Изобретение относится к замещенным тетразолам, которые пригодны в качестве промежуточных соединений в получении гипотензивных соединений.
В журнале J. Organometallic Chem. 33, 337, (1971 г.) и там же, 92, 303 (1975 г.) автор S. Kozima с сотр. описывает замещенные тетразолы формулы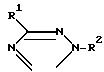
где
R1 низший алкил, бензил, низший алкенил или незамещенный или замещенный нитрогруппой, низшим алкилом, низшей алкоксигруппой или галогеном фенил и R2 SnR3.
B J. Amer. Chem. Soc. 80, 3909 (1958 г.) автор R. Lofquist с сотр. описывает замещенные тетразолы формулы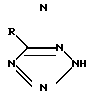
где
R низший алкил, бензил, циклоалкил с 4 атомами С, n- гептилперфторгруппа, -SR1, где R1 низший алкил, бензил; -(CH2)nR2, где R2 OH, CO2R1, OR1, SO3Na, и n 1 или 2; или незамещенный или замещенный аминогруппой, низшей алкоксигруппой, низшим алкилом, нитро- или цианогруппой фенил.
В Chem. Ber. 116, 2691 (1983 г.) автор W Beck с сотр. описывает получение 2-тритил-5-фенил-тетразола.
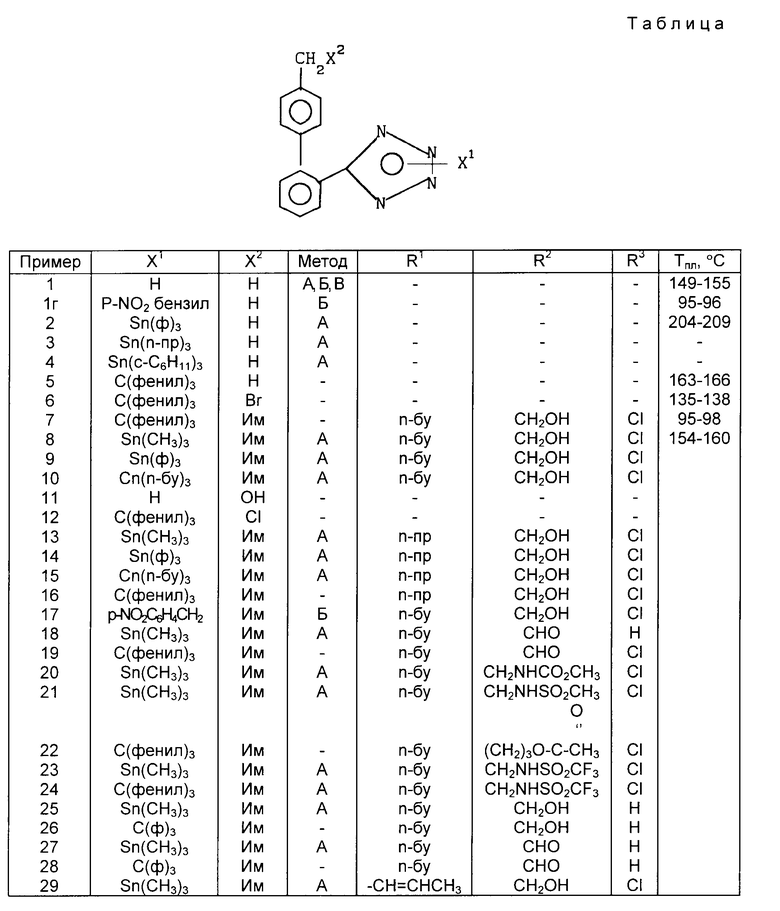
Согласно изобретению предлагаются новые соединения формулы 1, представляющие собой тетразолы, которые пригодны в качестве промежуточных соединений в получении гипотензивных соединений. Эти тетразолы имеют нижеследующую формулу: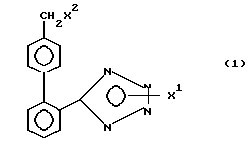 ,
,
где
X1 H, -Sn(метил)3, -Sn(фенил)3, -C(фенил)3, p-NO2-бензил или -CH2CH2CN,
X2 H, Br или группу формулы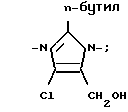
Новые соединения формулы 1 получают с использованием известной в этой области реакций и приемов. Эти реакции проводят в растворителе, соответствующем используемым реагентам и пригодным для осуществления превращения. Для специалиста в области органического синтеза ясно, что функциональность, находящаяся у имидазола, и другие части молекулы должны соответствовать осуществляемым предлагаемым химическим превращениям. В связи с этим специалисту часто приходится устанавливать порядок стадий синтеза, тип и число защитных групп, условия удаления защитных групп и активации бензильного положения в целях содействия присоединению азота к имидазольному ядру. Следует отметить, что не все соединения 1, подпадающие под данный класс соединений, обязательно могут быть получены с помощью всех методов, описанных для этого класса. Дело в том, что определенные заместители исходных веществ могут оказаться несовместимыми с определенными условиями реакции, необходимыми согласно некоторым из описанных методов. Специалист в данной области легко поймет подобного рода ограничения насчет заместителей и в таком случае может пользоваться другими методами.
Соединения 1, где X1 Sn(R)3 и R C1-6 или фенил и X2 Н или имидазоил, где R1 n-бутил, R3 хлор и R2 оксиметил, можно получить путем 1,3- диполярного циклоприсоединения триалкил-Sn- или трифенил-Sn-азидов до соответственно замещенного нитрила (II) (см. схему реакций 1 в конце текста). Пример этого метода описан автором S. Kozima с сотр. в J. Organometallic Chem. 33, 337, (1971 г.). Другие используемые в предлагаемых целях нитрилы и триалкил- или триарил-Sn-азиды представляют собой либо торговые продукты, либо соединения, которые можно получить с использованием известных в химической литературе приемов и методов (см. J. Luijten с сотр. Rec. Trav. Chem. 81, 202 /1962/).
Соединения формулы 1, где X1 и X2 означают Н, можно получить удалением подходящей защитной группы тетразольного ядра. К подходящим защитным группам тетразольного фрагмента относятся р- нитробензил, β - пропионитрил, трифенилметил и триалкилолово, которые получают по нижеприведенным способам. Защитный нитробензил присоединяется, как показано в схеме реакций II (см. в конце текста). Кислоту (IV) превращают в промежуточный хлорангидрид с помощью оксалилхлорида в обычных условиях реакции. Хлорангидрид затем переводят в амид (V) конденсацией с 4-нитробензиламингидрохлоридом в пиридине в присутствии каталитического количества 4-диметиламинопиридиниа (ДМАП). Амид (V) затем переводят в промежуточный иминоилхлорид, подвергая амид реакции с пентахлоридом фосфора в тетрахлорметане. Пример этого способа описан автором H. Ulrich, в The Chemistry of Imidoyl Halides, Plenum Press, Нью-Йорк, (1968 г. ). Промежуточный иминоилхлорид переводят в тетразол (VI) реакцией с азидом лития в диметилформамиде (ДМФ). Пример этого способа описан автором Elderfield, в Heterocyclic Compounds, John Wiley and Sons, (1967 г.). Защищенный тетразол (VI) гидрируют под давлением 50 psi в присутствии каталитического количества никеля Ранея W6 в этаноле, в результате чего получают соединение 1. Требуемую кислоту (IV) можно купить в готовом виде или ее можно получить по способам и приемам, описанным в химической литературе.
Трифенилметил присоединяется к тетразолу, как показано в схеме реакций III (см. в конце текста). Тетразол (1) реагирует с трифенилметилхлоридом в метиленхлориде, содержащем в качестве основания триэтиламин, в обычных условиях с получением защищенного тетразола (VII).
Пропионитрильная защитная группа присоединяется к тетразолу в соответствии со схемой IV (см. в конце текста). Бифенилкарбоновую кислоту IV превращают в хлорангидрид кислоты с помощью ряда реагентов, известных специалисту в данной области. Промежуточный хлорангидрид кислоты реагирует с b--аминопропионитрилом в присутствии акцептора кислоты, например, водного раствора гидроокиси натрия с получением амида VIII. Последний реагирует с пятихлористым фосфором или фосгеном с образованием промежуточного иминоилхлорида IX, который, в свою очередь, реагирует с гидразином с получением амидразона X. Последний легко реагирует с четырехокисью азота (N2O4), которую выгодно использовать в виде раствора в тетрахлорэтане с получением тетразола XI. Гидразины и гидразиды с помощью четырехокиси азота легко превратить в соответствующие азиды, как описано автором Y.H. с сотр. Kim Tetrahedron Letters, 27, 4749 (1986 г.). С защищенного тетразола XI удаляют защитную группу с помощью водного основания, например, 1-н. раствора едкого натра или без добавочного органического растворителя, например, ТГФ с получением тетразола 1. Амидразон X можно также перевести в тетразол XI с помощью азотистой кислоты или ее эквивалентов, как описано автором D. G. Neilson с сотр. в Chem. Rev. 70, 151 (1970 г.).
Предпочитаются такие защитные группы, где X1 Sn(R)3 и С(фенил)3 и R соответствует вышеуказанному описанию (схема реакций V в конце текста). Вышеуказанные группы можно удалить по выбору кислотным или щелочным гидролизом, каталитическим гидрированием, и облучением согласно описанию автора Greene в Protective Groups in Organic Synthesis, Wiley-Interscience, (1980 г.).
Соединения формулы 1, где X1 означает С(фенил)3 и X2 бром, получают путем радикального бромирования соединения VII N-бромсукцинимидом (NБС) и дибензоилперекисью (Bz2O) с получением соединения XII (см. схему реакций VI в конце текста). Пример этого превращения описан автором L. Horner в Angew. Chem. 71, 349 (1959 г.).
Соединения формулы 1, где X1 С(фенил)3 и X2 I, получают путем вытеснения бромистого фрагмента в соединении XII йодидом натрия в ацетоне в нормальных условиях с получением соединения XIII. Путем вытеснения бромида XII гидроксильным ионом получают замещенный бензильный спирт XIV. Последний можно превратить в хлорид XV реакцией с тетрахлорметаном и трифенилфосфином. Однако бензиловый спирт XIV можно превратить и в тозилат или мезилат XVI путем реакции в нормальных условиях с р- толуол- или метансульфонилхлоридом в пиридине (см. схему реакций VII в конце текста).
Соединения 1, где X1 С(фенил)3 и X2 имидазоил, где R1 n-бутил, R3 хлор и R2 оксиметил, можно получить алкилированием имидазола XVII, замещенным соответствующим образом бензилгалогенидом, с использованием этанолята натрия в качестве основания и последующим восстановлением формальдегидного фрагмента имидазола XVII до оксиметила с помощью борогидрида натрия, в результате чего получают соединение XVIII. Получение имидазола XVII по схеме реакций VIII (см. в конце текста) описано автором Furukawa с сотр. в патенте США N 4355040.
Предлагаемые соединения и способ их получения иллюстрируются нижеследующими примерами, которыми они однако не ограничиваются. Если нет других указаний, то все температуры указаны в градусах Цельсия и все части и проценты даны в пересчете на вес.
Пример 1 Метод А.
Часть А: N-триметилстаннил-5-[2-(4'-метилбифен-2-ил)]-тетразол
К раствору 19,30 г (0,100 моль) 2-циано-4'-метилбифенила в 110,0 мл толуола при комнатной температуре добавляют 24,60 г (0,120 моль) триметил-Sn-азида. Затем реакционную массу нагревают при температуре дефлегмации в течение суток, после чего ее охлаждают до комнатной температуры. Наконец, путем фильтрации выделяют 32,60 г (82) белого твердого вещества N-триметилстаннил-5-[2-(4'- метилбифен-ил)] -тетразола. Т.пл. 265oC (с разл.); 1Н-ЯМР (ДМСО-d6) δ: 7.50 (c. 4H); 7.00 (c. 4H); 2.25 (c. 3H); 0.35 (c. 3H).
Часть Б: 5-[2-(4'-метилбифенилил)]-тетразол
Через раствор 32,0 (0,080 моль) N-триметилстаннил-5-[2-(4'- метилбифенилил)] -тетразола в 230 мл толуола барботируют 15,0 мл ТГФ в достаточном количестве безводного хлористого водорода до получения прозрачного раствора при комнатной температуре. Из этого раствора выкристаллизовывается 19,1 г 5-[2-(4'-метилбифенилил)]-тетразола. Путем перекристаллизации из толуола получают 18,1 г (95%) целевого продукта. Тпл 149-152oC. 1Н-ЯМР (CDCl3/ДМСО-d6) δ: 7.50 (м. 4H); 7.07 (м. 4H); 2.35 (c. 3H).
Пример 1 Метод Б.
Часть А: 4'-метил-бифенил-2-карбонил-хлорид
Раствор 31,84 г (0,15 моль) 4'-метил-бифенил-2-карбоновой кислоты в 200 мл хлороформа при комнатной температуре по каплям добавляют к перемешиваемой системе 25 мл хлороформа, 25 мл оксалилхлорида и 1,0 мл ДМФ. После перемешивания смеси в течение суток при комнатной температуре раствор выпаривают в вакууме с получением 36,4 г сырого хлорангидрида кислоты. ИК: 1784.0 см-1 (COCl).
Часть Б: N-(4-нитробензил)-4'-метил-бифенил-2-карбоксамид.
Раствор 36,4 г вещества, полученного по части А, в сухом ацетонитриле, по каплям добавляют к охлаждаемой (на ледяной бане), перемешиваемой смеси 23,45 г (0,12 моль) 4- нитробензиламингидрохлорида, 0,5 г (0,0041 моль) 4-диметиламино-пиридина и 150,0 мл пиридина. Через 30 мин смеси дают нагреться до комнатной температуры, после чего ее перемешивают 16 ч при комнатной температуре. Затем ее выливают в перемешиваемую смесь 800,0 мл 3 н. HCl, 400,0 г льда и 400 мл дихлорметана. Органический слой промывают 2н. NaOH (два раза по 200 мл) и рассолом (100 мл), высушивают сульфатом магния и выпаривают в вакууме с получением 61,9 г сырого продукта. После перекристаллизации этого продукта из этилацетата получают 31,3 г (73%) целевого продукта с Т.пл. 153-154oC. 1Н-ЯМР (CDCl3) δ: 8.03 (д, 2H,ароматический); 7.65-7.69 (м. 1Н, ароматический); 7.12 -7.48 (м. 7Н, ароматический); 7.04 (д, 2H, ароматический); 5.77 5.79 (м. 1H, NH); 4.41 (д. 2H, J 6.0 Гц, CH2);2.39 (c. 3H, CH3). Масс-спектр m/z 347 (M + 1).
Часть В: N-(4-нитробензил)-4'-метил-бифен-2-ил-карбоиминоил- хлорид.
20,78 г (0,060 моль) продукта по части Б тремя порциями добавляют к охлаждаемому (на ледяной бане) перемешиваемому раствору 12,49 г (0,066 моль) пентахлорида фосфора в 200 мл четыреххлористого углерода. Смесь перемешивают 30 мин при 0oC, затем смеси дают нагреться до комнатной температуры, после чего ее еще раз перемешивают в течение 16 ч. Затем смесь выпаривают в вакууме и получают 21,3 г сырого продукта. ИК: 1691 см-1 (C=N). 1Н-ЯМР (CDCl3) δ: 4.79 (c. 2H, CH2).
Часть Г: 1-(4-нитробензил)-5-(4'-метил-бифен-2-ил)-тетразол.
3,67 г (0,75 моль) азида лития по порциям добавляют к охлаждаемому на ледяной бане раствору 21,3 г продукта из части В в 200,0 мл ДМФ. Затем полученной смеси дают нагреться до комнатной температуры в течение 16 ч. Потом реакционную смесь выпаривают в вакууме. Затем остаток разделяют по слоям с помощью воды и этилацетата (100 мл). Образовавшийся органический слой промывают 100 мл воды, высушивают сульфатом магния и выпаривают в вакууме с получением 19,5 г темного остатка. В результате хроматографии на кремнеземе (CHCl3) с последующей перекристаллизацией из метанола получают 5,37 г (24,1% ) вышеназванного продукта с Т.пл. 95,9-96,0oC. 1Н-ЯМР (CDCl3) δ: 7.98- 8.02 (м. 2H,ароматический); 7.55-7.70 (м. 2Н, ароматический); 7.37 -7.49 (м. 2Н, ароматический); 6.99-7.10 (м. 2H,ароматический); 4.87 (д. J 8.7 Гц, ароматический); 4.88 (c. 2H, CH2); 2.33 (c. 3H, CH3). Масс-спектр m/z 372 (M + 1).
Часть Д: 5-[2-(4'-метилбифен-2-ил)]-тетразол
Смесь 1,00 г (2,80 ммоль) продукта по части Г, 150,0 мл этанола и 5,0 г никеля Ранея W6 при комнатной температуре в течение 2 ч гидрируют в аппарате для взбалтывания типа Parr Parr® Shaker под давлением 50 psi. Затем фильтрацией отделяют катализатор, после чего фильтрат выпаривают в вакууме. Остаток разделяют по слоям с помощью воды и диэтилового эфира (100 мл), после чего органический слой промывают 50 мл 1 н. соляной кислоты, 50 мл рассола, высушивают сульфатом магния и выпаривают в вакууме с получением твердого остатка, который перекристаллизовывают из этанола с получением 0,19 г (28,7%) продукта с Т.пл. 154-155oC. 1Н-ЯМР (CDCl3) δ 11.5 (ш. с. 1Н, NH); 8.02 (д. 1H, ароматический); 7.38-7.61 (м. 3Н, ароматический); 7.16 (д. 2Н, J 8.0 Гц, ароматический); 7.04 (д. J 8.0 Гц); 2.35 (c. 3H, CH3). Масс-спектр m/z 237 (M + 1).
Пример 1 Метод В.
Часть А: 2-(b-цианоэтиламинокарбонил)-4'-метил-бифенил 50,0 г (0,236 моль) 4'-метилбифенил-2-карбоновой кислоты, 87,5 мл (1,20 моль) тионилхлорида и 500 мл хлороформа смешивают и нагревают при температуре дефлегмации в течении 4 ч. Затем удаляют тионилхлорид и растворитель в вакууме, а остаток взвешивают в 300 мл толуола. Смесь выпаривают в вакууме, а остаток еще раз взвешивают в толуоле и выпаривают для того, чтобы обеспечить полное удаление тионилхлорида. Полученный в результате хлорангидрид кислоты растворяют в 100 мл ТГФ. Полученный таким образом раствор и 236,0 мл (0,236 моль) 1,0 н. раствора NaOH пятью порциями попеременно при 0oC медленно с перемешиванием добавляют в раствор 30,3 г (0,236 моль) b-аминопропионитрилфумарата в 236,0 мл (0,236 моль) 1,0 н. раствора NaOH. Реакционной массе дают медленно нагреться до комнатной температуры. Через 24 ч добавляют 500 мл воды. Водную смесь экстрагируют три раза этилацетатом (по 500 мл). Затем собирают органические слои и высушивают их сульфатом магния, после чего удаляют растворитель в вакууме и получают сырое твердое вещество. Перекристаллизацией этого вещества из смеси метилциклогексана и бутилхлорида получают 53,5 г белого твердого вещества. Т. пл. 102,0-103,5oC. 1Н-ЯМР (200 MH2 CDCl3) d: 7,68 (д. 1H, J 7 Гц); 7.56-7.19 (м. 7H); 5.65 (ш. м. 1H); 3.43 (д. т- та, 2Н); 2.39 (т. 2Н J 7 Гц). Анализ C17H16N2O: вычисл: C 77,25; H 6,10; N 10,60. Найдено: C 77,42; H 6,40; N 10,68.
Часть Б: N3-(β-цианоэтил)-4'-метилбифенил-2-ил- амидразон.
33,48 г (0,127 моль) 2-(b-цианоэтиламинокарбонил)-4'-метил-бифенила и 29,01 (0,139 моль) пятихлористого фосфора смешивают друг с другом в круглодонной колбе, которую затем присоединяют к водоструйному насосу через ловушку, наполненную хлористым кальцием. Колбу осторожно нагревают с помощью фена до расплавления твердых веществ. Колбу периодически нагревают в течение 15 20 мин.
Сырой иминоилхлорид растворяют в 100 мл сухого диоксана и по каплям добавляют к перемешанной смеси 20,1 мл (0,634 моль) гидразина в 200 мл сухого диоксана. Через сутки избыток гидразина и растворитель удаляют в вакууме. Потом добавляют 300 мл воды и полученную водную смесь три раза экстрагируют этилацетатом (по 300 мл). Потом собирают органические слои, высушивают сульфатом магния и удаляют растворитель в вакууме, чем получают масло. Это масло обрабатывают 30 50 мл раствора гексана и этилацетата (1:1), в результате чего выпадает в осадок твердое вещество. Последнее собирают и высушивают с получением 16,14 г светло-розового твердого вещества. Т.пл. 146,5-147,5oC. Масс-спектр на основе химической ионизации обнаруживает для C17H19N4: (M + Н)+ 279. Анализ C17H18N4• (N2H4)>0.1: вычисл: C 72,52; H 6,44; N 20,89. Найдено: C 72,50; H 6,54; N 21,13. ЯМР указывает на наличие смеси таутомеров.
Часть В: 2-[1-((β--цианоэтил)-1-Н-тетразол-5-ил]-4'- метилбифенил.
0,73-м. раствор (19,6 мл) газа N2О4 (14,3 ммоль) в тетрахлорметане добавляют к перемешиваемой взвеси N3-(β- цианоэтил)-4'-метилбифенил-2-иламидразона (2,00 г, 7,2 ммоль) в безводном ацетонитриле (40 мл) при комнатной температуре. Затем реакционную массу нагревают до комнатной температуры и перемешивают в течение ночи. Затем удаляют растворитель в вакууме, в результате чего получают сырое твердое вещество. Последнее смешивают с бутилхлоридом, после чего нерастворимое вещество отделяют фильтрацией. Фильтрат выпаривают и остаток подвергают БЖХ (flas- chromatography) на кремнеземе с использованием в качестве растворителя смеси гексана и этилацетата (1:1) с получением 1,10 г светло-желтого масла, которое медленно кристаллизуется. После перекристаллизации из смеси гексана и бутилхлорида получают 910 мг светло-желтых кристаллов. Т.пл. 90,0-92,0oC. ЯМР (200 MГц CDCl3) d 7.76-7.50 (м. 4H); 7.17 (д. 2H, J 10 Гц); 7.04 (д. 2H, J 10 Гц); 3.80 (т. 2H, J 7 Гц); 2.37 (c. 3H); 2.24 (ш.т. 2H, J 7 Гц). Анализ C17H15N5: вычисл: C 70,57; H - 5,23; N 24,20. Найдено: C 70,49; H 5,45; N 24,44.
Часть Г: 5-(4'-метилбифенил-2-ил)тетразол.
689 мг (2,38 ммоль) 2-[1-((β--цианоэтил)-1-Н-тетразол-5-ил]-4'- метилбифенила, 2,62 мл (2,62 ммоль) 1,0 н. раствора NaOH и 15 мл ТГФ смешивают друг с другом и перемешивают при комнатной температуре. Через 15 мин добавляют 100 мл воды и с помощью конц. HCl доводят значение pH до 3,0. Водную смесь экстрагируют три раза этилацетатом (по 100 мл). Затем собирают органические слои, высушивают их сульфатом магния и выпаривают в вакууме с получением 550 мг белого порошка. Т.пл. 148,5-150,0oC. Данные спектрального анализа соответствуют данным, полученным с пробой продукта, приготовленного по методу А.
Пример 5.
N-трифенилметил-5-[2-(4'-метилбифенилил)]-тетразол.
К раствору 17,0 г (0,072 моль) 5-[2-(4'-метилбифенилил)] тетразола в 260 мл метиленхлорида добавляют 21,20 г (0,076 моль) трифенилметилхлорида при комнатной температуре. При этой же температуре добавляют 12,0 г (0,086 моль) триэтиламина. Полученный раствор нагревают 2,5 часа при температуре дефлегмации. Затем охлаждают раствор до комнатной температуры, промывают его два раза водой (по 50 мл), высушивают его сульфатом магния и выпаривают его в вакууме. Остаток выкристаллизовывают из 80 мл толуола и получают N-трифенилметил-5-[2-(4'-метилбифенилил)]-тетразол (31,2 г, 90%) с т.пл. 163-166oC. 1Н-ЯМР (CDCl3) δ: 8.10-6,80 (комплекс, 23 Н); 2,28 (с. 3Н).
Пример 6.
N-трифенилметил-5-[2-(4'-бромметил-бифенилил)]-тетразол.
К раствору 31,0 г (0,065 моль) N-трифенилметил-5-[2-(4'- метилбифенилил)] -тетразола в 390,0 мл тетрахлорметана при комнатной температуре добавляют 11,50 г (0,065 моль) N-бромсукцинимида и 1,10 г (0,0045 моль) дибензоилперекиси. Полученную реакционную смесь нагревают в течение 3 ч при температуре дефлегмации, после чего ее охлаждают до 40oC и фильтруют. В результате выпаривания фильтрата в вакууме с последующим растиранием полученного остатка со 100 мл простого изопропилового эфира получают 33,10 г (92% ) N- трифенилметил-5-[2-(4'-бромметил-бифенилил)] -тетразола с т.пл. 135-138oC. 1Н-ЯМР (CDCl3) δ: 8.20-6,70 (комплекс, 23 Н); 4,33 (с. 2Н).
Пример 7.
1-{ [2'-(N-трифенилметил-тетразол-5-ил)-бифенил-4-ил]метил}-2- бутил-4-хлор-5-оксиметил-имидазол
К раствору 1,24 г (0,007 моль) 2-бутил-4-хлор-5-формилимидазола в 10 мл ДМФ добавляют 0,45 г (0,0066 моль) этанолята натрия, после чего полученную реакционную массу охлаждают до 5oC. Затем добавляют 3,70 г (0,0066 моль) N-трифенилметил-5-[2-(4'-бромметил- бифенилил)] -тетразола, после чего полученную реакционную массу нагревают до комнатной температуры. Через трое суток реакционную массу разбавляют 25 мл воды и экстрагируют три раза этилацетатом (по 25 мл) и три раза рассолом (по 25 мл), высушивают сульфатом магния и выпаривают в вакууме с получением масла. Последнее растворяют в 20 мл метанола, после чего при комнатной температуре добавляют 0,24 г (0,0063 моль) боргидрида натрия. Затем реакционную массу перемешивают в течение 1,5 ч, разбавляют 40 мл воды и два раза экстрагируют эталацетатом (по 50 мл). Органический слой промывают 25 мл воды, высушивают сульфатом магния и выпаривают в вакууме. Полученный остаток выкристаллизовывают сначала из смеси толуола и гептана, потом из толуола и наконец из метанола с получением 0,98 г (21% ) 1-{[2'-(N-трифенилметил-тетразол-5-ил)-бифенил-4-ил]метил}-2- бутил-4-хлор-5-оксиметил-имидазола с Т.пл. 95-98oC; 1Н-ЯМР (CDCl3) δ: 8.20-6,60 (комплекс, 23 Н); 5,16 (с. 3Н); 4,40 (с. 3Н); 2.85 (ш.c. 1H); 2.54 (т. 3H); 1.9-1.1 (м. 4H); 0.88 (т. 3H).
Пример 8.
1-{ [2'-(N-триметилстаннил-тетразол-5-ил)-бифенил-4-ил]метил}-2- бутил-4-хлор-5-оксиметил-имидазол.
К раствору 4,40 г (0,011 моль) 1-[2'- циано-бифенил-4-ил)метил]-2-бутил-4-хлор-5-оксиметил- имидазола в 40,0 мл ксилола добавляют 2,80 г (0,014 моль) триметил- Sn-азида. Реакционную массу нагревают 40 ч при температуре 115- 120oC. Затем массу охлаждают до 50oC и фильтруют с получением 6,55 (99% ) 1-{ [2'-(N-триметилстаннил-тетразол-5-ил)-бифенил-4-ил]метил}-2-бутил-4-хлор-5-оксиметил-имидазола с Т. пл. 154-160oC; 1Н-ЯМР (CDCl3/ДМСО-d6) δ: 7.80-7,30 (м. 4Н); 7,03 (кв. 4Н); 5,23 (с. 3Н); 4.43 (c. 3H); 2.54 (т. 3H); 2.00 (c. 1H); 1.80-1.10 (м. 4H); 0.85 (т. 3H); 0.40 (c. 9H).
Соединения по примерам 1, 5, 6, 7, 8 и другие соединения, которые получали или можно получать по приемам вышеупомянутых примеров, приведены в табл. 1.
Пример 9, иллюстрирующий получение целевого продукта.
Получение 2-бутил-4-хлор-5-оксиметил-1-/(2'-(1Н-тетразол-5-ил)-бифенил-4-ил)метил/имидазола.
В круглодонную колбу емкостью 12 л, снабженную механической мешалкой, холодильником с подводом азота и термометром, погружают 920 г 1-/(2'-(трифенилметилтетразол-5-ил)-бифенил-4-ил)метил/-2- бутил-4-хлор-5-оксиметилимидазола и 2,10 л метанола. Взвесь охлаждают до ок. 10oC, после чего в течение 10 мин добавляют 700 мл 3,4 н. соляной кислоты. После перемешивания при 10-20oC в течение 2 ч густую взвесь разбавляют 500 мл метанола и нагревают до 30oC. Смесь выдерживают при 30oC в течение 1 ч, после чего добавлением 420 мл 10 н. гидроокиси натрия нейтрализуют реакцию с установлением pH 13. Растворитель (главным образом метанол, 2,3 л) выпаривают, и добавляют еще 700 мл дистиллированной воды. Прекращают нагревание и добавляют еще 700 мл дистиллированной воды и 1,40 л толуола. После охлаждения смеси до ок. 30oC удаляют органическую фазу. Водную фазу подвергают второму экстрагированию 700 мл толуола. В сосуд, содержащий водную фазу, добавляют 1,20 л этилацетата. Смесь перемешивают 10 мин, а затем добавляют 130 мл уксусной кислоты. Смесь перемешивают еще 1 ч, а затем оставляют в течение ночи, после чего снова ее перемешивают. Взвесь охлаждают до около 5oC. Продукт выделяют вакуумной фильтрацией, снова взвешивают в 1,50 л дистиллированной воды и подвергают отсасыванию до полусухого состояния. Мокрый шлам погружают в круглодонную колбу емкостью 12 л и снова взвешивают в 4,0 л этилацетата при температуре окружающей среды в течение 0,5 ч. Продукт выделяют вакуумной фильтрацией, промывают 200 мл этилацетата и сушат в вакууме при 50oC в течение ночи.
Выход: 518 г (88,5%) продукта с т.пл. 184-185oC. Чистота по данным ВЖХ: 98,8%
ЯМР-спектр (200 MГц DMSO-d6) :δ 7.61 (м. 4H); 7.05 (м. 4H); 5.24 (c, 2H), 4.32 (c, 2H), 3.35 (шс, 1Н), 2.46 (т, 2H, J 7,8 Гц); 1.44 (м. 2H); 1.23 (м. 2H); 0.79 (т, 3H, J 7,2 Гц).
Пример 10, иллюстрирующий применение азо(бисизобутиронитрила) вместо дибензоилперекиси на стадии (4).
Получение 4'-бромметил-2-(трифенилметилтетразол-5-ил)-бифенила.
В круглодонную колбу емкостью 100 мл, снабженную термометром, холодильником и подводом азота, погружают 9,0 г (0,0188 моль) 2- /(трифенилметилтетразо-5-ил)/-4'-метил-(1,1'-бифенила), 4,0 г (0,0225 моль) N-бромсукцинимида, 0,1 г (0,0061 моль) азо(бисизобутиронитрила) и 40 мл четыреххлористого углерода. Реакционную смесь нагревают до температуры дефлегмации и выдерживают при этой температуре около 3 ч или пока по данным ЯМР реакция не завершена. После завершения реакции реакционную массу охлаждают до комнатной температуры, разбавляют 30 мл хлористого метилена и промывают 30 мл воды. Водную фазу сбрасывают.
| название | год | авторы | номер документа |
|---|---|---|---|
| АРАЛКИЛЗАМЕЩЕННЫЕ ИМИДАЗОЛЫ, ОБЛАДАЮЩИЕ ПРОТИВОГИПЕРТОНИЧЕСКИМ ДЕЙСТВИЕМ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЙ СОСТАВ, ОБЛАДАЮЩИЙ ИНГИБИРУЮЩЕЙ АКТИВНОСТЬЮ В ОТНОШЕНИИ АНГИОТЕНЗИНА II | 1990 |
|
RU2067581C1 |
| ПРОИЗВОДНЫЕ ИМИДАЗОЛА | 1992 |
|
RU2017733C1 |
| ЗАМЕЩЕННЫЕ ИМИДАЗОЛЫ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1991 |
|
RU2099330C1 |
| ПРОИЗВОДНЫЕ СЕМИКАРБАЗОНОВ, СПОСОБ УНИЧТОЖЕНИЯ АРТРОПОДОВ, АРТРОПОДИЦИДНАЯ КОМПОЗИЦИЯ | 1989 |
|
RU2067092C1 |
| СПОСОБ ЗАЩИТЫ СЕЯНЦЕВ ИЛИ РАСТЕНИЙ, ВЫРОСШИХ ИЗ НИХ, ОТ НАСЕКОМЫХ, КОМПОЗИЦИЯ ДЛЯ КОНТРОЛЯ ЗА НАСЕКОМЫМИ | 2002 |
|
RU2292138C2 |
| ЗАМЕЩЕННЫЕ 1,2,3-ТРИАЗОЛЫ | 1992 |
|
RU2076102C1 |
| ФТОРОАЛКОКСИАМИНОТРИАЗИНЫ | 1992 |
|
RU2047607C1 |
| Способ получения производных имидазола или их фармацевтически приемлемых солей | 1987 |
|
SU1694062A3 |
| КАРБОКСАНИЛИДЫ, АРТРОПОЦИДНАЯ КОМПОЗИЦИЯ И СПОСОБ БОРЬБЫ С АРТРОПОДАМИ | 1991 |
|
RU2096409C1 |
| Способ получения производных имидазола | 1989 |
|
SU1814646A3 |
Использование: в качестве промежуточных соединений в синтезе гипотензивных соединений. Описываются производные тетразола общей формулы 1: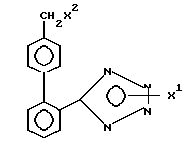
где
X' - H, -Sn(метил)3, -C(фенил)3, p- NO2-бензил или -CH2CH2CN, X2 - H, Br или группа формулы: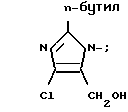
1 табл.
Производные тетразола общей формулы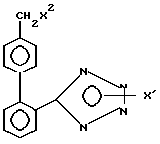
где Х1 Н, Sn(метил)3, С(фенил)3, р-нитробензил или -СН2СН2СN;
Х2 Н, Br или 2-бутил-4-хлор-5-гидроксиметилимидазолил, при условии, что когда Х1 водород, Х2 также водород.
| K.Sisido | |||
| Formation of organotic - Nitrogen Bonds III | |||
| Кипятильник для воды | 1921 |
|
SU5A1 |
| Способ сопряжения брусьев в срубах | 1921 |
|
SU33A1 |
| Ленточный тормозной башмак | 1922 |
|
SU337A1 |
| Способ получения сложных эфиров карбаминовой кислоты | 1960 |
|
SU147973A1 |
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| СПОСОБ ПОЛУЧЕНИЯ НЕЙТРАЛЬНОГО ГИПОХЛОРЙТАКАЛЬЦИЯ | 0 |
|
SU194064A1 |
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| Устройство для выпрямления опрокинувшихся на бок и затонувших у берега судов | 1922 |
|
SU85A1 |
| Аппарат для нагревания окружающей его воды | 1920 |
|
SU257A1 |
