Изобретение касается ингибиторов тромбина, которые являются антикоагулянтами у человека и животных. В частности, оно касается производных дипептида L-пролин-L-аргинин-альдегида, обладающих высокой антитромбовой активностью.
Ингибирование тромбина в настоящее время достигается введением гепаринов и кумаринов. Механизм действия этих препаратов изучается. Гепарины вводятся только парентерально, и уровни их должны точно контролироваться. Кумарины действуют блокированием или ингибированием образования протромбина и требуют некоторого времени для достижения максимальной эффективности.
Хотя и гепарины, и кумарины являются эффективными антикоагулянтами, существует необходимость в антитромбовых препаратах, которые быстро предотвращают образование тромбов, и которые не мешают действию плазмина по растворению существующих тромбов.
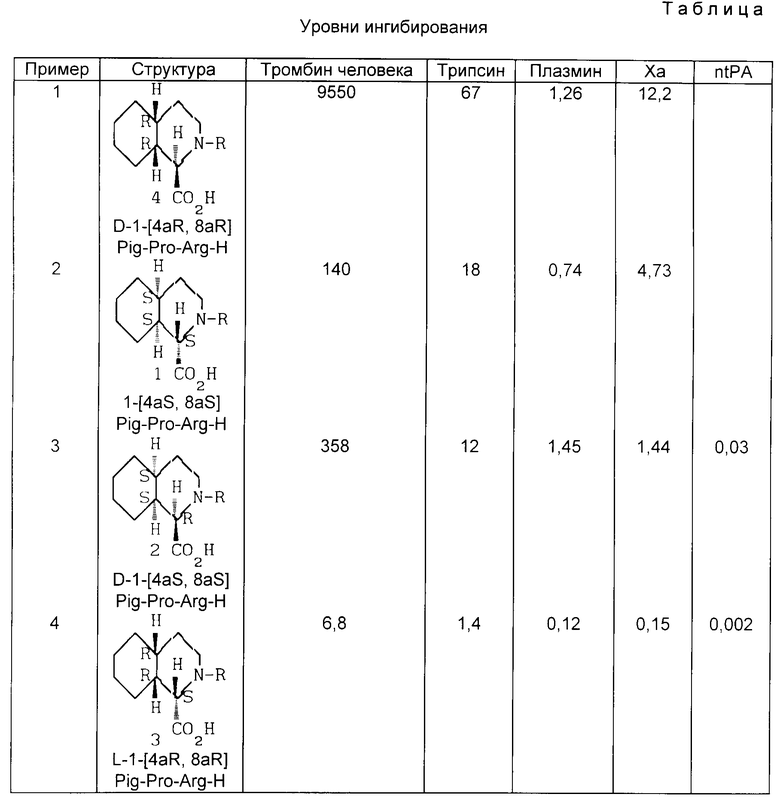
Настоящее изобретение неожиданно обнаруживает, что соединение настоящего изобретения является значительно более сильным ингибитором тромбина, чем соответствующий 4aS, 8aS энантиомер.
Соответственно, основной целью настоящего изобретения является (1R, 4aR, 8aR)-1,2,3,4,5,6,7,8-пергидроизохинолин-1-карбонил-(L)-пролинил-(L)-аргинин-альдегид, его фармацевтически приемлемые соли и сольваты, которые являются неожиданно более сильными ингибиторами тромбина и пригодны в качестве антикоагулянтов и препаратов против тромбоза с эмболией.
Предложенное этим изобретением соединение выражается следующей формулой I.
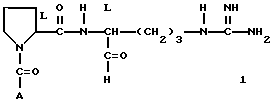
где A представляет собой
и его фармацевтически приемлемые нетоксичные соли и сольваты.
Пептид, выраженный формулой I, пригоден как антитромбовый препарат и может быть использован как вспомогательный препарат для активатора плазминогена ткани (tPA), стрептокиназа или урокиназа терапии.
Соединение получают обычными методами получения [1-3]. Например, при взаимодействии Cbz-1,2,3,4,5,6,7,8-пергидро-гидро-1-изохинолинкарбоновой кислоты с эфиром L-пролина образуется эфир Cbz-1,2,3,4,5,6,7,8-пергидроизохинолин-1-карбонил-Pro. Эфирную группу удаляют и Cbz-1,2,3,4,4,5,6,7,8-пергидроизохинолин-1-карбонил-Pro взаимодействует с лактамом 1-аргинина до образования Cbz-1,2,3,4,5,6,7,8-пергидроизохинолин-1-карбонил-Pro-Arg лактама в защищенной по аминогруппе форме. Arg лактамовое кольцо раскрывают восстановлением и снимают защитные группы с аргинин амино и пергидроизохинолинового азота и получают альдегид 1,2,3,4,5,6,7,8-пергидроизохинолин-1-карбонил-Pro-Arg. Пептид превращают в подходящую соль, такую как ацетат и сульфат.
1,2,3,4,5,6,7,8-пергидро-1-изохинолинкарбоновую кислоту легко получают гидрированием 1-изохинолинкарбоновой кислоты в эталоне или другом пригодном спирте в присутствии 5N соляной кислоты или другой подходящей сильной неорганической кислоты над 5% Rh/A12O3 или на другом подходящем катализаторе при давлении приблизительно от 500 до 1000 фунтов/дюйм2 и при температуре от приблизительно 30oC до приблизительно 80oC.
Эта методика дает возможность получить (1R, 4aS, 8aS)-1,2,3,4,5,6,7,8-пергидро-1-изохинолинкарбоновую кислоту и (1S, 4aR, 8aR)-1,2,3,4,5,6,7,8-пергидро-1-изохинолинкарбоновую кислоту как рацемическую смесь. Индивидуальные энантиомеры могут быть получены разделением рацемата классическими методами. Такие методы включают образование солей с оптически активными кислотами и также разделение с помощью жидкостной хроматографии высокого давления рацемата на хиральной колонке.
Термодинамические изомеры, (1R, 4aR, 8aR)-1,2,3,4,5,6,7,8-пергидро-1-изохинолинкарбоновую кислоту и (1S, 4aS, 8aS)-1,2,3,4,5,6,7,8-пергидро-1-изохинолинкарбоновую кислоту, выделяют получением эфиров рацемической смеси с помощью указанных выше методик и реакцией этих эфиров с этилатом натрия в эталоне и затем деэтерификацией результирующей рацемической смеси термодинамических изомеров.
Иначе, рацемическая смесь изомеров и рацемическая смесь термодинамических изомеров, в качестве кислот, защищается по аминогруппе, взаимодействует с пролином, защищенным по карбоксилу, с образованием дипептида. Диастереоизомеры, образующиеся при взаимодействии с L-пролином, разделяют кристаллизацией.
Эфирную группу, защищающую карбоксигруппу пролиновой части дипептида, затем удаляют (деблокированием или деэтерификацией) и дипептид в форме свободной кислоты взаимодействует с лактамом аргинина. Данный продукт (лактам аргининовый продукт) реагирует с гидридным восстановителем, предпочтительно алюмогидрид лития или три-трет-бутоксиалюмогидрид лития, в инертном растворителе или смеси растворителей, для восстановления лактамового кольца и получения трипептида в аргининальдегидной форме. Затем удаляют защитные группы по известным методикам, таким как гидрирование на металлическом катализаторе.
Изобретение также предлагает метод для предупреждения образования тромбов у млекопитающих и фармацевтические препараты, пригодные для использования в этом методе.
Как показано в формуле I, асимметрическим центром пролиновой и аргининовой частей является L.
Пергидропроизводные, включающие D- 1,2,3,4,4а,5,6,7,8,8а-дека- гидроизохинолин-1-карбонил(пергидроизохинолин-1-карбонил или I-Pig) и D-1,2,3,4,4а, 5,6,7,8,8а-декагидроизохинолин-3-карбонил (пергидроизохинолин-3-карбонил или 3-Pig) производные Pro-Arg-H приведены ниже.
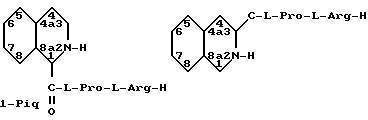
Пергидропроизводные могут существовать как цис- или трансстереоизомеры. Например, следующая пара стереоизомеров, идентифицируемая как цис-, может быть образована для D-пергидроизохинолин-1-карбонил-L-пролил-L-аргинин-альдегида: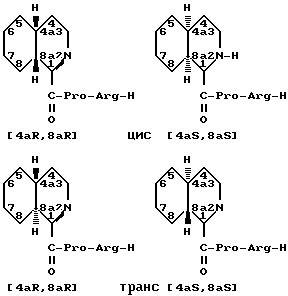
Фармацевтически приемлемые соли пептидов изобретения включают соли, образованные с неорганическими или карбоновыми кислотами. Примеры неорганических кислот, образующих соли, включают галогенводородные кислоты, такие как соляная, бромистоводородная кислоты, фосфорная кислота, серная кислота. Соли карбоновых кислот образуются с такими кислотами, как уксусная, пропионовая, малоновая, малеиновая, лимонная, янтарная, яблочная, бензойная, фумаровая и другие подобные карбоновые кислоты. Соли с кислотами получают обычными методами, например, с помощью нейтрализации соединения I в форме свободного основания с кислотой. Предпочтительными солями являются сульфат и гидрохлорид.
Как указано выше, настоящее изобретение включает сольваты соединений этого изобретения и их фармацевтически приемлемые соли. Соединение настоящего изобретения или его фармацевтически приемлемые соли могут образовывать сольваты с водой или обычными органическими растворителями. Такие сольваты включаются в область настоящего изобретения.
Соединение, представленное формулой I, получают известными методами пептидного синтеза. Согласно одному такому методу, кислота A-COOH, где A имеет те же значения, что приведены для формулы I, а атом азота защищен подходящей защитной группой для аминогруппы, взаимодействует с пролином, защищенным по карбокси, с образованием дипептида. Эфирная группа, защищающая карбоксигруппу пролиновой части продукта, удаляется, и дипептид в форме свободной кислоты взаимодействует с лактамной формой аргинина. Приведенная выше последовательность реакций иллюстрируется следующей схемой.

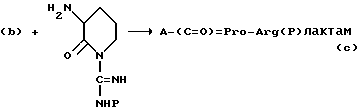
где P представляет собой защитную группу по аминогруппе.
Полученный Arg(P)лактам, продукт (c) восстанавливают алюмогидридом лития в инертном растворителе до расщепления лактамного кольца и получения трипептида в аргининальдегидной форме формулы:
A(C=O)-Pro-Arg(P)-H
где Arg(P)-H представляет собой защищенный по аминогруппе аргинин-альдегид.
Лактамную форму аргинина получают внутримолекулярным связыванием аминозащищенного аргинина [Arg-OH]. Например, Boc-Arg(Cbz)OH, имеющий формулу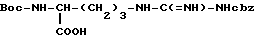
сначала превращается в активную эфирную форму, такую как смешанный ангидрид, с хлорформиатами, например этилхлорформиат до изобутилхлормиата. Образование эфира выполняется в присутствии третичного амина, такого как N-метилморфолин. Добавление сильного третичного амина, как основания, такого как триэтиламин, осуществляет внутреннее ацилирование до получения лактамной формы диаминозащищенного аргинина, как показано ниже: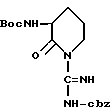
До проведения взаимодействия с A(C=O)-Pro-OH, как показано на приведенной выше схеме, Boc защитную группу селективно удаляют с трифторуксусной кислотой до получения требуемой свободной аминогруппы.
Связывание ACOOH соединения с пролиновым эфиром выполняется после защиты аминогруппы аминокислоты. Для временной защиты или блокировки аминогруппы обычно используют стандартные защитные группы для аминогрупп. Примерами таких защитных групп являются алкокси, алкенилокси, циклоалкокси и арилоксикарбонил группы, такие как этоксикарбонил, трет-бутилоксикарбонил (Boc), циклогексилоксикарбонил, адамантилоксикарбонил, трихлорэтоксикарбонил, бензилоксикарбонил (Cbz), дифенилметоксикарбонил и другие подобные группы. Эфирные группы, пригодные для защиты карбоксигруппы пролина при проведении реакции, могут быть любыми из обычно используемых, легко удаляемых групп, таких как трет-бутил, бензил, n-нитробензил, n-метоксибензил, дифенилметил, трихлорэтил, фенацил или триалкилсилил эфиры. При выполнении реакции взаимодействия используют такую эфирную группу для пролина, которая удаляется в условиях, при которых защитная группа аминогруппы остается не тронутой. Защитная группа аминогруппы ацилирующей кислоты ACOOH, таким образом, остается на месте для защиты аминогруппы при последующем превращении с аргининлактамным соединением с образованием формы с.
Пергидробицикло группы получают гидрированием или частично восстановленных или ненасыщенных кислот по обычным методикам. Например, 1,2,3,4-тетрагидроизохинолин-1-карбоновая кислота гидрируется над окисью платины в растворителе, таком как этанол или уксусная кислота, до образования пергидро(декагидро)изохинолин-1- карбоновой кислоты. Пергидрокислоты затем используются, как описано выше, при ацилировании пролинового эфира. Примеры таких пергидропроизводных, представленных формулой I, представляют собой N-(D-декагидроизохинолин-1-карбонил)-L-пролил-L-аргинин-альдегид и N-(D-декагидроизохинолин-3-карбонил)-L-пролил-L-аргинин.
Описанный выше процесс гидрирования приводит к образованию смеси цис- и трансстереоизомеров, что обсуждалось выше здесь, причем цисстереоизомеры образуются в большем количестве. Например, соединения включают D-I-(4aS, 8aS)-Pig-(L)-Pro-(L)-Arg-H, D-I-(4aR, 8aR)-Pig-(L)-Pro-(L)-Arg-H, D-I-(4aS, 8aR)-Pig-(L)-Pro-(L)-Arg-H, D-1-(4aR, 8aS)-Pig-(L)-Pro-(L)-Arg-H, D-3-(4aS, 8aS)-Pig-(L)-Pro-(L)-Arg-H, D-3-(4aR, 8aR)-Pig-(L)-Pro-(L)-Arg-H, D-3-(4aS, 8aR)-Pig-(L)-Pro-(L)-Arh-H, и D-3-(4aR, 8aS)-Pig-(L)-Arg-H.
Реакции, описанные выше, предпочтительно выполняются при низких температурах от -20oC до 15oC. Реакции выполняются в инертном органическом растворителе, таком как диметилформамид, диметилацетамид, тетрагидрофуран, хлористый метилен, хлороформ и другие подобные обычные растворители. В основном, применяют безводные условия, когда в реакции взаимодействия активный эфир ацилирующей кислоты используется.
Соединения изобретения лучше выделяются в форме соли с кислотой. Соли соединений формулы I, образованные с кислотами, как названные выше, пригодные как фармацевтически приемлемые для введения антитромбовых препаратов и для получения препаративных форм этих агентов. Другие соли с кислотами могут быть получены и использованы при выделении и очистке пептидов. Например, соли, образованные с сульфокислотами, такими как метансульфокислота, н-бутансульфокислота, n- толуолсульфокислота и нафталинсульфокислота, могут использоваться.
Предпочтительный метод выделения и очистки соединений формулы I, хотя за то же время получающий требуемую стабильную соль, описан в пат. США 5250660, включает препаративную очистку C18 обращенно-фазовой хроматографией. Водная фаза включает серную кислоту или соляную кислоту в концентрациях между приблизительно 0,01 и приблизительно 0,05%; органическая фаза - ацетонитрил, ТГФ, метанол или другие подходящие растворители. Величина pH кислотного элюата регулируется между приблизительно pH 4 и приблизительно pH 6, точное pH, являющееся функцией отдельного пептида, с основной смолой, например Bio-Rad AG-IX8 смолой в гидроксильной форме. После регулирования pH раствора солей трипептида, например сульфата или гидрохлорида, проводят лиофилизацию до получения очищенной соли в форме сухого порошка. В примере способа сырой 1-(1R, 4aR, 8aR)-Pig-L-Pro-L-Arg-H сульфат, загрязненный эпимерным D-Arg-H сульфатом, растворяют в воде и раствор вводят на колонку 5 х 50 см Vydac C18 RPHPLC. Используют градиент 2-20% B (A = 0,01% H2SO4; B = ацетонитрил) в течение 10 часов. Фракции собирают и фракции, содержащие требуемый продукт, по определению с помощью аналитической RP-HPLC (обращенно-фазовая жидкостная хроматография высокого давления), объединяют. Величина pH у объединенных фракций регулируется приблизительно от 4,0 до 4,5 со смолой Bio-Rad AG-IX8 в гидроксильной форме. Раствор после фильтрации лиофилизируют до чистого 1-(1R, 4aR, 8aR)-Pig-L-Pro-L-Arg-H сульфата.
Соединение изобретения предлагают для селективного ингибирования тромбина в сравнении с другими протеиназами и неферментными протеинами при коагуляции крови без заметного препятствия способности растворять природные сгустки в крови (соединения имеют низкий ингибирующий эффект на растворение кровяных сгустков). Кроме того, такая селективность допускает использование с препаратами, растворяющими тромбы, без значительного вмешательства в процессы растворения тромбов и сгустков крови.
Соединение, предложенное изобретением (формула I) селективно ингибирует действие тромбина у человека и животных (млекопитающие). Ингибирование тромбина показано с помощью ингибирования активности амидазы тромбина. Следующая таблица I перечисляет кажущиеся константы равновесия (Kass) для взаимодействия между тестируемым соединением (ингибитор) и тромбином. Данные в таблице получены анализом, в котором тромбин гидролизует хромогенный субстрат, N-бензоил-D-фенилаланил-L-валил-L-аргинин-n-нитроанилид.
Анализ выполняют в 50 мкл буфера (0,03 М трис-буфера, 0,15 M NaC1, pH 7,4) с 25 мкл раствора тромбина человека (очищенный тромбин человека, Enzyme Research Laboratories, South Bend, Indiana, при 8 NIH единиц/мл) и 25 мкл тестируемого соединения в растворителе (50% водного метанола (об:об). Затем вносят 150 мкл водного раствора хромогенного субстрата (при 0,25 мг/мл) и измеряют скорость гидролиза субстрата с помощью мониторинга реакций при 405 нм на высвобождение n-нитроанилина. Стандартные кривые получают построением зависимости свободной концентрации тромбина от скорости гидролиза. Скорость гидролиза, наблюдаемая для тестируемых соединений, затем преобразуется до величины "свободного тромбина" в соответствующем анализе с помощью использования стандартных кривых. Количество связанного тромбина (связанного с тестируемым соединением) рассчитывается вычитанием количества свободного тромбина, наблюдаемого в каждом анализе, из известного начального количества тромбина, наблюдаемого в каждом анализе. Количество свободного ингибитора в каждом анализе рассчитывается вычитанием количества молей связанного тромбина из количества молей добавленного ингибитора (тестируемое соединение).
Величина Kass является константой гипотетического равновесия для реакции между тромбином и тестируемым соединением (1).
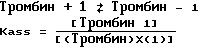
Kass рассчитывается для ряда концентраций тестируемых соединений и средняя величина приводится в единицах литр на моль.
С помощью, в основном, следующих методик, описанных выше для тромбина человека, использующих другие сериновые протеиназы системы коагуляции крови человека и использующих сериновые протеиназы фибринолитической системы, с соответствующими хромогенными субстратами, идентифицированными ниже, селективность соединений настоящего изобретения в соответствии с сериновыми протеиназами свертывания крови и фибронолитическими сериновыми протеиназами оценивают так же, как и отсутствие вмешательства в растворение сгустков в плазме человека.
Фактор Xa человека покупают у Enzyme Research Laboratories, South Bend, Indiana. Хромогенный субстрат: N - бензоил - Ile - Glu - Gly - Arg - n -нитроанилид (для фактора Xa) покупают у Kabi Vitrum, Stockholm, Sweden или у Midwest Biotech, Fishers, Indiana. Бычий трипсин покупают у Worthington Biochemicals, Freehold, New Jersey. Хромогенный субстрат, N - бензоил -Phe - Val - Arg - n - нитроанилид, субстрат для тромбина человека и для трипсина, синтезируют по методикам, описанным выше, для соединений настоящего изобретения с использованием известных методов пептидного синтеза из коммерчески доступных реагентов или покупают у Midwest Biotech, Fishers, Indiana.
Плазмин человека покупают у Boehringer Mannheim, Indianapolis, Indiana; nt-PA покупают как единичную покупку у American Diagnostica, Greenwich, Connecticut. Хромогенный субстрат плазмина, H-D-Val-Leu-Lys-n-нитроанилид и субстрат активатора плазминогена ткани (t-PA), H-D-Ile-Pro-Arg-n-нитроанилид, покупают у Kabi Vitrum, Stockholm, Sweden.
В хромогенных субстратах, описанных выше, используют трехбуквенные символы - Ile, Glu, Gly, Pro, Arg, Phe, Val, Leu и Lys, они показывают группы соответствующих аминокислот, изолейцина, глутаминовой кислоты, глицина, пролина, аргинина, фенилаланина, валина, лейцина и лизина соответственно.
Таблица представляет перечень величин Kass, полученных с указанными соединениями.
Соединение изобретения селективно ингибирует образование сгустков без заметного вмешательства в способность растворять природные сгустки, например соединения имеют низкую ингибирующую растворение фибрина способность.
Изобретение в одном из своих аспектов предлагает метод для ингибирования образования сгустков крови у человека и животных (млекопитающих), который включает введение человеку или животным соединения формулы I в дозе, эффективно ингибирующей образование сгустков и нетоксичной. Соединение-антикоагулянт вводят орально, парентерально, например при внутривенном вливании (в/в), внутримышечном введении (в/м) или подкожно (п/к).
Доза, эффективно ингибирующая образование сгустков, составляет между приблизительно 5 мг и приблизительно 1000 мг. Режим доз может изменяться, например, при профилактическом применении могут вводиться или единичная дневная доза, или многократные дозы 3 или 5 раз в день соответственно. В критических ситуациях соединение изобретения вводят в/в вливанием в дозе между приблизительно от 0,1 мг/кг/час и до приблизительно 1,0 мг/кг/час.
При оральном введении доза соединения настоящего изобретения составляет от приблизительно 0,1 мг/кг до приблизительно 20 мг/кг и может вводиться один или более раз в течение 24 часов. Предпочтительно, доза составляет от приблизительно 0,5 мг/кг до приблизительно 5 мг/кг и вводится 4 раза за 24 часа. Настоящие соединения могут вводиться орально в стандартных формах, таких как таблетки, капсулы и другие подобные, как показано ниже.
Метод этого изобретения также практикуется в сочетании с агентами, растворяющими сгустки, например, активатор плазминогена ткани (tPA), модифицированный tPA, стрептокиназа или урокиназа. В случае, когда происходит образование сгустков и блокируется артерия или вена, или частично, или полностью, обычно используется агент, растворяющий сгустки. Соединение изобретения может вводиться вместе с растворяющим агентом или после его использования для предотвращения повторного образования сгустков.
Это изобретение также предлагает фармацевтические препаративные формы для использования в описанных выше терапевтических методах. Фармацевтические препаративные формы включают эффективное для ингибирования тромбина количество соединения формулы I в сочетании с фармацевтически приемлемым носителем, эксципиентом или разбавителем. Для орального введения антитромбовые препараты формулируют в желатиновые капсулы или таблетки, которые могут включать эксципиенты, такие как связующее вещество, смазывающее вещество, дезинтегрирующий агент и другие подобные. Для парентерального введения антитромбовое вещество формулируется в фармацевтически приемлемый разбавитель, такой как физиологическая соль (0,9%), 5% декстроза, раствор Рингера и другие подобные.
Соединение настоящего изобретения может формулироваться в препаративную форму, включающую единичную дозу, составляющую между приблизительно 0,1 мг и приблизительно 1000 мг. Предпочтительно, соединение используют в форме фармацевтически приемлемой соли, такой как, например, сульфат, ацетат или фосфат. Например, препаративная форма в единичной дозе включает 5 мг соединения настоящего изобретения как фармацевтически приемлемую соль в 10 мл стерильной стеклянной ампуле. Другим примером препаративной формы единичной дозы является форма, включающая 10 мг соединения настоящего изобретения как фармацевтически приемлемую соль в 20 мл изотонического солевого раствора, помещенных в стерильную ампулу.
Соединения могут вводиться различными путями, включая оральный, ректальный, трансдермальный, подкожный, внутривенный, внутримышечный и внутрь носа. Соединения настоящего изобретения предпочтительно формулируются до их введения. Другим воплощением настоящего изобретения является фармацевтическая форма применения, включающая эффективное количество соединения формулы I или его фармацевтически приемлемой соли или сольвата в сочетании с фармацевтически приемлемым носителем, разбавителем или эксципиентом.
Активный ингредиент в таких формах составляет от 0,1% до 99,9% весовых от формы. Термин "фармацевтически приемлемый" означает носитель или эксципиент, совместимые с другими ингредиентами препаративной формы и не оказывающие вредного действия на пациентов.
Настоящие препаративные фармацевтические формы получают по известным методикам и из легко доступных ингредиентов. Составы этого изобретения могут формулироваться с помощью известных методов так, чтобы обеспечить после введения пациенту быстрое, поддерживаемое или замедленное высвобождение активного ингредиента. При приготовлении составов настоящего изобретения активный ингредиент обычно смешивают с носителем или разбавляют носителем, или включают внутрь носителя, который может быть в форме капсул, мешков, бумаги или других контейнеров. Когда носитель служит как разбавитель, он может быть твердым, полутвердым или жидким веществом, которое действует как связующее вещество, экспициент или среда для активного ингредиента. Таким образом, составы могут быть в форме таблеток, пиллет, порошка, лепешек, мешочков, облаток, эликсиров, суспензий, эмульсий, растворов, сиропов, аэрозолей (как твердых, или в жидкой среде), мягких или твердых желатиновых капсул, суппозиториев, стерильных растворов для инъекций, стерильно упакованных порошков и т.п.
Следующие примеры препаративных форм иллюстрируют, но не ограничивают область изобретения. "Активный ингредиент", конечно, означает соединение формулы I или его фармацевтически приемлемую соль, или сольват.
Препаративная форма 1
Твердые желатиновые капсулы готовят при использовании следующих ингредиентов (мг/капсула):
Активный ингредиент - 250
Крахмал, высушенный - 200
Стеарат магния - 10
Общее количество - 460
Препаративная форма 2
Таблетки получают с использованием ингредиентов, приведенных ниже (мг/капсула):
Активный ингредиент - 250
Целлюлоза, микрокристаллическая - 400
Двуокись кремния, пыль - 10
Стеариновая кислота - 5
Общее количество - 665
Компоненты смешивают и прессуют в таблетки, каждая весом 665 мг.
Препаративная форма 3
Аэрозольный раствор получают из следующих компонентов:
Активный ингредиент - 0,25
Этанол - 25,75
Пропеллент 22 (Хлордифторметан) - 70,00
Общее количество - 100,00
Активное соединение смешивают с этанолом и смесь добавляют к части пропеллента 22, охлаждают до -30oC и переносят в наполняющее устройство. Требуемое количество затем вносят в контейнер из нержавеющей стали, разбавляют оставшейся частью пропеллента. Затем к контейнеру крепят вентиль.
Препаративная форма 4
Таблетки, каждая из которых содержит 60 мг активного ингредиента, получают следующим образом (мг):
Активный ингредиент - 60
Крахмал - 45
Микрокристаллическая целлюлоза - 35
Поливинилпирролидон (как 10%-ный раствор в воде) - 4
Карбоксиметилкрахмал натрия - 4,5
Стеарат магния - 0,5
Тальк - 1
Общее количество - 150
Активный ингредиент, крахмал и целлюлозу пропускают через сита с размером отверстий N 45 США и тщательно смешивают. Водный раствор, содержащий поливинил-пирролидон, смешивают с полученным порошком, смесь пропускают через сита N 14. Гранулы, полученные таким образом, сушат при 50oC и пропускают через сита N 18. Карбоксиметилкрахмал натрия, стеарат магния и тальк, предварительно пропущенные через сита N 60, прибавляют к гранулам, которые после перемешивания прессуют на специальном устройстве для получения таблеток, и получают таблетки, каждая из которых весит 150 мг.
Препаративная форма 5
Капсулы, каждая содержащая 80 мг активного ингредиента, получают следующим образом (мг):
Активный ингредиент - 80
Крахмал - 59
Микрокристаллическая целлюлоза - 59
Стеарат магния - 2
Общее количество - 200
Активный ингредиент, целлюлоза, крахмал и стеарат магния смешивают, пропускают через сита N 45 и смесью наполняют капсулы твердого желатина количеством в 200 мг.
Препаративная форма 6
Суппозитории, каждая из которых содержит 225 мг активного ингредиента, готовят следующим образом (мг):
Активный ингредиент - 225
Глицириды насыщенных кислот - 2000
Общее количество - 2225
Активный ингредиент пропускают через сита N 60, суспендируют в глициридах насыщенных жирных кислот, предварительно расплавленных при использовании минимального нагревания. Смесь затем выливают в формы для суппозиториев с номинальной емкостью 2 г и охлаждают.
Препаративная форма 7
Суспензии, содержащие 50 мг активного ингредиента на 5 мл дозу, получают следующим образом (мг):
Активный ингредиент - 50
Карбоксиметилцеллюлоза натрия - 50
Сироп - 1,25
Раствор бензойной кислоты - 0,10
Отдушка - по виду
Краситель - по виду
Очищенная вода до общего количества - 5
Активный ингредиент пропускают через сито N 45, смешивают с карбоксиметилцеллюлозой натрия и сиропом до образования однородной пасты, прибавляют при перемешивании раствор бензойной кислоты, отдушку и краситель, разбавленные частью воды, и затем вносят необходимое количество воды до получения требуемого объема.
Препаративная форма 8
Препаративную форму для внутривенного введения получают следующим образом (мг):
Активный ингредиент - 100
Изотонический солевой раствор - 1000
Раствор из вышеприведенных ингредиентов, в основном, вводят внутривенно со скоростью 1 мл в минуту.
Пример препаративной формы единичной дозы включает 5 мг сульфата (1R, 4aR, 8aR)-пергидроизохинолин-1-карбонил-L-пролил-L-аргинин-альдегида в 10 мл стерильной стеклянной ампуле. Другим примером препаративной формы единичной дозы является 10 мг сульфата (1R, 4aR, 8aR)-пергидроизохинолин-1-карбонил-L-пролил-L-аргинин-альдегида в 20 мл изотонического солевого раствора, помещенные в стерильную ампулу.
Предпочтительной препаративной формой является форма единичной дозы, включающая между 5 мг и 50 мг сульфата (1R, 4aR, 8aR)-пергидроизохинолин-1-карбонил-L-пролил-L-аргинин-альдегида в стерильной ампуле.
Следующие примеры предлагают дальнейшее описание изобретения и не ограничивают область изобретения.
Величина Rf в следующих примерах определяется тонкослойной хроматографией на силикагеле при использовании кизельгеля 60F-254 (Merck, Darmstradt) в следующих системах растворителей:
(A) хлороформ-метанол-уксусная кислота, 135:15:1, об:об:об
(B) этилацетат - уксусная кислота - абсолютный этанол, 90:10:10, об:об: об
(C) хлороформ : метанол : уксусная кислота, 90:30:5, об:об:об
(D) этилацетат : гексан, 30:70, об:об
В примерах используются следующие методы аналитической HPLC (жидкостная хроматография высокого давления).
Метод 1. Waters 600 E с использованием Vydac C18 обращенно-фазовой колонки 0,46 х 10 см. Хроматограмма контролируется на LDC при 220 нM при использовании градиента А = 0,01 М ацетата аммония и B = ацетонитрила.
Метод 2. Pharmacia FPLC при использовании Pep RPC размером 0,5 х 5,0 см. Контроль выполняют на Pharmacia UV-M при 214 нM при использовании градиента или A = 0,01 M ацетата аммония, или B = ацетонитрил.
Обозначения, используемые здесь, имеют следующие значения.
Аминокислоты: Arg = аргинин, Pro = пролин, Phg = фенилглицин
Boc = трет-бутилоксикарбонил
Bz1 = бензил
Cbz (или z)=бензилоксикарбонил
DCC = дициклогексилкарбодиимид
DMF = диметилформамид
DMSO = диметилсульфоксид
FAB-MS = масс-спектр при бомбардировке устойчивыми атомами
FD-MS = масс-спектр полевой десорбции
THF = тетрагидрофуран
TLC = тонкослойная хроматография
Пример 1. Получение (1R, 4aR, 8aR)-пергидроизохинолин-1-карбонил-L-пролил-L-аргинин-альдегида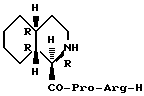
Метилкарбамат-фенэтиламин (I)
К раствору фенэтиламина (75,22 мл, 0,6 молей) и триэтиламина (83 мл, 0,6 молей) в ТГФ (500 мл) при перемешивании медленно прибавляют метилхлорформиат (46,2 мл, 0,6 молей) в ТГФ (50 мл), перемешивают 1 час при комнатной температуре и вносят диэтиловый эфир (2 л) и I N HCl (800 мл). Органический слой промывают водой, сушат (MgSO4), фильтруют, фильтрат упаривают в вакууме и получают прозрачное масло из чистого названного соединение (102 г, 95%).
Метилкарбамат-DL-1,2,3,4-тетрагидроизохинолин-1-карбоновая кислота (2)
К раствору метилкарбамат-фенэтиламина (1) (102 г, 0,57 молей) в трифторуксусной кислоте (300 мл) прибавляют глиоксалевую кислоту (63 г, 0,68 молей), кипятят 4 часа, охлаждают до комнатной температуры, растворитель упаривают в вакууме. Остаток обрабатывают диэтиловым эфиром (800 мл) / водой (100 мл), pH реакционной смеси доводят до 12 добавлением 5N NaOH. Водный слой отделяют, прибавляют к нему диэтиловый эфир (500 мл), подкисляют до pH 2,5 5N HCl. Органический слой отделяют, сушат (MgSO4), фильтруют, упаривают в вакууме и получают чистый названный продукт в виде масла (107 г, 80%); FAB-MS 236 (MH+).
Трет-бутиловый эфир метилкарбамат-DL-1,2,3,4-тетрагидроизохинолин-1-карбоновой кислоты (3)
К охлажденному (0oC) раствору метилкарбамат-DL-1,2,3,4- тетрагидроизохинолин-1-карбоновой кислоты (2) (105 г, 0,45 молей) в CH2Cl2 (200 мл) при перемешивании прибавляют третбутанол (52 мл, 0,54 молей), 4-диметиламино-пиридин (10 г, 0,08 молей) и DCC (92 г, 0,45 молей), перемешивают 2 часа при 0oC, 24 часа при комнатной температуре, упаривают растворитель в вакууме и добавляют к остатку этилацетат (800 мл)/IN NaHCO3(300 мл). Органический слой отделяют, последовательно промывают водой, 1,5N лимонной кислотой и водой. Органический слой сушат (MgSO4), фильтруют, фильтрат упаривают в вакууме и получают чистое названное соединение в виде масла (106 г, 81%); FAB-MS 292 (МН+); ТСХ Rf(A) 0,61;
Элементный анализ для C16H21NO4:
Вычислено: C 65,96 H 7,27 N 4,81
Найдено: C 66,24 H 7,28 N 4,73
Трет-бутиловый эфир метилкарбамат-DL-1,2,3,4,6,7,8-пергидроизохинолин-I-карбоновой кислоты (4)
Раствор трет-бутилового эфира метилкарбамата-DL-1,2,3,4-тетрагидроизохинолин-I-карбоновой кислоты (3) (105 г, 0,36 молей) в третбутаноле (800 мл) гидрируют на 5% Rh/Al2O3 (52,5 г) при 800 фунтов/дюйм2 на аппаратуре высокого давления при 50oC в течение 24 часов. Реакционную смесь фильтруют через прокладку из целита®, фильтрат упаривают в вакууме. Полученное масло сушат и получают чистое названное соединение (96,5 г, 90%); FD-MS 298 (МН+); ТСХ Rf(C) 0,63.
Этиловый эфир метилкарбамат-1,2,3,4,4aS,6,7,8,8aS-пергидроизохинолин-S-I-карбоновой кислоты + Этиловый эфир метилкарбамат-1,2,3,4,4aR, 6,7,8,8aR-пергидроизохинолин-R-I-карбоновой кислоты (5)
К раствору трет-бутилового эфира метилкарбамат-DL-I-2,3,4,6,7,8-пергидроизохинолин-I-карбоновой кислоты (4) (81,2 г, 273 ммолей) в EtOH (500 мл) прибавляют этилат натрия (21% в этаноле) (88,4 мл, 273 ммолей), реакцию кипятят 24 часа. Органический растворитель упаривают в вакууме, остаток обрабатывают этилацетатом (400 мл) и водой (100 мл). Органический слой отделяют, промывают водой, сушат (MgSO4), фильтруют, фильтрат упаривают в вакууме и выделяют чистое названное соединение в виде масла (70 г, 95%); FAB-MS 270 (МН+); ТСХ Rf (A) 0,61.
Метилкарбамат-1,2,3,4,4aS, 6,7,8,8aS-пергидроизохинолин-S-I-карбоновая кислота + Метилкарбамат-1,2,3,4,4aR,6,7,8,8aR-пергидроизохинолин-R-I-карбоновая кислота (6)
К раствору 5 (50 г, 260 ммолей) в ТГФ (250 мл) прибавляют 2 N NaOH (156 мл, 312 ммолей), реакцию перемешивают 30 часов при комнатной температуре. Органический слой упаривают в вакууме, остаток обрабатывают диэтиловым эфиром (400 мл) и водой (100 мл). Водный слой отделяют, добавляют этилацетат (400 мл), раствор подкисляют 5N HCl до pH 2,0. Органический слой сушат (MgSO4), фильтруют, фильтрат упаривают в вакууме и получают прозрачное масло, которое кристаллизуют из гексана (200 мл) и получают чистое названное соединение (46,4 г, 74%); FAB-MS 242 (MH+); ТСХ Rf (A) 0,36;
Элементный анализ для C12H19NO4:
Вычислено: C 59,74 H 7,94 N 5,81
Найдено : C 59,95 H 7,88 N 5,54
Cbz-1,2,3,4,4aS, 6,7,8,8aS-пергидроизохинолин-S-I-карбоновая кислота + Cbz-1,2,3,4,4aR,6,7,8,8aR-пергидроизохинолин-R-I-карбоновая кислота (7)
К раствору 6 (46 г, 191 ммолей) в безводном CH3CN (200 мл) при перемешивании при комнатной температуре в инертной атмосфере прибавляют раствор иодтриметилсилана (62,4 мл, 440 ммолей) в CH3CN (60 мл). Реакционную смесь перемешивают 30 минут при 55oC, охлаждают до комнатной температуры. Реакцию останавливают добавлением воды (100 мл) с последующей обработкой метабисульфитом натрия (1 г). Смесь подщелачивают 5N NaOH до pH 10,0, прибавляют по каплям бензилхлорформиат (27,3 мл, 191 ммолей), в то время, как pH поддерживают на уровне 10 с помощью 2N NaOH. Затем реакцию перемешивают 30 минут при комнатной температуре, органический растворитель упаривают в вакууме, вносят диэтиловый эфир (200 мл). Реакцию оставляют на 2 часа при комнатной температуре и добавляют этилацетат (200 мл). Водный раствор подкисляют 5N HCl до pH 2,5, органический слой отделяют, сушат (MgSO4), фильтруют, фильтрат упаривают в вакууме и получают чистое названное соединение в виде масла (39,5 г, 65%); FAB-MS 318 (МН+);
Элементный анализ для C18H23NO4:
Вычислено: C 68,12 H 7,30 N 4,41
Найдено: C 66,37 H 7,52 N 4,37
Cbz-1,2,3,4,4aS, 6,7,8,8aS-пергидроизохинолин-S-I-карбонил-Pro-трет-Bu + Cbz-1,2,3,4,4aR,6,7,8,8aR-пергидроизохинолин-R-I-карбонил-Pro-трет-Bu (8)
К раствору 7 (39 г, 123 ммолей) в ДМФА (200 мл) при перемешивании и при охлаждении (0oC) прибавляют трет-бутиловый эфир пролина (21,1 г, 123 ммолей), I-гидроксибензотри-азол (16,6 г, 123 ммолей) и DCC (25,3 г, 123 ммолей), перемешивают 2 часа при 0oC и 24 часа при комнатной температуре. Выпавший осадок отфильтровывают, фильтрат упаривают в вакууме до масла. Масло растворяют в EtOAc (200 мл) и воде (100 мл). Органический слой последовательно промывают IN NaHCO3, водой, 1,5 N лимонной кислотой и водой. Органический слой отделяют, сушат (MgSO4), фильтруют, фильтрат упаривают до аморфного твердого вещества и получают названное соединение (52,7 г, 91%), FAB-MS 471 (МН+).
Cbz-1,2,3,4,4aR,6,7,8,8aR-пергидроизохинолин-(IR)-карбонил- Pro-OH (9)
К раствору 8 (52,4 г, III ммолей) в CH2Cl2 (20 мл) прибавляют трифторуксусную кислоту (70 мл) и анизол (5 мл), перемешивают 1 час при комнатной температуре, упаривают в вакууме без нагревания, разбавляют диэтиловым эфиром (400 мл), водой (100 мл) и pH раствора доводят до pH 10,0 с помощью 5N NaOH. Водный слой отделяют, добавляют этилацетат (300 мл), подкисляют 5N HCl до pH 2,5. Органический слой отделяют, сушат (MgSO4), фильтруют, фильтрат упаривают в вакууме и получают прозрачное масло. Масло растворяют в диэтиловом эфире (500 мл) и прибавляют L(-)-[α]D-метилбензиламин, выдерживают 24 часа при комнатной температуре, образовавшийся осадок отфильтровывают, промывают диэтиловым эфиром, сушат. Твердый осадок суспендируют в этилацетате, промывают 1,5 N лимонной кислотой и водой. Органический слой сушат (MgSO4), фильтруют, фильтрат упаривают и получают названное соединение в виде масла (20,2 г, 44%); FAB-MS 415 (МН+); [α]D=3,2o (C, 0,5 MeOH);
Элементный анализ для C23H30N2O5:
Вычислено: C 66,65 H 7,30 N 6,76
Найдено : C 66,38 H 7,36 N 6,63
(A) Boc-L-Arg(Cbz)-OH
N-Boc-аргинин гидрохлорид (Boc-Arg-OH х HСl) (82,1 г, 250 ммолей) растворяют в 240 мл 5 N NaOH в круглодонной трехгорлой колбе, охлаждают до -5oC и прибавляют по каплям в течение 56 минут бензилхлорформиат (143 мл, 1,0 молей, 4 экв.), поддерживания с помощью добавления 5 N NaOH (250 мл) pH на уровне 13,2-13,5. Реакционную массу перемешивают 1 час при температуре -5oC, разбавляют 100 мл воды и 500 мл диэтилового эфира. Водный слой отделяют, экстрагируют дважды 40 мл порциями диэтилового эфира. Водный слой подкисляют 3 N H2SO4 (560 мл) до pH 3,0 и экстрагируют 550 мл этилацетата. Отделенный водный слой экстрагируют только этилацетатом, экстракт объединяют с предыдущим этилацетатным экстрактом. Объединенные экстракты промывают водой, сушат над MgSO4, упаривают в вакууме досуха. Остаток затирают с эфиром, выпавший продукт отфильтровывают, сушат и получают 66,1 г (3) Boc-Arg(Cbz)-OH (65% от теории): ТСХ Rf (C) 0,43; FD-MS 408(M+).
1H ЯМР (CDCl3), δ 1,42 (с, 9H), 1,61-1,91 (м, 4H), 3,23-3,41 (м, 2H), 4,17 (д, IH), 5,21 (с, 2H), 5,62 (д, IH), 7,30-7,42 (м, 6H), 8,37 (м, 1H).
(B) Boc-Arg(Cbz)-лактам
Раствор Boc-Arg(Cbz)-OH (A), полученного по методике, описанной ранее, (66,0 г, 0,162 молей) в 230 мл сухого ТГФ охлаждают на бане лед-ацетон до -10oC, к охлажденному раствору прибавляют N-метилморфолин (18,7 мл, 1,05 экв.) с последующим внесением изобутилхлорформиата (22,5 мл, 1,05 экв.), перемешивают 5 минут при -10oC, затем, добавляют триэтиламин (23,5 мл, 1,05 экв. ), перемешивают 1 час при -10oC и 1 час при комнатной температуре. Реакционную смесь выливают в 1 л смеси лед-вода и выпадает названное соединение, которое отфильтровывают, промывают холодной водой, сушат в вакууме, кристаллизуют из этилацетата и получают 38,05 г (60% от теории) названного соединения, Boc-Arg(Cbz)-лактам; ТСХ Rf (A) 0,77; FD-MS 291 (MH+).
1H-ЯМР (CDCl3), δ 1,48 (с, 9H), 1,78-1,98 (м, 2H), 2,50 (м, IH), 3,41 (м, IH), 4,43 (м, IH), 4,90 (м, IH), 4,16 (с, 2H), 5,27 (м, IH), 7,28-7,45 (м, 6H), 9,41 (м, IH), 9,68 (м, IH).
Cbr-1,2,3,4,4аR, 6,7,8,8аR-пергидроизохинолин-R-I-карбонил-Pro-Arg(Cbz) лактам (II)
Колба 1. К соединению 9 (13,9 г, 33,5 ммолей) в ДМФА (50 мл), охлажденному до -15oC прибавляют N-метилморфолин (3,7 мл, 33,5 ммолей), затем, вносят изобутилхлорформиат (4,4 мл, 33,5 ммолей), перемешивают 1 минуту при -15oC.
Колба 2. К HCl х Arg(Cbz)-лактаму (10,9 г, 33,5 ммолей), растворенному в ДМФА (50 мл) и охлажденному до 0oC, прибавляют диизопролилэтиламин (14,6 мл, 83,8 ммолей), перемешивают 1 минуту при 0oC.
Содержимое колбы 2 прибавляют к содержимому колбы 1, реакционную смесь перемешивают 2 часа при -15oC, затем, 2 часа при комнатной температуре, вносят I N NaHCO3(10 мл) и упаривают в вакууме растворитель до масла. Остаток растворяют в EtOAc (200 мл), промывают 1,5 N лимонной кислотой, водой, I N NaHCO3 (100 мл) и водой. Органический раствор сушат (MgSO4), фильтруют, упаривают в вакууме до сырого твердого остатка, который очищают хроматографически на силикагеле при использовании ступенчатого градиентного элюирования (гексан 100 до гексан-EtOAc 30:70), и выделяют чистое соединение 10. (9,0 г, 39%); FAB-MS 687 (MH+);
Элементный анализ для C37H46N6O7:
Вычислено: C 64,71 H 6,75 N 12,24
Найдено: C 64,23 H 6,69 N 11,88
Cbr-1,2,3,4,4аR, 6,7,8,8aR-пергидроизохинолин-R-I-карбонил-Pro-Arg (Cbz)-H (12)
К раствору II (9,0 г, 13,1 ммолей) в безводном ТГФ (100 мл) при перемешивании при охлаждении до -70oC в атмосфере азота прибавляют алюмогидрид лития I M в ТГФ (13,1 мл, 13,1 ммолей), перемешивают 30 минут при этой температуре, вносят по каплям 5 мл ТГФ и 5 мл 0,5 N H2SO4, разбавляют EtOAc (175 мл) и водой (100 мл). Органический слой отделяют, сушат (MgSO4), фильтруют, упаривают в вакууме органический растворитель и получают аморфный твердый продукт названного соединения (8,2 г, 91%); FAB-MS 689 (MH+).
1,2,3,4,4аR, 6,7,8,8аR-пергидроизохинолин-R-I-карбонил-Pro-Arg-H• H2SO4 (12)
Соединение 12 (8,2 г, 11,9 ммолей), растворенное в этаноле (50 мл), воде (10 мл) и I N H2SO4 (30 мл, 29,7 ммолей), гидрируют в присутствии 5% Pd/c катализатора (4,0 г) при комнатной температуре и давлении. После окончания реакции, катализатор удаляют фильтрацией. Фильтрат упаривают в вакууме до 40 мл и добавляют 50 мл воды. Величину pH раствора доводят до 4,2 с помощью смолы BioRad AGI-X8 (гидроксидная форма). Смолу удаляют фильтрацией, раствор лиофилизируют и получают сырое названное соединение (5,46). Твердый осадок (5,46 г) растворяют в 0,01% H2SO4 и сажают на колонку 5 х 25 см с Vydac C18 смолой. Для элюирования пептида из колонки используют градиент c увеличивающейся концентрацией CH3CN (1% до 5%). Фракции собирают и объединяют по данным аналитической RP-HPLC (обращенно-фазовая жидкостная хроматография высокого давления). Величину pH объединенных фракций доводят до 4,2 с помощью AGI-X8 смолы (Bio-Rad аналитическая анионобменная смола с 50 - 100 отверстиями) в гидроксидной форме. Раствор фильтруют, фильтрат лиофилизируют и получают чистое названное соединение (2,4 г, 39%); FAB-MS 421 (MH+; [α]D = -102,8o (C, 0,5/0,01 N H2SO4);
Элементный анализ для C21H36N6O3 • H2SO4 • 2H2O:
Вычислено: C 45,47 H 7,63 N 15,16
Найдено: C 45,05 H 7,44 N 15,02R
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ТРИПЕПТИДОВ В ВИДЕ R- ИЛИ RS-ФОРМЫ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ НЕТОКСИЧНЫЕ СОЛИ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1991 |
|
RU2077538C1 |
| ПРОИЗВОДНЫЕ ПЕПТИДОВ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМКОМПОЗИЦИЯ И СПОСОБ ИНГИБИРОВАНИЯ ТРОМБИНА У МЛЕКОПИТАЮЩЕГО | 1995 |
|
RU2148585C1 |
| ПРОИЗВОДНЫЕ ЦЕФАЛОСПОРИНА И ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1992 |
|
RU2104280C1 |
| ЦИС-(-)-4[(1(2)Н-ТЕТРАЗОЛ-5-ИЛ)МЕТИЛ]-2-ПИПЕРИДИНКАРБОНОВАЯ КИСЛОТА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ПРОЯВЛЯЮЩАЯ АКТИВНОСТЬ АНТАГОНИСТА ВОЗБУДИТЕЛЬНЫХ АМИНОКИСЛОТНЫХ РЕЦЕПТОРОВ | 1991 |
|
RU2089546C1 |
| МАКРОЛИДНЫЕ СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1992 |
|
RU2086560C1 |
| ПРОИЗВОДНЫЕ ГЛИКОПЕПТИДОВ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ | 1991 |
|
RU2053240C1 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛА ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ | 1991 |
|
RU2076100C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 1,2,3,4-ТЕТРАГИДРОНАФТАЛИНОВ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМОЙ КИСЛОТНО-АДДИТИВНОЙ СОЛИ | 1989 |
|
RU2014330C1 |
| БЕНЗО [F] ХИНОЛИНОН, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ | 1995 |
|
RU2172312C2 |
| ИНГИБИТОРЫ СЕРИНПРОТЕАЗЫ | 1997 |
|
RU2191193C2 |
Использование: изобретение предлагает (1R, 4aR, 8aR)-1,2,3,4,5,6,7,8-пергидроизохинолин-1-карбонил-(L)-пролинил-(L)-аргинин альдегид и его фармацевтически приемлемые соли и сольваты, фармацевтические препаративные формы, содержащие вышеназванные соединения, и методы их использования в качестве ингибиторов тромбина, ингибиторов коагуляции и препаратов для лечения тромбоза с эмболией. 2 с. и 2 з.п. ф-лы, 1 табл.
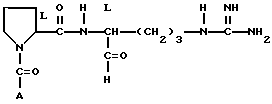
где А представляет собой 
или его фармацевтически приемлемые соли или сольваты.
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| US, патент, 5250660, кл | |||
| Устройство для сортировки каменного угля | 1921 |
|
SU61A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| US, патент, 5252566, кл | |||
| Устройство для сортировки каменного угля | 1921 |
|
SU61A1 |
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| EP, 0479489, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |

