Изобретение относится к борсодержащим пептидам, новым биологически активным соединениям, которые могут найти применение в биохимии в качестве ингибиторов трипсинподобных сериновых протеаз, таких как тромбин, калликреин плазмы и плазмин.
Известны ингибиторы тромбина - высокоэффективные пептидные хлорметилкетоны (Ki = 37 нМ), которые являются эффективными в предотвращении коронарного тромбоза в модели кролика [1].
Замещенные аргининамиды, состоящие из вторичных аминов, например (2R, 4R)-4-метил-1 {N2-[(3-метил-1,2,3,4-тетрагидро-8-хинолинил)-сульфонил]-L-аргин- ил}- 2-пиперидинкарбоновая кислота [2], являются ингибиторами тромбина (Ki = 19 нМ).
Этот ингибитор повышает протромбиновые сроки плазмы в анализах на свертывание крови in vitro в 2 раза при 1 мкМ и является фибринолитически усиливающим средством для использования в сочетании с тканевым активатором плазминогена.
Наиболее эффективным из известных ингибиторов тромбина является N- α -/2-нафтилсульфонилглицил/-4-амидинофенил -аланин-пиперидин [3] (Ki = 6 нМ). Его эффективность определена у мышей и крыс in vivo.
Цель настоящего предложения - изыскание новых производных пептидной природы - малотоксичных, обладающих более высокой ингибирующей активностью и более широким спектром действия, растворы которых могут быть приготовлены без использования солюбилизатора (органического растворителя).
Поставленная цель достигается описываемыми производными борсодержащих пептидов общей формулы
R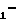 Z
Z NH
NH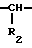 BY·HW, (I) где R1 = H, Ac, Bz, Boc, Boc-Leu;
BY·HW, (I) где R1 = H, Ac, Bz, Boc, Boc-Leu;
R2 = (CH2)nNHC(NH)NH2;
n = 3, 4;
Y = остаток пинаконового или пинандиолового эфира;
HW:C6H5SO3H, HCl, HBr;
Z = D или L-Phe, DPhe-Pro, DPhe-Phe, Ala-Lys, Pro-Phe, Ala-Phe, Glu-Phe, Ala-Glu, Glu-Gly, Gly-Leu-Ala, Pyro-Glu-Phe, Val-Val, DVal-Leu, Lys-Pro, Leu-Thr. Их получают, например, взаимодействием амина формулы
H2N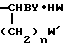
(II) где n = 3, 4;
W и WI = Cl, Br;
Y - остаток пинаконового или пинандиолового эфира, вводят во взаимодействие с аминокислотой или пептидом формулы
R1-Z-OH, (III) где значения R1 и Z указаны выше, и полученный продукт формулы
R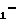 Z
Z N
N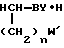 W
W
(IV) где значения R1, Z, Y, W, WI и n - указаны выше, обрабатывают азидом щелочного металла с образованием соединения формулы
R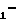 Z
Z NH
NH W
W
(V) где значения R1, Z, Y, W и n - указаны выше, которое затем восстанавливают в присутствии органической или минеральной кислоты, предпочтительно бензолсульфокислоты, с образованием соединения
R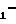 Z
Z NH
NH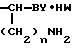
(VI) значения R1, Z, Y, W и n - указаны выше, и последнее подвергают взаимодействию с цианамидом в низшем спирте при температуре 50-150оС.
В описании изобретения используются следующие сокращения для аминокислотных остатков:
Ala = L-аланин
Arg = L-аргинин
Asn = L-аспарагин
Asp = L-аспарагиновая кислота
Gys = L-цистеин
Gln = L-глутамин
Glu = L-глутаминовая кислота
Gly = глицин
His = L-гистидин
Ile = L-изолейцин
Leu = L-лейцин
Lys = L-лизин
Met = L-метионин
Phe = L-фенилаланин
Pro = L-пролин
Ser = L-серин
Thr = L-треонин
Trp = L-триптофан
Tyr = L-тирозин
Val = L-валин
Там, где прибавлен префикс D, упомянутые выше сокращения обозначают аминокислоту D-конфигурации. Там, где прибавлен префикс D или L, упомянутые выше сокращения обозначают то, что аминокислота может иметь или конфигурацию D, или конфигурацию L.
Используемый термин "N-концевая защитная группа" относится к аминоконцевым защитным группам, используемым в синтезе пептидов. Пригодные группы включают ацильную защитную группу, ацетил (Ас), бензоил (Bz) или алифатические уретановые защитные группы, например третбутоксикарбонил (Вос).
Приводимые далее в описании примеры иллюстрируют конкретные варианты осуществления изобретения. Все указанные точки плавления не откорректированы. Все части приведены в массовых частях, а температура указана в оС. Химические сдвиги в результате спектроскопии протонного ядерного магнитного резонанса (ЯМР или 1НЯМР) указаны в дельта-единицах, частях на млн., исходя из внутреннего тетраметилсиланового стандарта. Многочисленные сокращения, используемые в указанных примерах, имеют следующие значения: ТFA - трифторуксусная кислота; DMF - -N,N-диметилформамид; МС - масс-спектрометрия; ТСХ - тонкослойная хроматография; оф-ТСХ - тонкослойная хроматография с обращенной фазой. Сложноэфирные защитные группы борных кислот имеют аббревиатуры: -C6H12 = пинаконовая группа и -C10H16 = пинандиоловая группа.
Все аминокислотные остатки представлены в L-конфигурации, если не оговорено особо.
ТСХ и оф-ТСХ осуществляют на пластинках из силикагеля 60 фирмы "Е.Мерк" и пластинках КI18F для оф-ТСХ фирмы Whatman. Нейтральные соединения наблюдали в УФ-области и после экспонирования парами йода. Соединения, содержащие свободные аминогруппы, подкрашивали нин гидрином, а соединения с гуанидиногруппами подкрашивали красителем Sakaguchi. Краситель Sakaguchi проявляет значительную специфичность к монозамещенным гуанидинам, например которые содержатся в бораргининовых пептидах.
П р и м е р 1а. 1-Амино-4-бром-бутилборонат пинандиол ˙HCl.
NH2-CH[(CH2)3Br]BO2-C10H16 ˙HCl
4-Бром-1-хлорбутилборонатпинандиол получают по методике, описанной Matte Son и др. В обычном эксперименте осуществляют гидроборирование аллилбромида (173 мл, 2,00 моль) бораном пирокатехина (240 мл, 2,00 моль) путем прибавления борана к аллилбромиду с последующим нагреванием реакционной смеси в течение 4 ч до 100оС в атмосфере азота. Полученный продукт, пирокатехин 3-бромпропилбороната (т. кип. 95-102оС, 0,25 мм), выделяют перегонкой с выходом 49% . Указанный пирокатехиновый эфир (124 г, 0,52 моль) трансэтерифицируют (+) α-пинандиолом (88 г, 0,52 моль) при смешивании их в 50 мл тетрагидрофурана (THF), интенсивно перемешивая указанную смесь в течение 0,5 ч при 0оС, а затем 0,5 ч при комнатной температуре. Растворитель упаривают и прибавляют 250 мл гексана. Пирокатехин удаляют в виде кристаллического твердого вещества. Количественный выход получают путем последовательного разбавления гексаном до объема 500 и 1000 мл с выделением кристаллов при каждом разведении. При упаривании растворителя получают продукт (147 г) в виде масла.
C13H22O2BrB.
Вычислено, %: C 51,85; H 7,38; Br 26,54.
Найдено, %: C 52,85; H 7,30; Br 26,58.
4-Бром-1-хлорбутилбороната пинандиол получают гомологизацией соответствующего пропилбороната. Хлористый метилен (34,8 мл, 0,540 моль) растворяют в 500 мл THF и затем медленно прибавляют к 1,54М н-бутиллития в гексане (350 мл, 0,540 ммоль) при температуре -100оС. Пинандиол 3-бромпропилбороната (148 г, 0,490 моль) растворяют в 500 мл THF, охлаждают до точки замерзания указанного раствора и вводят в реакционную смесь. Хлорид цинка (33,5 г, 0,246 моль) растворяют в 250 мл THF, охлаждают до 0оС и прибавляют несколькими порциями к реакционной смеси. Реакционную смесь при одновременном перемешивании нагревают постепенно до комнатной температуры в течение ночи. Затем растворитель упаривают, а полученный осадок растворяют в гексане и промывают водой. После просушивания над безводным сульфатом магния и фильтрования, растворитель удаляют с выходом целевого продукта (140 г).
Пинандиол 1-амино-4-бромбутилбороната получают путем растворения гексаметилдисилизана (28,0 г, 80,0 ммоль) в 30 мл THF, охлаждения полученного раствора до -78оС и прибавления к нему 1,62 н. н-бутиллития в гексане (49,4 мл, 80,0 моль). Затем раствор медленно нагревают до комнатной температуры, повторно охлаждают до -78оС и прибавляют пинандиол 4-бром-1- -хлорбутилбороната (28,0 г, 80,0 моль) в 20 мл THF. Реакционную смесь медленно нагревают до комнатной температуры и перемешивают в течение ночи. Растворитель удаляют упариванием, вводят сухой гексан (400 мл) и получают осадок, который выделяют фильтрованием в атмосфере азота. Фильтрат охлаждают до -78оС и добавляют 4н. HCl в диоксане (60 мл, 240 моль). Затем реакционную смесь медленно нагревают до комнатной температуры и при этой температуре перемешивают ее 2 ч. Полученный продукт (20 г) выделяют фильтрованием в виде твердого вещества. После просушивания в вакууме неочищенный продукт растворяют в хлороформе и нерастворившееся вещество выделяют фильтрованием. Фильтрат упаривают и осадок растворяют в этилацетате. Целевой продукт выкристаллизовывают из этилацетата с выходом его 15,1 г (т.пл. 142-144,5оС, [α] D-25 = +16,7 ± 0,80; c = 1,0, в абсолютном метаноле.
C14H26NO2BrClB.
Вычислено, %: C 45,87; H 7,16; N 3,82; B 2,95.
Найдено, %: C 45,76; H 7,21; N 3,79; B 3,04.
П р и м е р 1b.
(D,L) 1-амино-4-бромбутилборонатпинакон ˙HCl.
(D,L) NH2-CH[(CH2)3Br]BO2-C6H12 ˙HCl.
Пинакон 4-бром-1-хлорбутилбороната получают по методу, описанному для получения соответствующего пинандиола (пример Ia), за исключением того, что вместо пинандиола используют пинакон, и пинакон 3-бром-пропилбороната (т. кип. 60-64оС, 0,35 мм) и 4-бром-1-хлорбутилбороната пинакон (т.кип. 110-112оС, 0,20 мм) перегоняют.
C10H19O2BrClB.
Вычислено, %: C 40,38; H 6,45.
Найдено, %: C 40,70; H 6,37.
Хлористоводородную соль пинакона 1- -амино-4-бромбутилбороната также получают по методике примера 1а. Целевой продукт выкристаллизовывают из смеси этилацетат:гексан с выходом его 52%.
C10H22NO2BrClB.
Вычислено, %: C 38,19; H 7,05; N 4,45; Cl 11,27; Br 25,41.
Найдено, %: C 38,28; H 7,39; N 4,25; Cl 11,68; Br 26,00.
П р и м е р 1с. Хлористоводородная соль пинакона 1-амино-4-хлорбутилборонат.
(D,L)NH2-CH[(CH2)3Cl]BO2-C6H12 ˙HCl.
Пирокатехин 3-хлорпропилборонат (т. кип. 80-85оС, 0,30 мм) и пинакон 3-хлор- пропилового эфира бороновой кислоты (т.кип. 63оС, 0,20 мм) получают по методике примера 1а, за исключением того, что вместо аллилбромида используют аллилхлорид, а вместо пинандиола берут пинакон.
C9H18O2ClB.
Вычислено, %: C 52,85; H 8,89; Cl 17,33.
Найдено, %: C 53,41; H 8,15; Cl 16,81.
Гомологизацию также осуществляют по методике примера 1а, а полученный продукт выделяют перегонкой (т.кип. 95оС, 0,25 мм) с 65%-ным выходом.
С10Н19О2Cl2B.
Вычислено, %: С 47,47; Н 7,58; Cl 28,02.
Найдено, %: C 47,17; H 7,45; Cl 27,75.
Хлористоводородную соль пинакона 1- -амино-4-хлорбутилового эфира бороновой (борной) кислоты получают по методике, аналогичной в примере 1а. Целевой продукт выкристаллизовывают из этилацетата с получением 8,8 г продукта (т.пл. 132-135,5оС) и 2,2 г продукта (т.пл. 145-147оС). Продукт с т.пл. 145-147оС используют для анализа.
C10H22NO2ClB.
Вычислено, %: C 44,47; H 8,23; N 5,19; B 4,00.
Найдено, %: C 44,01; H 8,23; N 4,77; B 3,80.
П р и м е р 1d (D,L) 1-амино-5-бромпентилового эфира бороновой кислоты пинакон ˙HCl.
(D,L) NH2-CH[(CH2)4Br]BO2C6H12 ˙HCl.
Пинакон 4-бромбутилового эфира бороновой кислоты получают по методу, описанному для получения пинандиола 3-бромпропилового эфира борной кислоты (пример 1а), за исключением того, что аллилбромид заменяют на 4-бром-1-бутен, а пинандиол - на пинакон. Продукт выделяют в виде масла (т.кип. 77оС, 0,3 мм). После гомологизации получают 5-бром-1-хлорпентилового эфира бороновой кислоты пинакон.
Масс-спектроскопия (Cl): C11H21O2BrClB.
Вычислено, -Н: 310,47.
Найдено, -Н: 310.
Целевой продукт хлористоводородную соль пинакона 1-амино-5-бромпентилового эфира бороновой кислоты получают по методике примера 1а при выходе 35%.
С11H24NO2BrBCl.
Вычислено, %: C 40,22; H 7,36; N 4,26; Cl 10,79; Br 24,32; B 3,29.
Найдено, %: C 39,23; H 7,18; N 4,04; Cl 15,21; Br 25,66; B 3,75.
П р и м е р 2. Boc-(D)Phe-Pro-NH- -CH[(CH2)3Br]BO2-C10H16.
Синтез дипептида Boc-(D)Phe-Pro-OH осуществляют сначала с получения дипептидного бензилового эфира, а затем удаления указанного сложного эфира каталитическим гидрированием. Boc-(D) -Phe-OH (10,0 г, 37,7 ммоль) растворяют в 50 мл THF и к полученному раствору прибавляют N-метилморфолин (4,14 мл, 37,7 ммоль). Раствор охлаждают до -20оС, а затем к нему прибавляют изобутиловый эфир хлормуравьиной кислоты (4,90 мл, 37,7 ммоль). Через 5 мин в реакционную смесь вводят Н-Pro-OBr 1-.HCl (9,11 г, 37,7 ммоль), растворенный в 50 мл хлороформа и охлажденный до -20оС. Прибавляют триэтиламин (5,25 мл, 37,7 ммоль) и полученную смесь перемешивают 1 ч при -20оС и 2 ч при комнатной температуре. Реакционную смесь фильтруют и фильтрат упаривают. Осадок растворяют в этилацетате с последующим промыванием 0,2н. HCl, 5% -ным водным раствором бикарбоната натрия и насыщенным водным раствором хлористого натрия. Органический слой сушат над безводным сульфатом натрия, фильтруют и упаривают с получением 15,2 г Boc-(D) Phe-Pro-OBz 1 в виде масла. Полученный бензиловый эфир (15,2 г) растворяют в 100 мл метанола и затем гидрируют при начальном давлении 40 фунтов/дюйм2 (2,812 кг/см2) в аппарате Парра в присутствии 0,5 г 10% палладиевой черни (Pd/C). Реакционный раствор фильтруют через цеолиттм и упаривают с выходом твердого вещества. Указанный твердый продукт выделяют и промывают этилацетатом, а затем простым эфиром с получением 10,0 г требуемого продукта (т.пл. 176,5-177оС).
C19H26N2O5
Вычислено, %: C 62,95; H 7,24; N 7,73.
Найдено, %: C 62,91; H 7,15; N 7,53.
Boc-(D)Phe-Pro-NH-CH[(CH2)3Br] BO2-C10H16 получают связыванием указанного дипептида с соответствующим амином по методике смешанного ангидрида. Смешанный ангидрид Boc- -(D)Phe-Pro-OH получают при растворении указанной кислоты (4,94 г, 13,6 ммоль) в 30 мл THF c последующим прибавлением N-метилморфолина (1,50 мл, 13,6 ммоль). Затем реакционный раствор охлаждают до -20оС и добавляют изобутиловый эфир хлормуравьиной кислоты (1,77 мл, 13,6 ммоль). После перемешивания в течение 5 минут при -20оС, в реакционную смесь вводят амин, который получен в примере 1а, NH2- -CH[(CH2)3Br]BO2-C10H16˙HCl (5,0 г, 13,6 ммоль), растворенный в 10 мл охлажденного хлороформа. Охлажденный тетрагидрофуран (10 мл) и триэтиламин (1,90 мл, 13,6 ммоль) прибавляют к реакционной смеси, и перемешивают смесь в течение 1 ч при -20оС и примерно 2 ч при комнатной температуре. Смесь фильтруют и оставшуюся жидкость в фильтрате упаривают. Осадок растворяют в этилацетате и промывают 0,2н. HCl, 5%-ным водным раствором бикарбоната натрия и насыщенным водным раствором натрийхлорида. Органическую фазу сушат над безводным сульфатом натрия, фильтруют, а растворитель упаривают с выходом 9,0 г продукта в виде масла. Указанный продукт растворяют в метаноле и хроматографируют на колонке 2,5 х 50 см с LH-20. Фракции, содержащие целевой продукт, объединяют и упаривают с получением 5,8 г твердого вещества. ТСХ в смеси метанол/хлороформ (1:9) показывает единичное пятно; Rf 0,70.
C33H49N3O6BBr.
Вычислено, +Н: 674,30.
Найдено, +Н: 674,30.
П р и м е р 3. Boc-(D)Phe-Pro-NH- -CH[(CH2)3N3]BO2-C10H16.
Продукт примера 2, Boc-(D)Phe-Pro-NH--CH[(CH2)3Br]BO2-C10H16 (4,4 г, 6,54 ммоль) растворяют в 7 мл диметилформамида (DMF) и азид натрия (0,919 г, 14,1 ммоль) прибавляют к указанному раствору. Реакционную смесь нагревают при 100оС в течение 3 ч. Прибавляют этилацетат (100 мл) к реакционной смеси и затем промывают водой и насыщенным водным раствором хлористого натрия. Полученную органическую фазу сушат над безводным сульфатом натрия, фильтруют и упаривают. Получают 4,1 г твердого продукта. Указанный продукт хроматографируют на колонке 2,5 х 50 см из LH-20 в метаноле. Фракции, содержащие целевой продукт, объединяют, оставшуюся жидкость упаривают с выходом 2,3 г названного азида. ТСХ в системе метанол: хлороформ (1:9) показывает единичное пятно; Rf 0,76.
C33H48N6O6B.
Вычислено, %: C 62,35; H 7,63; N 13,33; B 1,70.
Найдено, %: C 63,63; H 8,02; N 11,58; B 1,80.
Масс-спектроскопия (FAB) C33H48N6O6B.
Вычислено, %: +H 637,39.
Найдено, %: +H 637,49.
П р и м е р 4. Boc-(D)Phe-Pro-NH- -CH[(CH2)3NH2]BO2-C10H16 ˙Бензолсульфокислота.
Азид примера 3 (8,80 г, 13,8 ммоль) растворяют в 150 мл метанола и гидрируют на установке Парра при давлении 40 фунтов/дюйм2 (2,812 кг/см2) в присутствии 0,50 г 10% палладиевой черни и бензолсульфокислоты (2,19 г, 13,8 ммоль). Через час катализатор удаляют, и после упаривания раствора получают 9,9 г требуемого продукта. оф-ТСХ в системе растворителей метанол: вода (85: 15) показывает пятно в УФ области; Rf 0,91 и пятно, позитивное к нингидрину, Rf 0,52.
П р и м е р 5. Boc-(D)Phe-Pro-NH- -CH[(CH2)3-NH-C(NH)NH2]BO2-C10H16˙ бензолсульфокислота.
Boc-(D)Phe-Pro-бороArg-C10H16 ˙бензол- сульфокислота.
Boc-(D)Phe-Pro-бороArg-C10H16 ˙бензолсульфокислоту, полученную в примере 4 (4,6 г, 6,11 ммоль), нагревают с обратным холодильником при 100оС в 20 мл абсолютного этанола, содержащего цианамид (50 мг/мл). Ход реакции регулируют оф-ТСХ в системе растворителей метанол:вода (85:15), где наблюдается исчезновение нин- гидринового пятна из исходного материала амина (Rf 0,54) и появление красителя Sakaguchi на полученном продукте (Rf 0-0,13). Указанный продукт можно обнаружить после нагревания с обратным холодильником в течение 18 ч, причем его уровень постепенно повышается. Через 7 дней, когда амин нельзя обнаружить, реакционный раствор концентрируют до примерно 50% объема пассивным упариванием. Затем реакционный раствор фильтруют, сгущают и хроматографируют на 2,5 х 100 см колонке с LH-20 в метаноле. Фракции, содержащие требуемый продукт, объединяют и после упаривания получают 3,7 г требуемого продукта. Порцию продукта (2,3 г) кристаллизуют смесью этилацетат:гексан, с выходом 0,89 г вещества, а остаток (1,2 г) получают порошкованием простым эфиром в виде твердого продукта.
Масс-спектроскопия (FAB) для С34H53N6O6SB.
Вычислено, % : +H 653,42.
Найдено, %: +H 653,38.
Элементный анализ для С40Н59N6O9SB. H2O, %.
Вычислено, %: C 57,95; H 7,43; N 10,14; B 1,30.
Найдено, %: C 57,20; H 7,14; N 10,94; B 1,01.
П р и м е р 6. Н-(D)Phe-Pro-бороArg- -C10H16 ˙2HCl.
Продукт примера 5, Boc-(D)Phe-Pro-бороArg-C10H16, бензолсульфокислота (1,17 г, 1,54 ммоль), подвергают взаимодействию с 5 мл 4н. хлористого водорода в диоксане в течение 15 мин при комнатной температуре. Полученный продукт осаждают при добавлении простого эфира, выделяют и промывают простым эфиром с просушиванием в вакууме. Затем продукт растворяют в 10 мл воды и наносят на анионно-обменную колонку 5 мл анионно-обменную колонку (Cl--форма Bио-RAD AGI X8тм Richmond, CA) c последующим промыванием колонки водой (примерно 30 мл). Элюент упаривают в вакууме, а осадок порошкуют простым эфиром с выходом требуемого продукта (0,80 г).
Масс-спектроскопия (FAB) для C29H45N6O4B:
Вычислено, %: +H 553,37.
Найдено, %: + H 553,40 и 538,40 (не идентифицирован).
Элементный анализ H-(D)Phe-Pro-бороArg-C10H16˙ I BSA.TFA:
Найдено, %: 553,4.
П р и м е р ы 7-8. Ac-(D)Phe-Pro-бороArg--C10H16˙ HCl (пример 7).
Ac-(D)Phe-Pro-бороArg-OH ˙ HCl (пример 8).
Boc-(D)Phe-Pro-бороArg-C10H16 ˙ бензолсульфокислота, продукт примера 5 (0,86 г, 1,13 ммоль), подвергают взаимодействию с безводной трифторуксусной кислотой (TFA) (примерно 5 мл) в течение 15 мин при комнатной температуре. Избыток TFA удаляют упариванием, а полученный осадок порошкуют простым эфиром с выходом 0,76 продукта. Указанный продукт (0,70 г, 0,91 ммоль) растворяют в смеси, состоящей из 2 мл диоксана и 1 мл воды. Уксусный ангидрид (0,47 мл, 5,0 ммоль) и бикарбонат натрия (0,42 г, 5,0 ммоль) прибавляют к реакционному раствору. Затем полученную смесь перемешивают в течение 20 мин при комнатной температуре. Добавляют этилацетат (50 мл) и воду (5 мл). Образовавшиеся фазы разделяют и органический слой сушат над безводным сульфатом натрия. После фильтрования и удаления растворителя упариванием получают 0,56 г полутвердого вещества.
Полученную пробу растворяют в 4 мл ледяной уксусной кислоты с последующим разбавлением раствора 16 мл воды. Затем сразу же наносят на колонку с 15 мл SP-сефадексатм (Н+-форма) и уравновешивают 20%-ным раствором уксусной кислоты. Колонку промывают 300 мл 20%-ной уксусной кислоты и устанавливают линейный градиент концентраций от 100 мл 20%-ной уксусной кислоты, до 100 мл 20%-ной уксусной кислоты, хлористоводородную кислоту. Фракции, собранные в градиенте концентрации от 0,08 до 0,17н. HCl, содержат N-ацетиловый пептид (0,29 г) в виде смеси из свободной бороновой кислоты и сложного пинандиолового эфира.
Сложный эфир пинандиола и свободную бороновую кислоту разделяют хроматографией на 2,5 х 100 см колонке, содержащей 1Н-20 в метаноле. Размер фракций составляет 8,2 мл. Пинандиоловый эфир (102 мг) элюируют в фракциях 41-43, в то время как свободную борную кислоту (131 мг) постепенно вымывают в фракциях 45-129.
Масс-спектроскопия (FAB) (пример 7):
Ac-(D)Phe-Pro-бороArg-C10H6 для C31H47N6O5B:
Вычислено, +Н: 595,33.
Найдено, +Н :595,33.
Масс-спектроскопия (FAB) (пример 8):
Ac-(D)Phe-Pro-бороArg-OH˙ HCl для C21H33N6O5:
Вычислено, +Н: 449,60.
Найдено, +НI: 579,24-581,24.
Последние данные нельзя объяснить. Тем не менее, данные ЯМР совпадают со структурой свободной бороновой кислоты, так как заданные полосы испускания для пинандиоловой группы, например как синглеты метильных групп при δ 0,85 (3Н), 1,30 (3Н) и 1,36 (3Н), не наблюдались. В качестве дополнительного доказательства этой структуры, образец продукта свободной бороновой кислоты переэтерифицируют с выходом материала примера 7. Аналитическую пробу (20 мг) обрабатывают двукратным избытком пинандиола (14 мг) в 3 мл метанола в течение 5 мин. Растворитель упаривают, а избыток пинандиола удаляют порошкованием простым эфиром указанного образца с выходом требуемого продукта (26 мг).
Масс-спектроскопия (FAB) (найдено: 595,38) и данные ЯМР совпадают со структурой, предполагаемой для этерифицированного продукта и почти идентичны пинандиоловому продукту примера 7.
П р и м е р 9. Ac-Phe-бороArg-C10H16˙ ˙HCl.
По методике, описанной в примере 2, получают Ac-Phe-NH-CH[CH2)3Br]BO2- -C10H16. Затем получают смешанный ангидрид Ac-Phe-OH (0,565 г, 2,73 ммоль) в 10 мл TFA, который связывают с NH2-CH[(CH2)3Br]BO2-C10H16˙HCl (продукт примера 1а, 1,00 г, 2,73 ммоль), растворенного в 10 мл охлажденного THF c выходом 1,47 г белой пены. Указанный продукт перемешивают в течение ночи вместе с гексаном. Получают твердое вещество 1,01 г (т.пл. 106,5-109оС).
Элементный анализ для C25H36N2O4BrB, %:
Вычислено, %: C 57,81; H 7,00; N 5,40; Br 15,40; B 2,08.
Найдено, %: C 58,33; H 7,33; N 4,76; Br 14,18; B 1,80.
Масс-спектроскопия (FAB) для С25Н36N2O4BrB.
Вычислено, +Н: 519,20.
Найдено, +Н: 519,23.
Ac-Phe-NH-CH[(CH2)3N3] BO2-C10H16 получают при обработке Ac-Phe-NH- -CH[(CH2)3Br]BO2-C10H16 (3,22 г, 6,20 ммоль) натрийазидом по методике, описанной в примере 3. Полученный продукт (3,03 г) хроматографируют на колонке с LH-20. Фракции, содержащие требуемый продукт, объединяют и упаривают. Осадок порошкуют гексаном с выходом 2,21 г указанного азида.
Бензолсульфонат Ac-Phe-бороОrn-C10H16 получают из указанного азида Ac- -Phe-NH-CH[(CH2)3N3] BO2-C10H16 (2,21 г, 4,59 ммоль) по методике примера 4, за исключением того, что гидрирование осуществляют при атмосферном давлении. После фильтрования и упаривания растворителя путем порошкования простым эфиром получают требуемый продукт (2,22 г).
Ac-Phe-бороArg-C10H16 ˙бензолсульфонат получают путем обработки соединения Ac-Phe-бороOrn-C10H16˙ ˙бензолсульфокислота (2,0 г, 3,26 ммоль) 10 мл раствора цианамида (100 мг/мл) в этаноле. Реакцию гуанидирования осуществляют по методике примера 5, за исключением того, что реакционное время составляет 3 дня, а реакционная смесь содержит смесь из исходного материала и полученного продукта. Указанная смесь требует дополнительной очистки, которую по всей вероятности можно не проводить, продлив продолжительность реакции. Реакционный раствор далее концентрируют и хроматографируют на колонке 2,5 х 100 см из LH-20 в метаноле. Фракции, содержащие требуемый продукт, обнаруженные при помощи подкрашивающего агента Sakaguchi, объединяют и упаривают с выходом 1,4 г продукта. Полученный продукт (1,2 г) растворяют в 6 мл уксусной кислоты, разбавляют 30 мл воды и получают раствор молочного цвета. Этот раствор наносят на колонку объемом 30 мл с SP-Сефадексомтм С-25 (Н+-форма), доведенного до равновесного состояния 20%-ным водным раствором уксусной кислоты. Далее колонку промывают 240 мл 20%-ной уксусной кислоты и устанавливают линейный градиент концентрации от 250 мл 20%-ной уксусной кислоты до 250 мл 20%-ной уксусной кислоты, содержащей 0,30 н. соляной кислоты. Фракции, элюированные из колонки в интервале от 0,12 до 0,16 н. соляной кислоты, объединяют с выходом 0,42 г требуемого пептида в виде смеси свободной борной кислоты и сложного эфира пинандиола. Указанную смесь растворяют в метаноле (10 мл) и для этерификации свободной борной кислоты прибавляют 80 г пинандиола. После перемешивания в течение 30 мин растворитель упаривают, а после порошкования осадка простым эфиром получают 0,28 г требуемого продукта.
Элементный анализ для C26H40N5O4B˙ ˙HCl ˙2H2O, %:
Вычислено: C 54,78; H 8,15; N 12,30; B 1,90.
Найдено: C 55,34; H 7,83; N 11,66; B 1,99.
Масс-спектроскопия (FAB) для C26H40N5O4B:
Вычислено, +Н: 498,32.
Найдено, +Н: 498,31.
П р и м е р 10. Ac-(D,L)Phe-(D,L)-бороArg--C6H12.
Промежуточное соединение Ac- -(D,L)Phe-(D,L)-NH-CH-[(CH2)3Br]BO2-C6H12 получают модифицированием методов, описанных в примере 1 и 2. Хлорангидрид Ac- -Phe-OH получают при взаимодействии Ac-Phe-OH (30 г, 0,145 моль) с пентахлоридом фосфористой кислоты (30 г, 0,144 моль) в 175 мл THF при -10оС. Реакционную смесь перемешивают при 0оС приблизительно в течение 1 ч, затем разбавляют до объема 350 мл охлажденным простым эфиром, продукт выделяют в виде твердого вещества, промывают холодным простым эфиром и после высушивания в вакууме получают 21 г продукта. Активированное ацетилфенилпроизводное (14,8 г, 65,6 ммоль) растворяют в 40 мл THF и прибавляют к продукту взаимодействия пинакона 4-бром-1-хлорбутилбороната и гексаметилдисилизана (приготовленных в 20 ммольной концентрации) при -78оС. Затем реакционную смесь нагревают до комнатной температуры с последующим перемешиванием ее в течение ночи. Растворитель удаляют путем упаривания. Осадок растворяют в этилацетате и промывают последовательно водой, 5%-ным раствором бикарбоната натрия и насыщенным водным раствором хлористого натрия. Органическую фазу полученной смеси сушат над безводным сульфатом натрия и после концентрирования получают требуемый продукт в виде кристаллического твердого вещества (1,37 г; т.пл. 146,5-148оС). При исследовании структуры кристалла получен следующий химический состав.
Элементный анализ для C21H32N2O4BrB, %:
Вычислено: C 53,98; H 6,92; N 6,00; Br 17,10; B 2,31.
Найдено: C 54,54; H 6,78; N 5,89; Br 16,46; B 3,40.
Алкилбромид превращают в соответствующий азид по методике примера 3. Продукт выкристаллизовывают из этилацетата (т.пл. 143-144оС).
Элементный анализ для C21H32N5O4B, %:
Вычислено: C 58,74; H 7,53; N 16,31; B 2,53.
Найдено: C 58,85; H 7,48; N 16,53; B 2,93.
Полученный азид превращают в соль бензолсульфокислоты Ac-(D,L)Phe-(D, L)-бороOrn-C6H12 по методике примера 4, за исключением того, что гидрирование проводят при атмосферном давлении.
Ac-(D, L)Phe-(D, L)бороOrn-C6H12 ˙бензолсульфокислота (0,243 г, 0,433 ммоль) подвергают взаимодействию с цианамидом (0,20 г, 0,476 ммоль) при 100оС в 2 мл абсолютного этанола в течение ночи. Раствор концентрируют и после порошкования простым эфиром получают 0,21 г белого вещества. При оф-ТСХ отмечено характерное положительное окрашивание полосы при использовании красителя Sakaguchi в случае бороаргининовых пептидов, Rf 0-0,55, и дискретное пятно, Rf 0,68, соответствующее непрореагированному исходному материалу. Полученный продукт (81 мг) повторно обрабатывают 2 мл раствора цианамида (10 мг/мл) в течение ночи по вышеуказанной методике и после порошкования простым эфиром получают 71 мг целевого продукта.
Масс-спектроскопия (FAB) для C22H37N5O4B:
Вычислено, +H: 446,30.
Найдено, +Н: 446,23 и 404,19 (соответствующий непрореагированному боронитиновому пептиду) .
Следует иметь в виду, что методика по примеру 5, представляет собой наилучший способ получения бороаргининовых пептидов и отличается в том, что используют больший избыток цианамида и более продолжительное время взаимодействия.
П р и м е р 11. Boc-(D)Phe-бороArg- -C10H16-бензолсульфонат.
Boc-(D)Phe-Phe-OH получают по методике, описанной для получения дипептида Boc-(D)Phe-Pro-OH в примере 2. После гидрирования сложного бензилового эфира, полученный продукт выкристаллизовывают из смеси хлороформ:гексан с выходом требуемого пептида (т.пл. 133-133,5оС).
Элементный анализ для C23H28N2O5, %:
Вычислено: C 66,96; H 6,86; N 6,79.
Найдено: C 66,75; H 6,79; N 6,56.
Boc-(D)Phe-Phe-NH-CH[(CH2)3Br] BO2-C10H16 получают путем смешивания Boc-(D)Phe-Phe-OH (6,00 г, 14,5 ммоль) с NH2-CH[(CH2)3Br]BO2-C10H16˙ ˙HCl (пример 1а, 5,33 г, 14,5 ммоль) по методике, описанной в примере 2, за исключением того, что не выполняют хроматографии на колонке с LH-20. Полученный продукт выкристаллизовывают из этилацетата с выходом в первом погоне 2,47 г продукта (т. пл. 132-134оС и 5,05 г продукта (т.пл. 133-135оС) во втором погоне. При оф-ТСХ в системе метанол:вода (85:15) отмечено единичное пятно, Rf 0,29.
Элементный анализ для C37H51N3O6BrB, %:
Вычислено: C 61,32; H 7,11; N 5,80; Br 11,03.
Найдено: C 61,21; H 7,02; N 5,59; Br 10,22.
Boc-(D)Phe-Phe-NH-CH[(CH2)3N3]BO2-C10H16 получают при обработке соответствующего алкилбромида (7,15 г, 9,87 ммоль) азидом натрия по методике, описанной в примере 3, за исключением того, что для очистки не нужно проводить хроматографического разделения на колонке с LH-20. Требуемый продукт элюируют из смеси растворителей этилацетат:гексан в виде геля и после выделения и промывания гексаном получают 3,0 г требуемого продукта в первом погоне и 2,9 г во второй порции. Бензолсульфонат Boc-(D)Phe-Phe-бороOrn-C10H16 получают из указанного азида (5,37 г, 7,82 ммоль) по методике, описанной в примере 4 с выходом 5,33. При оф-ТСХ в смеси метанол:вода (85: 15) отмечается интенсивное положительное окрашивание нингидрином, Rf 0,42 и слабое пятно в УФ-области, 0,92 (образование пятна в УФ-области при Rf 0,92 характерно для аминов или гуанидиновых соединений, которые являются солями бензолсульфокислоты).
Масс-спектроскопия (FAB) для С37H53N4O6B:
Вычислено, +Н: 661,76.
Найдено, +H: 661,14.
Boc-(D)Phe-Phe-бороArg-C10H16 получают по методике, описанной в примере 5. Бороорнитиновый пептид (4,83 г, 5,90 ммоль) обрабатывают раствором цианамида (50 мг/мл) в 20 мл абсолютного этанола в течение 7 дней. Из реакционной смеси выделяют порцию продукта, соответствующую 1,0 г исходного материала, и нагревают отдельно без обратного холодильника в течение ночи с полным превращением амина в гуанидино-соединение. После хроматографии на колонке с LH-20 и порошкования продукта простым эфиром получают 0,52 г требуемого бонила.
Элементный анализ для C44H61N6O9SB, %:
Вычислено: C 61,38; H 7,16; N 9,76; B 1,25.
Найдено: C 59,69; H 7,41; N 9,82; B 1,26.
Масс-спектроскопия (FAB) для C38H55N6O6B.
Вычислено, +Н: 703,43.
Найдено, +Н: 703,49.
П р и м е р 12. H-(D)Phe-Phe-бороArg- -C10H16 ˙2HCl.
Бензолсульфонат Boc-(D)Phe-Phe-бороArg-C10H16 (полученный по примеру 11, 0,59 г, 1,25 ммоль) деблокируют по методике примера 6, за исключением того, что пробу наносят на ионнообменную колонку в 20%-ном этаноле с последующим промыванием указанной колонки 20%-ным раствором этанола. Продукт получают (0,424 г) в виде белого твердого вещества.
Масс-спектроскопия (FAB) для C33H47N6O4B:
Вычислено, +Н: 603,38.
Найдено, +Н: 603,41.
П р и м е р 13. Бензолсульфат Ac-Ala-Lys(Boc)-бороArg-C10H16.
Ac-Ala-Lys(Boc)-OH получают путем сопряженной реакции N-оксиянтарной кислоты имидоэфира Ac-Ala-OH (полученного по методу Anderson) c H-Lys(Boc)-OH. N-оксисукцинимид Ac-Ala-OH (6,25 г, 27,4 ммоль) растворяют в 30 мл диоксана и прибавляют к раствору H-Lys(Boc)-OH (7,50 г, 30,4 ммоль), который растворен в смеси, состоящей из 30 мл 1,0 н. NaOH и триэтиламина (2,12 мл, 15,0 ммоль). Реакционную смесь перемешивают в течение ночи, а затем подкисляют соляной кислотой. Для получения требуемой насыщаемости раствора добавляют нужное количество сухого хлорида натрия. Продукт экстрагируют в этилацетате и затем промывают 0,2 н. HCl, полученной в насыщенном водном растворе хлористого натрия. Растворитель удаляют путем упаривания. После кристаллизации продукта из смеси этилацетат:гексан получают 7,3 г требуемого продукта, (т.пл. 86-89оС).
Ac-Ala-Lys(Boc)-NH-CH[(CH2)3Br] BO2-C10H16 получают по методике примера 2 за исключением того, что получаемый продукт очищают фракционированной кристаллизацией из этилацетата. В продукте (1,13 г), полученном во второй и третьей фракции, отмечается единичное пятно при оф-ТСХ в смеси растворителей метанол: вода (85: 15) при значении Rf 0,51. Тонкослойную пластинку подвергают воздействию паров хлористоводородной кислоты, на которой после прибавления нингидринового индикатора обнаруживают целевой амин.
Ac-Ala-Lys(Boc)-NH-CH[(CH2)3N3] BO2-C10H16 получают из соответствующего алкилбромида (1,95 г, 2,90 ммоль) по методике описанной в примере 3, за исключением того, что очистку продукта осуществляют кристаллизацией его из этилацетата, а не хроматографией на LH-20. После кристаллизации неочищенного продукта (1,60 г) получают 0,55 г чистого продукта (т.пл. 79-84оС) и 0,96 г осадка. Ниже представлен химический состав кристаллического продукта.
Элементный анализ для C30H52N7O7B, %:
Вычислено: C 56,86; H 8,29; N 15,48; B 1,71.
Найдено: C 56,76; H 8,26; N 15,89; B 1,65.
Бензолсульфонат Ac-Ala-Lys(Boc)-бороOrn-C10H16 получают из соответствующего алкилазида (0,433 г, 0,683 ммоль) по методике, описанной в примере 4. После удаления кристаллизатора и растворителя получают названный продукт (0,45 г) при порошковании простым эфиром.
Масс-спектроскопия (FAB) для C30H54N5O7B:
Вычислено, +Н: 608,42.
Найдено, 608,49.
Бензолсульфонат Ac-Ala-Lys(Boc)-бороArg-C10H16 получают при взаимодействии соответствующего бороорнитинового пептида с цианамидом по методике, описанной в примере 5. Хроматографические фракции, содержащие требуемый продукт, порошкуют простым эфиром с выходом 0,83 г продукта в виде белого твердого вещества.
Элементный анализ для C37H62N7O10BS, %:
Вычислено: C 55,00; H 7,75; N 12,14; B 1,34.
Найдено: C 54,09; H 7,53; N 12,22; B 1,34.
П р и м е р 14. Ac-Ala-Lys-бороArg-C10H16˙ ˙2HCl.
Бензолсульфонат Ac-Ala-Lys(Boc)-бороArg-C10H16 (0,200 г, 0,248 ммоль) деблокируют по методике примера 6. После ионнообменной хроматографии, упаривания растворителя, просушивания в вакууме и порошкования простым эфиром получают 0,14 г требуемого продукта.
Масс-спектроскопия (FAB) для C26H48N7O5B:
Вычислено, +H: 550,39.
Найдено, +Н: 550,42.
П р и м е р 15. Бензолсульфонат Boc- -Leu-Gly-Leu-Ala-бороArg-C10H16.
Boc-Leu-Ala-OBzL получают по методике синтеза дипептидов, описанной в примере 2. Boc-Leu-Ala-OBzL (23,7 г, 57,7 ммоль) растворяют в 40 мл безводной трифторуксусной кислоты. Через 15 минут удаляют избыток трифторуксусной кислоты путем упаривания и после обработки полученного осадка простым эфиром получают трифторацетат H-Leu-Ala-OBzL в виде кристаллического продукта (22,8 г).
Элементный анализ для C18H25N2O5F3, %:
Вычислено: C 53,19; H 6,21; N 6,89.
Найдено: C 53,37; H 5,68; N 6,84.
Boc-Gly-Leu-Ala-OBzL получают реакцией сопряжения Вос-Gly-OH (5,70 г, 32,6 ммоль) с H-Leu-Ala-OBzL по методике смешанного ангидрида, описанной в примере 2. Продукт (13,8 г) получают в виде аморфного твердого вещества. Boc-Gly-Leu-Ala-OBzL деблокируют трифторуксусной кислотой по методике, описанной для приготовления H--Leu-Ala-OBzL, за исключением того, что трифторацетатную соль может быть растворена в простом эфире. Продукт растворяют в этилацетате и очищают безводным хлористым водородом. После осаждения полученного продукта при добавлении простого эфира получают 7,7 г хлорида H-Gly-Leu-Ala--OBzL в первом сборе.
Boc-Leu-Gly-Leu-Ala-OBzL получают сопряженной реакцией Boc-Leu-OH (2,62 г, 10,5 ммоль) с H-Gly-Leu-Ala-OBzL по методике получения смешанного ангидрида, описанной в примере 2. После выкристаллизации полученного продукта из смеси этилацетат:гексан получают в первом сборе 2,7 г названного продукта (т.пл. 95-96оС).
Элементный анализ для C29H46N4O7, %:
Вычислено: C 61,89; H 8,26; N 9,96.
Найдено: C 62,00; H 8,40; N 9,83.
Boc-Leu-Gly-Leu-Ala-OH получают каталитическим гидрированием сложного бензилового эфира (2,6 г, 4,62 ммоль) по методике, описанной в примере 2, с выходом 2,1 г требуемого продукта. После выкристаллизации полученного продукта из горячего раствора этилацетата получают 1,4 г вещества.
Элементный анализ для C22H40N4O7, %:
Вычислено: С 55,90: H 8,55; N 11,86.
Найдено: C 55,42; H 8,47; N 11,73.
Boc-Leu-Gly-Leu-Ala-NH-CH[(CH2)3Br] BO2--C10H16 получают реакцией сопряжения Boc--Leu-Gly-Leu-Ala-OH (1,40 г, 2,96 ммоль) с амином из примера 1а по методике, описанной в примере 2, за исключением того, что не проводят хроматографического разделения. После кристаллизации продукта из смеси этилацетат: гексан получают 1,17 г требуемого продукта. При ТСХ в системе растворителей метанол:хлороформ (1:9) отмечено единичное пятно при значении Rf 0,68.
Элементный анализ для C36H63N5O8BrB, %:
Вычислено: C 55,10; H 8,11; N 8,93; B 1,38.
Найдено: C 55,96; H 8,30; N 8,74; B 1,33.
Соответствующий азид получают по методике примера 3 с 97% выходом, который превращают в Boc-Leu-Gly-Leu-Ala-бороОrn-С10Н16, согласно методу в примере 4. Аналатическую пробу приготавливают путем осаждения указанного продукта простым эфиром и хроматографического разделения его на колонке с LH-20 с последующим элюированием из смеси хлороформа с гексаном.
Масс-спектроскопия (FAB) для C36H65N6O8B:
Вычислено, +Н: 721,50.
Найдено, +H: 721,55.
Бензолсульфонат Boc-Leu-Gly-Leu-Ala- -бороArg-C10H16 получают по методике, описанной в примере 5. Соответствующий бороорнитиновый пептид (0,695 г, 0,791 ммоль) подвергают взаимодействию с 5 мл раствора цианамида (50 мг/мл) в абсолютном этаноле. После хроматографирования вышеуказанной смеси и порошкования простым эфиром получают 0,41 г требуемого продукта.
Масс-спектроскопия (FAB) для C37H67N6O8B:
Вычислено: +H: 763,53.
Найдено: 763,8.
П р и м е р 16. Бензолсульфонат H-Leu--Gly-Leu-Ala-бороArg-C10H16 ˙HCl.
Бензолсульфонат Boc-Leu-Gly-Leu-Ala- -бороArg-C10H16 (полученный в примере 15, 0,050 г, 0,0543 ммоль) подвергают взаимодействию с 2 мл 4Н хлороводорода в диоксане в течение 5 мин при комнатной температуре. Растворитель и избыток HCl удаляют упариванием. Полученную пробу высушивают над гидроксидом калия в вакууме в течение ночи, а затем после порошкования простым эфиром получают требуемый продукт (46 мг) в виде смешанной соли.
Масс-спектроскопия (FAB) для C32H59N8O6B:
Вычислено, +Н: 663,47.
Найдено, +Н: 663,50.
П р и м е р 17. Бензолсульфонат Bz-Glu--(OBu)-Gly-boroArg-C10H16.
Bz-Glu-(OBu)-Gly-NH-CH-[(CH2)3Br]BO2-C10 H16 получают сопряженной реакцией Bz-Glu-(OBu)--Gly-OH c указанным амином по методике, описанной в примере 2. Соответствующий азид получают в соответствии с методом, описанным в примере 3, а бороорнитиновый пептид получен по методике примера 4.
Масс-спектроскопия (FAB) для С32H49N4O7B:
Вычислено, +Н: 613,38.
Найдено, +Н: 613,60.
Конечный продукт получают по методике, описанной в примере 5.
Масс-спектроскопия (FAB) для C33H51N6O7B:
Вычислено, +Н: 655,40.
Найдено: 655,37.
Элементный анализ для C39H5N6O10SB, %:
Вычислено: C 57,62; H 7,08; N 10,34; B 1,33.
Найдено: C 57,43; H 7,25; N 9,91; B 1,23.
П р и м е р 18. Бензолсульфонат Bz-Glu--Gly-бороArg-C10H16.
Бензолсульфонат Bz-Glu-(OBu)-Gly-бороArg-C10H16 (0,13 г, 0,16 ммоль) растворяют в 5 мл диоксана, прибавляют бензолсульфокислоту (0,10 г, 0,66 ммоль) и реакционный раствор перемешивают в течение ночи при комнатной температуре. После концентрации раствора до объема примерно 1 мл путем упаривания с последующим порошкованием простым эфиром получают твердое вещество (0,14 г). Полученный продукт хроматографируют на 2,5 х 50 см колонке с LH-20 в метаноле. Фракции, содержащие требуемый продукт, упаривают, а осадок порошкуют простым эфиром с выходом 53 г названного продукта.
Масс-спектроскопия (FAB) для C29H43N6O7B:
Вычислено, +Н: 599,34.
Найдено, +H: 599,35; 613,36 (не идентифицирован).
П р и м е р 18а. Бензолсульфонат Bz-Glu--Gly-бороArg-C10H16.
Бензолсульфонат Bz-Glu-(OBu)-Gly-бороArg-C10H16 (пример 17, 0,20 г, 0,246 ммоль) обрабатывают безводным хлористым водородом по методике, описанной в примере 6, в течение 45 мин. После порошкования указанного продукта, данные ЯМР показывают, что примерно 30% трет-бутиловой защитной группы все еще находится в указанном продукте. Затем продукт подвергают взаимодействию с безводной трифторуксусной кислотой в течение 45 мин при комнатной температуре. TFA удаляют упариванием и после порошкования полученного осадка простым эфиром получают 143 мг названного продукта.
Масс-спектроскопия (FAB) для C29H43N6O7B:
Вычислено, +Н: 599,34.
Найдено, +Н: 599,35.
П р и м е р 19. Бензолсульфонат Bz-Pro--Phe-бороArg-C10H16.
Bz-Pro-Phe-OH (т.пл. 200-201оС) получают по методике, описанной в примере 2, используемой для синтеза дипептидов.
Элементный анализ для C21H22N2O4, %:
Вычислено: C 68,82; H 6,06; N 7,65.
Найдено: C 68,91; H 6,09; N 7,47.
Bz-Pro-Phe-NH-CH[(CH2)3Br] BO2-C10H16 получают сопряженной реакцией Bz-Pro- -Phe-OH c указанным амином по общему методу, описанному в примере 2, за исключением того, что хроматографическое разделение не осуществляют. При ТСХ в смеси растворителей метанол: хлороформ (1:9) наблюдается обширное пятно при Rf 0,72 и следы при Rf 0,86.
Масс-спектроскопия (FAB) для C35H45N3O5BBr:
Вычислено, +Н: 678,27.
Найдено, +Н: 677,95.
Полученный алкилгалогенид превращают в азид и бороорнитиновый пептид по методике, описанной в примерах 3 и 4.
Масс-спектроскопия (FAB) для (Bz-Pro- -Phe-бороOrn-C10H16)-C35H47N4O5B:
Вычислено, +Н: 615,37.
Найдено, +Н: 615,42.
Целевой бензолсульфонат Bz-Pro-Phe- -бороArg-C10H16 получают по методике, описанной в примере 5.
Масс-спектроскопия (FAB) для C36H49N6O5B:
Вычислено, +Н: 657,39.
Найдено, +Н: 657,13.
Элементный анализ для C42H55N6O8SB, %:
Вычислено: C 61,90; H 6,82; N 10,31; B 1,33.
Найдено: C 60,16; H 7,27; N 9,79; B 1,44.
П р и м е р 20. Хлорид Bz-Pro-Phe-бороArg-OH.
Бензолсульфонат Bz-Pro-Phe-бороArg- -C10H16 (соединение примера 19, 0,64 г, 0,79 ммоль) растворяют в 4 мл хлористого метилена и охлаждают до -78оС. Реакционную смесь сливают в колбу, содержащую 4 мл 0,50 н. трихлорида бора, полученного путем разбавления 1,0 н. трихлорида бора (Aldrich Chemical Co., Milwankee, WI) до 50% сухим хлористым метиленом и помещенную на баню с сухим льдом. Раствор перемешивают при -78оС в течение 5 мин, затем колбу помещают на баню с льдом (0оС), где раствор перемешивают в течение 15 мин. В реакционный раствор медленно прибавляют (5 мл) холодной воды с последующим разбавлением его до 120 мл 20%-ной уксусной кислотой. Органическую фазу отделяют и извлекают продукт. Водную фазу наносят на колонку объемом 20 мл на SP-Сефадексетм, уравновешенную 20%-ной уксусной кислотой. Колонку промывают примерно 150 мл раствора 20%-ной уксусной кислотой с последующим установлением линейного градиента концентраций от 200 мл 20% -ной уксусной кислоты до 200 мл 20%-ной уксусной кислоты, содержащей 0,30 н. HCl. Продукт элюируют при концентрации HCl в интервале 0,08-0,15 н. После упаривания растворителя, высушивания в вакууме и порошкования простым эфиром, получают требуемый продукт (0,19 г).
Масс-спектроскопия (FAB) для C26H35N6O5B:
Вычислено, +Н: 523,29.
Найдено, +Н: 579,34 (не идентифицирован).
Элементный анализ для C26H36N6O5˙ClB, %:
Вычислено: C 53,29; H 6,55; N 14,34; B 1,84.
Найдено: C 53,27; H 6,58; N 13,25; B 1,89.
При этерификации названного продукта пинандиолом по методике, описанной в примере 8а, получают продукт, свойства которого при ЯМР и МС совпадают с исходным сложным эфиром примера 19.
Масс-спектроскопия (FAB) для C36H49N6O5B:
Вычислено, +Н: 657,40.
Найдено, +Н: 657,39.
П р и м е р 21. Хлористоводородная соль Bz-Pro-Phe-бороArg-F
Bz-Pro-Phe-NH-CH[(CH2)3NH-C(NH)NH2]- BF2 ˙HCl
Свободную бороновую кислоту (соединение примера 20, 0,100 г, 0,179 ммоль) растворяют в 2 мл воды. К полученному раствору прибавляют 0,040 мл 48% -ной фтористоводородной кислоты при комнатной температуре. Почти сразу же образуется смолистообразный преципитат. Реакционную смесь перемешивают в течение 10 мин, затем замораживают и в вакууме удаляют избыток фтористоводородной кислоты и воду. Остаток растворяют в метаноле, концентрируют и порошкуют простым эфиром. Выход составляет 0,093 г требуемого продукта.
Масс-спектроскопия (FAB) для C26H33N6O3BF2:
Вычислено, +Н: 527,29.
Найдено, 527,31 и дополнительные значения масс, характерных для свободной бороновой кислоты.
Элементный анализ для C26H34N6O3BF2Cl˙ H2O, %:
Вычислено: C 53,47; H 6,25; N 14,47; B 1,86; F 6,54.
Найдено: C 54,00; H 6,40; N 13,48; B 1,95; F 7,06.
П р и м е р 22. Boc-Ala-Phe(D,L)бороLys--C6H12 ˙HCl.
Boc-Ala-Phe-NH-CH[(CH2)4NH2]BO2- -C6H12 ˙бензолсульфокислота.
Продукт - Boc-Ala-Phe-NH- -CH[(CH2)4Br]BO2-C6H12, превращают в алкилазид по методике, описанной в примере 3, за исключением того, что для очистки полученного продукта не требуется хроматография на колонке с LH-20. Полученный азид гидрируют по методике примера 4, кроме того, что берут 2 экв. бензолсульфокислоты, а гидрирование проводят в течение 2 ч. Получают конечный продукт при 40% выходе (т.пл. 154-160оС, разложение).
Масс-спектроскопия (FAB) для C28H46N4O6B:
Вычислено, +Н: 547,38.
Найдено, +Н: 547,43.
П р и м е р 23. Трифторацетатбензолсульфонат H-Ala-Phe-(D,L)бороLys-C6H12.
Бензолсульфонат Boc-Ala-Phe-(D, L)бороLys-C6H12 подвергают взаимодействию с трифторуксусной кислотой в течение 1 ч при комнатной температуре. После упаривания растворителя и порошкования осадка простым эфиром получают требуемый продукт в виде твердого вещества.
Масс-спектроскопия (FAB) для C23H39N4O4B:
Вычислено, +Н: 447,31.
Найдено, +Н: 447,31.
Элементный анализ для C31H46N4O9SF3B ˙2H2O, %:
Вычислено: C 49,34; H 6,68; N 7,42; B 1,43.
Найдено: C 49,26; H 5,94; N 7,12; B 1,34.
П р и м е р 24. Бензолсульфонат Boc- -(D)Yal-Leu-бороLys-C6H12.
Boc-(D)Yal-Leu-OH получают по методике, описанной в примере 2. Сложный бензиловый эфир получен с 76% выходом.
Масс-спектроскопия (FAB) для C23H36N2O5:
Вычислено, +Н: 421,27.
Найдено, +Н: 421,38.
После гидрирования, получают свободную борную кислоту с 100% выходом в виде кристаллического твердого вещества белого цвета.
Элементный анализ для C16H29N2O5, %:
Вычислено: C 59,34; H 8,87; N 8,50.
Найдено: C 59,34; H 8,87; N 8,50.
Boc-(D)-Yal-Leu-OH связывают с амином, полученным в примере 1d, с выходом Boc-(D)Yal-Leu-NH-CH[(CH2)4Br]BO2-C6H12 97%.
Масс-спектроскопия (FAB) для C27H51N3O6BBr:
Вычислено, +Н: 604,31.
Найдено, +Н: 604,31.
Полученный алкилбромид превращают в соответствующий азид при 85% выходе по методике, описанной в примере 3, с последующим его гидрированием. Конечный продукт получают в виде белого твердого вещества с 62% выходом.
Масс-спектроскопия (FAB) для C27H53N4O6B:
Вычислено, +Н: 541,41.
Найдено, +Н: 541,46.
Элементный анализ для C33H59N4O9SB˙ ˙1,5H2O, %:
Вычислено: C 54,62; H 8,61; N 7,73; B 1,49.
Найдено: C 54,58; H 8,59; N 7,92; B 1,98.
П р и м е р 25. Бензолсульфонат Ac-Phe--бороLys-C6H12.
Продукт примера 25 получают по методике, описанной в примере 22.
Ac-Phe-NH-CH[(CH2)4Br]BO2-C6H12 получен с 72% выходом.
Масс-спектроскопия (FAB) для С22H34N2O4BBr:
Вычислено, +Н: 481,00.
Найдено, +Н: 481,21.
Азид получен с 57%-ным выходом. Конечный продукт получают при 50% его выходе.
Масс-спектроскопия (FAB) для C22H37N3O4B:
Вычислено, +Н: 418,29.
Найдено, +Н: 418,31.
Элементный анализ для C29H42N3O7SB˙ ˙H2O, %:
Вычислено: C 56,66; H 7,47; N 7,08; B 1,82.
Найдено: C 56,88; H 7,43; N 7,22; B 1,53.
П р и м е р 26. Bz-(D,L)borolg-C6H12 ˙HBr.
Bz-(D, L)NH-CH[(CH2)3Br] BO2-C6H12 получают путем взаимодействия амина (полученного в примере 16, (5,0 г, 15,9 ммоль) с эквивалентным количеством бикарбоната натрия в смеси, состоящей из 4 мл диоксана и 4 мл воды при 0оС. После первоначального перемешивания реагентов реакционную смесь разбавляют 6 мл раствора из 50% диоксана с водой и нагревают до комнатной температуры. Реакционную смесь перемешивают в течение 30 мин при комнатной температуре с последующим экстрагированием ее в этилацетате и промыванием водой, 0,2 н. HCl, 5% -ным водным раствором бикарбоната натрия и насыщенным водным раствором хлористого натрия. Органический слой высушивают над безводным сульфатом натрия, и после фильтрования и упаривания получают кристаллический продукт. После выделения и промывания этилацетатом получают 3,26 г соединения, т.пл. 176-177оС.
Элементный анализ для C17H25NO3BrB, %:
Вычислено: C 53,44; H 6,59; N 3,67; B 2,83.
Найдено: C 54,50; H 6,76; N 3,68; B 2,84.
Полученный алкилогалогенид (1,00 г, 2,62 ммоль) превращают в соответствующую соль изотиорония. Требуемый продукт 0,84 г получится в виде твердого вещества белого цвета.
Масс-спектроскопия (FAB) для C18H28N3O3SB:
Вычислено, +Н: 378,20.
Найдено, +Н: 378,21.
Элементный анализ для C18H29N3O3SBBr, %:
Вычислено: C 47,18; H 6,38; N 9,17; B 2,36.
Найдено: C 46,11; H 6,71; N 8,97; B 2,22.
П р и м е р 27. Бензолсульфонат Bz(D,L)бороArg-C6H12.
Алкилгалогенид (полученный в примере 26, 2,0 г, 5,25 ммоль) превращают в азид (0,97 г, т.пл. 138-139оС) по методике, описанной в примере 3. Полученный азид превращают бензолсульфонат Bz-бороOrn-C6H12 по методике примера 4 при почти количественном выходе.
Масс-спектроскопия (FAB) для C18H27N2O3B:
Вычислено, +Н: 319,22.
Найдено, +Н: 319,26.
Бензолсульфонат Bz-бороOrn-C6H12 (0,90 г, 1,84 ммоль), подвергают взаимодействию с цианамидом по методике, описанной в примере 5, с выходом 0,65 г кристаллического продукта, т.пл. 242-244оС.
Масс-спектроскопия (FAB) для C18H29N4O3B:
Вычислено, +Н: 361,24.
Найдено, +Н: 361,24.
Элементный анализ для C24H35N4O6SB, %:
Вычислено: C 55,59; H 6,82; N 10,81; B 2,08.
Найдено: C 54,60; H 6,70; N 11,24; B 1,87.
П р и м е р 28. Бензолсульфонат Ac-Leu--Thr(OBu)-бороArg-C10H16.
Ac-Leu-Thr(OBu)-OH получают путем связывания Ac-Leu-OSu c H-Thr(OBu)-OH по методике, описанной в примере 13, для дипептидного синтеза, за исключением того, что конечный продукт получают в виде белого аморфного твердого вещества после хроматографии на колонке с LH-20. Ac-Leu-Thr(OBu)-OH (3,29 г, 9,90 ммоль) подвергают сопряженному взаимодействию с амином (примера 1а) по методике получения смешанного ангидрида, описанной в примере 2, за исключением того, что не нужно проводить хроматографию на колонке с LH-20.
Ac-Leu-Thr(OBu)-NH-CH[(CH2)3Br] BO2- -C10H16 получают в виде аморфного твердого вещества белого цвета с выходом 5,39 г. Полученный алкилгалогенид превращают в соответствующий азид с 82%-ным выходом по методике примера 3, за исключением того, что для последующей очистки нет необходимости в проведении хроматографии. Полученный азид (3,88 г, 6,42 ммоль) гидрируют по методике, описанной в примере 4. Бензол- сульфонат Ас-Leu-Thr-(OBu)-бороOrn-C10H16 получают с 74% выходом после хроматографии на колонке с LH-20 и порошкования простым эфиром.
Масс-спектроскопия (FAB) для C30H55N4O6B:
Вычислено, +Н: 579,43.
Найдено, +Н: 579,48.
Бороорнитиновый пептид превращают в конечный продукт с выходом его 86% по методике примера 5.
Масс-спектроскопия (FAB) для C31H57N6O6B:
Вычислено, +Н: 621,45.
Найдено, +Н: 621,50.
Элементный анализ для C37H63N6SO9B, %:
Вычислено: C 57,05; H 8,17; N 10,79; B 1,39.
Найдено: C 56.47; H 8,01; N 10,93; B 1,34.
П р и м е р 29. Бензолсульфонат Ac-Leu--Thr-бороArg-C10H16.
Бензолсульфонат Ac-Leu-Thr-(OBu)-бороArg-C10H16 (пример 28, 0,200 г, 0,257 ммоль) растворяют в смеси, состоящей из 2 мл хлористого метилена и 2 мл 4 н. HCl:диоксан, и затем полученную реакционную смесь перемешивают в течение 30 минут при комнатной температуре. Растворитель упаривают в вакууме, а полученный осадок сушат в высоком вакууме. Требуемый продукт получают в виде белого твердого вещества с 97% выходом после порошкования простым эфиром.
Масс-спектроскопия (FAB) для C27H49N6O6B:
Вычислено, +Н: 565,39.
Найдено, +Н: 565,48.
П р и м е р 30. Бензолсульфонат Ac- -Lys(Boc)-Pro-бороArg-C10H16.
Дипептид Ac-Lys(Boc)-Pro-OH получают по методике, описанной в примере 13. После кристаллизации из этилацетата получают указанный продукт в виде белого твердого вещества (темп. 160-161,5оС). Указанный дипептид, Ac-Lys(Boc)-Pro-OH (3,15 г, 8,18 ммоль) подвергают сопряженной реакции с амином (пример 1а) по методике, описанной в примере 2. Полученный продукт (5,8 г) используют без дополнительной очистки. Полученное соединение превращают в азид по методике примера 3 с выходом его 73% после хроматографии на колонке с LH-20. После гидрирования по методике примера 4, хроматографии на колонке с LH-20 и порошкования простым эфиром получают бензолсульфонат Ac-Lys(Boc)-Pro-бороOrn-C10H16 с выходом 81%.
Масс-спектроскопия (FAB) для C32H55N5O7B:
Вычислено, +Н: 634,43.
Найдено, +Н: 634,46.
Бороорнитиновый пептид (2,0 г, 2,53 ммоль) подвергают взаимодействию с цианамидом по методике примера 5 с выходом 1,8 г требуемого продукта в виде твердого вещества белого цвета.
Масс-спектроскопия (FAB) для C33H57N7O7B:
Вычислено, +Н: 676,46.
Найдено, +Н: 676,41.
Элементный анализ для C39H63N7O10BS, %:
Вычислено: C 56,23; H 7,64; N 11,77; B 1,30.
Найдено: C 56,06; H 7,48; N 11,75; B 1,22.
П р и м е р 31. Ac-Lys-Pro-бороArg-C10H16 ˙2HCl.
Бензолсульфонат Ac-Lys(Boc)-Pro-бороArg-C10H16 (пример 30, 0,30 г, 0,360 ммоль) подвергают взаимодействию со смесью, состоящей из ледяной уксусной кислоты с 4 н. HCl и диоксана в соотношении 50:50 (%) в течение 15 мин при комнатной температуре. Растворитель упаривают, а остаток сушат в вакууме. Полученный осадок растворяют в воде и пропускают через 5 мл колонку на AGI-X8 (Cl--форма). Полученную пробу упаривают и после порошкования остатка простым эфиром получают требуемый продукт в виде твердого вещества белого цвета (230 мг).
Масс-спектроскопия (FAB) для C28H49N7O3B:
Вычислено, +H: 576,40.
Найдено, +Н: 576,45.
П р и м е р 32. Бензолсульфонат Ac-Ala--Glu(OBu)-бороArg-C10H16.
Ac-Ala-Glu(OBu)-OH получают путем связывания Ac-Ala-OSu c H-Glu(OBu)-OH по методике примера 13. Продукт выкристаллизовывают из смеси этилацетат:гексан (т.пл. 147,5-148оС).
Элементный анализ для C14H24N2O6, %:
Вычислено: C 53,14; H 7,66; N 8,85.
Найдено: C 53,28; H 7,53; N 9,08.
Ac-Ala-Glu(OBu)-NH-CH[(CH2)3Br]BO2- -C10H16 получают по методике примера 2, за исключением того, что вместо этилацетата используют хлороформ для отделения органического слоя на начальной стадии реакции и не проводят хроматографию на колонке с LH-20. После упаривания органического слоя получают 87% выхода требуемого продукта в виде частично кристаллического твердого вещества. Алкилбромид превращают в азид по методике, описанной в примере 3. Целевой продукт (т.пл. 165-166оС) получен с 50%-ным выходом после кристаллизации неочищенного продукта из хлороформа.
Элементный анализ для C28H47N6O7B, %:
Вычислено: C 53,51; H 7,55; N 6,69; B 1,73.
Найдено: C 55,51; H 7,50; N 6,50; B 1,66.
Бороорнитиновый пептид получают по методике примера 4 с 79%-ным выходом требуемого продукта.
Масс-спектроскопия (FAB) для C28H49N4O7B:
Вычислено, +Н: 565,38.
Найдено, +Н: 565,51.
Конечный продукт получают в виде твердого аморфного вещества белого цвета с выходом 70% по методике примера 5.
Масс-спектроскопия для C29H51N6O7B:
Вычислено, +Н: 607,40.
Найдено, +Н: 607,41.
Элементный анализ для C35H57N6O10BS, %:
Вычислено: C 54,96; H 7,53; N 10,99; B 1,41.
Найдено: C 54,36; H 7,71; N 11,27; B 1,21.
П р и м е р 33. Бензолсульфонат Ac-Ala--Glu-бороArg-C10H16.
Бензолсульфонат Ac-Ala-Glu(Bu)-бороArg-C10H16 (пример 32, 0,10 г, 0,131 ммоль) растворяют в 10 мл уксусной кислоты, через полученный раствор в течение 20 мин пропускают хлороводород. Затем раствор перемешивают в течение 1,5 ч при комнатной температуре, и после упаривания растворителя, получают масло. Требуемый продукт получают в виде твердого белого вещества (82 мг) после просушивания в вакууме и порошкования простым эфиром.
Масс-спектроскопия (FAB) для C25H43N6O7B:
Вычислено, +Н: 551,34.
Найдено, +Н: 551,41.
Используя аналогичные методы, которые описаны в примерах 22 и 23, получены дополнительно следующие соединения, представленные в табл. 1.
Проведены биологические испытания соединений, полученных описываемым способом.
Показано, что все эти соединения могут быть отнесены к категории малотоксичных веществ.
Ингибиция калликреина плазмы человеческой крови
Калликреин плазмы человека получают из протогена АГ (AG) поставляемого из Швейцарии. Специфическая активность, как описывает поставщик, составляет 15 единиц на мг. 1 единица определена как количество фермента, необходимого для гидролиза 1 мкмоль субстрата, H-(D)Pro- -Phe-Arg-пара-нитроанилида, (Kabi S2302), за 1 мин при концентрации субстрата 0,50 мМ при 25оС в 50мМ калий-фосфатном буфере, рН 8,0.
Маточный раствор фермента 1 ед/мл приготавливают в 50%-глицерин - 0,1 М натрийфосфатном буфере, рН 7,5, содержащем 0,20 М хлористого натрия и 0,1% полиэтиленгликоля 6000 (PEG). В стандартных испытаниях, 10 мкл основного раствора калликреина прибавляют к 990 мкл раствора, содержащего 0,20 мМ H-(D)Pro-Phe-Arg--пара-нитроанилида S2302 в 0,10 мМ натрийфосфатного буфера, рН 7,5, в состав которого входит 0,20 М хлорида натрия и 0,1% PEG, при 25оС. Действие ингибиторов оценивают при регистрации ферментативной активности, определяемой при измерении повышения интенсивности поглощения при 405 нм по времени как в присутствии, так и в отсутствии ингибиторов. В табл. 2 приведены уровни ингибиторов и активность, замеренные в интервале от 10 до 20 мин после стимуляции активности. Активность контрольных ингибиторов составляет 0,0092 + 0,0095 мин-1.
В табл. 2 показана биологическая активность ингибиторов калликреина плазмы крови человека.
Из таблицы следует, что указанные ингибиторы имеют значения Ki < 0,7 мкм. Наиболее эффективным синтетическим обратимодействующим ингибитором является 6-амидино-2-(4-амидинофенил)-бензо/ β /тиофен, имеющий значение Ki = 0,7 мкм.
Таким образом, приведенные в табл. 2 соединения являются более активными ингибиторами каллекреина, чем известное производное бензо/β /тиофена.
Кроме участия в коагуляции крови, калликреин высвобождает брадикинин из белковых субстратов. Брадикинин же повышает васкулярную проницаемость, оказывает хемотактическое действие на лейкоциты и вызывает боль. Первые два вида активности связаны с воспалением. Поэтому предполагается, что ингибирование калликреина имеет противовоспалительный и обезболивающий эффект.
Соединения по изобретению были испытаны также в отношении ингибирования тромбина.
Ингибиция тромбина (Эстеразная активность).
Тромбин человека (специфическая активность 2345 N1H ед/мг получают из R. Q.P.Laboratories, South Bend, IN /Lot HT 102/. Маточный раствор тромбина готовят в 0,01 М PIPES-буфере, рН 6,0, содержащем 0,75 М хлорида натрия. Анализ тромбина проводят в натрий-фосфатном буфере, рН 7,5, содержащем 0,20М натрийхлорида и 0,1% PEG-6000. Исходная концентрация субстрата составляет 0,10 мМ, а концентрация тромбина составляет 1,0 нМ в расчете на массу. В табл. 3 приведены уровни ингибиторов и активность, измеренная в интервале от 10 до 20 мин после стимуляции реакции. Активность тромбина для контрольной группы составляет 0,0076-0,0005 мин-1.
Результаты приведены в табл. 3.
Биологические данные, представленные в табл. 3, подтверждают превосходство соединений в соответствии с описываемым изобретением над известным соединением N- α -(2-нафтил-сульфонилглицил)-4-амидинофенилаланинпиперидин. Хотя и данные в табл. 3 не выражены в виде значений Ki, но все-таки возможно определить значения Ki на основе данных, представленных в табл. 3. Значение Ki определяется как концентрация ингибитора, требуемая для 50% ингибирования фермента в отсутствии субстрата. Присутствие субстрата, например тромбина, имеет защитный эффект на фермент. Из данных, представленных в табл. 3, следует, что все испытанные соединения имеют значение Ki ниже 6 нМ.
Для следующих соединений значения Ki были определены методом Лайнуивера и Брука после установления равновесия фермента и ингибитора при рН 7,5 (см. табл. 4).
Эти соединения, включающие Ac-Ala-Glu-boroArg-C10H16, которое обладает значением Ki (конечной концентрации) 10 нМ, т.е. значением которое считается примерно равным Ki 6, превосходят сравнительное соединение еще по той причине, что их можно вводить без применения органического растворителя - солюбилизатора.
В случае пептидов - производных бороновой кислоты, растворимость которых 1-50 мг/мл, органический растворитель - солюбилизатор, не требуется.
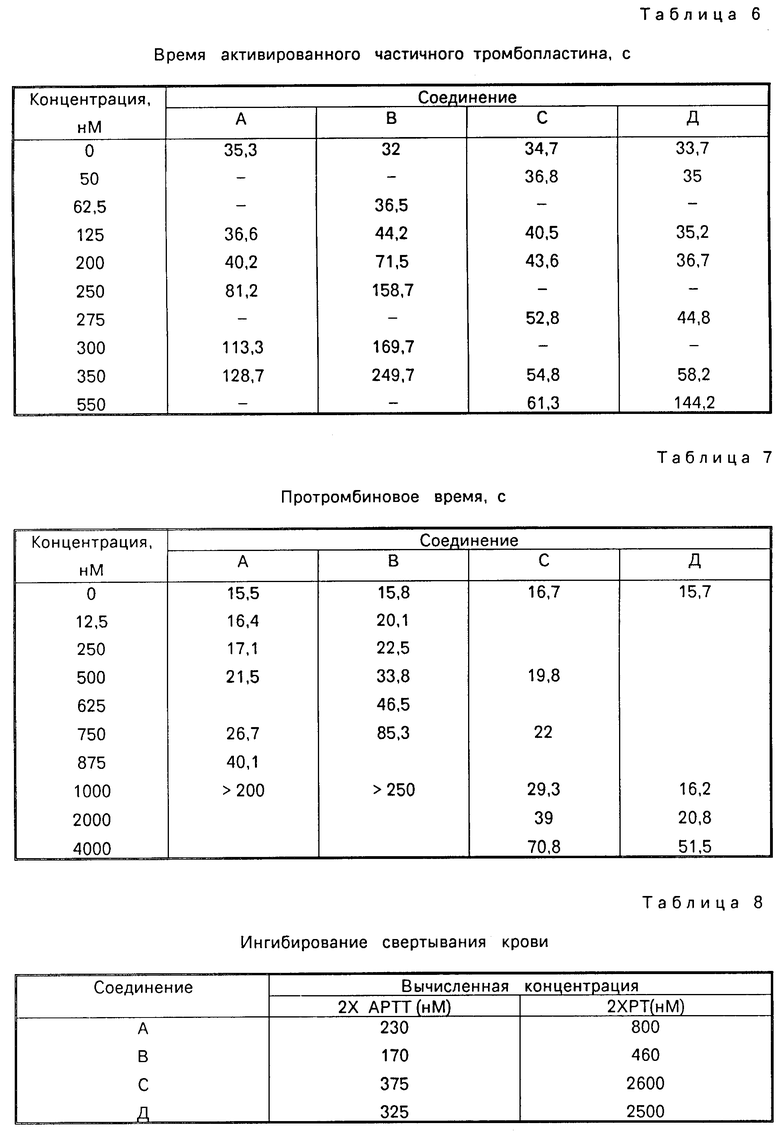
Ингибиция свертываемости крови, проявляемая при определении АРТТ и РТ.
Эффект ингибиторов протеазы на свертываемость крови in vitro определяют путем измерения их действия по двум разным клиническим параметрам, времени частичного активированного тромбопластина (АРТТ) и времени протромбина (РТ). Реагенты для каждого из указанных анализов поставляет GeneraI Piagnostics, Jessup MD. Маточные растворы ингибиторов готовят в 25 мМ HEPES-буфере, рН 7,5, содержащем 0,10 М хлорида натрия. Для АРТТ-анализа, раствор ингибитора (0,100 мл) инкубируют обычной человеческой плазмой крови (0,100 мл) и спонтанным реагентом АРТТ (0,100 мл). После инкубирования в течение 5,0 мин при температуре 37оС к указанному раствору прибавляют хлорид кальция (0,100 мл) и время свертываемости, измеренное в секундах, определяют на фиброскопе. В табл. 5 приведены данные влияния различных концентраций ингибитора на время свертываемости крови по сравнению с временем свертывания крови контрольных групп, установленного при отсутствии ингибитора.
Для РТ-анализа, растворы ингибитора (0,100 мл) инкубируют обычно плазмой крови человека (0,100 мл) в течение 2 мин при температуре 37оС. Затем к инкубированному раствору прибавляют симпластиновый реагент (0,200 мл) и определяют время свертываемости крови, приведенного в табл. 6.
В табл. 8 приведены суммарные данные результатов, представленных в табл. 5 и 6, показывающие расчетные концентрации ингибитора, необходимого для увеличения времени частично активированного тромбопластина (2хАРТТ) и времени протромбина (2хРТ) в два раза.
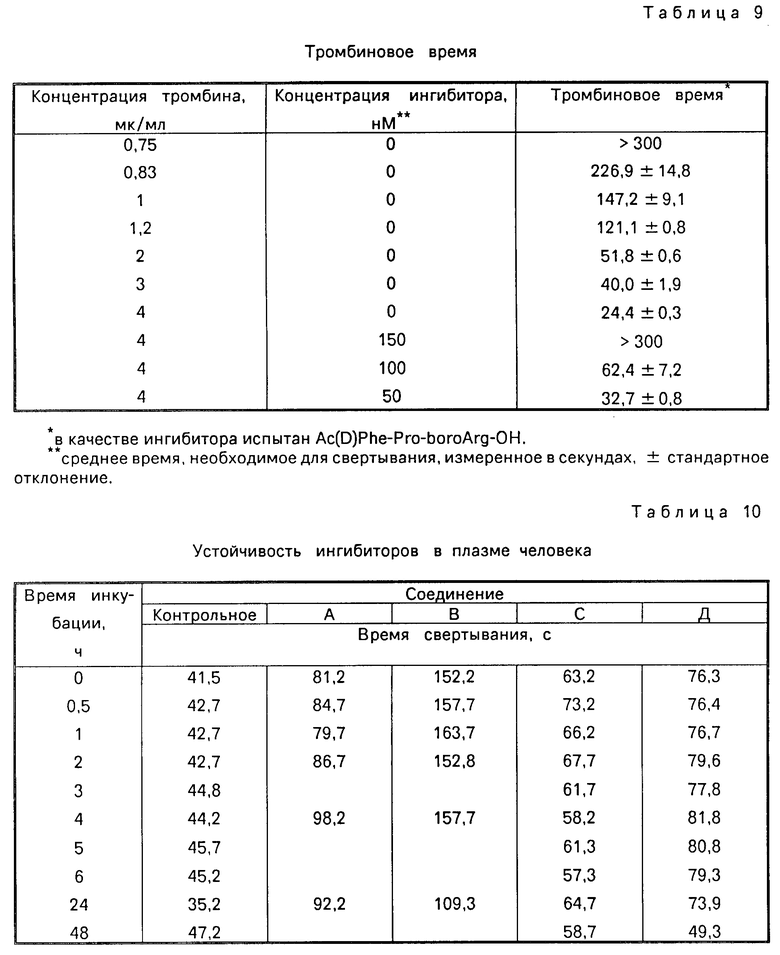
Ингибиция свертываемости крови, установленная при определении (ТТ) (времени тромбина).
Действие ингибитора протеазы Ac-(D)- -Phe-Pro-бороArg-OH на свертывание крови in vitro определяют путем измерения его влияния на тромбиновое время (ТТ). Смесь, состоящую из 0,2 мл обычной кроличьей плазмы и 0,05 мл буфера, содержащего ингибитор в 6-кратном количестве требуемой конечной концентрации, нагревают до 37оС. Свертываемость крови стимулируют путем введения тромбина (0,05 мл в 6-кратном количестве конечной концентрации). Используемый тромбин закупают от Sigma Chemical Comp. (NT-6634, активность 1190:11H ед/на мг белка) и готовят в буфере. Используемый как для ингибитора, так и тромбина, буфер представляет собой 0,1 М трис-буфер (12,10 г/л), содержащий 0,154 М NaCl (8,84 г/л) и 2,5 мг/мл сывороточного бычьего альбумина, рН 7,4. Тромбиновое время, измеряемое в секундах, определяют на фиброскопе. В табл. 9 представлены данные влияния ингибитора на время свертывания крови по сравнению со временем свертываемости контрольной группы без использования ингибиторов. Показатели эффекта представляют усредненное значение как минимум трех замеров. Если реакция свертываемости не происходит в течение 300 с, то ее прекращают.
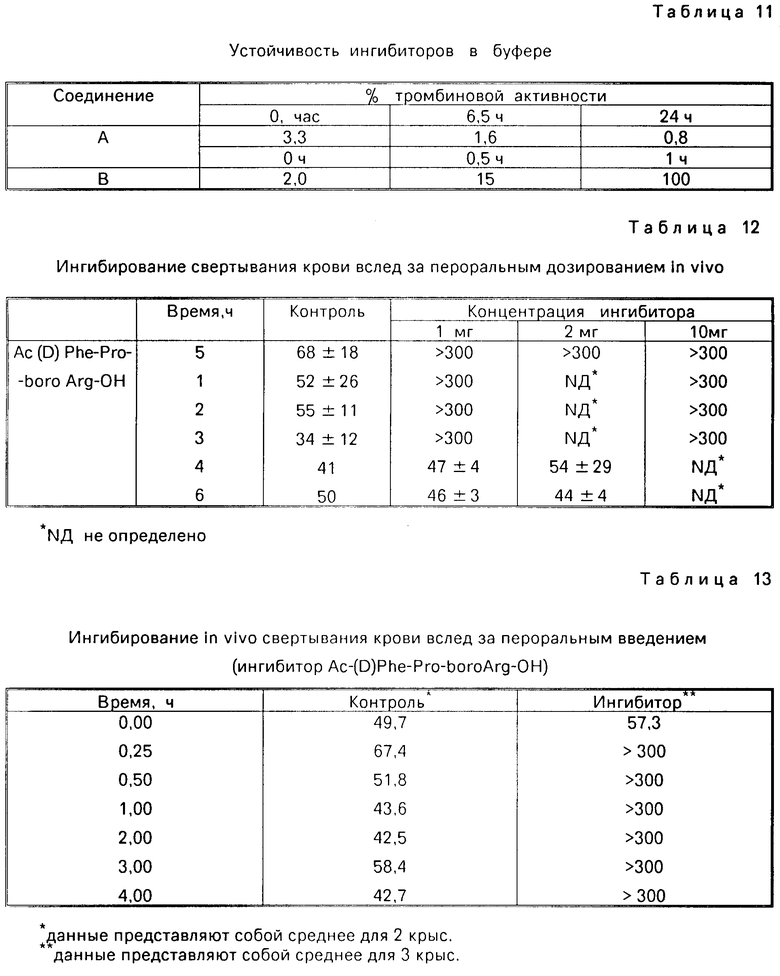
Стабильность ингибиторов в человеческой плазме, определенная при АРТТ
Стабильность ингибиторов в плазме оценивают по их способности ингибировать свертываемость крови. В первую очередь, основные растворы (1,0 мкМ) ингибиторов, подлежащих испытанию, которые приготовлены в 25 мл HEPES-буфере, рН 7,5, содержащем 0,10 М хлорида натрия, разбавляют до 50% обычной плазмой крови человека. Смеси готовят при 0оС, затем аликвоты (0,200 мл) отбирают и инкубируют в течение 2 мин при 37оС. Прибавляют одинаковый объем спонтанно приготовленного реагента АРТТ и замеряют время свертывания крови, как описано. Конечная концентрация ингибитора в течение анализа свертывания крови составляет 250 нМ. Инкубационное время (приведенное в часах) и время свертывания (замеряемое в секундах) для каждого ингибитора приведены в табл. 10. Показатели стабильности для соединений Е и F определены одновременно с контрольной группой. Показатели для соединений А, В получены на другой день.
Стабильность ингибиторов в буферном растворе
Ингибиторы, каждый при концентрации 1,0 мкМ, инкубируют при комнатной температуре в 0,20 М натрийфосфатном буфере, рН 7,5, содержащем 0,20 М хлорида натрия и 0; 10% PEG. Затем отбирают аликвоты (4,0 мкл) и анализируют на тромбиновую пробу, как описано в примерах 72-110. В таблице 8 приведены данные тромбиновой активности в (%), сохраняющейся после инкубации и продолжительность нахождения в натрийфосфатном буфере испытуемых ингибиторов. В случае использования ингибиторе А имеется незначительная потеря ингибиторной активности. Ингибитор В теряет свою биологическую активность в течение часа.
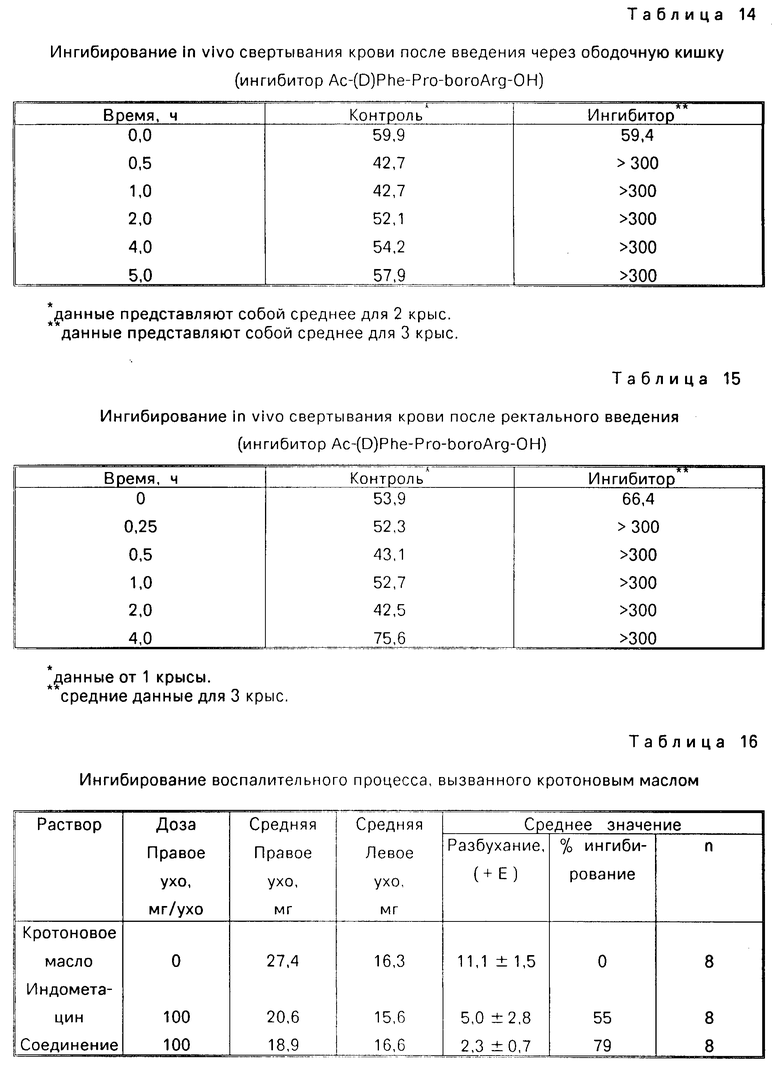
Ингибирование свертывания крови после перорального приема ингибиторов in vivo.
Самки крыс (Sprague Dawley CD Rats, массой 130-140 г, предоставленные Charles River Labs, Inc, Wilmington MA) анестезируют пентобарбиталом натрия (50 мг/кг, внутрибрюшинно). На вентральной поверхности шеи выполняют срединный надрез и в одну из каротидных артерий вводят полиэтиленовый катетер с выведением его у тыльной части шеи. После выведения крысы из состояния наркоза, контрольные пробы крови берут из введенного в каротидную артерию катетера, производят антикоагуляцию цитратом натрия и центрифугируют (2000 х об/м, 10 мин). Плазму переносят в пластиковые пробирки и выдерживают на льду до их анализа. Тромбиновое время определяют в фиброскопе.
Крысы получают протеазный ингибитор Ac-(D)Phe-Pro-бороArg-OH в носителе, либо только носитель, через желудочный зонд в объеме менее 4 мл. В качестве носителя используют 5%-ный диметилсульфоксид в физиологическом растворе. Пробы крови берут в разное время после орального приема, которые анализируют как описано выше. В представленной ниже табл. 12 приведены полученные данные времени свертывания крови в секундах. В случае, когда время свертывания крови превышает 300 с, в таблице указано как >300. Остальные данные показывают среднее время, необходимое для коагуляции, измеренное в секундах, ± означает стандартное отклонение от нормы.
Ингибиция in vivo коагулирования крови после перорального приема ингибитора.
Для дополнительной демонстрации способности данного соединения ингибировать свертываемость крови in vivo, крыс анестезируют пентобарбиталом натрия (50 мг/кг, внутрибрюшинно), после чего в яремную вену вставляют катетер и отверстие закрывают. После выведения крыс из наркоза перорально вводят либо 5 мг/кг ингибитора протеазы, Ac-(D)Phe-Pro-бороArg-OH, растворенный в воде либо равный объем воды. Через 30-60 мин, всем крысам вливают 500 ед/кг тромбина в течение 1 мин. Все 14 крыс, получающие только воду, погибли в течение 10 мин, после вливания тромбина. В противоположность этому только 8 из 17 крыс, получившие ингибиторсодержащую воду, погибли в течение 10 мин, а оставшиеся жили 1 ч, и в это время их усыпили.
Ингибиция in vivo свертываемости крови после перорального, местного и ректального приема.
Общая методика:
Самцы крыс Lewis массой 300-350 г анестезируют пентобарбиталом натрия (50 мг/кг, внутрибрюшинно) и в яремную вену вставляют силастиковую канюлю, прикрепленную к полиэтиленовой трубке. Указанную трубку выводят наружу у тыльной части шеи и прикрепляют к шприцу через стопорный вентиль. Пробы крови (0,5 мл) набирают в шприц, который промыт цитратным буфером до каждого забора крови перед и через разные промежутки времени после приема ингибитора протеазы Ac-(D)Phe-Pro-бороArg-OH. Затем пробы крови переносят в герметичные емкости, содержащие цитратный буфер. Кроме того, после каждого забора крови, канюлю промывают физиологическим раствором. Затем пробы крови центрифугируют (2500 об/мин в течение 15 мин) и для измерения времени коагуляции используют 0,2 мл проб плазмы. Измерения времени свертываемости крови проводят на фиброскопе, как описано ниже. Прежде всего, плазму (0,2 мл) помещают в фибростакан и прибавляют трис-буфер (50 мкл), рН 7,4. Буферный раствор, содержащий плазму, инкубируют при 37оС в течение 1мин, затем добавляют 50 мкл 24 мк/мл тромбинового раствора в трис-буфере, и замеряют время коагуляции в секундах. Если время коагулирования превышает 300 с, далее указывают, как > 300.
Пероральное введение:
Крыс, с введенным в яремную вену катетером, выводят из состояния наркоза до перорального приема ингибитора. Водный раствор ингибитора протеазы Ac-(D)Phe- -Pro-бороArg-OH, содержащий 3 мг ингибитора на кг массы крысы (примерно 1 мг на крысу) в 0,75 мл воды на 1 кг массы крысы, вводят через зонд. В представленной ниже табл. 13 приведены полученные данные.
Местное применение:
В брюшной полости крыс с введением в яремную вену катетером, выполняют разрез 3 см, пока они все еще находятся в наркотизированном состоянии. Находят толстую кишку и разъединяют ее восходящую и нисходящую части. Водный раствор ингибитора протеазы Ac-(D)Phe-Pro-бороArg-OH, содержащий 3 мг ингибитора на 1 кг массы крысы (примерно 1 мг на крысу) в 1 мл воды на 1 кг массы крысы, инъецируют в восходящую часть полости толстой кишки. Разрез закрывают, используя скобки для наложения шва. В представленной ниже таблице приведены полученные данные.
Ректальное применение:
В качестве методики для ректального введения ингибитора крысам с вставленным в яремную вену катетером используют методику, описанную Камию в J. Pharm. Sci., 71: 621 (1982).
Изготавливают приспособление, состоящее из 0,89 см и 0,71 см силиконовых прокладок, соединенных в длину на 2 см проволочкой. Указанное приспособление вставляют в ректальное отверстие крысы, причем сначала прокладку большего размера и затем приклеивают к анальному проходу соответствующим клеем. Введение ингибитора осуществляют путем инъекции через расположенную снаружи прокладку. Доза для ректального приема составляет 3 мг ингибитора протеазы Ac-(D)Phe-Pro-бороArg-OH на 1 кг массы крысы (примерно 1 мг на крысу) в 0,6 мл воды на 1 кг массы крысы. В представленной ниже табл. 15 приведены полученные данные.
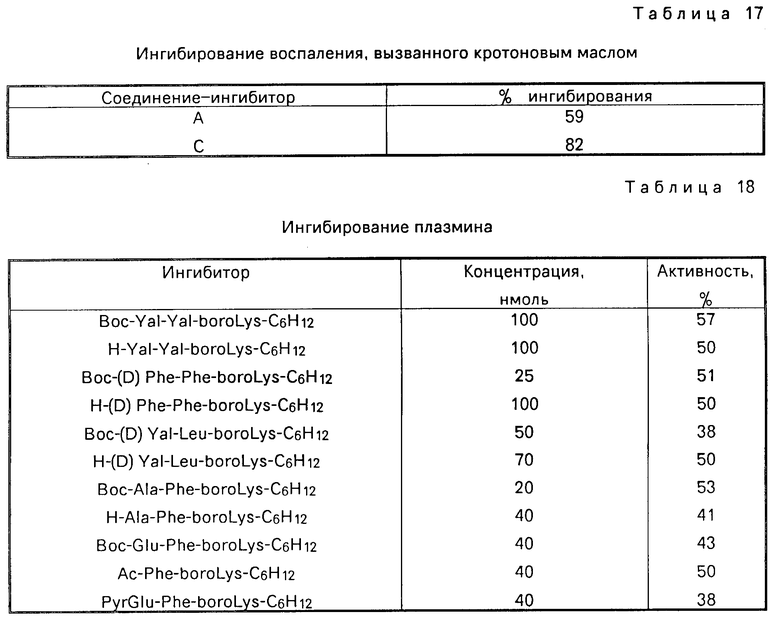
Ингибиция in vivo воспалительного процесса, вызванного кротоновым маслом
Получают два раствора: в первом содержится 5%-ное кротоновое масло, известное как агент, вызывающий воспаление, в ацетоновом носителе (кротоновый раствор), а второй раствор содержит 5%-ное кротоновое масло в ацетоновом носителе, к которому добавлено 10 мг/мл предлагаемого соединения (испытуемый раствор). Кротоновый раствор (10 мкл) или в альтернативном варианте испытуемый раствор наносят на правое ухо каждого животного (крысы Sprague Dawley CD, массой 130-140 г, предоставленные Charles River Labs, Inc, Wilmington, MA). Ацетоновый носитель в чистом виде (ацетоновый раствор) (10 мкл) наносят на левое ухо каждого животного. Через час после обработки животных усыпляют, их уши отрезают, штампуют на диски размером 1/4 дюйма в диаметре и взвешивают. Разбухание измеряют как разницу в массе между кротоновым раствором, которым обработано правое ухо и ацетоновым раствором - левое ухо. Полученные данные сравнивают с индометацином, который известен в качестве нестероидного противовоспалительного средства, приготовленного и наносимого аналогичным образом как и испытуемое соединение. В табл. 16 приведены усредненные значения для соединения F, Ac-Phe-бороArg- -C10H16. Используемый ниже термин "доза" означает количество активного противовоспалительного ингредиента в мкг (соединения A, C, D, E, F или G, или индометацина) в растворе, нанесенном на правое ухо, а n означает количество крыс, которое используют в каждом тесте. "SE" означает стандартную погрешность.
В табл. 17 показана антивоспалительная активность соединений А и С, которые взяты при одних и тех же условиях (доза - 100 мкг).
Ингибирование плазмина боролизиновыми пептидами.
Плазмин плазмы человека получают от фирмы American Diagnostics, Greenwich. Концентрация активных центров в препарате определяют методом, аналогичным методу, описанному Coleman и др. Анализ ферментов осуществляют на спектрофотометре Perkin Elmer Lambda 4C c использованием вычислительной машины типа Perkin-Elmer 7300. В качестве буфера используют 50 ммоль трис-буфера, рН 7,40, содержащего 110 ммоль хлорида натрия, 0,1% ПЭГ 6000 и 0,30 ммоль H-(D)Val-Leu-Lys-п-нитроанилида. К этой смеси прибавляют соответствующие количества ингибитора и затем плазмина. При 25оС измеряют повышение интенсивности поглощения при 405 нм по времени. Поскольку ингибиторы связываются медленно, скорость реакции определяется по касательной, проводимой к кривой реакции, по истечении 10 мин от начала реакции. В каждом случае осуществляют контрольное испытание без применения ингибитора. Во всех случаях концентрация плазмина составляет менее 10%-ной концентрации ингибитора.
Результаты приведены в табл. 18.
Проведенные испытания показали, что соединения по изобретению малотоксичны, обладают высокой ингибирующей активностью с широким спектром действия, кроме того, все эти соединения могут быть введены в организм без применения растворителя-солюбилизатора.
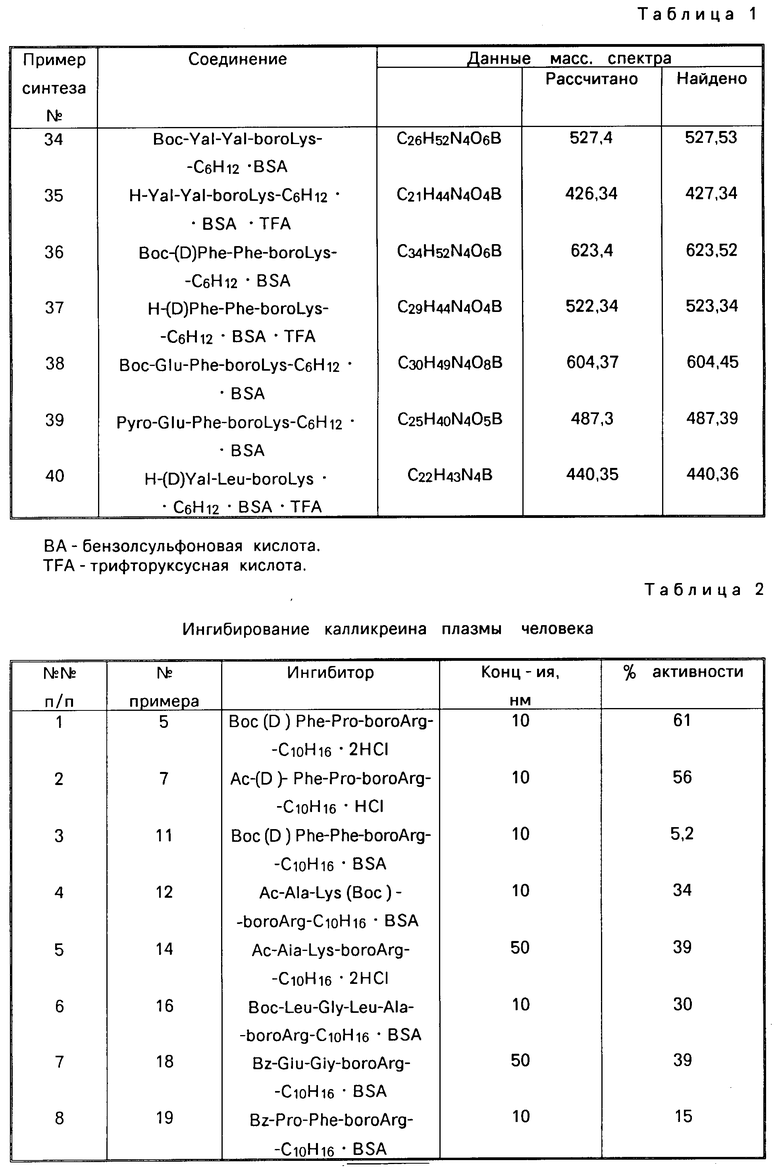
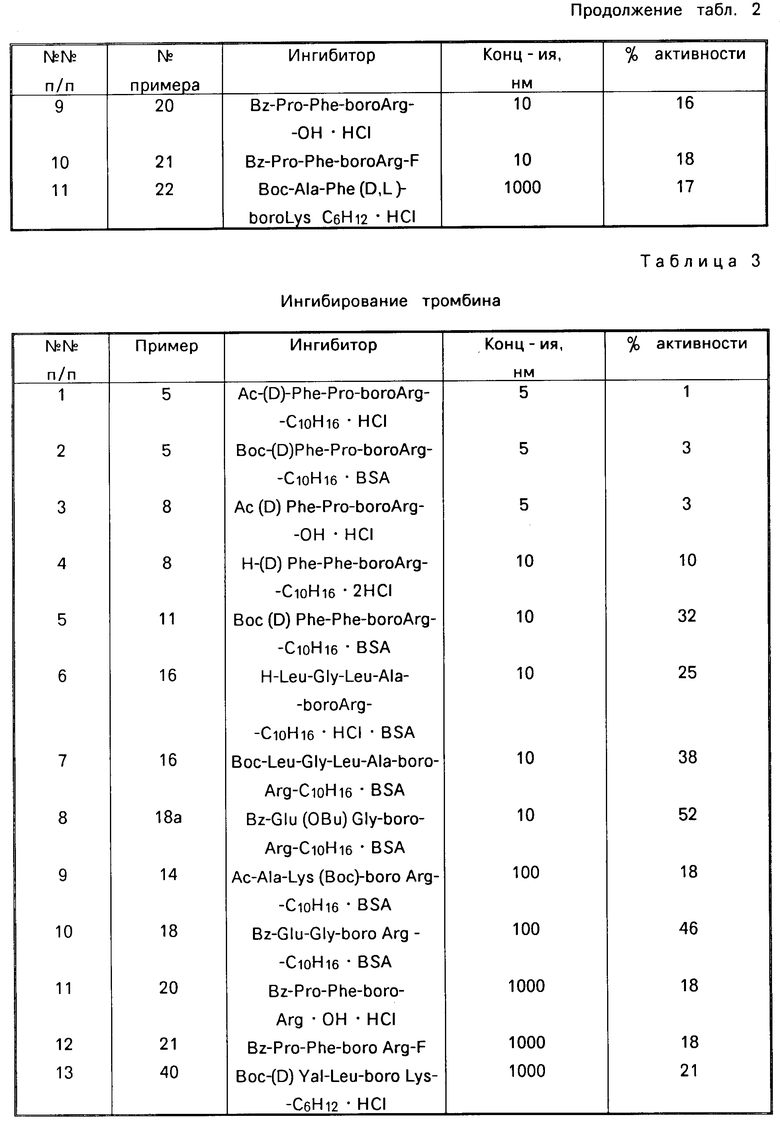
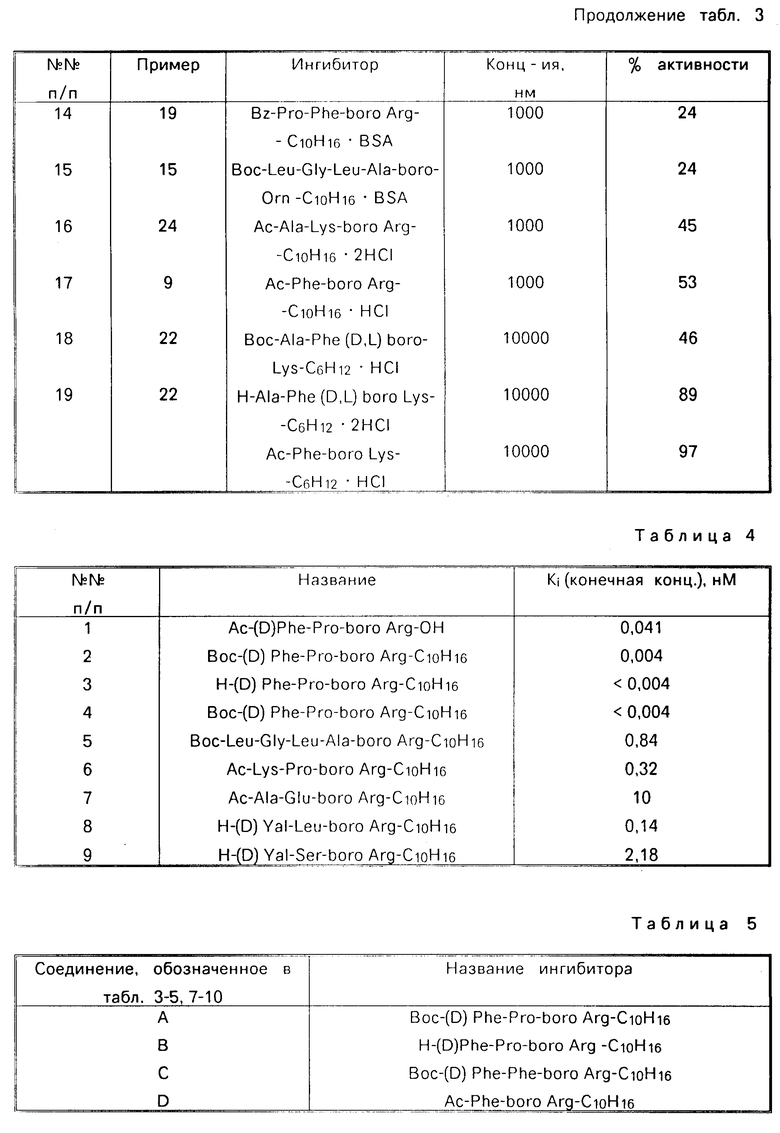
Назначение: в биохимии и медицине, в качестве ингибитора трипсиноподобных сериновых протеаз, таких как тромбин, калликреин плазмы и плазмин. Сущность изобретения: производные борсодержащих пептидов. Приведена ф-ла. Реагент 1: соответствующий амин. Реагент 2: R - Z - OH. Конденсацию ведут в растворителе в присутствии триэтиламина. Полученный продукт обрабатывают азидом щелочного металла с последующим восстановлением в присутствии органической или минеральной кислоты, предпочтительно бензолсульфокислоты и взаимодействием с цианамидом в низшем спирте при 50 - 150°С. 18 табл. Формула: 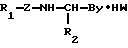 , где R1 = H, Ac, Br, Boc, BocHu; R2=(CH2)nNHC(NH)NH2; n= 3,4; y - остаток пинаконового и пинандиолового эфира; HW = C6H5SO3, HCl, HBr; Z = D- или L-Phe, DPhe - Pro, DPhe - Phe, Ala - huS, Pro - Phe, Ala - Phe, Glu - Phe, Ala - Ghe, Glu - Gly, Gly - Leu - Ala, Pyro - Glu - Phe, Val - Val, (D)Val - Leu, Lys - Pro, Leu - Thr. 18 табл.
, где R1 = H, Ac, Br, Boc, BocHu; R2=(CH2)nNHC(NH)NH2; n= 3,4; y - остаток пинаконового и пинандиолового эфира; HW = C6H5SO3, HCl, HBr; Z = D- или L-Phe, DPhe - Pro, DPhe - Phe, Ala - huS, Pro - Phe, Ala - Phe, Glu - Phe, Ala - Ghe, Glu - Gly, Gly - Leu - Ala, Pyro - Glu - Phe, Val - Val, (D)Val - Leu, Lys - Pro, Leu - Thr. 18 табл.
ПРОИЗВОДНЫЕ БОРСОДЕРЖАЩИХ ПЕПТИДОВ общей формулы I
R1-Z-NH-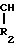 -Bу-HW
-Bу-HW
где R1 - H, Ac, Bz, Boc, Boc - Leu;
R2 - (CH2)n, NHC(NH)NH2;
n = 3,4;
y - остаток пинаконового или пинандиолового эфира;
HW = C6H5SO3H, HCl, HBr;
Z = D или L - Phe, DPhe - Pro, DPhe - Phe, Ala - Lys, Pro - Phe, Ala - Phe, Glu - Phe, Ala - Glu, Glu - Gly, Gly - Leu - Ala, pyro - Glu - Phe, Val - Val, (D)Val - Leu, Lys -Pro, Leu - Thr.
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| B.Kajser, F.Markwardt "Antitrowbetic and Haemorrhagic Effects of Synthetic and Naturally occurring Throwbin inhibitors" Throwboris Rescareh 43, 613-620, 1986. | |||
