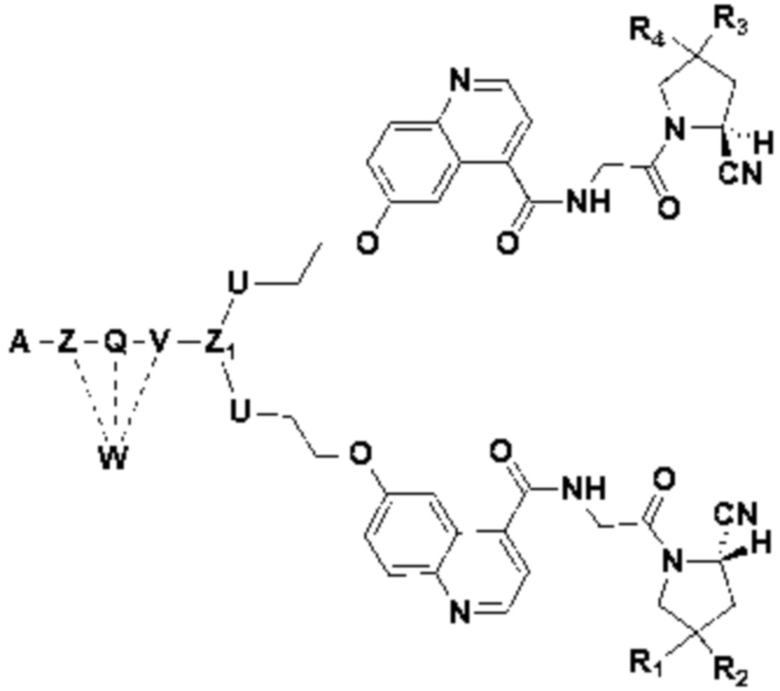 (VI)
(VI)
где W представляет собой фрагмент с нуклидной хелатной группой и получен из 1,4,7,10-тетраазациклододекан-N,N',N,N'-тетрауксусной кислоты (DOTA) или 1,4,7-триазациклононан-1,4,7-триуксусной кислоты (NOTA); или W представляет собой 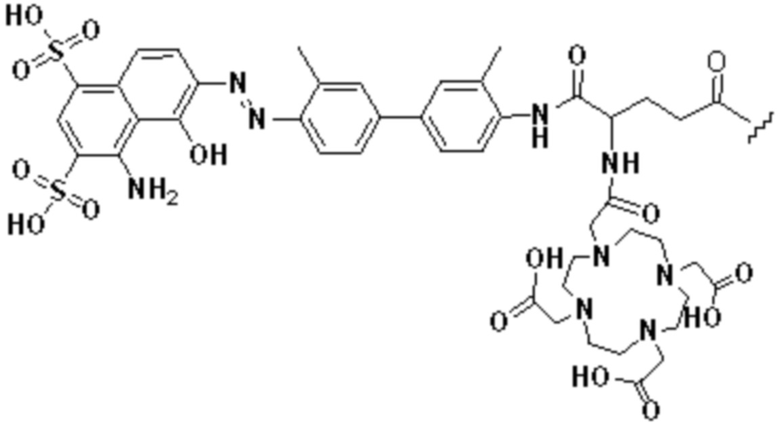 ,
,
при этом А представляет собой лигандную структуру, специфически связанную с интегрином αvβ3, причем эта структура показана в формуле (III) или формуле (IV):
(III) или (IV),
G в формуле (IV) представляет собой ;
M в формуле (IV) представляет собой -NH-CH2-(CH2-O-CH2)4-CH2-(CO)-;
P в формуле (IV) представляет собой -NH-CH2-(CH2-O-CH2)4-CH2-(CO)-;
Z в формуле (V) или формуле (VI) представляет собой -NH-CH2-(CH2-O-CH2)2-CH2-(CO) or -(CH2)0-;
Z1 в формуле (VI) представляет собой ;
Q в формуле (V) или формуле (VI) представляет собой или; и
V в формуле (V) или формуле (VI) представляет собой -NH-CH2-(CH2-O-CH2)2-CH2-(CO) или -(CH2)0-;
U в формуле (V) или формуле (VI) представляет собой .
3. Соединение двойного нацеливания по п. 2, где соединение формулы (V) имеет любую из следующих структур, как показано в формулах (V-1)-(V-14), (V-16)-(V-23) и (V-25)-(V-40):
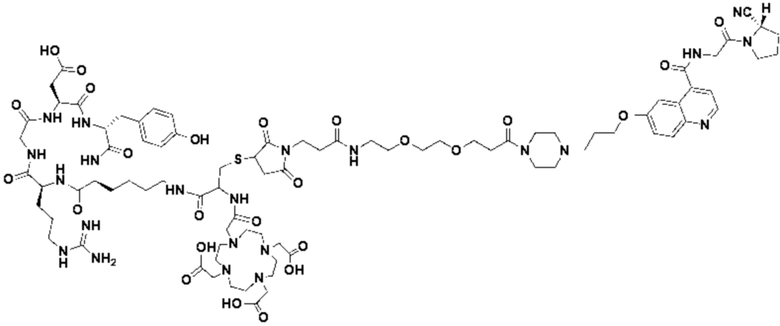
Формула (V-1),
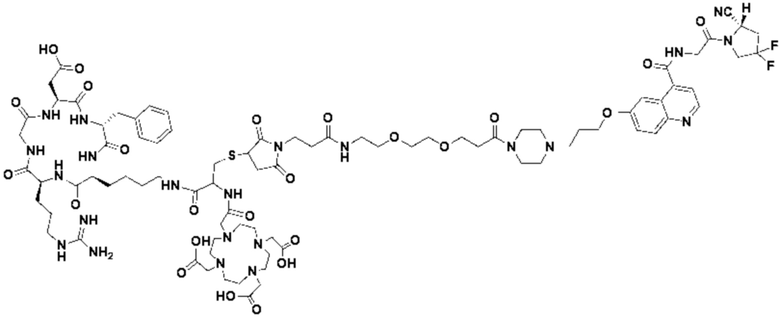
Формула (V-2),
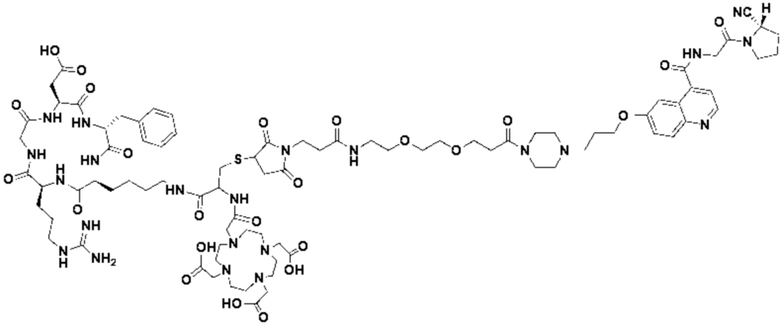
Формула (V-3),
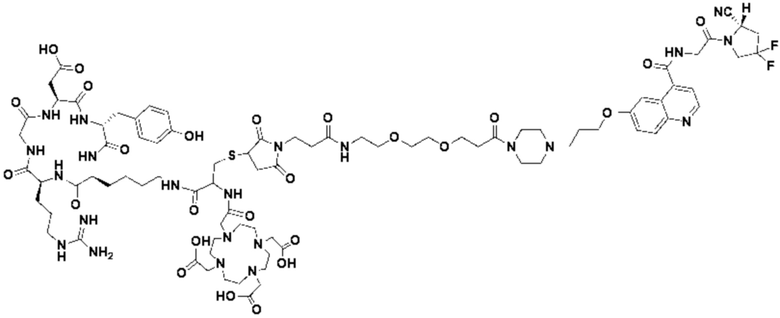
Формула (V-4),
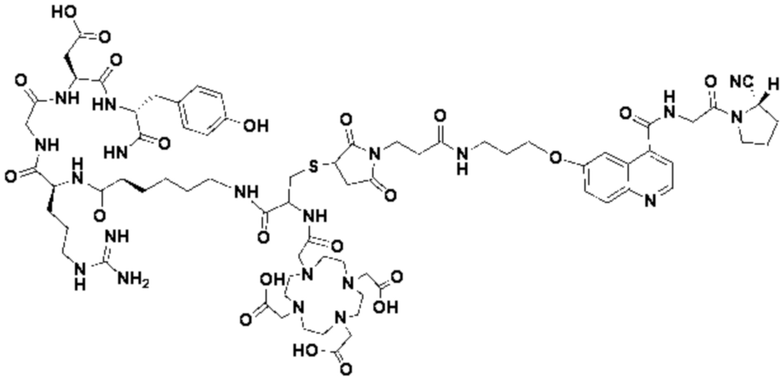
Формула (V-5),
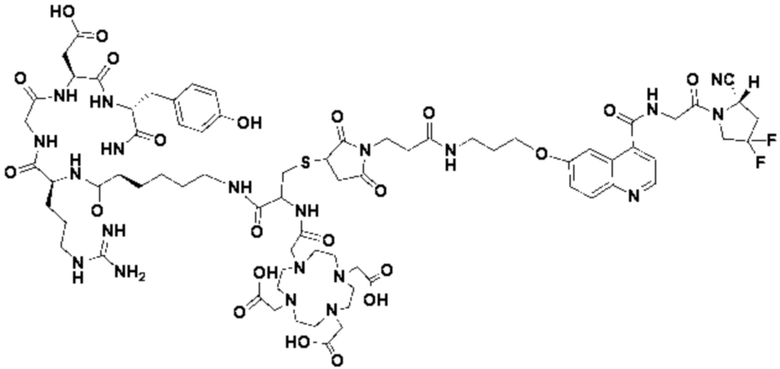
Формула (V-6),
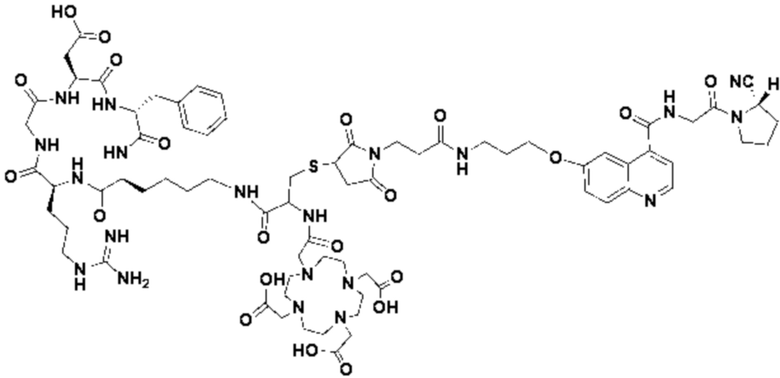
Формула (V-7),
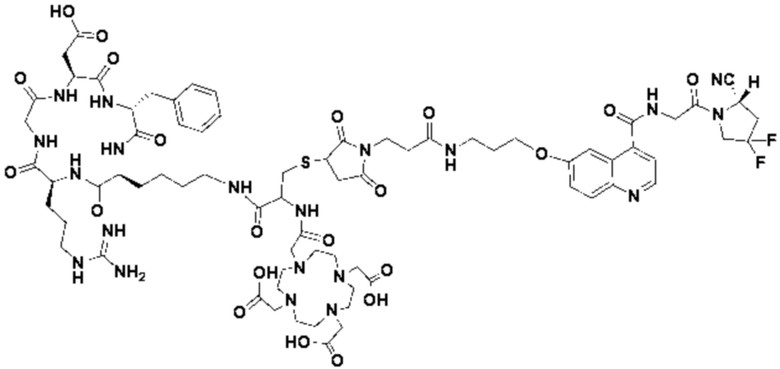
Формула (V-8),
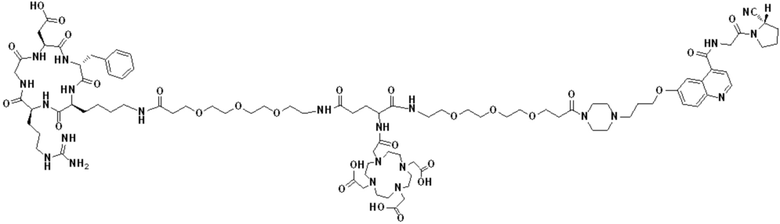
Формула (V-9),
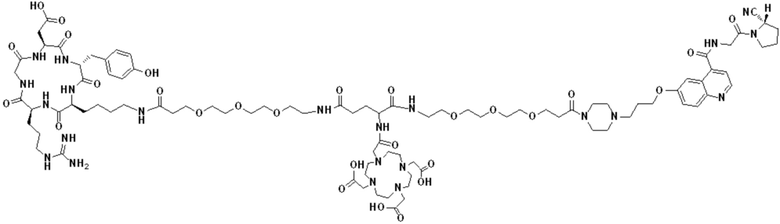
Формула (V-10),
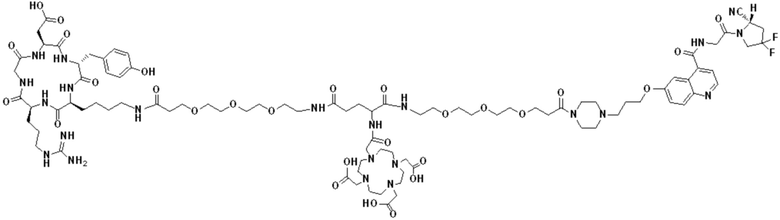
Формула (V-11),
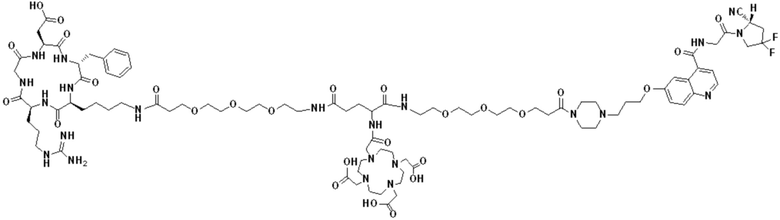
Формула (V-12),
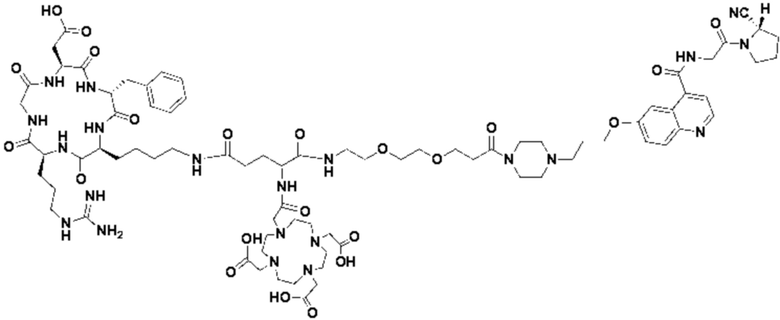
Формула (V-13),
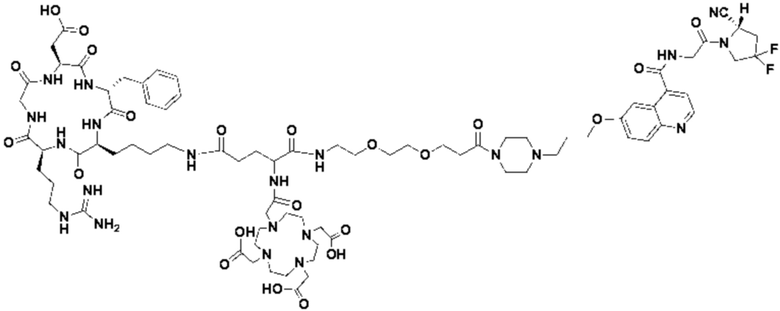
Формула (V-14),
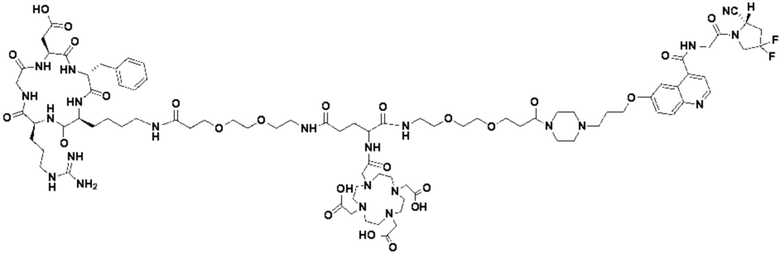
Формула (V-16),
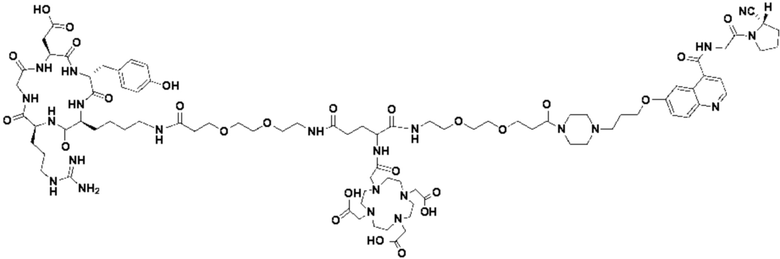
Формула (V-17),
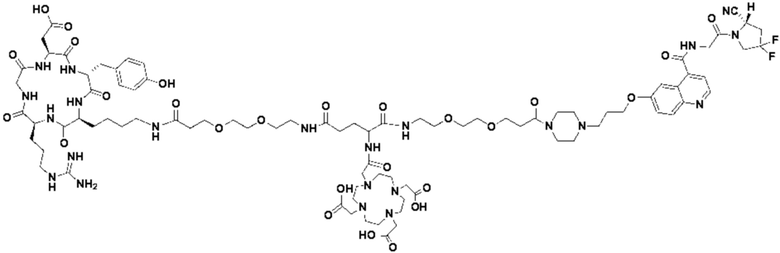
Формула (V-18),
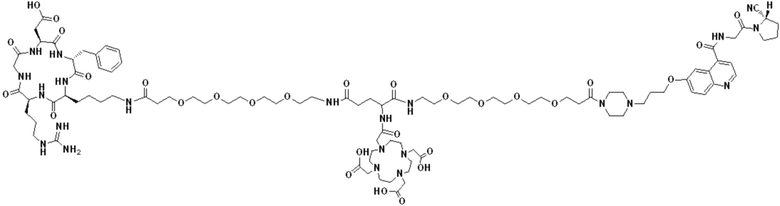
Формула (V-19),
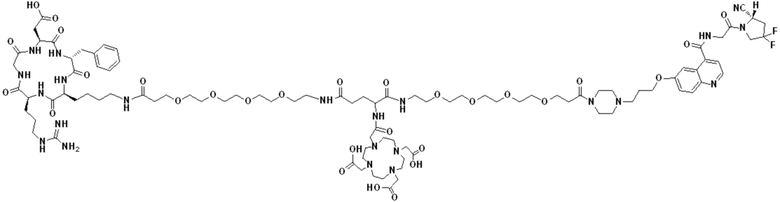
Формула (V-20),
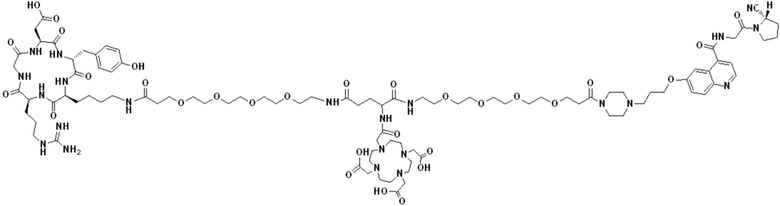
Формула (V-21),
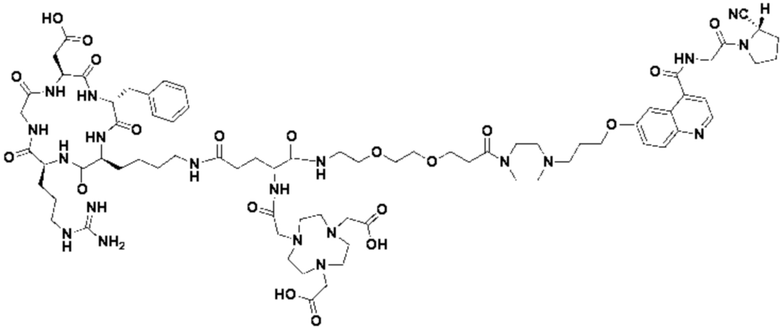
Формула (V-22),
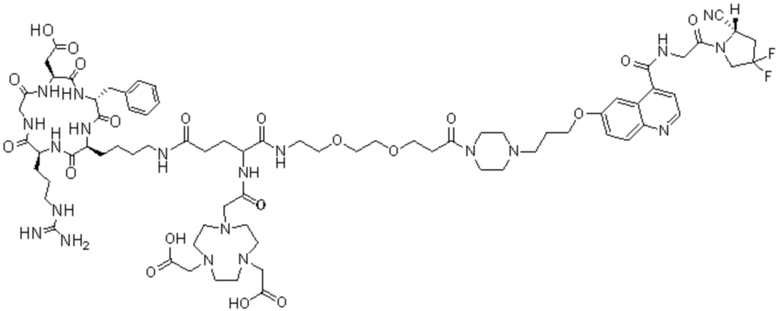
Формула (V-23),
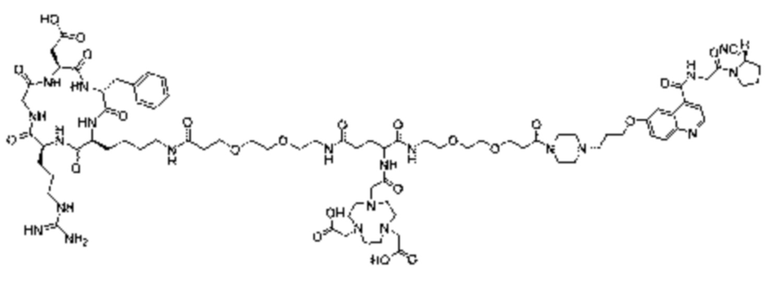
Формула (V-25),
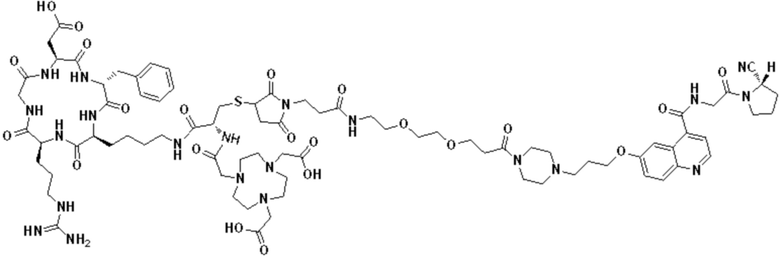
Формула (V-26),
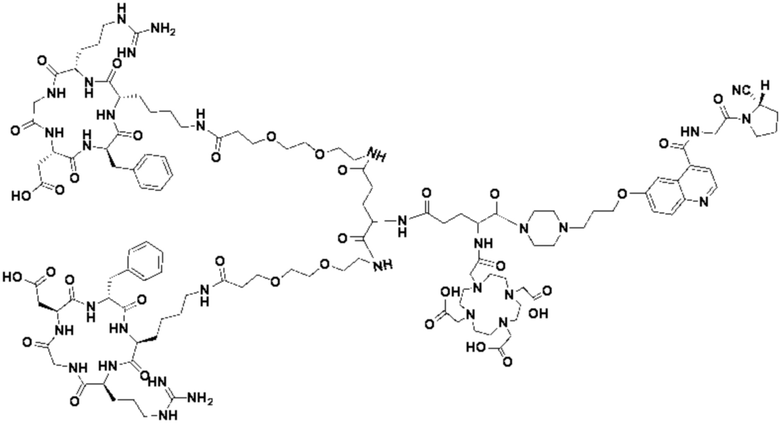
Формула (V-27),
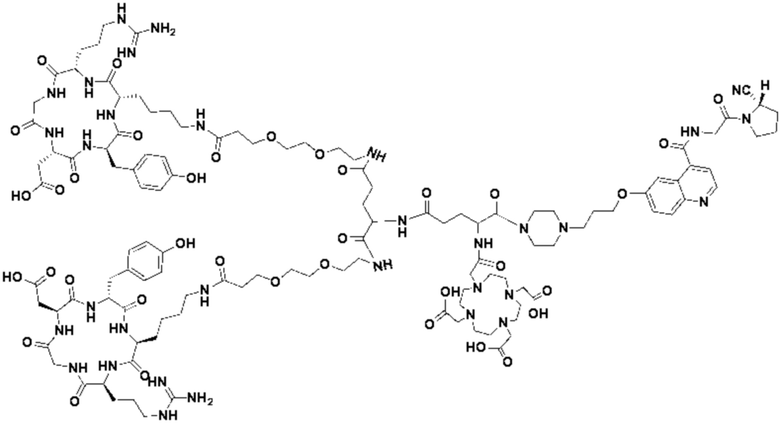
Формула (V-28),
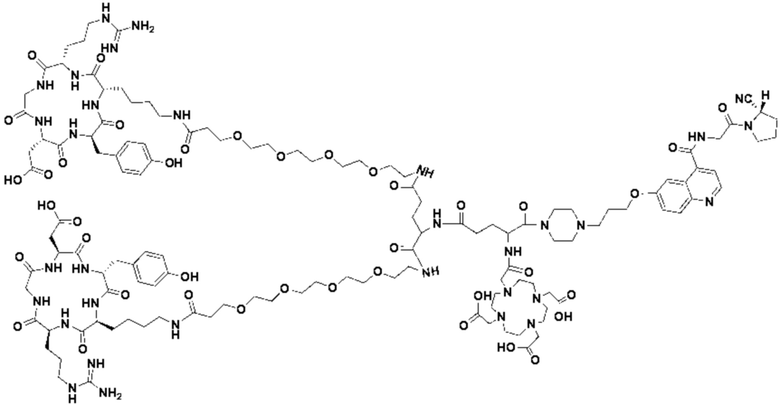
Формула (V-29),
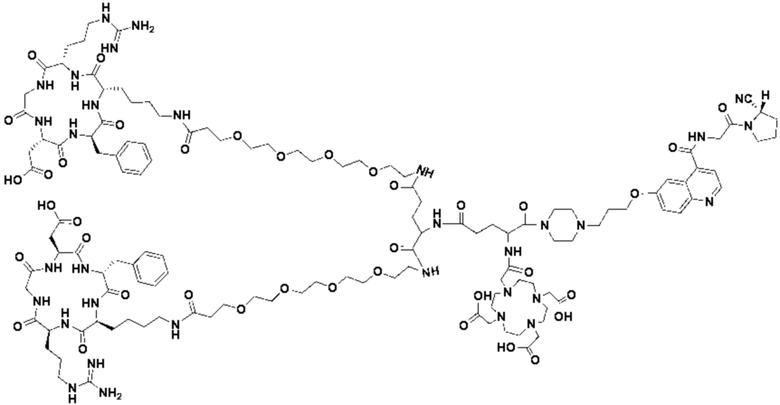
Формула (V-30),
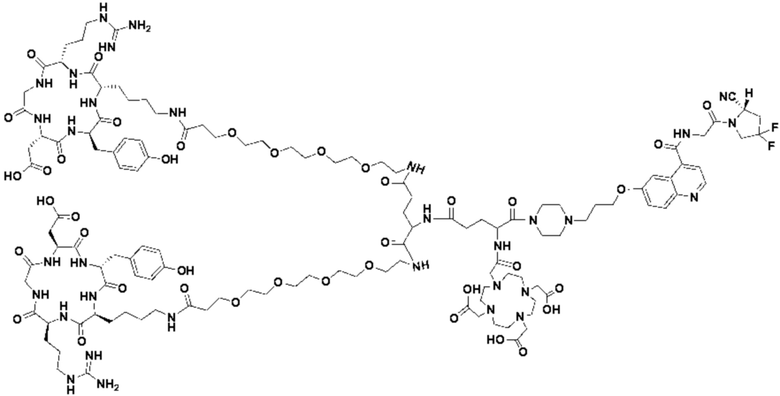
Формула (V-31),
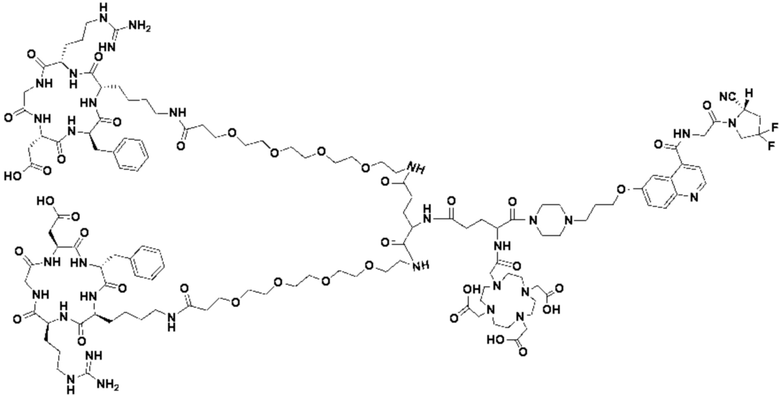
Формула (V-32),
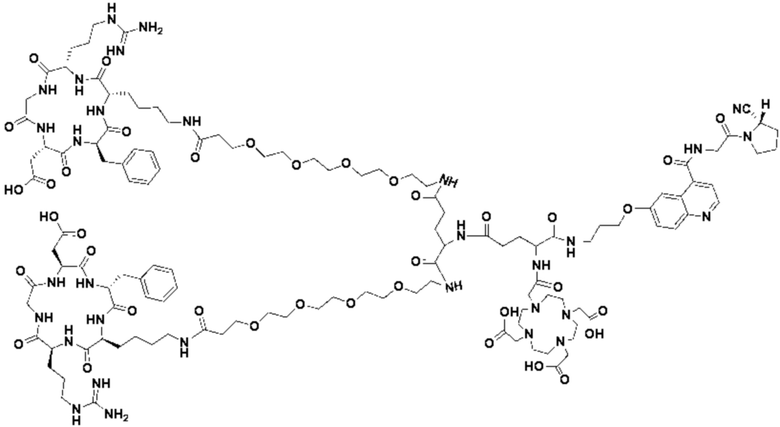
Формула (V-33),
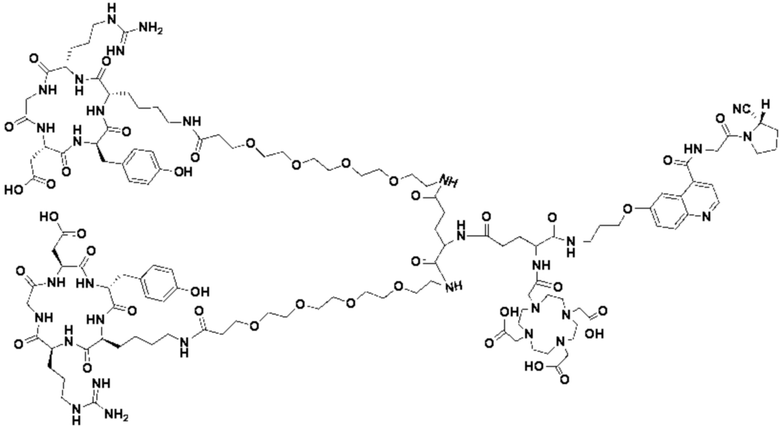
Формула (V-34),
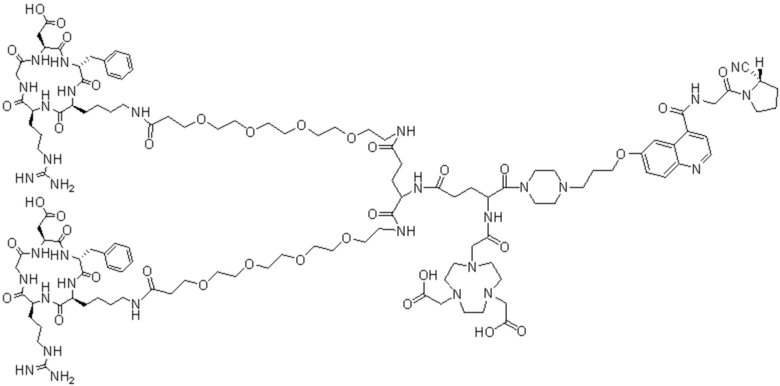
Формула (V-35),
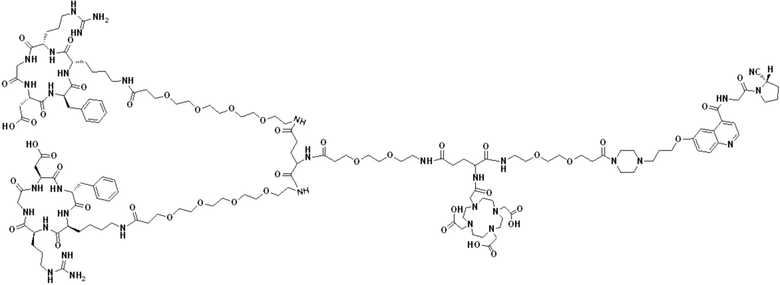
Формула (V-36),
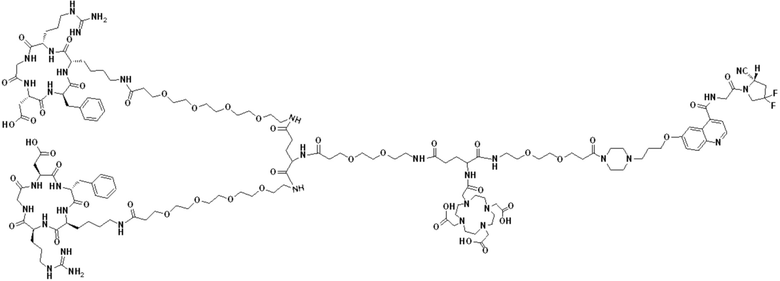
Формула (V-37),
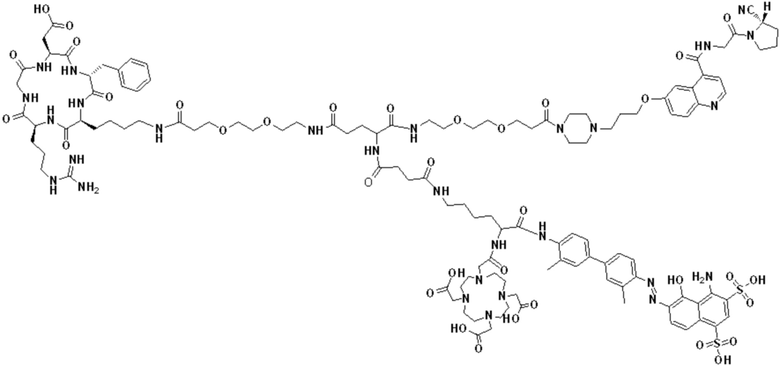
Формула (V-38),
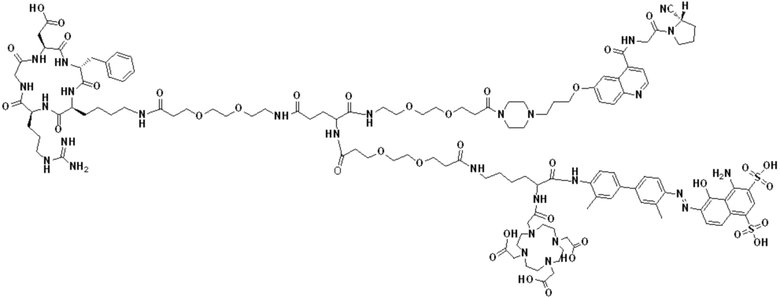
Формула (V-39) или
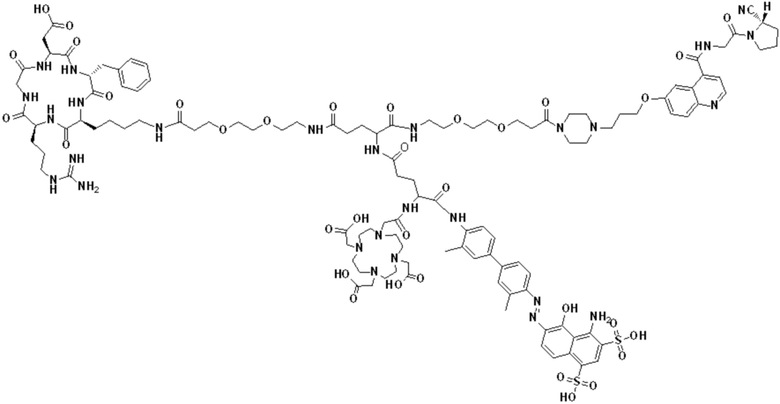
Формула (V-40).
4. Соединение двойного нацеливания по п. 2, где соединение формулы (VI) имеет любую из следующих структур, как показано в формулах (VI-1)-(VI-8):
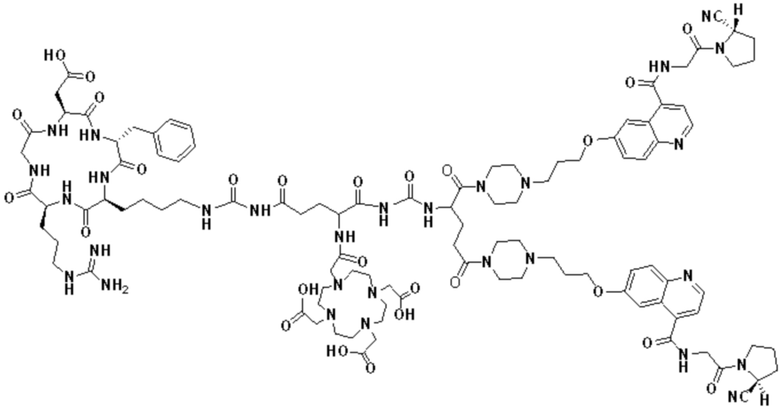
Формула (VI-1),
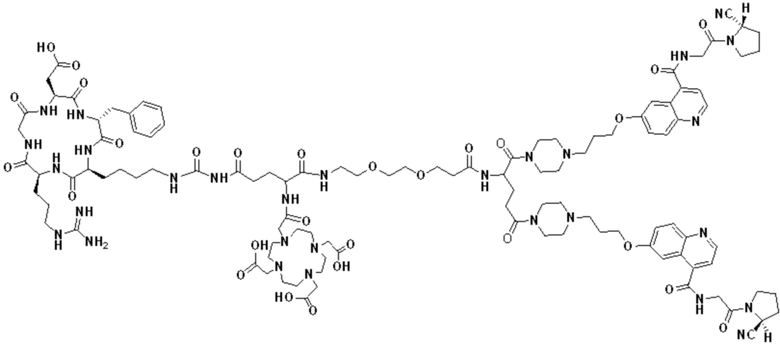
Формула (VI-2),
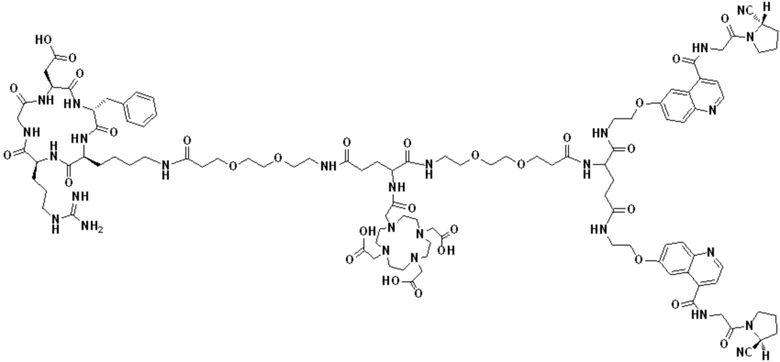
Формула (VI-3),
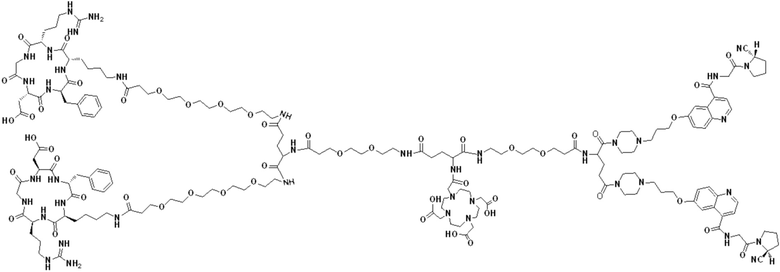
Формула (VI-4),
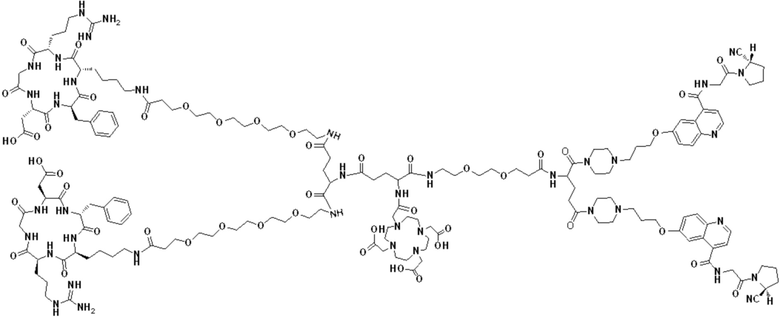 Формула (VI-5),
Формула (VI-5),
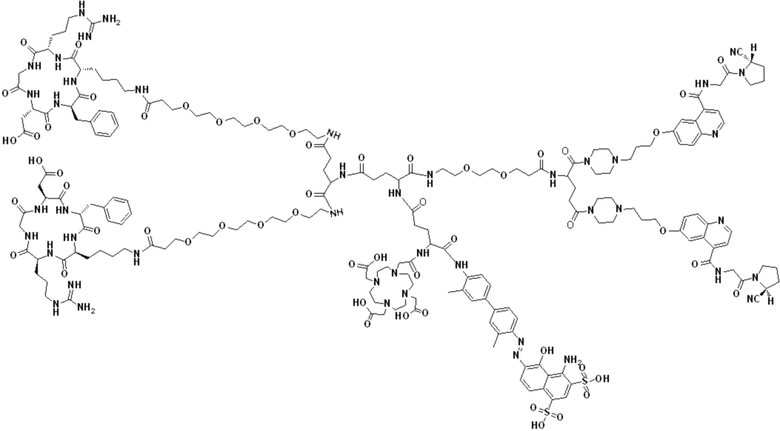
Формула (VI-6),
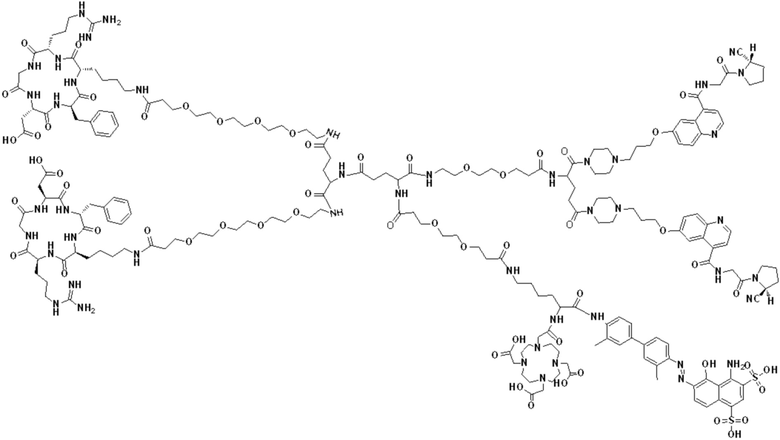
Формула (VI-7) или
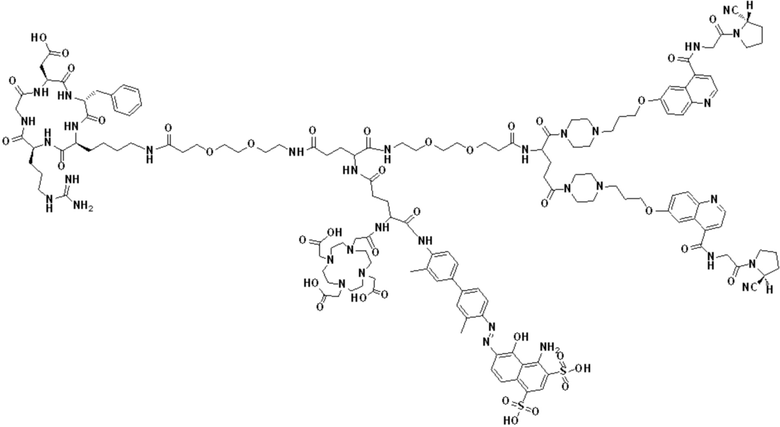
Формула (VI-8).
5. Меченное радионуклидом соединение двойного нацеливания, представляющее собой соединение двойного нацеливания по п. 2, содержащее радионуклид, где радионуклид выбран из изотопов, испускающих α-лучи, изотопов, испускающих β-лучи, изотопов, испускающих γ-лучи, изотопов, испускающих оже-электроны, или изотопов, испускающих рентгеновские лучи.
6. Способ получения соединения двойного нацеливания, которое можно метить радионуклидом, представляющего собой соединение формулы (V) по п. 2 или 3, включающий в себя следующие этапы: сначала проводят реакцию амидной конденсации между карбоксилом 6-гидроксихинолин-4-карбоновой кислоты и амино-трет-бутиловым эфиром глицина; затем соединение Boc-защищенного пиперазинила с помощью алкильной цепи в гидроксильном положении продукта конденсации амида; удаление Boc- и трет-бутильных защитных групп в кислых условиях, а затем введение Boc-защитной группы в пиперазиновое кольцо; после чего проводят реакцию конденсации амида с гидрохлоридом (S)-пирролидин-2-карбонитрила или гидрохлоридом (S)-4,4-дифторпирролидин-2-карбонитрила; после удаления Boc-защитной группы проводят реакцию конденсации с N-Boc-3-[2-(2-аминоэтокси)этокси]пропионовой кислотой; затем проводят удаление Boc-защитной группы и последовательное проведение реакции с пропионатом малеимида и цистеином с защитной группой либо затем проводят реакцию с глутаминовой кислотой или лизином с защитной группой; затем вводят c(RGDyK), c(RGDfK) или c(RGDyK)/c(RGDfK) с короткой цепью PEG посредством реакции активированного сложного эфира с получением соединения двойного нацеливания; и, наконец, проводят реакцию между соединением двойного нацеливания и нуклидным хелатирующим агентом с получением соединения двойного нацеливания, которое можно метить радионуклидом.
7. Способ получения меченного радионуклидами соединения двойного нацеливания по п. 5, включающий в себя следующие этапы: сначала проводят реакцию амидной конденсации между карбоксилом 6-гидроксихинолин-4-карбоновой кислоты и амино-трет-бутиловым эфиром глицина; затем соединение Boc-защищенного пиперазинила с помощью алкильной цепи в гидроксильном положении продукта конденсации амида; удаление Boc- и трет-бутильных защитных групп в кислых условиях, а затем введение Boc-защитной группы в пиперазиновое кольцо; после чего проводят реакцию конденсации амида с гидрохлоридом (S)-пирролидин-2-карбонитрила или гидрохлоридом (S)-4,4-дифторпирролидин-2-карбонитрила; после удаления Boc-защитной группы проводят реакцию конденсации с N-Boc-3-[2-(2-аминоэтокси)этокси]пропионовой кислотой; затем проводят удаление Boc-защитной группы и последовательное проведение реакции с пропионатом малеимида и цистеином с защитной группой либо затем проводят реакцию с глутаминовой кислотой или лизином с защитной группой; затем вводят c(RGDyK), c(RGDfK) или c(RGDyK)/c(RGDfK) с короткой цепью PEG посредством реакции активированного сложного эфира с получением соединения двойного нацеливания; затем проводят реакцию между соединением двойного нацеливания и нуклидным хелатирующим агентом с получением соединения двойного нацеливания, которое можно метить радионуклидом; и проведение реакции между соединением, которое можно метить радионуклидом, и соединением, содержащим радионуклид, с помощью существующего способа мокрого мечения или способа лиофилизационной сушки для подготовки и получения меченного радионуклидами целевого соединения.
8. Применение соединения двойного нацеливания по п. 1, соединения двойного нацеливания, которое можно метить радионуклидом, по любому из пп. 2-4 или меченного радионуклидами соединения двойного нацеливания по п. 5 при получении лекарственных средств для диагностики или лечения заболеваний, характеризующихся сверхэкспрессией белка активации фибробластов (FAP) и /или интегрина αvβ3 у животных или людей.
9. Применение по п. 8, отличающееся тем, что заболевания, характеризующиеся сверхэкспрессией белка активации фибробластов (FAP) и/или интегрина αvβ3, содержат, помимо прочего: рак, хроническое воспаление, атеросклероз, фиброз, ремоделирование тканей и рубцовые заболевания; и предпочтительно рак дополнительно выбирают из рака молочной железы, рака поджелудочной железы, рака тонкой кишки, рака толстой кишки, рака прямой кишки, рака легких, рака головы и шеи, рака яичников, гепатоцеллюлярной карциномы, рака пищевода, карциномы гортаноглотки, карциномы носоглотки, карциномы гортани, клеток миеломы, рака мочевого пузыря, холангиоцеллюлярной карциномы, светлоклеточного почечноклеточного рака, нейроэндокринного новообразования, канцерогенной остеомаляции, саркомы, рака неизвестной первичной формы (CUP), карциномы тимуса, глиомы, нейроглиомы, астроцитомы, рака шейки матки или рака простаты.
10. Набор для диагностики заболевания, характеризующегося сверхэкспрессией белка активации фибробластов (FAP) и/или интегрина αvβ3, содержащий или состоящий из: (1) соединения двойного нацеливания по п. 1, соединения двойного нацеливания, которое можно метить радионуклидом по любому из пп. 2-4, меченное радионуклидами соединение двойного нацеливания по п. 5 или любых фармацевтически приемлемых таутомеров, рацематов, гидратов, сольватов или их солей; и (2) инструкции по диагностике заболеваний.
| CN 114984255 A, 02.09.2022 | |||
| CN 113880810 A, 04.01.2022 | |||
| CN113621021 A, 09.11.2021 | |||
| Liang Zhao et al, "Fibroblast activation protein-based theranostics in cancer research: A state-of-the-art review", Theranostics, Jan 2022, 12(4), pages 1557-1569 | |||
| CN 110420337 A, 08.11.2019 | |||
| RU 2020126278 A, 09.03.2022 | |||
| US 20210369877 A1, 02.12.2021 | |||
| CN 113292538 |

