Данная заявка является частичным продолжением заявки США N 08/057035, зарегистрированной 5 мая 1993 года.
Настоящее изобретение относится к соединениям, включая 5-хлор-2-[2-[[(4-метилфенил)сульфонил] окси] этил] -7-[2,4,6- триметилфенил)метокси]антра[1,9-cd] пиразол-6(2H)он и его аналоги, которые используют в качестве промежуточных продуктов синтеза антрапиразолонов, обладающих противоопухолевой активностью, в том числе лозоксантрона. Изобретение также относится к способам синтеза антрапиразолонов, обладающих противоопухолевой активностью, включая лозоксантрон.
Лозоксантрон является активным ингредиентом препарата для лечения злокачественной опухоли груди. Известен способ получения лозоксантрона (Showalter и др. J.Med. Chem., вып. 30, стр. 121-131, 1987; J.Heterocyclic Chem., вып. 26, стр. 85, 1989), где необходимо использование 2-[(гидразиноэтил)амино] этанола в качестве исходного материала. Ограниченный доступ к этому материалу не позволяет осуществить практическую реализацию этого известного способа получения лозоксантрона. Кроме того, в этом способе для выделения целевого региоизомера необходимо проведение дорогостоящего и вредного для окружающей среды хроматографического разделения. Таким образом, существует потребность в улучшении способов синтеза лозоксантрона и родственных соединений, которые исключают необходимость использования 2-[(гидразиноэтил)амино] этанола и проведения хроматографического выделения целевого продукта.
В патенте США 4556654, опубликованном 12.03.1985 г., Showalter и др. раскрывают способ синтеза производных антра[1,9-cd]пиразол-6(2H)-она формулы 1 и 2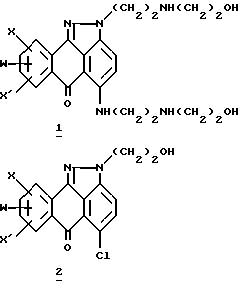
где X, X' и могут означать H, OH, алкоксильную группу или Cl. Showalter и др. также раскрывают способы синтеза лозоксантрона по нижеприведенным реакционным схемам A и B.
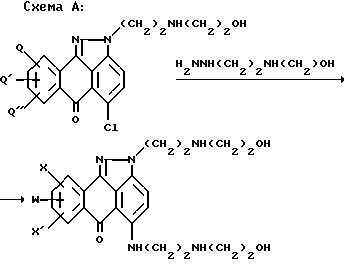
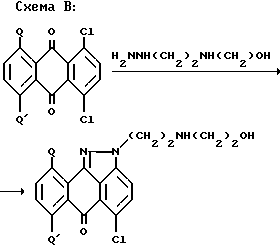
где Q, Q' и Q'' означают H, алкил, бензилокси, парахлорбензилокси или параметоксибензилокси, а X, X' и W имеют вышеуказанные значения.
В патенте США 4608439, опубликованном 26.08.1986 г., Johnson и Showalter раскрывают способ получения антра[1,9-cd]пиразол-6(2H)-онов из 1,2-дихлор-5,8-двузамещенных-9,10-антрацендионов и гидразина в соответствии с реакционной схемой C.
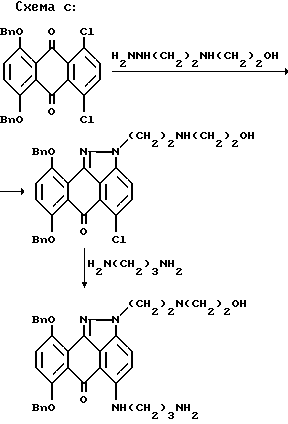
В патенте США 4672129, опубликованном 06.09.1987 г. Beylin и др. раскрывают улучшенный способ получения антра[1,9-cd]пиразол-6(2H)-онов формулы из 1,2-дихлор-5,8-двузамещенных-9,10-антрацендионов через хроматографическое разделение изомеров формулы 3 и 4 по схеме D, приведенной ниже.
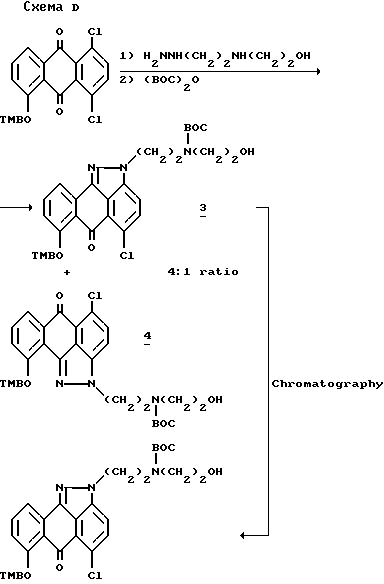
Ни одна из противопоставленных выше ссылок не раскрывает предлагаемые в настоящем изобретении способы синтеза антрапиразолонов, обладающих противоопухолевой активностью или соединения, которые можно использовать в качестве промежуточных продуктов синтеза таких антрапиразолонов.
Данное изобретение предлагает соединения формулы (I), включая 5-хлор-2-[2-[[(4-метилфенил)сульфонил] окси] этил]-7-[2,4,6- триметилфенил)метокси] антра[1,9-cd] пиразол-6(2H)-он и его аналоги, которые можно использовать в качестве промежуточных продуктов синтеза антрапиразолонов, обладающих противоопухолевой активностью, в том числе лозоксантрона. Изобретение также относится к способам синтеза антрапиразолонов, обладающих противоопухолевой активностью, в том числе лозоксантрона.
Настоящее изобретение предлагает новые производные антрапиразолона, пригодные в качестве промежуточных продуктов синтеза антрапиразолонов, обладающих противоопухолевой активностью, и представленных формулой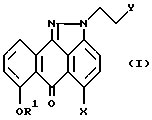
или их фармацевтически приемлемые соли,
где R1 означает H или защитную группу гидроксила;
X выбирают из группы, содержащей:
(a) F, Cl, Br, I,
(b) метансульфонилокси,
(c) толуолсульфонилокси,
(d) трифторметансульфонилокси или
(e) -OH;
Y означает подходящую уходящую группу, выбранную, но не ограничивая ее объема, например из группы, содержащей:
(a) Cl, Br, I,
(b) -OSO2R2 или
(c) -OH;
R2 выбирают из группы, содержащей:
(a) C1-C4 алкил,
(b) CvF2v+1, где v равно 1-4, или
(c) фенил или фенил, возможно замещенный 1-3 заместителями, выбранными из группы Cl, F, Br, NO2, -OR6 или C1-C4 алкила;
R6 выбирают из группы, содержащей: H, C1-C8 алкил, C2-C6 алкенил, C3-C8 циклоалкил, C4-C8 циклоалкилметил, C6-C10 арил или C7-C11 арилалкил;
при условии что, если x означает Cl, Y не может быть - OH.
К предпочтительным соединениям настоящего изобретения относятся соединения формулы (I), где:
R1 выбирают из группы, содержащей:
(a) бензил, замещенный 0-3 группами R5;
(a) бензил, замещенный 0-3 группами R5;
(b) нафтилметил, замещенный 0-3 группами R5;
(c) антрилметил, замещенный 0-3 группами R5;
(d) C1-C4 алкил; или
(e) H;
R5 независимо означает C1-C4 алкил, галоген, OR6, NO2;
R6 независимо означает H, C1-C8 алкил, C2-C6 алкенил, C3-C8 циклоалкил, C4-C8 циклоалкилметил, C6-C10 арил или C7-C11 арилалкил;
X выбирают из группы, содержащей:
(a) F, Cl, Br, I,
(b) метансульфонилокси,
(c) толуолсульфонилокси,
(d) трифторметансульфонилокси или
(e) -OH;
Y выбирают из группы, содержащей:
(a) Cl, Br, I,
(b) -OSO2R2 или
(c) -OH;
R2 выбирают из группы, содержащей:
(a) C1-C4 алкил,
(b) CvF2v+1, где v равно 1-4, или
(c) фенил или фенил, возможно замещенный 1-3 заместителями, выбранными из группы Cl, F, Br, NO2, -OR6 или C1-C4 алкила;
при условии, что если X означает Cl; R1 означает H, C1-C4 алкил, бензил, парахлорбензил или параметоксибензил-, Y не может быть -OH.
В объем настоящего изобретения входят также соединения формулы (I), имеющие вышеуказанные значения, при условии, что если X означает Cl, то Y не может быть -OH.
К предпочтительным соединениям настоящего изобретения относятся соединения формулы (I), где:
R1 выбирают из группы, содержащей:
(a) бензил;
(b) параметоксибензил;
(c) 2,4,6-триметилбензил;
(d) C1-C4 алкил; или
(e) H;
X выбирают из группы, содержащей:
(a) F, Cl, Br, I,
(b) метансульфонилокси,
(c) толуолсульфонилокси,
(d) трифторметансульфонилокси или
(e) -OH;
Y выбирают из группы, содержащей:
(a) Cl, Br, I,
(b) -OSO2R2 или
(c) -OH;
R2 выбирают из группы, содержащей:
(a) C1-C4 алкил,
(b) CvF2v+1, где v равно 1-4, или
(c) фенил или фенил, возможно замещенный 1-3 заместителями, выбранными из группы Cl, F, Br, NO2, или CH3;
при условии, что если X означает Cl; R1 означает H, C1-C4 алкил, бензил или параметоксибензил, то Y не может быть -OH.
К предпочтительным соединениям относятся соединения формулы (I), где
X означает галоген и
Y -OSO2R2.
Наиболее предпочтительны соединения формулы (I), где
X означает Cl, а
Y означает толуолсульфонилокси.
В объем предлагаемого изобретения также входят соединения формулы (I)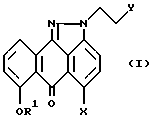
где R1 выбирают из группы, содержащей:
(a) бензил;
(b) парахлорбензил;
(c) параметоксибензил;
(d) C1-C4 алкил; или
(e) H;
X означает Cl;
Y означает -OH.
Используемый в данном описании термин "гидроксилзащищающая группа" означает любую группу, известную в органическом синтезе, пригодную для защиты гидроксильных групп. В качестве таких защитных групп можно указать, например, но не ограничивая их объема, группы, упоминаемые в монографии Greene и Wuts "Protective Groups in Organic Synthesis", John Wiley & Sons, Нью-Йорк, 1991 г., которые включены в описание настоящего изобретения в качестве ссылки. К гидроксилзащищающим группам могут также относиться, но не ограничивая объема, ацильные группы, например типа ароматических карбаматов и типа алкилов. В качестве примера можно указать метил, метоксиметил, метилтиометил, бензилоксиметил, трет-бутоксиметил, 2-метоксиэтоксиметил, 2,2,2-трихлорэтоксиметил, 2-(триметилсилил)этоксиметил, тетрагидропиранил, тетрагидрофуранил, трет-бутил, трифенилметил, триметилсилил, триэтилсилил, трет-бутилдиметилсилил, трет-бутилдифенилсилил, пивалоат, N-фенилкарбамат, арилметил, замещенный арилметил, бензил, замещенный бензил. Приводимые ниже термины и условные обозначения, используемые в описании, имеют следующие значения. Используемая в данном описании аббревиатура "DMF" означает диметилформамид. "DMAC" означает диметилацетамид, "DMSO" означает диметилсульфоксид. "TMBO" означает триметилбензилокси. "OTs" означает толуолсульфонилокси и "BnO" означает метилбензилокси.
Соединения, раскрываемые в данном описании, могут иметь асимметричные центры. Если не оговорено особо, все хиральные, диастереоизомерные и рацемические формы входят в объем предлагаемого изобретения. Многие геометрические изомеры олефинов, двойных связей C=N и тому подобное могут также входить в состав соединений, раскрываемых в данном описании, и все такие устойчивые изомеры рассматриваются в пределах объема настоящего изобретения. Необходимо особенно подчеркнуть, что соединения предлагаемого изобретения содержат асимметрически замещенные атомы углерода, и их можно выделить в виде оптически активных изомерных и рацемических форм. Известны способы получения оптически активных изомерных соединений, например, путем разложения рацематов или синтезом из оптических изомеров, используемых в качестве исходных соединений. Все хиральные, диастереоизомерные, рацемические формы и все геометрические изомеры любого строения предназначены для цели настоящего изобретения, если не оговорена особо их стереохимия или изомерная форма.
Если любая переменная (например, но не ограничивая объема, R5 и R6) повторяется более одного раза в любой составляющей или любой формуле, то ее значение определяют в каждом случае независимо от ее значения в каждом другом случае. Так, например, если группа, как дано условие, замещена 0-2 группами R5, то указанная группа необязательно может иметь до 2 заместителей R5, и значение R5 в каждом случае выбирают независимо из указанного перечня возможных значений R5.
Комбинации заместителей и/или переменных допустимы только при условии, что такие комбинации приводят к образованию устойчивых соединений. Под устойчивым соединением или устойчивой структурой в данном описании имеется в в виду любое соединение, которое достаточно стабильно, чтобы выдержать стадию выделения его с соответствующей степенью чистоты из реакционной смеси.
Используемый в данном описании термин "замещенный" означает, что один или несколько атомов водорода у заданного атома имеет заместитель, выбранный из указанной группы, при условии, что главная валентность обозначенного атома не превышает заданного значения и что реакция замещения приводит к образованию устойчивого соединения.
Используемый в данном описании термин "алкил" означает насыщенные алифатические углеводородные радикалы как с разветвленной, так и неразветвленной цепью, содержащие определенное количество атомов углерода; термин "галогензамещенный алкил" означает как разветвленные, так и неразветвленные насыщенные алифатические углеводородные радикалы с определенным количеством атомов углерода, замещенные одним или несколькими атомами галогена (например, -CvFw, где v = 1-3, a w = от 1 до (2v+1); термин "алкокси" означает алкильную группу с определенным количеством атомов углерода, связанных через кислородный мостик; термин "циклоалкил" означает в данном описании циклоалифатические насыщенные углеводородные радикалы, в том числе одно-, дву- или многоциклические кольцевые системы, например циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклооктил и адамантил. Используемый в данном описании термин " алкенил " означает углеводородные цепи, как разветвленной, так и неразветвленной конфигурации, и одну или несколько ненасыщенных углерод-углеродных связей, которые могут возникать в любой устойчивой точке основной цепи, например этенил, пропенил и тому подобное; термин "алкинил" означает углеводородные цепи, как разветвленной, так и неразветвленной конфигурации и одну или несколько ненасыщенных углерод-углеродных связей, которые могут возникать в любой устойчивой точке основной цепи, например этинил, пропинил и тому подобное.
Используемый в данном описании термин "галоидзамещенный" или "галогензамещенный "означает фтор-, хлор-, бром- и йодзамещенную группу.
Используемый в данном описании термин "арил" или "ароматический остаток" означает фенил или нафтил; термин "арилалкил" в данном описании означает арильную группу, прикрепленную к соседнему атому через алкильный мостик.
Выражение "фармацевтически приемлемые" используют в данном описании для обозначения указанных соединений, реагентов, составов и/или лекарственных форм, которые, в пределах объема разумной оценки с точки зрения медицины, пригодны для использования при контакте с кожной тканью людей или животных, не оказывая чрезмерного токсичного действия, не вызывая раздражение или аллергическую реакцию, либо другие проблемы или осложнения, и при этом сохраняя разумное соотношение терапевтического эффекта и степени риска.
Химические реакции при использовании заявленных методов синтеза проводят в любом подходящем растворителе, причем в качестве любого подходящего растворителя обычно используют любой растворитель, который не вступает в химическую реакцию (за исключением случаев, где растворитель выступает также в качестве соответствующего основания, рассматриваемого далее в описании) с исходными материалами (реагентами), полупродуктами или продуктами при таких температурах, при которых проводят взаимодействия реагентов, то есть в диапазоне температуры с точки замерзания растворителя до точки кипения растворителя. В качестве растворителей можно использовать апротонные растворители, в том числе, но не ограничивая объема, полярные апротонные органические растворители. В качестве предпочтительных растворителей, используемых в соответствии с предлагаемым изобретением, можно указать, например, но не ограничивая объема, толуол, пиридин, диметилсульфоксид (DMSO), N,N-диметилформамид (DMF), N,N-диметилацетамид (DMAC), DMSO, диэтиловый эфир, бензол или тетрагидрофуран. При желании, такой растворитель может также действовать в качестве подходящего основания в методах синтеза предлагаемого изобретения, например, где в качестве подходящего растворителя/основания используют пиридин.
Химические реакции при использовании заявленных методов синтеза предпочтительно осуществляют в присутствии любого подходящего основания, причем в качестве подходящего основания можно использовать любое, выбранное из широкого ряда оснований, участие которого в химической реакции облегчает синтез требуемого продукта. Специалисты в области органического синтеза могут подобрать подходящие основания. В качестве оснований можно использовать, например, но не ограничивая объема, органические основания. В качестве примера подходящих оснований можно указать, но не ограничивая объема, например, гидроксиды, алкоксиды, фосфаты и карбонаты щелочного металла, щелочноземельного металла, таллия и аммония, например гидроокись натрия, гидроокись калия, карбонат калия, карбонат натрия, карбонат цезия, гидроокись таллия, карбонат таллия, тетра-н-бутиламмонийкарбонат, гидроокись аммония. В качестве подходящих оснований в пределах объема предлагаемого изобретения можно использовать органические основания, но не ограничивая объема, например ароматические и алифатические амины, например пиридин, N,N-диметиламинопиридин, триалкилзамещенный амин, например триэтиламин, N,N-диизопропилэтиламин, 1,5-диазабицикло[4.3.0]нон-5-ен (DBN), 1,4-диазабицикло[2.2.2]октан (DABCO), 4-диметиламинопиридин (DMAP), 1-8-диазабицикло[5.4.0]ундец-7-ен (DBU) или тетраметилэтилендиамин (TMEDA).
Стадия 3 синтеза.
Настоящее изобретение также предлагает способ синтеза соединения формулы (I), включающий взаимодействие соединения формулы (II) или смеси соединения формулы (II) или формулы (III)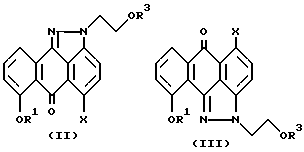
где R1 и X имеют значения, указанные выше для соединения формулы (I), а R3 означает водород;
с подходящим бромирующим, хлорирующим или йодирующим реагентом, при этом реакцию проводят в любом подходящем растворителе, возможно в присутствии соответствующего основания с образованием соединения формулы (I), где Y означает Br, Cl или I.
Соответствующий бромирующий, хлорирующий или йодирующий реагент может превратить гидроксильную группу в соединении (II) в группу Br, Cl или I. Специалистам в области органического синтеза хорошо известны такие бромирующие, хлорирующие или йодирующие реагенты. В качестве примера таких бромирующих реагентов можно указать, но без ограничения объема, например трифенилфосфин/тетрабромид углерода, HBr, дифос-Br2N-бромсукцинимид (NBS) и тионилбромид. В качестве примера таких хлорирующих реагентов можно указать, но без ограничения объема, например трифенилфосфин/тетрахлорид углерода, HCl, дифос-Cl2N-хлорсукцинимид (NCS) и тионилхлорид.
В соответствии с общей методикой, в зависимости от выбранных растворителя, основания и бромирующего, хлорирующего или йодирующего реагентов, реакцию можно проводить при температуре примерно от - 10 до + 60oC в течение примерно от 0,1 до 72 часов с образованием соединения формулы (I).
Изобретение также предлагает способ синтеза соединения формулы (I), включающий взаимодействие соединения формулы (II) или смеси соединения формулы (II) или формулы (III)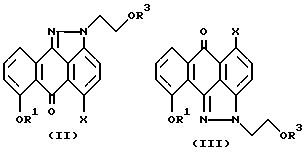
где R1 и X имеют значения, указанные выше для соединения формулы (I), а R3 означает водород;
с любым реагентом, который может превратить гидроксил в уходящую группу. Указанный реагент, пригодный для превращения гидроксила в уходящую группу, можно подобрать из широкого ряда реагентов, которые хорошо известны специалистам в области органического синтеза. Такие реагенты можно выбрать из реагентов, но не ограничивая объема, например из группы формулы ClSO2R2, где R2 имеет вышеуказанные значения для соединения формулы (I), например бензолсульфонилхлорид, диметилбензолсульфонилхлорид, триметилбензолсульфонилхлорид, хлорбензолсульфонилхлорид, дихлорбензолсульфонилхлорид, трихлорбензолсульфонилхлорид, толуолсульфонилхлорид, предпочтительно толуолсульфонилхлорид (хлористый тозил). Вышеуказанную реакцию проводят в подходящем растворителе, возможно в присутствии соответствующего основания с образованием соединения формулы (I).
В соответствии с общей методикой, в зависимости от выбранных растворителя, основания и реагента, реакцию можно проводить при температуре примерно от - 10 до + 50oC в течение примерно от 2 до 72 часов с образованием соединения формулы (I). После завершения реакции, целевое соединение можно выделить из смеси изомеров, участвующих во взаимодействии, например, осаждением требуемого соединения формулы (I) при добавлении спирта в реакционную смесь.
Реакцию на стадии 3 необязательно, но предпочтительно проводят в присутствии подходящего основания. В предпочтительном варианте в качестве основания можно использовать пиридин и 1,8-диазабицикло[5.4.0]ундец-7-ен (DBU). Температура реакционной смеси обычно варьирует в диапазоне примерно от -15oC до +20oC, предпочтительно использование охлажденного раствора. Например, реакцию можно проводить в присутствии (DBU) при температуре в диапазоне примерно от -10oC до +10oC в течение примерно от 3 до 5 часов. Реакцию на стадии 3 в альтернативном варианте можно проводить в присутствии, например, пиридина при температуре в интервале примерно от -10oC до +25oC в течение примерно от 24 до 60 часов. Можно также использовать другие основания, например, 1,5-диазабицикло[4.3.0]нон-5-ен (DBN), 1,4-диазабицикло[2.2.2]октан (DABCO), 4-диметиламинопиридин (DMAP) или тетраметилэтилендиамин (TMEDA).
Стадия 2 синтеза.
Настоящее изобретение также предлагает способ синтеза соединения формулы (II) и формулы (III), имеющие вышеуказанные значения, включающий взаимодействие 2-гидроксиэтилгидразина с соединением формулы (IV):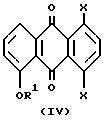
где R1 и X имеют вышеуказанные значения,
при этом реакцию проводят в любом подходящем растворителе, возможно в присутствии соответствующего основания с образованием соединения формулы (II) или (III). В качестве растворителя предпочтительно использование DMF, DMAC или DMSO, и наиболее предпочтительно DMAC. В качестве основания предпочтительно использование карбоната калия, карбоната натрия, бикарбоната калия, бикарбоната натрия, N,N-диметиламинопиридина или триалкиламина и особенно предпочтительно N,N-диизопропилэтиламина. В соответствии с общей методикой, в зависимости от выбранных растворителя и основания реакцию можно проводить при температуре в интервале примерно от 20 до 160oC в течение примерно от 1 до 20 часов с образованием соединения формулы (II) или (III).
Стадия 4 синтеза.
Настоящее изобретение также предлагает способ синтеза соединения формулы (V):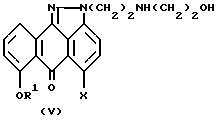
где R1 и X имеют вышеуказанные значения,
заключающийся в том, что осуществляют взаимодействие соединения формулы (I) с этаноламином в соответствующем растворителе, возможно в присутствии подходящего основания с образованием соединения формулы (V). В соответствии с общей методикой, в зависимости от выбранных растворителя и основания реакцию можно проводить в течение примерно от 6 до 24 часов при температуре в интервале примерно от 20 до 100oC. В качестве основания предпочтительно использование карбоната калия.
Хлористоводородную соль соединения формулы (V), обозначаемую как соединение (Vb), можно получить взаимодействием соединения формулы (V) с хлористым водородом в смеси растворителей, например, смеси спирта и соответствующего растворителя. В соответствии с общей методикой реакцию можно проводить при температуре в интервале примерно от -10 до 30oC в течение примерно 1-24 часов с выходом соединения формулы (Vb) (хлористоводородная соль соединения формулы (V)). Можно также получить другие формы фармацевтически пригодной соли соединения формулы (V).
Стадия 5 синтеза.
Настоящее изобретение также предлагает способ синтеза соединения формулы (VI), обладающего противоопухолевой активностью, например лозоксантрона (R1= H), включающий взаимодействие соединения формулы (V) или его фармацевтически приемлемой соли, например (Vb) с 2-(2-аминоэтиламино)этанолом в подходящем растворителе, при желании в присутствии соответствующего основания, с образованием соединения формулы (VI) или его фармацевтически приемлемой соли, например (Vb). В качестве основания и растворителя предпочтительно использование пиридина. В соответствии с общей методикой, в зависимости от выбранных растворителя и основания реакцию можно проводить при температуре в интервале примерно от 60oC до точки кипения растворителя в течение примерно от 6 до 24 часов с получением соединения формулы (VI) или его фармацевтически приемлемой соли.
Изобретение также предлагает способ получения соединения формулы (V):
или его фармацевтически приемлемой соли, где R1 означает 2,4,6-триметилбензил, а X означает Cl; включающий стадии:
(1) взаимодействия соединения формулы (II):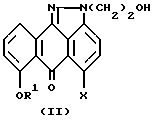
или его фармацевтически приемлемой соли, где R1 и X имеют вышеуказанные значения,
с подходящим бромирующим, хлорирующим или йодирующим реагентом в любом подходящем растворителе; и
(2) взаимодействие продукта, полученного из стадии (1) с этаноламином с образованием соединения формулы (V).
Бромирующий, хлорирующий или йодирующий реагент для использования в вышеуказанной стадии (1) можно выбрать из любого из множества различных реагентов, которые общеизвестны в области органического синтеза, например, но не ограничивая объема, трифенилфосфин/тетрабромида углерода, Hbr, дифос-Br2N-бромсукцинимида (NBS) и тионилбромида, трифенилфосфин/тетрахлорида углерода, и тионилхлорида.
В соответствии с общей методикой, в зависимости от выбранных растворителя, основания и бромирующего, хлорирующего или йодирующего реагентов, реакцию можно проводить при температуре примерно от 10 до 60oC в течение примерно от 10 до 120 мин. Взаимодействие с этаноламином стадии (2) можно проводить при температуре от примерно 20 до 100oC в течение от примерно 2 до 48 часов с выходом соединения формулы (V).
Фармацевтически приемлемые соли соединений, полученных в соответствии со способами настоящего изобретения, обычно можно получить путем взаимодействия указанных соединений в виде свободного основания со стехиометрическим количеством соответствующей кислоты в любом органическом растворителе. Перечень подходящих солей можно найти в справочнике "Remington's Pharmaceutical Sciences, Mack Publishing Company, 17 издание, Easton PA, стр. 1418, 1985, которые включены в данное описание в качестве ссылки на предшествующий уровень техники.
В приводимой ниже схеме (1) раскрывается вся реакционная цепь получения целевого соединения формулы (VI) и его фармацевтически приемлемых солей.
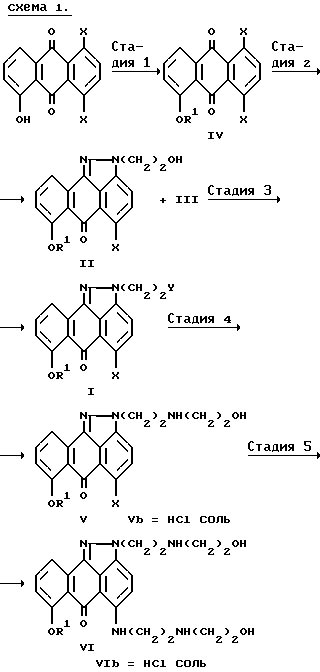
где R1, X и Y имеют вышеуказанные значения.
Таким образом, цель предлагаемого изобретения - создание улучшенного способа синтеза изоксантрона и других противоопухолевых соединений формулы (VI).
Стадия 1:
В стадии 1 реакционной схемы, показанной выше, в качестве исходного материала используют соединение формулы: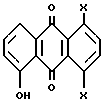
где X имеет значения, указанные выше для соединения формулы (I). Взаимодействие этого исходного материала с реагентом, пригодным для защиты гидроксильной группы, в подходящем растворителе, возможно в присутствии основания, обеспечивает выход соединения формулы (IV), где R1 означает гидроксилзащищающую группу. Например, при взаимодействии исходного материала с галоидным бензилом, например 2,4,6-триметилбензилхлоридом, получают соединение формулы (IV), где R1 означает бензил или замещенный бензил. Как указывалось выше, R1 может быть представлен любой группой, выбранной из большого разнообразия других групп, общеизвестных специалистам в области органического синтеза для защиты гидроксильной группы. Таким образом, для выбора гидроксильной защитной группы из широкого ряда других подходящих защитных групп, которые можно использовать для цели настоящего изобретения, специалисты могут обратиться к изданию "Protective Groups in Organic Synthesis", под редакцией Greene и Wuts, John Wiley & Sons, 1991.
Реакция на стадии 1 может проходить в любом подходящем растворителе или смеси растворителей, например в смеси ацетона и диметилформамиде или ацетона и диметилацетамида. В соответствии с общей методикой, в зависимости от растворителя и группы, выбранной для защиты гидроксила, реакцию можно проводить при температуре в интервале примерно от 22 до 80oC, предпочтительно при температуре примерно 65oC. Реакцию проводят в среде индифферентного газа, например азота. После охлаждения соединение формулы (IV) можно выделить из реакционной смеси и затем использовать его без дальнейшей очистки в стадии 2.
На стадии 2 синтеза соединения формулы (II) и формулы (III) получают в виде смеси региоизомеров (соотношение примерно 4:1 и выход 85%) при взаимодействии соединения формулы (IV) с 2-гидроксиэтилгидразином в любом подходящем апротонном растворителе, например, тетрагидрофуране, N,N-диметилформамиде, диметилсульфоксиде или N,N-диметилацетамиде, предпочтительно N,N-диметилацетамиде. Реакцию можно проводить при температуре в интервале от примерно 20 до 160oC, предпочтительно около 80oC. Реакцию предпочтительно проводят в присутствии подходящего основания, например, карбоната калия, карбоната натрия, N, N-диметиламинопиридина или триэтиламина, диизопропилэтиламина, предпочтительно диизопропилэтиламина. Соединения формулы (II) или (III) можно выделить при добавлении к реакционной смеси воды. Выпавший осадок можно собрать фильтрованием и после промывания фильтрата водой, этилацетатом и гексаном получают смесь соединений формулы (II) и (III) (в виде региоизомеров), которую используют в следующей стадии синтеза без дальнейшей очистки.
На стадии 3 новые соединения формулы (I) получают путем взаимодействия соединения формулы (II) или смеси региоизомеров формулы (II) и формулы (III) с реагентом, пригодным для превращения гидроксильной группы в уходящую группу, например, группу формулы ClSO2R2. Такой реагент, пригодный для превращения гидроксила в уходящую группу, можно выбрать из широкого ряда реагентов, которые хорошо известны специалистам в области органического синтеза. Такие реагенты можно выбрать из группы, например, бензолсульфонилхлорида, диметилбензолсульфонилхлорида, триметилбензолсульфонилхлорида, хлорбензолсульфонилхлорида, дихлорбензолсульфонилхлорида, трихлорбензолсульфонилхлорида, толуолсульфонилхлорида, предпочтительно толуолсульфонилхлорида (хлористого тозила).
Как указывалось выше, хлорирующий агент, например хлористый тионил, можно также использовать в качестве такого реагента, обеспечивая выход соединения формулы (I), где Y=Cl. Практически изомерно чистую форму соединения формулы (I) (99,5%) можно выделить осаждением его из системы растворителей, состоящей, например, из метанола и хлористого метилена. Выделенное соединение формулы (I) можно использовать в следующей реакции без хроматографического разделения.
Хроматографическое разделение обязательно для соединения формулы (I), где Y означает хлор, бром, йод или метансульфонилоксигруппу, для получения формы изомерной чистоты.
Указанную реакцию на стадии 3 необязательно, но предпочтительно проводят в присутствии подходящего основания. В предпочтительном варианте в качестве основания можно использовать пиридин и 1,8-диазабицикло[5.4.0]ундец-7-ен (DBU). Например, DBU способствует получению значительно обогащенных форм в целевом тозилзамещенном продукте формулы (I), полученном из основного изомерного спирта формулы (II). DBU также обеспечивает выход обогащенной формы за примерно 3-5 часов реакции. Основание и реагент, пригодный для превращения гидроксила в уходящую группу, например, толилсульфонильный реагент, можно использовать примерно в 2-4-кратном молярном избытке относительно спирта формулы (II); температура реакционной смеси обычно варьирует в диапазоне примерно от -15oC до +30oC, предпочтительно использование охлажденного раствора.
Можно также использовать другие основаняи, например, 1,5-диазабицикло[4.3.0]нон-5-ен (DBN), 1,4-диазабицикло[2.2.2]октан (DABCO), 4-диметиламинопиридин (DMAP), или тетраметилэтилендиамин (TMEDA). Как правило, использование таких оснований не только снижает продолжительность реакции в отношении пиридина, но может привести к образованию защищенных изомерных спиртов.
Реакцию на стадии 3 можно также проводить с использованием хлористого тионила (SOCl2) в соответствующем растворителе в присутствии любого подходящего основания с образованием соединения формулы (I), где Y означает Cl. Эту реакцию можно проводить при температуре в интервале примерно от -10oC до +50oC в течение примерно от 1 до 24 часов с образованием соединения формулы (I), где R1 имеет вышеуказанные значения, а X и Y означают Cl.
На стадии 4 соединения формулы (Vb), в виде хлористоводородной соли, можно получить путем взаимодействия соединения формулы (I) с этаноламином с образованием соединения формулы (V) с последующей ее обработкой соляной кислотой.
На стадии 5 соединение формулы (V) или (Vb) взаимодействует с 2-(2-аминоэтиламино)этанолом в подходящем растворителе, например пиридине, с последующим превращением полученного промежуточного продукта в форму дихлоргидрата для получения соединения формулы (VI), например, лозоксантрона (где R1=H).
Способы синтеза предлагаемого изобретения можно также использовать для получения других противоопухолевых веществ, раскрываемых Zagotto и др., в журнале Bioorganic and Medical Chemistry, 2(7), 1992, стр. 659; H.D.H. Showalter и др. , J.Med.Chem., 27, 1984, стр. 255; H.D.H. Showalter и др., J. Med.Chem., 30, 1987, стр. 121; W.R. Leopold, Cancer Research, 45, 1985, стр. 5532; V.G. Beylin и др., J.Heterocyclic Chem., стр. 85; в описании к патенту США N 4556654 и Европатенту N 0103381, каждое из которых включено в данное описание изобретения в качестве ссылки.
Настоящее изобретение более подробно иллюстрируется нижеследующими примерами, которые ни в коем случае не должны ограничивать объем предлагаемого изобретения.
В соответствии с методами синтеза, описанными выше и показанными в схеме 1, получены следующие соединения формулы (I).
Пример 1
Получение 1,4-Дихлор-5-[(2,4,6-триметилфенил)метокси] -9,10- антрацендиона (формулы (IV), где X=Cl, а R1=2,4,6-триметилбензил).
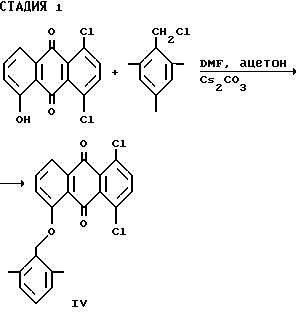
Четырехгорлую круглодонную колбу (емкостью 1 л), оснащенную механической мешалкой, термометром и холодильником с азотным барботером, продувают азотом в течение 10 мин. В колбу помещают 1,4-дихлор-6-гидрокси-9,10-антрацендион (220,0 г, 0,75 моль), карбонат цезия (164,0 г, 0,5 моль), 2,4,6-триметилбензилхлорид (180,0 г, 1,0 моль), ацетон (2,8 л) и N,N-диметилформамид (0,9 л). Полученный реакционный раствор нагревают с обратным холодильником (65oC) в атмосфере азота (при давлении 1 атм) в течение 10 часов, а затем охлаждают до комнатной температуры (22oC) и перемешивают в течение 16 часов. Полученную реакционную массу охлаждают до 0oC и выдерживают при этой температуре в течение 2 часов. Осадок собирают фильтрованием, промывают теплой водой (50oC, 2х400 мл), метанолом (2х120 мл) и сушат в вакуум-сушилке (50oC, 10 мм) в течение 20 часов с выходом целевого соединения IV (311,8 г, 98% выход), т.пл. 220-222oC. Показатель m/e масс-спектра Cl 425 (M+1).
Пример 2
Получение 5-Хлор-2-(2-гидроксиэтил)-7-[(2,4,6-триметилфенил) метокси]-антра-[1,9cd] -пиразол-6-(2H)она формулы (II) и 5-Хлор-2-(2-гидроксиэтил)-10-[(2,4,6-триметилфенил)метокси] -антра- [1,9cd]-пиразол-6-(2H)она формулы (III).
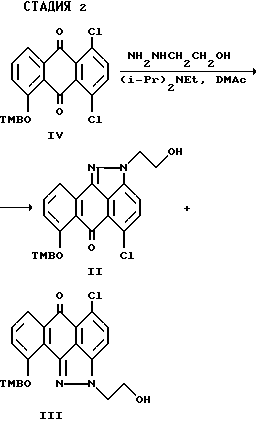
В четырехгорлую круглодонную колбу (емкостью 12,0 л), оснащенную механической мешалкой, трубкой для подачи азота и холодильником с барботером, загружают соединение IV (304,0 г, 0,72 моль) диметилацетамид (1,8 л). Полученную реакционную смесь нагревают до температуры 80oC) и в течение 3 часов прибавляют раствор, содержащий 2-гидроксиэтилгидразин (175,0 г, 2,23 моль), N,N-диизопропилэтиламин (182,0 г, 1,4 моль) и диметилацетамид (1,7 л). Полученную реакционную массу перемешивают при температуре 80oC еще в течение 4 часов, а затем при температуре 30oC в течение 16 часов. Реакционную массу охлаждают до комнатной температуры, а затем медленно выливают в воду (7,2 л). Осадок собирают фильтрованием, промывают водой (2х1,0 л), охлажденным этилацетатом (2х1,0 л) и гексаном (1,0 л). Полученный твердый продукт сушат в вакуум-сушилке (40oC, 10 мм) в течение 24 часов с выходом смеси требуемых соединений II и III (269,0 г, соотношение II к III - 80:20, 85% выход), т. пл. 200-203oC. Показатель m/e масс-спектрометрии Cl в комбинации с ВЭЖХ 447 (M+1).
Пример 3
Получение 5-Хлор-2-[2-[[(4-метилфенил)сульфонил]окси]этил])-7- [(2,4,6-триметилфенил)метокси]-антра-[1,9cd]-пиразол-6-(2H)она (формулы (I).
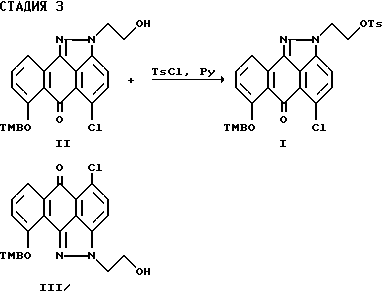
Четырехгорлую круглодонную колбу (емкостью 2,0 л), оснащенную механической мешалкой, термометром и холодильником с азотным барботером, продувают азотом в течение 10 мин. В колбу загружают смесь соединений II и III (100,0 г, 0,22 моль), метиленхлорид (1,0 л) и пиридин (Py) (54,0 г,0,68 моль). Полученную реакционную массу охлаждают до температуры 0oC и в течение 25 мин. к ней медленно прибавляют паратолуолсульфонилхлорид (TsCl) (84,0 г, 0,44 моль). Смесь перемешивают при комнатной температуре в течение 48 часов, а затем прибавляют дополнительную порцию паратолуолсульфонилхлорида (21,0 г, 0,11 моль). Полученную реакционную массу перемешивают при комнатной температуре в течение 10 часов, а затем прибавляют к ней метиленхлорид (2,0 л). Смесь отфильтровывают и полученный фильтрат промывают водой (2х0,8 л) и солевым раствором (0,5 л). Органический слой сушат (K2CO3), а растворитель отгоняют при пониженном давлении. Остаток кристаллизуют при добавлении смеси растворителей из метанола и метиленхлорида (0,6 л, 2:1), и полученный раствор охлаждают до температуры 4oC в течение 4 часов. Осадок отфильтровывают, промывают смесью растворителей из метанола и метиленхлорида (3х20 мл, 2:1) и после сушки продукта в вакуум-сушилке (40oC, 10 мм) в течение 20 часов получают целевое соединение I (89,4 г, максимальная степень чистоты спирта - 99,5%, 83% выход в расчете на 80% чистоту требуемого изомера в исходном материале), т.пл. 180-183oC.
Пример 3A
Этот пример относится к стадии 3, где реакцию проводят по методике примера 3, за исключением того, что вместо пиридина используют 1,8-диазабицикло[5.4.0] ундец-7-ен (DBU). В трехгорлый круглодонный реакционный сосуд емкостью 1,0 л загружают 14 г смеси спиртов формулы (II)) и (III) (74% спирта составляет основной региоизомер, 0,74 х 31,22 ммоль = 23,1 ммоль). Затем в колбу добавляют 1,8-диазабицикло[5.4.0]ундец-7-ен (3,5х31,22 ммоль DBU фирмы Aldrich, 98% чистоты = 16,974 г), обработанный 140 мл метиленхлорида. Полученную суспензию перемешивают механической мешалкой, а затем охлаждают постепенно до температуры - 12oC в кювете с метанолом и льдом. После этого к реакционной массе прибавляют паратолуолсульфонилхлорид (фирмы Aldrich, 98% степени чистоты, 3,5 х 31,22 ммоль = 21,26 г.). Температуру реакционной смеси быстро повышают до 7oC, а затем понижают до 0oC (суммарно в течение 3 мин) Кювету для охлаждения убирают и реакционную массу доводят до комнатной температуры в течение 50 мин и выдерживают при комнатной температуре еще 3 часа 11 мин. Затем к реакционной массе прибавляют 560 мл метиленхлорида и перемешивают в течение 0,5 часа и фильтруют через воронку с пористым стеклянным фильтром. Полученный осадок и реакционный сосуд промывают 140 мл метиленхлорида. Полученное твердое вещество сушат в глубоком вакууме с выходом 1,7 г ключевого спирта (III) (в форме неосновного изомера), идентифицированного при проведении ВЭЖХ (12,1% выход из исходного материала). Фильтрат переносят в промытый реакционный сосуд, куда добавлено 140 мл метанола. Полученную реакционную смесь кипятят с отгонкой метиленхлорида до тех пор, пока сосуд не нагреется до 52oC, приводя к осаждению продукта. Осадок отфильтровывают через фильтр из пористого стекла, промывают и ополаскивают 280 мл метанола при комнатной температуре, а затем сушат сначала методом отсасывания влаги, а затем в глубоком вакууме при температуре в интервале 45-50oC. Целевое тозилированное соединение (I) в форме изомера весит 10,44 г (75,2% выход выделенного продукта в расчете 74% степень чистоты требуемого изомера в исходном материале) и не содержит, как показывают данные ВЭЖХ, неосновного тозилсодержащего изомера (максимальная степень чистоты изоспирта - 98,69%). В маточных растворах не обнаружено никакого целевого тозилированного изомера.
Пример 4
Получение 5-хлор-2-[2-[(2-гидроксиэтил)амино]этил]-7-[(2,4,6- триметилфенил)метокси]-антра-[1,9cd]-пиразол-6-(2H)она формулы Va.
Стадия 4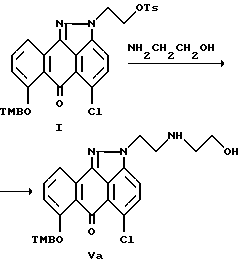
Четырехгорлую круглодонную колбу (емкостью 1 л), оснащенную механической мешалкой, термометром и холодильником с азотным барботером, продувают азотом в течение 10 мин. В колбу помещают соединение формулы 1 (75,0 г, 0,12 моль), диметилацетамид (0,7 л), этаноламин (30,0 г, 0,49 моль), и карбонат калия (26,0 г, 0,19 моль). Полученную реакционную смесь перемешивают при температуре 45oC в течение 18 часов, охлаждают до комнатной температуры, а затем прибавляют воду со льдом (4oC, 2 л). Реакционную массу выдерживают при комнатной температуре в течение 18 часов, а затем фильтруют. Осадок промывают водой (2х0,2 л), гексаном (0,2 л) и сушат в вакуум-сушилке (40oC 20 мм) в течение 20 часов с выходом целевого соединения Va (55,5 г, 91% выход), т.пл. 177-179oC. Показатель m/e масс-спектра Cl 490 (M+1).
Пример 5
Получение гидрохлорида 5-хлор-2-[2-[(2-гидроксиэтил)амино] этил] - 7-[(2,4,6-триметилфенил)метокси]-антра-[1,9cd]-пиразол-6-(2H) она (хлористоводородная соль соединения формулы Vb).
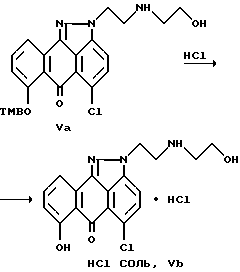
В четырехгорлую круглодонную колбу (емкостью 1,0 л), оснащенную механической мешалкой, термометром, трубкой для подачи хлористого водорода и холодильником, загружают соединение Va (54,5 г, 0,11 моль) и смесь растворителей метанола и метиленхлорида (0,51 л, 1:4). Реакционный раствор охлаждают до 0oC, а затем через раствор пропускают хлористый водород. Как только температура реакционной массы поднимется до 20oC, барботирование хлористым водородом прекращают и смесь охлаждают до температуры 5oC. После трехкратного повторения этой процедуры, реакционную массу закрывают герметично резиновыми прокладками, доводят до комнатной температуры и перемешивают в течение 16 часов. Через реакционную массу пропускают азот в течение 20 минут, и выпавший осадок собирают фильтрованием, промывают метиленхлоридом (2х0,4 л) и гексаном (0,8 л) с выходом целевого продукта (43,3 г, 100% выход), т.пл. 245-250oC.
Пример 6
Получение лозоксантрона.
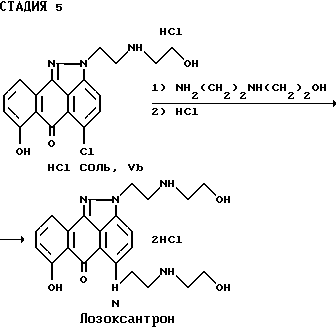
Четырехгорлую круглодонную колбу (емкостью 1 л), оснащенную механической мешалкой, термометром и холодильником с азотным барботером, продувают азотом в течение 10 мин. В колбу загружают гидрохлорид соединения Vb (40,0 г, 0,10 моль), 2-(2-аминоэтиламино)этанол (104,0 г, 1,0 моль) и пиридин (0,2 л). Полученную смесь перемешивают в атмосфере азота (1 атм) при температуре 82oC в течение 18 часов, а затем охлаждают до комнатной температуры. После этого в реакционную массу добавляют 2-пропанол (0,3 л) и полученную смесь перемешивают в течение 4 часов при температуре 5oC. Выпавший осадок отфильтровывают, промывают охлажденным изопропанолом (2х0,2 л) и гексаном (2х0,2 л). Твердый продукт растворяют в метаноле (1,0 л), а затем прибавляют раствор, содержащий HCl и изопропанол (6 н, 50 мл). Реакционную массу перемешивают при комнатной температуре в течение 10 мин, а затем нагревают с обратным холодильником (65oC) в течение 10 мин. Затем реакционную массу охлаждают и перемешивают при 0oC в течение 2 часов. Выпавший красно-оранжевый осадок отфильтровывают, промывают холодным метанолом (4х150 мл), гексаном (2х250 мл) и сушат в вакуум-сушилке (60oC, 20 мм) с выходом целевого лозоксантрона (27,7 г, 55% выход), т.пл. 268-270oC.
Пример 7
Одностадийное получение продуктов реакции PPh3/Br4, этаноламина и 5-хлор-2-[2-[(2-гидроксиэтил)амино]этил]-7-[(2,4,6- триметилфенил)метокси]-антра-[1,9cd]-пиразол-6-(2H)она, формулы (V).
В трехгорлую круглодонную колбу (емкостью 500 мл), оснащенную магнитной мешалкой, азотным барботером и холодильником, загружают смесь региоизомеров триметилбензилгидроксиэтилпиразола II и III (10,0 г, 94% чистоты, 21,0 ммоль), трифенилфосфин (99%, 11,4 г, 43,0 ммоль) и дихлорметан (сухой, 200 мл). Реакционный раствор перемешивают в атмосфере азота (1 атм) при комнатной температуре в течение 10 мин и прибавляют одной порцией тетрабромид углерода (7,2 г, 99%, 21,5 ммоль), при этом температура реакции повышается примерно на 10oC. Полученную реакционную массу перемешивают в атмосфере азота (1 атм) при комнатной температуре в течение 1 часа, а затем при температуре 40oC в течение 10 мин (при этой температуре все исходные материалы должны превратиться в их соответствующие бромиды). Затем реакционную массу охлаждают опять до комнатной температуры и прибавляют одну порцию этаноламина (10,0 г, 99%, 162,1 ммоль). Полученную смесь перемешивают в атмосфере азота (1 атм) при температуре 40oC в течение 24 часов. Растворитель отгоняют при пониженном давлении и осадок промывают этилацетатом (100,0 мл) и отфильтровывают. Полученный твердый продукт промывают гексаном и сушат в атмосфере азота в сушилке (60oC, 20 мм) в течение 60 часов с получением целевого продукта Va и его региоизомера (100% выход).
Пример 8
Получение 5-хлор-2-(2-хлортил)-7-[(2,4,6-триметилфенил)метокси] - антра-[1,9cd]-пиразол-6-(2H)она, формулы (I).
В трехгорлую круглодонную колбу (емкостью 250 мл), оснащенную магнитной мешалкой, трубкой для подачи азота и холодильником с барботером, загружают смесь региоизомеров 5-хлор-2-(2-гидроксиэтил)-7-[(2,4,6-триметилфенил)метокси] -антра- [1,9cd]-пиразол-6-(2H)она (II) и 5-хлор-2-(2-гидроксиэтил)-10-[(2,4,6- триметилфенил)метокси]-антра-[1,9cd]-пиразол-6-(2H)она III (5,0 г, 94% чистоты, 10,1 ммоль), пиридин (1,6 г, 20,0 ммоль) и ацетонитрил (сухой, 100 мл). Полученную реакционную массу охлаждают до 0oC. Затем к реакционной массе прибавляют раствор тионилхлорида в дихлорметане (2 М, 7 мл). Полученную смесь перемешивают при комнатной температуре в течение 6 часов и затем реакционный процесс регулируют с помощью ВЭЖХ каждые три часа. После завершения, реакцию гасят водой (100 мл), доводя температуру до 0oC. Осадок растворяют в этилацетате (300 мл) и образовавшиеся слои разделяют. Органический слой промывают водой (50 мл), бикарбонатом натрия (5% раствор, 50 мл), солевым раствором (50 мл) и сушат (MgSO4). Растворитель отгоняют при пониженном давлении и после флэш-хроматографии на силикагеле (400 г, элюирование из смеси EtOAc/гексан, соотношение от 20:80 до 50:50) получают целевой региоизомер I (0,7 г). Показатель m/e Масс-спектра (Cl) 465 (M+1).
Пример 9
Получение 5-хлор-2-[2-[(2-хлорбензол)сульфонил] окси]этил]-7- [(2,4,6-триметилфенил)метокси]-антра-[1,9cd]-пиразол-6-(2H)она, формулы (I).
В соответствии с методикой примера 3, используя в качестве исходных материалов 2-хлорбензолсульфонилхлорид и смесь региоизомеров 5-хлор-2-(2-гидроксиэтил)-7-[(2,4,6-триметилфенил)метокси] -антра- [1,9cd] -пиразол-6-(2H)она (II) и 5-хлор-2-(2-гидроксиэтил)-10-[(2,4,6- триметилфенил)метокси] -антра-[1,9cd]-пиразол-6-(2H)она (III), получают названное соединение.
Пример 10
Получение 5-хлор-2-[2-[(3-хлорбензол)сульфонил] окси]этил]-7- [(2,4,6-триметилфенил)метокси]-антра-[1,9cd]-пиразол-6-(2H)она, формулы (I).
В соответствии с методикой примера 3, используя в качестве исходных материалов 3-хлорбензолсульфонилхлорид и смесь региоизомеров 5-хлор-2-(2-гидроксиэтил)-7-[(2,4,6-триметилфенил)метокси] -антра- [1,9cd] -пиразол-6-(2H)она (II) и 5-хлор-2-(2-гидроксиэтил)-10-[(2,4,6- триметилфенил)метокси] -антра-[1,9cd]-пиразол-6-(2H)она (III), получают названное соединение.
Пример 11
Получение 5-хлор-2-[2-[(бензолсульфонил)оксиэтил] -7-[(2,4,6- триметилфенил)метокси]-антра-[1,9cd]-пиразол-6-(2H)она, формулы (I).
В соответствии с методикой примера 3, используя в качестве исходных материалов хлорбензолсульфонилхлорид и смесь региоизомеров 5-хлор-2-(2-гидроксиэтил)-7-[(2,4,6-триметилфенил)метокси]-антра- [1,9cd]-пиразол-6-(2H)она (II) и 5-хлор-2-(2-гидроксиэтил)-10-[(2,4,6- триметилфенил)метокси]-антра-[1,9cd]-пиразол-6-(2H)она (III), получают названное соединение.
Пример 12
Получение 5-хлор-2-[2-[(метансульфонил)оксиэтил] -7-[(2,4,6- триметилфенил)метокси]-антра-[1,9cd]-пиразол-6-(2H)она, формулы (I).
В соответствии с методикой примера 3, используя в качестве исходных материалов метансульфонилхлорид и смесь региоизомеров 5-хлор-2-(2-гидроксиэтил)-7-[(2,4,6-триметилфенил)метокси] -антра- [1,9cd]-пиразол-6-(2H)она (II) и 5-хлор-2-(2-гидроксиэтил)-10-[(2,4,6- триметилфенил)метокси]-антра-[1,9cd] -пиразол-6-(2H)она (III), получают названное соединение.
Хотя данное изобретение описано на конкретных примерах его осуществления, следует подразумевать, что изобретение не ограничивается изложенными деталями этих примеров и что в него могут быть внесены различные эквивалентные варианты и изменения, не выходящие за рамки существа и объема настоящего изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПОЛИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, ИХ ПРОИЗВОДНЫЕ, ФАРМАЦЕВТИЧЕСКИЕ ПРЕПАРАТЫ КАК УСИЛИТЕЛИ И МЕТОД ЛЕЧЕНИЯ НАРУШЕНИЯ МЫСЛИТЕЛЬНОЙ ДЕЯТЕЛЬНОСТИ | 1994 |
|
RU2152944C1 |
| СПОСОБ ПОЛУЧЕНИЯ ИМИДАЗОЛОВ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМОЙ СОЛИ | 1989 |
|
RU2028293C1 |
| 1-N-АЛКИЛ-N-АРИЛПИРИМИДИНАМИНЫ, СПОСОБ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1994 |
|
RU2153494C2 |
| ИЗОКСАЗОЛИНЫ И ИЗОКСАЗОЛЫ, СПОСОБ ПОДАВЛЕНИЯ АГРЕГАЦИИ ТРОМБОЦИТОВ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ПОДАВЛЯЮЩАЯ АГРЕГАЦИЮ ТРОМБОЦИТОВ | 1994 |
|
RU2149871C1 |
| ЦИКЛИЧЕСКИЕ МОЧЕВИНЫ, СПОСОБ ИНГИБИРОВАНИЯ РОСТА РЕТРОВИРУСОВ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1992 |
|
RU2131420C1 |
| АЗОТСОДЕРЖАЩИЕ ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ФЛУОРЕНА | 1992 |
|
RU2051151C1 |
| КАРБОЦИКЛИЧЕСКИЕ И ГЕТЕРОЦИКЛИЧЕСКИЕ КОНДЕНСИРОВАННЫЕ СОЕДИНЕНИЯ ХИНОЛИНКАРБОНОВОЙ КИСЛОТЫ, ПРИГОДНЫЕ В КАЧЕСТВЕ ИММУНОДЕПРЕССАНТОВ | 1994 |
|
RU2133740C1 |
| ЦИКЛИЧЕСКИЕ ПЕПТИДЫ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1992 |
|
RU2096415C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ИМИДАЗОЛА ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫХ СОЛЕЙ | 1991 |
|
RU2026292C1 |
| ФУНГИЦИДНЫЕ ПИРАЗОЛЫ | 2010 |
|
RU2577247C2 |
Настоящее изобретение относится к соединениям, включая 5-хлор-2-[2- [[(4-метилфенил)сульфонил] окси] этил] -7-[2,4,6-триметилфенил)метокси] антра[1,9-сd]пиразол-6(2Н)он и его аналоги формулы I, которые используют в качестве промежуточных продуктов синтеза антрапиразолонов, обладающих противоопухолевой активностью, в том числе лозоксантрона. Изобретение также относится к способам получения и выделения соединений формулы I, а также к способам синтеза противоопухолевых производных антрапиразолона (включая лозоксантрон). Полученные соединения расширяют арсенал противоопухолевых средств. 8 с. и 17 з.п. ф-лы.
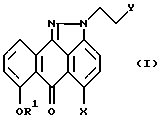
или их фармацевтически приемлемые соли,
где R1 означает H или гидроксилзащищающую группу;
X выбирают из группы, содержащей: (а) F, Cl, Br, I, (b) метансульфонилокси-, (c) толуолсульфонилокси-, (d) трифторметансульфонилокси- или (e) -OH;
Y выбирают из группы, содержащей: (а) F, Cl, Br, I, (b) -OSO2 R2 или (c) -OH;
R2 выбирают из группы, содержащей: (a) C1 - C4-алкил, (b) CvF2v+1, где v = 1 - 4, или (c) фенил или фенил, возможно замещенный 1 - 3 заместителями, выбранными из группы Cl, F, Br или C1 - C4-алкила;
при условии, что если X означает Cl, R1 означает H, C1 - C4-алкил, бензил, парахлорбензил или параметоксибензил, то Y не может быть группой -OH.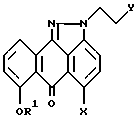
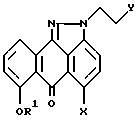
или его фармацевтически приемлемая соль,
где R1 выбирают из группы, содержащей: (а) бензил, замещенный 0 - 3 группами R5; (b) нафтилметил, замещенный 0 - 3 группами R5; (c) антрилметил, замещенный 0 - 3 группами R5; (d) C1 - C4-алкил или (e) H, и R5 независимо выбирают из группы, содержащей C1 - C4-алкил;
X и Y имеют значения, указанные в п.1.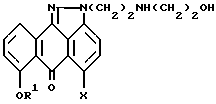
или его фармацевтически приемлемой соли,
где X выбирают из группы, содержащей (а) F, Cl, Br, I, (b) метансульфонилокси-, (c) толуолсульфонилокси-, (d) трифторметансульфонилокси- или (e) -OH; R1 выбирают из группы, содержащей (а) бензил, замещенный 0 - 3 группами R5; (b) нафтилметил, замещенный 0 - 3 группами R5; (c) антрилметил, замещенный 0 - 3 группами R5; (d) C1 - C4-алкил или (e) H;
R5 независимо выбирают из группы, содержащей C1 - C4-алкил,
отличающийся тем, что осуществляют следующие стадии: (1) взаимодействие соединения формулы IV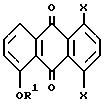
где R1 и X имеют указанные значения,
с 2-гидроксиэтилгидразином в подходящем растворителе в присутствии основания с образованием смеси региоизомеров формулы II и формулы III при соотношении II : III, равном 4 : 1,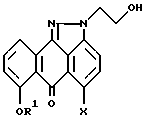
где R1 и X имеют указанные значения,
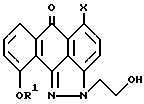
(2) взаимодействие смеси региоизомеров формулы II и формулы III с ClSO2R2, где R2 выбирают из группы, содержащей (a) CvF2v+1, где v = 1 - 4, или (b) фенил или фенил, возможно замещенный 1 - 3 заместителями, выбранными из группы Cl, F, Br или CH3;
при этом реакцию проводят в подходящем растворителе в присутствии подходящего основания с последующим осаждением из органического растворителя для получения простого изомера формулы I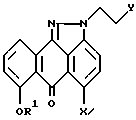
где Y означает -OSO2R2;
R1 и X имеют указанные значения,
(3) взаимодействие соединение формулы I с этаноламином в подходящем растворителе в присутствии подходящего основания с образованием соединения формулы V.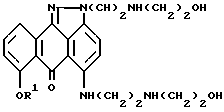
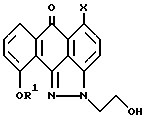
или его фармацевтически приемлемой соли,
где R1 выбирают из группы, содержащей: (а) бензил, замещенный 0 - 3 группами R5; (b) нафтилметил, замещенный 0 - 3 группами R5; (c) антрилметил, замещенный 0 - 3 группами R5; (e) H;
R5 независимо выбирают из группы, содержащей C1 - C4-алкил,
отличающийся тем, что осуществляют взаимодействие соединения формулы V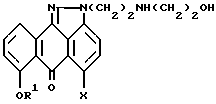
с 2-(2-аминоэтиламино)этанолом в подходящем растворителе в присутствии подходящего основания.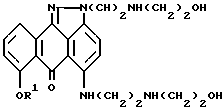
где R1 выбирают из группы, содержащей: (а) бензил, замещенный 0 - 3 группами R5; (b) нафтилметил, замещенный 0 - 3 группами R5; (c) антрилметил, замещенный 0 - 3 группами R5; (e) H;
R5 независимо выбирают из группы, содержащей C1 - C4-алкил,
отличающийся тем, что осуществляют взаимодействие соединения формулы V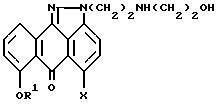
с хлористым водородом в смешанном растворителе из спирта и подходящего растворителя с образованием гидрохлорида соединения формулы V (соединение Vb), и взаимодействие соединения формулы Vb с 2-(2-аминоэтиламино) этанолом в подходящем растворителе в присутствии подходящего основания.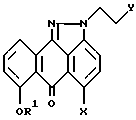
где X выбирают из группы, включающей: (а) F, Cl, Br, I, (b) метансульфонилокси-, (c) толуолсульфонилокси-, (d) трифторметансульфонилокси- или (e) -OH;
Y означает -OSO2R2;
R2 выбирают из группы, содержащей: (a) CvF2v+1, где v = 1 - 4, или (b) фенил или фенил, возможно замещенный 1 - 3 заместителями, выбранными из группы Cl, F, Br или CH3;
R1 выбирают из группы, содержащей: (а) бензил, замещенный 0 - 3 группами R5; (b) нафтилметил, замещенный 0 - 3 группами R5; (c) антрилметил, замещенный 0 - 3 группами R5;(d) C1 - C4-алкил или (e) H;
R5 независимо выбирают из группы, содержащей C1 - C4-алкил,
отличающийся тем, что осуществляют взаимодействие смеси, состоящей из соединения формулы II и соединения формулы III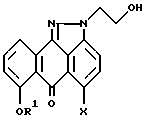
где R1 и X имеют указанные значения,
с ClSO2R2, где R2 имеет указанные значения, при этом реакцию проводят в подходящем растворителе в присутствии подходящего основания с последующим осаждением из органического основания для получения отдельного изомера соединения формулы I.
где R1 выбирают из группы, содержащей: (а) бензил, замещенный 0 - 3 группами R5; (b) нафтилметил, замещенный 0 - 3 группами R5; (c) антрилметил, замещенный 0 - 3 группами R5;(d) C1 - C4-алкил или (e) H;
R5 независимо выбирают из группы, содержащей C1 - C4-алкил;
X означает F, Cl, Br, I,
отличающийся тем, что осуществляют следующие стадии: (1) взаимодействие соединения формулы II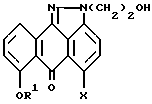
где R1 и X имеют указанные значения,
с любым хлорирующим, бромирующим или йодирующим агентом в подходящем растворителе, и (2) взаимодействие продукта реакции, полученного на стадии (1) с этаноламином с образованием соединения формулы V.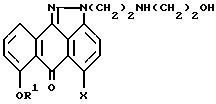
где R1 означает 2,4,6-триметилбензил;
X означает Cl,
отличающийся тем, что осуществляют стадии: (1) взаимодействие соединения формулы II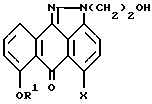
или любой его фармацевтически приемлемой соли,
где R1 и Х имеют вышеуказанные значения,
с бромирующим агентом в подходящем растворителе, и (2) взаимодействие продукта реакции, полученного на стадии (1), с этаноламином с образованием соединения формулы V.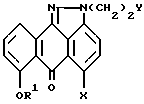
где R1 выбирают из группы, содержащей: (а) бензил, замещенный 0 - 3 группами R5; (b) нафтилметил, замещенный 0 - 3 группами R5; (c) антрилметил, замещенный 0 - 3 группами R5;(d) C1 - C4-алкил или (e) H;
R5 независимо выбирают из группы, содержащей C1 - C4-алкил;
X означает F, Cl, Br, I;
Y означает Cl, Br, I,
отличающийся тем, что осуществляют взаимодействие соединения формулы II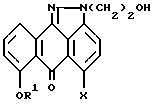
где R1 и X имеют указанные значения,
с хлорирующим, бромирующим или йодирующим агентом в подходящем растворителе в присутствии подходящего основания с образованием соединения формулы I.
где R1 означает 2,4,6-триметилбензил;
X и Y означают Cl,
отличающийся тем, что осуществляют взаимодействие соединения формулы II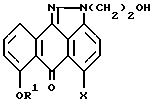
где R1 и X имеют указанные значения,
с хлорирующим агентом, выбранным из тионилхлорида или четыреххлористого углерод/трифенилфосфина, в подходящем растворителе в присутствии подходящего основания с образованием соединения формулы I.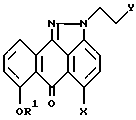
где R1 выбирают из группы, содержащей: (а) бензил, (b) параметоксибензил, (c) 2,4,6-триметилбензил, (d) C1 - C4-алкил или (e) H;
X выбирают из группы, содержащей: (a) F, Cl, Br, I, (b) метансульфонилокси, (c) толуолсульфонилокси, (d) трифторметансульфонилокси- или (e) -OH;
Y выбирают из группы, содержащей: (a) -OSO2R2 или (b) -OH;
R2 выбирают из группы, содержащей: (a) CvF2v+1, где v = 1 - 4, или (b) фенил или фенил, возможно замещенный 1 - 3 заместителями, выбранными из группы Cl, F, Br, CH3,
из смеси региозомеров путем осаждения соединения формулы I из смеси любого спирта и подходящего растворителя.
Приоритет по признакам:
05.05.93 при R1 - H, бензил, параметоксибензил, C1 - C4-алкил или 2,4,6-триметилбензил, X - F, Cl, Br, I, метансульфонилокси, толуолсульфонилокси-, трифторметансульфонилокси- или -OH группа, Y = Cl, Br, I, -OSO2R2 и -OH-группа, R2 - C1 - C4-алкил, CvF2v+1, где v = 1 - 4, или фенил, или фенил, возможно замещенный Cl, F или CH3;
25.10.93 при R1 - гидроксилзащищающая группа, R2 - фенил, возможно замещенный атомом брома, или C1 - C4-алкил, при условии, что, если X = Cl, R1 - H или C1 - C4-алкил, бензил, парахлорбензил или параметоксибензил, то Y не может быть группой -OH.
| Способ получения конденсированных производных пиразола или их фармацевтически приемлемых солей | 1988 |
|
SU1676453A3 |
| US 4556654 A, 1985 | |||
| Плотномер для определения твердости почвы | 1955 |
|
SU103381A2 |
| 0 |
|
SU244819A1 | |
| СПОСОБ ПОЛУЧЕНИЯ 2-ФЕНИЛТИОНАФТЕНА ИЛИ ЕГО ПРОИЗВОДНЫХ | 0 |
|
SU203697A1 |
