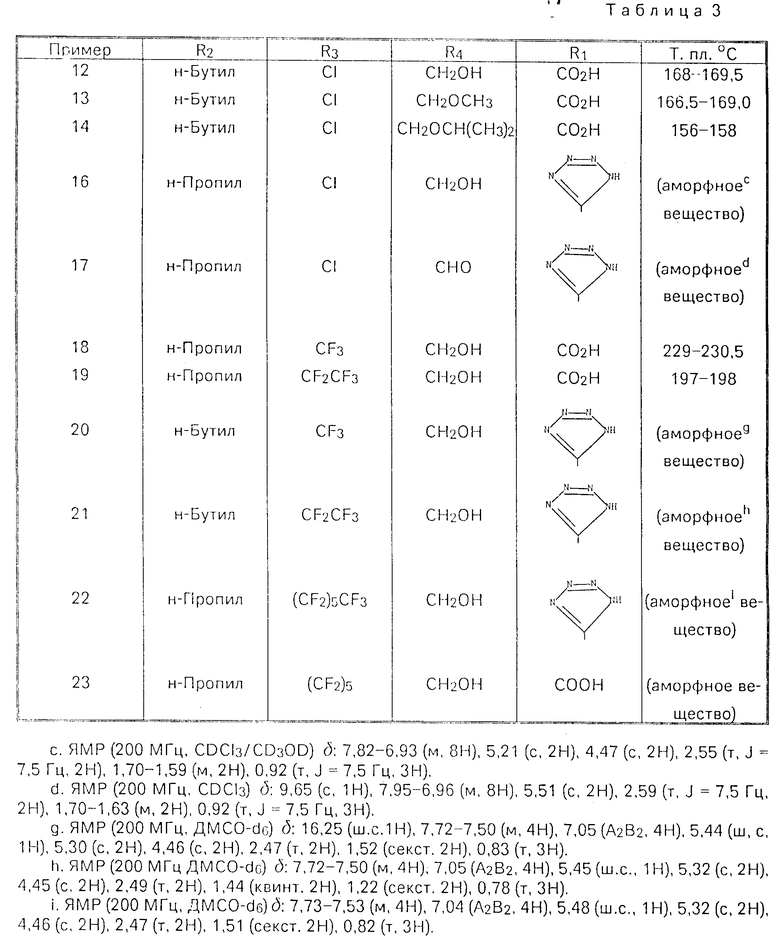
Изобретение относится к новым производным имидазола общей формулы I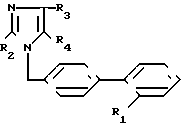 где R1-COOH или группа
где R1-COOH или группа 
R2=H-C3H7 или н-С4Н9;
R3-Cl, CF3, C2F5, C6H5 или СООН;
R4-COOH, CHO или СН2ОН, при условии, что
а) когда R4-CH2OH, то R3-C2F5 и R2-н-С3Н7,
б) когда R3-СООН, то R4 тоже является СООН,
в) когда R2-н-С3Н7, R3-C2F5 и R4-COOH, то R1 является группой  которые ингибируют действие гормона ангиотензина и могут быть использованы в медицине.
которые ингибируют действие гормона ангиотензина и могут быть использованы в медицине.
Известно, что N замещенные производные имидазола могут быть получены реакцией алкилирования (I).
Известно соединение формулы II, являющееся структурным аналогом соединений I,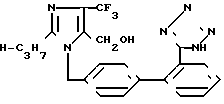 которое также обладает активностью в качестве антагониста к рецептoрам ангиотензина и противогипертонической активностью.
которое также обладает активностью в качестве антагониста к рецептoрам ангиотензина и противогипертонической активностью.
Целью изобретения является представление новых производных имидазола, обладающих более высокой биологической активностью, чем структурный аналог с использованием известного метода алкилирования имидазолов.
Новые производные имидазола формулы I получаются взаимодействием производного имидазола формулы III: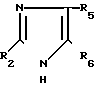 где R2 принимает указанные значения,
где R2 принимает указанные значения,
R5-Cl, CF3, C2F5, C6Н5 или СОО (С1-4-алкил)
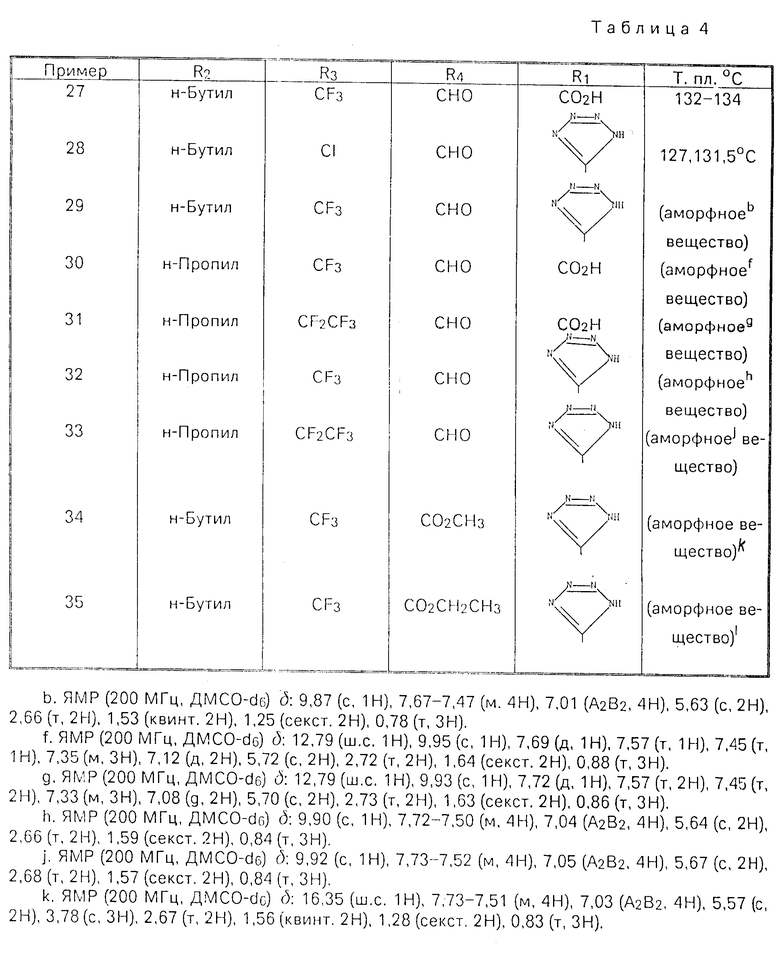
R6-CO2 (C1-C4-алкил), -СН2ОН или -СНО с производным дифенилметила формулы IV: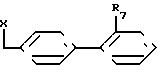 где Х галоген, п толуолсульфонилокси или метилсульфонилокси,
где Х галоген, п толуолсульфонилокси или метилсульфонилокси,
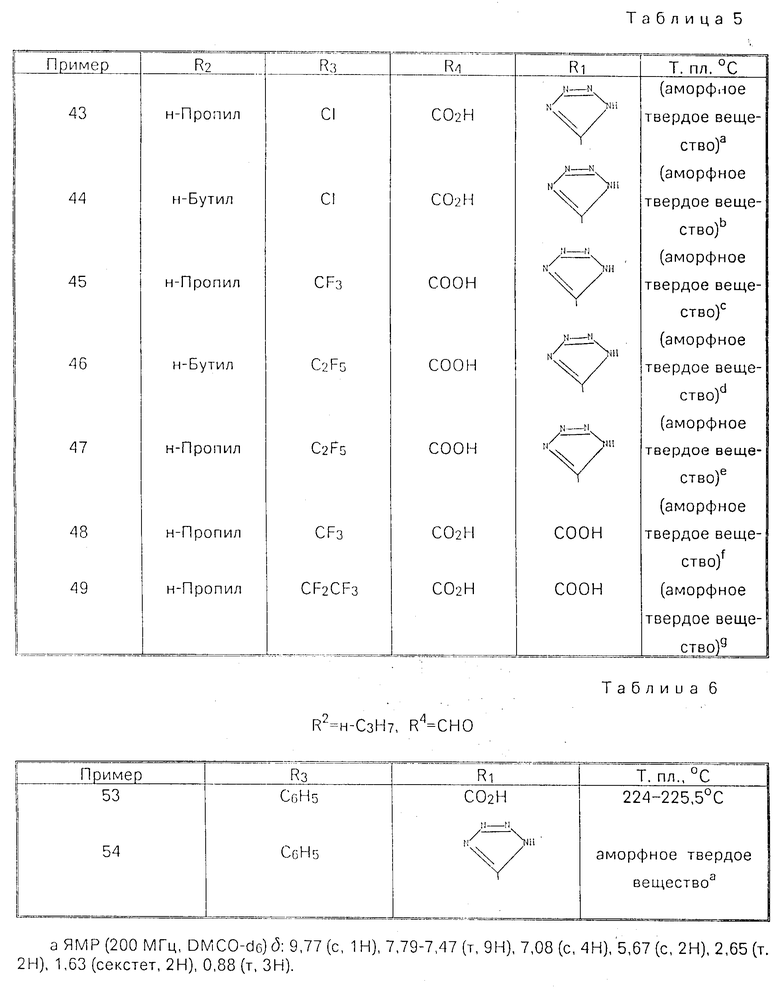
R7-COO (C1-C4-алкил), CN или  C(C6H5)3 в растворителе в присутствии основания в течение 1-10 ч при температуре от 20оС до температуры кипения растворителя и в случае, когда R5, R6 или R7 сложноэфирная группа, ее деэтерифицируют и выделяют целевой продукт или в случае, когда R6-CH2OH, его подвергают окислению с получением целевого продукта, где R4-CHO или СООН, или в случае, когда R6-СНО возможно после алкилирования с производным формулы IV восстановить его с получением целевого продукта, где R6-CH2OH, или в случае, когда R7-CN, его переводят в целевое соединение, где R1-
C(C6H5)3 в растворителе в присутствии основания в течение 1-10 ч при температуре от 20оС до температуры кипения растворителя и в случае, когда R5, R6 или R7 сложноэфирная группа, ее деэтерифицируют и выделяют целевой продукт или в случае, когда R6-CH2OH, его подвергают окислению с получением целевого продукта, где R4-CHO или СООН, или в случае, когда R6-СНО возможно после алкилирования с производным формулы IV восстановить его с получением целевого продукта, где R6-CH2OH, или в случае, когда R7-CN, его переводят в целевое соединение, где R1- 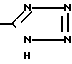 или в случае, когда R7
или в случае, когда R7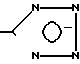 _
_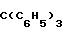 его переводят в целевое соединение, где R1-
его переводят в целевое соединение, где R1- 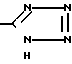
П р и м е р 1. Раздел А:
Получение метил-4'-метилбифенил-3-карбоксилата.
К перемешиваемому раствору 25,2 г метил-3-иодбензоата и 21,0 г 4-иодтолуола при 180-190оС в атмосфере азота прибавляют 30,3 г медного порошка порциями в течение 1 ч. Когда была прибавлена примерно одна треть порошка, начинается реакция и температура самопроизвольно повышается до 240оС. Смеси дают остыть до 210оС, затем выдерживают при 210оС во время прибавления остальной меди в течение дополнительного часа. Смеси дают остыть до комнатной температуры и фильтруют, используя бензол в качестве растворителя, полученный в результате фильтрат концентрируют в вакууме, чтобы получить сырой продукт.
Хроматографией на колонке с силикагелем (элюирование 50-100% бензол/гексан) с последующей дистилляцией 7,60 г метил-4' -метилбифенил-3-карбоксилата (т. кип. 114-115оС/0,025 торр) в виде бесцветного масла: ЯМР (200 МГц, CDCl3), δ: 8,27 (шир.с. 1Н), 7,99 (д, 1Н), 7,77 (д, 1Н), 7,50 (т, 1Н), 7,39 (А2В2, 4Н), 3,94 (с, 3Н), С, 2,41 (с, 3Н).
Раздел В.
Получение метил-4' -бромметилбифенил-3-карбоксилата.
Раствор 7,31 г метил-4' -метилбифенил-3-карбоксилата, 5,75 г N-бромсукцинимида, 0,125 г азо(бисизобутиронитрила) и 500 мл четыреххлористого углерода кипятят с обратным холодильником в течение 3 ч. После охлаждения до комнатной температуры полученной суспензии ее фильтруют, а затем концентрируют в вакууме, получают 9,90 г сырого метил-4' -бромметилбифенил-3-карбоксилата, который используют в последующей реакции без дополнительной очистки.
ЯМР (200 МГц, CDCl3): δ 8,28 (c, 1H), 8,05 (д, 1Н), 7,79 (д, 1Н), 7,67-7,48 (м, 5Н), 4,55 (с, 2Н), 3,98 (с, 3Н).
Следующие броммeтилбифенильные промежуточные продукты получают при использовании указанной выше процедуры.
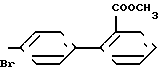 ЯМР (200 МГц, CDCl3) δ: 7,82 (д, 1Н), 7,59-7,23 (м, 7Н), 4,52 (с, 2Н), 3,62 (с, 3Н)
ЯМР (200 МГц, CDCl3) δ: 7,82 (д, 1Н), 7,59-7,23 (м, 7Н), 4,52 (с, 2Н), 3,62 (с, 3Н)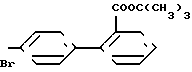 δ 7,79 (д, 1Н), 7,56-7,24 (м, 7Н), 4,51 (с, 2Н), 1,25 (с, 9Н).
δ 7,79 (д, 1Н), 7,56-7,24 (м, 7Н), 4,51 (с, 2Н), 1,25 (с, 9Н).
Раздел С:
Получение 1-[(3' -карбометоксибифенил-4-ид)метил]-2-бутил-4-хлор-5-оксимети- лимидазола.
К суспензии 1,43 г метоксида натрия в 20 мл диметилформамида при 25оС прибавляют раствор 5,00 г 2-бутил-4(5)-хлор-5(4)-оксиметилимидазола в 15 мл ДМФ. Полученную в результате смесь перемешивают при 25оС в течение 0,25 ч, затем к этой смеси прибавляют по каплям раствор 9,90 г метил-4' -бромметилбифенил-3-карбоксилата в 15 мл ДМФ. Наконец, реакционную смесь перемешивают при 40оС в течение 4 ч. После охлаждения до 25оС растворитель удаляют в вакууме. Остаток растворяют в этилацетате и этот раствор промывают водой и рассолом, сушат над безводным сульфатом натрия, фильтруют и концентрируют. Сырой продукт содержит два стереоизомера, быстрее движущийся при ТСХ является более мощным изомером. Колоночная хроматография на силикагеле (элюация 10-25% этилацетат (бензол) приводит к 3,85 г 1-[(3' -карбометоксибифенил-4-ил)-метил]-2-бу- тил-4-хлор-5-оксиметилимидазола (т.пл. 162-163оС).
Стереизомер с более высоким Rf: ЯМР (200 МГц, CDCl3), δ 8,24 (с, 1Н), 8,03 (д, 1Н), 7,76 (д, 1Н), 7,52 (т, 1Н), 7,33 (А2В2, 4Н), 5,27 (с, 2Н), 4,52 (д, 2Н), 3,93 (С, 3Н), 2,60 (т, 2Н), 1,89 (т, 1Н), 1,67 (квинтет, 2Н), 1,35 (секстет, 2Н), 0,88 (т, 3Н).
Раздел D.
Получение 1-[(3' -карбометоксибифенил-4-ил)метил]-2-бутил-5-оксиметилимида- зола.
Смесьь 1,00 г 10%-ного палладия на угле и 1,00 г 1-[(3' -карбометоксибифенил-4-ил)метил]-2-бутил-4-хлор-5-оксиметилими- дазола в 20 мл метанола перемешивают при 25оС в течение 5 мин. Водород барботируют в раствор и смесь перемешивают в атмосфере Н2 (г) (1 атм.) при 25оС в течение 3,5 ч. Смесь фильтуют и полученный раствор концентрируют в вакууме. Колоночная хроматография (элюация: 0-5% метанола (хлороформ) приводит к 0,33 г 1-[(3 -карбометоксибифенил-4-ил)метил]-2-бутил-5-окси-метилимидазола.
ЯМР (200 МГц, ДМСО-d6 ) δ 8,20 (с, 1Н), 7,98 (д, 2Н), 7,65 (т, 1Н), 7,41 (А2М2, 4Н), 6,80 (с, 1Н), 5,30 (с, 2Н), 5,12 (т; 1Н), 4,37 (д, 2Н), 3,90 (с, 3Н), 2,52 (т, 2Н), 1,51 (квинтет, 2Н), 1,27 (секстет, 2Н), 0,80 (т, 3Н).
Следующие промежуточные продукты, приведенные ниже в табл. 1, также получены по методикам, описанным в примере 1.
П р и м е р 4. Часть А.
Получение 1-[(3' -карбметоксидифенил-4-ил)-метил]-2-бутил-4-хлор-5-метоксимети- лимидазола.
Раствор 5,00 г 1-[(3' -карбметоксидифенил-4-ил)-метил]-2-бутил-4-хлор-5-оксиме-тилимидазола и 1 мл концентрированной серной кислоты в 200 мл метанола кипятят с обратным холодильником в течение 20 ч. После охлаждения растворитель удаляют в вакууме и остаток выливают в насыщенный раствор бикарбоната натрия. Образующуюся смесь экстрагируют метиленхлоридом, соединенные вместе органические фазы промывают водой и рассолом, сушат над безводным сульфатом натрия, фильтруют и концентрируют в вакууме. После хроматографирования на колонке с силикагелем (элюирование: 0-20% этилацетат (бензол) получают 5,35 г 1-[(3' -карбметоксидифенил-4-ил)метил]-2-бутил-4-хлор-5-метоксимети-лимидазола.
ЯМР (200 МГц, CDCl3), δ 8,26 (т, 1Н), 8,03 (д, т, 1Н), 7,76 (д т, 1Н), 7,51 (т, 1Н). 7,33 (А2М2, 4Н), 5,20 (с, 2Н), 4,31 (с, 2Н), 3,94 (с, 3Н), 3,27 (с, 3Н), 259 (т, 2Н), 1,68 (квинт, 2Н), 1,34 (секст. 2Н), 0,87 (т, 3Н).
Следующие интермедиаты получают с использованием вышеописанной методики.
Характеристики промежуточных соединений приведены в табл. 2.
П р и м е р 7 (известное соединение). Часть А.
Получение 4' -метилдифенил-2-карбоновой кислоты.
Метил-4' -метилдифенил-2-карбоксилат (10,0 г, 44,2 ммоль, 1 экв) 0,5 н. КОН в метаноле (265,5 мл, 133 ммоль, 3 экв.) и воду (50 мл) смешивают и кипятят с обратным холодильником в атмосфере N2. Через 5 ч растворитель удаляют в вакууме и добавляют воду (200 мл ) и этилацетат (200 мл). Водный слой подкисляют концентрированной соляной кислотой до рН 3 и слои разделяют. Водную фазу экстрагируют этилацетатом (2х200 мл), органические слои объединяют, сушат (MgSO4) и растворитель отгоняют в вакууме, получая 8,71 г белого твердого вещества, т.пл. 140,0-145,0.
ЯМР (200 МГц, DMCO-d6) δ 7,72 (д, 1Н, J=7 Гц), 7,56 (т, 1Н, J=7 Гц), 7,45 (д, 1Н, J=7 Гц), 7,40 (т, 1Н, J=7Гц), 7,25 (с, 4Н), 2,36 (с, 3Н).
Вычислено, С 79,23; Н 5,70.
С14Н12О2:
Найдено, С 79,22; Н 5,47.
Часть В.
Получение 4' -метил-2-цианодифенила.
4' -Метилдифенил-2-карбоновую кислоту (8,71 г, 41 ммоль, 1 экв.) и тионилхлорид (30,0 мл, 411 ммоль, 10 экв) cмешивают и кипятят с обратным холодильником в течение 2 ч. Избыток тионилхлорида отгоняют в вакууме и остаток растворяют в толуоле. Толуол удаляют роторным испарением и эту методику испарения толуола повторяют, чтобы убедиться, что весь тионилхлорид удален. Неочищенный кислый хлорид затем медленно добавляют к холодному (ОоС) концентрированному NH4OH (50 мл), так чтобы температура держалась ниже 15оС. После перемешивания в течение 15 мин добавляют воду (100 мл) и осаждают твердое вещество. Это вещество собирают вместе, хорошо промывают водой и сушат в высоком вакууме над Р2О5 в эксикаторе в течение ночи, получая 7,45 г белого твердого вещества, т.пл. 126,0-128,5оС.
ЯМР (200 МГц, DMCO-d6) δ 7,65-7,14 (м, 10Н), 2,32 (с, 3Н).
Вычислено, С 79,59; Н 6,20; N 6,63.
С14Н13NO
Найдено, С 79,29; Н 6,09; N 6,52.
Полученный выше амид (7,45 г, 35 ммоль, 1 экв.) и тионилхлорид (25,7 мл, 353 ммоль, 10 экв.) смешивают и кипятят с обратным холодильником в течение 3 ч. Тионилхлорид удаляют, используя ту же самую методику, как описано выше. Остаток промывают небольшим количеством гексана, который частично солюбилизирует продукт, но также удаляет примесь, получая 6,64 г белого твердого вещества.
Т.пл. 44,0-47,0оС.
ЯМР (200 МГц, DMCO-d6) δ 7,95 (д, 1H, J=8 Гц), 7,78 (т, 1Н, J=7 Гц), 7,69-7,32 (м, 6Н), 2,39 (с, 3Н).
Вычислено, C 87,01; Н 5,74.
С14Н11N
Найдено, С 86,44; Н 5,88.
Часть С.
Получение 4'-бромметил-2-циандифенила.
4' -Метил-2-циандифенил (5,59 г) бромируют в бензильное положение по методике примера 1, часть В, используя перекись бензоила в качестве инициатора. Продукт перекристаллизовывают из эфира, получая 4,7 г продукта, т.пл. 114,5-120,0о.
ЯМР (200 МГц, CDCl3) δ 7,82-7,37 (м, 8Н), 4,50 (с, 2Н).
Вычислено, С 61,79; Н 3,70; N 5,15.
С14Н10BrN.
Найдено, С 62,15; Н 3,45; N 4,98.
Часть D.
Получение 2-н-бутил-4-хлор-1-(2' -циандифенил-4-ил/метил)-5-(оксиметил)-имида- зола.
4'-Бромметил-2-циандифенил (4,6 г) алкилируют в 2-н-бутил-4-хлор-5-(оксиметил)имидазол по известной методике.
Часть А.
Обработка и мгновенная хроматография в 1:1 гексан (этилацетате на силикагеле с целью разделения изомерных продуктов, дают 2,53 г быстрее экстрагирующегося изомера. Перекристаллизация из ацетонитрила дает 1,57 г аналитически чистого продукта, т.пл. 153,5-155,5оС.
ЯМР (200 МГц, CDCl3) δ 7,82-7,43 (м, 6), 7,12 (д.2, J=8 Гц), 5,32 (с, 2), 4,52 (с, 2), 2,62 (т, 2 J=7 Гц), 1,70 (т т,2, J 7,7 Гц), 1,39 (т к, 2, J=7,7 Гц), 0,90 (т, 3, J=7 Гц).
Вычислено, С 69,56; Н 5,84; N 11,0.
С22Н22СlN3O
Найдено, С 69,45; Н 5,89; N 10,79.
Часть Е.
Получение 2-н-бутил-4-хлор-5-оксиметил-1-[(2' -(1Н-тетразол-5-ил)дифенил-4-ил)метил] имидазола. 2-н-бутил-4-хлор-1-(2' -циандифенил-4-ил)метил]-5-(оксиметил) имидазол (11,93 г) превращают в описанный продукт по методике, описанной в примере 8, часть С. Продукт очищают мгновенной хроматографией в 100% этилацетата до 100% этанола на силикагеле, получая 5,60 г светложелтого твердого вещества. Перекристаллизацией из ацетонитрила получают 4,36 г светложелтых кристаллов, которые все же плавятся в широком интервале температур. Кристаллы помещают в 100 мл горячего ацетонитрила. Твердое вещество, которое не растворилось, отфильтровывают, получая 1,04 г продукта в виде светложелтого твердого вещества, т.пл. 183,5-184,5оС. После охлаждения маточная жидкость дает дополнительно 1,03 г продукта в виде светло-желтого твердого вещества, т.пл. 179,0-180,0оС.
ЯМР (200 МГц, DMCO-d6) δ 7,75-7,48 (м, 4Н), 7,07 (д, 2Н, J=9 Гц), 7,04 (д, 2Н, J=9 Гц), 5,24 (с, 2Н), 5,24 (ш.с, 1Н), 4,34 (с, 2Н), 2,48 (т, 2Н, J= 7 Гц), 1,48 (т.т, 2Н, J=7,7 Гц), 1,27 (т.к, 2Н, J=7,7 Гц), 0,81 (т, 3Н, J=7 Гц).
Рассчитано для С22Н23ClN6O,
С 62,48; Н 5,48, Cl 8,38.
Найдено для твердых веществ, которые не растворяются в 100 мл ацетонитрила, С 62,73; Н 5,50; Cl 8,26.
Найдено для твердых веществ, выведенных из маточной жидкости, С 62,40; Н 5,23; Cl 8,35.
П р и м е р 8.
Часть А.
Получение 2-н-бутил-4-хлор-5-хлорметил-1-[(2'-циандифенил-4-ил)метил] имида- зол. HCl соли.
2-н-Бутил-4-хлор-5-оксиметил-1-[(2'-ци- андифенил-4-ил)-метил]имидазола (15,00 г, 39,3 ммоль, 1 экв.) превращают в хлорид. Время реакции 5 ч.
Неочищенный твердый продукт промывают эфиром до исчезновения желтого цвета. Твердый белый порошкообразный продукт затем сушат в высоком вакууме, выход 10,02 г, т.пл. 152,0-154,0оС.
ЯМР (200 МГц, CDCl3) δ 7,85-7,46 (м, 6Н), 7,20 (д, 2Н, J=10 Гц), 5,47 (с, 2Н), 4,50 (с, 2Н), 3,06 (т, 2Н, J=7 Гц), 1,82 (т.т, 2Н, J=7,7 Гц), 1,45 (т к, 2Н, J=7,7 Гц), 0,94 (т, 3Н, J=7 Гц). Масса, рассчитанная для С22Н21Сl2N3, 397,1113.
Найдено, 397, 1105.
Часть В.
Получение 2-н-бутил-4-хлор-1-[(2'-циандифенил-4-ил)метил]-5-(метоксиметил)ими- дазола.
2-н-Бутил-4-хлор-5-хлорметил-1-[(2'-ци- андифенил-4-ил)-метил]имидазол. HCl соль (5,00 г, 11,5 ммоль, 1 экв), метоксид натрия (1,37 г, 25,3 ммоль, 2,2 экв. ) и метанол (100 мл) смешивают и перемешивают в течение 3 дней. Растворитель удаляют в вакууме и добавляют этилацетат (200 мл) и воду (200 мл). Слои разделяют и водный слой экстрагируют этилацетатом (2х200 мл). Органические слои сушат (MgSO4), растворитель удаляют в вакууме и остаток мгновенно хроматографируют на силикагеле в 1:1 гексан/этилацетате, получая 4,08 г прозрачного светло-желтого масла.
ЯМР (200 МГц, CDCl3) δ 7,82-7,43 (м, 6), 7,10 (д, 2Н, J=7 Гц), 5,23 (с, 2Н), 4,32 (с, 2Н), 3,30 (с, 3Н), 2,60 (т, 2Н, J=7 Гц), 1,70 (т т, 2Н, J=7,7 Гц), 1,38 (т к, 2Н, J=7,7 Гц), 0,89 (т, 3Н, J=7 Гц).
Вычислено, С 68,11; Н 6,54; Cl 9,58.
С23Н24ClN3O
Найдено, С 68,70; Н 6,11; Cl 9,51.
Масса, рассчитанная для C23H24ClN3O, 393, 1607.
Найдено: 393,1616.
Часть С.
Получение 2-н-бутил-4-хлор-5-метоксиметил-1-[(2'-(1Н-тетразол-5-ил)дифенил-4-метил]ими дазо
2-н-Бутил-4-хлор-1-[2'-циандифенил-4- ил-(метил)-5-метоксиметил]имидазол (3,9 г, 10 ммоль, 1 экв.) азид натрия (1,95 г, 30 ммоль, 3 экв).
Хлорид аммония (1,60 г, 30 ммоль, 3 экв.) смешивают и перемешивают в ДМФ (150 мл) в круглодонной колбе, соединенной с обратным холодильником, в атмосфере N2. Затем используют масляную баню с регулятором температуры для того, чтобы осуществлять реакцию при 100оС в течение 2 дней, после чего температуру поднимают до 120оС в течение 6 дней. Реакционную сместь охлаждают и добавляют более 3 эквивалентов каждого из хлорида аммония и азида натрия. Реакционную смесь нагревают снова в течение еще 5 дней при 120оС. Реакционную смесь охлаждают, отфильтровывают неорганические соли и отфильтрованный растворитель удаляют в вакууме. К остатку добавляют воду (200 мл) и этилацетат (200 мл) и слои разделяют. Водный слой экстрагируют этилацетатом -(2х200 мл), органические слои объединяют вместе, сушат (MgSO4) и растворитель удаляют в вакууме, получая темно-желтое масло. Быстрой хроматографией в 100% этилацетате получают 3,54 г белого стекла.
ЯМР (200 МГц, CDCl3) δ 7,83 (д, 1Н, J=7 Гц), 7,59 (т, 1Н, J=7 Гц), 7,50 (т, 1Н, J=7 Гц), 7,39 (д, 1Н, J=7 Гц), 7,03 (д, 2Н, J=8 Гц), 6,73 (д, 2Н, J= 8 Гц), 5,08 (с, 2Н), 4,12 (с, 2Н), 3,18 (с, 3Н), 2,32 (т, 2Н, J=7 Гц), 1,52 (т т, 2Н, J=7,7 Гц), 1,28 (т к, 2Н, J=7,7 Гц), 0,83 (т, 3Н, J=7 Гц). Масса, рассчитанная для С23Н25ClN6O: 436,1178.
Найдено: 436,1750.
П р и м е р 9. Часть А.
Получение 1-[(2'-трет-бутоксикарбонил-дифенил-4-ил)метил]-2-бутил-4-иод-5-(2-ме- токсиэтоксиметоксиметил]-имидазола.
К раствору 5,56 мл (н-бутиллитий)гексан в 80 мл тетрагидрофурана при 0оС добавляют по каплям 1,15 мл трет-бутанола. К раствору добавляют 3,28 г 1-[(2-трет-бутоксикарбонилдифенил-4-ил) метил]-2-бутил-5-оксиметил-4-иодимидазо- ла с последующим добавлением 1,15 мл 2-метоксиэтоксиметилхлорида. Полученный раствор перемешивают при 25оС в течение 16 ч. Смесь разбавляют диэтиловым эфиром, промывают водой и рассолом, сушат над безводным сульфатом натрия, фильтруют и концентрируют. Хроматографированием на колонках получают 2,61 г 1-(2' -трет-бутоксикарбонилдифенил-4-ил-метил)] 2-бутил-4-иод-5-(2-метоксиэтоксиметоксиметил)имидазола.
ЯМР (200 МГц, CDCl3) δ: 7,78 (д, 1Н), 7,43 (м, 2Н), 7,28 (м, 3Н), 6,98 (д, 2Н), 5,26 (с, 2Н), 4,69 (с, 2Н), 4,45 (с, 2Н), 368 (м, 2Н), 3,57 (м, 2Н), 3,37 (с, 3Н), 2,58 (т, 2Н), 1,67 (квинт, 2Н), 1,34 (секст, 2Н), 1,26 (с, 9Н), 0,87 (т, 3Н).
Часть В.
Получение 1-[(2' -трет-бутоксикарбонилдифенил-4-ил)-метил]-2-бутил-5-(2-метокси- этоксиметоксиметил)-4-трифторметилими- дазола.
К суспензии 22,4 г порошка кадмия в 50 мл диметилформамида при 25оС добавляют по каплям 8,60 мл бромхлордифторметана.
Полученную смесь перемешивают при 25оС в течение 2 ч и затем фильтруют через фильтр Шленка со средним размером пор под давлением азота, получая темно-коричневый раствор трифторметилкадмий реагента.
К смеси 15 мл указанного раствора и 20 мл триамида гексаметилфосфoрной кислоты при 0оС добавляют 2,10 г бромида меди (1), с последующим добавлением 2,61 г 1-[(2' -трет-бутоксикарбонилфенил-4-ил)метил]-2-бутил-4-иод-5-(2-метоксиэтоксиметок симетил) имидазола в 5 мл диметилформамида. Реакционную смесь перемешивают при 70-75оС в течение 6 ч. После охлаждения смесь разбавляют водой и затем экстрагируют метиленхлоридом. Объединенные вместе органические фазы промывают водой и рассолом, сушат над безводным сульфатом натрия, фильтруют и концентрируют. Хроматографированием на колонках (элюирование: этилацетат/гексан) получают 2,30 г 1-[(2'-трет-бутоксикарбонилдифенил-4-ил-метил] -2-бутил-5-(2-метоксиэтоксимет оксиметил)-4-трифтор-метилимидазола.
ЯМР (200 МГц, CDCl3) δ 7,79 (д, 1Н), 7,46 (м, 2Н), 7,28 (м, 3Н), 7,00 (д, 2Н), 5,28 (с, 2Н), 4,71 (с, 2Н), 4,58 (с, 2Н), 3,66 (м, 2Н), 3,54 (м, 2Н), 3,38 (с, 3Н), 2,62 (т, 2Н), 1,70 (квинт, 2Н), 1,36 (секст. 2Н), 1,27 (с, 3Н), 0,88 (т, 3Н).
Часть С.
Получение 1-[(2'-карбоксидифенил-4-ил)-метил] -2-бутил-5-оксиметил-4-трифто-рметилимида зола
Раствор 2,30 г 1-[(2' -трет-бутоксикарбонилдифенил-4-ил)-метил]-2-бутил-5-(2-мето- ксиэтоксиметоксиметил)-5-трифтормети- лимидазола в 200 мл 1,5 М водной тетрафторборной кислоты/ ацетонитрила перемешивают при 25оС в течение 18 ч, затем смесь выливают в воду. Получающийся водный раствор доводят до рН 3, применяя насыщенный раствор бикарбоната натрия и затем экстрагируют хлороформом. Объединенные вместе органические фазы промывают рассолом, сушат над безводным сульфатом натрия, фильтруют и концентрируют. Хроматографированием на колонках (элюирование: метанол/хлороформ) получают 1,38 г 1-[(2' -карбоксидифенил-4-ил)метил] -2-бутил-5-оксиметил-4- трифторметилимидазола (т.пл. 198-199,5оС).
ЯМР (200 МГц, DMCO-d6) δ: 7,75 (д, 1Н), 7,54 (т,1Н), 7,43 (т. 1Н), 7,32 (м, 3Н), 7,10 (д, 2Н), 5,36 (с, 2Н), 4,51 (с, 2Н), 2,56 (т, 2Н), 1,56 (квинт, 2Н), 1,30 (секст. 2Н), 0,83 (т, 3Н).
П р и м е р 10. Часть А.
Получение 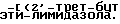 илоксикарбонилдифенил-4-ил-метил-2-бутил-5-)2-мето-ксиэтоксим еток
илоксикарбонилдифенил-4-ил-метил-2-бутил-5-)2-мето-ксиэтоксим еток
К 20 мл трифторметиликадмий реагента, полученного в примере 9, часть В, добавляют 2,80 г бромида меди (1) и полученный раствор перемешивают при 25оС в течение 14 ч. На данном этапе добавляют 20 мл триамида гексаметилфосфорной кислоты, с последующим добавлением 1,90 г 1-[(2' -трет-бутоксикарбонилдифенил-4-ил)-метил] -2-бутил-4-иод-5-(2-метоксиэтоксиметокси- метил)-имидазола в 5 мл диметилформамида. Затем реакционную смесь перемешивают при 70-75оС в течение 6 ч. После охлаждения смесь разбавляют водой и затем экстрагируют метиленхлоридом. Объединенные вместе органические фазы промывают водой и рассолом, сушат над безводным сульфатом натрия, фильтруют и концентрируют. Хроматографией на колонках (элюирование: этилацетат:бензол) получают 1,71 г 1-[2' третбутоксикарбонилдифенил-4-ил)метил]-2-бутил-5-(2-метокси-этоксиметоксимет ил)дазола.
ЯМР (200 МГц, CDCl3) δ: 7,77 (д, 1Н), 7,55-7,35 (м, 2Н), 7,27 (м, 3Н), 6,97 (д, 2Н), 5,28 (с, 2Н), 4,69 (с, 2Н), 4,55 (с, 2Н), 3,65 (м, 2Н), 3,53 (м, 2Н), 3,33 (с, 3Н), 2,63 (т, 2Н), 1,68 (квинт. 2Н), 1,35 (секст. 2Н), 1,26 (с, 9Н), 0,87 (т, 3Н).
Часть В.
Получение 1-[(2-карбоксидифенил-4-ил)-метил]-2-бутил-5-оксиметил-4-пента- фторэтилимидазола.
Это соединение получают согласно методике, описанной в примере 9, часть С. Из 1,71 г 1-[(2' -трет-бутоксикарбонилдифенил-4-ил)метил]2-бутил-5-(2-метоксиэтоксиме- токсиметил)-4-пентафторэтилимидазола получают 0,72 г 1-[(2' -карбоксидифенил-4-ил)метил] -2-бутил-5-оксиметил-4-пентафто- рэтилимидазола (т.пл. 190-191оС).
ЯМР (200 МГц, DMCO-d6) δ: 7,72 (д, 1Н), 7,61-7,42 (м, 2Н), 7,34 (м, 3Н), 7,11 (д, 2Н), 5,50 (щ.с. 2Н), 5,39 (с, 2Н), 4,50 (с, 2Н), 2, 55 (т, 2Н), 1,50 (квинт, 2Н), 1,25 (секст. 2Н), 0,80 (т, 3Н).
П р и м е р 11.
Часть А.
Получение 2-бутил-1-[(2' -циандифенил-4-ил)-метил]5-оксиметил-4-трифторметили- мидазола.
Это соединение получают согласно методике, описанной в примере 9, части А-С. Из 2-бутил-1-[(2 -циандифенил-4-ил)-метил]-5-оксиметил-4-иодимидазола получают 2-бутил-1-[(2-циандифенил-4-ил)-метил]-5-ок- симетил-4-трифторметилимидазол (т.пл. 136,5-137,5оС).
ЯМР (200 МГц, CDCl3) δ: 7,76 (д, 1Н), 7,64 (т, 1Н), 7,56-7,42(м, 4Н), 7,08 (д, 2Н), 5,33 (с, 2Н), 4,65 (д, 2Н), 3,65 (т, 2Н), 1,97 (ш.т. 1Н), 1,69 (квинт. 2Н), 1,38 (секст. 2Н), 0,89 (т, 3Н).
Часть В.
Получение 2-бутил-5-оксиметил-4-трифторметил-1-[(2' -(трифенилметилтетразол-5-ил)дифенил-4-ил)-метил]имидазола.
Раствор 6,45 г 2-бутил-1-[(2' -циандифенил-4-ил)-метил]-5-оксиметил-4-трифторме- тилимидазола и 4,00 г триметилстаннилазида в 65 мл ксилола перемешивают при 115-120оС. Через 24 ч и через 48 ч в реакции добавляют порции по 1,00 г триметилстаннилазида. После в общей сложности 64 ч реакции при 115-120оС смесь охлаждают до 80оС и фильтруют, получая 10,22 г небелого твердого вещества.
К суспензии этого твердого вещества в 60 мл метиленхлорида и 10 мл ТГФ при 25оС добавляют по каплям в течение нескольких минут 1,65 мл 10 н. водного раствора гидроксида натрия и смесь перемешивают при 25оС в течение 15 мин. Затем к реакционной смеси добавляют 4,60 г трифенилметилхлорида и полученную смесь перемешивают при 25оС в течение 2 ч. В конце концов смесь выливают в воду и затем экстрагируют метиленхлоридом. Объединенные вместе органические фазы промывают водой и рассолом, сушат над безводным сульфатом натрия, фильтруют и концентрируют. Перекристаллизацией неочищенного продукта из смеси толуол/гексан получают 7,59 г 2-бутил-5-оксиметил-4-трифторметил-1-[(2'- (трифенилметилтетразол-5-ил)дифенил-4- ил)метил]имидазола.
ЯМР (200 МГц, CDCl3) δ: 7,93 (д д, 1Н) 7,46 (м, 2Н), 7,35-7,08 (м, 12Н), 6,90 (д, 6Н), 6,71 (д, 2Н), 5,13 (с, 2Н), 4,39 (д, 2Н), 2,53 (т, 2Н), 1,63 (квинт, 2Н), 1,30 (секст. 2Н), 0,85 (т, 3Н).
Часть С.
Получение 2-бутил-5-оксиметил-1-[(2' -(1Н-тетразол-5-ил)дифенил-4-ил)метил]-4-три- фторметилимидазола.
Раствор 4,06 г 2-бутил-5-оксиметил-4-трифторметил-1-[(2'-(трифенилметилтетра-зол-5-ил) дифенил-4-ил)метил]имидазола в 40 мл 10%-ной соляной кислоты и 80 мл тетрагидрофурана перемешивают при 25оС в течение 2 ч и затем выливают в воду, содержащую избыток гидроксида натрия. Водный раствор промывают диэтиловым эфиром, доводят до рН 3 с помощью 10%-ной соляной кислоты, и затем экстрагируют хлороформом. Объединенные вместе экстракты в хлороформе промывают рассолом, сушат над безводным сульфатом натрия, фильтруют, и концентрируют. Хроматографирование на колонках (элюирование: 10% метанол/хлороформ) дает 2,04 г 2-бутил-5-оксиметил-1-[(2' -(1Н-тетразол-5-ил)дифенил-4-ил)метил]-4-трифторметилимидазола в виде аморфного твердого вещества.
ЯМР (200 МГц, DMCO-d6) δ: 7,68-7,47 (м, 4Н), 7,02 (А2В2 4Н), 5,43 (ш.с. 1Н), 5,27 (с, 2Н), 4,44 (с, 2Н), 2,47 (т, 2Н), 1,47 (квинт. 2Н), 1,22 (секст. 2Н), 0,77 (т. 3Н).
В табл. 3 представлены соединения 1, которые получают по методикам примеров 7-11.
П р и м е р 24. Получение 1-[(2' -карбоксидифенил-4-ил)метил]-2-бутил-4-хлорими- дазол-5-карбоксальдегида.
Смесь 1,46 г 1-[(2' -карбоксидифенил-4-ил)-метил]-2-бутил-4-хлор-5-оксиметилими- дазола и 7,30 г активированной двуокиси марганца в 40 мл тетрагидрофурана перемешивают при 25оС в течение 5 дней. Смесь фильтруют через Celite и фильтрат концентрируют в вакууме. Хроматографирование на колонках с силикагелем (элюирование: 2-10% метанол-(хлороформ) с последующей перекристаллизацией из этиланетата дает 0,71 г 1-[(2' -карбоксидифенил-4-ил)-метил]-2-бутил-4-хлоримидазол-5-карбоксальдегида (т.пл. 154-158оС/разл).
и фильтрат концентрируют в вакууме. Хроматографирование на колонках с силикагелем (элюирование: 2-10% метанол-(хлороформ) с последующей перекристаллизацией из этиланетата дает 0,71 г 1-[(2' -карбоксидифенил-4-ил)-метил]-2-бутил-4-хлоримидазол-5-карбоксальдегида (т.пл. 154-158оС/разл).
ЯМР (200 МГц, DMCO-d6) δ: 1 2,85 (ш.с. 1Н), 9,77 (с, 1Н), 7,77 (д, 1Н), 7,62 (т, 1Н), 7,50 (т, 1Н), 7,40 (д, 1Н), 7,26 (А2В2, 4Н), 5,67 (с, 2Н), 2,70 (т, 2Н), 1,56 (квинт. 2Н), 1,28 (секст. 2Н), 0,83 (т, 3Н).
П р и м е р 25. Получение метил 1-[(2' -карбоксидифенил-4-ил)метил]-2-бутил-4- хлоримидазол-5-карбоксилата.
К смеси 1,45 г 1-[(2' -карбоксидифенил-4-ил)-метил]-2-бутил-4-хлоримидазол-5-ка-рбоксальдегида и 0,91 г цианида натрия в 20 мл метанола при 25оС добавляют 0,32 мл уксусной кислоты с последующим добавлением 7,25 г двуокиси марганца. Полученную смесь перемешивают при 25оС в течение 40 ч. Реакционную смесь фильтруют через Celite и фильтрат разбавляют водой. Водный раствор доводят до рН 3, используя соляную кислоту и экстрагируют метиленхлоридом. Объединенные вместе органические фазы промывают рассолом, сушат над безводным сульфатом натрия, фильтруют и концентрируют. Неочищенный продукт перекристаллизовывают из серного эфира, получaя 0,90 г метил-[(2' -карбоксидифенил-4-ил)метил]-2-бутил-4-хлоримидазол-5-ка-рбоксилата (т.пл. 154-155оС).
и фильтрат разбавляют водой. Водный раствор доводят до рН 3, используя соляную кислоту и экстрагируют метиленхлоридом. Объединенные вместе органические фазы промывают рассолом, сушат над безводным сульфатом натрия, фильтруют и концентрируют. Неочищенный продукт перекристаллизовывают из серного эфира, получaя 0,90 г метил-[(2' -карбоксидифенил-4-ил)метил]-2-бутил-4-хлоримидазол-5-ка-рбоксилата (т.пл. 154-155оС).
ЯМР (200 МГц, DMCO-d6) δ: 12,75 (ш.с. 1Н), 7,73 (д: 1Н), 7,58 (т, 1Н), 7,46 (т. 1Н). 7,34 (м, 3Н), 7,07 (д, 2Н), 5,63 (с, 2Н), 3,78 (с, 3Н), 2,67 (т, 2Н), 1,56 (квинт, 2Н), 1,29 (секст. 2Н), 0,83 (т, 3Н).
П р и м е р 26. Часть А.
Получение 1-[(2' -карбметоксидифенил-4-ил)метил]-2-бутил-4-хлоримидазол-5-ка-рбоксальдегида.
Смесь 2,06 г 1-[(2' -карбметоксидифенил-4-ил)-метил]-2-бутил-4-хлор-5-оксиме-тилимидазола и 3,08 г активированной двуокиси марганца в 20 мл метиленхлорида при 25оС перемешивают в течение 40 ч. Реакционную смесь фильтруют через Celite ® и фильтрат концентрируют в вакууме. Хроматографирование на колонках (элюирование: этилацетат/бензол) дает 1,15 г 1-[(2' -карбметоксидифенил-4-ил)метил-]2-бутил-4-хло- римидазол-5-карбоксальдегида.
ЯМР (200 МГц, CDCl3) δ: 9,76 (с, 1Н), 7,83 (д, д, 1Н), 7,52 (т, д, 1Н), 7,40 (т д, 1Н), 7,31 (д д, 1Н), 7,17 (А2В2, 4Н), 5,58 (с, 2Н), 3,63 (с, 3Н), 2,67 (т, 2Н), 1,70 (квинт, 2Н), 1,38 (секст. 2Н), 0,90 (т, 3Н).
В табл. 4 представлены соединения I, которые получают по методикам примеров 24-26.
П р и м е р 36. Часть А.
Получение 2-пропил-4-хлоримидазол-5-карбоксальдегида.
В этом примере иллюстрируется предпочтительнаяя методика получения соединения примера 17.
К раствору 2-пропил-4-хлор-5-гидроксиметилимидазола (полученного согласно патенту США N 4355040) т.пл. 110,5-114оС, 32,0 г (0,18 моля) в дихлорметане (1 л) добавляли активированную двуокись марганца (207 г, 2,38 моля, 13 экв. ). Полученную смесь перемешивали в течение 4-18 ч при комнатной температуре и после этого фильтровали через Целит® Целит® промывали 500 мл смеси дихлорметан/метанол (1,1 об/об.) и фильтрат концентрировали в вакууме с получением 24,7 г бледно-желтого твердого вещества. Перекристаллизацией из этилацетата получали 16,6 г (53%) чистого продукта, т.пл. 139-141,5. ЯМР (200 МГц, CDCl3, CD3OD, TMC) δ: 9,61 (синглет, 1Н), 2,66 (триплет J=7,5 Гц, 2Н), 1,83-1,67 (мультиплет, 2Н), 0,98 (триплет, J=7 Гц, 3Н).
Часть В.
Получение 2-пропил-4-хлор-1[(2' -(1-трифенилметилтетразол-5-ил)бифенил-4-илме- тил]имидазол-5-карбоксальдегида. К смеси 2-пропил-4-хлоримидазол-5-карбоксальде-гида (15,0 г, 86,9 ммоля) и карбоната калия (13,2 г, 95,6 ммоля) в N,N-диметилформамиде (800 мл) добавляли и 4' -бромметил-2-(1-трифенилметилтетразол-5-ил)бифенил, (полученный согласно части В примера 38 (53,3 г, 4 95,6 ммоля). Смесь нагревали до 75-80оС в течение 4-18 ч, охлаждали до комнатной температуры и переливали в делительную воронку, содержащую 1 л воды и 1 л этилацетата. Водную фазу дважды экстрагировали этилацетатом (250 мл) объединенную органическую фазу промывали водой (4х500 мл) и насыщенным водным раствором хлористого натрия (500 мл), сушили над безводным сульфатом магния, фильтровали и концентрировали в вакуме с получением сырого продукта.
В результате очистки методом мгновенной хроматографии на силикагеле (1 кг, 10-20% EtOAc (гексан) получали 27,5 г (49%) целевого соединения в виде бледно-желтого твердого вещества, т.пл. 55-62оС.
ЯМР (200 МГц, CDCl3, TMC) δ: 9,73 (синглет, 1Н), 7,95-6,81 (мультиплет, 23Н), 5,45 3 (синглет, 2Н), 2,49 (триплет, J= 7,5 Гц, 2Н), 1,75-1,64 (мультиплет, 2Н), 0,89 (триплет, J=7 Гц, 3Н).
Часть С.
Получение 2-пропил-4-хлор-1-[(2'-(1Н-тетразол-5-ил)бифенил-4-ил)метил/имида-зол-5-карб оксальдегида
К шламму 2-пропил-4-хлор-1-[(2' -(1-трифенилметилтетразол-5-ил)бифенил-4-ил)ме- тил] имидазол-5-карбоксальдегида (26,5 г 40,8 ммоля) в воде (100 мл) по каплям добавляли в течение 15 минут 50%-ный водный раствор трифторуксусной кислоты (об/об, 200 мл). Еще через 15 мин смесь подщелачивали 4 н. раствором NaOH (350 мл). Полученную в результате смесь экстрагировали эфиром (2х100 мл) и водную фазу подкисляли до рН 4-5 4 н. раствором HCl и полученный осадок экстрагировали этилацетатом (2х100 мл). Объединенные этилацетатные слои сушили над безводным сульфатом магния и затем фильтровали и концентрировали в вакууме с получением 16 г сырого продукта.
В результате очистки методом мгновенной хроматографии на силикагеле (100 г, 50% EtOAc (гексан) получали 13,7 г (83%) очищенного целевого соединения, т.пл. 165-167 об.
ЯМР (200 МГц, CDCl3, ТМС) δ: 9,65 (синглет, 1Н), 7,95-6,96 (мультиплет, 8Н), 5,51 (синглет, 2Н), 2,59 (триплет, J=7,5 Гц, 2Н), 1,70-1,63 (мультиплет, 2Н), 0,92 (триплет, J=7 Гц, 3Н).
П р и м е р 37. Этот пример иллюстрирует методику получения вещества примера 4, часть Е и его калиевой соли, которая является предпочтительным веществом этого изобретения.
Часть А.
Приготовление 1-[(2'-(триметилстаннилтетразол-5-ил)дифенил-4-ил)метил] -2-бутил-4- хлор-5-гидроксиметилимидазола
1-[(2' -Цианодифенил-4-ил)метил] -2-бутил-4-хлор-5-гидровоксиметилимидазол (766 г), азид триметилолова (766 г) и 7,90 л ксилолов помещают в 12-литровую круглодонную колбу, снабженную механической мешалкой, холодильником с введением азота и термометром и заключенную в обогревающую рубашку. Суспензию нагревают до 115оС и получают прозрачный раствор, который выдерживают в течение 41 ч. Образовавшуюся суспензию охлаждают до комнатной температуры и неочищенный продукт выделяют путем вакуумной фильтрации, промывают его толуолом (800 мл) и сушат в вакууме примерно при 50оС в течение ночи. Неочищенный продукт (1202 г) загружают в 12-литровую круглодонную колбу и суспендируют его при 105оС в толуоле (7,0 л). Эту суспензию охлаждают до 50оС и продукт выделяют путем вакуумной фильтрации, промывают его одним литром толуола и сушат в вакууме при 50оС в течение ночи.
Выход 1071 г, 94%
Т.пл. 211-214оС.
Часть В.
Приготовление 1-[(2' -(трифенилметилтетразол-5-ил)-дифенил-4-ил)метил] -2-бутил-4-хлор-5-гидроксим етилимидазола.
1-[(2' -(Триметилетаннилтетразол-5-ил)-дифенил-4-ил)метил)]-2-бутил-4-хлор-5 -гидроксиметилимидазол (1,046 кг) хлористый метилен (5,00 л) тетрагидрофуран (0,85 л) и 10 н. раствор гидроксида натрия (192 мл) помещают в 12-литровую круглодонную колбу, оборудованную механической мешалкой, холодильником с введением азота и термометром. После перемешивания в течение 5 мин при комнатной температуре добавляют хлористый трифенилметил (0,530 кг) и смесь перемешивают 3 ч. Добавляют 20 мл 10 н. раствора гидроксида натрия и дополнительное количество (50 г) хлористого трифенилметила и смесь перемешивают в течение ночи. Добавляют 3,70 л деионизированной воды и 30 мл 10 н. раствора гидроксида натрия и дают разделиться фазам. Органическую фазу дважды промывюат 2,0-литрoвыми порциями воды, сушат сульфатом натрия (100 г) и фильтруют в 12-литровую круглодонную колбу, оборудованную для перегонки. Отгоняют примерно 2,0 л хлористого метилена. Нагревание прекращают и добавляют 5,0 л гептана. Образовавшуюся суспензию перемешивают при комнатной температуре (примерно 68 ч). Смесь охлаждают до примерно 5оС и продукт, выделенный посредством вакуумной фильтрации, промывюат гептаном (1,0 л), сушат в течение 48 ч в вакууме при 40-50оС.
Выход 959,5 г, 80%
Т. пл. 167-169оС. Чистота 99,8% по данным жидкостной хроматографии высокого давления.
Часть С.
Приготовление 2-бутил-4-хлор-5- гидроксиметил-1- [(2'(1H-тетразол-5-ил)-дифенил-4-ил)метил] имидазола 1-[(2' -(трифенилметилтетразол-5-ил)дифенил-4-ил)метил] -2-бутил-4-хлор-5- гидроксиметилимидазолa (920 г) и 2,10 л метанола помещают в 12-литровую круглодонную колбу, снабженную механической мешалкой, холодильником с введением азота и термометром. Суспензию охлаждают примерно до 10оС и добавляют 700 мл 3,4 н. соляной кислоты (в течение 10 мин). После перемешивания 2 ч при 10-20оС густую суспензию разбавляют метанолом (500 мл) и нагревают до 30оС.
Спустя 1 ч при 30оС реакционную смесь нейтрализуют до рН 13 с помощью 10 н раствора гидроксида натрия (420 мл). Растворитель (в значительной степени метанол 2,3 л) отгоняют при добавлени 2,3 л деионизированной воды. Нагревание прекращают и добавляют 700 мл деионизированной воды и 1,40 л толуола. После охлаждения примерно до 30оС органическую фазу удаляют. Водную фазу повторно экстрагируют толуолом (700 мл). В колбу, содержащую водную фазу, добавляют 1,20 л этилацетата. После перемешивания 10 мин добавляют 130 мл уксусной кислоты. Смесь перемешивают 1 ч, затем оставляют выдерживаться в течение ночи. Возобновляют перемешивание и смесь охлаждают до примерно 5оС. Продукт выделяют путем вакуумной фильтрации, повторно суспендируют в 1,50 л деионизированной воды и отсасывают воду до полусухого состояния. Влажную лепешку помещают в 12-литровую круглодонную колбу и повторно суспендируют 0,5 ч при комнатной температуре 4,0 л этилацетата. Продукт выделяют путем вакуумной фильтрации, промывают 200 мл этилацетата и сушат в вакууме в течение ночи при 50оС.
Выход 518 г, 88,5% Т.пл. 184-185оС.
Чистота по данным жидкостной хроматографии высокого давления: 98,8%
Спектр ЯМР (200 МГц, пердейтеродиметилсульфоксид): б-мультиплет (хим. сдвиг) 7,61 (м, 4Н), 7,05 (м, 4Н), 5,24 (с, 2Н), 4,32 (синглет, 2Н), 3,35 (шир. синглет, 1Н), 2,46 (триплет, 2Н, J=7,8 Гц), 1,44 (мультиплет, 2Н), 1,23 (мультиплет, 2Н), 0,79 (триплет, 3Н, J=7,2 Гц).
Часть D.
Приготовление калиевой соли 2-бутил-4-хлор-5-гидрооксиметил-1-[(2'-(1Н-тетра- зол-5-ил)дифенил-4-ил)метил]имидазола.
Продукт из части С (11,00 г) и 30 мл изопропилового спирта загружают в 100-миллилитровую круглодонную колбу, снабженную магнитной мешалкой, термометром и ловушкой Дина-Старка в атмосфере азота. Суспензию нагревают до 40оС. Добавляют раствор 87% гидроксида калия (2,00 г) (изопропанол/20 мл) вода (1,0 мл) до значения рН 11 (18,5 мл). Большую часть воды удаляют путем азеотропной перегонки изопропанола (отгоняется 20 мл). Добавляют 25 мл гептана и суспензию охлаждают до комнатной температуры. Добавляют дополнительное количество (15 мл) гептана и смесь перемешивают в течение 1/2 ч. Продукт выделяют путем вакуумной фильтрации, промывают 1 раз гептаном (20 мл) и сушат в течение ночи при 60оС в вакууме.
Выход: 10,33 г, 86% Т.пл. выше 250оС.
П р и м е р 38. Этот пример иллюстрирует другой предпочтительный способ получения вещества примера 7, часть Е.
Часть А.
Приготовление 2-(трифенилметилтетразол-5-ил)-4' -метилдифенила.
2-(пара-Толил)бензонитрил (9,00 г), азид натрия (3,00 г), толуол (35 мл) и хлорид трибутилолова (16,4 г) загружают в 250-миллилитровую круглодонную колбу, оборудованную механической мешалкой, холодильником с введением азота и термометром в обогревающей рубашке. Эту смесь нагревают до 110оС и выдерживают в течение 70 ч. Смесь разбавляют 35 мл толуола и охлаждают до комнатной температуры. Добавляют 5,5 мл 10 н. раствора гидроксида натрия и 13,5 г трифенилметиленхлорида и смесь перемешивают 3 ч при комнатной температуре. Добавляют 35 мл деионизированной воды и 70 мл гептана и образовавшуюся суспензию охлаждают в ледяной бане 0,5-1 ч. Смесь фильтруют в вакууме, промывают 2 раза водой (по 50 мл) и один раз 50 мл смеси (3:2 по объему) гептан/толуол и сушат в вакууме в течение ночи при 40оС. Выход сырого вещества равен 18,32 г (82% ). Неочищенный продукт растворяют в хлористом метилене (200 мл) и промывают 1 раз 0,4 н. раствором гидроксида натрия (52 мл). Органическую фазу фильтруют под действием гравитации, упаривают на роторном испарителе и продукт повторно суспендируют в гептане (100 мл), фильтруют и сушат в вакууме при 40оС в течение ночи. Общий вид 15,1 г, 68% Т.пл. 161-612оС.
Часть В.
Приготовление 4' -бромметил-2-(трифенилметилтетразол-5-ил)дифенила.
В круглодонную колбу емкостью 100 мл, снабженную термометром, холодильником, в атмосфере азота вводят 9,0 г [2-((-трифенилметилтетразол-5-ил))-4' -метил-1,1' -дифенил (18,8 ммоль), 4,0 г N-бромсукцинимида (22,5 ммоль) 0,1 г азо(биизобутиронитрила) (0,61 ммоль) и 40 мл четыреххлористого углерода. Реакционную массу охлаждают до комнатной температуры, разбавляют хлористым метиленом (30 мл) и промывают водой (30 мл). Водную фазу выбрасывают.
Часть С.
Приготовление 2-н-бутил-4-хлоримидазол-5-карбоксальдегида.
2-н-Бутил-4-хлор-5-гидроксиметилими- дазол (50,0 г, 265 ммоль. 1 экв.) растворяют в ледяной уксусной кислоте (150 мл). Затем к перемешиваемому раствору имидазола по каплям добавляют 1 н. раствор церий (IV) аммоний нитрата (575 мл, 595 ммоль, 2,25 экв.), поддерживая температуру смеси при 20-30оС. После завершения добавления еще добавляют 10 мл 1 н. раствoра церийаммонийнитрата, так, чтобы смесь оставалась оранжевой. Спустя 3 ч реакционную смесь охлаждают на льду и добавляют 50%-ный раствор гидроксида натрия (210 мл), чтобы нейтрализовать уксусную кислоту.
Продукт выпадает в осадок. Значение рН доводится до 6 и твердое вещество отфильтровывается, промывается водой (3 раза по 500 мл) и сушится при высоком вакууме. Получают 38,13 г белого порошка с т.пл. 92,5-93,5оС.
Спектр ЯМР (200 МГц, пердейтерохлороформ), δ; 11,83 (мультиплет, 1Н), 9,64 (синглет, 1Н), 2,85 (триплет 2Н, J=7 Гц), 1,78 (триплет триплетов, 2Н, J= 7,7 Гц), 1,38 (триплет квартетов, 2Н, J=7,7 Гц), 0,93 (триплет, 3Н, J=7 Гц).
Вычислено, С 51,48; Н 5,94; Cl 19,00; N 15,01.
С8Н11ClN2O
Найдено, С 51,75; Н 5,82; Cl 18,73; N 14,87.
Часть D.
Приготовление 1-[(2' -(трифенилметилтетразол-5-ил)-дифенил-4-ил)метил] -бутил-4-хлор-5-гидроксиметилимидазола
Органическую фазу загружают в круглодонную колбу емкостью 100 мл, снабженную холодильником, термометром в атмосфере азота. Кроме того в колбу загружают 2,56 г 2-бутил-4-хлоримидазол-5-карбоксальдегида (13,7 ммоль), 9,5 мл воды, 2,8 мл 10 н. раствора гидроксида натрия и 1,2 мл аликвоты 336, эту двухфазную систему перемешивают в течение ночи при комнатной температуре. В эту реакционную массу добавляют 0,48 г боргидрида натрия (12,7 ммоль), и реакционную массу снова перемешивают в течение ночи при комнатной температуре. По завершении реакции эту массу промывают 30 мл воды и водную фазу выбрасывают.
Органическую фазу вводят в круглодонную колбу емкостью 100 мл, снабженную термометром, конденсатором флегмы, сборником и капельной воронкой. Хлористый метилен и четыреххлористый углерод отгоняют и реакционный объем заполняют 25 мл толуола. Перегонку продолжают, пока температура колбы не достигнет примерно 110оС. Реакционную массу охлаждают примерно до 40оС и затем разбавляют 15 мл этилацетата и 20 мл н-гептана. Добавляют затравочный кристалл и реакционную массу дополнительно охлаждают до 0-10оС и перемешивают в течение 1,0-2,0 ч. Суспензию фильтруют через воронку Бюхнера, причем твердое вещество промывают небольшим количеством холодной смеси толуола и этилацетата. Твердое вещество сушат в вакуумном шкафу в течение ночи, получая 5,91 г. Выход 51,7% в расчете на 2-бутил-4-хлор-имидазол-5-карбоксальдегид или 47,2% (в расчете на 2-(трифенилметилтетразол-5-ил)-4' -метилдифенил).
Неочищенный материал подвергают перекристаллизации из 30 мл толуола, получaя 4,57 г продукта (выделение на 77,33%) с температурой плавления 161-162,5оС.
Часть Е.
Приготовление 2-бутил-4-хлор-5-гидроксиметил-1-[(2' -(1Н-тетразол-5-ил)-дифенил-4-ил)метил]имидазола и его калиевой соли.
Продукт части С превращают в указанное в заготовке соединение и его калиевую соль по методике примера 37, части С и D.
П р и м е р 39.
Часть А.
Приготовление 2-бутил-4-хлор-1[(2' -N-трифенилметил-(1Н-тетразол-5-ил) дифенил-4-ил)метил]имидазол-5-карбоксальдегида.
2-н-Бутил-4-хлоримидазол-5-карбокса-льдегид (26,78 г 143,0 ммоль, 1 экв. ) алкилируют 4' -бромметил-2-(N-трифенилметил-(1Н-тетразол-5-ил)дифенилом (80,0 г, 143 ммоль, 1 экв.) (выделен из примера 38, часть В) по известной методике.
После хроматографирования и перекристаллизации из смеси гексан-тетрагидрофуран одной трети неочищенного материала получают 19,63 г белого порошка с т.пл. 86,0-88,0оС.
Спектр ЯМР (в пердейтерохлороформе): 9,76 (синглет, 1Н), 7,96 (дублет, 1Н, J=8 Гц), 7,56-6,80 (мультиплет, 22Н), 5,47 (синглет, 2Н), 2,53 (триплет, 2Н), 1,65 (триплет триплетов, 2Н, J=7,7 Гц), 1,30 (триплет квартетов, 2Н, J= 7,7 Гц, 0,83 (триплет, 3Н, J=7 Гц).
Вычислено, С 73,5; Н 5,89; N 11,43.
С41Н35ClN6O.
Найдено, С 73,32; Н 5,88; N 11,84.
П р и м е р 40. Часть А.
Приготовление 4-карбометокси-5-гидроксиметил-2-н-пропил-1-[(2' -N-трифенилметил(1Н-тетразол-5-ил)дифенил-4-ил)ме- тил]имидазола.
4,5-Дикарбометокси-2-н-пропил-1-[(2' -N-трифенилметил(1Н-тетразол-5-ил)дифе-нил-4-ил)метил] имидазол (см. пример 59) (10,0 г, 14,0 ммоль, 1 экв) растворяют в 50 мл тетрагидрофурана и туда же добавляют раствор три-трет-бутоксиалюминийгидрида лития в тетрагидрофуране (7,2 г, 28,0 ммоль/,2 экв). Спустя 24 ч добавляют еще 0,5 экв. восстанавливающего агента. Спустя еще 24 ч реакцию прерывают путем добавления 10 мл метанола, а растворитель удаляют в вакууме. При хроматографии в смеси 1:1 гексан/этилацетат до смеси 9:1 этилацетат/изопропанол получают 2,16 г белой стеклообразной массы. По данным ЯМР она представляет собой 6:1 региоизомеров при 4,5 положениях имидазола.
Спектр ЯМР (в дейтерохлороформе, основной изомер) δ: 7,96 (мультиплет, 1Н), 7,80 (мультиплет, 2Н), 7,39-7,18 (мультиплет, 10Н), 7,13 (дублет, 2Н, J= 9 Гц), 6,95 (мультиплет, 6Н), 6,71 (дублет, 2Н, J=9 Гц), 5,08 (синглет, 2Н), 4,57 (дублет, 2Н, J=6 Гц), 3,95 (синглет, 3Н), 3,50 (мультиплет, 1Н), 2,55 (триплет, 2Н, J=7 Гц), 1,65 (трипл. кварт. 2Н, J=7,7 Гц), 1,62 (вода) 0,89 (триплет, 3Н), J=7 Гц). Ключевые пики примесного изомера (ЯМР) δ: 5,45 (синглет, 2Н), 4,84 (мультиплет, 2Н), 3,84 (мультиплет, 1Н), 3,72 (синглет, 3Н).
Вычислено, С 73,77; Н 5,74; N 12,29.
С42Н38N6О3 (H2O)0,5
Найдено, С 73,54; Н 5,76; N 12,59.
Часть В.
Приготовление 4-карбометокси-5-гидроксиметил-2-н-пропил-1-[(2' -(1Н-тетразол-5-ил)дифенил)метил]имидазола.
Продукт из части 4 подвергают деметилированию по методике примера 59 и получают стеклообразную массу. Ее кристаллизацию осуществляют путем перемешивания в этилацетате. Т.пл. 113-210оС (наблюдается разложение). Спектр ЯМР (в пердейтеродиметилсульфоксиде) δ: 7,54 (мультиплет, 1Н), 7,43-7,28 (мультиплет, 3Н), 7,08 (дублет, 2Н, J=9 Гц), 6,88 (дублет, 2Н, J=9 Гц), 5,30 (синглет, 2Н), 4,72 (синглет, 2Н), 3,73 (синглет, 3Н), 2,48 (триплет, 2Н, J= 7 Гц), 1,56 (триплет, кварт.2Н, J=7,7 Гц), 0,87 (триплет, 2Н, J=7 Гц).
ИК-спектр (в нудколе), 3206 (шир.) 1702, 761 см-1.
Вычислено, С 55,75; H 6,30; N 16,96.
С23Н24N6O3: (H2O)3,5
Найдено, С 55,83; Н 5,71; N 16,86.
П р и м е р 41.
Получение 5-гидроксиметил-2-н-пропил-1-[(2' -(1Н-тетразол-5-ил)дифенил)метил]имидазол-4-карбоновой кислоты.
Продукт из примера 40, часть А, подвергают взаимодействию с трифторуксусной кислотой, как описано в примере 36, часть С. После подкисления водной фазы соляной кислотой образовавшееся клейкое вещество перемешивают в водной смеси, в которую был добавлен этилацетат. Образуется белый кристаллический продукт, нерастворимый в обеих фазах. Этот продукт отфильтровывают и сушат. Т. пл. 250оС (потемнение, выше 275оС). Спектр ЯМР (в пердейтеродиметилсульфоксиде),δ
7,73-7,47 (мультиплет, 4H), 7,07 (дублет, 2Н, J=9 Гц), 6,98 (дублет 2Н, J= 9 Гц), 5,30 (синглет, 2Н), 4,72 (синглет, 2Н), 3,5 (вода) 2,44 (триплет, 2Н, J=7 Гц), 1,52 (триплет, кв. 2Н, J=7,7 Гц) 0,85 (триплет 3Н, J=7 Гц).
Вычислено, С 62,47; Н 5,36; N 19,87.
С22Н22N6O3 (H2O)0,25
Найдено, С 62,63; Н 5,25; N 19,51.
П р и м е р 42. Получение 2-бутил-1-[(2' (1Н-тетразол-5-ил)-бифенил-4-ил)метил]-4-три- фторметилимидазол-5-карбоновой кислоты.
Смесь 4,00 г 2 утил-5-гидроксиметил-4- трифторметил-1-[(2'-трифенилметилтетразол-5-ил)-бифенил- 4-ил) метил/ имидазола и 8,00 г активированного диоксида марганца в 50 мл хлористого метилена перемешивали при 25оС. Через 24 ч в реакционную смесь добавляли 2,00 г диоксида марганца. Через 100 ч реакционную смесь фильтровали с использованием хлористого метилена. Затем твердые вещества промывали метанолом и метанольный фильтрат концентрировали. Остаток растворяли в воде. Полученный в результате водный раствор доводили до рН 3 с использованием 10%-ного раствора хлористоводородной кислоты и затем экстрагировали смесью хлороформ/изопропанол. Объединенные органические фазы промывали рассолом, сушили над безводным сульфатом натрия, фильтровали и концентрировали. В результате очистки методом колонной хроматографии (элюирование смесью хлороформ/метанол/уксусная кислота в соотношении 95:5: 0,5) получали 0,25 г 2-бутил-1-[(2' -(1Н-тетразол-5-ил)-бифенил-4-ил)метил] -4-трифторметилимида- зол-5-карбоновой кислоты в виде аморфного твердого вещества.
ЯМР (200 МГц, DMCO-d6) δ: 7,70-7,48 (мультиплет, 4Н), 7,00 (А2В2, 4Н), 5,58 (синглет, 2Н), 2,59 (триплет, 2Н), 1,51 (квинтет, 2Н), 125 (секстет, 2Н), 0,79 (триплет, 3Н).
а. ЯМР (200 МГц, CDCl3) CD3OD, ТМС/δ 7,88-6,90 (мультиплет, 8Н), 5,52 (синглет, 2Н), 2,63 (триплет, J=7,5 Гц, 2Н), 1,77-1,66 (мультиплет, 2Н), 0,95 (триплет, J=7 Гц, 3Н).
b. ЯМР (200 МГц, DMCO-d6) δ: 7,46-7,63 (мультиплет, 4Н), 7,05 (дублет, 2Н, J= 8 Гц), 6,93 (дублет, 2Н, J=8 Гц), 5,56 (синглет, 2Н), 4,1 (синглет, 12Н), 2,55 (триплет, 2Н, J=7,5 Гц), 1,44-1,52 (мультиплет, 2Н), 1,17-1,28 (мультиплет, 2Н), 0,78 (триплет, 3Н, J=7 Гц).
с. ЯМР (200 МГц, DMCO-d6) δ: 7,71-7,50 (мультиплет, 4Н), 7,02 (А2В2, 4Н), 5,60 (синглет, 2Н), 2,59 (триплет, 2Н), 1,57 (секcтет, 2Н), 0,84 (триплет, 3Н).
d. ЯМР (200 МГц, DMCO-d6) δ: 7,74-7,52 (мультиплет, 4Н), 7,05 (А2В2, 4Н), 5,58 (синглет, 2Н), 2,62 (триплет, 2Н), 1,51 (квинтет, 2Н), 1,25 (секстет, 2Н), 0,80 (триплет, 3Н).
е. ЯМР (200 МГц, DMCO-d6) δ: 7,73-7,53 (мультиплет, 4Н), 7,04 (А2В2, 4Н), 5,58 (синглет, 2Н), 2,60 (триплет, 2Н), 1,56 (секстет, 2Н), 0,84 (триплет, 3Н).
f. ЯМР (200 МГц, DMCO-d6) δ: 13,78 (ш.с. 1Н), 12,82 (ш.с. 1Н), 7,75 (дублет, 1Н), 7,59 (триплет, 1Н), 7,47 (триплет, 1Н), 7,35 (мультиплет, 3Н), 7,08 (дублет, 2Н), 5,63 (синглет, 2Н), 2,66 (триплет, 2Н), 1,61 (секстет, 2Н), 0,86 (триплет 3Н).
g. ЯМР (200 МГц, DMCO-d6): δ: 13,73 (ш.с. 1Н), 12,80 (ш.с. 1Н), 7,74 (дублет, 1Н), 7,59 (триплет, 1Н), 7,46 (триплет, 1Н), 7,33 (мультиплет, 3Н), 7,07 (дублет, 2Н), 5,65 (синглет, 2Н), 2,65 (триплет, 2Н), 1,62 (секстет, 2Н), 0,85 (триплет, 3Н).
Вещества примеров в табл. 5 получали с использованием методов, описанных в примере 42.
Получено также соединение 1, где R1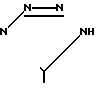 R2-н-бутил, R3-Cl и R4-CH2OH (пример N 50).
R2-н-бутил, R3-Cl и R4-CH2OH (пример N 50).
П р и м е р 51. Часть А.
Приготовление 4(5)-метил-2-пропилимидазола.
В хорошо перемешиваемую смесь 72,0 мл масляного альдегида и 240 г моногидрата ацетата меди (II) в 1000 мл 25%ного водного аммиака при 0оС добавляют 32,8 мл ацетона по каплям, в течение 0,25 ч. Затем смесь нагревают до 80-100оС в течение 0,5 ч. После того, как смеси дадут остыть, образовавшийся серо-зеленый осадок выделяют посредством фильтрации.
В суспензию этого твердого вещества в воде при 80оС барботируют газообразный сероводород в течение 0,5 ч. Затем фильтруют в горячем состоянии для того, чтобы удалить твердый сульфид меди (I). После охлаждения до 25оС смесь экстрагируют хлористым метиленом. Затем объединенные органические фазы промывают рассолом, сушат над безводным сульфатом натрия, фильтруют и концентрируют, получая 26,4 г 4(5)-метил-2-пропилимидазола в виде вязкого оранжевого масла.
ЯМР- спектр (200 МГц, в пердейтерохлороформе) δ: 10,15 (ш.с. 1Н), 6,61 (синглет, 1Н), 2,64 (триплет, 2Н), 2,20 (синглет, 3Н), 1,72 (секстет, 2Н), 0,92 (триплет, 3Н).
Часть В.
Приготовление 4(5)-гидроксиметил-5(4)-метил-2-пропилимидазола.
Раствор 21,0 г 4(5)-метил-2-пропилимидазола, 14,0 г 37%-ного водного формальдегида, 76,0 г концентрированной соляной кислоты и 100 мл воды кипятят с обратным холодильником в течение 62 ч. После охлаждения смесь разбавляют водой. Образовавшийся водный раствор подщелачивают до рН 10, используя 10%-ный водный раствор гидроксида натрия и затем экстрагируют смесью 4: 1 хлороформ/изопропанол. Объединенные органические фазы промывают рассолом, сушат над безводным сульфатом натрия, фильтруют и концентрируют. После хроматографирования на колонке (элюирование смесью 10% метанола) хлороформ с 0,2% концентрированного аммиака) с последующей перекристаллизацией из этилацетата дает 13,9 г 4(5)-гидроксиметил-5(4)-метил-2-пропилимидазола с температурой плавления 138,5-139,5оС.
ЯМР-спектр (200 МГц, в пердейтеродиметилсульфоксиде) δ: 11,30 (ш.с. 1Н), 4,68 (ш.с. 1Н), 4,26 (синглет, 2Н), 2,46 (триплет, 2Н), 2,06 (синглет, 3Н), 1,60 (секстет, 2Н), 0,88 (триплет, 3Н).
Часть С.
Приготовление 4(5)-метил-2-пропилимидазол-5(4)-карбоксальдегида.
К раствору 12,1 г 4(5)-гидроксиметил-5(4)-метил-2-пропилимидазола в 200 мл уксусной кислоты при 25оС добавляют 170 мл 1,0-нормального раствора церийаммоний нитрата в воде по каплям в течение 1 ч. Образовавшийся раствор перемешивают в течение часа при 25оС и затем выливают в воду. Этот раствор доводят до значения рН 4, используя 10%-ный водный раствор гидроксида натрия и затем экстрагируют хлороформом.
Объединенные органические фазы промывают водой и рассолом, сушат над безводным сульфатом натрия, фильтруют и концентрируют. Неочищенный продукт подвергают перекристаллизации, получая 9,66 г 4(5)-метил-2-пропилимидазол-5(4)-карбоксальдегида с т.пл. 128-128,5оС.
ЯМР-спектр (200 МГц, в пердейтеродиметилсульфоксиде) δ: 12,49 (ш.с. 1Н), 9,69 (синглет, 1Н), 253 (триплет, 2Н), 2,38 (синглет, 3Н), 1,65 (секстет, 2Н), 0,87 (триплет, 3Н).
Часть D.
Приготовление 1-[(2'-трет-бутоксикарбонилдифенил-4-ил)метил] -4-метил-2-пропилимидазол-2-карбоксальдегида.
Раствор 3,60 г 4(5)-метил-2-пропилимидазол-5(4)-карбоксальдегида, 8,64 г трет-бутил-4' -бромметилдифенил-2-карбоксилата, 6,54 г безводного карбоната калия и 60 мл диметилформамида перемешивают 18 ч при 25оС. Реакционную смесь фильтруют, и фильтрат разбавляют водой и затем экстрагируют этилацетатом. Объединенные органические экстракты фильтруют и концентрируют. После хроматографической очистки на колонке, элюируемой смесью этилацетат/бензол, получают 6,31 г 1-[(2' трет-бутоксикарбонилдифенил-4-ил)метил] -4-метил-2-пропилимидазол-5-карбоксальдегида.
ЯМР-спектр (200 МГц, в пердейтерохлороформе) δ: 9,77 (синглет, 1Н), 7,78 (дублет, 1Н), 7,51-7,35 (мультиплет, 2Н), 7,27 (мультиплет, 3Н), 7,05 (дублет, 2Н), 5,59 (синглет, 2Н), 2,64 (триплет, 2Н), 2,50 (синглет, 3Н), 1,78 (секстет, 2Н), 1,20 (синглет, 9Н), 0,97 (триплет, 3Н).
Часть Е.
Приготовление 1-[(2' -карбоксидифенил-4-ил)метил]-4-метил-2-пропилимида- зол-5-карбоксальдегида.
Это вещество получают в соответствии с методикой, описанной в примере 9, часть С, из 4,20 г 1-[(2' -трет-бутоксикарбонилдифенил-4-ил)метил]-4-метил-2-пропилимида- зол-5-карбоксальдегида получают 0,92 г 1-[(2'-карбоксидифенил-4-ил)-метил)-4-метил -2-пропилимидазол-5-карбоксальдегида с т.пл. 243-245оС.
ЯМР-спектр (200 МГц, в пердейтеродиметилсульфоксиде), δ 12,77 (ш.с. 1Н), 9,75 (синглет, 1Н), 7,71 (дублет, 2Н), 7,55 (триплет, 1Н), 7,43 (триплет, 1Н), 7,36-7,27 (мультиплет, 3Н), 7,06 (дублет, 2Н), 5,59 (синглет, 2Н), 2,60 (триплет, 2Н), 2,41 (синглет, 3Н), 1,62 (секстет, 2Н), 0,86 (триплет, 3Н).
П р и м е р 52. Часть А.
Приготовление 1-[(2'-трет-бутоксикарбонилдифенил-4-ил)-метил] -5-гидроксиметил-4-метил-2-пропилимидазола.
К раствору 3,43 г -[(2'-трет-бутоксикарбонилдифенил-4-ил)-метил] -4-метил-2-пропилимидазол-5-карбоксальдегида (из примера 51, часть D) в 22 мл метанола и 22 мл тетрагидрофурана при 25оС добавляют несколькими порциями 3,09 г боргидрида натрия. Реакционную смесь перемешивают при 25оС в течение 1,5 ч и затем выливают в разбавленный водный раствор гидроксида натрия. После перемешивания в течение 0,2 ч при 25оС этот раствор экстрагируют хлороформом. Объединенные органические фазы промывают водой и рассолом, сушат над безводным сульфатом натрия, фильтруют и концентрируют. Хроматографирование на колонке, элюируемой смесью этилацетат/бензол, обеспечивает 3,32 г 1-[(2' -трет-бутоксикарбонилдифенил-4-ил)-метил] -5-гидроксиметил-4-метил-2-пропилимидазола.
ЯМР-спектр (200 МГц, в пердейтерохлороформе) δ: 7,76 (д, 1Н), 7,42 (м, 2Н, 7,28-7,24 (мультиплет, 3Н), 6,96 (дублет, 2Н), 5,24 (синглет, 2Н), 4,47 (синглет, 2Н), 2,56 (триплет, 2Н), 2,21 (синглет, 3Н), 1,71 (секстет, 2Н), 1,25 (синглет, 9Н), 0,95 (триплет, 3Н).
Часть В.
Приготовление 1-[(2'-карбоксидифенил-4-ил)метил] -5-гидроксиметил-4-метил-2-пропилимидазол гидрохлорида.
Раствор 3,32 г 1-[(2'-трет-бутоксикарбонилдифенил-4-ил)-метил] -5-гидроксиметил-4-метил-2-пропилимидазола в 100 мл 10%-ной водной соляной кислоты перемешивают 16 ч при 25оС. Затем растворитель и избыток соляной кислоты удаляют в вакууме, получая 2,22 г 1-[(2' -карбоксидифенил-4-ил)метил]-5-гидроксиметил-4-метил-2-про- пилимидазол гидрохлорида с т. пл. 208-210оС. (разлагается).
ЯМР-спектр (200 МГц, в пердейтеродиметилсульфоксиде) δ: 12,92 (ш.с. 1Н), 7,74 (дублет, 1Н), 7,58 (триплет, 1Н), 7,47 (триплет, 1Н), 7,34 (мультиплет, 3Н), 7,26 (дублет, 2Н), 5,67 (ш.с. 1Н), 5,53 (синглет, 2Н), 4,42 (синглет, 2Н), 2,86 (триплет, 2Н), 2,30 (синглет, 3Н), 1,54 (секстет, 2Н), 0,83 (триплет, 3Н).
Вещества табл. 5 были получены или могут быть получены по методикам примеров 51-52.
П р и м е р 55.
Часть А.
Приготовление 2-н-пропил-4,5-дикарбометоксиимидазола-2-н-пропилимидазол-4,5-дикарбоновую кислоту.
Т. пл. 257оС (с разл.) (17,14 г, 86,6 ммоль, 1 экв.) 400 мл метанола и хлористый ацетил (38,1 мл, 534 миллимоля, 6 экв.) осторожно смешивают (добавление хлористого ацетила к метанолу протекает весьма экзотермично) и кипятят с обратным холодильником в течение ночи. Растворитель удаляют в вакууме и добавляют 100 мл воды и 10 н. раствор гидроксида натрия до установления рН 7. Водную смесь экстрагируют трижды этилацетатом, органические слои объединяют, сушат сульфатом магния и растворитель удаляют в вакууме, получая 12 00 г белого твердого вещества. При перекристаллизации из смеси гексан/этилацетат получают 11,41 г белого твердого вещества. Т.пл. 162-164,5оС.
ЯМР-спектр (в пердейтерохлороформе) δ 3,95 (синглет, 6Н), 2,78 (триплет, 2Н), 1,83 (триплет трипл. 2Н, J=7,7 Гц) 0,87 (триплет 3Н, J=7 Гц).
Вычислено, С 52,06; Н 6,28; N 12,14.
С10Н14N2О4 (Н2О)0,25
Найдено, С 52,06; Н 6,17; N 12,49.
Часть В.
Приготовление 1-[(2'-карбометоксидифенил-4-ил)метил] -4,5-дикарбометокси-2-н-пропилимидазола. 2-н-Пропил-4,5-дикарбометоксиимидазол (2,00 г, 8,8 ммоль, 1 экв.) алкилируют 4' -бромметил-2-карбометоксидифенилом (2,70 г, 8,8 ммоль, 1 экв.) по известной методике.
Получают 3,87 г желтого масла, которое пригодно для дополнительного превращения.
ЯМР-спектр (в пердейтеродиметилсульфоксиде) δ: 7,84-7,22 (мультиплет, 4Н), 7,22 (дублет, 2Н, J=9 Гц), 7,13 (дублет, 2Н, J=9 Гц), 5,50 (синглет, 2Н), 3,77 (синглет, 3Н), 3,75 (синглет, 3Н), 3,55 (синглет, 3Н), 2,67 (триплет, 2Н, J=7 Гц), 1,67 (триплет, кварт. 2Н, J=7,7 Гц), 0,88 (триплет, 3Н, J=7 Гц).
Часть С.
Приготовление 1-[(2' -карбоксидифенил-4-ил)метил]-имидазол-4,5-дикарбоно-вой кислоты.
Триэфир из части В омыляют по известной методике.
Образовавшуюся стеклообразную массу кристаллизуют из хлороформа. Т.пл. 143оС (усадка) 152,0оС (разложение).
ЯМР-спектр (в пердейтеродиметилсульфоксиде) δ: 12,74 (мультиплет, 1Н), 7,72 (дублет, 1Н, J=9 Гц), 7,56 (триплет, 1Н, J=9 Гц), 7,46 (триплет, 1Н), J=9 Гц), 7,36 (дублет, 1Н, J=9 Гц), 7,30 (дублет, 2Н, J=9 Гц), 7,20 (дублет, 2Н, J=9 Гц), 5,99 (синглет, 2Н), 2,89 (триплет, 2Н, J=7 Гц), 1,48 (триплет, кварт. 2Н, J=7,7 Гц), 0,80 (триплет, 3Н, J=7 Гц).
Вычислено, С 60,68; Н 5,32; N 6,43.
С22Н20N2О6 (Н2О)1,5
Найдено, С 60,99; Н 5,71; N 6,50.
Вещества табл. 7 могут быть получены по методу, описанному в примере 55.
П р и м е р 59. Приготовление 4,5-дикарбометокси-2-н-пропил-1-[(2' -(1Н-тетразол-5-ил)дифенил-4-ил)метил] имидазол. 4,5-Дикарбометокси-2-н-пропил-1-[(2'-N -трифенилметил(1Н-тетразол-5-ил)дифе- нил-4-ил)метил]имидазол приготовлен по методике примера 55. Часть В. т.пл. 124-125,5оС, из 4' -бромметил-2-(N-трифенилметил(1Н-тетразол-5-ил)дифенил) (3,00 г) смешивают и кипятят с обратным холодильником в 50 мл метанола в течение 4 ч. Растворитель удаляют в вакууме, и остаток сразу же подвергают флеш-хроматографированию на силикагеле в смеси 1: 1 гексан (этилацетат и в 100%-ном этаноле. Получают 1,30 г белой стеклообразной массы, которую затем перемешивают с эфиром, что дает 0,92 г белого твердого вещества с т.пл. 100оС (медленно разлагается).
ЯМР-спектр (в пердейтеродиметилсульфоксиде) 7,68-7,43 (мультиплет, 4Н), 7,08 (дублет, 2Н, J=9 Гц), 6,96 (дублет, 2Н, J=9 Гц), 5,41 (синглет, 2Н), 3,80 (синглет, 3Н), 3,74 (синглет, 3Н), 2,63 (триплет, 2Н, J=7 Гц), 1,62 (триплет кварт. 2Н, J=7,7 Гц), 0,88 (триплет, 3Н, J=7 Гц).
Вычислено, C 59,13; Н 5,58; N 17,23.
С24Н24N6О4 (Н2О)1,5.
Найдено, C 59,27; Н 5,31; N 17,11.
Полезность.
Гормональный ангиотенсин П ( АП) производит многочисленные биологические отклики (например, сжатие сосудов) путем стимулирования его рецепторов на клеточных мембранах. С целью идентификации соединений, таких, как антагонисты АП, которые способны взаимодействовать с рецептором АП, для начального отбора было использовано испытание на лиганд-рецепторное связывание. Это испытание проводили по методу, описанному Глоссманном и сотр. но с некоторыми изменениями. Реакционная смесь содержала адренально-кортикальные микросомы крысы (источник рецептора А11) в буферном растворе Трис и 2 наномоля 3Н-АП с потенциальным АП антагонистом или без него. Эту смесь выдерживали 1 ч при комнатной температуре, причем, реакцию последовательно прерывали путем быстрой фильтрации и промывания на стеклянном микроволокнистом фильтре. Рецепторно связанный 3Н-АП, уловленный на фильтре, был определен количественно с помощью сцинтилляционного счетчика. Ингибирующая концентрация (ИК50) потенциального АП антагониста, которая дает 50%-ное замещение всего специфически связанного 3Н-АП, представлена в качестве меры сродства такого соединения для АП-рецептора (см. таблицу 7).
Потенциальные противогипертонические эффекты соединений этого изобретения могут быть продемонстрированы путем введения соединений бодрствующим крысам, которые сделаны гипертоническими посредством связывания левой почечной артерии. По этой методике давление крови увеличивается за счет увеличения производства ренина с последующим увеличением уровня АП. Вещества вводились перорально в дозе 100 мг/кг и/или внутривенно, через полую трубочку в яремную вену в дозе 10 мг/кг. Артериальное давление крови непрерывно измеряли непосредственно через полую трубку из каротидной артерии и записывали, используя преобразователь давления и многоканальный самописец. Уровень давления крови после введения вещества сравнивался с уровнем до введения, чтобы определить противогипертонический эффект вещества (см. табл. 8).
Хотя многие из испытанных веществ не были активны перорально, они обладали активностью при внутривенном введении. Некоторые вещества не давали значительного уменьшения давления крови при внутривенной дозировке 10 мг/кг, но обеспечивали некоторое уменьшение давления при такой дозе, и поэтому можно ожидать, что они будут активными внутривенно при повышенной дозе, например, 30 мг/кг.
NT не испытывалось.
Вещества, перечисленные в табл. 9, были испытаны таким же образом, как описано для табл. 8, с тем исключением, что при испытании на противогипертоническое действие в почках гипертонических крыс вещества вводились перорально в дозе 30 мг/кг и внутривенно в дозе 3 мг/кг.
В табл. 10 даны значения ЭД30 для некоторых соединений. Значения ИК50 и/или ЭД30 большинства соединений превышают соответствующие значения известных соединений, полученных в примерах 7 и 20.
Таким образом, соединения формулы I обладают высокой активностью в качестве антагонистов к рецепторам ангиотензина П и противогипеpтонической активностью и могут быть использованы в медицине.
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения производных имидазола | 1989 |
|
SU1814646A3 |
| Способ получения азолов | 1989 |
|
SU1709907A3 |
| ЗАМЕЩЕННЫЕ 1,2,3-ТРИАЗОЛЫ | 1992 |
|
RU2076102C1 |
| АРАЛКИЛЗАМЕЩЕННЫЕ ИМИДАЗОЛЫ, ОБЛАДАЮЩИЕ ПРОТИВОГИПЕРТОНИЧЕСКИМ ДЕЙСТВИЕМ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЙ СОСТАВ, ОБЛАДАЮЩИЙ ИНГИБИРУЮЩЕЙ АКТИВНОСТЬЮ В ОТНОШЕНИИ АНГИОТЕНЗИНА II | 1990 |
|
RU2067581C1 |
| ЗАМЕЩЕННЫЕ ИМИДАЗОЛЫ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1991 |
|
RU2099330C1 |
| ПРОИЗВОДНЫЕ ТЕТРАЗОЛА | 1992 |
|
RU2091376C1 |
| Способ получения @ , @ -дизамещенных ароматических и гетероароматических соединений | 1988 |
|
SU1750425A3 |
| ФТОРОАЛКОКСИАМИНОТРИАЗИНЫ | 1992 |
|
RU2047607C1 |
| Способ получения 2-замещенных 1-нафтолов | 1986 |
|
SU1600627A3 |
| Способ получения производных имидазола или их фармацевтически приемлемых солей | 1987 |
|
SU1694062A3 |
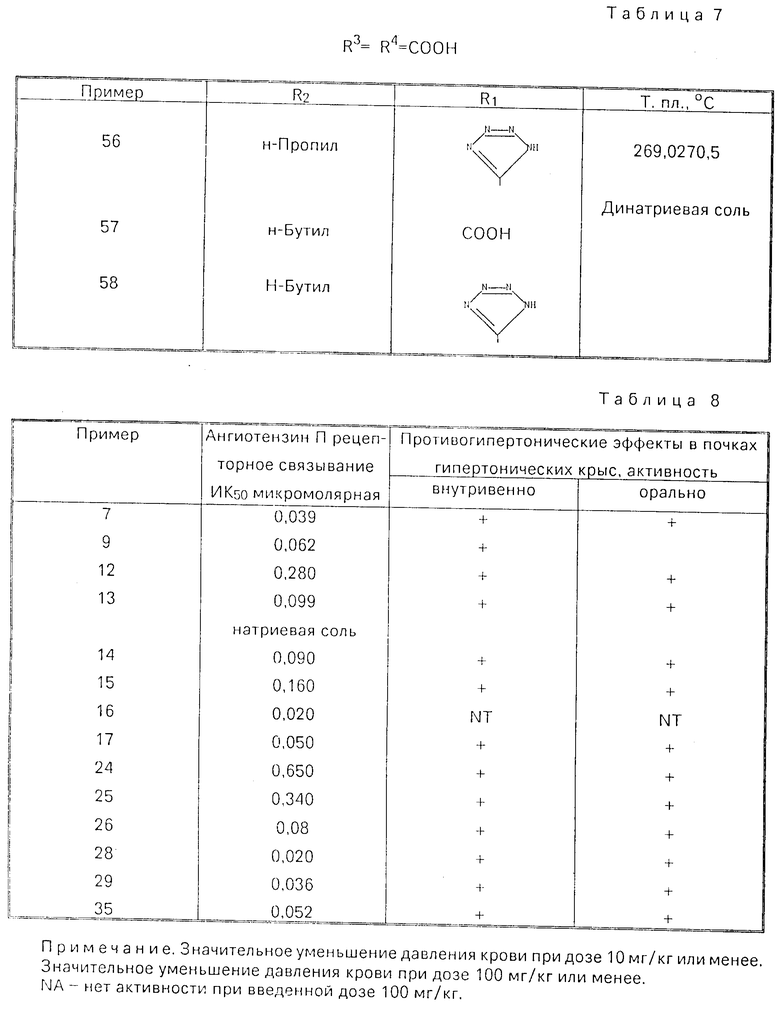
Использование: в медицине в качестве ингибиторов действия гормона ангиотензина. Сущность изобретения: продукты - производные имидазола ф-лы I, где R1 -COOH, или  , R2 - H-C3H7 или H-C4H9 , R3- Cl,CF3,C2F5,C6H5 или COOH, R4 - COOH, CHO или CH2OH , при условии, что а) когда R4-CH2OH , то R3-C2F5 и R2-H-C3H7 , б) когда R3 - COOH, то R4 - тоже является COOH, в) когда R2-H-C3H7 , R3-C2F5 и R4 - COOH, то R1 - является группа
, R2 - H-C3H7 или H-C4H9 , R3- Cl,CF3,C2F5,C6H5 или COOH, R4 - COOH, CHO или CH2OH , при условии, что а) когда R4-CH2OH , то R3-C2F5 и R2-H-C3H7 , б) когда R3 - COOH, то R4 - тоже является COOH, в) когда R2-H-C3H7 , R3-C2F5 и R4 - COOH, то R1 - является группа  Реагент 1: соединение ф-лы 2, где R2 - имеет указанные значения, R5-Cl, CF3,C2F5, C6H5 или COO( C1-C4 -алкил), R6-COO(C1-C4) - алкил), CH2OH или CHO Реагент 2: соединение ф-лы 3, где Х -галоген, п-толуолсульфонилокси или метилсульфонилокси, R7-COO(C1-C4) -алкил), CN или
Реагент 1: соединение ф-лы 2, где R2 - имеет указанные значения, R5-Cl, CF3,C2F5, C6H5 или COO( C1-C4 -алкил), R6-COO(C1-C4) - алкил), CH2OH или CHO Реагент 2: соединение ф-лы 3, где Х -галоген, п-толуолсульфонилокси или метилсульфонилокси, R7-COO(C1-C4) -алкил), CN или 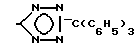 Условия реакции: в среде растворителя в присутствии основания при температуре от 20°С до температуры кипения растворителя. 10 табл.
Условия реакции: в среде растворителя в присутствии основания при температуре от 20°С до температуры кипения растворителя. 10 табл.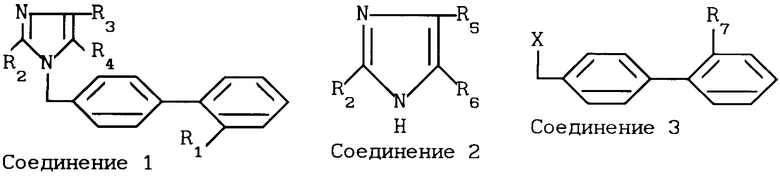
ПРОИЗВОДНЫЕ ИМИДАЗОЛА общей формулы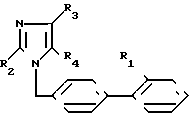

где R1 CO2H или группа ,
,
R2 н-пропил или н-бутил;
R3-Cl, -CF3, -C2F5, фенил или CO2H;
R4-CO2H, -CHO или -CH2OH,
при условии, что: а) когда R4-CH2OH, то R3-C2F5, R2-н-пропил;б) когда R3-CO2H, то R4 тоже CO2H; в) когда R2-н-пропил, R3-C2F5 и R4-CO2H, то R1
группа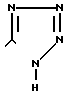
Приоритет по признакам:
07.01.88 при R1 CO2H или группа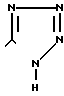
R2 H-пропил или н-бутил,
R3-Cl, -CF3 или -C2F5,
R4-CO2H, -CHO или -CH2OH;
06.12.88 при R3-CO2H или фенил.
R1, R2 и R4 имеют указанные значения.
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Способ получения производных имидазола или их солей | 1980 |
|
SU999966A3 |
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| Гребенчатая передача | 1916 |
|
SU1983A1 |
