Область техники
Изобретение относится к области ядерной медицины и молекулярной визуализации, и в частности оно относится к ингибитору белка активации фибробластов, модифицированному усеченным синим Эванса, его получению, мечению и применению.
Уровень техники
Белок активации фибробластов (FAP) представляет собой мембранную сериновую пептидазу, экспрессируемую на поверхности фибробластов, активированных в опухолевой строме, и играющую важную роль в возникновении и развитии опухолей. Более ранние исследования показали, что FAP, как правило, не экспрессируется в нормальных тканях человека, однако избирательно сверхэкспрессируется на поверхности стромальных фибробластов более чем 90% эпителиальных злокачественных опухолей, включая рак молочной железы, рак яичников, рак легкого, колоректальный рак, рак желудка и рак поджелудочной железы. Учитывая широкое распространение FAP и его важную роль в опухолях, он стал важной мишенью для визуализации и лечения опухолей.
На сегодняшний день значительный прогресс в области точной визуализации опухолей был достигнут благодаря радиоактивно меченным ингибиторам белка активации фибробластов (FAPI), представленным производными хинолиновой кислоты. Например, с помощью средств визуализации PET/CT, например FAPI-02 и FAPI-04, удалось добиться специфической визуализации более 30 различных типов опухолей. По сравнению с визуализацией FDG, визуализация FAPI имеет более низкий фон в головном мозге, печени и слизистой оболочке ротоглотки и обладает более высоким уровнем обнаружения опухолевых поражений. FAPI, о котором сообщается в настоящее время, быстро выводится из кровотока и в то же время быстро элюируется из места опухоли. Эта метаболическая характеристика полезна для визуализации, поскольку она может обеспечить более чистый фон. Однако для лечения это чрезвычайно неблагоприятно, поскольку быстрые метаболизм и элюирование приводят к снижению эффективных доз и более короткому времени удерживания в месте опухоли, что требует использования высоких доз или более частого введения для удовлетворения потребностей в лечении и увеличивает вероятность побочных реакций.
Если взять в качестве примера FAPI-02, то он полностью выводится из кровотока в течение одного часа, а через 24 часа удерживаемая в месте опухоли доза снижается приблизительно на 75%. Хотя недавние исследования оптимизировали нефармакофорную часть структуры FAPI, улучшение дозы поглощения опухолью и времени удерживания FAPI очень ограничено и не может удовлетворить потребности терапевтических целей. Специалисты в данной области знают, что если низкомолекулярное лекарственное средство циркулирует в кровеносных сосудах слишком короткое время или быстро выводится из организма, то это приведет к недостаточному связыванию лекарственного средства с мишенью. Следовательно, при подготовке зондов FAPI можно увеличить дозу поглощения и время удерживания зонда в целевом участке, если период полураспада зонда можно соответствующим образом продлить.
Следовательно, необходимы новые стратегии для продления периода полураспада зондов FAPI, что позволит им иметь соответствующую метаболическую кинетику, более высокую дозу поглощения опухолью и более длительное время удерживания в опухоли для удовлетворения потребностей радионуклидной терапии и визуализации.
Сущность изобретения
На основании вышеизложенного основной задачей настоящего изобретения является разработка конъюгата на основе усеченного синего Эванса (tEB) и ингибитора белка активации фибробластов (FAPI), характеризующегося эффективным связыванием усеченного синего Эванса с сывороточным альбумином, что позволяет использовать альбумин в качестве носителя для доставки FAPI, тем самым продлевая период его полураспада в периферической крови и улучшая поглощение, накопление и время удерживания в опухолях. Конъюгат tEB-FAPI, разработанный согласно настоящему изобретению, может преодолевать недостатки быстрого метаболизма низкомолекулярного FAPI и короткого времени удерживания в органах-мишенях, улучшать терапевтические и визуализирующие эффекты целевого белкового нуклида FAP и имеет потенциал для клинического внедрения и применения.
Другой целью настоящего изобретения является обеспечение радиоактивно меченного ингибитора белка активации фибробластов, модифицированного усеченным синим Эванса (tEB-FAPI), с длительным периодом полураспада;
еще одной целью настоящего изобретения является обеспечение способа получения радиоактивно меченного комплекса tEB-FAPI;
еще одной целью настоящего изобретения является обеспечение применения комплекса в радионуклидной визуализации и лечении опухолей с нацеливанием на белок FAP.
Технические решения для реализации вышеупомянутых основных целей настоящего изобретения имеют следующие два аспекта: синтез лиганда и радиоактивное мечение.
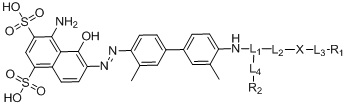
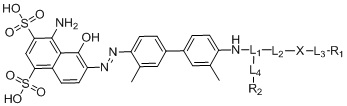
В первом аспекте согласно настоящему изобретению предложен ингибитор белка активации фибробластов (FAPI), модифицированный усеченным синим Эванса (tEB), при этом структура соединения представлена следующей формулой (I) и обозначена как «tEB-FAPI»,
 формула (I),
формула (I),
где L1 представляет собой структуру на основе лизина или глутаминовой кислоты или структуру на основе производного соединения, предусматривающую структуру на основе лизина или глутаминовой кислоты;
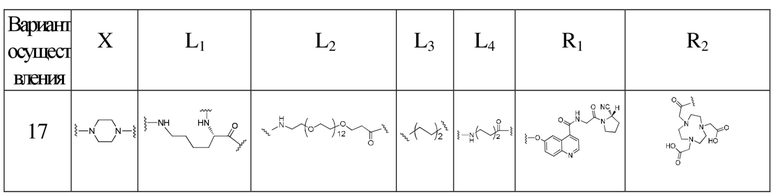
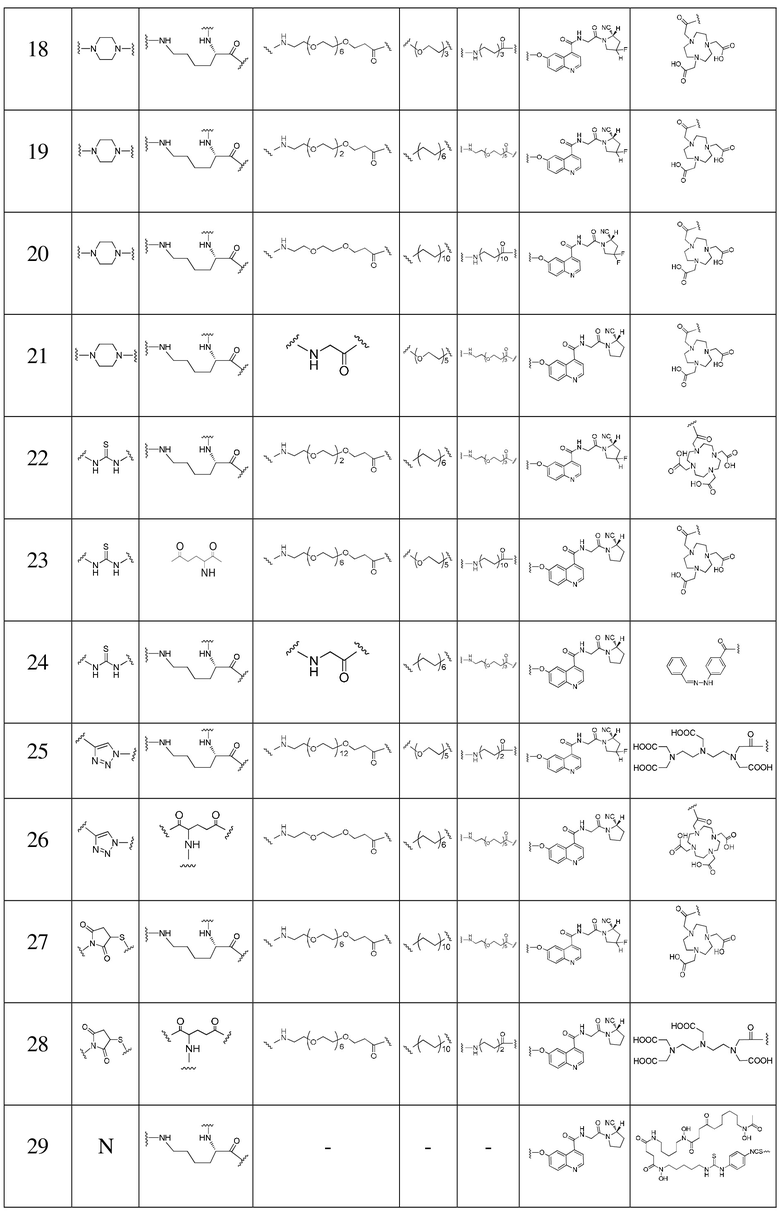
L2 представляет собой -(CH2)n-, где n представляет собой целое число от 0 до 30, где каждая CH2 отдельно может быть замещена с помощью -O-, -NH-, -(CO)-, -NH(CO)- или -(CO)-NH- или не замещена, при условии, что отсутствует замещение двух смежных групп CH2;
L3 представляет собой -(CH2)m-, где m представляет собой целое число от 0 до 30, где каждая CH2 отдельно может быть замещена с помощью -O- или -(CO)- или не замещена, при условии, что отсутствует замещение двух смежных групп CH2;
L4 представляет собой -(CH2)p-, где p представляет собой целое число от 0 до 30, где каждая CH2 отдельно может быть замещена с помощью -O-, -NH-, -(CO)-, -NH(CO)- или -(CO)-NH- или не замещена, при условии, что отсутствует замещение двух смежных групп CH2;
X выбран из N, C, O, S или любой из следующих структур:
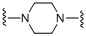 ,
,  ,
,  ,
, 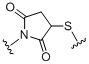 ;
;
R1 представляет собой следующую структуру ингибитора белка активации фибробластов:
 ;
;
R2 представляет собой нуклидную хелатирующую группу, выбранную из любой из следующих структур:
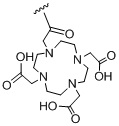 ,
, 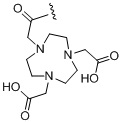 ,
, 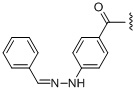 ,
, 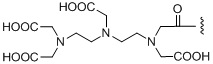 ,
,  ;
;
R3 - R4 являются одинаковыми или разными, при этом все из них независимо выбраны из H или F.
В предпочтительном варианте осуществления настоящего изобретения L2 в формуле (I) представляет собой -(CH2)n-; n представляет собой целое число от 0 до 16, более предпочтительно целое число от 0 до 12, еще более предпочтительно 0, 3 или 10; где каждая -CH2- отдельно может быть замещена с помощью -O-, -NH- или -(CO)- или не замещена, при условии, что отсутствует замещение двух смежных групп -CH2-.
В предпочтительном варианте осуществления настоящего изобретения L3 в формуле (I) представляет собой -(CH2)m-; m представляет собой целое число от 0 до 20, более предпочтительно целое число от 1 до 6, еще более предпочтительно 2 или 3; где каждая -CH2- отдельно может быть замещена с помощью -O- или не замещена, при условии, что отсутствует замещение двух смежных групп -CH2-.
В предпочтительном варианте осуществления настоящего изобретения L4 в формуле (I) представляет собой -(CH2)p-; p представляет собой целое число от 0 до 20, более предпочтительно целое число от 0 до 12, еще более предпочтительно 3, 4, 9 или 12, наиболее предпочтительно 3; где каждая -CH2- отдельно может быть замещена с помощью -O-, -NH-, -(CO)-, -NH(CO)- или -(CO)-NH- или не замещена, при условии, что отсутствует замещение двух смежных групп -CH2-.
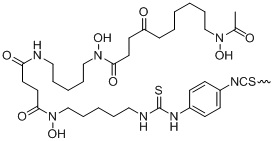
В предпочтительном варианте осуществления настоящего изобретения X в формуле (I) представляет собой , L3 представляет собой -(CH2)3-, L4 представляет собой -(CH2)0-, R2 представляет собой DOTA, то есть tEB-FAPI, как предпочтительный вариант осуществления согласно настоящему изобретению, имеет структуру, представленную следующей формулой II:
 формула (II),
формула (II),
где R3 и R4 одновременно представляют собой атомы H или F, L1 представляет собой структуру на основе глутаминовой кислоты или лизина, L2 представляет собой -(CH2)0, -NH-CH2-(CO)-, -NH-CH2-(CH2OCH2)2-CH2-(CO)-, -NH-CH2-(CH2OCH2)4-CH2(CO)-, -(CO)-CH2-(CO)-, -(CO)-(CH2)2-(CO)-, -(CO)-CH2-(CH2OCH2)2-CH2(CO)- или -(CO)-CH2-(CH2OCH2)4-CH2(CO)-.
В более предпочтительном варианте осуществления настоящего изобретения X в формуле (I) представляет собой , L1 представляет собой структуру на основе глутаминовой кислоты, L2 представляет собой -(CH2)0-, -NH-CH2-(CO)-, -NH-CH2-(CH2OCH2)2-CH2-(CO)- или -NH-CH2-(CH2OCH2)4-CH2(CO)-, L3 представляет собой -(CH2)3-, L4 представляет собой -(CH2)0-, R2 представляет собой DOTA, R3 и R4 одновременно представляют собой атомы H или F.
В другом более предпочтительном варианте осуществления настоящего изобретения X в формуле (I) представляет собой , L1 представляет собой структуру на основе лизина, L2 представляет собой -(CO)-CH2-(CO)-, -(CO)-(CH2)2-(CO)-, -(CO)-CH2-(CH2OCH2)2-CH2(CO)- или -(CO)-CH2-(CH2OCH2)4-CH2(CO)-, L3 представляет собой -(CH2)3-, L4 представляет собой -(CH2)0-, R2 представляет собой DOTA, R3 и R4 одновременно представляют собой атомы H или F.
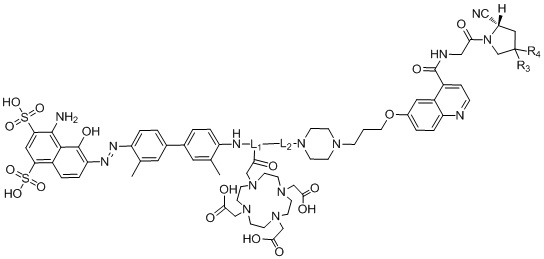
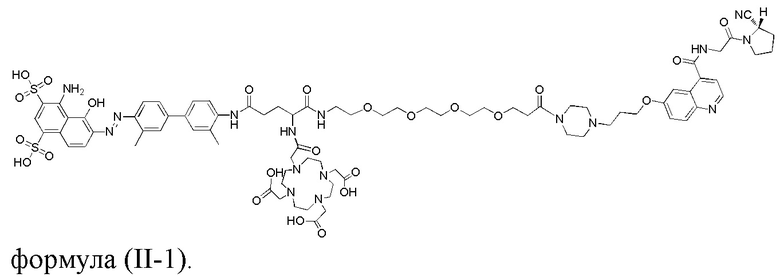
В дополнительном предпочтительном варианте осуществления настоящего изобретения структура соединения tEB-FAPI представлена любой из следующих формул (II-1) - (II-16):
 формула (II-1),
формула (II-1),
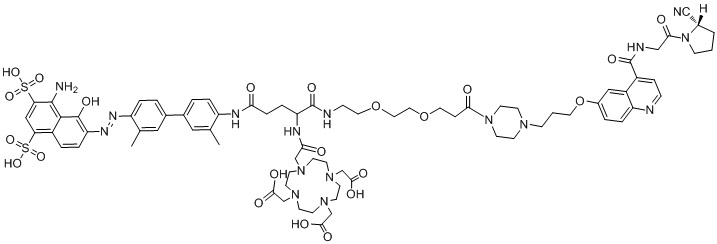 формула (II-2),
формула (II-2),
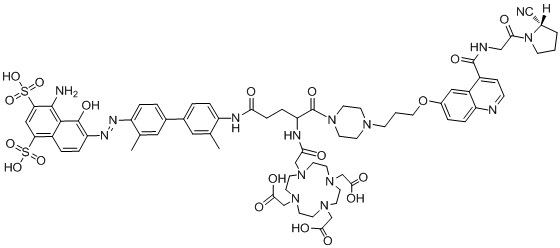 формула (II-3),
формула (II-3),
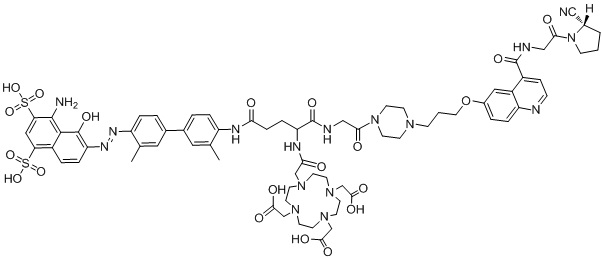 формула (II-4),
формула (II-4),
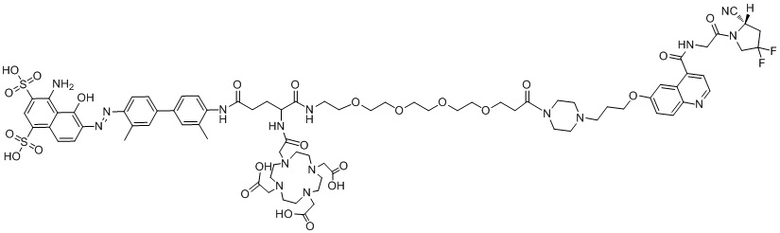 формула (II-5),
формула (II-5),
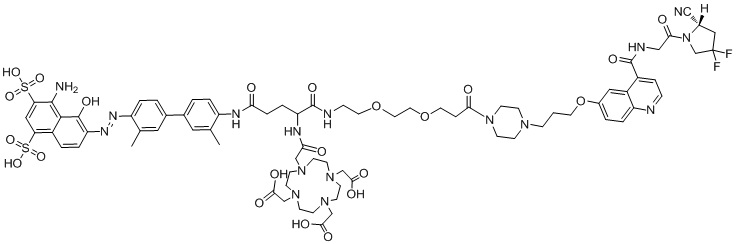 формула (II-6),
формула (II-6),
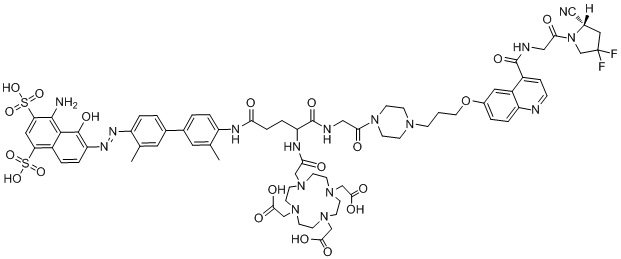 формула (II-7),
формула (II-7),
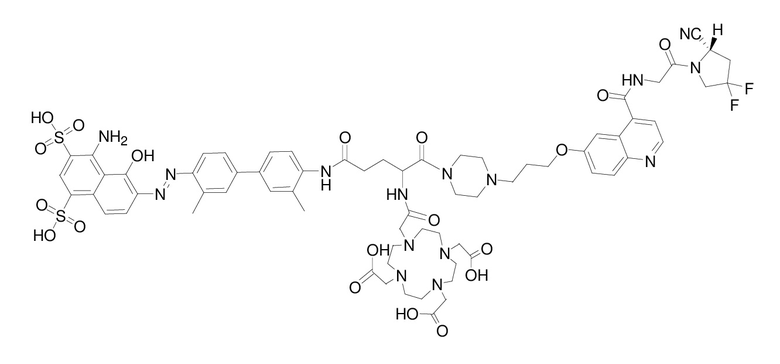 формула (II-8),
формула (II-8),
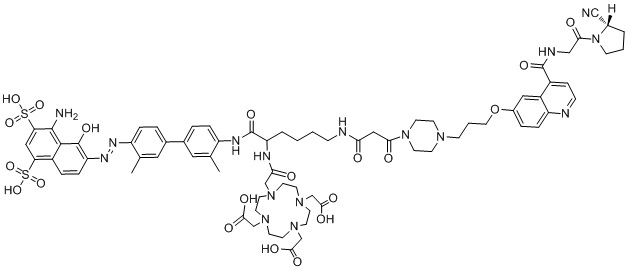 формула (II-9),
формула (II-9),
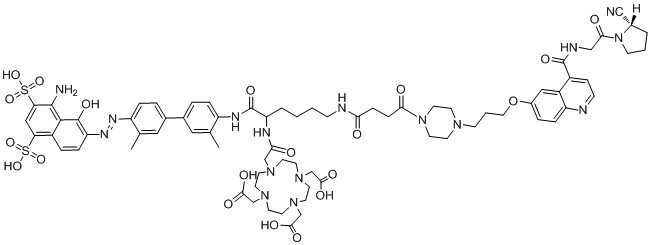 формула (II-10),
формула (II-10),
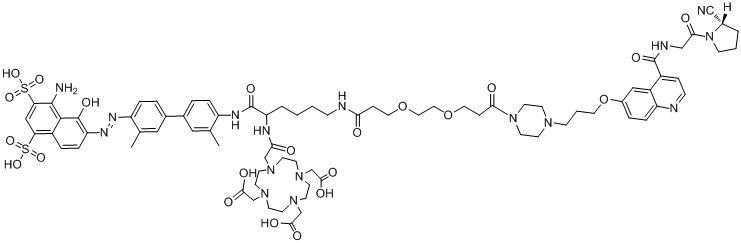 формула (II-11),
формула (II-11),
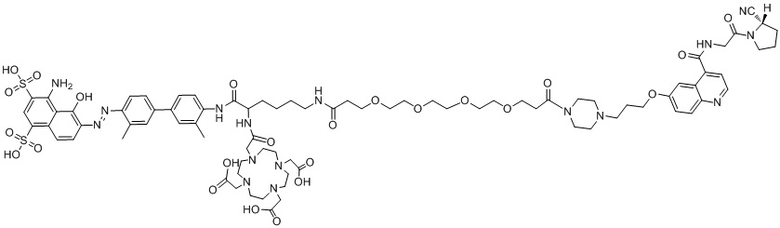 формула (II-12),
формула (II-12),
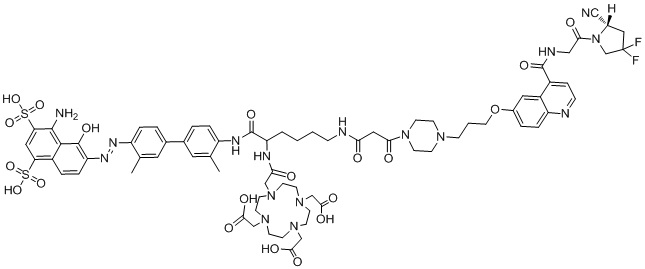 формула (II-13),
формула (II-13),
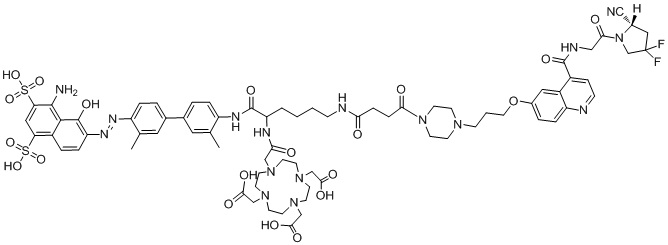 формула (II-14),
формула (II-14),
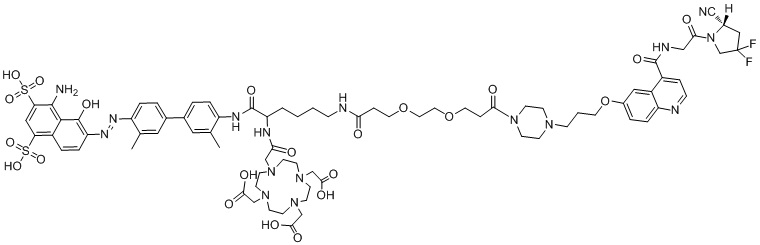 формула (II-15)
формула (II-15)
или
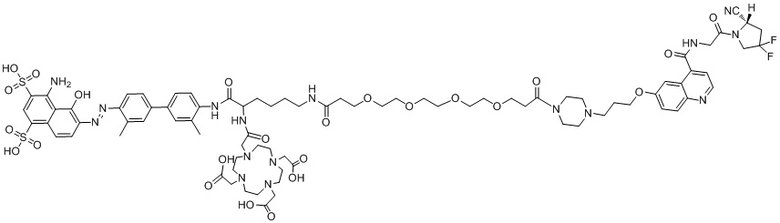 формула (II-16).
формула (II-16).
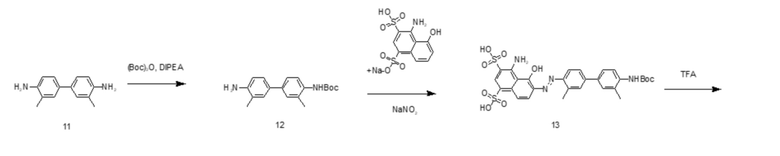
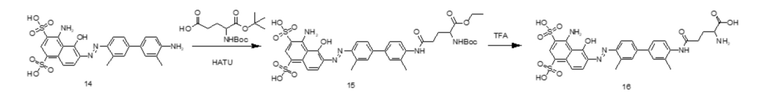
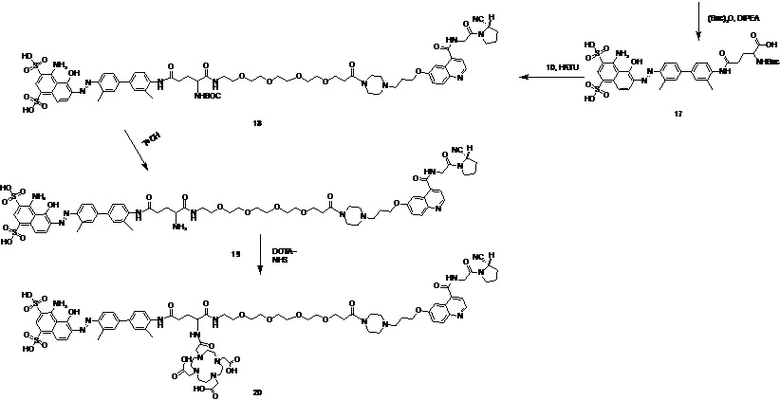
На основании изложенного настоящее изобретение дополнительно предусматривает способ получения соединения tEB-FAPI, представленного формулой (II-1), включающий следующие стадии, на которых:
- проводят реакцию амидной конденсации 6-гидрокси-4-хинолинкарбоновой кислоты и сложного трет-бутилового эфира глицина; затем последовательно вводят в реакцию 1-бром-3-хлорпропан и 1-трет-бутилоксикарбонилпиперазин; затем удаляют защитные Boc- и трет-бутильную группы под воздействием трифторуксусной кислоты (TFA); затем вводят защитную Boc-группу при аминогруппе; затем проводят реакцию амидной конденсации с гидрохлоридом (S)-пирролидин-2-формонитрила; используют п-толуолсульфоновую кислоту для удаления защитной Boc-группы; затем проводят реакцию конденсации со сложным 1-трет-бутиловым эфиром 5,8,11,14-тетраокса-2-азагептадекандиовой кислоты; затем снова удаляют защитную Вос-группу под воздействием п-толуолсульфоновой кислоты с получением промежуточного соединения А;
- в 4,4'-диамино-3,3'-диметилбифенил на один конец вводят защитную Boc-группу, после чего проводят реакцию с мононатриевой солью 1-амино-8-нафтол-2,4-дисульфоновой кислоты с получением производного усеченного синего Эванса; удаляют защитную Вос-группу и затем проводят реакцию амидной конденсации со сложным 1-трет-бутиловым эфиром N-трет-бутилоксикарбонил-L-глутаминовой кислоты; затем удаляют защитные Boc- и трет-бутильную группы под воздействием TFA; затем проводят реакцию с ди-трет-бутилдикарбонатом с введением защитной Boc-группы при аминогруппе с получением промежуточного соединения B;
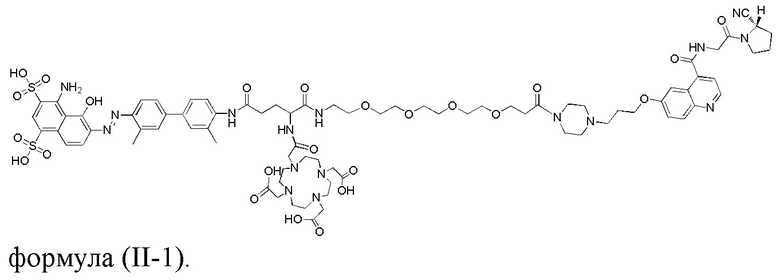
- промежуточное соединение A и промежуточное соединение B подвергают реакции амидной конденсации; затем используют п-толуолсульфоновую кислоту для удаления защитной Boc-группы; наконец, проводят реакцию со сложным эфиром DOTA-NHS, чей номер CAS составляет 170908-81-3 (DOTA-NHS), с получением соединения, представленного формулой (II-1).
Предпочтительный способ получения соединения tEB-FAPI, представленного формулой (II-1), согласно настоящему изобретению, в частности, включает следующие стадии, на которых:
6-гидрокси-4-хинолинкарбоновую кислоту (соединение 1) и сложный трет-бутиловый эфир глицина растворяют в N,N-диметилформамиде, добавляют HATU с получением соединения 2; растворяют соединение 2 в N,N-диметилформамиде, добавляют 1-бром-3-хлорпропан и карбонат калия, в течение некоторого периода времени нагревают реакционную систему до 60°С с получением соединения 3; растворяют соединение 3 в N,N-диметилформамиде, добавляют 1-трет-бутилоксикарбонилпиперазин и йодид калия и обеспечивают протекание реакции с получением соединения 4; соединение 4 растворяют в растворе трифторуксусной кислоты для удаления защитной группы с получением соединения 5; соединение 5 растворяют в N,N-диметилформамиде и добавляют ди-трет-бутилдикарбонат и кислотосвязывающее вещество с получением соединения 6; проводят реакцию конденсации соединения 6 с гидрохлоридом (S)-пирролидин-2-карбонитрила под воздействием HATU и DIPEA с получением соединения 7; у соединения 7 удаляют защитную группу под воздействием п-толуолсульфоновой кислоты с получением соединения 8; проводят реакцию конденсации соединения 8 со сложным 1-трет-бутиловым эфиром 5,8,11,14-тетраокса-2-азагептадекандиовой кислоты под воздействием HATU и DIPEA с получением соединения 9; у соединения 9 удаляют защитную группу под воздействием п-толуолсульфоновой кислоты с получением соединения 10 (промежуточное соединение А);
проводят реакцию 4,4'-диамино-3,3'-диметилбифенила (соединение 11) с ди-трет-бутилдикарбонатом с получением соединения 12; проводят реакцию соединения 12 с мононатриевой солью 1-амино-8-нафтол-2,4-дисульфоновой кислоты и нитритом натрия с получением производного усеченного синего Эванса (соединение 13); у соединения 13 удаляют защитную Boc-группу с получением соединения 14; проводят реакцию конденсации соединения 14 со сложным 1-трет-бутиловым эфиром N-трет-бутилоксикарбонил-L-глутаминовой кислоты под воздействием HATU и DIPEA с получением соединения 15; соединение 15 растворяют в растворе трифторуксусной кислоты с удалением защитной группы с получением соединения 16; соединение 16 растворяют в N,N-диметилформамиде и добавляют ди-трет-бутилдикарбонат и кислотосвязывающее вещество с получением соединения 17 (промежуточное соединение B);
проводят реакцию конденсации соединения 17 и соединения 10 под воздействием HATU и DIPEA с получением соединения 18; у соединения 18 удаляют защитную группу под воздействием п-толуолсульфоновой кислоты с получением соединения 19; проводят реакцию соединения 19 с DOTA-NHS с получением конечного соединения 20, представленного формулой (II-1);
путь синтеза для вышеуказанных конкретных стадий является следующим:
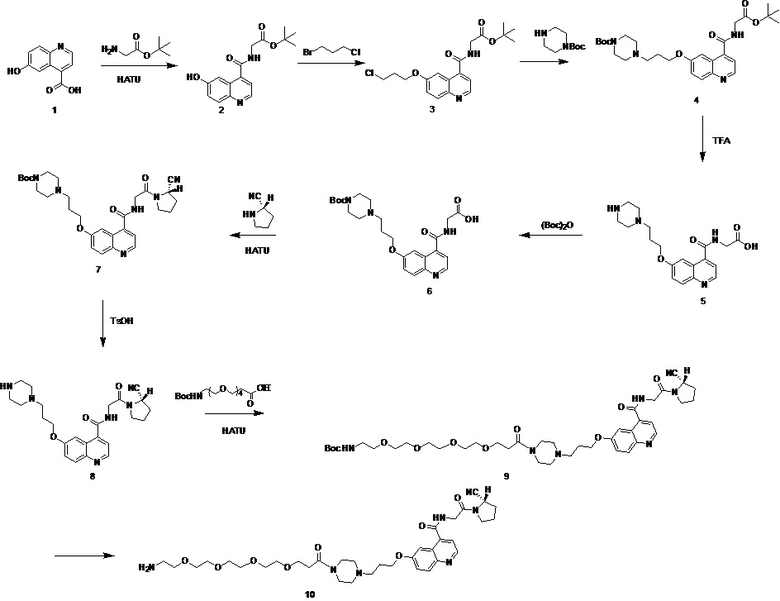
 .
.
Способ получения других соединений tEB-FAPI со схемы согласно настоящему изобретению аналогичен способу получения соединения 20, при этом получение в целом может быть осуществлено на основании существующих общепринятых средств со ссылкой на путь синтеза соединения 20.
С другой стороны, настоящее изобретение дополнительно предусматривает радиоактивно меченный комплекс на основе tEB-FAPI, который представляет собой комплекс, полученный путем мечения радионуклидом соединения формулы (I), описанного в настоящем изобретении, в качестве лиганда. Радиоактивно меченный комплекс может быть использован в качестве нового типа зонда для лучевой диагностики и лечения опухолей, т.е. его можно использовать в качестве радионуклидного диагностического зонда или радионуклидного терапевтического зонда. Нуклиды могут быть выбраны из любого из 177Lu, 90Y, 18F, 64Cu, 68Ga, 62Cu, 67Cu, 86Y, 89Zr, 99mTc, 89Sr, 153Sm, 149Tb, 161Tb, 186Re, 188Re, 212Pb, 213Bi, 223Ra, 225Ac, 226Th, 227Th, 131I, 211At или 111In; предпочтительно из 68Ga, 177Lu или 90Y.
Предпочтительный комплекс согласно настоящему изобретению имеет структуру, представленную следующей формулой (IV):
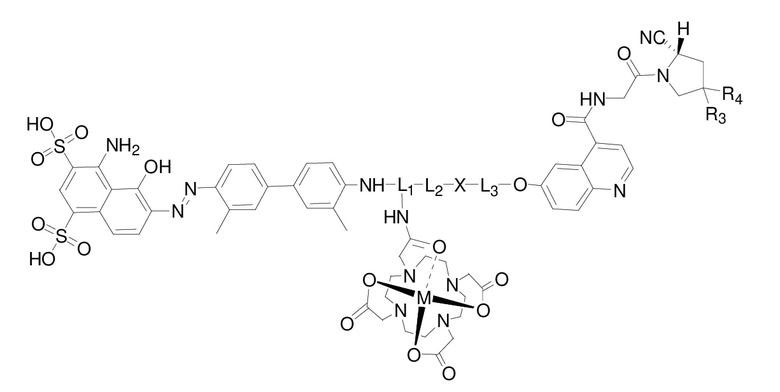 формула (IV),
формула (IV),
где L1 представляет собой структуру на основе лизина или глутаминовой кислоты или структуру на основе производного соединения, предусматривающую структуру на основе лизина или глутаминовой кислоты;
L2 представляет собой -(CH2)n-, где n представляет собой целое число от 0 до 30, где каждая CH2 отдельно может быть замещена с помощью -O-, -NH-, -(CO)-, -NH(CO)- или -(CO)-NH- или не замещена, при условии, что отсутствует замещение двух смежных групп CH2;
L3 представляет собой -(CH2)m-, где m представляет собой целое число от 0 до 30, где каждая CH2 отдельно может быть замещена с помощью -O- или -(CO)- или не замещена, при условии, что отсутствует замещение двух смежных групп CH2;
X выбран из N, C, O, S или следующих структур:
, , , ;
R3 и R4 являются одинаковыми или разными, при этом оба независимо выбраны из H или F;
М представляет собой радионуклид, выбранный из любого из 68Ga, 177Lu или 90Y.
В предпочтительном варианте осуществления комплекса согласно настоящему изобретению L2 в формуле (IV) представляет собой -(CH2)n-; n представляет собой целое число от 0 до 16, более предпочтительно целое число от 0 до 12, еще более предпочтительно 0, 3 или 10; где каждая -CH2- отдельно может быть замещена с помощью -O-, -NH- или -(CO)- или не замещена, при условии, что отсутствует замещение двух смежных групп -CH2-. Более предпочтительно L2 представляет собой -(CH2)0, -NH-CH2-(CO)-, -NH-CH2-(CH2OCH2)2-CH2-(CO)-, -NH-CH2-(CH2OCH2)4-CH2(CO)-, -(CO)-CH2-(CO)-, -(CO)-(CH2)2-(CO)-, -(CO)-CH2-(CH2OCH2)2-CH2(CO)- или -(CO)-CH2-(CH2OCH2)4-CH2(CO)-.
В предпочтительном варианте осуществления комплекса согласно настоящему изобретению L3 в формуле (IV) представляет собой -(CH2)m-; m представляет собой целое число от 0 до 20, более предпочтительно целое число от 1 до 6, еще более предпочтительно 2 или 3; где каждая -CH2- отдельно может быть замещена с помощью -O- или не замещена, при условии, что отсутствует замещение двух смежных групп -CH2-. Более предпочтительно L3 представляет собой -(CH2)3-.
Радиоактивно меченный комплекс согласно настоящему изобретению может быть получен из соединения, содержащего радионуклид, и соединения формулы (I), описанного в настоящем изобретении, в соответствии с различными существующими способами мечения; предпочтительными способами мечения согласно настоящему изобретению являются описанные ниже мечение мокрым способом или мечение способом лиофилизации:
схема мечения мокрым способом включает: растворение соответствующего количества соединения формулы (I), описанного в настоящем изобретении, в буферном растворе или деионизированной воде; добавление к полученному раствору раствора радионуклида и обеспечение реакции в закрытых условиях в течение 5-40 мин с образованием меченного радиоактивным нуклидом комплекса;
в качестве альтернативы схема мечения способом лиофилизации включает: растворение соответствующего количества соединения формулы (I) согласно настоящему изобретению в буферном растворе или деионизированной воде; распределение полученного после стерильной фильтрации раствора по контейнерам, лиофилизацию, укупоривание пробкой с получением набора контейнеров с лиофилизатом; добавление в набор контейнеров с лиофилизатом соответствующего количества раствора уксусной кислоты или буферного раствора для растворения, а затем добавление соответствующего раствора радионуклида и обеспечение протекания реакции в закрытых условиях в течение 5-40 мин с образованием меченного радионуклидом комплекса. При этом контейнеры для распределения предпочтительно представляют собой пробирки для криоконсервации или флаконы с контрольным антибиотиком. Также в набор контейнеров можно добавить вспомогательные вещества, как, например, маннит, аскорбиновая кислота и т.д., в соответствии с условиями формирования лиофилизированного порошка в наборе контейнеров, и путем регулирования дозировки соединения формулы (I) согласно настоящему изобретению и вспомогательных веществ добиться оптимального формования набора контейнеров.
Продукты, полученные по схеме мечения мокрым способом и схеме мечения способом лиофилизации, могут подвергаться общепринятой обработке (например, хроматографическому разделению и очистке, удалению растворителя ротационным выпариванием, растворению остатка в PBS, или воде, или физиологическом растворе, стерильной фильтрации и т.д.) для дальнейшего приготовления растворов для инъекций.
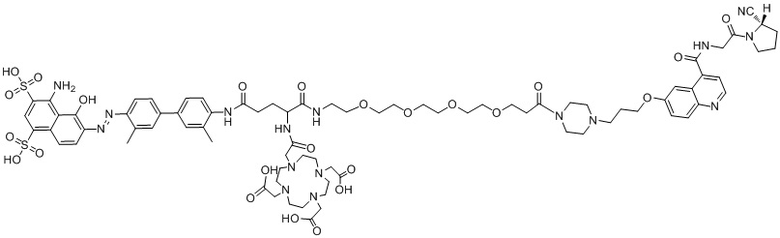
В предпочтительном варианте осуществления настоящего изобретения при использовании соединения 20, представленного формулой (II-1), в качестве лиганда предпочтительным способом получения радиоактивно меченного соединения 20 является способ мокрого мечения, который включает следующие стадии, на которых: соединение 20 растворяют в буферном растворе или деионизированной воде; добавляют к нему свежеполученный радиоактивный раствор, герметично закрывают, обеспечивают протекание реакции при температуре 37-90°C в течение 5-40 мин и охлаждают; реакционный раствор разбавляют водой, после чего разделяют и очищают на хроматографической колонке Sep-Pak C18, промывают хроматографическую колонку буферным раствором или водой для удаления непрореагировавших радиоактивных ионов, промывают этанольным раствором соляной кислоты или этанольным раствором, а затем разбавляют физиологическим раствором или PBS и проводят стерильную фильтрацию с получением раствора для инъекций на основе радиоактивно меченного комплекса со структурой, описанной в формуле (IV-1); при этом радионуклид М представляет собой 68Ga, 177Lu, 90Y и т.д.
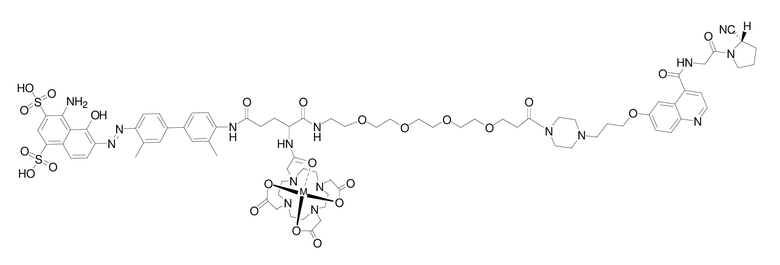 Формула (IV-1)
Формула (IV-1)
Другим предпочтительным способом получения радиоактивно меченного соединения 20 согласно настоящему изобретению является способом мечения путем лиофилизации, включающий стадии, на которых: в буферном растворе растворяют соединение 20 и другие необходимые реагенты, полученный раствор стерильно фильтруют, затем распределяют по пробиркам для криоконсервации и после лиофилизации герметично закрывают с получением набора контейнеров с лиофилизатом; добавляют соответствующее количество буферного раствора в набор контейнеров с лиофилизатом для растворения, затем добавляют свежеприготовленный радиоактивный раствор, герметично закрывают, обеспечивают протекание реакции при температуре 37-120°C в течение 5-40 мин и охлаждают; реакционный раствор разбавляют водой, после чего разделяют и очищают на хроматографической колонке Sep-Pak C18, промывают хроматографическую колонку буферным раствором или водой с удалением непрореагировавших радиоактивных ионов, промывают этанольным раствором соляной кислоты или этанольным раствором, а затем разбавляют физиологическим раствором или PBS и проводят стерильную фильтрацию с получением раствора для инъекций на основе радиоактивно меченного комплекса со структурой, показанной в формуле (IV-1); при этом радионуклид M представляет собой 68Ga, 177Lu, 90Y и т.д.
Другие химические вещества, используемые на вышеуказанных стадиях синтеза, являются коммерчески доступными.
Буферный раствор представляет собой вещество для стабилизации значения pH реакционного раствора, которое может представлять собой ацетат, лактат, тартрат, малат, малеат, сукцинат, аскорбат, карбонат и фосфат, а также их смеси и др.
В другом аспекте согласно настоящему изобретению также предложено применение соединения tEB-FAPI, представленного формулой (I), или его фармацевтически приемлемой соли в получении лекарственных средств для радионуклидной терапии или визуализации опухолей с высоким уровнем экспрессии белка FAP.
Согласно настоящему изобретению также предложено применение радиоактивно меченного комплекса на основе tEB-FAPI, представленного формулой (IV), в радионуклидной терапии и визуализации опухолей с высоким уровнем экспрессии белка FAP.
В предпочтительном применении настоящего изобретения комплекс готовят в виде инъекционного раствора и вводят внутривенно пациентам с опухолями с высоким уровнем экспрессии белка FAP.
При применении настоящего изобретения опухоли с высоким уровнем экспрессии белка FAP включают без ограничения рак молочной железы, рак яичников, рак легкого, колоректальный рак, рак желудка или рак поджелудочной железы.
Согласно настоящему изобретению предложен ингибитор белка активации фибробластов, модифицированный усеченным синим Эванса, tEB-FAPI, и меченный радионуклидом комплекс на его основе, а также способ получения и способ мечения таких соединений. Результаты биологических испытаний показывают, что он обладает значительно увеличенным периодом полураспада в кровотоке, увеличенным накоплением за счет поглощения опухолью и временем удерживания. На данный момент такая новая особенность отсутствует у других средств визуализации FAPI, и он подходит для радионуклидной терапии и визуализации опухолей с высоким уровнем экспрессии белка FAP.
Описание прилагаемых графических материалов
Фиг. 1 представляет собой масс-спектрограмму соединения 2 в варианте осуществления 1 настоящего изобретения.
Фиг. 2 представляет собой спектр ядерного магнитного резонанса на ядрах водорода для соединения 2 в варианте осуществления 1 настоящего изобретения.
Фиг. 3 представляет собой спектр ядерного магнитного резонанса на ядрах углерода для соединения 2 в варианте осуществления 1 настоящего изобретения.
Фиг. 4 представляет собой масс-спектрограмму соединения 3 в варианте осуществления 1 настоящего изобретения.
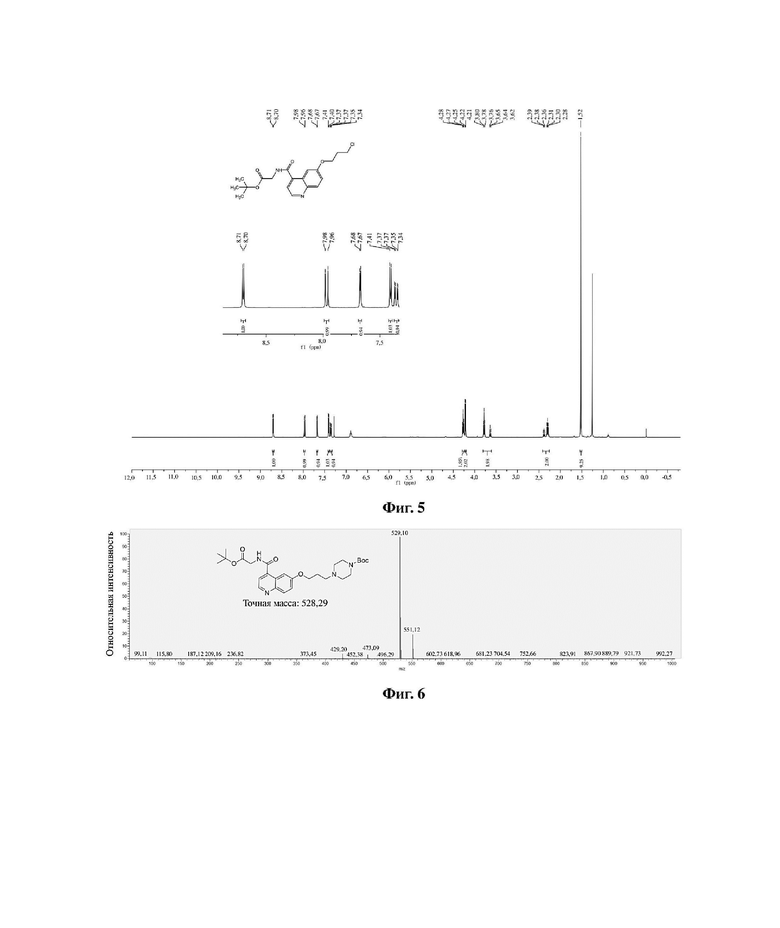
Фиг. 5 представляет собой спектр ядерного магнитного резонанса на ядрах водорода для соединения 3 в варианте осуществления 1 настоящего изобретения.
Фиг. 6 представляет собой масс-спектрограмму соединения 4 в варианте осуществления 1 настоящего изобретения.
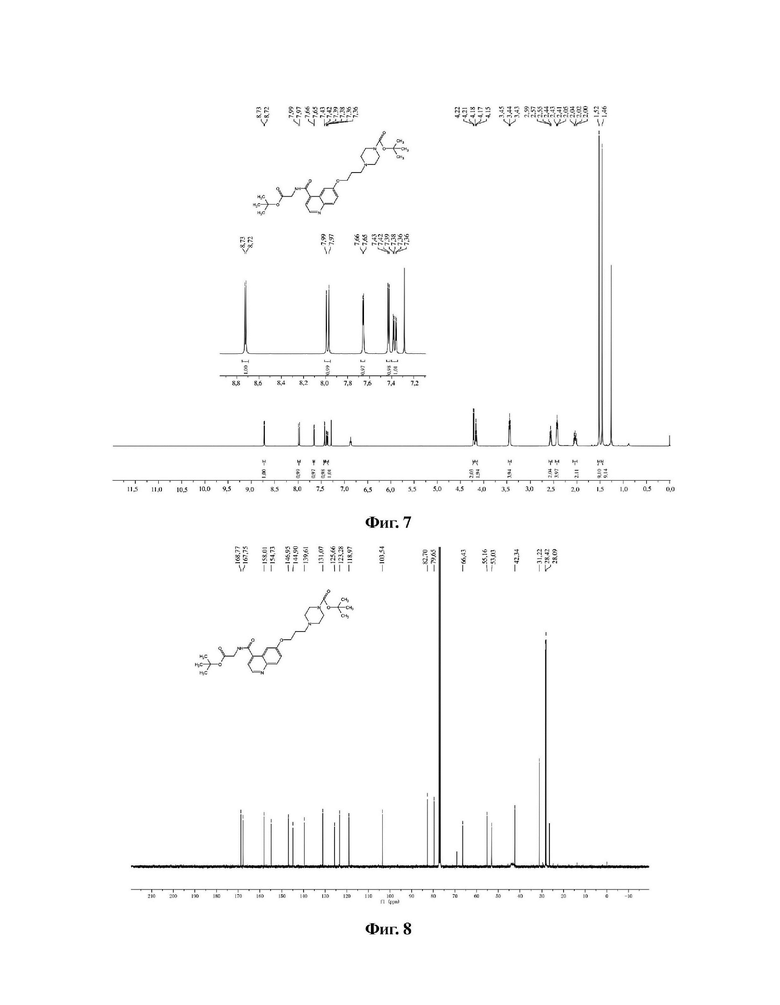
Фиг. 7 представляет собой спектр ядерного магнитного резонанса на ядрах водорода для соединения 4 в варианте осуществления 1 настоящего изобретения.
Фиг. 8 представляет собой спектр ядерного магнитного резонанса на ядрах углерода для соединения 4 в варианте осуществления 1 настоящего изобретения.
Фиг. 9 представляет собой масс-спектрограмму соединения 7 в варианте осуществления 1 настоящего изобретения.
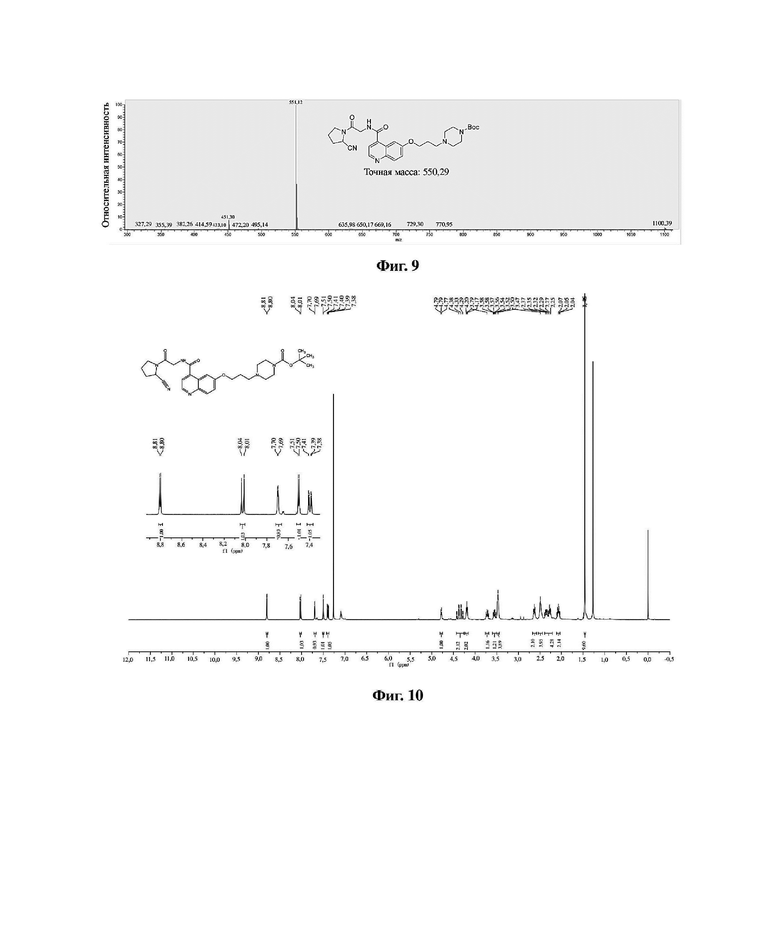
Фиг. 10 представляет собой спектр ядерного магнитного резонанса на ядрах водорода для соединения 7 в варианте осуществления 1 настоящего изобретения.
Фиг. 11 представляет собой спектр ядерного магнитного резонанса на ядрах углерода для соединения 7 в варианте осуществления 1 настоящего изобретения.
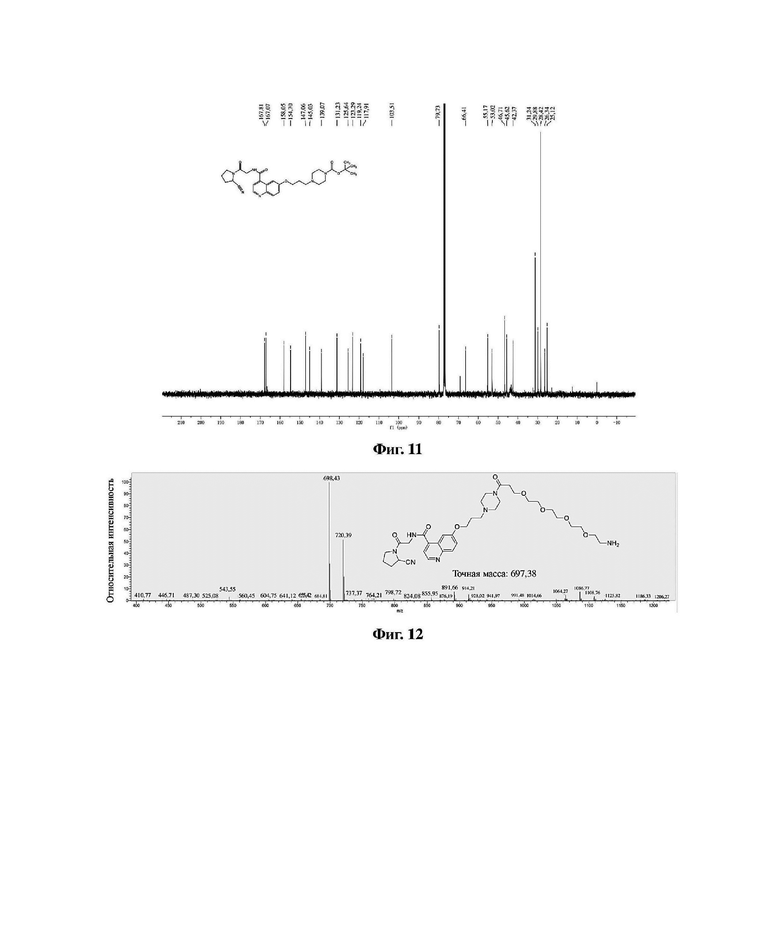
Фиг. 12 представляет собой масс-спектрограмму соединения 10 в варианте осуществления 1 настоящего изобретения.
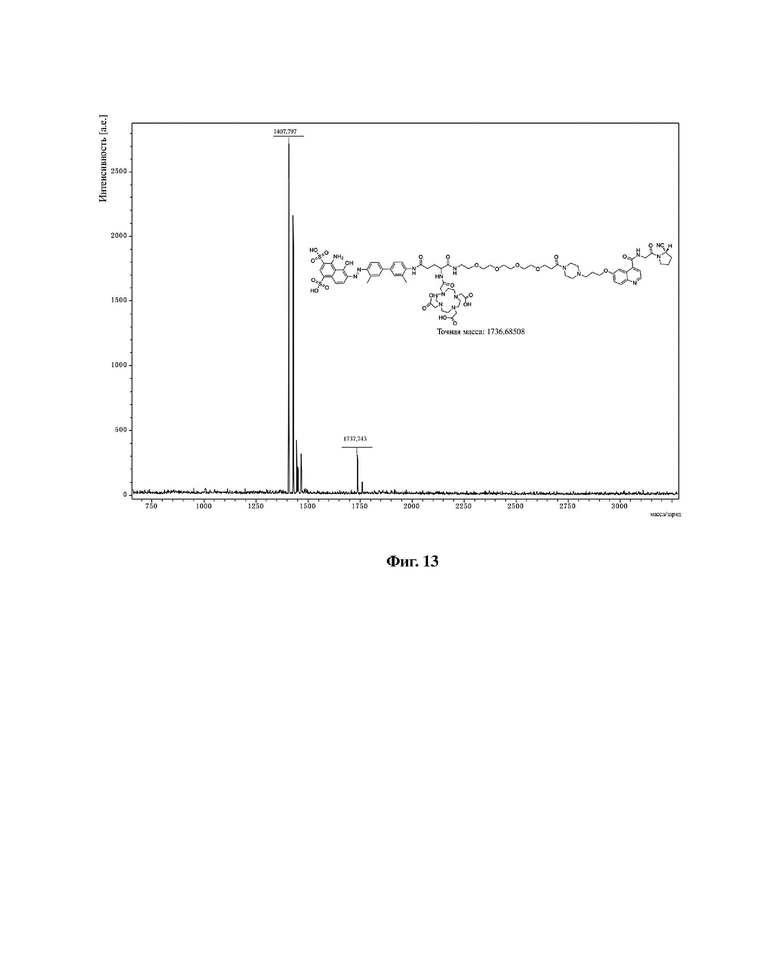
Фиг. 13 представляет собой масс-спектрограмму соединения 20 в варианте осуществления 1 настоящего изобретения.
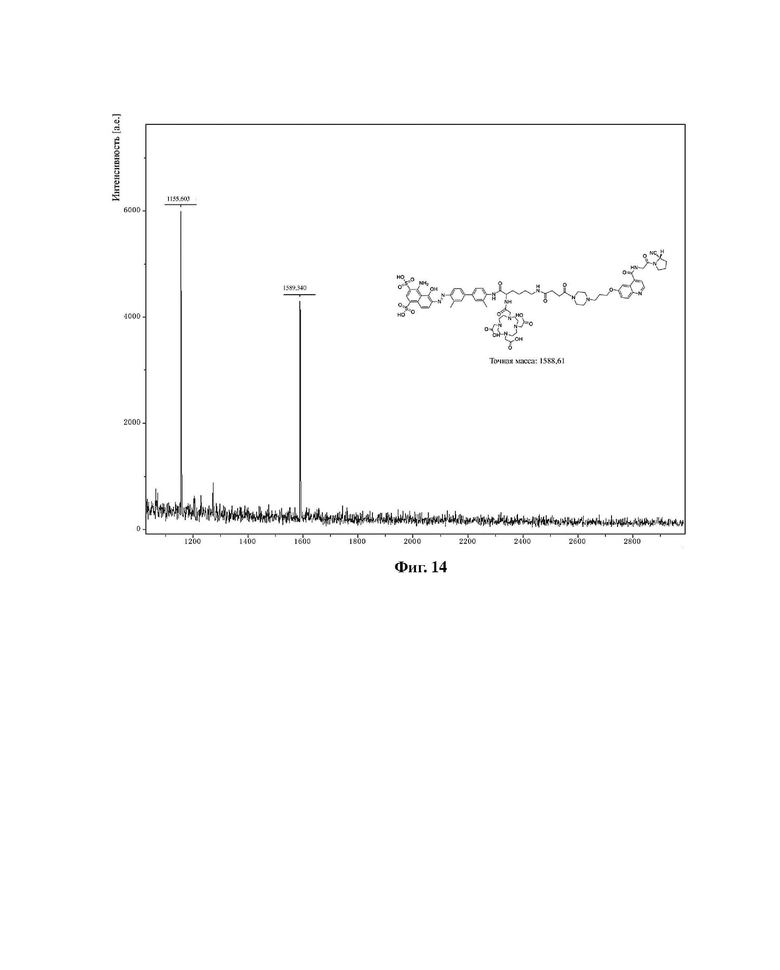
Фиг. 14 представляет собой масс-спектрограмму соединения в варианте осуществления 10 настоящего изобретения.
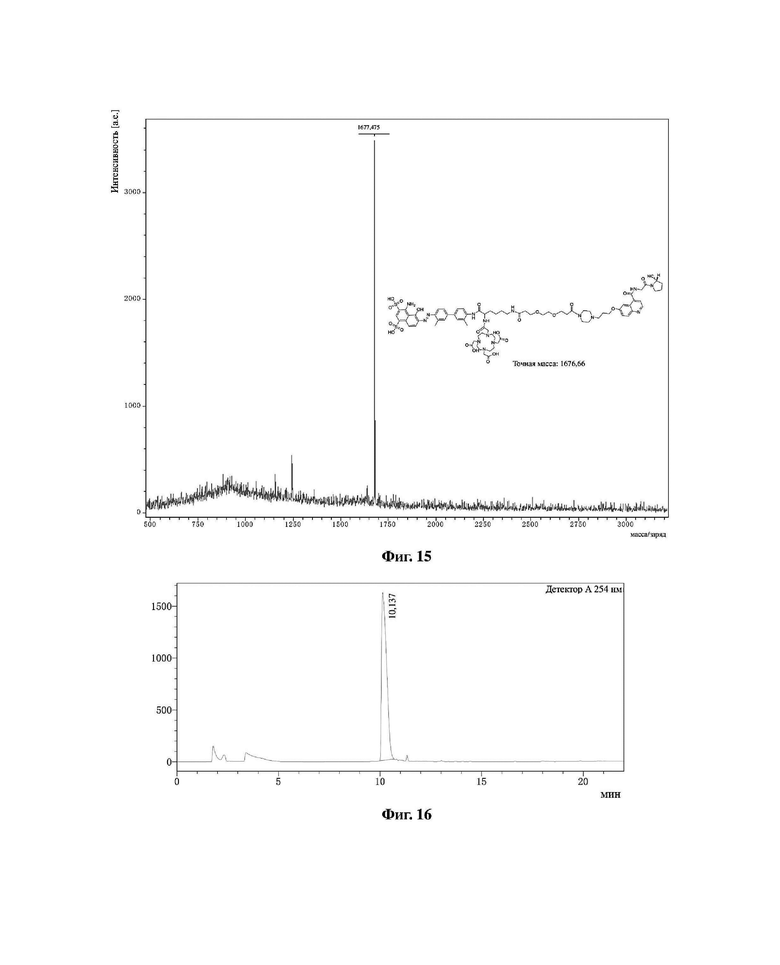
Фиг. 15 представляет собой масс-спектрограмму соединения в варианте осуществления 11 настоящего изобретения.
Фиг. 16 представляет собой хроматограмму HPLC соединения 10 в варианте осуществления 1 настоящего изобретения.
Фиг. 17 представляет собой хроматограмму HPLC соединения 17 в варианте осуществления 1 настоящего изобретения.
Фиг. 18 представляет собой хроматограмму HPLC реакционной системы соединения 17 и соединения 10 в варианте осуществления 1 настоящего изобретения.
Фиг. 19 представляет собой хроматограмму HPLC соединения 19 в варианте осуществления 1 настоящего изобретения.
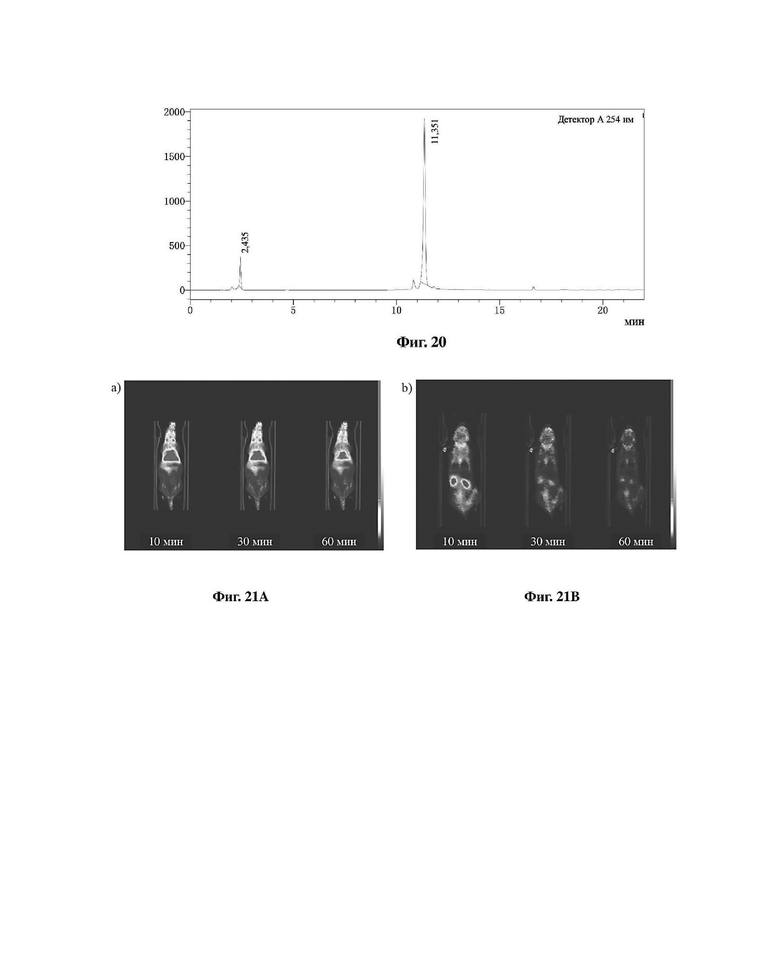
Фиг. 20 представляет собой хроматограмму HPLC реакционной системы соединения 19 и DOTA-NHS в варианте осуществления 1 настоящего изобретения.
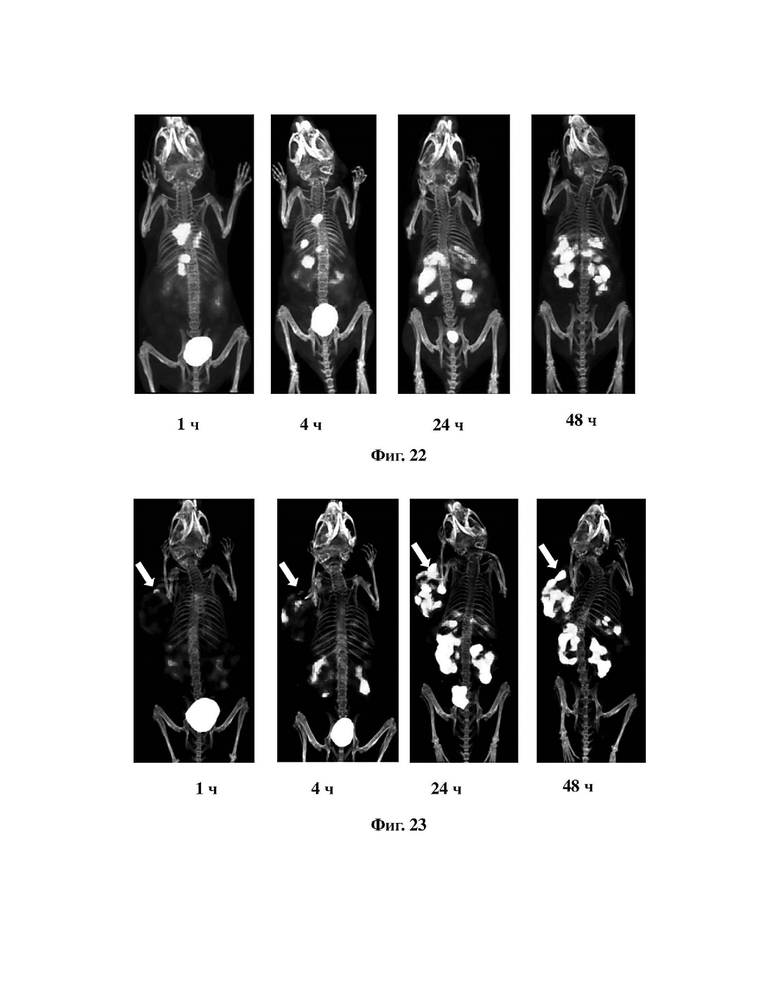
На фиг. 21A и фиг. 21B проиллюстрирована визуализация MicroPET комплексов на основе tEB-FAPI, меченных 68Ga, согласно настоящему изобретению и FAPI-02, меченных 68Ga, у нормальных мышей.
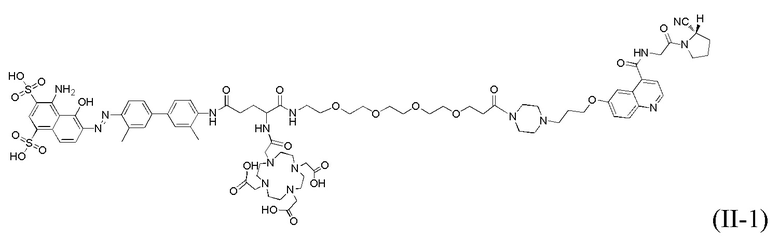
На фиг. 22 показана визуализация SPECT 177Lu-tEB-FAPI, полученного в варианте осуществления 40 согласно настоящему изобретению, у нормальных мышей в различные моменты времени.
На фиг. 23 показана визуализация SPECT 177Lu-tEB-FAPI, полученного в варианте осуществления 40 настоящего изобретения, на мышах с моделью ксенотрансплантата рака поджелудочной железы человека в различные моменты времени.
Конкретные способы осуществления
Технические решения настоящего изобретения дополнительно проиллюстрированы и описаны ниже посредством конкретных вариантов осуществления со ссылкой на прилагаемые графические материалы.
Вариант осуществления 1. Получение конъюгата tEB-FAPI (соединение 20)
Синтез соединения 2.
В колбу емкостью 100 мл к 30 мл N,N-диметилформамида последовательно добавляли в указанном порядке соединение 1 (6-гидрокси-4-хинолинкарбоновая кислота, 1,89 г, 10,0 ммоль), сложный трет-бутиловый эфир глицина (1,89 г, 10,0 ммоль), HATU (3,8 г, 10,0 ммоль) и N,N-диизопропилэтиламин (2,6 г, 20,0 ммоль). Реакционную смесь перемешивали в течение ночи, растворитель отгоняли перегонкой при пониженном давлении с получением неочищенного продукта. На колонке с силикагелем выполняли очистку (дихлорметан/метанол = 30:1) с получением соединения 2 в виде белого твердого вещества с выходом 87%; при этом фиг. 1 представляет собой масс-спектрограмму соединения 2, фиг. 2 представляет собой спектр ядерного магнитного резонанса на ядрах водорода для соединения 2, фиг. 3 представляет собой спектр ядерного магнитного резонанса на ядрах углерода для соединения 2.
Синтез соединения 3.
В колбу емкостью 100 мл к 50 мл N,N-диметилформамида последовательно добавляли в указанном порядке соединение 2 (1,51 г, 5,0 ммоль), 1-бром-3-хлорпропан (1,55 г, 10,0 ммоль) и карбонат калия (1,38 г, 10,0 ммоль). Температуру системы повышали до 60°С, систему перемешивали в течение ночи при поддержании температуры 60°С, растворитель отгоняли перегонкой при пониженном давлении с получением неочищенного продукта. На колонке с силикагелем выполняли очистку (дихлорметан/метанол = 50:1) с получением соединения 3 в виде белого твердого вещества с выходом 63%; при этом фиг. 4 представляет собой масс-спектрограмму соединения 3, а фиг. 5 представляет собой спектр ядерного магнитного резонанса на ядрах водорода для соединения 3.
Синтез соединения 4.
В колбу емкостью 100 мл к 30 мл ацетонитрила последовательно добавляли в указанном порядке соединение 3 (0,76 г, 2,0 ммоль), 1-трет-бутилоксикарбонилпиперазин (0,55 г, 3,0 ммоль) и йодид калия (0,49 г, 3,0 ммоль). Систему нагревали до 60 градусов Цельсия и перемешивали при 60 градусах Цельсия в течение ночи, растворитель отгоняли перегонкой при пониженном давлении с получением неочищенного продукта. Очистку проводили на колонке с силикагелем (дихлорметан/метанол = 30:1) с получением соединения 4 в виде белого твердого вещества с выходом 58%. MS (ESI) масса/заряд: расчетное значение для [C28H40N4O6]: 528,29; найденное значение: 529,10 [M+H]+; фиг. 6 представляет собой масс-спектрограмму соединения 4, фиг. 7 представляет собой спектр ядерного магнитного резонанса на ядрах водорода для соединения 4, а фиг. 8 представляет собой спектр ядерного магнитного резонанса на ядрах углерода для соединения 4.
Синтез соединения 5.
На ледяной бане соединение 4 (0,52 г, 1,0 ммоль) растворяли в 10 мл смешанного раствора дихлорметана и трифторуксусной кислоты (объемное соотношение 9:1), систему нагревали до комнатной температуры и обеспечивали протекание реакции в течение 2 ч, после этого растворитель выпаривали при пониженном давлении, а оставшееся растворяли в 10 мл N,N-диметилформамида для дальнейшего использования.
Синтез соединения 6.
К N,N-диметилформамиду соединения 5 добавляли в указанном порядке ди-трет-бутилдикарбонат (0,22 г, 1,0 ммоль) и N,N-диизопропилэтиламин (0,39 г, 3,0 ммоль), смесь перемешивали при комнатной температуре в течение ночи и растворитель выпаривали при пониженном давлении с получением неочищенного продукта. Очистку проводили на колонке с силикагелем (дихлорметан/метанол = 10:1) с получением соединения 6 в виде белого твердого вещества с выходом 72%.
Синтез соединения 7.
В колбу емкостью 100 мл к 10 мл N,N-диметилформамида последовательно добавляли в указанном порядке соединение 6 (0,47 г, 1,0 ммоль), гидрохлорид (S)-пирролидин-2-карбонитрила (0,13 г, 10,0 ммоль), HATU (0,38 г, 1,0 ммоль) и N,N-диизопропилэтиламин (0,26 г, 2,0 ммоль). Реакционную смесь перемешивали при комнатной температуре до завершения реакции, и растворитель отгоняли перегонкой при пониженном давлении с получением неочищенного продукта. Очистку проводили на колонке с силикагелем (дихлорметан/метанол = 50:1) с получением соединения 7 в виде белого твердого вещества с выходом 85%. Фиг. 9 представляет собой масс-спектрограмму соединения 7, фиг. 10 представляет собой спектр ядерного магнитного резонанса на ядрах водорода для соединения 7, фиг. 11 представляет собой спектр ядерного магнитного резонанса на ядрах углерода для соединения 7.
Синтез соединения 8.
В колбу емкостью 100 мл к 10 мл ацетонитрила добавляли в указанном порядке соединение 7 (0,55 г, 1,0 ммоль) и моногидрат п-толуолсульфоновой кислоты (0,27 г, 1,5 ммоль). Реакционную систему нагревали до 60 градусов Цельсия и перемешивали до завершения реакции, растворитель отгоняли перегонкой при пониженном давлении с получением неочищенного продукта.
Синтез соединения 9.
В реакционную колбу с указанным выше соединением 8 добавляли в указанном порядке сложный 1-трет-бутиловый эфир 5,8,11,14-тетраокса-2-азагептадекандиовой кислоты (0,19 г, 1,0 ммоль), HATU (0,38 г, 1,0 ммоль), N,N-диизопропилэтиламин (0,26 г, 2,0 ммоль) и 10 мл N,N-диметилформамида. Реакционную смесь перемешивали в течение ночи, растворитель отгоняли перегонкой при пониженном давлении с получением неочищенного продукта. Очистку проводили на колонке с силикагелем (дихлорметан/метанол = 50:1) с получением соединения 9 в виде белого твердого вещества с выходом 64%.
Синтез соединения 10.
В колбу емкостью 100 мл к 10 мл ацетонитрила добавляли в указанном порядке соединение 9 (0,61 г, 1,0 ммоль) и моногидрат п-толуолсульфоновой кислоты (0,27 г, 1,5 ммоль). Реакционную систему нагревали до 60 градусов Цельсия и перемешивали до завершения реакции, растворитель отгоняли перегонкой при пониженном давлении с получением неочищенного продукта. Очистку проводили на колонке с силикагелем (дихлорметан/метанол = 10:1) с получением соединения 10 в виде белого твердого вещества с выходом 59%. MS (ESI) масса/заряд: расчетное значение для [C35H51N7O8]: 697,38; найденное значение: 698,43 [M+H]+; фиг. 12 представляет собой масс-спектрограмму соединения 10.
Путь синтеза на вышеуказанных стадиях следующий:
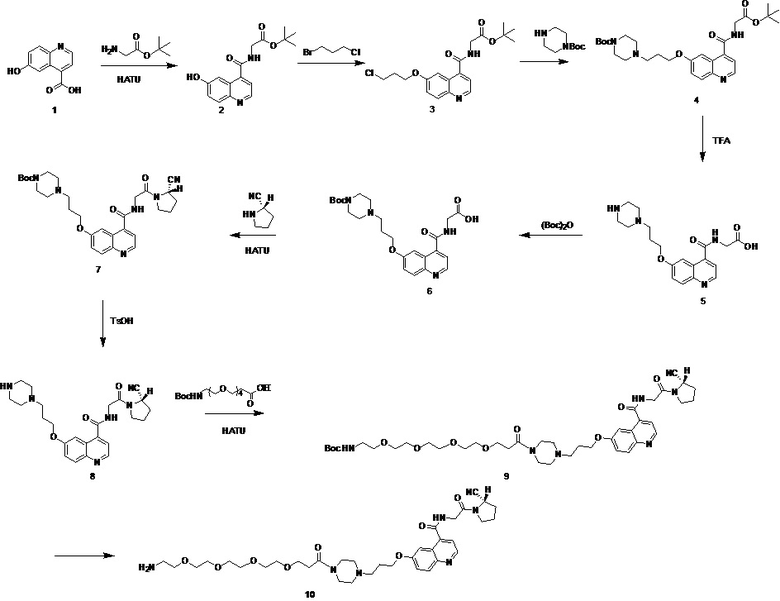 .
.
Синтез соединения 12.
В колбу емкостью 100 мл добавляли в указанном порядке 4,4'-диамино-3,3'-диметилбифенил (соединение 11) (2,12 г, 10,0 ммоль), ди-трет-бутилдикарбонат (2,2 г, 10,0 ммоль), N,N-диизопропилэтиламин (1,3 г, 10,0 ммоль) и 20 мл дихлорметана, перемешивали при комнатной температуре в течение ночи, контролировали завершение реакции с помощью HPLC (к. т. 10,13 мин), растворитель отгоняли перегонкой при пониженном давлении с получением неочищенного продукта, очищение проводили на колонке с силикагелем (петролейный эфир/этилацетат = 5:1) с получением соединения 12 в виде белого твердого вещества с выходом 59%.
Синтез соединения 13.
Соединение 12 (0,31 г, 1,0 ммоль) и 4 мл ацетонитрила помещали в указанном порядке в колбу емкостью 50 мл, ставили на ледяную баню, в реакционную колбу по каплям добавляли 1,5 мл 2 М хлористоводородной кислоты и обеспечивали протекание реакции в течение 15 мин, нитрит натрия (0,068 г, 1,0 ммоль) растворяли в 2 мл воды, а затем добавляли по каплям в реакционную колбу, обеспечивали протекание реакции в течение получаса и оставляли полученное в качестве раствора А для дальнейшего использования. Отдельно подготавливали реакционную колбу емкостью 50 мл, добавляли мононатриевую соль 1-амино-8-нафтол-2,4-дисульфоновой кислоты (0,33 г, 1,0 ммоль), карбонат натрия (0,105 г, 1,0 ммоль) и 5 мл воды; на ледяной бане медленно по каплям добавляли раствор А к раствору В и перемешивали на ледяной бане в течение 2 часов. Чистое соединение 13 получали путем хроматографии с обращенной фазой и лиофилизации, причем выход составлял 47%.
Синтез соединения 14.
На ледяной бане соединение 13 (0,52 г, 1,0 ммоль) растворяли в трифторуксусной кислоте, систему нагревали до комнатной температуры и обеспечивали протекание реакции в течение 2 ч, по завершении реакции растворитель выпаривали при пониженном давлении с получением неочищенного продукта. Неочищенный продукт подвергали хроматографии с обращенной фазой и лиофилизировали с получением чистого соединения 14 с выходом 73%.
Синтез соединения 15.
В колбу емкостью 100 мл добавляли в указанном порядке соединение 14 (0,54 г, 1,0 ммоль), сложный 1-трет-бутиловый эфир N-трет-бутилоксикарбонил-L-глутаминовой кислоты (0,30 г, 1,0 ммоль), HATU (0,38 г, 1,0 ммоль), N,N-диизопропилэтиламин (0,26 г, 2,0 ммоль) и 10 мл N,N-диметилформамида. Реакционную смесь перемешивали до завершения реакции, и растворитель отгоняли перегонкой при пониженном давлении с получением неочищенного продукта. Неочищенный продукт подвергали хроматографии с обращенной фазой и лиофилизировали с получением чистого соединения 15 с выходом 52%.
Синтез соединения 16.
Проводили отщепление сложного трет-бутилового эфира и удаление защитной Boc-группы с применением смеси тиоанизол:1,2-этандитиол:анизол:TFA (5:3:2:90) при комнатной температуре. После окончания реакции TFA удаляли с помощью потока аргона, после чего полученное растворяли в 10 мл N,N-диметилформамида для дальнейшего использования.
Синтез соединения 17.
К N,N-диметилформамиду соединения 16 добавляли последовательно ди-трет-бутилдикарбонат (0,22 г, 1,0 ммоль) и N,N-диизопропилэтиламин (0,39 г, 3,0 ммоль), перемешивали при комнатной температуре в течение ночи и завершение реакции контролировали с помощью HPLC (к. т. 10,84 мин). Растворитель отгоняли перегонкой при пониженном давлении с получением неочищенного продукта. Неочищенный продукт подвергали хроматографии с обращенной фазой и лиофилизировали с получением чистого соединения 17, причем выход за две стадии составлял 43%.
Синтез соединения 18.
В колбу емкостью 50 мл последовательно добавляли соединение 17 (0,77 г, 1,0 ммоль), соединение 10 (0,51 г, 1,0 ммоль), HATU (0,38 г, 1,0 ммоль), N,N-диизопропилэтиламин (0,26 г, 2,0 ммоль) и 10 мл N,N-диметилформамида. Реакционную смесь перемешивали и завершение реакции контролировали с помощью HPLC (к. т. 12,16 мин). Растворитель отгоняли перегонкой при пониженном давлении с получением неочищенного продукта. Неочищенный продукт подвергали хроматографии с обращенной фазой и лиофилизировали с получением чистого соединения 18 с выходом 55%.
Синтез соединения 19.
В колбу емкостью 25 мл к 5 мл ацетонитрила в указанном порядке добавляли соединение 15 (0,13 г, 0,1 ммоль) и моногидрат п-толуолсульфоновой кислоты (0,05г, 0,3 ммоль). Реакционную систему нагревали до 60 градусов Цельсия и реакционную смесь перемешивали, процесс удаления защитной группы контролировали с помощью HPLC до окончания реакции (к. т. 10,47 мин), и растворитель отгоняли перегонкой при пониженном давлении с получением неочищенного продукта. Неочищенный продукт подвергали хроматографии с обращенной фазой и лиофилизировали с получением чистого соединения 19 с выходом 61%.
Синтез соединения 20.
В колбу емкостью 25 мл к 5 мл N,N-диметилформамида последовательно добавляли соединение 19 (0,12 г, 0,1 ммоль), DOTA-NHS (0,05 г, 0,1 ммоль) и N,N-диизопропилэтиламин (0,04 г, 0,3 ммоль). Реакционную систему перемешивали при комнатной температуре и контролировали с помощью HPLC до завершения реакции (к. т. 11,35 мин), а растворитель отгоняли перегонкой при пониженном давлении с получением неочищенного продукта. Неочищенный продукт подвергали хроматографии с обращенной фазой и лиофилизировали с получением чистого соединения 20 с выходом 53%. MS (ESI) масса/заряд: расчетное значение для [C80H104N16O24S2]: 1736,69; найденное значение: 1737,743 [M+H]+; фиг. 13 представляет собой масс-спектрограмму соединения 20.
Путь синтеза на вышеуказанных стадиях следующий:
.
Варианты осуществления 2-16
Структуры соединений согласно вариантам осуществления 2-16 представлены соответственно формулами (II-2) - (II-16); способы их получения могут ссылаться на вариант осуществления 1, при этом у них структуру на основе глутаминовой кислоты, вступающую в реакцию с соединением 14, заменяли структурой на основе лизина; или сложный 1-трет-бутиловый эфир 5,8,11,14-тетраокса-2-азагептадекандиовой кислоты, вступающий в реакцию с соединением 8, заменяли сложным 1-трет-бутиловым эфиром 5,8,11-триокса-2-азатридекандиовой кислоты, сложным трет-бутиловым эфиром 9-амино-4,7-диоксанонановой кислоты, сложным трет-бутиловым эфиром глицина или другими подходящими соединениями; или гидрохлорид (S)-пирролидин-2-карбонитрила, вступающий в реакцию с соединением 6, заменяли гидрохлоридом 3,3-дифторпирролидина; или выполняют одновременную замену с получением следующих соответствующих структур:
формула (II-2),
формула (II-3),
формула (II-4),
формула (II-5),
формула (II-6),
формула (II-7),
 формула (II-8),
формула (II-8),
формула (II-9),
формула (II-10),
формула (II-11),
формула (II-12),
формула (II-13),
формула (II-14),
формула (II-15)
или
формула (II-16).
Масс-спектрограмма соединения (II-10) из варианта осуществления 10 показана на фиг. 14, масс-спектрограмма соединения (II-11) из варианта осуществления 11 показана на фиг. 15.
Варианты осуществления 17-38
Обращаясь к способам получения согласно вариантам осуществления 1-16, получали соединение tEB-FAPI, выраженное следующей формулой (I):
 формула (I).
формула (I).
Вариант осуществления 39. Получение комплекса tEB-FAPI, меченного радиоактивным Ga-68
Мокрый способ: раствор хлористоводородной кислоты (элюированный из германий-галлиевого генератора), приблизительно 18,5-1850 мегабеккерелей (МБк) 68GaCl3, добавляли в центрифужную пробирку с уксусно-ацетатным раствором (1,0 г/л), содержащим 0,5 мл соединения 20, полученного в варианте осуществления 1, и помещали в условия температуры 37°С на 20 мин. Брали разделительную колонку C18, сначала медленно промывали с помощью 10 мл абсолютного этанола, после чего промывали с помощью 10 мл воды. После разбавления раствора для мечения с помощью 10 мл воды, образец загружали в разделительную колонку; сначала использовали 10 мл воды для удаления немеченных 68Ga ионов, а затем проводили элюирование с помощью 0,3 мл 10 мМ этанольного раствора HCl с получением комплекса tEB-FAPI, меченного 68Ga. Элюент разбавляли физиологическим раствором и фильтровали в стерильных условиях с получением раствора для инъекций на основе комплекса tEB-FAPI, меченного 68Ga.
Способ лиофилизации: раствор хлористоводородной кислоты (элюированный из германий-галлиевого генератора), приблизительно 18,5-1850 мегабеккерелей (МБк) 68GaCl3, добавляли в набор контейнеров с лиофилизатом, содержащий соединение 20, перемешивали и обеспечивали протекание реакции при 37°C в течение 20 мин. Брали разделительную колонку C18, сначала медленно промывали с помощью 10 мл абсолютного этанола, после чего промывали с помощью 10 мл воды. После разбавления раствора для мечения с помощью 10 мл воды, его загружали в разделительную колонку; сначала использовали 10 мл воды для удаления немеченных 68Ga ионов, а затем проводили элюирование с помощью 0,3 мл 10 мМ этанольного раствора HCl с получением элюента, представляющего собой комплекс. Элюент разбавляли физиологическим раствором и фильтровали в стерильных условиях с получением раствора для инъекций на основе комплекса tEB-FAPI, меченного 68Ga.
Вариант осуществления 40. Получение комплекса tEB-FAPI, меченного Lu-177
Мокрый способ: раствор ацетата натрия, приблизительно 18,5-1850 МБк 177LuCl3, добавляли в три центрифужные пробирки со сложным эфиром уксусной кислоты (1,0 г/л), содержащим 0,5 мл соединения 20 согласно варианту осуществления 1, варианту осуществления 2 (соединение формулы II-2) и варианту осуществления 3 (соединение формулы II-3), и помещали в условия температуры 90°С на 20 мин. Брали разделительную колонку C18, сначала медленно промывали с помощью 10 мл абсолютного этанола, после чего промывали с помощью 10 мл воды. После разбавления раствора для мечения с помощью 10 мл воды, его загружали в разделительную колонку; сначала использовали 10 мл воды для удаления немеченных 177Lu ионов, а затем проводили элюирование с помощью 0,3 мл 10 мМ этанольного раствора HCl с получением трех комплексов tEB-FAPI, меченных 177Lu. Элюент разбавляли физиологическим раствором и фильтровали в стерильных условиях с получением трех растворов для инъекции на основе комплекса tEB-FAPI, меченного 177Lu.
Способ лиофилизации: раствор ацетата натрия, приблизительно 18,5-1850 МБк 177LuCl3, добавляли в три набора контейнеров с лиофилизатом, содержащие соединение 20 согласно варианту осуществления 1, варианту осуществления 2 (соединение формулы II-2) и варианту осуществления 3 (соединение формулы II-3) соответственно, после перемешивания обеспечивали протекание реакции при 90°C в течение 20 мин. Брали разделительную колонку C18, сначала медленно промывали с помощью 10 мл абсолютного этанола, после чего промывали с помощью 10 мл воды. После разбавления раствора для мечения с помощью 10 мл воды, его загружали в разделительную колонку; сначала использовали 10 мл воды для удаления немеченных 177Lu ионов, а затем проводили элюирование с помощью 0,3 мл 10 мМ этанольного раствора HCl с получением трех комплексов tEB-FAPI, меченных 77Lu, в качестве элюента. Элюент разбавляли физиологическим раствором и фильтровали в стерильных условиях с получением трех растворов для инъекции на основе комплекса tEB-FAPI, меченного 177Lu.
Экспериментальный пример. Анализ и эффект применения
1. Аналитическая оценка посредством HPLC
Система HPLC является следующей: для анализа использовали хроматографическую колонку SHIMADZULC-20A, C18 (YMC, 3 мкм, 4,6×150 мм). Длина волны обнаружения 254 нм, скорость потока 1 мл/мин, градиент элюирования: 0-3 мин: 10% ацетонитрила, 0 и 90% воды (50 мМ ацетата аммония) остаются неизменными; 3-16 мин: увеличение до 90% ацетонитрила и 10% воды (50 мМ ацетата аммония); 16-18 мин: поддержание 90% ацетонитрила и 10% воды (50 мМ ацетата аммония); 18-20 мин: уменьшение до 10% ацетонитрила и 90% воды (50 мМ ацетата аммония); 20-22 мин: поддержание 10% ацетонитрила и 90% воды (50 мМ ацетата аммония).
В соответствии с вышеуказанной системой были идентифицированы и проанализированы соединение 10, соединение 17, реакционная система соединения 10 и соединения 17, соединение 19 и реакционная система соединения 19 и DOTA-NHS в варианте осуществления 1; полученные результаты показаны на фиг. 16-20.
В качестве экспериментальных средств использовали следующие два радиоактивно меченных зонда, полученные в варианте осуществления 39 и варианте осуществления 40, и измерение их эффективности происходило, как представлено ниже.
2. Визуализация MicroPET комплекса на основе tEB-FAPI, меченного 68Ga, у нормальных мышей
В соответствии со способом, описанным в варианте осуществления 39, получали 68Ga-tEB-FAPI с чистотой более 95%, вводили 3,7 МБк 68Ga-tEB-FAPI или 68Ga-FAPI-02 (в качестве контроля) нормальным мышам FVB через хвостовую вену, затем через 0-120 мин после введения под анестезией изофлураном выполняли визуализацию MicroPET, результаты показаны на фиг. 21А и фиг. 21В. Результаты показали, что комплекс 68Ga-tEB-FAPI согласно варианту осуществления 39 имеет высокую степень поглощения в пуле сердечной крови мышей (фиг. 21А), в то время как 68Ga-FAPI-02 почти полностью выводится в течение времени проведения испытания (фиг. 21В), что указывает на то, что введение усеченного синего Эванса могло значительно продлить период полураспада в крови.
3. Эксперимент на поглощение опухолями комплекса tEB-FAPI, меченного 177Lu, на мышах с ксенотрансплантатной моделью рака поджелудочной железы человека.
177Lu-tEB-FAPI с чистотой более 95% (в расчете на соединение 20 из варианта осуществления 1) получали согласно способу из варианта осуществления 40; нормальным мышам и мышам с ксенотрансплантатной моделью рака поджелудочной железы человека вводили через хвостовую вену 1,3 МБк 177Lu-tEB-FAPI. После инъекции в различные моменты времени проводили визуализацию SPECT, результаты визуализации SPECT нормальных мышей показаны на фиг. 22, а результаты визуализации SPECT мышей с ксенотрансплантатной моделью рака поджелудочной железы человека показаны на фиг. 23. Результаты показывают, что 177Lu-tEB-FAPI характеризуется хорошей фармакокинетикой у нормальных мышей и может непрерывно поглощаться опухолевыми тканями и сохраняться в течение более 48 часов у мышей с ксенотрансплантатной моделью рака поджелудочной железы человека, что указывает на то, что tEB-FAPI характеризуется значительно увеличенными поглощением и временем удерживания в опухоли и может использоваться в качестве терапевтического средства для лечения опухолей и средства визуализации.
Таким образом, ингибитор белка активации фибробластов, модифицированный усеченным синим Эванса, предлагаемый в настоящем изобретении, позволяет значительно продлить его период полураспада и позволяет увеличить поглощение, накопление и время удерживания в опухоли; это новое свойство в настоящее время недоступно у других средств визуализации FAPI. Благодаря дальнейшим доклиническим исследованиям на животных и клиническим исследованиям было подтверждено, что он может использоваться в радионуклидной терапии и визуализации опухолей с высоким уровнем экспрессии белка FAP.
Хотя настоящее изобретение было подробно описано с общими пояснениями, конкретными вариантами осуществления и испытаниями, специалистам в данной области техники очевидно, что на основе настоящего изобретения могут быть сделаны некоторые модификации или улучшения. Следовательно, все модификации или улучшения, сделанные без отклонения от сущности настоящего изобретения, подпадают под объем охраны, заявленный настоящим изобретением.
| название | год | авторы | номер документа |
|---|---|---|---|
| СОЕДИНЕНИЕ ДВОЙНОГО НАЦЕЛИВАНИЯ ДЛЯ БЕЛКА АКТИВАЦИИ ФИБРОБЛАСТОВ (FAP) И ИНТЕГРИНА αβ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2023 |
|
RU2838403C1 |
| СОЕДИНЕНИЕ, ЦЕЛЕНАПРАВЛЕННО ВОЗДЕЙСТВУЮЩЕЕ НА ПРОСТАТСПЕЦИФИЧЕСКИЙ МЕМБРАННЫЙ АНТИГЕН, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2022 |
|
RU2832300C2 |
| СОЕДИНЕНИЕ ДВОЙНОГО НАЦЕЛИВАНИЯ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ | 2022 |
|
RU2838179C2 |
| ИНГИБИТОР FAP | 2019 |
|
RU2797409C2 |
| НОВЫЕ ПСА-СВЯЗЫВАЮЩИЕ АГЕНТЫ И ИХ ПРИМЕНЕНИЕ | 2018 |
|
RU2787105C2 |
| КОНЪЮГИРОВАННЫЕ БИСФОСФОНАТЫ ДЛЯ ДИАГНОСТИКИ И ТЕРАПИИ ЗАБОЛЕВАНИЙ КОСТЕЙ | 2015 |
|
RU2742660C2 |
| МЕЧЕННЫЙ РАДИОАКТИВНЫМ ИЗОТОПОМ АНТАГОНИСТ GRPR ДЛЯ ПРИМЕНЕНИЯ В КАЧЕСТВЕ ЛЕЧЕБНО-ДИАГНОСТИЧЕСКОГО СРЕДСТВА | 2020 |
|
RU2839888C1 |
| НОВЫЕ СВЯЗЫВАЮЩИЕ ОПУХОЛЕВЫЙ АНТИГЕН АГЕНТЫ И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2831681C2 |
| КОНЪЮГАТЫ АНТАГОНИСТА ПЕПТИДА АНАЛОГА БОМБЕЗИНА | 2009 |
|
RU2523531C2 |
| АНТАГОНИСТЫ GRPR ДЛЯ ОБНАРУЖЕНИЯ, ДИАГНОСТИКИ И ЛЕЧЕНИЯ GRPR-ПОЗИТИВНОГО ОНКОЛОГИЧЕСКОГО ЗАБОЛЕВАНИЯ | 2013 |
|
RU2693465C2 |

Настоящее изобретение относится к соединению, представляющему собой ингибитор белка активации фибробластов, модифицированный усеченным синим Эванса, или его фармацевтически приемлемой соли, при этом его структура представлена формулой (II-1). Также предложены способ получения соединения формулы (II-1), способы получения радиоактивно меченного комплекса на основе соединения формулы (II-1) и применение формулы (II-1) в получении лекарственных средств для радионуклидной терапии или визуализации опухолей с высоким уровнем экспрессии белка FAP. Предложенное соединение и радиоактивная метка на основе структуры указанного соединения характеризуются значительно продленным периодом полураспада в кровотоке, а также увеличенным поглощением, накоплением и временем удерживания в опухоли, и являются подходящими для нуклидной терапии и визуализации опухолей с высоким уровнем экспрессии FAP. 5 н. и 1 з.п. ф-лы, 23 ил., 1 пр.
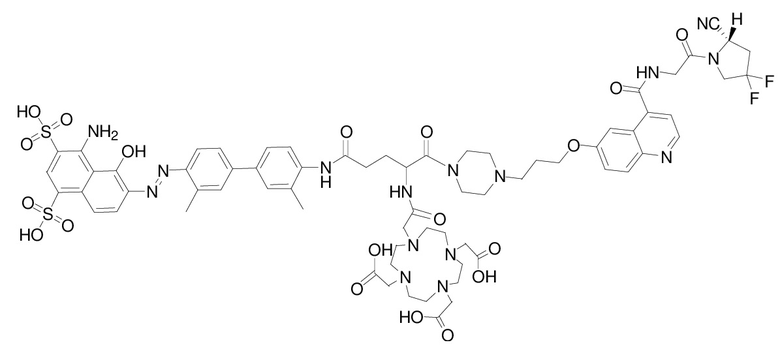
1. Соединение, представляющее собой ингибитор белка активации фибробластов, модифицированный усеченным синим Эванса, или его фармацевтически приемлемая соль, при этом его структура представлена следующей формулой (II-1):
2. Способ получения ингибитора белка активации фибробластов, модифицированного усеченным синим Эванса, отличающийся тем, что включает следующие стадии, на которых:
1) проводят реакцию амидной конденсации 6-гидрокси-4-хинолинкарбоновой кислоты и сложного трет-бутилового эфира глицина; затем последовательно вводят в реакцию 1-бром-3-хлорпропан и 1-трет-бутилоксикарбонилпиперазин; затем удаляют защитные Boc- и трет-бутильную группы под воздействием трифторуксусной кислоты (TFA); затем вводят защитную Boc-группу при аминогруппе; затем проводят реакцию амидной конденсации с гидрохлоридом (S)-пирролидин-2-формонитрила; используют п-толуолсульфоновую кислоту для удаления защитной Boc-группы; затем проводят реакцию конденсации со сложным 1-трет-бутиловым эфиром 5,8,11,14-тетраокса-2-азагептадекандиовой кислоты; затем снова удаляют защитную Вос-группу под воздействием п-толуолсульфоновой кислоты с получением промежуточного соединения А;
2) в 4,4'-диамино-3,3'-диметилбифенил на один конец вводят защитную Boc-группу, после чего проводят реакцию с мононатриевой солью 1-амино-8-нафтол-2,4-дисульфоновой кислоты с получением производного усеченного синего Эванса; удаляют защитную Вос-группу и затем проводят реакцию амидной конденсации со сложным 1-трет-бутиловым эфиром N-трет-бутилоксикарбонил-L-глутаминовой кислоты; затем удаляют защитные Boc- и трет-бутильную группы под воздействием TFA; затем проводят реакцию с ди-трет-бутилдикарбонатом с введением защитной Boc-группы при аминогруппе с получением промежуточного соединения B; при этом химическая структура усеченных производных синего Эванса представляет собой 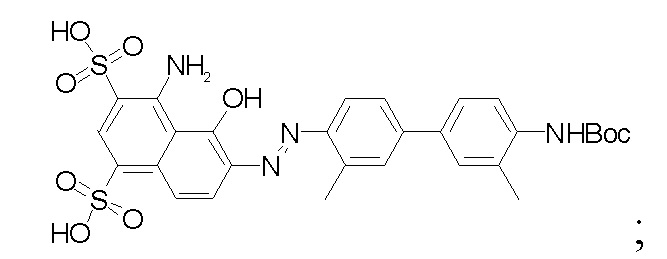
3) промежуточное соединение A, полученное на стадии 1, и промежуточное соединение B, полученное на стадии 2, подвергают реакции амидной конденсации, затем используют п-толуолсульфоновую кислоту для удаления защитной Boc-группы; наконец, проводят реакцию со сложным эфиром DOTA-NHS, чей номер CAS составляет 170908-81-3 (DOTA-NHS), с получением соединения, представляющего собой ингибитор белка активации фибробластов, модифицированный усеченным синим Эванса, со структурой, представленной следующей формулой (II-1):
3. Способ получения радиоактивно меченного комплекса на основе ингибитора белка активации фибробластов, модифицированного усеченным синим Эванса, включающий следующие стадии: растворение соединения формулы (II-1) по п. 1, в буферном растворе или деионизированной воде; добавление к полученному раствору раствора радионуклида и обеспечение протекания реакции в закрытых условиях в течение 5-40 мин с образованием меченного радионуклидом комплекса.
4. Способ получения радиоактивно меченного комплекса на основе ингибитора белка активации фибробластов, модифицированного усеченным синим Эванса, включающий следующие стадии: растворение соединения формулы (II-1) по п. 1 в буферном растворе или деионизированной воде; распределение полученного после стерильной фильтрации раствора по контейнерам, лиофилизацию, укупоривание пробкой с получением набора контейнеров с лиофилизатом; добавление в набор контейнеров с лиофилизатом соответствующего количества раствора уксусной кислоты или буферного раствора для растворения, а затем добавление соответствующего раствора радионуклида и обеспечение протекания реакции в закрытых условиях в течение 5-40 мин с образованием меченного радионуклидом комплекса.
5. Применение соединения по п. 1 или его фармацевтически приемлемой соли в получении лекарственных средств для радионуклидной терапии или визуализации опухолей с высоким уровнем экспрессии белка FAP.
6. Применение по п. 5, где соединение готовят в виде инъекционного раствора, вводят посредством внутривенной инъекции и применяют в отношении пациентов с опухолями с высоким уровнем экспрессии белка FAP; при этом опухоли с высоким уровнем экспрессии белка FAP включают без ограничения рак молочной железы, рак яичников, рак легкого, колоректальный рак, рак желудка или рак поджелудочной железы.
| WO 2019154886 A1, 15.08.2019 | |||
| LAU J | |||
| et al., Bench to Bedside Albumin Binders for Improved Cancer Radioligand Therapies, BC Bioconjugate Chemistry, 2019, v | |||
| Способ обработки медных солей нафтеновых кислот | 1923 |
|
SU30A1 |
| Кренометр | 1923 |
|
SU487A1 |
| JACOBSON O | |||
| et al., Albumine-Binding Evans Blue Derivatives for Diagnostic Imaging and Production of Long-Acting Therapeutics, BC Bioconjugate Chemistry, 2016, v | |||
| Прибор с двумя призмами | 1917 |
|
SU27A1 |

