Изобретение относится к способам получения новых имидазолов, действующих как ингибиторы активности ацил-СоА: холестеринацилтрансферазы (АСАТ). Они могут быть использованы в качестве антигиперхолестеринематических препаратов.
Известны соединения общей формулы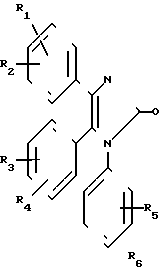 - (CH2)n- COOR7 где R1, R2, R3, R4, R5 и R6 независимо друг от друга представляют собой H, F, Cl, Br, I, алкил, алкокси, при условии, что одна или несколько пар R1 и R2, R3, и R4 или R5 и R6 взятые вместе, представляют собой метилендиокси; R7 паредставляет собой Н, ион щелочного металла, алкил с 1-6 атомами углерода или бензил, и n представляет собой число от 0 до 10, которые используются при лечении тромбоэмболии, воспалительных и/или атеросклеротических заболеваний.
- (CH2)n- COOR7 где R1, R2, R3, R4, R5 и R6 независимо друг от друга представляют собой H, F, Cl, Br, I, алкил, алкокси, при условии, что одна или несколько пар R1 и R2, R3, и R4 или R5 и R6 взятые вместе, представляют собой метилендиокси; R7 паредставляет собой Н, ион щелочного металла, алкил с 1-6 атомами углерода или бензил, и n представляет собой число от 0 до 10, которые используются при лечении тромбоэмболии, воспалительных и/или атеросклеротических заболеваний.
Цель изобретения - синтез новых производных имидазола, по своим свойствам превосходящих структурный аналог.
Поставленная цель достигается предлагаемым способом получения новых соединений общей формулы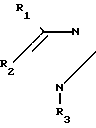



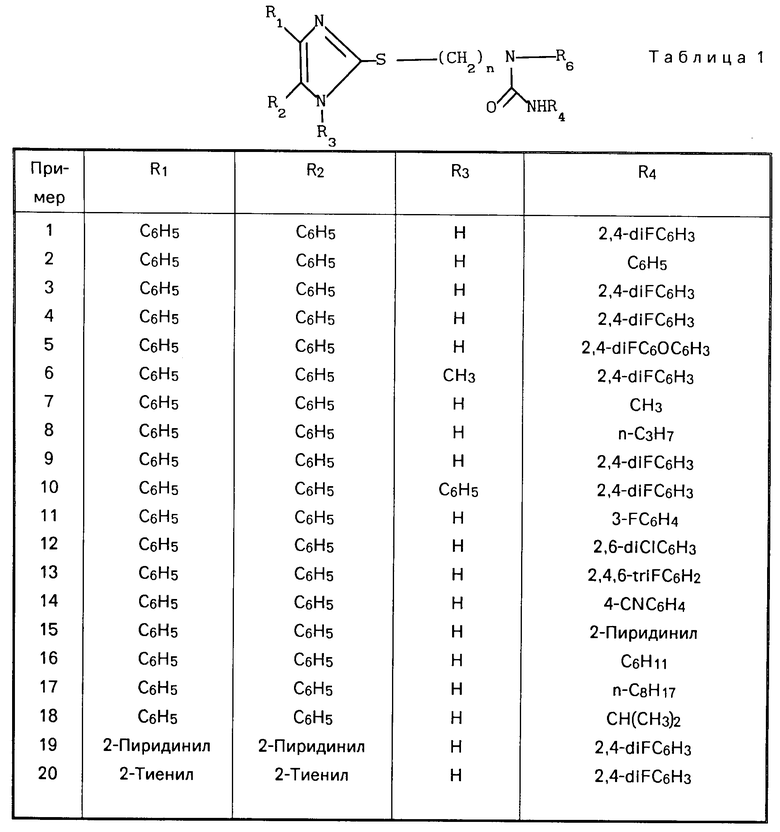
(I) где R1 и R2 независимо друг от друга выбраны из ряда, включающего Н, С1-С8-алкил, С3-С8-алкил с разветвленной цепью, С3-С7-циклоалкил, 2-, 3- или 4-пиридинил, 2-тиенил, 2-фуранил, фенил, в определенных случаях замещенный 1-3 группами, выбранными из ряда, включающего F, Cl, Br, OH, С1-С4-алкокси, С1-С4-алкил, СН3S(O)2 CF3 или NR7R8, или R1 и R2 вместе
взятые, образуют группу формулы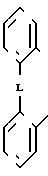
(II) где L представляет собой О, О(СН2), О;
R3 представляет собой Н, С1-С6-алкил или фенил; Z-NHR4, R4, OR4;
R4 представляет собой С1-С8-алкил с нормальной цепью, С3-С8-алкил с разветвленной цепью, С3-С7-циклоалкил, С4-С10-циклоалкилалкил, С7-С14-аралкил, где арильная группа в определенных случаях замещена 1/3 группами, выбранными из ряда, включающего С1-С4-алкил, фенил, в определенных случаях замещенный 1-3 группами, выбранными из ряда, включающего С1-С4-алкил, С1-С4-алкокси, F, Cl, CN; пентафторфенил, бензил, в определенных случаях замещенный 1-3 группами, выбранными из ряда, включающего С1-С4-алкил, F, Cl, 2-пиридинил;
R5 представляет собой Н, С1-С6-алкил, NR7R8, бензил, С3-С8-алкил с разветвленной цепью или фенил, в определенных случаях замещенный 1-3 группами, выбранными из ряда, включающего С1-С4-алкил или -алкокси, или
R7 и R8 независимо выбраны из группы, включающей Н и С1-С4-алкил;
Х представляет собой S(O)r или 0;
А представляет собой С2-С10-алкил, С3-С10-алкил с разветвленной цепью;
Y представляет собой O, S, H2;
r = 0-2,
или их фармацевтически приемлемых солей, заключающимся в том, что он включает стадию алкилирования соединения общей формулы (III)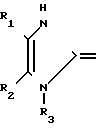 X где R1-R3 и Х имеют указанные значения, соединением формулы IV
X где R1-R3 и Х имеют указанные значения, соединением формулы IV
M(A) 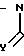
 R5
R5
(IV) где М - галоид, А, R5, Z имеют указанные значения, с выделением целевого продукта или в случае, когда Y означает 0, полученное соединение подвергают реакции, с восстановителем таким, как алюмогидрид лития или боргидрид натрия, в результате чего получают соединение формулы I, где Y-H2 или когда Х означает S, подвергают реакции подходящим окислителем, в результате чего получают либо сульфоксид SO, где r = 1, либо сульфон SO2, где r = 2.
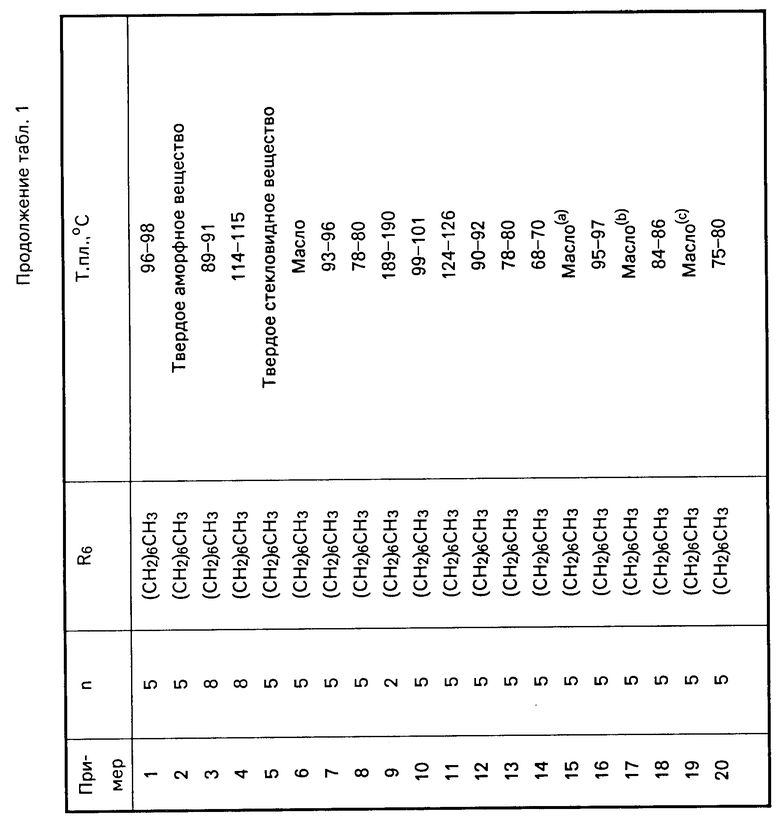
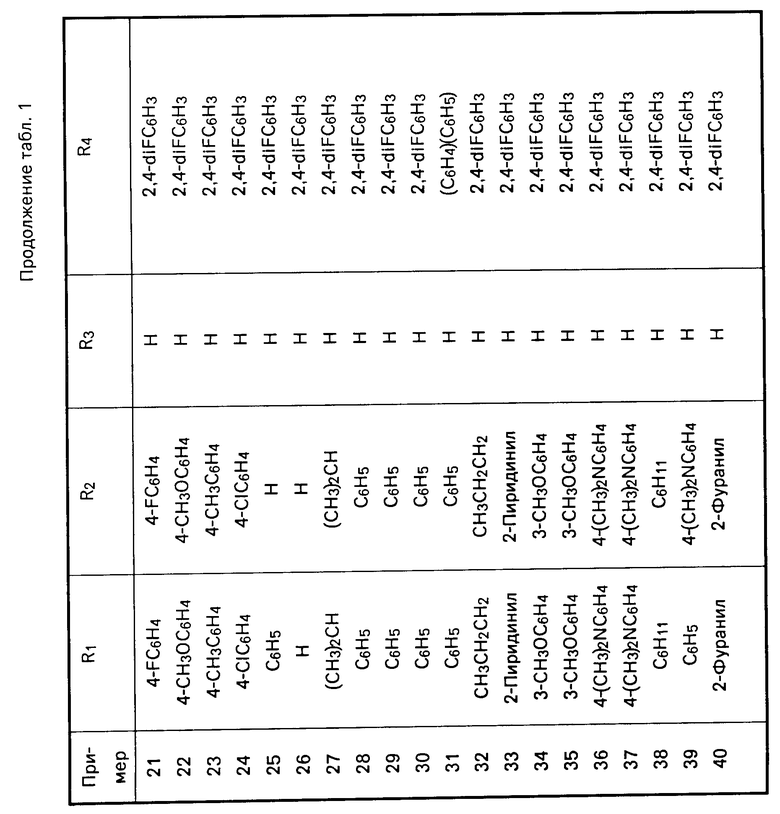
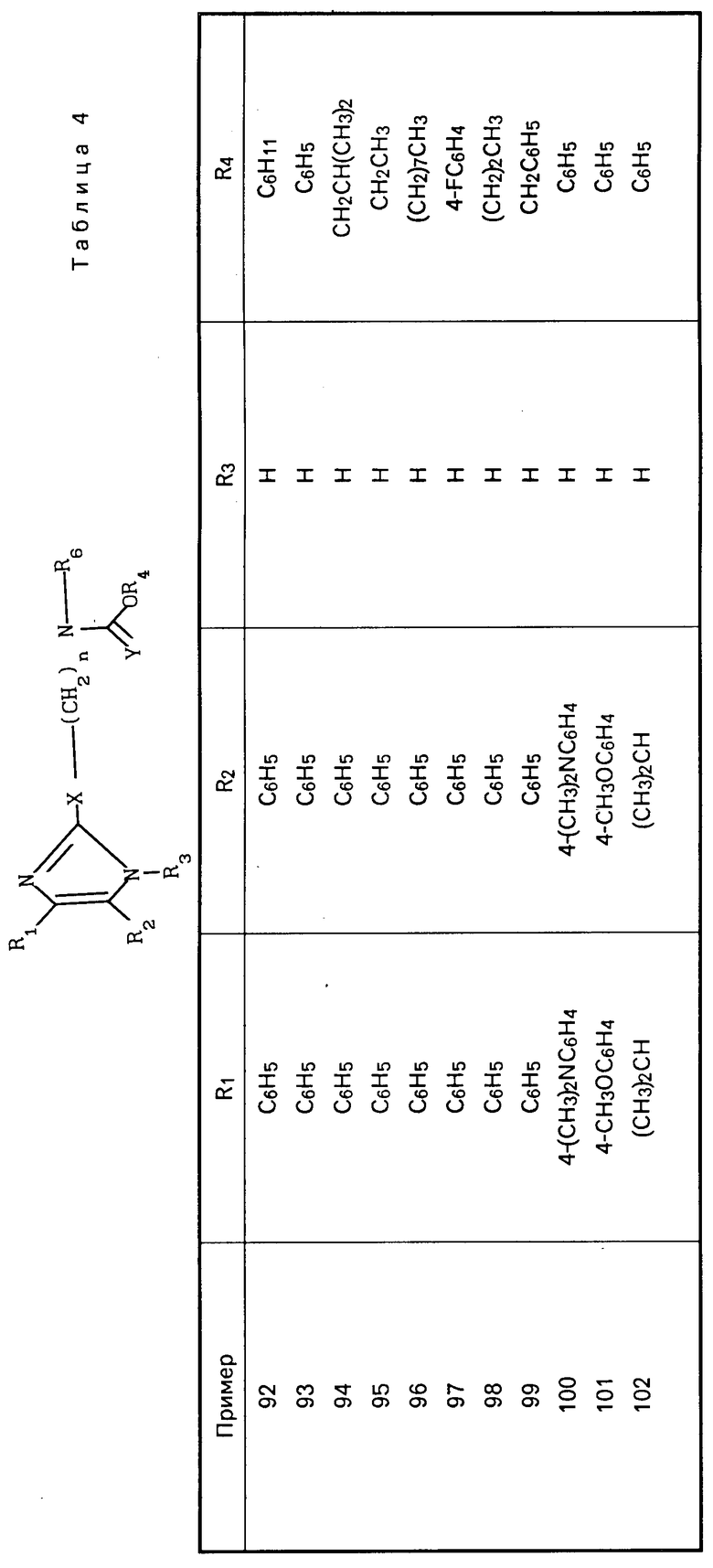
П р и м е р 1. Получение N-[5-[4,5-бис-(4-метоксифенил)-1Н-имидазол-2-илтио]- пентил]-N1-(2,4- дифторфенил)-N-гептилмочевины.
Часть А. Раствор гамма-валеролактона (2 г, 0,249 моль в толуоле 500 мл) в н-гептиламине (35,96 г, 0,312 моль) нагревали до температуры дефлегмации в течение 18 ч в атмосфере азота. Реакционной смеси дали остыть до температуры окружающей среды, разбавили этилацетатом (300 мл), промыли 1н. водным раствором соляной кислоты (50 мл), рассолом, обезводили над сульфатом магния и сконцентрировали, в результате чего получили твердое белое вещество. Полученный продукт подвергли кристаллизации из смеси этиловый эфир-гексан и получили N-гептил-5-гидроксипентанамид (41,8, 0,194 моль) в виде белых пластин; т.пл. 55-56о.
1Н ЯМР (СDCl3): 6,06 (шир.с., 1Н), 3,61 (Т, 2Н), 3,24 (кв. 2Н) 3,19 (шир.с. 1Н), 2,19 (т, 2Н), 1,80-1,23 (м, 14Н), 0,866 (т, 3Н).
Часть В. К раствору алюмогидрид лития (6,7 г, 0,176 моль) в безводном тетрагидрофуране (300 мл) по каплям в атмосфере азота добавили раствор гептил-5-гидроксипентанамида (19,0 г, 0,088 моль) в безводном тетрагидрофуране (100 мл). Реакционная смесь была нагрета до температуры дифлегмации в течение 18 ч, ей дали остыть до комнатной температуры и медленно влили в перемешиваемую смесь 10%-ного водного раствора сульфата натрия (400 мл) со льдом (200 мл). Полученную смесь профильтовывали через цеолит и фильтрат экстрагировали этилацетатом (2 х 500 мл). Органические слои объединили, промыли водой, рассолом, обезводили над сульфатом магния, сконцентрировали и получили вязкое жидкое масло. Продукт подвергли кристаллизации из гексана и получили N-(5-гидроксипентил)-N-гептиламин (15,2 г, 0,075 моль) в виде белого порошка; т.пл. 47-48о.
1Н ЯМР (CDCl3) δ: 3,63 (т, 2Н), 2,63 (кв, 4Н), 2,39 (шир.с. 2Н), 1,66-1,24 (м, 16Н), 0,905 (т, 3Н).
Часть С. К раствору N-(5-гидроксипентил)-N-гептиламина (11,65 г, 0,0578 моль) в хлористом метилене (75 мл) в атмосфере азота, охлажденному до 0о, медленно добавили 2,4-дифторфенилизоцианат (8,97 г, 0,579 моль). Реакционную смесь перемешивали в течение 1 ч, затем влили в 1н. водный раствор соляной кислоты (200 мл) и экстрагировали этилацетатом (300 мл). Объединенные органические слои промыли водой, рассолом, обезводили над сульфатом магния, сконцентрировали и получили N`-(2,4-дифторфенил)-N-гептил-N-5-гидроксипен-тилмочевину в виде светло-желтого масла (20,0 г, 0,056 моль).
1Н ЯМР (CDCl3): 8,03 (м, 1Н), 6,88-6,59 (м, 2Н), 6,45 (шир.с., 1Н), 3,68 (т, 2Н), 3,33 (м, 4Н), 1,81-1,22 (м, 16Н), 0,907 (т, 3Н).
Часть D. К раствору N'-(2,4-дифторфенил)-N-гептил-N-5-гидроксипентилмочевины (15,0 г, 0,042 моль) и тетрабромметана (16,75 г, 0,051 моль) в хлористом метилене (350 мл) в атмосфере азота при температуре окружающей среды медленно добавили раствор трифенилфосфина (13,24 г, 0,051 моль) в хлористом метилене (100 мл). Реакционную смесь перемешивали в течение 3 ч, сконцентрировали в вакууме и получили сырое вязкое масло. Этот продукт подвергли очистке с помощью быстродействующей хроматографии (БХ) на силикагеле (400 мл), элюируя смесью гексан-этилацетат (90:10 по объему), и получили N-(5-бромпентил)-N -(2,4-дифторфенил)-N-гептилмочевину в виде вязкого бесцветного масла (17,5 г, 0,042 моль).
1Н ЯМР (CDCl3): 8,14-8,0 (м, 1Н), 6,92-6,79 (м, 2Н), 6,35 (шир. с, 1Н), 3,49-3,25 (м, 6Н), 1,99-1,26 (м, 16Н), 0,915 (т, 3Н).
Часть Е. В суспензию гидрида натрия (0,88 г, дисперсия в минеральном масле, 0,0022 моль) (промыта от минерального масла гексаном) в N,N-диметилформамиде (15 мл) в атмосфере азота при охлаждении до 0оС медленно добавили раствор 4,5-[бис-(4-метоксифенил)-1Н-имидазол] -2-тиона (0,63 г, 0,002 моль) в N,N-диметилформамиде (5 мл). Реакционную смесь перемешивали в течение 2 ч и затем добавили к ней раствор N-(5-бромпентил)-N`-(2,4-дифторфенил)-N-гептилмочевины (0,845 г, 0,002 моль) в N,N-диметилформамиде (3 мл). Реакционной смеси дали нагреться до температуры окружающей среды, перемешивали еще 2 ч, влили в воду (50 мл) и экстрагировали этилацетатом (2 × 50 мл). Объединенные органические экстракты промыли водой, рассолом, обезводили над сульфатом магния, сконцентрировали и получили вязкое масло. Этот продукт очистили с помощью БХ на силикагеле (100 мл), элюируя смесью гексан-этилацетат (70:30 по объему) и получили указанное в заголовке соединение в виде чистой желтой пены (0,98 г, 0,0015 моль).
1Н ЯМР (CDCl3): 10,15 (шир., с. 1Н), 7,87-7,76 (м, 1Н), 7,51 (д, 2Н), (д, 2Н), 6,86-6,6 (м, 6Н), 6,42 (д, 1Н), 3,8 (с, 6Н), 3,4 (т, 2Н), 3,26 (т, 2Н), 2,99 (т, 2Н), 1,84-1,25 (м, 16Н), 0,89 (т, 3Н).
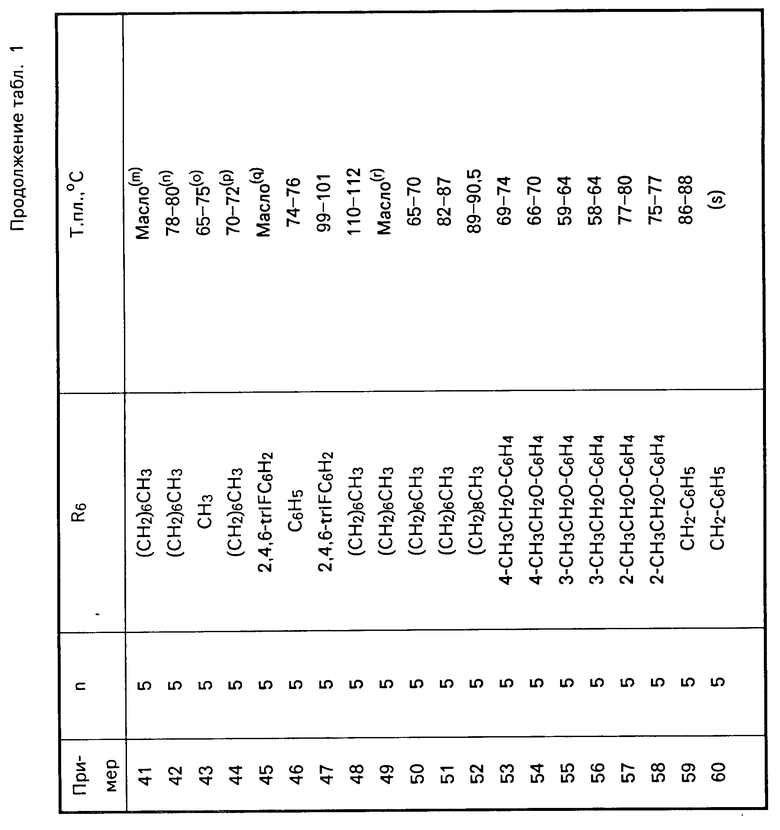
П р и м е р 2. Получение N-[5-[4,5-бис-(4-гидроксифенил)-1Н-имидазол-2-илтио]-пентил]-N'-(2,4-дифторф енил
В перемешиваемый раствор N-[5-[4,5-бис-(4-метоксифенил)-1Н-имидазол-2-илтио] -пентил] -N1-(2,4- дифторфенил)-N-гептилмочевины (0,78 г, 0,0012 моль) в хлористом метилене (30 мл), охлажденный до -78оС в атмосфере азота добавили 1М раствор трехбромистого бора в хлористом метилене (3,6 мл). Реакционную смесь перемешивали в течение 1 ч при температуре 0о, затем влили в лед (100 мл) и экстрагировали этилацетатом (2 х 50 мл). Объединенные органические слои промыли 10%-ным водным раствором NaHCO3 (50 мл), водой, рассолом, обезводили над сульфатом магния, сконцентрировали в вакууме и получили сырое масло. Продукт очистили с помощью БХ на силикагеле (100 мл), используя в качестве элюента смесь гексан-этилацетат (40:60 по объему), и получили белую пену; т.пл. 110-112о (0,5 г, 0,00008 моль).
1Н ЯМР (DMCO-d6) δ: 12,22 (шир. с.1Н), 9,55 (шир. с. 1Н), 9,32 (шир. с. 1Н), 7,92 (с, 1Н), 7,45-6,6 (м, 11Н), 3,24 (м, 4Н), м 3,06 (т, 2Н), 1,77-1,17 (м, 16Н), 0,88 (т, 3Н).
П р и м е р 3. Получение N-[5-(1Н, 9Н-дибенз-[4,5:8,9][1,3] диоксонино-[6,7-d]имидазол-2-илтио) пентил]-N`-(2,4-дифторфенил)-N-гептилмочевины.
Часть А. К суспензии гидрида натрия (промыли от минерального масла гексаном) (2,45 г масляной дисперсии, 0,081 моль) в безводном N,N-диметилформамиде (50 мл) в атмосфере азота при охлаждении до 0оС медленно добавили раствор салисальдегида (10,0 г, 81,9 ммоль) в безводном N,N-диметилформамиде (10 мл). Реакционную смесь перемешивали при 0о в течение 2 ч и добавили к ней дийодометан (11,3 г, 0,041 моль). Реакционной смеси дали нагреться до температуры окружающей среды в течение 18 ч, затем нагревали до 60оС в течение 20 ч. Реакционной смеси дали остыть до температуры окружающей среды, влили в 1н. водный раствор соляной кислоты (100 мл) и экстрагировали этилацетатом (2 х 100 мл). Объединенные органические экстракты промыли водой, рассолом, обезводили над сульфатом магния, сконцентрировали и получили твердое вещество. Продукт очистили БХ на силикагеле (300 мл), элюируя хлористым метиленом (100%) и получили 2,2 -(метилендиокси)-бис-(2-бензальдегид) в виде белого твердого кристaллического вещества, т.пл. 131-133оС (5,1 г, 0,0199 моль).
1Н ЯМР (CDCl3) δ: 10,47 (с, 2Н), 7,87 (д, 2Н), 7,68-7,54 (м, 2Н), 7,21 (д, 2Н), 7,15 (т, 2Н), 6,02 (с, 2Н).
Часть В. Смесь 2,2'-(метилендиокси)-бис-(2-бензальдегида) (5,0 г, 0,0195 моль), цианида калия (0,63 г, 0,0975 моль) в эталоне (75 мл) и воде (50 мл) нагревали до температуры дефлегмации в течение 6 ч. Реакционной смеси дали остыть до температуры окружающей среды, сконцентрировали в вакууме и полученный водный остаток разделили между этилацетатом и водой. Органический слой промыли водой, рассолом, обезводили над сульфатом магния, сконцентрировали и получили вязкое масло. Продукт очистили БХ на силикагеле (250 мл), элюируя смесью гексан-этилацетат (80:20 по объему) и получили 13-гидроксидибензо- [d,h] [1,3]-диоксонино-12 (13Н)-она в виде кристаллического твердого вещества; т.пл. 129-130o (2,5 г, 0,0975 моль).
1Н ЯМР (DMCO-d6): 7,49 (т, 2Н), 7,29-7,08 (м, 6Н), 6,40 (д, 1Н), 5,97 (д, 1Н), 5,92 (д, 1Н), 5,24 (д, 1Н).
Часть С. Раствор 13-гидроксидибензо- [d,h][1,3]-диоксонино-12 (13Н)-она (2,0 г, 0,0078 моль), тиомочевины (0,82 г, 0,0108 моль) и гексанола (25 мл), в колонке с ситами 4А и конденсатором нагревали до 160o в течение 20 ч в атмосфере азота. Реакционной смеси дали охладиться до температуры окружающей среды и разбавили ее этиловым эфиром (100 мл), в результате чего получили твердое вещество. Это твердое вещество промыли этиловым эфиром, обезводили и получили N-(1Н, 9Н-дибенз-[4,5:8,9][1,3 диоксонино-[6,7-d] имидазол-2-тион в виде белого кристаллического порошка (1,6 г, 0,00539 моль); т.пл. > 260оС.
1Н ЯМР (DMCO-d6) δ: 12,5 (с, 2Н), 7,43-7,08 (м, 8Н), 6,2-5,0 (шир. д., 2Н).
Часть D. Используя метод примера 22, часть Е, из N-(1Н, 9Н-дибенз-[4,5: 8,9][1,3]-диоксонино-[6,7-d]имидазол)-2-тиона получили указанное в заголовке соединение в виде белой пены, т.пл. 65-70оС (0,85 г, 0,00134 моль).
1Н ЯМР (CDCl3): 10,35-10,10 (шир. с. 1Н), 7,56) (м, 1Н), 7,30-6,9 м, 10Н), 6,4 (д, 1Н), 5,70-5,20 (шир. с. 2Н), 3,40-3,19 (м, 4Н), 3,08 (т, 2Н), 1,85-1,23 (м, 16Н), 0,88 (т, 3Н).
П р и м е р 4. Получение N'-[5-(1Н-дибенз[2,3:6,7] окседин [4,5-d]имидазол-2-илтио)-пентил-N-(2,4-дифторфенил)-N-гептил- мочевины.
По способу примера 22, часть Е, используя 1Н-дибенз-[2,3:6,7]окседин[4,5-d]имидазол-2-тион, выделили указанное в заголовке соединение в виде белого порошка; т.пл. 82-87о (0,36 г, 0,00059 моль).
1Н ЯМР (CDCl3) δ: 9,75-8,5 (шир. с.2Н), 7,84-7,59 (м, 3Н), 7,43-7,05 (м, 6Н), 5,13-6,53 (м, 3Н), 3,43-3,13 (м, 6Н), 1,75-1,20 м (16Н), 0,88 (т, 3Н).
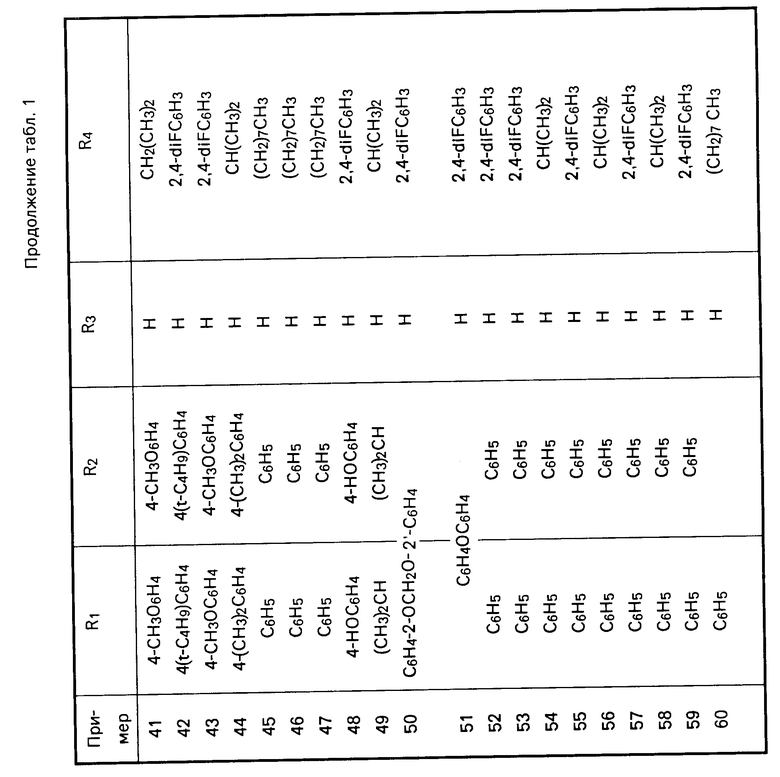
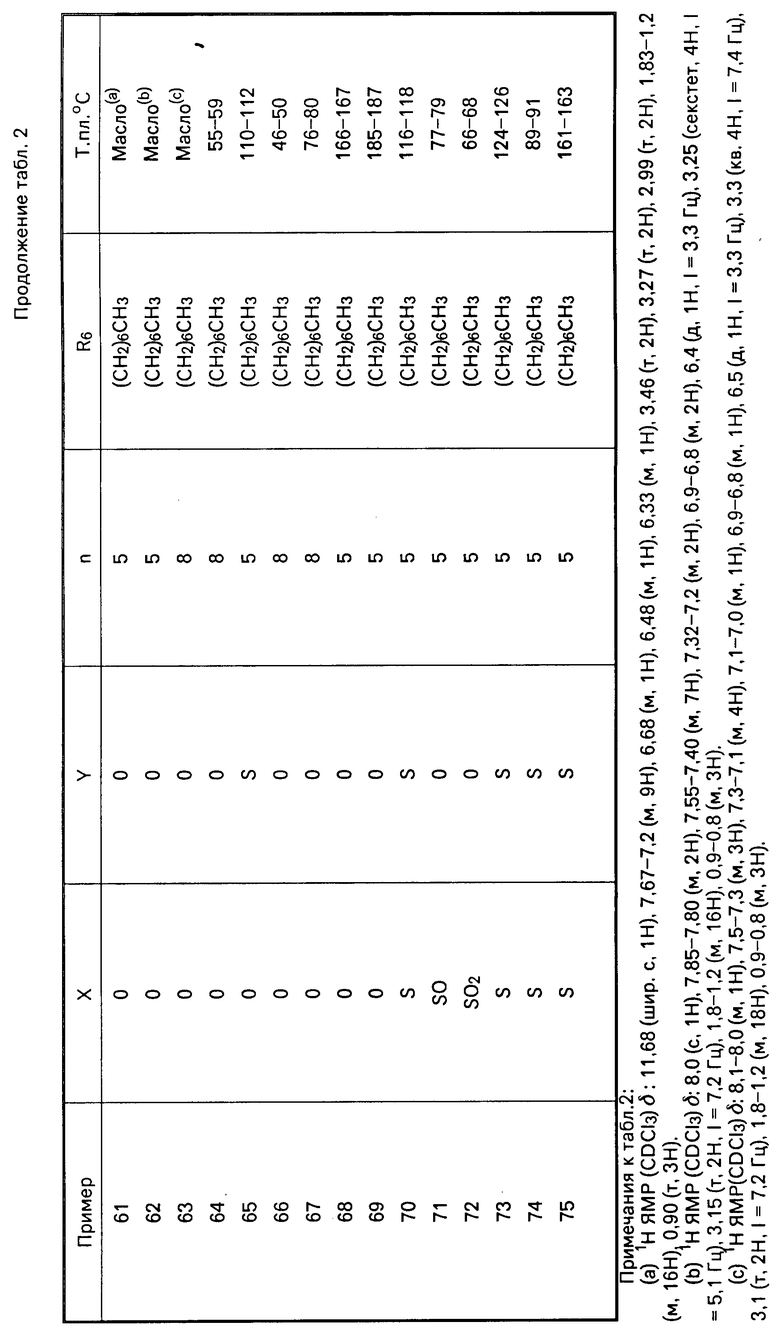
Перечисленные в табл.1 и 2 мочевины были получены аналогично описанным методикам.
П р и м е р 5. Получение N' -(2,4-дифторфенил)-N-[5-(4,5-дифенил-1Н-имидазол-2- илсульфонил)пентил]-N-гептил- мочевины.
В раствор N'-(2,4-дифторфенил)-N-[5-(4,5-дифенил-1Н-имидазол-2-илтио)-пен- тил] -N-гептилмочевины (0,59 г, 0,001 моль) в хлористом метилене (50 мл), охлажденный до -78оС по каплям добавили раствор м-хлорпербензойной кислоты (0,286 г, 0,0017 моль) в хлористом метилене (10 мл). Реакционную смесь перемешивали при температуре - 78оС в течение 1 ч и затем дали ей нагреться до температуры окружающей среды. Затем реакционную смесь охладили до 0оС и добавили в нее по каплям раствор насыщенного бисульфита натрия. Слои разделили и органический слой промыли насыщенным раствором бисульфита натрия. Слои разделили и раствор хлористого натрия обезводили над сульфатом натрия и сконцентрировали в вакууме. Остаток (0,76 г) хроматографировали, используя смесь 1:1 гексан-этилацетат, и получили целевой продукт -соединение (0,43 г, 0,00071 моль) в виде желтого твердого вещества; т.пл. 77-79оС.
1 Н ЯМР (CDCl3) δ:8,1-7,9 (м, 1Н), 7,6-7,2 (м, 10Н), 6,9-6,7 (м, 2Н), 6,4 (д, 1Н, I = 3,3 Гц), 3,4-3,1 (м, 6Н), 2,0-1,1 (м, 18Н), 0,9 (т, 3Н, I = 6,4 Гц).
П р и м е р 6. Получение N '-(2,4-дифторфенил)-N-[(5-[(4,5-дифенил-1Н-имидазол-2-ил) сульфонил)пентил]-N-гептилмочевины.
В раствор N' -(2,4)-дифторфенил)-N-[(5-(4,5-дифенил-1Н-имидазол-2-илтио)-пентил] -N-гептил моче(0,11 г, 0,00019 моль) в метаноле (5 мл) добавили по частям твердое вещество ® (0,234 г, 0,00038 моль) и реакционную смесь перемешивали при температуре окружающей среды в течение 7 ч. Твердые частицы отфильтровали и промыли метанолом. Фильтрат сконцентрировали в вакууме и остаток хромотографировали смесью 6:4 гексанэтилацетат и получили указанное в заголовке соединение (0,06 г, 0,000096 моль) в виде стеклообразного бесцветного твердого вещества; т.пл. 66-68оС.
1Н ЯМР (CDCl3)δ : 7,85-7,75 (м, 1Н), 7,6-7,1 (м, 11Н), 6,8-6,6 (м, 2Н), 6,4 (с, 1Н), 3,4 (т, 4Н, I = 10 Гц), 3,25 (т, 2Н, I = 7 Гц), 1,9-1,75 (м, 2Н), 1,75-1,4 (м, 6Н), 1,4-1,1 (м, 8Н), 0,9 (т, 3Н, I = 8 Гц).
П р и м е р 7. Получение N-[5-[(4,5-бис(4-метоксифенил)-1Н-имидазол-2-илтио]-пен-тил]-2,4-дифтор-N-ге птилбензолэтанамина.
В раствор алюмогидрида лития (1н. в тетрагидрофуране, 2 мл в безводном тетрагидрофуране - 30 мл) медленно добавили раствор N-[5-[4,5-бис-(4-метоксифенил)-1Н-имидазол-2-илтио] -пентил] -2,4-дифтор-N- гептилбензолацетамида (0,70 г, 0,00107 моль) в безводном тетрагидрофуране (15 мл). Реакционную смесь нагревали до температуры дефлегмации в течение 5 ч и затем оставили охлаждаться до температуры окружающей среды.
Реакционную смесь влили в смесь 10н. водного раствора сульфата натрия (150 мл) и льда (150 мл). Полученную эмульсию профильтровали через цеолит и фильтрат экстрагировали этилацетатом (3х100 мл). Органический слой промыли водой, рассолом, обезводили над сульфатом магния, сконцентрировали в вакууме и получили сырое масло. Продукт очистили БХ на силикагеле (100 мл), элюируя смесью метанол: хлористый метилен (5:95 по объему) и получили указанное в заголовке соединение в виде вязкого бесцветного масла (0,46 г, 0,000723 моль).
1Н ЯМР (CDCl3) δ : 9,2-9,15 (шир. с, 1Н), 7,56-7,25 (м, 4Н), 7,11 (м, 1Н), 6,94-6,70 (м, 6Н), 3,81 (м, 6Н), 3,07 (т, 2Н), 2,74-2,58 (м, 4Н), 2,43 (м, 4Н), 1,71 (м, 2Н), 1,53-1,20 (м, 14Н), 0,91 (т, 3Н).
П р и м е р 8. Получение N-[5-[4,5-бис(4-метоксифенил)-1Н-имидазол-2-илтио]-пен-тил]-N-гептилциклогек сана
Часть А. По методике примера 22, часть С, используя 2-циклогексанацетилхлорид, получили N-гептил-N-(5-гидроксипентил)- циклогексанацетамид в виде масла (1,5 г, 0,0046 моль).
1Н ЯМР (СDCl3) δ : 3,70-3,61 (м, 2Н), 3,37-3,18 (м, 4Н), 2,02 (д, 2Н), 1,97-1,08 (м, 26Н), 1,02-0,86 (м, 4Н).
Часть В. По методике примера 22, часть D, используя N-гептил-N-(5-гидроксипентил)-циклогексанацетамид, получили N-(5-бромпентил)-N-гептилциклогексанацета- мид в виде масла (1,3 г, 0,00334 моль).
1 Н ЯМР (CDCl3) δ: 3,47-3,39 (м, 2Н), 3,36-3,18 (м, 4Н), 2,17 (д, 2Н), 1,96-0,86 (м, 30Н).
Часть С. По методике примера 22, часть Е, используя N-(5-бромпентил)-N-гептилциклогексанацетамид, получили указанное в заголовке соединение в виде масла (0,47 г, 0,00075 моль).
1Н ЯМР (DMCO-d6) δ: 12,34 (с, 1Н), 7,29 (д, 2Н), 6,95 (д, 2Н), 6,85 (д, 2Н), 3,77 Эс, 3Н), 3,73 (с, 3Н), 3,18 (м, 4Н), 3,07 (м, 2Н), 2,09 (д, 2Н), 1,73-0,81 (м, 30Н).
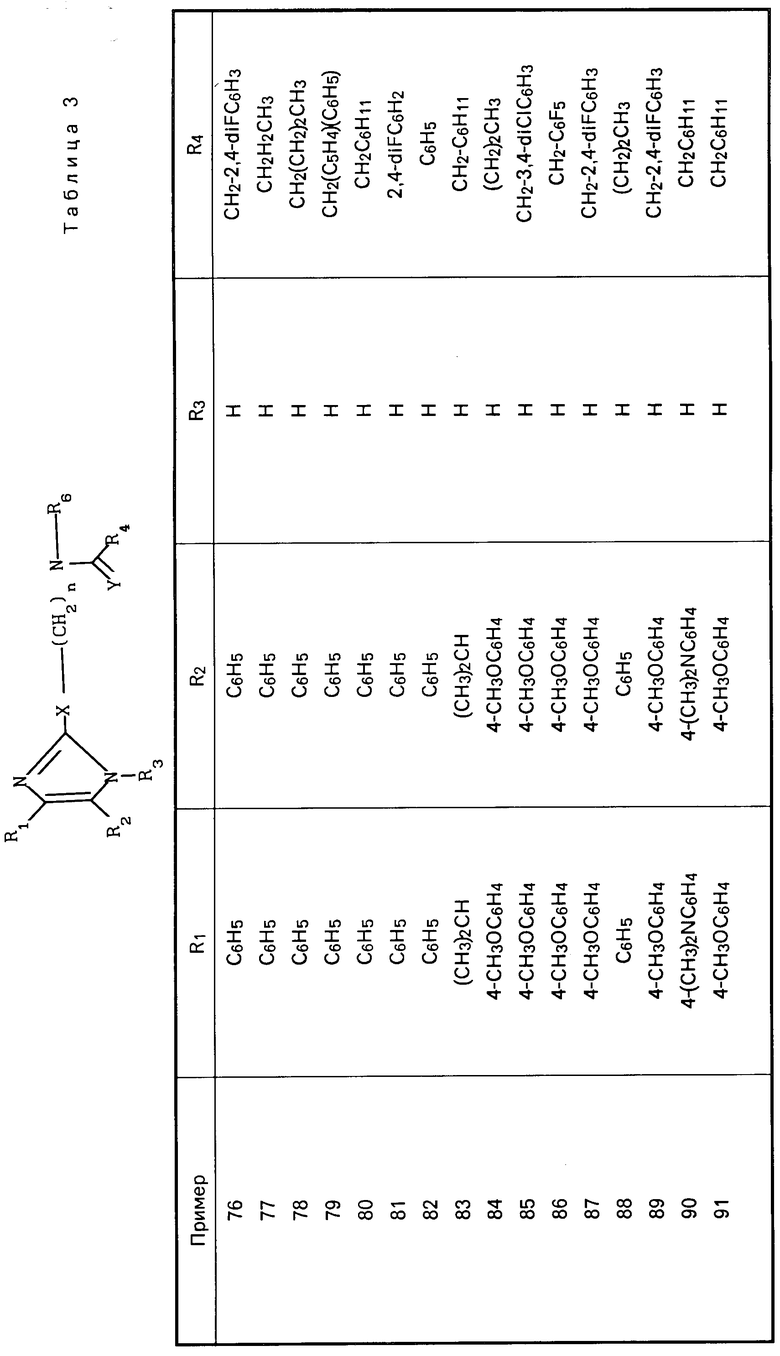
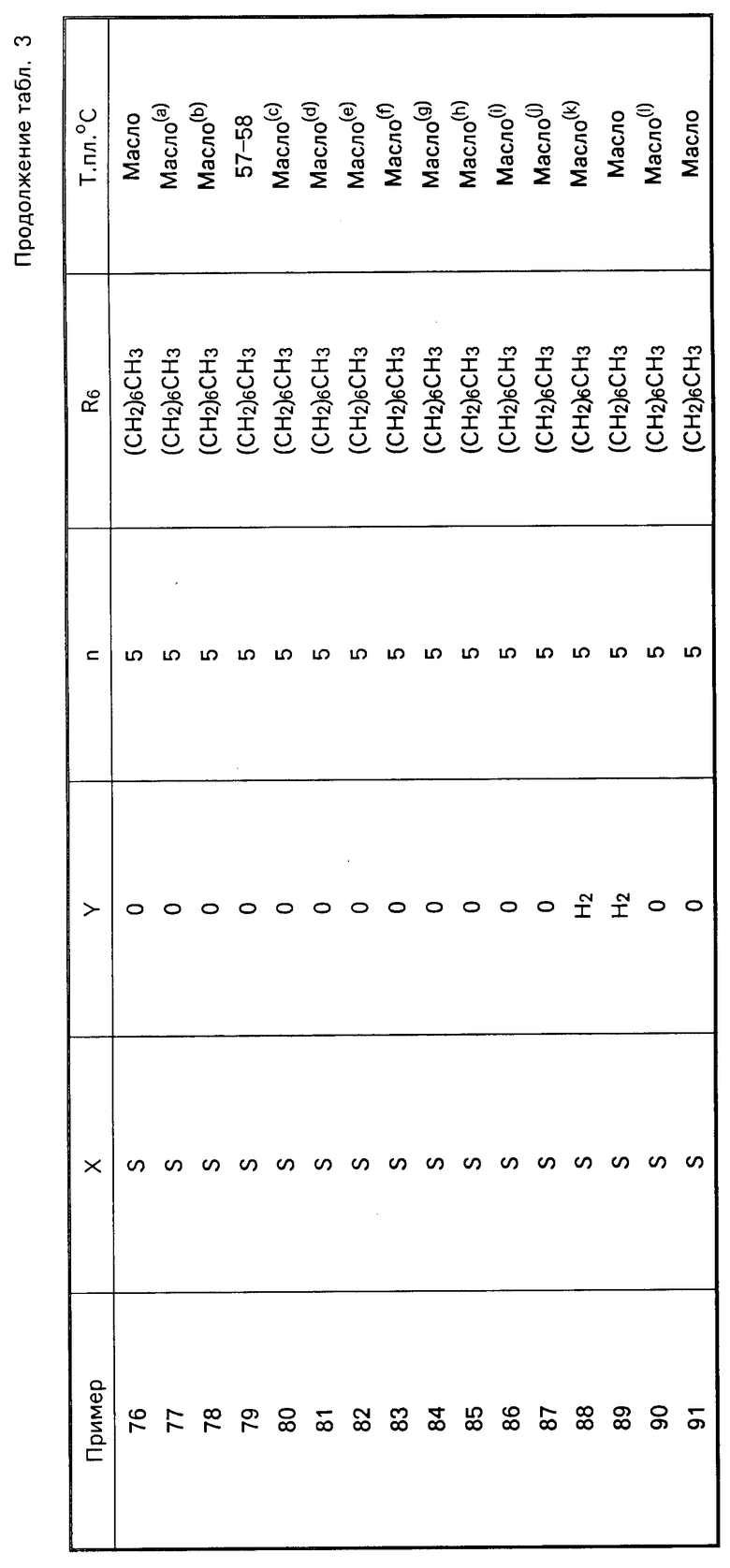
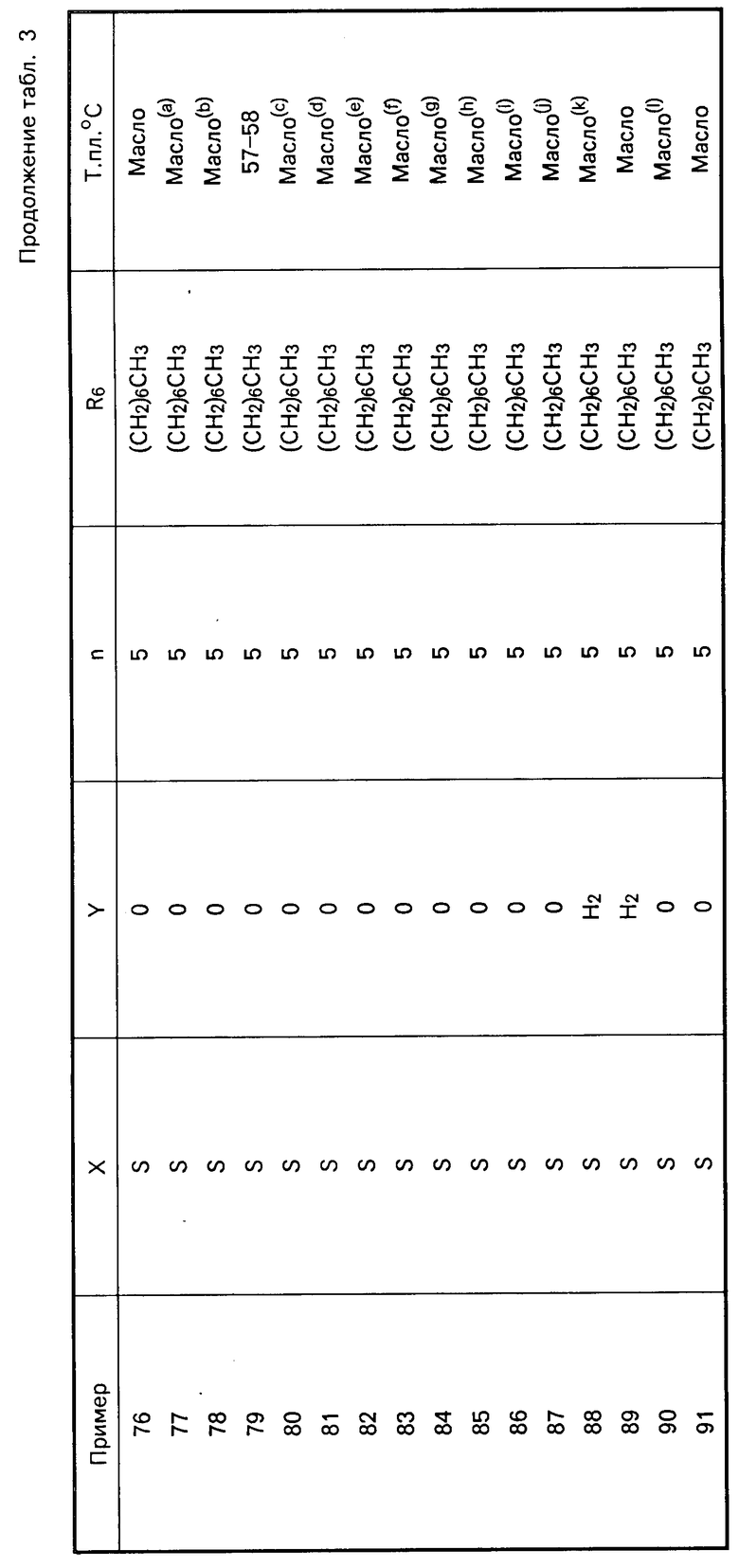
Перечисленные в табл. 3 амиды получены в соответствии с методиками, описанными в примерах 7 и 8.
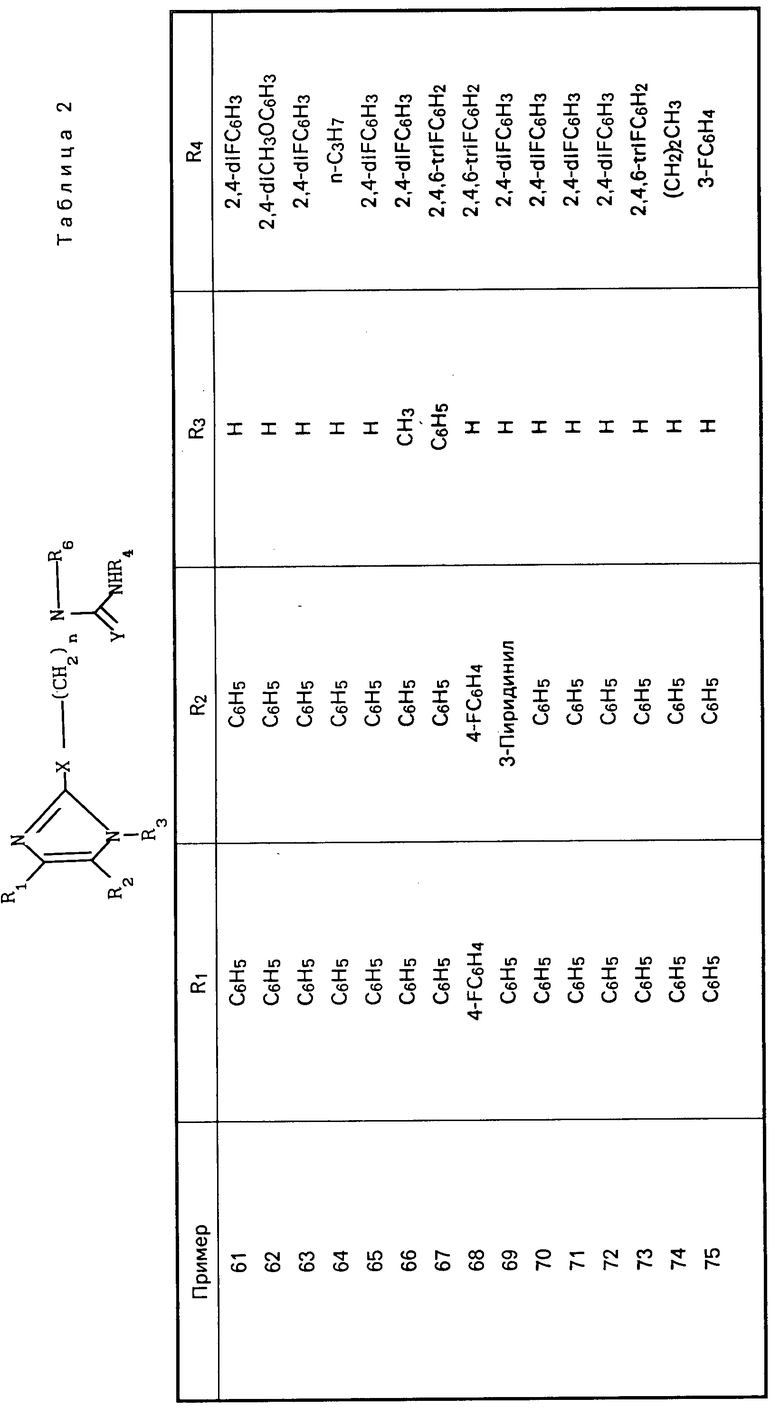
П р и м е р 9. Получение фенил-N-[5-[4,5-бис-(1-метилэтил)-1Н-имидазол-2-илтио]- пентил-]-N-гептилкарбамата.
Часть А. По способу примера 22, часть В, используя фенилхлорформиат и триэтиламин, получили фенил-N-гептил-N-(5-гидроксипентил)-карбамат в виде масла (3,18 г, 0,00989 моль).
1Н ЯМР (CDCl3) δ: 7,40-7,06 (м, 5Н), 3,68-3,63 (м, 2Н), 3,42-3,27 (м, 4Н), 2,08-1,95 (шир. с, 1Н), 1,75-1,26 (м, 16Н), 0,90 (т, 3Н).
Часть В. По способу примера 22, часть С, используя фенил-N-гептил-N-(5-гидроксипентил) карбамат, получили фенил-N-(5-бромпентил)-N-гептилкарбамат в виде масла (3,8 г, 0,0099 моль).
1 Н ЯМР (CDCl3) δ: 7,39-7,07 (м, 5Н), 3,47-3,25 (м, 6Н), 1,97-1,89 (м, 2Н), 1,75-1,26 (м, 14Н), 0,87 (т, 3Н).
Часть С. По способу примера 22, часть D, используя фенил-N-(5-бромпентил)-N-гептилкарбамат, получили указанное в заголовке соединение в виде масла (0,3 г, 0,000615 моль).
1 Н ЯМР (DMCO-d6) δ: 11,07 (с, 1Н), 7,35 (м, 2Н), 7,18 (т, 1Н), 7,05 (д, 2Н), 3,31 (м, 2Н), 3,20 (м, 2Н), 2,95 (м, 3Н), 2,8 (м, 1Н), 1,67-1,06 (м, 2Н), 0,86 (м, 3Н),
Карбаматы представлены в табл.4 и далее.
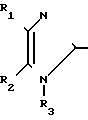 S
S  (CH2)
(CH2)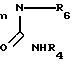 где R1-4-CH3OC6H4;R2-4-CH3OC6H4; R3-H; X-O; A-(CH2)3CH(CH3)2CH2; Y-H2; Z-NHCH(CH3)2; R6-(CH2)5CH3. (т. пл. 138-139оС)
где R1-4-CH3OC6H4;R2-4-CH3OC6H4; R3-H; X-O; A-(CH2)3CH(CH3)2CH2; Y-H2; Z-NHCH(CH3)2; R6-(CH2)5CH3. (т. пл. 138-139оС)
Биологические испытания
Соединения по изобретению являются ингибиторами фермента ацил-СоА: холестеринацилтрансферазы и поэтому являются эффективными для замедления этерификации и транспорта холестерина через стенки кишечника.
А. Исследование ингибирования ацил-СоА: холестеринацилтрансферазы (АСАТ) в печеночных микросoмах иллюстрируется ниже.
Способность предлагаемых соединений ингибировать АСАТ, фермента, ответственного за внутриклеточный синтез холестериловых эфиров, исследовалась следующим образом. Крыс Sprague Dawley мужских особей массы 150-300 г кормили по потребности. Затем животные голодали в течение 24 ч перед тем, как их умертвили путем декапитирования. Печень подвергли перфузии на месте 50 мл холодного 0,25 М раствора сахарозы, извлекли и гомогенизировали в трех объемах 0,1М фосфатного буфера , рН 7,4, который содержал 0,5 ммоль ЭДТК (этилендиаминтетрауксусная кислота), 1,0 ммоль глютатиона, 0,25 ммоль сахарозы и 20 ммоль лейпептина. Микросомы получили путем дифференциального центрифугирования; надосадочную жидкость центрифугировали сначала при 15000 g в течение 15 мин и затем при 105000 g в течение 1 ч для осаждения микросхем. Затем эти микросомы суспендировали в гомогенизирующем буфере, снова выделили путем центрифугирования и хранили при температуре -70оС. Микросомы использовали в течение одного месяца после получения.
Контрольный текст провели при конечном объеме 200 мкл, который содержал 200 мкг микросомного протеина, 75 мкмоль С-олеоил-СоА (10000 распадов в мин/нмоль) в 0,1М фосфате, рН 7,4, который включал 1 ммоль глютатиона. Соединения добавили в 5-10 мкл ДМСО (диметилсульфоксид) и провели дополнительные контрольные испытания только с ДМСО. Все компоненты, за исключением олеоил-СоА, предварительно инкубировали в течение 15 мин при 37оС перед тем, как инициировать реакцию путем добавления олеоил-СоА. Тест закончили через 10 мин путем добавления 500 мкл смеси гексан-изопропанол (3:2 по объему). 20000 распадов в минуту Н-холестерилолеата и 10 мкг немаркированного холестерилолеата и олеиновая кислота были добавлены как внутренний стандарт и носитель соответственно. После выдержки в течение 10 мин для экстрагирования липидов образцы центрифугировали при 1000 g в течение 10 минут для разделения слоев растворителя. 200 мкл верхнего (гексанового) слоя, содержащего нейтральные липиды, нанесли на Baker S 1250-Ра силикагелевую пластину для ТСХ и эту пластину обработали, используя смесь гексан-диэтиловый эфир-уксусная кислота (170:30:1 по объему) в качестве элюента. Липиды визуализировали, введя их во взаимодействие с парами йода и холестериловое эфирное пятно поместили в сцинтилляционный сосуд и просчитали. Удельная активность АСАТ в контрольном замере равна в среднем 260 нмоль/мин/мг микросомного протеина. Подавление активности АСАТ соединениями изобретения представлено ниже, данные представлены как концентрация (С), при которой активность АСАТ подавляется на 50% (ИК50).
В. Исследование ингибирования этерификации холестерина в клетках молочной железы.
Этерификацию холестерина наблюдали в макрофагоподобных клетках линии I774A. Клетки высевали в 35 мм ячейки с плотностью 300000 клеток в ячейку, в 2 мл среды Dulbeccos Еagle Medium (DMEM), в которую добавили 10 мл фетальной бычьей сыворотки (ФБС). Клетки инкубировали при температуре 37оС в атмосфере СО и при 93%-ной влажности. Через 24 ч среду заменили на 0,68 мл 10% ФБС-DMEM, содержащую 34 мкг ацетилированного низкой плотности липопротеина человека (ac-LDL) для увеличения внутриклеточной концентрации холестерина и способствования этерификации. На 41 часу различные ингибиторы добавляли в раствор клеток в DMCO (10 мкл/мл максимум). На 43 часу клетки обработали 0,1 ммоль С-олеиновой кислоты (10000 распадов в минуту-нмоль) в комплексе с альбумином бычьей сыворотки ЭАБС) для отслеживания образования эфира холестерина. Эксперимент завершили на 45 часу путем промывки монослоев 3 раза 3 мл трис-забуференного солевого раствора при 4оС. Липиды экстрагировали путем инкубирования монослоев 1,5 мл смеси гексан-изопропанол (3: 2 по объему) в течение 30 мин при осторожном перемешивании. В течение этого времени 1000 распадов в минуту Н-холестериллинолеата и 10 мкг холестерилолеата добавили в качестве внутреннего стандарта и носителя соответственно. Органический слой удалили и клетки промыли дополнительным 1,0 мл смеси гексан - изопропанол, которую объединили с первоначальными экстрактами. Клеткам дали высохнуть в течение ночи, обработали 1,5 мл 0,2 н.раствором едкого натра в течение 1 ч и использовали аликвоту растворенного протеина для определения протеина по методу Лоури. Органический экстракт высушили досуха, остаток снова суспендировали в 100 мкл хлороформа и липиды отделили на пропитанных силикагелем пластинах из стекловолокна, используя систему растворителей гексан-диэтиловый эфир-уксусная кислота (170:30:1 по объему). Индивидуальные липиды были визуализированы с помощью йода и пятна холестерилового эфира были вырезаны и помещены в сцинтилляционный сосуд для определения радиоактивности. Превращение олеиновой кислоты в холестериловый эфир в контрольном опыте составило 0,54 ммоль/ч/мг протеина и возосло при добавлении до 10,69 ± 0,69 ммоль/ч/мг протеина. Замедление этерификации соединениями, полученным способом настоящего изобретения, показано ниже, данные выражены как концентрация, при которой активность АСАТ снижается на 50% (ИК50). Следует отметить, что множество промежуточных веществ обладали подавляющей активностью in vitro в АСАТ-исследовании и в исследовании с макрофагами. Например, N-[5-(4,5-дифенил-1Н-имидазол-2-илтио)- пентил]-1-гептанамингидридрохлорид имел ИК50 100 нмоль и 6 мкмоль соответственно, в in vitro АСАТ-исследовании и в исследовании с макрофагами.
С. Исследование антигиперхолестеринемической активности у хомяков, кормленных холестерином.
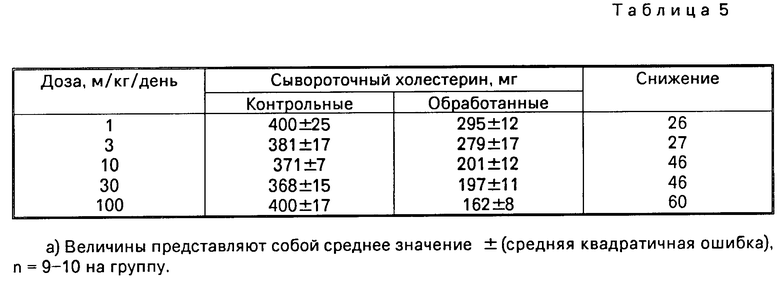
Ингибирование активности АСАТ в кишке снижает абсорбцию холестерина у кормленных холестерином животных. Хомяков массой примерно 100 г держали на диете с 0,8 г холестерина. Подопытные животные получали 1-100 мг/кг/день в рот испытываемого соединения, растворенного в 500 мкл кукурузного масла в течение 2 недель. Контрольную группу кормили как подопытную, но ей давали только 500 мкл кукурузного масла. Хомяков умертвили, сделав анестезию с помощью СО, и отобрали кровь проколом сердца. Определили общее содержание холестерина на Du Pont. Данные представлены как количество мг холестерина на 100 мл сыворотки (мг %). Антигиперхолестеринемическая активаность соединения примера 1 представлена в табл.5.
Ингибирование in vitro активности почечной АСАТ различными соединениями приведено ниже. Соединение по примеру, C(ИК50) 1 13 2 23 3 8 4 60 5 12 6 3600 7 41 8 10 9 930 10 20 11 17 12 30 13 16 14 60 15 60 16 10 17 25 18 20 19 1000 20 60 21 40 22 170 23 80 25 490 26 2850 27 20 28 70 29 30 30 400 31 70 32 60 33 1999 34 40 35 300 36 119 37 40 38 20 39 20 40 200 41 220 43 500 44 74 45 40 46 9 47 20 48 1400 49 10 50 32 51 60 52 53 80 54 110 55 130 56 30 57 160 58 370 59 200 60 150 61 40000 64 80 65 200 66 40 67 810 68 230 69 230 70 58 71 8 72 16 73 30 74 140 75 130 76 25 77 3 78 30 79 160 80 30 83 30 84 700 85 200 86 605 87 250 88 300 89 240 93 20 94 35 95 33 96 500 97 10 98 40 99 9 100 120 103 70 Ингибирование этерификации холестерина в макрофагах различными соединениями приведено ниже. Соединение примера, Этерификация холестерина ИК50, мкмоль 1 1,0 2 0,8 3 17,5 4 4,6 5 2,5 6 3,8 7 7,5 8 0,5 9 11,2 10 54,5 11 0,4 12 0,6 13 1,9 14 3,1 15 0,9 16 0,1 17 0,7 18 0,3 19 2,3 20 0,9 21 3,5 22 0,1 23 0,3 25 1,6 26 6,2 27 0,9 28 2,2 30 2,4 31 2,0 32 2,7 33 4,1 34 0,4 35 1,4 36 0,1 38 0,6 41 0,004 42 50,0 44 0,003 45 0,4 46 0,6 48 4,8 49 0,8 50 0,7 51 1,7 52 - 53 3,11 54 0,26 55 2,44 56 0,25 57 0,94 58 0,73 59 2,48 60 1,83 61 25,0 62 0,9 63 6,0 67 0,1 68 0,86 70 6,1 71 1,2 72 3,5 73 2,5 74 1,2 75 0,9 76 0,2 77 0,1 78 1,6 79 1,1 83 0,3 84 0,2 93 0,4 94 0,5 95 0,5 96 3,9 97 0,6 98 0,8 99 1,3 103 0,67
Реакция на введение соединения по примеру 1 у хомяков с гиперхолестеринемией дана в табл. 5.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ИМИДАЗОЛОВ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМОЙ СОЛИ | 1989 |
|
RU2028293C1 |
| ПРОИЗВОДНЫЕ МАСЛЯНОЙ КИСЛОТЫ | 2002 |
|
RU2293726C2 |
| 6-(ПОЛИЗАМЕЩЕННЫЙ АРИЛ)-4-АМИНОПИКОЛИНАТЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ГЕРБИЦИДОВ | 2007 |
|
RU2428416C2 |
| N-ФЕНИЛАМИДНЫЕ И N-ПИРИДИЛАМИДНЫЕ ПРОИЗВОДНЫЕ, СПОСОБ ИХ ПОЛУЧЕНИЯ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 1998 |
|
RU2214409C2 |
| АЗОТСОДЕРЖАЩИЕ ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ФЛУОРЕНА | 1992 |
|
RU2051151C1 |
| СПОСОБ ПОЛУЧЕНИЯ СТЕРЕОСПЕЦИФИЧЕСКОГО СОЕДИНЕНИЯ | 1989 |
|
RU2024528C1 |
| ИНГИБИТОРЫ ТИРОЗИНФОСФАТАЗЫ БЕЛКА ЧЕЛОВЕКА И СПОСОБЫ ПРИМЕНЕНИЯ | 2007 |
|
RU2435763C2 |
| ТРИЦИКЛИЧЕСКИЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1992 |
|
RU2072997C1 |
| ПРОИЗВОДНЫЕ ДИБЕНЗ(B, F) (1,4)ОКСАЗЕПИН-11-ОНА И ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ КИСЛОТНО-АДДИТИВНЫЕ СОЛИ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ УМЕНЬШЕНИЯ МУЛЬТИЛЕКАРСТВЕННОЙ РЕЗИСТЕНТНОСТИ РАКОВЫХ КЛЕТОК К ЦИТОТОКСИЧЕСКОМУ ЛЕКАРСТВУ | 1992 |
|
RU2086545C1 |
| ПРОИЗВОДНЫЕ ТЕТРАЗОЛА | 1992 |
|
RU2091376C1 |




Использование: в качестве препарата для лечения тромбоэмболин, воспалительных и/или атеросклеротических заболеваний. Сущность изобретения: продукт - соединение ф-лы I, где R1 и R2 - H, C1- C8 - алкил, C3- C8 - алкил с разветвленной цепью, C3- C7 - циклоалкил, 2-, 3- или 4-пиридинил, 2-тиенил, 2 фуранил, фенил, в определенных случаях замещенный 1-3 группами, выбранными из F, Cl, Br, Oh, C1- C4 -алкокси, C1- C4 -алкил, CH3S(O)2, CF3 или NR7R8 или R1 и R2 , вместе взятые, образуют группу ф-лы II, где L-O, O(CH2)O; R3-H, C1-C6 -алкил или фенил; Z-NHR4, R4 , OR4, R4-C1-C8 - алкил с нормальной цепью, C3-C8 -алкил с разветвленной цепью, C3-C7 - циклоалкил, C4-C10 -циклоалкилалкил, C7-C14 -аралкил, где арильная группа в определенных случаях замещена 1-3 группами, выбранными из ряда, включающего C1-C4 -алкил, фенил, в определенных случаях замещенный 1-3 группами, выбранными из ряда, включающего C1-C4 -алкил, фенил, в определенных случаях замещенный 1-5 группами, выбранными из ряда, включающего C1-C4 -алкил, C1-C4 -алкокси, F, CN, C1, пентафторфенил, бензил, в определенных случаях замещенный 1-3 группами, выбранными из ряда, включающего C1-C4 -алкил, F, Cl, 2-пиридиния; R5-H , C1-C9 -алкил, NR7R8 , бензил, C3-C8 -алкил с разветвленной цепью, или фенил, в определенных случаях, замещенный 1-3 группами, выбранными из ряда, включающего C1-C4 -алкил или -алкокси, или F; R7 и R8, независимо, выбраны из группы, включающей H и C1-C4 -алкил; X-S(O)r или 0; A-C2-C10 -алкил, C3-C10 -алкил с разветвленной цепью Y-0, S, H2; r-0-2. Реагент 1: соединение ф-лы III. Реагент 2: соединение ф-лы IV, где M-галоид, остальные радикалы имеют вышеуказанные значения.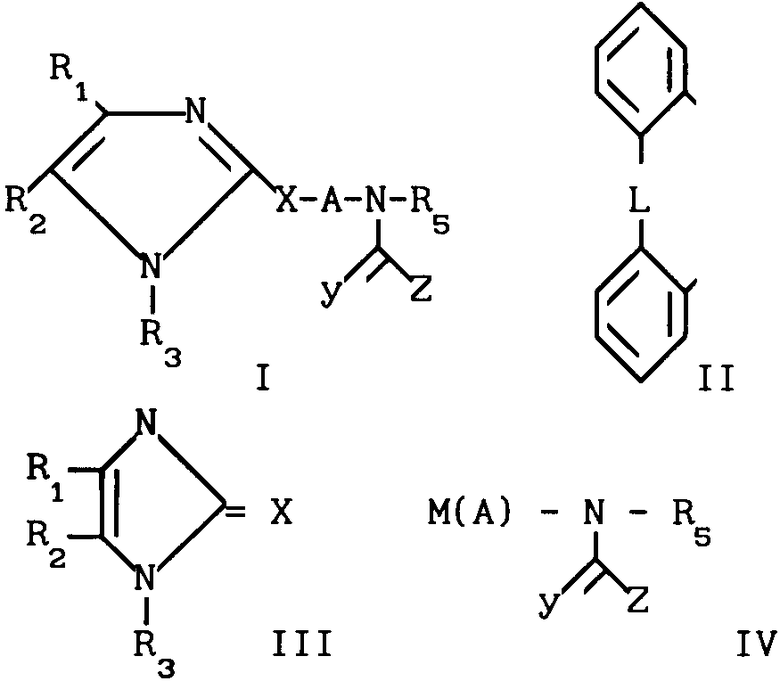
СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ИМИДАЗОЛА ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫХ СОЛЕЙ.
Способ получения производных имидазола общей формулы I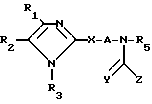
где R1 и R2 независимо друг от друга, выбраны из ряда, включающего Н, С1 - С8-алкил, С3 - С8-алкил с разветвленной цепью, С3 - С7-циклоалкил, 2-, 3- или 4-пиридинил, 2-тиенил, 2-фуранил, фенил, в определенных случаях замещенный 1 - 3 группами, выбранными из ряда, включающего F, Cl, Br, OH, С1 - С4-алкокси, С1 - С4-алкил, CF3, CH3S(O)r или NR7R8 или R1 и R2 вместе образуют группу общей формулы II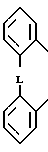
где L - О, О(СН2)О;
R3 - Н, С1 - С6-алкил, или фенил;
Z - NHR4, R4, OR4;
R4 - С1 - С8-алкил с нормальной цепью, С3 - С8-алкил с разветвленной цепью, С3 - С7-циклоалкил, С4 - С10-циклоалкилалкил, С7 - С14-аралкил, где арильная группа в определенных случаях замещена 1 - 3 группами, выбранными из ряда, включающего С1 - С4-алкил, фенил, в определенных случаях замещенных 1 - 3 группами, выбранными из ряда, включающего С1 - С4-алкил, С1 - С4-алкокси, F, CN, Cl, пентафторфенил, бензил, в определенных случаях замещенный 1 - 5 группами, выбранными из ряда, включающего С1 - С4-алкил, F, Cl, 2-пиридинил, бифенил;
R5 - Н, С1 - С9-алкил, NR7R8, бензил, С3 - С8-алкил с разветвленной цепью, или фенил, в определенных случаях замещенный 1 - 3 группами, выбранными из ряда, включающего С1 - С4-алкил или алкокси, или F;
R7 и R8 независимо, выбраны из группы, включающей Н и С1 - С4-алкил;
X - S(O)r или O,NR5, CH2;
A - С2 - С10-алкил, С3 - С10-алкил с разветвленной цепью;
Y - O, S, H; r -0-2,
или их фармацевтически приемлемых солей, отличающийся тем, что он включает стадию алкилирования соединения общей формуды III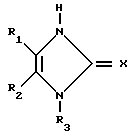
где R1 - R3, Х имеют указанные значения,
соединением общей формулы IV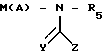
где М - галоид;
А, R5 и Z имеют указанные значения, с выделением целевого продукта или в случае, когда Y - 0, полученное соединение подвергают релаксации с восстановителем, таким, как алюмогидрид лития или боргидрид натрия, в результате чего получают соединение общей формулы I, где Y - H2, или, когда Х - S, подвергают реакции с подходящим окислителем, в результате чего получают либо сульфоксид SO, где r = 1, либо сульфон SO2, где r = 2.
Приоритет по признакам
05.12.88 при R1 и R2 - C6H5; R3 - H; R4 - CH3, C3H7, (CH2)2CH3, CH(CH3)2, C8H17, C6H5, 2,4-ди-CH3OC6H3, 3-FC6H4, 2,4-ди-FC6H3, 2,6-ди-Cl C6H3, 4-CNC6H4, 2,4,6-три-FC6H2; R6 - (CH2)2CH3, (CH2)3CH3, (CH2)6CH3; X - S, SO2; A - (CH2)2, (CH2)6, (CH2)8; Y - O, S; Z - NHR4; n = 0; 2.
| Патент США N 4460598, кл | |||
| СПОСОБ ПОЛУЧЕНИЯ ПРОДУКТОВ КОНДЕНСАЦИИ ФЕНОЛОВ С ФОРМАЛЬДЕГИДОМ | 1925 |
|
SU514A1 |
