Изобретение относится к 4-замещенным антрациклинонам, используемым в качестве промежуточных соединений и получаемым из них антрациклингликозиды.
4-Замещенные антрациклиноны имеют формулу I в которой R представляет алкенильную или алкинильную группу с прямой или разветвленной цепью, имеющую до 4 атомов углерода.
в которой R представляет алкенильную или алкинильную группу с прямой или разветвленной цепью, имеющую до 4 атомов углерода.
Антрациклиноны формулы I представляют промежуточные соединения при получении противоопухолевых ант- рациклингликозидов. В изобретении прдеусматриваестя антрациклингликозидом формулы IХ где R имеет указанные значения;
где R имеет указанные значения;
R1 - атом водорода или гидроксильная группа, и их фармацевтически приемлемые солей. Предпочтительными кислыми аддитивными солями являются хлористоводородные соли.
Предпочтительными группами, которые могут быть выражены символом R, являются винильная (этильная), аллильная (пропенильная, например, 2' -пропенил) группа.
Предпочтительные соединения формулы I выбирают из группы, включающей 4-деметокси-4-этенил-дауномицинон, 4-диметокси-4-(2-пропенил)-дауномицинон. Предпочтительными соединениями формулы IХ являются 4-деметокси-4-этенил-дауномицин и его хлористоводородная соль.
Соединения общей формулы I получают из 4-сульфонилантрациклинов формулы II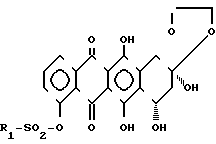 в которой R представляет алкильную группу, имеющую 1-10 атомов углерода, которая произвольно азмещена одним или несколькими атомами галогена или арильной группой, произвольно замещенной атомом галогена, алкильной, алкоксильной или нитрогруппой. Предпочтительными группами, которые могут быть выражены символами R ', являются трифторметил, 4-фторфенил и 4-толил. Соединения формулы I получают путем введения атома углерода или углеродной цеп в положение у атома С-4 в мягких условиях, которые в противном случае можно получить только в результате полного химического синтеза. Кроме того необходимо отметить, что ни одна из оставшихся функциональных групп не подвергается воздействию в ходе осуществления этой реакции и полностью сохраняется стереохимия в положениях у атомов С-7 и С-9.
в которой R представляет алкильную группу, имеющую 1-10 атомов углерода, которая произвольно азмещена одним или несколькими атомами галогена или арильной группой, произвольно замещенной атомом галогена, алкильной, алкоксильной или нитрогруппой. Предпочтительными группами, которые могут быть выражены символами R ', являются трифторметил, 4-фторфенил и 4-толил. Соединения формулы I получают путем введения атома углерода или углеродной цеп в положение у атома С-4 в мягких условиях, которые в противном случае можно получить только в результате полного химического синтеза. Кроме того необходимо отметить, что ни одна из оставшихся функциональных групп не подвергается воздействию в ходе осуществления этой реакции и полностью сохраняется стереохимия в положениях у атомов С-7 и С-9.
Способ получения 4-замещенного антрациклинона формулы I, включает взаимодействие 4-диметил-4-сульфони-13-диоксиланил-дауно- мицинона формулы II с:
(I) ненасыщенным соединением формулы IIIа
Н-R" в которой R" представляет алкенильную или алкинильную группу, имеющую до 16 атомов углерода, произвольно замещенную указанной группой от (а) до (е), или (II) металлоорганическим соединением общей формулы III b
М RnYm,
где М - атом металла; R имеет указанные значения; n и m могут иметь различные значения от 0 до 4, но не является О и Y может представлять атом галогена или алкильную группу с прямой или разветвленной цепью, имеющую 1-6 атомов углерода, в присутствии каталитического количества соединения формулы (IV) (далее определяется как катализатор):
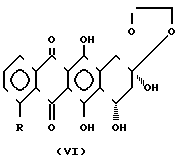
М' LрL'q (IV) в котором М ' представляет атом переходного металла, L и L' которые могут иметь одинаковые или различные значения, представляют такой анион, как Сl- или СН3СОО- , или такую нейтральную молекулу, как молекула растворителя, моно- или дифосфин, фосфит или диамин, а р и q могут иметь различные значения от 0 до 4, что позволяет получить соединение формулы VI где R имеет указанные значения, и удаление защитной 13-оксогруппы с помощью кислотного гидролиза.
где R имеет указанные значения, и удаление защитной 13-оксогруппы с помощью кислотного гидролиза.
В соединении формулы IV предпочтительными атомами переходного металла, которые могут быть выражены символом М' , являются атомы палладия или никеля. Предпочтительными группами, которые могут быть выражены символами L и/или L' , являются хелатообразующие дифосфины, такие как 1,3-дифенилфосфинопропан, 1,1' -бис-(дифенилфосфино)ферроцен и 1,2-бис[N-(1-фенилэтил), N-(дифенилфосфино)амино]этан. Обычно сумма m+n равняется по крайней мере 1, например 1,2,3 или 4. Поэтому соединение формулы IV представляет комплекс переходного металла, который включает атом прееходного металла, предпочтительно такой как атом палладия или никеля, и хелатообразователь, указанный выше. Молярное отношение атома переходного металла к хелатному лиганду обычно составляет от 1:1 до 1:4.
Соединения формулы (IIIа) представляют ненасыщенные соединения, способные вступать в реакцию Хека. Предпочтительными ненасыщенными молекулами, которые можно использовать при осуществлении этого способа, являютcя, молекулы, вступающие в реакцию Хека, в частности триметилсилилацетилен, фенилацетилен, алкилакрилаты и винилтриметилсилан.
Для соединений формулы (IIIb) предпочтительными атомами металлов, которые могут быть выражены символом М, являются атомы олова, цинка, кадмия и магния. Обычно n равняется 1,2,3 или 4, в то время как m может равняться 0,1,2,3 или 4. Сумма m и n зависит от валентности элемента М. Если y является атомом галогена, он может представлять атом хрома, брома или йода. Если y является алкильной группой, он может представлять метил.
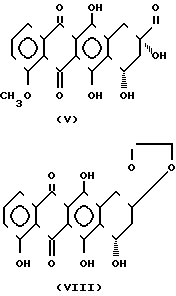
Соединения формулы II можно получить из природного дауномицинона V Таким образом настоящее соединение позволяет синтезировать соединения формулы I с хорошим выходом и высокой оптической и химической чистотой непосредственно из дауномицинона, как это суммируется в схеме реакции 1.
Таким образом настоящее соединение позволяет синтезировать соединения формулы I с хорошим выходом и высокой оптической и химической чистотой непосредственно из дауномицинона, как это суммируется в схеме реакции 1.

Способ по изобретению может быть осуществлен следующим образом.
Соединения формулы II растворяют в соответствующем полярном растворителе и добавляют в инертной атмосфере к раствору катализатора, который готовят заранее или "на месте" из соответствующих исходных веществ в присутствии ненасыщенного соединения IIIа, способного вступать в реакцию Хека, или металлорганического соединения формулы IIIb и вариантно в присутствии соответствующего основания, такого как триалкиламин. Температура реакции составляет 0-150оС, предпочтительно 30-100оС, и катализатор используется в молярном отношении к соединению II от 1:1 до 1:10000, предпочтительно от 1: 20 до 1:1000.
Полученные таким образом соединения общей формулы VI легко превращаются в конечные продукты I посредством кислотного гидролиза защитной группы у С-13 карбонила, например, с помощью трифторуксусной кислоты при 0оС в течение 45 мин. Сырое соединение формулы 1 можно очищать при помощи хроматографии на колонке из силикагеля с использованием в качестве системы для элюирования смеси хлороформа и ацетона (с процентным соотношением объемов 95: 5).
 _____→
_____→  H
H
Давно известен процесс арилирования ненасыщенных соединений на палладиевом катализаторе (реакция Хека) с использованием арилгалогенидов, и по этому вопросу было опубликовано много статей и патентов, касающихся применения этого процесса в органической химии.
В научной литературе сообщается о гороздо меньшем числе работ, посвященных применению арилсульфонатов в качестве арилирующих соединений. В течение несколькаих лет также известны способы сочетания с применением палладиевых или никелевых катализаторов арилгалогенидов или сульфонатов с металлорганическими соединениями.
Однако нигде не сообщалось о применении реакции обоих типов в химии антрациклинов, вероятно из-за наличия других интерферирующих функциональных групп. Проблемы, возникающие из-за наличия указанных групп, а именно ароматизация кольца А, образованные 7-деоксипроизводных, гидролиз 4-сульфонильных производных и/или модицикации хиноновой части, можно устранить в условиях осуществления настоящего изобретения.
Способ получения антрациклингликозида формулы IХ или его фармацевтически приемлевой соли, включает:
I взаимодействие 4-замещенного антрациклинона формулы I с галоидзамещенным сахаром формулы Х: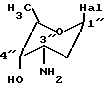 в которой Наl представляет атом галогена, 3" -аминогруппа является защищенной или незащищенной и 4" -гидроксильная группа является защищенной или незащищенной, и в случае необходимости удаление каждой защитной группы из полученного таким образом продукта, в результате чего достигается получение антрациклингликозида формулы (IХ), в которой R1представляет атом водорода,
в которой Наl представляет атом галогена, 3" -аминогруппа является защищенной или незащищенной и 4" -гидроксильная группа является защищенной или незащищенной, и в случае необходимости удаление каждой защитной группы из полученного таким образом продукта, в результате чего достигается получение антрациклингликозида формулы (IХ), в которой R1представляет атом водорода,
(II) превращение при желании полученного таким образом указанного гликозида офрмулы (IХ) в его фармацевтически приемлемую соль,
(III) бромирование при желании указанного гликозида формулы (IХ) или его фармацевтически приемлемых солей и гидролиз полученного таким образом 14-бромзамещенного производного с образованием соответствующего гликозида формулы (IХ), в которой R1 представляет гидроксильную группу и,
IV превращение при желании указанного гидрогликозида формулы IХ, в которой R1 представляет гидроксильную группу, в его фармацевтически приемлемую соль.
Антрациклинон формулы I предпочтительно подвергают взаимодействию с галоидзамещенным сахаром формулы Х на стадии I в инертном органическом растворителе в инертной атмосфере при 5-30оС и в присутствии трифторметансульфоната серебра, полученный антрациклингликозид при желании выделяют на стадии II в виде его хлористоводородной соли путем обработки раствором хлороводорода в метаноле, при желании осуществляют стадию III, в процессе которой производится бромирование и гидролиз полученного 14-бромзамещенного производного, после чего при желании образующийся антрацикликозид выделяют на стадии IV в виде его хлористоводородной соли путем обработки раствором хлористоводорода в метаноле.
Антрацикликозид формулы IХ, в которой R1 представляет атом водорода, получают в результате взаимодействия антрациклинона формулы I с галоидзамещенным сахаром формулы Х В формуле Х Наl обычно означает атом хлора. Если 3" -аминогруппа защищена, то защитной группой может быть трифторацетильная группа. Если 4" -гидроксильная группа защищена, то защитной группой также может быть трифторацетильная группа. Конденсация антрациклинона формулы I и галоидзамещенного сахара формулы Х обычно происходит в присутствии трифторметансульфоната серебра (трифлат).
Антрациклинон может растворяться в инертном органическом раствореле, таком, как метилендихлорид, при этом реакция осуществляется в инертной атмосфере, такой как раргон, при темпераутре 5-30оС, обычно при комнатной температуре. Любые защитные группы можно удалить с помощью мягкого щелочного гидролиза, например, путем обработки 0,1 н. водным раствором гидроокиси натрия. Антрациклингликозид предпочтительно выделяют в виде его хлористоводородной соли путем обработки свободного основания с помощью раствора хлороводорода с метаноле.
Антрациклингликозид формулы IХ, в которой R1 представляет атом водорода, или одну из его солей можно превратить в соответствующее доксорубициновое производное, в котором R1 представляет гидроксильную группу, путем бромирования в положении у 14-го атома и гидролиза 14-бромзамещенного производного водным растором формиата натрия.
В частности, гликозид формулы IХ, в которой R1 представляет атом водорода, или одну из его солей можно подвергать взаимодействию с бромом в хлороформе, что позволяет получить 14-бромзамещенное производное, из которого после гидролиза, осуществляемого при комнатной температуре, в течение 48 ч в атмосфере азота при использовании водного раствора формиата натрия, можно получить соединение формулы IХ, в которой R1представляет гидроксильную группу, в виде свободного основания, а затем выделить в виде его хлористоводородной соли путем обработки раствором НСl в безводном метаноле.
Соединения по изобретению являются полезными при терапевтическом лечении людей или животных. Они оказываются эффективными в качестве противоопухолевых средств. Больному вводится эффективное в терапевтическом отношении количество препарата. Можно вводить количество препарата, достаточное для ингибирования роста опухоли. Опухоль может представлять аденокарциному Колона или лейкоз Гросса.
П р и м е р 1. 4-Деметокси-4-этенилдауномицинон [(1), R=СН=СН2].
К раствору 1 г 4-диметил-4-трифторметансульфонил-13-диоксоланилдауномици-нона ((II), R =СF3) (1,78 ммоля) в 50 мл диоксана в инертной атмосфере последовательно добавляли 1,55 мл диизопропилэтиламина, 0,3 мл уксусной кислоты, 55 мг 1,1' -бис-(дифенилфосфино)-ферроцена (0,097 ммоля), 20 мг ацетата палладия (0,089 ммоля) и 3,52 г винилтриметил-силана (35,2 ммоля). Реакционную смесь перемешивали при 60оС в течение ночи, затем охлаждали до 0оС, подкисляли 10%-ной хлористоводородной кислотой и экстрагировали метиленхлоридом. Органическую фазу выпаривали до сухого состояния, в результате чего был получен сырой 4-диметокси-4-(2' -триметилсилил)-этенил-13-диоксоланилдавуномицинон [(VI), R=СН=СН-Si-(СН3)3)].
Спектр Н-ЯМР 300 МГц (в CDCl3): δ=0,24(9Н, с), 1,47 (3Н, с), 1,95 (1Н, двойной дублет, J= 5,0, 14,6 Гц), 2,44 (1Н, д J=14,6 Гц), 2,73 (1Н, д, J= 18,9), 3,18 (1Н, двойной дублет, J=2,5; 18,9), 3,29 (1Н, с), 3,8 (1Н, д, J= 6,6 Гц), 4,08 (4Н, С), 5,2 (1Н, т, J==-5,0 Гц), 6,32 (1Н, д, J=18,9 Гц), 7,69 (1Н, т J=7,7 Гц), 7,80 (1Н, двойной дублет J=1,0, 7,7 Гц), 7,96 (1Н, д, J= 18,9 Гц), 8,24 (1Н, двойной дублет, J=1,3, 7,7 Гц), 13,24 (1Н, с), 13,75 (1Н, с).
Ультрафиолетовый спектр (в этиловом спирте): λ =526,492, 359, 256, 214 нм, λмакс=256 нм.
Инфракрасный спектр (гранула КВr): ν=3480, 1612, 1585, 1575 см-1.
[α ]20D (с=0,1 в диоксане) = +179
Масс-спектр m/Z=510 (М+, основной пик).
Тонкослойная хроматография на пластине из кизельгеля F 254 (Мерк) с использованием смеси хлороформа и ацетона (9:1 по объему) Rf=0,72.
Сырой продукт [(VI), R=СН=СН-Si(СН3)3] перемешивали при температуре 0оС в 6 мл трифторуксусной кислоты и 0,4 мл воды в течение 45 минут. Реакционную смесь разбавляли 150 мл воды и экстрагировали метиленхлоридом. Органический слой промывали насыщенным раствором бикарбоната натрия и водой до достижения нейтрального состояния высушивали над сульфатом натрия и выпаривали до сухого состояния. Остаток хроматогафировали на силикагеле (с использованием смеси хлороформа и ацетона с объемным соотношением 95:5 в качестве элюента), что позволило получить 0,45 г [64% от соединения (II) R' = СF3] , 4-деметокси-4-этенилдауномицинона [(1), R=СН=СН2), высокоэффективная жидкостная хроматография 97,8%.
Анализ с помощью высокоэффективной жидкостной хроматографии:
Колонка: Мерк RР 18/7 мкм (250х4,2 мм)
Подвижная фаза:
А-0,01 М раствор гептансульфоната натрия и 0,02 М раствор фосфорной кислоты 6
Ацетонитрил 4
В-Метанол 7
Ацетонитрил 3
Градиент: от 20% В до 70% В в течение 25 минут.
Скорость растяжения: 1,5 мл/мин.
Детектор: ультрафиолетовый с длиной волны 254 нм.
Спектр 1Н - ЯМР 300 МГц (в СDСl3): δ=2,15/1Н, двойной дублет, J=4,8, 14,5 Гц), 2,35 (1Н, двойной триплет, J=2,0 14,5 Гц), 2,42 (3Н, с), 2,95 (1Н, д J=18,6 Гц), 3,20 (1Н, двойной дублет, J=2,0 18,6 Гц), 3,75 (1Н, д J= 5,7 Гц), 4,53 (1Н, с) 5,32 (1Н, м), 5,51 (1Н, двойной дублет, J=1,4, 11,0 Гц) 5,64 (1Н, двойной дублет, J=1,4 17,3 Гц) 7,74-7,92 (3Н, м), 8,37 (1Н, двойной дублет, J=2,0, 7,5 Гц), 13,28 (1Н, с), 13,71 (1Н, с).
Ультрафиолетовый спектр (в этиловом спирте): λ =525491, 356, 256, 212 нм, λмакс =256 нм.
Инфракрасный спектр (гранула КВr): ν=3480, 1712, 1610, 1575 см-1[α ] 20D/с=0,1 в диоксане)= +190. Масс-спектр m/Z=394 (М+, основной пик).
Тонкослойная хроматография на пластине из кизельгеля F254 (Мерк) с использованием смеси хлороформа и ацетона (9:1) по объему Rf=0,67.
П р и м е р 2. 4-Деметокси-4-этинилдауномицинон [(1), R=СН=СН2].
Эту реакцию осуществляли так же, как в примере 1, за исключением того, что в качестве лиганда для палладия использовали дифенилфосфинпропан (40 мг, 0,097 ммоля), что позволило получить сырой 4-деметокси-4-(2' -триметилсилил)этенил-13-диоксоланилдауномицинон (VI) R=СН=СН-Si(СН3)3].
Сырое соединение [(VI), R=СН-СН-Si(СН3)3], обрабатывали трифторуксусной кислотой, как это описывалось в примере 1, в результате чего после хроматографии на силикагеле (в качестве элюента использовали смесь хлороформа и ацетона с соотношением объемов 95:5) было получено 0,41 г [58,3% от соединения(II), R' =CF3]4-деметокси-4-этенилдауномицинона [(1), R=СН-СН2] высокоэффективная жидкостная хроматография 98,2%.
П р и м е р 3. 4-Деметокси-4-(2-метоксикарбонил)этенилдауномицинон [(1), R=СН-СН-СООСН3].
Эту реакцию осуществляли, так, как описывалось в примере 16, за исключением того, что в качестве реагента использовали метилакрилат (3,17 мл, 35,2 ммоля), что позволило получить сырой 4-деметокси-4-(2' -метоксикарбонил)этенил-13-диоксоланил дауномицинон [(VI) R=СН-СН1-СООСН3].
Спектр 1Н - ЯМР 300 МГц (В СDСl3): δ=1,48 (3Н, с), 1,98 (1Н: двойной дублет, J= 5,1 14,7 Гц), 2,46 (1Н, двойной триплет, J=2,0, 14,7 Гц), 2,79 (1Н, д, J=18,9 Гц), 3,24 (1Н; двойной дублет, J=2,1, 18,9, Гц), 3,24 (1Н; двойной дублет, J=2,1 18,9 Гц), 3,34 (1Н, с), 3,87 (4Н; с), 4,08 (4Н, с), 5,26 (1Н, двойной дублет, J=1,5; 4,9 Гц), 6,24 (1Н, д, J=15,9 Гц) 7,75-7,80 (2Н, м), 8,36-8,44 (1Н, м), 8,72 (1Н, д, J=15,9 Гц) 13,35 (1Н, с), 13,54 (1Н, с).
Ультрафиолетовый спектр (в этиловом спирте): λ=527, 492, 347, 264, 213 нм, λмакс=264 нм.
Инфракрасный спектр (гранула КВr): ν=3470, 1716, 1610, 1575 см-1.
[α ]20D/с=0,1 в диоксане)=+195.
Масс-спектр m/Z=496 (М+, основной пик).
Тонкослойная хроматография на пластине из кизельгеля F=254 (Мерк) с использование смеси хлороформа и ацетона (9:1 по объему), Rf=0,44.
Сырое соединение VI (R=СН=СН-СООСН3) обрабатывали трифторуксусной кислотой так, как описывалось в примере 1, в результате чего после хроматографии на силикагеле (в качестве элюента использовалась смесь хлороформа и ацетона с соотношением объемов 95:5) было получено 0,41 г (50,8% от соединения (II), R' =СF3 (4-деметокси-4-)2' -метоксикарбонил)этенилдауномицинона [(1), R=СН=СН-СООСН3]= (высокоэффективная жидкостная хроматография 97,8%).
Спектр 1Н-ЯМР 300 МГц (в (СDCl3): δ=2,16 (1Н, двойной дублет, J=5,0, 14,8 Гц), 2,36 (1Н, двойной триплет, J=2,2, 14,8 Гц) 2,43 (3Н, с), 2,96 (1Н, д, J=18,8 Гц), 3,21 (1Н, двойной дублет J=2,2, 18,8 Гц), 3,75 (1Н, д J= 1,2 Гц), 3,87 (3Н, с), 4,55 (1Н, с), 5,34 (1Н, широкий синглет), 6,26 (1Н, д, J=15,8 Гц) 7,80-7,90 (2Н, м), 8,46 (1Н, двойной дублет, J=2,9, 6,2 Гц) 8,75 (1Н, д, J=15,8 Гц), 13,24 (1Н, с), 13,53 (1Н, с).
Ультрафиолетовый спектр (в этиловом спирте): λ=493, 348, 265, 214 нм, λмакс=265 нм.
Инфракрасный спектр (гранула КВr): ν 3390, 1713, 1690, 1615, 1575 см-1.
[ α]20D (с=0,1 в диоксане)= +188.
Масс-спектр m/Z=452/ М+, основной пик.
Тонкослойная хроматография на пластине из кизельгеля = D 254 (Мерк) с использованием смеси хлороформа и ацетона (9:1 по объему) Rf=0,61.
П р и м е р 4. 4-Деметокси-4-(2' -метоксикарбонил)этенилдауномицинон [(1), R=СН=СН-СООСН3].
Эту реакцию осуществляли так, как описывалось в примере 3, за исключением того, что в качестве растворителя использовали диметилформамид (50 мл) и в качестве лиганда для палладия использовали 1,2-бис-[N-(1-фенилэтил), N-(дифенилфосфи- но)амино]этан (62 мг, 0,097 ммоля), в результате чего после хроматографии на силикагеле (в качестве элюента использовали смесь хлороформа и ацетона с соотношением объемов 95:5 (было получено 0,34 г [42% от соединения (II) R '=CF3] 4-деметокси 4-(2' - метоксикарбонил)этенилдауномицинона (I), R= СН-СН-СООСН3(высокоэффективная жидкостная хроматография 98,2%).
П р и м е р 5. 4-Деметокси-4-триметилсилилэтинилдауномицинон [(I), R=С= С-Si(СН3)3].
Эту реакцию осуществляли так, как описывалось в примере 1, за исключением того, что в качестве реагента использовали триметилсилилацетилен (5,9 мл 35,2 ммоля), что позволило получить сырой 4-деметокси-4-триметилсилилэтинил-13-диоколанилдаун- омицинон (VI) R=С=С-Si-(СН3)3.
Спектр 1Н-ЯМР 300 Гц (в СDCl3): δ =0,35 (9Н, с), 1,48 (3Н, с), 1,99 (1Н, двойной дублет, J=5,0 14,6 Гц) 2,47 (1Н, д, J=14,6 Гц), 2,78 (1Н, д, J= 19,0), 3,15 (1Н, широкий синглет), 3,23 (1Н, двойной дублет, J=2,0, 19,0 Гц), 3,82 (1Н, широкий синглет), 4,08 (4Н, с), 5,28 (1Н, д, J=3,7 Гц), 7,72 (1Н, т. J=7,7 гц), 7,94 (1Н, двойной дублет J=1,4, 7,7 Гц) 8,34 (1Н, двойной дублет, J=7,7 Гц), 13,22 (1Н, с), 13,80 (1Н, с).
Ультрафиолетовый спектр (в этиловом спирте); λ=528, 494, 363, 269, 247, 214 нм, λмакс=269 нм.
Инфракрасный спектр (гранула КВr): ν=3540, 3470, 1615, 1565 см-1.
[ α]20D (с=0,1, в диоксане) = +183.
Масс-спектр m/Z=508 (М+, основной пик).
Тонкослойная хроматография на пластине из кизельгеля F 254 (Мерк) с использованием смеси хлороформа и ацетона (9:1 по объему) Rf=0,65.
Сырое соединение [(VI), R=СН=С-Si(СН3)3 обрабатывали трифторуксусной кислотой, как описывалось в примере 1, в результате чего после хроматографии на силикагеле (в качестве элюента использовали смесь хлороформа и ацетона с соотношением объемов 95:5) было получено 0,12 г [15% от соединения (II), R' =СF3] 4-деметокси-4-триметилсилилэтинилдауномицинона [(I), R=С= СSi(СН3)3], высокоэффективная жидкостная хроматография 96,4%.
Спектр 1 Н-ЯМР 300 МГц (в СDCl3): δ=0,30 (9Н, с), 2,04 (1Н, двойной дублет, J=4,8, 14,6 Гц) 2,25 (1Н, д, J=14,6 Гц), 2,34 (3Н, с), 2,69 (1Н, д, J= 18,7 Гц) 2,91 (1Н, двойной дублет, J=1,4, 18,7 Гц), 4,06 (1Н, д, J=5,6), 4,71 (1Н, с), 5,11 (1Н, т, J=4,2 Гц), 7,60 (1Н, т, J=7,8 Гц), 7,80 (1Н, двойной дублет, J= 1,3, 7,7 Гц) 8,03 (1Н, двойной дублет J=1,3, 7,7 Гц) 12,82 (1Н, с), 13,29 (1Н, с).
Ультрафиолетовый спектр (в этиловом спирте): λ=493, 366, 269, 246, 222, 204 нм, λмакс=269 нм.
Инфракрасный спектр (гранула КВr: ν=3490, 1715, 1615, 1565 см-1.
Масс-спектр m/Z=464 (М+, основной пик).
Тонкослойная хроматография на пластине из кизельгеля F254 (Мерк) с использованием смеси хлороформа и ацетона (9:1 по объему) Rf=0,44.
П р и м е р 6. 4-Деметокси-4-(2 -пропенил)дауномицинон [(I), R=СН2-СН= СН2].
К раствору 1 г 4-диметил-4-трифторметансульфонил-13-диоксоланилдауномици-нона [(II), R' =CF3] 1,78 ммоля) в 50 мл диоксана в инертной атмосфере последовательно добавляли 55 мг (0,097 ммоля) 1,1 -бис(дифенилфосфино)ферроцена, 20 мг (0,089 ммоля) ацетата палладия и 1,1 мл (3,56 ммоля) аллилтриметилолова. Реакционную смесь перемешивали при температуре 70оС в течение ночи, затем охлаждали до 0оС и обрабатывали так, как описывалось в примере 1, что позволило получить сырой 4-деметокси-4-(2-пропенил)-13-диоксоланилдау- номицинон VI (R=СН2-СН=СН2).
Спектр 1Н-ЯМР 300 МГц (в CDCl3): δ=1,47 (3Н, с), 1,98 (1Н, двойной дублет, J=5,0 14,7 Гц), 2,45 (1Н, двойной триплет, J=2,0; 14,7 Гц) 2,74 (1Н, д, J=18,9 Гц) 3,14 (1Н, с), 3,24 (1Н, двойной дублет, J=2,2; 18,9 Гц) 3,78 (1Н, д, J=6,8 Гц) 4,06-4,12 (6Н, м), 5,00-5,21 (2Н, м), 5,23-5,30 (1Н, м), 6,00-6,18 (1Н, м), 7,60-7,75 (2Н, м), 8,31 (1Н, двойной дублет, J=1,9, 7,4 Гц) 13,36 (1Н, с), 13,85 (1Н, с).
Ультрафиолетовый спектр (в этиловом спирте): λ=523, 489, 339, 287, 254, 207 нм, λмакс=254 нм.
Инфракрасный спектр (гранула КВr): 3470, 335, 1615, 1575 см-1 [ α]20D (с=0,1 в диоксане)= +115.
Масс-спектр m/Z=452/М+, основной пик).
Тонкослойная хроматография на пластине из кизельгеля F 254 (Мерк) с использованием смеси хлороформа и ацетона (9:1 по объему) Rf=0,68.
Сырое соединение VI (R=СН2-СН=СН2) обрабатывали трифторуксусной кислоты так, как описывалось в примере 1, в результате чего после хроматографии на силикагеле (в качестве элюента использовали смесь хлороформа и ацетона с соотношением объемов 95:5) было получено 0,51 г [70% от соединения II, R' = CF3] 4-деметокси-4-(2' -пропенил)дауномицинона [(I), RF= СН2-СН= СН2] (высокоэффективная жидкостная хроматография 97,9%).
Спектр Н-ЯМР 300 МГц (в СDCl3):S 2,17(1Н, двойной дублет, J=4,8, 14,5 Гц), 2,35 (1Н, двойной триплет, J=2,0, 14,5 Гц) 2,43 (3Н, с), 2,94 (1Н, д, J= 18,7 Гц), 3,19 (1Н, двойной дублет, J=2,2, 18,7 Гц)= 3,77 (1Н, шир. синглет, (4,08-4,12) 2Н, м), 4,55 (1Н, широкий синглет), 5,02-5,13 (2Н, м, 5,30-5,35 (1Н, м) 6,04-6,19 (1Н, м), 7,65 (1Н, двойной дублет, J=1,8, 7,7 Гц) 7,74 (1Н, т, J=7,7 Гц), 8,33 (1Н, двойной дублет, J=1,8, 7,4 Гц) 13,28 (1Н, с), 13,82 (1Н, с).
Ультрафиолетовый спектр (в этиловом спирте): λ=489, 339, 286, 254, 208 нм, λмакс=254 нм.
Инфракрасный спектр (гранула КВr): ν=3410, 1710, 1618, 1575 см-1.
[α]20D (с=0,1 в диоксане)= +152.
Масс-спектр m/Z=408 (М+, основной пик).
Тонкослойная хроматография на пластине из кизельгеля F 254 (Мерк) с использованием смеси хлороформа и ацетона (9:1 по объему) Rf=0,78.
П р и м е р 7. Получение хлористоводородной соли 4-этенил-(4-деметокси)-дауномицина.
К перемешанному раствору 4-этенил-(4-деметокси)дауномицинона (0,468 г, 1,2 ммоля) в СН2Сl2 80 мл при комнатной температуре в атмосфере аргона в течение десяти минут одновременно добавляли раствора хлордауносаммина (0,536 г, 1,5 ммоля), в СН2Сl2 10 мл) и раствор АgCF3SO3 (0,398 г, 1,5 ммоля) в простом этиловом эфире (12 мл). Через 30 мин добавляли 0,144 мл пиридина, после чего реакционную смесь фильтровали на дикалите. Этот раствор последовательно промывали 1%-ным раствором НCl, водой, сушили (Na2SO4) и выпаривали в условиях вакуума.
Остаток растворяли в ацетоне (20 мл), охлаждали до 0оС и обрабатывали 0,075 М раствором NаОН (100 мл). Через час добавляли СН2Сl2и воду, при этом показатель рН доводили до 4 с помощью 3% НСl. Водную фазу отделяли, обрабатывали 1% раствором NН4ОН до достижения показателя рН 8 и экстрагировали СН2Сl2 (3х100). Собранные органические фазы сушили над Nа2SO4 и выпаривали в условиях вакуума. Полученный продукт очищали посредством хроматографии на колонке из SiО2(СН2Сl2/МеОН/СН3СООН/Н2O= =180/25/2/3). Собранные фракции разбавляли водой и показатель рН доводили до 8 с помощью 1% раствора NН4ОН. Органическую фазу отделяли, сушили и выпаривали в условиях вакуума, что позволило получить 0,175 г свободного основания.
К раствору свободного основания в минимальном количестве СНСl3добавляли 0,11 мл 3 н. раствора НСl и метилового спирта. Осадок фильтровали, промывали простым эфиром и сушили с образованием 0,170 г целевого соединения. (Высокоэффективная жидкостная хроматография = 96,17%).
Спектр 3Н-ЯМР 200 МГц (ДМСО-d6/: δ/ частей на миллион (1,16) 3Н, д. J= 6,6 Гц), 1,81 (2Н, м), 2,12 (2Н, м), 2,27 (3Н, с), 2,95 (2Н, широкий синглет)= 3,39 (1Н, м), 3,58 (1Н, широкий синглет), 4,21 (1Н, кв. J=6,6 Гц) 4,92 (1Н, широкий синглет), 5,30 (1Н, широкий синглет), 5,52 (3Н, м), 5,73 (1Н, д. J=17,265 Гц), 7,85 (3Н, м), 8,28 (1Н, двойной дублет, J=7,05 Г ц, J=1,87 Гц), 13,40 (2Н, широкий синглет).
Ультрафиолетовый спектр (этиловый спирт): λ=523,6, 489,6, 354,8, 258,4, 213,2 нм, λмакс=258,4 нм.
Тонкослойная хроматография на пластине из кизельгеля F 254 (Мерк) с использованием смеси СН2l2((МеОН)СН3СООН/Н2О (8:2:0,7:0,3 по объему) Rf= 0,83.
П р и м е р 8. Получение хлористоводородной соли 4-этенил-4-(деметокси)доксорубицина.
Целевое соединение можно получить из хлористоводородной соли 4-этенил-4-(деметокси)-дауномицина, 0,2 г хлористоводородной соли 4-этенил-4-((деметокси)-дауномицина растворяли в смеси безводного метанола и диоксана. Добавляли раствор 1 г брома в 10 мл метиленхлорида, в результате чего было получено 14-бромзамещенное производное. 14-Бромзамещенное производное гидролизовали при комнатной температуре в течение 48 часов в атмосфере азота водным раствором формиата натрия. Таким образом получали 4-этил-4-(деметокси)доксорубицин, который в результате обработки хлороводородом в безводном метаноле выделяли в виде его хлористоводородной соли.
Данные о цитотоксичности.
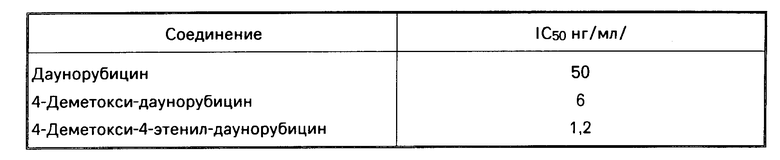
Цитотоксический эффект оценивался на линии клеток типа Лово (LоVо) (аденокарцинома толстой кишки человека), рост колонии оценивался после четырехчасового воздействия вещества на колонию и последующего выращивания культуры в течение 8 дней. Для исследуемого соединения 4-деметокси-4-этенил-даунорубицина и двух сравниваемых с ним веществ: 4-деметоксидаунорубицина и даунорубицина, производилась оценка концентрации вещества, требуемой для 50%-ного подавления роста колонии (IС50) по сравнению с контрольной. Данные приведены в таблице.
Низкое значение концентрации IС50 для предлагаемого в настоящем изобретении соединения показывает его эффективность по сравнению с остальными исследованными соединениями.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ 4-ЗАМЕЩЕННЫХ АНТРАЦИКЛИНОНОВ | 1989 |
|
RU2071463C1 |
| АНТРАЦИКЛИНОВЫЙ ГЛИКОЗИД И СПОСОБЫ ЕГО ПОЛУЧЕНИЯ | 1990 |
|
RU2073681C1 |
| СПОСОБ ПОЛУЧЕНИЯ АНТРАЦИКЛИНОНОВ | 1990 |
|
RU2077526C1 |
| СОЕДИНЕНИЕ ЦЕФЕМА И ЕГО ФАРМАЦЕВТИЧЕСКИ ИЛИ ВЕТЕРИНАРНО ПРИЕМЛЕМЫЕ СОЛИ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 1990 |
|
RU2074186C1 |
| 8-ФТОРАНТРАЦИКЛИНГЛИКОЗИДЫ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ ПРИСОЕДИНЕНИЯ КИСЛОТ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1991 |
|
RU2095365C1 |
| АНТРАЦИКЛИН ГЛИКОЗИД И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 1992 |
|
RU2081878C1 |
| Способ получения антрациклиновых гликозидов | 1988 |
|
SU1614764A3 |
| АНТРАЦИКЛИН ГЛИКОЗИДЫ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, ОБЛАДАЮЩИЕ ПРОТИВООПУХОЛЕВЫМИ СВОЙСТВАМИ, И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1990 |
|
RU2043360C1 |
| Способ получения антрациклиновых гликозидов | 1986 |
|
SU1553015A3 |
| Способ получения антрациклингликозидов | 1983 |
|
SU1378784A3 |
Использование: в медицине в качестве противоопухолевых средств. Сущность изобретения: продукт - 4-замещенные антрациклиноны ф-лы 1. Гликозид антрациклина ф-лы 2. Реагент 1: 4-деметил-4-сульфонил-13-диоксоланилдауномицинон. Реагент 2: ненасыщенные соединения ф-лы H-R'' , где R'' - алкенил, алкинил. Реагент 3: металлоорганическое соединение ф-лы MRnYm, где M-атом металла, R-линейный или разветвленный алкенил или алкинил, содержащий до 4 атомов углерода. Условия реакции: каталитическое количество соединения ф-лы 3: 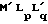 , где M' -атом переходного металла, L,L' -анион или нейтральная молекула; p и q - значения от 0 до 4. 2 с. и 2 з.п. ф-лы. 1 табл. Структура соединения ф-лы 1 и 2 (чертеж) .
, где M' -атом переходного металла, L,L' -анион или нейтральная молекула; p и q - значения от 0 до 4. 2 с. и 2 з.п. ф-лы. 1 табл. Структура соединения ф-лы 1 и 2 (чертеж) .

где R - линейная, или разветвленная алкенильная, или алкинильная группа, содержащая до 4 атомов углерода.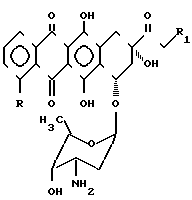
где R - алкенильная или алкинильная группа, содержащая до 4 атомов углерода;
R1 - водород или гидроксигруппа,
и его фармацевтически приемлемые соли.
| ТОПЛИВНЫЙ НАСОС | 1997 |
|
RU2169284C2 |
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
