Изобретение относится к аналитическому контролю жидкофазных материалов, в частности к количественному и качественному анализу элементного состава примесей в жидких органических и неорганических веществах, используемых в технологии силовых полупроводниковых приборов и электротехнических изделий.
Методами оптической спектроскопии можно анализировать твердые объекты при условии их предварительного перевода в растворенное состояние и таким образом подготовленная проба после ряда последующих операций пробоподготовки подвергается анализу элементного и примесного состава [1].
Наиболее близким к изобретению является способ определения примесей в жидких пробах методом твердотельного анализа [2], включающий осаждение примеси из раствора на твердую подложку из пористого кремния путем погружения в анализируемый раствор и проведение анализа масс-спектрометрическими методами с различными источниками возбуждения.
Время погружения пористого кремния в анализируемый раствор составляет 1-2 мин. За это время происходит сорбция примеси из раствора, и концентрация ее в слое пористого кремния (СПК) примерно равна концентрации примесей в растворе. Поэтому данный способ не пригоден для анализа сверхразбавленных проб с концентрацией примеси С ≅10-5%, так как это ниже предела обнаружения большинства твердотельных методов анализа.
Целью изобретения является повышение чувствительности способа за счет использования ионно-обменного концентрирования примесей из сильно разбавленных растворов.
Это достигается тем, что в способе определения примесей в жидких пробах методом твердотельного анализа, включающем осаждение примеси из раствора на твердую подложку из пористого кремния путем погружения в анализируемый раствор и проведение анализа масс-спектрометрическими методами с различными источниками возбуждения подложку погружают в пробу анализируемого раствора на 1,5-2 ч.
Предлагаемый способ анализа жидких проб позволяет без использования классических способов пробоподготовки и концентрирования проводить анализ жидких проб в широком диапазоне концентраций с одновременной регистрацией всего спектра неорганических примесей на оборудовании, предназначенном для анализа твердофазных объектов. Режим длительной экспозиции (1,2-2 ч) подложки с СПК позволяет анализировать растворы с концентрацией (10%-10-7%) примесных элементов ниже предела обнаружения методов анализа.
Сущность изобретения заключается в следующем. При электрохимическом анодировании кремниевых подложек в растворах HF плотностями тока i = 5-150 мА/см2 на их поверхности формируется СПК, толщиной 3-200 мкм и пористостью 10-75% , которые определяются типом проводимости, степенью легирования, i и временем анодирования (τ ). Высокопористые (40-75%) СПК с удельной поверхностью микропор 100-200 м2/см3, диаметром однородно распределенных и сообщающихся микропор 30-300  и их поверхностной плотностью 1010-1011 см-2 формируются на р- и n+-кремнии. При погружении СПК в анализируемый раствор, имеющиеся в растворе примеси практически всех металлов и неметаллов могут сорбироваться СПК в количестве, соответствующем абсорбированному микрообъему раствора, а также ионообменной адсорбции примесей на гидроксилированной поверхности микропор. Гидроксилирование (гидратация) происходит благодаря реакции гетеролитического расщепления дисилоксановых связей Si-O-Si тонкой окисленной пленки (20-30
и их поверхностной плотностью 1010-1011 см-2 формируются на р- и n+-кремнии. При погружении СПК в анализируемый раствор, имеющиеся в растворе примеси практически всех металлов и неметаллов могут сорбироваться СПК в количестве, соответствующем абсорбированному микрообъему раствора, а также ионообменной адсорбции примесей на гидроксилированной поверхности микропор. Гидроксилирование (гидратация) происходит благодаря реакции гетеролитического расщепления дисилоксановых связей Si-O-Si тонкой окисленной пленки (20-30  ) на стенках микропор:
) на стенках микропор: =
=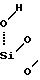 Si = ___→
Si = ___→  i-O-
i-O- i (1) Обменная адсорбция катионов примеси в СПК, как и в случае силикагелей, протекает с участием поверхностных силанольных групп Si-OH
i (1) Обменная адсорбция катионов примеси в СПК, как и в случае силикагелей, протекает с участием поверхностных силанольных групп Si-OH
≡ Si-OH ≡ Si - O-+ H++ Men+__→ ≡ (Si-O-)nM+n +nH+ (2)
≡ Si - O-+ H++ Men+__→ ≡ (Si-O-)nM+n +nH+ (2)
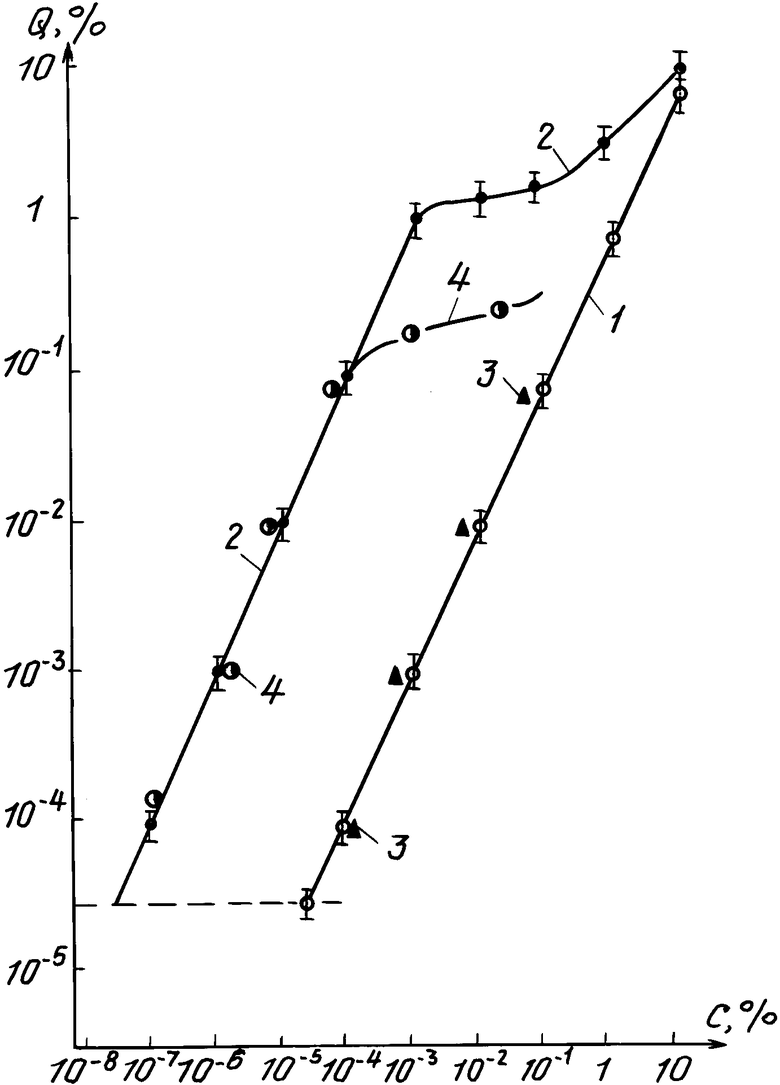
На чертеже приведена зависимость содержания примеси в СПК относительно его массы (Q, %) от концентрации примеси в растворе (С, %).
Кривая 1 - при сорбционной окклюзии (поглощении) микрообъема раствора меди в условиях кратковременной экспозиции СПК в растворе в течение 1-1,5 мин (известный способ).
Кривая 2 - при ионообменном адсорбционном концентрировании меди в условиях длительной выдержки СПК в растворе меди в течение 1,5-2 ч (предлагаемый способ).
Кривая 3 - условия экспозиции соответствуют кривой 1, раствор содержит примеси Na, K, Ca, Mg, Al, Ni, Fe, Mn, Cd в количестве 0,1% каждого элемента (суммарная концентрация всех примесей 1%) - по известному способу.
Кривая 4 - условия экспозиции соответствуют кривой 2, состав раствора содержит те же примеси, что и на кривой 3 (по предлагаемому способу).
Кривые 1 и 3 аналогичны кривым, полученным по известному способу из [2] .
Проверка и экспериментальное обоснование предлагаемого способа были проведены двумя методами: лазерным масс-спектрометрическим и атомно-абсорбционным путем анализа растворов при раздельном (медь) или одновременном присутствии примесей (Na, Cu, Mg, Fe) в интервале концентраций C = 10-7 - 10% в условиях кратковременной сорбции СПК микрообъема раствора (С = 10-4 - 10%) и длительного адсорбционного концентрирования примеси (примесей) в СПК (С = 10-8 - 10-5%), где С - концентрация примеси в растворе.
Для этого брали 10%-ный раствор меди и десятикратным разбавлением аликвоты приготовляли растворы с концентрацией меди С = 10-7 - 10%. Приемлемыми для сорбции и концентрирования примеси из растворов (в общем случае для любых примесей) являются СПК с пористою Р = 0,4-0,7 и h = 30-200 мкм, но наиболее оптимальные и надежные результаты получаются с СПК с Р = 0,6 и h = 100 мкм благодаря тому, что такой СПК обладает достаточно высокой сорбционной и адсорбционной емкостью и вследствие этого позволяет охватить широкий интервал концентрации примеси. Введение меди в СПК с Р = 0,6 и h = 100 мкм осуществляли двумя режимами.
По известному способу [2] в каждый из растворов с любым объемом с С = 10-4 - 10% с помощью фторопластового держателя погружали на 1-2 мин пластины с СПК размером 10х10 мм, которые предварительно были просушены на воздухе. После быстрого извлечения и осушения фильтром проводили количественный анализ меди в СПК указанными выше методами по общепринятой методике.
В случае атомно-абсорбционного метода анализа пробоподготовка заключалась в растворении 10-15 мкм СПК в малом объеме (1 мл) травителя HF : HNO3 (1: 17 об.ч.), нейтрализации, ИК-сушке досуха и приливании в фторопластовую чашечку 1-2 мл деионизованной воды. Регистрируемые аналитические сигналы сопоставлялись с калибровочными кривыми. При непосредственном анализе СПК на лазерном масс-анализаторе ЭМАЛ-2 калибровочная кривая была построена с использованием СПК с такими же параметрами с точно заданными в СПК количествами меди, что исключило введение поправочного коэффициента, связанного с матричным эффектом. Приведенная на фигуре зависимость содержания меди Q в СПК от концентрации С детектируемой примеси в растворе в интервале n˙10-5 - 10% (кривая 1) показывает, что нижний предел С и соответствующее значение Q совпадает с пределом чувствительности масс-спектрометра ЭМАЛ-2 и в силу этого анализ растворов с С ≥ n˙10-5% (n < 5) без операции длительного концентрирования не имеет смысла.
Вторая серия анализов была проведена с образцами СПК по предлагаемому способу, когда образцы выдерживались в подкисленной до рН 3-4 деионизованной воде для гидроксилирования окисленных стенок микропор по реакции (1). Образцы (10х10 мм) погружали в 10 мл растворов и выдерживали не менее τ = 1,5-2 ч, в течение которых происходит полное адсорбционное насыщение активных силанольных групп по реакции (2). При τ < 1-1,5 ч процесс концентрирования полностью не завершается, особенно в сильно разбавленных растворах, а при tau>> 2 ч все связи полностью заняты. Результаты анализа представлены кривой 2.
В интервале С = 10-7 - 10-3%, как и в случае кратковременной окклюзионной сорбции (кривая 1), наблюдается линейная зависимость между Q и С, причем концентрация раствора, соответствующая пределу чувствительности масс-анализатора ЭМАЛ-2 (Q = n˙10-5%), составляет C≈n˙ 10-8%.
Следовательно, в условиях длительной экспозиции СПК в 10 мл жидкой пробы нижний предел концентрации примеси в растворе на три порядка ниже, чем при кратковременной окклюзионной сорбции СПК микрообъема раствора. Однако, начиная с величины С ≈10-3%, которая соответствует максимальной адсорбционной емкости СПК, количество меди Q в СПК значительно слабее возрастает при дальнейшем увеличении С, и лишь при С > 10-1% рост Q для двух серий опытов (кривые 1 и 2) с различными режимами концентрирования практически совпадают благодаря сравнимости количеств меди, введенного в СПК по механизму простой сорбции и ионообменной адсорбции.
В случае группового содержания режимы введения примесей в СПК были такими же, как и при индивидуальном содержании меди. Исходный раствор содержал Na, K, Ca, Mg, Al, Ni, Fe, Mn, Cd в количестве 0,1% каждого элемента (суммарная концентрация 1%). Разбавлением аликвоты этого раствора готовились растворы с концентрацией каждой примеси в интервале 10-7 - 10-1%. Результаты анализа на лазерном масс-спектрометре ЭМАЛ-2 с использованием образцов сравнения на основе СПК с содержанием каждой из указанных примесей 10-1, 10-3, 10-4% при их совместном присутствии в СПК представлены кривыми 3 и 4. В согласии с данными, приведенными на фигуре для меди, зависимость Q от С каждой детектируемой примеси при кратковременном контакте пластинки с СПК с раствором прослеживается в интервале С = 10-4 - 10-1%, и практически при тех же величинах Q и С, как и в случае наличия в растворе только примеси меди. В условиях же длительной экспозиции СПК в 10 мл растворов прямая зависимость между Q и С имеет место в интервале С ≅ 10-4%, а при С > 10-4% содержание каждой примеси при их групповом присутствии слабо зависит от С. Смещение верхнего предела концентрации С каждой примеси с 10-3 до 10-4% по сравнению с условиями, когда в растворе имеется только одна примесь, обусловлено тем, что общая концентрация всех примесей с С = 10-4% каждой из них, составляет 10-3%, при которой происходит насыщение адсорбируемости СПК при полной ионнообменной адсорбции из раствора всех примесей.
С изменением параметров СПК (h, Р) в сторону увеличения или уменьшения от h = 100 мкм и Р = 0,6 кривая 4 будет смещаться влево-вправо вдоль оси С.
П р и м е р. Ход анализа раствора с неизвестными примесями и их содержанием заключается в следующем. Первоначально для определения спектра примесей и получения информации о концентрационном интервале одну просушенную пластину (10х10 мм) с СПК (Р = 0,6, h = 100 мкм) погружают на 1-2 мин в пробу раствора с нефиксированным объемом и затем проводят описанные операции. Вторую пластину с такими же параметрами СПК извлекают из подкисленной до рН 3-4 деионизованной воды и приводят в контакт с 10 мл анализируемого раствора на 1,5-2 ч и по истечении этого срока повторяют указанные выше операции. Оба образца анализируют на лазерном масс-спектрометре ЭМАЛ-2 по общепринятой методике.
По результатам расшифровки масс-спектров делают заключение о числе детектируемых примесей в обоих образцах. Что касается количественной стороны при раздельном или групповом присутствии примесей в растворе, то анализ основывается на данных рассмотренной фигуры, исходя из следующих возможных случаев.
Примесь (примеси) детектируются в СПК при двух режимах его контакта с раствором в таких количественных соотношениях, что величина Q-содержания в СПК в случае длительной экспозиции образца в растворе практически на три порядка больше, чем при кратковременном погружении. Это свидетельствует о том, что концентрация индивидуальной примеси или суммарная концентрация примесей в растворе С ≅10-3% и в силу этого в основу количественной расшифровки результатов следует положить данные, представленные кривыми 2 и 4 и соответствующие условиям полного ионнообменного концентрирования примеси при длительном контакте СПК с 10 мл анализируемого раствора.
Количество примеси (примесей) Q в СПК для двух режимов различаются только на порядок - два, что возможно только в случае, когда концентрация примеси (примесей) в растворе С ≥10-3%. На этом основании количественный анализ пробы независимо от его объема проводят с учетом данных, представленных кривыми 1 и 3 и полученных в условиях кратковременного (1-2 мин) погружения СПК в анализируемый раствор.
Содержание примеси (примесей) детектируемой в соответствии с пределом чувствительности лазерного масс-спектрометра (n ˙10-5%) только в случае длительной экспозиции образца в растворе, что реализуется согласно кривой 2 и 4 при концентрации примеси (примесей) в растворе в интервале n ˙ 10-8 ≅ С ≅ (10-4 - 10-3)%.
В качестве конкретного объекта для анализа использовалась дистиллированная вода до ее поступления в систему окончательной очистки ионообменными смолами. Пластину с СПК (10х10 мм, Р = 0,6, h = 100 мкм) извлекали из стакана с деоинизованной водой (рН 3-4), которая была предварительно подвергнута тщательной очистке от примесей путем длительного контакта с пластиной кремния с пористым слоем площадью 5-6 см2, опускали в тефлоновый стаканчик с 10 мл анализируемой воды и выдерживали 1,5-2 ч. Затем пластину переносили на фильтр, осушали и поверхностный СПК-адсорбент подвергали анализу методом лазерной масс-спектрометрии (ЛМС). Параллельно проводился атомно-абсорбционный анализ (ААС) с предварительной операцией концентрирования путем упаривания 0,5 л воды до 5-10 мл в условиях относительно длительного ИК-нагрева.
Для построения калибровочных кривых для количественного определения содержания примесей лазерным масс-спектрометром ЭМАЛ-2 были использованы образцы сравнения на основе СПК с заданным содержанием каждой из детектируемых примесей (Na, K, Ca, Mg, Al, Fe, Co, Mn, Cr, Ni) в пористом слое 10-4, 10-3, 10-2%. Результаты анализа обнаруженных в дистиллированной воде примесей указанными методами приведены в табл.1.
Результаты в допустимых пределах хорошо согласуются.
Вторым объектом анализа был травитель на основе смеси HF (48%) - HNO3 (70% ) - СН3СООН (96%) в объемном соотношении 1:6:1 после нескольких операций мезатравления структур (число структур - 100, объем травителя в ванне - 8 л). Для анализа методом ЛМС отбирали в тефлоновый стаканчик аликвоту травителя объемом 5-10 мл и нейтрализовали NH4OH марки "ос.ч" до слабокислой реакции (рН 3-4), затем в 10 мл такого раствора погружали образец с СПК на 1,5-2 ч. В случае метода ААС - 150-200 мл травителя концентрировали упариванием досуха с последующим растворением сухого осадка в 2-3 мл деионизованной воды. Результаты анализа примесей в травителе после 3-4-х операций травления этими методами приведены в табл. 2.
Из данных таблицы видно, что, как и в случае анализа дистиллированной воды, результаты, полученные двумя методами, в пределах погрешности достаточно хорошо согласуются.
Необходимо отметить, что при анализе дистиллированной воды и травителя известным способом, когда пластину с СПК погружали в пробу на 1-2 мин, последующий анализ на ЭМАЛ-2 показал, что примесные элементы, содержащиеся в растворе, просто не детектировались. Это является отражением того факта, что концентрация сорбированных примесей в СПК в данном случае ниже предела обнаружения прибора.
Достоинство предлагаемого способа заключается в следующем:
расширение возможности в плане анализа жидких объектов методами, предназначенными для анализа твердофазных материалов;
возможность управляемого варьирования режимов экспозиции СПК для расширения анализируемых пределов концентрации примеси, выбора режима в зависимости от интервала концентрации примеси;
упрощение или полное исключение традиционных операций пробоподготовки;
возможность анализа широкого спектра элементов примесей при их индивидуальном или совместном присутствии.

Использование: изобретение относится к аналитическому контролю жидкофазных материалов, в частности к количественному и качественному анализу элементного состава примесей в жидких органических и неорганических веществах, используемых в технологии силовых полупроводниковых приборов и электромеханических изделий. Сущность изобретения: проводят осаждение примеси на твердую подложку из раствора путем ионообменного адсорбционного концентрирования. Подложку погружают в пробу анализируемого раствора на 1,5 - 2 ч. В качестве подложки используют монокристаллический кремний с поверхностным пористым слоем. После извлечения подложки из раствора проводят анализ примесей масс-спектрометрическими методами с различными источниками возбуждения. 1 ил, 2 табл.
СПОСОБ ОПРЕДЕЛЕНИЯ ПРИМЕСЕЙ В ЖИДКИХ ПРОБАХ методом твердотельного анализа, включающий осаждение примеси из раствора на твердую подложку из пористого кремния путем погружения в анализируемый раствор и проведение масс-спектрометрического анализа, отличающийся тем, что, с целью повышения чувствительности способа, при осаждении примеси подложку погружают в пробу анализируемого раствора на 1,5 - 2,0 ч.
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Способ создания образцов сравнения для лазерного и вторично-ионного масс-спектрометрического количественного анализа примесей на поверхности кремния и структур на его основе | 1990 |
|
SU1756827A1 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
