Изобретение относится к имидазолам как ингибиторам активности ацил-СоА; холестеринацилтрансферазы (АСАТ), способу их получения и их использования в качестве антигиперхолестеринемических препаратов.
Гиперхолестеринемия представляет собой установленный фактор риска развития атеросклероза. Терапевтические препараты, которые регулируют уровень сывороточного холестерина, являются эффективными при лечении заболеваний коронарных сосудов. Эти препараты могут модулировать уровни несущих холестерин липопротеинов в крови, но они оказывают очень незначительное или не оказывают вовсе влияния на абсорбцию холестерина в кишечнике. Поступающий с пищей холестерин может повысить уровень сывороточного холестерина до таких уровней, которые приводят пациента на грань повышенного риска развития или обострения атеросклероза. Поскольку большое количество свободного или неэтерифицированного холестерина, который впитывается клетками слизистой кишечника, должен сначала быть этерифицирован АСАТ до его включения и секреции в кровь в больших частицах липопротеина, называемых хиломикронами, ингибирование выработки АСАТ может уменьшить поглощение пищевого холестерина. Кроме того, аккумулирование и хранение холестериновых эфиров в стенках артерий связано с повышенной активностью АСАТ. Ингибирование выделения фермента, как ожидается, приведет к замедлению образования или увеличения атеросклеротических поражений у млекопитающих.
В литературе имеются описания незначительного количества случаев, при которых указаны препараты, полезные в качестве ингибиторов АСАТ, в частности и атеросклеротических препаратов вообще. Например, в патенте США N 4623662, выданном De Vries 18 ноября 1986 г., описаны мочевины и тиомочевины как ингибиторы АСАТ, полезные для уменьшения содержания холестериновых эфиров в стенках артерий, замедляющих образование атеросклеротических поражений, и/или лечение гиперлипидемии у млекопитающих. В патенте США N 4722927, выданном 2 февраля 1988 г., описаны двузамещенные пиримидинамиды олеиновой и линолевой кислот как ингибиторы АСАТ, полезные для замедления всасывания холестерина в кишечнике.
В патенте США N 4460598, выданном Lautenschlager et al., 17 июля 1984 г. раскрыты соединения формулы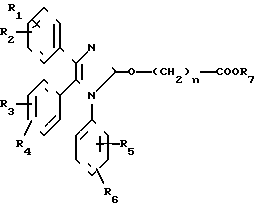 где R1, R2, R3, R4, R5 и R6, независимо выбраны из ряда, включающего H, F, Cl, Br, I, алкил, алкокси, или CF3 при условии, что одна или несколько пар R1 и R2, R3 и R4 или R5 и R6, взятые вместе, представляют метилендиокси;
где R1, R2, R3, R4, R5 и R6, независимо выбраны из ряда, включающего H, F, Cl, Br, I, алкил, алкокси, или CF3 при условии, что одна или несколько пар R1 и R2, R3 и R4 или R5 и R6, взятые вместе, представляют метилендиокси;
R7 - H, ион щелочного металла, алкил с 1-6 атомами углерода или бензил, и n = 0-10.
Описывается использование этих соединений при лечении тромбоэмболии, воспалительных и/или этеросклеротических заболеваний.
В патенте США N 4654358, выданном Lautenshlager et al. 31 марта 1987 г. , описаны соединения формулы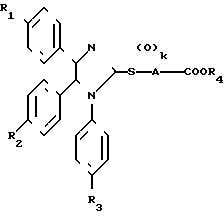 где k = 0,1 или 2, R1, R2 и R3, независимо, выбраны из ряда, включающего Н, F, Cl, CH3, CH3O или CF3;
где k = 0,1 или 2, R1, R2 и R3, независимо, выбраны из ряда, включающего Н, F, Cl, CH3, CH3O или CF3;
R4 - H, Na, K, CH3, CH3CH2, (CH3)2CH, CH3(CH2)2 или бутил; А - С(СН3)2, СН(СН2)mCH3, (CH2)n или (СН2)n-2CH(CH3); m = 0-8 и n = 2-10.
Описаны синтез и использование этих соединений при лечении воспалительных заболеваний, расстройств липидного метаболизма и/или гиперлипидемии.
В выложенной заявке на патент ФРГ N DE 3504679, поданной Lautenshlager et. al. и опубликованной 14 августа 1986 г., раскрыты соединения формулы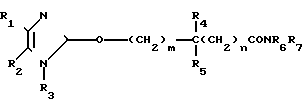 где R1, R2 и R3, независимо, выбраны из ряда, включающего Н, алкил с 1-6 атомами углерода, циклоалкил с 1-6 атомами углерода или
где R1, R2 и R3, независимо, выбраны из ряда, включающего Н, алкил с 1-6 атомами углерода, циклоалкил с 1-6 атомами углерода или R4 и R5 независимо, выбраны из ряда, включающего Н, С6Н5 или алкил с 1-9 атомами углерода; R6 и R7, независимо, выбраны из ряда, включающего Н, ОН, насыщенный или ненасыщенный алкил, циклоалкил или гидроксиалкил с 1-10 атомами углерода,
R4 и R5 независимо, выбраны из ряда, включающего Н, С6Н5 или алкил с 1-9 атомами углерода; R6 и R7, независимо, выбраны из ряда, включающего Н, ОН, насыщенный или ненасыщенный алкил, циклоалкил или гидроксиалкил с 1-10 атомами углерода,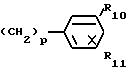 ,
,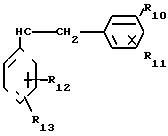 ,or
,or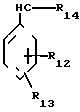
R8, R9, R10, R11, R12 и R13, независимо, друг от друга выбраны из ряда, включающего Н, F, Cl, Br, NO2, CH3CONH, OH, алкил с 1-3 атомами углерода, CF3 и алкокси с 1-3 атомами углерода, при условии, что R8 и R9, R10 и R11 или R12 и R13, взятые вместе, представляют собой метилендиокси;
R14 - алкил с 1-2 атомами углерода,
m и n, взятые вместе, представляют собой целое число от 0 до 9, p = 0-2, S = 0-2 и t представляет собой 0 или 2.
Описаны синтез и использование этих препаратов для лечения тромбоэмболии, воспалительных и атеросклеротических заболеваний и вообще расстройств липидного метаболизма.
В выложенной заявке на патент ФРГ N DE 3504680, поданной Lautenschlager et al. и опубликованной 14 августа 1986 г., описаны соединения формулы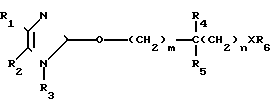 где R1, R2 и R3, независимо, выбраны из ряда, включающего Н, алкил с 1-6 атомами углерода, циклоалкил с 1-6 атомами углерода или
где R1, R2 и R3, независимо, выбраны из ряда, включающего Н, алкил с 1-6 атомами углерода, циклоалкил с 1-6 атомами углерода или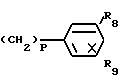 R1 и R2 могут быть взяты вместе с атомами углерода в 4- и 5-положении имидазольного кольца и тогда представят карбоциклические 5- или 6-членное ароматическое или частично гидрогенизированное кольцо, которое может быть замещено R8 или R9; R4 и R5 независимо друг от друга представляют собой Н, С6Н5 или алкил с 1-9 атомами углерода; R6 представляет собой алкил, циклоалкил или гидроксиалкил с 1-20 атомами углерода, Н, щелочной металл, если Х представляет собой -СОО-, 1-фенетил, или
R1 и R2 могут быть взяты вместе с атомами углерода в 4- и 5-положении имидазольного кольца и тогда представят карбоциклические 5- или 6-членное ароматическое или частично гидрогенизированное кольцо, которое может быть замещено R8 или R9; R4 и R5 независимо друг от друга представляют собой Н, С6Н5 или алкил с 1-9 атомами углерода; R6 представляет собой алкил, циклоалкил или гидроксиалкил с 1-20 атомами углерода, Н, щелочной металл, если Х представляет собой -СОО-, 1-фенетил, или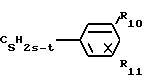 R7 представляет собой Н, ОН, если Х представляет собой -CONR7-, или алкил с 1-4 атомами углерода, R8, R9, R10 и R11 независимо друг от друга представляют собой Н, Cl, F, Br, NO2, CH3CONH, OH, алкил, с 2-3 атомами углерода, CF3; или алкокси с 1-3 атомами углерода или R8 и R9 или R10 и R11, взятые вместе, представляют собой метилендиокси; Х представляет собой связь, О, ОС(=О)О, С(=О)О, CONR7, OC(=O) или ОС(=О)NR7, m и n, взятые вместе, представляют собой целое число от 0 до 9, p = 0-2, S = 0-2 и t представляет собой 0 или 2.
R7 представляет собой Н, ОН, если Х представляет собой -CONR7-, или алкил с 1-4 атомами углерода, R8, R9, R10 и R11 независимо друг от друга представляют собой Н, Cl, F, Br, NO2, CH3CONH, OH, алкил, с 2-3 атомами углерода, CF3; или алкокси с 1-3 атомами углерода или R8 и R9 или R10 и R11, взятые вместе, представляют собой метилендиокси; Х представляет собой связь, О, ОС(=О)О, С(=О)О, CONR7, OC(=O) или ОС(=О)NR7, m и n, взятые вместе, представляют собой целое число от 0 до 9, p = 0-2, S = 0-2 и t представляет собой 0 или 2.
Описаны синтез и использование этих соединений при лечении тромбоэмболии, воспалительных и атеросклеротических заболеваний и расстройств липидного метаболизма.
Неизвестны источники, в которых описаны имидазолы предлагаемого изобретения и их использование в качестве ингибиторов АСАТ или их использование для лечения атеросклероза.
Соединения предлагаемого изобретения представляют собой очень сильные ингибиторы АСАТ. Как показывают данные, приведенные в табл. 4, соединения предлагаемого изобретения снижают активность АСАТ in vitro по меньшей мере в 10 раз сильное любого другого описанного в литературе ингибитора АСАТ. Как показывают данные, приведенные в табл. 6, предлагаемые соединения вызывают снижение уровня сывороточного холестерина у хомяков, кормленых холестериновой пищей. Таким образом ожидается, что соединения будут полезны в фармацевтических композициях для лечения атеросклероза. Было показано, что описываемые соединения понижают уровень сывороточного холестерина, и предлагаемое изобретение не следует рассматривать как ограниченное каким-то определенным антигиперхолестеринемическим механизмом действия.
Предлагаемое изобретение предлагает новые соединения формулы (1), способ их получения, фармацевтические композиции, включающие такие имидазолы, и терапевтические способы их использования в качестве антигиперхолестеринемических препаратов.
Описываемое изобретение предлагает соединения формулы (1)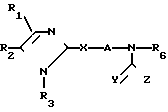 где R1 и R2 независимо друг от друга выбраны из ряда, включающего Н, С1-8-алкил, С3-8-разветвленный алкил, С3-С7-циклоалкил, С4-С10-циклоалкилалкил, С7-С14-аралкил, 2,3- или 4-пиридинил, 2-тиенил, 2-фуранил, фенил, в определенных случаях замещенный 1-3 группами, выбранными из ряда, включающего F, Cl, Br, OH, C1-C4-алкокси, С1-С4-алкил, С3-С8-разветвленный алкил, CH3S(O)r, NO2, CF3 или NR7R8; или R1 и R2 могут
где R1 и R2 независимо друг от друга выбраны из ряда, включающего Н, С1-8-алкил, С3-8-разветвленный алкил, С3-С7-циклоалкил, С4-С10-циклоалкилалкил, С7-С14-аралкил, 2,3- или 4-пиридинил, 2-тиенил, 2-фуранил, фенил, в определенных случаях замещенный 1-3 группами, выбранными из ряда, включающего F, Cl, Br, OH, C1-C4-алкокси, С1-С4-алкил, С3-С8-разветвленный алкил, CH3S(O)r, NO2, CF3 или NR7R8; или R1 и R2 могут
быть также взяты вместе как где L - О, О(СН2)m+1O или (CH2)m, где m = 0-4; R3 - H, C1-C6-алкил, аллил, бензил или фенил, в определенных случаях замещенный F, Cl, CH3, CH3O или CF3, R4 - C1-C8-алкил с нормальной цепью, в определенных случаях замещенный F, C3-C8-алкил с разветвленной цепью, С3-С7-циклоалкил, С4-С10-циклоалкилалкил, С7-С14-аралкил, где арильная группа в определенных случаях замещена 1-3 группами, выбранными из ряда, включающего С1-С4-алкил или алкокси, F, Br, Cl, NH2, OH, CN, CO2H, CF3NO2, C1-C4-карбоалкокси, NR7R8 или NCOR7; C3-C6-алкенил или алкинил, С1-С3-перфторалкил, фенил, в определенных случаях замещенный 1-3 группами, выбранными из ряда, включающего С1-С4-алкил, С1-С4-алкокси, F, Br, Cl, NH2OH, CN, CO2H, CF3, NO2, C1-C4-карбоалкокси, NR7R8 или NCOR7; пентафторфенил, бензил, в определенных случаях замещенный 1-3 группами, выбранными из ряда, включающего С1-С4-алкил или алкокси, F, Br, Cl, NH2, OH, CN, CO2H, CF3, NO2, C1-C4-карбоалкокси, NR7R8 или NCOR7, 2-, 3- или 4-пиридинил, пиримидинил или дифенил;
где L - О, О(СН2)m+1O или (CH2)m, где m = 0-4; R3 - H, C1-C6-алкил, аллил, бензил или фенил, в определенных случаях замещенный F, Cl, CH3, CH3O или CF3, R4 - C1-C8-алкил с нормальной цепью, в определенных случаях замещенный F, C3-C8-алкил с разветвленной цепью, С3-С7-циклоалкил, С4-С10-циклоалкилалкил, С7-С14-аралкил, где арильная группа в определенных случаях замещена 1-3 группами, выбранными из ряда, включающего С1-С4-алкил или алкокси, F, Br, Cl, NH2, OH, CN, CO2H, CF3NO2, C1-C4-карбоалкокси, NR7R8 или NCOR7; C3-C6-алкенил или алкинил, С1-С3-перфторалкил, фенил, в определенных случаях замещенный 1-3 группами, выбранными из ряда, включающего С1-С4-алкил, С1-С4-алкокси, F, Br, Cl, NH2OH, CN, CO2H, CF3, NO2, C1-C4-карбоалкокси, NR7R8 или NCOR7; пентафторфенил, бензил, в определенных случаях замещенный 1-3 группами, выбранными из ряда, включающего С1-С4-алкил или алкокси, F, Br, Cl, NH2, OH, CN, CO2H, CF3, NO2, C1-C4-карбоалкокси, NR7R8 или NCOR7, 2-, 3- или 4-пиридинил, пиримидинил или дифенил;
X - S(O)n, O, NR5, CH2;
R5 представляет собой Н, С1-С6-алкил или бензил;
R6 представляет собой Н, С1-С8-алкенил или алкинил, фенил, в определенных случаях замещенный 1-3 группами, выбранными из ряда, включающего С1-С4-алкил или алкокси, F, Br, Cl, NH2, OH, CN, CO2H, CF3, NO2, C1-C4-карбоалкокси, NR7R8 или NCOR7; пентафторфенил, бензил, в определенных случаях замещенный 1-3 группами, выбранными из ряда, включающего С1-С4-алкил или алкокси, F, Br, Cl, NH2, OH, CN, CO2H, CF3, NO2, C1-C4-карбоалкокси, NR7R8 или NCOR7;
R7 и R8 независимо друг от друга выбраны из группы, включающей Н и С1-С4-алкил;
А - С2-С10-алкил, С3-С10-алкил с разветвленной цепью, С3-С10-алкенил или С3-С10-алкинил;
Y - O, S; H2;
Z представляет собой NHR4,
r = 0-2, или их фармакологически приемлемые соли.
Предпочтительными являются соединения формулы (1), в которых R1 и R2 выбраны независимо друг от друга из группы, включающей С1-С8-алкил, С3-С8-алкил с разветвленной цепью, С3-С7-циклоалкил, С4-С10-циклоалкилаллил, С7-С14-араалкил, 2-, 3- или 4-пиридинил, 2-тиенил, 2-фуранил, фенил, в определенных случаях замещенный 1-2 группами, выбранными из ряда, включающего F, Cl, Br, OH, C1-C4-алкокси, С1-С4-алкил, С3-С8-алкил с разветвленной цепью, CH3S(O)r, NO2, CF3 или NR7R8 или R1 и R2
могут быть также взяты вместе как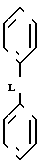 где L представляет собой O, O(CH2)m+1O или (CH2)m, где m = 0-4.
где L представляет собой O, O(CH2)m+1O или (CH2)m, где m = 0-4.
Более предпочтительными являются соединения формулы (1), в которых R3 представляет собой Н, СН3, фенил;
R6 представляет собой Н, С1-С8-алкил, С3-С8-алкил с разветвленной цепью, С3-С7-циклоалкил, фенил, в определенных случаях замещенный 1-3 группами, выбранными из ряда, включающего СН3, СН3О, F, Br, Cl, NH2, OH, CN, CO2H, CF3 или ди(С1-С4)алкиламино; или бензил, в определенных случаях замещенный 1-3 группами, выбранными из ряда, включающего СН3, СН3О, F, Br, Cl, NH2, OH, CN, CO2H, CF3 или ди(С1-С4)алкиламино;
Х представляет собой S(O)r, CH2,
A представляет собой С2-С10-алкил, С4-С9-алкил с разветвленной цепью.
Еще более предпочтительными из-за их биологической активности являются соединения формулы (1), в которых R1 и R2 независимо друг от друга выбраны из группы, включающей С1-С8-алкил, С3-С8-алкил с разветвленной цепью, С3-С7 циклоалкил, С4-С10-циклоалкилалкил, С7-С14аралкил, 2-, 3- или 4-пиридинил, 2-тиенил, или фенил, в определенных случаях замещенный 1-2 группами, выбранными из ряда, включающего F, Br, Cl, C1-C4-алкил, С3-С8-алкил с разветвленной цепью, CH3O, CH3S(O)r, NO2, CF3 или ди(С1-С4)алкиламино, или R1 и R2 могут быть также взяты вместе как где L представляет собой О или ОСН2О; R3 представляет собой Н, R4представляет собой С1-С8-алкил, С3-С8-алкил с разветвленной цепью, С3-С7-циклоалкил, С4-С10-циклоалкилалкил, С7-С14-аралкил, фенил, замещенный
где L представляет собой О или ОСН2О; R3 представляет собой Н, R4представляет собой С1-С8-алкил, С3-С8-алкил с разветвленной цепью, С3-С7-циклоалкил, С4-С10-циклоалкилалкил, С7-С14-аралкил, фенил, замещенный
1-3 группами, выбранными из ряда, включающего СН3, F, Cl, CH3O, CN; или бензил, в определенных случаях замещенный 1-3 группами, выбранными из ряда, включающего СН3, СН3О, F, Cl или CN; R6 представляет собой алкил или фенил, в определенных случаях замещенный 1-3 группами, выбранными из ряда, включающего СН3, СН3О, F, Cl, CN; A - C4-C9-алкил, Х - S(O)r, Y - O, H2.
N'-(2,4-дифторфенил)-N-[5-(4,5-дифенил- 1Н-имидазол-2-илтио)фенил] -N-гептилмочевина
N-[5-(4,5-дифенил-1Н-имидазол-2- илтио)пентил]-N-гептил-N'-фенилмочевина
N'-(2,4-дифторфенил)-N-[8-(4,5-дифенил- 1Н-имидазол-2-илтио)октил] -N-гептилмочевина
N-бутил-N'-(2,4-дифторфенил)-N-[8-(4,5- дифенил-1Н-имидазол-2-илтио) октил]мочевина
N'-(2,4-диметоксифенил)-N-[4,5- дифенил-1Н-имидазол-2-илтио)-пентил] -N-гептилмочевина
N-[5-(4,5-дифенил-1Н-имидазол-2- илтио)-пентил]-N-гептил-N'-метилмочевина
N-[5-(4,5-дифенил-1Н-имидазол-2- илтио)-пентил]-N-гептил-N'-пропилмочевина
N'-(2,4-дифторфенил)-N-{ 5-[(4,5- дифенил-1Н-имидазол-2-ил)сульфонил] пентил}-N-гептилмочевина
N-[5-(4,5-дифенил-1Н-имидазол-2- илтио)-пентил] -N'-(3-фторфенил)-N- гептилмочевина
N-[5-(4,5-дифенил-1Н-имидазол-2- илтио)-пентил] -N'-(3-фторфенил) -N-гептилмочевина
N'-(2,4-дифторфенил)-N-гептил-N-[5-(4- фенил-1Н-имидазол-2-илтио)пентил] мочевина
N-[5-(4,5-дифенил-1Н-имидазол-2- илтио)-пентил] -N-гептил-N'-(2,4,6- трифторфенил)-тиомочевина.
N'-(2,6-дихлорфенил)-N-[5-(4,5-дифенил- 1Н-имидазол-2-илтио)пентил]-N- гептилмочевина
N-[5-(4,5-дифенил-1Н-имидазол-2- илтио)пентил]-N-гептил-N'-(1-метилэтил)мочевина
N-[5-(4,5-дифенил-1Н-имидазол-2- илтио)пентил] -2,4-дифтор-N- гептилбензолацетамид
N-[5-(4,5-дифенил-1Н-имидазол-2- илтио)пентил]-N-гептил-N'-октилмочевина N'-циклогексил-N-[5-(4,5- дифенил-1Н-имидазол-2-илтио)пентил] -N-гептилмочевина
N'-(2,4-дифторфенил)-N-[5-(4,5- дифенил-1Н-имидазол-2-ил)сульфинил)-пентил]- N-гептилмочевина
N'-(2-4-дифторфенил)-N-[2-(4,5-дифенил -1Н-имидазол-2-илтио)этил]- N-гептилмочевина
N-[5-(4,5-дифенил-1Н-имидазол-2- илтио)пентил]-N-гептилбутанамид
N-{ 5-[4,5-бис(4-метоксифенил)-1Н- имидазол-2-илтио] -пентил}-N'-(2,4- дифторфенил)-N-гептилмочевина
N-{ 5-[4,5-бис(1-метилэтил)-1Н- имидазол-2-илтио]пентил}-N'-(2,4-дифторфенил)- N-гептилмочевина
N'-(2,4-дифторфенил)-N-[5-(4,5- дипропил-1Н-имидазол-2-илтио)-пентил]- N-гептилмочевина
N-{ 5-[4,5-бис(4-фторфенил)-1Н- имидазол-2-илтио]пентил}-N'-(2,4-дифторфенил)- N-гептилмочевина
N-[5-(1H-дибенз/2,3,6,7/окседино/4,5- d/имидазол-2-илтио)пентил] -N'- (2,4-дифторфенил)-N-гептилмочевина N-[5-(4,5-бис/2-тиенил/-1H- имидазол-2-илтио)пентил]-N'-(2,4-дифторфенил)- N-пентилмочевина
N-[5-(4,5-дифенил-1Н-имидазол-2- илтио)пентил]-N-гептилпентанамид
N-[5-(4,5-дифенил-1Н-имидазол-2-илтио) пентил] -N-гептил(1,1'-дифенил)-4-ацетамид
N-[5-(4,5-дифенил-1Н-имидазол-2- илтио)пентил] -N-гептил-N'-(2,4,6-трифтор- фенил) мочевина N-{5-(4,5-бис(2-пиридинил)-1Н-имидазол- 2-илтио] фенил}-N'-(2,4-дифторфенил)- N-гептилмочевина
N'-(2,4-дифторфенил)-N-[6-(4,5-дифенил-1Н-имидазол -2-ил)гексил]-N-гептил- мочевина
N-{ 5-[4,5-бис(4-метилфенил)-1Н- имидазол-2-илтио]пентил}-N'-(2,4-дифторфенил)- N-гептилмочевина
N-{ 5-[4,5-бис(4-метоксифенил)-1Н- имидазол-2-илтио]пентил}-N-гептилбутана- мид
N-{ 5-[4,5-бис(4-гидроксифенил)-1Н- имидазол-2-илтио] пентил}-N'-(2,4- дифторфенил)-N-гептилмочевина
N-{5-[4,5-бис(1-метилэтил)-1Н- имидазол-2-илтио]пентил}-N- гептилциклогексанацетамид
N-{ 5-[4,5-бис(3-метоксифенил)-1Н- имидазол-2-илтио]пентил}-N'-(2,4-дифторфенил)- N-гептилмочевина
N-{ 5-[4,5-бис(2-метоксифенил)-1Н- имидазол-2-илтио] пентил} -N'-(2,4- дифторфенил)-N-гептилмочевина
N'-[(1,1'-дифенил)-4-ил]-N-[5-(4,5- дифенил-1Н-имидазол-2-илтио)пентил] -N- гептилмочевина
N-(2,4-дифторфенил)-N-[5-(4,5- дифенил1Н-имидазол-2-илтио)пентил] -N'- октилмочевина
Пропил [5-(4,5-дифенил-1Н-имидазол -2-илтио)пентил]гептилкарбамат
Пентилметил [5-(4,5-дифенил-1Н-имидазол-2-илтио) пентил]гептилкарбамат
Пентил [5-(4,5-дифенил-1Н)-имидазол -2-илтио)пентил]гептилкарбамат
(2-Метилпропил)-[5-(4,5-дифенил-1Н- имидазол-2-илтио)пентил]гептилкарбамат
Этил-[5-(4,5-дифенил-1Н-имидазол-2- илтио)пентил]гептилкарбамат
Октил-[5-(4,5-дифенил-1Н-имидазол-2- илтио)пентил]гептилкарбамат N-[5-{ 4,5-бис[4-(диметиламино)фенил] - 1H-имидазол-2-илтио}пентил/-N'-(2,4- дифторфенил)-N-гептилмочевина N-[5-(4,5-дициклогексил-1Н-имидазол-2- илтио)пентил]-N'-(2,4-дифторфенил)- N-гептилмочевина
(4-Фторфенил)-[5-(4,5-дифенил-1Н- имидазол-2-илтио)пентил]гептилкарбамат
N-[5-(4,5-дифенил-1Н-имидазол-2- илтио)пентил]-N'-октил-N-фенилмочевина
N-[5-(1H, 9H-дибенз/4,5: 8,9//1,3/ диоксанино/6,7-d/имидазол-2-илтио)-пентил] - N'-(2,4-дифторфенил)-N-гептилмочевина N-(4-цианофенил)-N-[5-(4,5-дифенил -1Н-имидазол-2-илтио)пентил]- N-гептилмочевина
N-(2,4-дифторфенил)-N'-[5-(4,5- дифенил-1Н-имидазол-2-илтио)пентил] мочевина
N-{ 5-[4,5-бис(4-метоксифенил)-1Н- имидазол-2-илтио]пентил}-(2,4-дифтор)- N-гептилбензолацетамид
Фенил{ 5-[4,5-бис(4-диметиламино) фенил-1Н-имидазол-2-илтио] -пентил} гептил-карбамат,
или их фармацевтически приемлемые соли.
С и н т е з.
Новые соединения формулы (1) могут быть получены путем использования реакций и технологий, описанных ниже. Эти реакции проводят в растворителях, подходящих для материалов и реактивов, используемых в реакциях, и подходящих для материалов и реактивов, используемых в реакциях, и подходящих для осуществляемых преобразований. Специалистам понятно, что функциональные группы имидазола и другие части молекулы должны быть совместимыми с предлагаемыми реактивами и условиями реакции. Не все соединения формулы (1), входящие в данный класс, могут быть совместимы с условиями некоторых реакций, требуемыми в некоторых описанных способах. Такие ограничения на выбор заместителей, которые совместимы с условиями проведения реакций, сразу станут очевидными для специалиста, и в этом случае следует использовать альтернативный описанный способ.
Соединения формулы (1), где Х - О, S, СH2 или NH, можно получить способом, схематически представленным на схеме 1. Эфиры формулы (3), где Х - О или S, можно получить превращением 4-имидазолин-2-она (1), где Х - 0, или 4-имидазолил-2-тиона (1), где Х - S, в соответствующую щелочнометаллическую соль путем добавления основания такого, как гидрид натрия, затем соль алкилируют с помощью соединения формулы М-(А')CO2R, где R - СH3 или C2H5, М представляет собой галоген или тозилатную группу и A' - радикал, имеющий на одну метиленовую группу меньше, чем А, в полярном растворителе, таком как N,N-диметилформамид. Альтернативно, эфиры формулы (3), где Х - S, можно получить прямым алкилированием 4-имидазолин-2-тиона соединением формулы M-(A')CO2R, не прибегая к добавлению подходящего основания, в полярном растворителе, таком как N,N-диметилформамид, при температуре от комнатной до температуры дефлегмации растворителя. Эфиры формулы (3), где Х - NH, можно получить путем реакции 2-аминоимидазола формулы (2) с соединением формулы М-(A')CO2R, где R, M и A' имеют вышеуказанные значения, в подходящем растворителе, таком как N,N-диметилформамид. Соединения формулы (2), где R3 - H, предпочтительно алкилируют по атому азота кольца. Поэтому для того, чтобы получить соединения формулы (1), где Х - NH и R2 - Н, обычно необходимо защитить атом азота кольца. Предпочтительно, чтобы защитная группа была стабильна при основных условиях и легко отщеплялась при кислотных условиях, например, силил или тритилгруппы. Защищенный 2-аминоимидазол затем может быть использован для получения эфиров формулы (3), где R3 - защитная группа. Защитная группа может быть отщеплена на любой подходящей стадии процесса синтеза соединений формулы (1), где Х - NH и R3 - H.
Схема 1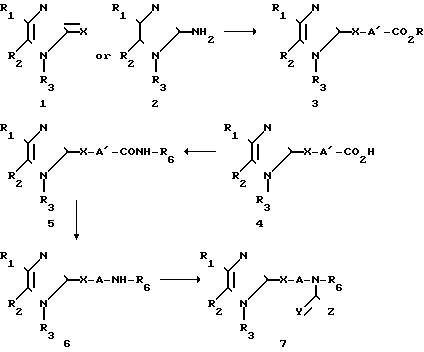
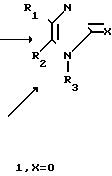
Эфиры формулы 3 гидролизуют до соответствующих карбоновых кислот формулы (4) хорошо известными из химической литературы способами. Например, гидролиз можно осуществить в результате реакции с гидроокисью щелочного металла в водных или органических растворителях, таких как вода, спирты, эфиры или их смеси, с последующим подкисливанием неорганической кислотой. Способы, используемые для получения соединений формулы (4), аналогичны способам, описанным, в патенте США 4654358, патенте США 4460598 и переданной заявке на патент США 244170 (патент Великобритании 6339), поданной 14 сентября 1988 г. , содержание которых включено в настоящую заявку в качестве ссылок. Соединения формулы (4), где R1 и R2 - фенил или замещенный фенил, R3 - H; X - S; A' - (CH2)n-1 и n = 8-21, заявлены как антигиперхолестеринемические препараты в переданной заявке на патент США 244170 (патент Великобритании 6339).
Амиды формулы (5) получают путем соединения карбоновых кислот формулы (4) с первичным амином с помощью образующих амидную связь реакций, которые хорошо известны из химической литературы. Одним способом образования амидной связи является использование сочетающего реагента, который вырабатывает реакционно способный интермедиат, такой как смешанный ангидрид или активный эфир. Примерами таких сочетающихся реагентов являются двузамещенные карбодиимиды, N,N'-карбонилдиимидазол, дифенилфосфорилазид и т.п. Например, реакцию сочетания можно провести с двузамещенным карбодиимидом, таким как дициклогексилкарбодиимид, в подходящем растворителе, таком как хлористый метилен, ацетонитрил, толуол или N,N-диметилформамид. В качестве катализаторов реакции могут быть добавлены нуклеофильные гидроксисоединения, такие как 1-гидрокси-1Н-бензотриазол, которые образуют высоко активные эфиры.
Известно несколько альтернативных подходов к получению амидов формулы (5). Например, реакция карбоновой кислоты формулы (4) с первичным амином, катализируемая этератом трифторида бора, с последующим азеотропным удалением воды дает амиды формулы (5). Другой подход включает превращение карбоновых кислот формулы (4), в соответствующий хлорид кислоты, используя тионилхлорид, оксалилхлорид или им подобное соединение, после чего осуществляют реакцию указанного хлорида с первичным амином в присутствии основания, такого как триэтиламин, в результате чего получают амиды формулы (5). Еще один подход, при котором эфиры формулы (3) могут быть непосредственно превращены в амиды формулы (5) путем аминирования эфира в присутствии сильного щелочно-металлического катализатора, такого как амид натрия, гидрид натрия, метилат натрия, реактивы Гриньяра или бутиллитий, или в присутствии более слабых катализаторов, таких как 2-пиридон трибромид бора или диметилалюминийамиды.
Амины формулы (6) можно получить восстановлением соответствующих амидов формулы (5) целым рядом разных способов, хорошо известных специалистам. Например, такие реагенты как литий алюминий гидрид, диборан, натрий бис(2-метоксиэтокси)алюминий гидрид (Red-Al ) и диизобутилалюминий гидрид могут быть использованы для восстановления амида до амина. Такие реакции обычно проводят в подходящем безводном апротонном растворителе, таком как диэтиловый эфир, толуол или тетрагидрофуран при температуре от комнатной до температуры кипения растворителя в течение 2-48 ч.
) и диизобутилалюминий гидрид могут быть использованы для восстановления амида до амина. Такие реакции обычно проводят в подходящем безводном апротонном растворителе, таком как диэтиловый эфир, толуол или тетрагидрофуран при температуре от комнатной до температуры кипения растворителя в течение 2-48 ч.
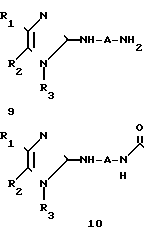
Альтернативно амины формулы (6), где Х представляет собой NH, можно получить в результате осуществления последовательности реакций, представленных на схеме 2. Первичные амины (9) можно получить в результате реакции 2-бромимидазолов формулы (8) с подходящим подобранным диамином в чистом виде при определенном температурном режиме или в подходящем растворителе, таком как N, N-диметилформамид, толуол, ацетонитрил или тетрагидрофуран при температуре кипения растворителя или ниже.
Схема 2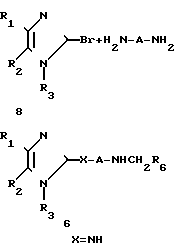


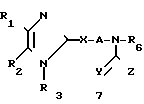
Соединения формулы (7), где Х представляет собой СН2, получают по схеме 6. Соединения формулы (18) получают путем превращения соответствующих имидазолов формулы (17), где R3 представляет собой алкил или подходящую защитную группу, в соответствующую щелочно-металлическую соль, путем добавления основания, такого как н-бутиллитий, с последующим алкилированием соответствующим галоидалкилом в растворителе, таком как тетрагидрофуран, в инертной атмосфере и при пониженных температурах. Соединения формулы (19) получают из соединений формулы (18) путем реакции с соответствующим образом замещенным амином в инертном растворителе, таком как толуол, ацетонитрил, тетрагидрофуран или N, N-диметилформамид, при температуре кипения растворителя или ниже. Имидазольные соединения формулы (20) получают реакцией вторичных аминов формулы (19) с соответствующим изоцианатом, хлорформатом, хлорангидридом или другим активированным производным карбоновой кислоты, как описано выше. Или имидазольные соединения формулы (20) можно получить реакцией щелочно-металлической соли соединений формулы (17) с подобранными соединениями формулы (16) в условиях, аналогичных описанным выше. Соединения формулы (7), где Х - СН2 и R3 - H, получают снятием защитной группы с соединений формулы (20), где R3 представляет собой защитную группу. Например, когда R3 представляет собой силиловую защитную группу, удаление этой группы с помощью фторида тетрабутиламмония в тетрагидрофуране при температуре дефлегмации приводит к получению соединений формулы (7), где Х представляет собой СН2.
Также соединения формулы (7), где Х - O, S, NH или СН2 и Y - H2, могут быть получены в результате реакции соединений, аналогичных соединениям формулы (18), с подходящим образом замещенным вторичным амином, HNCH2ZR6, в растворителе, таком как толуол, ацетонитрил, тетрагидрофуран или N,N-диметилформамид, при температурах кипения растворителя или ниже.
Схема 6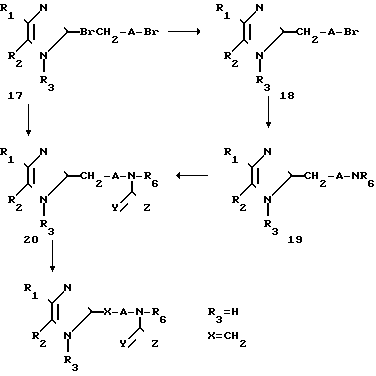
Сшитые фенильные соединения формулы (24) получены по схеме 7. Сшитые бис-бензоальдегидные соединения формулы (21) получены путем бисалкилирования дигалоидалкила с соответствующими функциональными группами замещенным салисальдегидом с использованием щелочного основания, такого как гидрид натрия, в инертном растворителе, таком как N,N-диметилформамид. α-гидроксикетоны формулы (22) получают с помощью известных бензиоинобразующих реакций, Walter S. Ide, Johannes S. Buck. Organireactions, vol. IV, p. 269, в которых используют цианид калия в водно-этанольном растворе при температуре дефлегмации.
Имидазолы формулы (23) получают с помощью хорошо известных из литературы способов, путем конденсации α-гидроксикетоновых соединений формулы (22) с тиомочевиной, или тиоцианатом аммония, или соответствующим образом замещенной тиомочевиной в подходящем растворителе, таком как N,N-диметилформамид, этанол или гексанол, при температуре кипения растворителя или ниже.
Соединения формулы (24) получают путем алкилирования щелочнометаллической соли имидазола (23) соединением формулы (16), как описано выше, и получают соединения формулы (24) непосредственно или с помощью соединения формулы M(A')CO2R, где R - СН3 или С2Н5, М представляет собой галоидную или тозилатную группу и А' представляет собой радикал, имеющий на одну метиленовую группу меньше, чем А, как описано на схеме 1.
Схема 7
Соединения формулы (1), схема 8, где Х представляет собой L, имеются на рынке или их можно получить вышеописанными способами.
Схема 8 -
-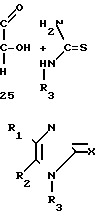
Альтернативно, соединения формулы (1), где Х представляет собой S, схема 8, можно получить из соответствующих 4-имидазолин-2-онов формулы (1), где Х представляет собой O, Org. S уп. CoII., Vol, II, p. 231, в результате реакции с ними реактивы Лавессона или дифосфорпентасульфида в подходящем растворителе, таком как толуол.
Как показано на схеме 9, 2-аминоимидазолы формулы (2) можно получить в результате реакции соответствующим образом замещенных α-аминокетонов формулы (27) с цианамидом (28). Соединения формулы (2) могут быть использованы для получения соединений формулы (1), как описано ранее в схеме 1.
Схема 9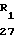 -
-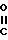 -
-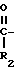 NHR3+H
NHR3+H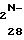 CN _____→
CN _____→ 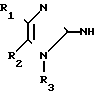
Как показано на схеме 10, соединения формулы (1), где Х - S(O)r и r представляет собой 1 или 2, можно получить путем окисления соединений формулы (29) известными из химической литературы способами. Например, окисление (29) одним эквивалентом перкислоты, такой как м-хлорпероксибензойная кислота, в подходящем растворителе, таком как хлористый метилен, при низких температурах дает сначала сульфоксиды формулы (30), а окисление (29) таким окислителем, как калий гидрогенперсульфат или Оксон в подходящем растворителе, таком как метанол, дает сульфоны формулы (31).
в подходящем растворителе, таком как метанол, дает сульфоны формулы (31).
Схема 10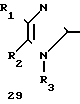 S-A-
S-A-
Альтернативно, соединения формулы (7), где R3 не является Н, схема 11, можно получить непосредственным алкилированием соединений формулы (7), где R представляет собой Н, в присутствии или в отсутствие основания, такого, как карбонат калия, пиридин, гидрид натрия, триэтиламин или т-бутокси калия, в соответствующем растворителе, таком как N,N-диметилформамид, тетрагидрофуран, пиридин или хлористый метилен.
Схема 11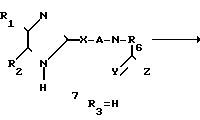
Фармацевтически приемлемые соли формулы (1) можно получить в соответствии с хорошо известными методиками получения солей. Физиологически приемлемые соли включают кислотно-аддитивные соли, например, хлористоводородные, серные, углекислые, трифторуглекислые, янтарной, лимонной бензолсульфокислоты соли.
П р и м е р 1. Получение N'-(2,4-дифторфенил)-N-[5-(4,5-дифенил-1Н-имидазол-2- илтио)пентил]-N- гептилмочевины.
Часть А. К раствору 4,5-дифенил-2-имидазолтиола (25,2 г, 0,1 моль) в N, N-диметилформамиде (250 мл) добавили по каплям раствор 5-бромпентаноата (23,73 мл, 31,35 г, 0,15 моль) в N,N-диметилформамиде (80 мл), реакционную смесь перемешивали при температуре дефлегмации в атмосфере азота в течение 18 ч. Реакционную смесь охладили, влили в 5%-ный бикарбонат натрия и лед и затем экстрагировали этилацетатом. Органические экстракты объединили, последовательно промыли 5% -ным бикарбонатом натрия, водой, насыщенным раствором хлористого натрия, высушили над сульфатом магния и сконцентрировали в вакууме. Остаток хроматографировали, используя смесь 7:3 гексан-этилацетат, полученное твердое вещество рекристаллизировали из ацетонитрила и перетерли с гексаном, в результате чего получили 5-(4,5-дифенил-1H-имидазол-2-илтио)-пентановой кислоты этиловый эфир (25, 95 г, 0,068 моль) в виде белого твердого вещества, т. пл. 87-89оС.
1H ЯМР (DМСО -d6) δ: 7,55 - 7,15 (м, 11H), 4,4 (к, 2H, J = 8Гц), 2,9 (т. 2H, J = 7 Гц), 2,3 (т, 2H, J = 7 Гц), 1,9 - 1,6 (м, 4H), 1,2 (т, 3H, J = 8 Гц).
Кроме того, эфиры, которые могут быть использованы как интермедиаты для получения соединений формулы (1), получают аналогично тому, как описано в заявке на патент США 244170 (ВР-6339).
Часть В. К раствору 5-(4,5-дифенил-1Н-имидазол-2-илтио)-пентановой кислоты этилового эфира (7,6 г, 0,02 моль) в этаноле (200 мл) добавили по каплям раствор едкого натра (7,6 г) в воде (200 мл) и реакционную смесь перемешивали при температуре дефлегмации в атмосфере азота в течение 3 ч. Затем реакционную смесь концентрировали до половины первоначального объема и затем экстрагировали диэтиловым эфиром. Эфирные экстракты удалили. Реакционную смесь подкислили до рН 1 с помощью 1 н. соляной кислоты и экстрагировали диэтиловым эфиром, объединенные органические экстракты обезводили над сульфатом магния и сконцентрировали в вакууме. Полученное твердое вещество рекристаллизовали из ацетонитрила и перетерли с гексаном, в результате чего получили 5-(4,5-дифенил-1Н-имидазол-2-илтио)-пента- новую кислоту (3,88 г, 0,011 моль) в виде белого твердого вещества, т.пл. 190-195оС.
1Н ЯМР (DMCO - d6) δ: 12,6 (c, 1H), 7,6-7,1 (м, 10Н), 3,3-3,1 (м, 2Н), 2,3-2,1 (м, 3Н), 1,8-1,6 (м, 4Н).
Дополнительные соли, которые можно использовать как интермедиаты для получения соединений формулы (1), получают аналогично, и они заявлены в одновременно поданной заявке на патент США N 244170.
Часть С. Способ 1. К раствору 5-(4,5-дифенил-1Н-имидазол-2-илтио)-пентановой кислоты (2,0 г, 0,0057 моль) в N,N-диметилформамиде (25 мл) добавили 1-гидроксибензотриазол гидрат (0,93 г, 0,0069 моль) и раствор гептиламина (1,10 мл, 0,86 г, 0,0074 моль) в N,N-диметилформамиде (10 мл). Реакционную смесь охладили до 0оС и к ней по частям в виде твердого вещества добавили дициклогексилкарбодиимид (1,42 г, 0,0069 моль). Реакционную смесь перемешивали в течение 2 ч при 0оС и затем перемешивали в течение 48 ч при комнатной температуре. Твердые частицы отфильтровывали и промывали N, N-диметилформамидом. Фильтрат сконцентрировали и остаток хроматографировали смесью 1:1 гексана с этилацетатом. Полученное твердое вещество рекристаллизовали из ацетонитрила и перетерли с гексаном, в результате чего получили 5-(4,5-дифенил-1Н-имидазол-2-илтио)-N-гептилпентамид (2,21 г, 0,0049 моль) в виде белого твердого вещества, т.пл. 104-106оС.
1Н ЯМР (CDCl3) δ: 11,6 (c, 1H), 7,6-7,1 (м, 10Н), 6,1-6,0 (м, 1Н), 3,1-2,8 (м, 4Н), 2,2 (т, 2Н, J = 7 Гц), 1,9-1,7 (м, 2Н), 1,7-1,5 (м, 2Н), 1,4-1,1 (м, 10Н), 0,9 (т, 3, J = 8 Гц).
Часть С. Способ 2. В раствор 5-(4,5-дифенил-1Н-имидазол-2-илтио)-пентановой кислоты (2,0 г, 0,0057 моль) в толуоле (35 мл) добавили гептиламин (1,63 мл, 1,27 г, 0,011 моль) и затем трифторэтерат бора (1,35 мл, 1,56 г, 0,011 моль) и реакционную смесь перемешивании при температуре дефлегмации в течение 120 ч, используя ловушку для влаги Дина-Старка. Реакционную смесь охладили, экстрагировали 0,1 н. едким натром, 0,1 н. соляной кислотой и водой и затем объединенные органические экстракты обезводили над сульфатом магния и сконцентрировали в вакууме. Остаток хроматографировали и обработали, как описано в части С, способ 1, в результате чего получили 5-(4,5-дифенил-1Н-имидазол-2-илтио)-N-гептилпентамид (2,35 г, 0,005 моль) в виде белого твердого вещества.
Часть D. К раствору литий алюминий гидрида (1,52 г, 0,04 моль) в безводном тетрагидрофуране (50 мл) добавили по каплям раствор 5-(4,5-дифенил-1Н-имидазол-2-илтио)-N-гептилпентамида (4,04 г, 0,009 моль) в тетрагидрофуране (25 мл) и реакционную смесь перемешивали при температуре дефлегмации в течение 18 ч. Реакционную смесь охладили до 0оС, погасили медленным, аккуратным и последовательным добавлением воды (1,52 мл), 15%-ного едкого натра (4,56 мл), воды (4,56 мл) и затем перемешивали при 0оС в течение 30 мин. Затем раствор обезводили над сульфатом магния и сконцентрировали в вакууме и остаток хроматографировали растворами 1:1, 3:1 и 1:1 этилацетат-метанол. Полученное желтое масло перетерли с холодным гексаном и получили N-/5-(4,5-дифенил-1Н-имидазол-2-илтио)-пентил/-1-гептанамид в виде белого твердого вещества. Раствор этого амина (0,80 г, 0,0018 моль) в диэтиловом эфире (20 мл) перетерли с достаточным количеством эфирной соляной кислоты (25 мл) для получения полного осаждения амина в виде соли соляной кислоты. Реакционную смесь перемешивали в течение 15 мин, недосадочную жидкость слили и получили смолообразную твердую массу, которую перетерли с горячим ацетонитрилом и затем с холодным гексаном и получили N-5-(4,5-дифенил-1Н-имидазол-2-илтио)-фенил/-1-гептанами- да хлоргидрат (0,82 г, 0,0017 моль) в виде белого твердого вещества, т.пл. 187-190оС.
1Н ЯМР (CDCl3) δ: 9,3 (с, 2Н), 7,7-7,3 (м, 10Н), 3,7-3,5 (м, 2Н), 3,0-2,7 (м, 4Н), 2,0-1,2 (м, 16Н), 0,9 (т, 3Н, J = 8 Гц).
Часть Е. К раствору N-[5-(4,5-дифенил-1Н-имидазол-2-илтио)-пентил]-1-гептана-мина (1,0 г, 0,0024 моль) в гексане (50 мл) добавили по каплям раствор 2,4-дифторфенилизоцианата (0,296 мл, 0,388 г, 0,0025 моль) в гексане (25 мл) и реакционную смесь перемешивали при температуре окружающей среды в течение 3 ч. Реакционную смесь сконцентрировали в вакууме и остаток хроматографировали, используя смесь 7: 3 гексан-этилацетат, и получили указанное в названии соединение (0,86 г, 0,0015 моль) в виде белого твердого вещества, т.пл. 96-98оС.
1Н ЯМР (CDCl3) δ: 10,8 (с, 1Н), 7,7-7,1 (м, 14Н), 3,4 (т, 2Н, J = 7 Гц), 3,2 (т, 2Н, J = 7 Гц); 3,0 (т, 2Н, J = 7 Гц), 1,9-1,4 (м, 16 Н), 0,9 (т, 3Н, J = 8 Гц).
П р и м е р 2. Получение N-[5-(4,5-дифенил-1Н-имидазол-2-илтио)пентил] -N- гептил-N'-фенилмочевины.
В раствор N-[5-(4,5-дифенил-1Н-имидазол-2-илтио)пентил]-1-гептанамида (1,0 г, 0,0024 моль) в гексане (50 мл) добавили по каплям раствор фенилизоцианата (0,27 мл, 0,298 г, 0,0025 моль) в гексане (25 мл) и реакционную смесь перемешивали при температуре окружающей среды в течение 4 ч. Реакционную смесь сконцентрировали в вакууме и остаток хроматографировали, используя смесь 6:4 гексан-этилацетат, и получили указанное в названии соединение (0,5 г, 0,009 моль) в виде желтого аморфного вещества.
1Н ЯМР (CDCl3) δ: 11,0 (C, 1H), 7,7-6,9 (м, 14Н), 6,4 (с, 1Н), 3,4 (т, 2Н, J = 7 Гц), 3,2 (т, 2Н, J = 7 Гц), 3,0 (т, 2Н, J = 7 Гц), 1,9-1,1 (м, 168), 0,9 (т, 3Н, J = 8 Гц).
П р и м е р 3. Получение N'-(2,4-дифторфенил)-N-[8-(4,5-дифенил-1Н-имидазол-2- илтио)октил]-N- гептилмочевины.
Часть А. В раствор 8-(4,5-дифенил-1Н-имидазол-2-илтио)-октановой кислоты (8,44 г, 0,02 моль) в хлористом метилене (100 мл) при 0оС добавили по частям в виде твердого вещества дициклогексилкарбодиимид (4,12 г, 0,02 моль) и реакционную смесь перемешивали при 0оС в течение 30 мин. В эту реакционную смесь добавили по каплям гептиламин (2,96 мл, 2,3 г, 0,02 моль) и реакционную смесь перемешивали при температуре дефлегмации в течение 72 ч. Реакционную смесь охладили, твердые частицы отфильтровали и промыли хлороформом. Фильтрат сконцентрировали в вакууме и остаток хроматографировали сначала смесью 7:3, а затем 1:1 гексан-этилацетат. Полученное твердое вещество рекристаллизовали из ацетонитрила и перетерли с гексаном, в результате чего получили 8-(4,5-дифенил-1Н-имидазол-2-илтио)-N- гептилоктанамид (3,28 г, 0,0067 моль) в виде белого твердого вещества, т.пл. 119-120оС.
1Н ЯМР (DMCO - d6) δ: 12,5 (c, 1H), 7,8-7,1 (м, 10Н), 3,2-2,9 (м, 4Н), 2,0 (т, 2Н, J = 7 Гц), 1,75-1,0 (м, 21Н), 1,0-0,8 (м, 3Н).
Часть В. К раствору литий алюминий гидрида (0,96 г, 0,025 моль) в безводном тетрагидрофуране (30 мл) добавили по каплям раствор 8-(4,5-дифенил-1Н-имидазол-2-илтио)-N-гептилоктанамида (2,82 г, 0,0057 моль) в тетрагидрофуране (15 мл) и реакционную смесь перемешивали при температуре дефлегмации в течение 18 ч. Реакционную смесь охлаждали до 0оС, погасили путем медленного аккуратного и последовательного добавления воды (0,96 мл), 15% -ного едкого натра (2,88 мл) и воды (2,88 мл) и затем перемешивали при 0оС в течение 30 мин. Затем раствор обезводили над сульфатом магния и остаток хроматографировали смесью 1:1 гексан-этилацетат, а затем использовали смеси 1: 0, 3:1 и 1:1 этилацетат-метанол и получили 8-(4,5-дифенил-1Н-имидазол-2-илтио-N-гептил-1-октан- амин (1,07 г, 0,0022 моль) в виде белого твердого вещества, т.пл. 87-89оС.
1Н ЯМР (CDCl3) δ: 7,6-7,2 (м, 11Н), 3,1 (т, 2Н, J = 7 Гц), 2,7-2,5 (м, 2Н), 1,8-1,1 (м, 25Н), 0,9 (т, 3Н, J = 8 Гц).
Часть С. К раствору 8-(4,5-дифенил-1Н-имидазол-2-илтио)-N-гептил-1-октанамина (0,5 г, 0,001 моль) в гексане (25 мл) добавили по каплям раствор 2,4-дифторфенилизоцианата (0,15 мл, 0,194 г, 0,00125 моль) в гексане (10 мл) и реакционную смесь перемешивали при температуре окружающей среды в течение 3 ч. Реакционную смесь сконцентрировали в вакууме и остаток хроматографировали, используя смесь 8:2 гексан-этилацетат, и получили твердое вещество, которое перетерли с холодными этилацетатом, затем гексаном и получили указанное в названии соединение (0,18 г, 0,00028 моль) в виде белого твердого вещества, т.пл. 89-91оС.
1Н ЯМР (DMCO - d6) δ: 12,5 (c, 1H), 7,9 (с, 1Н), 7,5-7,1 (м, 10Н), 3,3-3,1 (м, 5Н), 1,8-1,2 (м, 17Н), 0,9 (т, 3Н, J = 8 Гц).
П р и м е р 4. Получение N-бутил-N'-(2,4-дифторфенил)-N-[8-(4,5-дифенил-1Н -имидазол-2-илтио) октил]мочевины.
Часть А. В растворе 8-(4,5-дифенил-1Н-имидазол-2-илтио)-октановой кислоты (4,4 г, 0,0125 моль) в хлористом метилене (65 мл) при 0оС добавили по частям в виде твердого вещества дициклогексил карбодиимид (2,3 г, 0,011 моль) и реакционную смесь перемешивали при 0оС в течение 30 мин. В реакционную смесь добавили по каплям раствор бутиламина (1,24 мл, 0,92 г, 0,012 моль) в хлористом метилене (15 мл) и реакционную смесь перемешивали при температуре дефлегмации в течение 18 ч. Реакционную смесь охладили, твердое вещество отфильтровали и промыли хлористым метиленом. Фильтрат сконцентрировали в вакууме и остаток хроматографировали, используя смесь 7:3 и 1:1 гексан-этилацетат. Полученное твердое вещество рекристаллизовали из ацетонитрила и перетерли с гексаном, в результате чего получили N-бутил-8-(4,5-дифенил-1Н-имидазол-2-ил-тио)октанамид (1,43 г, 0,003 моль) в виде белого твердого вещества, т.пл. 136-137оС.
1Н ЯМР (DMCO-d6) δ: 12,5 (c, 1H), 7,8-7,7 (м, 1Н), 7,7-7,1 (м, 10Н), 3,2-2,9 (м, 4Н), 2,0 (т, 2Н, J = 7 Гц), 1,8-1,1 (м, 14Н), 0,9 (т, 3Н, J = 8 Гц).
Часть В. К раствору литий алюминий гидрида (0,46 г, 0,012 моль) в безводном тетрагидрофуране (15 мл) добавили по каплям раствор N-бутил-8-(4,5-дифенил-1Н-имидазол-2-илтио)-октанамида (1,20 г, 0,0027 моль) в тетрагидрофуране (8 мл) и реакционную смесь перемешивали при температуре дефлегмации в течение 18 ч. Реакционную смесь охладили до 0оС и погасили путем медленного, осторожного последовательного добавления воды (0,46 мл), 15%-ного едкого натра (1,38 мл) и воды (1,38 мл) и затем реакционную смесь перемешивали при 0оС в течение 30 мин. Раствор обезводили над сульфатом магния и сконцентрировали в вакууме, остаток хроматографировали, используя смесь 1:1 гексан-этилацетат и затем смесь 1:0, 8:2 и 1:1 этилацетатметанол. Оставшееся твердое вещество перетерли с гексаном и получили N-бутил-3-(4,5-дифенил-1Н-имидазол-2-илтио)-октанамин (0,45 г, 0,001 моль) в виде белого твердого вещества, т.пл. 75-78оС.
1Н ЯМР (CDCl3) δ: 7,6-7,1 (м, 10Н), 3,1 (т, 2Н, J = 7 Гц), 2,5 (т, 2Н, J = 7 Гц), 1,7-1,0 (м, 16Н), 0,9 (т, 3Н, J = 8 Гц).
Часть С. К раствору N-бутил-8-(4,5-дифенил-1Н-имидазол-2-илтио)-октанамин (0,2 г, 0,00045 моль) в гексане (15 мл) прибавили по каплям раствор 2,4-дифторфенилизоцианат (0,65 мл, 0,085 г, 0,0005 моль) в гексане (5 мл) и реакционную смесь перемешивали при температуре окружающей среды в течение 3 ч. Затем реакционную смесь сконцентрировали в вакууме и остаток хроматографировали смесью 7:3 гексан-этилацетат и оставшееся твердое вещество рекристаллизовали из ацетонитрила и перетерли с гексаном, в результате чего получили соединение, указанное в названии (0,138 г, 0,00023 моль) в виде белого твердого вещества, т.пл. 114-115оС.
1Н ЯМР (CDCl3) δ: 8,1-7,9 (м, 1Н), 7,6-7,2 (м, 11Н), 6,95-6,75 (м, 2Н), 6,5-6,4 (м, 1Н), 3,4-3,1 (м, 6Н), 1,8-1,3 (м, 16Н), 1,0 (т, 3Н, J = 8 Гц).
П р и м е р 5. Получение N'-(2,4-диметоксифенил)-N-[5-(4,5-дифенил-1Н-имидазол-2- илтио)пентил]-N- гептилмочевины.
К раствору N-/5-(4,5-дифенил-1Н-имидазол-2-илтио)-пентил/-1-гептанамина (0,75 г, 0,0017 моль), полученному в соответствии с методикой примера 1, часть D, в гексане (40 мл) прибавили по каплям раствор 2,4-диметоксифенилизоцианата (0,358 г, 0,002 моль) в гексане (20 мл) и реакционную смесь перемешивали при температуре окружающей среды в течение 4,5 ч. Реакционную смесь сконцентрировали в вакууме и остаток хроматографировали, используя смесь 7: 3 гексан-этилацетат. Полученное твердое вещество перетерли с гексаном и получили указанное в названии соединение (0,83 г, 0,0014 моль) в виде стекловидного твердого вещества.
1Н ЯМР (CDCl3) δ: 7,7-7,1 (м, 10Н), 6,8-6,1 (м, 3Н), 3,8 (с, 3Н), 3,7 (с, 3Н), 3,45 (с, 1Н), 3,4-3,3 (м, 2Н), 3,2 (т, 2Н, J = 7 Гц), 3,0 (т, 2Н, J = 7 Гц), 1,8-1,1 (м, 16Н), 0,9 (т, 3Н, J = 8 Гц).
П р и м е р 6. Получение N'-(2,4-дифторфенил)-N-гептил-N-[5-(1-метил-4,5-дифенил-1Н-имидазол- 2-илтио)пентил]мочевины.
К раствору карбоната калия (0,056 г, 0,00042 моль) в безводном тетрагидрофуране (10 мл) добавили по частям твердое вещество N'-(2,4-дифторфенил)-N-[5-(4,5-дифенил-1Н-имидазол-2-илтио)пентил] -N- гептилмочевину (0,25 г, 0,00042 моль) и реакционную смесь перемешивали при комнатной температуре в течение 10 мин. В эту реакционную смесь добавили по каплям иодистый метил (0,039 мл, 0,0895 г, 0,00063 моль) и реакционную смесь перемешивали в течение 18 ч при температуре окружающей среды. Реакционную смесь затем обработали N, N-диметилформамидом (1,0 мл) и иодистым метилом (0,1 мл) и полученную смесь перемешивали при температуре дефлегмации еще 24 ч. Реакционную смесь охладили, влили в воду и экстрагировали этилацетатом. Органические экстракты объединили, обезводили над сульфатом магния и сконцентрировали в вакууме. Остаток хроматографировали, используя смесь 3:7 гексан-этилацетат, и получили указанное в названии соединение (0,13 г, 0,00022 моль) в виде желтого масла.
1Н ЯМР (CDCl3) δ: 8,1-8,0 (м, 1Н), 7,5-7,1 (м, 1Н), 6,9-6,7 (м, 2Н), 6,4 (с, 1Н), 3,5 (с, 3Н), 3,4-3,2 (м, 5Н), 1,9-1,2 (м, 17Н), 0,9 (т, 3Н, J = 8 Гц).
П р и м е р 7. Получение N-[5-(4,5-дифенил-1Н-имидазол-2-илтио)пентил] -N- гептил-N'-метилмочевины.
К раствору N-[5-(4,5-дифенил-1Н-имидазол-2-илтио)пентил]-1-гептанамина (0,30 г, 0,0007 моль) в гексане (15 мл) добавили метилизоцианат (0,06 мл, 0,057 г, 0,001 моль) и реакционную смесь перемешивали при температуре окружающей среды в течение 18 ч. Реакционную смесь сконцентрировали в вакууме и остаток хроматографировали, используя смесь 1:1 гексан-этилацетат. Полученное масло перетерли с гексаном и получили указанное в названии соединение (0,23 г, 0,00047 моль) в виде белого твердого вещества, т.пл. 93-96оС.
1Н ЯМР (CDCl3) δ: 7,6-7,2 (м, 11Н), 4,35-2,7 (м, 9Н), 1,9-1,2 (м, 16Н), 0,9 (т, 3Н, J = 8 Гц).
П р и м е р 8. Получение N-[5-(4,5-дифенил-1Н-имидазол-2-илтио)пентил] -N- гептил-N'-пропилмочевины.
К раствору N-[5-(4,5-дифенил-1Н-имидазол-2-илтио)пентил] -1-пентамина (0,36 г, 0,0008 моль) в гексане (15 мл) добавили пропилизоцианат (0,094 мл, 0,085 г, 0,001 моль) и реакционную смесь перемешивали при температуре окружающей среды в течение 4 ч. Затем реакционную смесь обработали дополнительным количеством пропилизоцианата (0,094 мл, 0,085 г, 0,001 моль) и перемешивали ее при температуре окружающей среды в течение ночи и затем при температуре дефлегмации в течение 72 ч. Затем реакционную смесь сконцентрировали в вакууме и остаток хроматографировали, используя 2:8 смесь гексан-этилацетат. Полученное масло перетерли с гексаном и получили указанное в названии соединение (0,8 г, 0,00015 моль) в виде белого твердого вещества, т.пл. 78-80оС.
1Н ЯМР (СDCl3) δ: 7,6-7,2 (м, 10Н), 4,4 (т, 1Н, J = 7 Гц), 3,4-2,9 (м, 8Н), 1,9-1,1 (м, 19Н), 1,0-0,75 (м, 6Н).
П р и м е р 9. Получение N'-(2,4-дифторфенил)-N-[2-(4,5-дифенил-1Н-имидазол-2- илтио)этил]-N- пропилмочевины.
Часть А. К раствору бромацетилхлорида (25,51 мл, 48,67 г, 0,31 моль) в хлористом метилене (200 мл) при температуре - 15оС добавили по каплям раствор пропиламина (24,62 мл, 17,7 г, 0,3 моль) в хлористом метилене (100 мл) и реакционную смесь перемешивали при 0оС в течение 30 мин и затем перемешивали при температуре окружающей среды в течение 30 мин. Затем реакционную смесь влили в воду и экстрагировали хлористым метиленом. Органические остатки обезводили над сульфатом магния и сконцентрировали в вакууме. Остаток подвергли дистилляции и получили бром-N-пропилацетамид в виде прозрачной жидкости, т.пл. 138-142оС.
1Н ЯМР (CDCl3) δ: 7,1 (c, 1H), 3,9 (д, 2Н, J = 6 Гц), 3,3 (м, 2Н), 1,6 (м, 2Н), 0,9 (т, 3Н, J = 7 Гц).
Часть В. Часть гидрида натрия, 60% в минеральном масле (30,4 г, 0,01 моль) дважды промыли гексаном (10 мл) и затем гексан заменили N,N-диметилформамидом (100 мл). К этому раствору добавили по частям твердый иодистый натрий (0,4 г, 0,003 моль) и затем по каплям добавили раствор дифенилимидазола (2,52 г, 0,01 моль) в N,N-диметилформамиде (10 мл) и затем по каплям добавили раствор бром-N-пропилацетамида (1,80 г, 0,01 моль) в N,N-диметилформамиде (10 мл). Реакционную смесь перемешивали при температуре дефлегмации в течение 18 ч, затем охладили и осторожно влили в ледяную воду и затем экстрагировали этилацетатом. Объединенные органические экстракты промыли рассолом, обезводили над сульфатом магния и сконцентрировали в вакууме. Остаток хроматографировали, используя смесь 1:1 гексан-этилацетат и полученное твердое вещество рекристаллизовали из ацетонитрила, в результате чего получили 2-(4,5-дифенил-1Н-имидазол-2-илтио)-N-пропил- ацетамид в виде белого твердого вещества, т.пл. 183-185оС.
1Н ЯМР (DMCO - d6) δ: 12,6 (с, 1Н), 8,3 (с, 1Н), 7,5-7,1 (м, 10Н), 3,8 (с, 2Н), 3,0 (к, 2Н, J = 5 Гц); 1,4 (секстет, 2Н, J = 9 Гц), 0,8 (т, 3Н, J = 6 Гц).
Часть С. Используя методику примера 1, часть D, но из 2-(4,5-дифенил-1Н-имидазол-2-илтио)-N-пропилацетамида, получили N-/2-(4,5-дифенил-1Н-имидазол-2-илтио)-этил/-1-пропанамин (0,28 г, 0,00083 моль) в виде масла.
1Н ЯМР (CDCl3) δ: 7,9-7,6 (м, 2Н), 7,5-7,1 (м, 10Н), 3,1 (с, 4Н), 2,6 (т, 2Н, J = 6 Гц), 1,4 (секстет, 2Н, J = 12 Гц), 0,8 (т, 3Н, J = 9 Гц).
Часть D. Используя методику примера 1, часть Е, и N-/2-(4,5-дифенил-1Н-имидазол-2-илтио)этил/-1-пропанамин, получили указанное в названии соединение (0,20 г, 0,00045 моль) в виде белого твердого вещества, т.пл. 189-190оС.
1Н ЯМР (CDCl3) δ: 11,6-11,2 (c, 1H), 7,8-7,6 (c, 1H), 7,6-6,9 (м, 10Н), 6,8-6,6 (м, 2Н), 3,8 (т, 3Н, J = 7 Гц), 3,4 (т, 2Н, J = 6,5 Гц), 3,2 (т, 2Н, J = 6 Гц), 1,8-1,6 (м, 4Н), 1,0 (т, 3Н, J = 7,5 Гц).
П р и м е р 118. Получение N-{5-[4,5-бис(4-метоксифенил)-1Н-имидазол-2-ил- тио]-пентил}-N'-(2,4- дифторфенил)-N-гептилмочевины.
Часть А. Раствор γ-валеролактона (25,0 г, 0,249 моль) в толуоле (50 мл) и н-гептиламине (35,96 г, 0,312 моль) нагревали до температуры дефлегмации в течение 18 ч в атмосфере азота. Реакционной смеси дали остыть до температуры окружающей среды, разбавили этилацетатом (300 мл), промыли 1 н. водным раствором соляной кислоты (50 мл), водой, рассолом, обезводили над сульфатом магния и сконцентрировали, в результате чего получили твердое белое вещество. Полученный продукт подвергли кристаллизации из смеси этиловый эфир-гексан и получили N-гептил-5-гидроксипентанамид (41,8 г, 0,194 моль) в виде белых пластин, т.пл. 55-56оС.
1Н ЯМР (CDCl3) δ: 6,06 (шир.с, 1Н), 3,61 (т, 2Н), 3,24 (к, 2Н), 3,19 (шир.с., 1Н), 2,19 (т, 2Н, 1,80-1,23 (м, 14Н), 0,866 (т, 3Н).
Часть В. К раствору литий алюминий гидрида (6,7 г, 0,176 моль) в безводном тетрагидрофуране (300 мл) по каплям в атмосфере азота добавили раствор N-гептил-5-гидроксипентамида (19,0 г, 0,088 моль) в безводном тетрагидрофуране (100 мл). Реакционная смесь была нагрета до температуры дефлегмации в течение 18 ч, ей дали остыть до комнатной температуры и медленно влили в перемешиваемую смесь 10% водного раствора сульфата натрия (400 мл) и льда (200 мл). Полученную смесь профильтровали через Целит и фильтрат экстрагировали этилацетатом (2 х 500 мл). Органические слои объединили, промыли водой, рассолом, обезводили над сульфатом магния, сконцентрировали и получили вязкое желтое масло. Продукт подвергли кристаллизации из гексана и получили N-(5-гидроксипентид)-N-гептиламин (15,2 г, 0,075 моль) в виде белого порошка, т.пл. 47-48оС.
и фильтрат экстрагировали этилацетатом (2 х 500 мл). Органические слои объединили, промыли водой, рассолом, обезводили над сульфатом магния, сконцентрировали и получили вязкое желтое масло. Продукт подвергли кристаллизации из гексана и получили N-(5-гидроксипентид)-N-гептиламин (15,2 г, 0,075 моль) в виде белого порошка, т.пл. 47-48оС.
1Н ЯМР (CDCl3) δ: 8,03 (м, 1Н), 6,88-6,59 (м, 2Н), 6,45 (шир.с, 1Н), 3,68 (т, 2Н), 3,33 (м, 4Н), 1,81-1,22 (м, 16Н), 0,907 (т, 3Н).
Часть D. К раствору N'-(2,4-дифторфенил)-N-гептил-N-5-гидроксипентилмоче- вины (15,0 г, 0,042 моль) и тетрабромметана (16,75 г, 0,051 моль) в хлористом метилене (350 мл) в атмосфере азота при температуре окружающей среды медленно добавили раствор трифенилфосфина (13,24 г, 0,051 моль) в хлористом метилене (100 мл). Реакционную смесь перемешивали в течение 3 ч, сконцентрировали в вакууме и получили сырое вязкое масло. Этот продукт подвергли очистке с помощью пламенной хроматографии на силикагеле (400 мл), элюируя смесь гексан-этилацетат (90:10 по объему), и получили N-(5-бромпентил)-N'-(2,4-дифторфенил)-N-гептилмочевину в виде вязкого бесцветного масла (17,5 г, 0,042 моль).
1Н ЯМР (CDCl3) δ: 8,14-8,0 (м, 1Н), 6,92-6,79 (м, 2Н), 6,35 (шир. с, 1Н), 3,49-3,25 (м, 6Н), 1,99-1,26 (м, 16Н), 0,915 (т, 3Н).
Часть Е. В суспензию гидрида натрия (0,88 г, 60% дисперсия в минеральном масле, 0,0022 соль) (промыта от минерального масла гексаном) в N,N-диметилформамиде (15 мл) в атмосфере азота при охлаждении до 0оС медленно добавили раствор 4,5-/бис-(4-метоксифенил)-1Н-имидазол/-2- тиона (0,63 г, 0,002 моль) в N,N-диметилформамиде (5 мл). Реакционную смесь перемешивали в течение 2 часов и затем добавили к ней раствор N-(5-бромпентил)-N'-(2,4-дифторфенил)-N-гептилмочевины (0,845 г, 0,002 моль) в N,N-диметилформамиде (3 мл). Реакционной смеси дали нагреться до температуры окружающей среды, перемешивали еще 2 ч, влили в воду (50 мл) и экстрагировали этилацетатом (2 х 50 мл). Объединенные органические экстракты промыли водой, рассолом, обезводили над сульфатом магния, сконцентрировали и получили вязкое масло. Этот продукт очистили с помощью пламенной хроматографии на силикагеле (100 мл), элюируя смесью гексан-этилацетат (70:30 по объему), и получили указанное в названии соединение в виде чистой желтой пены (0,98 г, 0,0015 моль).
1Н ЯМР (CDCl3) δ: 10,15 (шир. с, 1Н), 7,87-7,76 (м, 1Н), 7,51 (д, 2Н), 7,3 (д, 2Н), 6,86-6,6 (м, 6Н), 6,42 (д, 1Н), 3,8 (с, 6Н), 3,4 (т, 2Н), 3,26 (т, 2Н), 2,99 (т, 2Н), 1,84-1,25 (м, 16Н), 0,89 (т, 3Н).
П р и м е р 191. Получение N-(2,4-дифторфенил)-N'-[5-(4,5-дифенил-1Н- имидазол-2-илтио)пентил]мочевины.
Часть А. Смесь 5-(4,5-дифенил-1Н-имидазол-2-илтио)пентановой кислоты (4,0 г, 0,011 моль) и мочевины (1,36 г, 0,023 моль) нагревали до 179-180оС в течение 5 ч. Охлажденную реакционную смесь разделили в карбонате натрия (5% ) и экстрагировали хлороформом. Органические слои промыли насыщенным раствором хлористого натрия и затем обезводили над сульфатом магния и сконцентрировали в вакууме. Остаток хроматографировали смесью 9:1 этил-ацетат и получили 5-(4,5-дифенил-1Н-имидазол-2-илтио)пентамид (0,73 г, 0,002 моль) в виде белого твердого вещества, т.пл. 136-138оС.
1ЯМР (CDCl3) δ: 10,65 (с, 1Н), 7,7-7,2 (м, 10Н), 5,9 (с, 1Н), 5,4 (с, 1Н), 3,0 (т, 2Н, J = =7,4 Гц), 2,3 (т, 2Н, J = 8 Гц), 2,0-1,6 (м, 4Н).
Часть В. Используя способ примера 1, часть D, из 5-(4,5-дифенил-1Н-имидазол-2-илтио)пентанамида (2,0 г, 0,0057 моль), получили 5-(4,5-дифенил-1Н-имидазол-2-илтио)-1-пентанамин (0,32 г, 0,00095 моль) в виде дубильного твердого вещества, т.пл. 111-113оС.
1ЯМР (DMCO - d6) δ: 7,5-7,2 (м, 12Н), 3,1 (т, 2Н, J = 7,2 Гц), 2,5 (т, 2Н, J = 6,2 Гц), 1,8-1,3 (м, 7Н).
Часть С. Раствор 5-(4,5-дифенил-1Н-имидазол-2-илтио)-1-пентанамина (0,34 г, 0,001 моль) и 2,4-дифторфенилизоцианата (0,24 мл, 0,31 г, 0,002 моль) в толуоле (10 мл) перемешивали при температуре окружающей среды в течение 120 ч. Раствор сконцентрировали в вакууме и получили остаток (0,53 г), который хроматографировали смесью 1:1 гексан-этилацетат. Полученное твердое вещество перетерли с холодным ацетонитрилом и получили указанное в названии соединение (0,13 г, 0,0026 моль) в виде белого твердого вещества, т.пл. 187-188оС.
1Н ЯМР (DMCO-d6) δ: 12,5 (с, 1Н), 8,2-8,0 (м, 2Н), 7,5-7,1 (м, 11Н), 7,0-6,9 (м, 1Н), 6,6-6,5 (м, 1Н), 3,2-3,0 (м, 4Н), 1,8-1,3 (м, 6Н).
П р и м е р 207. Получили N-[5-(4,5-дифенил-1Н-имидазол-2-илтио)пентил] -N-октил-N-фенилмочевины.
К раствору N-[5-(4,5-дифенил-1Н-имидазол-2-илтио)пентил] -бензоламина (0,41 г, 0,001 моль) в толуоле (25 мл) добавили н-октилизоцианат (0,23 г, 0,0015 моль). Реакционную смесь перемешивали при температуре дефлегмации в течение 18 ч и затем растворитель отогнали в вакууме. Остаток (1,0 г) хроматографировали, используя смесь 7: 3 гексан-этилацетат. Полученное твердое вещество перетерли с гексаном и получили указанное в названии соединение (0,32 г, 0,00056 моль) в виде белого твердого вещества, т.пл. 74-76оС.
1Н ЯМР (CDCl3) δ: 11,8 (c, 1H) 7,75-7,1 (м, 15Н), 4,3 (т, 1Н, J = 6,0 Гц), 3,8 (т, 2Н, J = 7,0 Гц), 3,0 (квинтет, 4Н, J = 6,0 Гц), 1,9-0,9 (м, 18 Гц), 0,8 (т, 3Н, J = 7 Гц).
П р и м е р 209. Получение N-{5-[4,5-бис(4-гидроксифенил)-1Н-имидазол-2-илтио]фенил}-N'-(2,4- дифторфенил)-N-гептилмочевины.
В перемешиваемый раствор N-{5-[4,5-бис(4-метоксифенил)-1Н-имидазол-2-илтио] пентил}-N'-2,4-дифторфенил)- N-гептилмочевины (0,78 г, 0,0012 моль) в хлористом метилене (30 мл), охлажденный до -78оС в атмосфере азота, добавили 1М раствор трехбромистого бора в хлористом метилене (3,6 мл). Реакционную смесь перемешивали в течение 1 ч при 0оС, затем влили в лед (100 мл) и экстрагировали этилацетатом (2 х 50 мл). Объединенные органические слои промыли 10%-ным водным раствором NaHCO3 (50 мл), водой, рассолом, обезводили над сульфатом магния, сконцентрировали в вакууме и получили сырое масло. Продукт очистили с помощью пламенной хроматографии на силикагеле (100 мл), используя в качестве элюента смесь гексан-этилацетат (40:60 по объему), и получили белую пену, т.пл. 110-112оС (0,5 г, 0,00008 моль).
1Н ЯМР (DMCO-d6) δ: 12,22 (шир.с, 1Н), 9,55 (шир.с, 1Н) 9,32 (шир.с, 1Н), 7,92 (с, 1Н), 7,45-6,6 (м, 11Н), 3,24 (м, 4Н), 3,06 (т, 2Н), 1,77-1,17 (м, 16Н), 0,88 (т, 3Н).
П р и м е р 211. Получение N-{5-(1H,9H,дибенз-/4,5: 8,9//1,3/диоксонин/6, 7-d/имидазол-2-илтио)пентил] -N'-(2,4-дифторфенил)-N-гептилмочевины.
Часть А. К суспензии гидрида натрия (промыли от минерального масла гексаном) (2,45 г, 80% масляной дисперсии, 0,081 моль) в безводном N,N-диметилформамиде (50 мл) в атмосфере азота при охлаждении до 0оС медленно добавили раствор салисальдегида (10,0 г, 81,9 моль) в безводном N,N-диметилформамиде (10 мл). Реакционную смесь перемешивали при 0оС в течение 2 ч и добавили к ней дииодометан (11,3 г, 0,041 моль). Реакционной смеси дали нагреться до температуры окружающей среды влили ее в 1 н. водный раствор соляной кислоты (100 мл) и экстрагировали этилацетатом (2 х 100 мл). Объединенные органические экстракты промыли водой, рассолом, обезводили над сульфатом магния, сконцентрировали и получили твердое вещество. Продукт очистили пламенной хроматографией на силикагеле (300 мл) элюируя хлористым метиленом (100% ), и получили 2,2'-(метилендиокси)-бис-(2-бензальдегид) в виде белого твердого кристаллического вещества, т.пл. 131-133оС (5,1 г, 0,0199 моль).
1Н ЯМР (CDCl3) δ: 10,47 (c, 2H), 7,87 (д, 2Н), 7,68-7,54 (м, 2Н), 7,21 (д, 2Н), 7,15 (т, 2Н), 6,02 (с, 2Н).
Часть В. Смесь 2,2'-(метилендиокси)-бис-(2-бензальдегида) (5,0 г, 0,0195 моль), цианида калия (0,63 г, 0,0975 моль) в этаноле (75 мл) и воде (50 мл) нагревали до температуры дефлегмации в течение 6 ч. Реакционной смеси дали остыть до температуры окружающей среды, сконцентрировали в вакууме и полученный водный остаток разделили между этилацетатом и водой. Органический слой промыли водой, рассолом, обезводили над сульфатом магния, сконцентрировали и получили вязкое масло. Продукт очистили пламенной хроматографией на силикагеле (250 мл), элюируя смесью гексан-этилацетат (80:20 по объему) и получили 13-гидроксидибензо/d/n//1,3/диоксонин-12(13Н)-он в виде кристаллического твердого вещества, т.пл. 129-130оС (2,5 г, 0,0975 моль).
1Н ЯМР (DMCO-d6) δ: 7,49 (т, 2Н), 7,29-7,08 (м, 6Н), 6,4 (д, 1Н, 5,97 (д, 1Н), 5,92 (д, 1Н), 5,24 (д, 1Н).
Часть С. Раствор 13-гидрокси-дибензо/d/п//1,3/-диоксонин-12(13Н)-она (2,0 г, 0,0078 моль), тиомочевины (0,82 г, 0,0108 моль) и гексанола (25 мл) в колонке с ситами 4А и конденсаром нагревали до 160оС в течение 20 ч в атмосфере азота. Реакционной смеси дали доохладиться до температуры окружающей среды и разбавили ее этиловым эфиром (100 мл), в результате чего получили твердое вещество. Это твердое вещество промыли этиловым эфиром, обезводили и получили N-(1H,9H-диабенз-/4,5:8,9//1,3/диоксонин/6,7-d/имидазол)-2-тион в виде белого кристаллического порошка (1,6 г, 0,00539 моль), т.пл. 250оС.
1Н ЯМР (DMCO-d6) δ: 12,5 (c, 2H), 7,43-7,08 (м, 8Н), 6,2-5,0 (шир.д, 2Н).
Часть D. Используя способ примера 118, часть Е, из N-(1H,9H-дибенз-/4,5-8,9//1,3/диоксонин/6,7-d/имидазол)-2-тиона получили указанное в названии соединение в виде белой пены, т.пл. 65-70оС (0,85 г, 0,00134 моль).
1Н ЯМР (CDCl3) δ: 10,35-10,10 (шир.с., 1Н), 7,56 (м,1Н), 7,30-6,95 (м, 10Н), 6,4 (д, 1Н), 5,70-5,20 (шир.с, 2Н), 3,40-3,19 (м, 4Н), 3,08 (т, 2Н), 1,85-1,23 (м, 16Н), 0,88 (т, 3Н).
П р и м е р 212. Получение N'-[5-(1H-дибенз/2,3,6,7/окседин/4,5-d/имидазол-2- илтио)пентил]-N-(2,4- дифторфенил)-N-гептилмочевины.
По способу примера 118, часть Е, используя 1Н-дибенз/2,3: 6,7/окседин/4,5-d/имидазол/-2-тион, выделили указанное в названии соединение в виде белого порошка, т.пл. 82-87оС (0,36 г, 0,00059 моль).
1Н ЯМР (CDCl3) δ: 9,75-8,5 (шир.с, 2Н), 7,84-7,59 (м, 3Н), 7,43-7,05 (м, 6Н), 5,13-6,53 (м, 3Н), 3,43-3,13 (м, 6Н), 1,65-1,20 (м, 16Н), 0,88 (т, 3Н).
Перечисленные в табл. 1-3 соединения были получены или могут быть получены аналогично по описанным выше методикам.
П р и м е р 267. Получение N'-(2,4-дифторфенил)-N-[5-(4,5-дифенил-1Н-имидазол-2-илтио)пентил]-N- гептилтиомочевины.
По способу примера 1, часть Е, используя 2,4-дифторфенилизотиоцианат (0,14 г, 0,0008 моль), получили указанное в названии соединение (0,19 г, 0,00031 моль) в виде белого твердого вещества, т.пл. 116-118оС.
1Н ЯМР (CDCl3) δ: 9,5-9,4 (c, 1H), 7,8-7,1 (м, 11Н), 7,06-6,7 (м, 3Н), 3,8 (т, 2Н, J = 7,7 Гц), 3,6 (т, 2Н, J = 7,8 Гц), 3,11 (т, 2Н, J = 7 Гц), 1,9-1,1 (м, 18Н), 0,9 (т, 3Н, J = 4 Гц).
П р и м е р 269. Получение N'-(2,4-дифторфенил)-N-[5-(4,5-дифенил-1Н-имида- зол-2-илсульфинил)-пентил] N-гептилмочевины.
В раствор N'-(2,4-дифторфенил)-N-[5-(4,5-дифенил-1Н-имидазол-2-илтио)пентил]- N- гептилмочевины (0,59 г, 0,001 моль) в хлористом метилене (50 мл), охлажденный до -78оС, по каплям добавили раствор мета-хлорандбензойной кислоты (0,286 г, 0,0017 моль) в хлористом метилене (10 мл). Реакционную смесь перемешивали при -78оС в течение 1 ч и затем дали ей нагреться до температуры окружающей среды. Затем реакционную смесь охладили до 0оС и добавили в нее по каплям раствор насыщенного бисульфита натрия. Слои разделили и органический слой промыли насыщенным раствором бисульфита натрия. Слои разделили и раствор хлористого натрия обезводили над сульфатом магния и сконцентрировали в вакууме. Остаток (0,76 г) хроматографировали, используя смесь 1:1 гексан-этилацетат, и получили указанное в названии соединение (0,43 г, 0,00071 моль) в виде желтого твердого вещества, т.пл. 77-79оС.
1Н ЯМР (CDCl3) δ: 8,1-7,9 (м, 1Н), 7,6-7,2 (м, 10Н), 6,9-6,7 (м, 2Н), 6,4 (д, 1Н, J = 3,3 Гц), 3,4-3,1 (м, 6Н), 2,0-1,1 (м, 18Н), 0,9 (т, 3Н, J = 6,4 Гц).
П р и м е р 272. Получение N'-(2,4-дифторфенил)-N-[5-(4,5-дифенил-1Н-имидазол-2-ил)-сульфонил/пентил]- N-гептилмочевины.
В раствор N'-(2,4-дифторфенил)-N-[5-(4,5-дифенил-1Н-имидазол-2-илтио)пентил] -N- гептилмочевины (0,11 г, 0,00019 моль) в метаноле (5 мл) добавили по частям твердое вещество ОксонТМ (0,234 г, 0,00038 моль) и реакционную смесь перемешивали при температуре окружающей среды в течение 7 ч. Твердые частицы отфильтровали и промыли метанолом. Фильтрат сконцентрировали в вакууме и остаток хроматографировали смесью 6:4 гексан-этилацетат и получили указанное в названии соединение (0,06 г, 0,000096 моль) в виде стеклообразного бесцветного твердого вещества, т.пл. 66-68оС.
1Н ЯМР (CDCl3) δ: 7,85-7,75 (м, 1Н), 7,6-7,1 (м, 11Н), 6,8-6,6 (м, 2Н), 6,4 (с, 1Н), 3,4 (т, 4Н, J = 10 Гц), 3,25 (т, 2Н, J = 7 Гц), 1,9-1,75 (м, 2Н), 1,75-1,4 (м, 6Н), 1,4-1,1 (м, 8Н), 0,9 (т, 3Н, J = 8 Гц).
П р и м е р 329. Получение N'-(2,4-дифторфенил)-N-[5-(4,5-дифенил-1Н-имидазол-2-иламино)пентил]- N-гептилмочевины.
Часть А. Раствор 2-бром-4,5-дифенил-1Н-имидазола (3,5 г, 0,0117 моль) в 1,5-диаминопентане (20 мл) нагревали до температуры дефлегмации в течение 48 ч. Реакционную смесь сконцентрировали в вакууме и получили вязкое масло, которое растворили в хлористом метилене (60 мл), промыли 10%-ным водным раствором NaHCO3, водой (2 х 50 мл), рассолом, обезводили над сульфатом магния, сконцентрировали в вакууме и получили 5-(4,5-дифенил-1Н-имидазол-2-иламино)-аминопентан в виде вязкого масла (3,5 г, 0,0109 моль).
1Н ЯМР (CDCl3) δ: 7,55-7,09 (м, 10Н), 4,79-3,79 (шир.с, 3Н), 3,14 (т, 2Н), 2,59 (т, 2Н), 1,79-1,22 (м, 6Н).
Часть В. В раствор 5-(4,5-дифенил-1Н-имидазол-2-иламино)аминопентана (1,7 г, 0,00531 моль) и триэтиламина (0,58 г, 0,0058 моль) в хлористом метилене, охлажденный до 0оС, в атмосфере азота медленно добавили хлористый гептаноил (0,788 г, 0,00531 моль). Реакционную смесь перемешивали в течение 1 ч при 0оС, вылили на воду и экстрагировали хлористым метиленом (2 х 50 мл). Объединенные органические экстракты промыли водой, рассолом, обезводили над сульфатом магния, сконцентрировали и получили N-/5-(4,5-дифенил-1Н-имидазол-2-иламино)-пентил/геп-танамид в виде вязкого масла. Продукт очистили пламенной хроматографией на силикагеле (250 мл), элюируя смесью хлористый метилен: метанол (95:5 по объему), и получили янтарную пену (1,3 г, 0,003 моль).
1Н ЯМР (CDCl3) δ: 7,43-7,15 (м, 10Н), 6,3 (м, 1Н), 3,24-3,1 (м, 4Н), 2,09 (т, 2Н), 1,6-1,16 (м, 14Н), 0,84 (т, 3Н).
Часть С. По способу примера 118, часть В: используя N-[5-(4,5-дифенил-1Н-имидазол-2-иламино)пентил] гептанамид, получили N-[5-(4,5-дифенил-1Н-имидазол-2-иламино) пентил]-N-гептиламин в виде янтарного масла (1,00 г, 0,00238 моль).
1Н ЯМР (CDCl3) δ: 7,56-6,85 (м, 10Н), 3,23 (м, 2Н), 2,49 (м, 4Н), 1,68-0,90 (м, 16Н), 0,88 (т, 3Н).
Часть D. По способу примера 118, часть С, используя N-[5-(4,5-дифенил-1Н-имидазол-2-иламино)пентил] N-гептиламин, получили указанное в названии соединение в виде желтой пены (0,395 г, 0,000688 моль).
1Н ЯМР (CDCl3) δ: 8,37-7,1 (м, 11Н), 6,9-6,67 (м, 2Н), 6,4 (д, 1Н), 4,53 (шир.с, 1Н), 3,27 (м, 6Н), 1,74-1,23 (м, 16Н), 0,89 (т, 3Н).
П р и м е р 330. Получение N'-(2,4-дифторфенил)-N-[6-(4,5-дифенил-1Н-имидазол- 2-ил)гексил]-N- гептилмочевины.
Часть А. В раствор 4,5-фенил-1-[(триметилсилил)этоксиметил]-1H-имидазола (2,5 г, 0,00734 моль) (B. hipsihutz B.Hubb, WHaz en, Tetraheoron Letter, 29, 3411-14, 1988) в безводном тетрагидрофуране (50 мл), охлажденный до -78оС, в атмосфере азота медленно добавили раствор н-бутиллития в гексане (2,5 М, 0,00734 моль). Реакционную смесь перемешивали в течение 1 ч и быстро добавили 1,6-дибромгексан (2,68 г, 0,0011 моль), перемешивали в течение 1/2 ч и дали нагреться до температуры окружающей среды и перемешивали еще 2 ч. Реакционную смесь влили в воду, экстрагировали этилацетатом (2 х 50 мл). Объединенный органический слой промыли водой, рассолом, обезводили над сульфатом магния, сконцентрировали и получили вязкое масло. Продукт очистили с помощью пламенной хроматографии на силикагеле (250 мл), элюируя смесью гексан: этилацетат (70: 30 по объему), и получили 6-бром-1-(4,5-дифенил-1-[(триметилсилил) этоксиметил] имидазол-2-ил)гексан в виде масла (2,18 г, 0,00424 моль).
1ЯМР (CDCl3) δ: 7,53-7,16 (м, 10Н), 5,10 (с, 2Н), 3,48 (т, 2Н), 3,34 (т, 2Н), 2,90 (т, 2Н) 1,99-1,5 (м, 8Н), 0,875 (т, 2Н), 0,008 (с, 9Н).
Часть В. Раствор 6-бром-1-(4,5-дифенил-1-[(триметилсилил)-этоксиметил] -1H- имидазол-2-ил)гексана (1,0 г, 0,00195 моль) и н-гептиламина (0,45 г, 0,00389 моль) в ацетонитриле (25 мл) нагревали до 60оС в течение 8 ч. Реакционную смесь влили в 10%-ный водный раствор бикарбоната натрия и экстрагировали этилацетатом (2 х 50 мл). Объединенный органический экстракт промыли водой, рассолом, обезводили над сульфатом магния, сконцентрировали и получили N-/6-(4,5-дифенил-/(триметилсилил)этоксиметил/-1H-имидазол-2-ил)- гексил/ -N-гептиламин в виде бесцветного вязкого масла (1,04 г, 0,00189 моль).
1Н ЯМР (CDCl3) δ: 7,52-7,2 (м, 10Н), 5,11 (с, 2Н), 4,7-4,2 (шир.с, 1Н), 3,3 (т, 2Н), 2,93-2,70 (м, 6Н), 1,95-1,34 (м, 18Н), 0,93 (т, 3Н), 0,86 (т, 2Н), 0,005 (с, 9Н).
Часть С. По способу примера 118, часть С, используя N-/6-(4,5-дифенил-1-[(триметилсилил)-этоксиметил-имидазол-2-ил)гексил] -N-гептиламин, выделили N'-(2,4-дифторфенил)-N-/6-(4,5-дифенил-1-/(триметил- силил)этоксиметил/-имидазол-2-ил)гексил/-N-гептилмочевину в виде вязкого масла (1,40 г, 0,00199 моль).
1Н ЯМР (CDCl3) δ: 8,12 (м, 1Н), 7,53-7,16 (м, 10Н), 6,88 (м, 2Н), 6,48 (д, 1Н), 5,1 (с, 2Н), 3,33 (м, 6Н), 2,90 (т, 2Н), 2,0-1,34 (м, 18Н), 0,88 (т, 3Н), 0,79 (т, 2Н), 0,055 (с, 9Н).
Часть D. В раствор N'-(2,4-дифторфенил)-N-[6-4,5-дифенил-1/(триметилсилил) этоксиметил-1Н- имидазол-2-ил)гексил]-N-гептилмочевины (0,60 г, 0,000853 моль) в безводном тетрагидрофуране (10 мл) в атмосфере азота добавили тетрабутиламмония фторид (1М раствор в тетрагидрофуране, 3,4 мл) и реакционную смесь нагревали до температуры дефлегмации в течение 7 ч. Реакционную смесь охладили, влили в воду (50 мл) и экстрагировали этилацетатом (2 х 50 мл). Объединенный органический слой промыли водой, рассолом, обезводили над сульфатом магния и сконцентрировали в вакууме. Продукт очистили пламенной хроматографией на силикагеле (75 мл), элюируя смесью гексан: этилацетат (60: 40 по объему), и получили указанное в названии соединение в виде бесцветного стекла (0,26 г, 0,000454 моль).
1Н ЯМР (CDCl3) δ: 9,5-9,0 (шир.с, 1Н), 7,87 (м, 1Н), 7,5-7,2 (м, 10Н), 6,83-6,7 (м, 2Н), 6,4 (д, 1Н), 3,28 (м, 4Н), 2,67 (т, 2Н), 1,75-1,26 (м, 18Н), 0,88 (т, 3Н).
БИОЛОГИЧЕСКИЕ ИСПЫТАНИЯ
Соединения предлагаемого изобретения являются ингибиторами фермента ацил-СоА: холестеринацилтрансферазы и поэтому являются эффективными для замедления этерификации и транспорта холестерина через стенки кишечника.
А. И с с л е д о в а н и е и н г и б и р о в а н и я А ц и л - С о А: х о л е с т р и н а ц и л т р а н с ф е р а з ы (АСАТ) в п е ч е н о ч н ы х м и к р о с о м а х.
Способность настоящих соединений ингибировать АСАТ, фермента, ответственного за внутриклеточный синтез холестриновых эфиров, исследовалась следующим образом Крыс S prague Dawley мужских особей массой 150-300 г кормили по потребности. Затем животные голодали в течение 24 ч перед тем, как их умертвили путем декапитирования. Печень подвергли перфузии на месте 50 мл холодного 0,25 М раствора сахарозы, извлекли и гомогенизировали в трех объемах 0,1 н фосфатного буфера, рН 7,4, который содержали 0,5 мМ ЭДТК (этилендиаминтетрауксусная кислота), 1,0 мМ глютатиона, 0,25 М сахарозы и 20 мМ лейпептина. Микросомы получили путем дифференциального центрифугирования: надосадочную жидкость центрифугировали сначала при 15000 g в течение 15 мин и затем при 105000 g в течение 1 ч для осаждения микросом. Затем эти микросомы суспендировали в гомогенизирующем буфере, снова выделили путем центрифугирования и хранили при -70оС. Микросомы использовали в течение одного месяца после получения.
Контрольный тест провели при конечном объеме 200 мкл, который содержал 200 мкг микросомного протеина, 75 мкМ 14С-олеил-СоА (10000 распадов в мин-ммоль) в 1М фосфате, рН 7,4, который включал 1 мМ глутатиона. Соединения добавили в 5-10 мкл DMCO (диметилсульфоксид) и провели дополнительные контрольные испытания только с DMCO. Все компоненты, за исключением олеоил-СоА, предварительно инкубировали в течение 15 мин при 37оС перед тем, как инициировать реакцию путем добавления олеоил-СоА. Тест закончили через 10 мин путем добавления 500 мкл смеси гексан-изопропанол (3:2 по объему). 20000 распадов в 1 мин 3Н-холестеринолеата и 10 мкг немаркированного холестеринолеата и олеиновая кислота были добавлены как внутренний стандарт и носитель соответственно. После выдержки в течение 10 мин для экстрагирования липидов образцы центрифугировали при 1000 g в течение 10 мин для разделения слоев растворителя. 200 мкл верхнего (гексанового) слоя, содержащего нейтральные липиды, нанесли на Bakers 1250-Pa силикагельную пластину для ТСХ и эту пластину обработали, используя смесь гексан-диэтиловый эфир-уксусная кислота (170:30:1 по объему) в качестве элюента. Липиды визуализировали, введя их во взаимодействие с парами иода, и холестериновое эфирное пятно поместили в суинцилляционный сосуд и просчитали. Удельная активность АСАТ в контрольном замере равна в среднем 260 пмоль/мин/мг микросомного протеина. Подавление активности АСАТ соединениями изобретения представлено в табл. 4; данные представлены как концентрация, при которой активность АСАТ подавляется на 500/ (ИК50).
В. И с с л е д о в а н и е и н г и б и р о в а н и я э т е р и ф и к а ц и и в к л е т к а х м о л о ч н о й ж е л е з ы
Этерификацию холестерина наблюдали в макрофагоподобных клетках линии I 774.A1. Клетки высеяли в 35 мм ячейки с плотностью 300000 клеток в ячейку в 2 мл среды Dublecco's Eagla Medium (DMEM), в которую добавили 10% сыворотки жеребой кобылы (СЖК). Клетки инкубировали при 37оС в атмосфере 5% СО2 и при 93% влажности. Через 24 ч среду заменили на 0,68 мл 10% СЖК-DMEM, содержащую 34 мкг ацетилированного низкой плотности липопротеина человека (ac-LDL) для увеличения внутриклеточной концентрации холестерина и способствования этерификации. На 41 часу различные ингибиторы добавляли в раствор клеток в DMCO (10 мкл/мл максимум). На 43 часу клетки обработали 0,1 мМ 14С-олеиновой кислотой (10000 распадов в минуту/нмоль) в комплексе с альбумином бычьей сыворотки (АБС) для отслеживания образования эфира холестерина. Эксперимент завершили на 45 часу промывки мотослоев 3 раза 3 мл трисзабуференного солевого раствора при 4оС. Липиды экстрагировали путем инкубирования монослоев 1,5 мл смеси гексан-изопропанол (3:2 по объему) в течение 30 мин при аккуратном перемешивании. В течение этого времени 10000 распадов в 1 мин 3Н-холестеринлинолеата и 10 мкг холестеринолеата добавили в качестве внутреннего стандарта и носителя соответственно. Органический слой удалили и клетки промыли дополнительными 1,0 мл смеси гексан-изопропанол, которую объединили с первоначальными экстрактами. Клеткам дали высохнуть в течение ночи, обработали 1,5 мл 0,2 н. растворам едкого натра в течение 1 ч и использовали аликвоту растворенного протеина для определения протеина по методу Лоури. Органический экстракт высушили досуха, остаток снова суспендировали в 100 мкл хлороформа и липиды отделили на пропитанных силикагелем пластинах из стекловолокна, используя систему растворителей гексан-диэтиловый эфир: уксусная кислота (170:30:1 по объему). Индивидуальные липиды были визуализированы с помощью иода, пятна эфира холестерины были вырезаны и помещены в суинцилляционный сосуд для определения радиоактивности. Превращение олеиновой кислоты в эфир холестерина в контрольном опыте составило 0,54 ммоль/час/мг протеина и возросло при добавлении ас-LDL до 10,69 + 0,69 ммол/час/мг протеина. Замедление этерификации соединениями настоящего изобретения показано в табл. 5; данные выражены как концентрация, при которой активность АСАТ снижается на 50% (ИК50). Следует отметить, что множество интермедиатов обладали подавляющей активностью in vitro в АСАТ исследовании и в исследовании с макрофагами. Например, N-/5-(4,5-дифенил-1Н-имидазол-2-илтио)-пентил/-1-гептанамин- гидрохлорид имел ИК50 100 мМ и 6 мкМ in vitro АСАТ исследовании и в исследовании с макрофагами соответственно.
С. И с с л е д о в а н и е а н т и г и п е р х о л е с т е р и н е м и ч е с к о й а к т и в н о с т и у х о м я к о в, к о р м л е н н ы х х о л е с т е р и н о м
Ингибирование активности АСАТ в кишке снижает абсорбцию холестерина у кормленых холестерином животных. Хомяков массой примерно 100 г держали на диете с 0,8% холестерина. Подопытные животные получали 1-100 мг/кг/день в рот испытуемого соединения, растворенного в 500 мкл кукурузного масла в течение 2 недель. Контрольную группу кормили, как подопытную, но ей давали только 500 мкл кукурузного масла. Хомяков умертвили, сделав анестезию с помощью CO2 и отобрали кровь проколом сердца. Определили общее содержание холестерина на Du Pont Aca IV.
IV.
Данные представлены как количество мг холестерина на 100 мл сыворотки (Мг %). Антигиперхолестеринемическая активность соединения примера 1 представлена в табл. 6.
ЛЕКАРСТВЕННЫЕ ФОРМЫ
Cоединения настоящего изобретения могут даваться пациенту в рот при использовании фармацевтически приемлемых лекарственных форм, известных в медицине для такого приема. Активный ингредиент может даваться в твердой лекарственной форме, такой как сухой порошок, гранулы, таблетки или капсулы, или в жидкой лекарственной форме, такой как сиропы или водные суспензии. Активный ингредиент может вводиться в чистом виде, но обычно его вводят с фармацевтически приемлемым носителе. Ценным руководством, относящимся к фармацевтическим лекарственным формам, является Remington's Pharmacentical Sciences, 16th Edition, 1980.
При терапевтическом применении кишечных ингибиторов АСАТ используемые соединения настоящего изобретения вводятся пациентам в дозах от 1 до 28 г в день. Для нормального взрослого мужчины массой примерно 70 кг это означает 14-400 мг на кг массы тела в день. Вводимые дозы, конечно, зависят от возраста, состояния, массы пациента, симптомов, вида сопутствующего лечения, частоты лечения и желаемого эффекта. Ниже проиллюстрированы полезные лекарственные формы соединений настоящего изобретения.
ТАБЛЕТКИ
Таблетки получают по обычным методикам таким образом, чтобы единица параметра содержала 500 мг активного ингредиента, 150 мг лактозы, 50 мг целлюлозы и 10 мг стеарата магния.
КАПСУЛЫ
Капсулы получают известными способами, таким образом, чтобы одна капсула содержала 500 мг активного ингредиента, 100 мг целлюлозы и 10 мг стеарата магния.
СИРОП, мас.% Активный ингредиент 10 Жидкий сахар 50 Сорбит 20 Глицерин 5 Отдушки, красители и консерванты Как требуется Вода Как требуется
Конечный объем доводят до 100% добавлением дистиллированной воды.
ВОДНАЯ СУСПЕНЗИЯ, мас. % Активный ингредиент 10 Сахарин натрия 0,01 Кетрол (пищевая ксантановая смола) 0,2 Жидкий сахар 5 Отдушки, красители, консерванты По требованию Вода По требованию
(пищевая ксантановая смола) 0,2 Жидкий сахар 5 Отдушки, красители, консерванты По требованию Вода По требованию
Ксантановую смолу медленно добавляют в дистиллированную воду перед введением активного ингредиента и остальных компонентов композиции. Полученную суспензию пропускают через гомогенизатор для получения тонкой консистенции целевого продукта.
СУСПЕНДИРУЕМЫЙ ПОРОШОК, мас.% Активный ингредиент 50,0 Лактоза 35,0 Сахар 10,0 Аравийская камедь 4,7
Карбоксиметилцеллю- лоза натрия 0,3
Каждый ингредиент тонко измельчали и затем получали однородную смесь всех ингредиентов. Альтернативно, порошок можно получить в виде суспензии и затем высушить ее при разбрызгивании.
ПОЛУТВЕРДЫЙ ГЕЛЬ, мас. % Активный ингредиент 10 Сахарин натрия 0,02 Желатин 2 Краситель, отдушка консервант По потребности Вода По потребности
Желатин приготовили в горячей воде. Тонко разбрызганный активный ингредиент суспендировали в растворе желатина и к этой смеси примешали остальные компоненты. Суспензию расфасовали в подходящие упаковочные емкости и охладили до получения геля.
ПОЛУТВЕРДАЯ ПАСТА, мас.%: Активный ингредиент 10
Гелкарин(R) (каррагее- ниновая смола) 1 Сахарин натрия 0,01
Краситель, отдушка, консервант По потребности Вода По потребности
Гелкарин(R) растворили в горячей воде (около 80оС) и в этом растворе суспендировали тонко измельченный активный ингредиент. В теплую суспензию добавили остальные ингредиенты композиции. Суспензию гомогенизировали и затем расфасовали в подходящие емкости.
ЭМУЛЬГИРУЕМАЯ ПАСТА, мас. % : Активный ингредиент 30 Твин(R) 80 и Спан(R) 80 6 Келтрол(R) 0,5 Минеральное масло 63,5
Все ингредиенты тщательно перемешали и получили гомогенную пасту.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ИМИДАЗОЛА ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫХ СОЛЕЙ | 1991 |
|
RU2026292C1 |
| АЗОТСОДЕРЖАЩИЕ ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ФЛУОРЕНА | 1992 |
|
RU2051151C1 |
| ЦИКЛИЧЕСКИЕ МОЧЕВИНЫ, СПОСОБ ИНГИБИРОВАНИЯ РОСТА РЕТРОВИРУСОВ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1992 |
|
RU2131420C1 |
| ИЗОКСАЗОЛИНЫ И ИЗОКСАЗОЛЫ, СПОСОБ ПОДАВЛЕНИЯ АГРЕГАЦИИ ТРОМБОЦИТОВ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ПОДАВЛЯЮЩАЯ АГРЕГАЦИЮ ТРОМБОЦИТОВ | 1994 |
|
RU2149871C1 |
| ПРОИЗВОДНЫЕ ИМИДАЗОЛА | 1992 |
|
RU2017733C1 |
| ПРОИЗВОДНЫЕ АНТРАПИРАЗОЛОНОВ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И СПОСОБЫ ПОЛУЧЕНИЯ ИЗ НИХ АНТРАПИРАЗОЛОНОВ, ОБЛАДАЮЩИХ ПРОТИВООПУХОЛЕВОЙ АКТИВНОСТЬЮ | 1994 |
|
RU2142456C1 |
| КОНДЕНСИРОВАННЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ | 2006 |
|
RU2408586C2 |
| ЦИКЛИЧЕСКИЕ ПЕПТИДЫ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1992 |
|
RU2096415C1 |
| 1-N-АЛКИЛ-N-АРИЛПИРИМИДИНАМИНЫ, СПОСОБ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1994 |
|
RU2153494C2 |
| ПОЛИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, ИХ ПРОИЗВОДНЫЕ, ФАРМАЦЕВТИЧЕСКИЕ ПРЕПАРАТЫ КАК УСИЛИТЕЛИ И МЕТОД ЛЕЧЕНИЯ НАРУШЕНИЯ МЫСЛИТЕЛЬНОЙ ДЕЯТЕЛЬНОСТИ | 1994 |
|
RU2152944C1 |
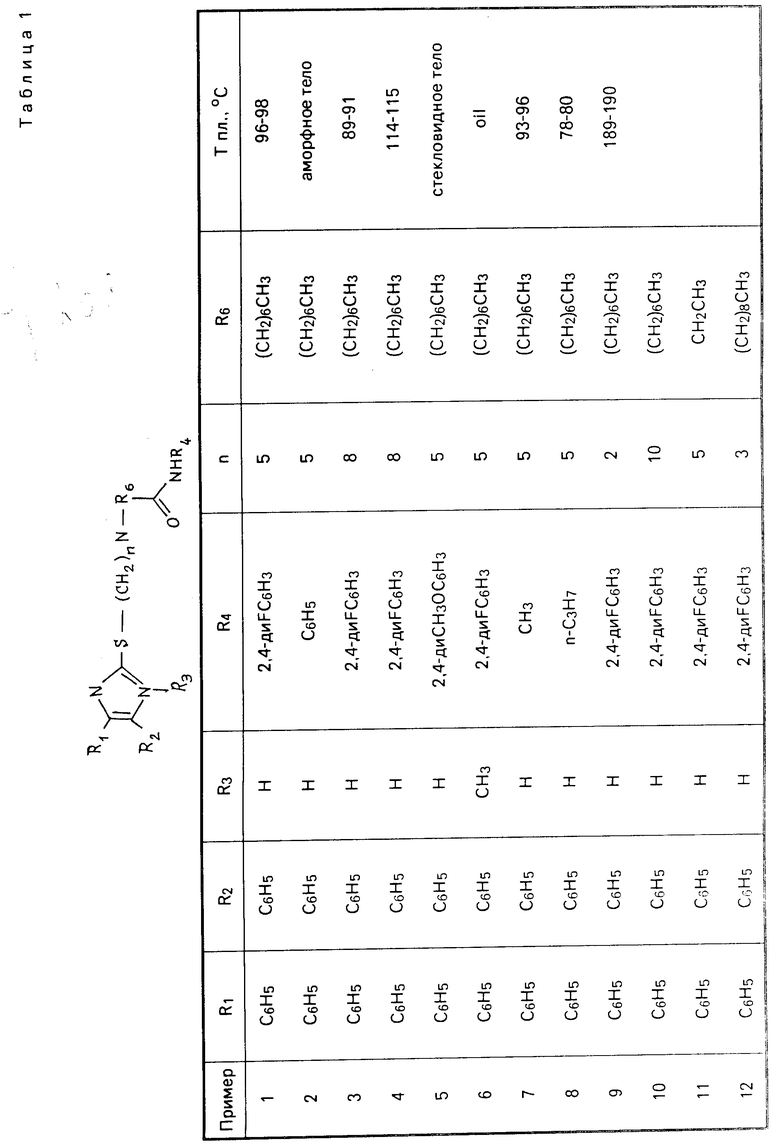
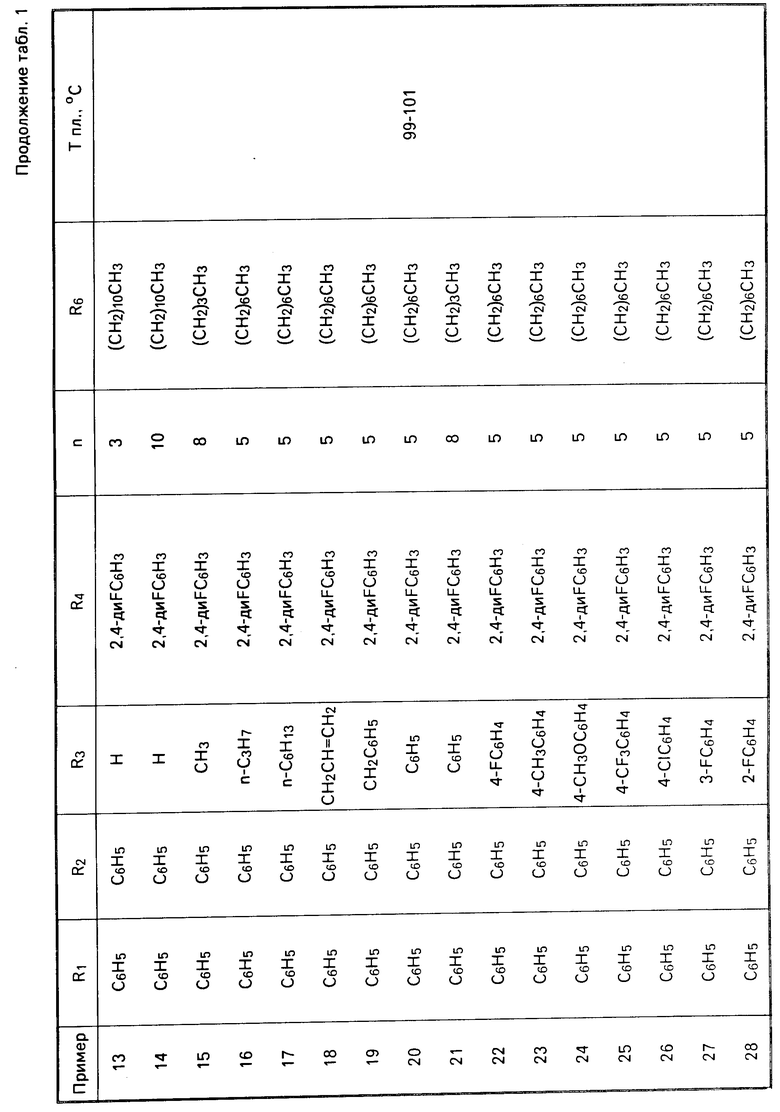
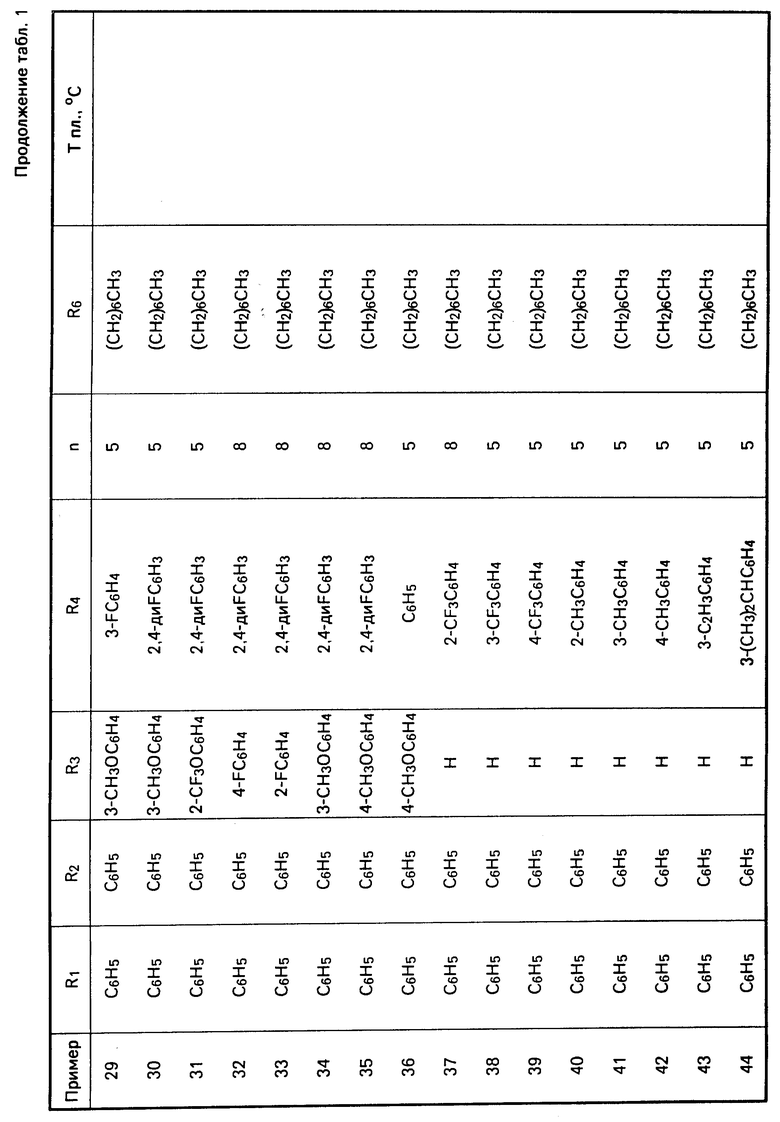
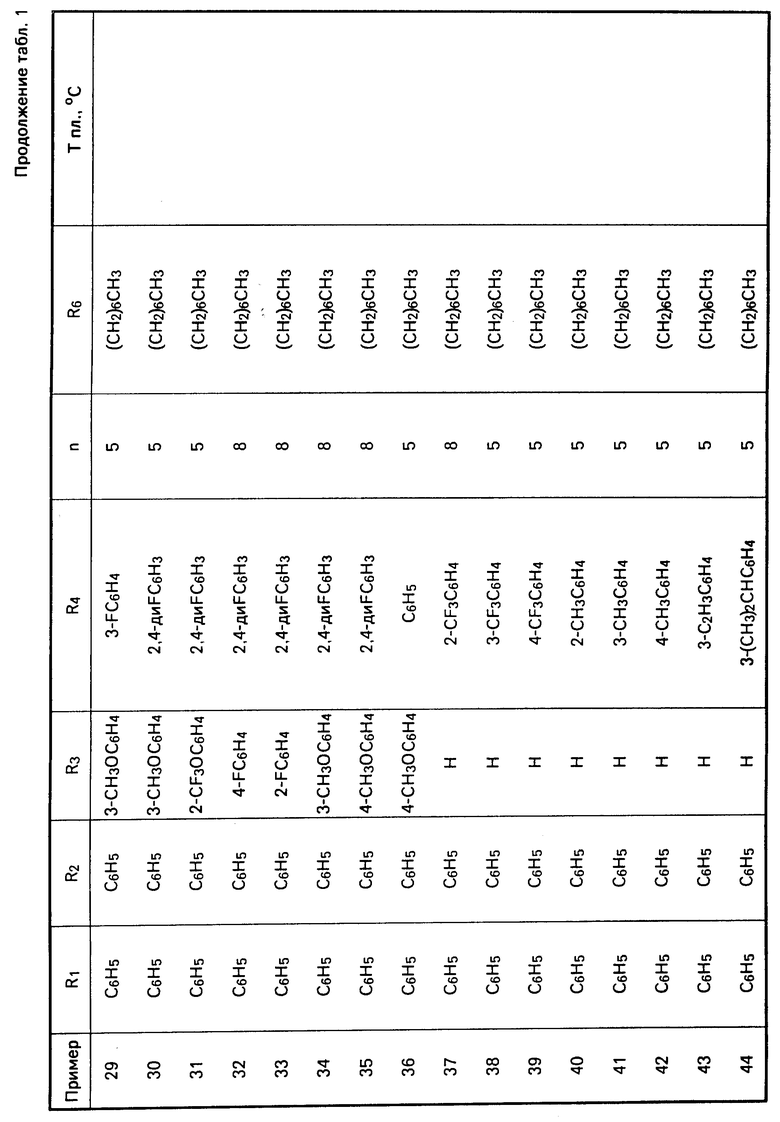

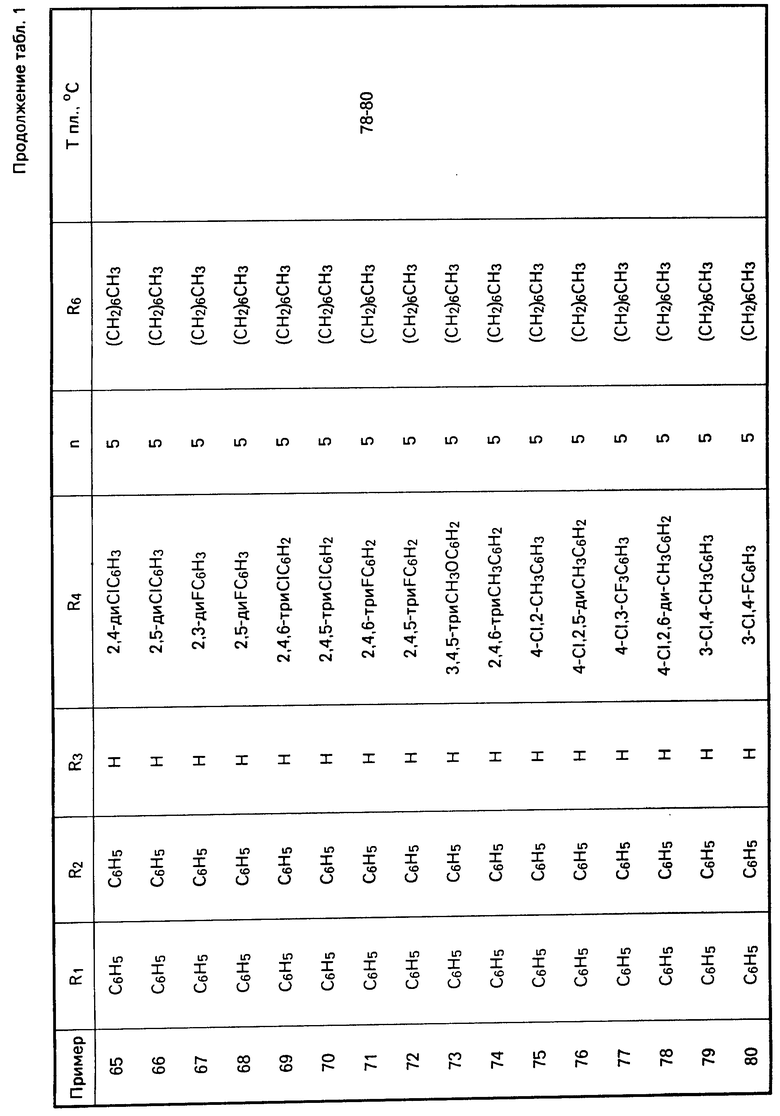
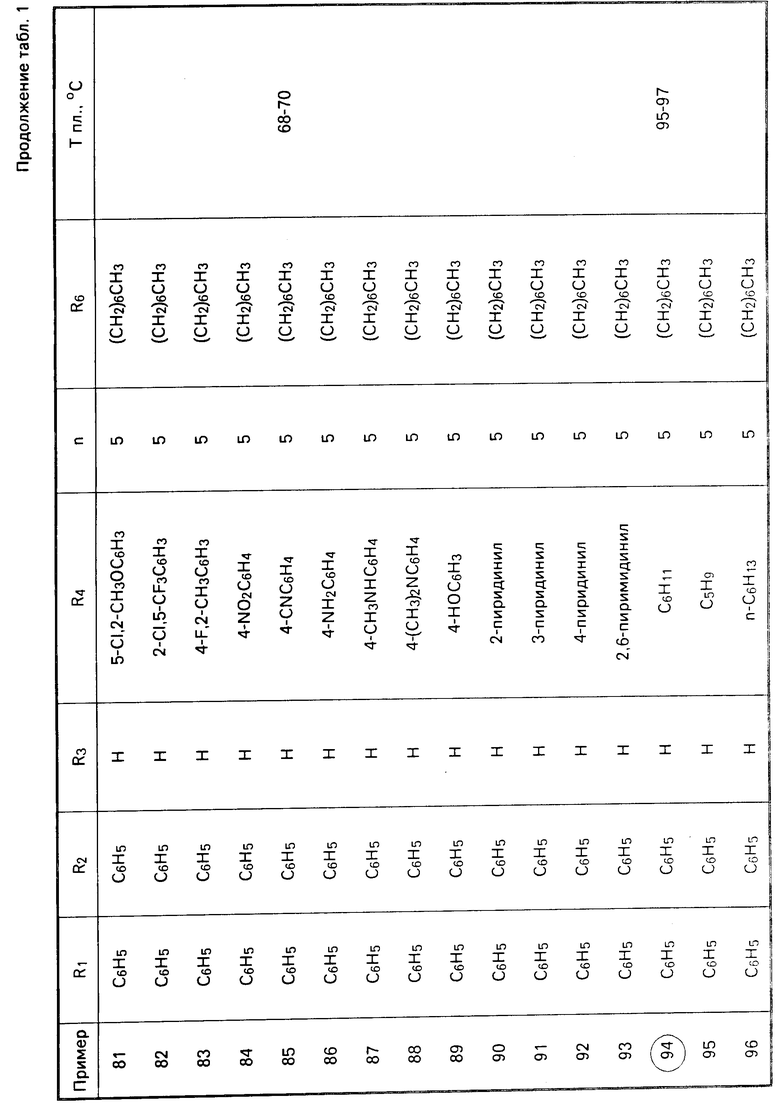
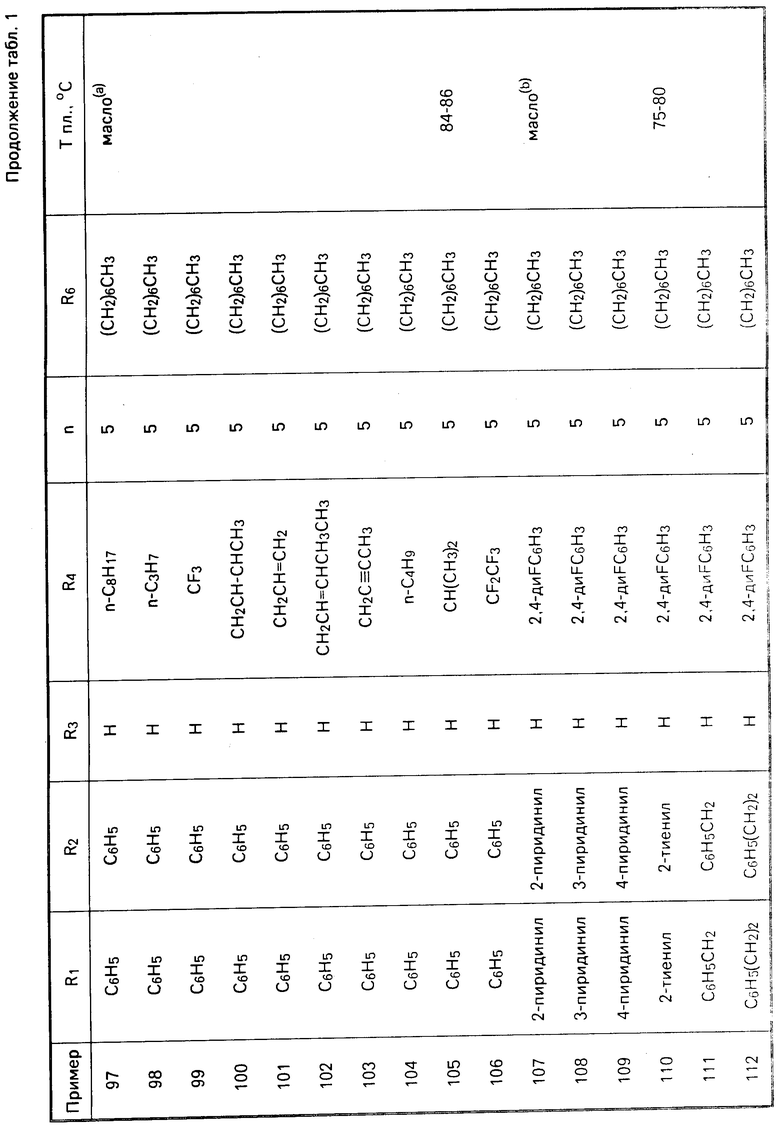
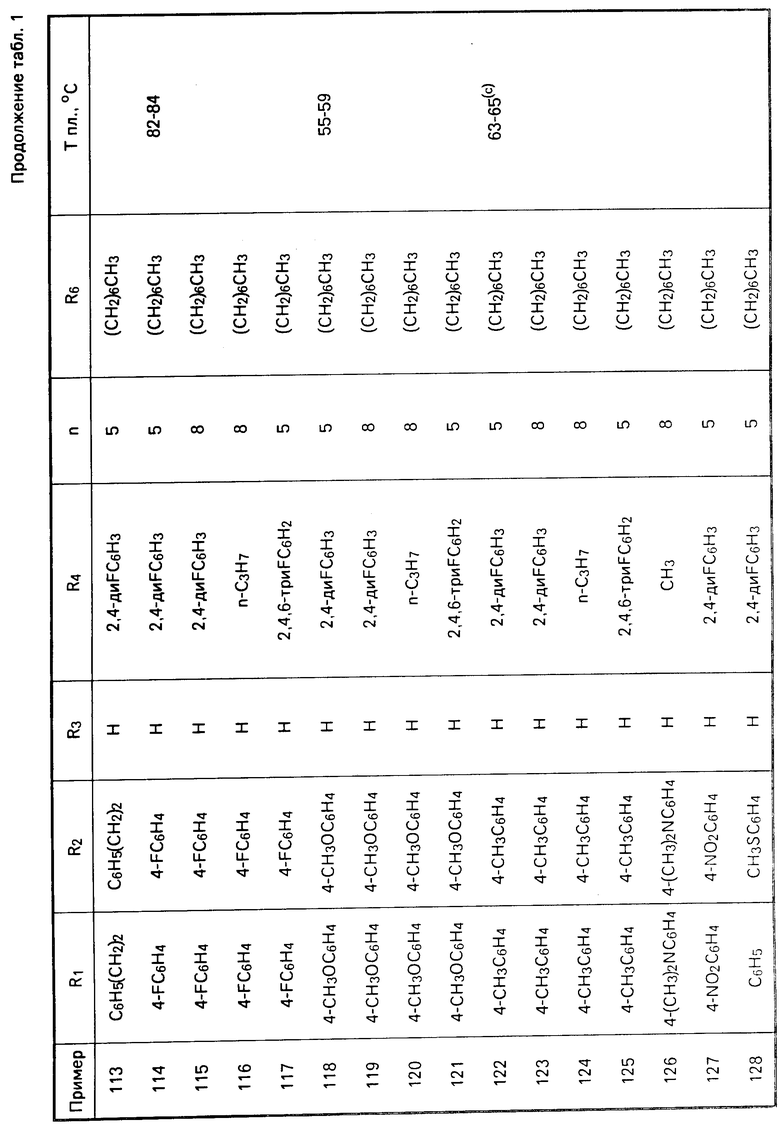
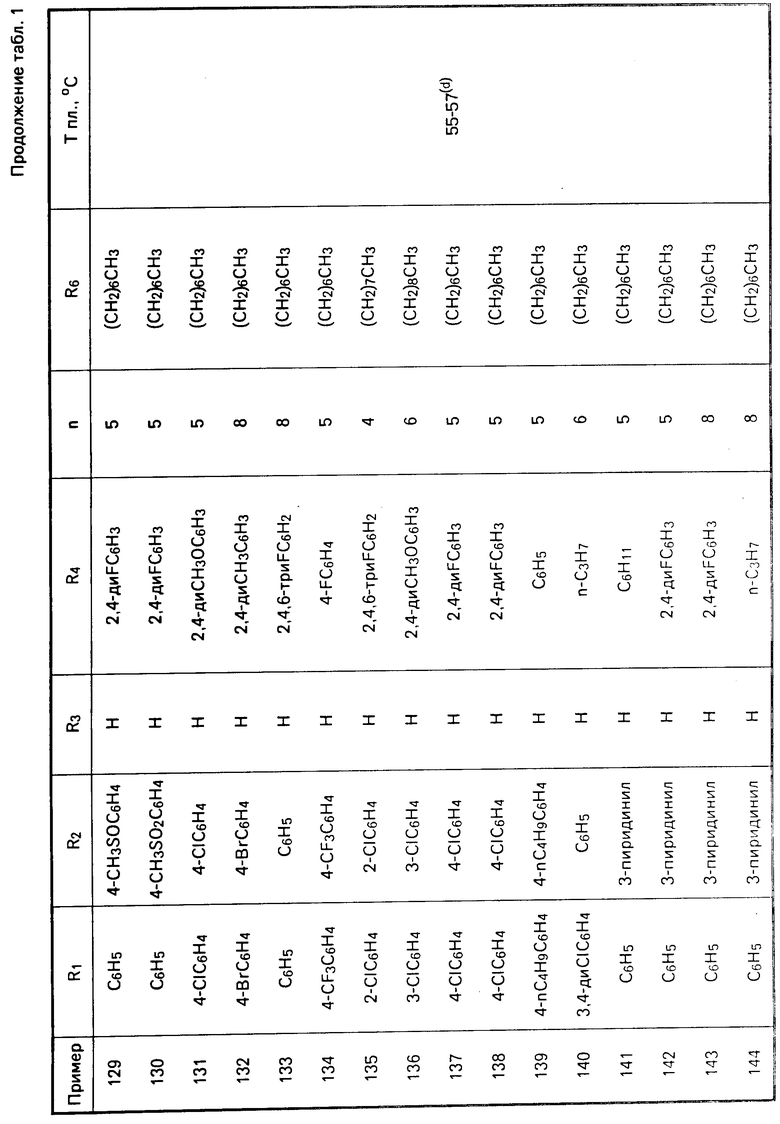
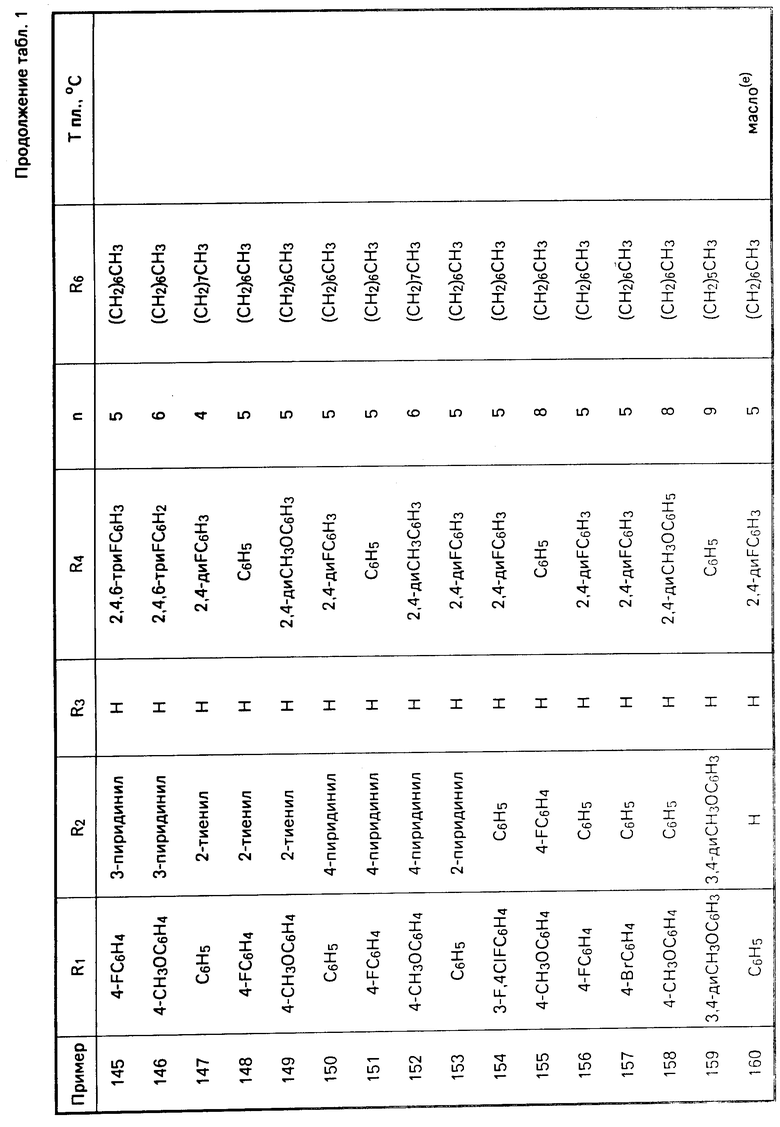
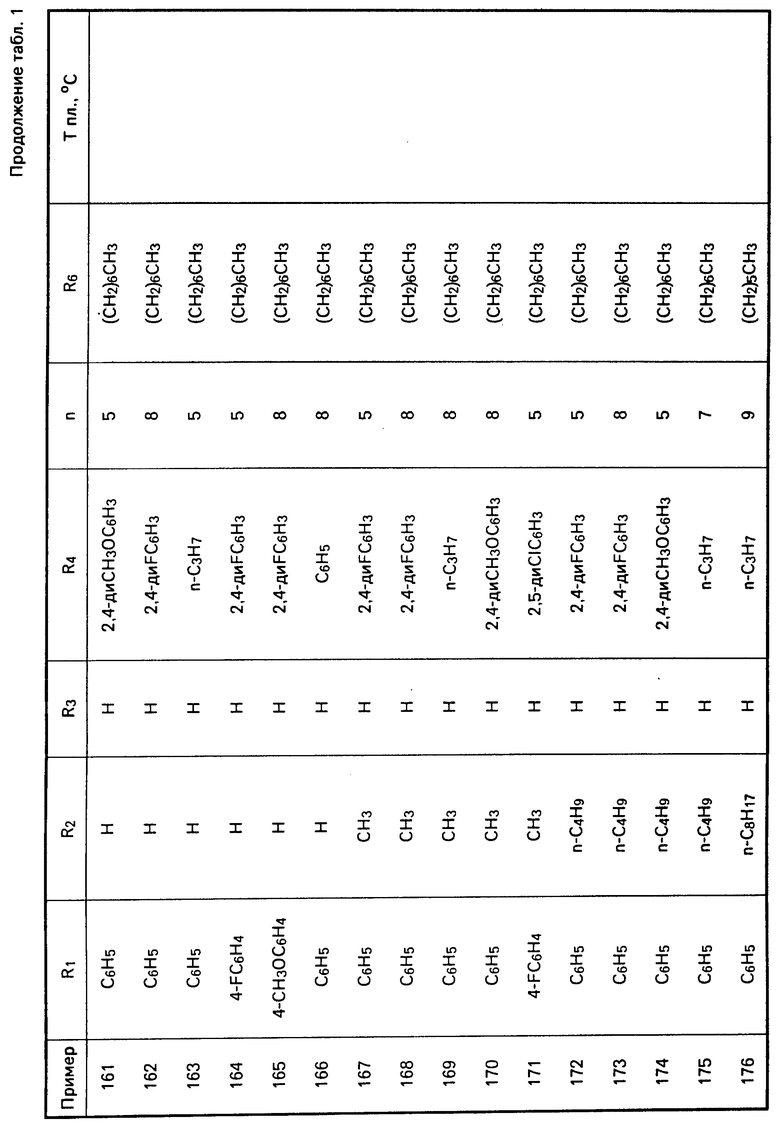
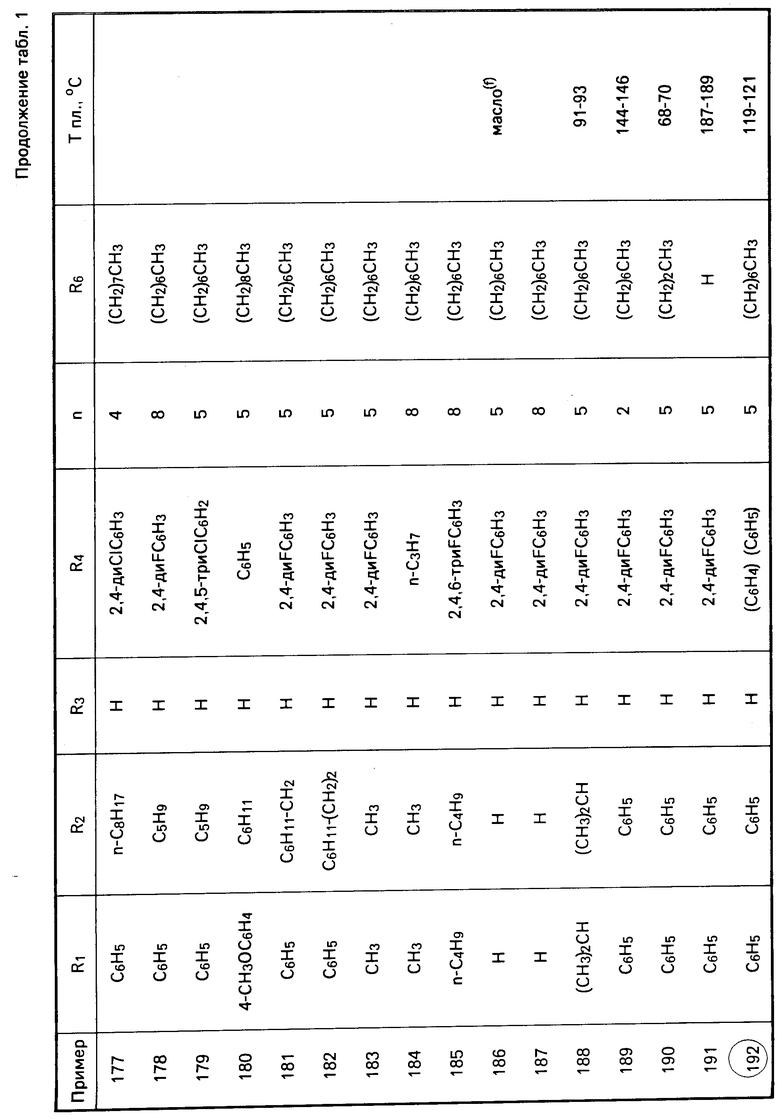
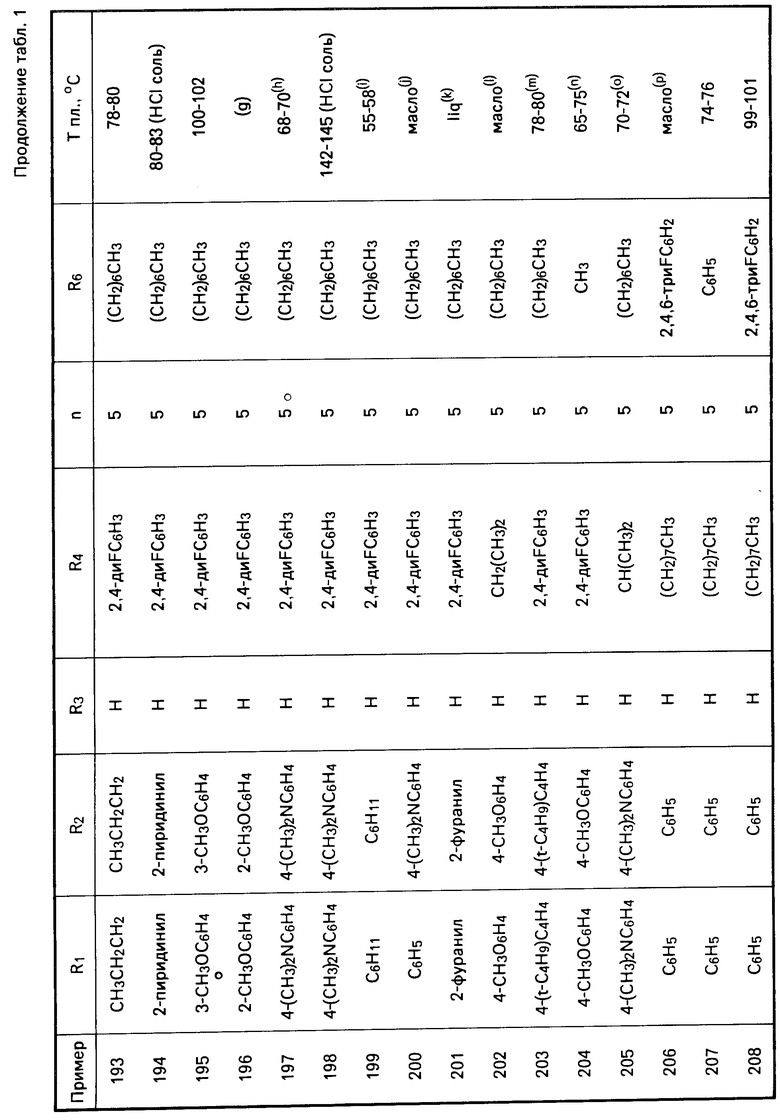
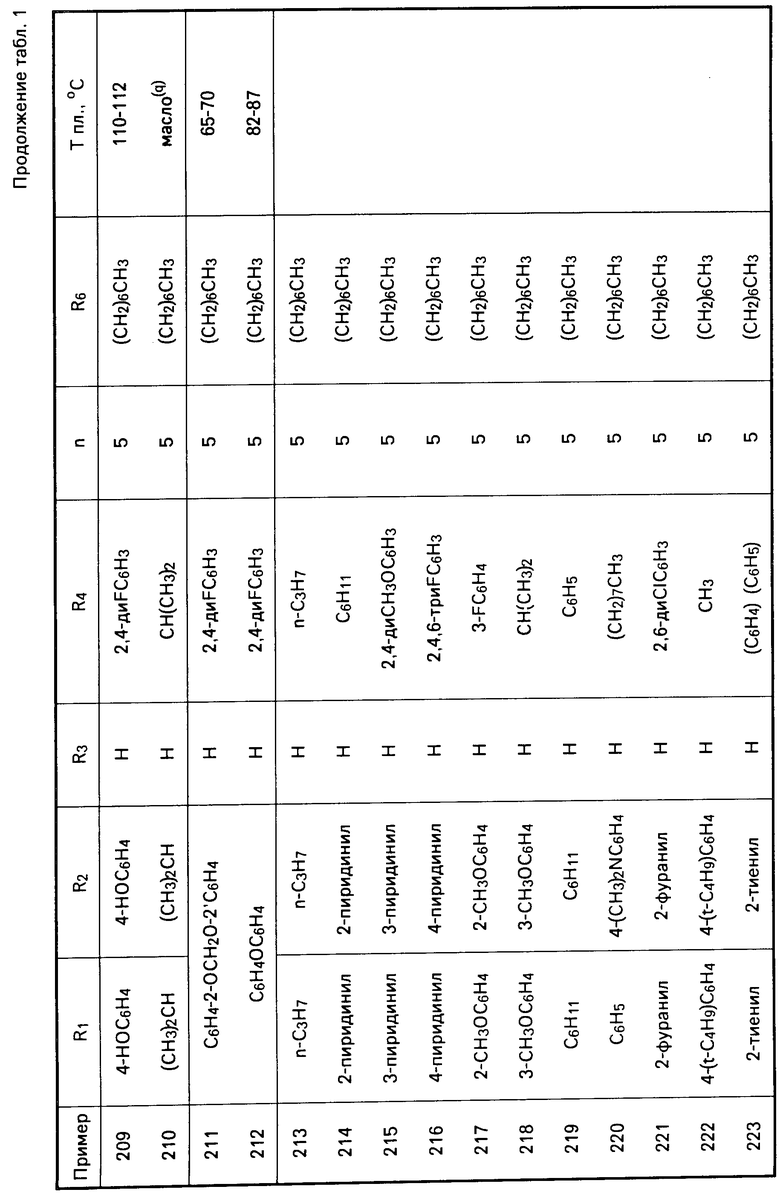
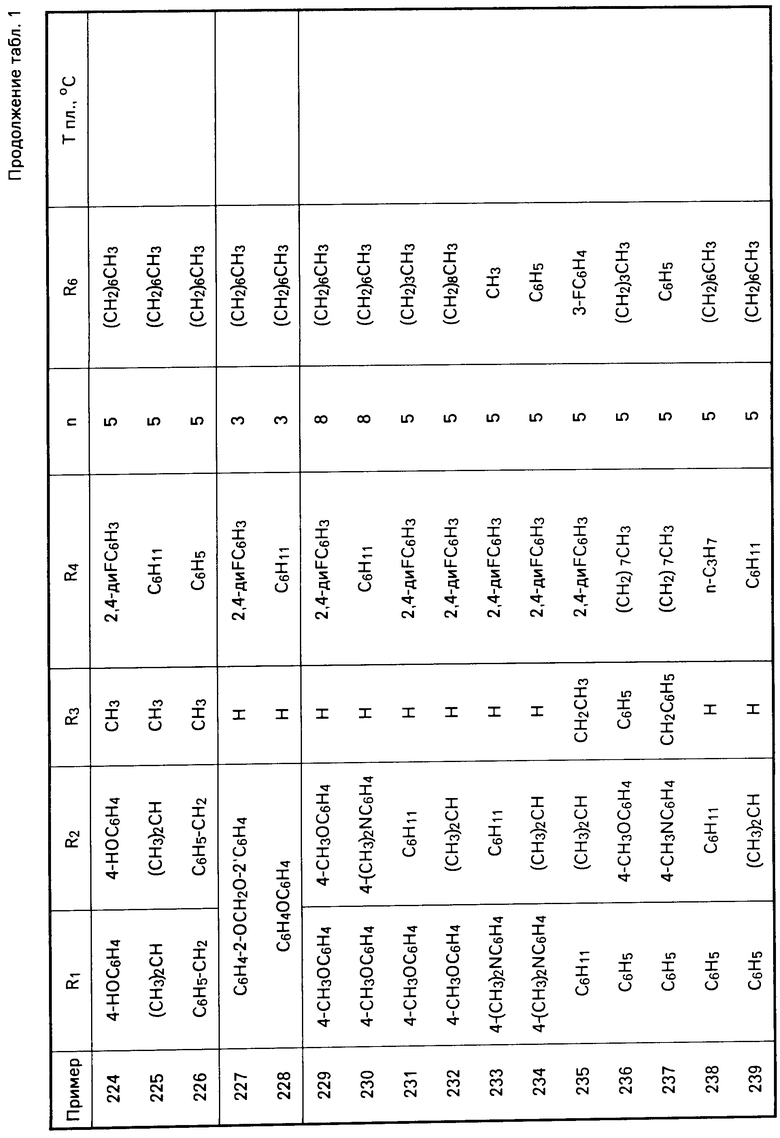
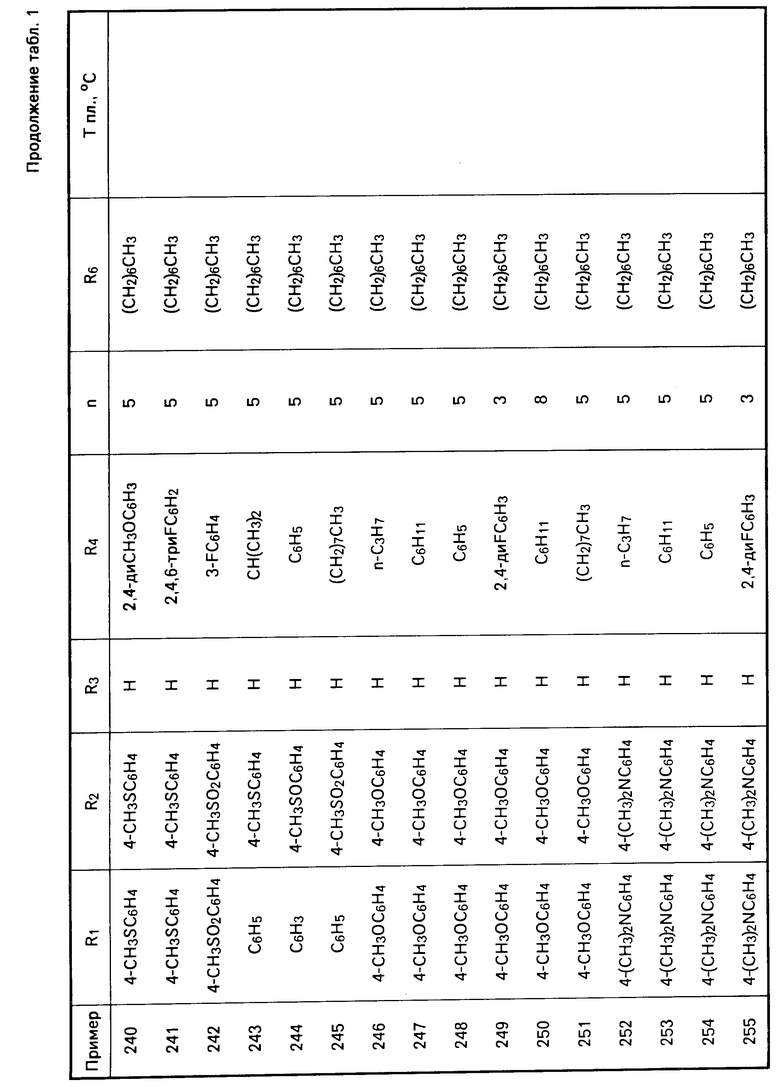
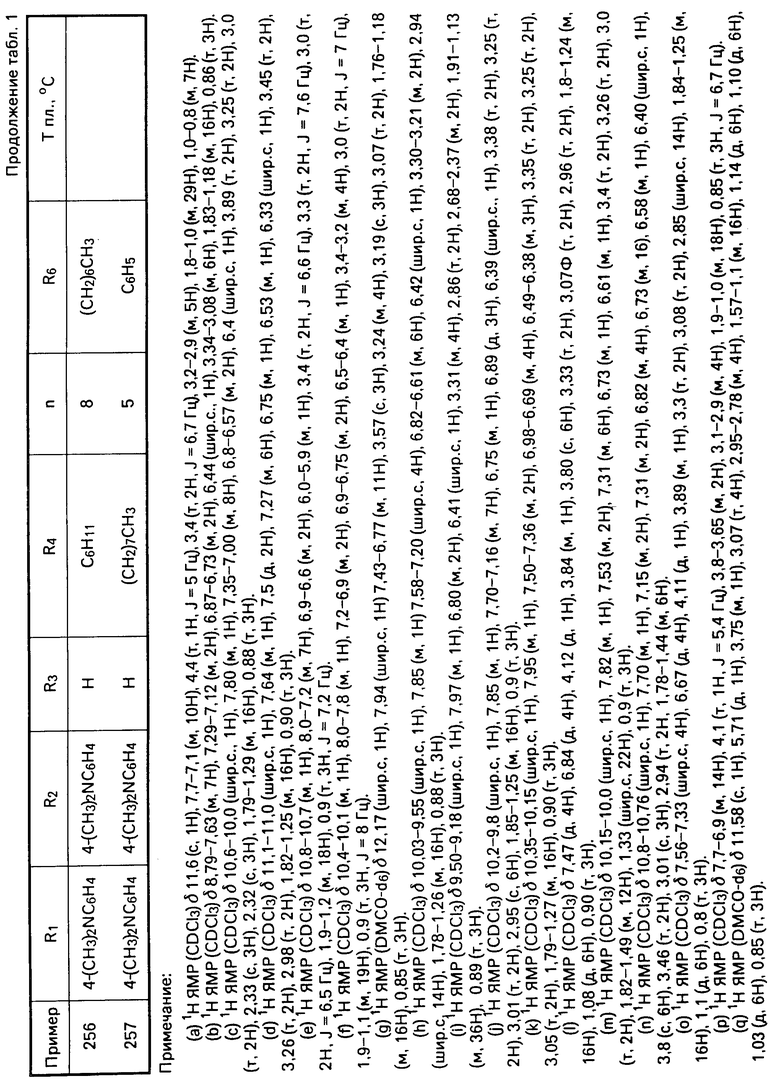
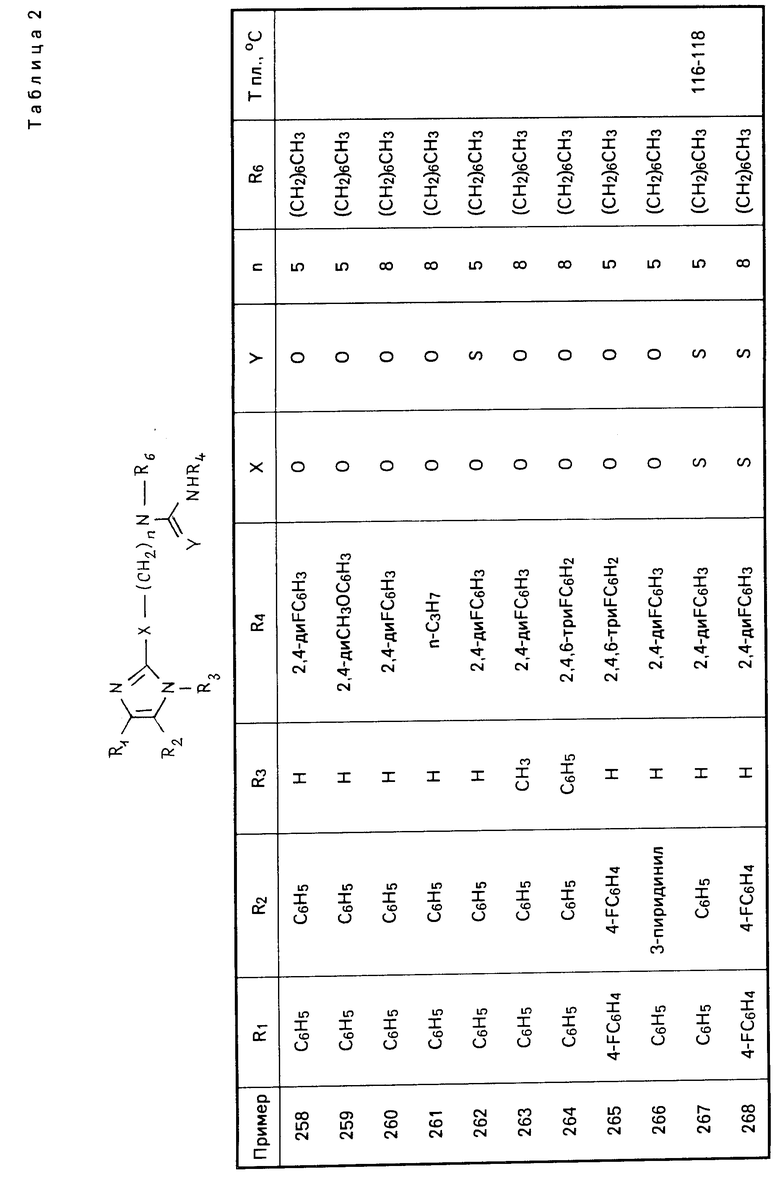
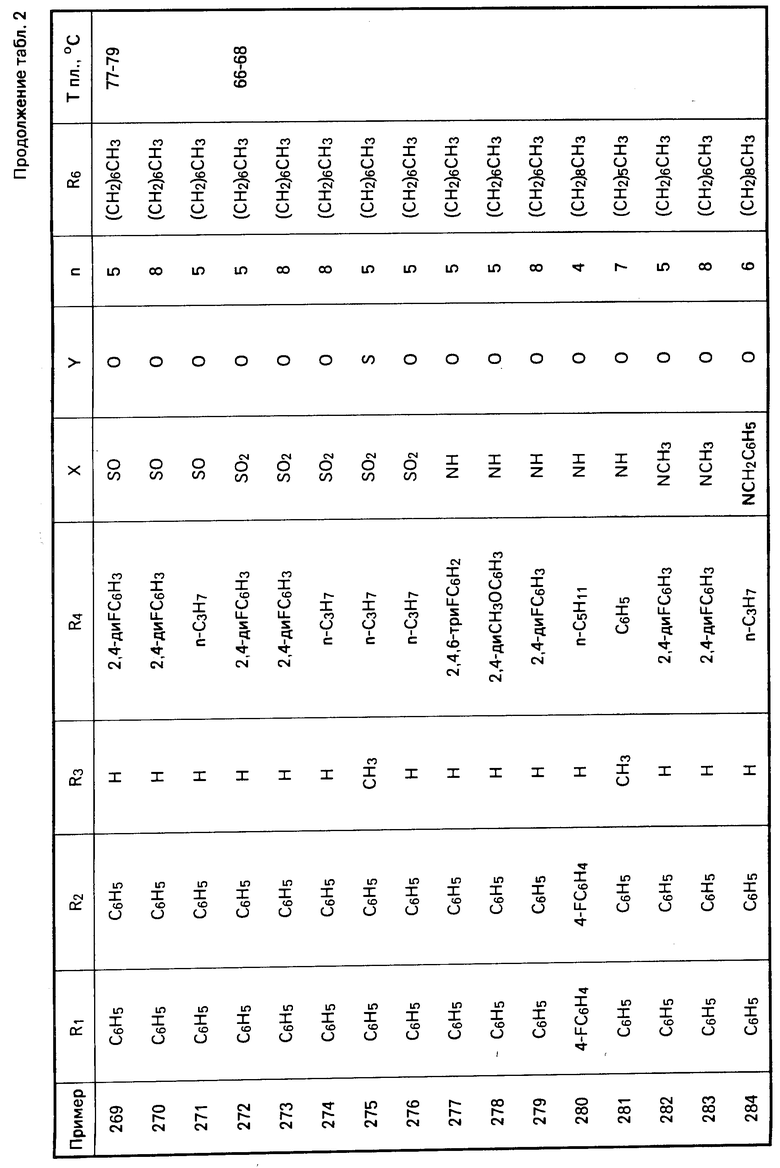
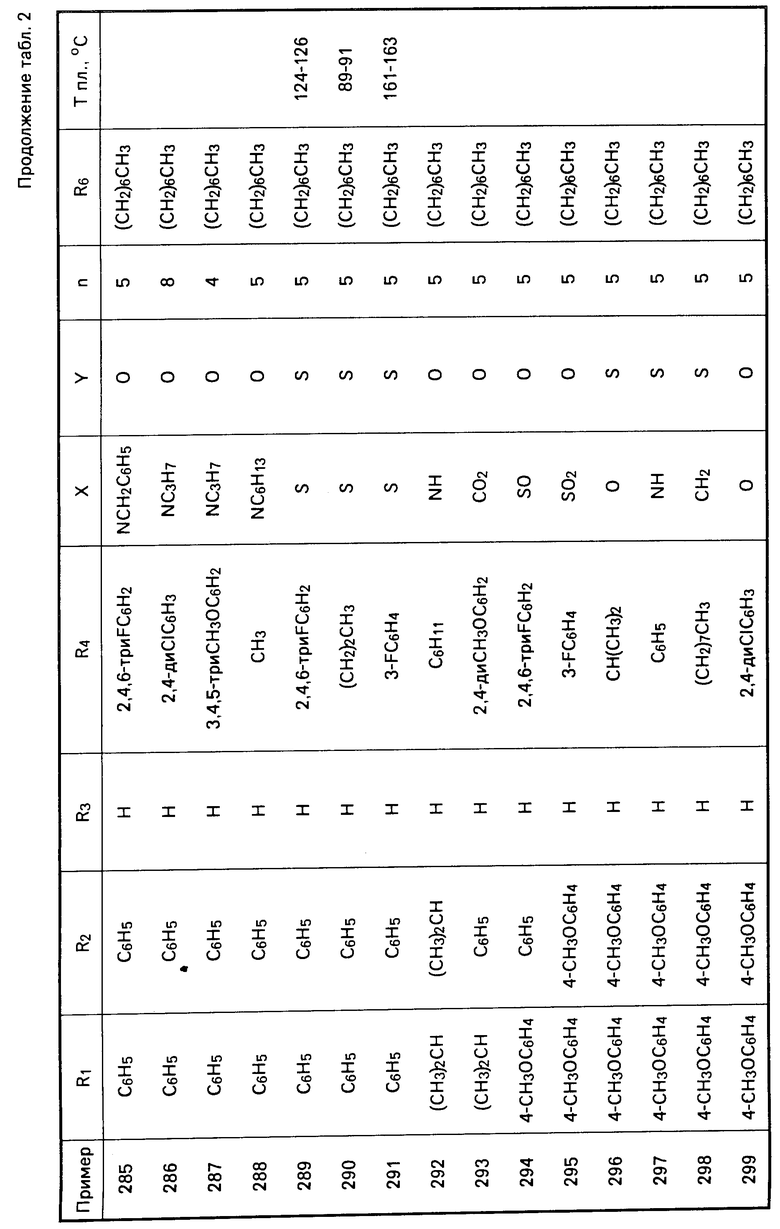
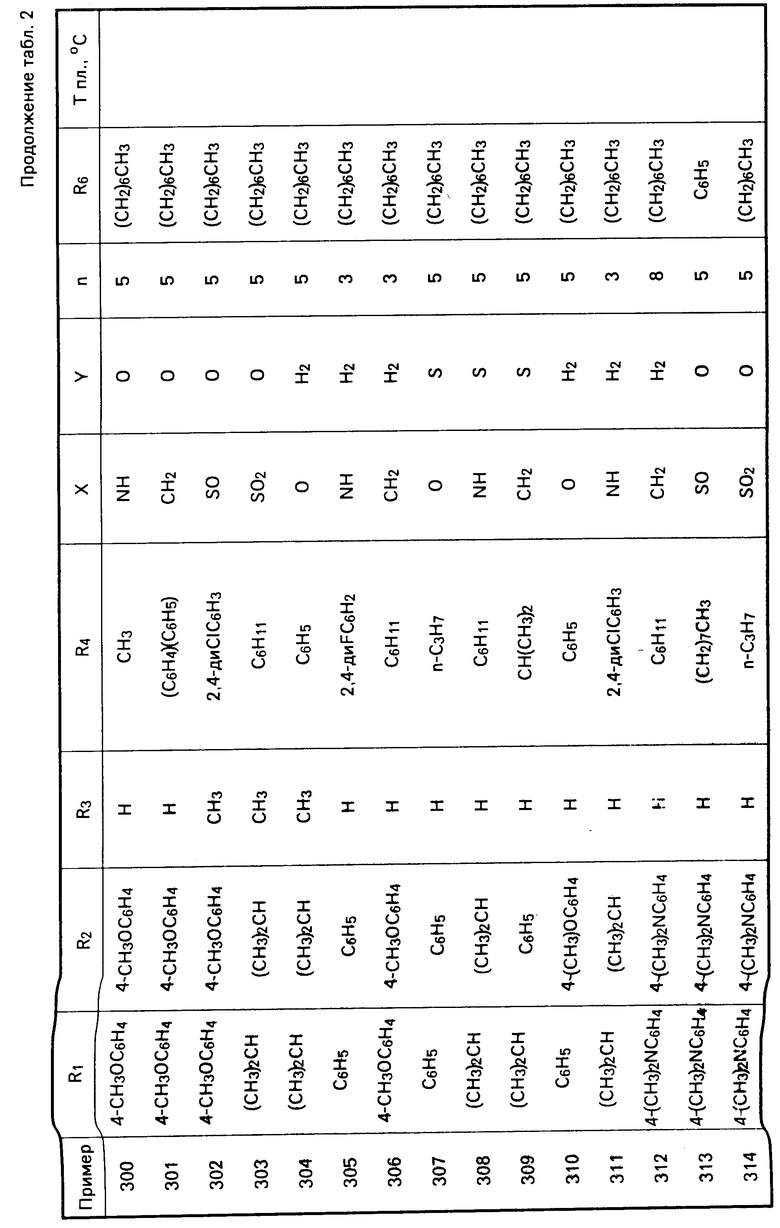
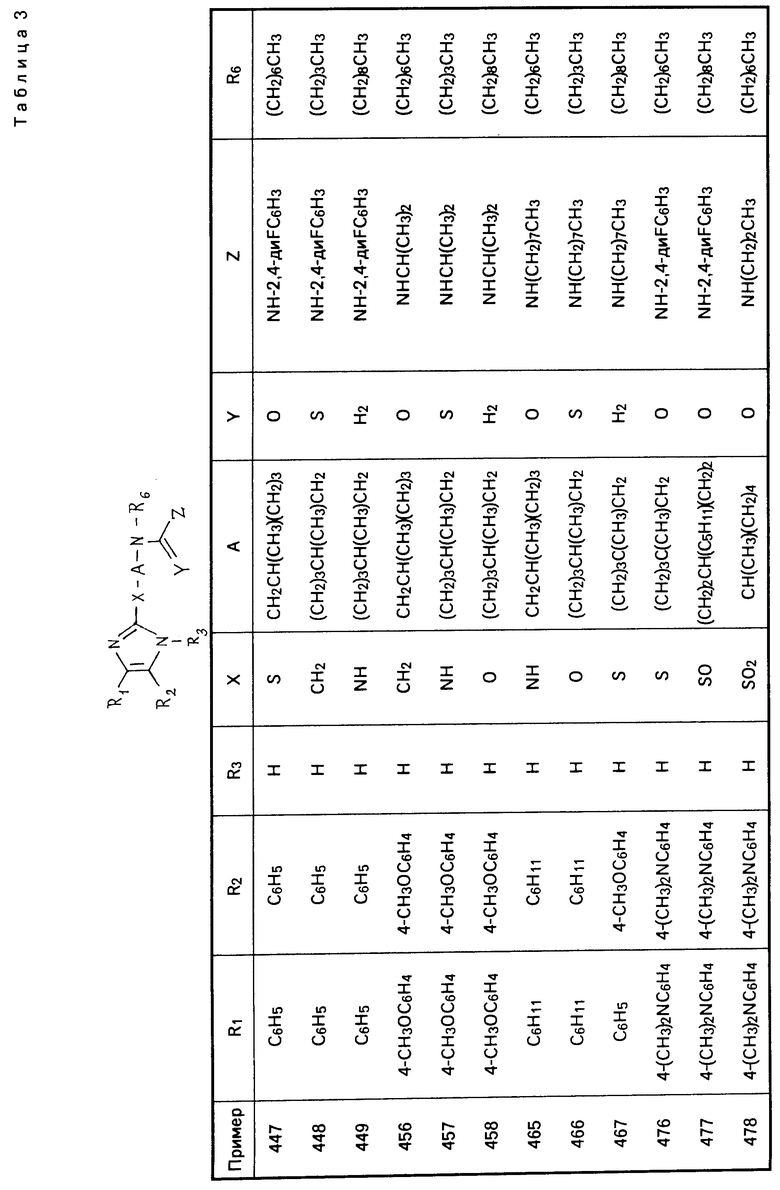
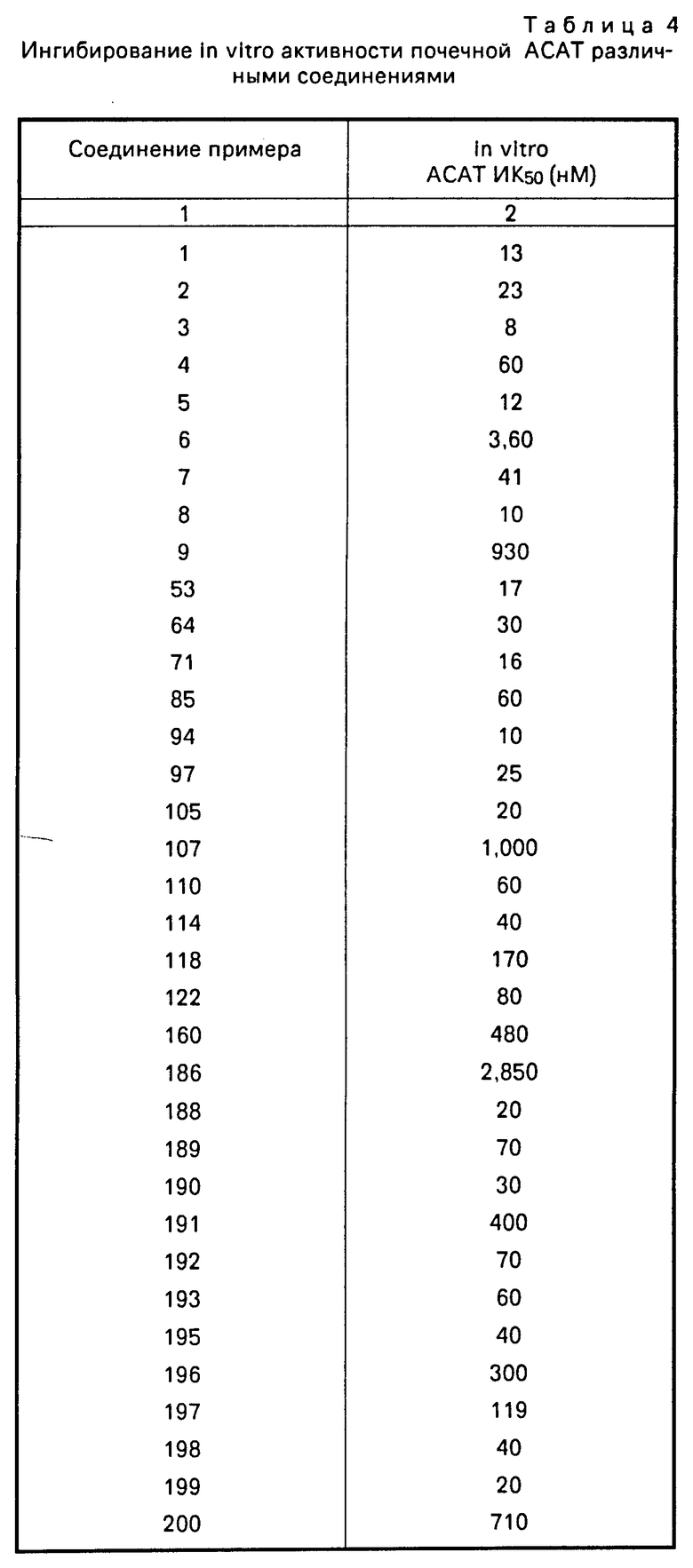
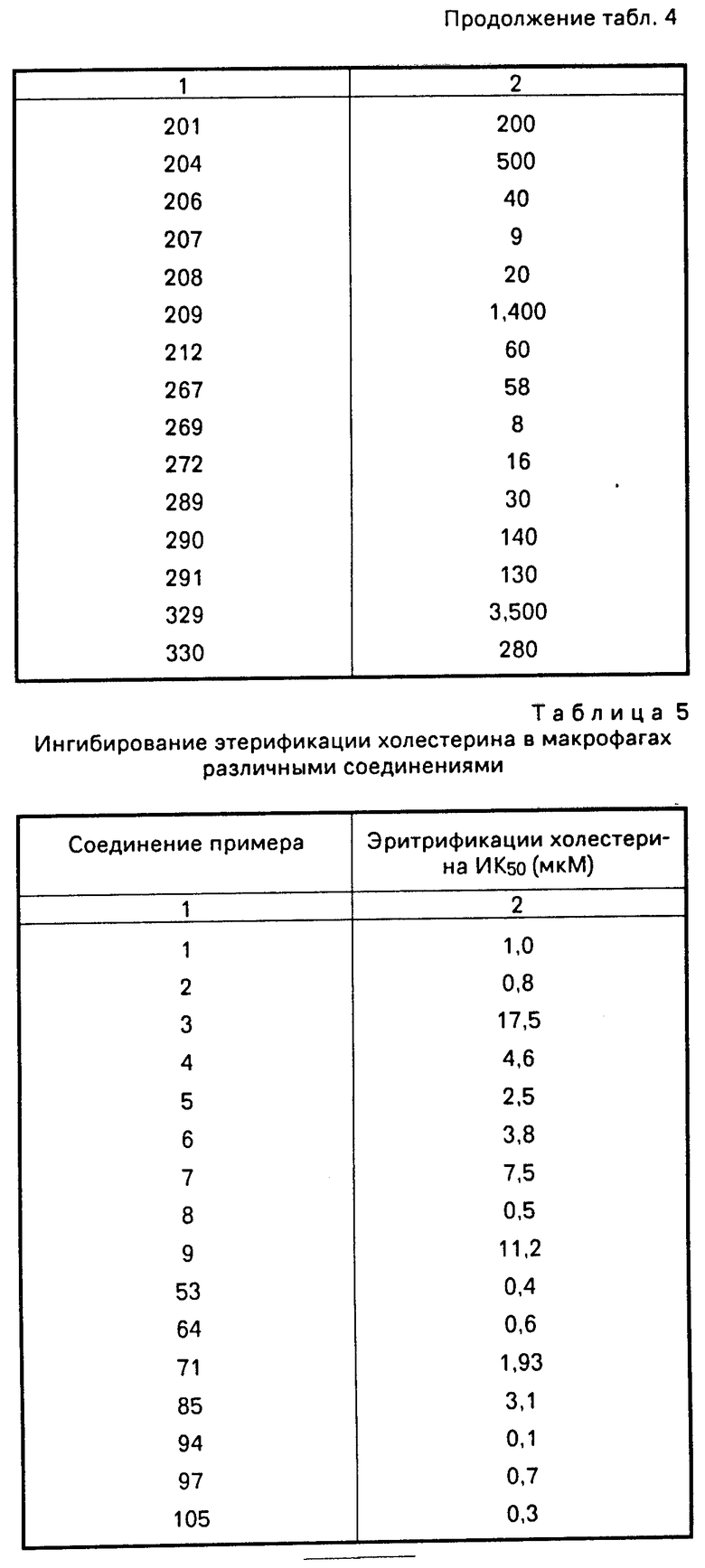
Использование: в качестве ингибиторов активности ацил-СоА: холестеринацилтрансферазы (АСАТ). Сущность изобретения: продукт общей формулы 1: 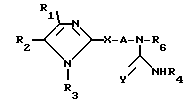 (I), где R1 и R2 независимо, выбраны из ряда, включающего H, C1-C8 -алкил, C3-C8 -разветвленный алкил, C3-C7- циклоалкил, C4-C10 -циклоалкилакил, C7-C14 -аралкил, 2-, 3- или 4-пиридинил, 2-тиенил, 2-фуранил, фенил, в определенных случаях замещенный 1-3 группами, выбранными из ряда, включающего F, Cl, Br, OH, C1-C4 -алкокси, C1-C4 -алкил, C3-C8 -разветвленный алкил, CH3S(O)nNO2, CF3 или NR7R8 или R1 и R2 ,взятые вместе, образуют группу (см.рисунок), где L - 0,0(CH2)m+1O или (CH2)m, где m = 0 - 4, R3 -Н, C1-C6 - алкил, аллил, бензил или фенил, в определенных случаях замещенный F, Cl, CH3, CH3O или CF3; R4-C1-C8 -алкил с неразветвленной цепью, в определенных случаях замещенный F, C3-C8 - алкил с разветвленной цепью, C3-C7 -циклоалкил, C4-C10 -циклоалкилалкил, C7-C14 -аралкил, где арильная группа в определенных случаях замещена 1 - 3 группами, выбранными из ряда, включающего C1-C4 -алкил или алкокси, F, Br, Cl, NH2 OH, CN, CO2H, CF3, NO2, C1-C4 -карбоалкокси, NR7R8 или NCOR7, C3-C6/ -алкенил или акинил, C1-C3 -перфторалкил, фенил, в определенных случаях замещенный 1 - 3 группами, выбранными из C1-C4 -алкила, C1-C4 -алкокси, F, Br, Cl, NH2, OH, CN, CO2H, CF3, NO2, C1-C4 - карбоалкокси, NR7R8 или NCOR7 , пентафторфенил, бензил, в определенных случаях замещенный 1 - 3 группами, выбранными из C1-C4 -алкила или алкокси, F, Br, Cl, NH2, OH, CN, CO2H, CF3, NO2, C1-C4 -карбоалкокси, NR7R8 или NCOR7 : 2 -, 3 - или 4-пиридинил, пиримидинил или бифенил; X-S(O)n,O,NR5,CH2, R5 -H, C1-C6 -алкил или бензил; R6 -H, C1-C8 - алкил, C3-C8 - алкил с разветвленной цепью, C3-C7 -циклоалкил, C3-C8 -алкенил или алкинил, фенил, в определенных случаях замещенный 1 - 3 группами выбранными из C1-C4 -алкила или алкокси, F, Br, Cl, NH2, OH, CN, CO2H, CF3, NO2, C1-C4 - карбоалкокси, NR7R8 или NCOR7 пентафторфенил, бензил, в определенных случаях замещенный 1-3 группами, выбранными из C1-C4 -алкила или алкокси, F, Br, Cl, NH2, OH, CN, CO2H,
(I), где R1 и R2 независимо, выбраны из ряда, включающего H, C1-C8 -алкил, C3-C8 -разветвленный алкил, C3-C7- циклоалкил, C4-C10 -циклоалкилакил, C7-C14 -аралкил, 2-, 3- или 4-пиридинил, 2-тиенил, 2-фуранил, фенил, в определенных случаях замещенный 1-3 группами, выбранными из ряда, включающего F, Cl, Br, OH, C1-C4 -алкокси, C1-C4 -алкил, C3-C8 -разветвленный алкил, CH3S(O)nNO2, CF3 или NR7R8 или R1 и R2 ,взятые вместе, образуют группу (см.рисунок), где L - 0,0(CH2)m+1O или (CH2)m, где m = 0 - 4, R3 -Н, C1-C6 - алкил, аллил, бензил или фенил, в определенных случаях замещенный F, Cl, CH3, CH3O или CF3; R4-C1-C8 -алкил с неразветвленной цепью, в определенных случаях замещенный F, C3-C8 - алкил с разветвленной цепью, C3-C7 -циклоалкил, C4-C10 -циклоалкилалкил, C7-C14 -аралкил, где арильная группа в определенных случаях замещена 1 - 3 группами, выбранными из ряда, включающего C1-C4 -алкил или алкокси, F, Br, Cl, NH2 OH, CN, CO2H, CF3, NO2, C1-C4 -карбоалкокси, NR7R8 или NCOR7, C3-C6/ -алкенил или акинил, C1-C3 -перфторалкил, фенил, в определенных случаях замещенный 1 - 3 группами, выбранными из C1-C4 -алкила, C1-C4 -алкокси, F, Br, Cl, NH2, OH, CN, CO2H, CF3, NO2, C1-C4 - карбоалкокси, NR7R8 или NCOR7 , пентафторфенил, бензил, в определенных случаях замещенный 1 - 3 группами, выбранными из C1-C4 -алкила или алкокси, F, Br, Cl, NH2, OH, CN, CO2H, CF3, NO2, C1-C4 -карбоалкокси, NR7R8 или NCOR7 : 2 -, 3 - или 4-пиридинил, пиримидинил или бифенил; X-S(O)n,O,NR5,CH2, R5 -H, C1-C6 -алкил или бензил; R6 -H, C1-C8 - алкил, C3-C8 - алкил с разветвленной цепью, C3-C7 -циклоалкил, C3-C8 -алкенил или алкинил, фенил, в определенных случаях замещенный 1 - 3 группами выбранными из C1-C4 -алкила или алкокси, F, Br, Cl, NH2, OH, CN, CO2H, CF3, NO2, C1-C4 - карбоалкокси, NR7R8 или NCOR7 пентафторфенил, бензил, в определенных случаях замещенный 1-3 группами, выбранными из C1-C4 -алкила или алкокси, F, Br, Cl, NH2, OH, CN, CO2H,  NO2, C1-C4 - карбоалкокси, NR7R8 или NCOR7 R7 и R8 независимо, H и C1-C4 - алкил, А- C2-C10 -алкил, C3-C10 - алкил с разветвленной цепью, C3-C10 -алкенил, C3-C10 -алкинил, Y - O, S, H2 r = 0 - 2. Реагент 1: соединение общей формулы:
NO2, C1-C4 - карбоалкокси, NR7R8 или NCOR7 R7 и R8 независимо, H и C1-C4 - алкил, А- C2-C10 -алкил, C3-C10 - алкил с разветвленной цепью, C3-C10 -алкенил, C3-C10 -алкинил, Y - O, S, H2 r = 0 - 2. Реагент 1: соединение общей формулы: 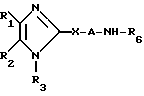 ,где R1,R2,R3,R6 имеют вышеуказанные значения. Реагент II: изоцианат формулы R4-N=C=O , где R4 имеет вышеуказанные значения. 6 табл.
,где R1,R2,R3,R6 имеют вышеуказанные значения. Реагент II: изоцианат формулы R4-N=C=O , где R4 имеет вышеуказанные значения. 6 табл.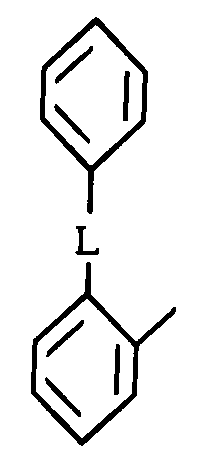
СПОСОБ ПОЛУЧЕНИЯ ИМИДАЗОЛОВ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМОЙ СОЛИ.
Способ получения имидазолов общей формулы I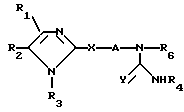
где R1 и R2 независимо выбраны из ряда, включающего H, C1 - C8-алкил, C3 - C8-разветвленный алкил, C3 - C7-циклоалкил, C4 - C10-циклоалкилалкил, C7 - C14-аралкил, 2-, 3- или 4-пиридинил, 2-тиенил, 2-фуранил, фенил, в определенных случаях замещенный 1-3 группами, выбранными из ряда, включающего F, Cl, Br, OH, C1 - C4-алкокси, C1 - C4-алкил, C3 - C8-разветвленный алкил, CH3S(O)r, NO2, CF3 или NR7R8, или R1 и R2, взятые вместе, образуют группу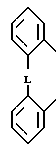
где L - O, O(CH2)m+1O или (CH2)m, где m = 0 - 4;
R3 - H, C1 - C6-алкил, аллил, бензил или фенил, в определенных случаях замещенный F, Cl, CH3, CH3O или CF3;
R4 - C1 - C8-алкил с неразветвленной цепью, в определенных случаях замещенный F, C3 - C8-алкил с разветвленной цепью, C3 - C7-циклоалкил, C4 - C10-циклоалкилалкил, C7 - C14-аралкил, где арильная группа в определенных случаях замещена 1 - 3 группами, выбранными из ряда, включающего C1 - C4-алкил или алкокси, F, Br, Cl, NH2, OH, CN, CO2H, CF3, NO2, C1 - C4-карбоалкокси, NR7R8 или NCOR7; C3 - C6-алкенил или алкинил, C1 - C3-перфторалкил, фенил, в определенных случаях замещенный 1 - 3 группами, выбранными из ряда, включающего C1 - C4-алкил, C1 - C4-алкокси, F, Br, Cl, NH2, OH, CN, CO2H, CF3, NO2, C1 - C4-карбоалкокси, NR7R8 или NCOR7; пентафторфенил, бензил, в определенных случаях замещенный 1 - 3 группами, выбранными из ряда, включающего C1 - C4-алкил или алкокси, F, Br, Cl, NH2, OH, CN, CO2H, CF3, NO2, C1 - C4-карбоалкокси, NR7R8 или NCOR7; 2-, 3- или 4-пиридинил, пиримидинил или бифенил;
X - S(O)r, O2NR5, CH2;
R5 - H, C1 - C6-алкил или бензил;
R6 - H, C1 - C8-алкил, C3 - C8-алкил с разветвленной цепью, C3 - C7-циклоалкил, C3 - C8-алкенил или алкинил, фенил, в определенных случаях замещенный 1 - 3 группами, выбранными из ряда, включающего C1 - C4-алкил или алкокси, F, Br, Cl, NH2, OH, CN, CO2H, CF3, NO2, C1 - C4-карбоалкокси, NR7R8 или NCOR7; пентафторфенил, бензил, в определенных случаях замещенный 1 - 3 группами, выбранными из ряда, включающего C1 - C4-алкил или алкокси, F, Br, Cl, NH2, OH, CN, CO2H, CF3, NO2, C1 - C4-карбоалкокси, NR7R8 или NCOR7;
R7 и R8 независимо друг от друга выбраны из ряда, включающего H и C1 - C4-алкил;
X - S(O)r, O, NR5, CH2;
A - C2 - C10-алкил, C3 - C10-алкил с разветвленной цепью, C3 - C10-алкенил, C3 - C10-алкинил;
Y - O, S, H2, r = 0 - 2,
или их фармацевтически приемлемой соли, отличающийся тем, что осуществляют реакцию соединения формулы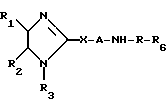
где R1, R2, X, A и R6 имеют указанные значения;
R3 имеет указанное значение или представляет собой защитную группу, такую, как силил или тритил,
с изоцианатом формулы
R4 - N = C = O,
где R4 имеет указанные значения,
в результате чего получают соединение общей формулы I, где Y - O, с выделением целевого продукта или, в случае необходимости, удалением защитной группы радикала R3 с получением соединения общей формулы I, где R3 - H, или соединение общей формулы I, где Y - O, подвергают взаимодействию с реактивом Лавессона или дифосфора пентасульфидом, в результате чего получают соединение общей формулы I, где Y = S, или соединение общей формулы I, где Y = O, подвергают взаимодействию с восстановителем, таким как литийалюмогидрид или борогидрид натрия, в результате чего получают соединение общей формулы I, где Y = H2, или соединение общей формулы I, где X - S, подвергают взаимодействию с окислителем с получением либо сульфоксида, где r = 1, либо сульфона, где r = 2, или соединение общей формулы I, где R3 - H, подвергают взаимодействию с алкилирующим агентом, таким как галоидалкил, в результате чего получают соединение общей формулы I, где R3 - C1 - C6-алкил, аллил или бензил, и выделяют целевой продукт в свободном виде или в виде его фармацевтически приемлемой соли.
Приоритет по признакам:
05.12.88 при X - S; Y - O; n = 5, 8, 2; R1 - C6H5, H; R2 - C6H5, H; R3 - R4 - H, 2,4-ди-FC6H3, CH3; C6H5 2,4-ди-CH3O6H3,C3H7, 3-FC6H4, 2,6-ди-Cl, 4-CNC6H4, C8H17, CH(CH3)2; R6 (CH2)6CH3, (CH2)3CH3, (CH2)2CH3, X = SO2, Y = S
10.10.89 при X = S, Y = O, R1 2-пиридинил, 2-тиенил, 4-FC6H4, 4-CH3OC6H4, 4-CH3C6H4, 4-ClC6H4, (CH3)2CH; CH3CH2CH2, 3-CH3OC6H4, 2-CH3OC6H4, 4-(CH3)2NC6H4, C6H11, 2-фуранил, 4-(t-C4H9)C6H4, 4-HOC6H4, (R1R2=)C6H4-2-OCH2-2, C6H4, (R1R2=)C6H4-O-C6H4; R2 2-пиридинил, 2-тиенил, 4-FC6H4, 4-CH3OC6H4, 4-CH3C6H4, 4-ClC6H4, (CH3)2CH, CH3CH2CH2, 3-CH3OC6H4, 2-CH3OC6H4, 4-(CH3)2NC6H4, C6H11, 2-фуранил, 4-(t-C4H9)C6H4, 4-HOC6H4, при Y = H2; R4-C6H5, (C6H4) - (C6H5), (CH2)7CH3, CH2-2,4-ди-FC6H3, CH2(CH2)2CH3, CH2(C6H4)(C6H5), CH2C6H11, CH2-3,4-ди-ClC6H3, CH2-C6F5; CH2CH(CH3)2, CH2CH3, 4-FC6H4, CH2C6H5; R6-H, CH3, C6H5.
| Вейганд Хильгетаг Методы эксперимента в органической химии, 1968, М.: Химия, с.377. |
