Изобретение относится к новым биологически активным соединениям 10-дигидро-10-дезоксо-11-азаэритронолида А и их фармацевтически приемлемым солям присоединения кислот, а также к способу их получения.
Известно, что многочисленные антибиотики, кроме их основной антибиотической активности, проявляют также противовоспалительные свойства. Однако, эта особенность очень часто не используется при лечении воспалительных процессов, которые не индуцируются патогенными микроорганизмами, для того, чтобы избежать слишком быструю устойчивость микроорганизмов и возможную получающуюся сверхчувствительность человеческого организма к ним.
Поэтому необходимы вещества с противовоспалительной активностью и без одновременных антибиотических свойств. Ими очень часто являются соединения, химическая структура которых не аналогична структуре антибиотиков или - в исключительных случаях они могут быть получены из антибиотиков посредством химических превращений.
Известен D-пеницилламин, получаемый из пенициллина (1).
Согласно известному способу, посредством методики Бекмановской перегруппировки оксима эритромицина А с последующим восстановлением полученного иминоэфира эритромицина А был синтезирован 10-дигидро-10-дезоксо-11-аза-эритромицин А (2). Путем восстановительного метилирования полученного амина согласно модифицированному методу Эйшвейлера-Кларка с помощью формальдегида в присутствии муравьиной кислоты был получен N-метил-11-аза-10-дезоксо-10-дигидроэритромицин А [3] , новый полусинтетический макролидный антибиотик с 15 членами азалактоновым циклом, который подвергали клиническим испытаниям под общим названием азитромицин. Кроме того, описан способ получения N-этил и N-(н-пропил)-производных 10-дигидро-10-дезоксо-11-азаэритромицина А, которые также являются эффективными антибактериальными веществами [4] .
Соединения 10-дигидро-10-дезоксо-11-азаэритронолида А и В и в особенности их N-алкил-производные, их соли и/или О- и/или N, О-замещенные алканоильные производные не описаны.
Объектом настоящего изобретения является также способ получения соединений 10-дигидро-10-дезоксо-11-азаэритронолида А формулы I  в которой R1 представляет алькильную группу или низшую алканоильную группу C1-C3, R2, R3 и R4 имеют одинаковые или различные значения и каждый представляет собой атом водорода или алконоильную С1-С3 группу, и необязательно их фармацевтически приемлемых солей присоединения кислот, который включает одно-стадийный гидролиз 10-дигидро-10-дезоксоазаэритромицина А формулы II
в которой R1 представляет алькильную группу или низшую алканоильную группу C1-C3, R2, R3 и R4 имеют одинаковые или различные значения и каждый представляет собой атом водорода или алконоильную С1-С3 группу, и необязательно их фармацевтически приемлемых солей присоединения кислот, который включает одно-стадийный гидролиз 10-дигидро-10-дезоксоазаэритромицина А формулы II  в которой R1 имеет упомянутое значение, R2' представляет дезосаминильную группу, R3' представляет кладинозильную группу, а R4' представляет атом водорода, в присутствии концентрированной соляной кислоты и хлороформа при температуре кипения с обратным холодильником в течение 16-48 ч или двухстадийный гидролиз сначала в присутствии разбавленной соляной кислоты при комнатной температуре в течение 10-20 ч с получением соединения формулы
в которой R1 имеет упомянутое значение, R2' представляет дезосаминильную группу, R3' представляет кладинозильную группу, а R4' представляет атом водорода, в присутствии концентрированной соляной кислоты и хлороформа при температуре кипения с обратным холодильником в течение 16-48 ч или двухстадийный гидролиз сначала в присутствии разбавленной соляной кислоты при комнатной температуре в течение 10-20 ч с получением соединения формулы  где R1' - водород или С1-С3 алкил, R2' - дезосаминильная группа, R3' и R4' - водород, а затем в присутствии смеси концентрированной соляной кислоты и хлороформа при температуре кипения с обратным холодильником и выделением продукта, где R1 - водород, С1-С3-алкил, R2, R3, R4 - водород, и в случае, когда R1 - водород, последующим восстановительным алкилированием путем кипячения с обратным холодильником с эквимолекулярным количеством формальдегида и 4,77-молочным избытком 98-100% -ной муравьиной кислоты в хлороформе или ацетоне в течение 2-8 ч, или путем взаимодействия с 10-молярным избытком ацетальдегида или пропиональдегида в присутствии водорода под давлением 20 бар и катализатора - 5% -ного палладия на угле при 18-25оС в течение 2-10 ч с последующим выделением целевого продукта 1, где R1 - С1-С3-алкил, R2, R3, R4-водород, или любой из полученных выше продуктов подвергают ацилированию ангидридом алифатической кислоты С1-С3 в среде пиридина при комнатной температуре в течение 2 ч - 7 дней с последующим выделением целевого продукта 1, где R1 - алкил С1-С3 или алканоил С1-С3, R2, R3, R4имеют одинаковые или различные значения и обозначают водород или алканоил С1-С3, при этом один из радикалов R1, R2, R3, R4 обязательно означает алканоил С1-С3, или переводом любого из полученных выше продуктов в его фармацевтически приемлемую соль.
где R1' - водород или С1-С3 алкил, R2' - дезосаминильная группа, R3' и R4' - водород, а затем в присутствии смеси концентрированной соляной кислоты и хлороформа при температуре кипения с обратным холодильником и выделением продукта, где R1 - водород, С1-С3-алкил, R2, R3, R4 - водород, и в случае, когда R1 - водород, последующим восстановительным алкилированием путем кипячения с обратным холодильником с эквимолекулярным количеством формальдегида и 4,77-молочным избытком 98-100% -ной муравьиной кислоты в хлороформе или ацетоне в течение 2-8 ч, или путем взаимодействия с 10-молярным избытком ацетальдегида или пропиональдегида в присутствии водорода под давлением 20 бар и катализатора - 5% -ного палладия на угле при 18-25оС в течение 2-10 ч с последующим выделением целевого продукта 1, где R1 - С1-С3-алкил, R2, R3, R4-водород, или любой из полученных выше продуктов подвергают ацилированию ангидридом алифатической кислоты С1-С3 в среде пиридина при комнатной температуре в течение 2 ч - 7 дней с последующим выделением целевого продукта 1, где R1 - алкил С1-С3 или алканоил С1-С3, R2, R3, R4имеют одинаковые или различные значения и обозначают водород или алканоил С1-С3, при этом один из радикалов R1, R2, R3, R4 обязательно означает алканоил С1-С3, или переводом любого из полученных выше продуктов в его фармацевтически приемлемую соль.
Фармацевтически приемлемые соли присоединения кислот соединений 10-дигидро-10-дезоксо-11-азаэритронолида А (1) получают путем взаимодействия соединений 10-дигидро-10-дезоксо-11-азаэритронолида А (1) с по меньшей мере эквимолярным количеством подходящей кислоты, например, хлористоводродной, бромистоводородной, серной, фосфорной, уксусной, пропионовой, лимонной, янтарной, бензойной кислот и так далее, необязательно в присутствии растворителя, инертного при условиях реакции. Соли присоединения кислот выделяются фильтрацией, при условии, что они не растворимы в применяемом инертном растворителе, или посредством осаждения, достигаемого путем добавления нерастворителя для соответствующей соли, или путем выпаривания растворителя, наиболее часто путем лиофилизации.
П р и м е р 1. Получение 10-дигидро-10-дезоксо-11-азаэритронолида А.
Смесь 10-дигидро-10-дезоксо-11-азаритромицина А (100 г, 136,06 ммоля), 6 М раствор HCl (750 мл) и СНСl3 (380 мл) кипятят при температуре кипения с обратным холодильником в течение 16 ч. После охлаждения до обычной температуры слои разделяются и водный слой экстрагируют хлороформом (2 х 100 мл).
Путем добавления натриевой щелочи рН водного раствора доводится до 5,0 и раствор повторно экстрагируется с помощью хлороформа (3 х 100 мл). Подобную процедуру повторяют при рН 8,5 (3 х 250 мл). Хлороформные экстракты с рН 8,5 высушивают над К2СО3 и выпаривают при пониженном давлении, получая 52,77 г (94,2% ) сырого 10-дигидро-10-дезоксо-11-азаэритронолида А. После кристаллизации из диэтилового эфира (300 мл) получают 34,45 г гомогенного продукта (ТСХ, С6Н6 : CHCl3 : CH3OH = 40 : 5 : 5, NH3, Rf 0,233) с физико-химическими константами, которые описаны в I. Chem. Soc. , Perkin Trans, 1, 1986, 1981.
П р и м е р 2. Получение 10-дигидро-10-дезоксо-11-метил11-азаэритронолида А.
Смесь азитромицина (II) (10 г, 13,35 ммоля), 6 М раствора HCl (75 мл) и хлороформа (38 мл) кипятят с обратным холодильником в течение 48 часов.
После охлаждения до комнатной температуры слои разделяют и водный слой экстрагируют хлороформом. Водный раствор доводят до рН 5,0 с помощью натриевого щелока и заново экстpагируют хлороформом.
Подобную процедуру повторяют при рН 8,5. Хлороформные экстракты получают при рН 8,5 и концентрируют под вакуумом до объема примерно 10 мл и оставляют кристаллизоваться. После фильтрации и высушивания получают 4,7 г (81,2% ) продукта, который необязательно перекристаллизовывают из хлороформа.
Т. пл. 208-210оС.
Вычислено, % : C 60,94; H 10,00; N 3,23.
C22H43NO7
Найдено, % : C 60,72; H 9,63; N 2,96.
П р и м е р 3. Получение 6-0-дезосаминил-10-дигидро-10-дезоксо-11-метил-11-азаэритромицина А.
Раствор азиромицина (10 г, 13, 35 ммолях в 0,25 М растворе HCl (500 мл) выдерживают в течение 15 ч при обычной температуре. После экстpакции хлороформом (3 х 75 мл) экстракты промывают с помощью 1 М раствора HCl и водой. Соединенные водные слои подщелачивают до значения рН, равной 10, с помощью натриевой щелочи и заново экстрагируют с помощью хлороформа.
Хлороформные экстракты высушивают над К2СО3 и впоследствии выпаривают досуха при пониженном давлении. После промывки сырого продукта диэтиловым эфиром получают 6,9 г (87,8% от теории) продукта реакции. Т. пл. 203-205оС.
Вычислено, % : C 60,99; H 9,90; N 2,96.
C30H58N2O9
Найдено, % : C 60,63; H 9,58; N 4,36.
П р и м е р 4. Согласно способу, описанному в примере 1, из 10 г продукта из примера 3 получают 6,56 г (89,4% ) продукта из примера 2.
П р и м е р 5. Получение 10-дигидро-10-дезоксо-11-метил-11-азаэритронолида А.
В раствор 10-дигидро-10-дезоксо-11-азаэритронолида А (1 г, 2,38 ммоля) в CHCl3 (20 мл) загружают 0,184 мл (2,38 ммоля) формальдегида (36% ) и 0,183 мл (4,77 ммоля) муравьиной кислоты (98-100% ) и реакционную смесь кипятят с обратным холодильником при перемешивании в течение 8 ч. Затем ее охлаждают до обычной температуры и оставляют стоять в течение 24 ч, после чего выпадающие в осадок кристаллы отфильтровывают, промывают хлороформом и высушивают, получая 1,0 г (96,5% ) сырого 10-дигидро-10-дезоксо-11-метил-11-азаэритронолида А. Продукт реакции необязательно кристаллизуют из хлороформа (ТСХ, Rf 0,306).
Т. пл. 208-210оС.
1Н ЯМР (СД3ОД): 2,351 м. д. (N-СН3).
П р и м е р 6. Получение 10-дигидро-10-дезоксо-11-этил-11-азаэритронолида А.
В раствор 10-дигидро-10-дезоксо-11-азаэритронолида А (5 г, 11, 92 ммоля) в 96% -ном этаноле (50 мл) загружают ацетальдегид (7 мл, 0,024 м) и 5% -ного палладия на древесном угле (2,5 г), после чего реакционную смесь подвергают гидрогенизации при перемешивании в течение 10 ч и при давлении 20 бар.
Катализатор отфильтровывают, промывают этанолом (20 мл) и объединенную жидкую фазу концентрируют путем выпаривания при пониженном давлении до объема примерно 30 мл. В реакционную смесь добавляют воду (100 мл) и CHCl3 (50 мл), рН доводят до 4,5 путем добавления 2 М раствора HCl, слои разделяют и водную фазу заново экстрагируют хлороформом (2 х 50 мл). Экстракцию хлороформом повторяют после подщелачивания водной фазы до рН-величины, равной 8,5 с помощью водного раствора натриевой щелочи (3 х 50 мл), объединенные хлороформные экстракты высушивают над К2СО3 и выпаривают при пониженном давлении, получая 4,65 г (87,2% ) сырого 10-дигидро-10-дезоксо-11-этил-11-азаэритронолида А. Полученный продукт суспендируют в диэтиловом эфире (10 мл), перемешивают в течение 1 ч при обычной температуре, фильтруют, осадок промывают диэтиловым эфиром и высушивают, получая 3,2 г хроматографически однородного продукта (ТСХ, Rf 0,390), т. пл. 204-206оС.
П р и м е р 7. Получение 10-дигидро-10-дезоксо-11/(н-пропил)-11-азаэритронолида А.
В раствор 10-дигидро-10-дезоксо-11-азаэритронолида А (6 г, 14,30 ммоля) в 96% -ном этаноле (60 мл) загружают пропионовый альдегид (11,4 мл, 157,31 ммоля) и 5% -ный палладий на древесном угле (3,0 г), после чего реакционную смесь гидрируют при перемешивании в течение 10 ч при давлении 22 бар. Катализатор отфильтровывают, фильтруют, концентрируют путем выпаривания при пониженном давлении в густой сироп, после чего продукт реакции выделяют путем рН-градиентной экстракции. В реакционную смесь добавляют воду (100 мл) и дихлорметан (50 мл), рН доводят до 4,6 с помощью 2 М раствора HCl, слои разделяют и водный слой заново экстрагируют дихлорметаном (2 х 50 мл). После подщелачивания с помощью водного раствора натриевой щелочи, экстракцию дихлорметаном повторяют при рН 8,5 (1 х 150 мл, 2 х 50 мл).
Объединенные органические экстракты при рН 8,5 фильтруют, фильтрат концентрируют путем выпаривания при пониженном давлении до образования густой суспензии, выделенные кристаллы отфильтровывают, промывают дихлорметаном и высушивают, чтобы получить хроматографически однородный продукт с вышеуказанным названием (ТСХ, Rf 0,415). Выход 4,3 г (65,2% ). Т. пл. 212-216оС.
П р и м е р 8. Получение 4-0-ацетил-10-дигидро-10-дезоксо-11-метил-11-азаритронолида А.
В раствор 10-дигидро-10-дезоксо-11-метил-11-азаэритронолида А (5 г, 11,53 ммоля) в пиридине (30 мл) добавляют ангидрид уксусной кислоты (30 мл) и реакционную смесь оставляют стоять в течение 2 часов при обычной температуре. Стадия ацилирования останавливается путем добавления льда и продукт реакции выделяется путем экстракции хлороформом при рН 9,0 (3 х 50 мл).
Объединенные органические экстракты высушивают над К2СО3 и выпаривают, получая 3,4 г сырого продукта. Суспендирование выпадающего осадка в диэтиловом эфире (10 мл), перемешивают суспензии в течение 1 часа при обычной температуре и фильтрация дает 1,75 г (53,2% ) названного выше продукта, т. пл. 187-189оС (ТСХ, Rf 0,564). ИК (KBr) : 1725 (С = 0 лактона и сложного эфира) и 1235 см-1 (ОАс), 1Н ЯМР (СД3ОД) : 2,343 (синглет, 3Н, N-СН3), 2,052 (синглет, 3Н, 4-ОАс) и 5,227 м. д. (дублет, 1Н, 4-Н).
С помощью подобного способа, за исключением того, что ангидрид уксусной кислоты заменяют ангидридом пропионовой кислоты, получают соответствующее 4-О-пропионильное производное.
П р и м е р 9. Получение 4,6, 13-0-триацетил-10-дигидро-10-дезоксо-11-метил-11-азаэритронолида А.
В раствор 10-дигидро-10-дезоксо-11-метил-11-азаэритронолида А (5 г, 11,53 ммоля) в пиридине (50 мл) загружают ангидрид уксусной кислоты (50 мл) и реакционную смесь оставляют стоять в течение 7 дней при обычной температуре. Реакцию останавливают путем добавления льда и продукт реакции выделяют путем экстракции хлороформом, как это описано в примере 8. Объединенные органические экстракты высушивают над К2СО3, выпаривают досуха и хроматографируют на силикагелевой колонке с помощью элюентной системы CH2Cl2(CH3OH) NH4OH = 90 : 9 : 1,5. Объединение фракций менее подвижного вещества, выпаривание растворителя и высушивание полученного аморфного продукта реакции дает 4,6, 13-0-ацетильного производное, т. пл. 180-182оС (Rf 0,337) ИК (KBr) : 1740 (C = О, сложный эфир) 1715 (С = О, лактон) и 1240 см-1 (О Р-Ас).
13С ЯМР (СДСl3): 43,1 (квартет, N-CH3), 173,5 (синглет), С = О лактона) и 170,2, 170,1 и 169,1 м. д. (синглеты, С = О ацетатов).
П р и м е р 10. Получение 11-N-4,6-0-триацетил-10-дигидро-10-дезоксо-11-азаэритронолида А.
Из 10-дигидро-10-дезоксо-11-азаэритронолида А (5,0 г, 11,9 ммоля) и ангидрида уксусной кислоты (50 мл) в пиридине (50 мл) получают путем ацилирования согласно способу, описанному в примере 8, сырой 11-N, 4,6-0-триацетил-10-дигидро-10-дезоксо-11-азаэритронолид А.
Реакцию ацетилирования останавливают спустя 24 часов, продукт реакции выделяют путем обычных методов экстракции хлороформом и полученный сырой осадок очищают путем суспендирования в диэтиловом эфире (50 мл), перемешивания реакционной суспензии в течение 1 ч при обычной температуре и фильтрации нерастворимого названного выше продукта реакции.
Выход: 4,08 г (68,3% ). Т. пл. 220-223оС.
Rf 0,417
ИК (KBr): 1715 (С = 0, лактон и сложный эфир), 1605 (N-Ас) и 1240 см-1 (ОАс).
1Н ЯМР (СД3ОД): 2,115 (синглет. 3Н, 4-Ас), 2,053 (синглет, 3Н, 4-ОАс) и 2,040 м. д. (синглет, 3Н, 6-ОАс).
П р и м е р 11. Получение хлоргидрата 10-дигидро-10-дезоксо-11-метил-11-азаэритронолида А.
В 25 мл воды суспендируют 10-дигидро-10-дезоксо-11-метил-11-азаэритронолида А (4,34 г, 10 ммоля) и при перемешивании, путем добавления по каплям 0,25 11 раствора HCl в течение 1 ч, доводят рН смеси до 5,8. Прозрачную реакционную смесь перемешивают в течение дополнительного часа при обычной температуре, фильтруют и лиофилизируют; выход 4,48 г (95,5% ) хлоргидрата 10-дигидро-10-дезоксо-11-метил-11-азаэритронолида А.
1Н ЯМР (СД3ОД): 3,03 м. д. (синглет, 3Н, N-СН3).
Анализ: Cl
Вычислено, % : 7,54
Найдено, % : 6,97
Аналогичным путем, заменой хлористоводородной кислоты бромистоводородной, уксусной, серной, фосфатной, лимонной, бензойной и другими кислотами получают соответствующие соли 10-дигидро-10-дезоксо-11-алкил-11-азаэритронолида А и N и/или N, О-замеденные производные его.
П р и м е р 12. Получение 10-дигидро-10-дезоксо-11-метил-11-азаэритронолида А.
Согласно способу примера 5, исходя из 10-дигидро-10-дезоксо-11-азаэритронолида А (1 г, 2,38 ммоля), формальдегида (36% -ного) (0,18 мл, 2,38 ммоля) и муравьиной кислоты (98-100% -ной) (0,183 мл, 4,77 ммоля) и реакции взаимодействия в ацетоне (20 мл) получают 0,93 г (89,8% ) названного продукта реакции.
П р и м е р 13. Получение 10-дигидро-10-дезоксо-11-этил-11-азаэритронолида А.
Согласно способу, описанному в примере 1, исходя из 10-дигидро-10-дезоксо-11-этил-11-азаэритромицина А (5 г, 6,55 ммоля) выделено 2,45 г (83,57% ) названного продукта реакции с одинаковыми физико-химическими константами, которые описаны в примере 6.
П р и м е р 14. Получение 10-дигидро-10-дезоксо-11-(Н-пропил)-11-азаэритронолида А.
Согласно способу, описанному в примере 1, исходя из 10-дигидро-10-дезоксо-11-(н-пропил)-11-азаэритромицина А (5 г, 6,44 ммоля) выделяют 2,4 г (80,7% ) названного выше продукта реакции с такими же физико-химическими константами, что и описанные в примере 7.
П р и м е р 15. Получение 6-0-дезосаминил-10-дезоксо-11-этил-11-азаэритромицина А.
В 0,25 М растворе HCl (50 мл) растворяют 10-дигидро-10-дезоксо-11-этил-11-азаэритромицин А (1 г, 1,31 ммоля) и составляют стоять в течение 16 часов при обычной температуре.
Затем реакционную смесь экстрагируют хлороформом (3 х 10 мл) и объединенные органические экстракты промывают 1 М раствором HCl и водой. Объединенный водный слой подщелачивают водным раствором натриевого щелока до рН 10 и заново экстрагируют хлороформом (3 х 20 мл).
Хлороформные экстракты высушивают над К2СО3 и выпаривают досуха. После промывки сырого продукта диэтиловым эфиром получают 0,7 г (88,4% ) названного выше продукта.
1Н ЯМР (СДСl3): 2,24 м. д. (синглет, 6Н, N(СН3)2.
С помощью подобного способа при гидролизе 6-0-дезосаминил-10-дигидро-10-дезоксо-11-(н-пропил)-11- азаэритромицина А получают соответствующее N-(н-пропил)-производное.
Найдено из ин витро и ин виво исследований, что соединения указанной формы (1) показывают сильную противовоспалительную активность. Их противовоспалительные свойства изучены ин витро, по сравнению с деклофенаком (ДИКЛ) и с D-пентицилламином (D-ПЕН), которые являются известными противовоспалительными веществами на модели внеклеточного освобождения лизосомных энзимов полиморфонуклеарными лейкоцитами человека.
Гидролиз азитромицина и получение соответствующего 6-О-дезосаминильного производного (ДЕЗАЗ) приводит к продукту с хорошей противовоспалительной активностью. При концентрации 10-5 ДЕЗАЗ показывает приблизительно равную активность, что и D-ПЕН при концентрации 10-7. Путем отщепления обеих сахарных групп и путем синтеза 10-дигидро-10-дезоксо-11-метил-11-азаэритронолида А (АЗЭР) получается соединение, которое значительно ингибирует внеклеточное освобождение лизосомных энзимов из полиморфонуклеарных лейкоцитов с активностью, подобной таковой D-ПЕН или при концентрации 10-7 - с более сильной активностью.
При ин-витро опытах ДИКЛ не влияет на внеклеточное освобождение энзимов. Ин-витро-активность N-этил-(АЭ) или N-(н-пропил)-производных иногда оказывается более низкой, по сравнению с АЗЭР. Однако ацилирование АЗЭР с помощью ангидрида уксусной кислоты и получение 4,6,13-триацетил-10-дигидро-10-дезоксо-11-метил-11-азаэритронолида А (АЛА-3) не приводит к существенному изменению в ин-витро активности.
Также были осуществлены ин-виво исследования на модели артрита у крыс, индуцированного стимулятором (вспомогательным средством). Установлено, что АЗЭР значительно понижает внеклеточное освобождение лиосомальных энзимов в синовиальную жидкость крыс с артритом, индуцированным стимулятором (вспомогательным средством), и что он проявляет уровень активности, который равен таковой D-РЕН и значительно выше чем уровень активности ДИКЛ. ДЕЗАЗ показывает относительно более низкую активность, чем D-ПЕН и ДИКЛ.
Из указанных результатов становится очевидным, что он виво метод сравним с ин-витро испытаниями. В этих опытах ин-витро и ин виво исследованные вещества незначительно влияют на освобождение энзимной лактат-дегидрогеназы (А), который доказывает, что клеточная мембрана повреждается незначительно.
Противососпалительная активность была также измерена на стек лапок у крыс, индуцированный каррагенином. Полученные результаты для соединений (1) (таблица) незначительно превышают таковые для D-ПЕН и ацетилсалициловой кислоты (Акисаля). Противовоспалительная активность N-этил и N-(н-пропил)-производных соединений (1) находится на уровне противовоспалительной активности АЗЭР. О- и N, О-замещенные производные соединений (1), например, 4,6,13-триацетил-10-дигидро-10-диоксо-11-метил-11-азаэритронолид А. гидрохлорид (АЗЭР. HCl) проявляют улучшенную активность в сравнении с D-ПЕН и почти такую же активность, как ацетилсалициловая кислота. (56) Abraham et de Nature, 1943, 151, c. 107.
Патент США N 4328334 кл. C 07 H 17/08, 1977.
Патент Великобритании N 2094293, кл. C 07 H 17/08, 1979. Патент США N 4464527, кл. A 61 K 31/71, 1979.

Сущность изобретения: продукт-10-дигидро-10-дезоксо-11-азаэритронолид А или его фармацевтически приемлемые соли 
 , где R1 C1-C3-алкил, C1-C3 -алканоил, R2, R3, R4 - Н, C1-C3-алканоил. Реагент 1: 10-дигидро-10-дезоксо-11-азаэритромицин А. Условия реакции: одностадийный гидролиз в присутствии конц. NCl и хлороформа при кипении с обратным холодильником в течен. 16 - 48 ч, или 2-стадийный гидролиз сначала в присутствии разб. NCl при комнатной температуре в течен. 10 - 20 ч, а затем в присутствии смеси конц. NCl и хлороформа при кипении с обратным холодильником и выделением 1, где R1 - C1-C3-алкил, R2, R3, R4 - Н или, когда R1 - Н, последующим кипячением с обратным холодильником с эквимолярным количеством НС(О)Н и 4,77-молярным избытком 98 - 100% -ной HCOOH в ХФ или ацетоне в течение 2 - 8 ч, или взаимодействием с 10-молярным избытком ацетальдегида или пропиональдегида в присут. H2 под давлением 20 бар в присутствии 5% -ного Pd на угле при 18 - 25С в течение 2 - 10 ч, с последующим выделением 1, где R2 - C1-C3-алкил, R2, R3, R4 - H2, или любой из полученных продуктов обрабатывают ангидридом алифатической кислоты C1-C3 в среде пиридина при комнатной температуре в течен. 2 ч. 7 дней с выделением 1, где R1 - алкил- C1-C3 или C1-C3 - алканоил, R2, R3, R4 - Н или C1-C3 - алканоил. 2 с. п. ф-лы, 1 табл.
, где R1 C1-C3-алкил, C1-C3 -алканоил, R2, R3, R4 - Н, C1-C3-алканоил. Реагент 1: 10-дигидро-10-дезоксо-11-азаэритромицин А. Условия реакции: одностадийный гидролиз в присутствии конц. NCl и хлороформа при кипении с обратным холодильником в течен. 16 - 48 ч, или 2-стадийный гидролиз сначала в присутствии разб. NCl при комнатной температуре в течен. 10 - 20 ч, а затем в присутствии смеси конц. NCl и хлороформа при кипении с обратным холодильником и выделением 1, где R1 - C1-C3-алкил, R2, R3, R4 - Н или, когда R1 - Н, последующим кипячением с обратным холодильником с эквимолярным количеством НС(О)Н и 4,77-молярным избытком 98 - 100% -ной HCOOH в ХФ или ацетоне в течение 2 - 8 ч, или взаимодействием с 10-молярным избытком ацетальдегида или пропиональдегида в присут. H2 под давлением 20 бар в присутствии 5% -ного Pd на угле при 18 - 25С в течение 2 - 10 ч, с последующим выделением 1, где R2 - C1-C3-алкил, R2, R3, R4 - H2, или любой из полученных продуктов обрабатывают ангидридом алифатической кислоты C1-C3 в среде пиридина при комнатной температуре в течен. 2 ч. 7 дней с выделением 1, где R1 - алкил- C1-C3 или C1-C3 - алканоил, R2, R3, R4 - Н или C1-C3 - алканоил. 2 с. п. ф-лы, 1 табл.

где R1 - C1 - C3-алкил или C1 - C3-алканоил;
R2, R3 и R4 - одинаковые или различные, каждый водород или C1 - C3-алканоил,
или их фармацевтически приемлемые соли, обладающие противовоспалительной активностью.
где R1 - C1 - C3-алкил или C1 - C3-алканоил;
R2, R3 и R4 - одинаковые или различные, каждый водород или C1 - C3-алканоил,
или их фармацевтически приемлемых солей, отличающийся тем, что 10-дигидро-10-дезоксо-11-азаэритромицин А общей формулы II
где R1 - водород или C1 - C3-алкил;
R2' - дезосаминильная группа;
R3' - кладинозильная группа;
R4' - водород,
подвергают одностадийному гидролизу в присутствии концентрированной соляной кислоты и хлороформа при температуре кипения с обратным холодильником в течение 16 - 48 ч или двустадийному гидролизу сначала в присутствии разбавленной соляной кислоты при комнатной температуре в течение 10 - 20 ч с получением соединения общей формулы III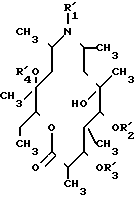
где R1' - водород или C1 - C3-алкил;
R2' - дезосаминильная группа;
R3' и R4' - водород,
а затем в присутствии смеси концентрированной соляной кислоты и хлороформа при температуре кипения с обратным холодильником и выделением продукта, общей формулы I, где R1 - водород, C1 - C3-алкил; R2, R3, R4 - водород, и в случае, когда R1 - водород, последующим восстановительным алкилированием путем кипячения с обратным холодильником с эквимолекулярным количеством формальдегида и 4,77-молярным избытком 98 - 100% -ной муравьиной кислоты в хлороформе или ацетоне в течение 2 - 8 ч или путем взаимодействия с 10-молярным избытком ацетальдегида или пропиональдегида в присутствии водорода под давлением 20 бар и катализатора - 5% -ного палладия на угле при 18 - 25oС в течение 2 - 10 ч с последующим выделением целевого продукта общей формулы I, где R1 - C1 - C3-алкил, R2, R3, R4 - водород, или любой из полученных выше продуктов подвергают ацилированию ангидридом алифатической кислоты C1 - C3 в среде пиридина при комнатной температуре в течение 2 ч - 7 дней с последующим выделением целевого продукта общей формулы I, где R1 - C1 - C3-алкил или C1 - C3-алканоил, R2, R3, R4 - одинаковые или различные, водород или C1 - C3-алканоил, при этом один из радикалов R1, R2, R3, R4 обязательно означает C1 - C3-алканоил, или переводом любого из полученных выше продуктов в его фармацевтически приемлемую соль.
