Изобретение относится к противомикробному соединению, используемому в качестве лекарственного средства для человека и животных, а также для рыб.
Относительно синтетических противомикробных агентов типа пиридонкарбоновой кислоты известно, что заместитель в положении 7 их хинолинового скелета (или в положении 10 пиридобензоксазинового скелета) оказывает влияние на противомикробную активность. В качестве заместителя в положении 7 были предложены различные группы. Известно, в частности, что производные пиридонкарбоновой кислоты, имеющие в положении 7 3-аминопирроли- динильную группу, обладают высокой противомикробной активностью и широким спектром противомикробного действия в отношении как грамотрицательных, так и грамположительных бактерий.
Вместе с тем считается, что производные пиридонкарбоновой кислоты, состоящие из 3-аминопирролидинильной группы и пиридобензоксазинового скелета, обладают плохими липофильными свойствами, и их фармакокинетические характеристики являются неудовлетворительными, возможно, вследствие плохого поглощения из кишечного тракта.
Целью изобретения является соединение 3-аминопирролидинилзамещенного производного пиридобензоксазина, обладающего улучшенными липофильными свойствами и одновременно проявляющего высокую противомикробную активность в отношении широкого спектра микроорганизмов, включая грамположительные бактерии.
В результате проведенных авторами изобретения исследований было найдено, что поставленная цель достигается использованием оптически активного производного пиридобензоксазина, содержащего 3-аминопирролидильный заместитель, в котором атом углерода, соседний с тем атомом углерода, который связан с аминогруппой, является дизамещенным алкильной группой, т.е. оптически активную 4-амино-3,3-диалкилзамещенную пирролидинильную группу, изображаемую формулой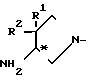 в которой каждая из групп R1 и R2, которые могут быть одинаковыми или разными, представляет собой алкильную группу с числом атомов углерода от 1 до 4.
в которой каждая из групп R1 и R2, которые могут быть одинаковыми или разными, представляет собой алкильную группу с числом атомов углерода от 1 до 4.
Само собой разумеется, что соединение согласно настоящему изобретению включает в себя все соответствующие диастереомеры, которые присутствуют в виде стереоизомеров, возникающих благодаря наличию асимметричных атомов углерода.
Настоящее изобретение обеспечивает оптически активные производные пиридобензоксазина, отвечающие нижеследующей формуле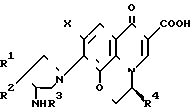 (I) где R1 и R2 имеют определенные выше значения;
(I) где R1 и R2 имеют определенные выше значения;
R3 представляет собой атом водорода;
R4 представляет собой метильную группу, находящуюся в β-конфигурации;
R5 представляет собой атом водорода или метильную группу;
Х представляет собой атом фтора или хлора, или соли этих производных.
В формуле I каждая из групп R1 и R2, которые могут быть одинаковыми или разными, предпочтительно представляет собой алкильную группу с числом атомов углерода 1-4, например метильную группу, этильную группу, пропильную группу и бутильную группу.
R4 представляет собой метильную группу. Атом углерода, с которым связана группа R4 является асимметрическим атомом углерода. Считается, что наиболее высокая противомикробная активность достигается в том случае, когда этот асимметрический атом углерода находится в β-конфигурации. В этом случае абсолютной конфигурацией является (S)-конфигурация, в которой R4 представляет собой метильную группу.
R5 представляет собой атом водорода или метильную группу.
Х - атом фтора или хлора, предпочтительно фтора.
Соединения согласно настоящему изобретению отличаются тем, что содержат в положении 10 пиридобензоксазинового скелета 4-амино-3,3-диалкилзамещенную пирролидинильную группу. Конкретные примеры 4-амино-3,3-диалкилпирролидинильных групп включают 4-амино-3,3-диметилпирролидинильную группу.
Стереоизомеры этих 4-амино-3,3-диалкилпирролидинильных групп появляются благодаря наличию асимметрического атома углерода, с которым связана аминогруппа. С точки зрения противомикробной активности, предпочтительной пространст- венной конфигурацией при атоме углерода, с которым связана аминогруппа, является (S)-конфигурация. С другой стороны, в пиридобензоксазиновом скелете асимметрическим является тот атом углерода, с которым связана группа R4. Для получения наиболее высокой противомикробной активности заместитель R4 при этом асимметрическом атоме углерода должен находиться в β-конфигурации.
Таким образом, каждое соединение настоящего изобретения содержит как минимум два асимметрических атома углерода. Следовательно, каждое соединение имеет как минимум четыре стереоизомера, включая диастереомеры. При использовании в качестве синтетического противомикробного агента во многих случаях соединение, содержащее один стереоизомер, предпочтительнее смеси этих стереоизомеров.
Соединения настоящего изобретения могут быть синтезированы реакцией 9,10-дифтор-2,3-дигидро-3-(S)-3-метил-7-оксо-7Н-пиридо/1,2,3-de//1,4/ бензоксазин-6-карбоновой кислоты или ее дифторбората (т.е. соединения, содержащего в положении 6 группу -С(=O)O-BF2 вместо группы СООН) с 4-амино-3,3-диалкилпирролидином в среде растворителя, в присутствии основания.
Дифторборат можно легко получить из соответствующей свободной кислоты и соответствующего соединения трехфтористого бора, например, эфирата трехфтористого бора, как описано в патенте США N 5053407.
Реакцию введения 4-амино-3,3-диалкилпирролидина обычно проводят в присутствии соединения, связывающего кислоту. В качестве этого соединения могут быть использованы органические и неорганические основания, причем обычно предпочтительным является использование органических оснований.
Предпочтительно органические соединения представляют собой третичные амины. Конкретные примеры подходящих третичных аминов включают триалкиламины, например триэтиламин, трипропиламин, N,N-диизопропилэтиламин, и трибутиламин; анилины, например N,N-диметиланилин и N,N-диэтиланилин, и гетероциклические соединения, например N-метилморфолин, пиридин и N,N-диметиламинопиридин.
Примеры подходящих неорганических оснований включают гидроксиды, карбонаты и бикарбонаты щелочных металлов, таких как литий, натрий и калий, например гидроокись лития, гидроксид натрия, гидроксид калия, безводный карбонат натрия, безводный карбонат калия, бикарбонат натрия и бикарбонат калия.
4-Амино-3,3-диалкилпирролидиновый реагент можно использовать в количестве, по крайней мере вдвое превышающем эквивалентное количество, требующееся для реакции, в этом случае указанный реагент служит также и соединением, связывающим кислоту.
В отношении растворителей, которые могут быть использованы для данной реакции, какие-либо специальные ограничения отсутствуют, если только они являются инертными по отношению к реакции, примеры включают ацетонитрил, амиды, например N,N-диметилформамид, N-метил-3- пирролидон, и N,N-диметилацетамид, ароматические углеводороды, например бензол, толуол и ксилол, полярные апротонные растворители, например диметилсульфоксид и сульфолан, низшие спирты, например метанол, этанол, пропанол, бутанол, амиловый спирт, изоамиловый спирт, циклогексиловый спирт, и 3-метоксибутанол, и эфиры, например диоксан, диметилцеллозольв, диэтилцеллозольв и диглим. В качестве водного растворителя могут быть использованы водорастворимые растворители. В этом случае в качестве соединения, связывающего кислоту, рекомендуется использовать органическое основание.
Температура реакции лежит в интервале от комнатной температуры до 180оС. Реакция проходит до конца за время от  10 мин до
10 мин до  48 ч, обычно от
48 ч, обычно от  30 мин до
30 мин до  30 ч.
30 ч.
Когда вводимый 4-амино-3,3-диалкилпирролидин содержит в пирролидиновом ядре защищенную группу, защитная группа может быть удалена после реакции обычным способом, выбираемым в зависимости от типа защитной группы.
4-Амино-3,3-диалкилпирролидин, например 4-амино-3,3-диметилпирролидин, можно синтезировать так, как это описано ниже в ссылочных примерах, или по схеме реакций, приведенной ниже: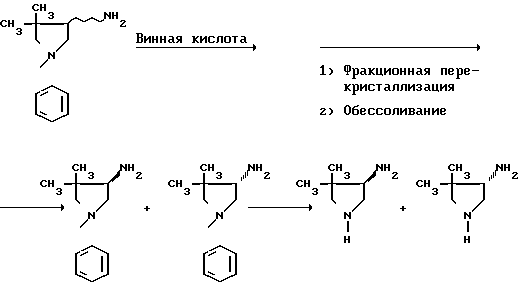
Когда в качестве исходного соединения используют дифторборатное соединение, в котором карбоксильная группа в положении 6 является борированной, получаемый продукт можно превратить в свободную кислоту, обрабатывая его протонным соединением, например водой или спиртами. Например, продукт обрабатывают этанолом в присутствии триэтиламина или в присутствии, или в отсутствие соединения, связывающего кислоту.
Полученное производное пиридонкарбоновой кислоты, имеющее формулу I, очищают перекристаллизацией, переосаждением, обработкой активированным углем, хроматографированием и им подобными методами, используемыми поодиночке или в сочетании друг с другом.
Содержащие 4-амино-3,3-диалкилпирролидинильную группу производные пиридондикарбоновой кислоты, согласно настоящему изобретению, обладают улучшенными липофильными свойствами, по сравнению с соответствующими соединениями, содержащими аминопирролидинильную группу без алкильных групп, поэтому можно ожидать, что они будут достаточно хорошо поглощаться при оральном введении и, вследствие этого, обладать более высокой противомикробной активностью.
Производные пиридонкарбоновой кислоты настоящего изобретения можно использовать в форме свободных соединений, в форме солей присоединения кислоты, и в форме солей, образуемых карбоксильной группой. Примеры солей присоединения кислоты включают соли с неорганическими кислотами, такие как гидрохлорид, сульфат, нитрат, гидробромид, гидройодид и фосфат, и соли с органическими кислотами, такие как ацетат, метансульфонат, бензолсульфонат, толуолсульфонат, цитрат, малеат, фумарат и лактат.
Примеры солей, образуемых карбоксильной группой, включают неорганические и органические соли, например соли щелочных металлов, такие как соли лития, натрия и калия, соли щелочноземельных металлов, такие как соли магния и кальция, аммонийные соли, соли триэтиламина, соли N-метилглюкамата и трис-гидроксиметиламинометана.
Эти свободные соединения, соли присоединения кислоты и соли карбоксильной группы производных пиридобензоксазин-6-карбоновой кислоты могут существовать в форме гидратов.
С другой стороны, производные пиридобензоксазин-6-карбоновой кислоты, в которых карбоксильные группы превращены в сложноэфирные группы, могут использоваться в качестве синтетических промежуточных соединений или пролекарств. Например, сложные алкиловые эфиры, бензоловые эфиры, алкоксиалкиловые эфиры, фенилалкиловые эфиры и фениловые эфиры используются в качестве синтетических промежуточных продуктов.
Сложными эфирами, используемыми в качестве пролекарств, являются те сложные эфиры, которые в живом организме легко расщепляются, давая свободную карбоновую кислоту. Примерами являются ацетоксиметиловый эфир, пивалоилоксимети- ловый эфир, этоксикарбонилоксиэфир, холиновый эфир, диметиламиноэтиловый эфир, 5-инданиловый эфир, фталидиниловый эфир, 5-замещенный-2-оксо-1,3-диоксо-4-ил-метиловый эфир и различные оксоалкиловые эфиры, например 3-ацетокси-2-оксобутиловый эфир.
Соединения, согласно настоящему изобретению, обладают высокой противомикробной активностью и, вследствие этого, могут использоваться в качестве лекарственных средств для людей и животных, рыб, а также в качестве сельскохозяйственных химикатов и пищевых консервантов.
Для людей доза соединения настоящего изобретения как активного ингредиента лекарственного средства составляет от 50 мг до 1 г, предпочтительно 100-300 мг в день (для взрослых). Для животных доза составляет 1-200 мг, предпочтительно 5-100 мг на 1 кг массы в день. Дневная доза должна регулироваться в зависимости от таких факторов, как предполагаемая цель использования (терапевтическая или профилактическая), вид, размер или возраст человека или животного, тип патогенных организмов, подлежащие лечению симптомы и т.д.
Вышеуказанная дневная доза может быть разделена на 1-4 части. Может возникнуть необходимость отклониться от вышеуказанных количеств, если этого требует конкретная ситуация (вызывающие заболевание микроорганизмы или степень тяжести проявляющихся симптомов).
Соединения согласно настоящему изобретению проявляют активность в отношении очень широкого спектра микроорганизмов, вызывающих различные инфекционные заболевания, и с их помощью можно предотвращать, смягчать и/или вылечивать заболевания, вызываемые этими патогенными микроорганизмами. Примеры бактерий или бактериоподобных микроорганизмов, в отношении которых соединения настоящего изобретения являются эффективными, включают Staphylococcus sp. , Streptococcus pyogenes, Streptococcus haemolyticus, Streptococcus fecalis, Streptococcus pneumoniae, Peptostreptococcus sp., Neisseria gonorrhoeae, Escherichia coli, Sitrobacter sp., Shigella sp., Klebsiella pneumoniae, Enterobacter sp., Serratia sp., Proteus sp., Pseudomonas aeruginosa, Haemophilus influenzae, Acinetobacter sp., Campylobacter sp., Chlamydozoon trachomatis.
Болезни, которые вызываются этими патогенными микроорганизмами, и которые могут быть предотвращены, смягчены или вылечены с помощью соединений настоящего изобретения, включают фолликулит, фурункул, фурункулез, карбункул, рожу, флегмону, лимфангит/лимфаденит, нагноение суставов пальцев, подкожный абсцесс, спираденит, фолликулярный дерматит, инфекционную атерому, перианальный абсцесс, мастаденит, поверхностные вторичные инфекции после травмы, ожога или хирургической травмы, фаринголарингит, острый бронхит, тонзиллит, хронический бронхит, бронхоэктаз, диффузный панбронхиолит, вторичные инфекции хронических респираторных заболеваний, пневмонию, пиелонефрит, цистит, простатит, эпидимит, гонококковый уретрит, негонококковый уретрит, холецистит, холангит, бактериальную дизентерию, энетерит, аднексит, внутриматочные инфекции, бартолинит, блефарит, ячмень, дакриоцистит, тарзаденит, изъязвление роговицы, отит, синусит, пародонтоз, перикоронит, воспаление челюсти, перитонит, эндокардит, септицемию, менингит и кожные инфекции.
Соединения настоящего изобретения эффективны также в отношении различных микроорганизмов, вызывающих болезни животных, например, принадлежащих к родам Escherichia sp., Salmonella sp., Pasteurella sp., Haemophilus sp., Bordetella sp., Staphylococcus sp., и Mycoplasma sp.
Иллюстративные примеры таких болезней животных включают болезни домашних птиц, такие как колибактериоз, птичий паратиф, холера домашних птиц, острый инфекционный ринит, стафиломикоз и микоплазмоз, болезни свиней, такие как колибактериоз, сальмонеллез, пастеррилез, гемофильные инфекции, атрофический ринит, эксудативный эпидермит, и микоплазмоз, болезни крупного рогатого скота, такие как колибактериоз, сальмонеллез, геморрагическая септицемия, микоплазмоз, бычья контагиозная плевропневмония и коровий мастит; болезни собак, такие как колисепсис, сальмонеллез, геморрагическая септицемия, пиометра и цистит; болезни кошек, такие как геморрагический плеврит, цистит, хронический ринит, и гемофильные инфекции, и болезни котят, такие как бактериальный энтрит и микоплазмоз.
Дозировочные формы фармацевтических препаратов, содержащих одно или несколько соединений настоящего изобретения в качестве активного ингредиента, могут быть выбраны в зависимости от предполагаемого способа введения, и могут быть приготовлены общеизвестными методами. Примерами дозировочных форм для орального введения являются таблетки, порошки, гранулы, капсулы, растворы, сиропы, эликсиры, а также масляные или водные суспензии.
Препараты для инъекций могут содержать наполнители, например стабилизаторы, консерванты и солюбилизаторы. Раствор для инъекций, который может содержать эти наполнители, может быть помещен в контейнер, например в ампулу или в пиалу, а может быть отвержден, например лиофилизацией, для приготовления твердого препарата, который растворяют перед употреблением, контейнер может содержать одну дозу или несколько доз.
Препараты для местного употребления включают растворы, суспензии, эмульсии, мази, гели, кремы, лосьоны и разбрызгиваемые составы.
Твердые препараты могут содержать фармацевтически приемлемые добавки, такие как наполнители, связывающие вещества, увлажнители, ускорители поглощения, смачивающие агенты, адсорбенты и смазывающие агенты.
Жидкие препараты включают растворы, суспензии и эмульсии. Они могут содержать наполнители, такие как суспендирующие агенты, эмульгаторы, стабилизаторы и консерванты.
Соединения настоящего изобретения можно также вводить животным как оральные или неоральные ветеринарные лекарственные средства. Такие лекарственные средства могут быть введены в виде смеси с пищей или водой. Составы для ветеринарных лекарственных средств или добавки можно приготовить известными способами, эти составы включают порошки, тонкодисперсные гранулы, растворимые порошки, сиропы, растворы и составы для инъекций.
Нижеприведенные примеры для препаратов являются чисто иллюстративными.
Пример А препаратов
Капсулы Соединение примера 1 100,0 мг Кукурузный крахмал 23,0 мг
Кальцийкарбоксиме- тилцеллюлоза 22,5 мг
Гидроксипропилметил- целлюлоза 3,0 мг Стеарат магния 1,5 мг
Всего 150,0 мг
(на капсулу)
Пример Б препаратов
Раствор Соединение примера 1 1-10 г
Уксусная кислота или гидроксид натрия 0,5-2 г Этил-п-гидроксибензоат 0,1 г Очищенная вода 88,9-98,4 г
Всего 100 г
Пример В для препаратов
Порошок для смешивания с пищей Соединение примера 1 1-10 г Кукурузный крахмал 98,5-89,5 г
Легкая безводная кремниевая кислота 0,5 г Всего 100 г
Испытания на противобактериальную активность проводили методом, установленным Японским Обществом Химиотерапии (Chemotherapy 29(1), 76 (1981). Таблицы противобактериальной активности следуют за схемами реакций синтеза различных циклических аминопроизводных, содержащих спироядра промежуточных соединений для синтеза хинолоновых ядер и синтеза различных спиросоединений.
Вспомогательный пример 1.
N-[1-(R)-фенилэтил]-3-оксобутанамид
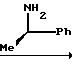
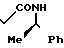
Раствор 21 г дикетена в 15 мл сухого хлористого метилена по каплям при перемешивании добавляют к охлаждаемому льдом раствору 31 г (R)-D-(+)-фенилэтиламина в 85 мл сухого хлористого метилена. Максимальная температура реакционной смеси при капельном добавлении составляет 15оС. Полученный раствор перемешивают при комнатной температуре в течение 15 ч. Реакционную смесь промывают последовательно 10%-ным водным раствором лимонной кислоты и насыщенным водным раствором бикарбоната натрия и сушат над безводным сульфатом натрия. Растворитель удаляют при пониженном давлении, получая 53,7 г целевого соединения.
1Н-ЯМР (CDCl3) δ: 1,48 (3H, д, J = 5,4 Гц), 2,24 (3Н, с), 3,40 (2Н, с), 5,14 (1Н, к, J = 5,4 Гц), 7,35 (5Н, с).
Вспомогательный пример 2.
2,2-Диметил-N-[1-(R)-фенилэтил]-3-ок-собутанамид
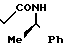 _______→
_______→ 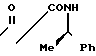
К раствору 48,5 г N-[1-(R)-фенилэтил]-3-оксобутанамида в 250 мл сухого N, N-диметилформамида добавляют 84 г йодистого метила, а затем при охлаждении льдом - 65,3 г карбоната натрия, после чего перемешивают полученную смесь в течение 1 недели при комнатной температуре. Через 72 ч после начала реакции к смеси добавляют 10 г йодистого метила и 35 г карбоната калия. Через 144 ч после начала реакции добавляют еще 10 г йодистого метила. Из реакционной смеси фильтрованием удаляют все нерастворимые продукты и при пониженном давлении удаляют из фильтрата растворитель. К остатку добавляют 100 мл воды и полученную смесь экстрагируют 400 мл этилацетата. Экстракт промывают водой и сушат над безводным сульфатом натрия. При пониженном давлении удаляют растворитель, и получают 50 г неочищенных кристаллов целевого соединения. Неочищенные кристаллы промывают изопропиловым спиртом, получая 33 г чистого продукта.
1Н-ЯМР (CDCl3) δ: 1,40 (3H, c), 1,42 (3H, c), 1,50 (3H, д, J = 5,4 Гц), 2,17 (3Н, с), 5,08 (1Н, к, J = 5,4 Гц), 6,18 (1Н, ш.с.), 7,32 (5Н, с)
Вспомогательный пример 3.
2-Метил-2-[1-метил-1-(N-(R)-1-фенил- этил]карбамоил]этил-1,3-диоксолан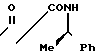 _______→
_______→ 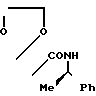
В 220 мл бензола растворяют 11,65 г 2,2-диметил-N-[1-(R)-фенилэтил]3-оксобута- намида и к полученному раствору добавляют 18 г этиленгликоля и 1 г п-толуолсульфокислоты. Полученную смесь кипятят с обратным холодильником в течение 3 дней, удаляя образующуюся воду с помощью прибора Дина-Старка. После охлаждения реакционную смесь выливают в 50 мл насыщенного раствора бикарбоната натрия в воде и встряхивают. Органический слой отделяют и сушат над безводным сульфатом натрия. При пониженном давлении удаляют растворитель, получая 15,2 г целевого соединения.
1Н-ЯМР (CDCl3) δ: 1,20 (3H, c), 1,22 (3H, c), 1,23 (3H, c), 1,50 (3Н, д, J = 5,4 Гц) 3,95 (4Н, с), 5,12 (1Н, к, J = 5,4 Гц), 7,12 (1Н, ш.с.), 7,32 (5Н, с).
Вспомогательный пример 4. 2-Бромметил-2-[1-метил-1-(N-(R)-1-фе-нилэтил] карбамоил]этил-1,3-диоксолан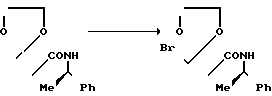
В 400 мл сухого 1,4-диоксана растворяют 37,68 г 2-метил-2-[1-метил-1-(N-(R)-1-фенилэтил)карбамоил]этил-1,3-диоксалана, к полученному раствору по каплям при комнатной температуре добавляют 22 г брома и перемешивают в течение 4 ч. При пониженном давлении удаляют из реакционной смеси растворитель, и добавляют к остатку 500 мл хлороформа. Слой хлороформа промывают последовательно насыщенным водным раствором бикарбоната натрия, 5%-ным водным раствором тиосульфата натрия и водой и сушат над безводным сульфатом натрия. При пониженном давлении удаляют растворитель, получая 45,25 г целевого соединения.
1Н-ЯМР (CDCl3) δ: 1,24 (3H, д, J = 3,6 Гц), 1,42 (3H, c), 1,54 (3H, c), 3,58 (2Н, ABк, J = 10,8 Гц), 3,90-4,50 (4Н, м), 5,05 (1Н, к, J = 3,6 Гц), 7,00 (1Н, гс), 7,30 (5Н, с).
Вспомогательный пример 5. 9,9-Диметил-8-оксо-7-[1-(R)-фенилэтил]-7-аза-1,4-диоксапиро/4,4/нонан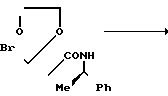
В 150 мл сухого N,N-диметилформамида растворяют 45,25 г 2-бромметил-2-[1-метил-1-(N-(R)-1-фенилэтил)карбамоил]этил- 1,3-диоксолана, к полученному раствору при охлаждении льдом добавляют 6,5 г 60%-ного гидрида натрия, после чего перемешивают при комнатной температуре в течение 18 ч. Реакционную смесь выливают в 300 мл ледяной воды и экстрагируют 600 мл бензола. Экстракт промывают водой и сушат над безводным сульфатом натрия. При пониженном давлении удаляют растворитель, и осадок очищают хроматографированием на колонке (заполнена 350 г силикагеля), элюируя смесью н-гексана и этилацетата состава 43: 1 (по объему). Объединяют фракции, содержащие целевой продукт, удаляют из них растворитель при пониженном давлении, получая 18,23 г целевого соединения в виде элюента.
1Н-ЯМР (CDCl3) δ: 1,18 (3H, д, J = 4,0 Гц), 1,50 и 1,58 (3Н, с, каждый), 3,04 (2Н, АБк, J = 10 Гц), 3,75-4,10 (4Н, м), 5,60 (1Н, к, J = 4 Гц), 7,32 (5Н, с).
Вспомогательный пример 6.
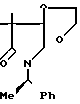
3,3-Диметил-1-[1-(R)-фенилэтил]пирро-лидин-2,4-дион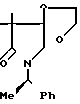 ________→
________→ 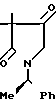
В 250 мл ацетона растворяют 18,23 г 9,9-диметил-8-оксо-7-(1-(R)-фенилэтил)-7- аза-1,4-диоксаспиро/4,4/нонана, и к полученному раствору добавляют 70 мл соляной кислоты и 20 г п-толуолсульфокислоты. Полученную смесь кипятят с обратным холодильником в течение 20 ч. Через 7 ч после начала реакции к реакционной смеси добавляют 20 г п-толуолсульфокислоты. По окончании реакции при пониженном давлении удаляют из реакционной смеси ацетон, и экстрагируют остаток 400 мл хлороформа. Слой хлороформа промывают насыщенным водным раствором бикарбоната натрия и сушат над сульфатом магния. При пониженном давлении удаляют растворитель, и остаток очищают хроматографированием на колонке с силикагелем (350 г), элюируя смесью н-гексана и этилацетата состава 4:1 (по объему). Объединяют фракции, содержащие целевое соединение, удаляют из них при пониженном давлении растворитель и получают 11,85 г целевого соединения.
1Н-ЯМР (CDCl3) δ: 1,20 и 1,26 (3Н, с, каждый), 1,60 (3Н, д, J = 7,2 Гц), 3,60 (2Н, АВк, J = 7,16 Гц), 5,80 (1Н, к, J = 7,2 Гц), 7,32 (5Н, с).
Вспомогательный пример 7.
3,3-Диметил-4-гидроксиимино-1-[1-(R)-фенилэтил]-пирролидин-2-он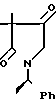 ________→
________→
В 100 мл этанола растворяют 11,85 г 3,3-диметил-1-[1-(R)-фенилэтил]-пирроли- дин-2,4-диона. К этому раствору добавляют 8 г солянокислого гидроксиламина в 45 мл триэтиламина, после чего кипятят с обратным холодильником в течение 1 ч. Растворитель удаляют при пониженном давлении и добавляют к остатку 300 мл хлороформа. Раствор в хлороформе последовательно промывают 10%-ным раствором лимонной кислоты и водой и сушат над безводным сульфатом натрия. Растворитель удаляют при пониженном давлении, получая 11,5 г целевого продукта.
1Н-ЯМР (CDCl3) δ: 1,30 и 1,34 (3Н, с, каждый), 1,58 (3Н, д, J = 7,2 Гц), 3,90 (2Н, АВк, J = 16,2 Гц), 5,65 (1Н, к, J = 7,2 Гц), 7,36 (5Н, с).
Вспомогательный пример 8.
4-Амино-3,3-диметил-1-[1-(R)-фенил- этил]-пирролидин-2-он
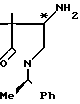
В 300 мл метанола растворяют 11,5 г 3,3-диметил-4-гидроксиимино-1-[1-(R)-фе-нилэтил] -пирролидин-2-он и к полученному раствору добавляют около 20 мл промытого метанолом никеля Ренея. Восстановление ведут при комнатной температуре в течение 16 ч в сосуде продолговатой формы (напоминающем баклажан). Катализатор удаляют фильтрованием и при пониженном давлении отгоняют из фильтрата растворитель. Остаток хроматографируют на силикагеле (400 г), элюируя смесью хлороформа и метанола состава 30:1 (по объему), получают 3,75 г фракции 1 (вещество с более низкой полярностью) и 10,2 г смеси фракции 1 и фракции 2 (вещество с более высокой полярностью).
1Н, ЯМР (CDCl3) δ (вещество с более низкой полярностью) 0,95 и 1,18 (3Н, с, каждый), 1,52 (3Н, д, J = 7,2 Гц), 2,40-2,55 (1Н, м), 3,00-3,50 (2Н, с), 5,52 (1Н, к, J = 7,2 Гц), 7,30 (5Н, с).
Вспомогательный пример 9.
4-Амино-3,3-диметил-1-[1-(R)-фенил- этил]-пирролидин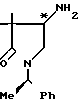 ________→
________→ 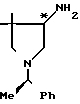
В 150 мл сухого тетрагидрофурана растворяют 3,1 г фракции 1, полученной во вспомогательном примере 8 (низкополярный 4-амино-3,3-диметил-1-[1-(R)-фенил]-пирролидин-2-он), к полученному раствору при охлаждении льдом медленно добавляют 2 г литийалюминийгидрида, и затем кипятят его с обратным холодильником в течение 13 ч. После охлаждения к реакционной смеси, охлаждаемой льдом, добавляют 2 мл воды, 2 мл 15%-ного водного раствора гидроксида натрия и 6 мл воды (в указанной последовательности), после чего перемешивают ее при комнатной температуре в течение 1 ч. Фильтрованием удаляют все нерастворимые продукты, из фильтрата при пониженном давлении удаляют растворитель, получая 3,69 г целевого соединения. Полученное таким образом соединение используют на следующей стадии без какой-либо дополнительной очистки.
Вспомогательный пример 10. 4-трет-Бутоксикарбониламино-3,3-ди-метил-1-[1-(R)-фенилэтил]пирролидин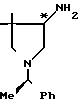 ________→
________→ 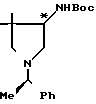
В 40 мл сухого тетрагидрофурана растворяют 3,69 г полученного во вспомогательном примере 9 4-амино-3,3-диме- тил-1-[1-(R)-фенилэтил]пирролидина и к полученному раствору при охлаждении льдом добавляют 4,92 г 2-(трет-бутоксикарбониламино)-2-фенилацетонитрила (BOC-ON). Полученную смесь перемешивают при комнатной температуре в течение 1 ч. Растворитель удаляют при пониженном давлении, и к остатку добавляют 100 мл этилацетата. Смесь трижды промывают 1 н. водного раствора гидроксида натрия. Этилацетатный слой сушат над безводным сульфатом натрия, и при пониженном давлении удаляют растворитель. Остаток очищают хроматографированием на колонке с силикагелем (200 г), используя в качестве элюента хлороформ, содержащий 1%-ный и 5%-ный метанол, получая 4,32 г целевого соединения.
1Н-ЯМР (СDCl3) δ: 1,00 и 1,16 (3Н, с), 1,40 (3Н, д, J = 7,2 Гц), 1,52 (9Н, с), 2,00-3,62 (5Н, м), 3,85-4,10 (1Н, м), 4,90 (1Н, ш.с.), 7,38 (5Н, с).
Вспомогательный пример 11.
4-трет-Бутоксикарбониламино-3,3-диметилпирролидин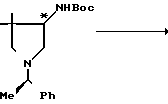
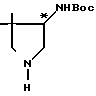
В 90 мл этанола растворяют 4,32 г 4-трет-бутоксикарбониламино-3,3-диметил-1-[1-(R)-фенилэтил]-пирролидина, к полученному раствору добавляют 4 г 10% палладия на угле и ведут реакцию восстановления при давлении водорода 4 атм, осуществляя нагрев облучением с помощью вольфрамовой лампы. Спустя примерно 7 ч катализатор удаляют фильтрованием и при пониженном давлении отгоняют из фильтрата растворитель. К остатку добавляют 200 мл этилацетата, после чего дважды промывают 10%-ным водным раствором лимонной кислоты. Содержащий раствор лимонной кислоты водный слой делают щелочным (рН около 10) добавлением водного раствора гидроокиси натрия, после чего экстрагируют 200 мл хлороформа. Экстракт сушат над безводным сульфатом натрия, растворитель удаляют при пониженном давлении, и получают 2,5 г целевого соединения.
1Н-ЯМР (CDCl3) δ: 0,98 и 1,08 (3Н, с, каждый), 1,48 (9Н, с), 2,30-4,00 (6Н, м), 4,50 (1Н, ш.с.).
П р и м е р 1. 10-(4-Амино-3,3-диметил-1-пирролидинил)-9-фтор-2,3-дигидро-3-(S)-3-метил- 7-оксо-7Н-пиридо/1,2,3-de//1,4/-бензоксазин-6-карбоновая кислота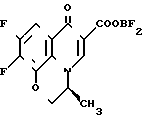
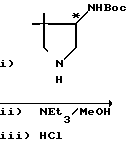
В 3 мл сухого диметилсульфоксида суспендируют 493,5 мг дифторборхелата 9,10-дифтор-2,3-дигидро-3-(S)-3-метил-7-оксо-7Н-пиридо/1,2,3-de//1,4/ безоксазин-6-карбоновой кислоты. К полученной суспензии добавляют 800 мг 4-трет-бутоксикарбониламино-3,3-диметилпирролидина, полученного во вспомогательном примере 11, и 0,5 мл триэтиламина, добавление проводят при комнатной температуре, после чего перемешивают полученную смесь в течение 30 мин. К реакционной смеси при охлаждении льдом добавляют воду, выпавшие кристаллы отфильтровывают и промывают водой. Кристаллы растворяют в 30 мл 90% -ного метанола, добавляют к полученному раствору 4 мл триэтиламина, после чего кипятят с обратным холодильником около 6 ч. При пониженном давлении удаляют из реакционной смеси растворитель. К остатку добавляют 5 мл концентрированной соляной кислоты, после чего перемешивают смесь при комнатной температуре в течение примерно 1 часа. Реакционную смесь промывают хлороформом, рН водного слоя доводят до 7 добавлением 50%-ного и 1 н. водных растворов гидроксида натрия, после чего экстрагируют 100 мл хлороформа. Слой хлороформа сушат над безводным сульфатом натрия и при пониженном давлении удаляют растворитель, получая около 500 мл неочищенных кристаллов целевого соединения. Неочищенный продукт перекристаллизовывают из этанола, содержащего небольшое количество водного аммиака, и одновременно осуществляют обработку активированным углем, получая 337 мг кристаллов.
Температура плавления: 263-268оС (разл.)
[α]D + 147,6o (c = 1,085; 1 н. NaOH).
Вычислено, %: С 60,79; Н 5,91; N 11,19.
C19H22N3O4F.
Найдено, %: C 60,59; H 5,84; N 10,99.
Вспомогательный пример 12.
Этил-2-(2,3,4,5-тетрафтор-6-метилбенз- оил)-3-[2-гидрокси-1-(S)-метиламино] акрилат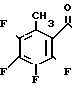
 ________→
________→ 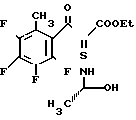
Смесь 8,00 г этил-2-(2,3,4,5-тетрафтор-6-метилбензоил)-акрилата, 7,2 мл триэтилортоформиата и 20 мл уксусного ангидрида нагревают до 120оС при перемешивании в течение 2 часов. После охлаждения смесь концентрируют досуха, получая желтоватый маслянистый осадок. Маслянистый осадок растворяют в 15 мл этанола. Раствор охлаждают на ледяной бане и добавляют к нему раствор 2,60 г 2-(S)-аминопропанола в 15 мл этанола. Смесь перемешивают при комнатной температуре в течение 1,5 ч. Затем смесь концентрируют досуха с получением 10,3 г маслянистого желтоватого целевого соединения. Данное соединение используют в следующих примерах без какой-либо очистки.
Вспомогательный пример 13.
Этил 9,10-дифтор-2,3-дигидро-3-(S)-метил-8-метил-7-оксо-7Н-пиридо/1,2,3-de/ /1,4/бензоксазин-6-карбоксилат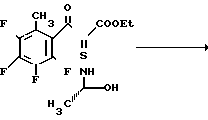
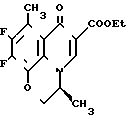
Cмесь из 7,9 г этил 2-(2,3,4,5-тетрафтор-6-метилбензоил)-3-[2-гидрокси- 1-(S)-метилэтиламино] акрилата, полученного в предыдущем примере, 50 мл безводного диметилсульфоксида и 8,0 г фторида калия нагревают при перемешивании в течение 4 ч. После охлаждения смесь концентрируют досуха. К остатку добавляют 100 мл воды и смесь экстрагируют из трех порций дихлорметана по 80 мл. Экстракт сушат над безводным сульфатом натрия, растворитель удаляют при пониженном давлении. Остаточную смесь очищают колоночной хроматографией на силикагеле (элюент, этилацетат, затем смесь хлороформа и метанола (20:1 по объему) получая 4,7 г целевого соединения в виде слегка желтоватых бесцветных кристаллов; т.пл. 205-207,5оС (разлож.).
Вспомогательный пример 14.
9,10-Дифтор-2,3-дигидро-3-(S)-метил-7-оксо-7Н-пиридо/1,2,3-de//1,4/ бензоксазин-6-карбоновая кислота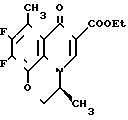 ________→
________→ 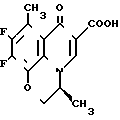
Смесь 3,50 г этил 9,10-дифтор-2,3-дигидро-3-(S)-метил-8-метил-7-оксо-7Н-пири-до/1,2,3,4-de/ /1,4/-бензоксазин-6-карбоксилата, 13 мл уксусной кислоты, 1,1 мл концентрированной серной кислоты и 10 мл воды нагревают с обратным холодильником в течение 3,5 часов. После охлаждения смесь выливают в 50 мл смеси льда с водой. Полученное кристаллическое вещество собирают с помощью фильтрования и промывают водой и этанолом. Сырые кристаллы перекристаллизовывают из этанола с получением 2,61 г целевого соединения в виде бесцветных кристаллов; т.пл. 253-255оС (разлож.).
П р и м е р 2. 10/4-(S)-амино-3,3-диметил-1-пирролидинил/-9-фтор-3-(S)-метил-8-метил-2,3- дигидро-7-оксо-7Н-пиридо /1,2,3-de/ /1,4/бензоксазин-6-карбоновая кислота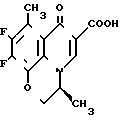 ________→
________→ 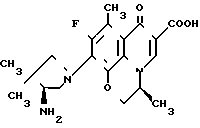
Смесь 300 мг 9,10-дифтор-2,3-дигидро-2,3-(S)-метил-8-метил-7-оксо-7Н-пиридо /1,2,3-de/ /1,4/-бензоксазин-6-карбоновой кислоты, 437 мг 4-(S)-трет-бутоксикарболамино-3,3-диметилпирролидина и 7,5 мл диметилсульфоксида нагревают при 90оС в течение 1 ч. После охлаждения смесь концентрируют досуха и к остатку добавляют воду. Полученные кристаллы собирают фильтрованием и промывают водой, а затем сушат. Кристаллы добавляют к 7,5 мл трифторуксусной кислоты, охлажденной до 0оС, и смесь перемешивают при комнатной температуре в течение 30 мин.
Смесь концентрируют досуха, а остаток растворяют в 1 н. NaOH. Нерастворившееся вещество удаляют фильтрованием, фильтрат нейтрализуют 1 н. соляной кислотой. Смесь экстрагируют из трех 50 мл порций хлороформа, экстракт сушат над безводным сульфатом натрия. Растворитель удаляют при пониженном давлении. Полученное сырое кристаллическое вещество перекристаллизовывают из смеси этанола и 28%-ного водного аммиака. Кристаллическое вещество собирают и промывают этанолом, затем сушат при пониженном давлении при 60оС в течение ночи с получением 121 мг целевого соединения в виде желтоватого порошка; т.пл. 199-201оС (разлож.).
[α]D19 - 141,77o (c = 0,808; 1 н. NaOH)
Вычислено, %: C 59,62; H 6,38; N 10,43.
C20H24FN3O4 3/4H2O
Найдено, %: C 59,68; H 6,18; N 10,11.
Ниже приводятся данные токсичности и активности заявленных соединений.
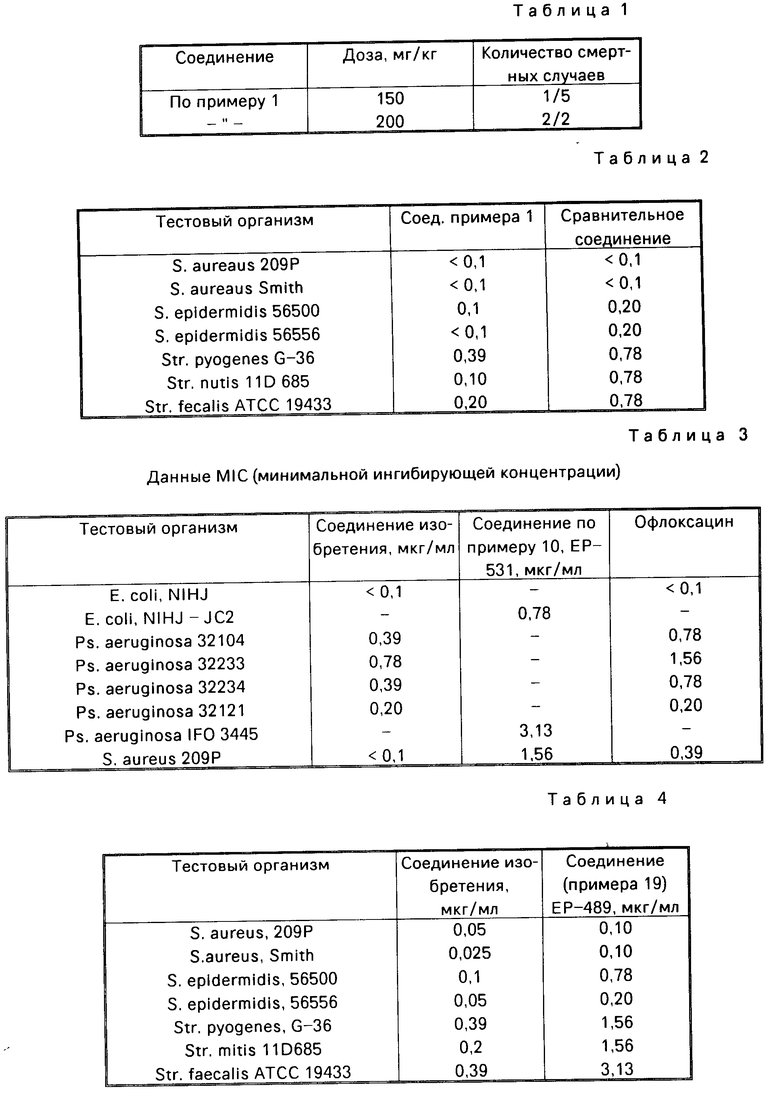
Острая токсичность
Соединение примера 1 растворяют в растворе 0,1 н. NaOH. Полученный раствор вводят внутривенно в хвостовую вену мыши в дозе 10 мг/кг со скоростью 0,1 мл/30 c. Каждой мыши вводят одну дозу. После этого в течение 14 дней, включая день инъекции, регистрируют количество смертей и симптомы у подопытных мышей.
Результаты представлены в табл. 1.
Липофильные свойства
Коэффициент распределения и концентрацию в крови определяли соответственно следующими способами:
(1) Коэффициент распределения
Измерения проводили с использованием хлороформа и фосфатного буфера (рН = 7,4). Соединение изобретения растворяли в хлороформе, а затем смешивали с эквивалентным объемом фосфатного буфера, который насыщали хлороформом, после чего раствор встряхивали. После разделения полученной смеси на две части измеряли концентрацию соединения в фосфатном буфере для определения коэффициента распределения; он составил 10,67.
(2) Концентрация в крови
Соединение настоящего изобретения вводили подопытным крысам орально в дозе 20 мг/кг. Пробы крови отбирали через 15, 30, 60, 120, 180, 240 и 360 мин после введения, после чего измеряли концентрацию сыворотки в каждой пробе. Максимальные уровни сыворотки (С макс.) достигались через 30 мин после введения, и они составили 3,0 мкг/мл.
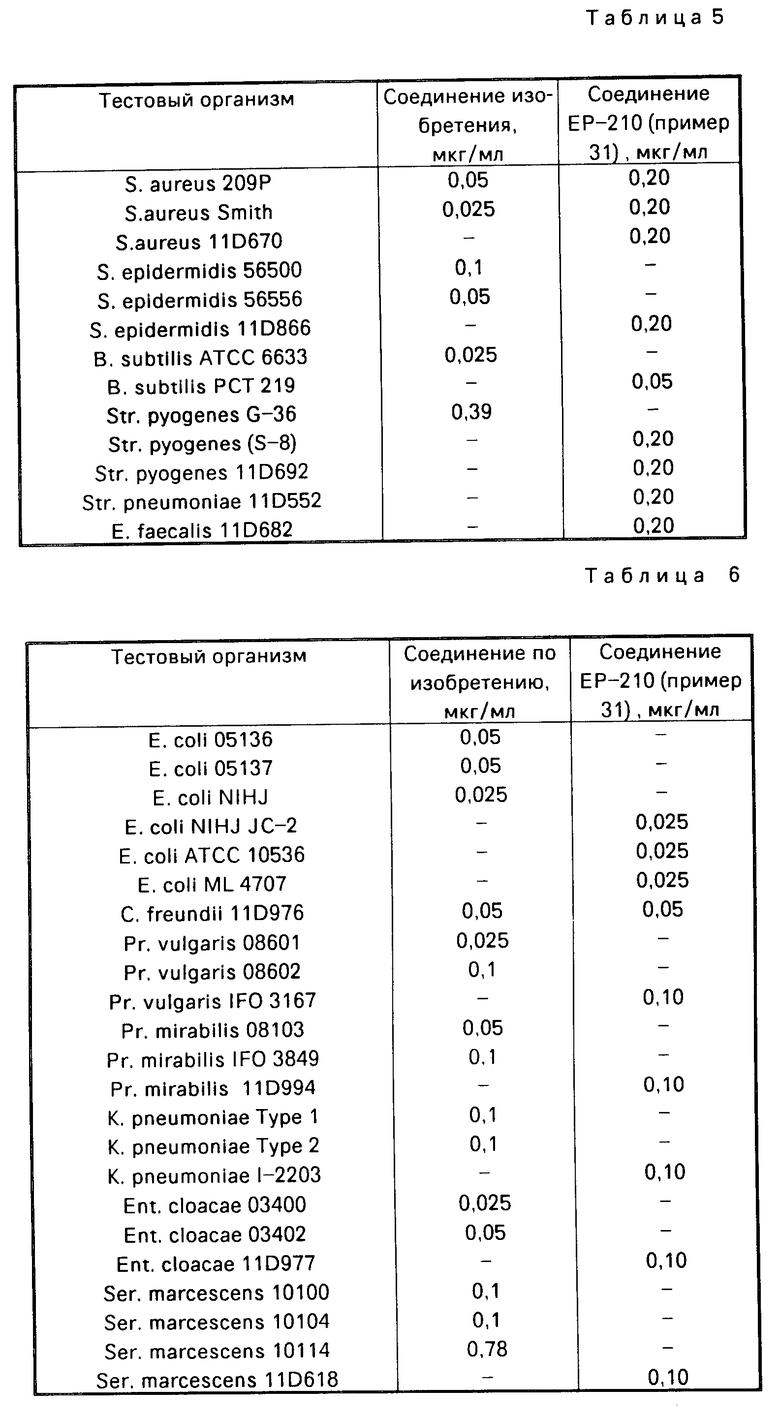
Противомикробная активность
Противомикробную активность соединения примера 1 и сравнительного соединения - 10-(3-амино-1-пирролидинил)-9- фтор-2,3,дигидро-3-(S)-3-метил-7-оксо-7Н-пиридо /1,2,3-de/ /1,4/-бензоксазин-6-карбоновой кислоты в отношении различных тестовых организмов, перечисленных ниже, определяли в соответствии с методом, установленным Японским обществом Химотерапии (Chemotherapy, 29 (1), 76 (1981). В табл. 2 приведены полученные в этих тестах минимальные ингибирующие концентрации (в мкг/мл). Из табл. 2 видно, что предлагаемое соединение проявляет неожиданно высокую противомикробную активность, превосходящую активность сравнительного соединения против S. epidermidis 56500, S. epidermidis 56556 и Str. pyogenes G-36 в 2 раза выше, против Str. fecalis, ATCC 19433 - в 4 раза и против Str. mitis, IID-685 в 8 раз. Поэтому, следует признать, что соединение по изобретению проявляет неожиданно высокую противомикробную активность, которой не обладают обычные соединение.
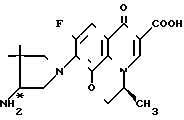
Соединения по ЕР N 0373531 (далее ЕР-531) являются соединениями имеющими пиридо/3,2,1-ij/ /3,1/-бензоксазиновое кольцо в качестве основного скелета, которое отличается от офлоксацина (9-фтор-3-метил-10-(4-метил-1-пиперазинид)-7-оксо- 2,3-дигидро-7Н-пиридо- /1,2,3-de/ /1,4/-бензоксазин-6-карбоновая кислота) положением эфирного атома кислорода в бензоксазиновом кольце. С другой стороны, соединение по настоящему изобретению, как и офлоксацин, имеет тот же основной скелет - пиридо/1,2,3-de/ /1,4/-бензоксазиновое кольцо. Следовательно, соединение настоящего изобретения структурно отличается от соединения ЕР-531 основным скелетом.
Соединение примера 10 Офлоксацин
ЕР-531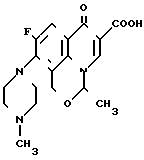
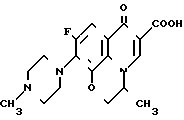
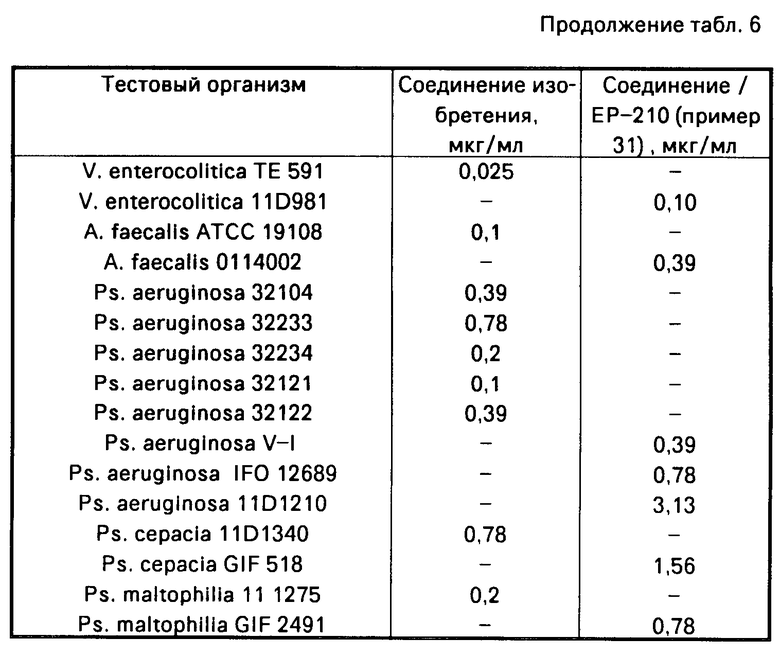
Противомикробная активность соединения примера 10 ЕР-531, офлоксацина и соединения настоящего изобретения показана в табл. 3.
Среди соединений, описанных в ЕР-531, которые могут рассматриваться как обладающие противомикробной активностью и химическая структура которых может быть конкретизирована, находится только соединение примера 10.
Как видно из результатов, приведенных в табл. 3, противомикробная активность соединения примера 10 ЕР-531 хуже, чем противомикробная активность офлоксацина. Это означает, что производное пиридо /1,2,3-de/ /1,4/-бензоксазина превосходит по активности производное пиридо /3,2,1-ij/ /3,1/-бензоксазина ЕР-531. Далее, из табл. 3 видно, что соединение настоящего изобретения, имеющее тот же основной скелет, что и офлоксацин, проявляет более высокую противомикробную активность, чем офлоксацин. Следует признать также, что противомикробная активность заявленного соединения является выражением превосходного эффекта, который нельзя ожидать от соединения ЕР-531.
Противомикробная активность соединения по ЕР - N 106489 (далее ЕР-489) определялась в соответствии с методом, описанным в Antimicr. Agents and Chemoth. 6, 124 (1974), в то время, как противомикробная активность соединения по изобретению определялась в соответствии с методом, рекомендованным Японским обществом Химиотерапии. Таким образом, для определения противомикробной активности соединения по настоящему изобретению и соединения по ЕР-489 использовали различные методы.
Чтобы провести надлежащее сравнение противомикробной активности соединения изобретения и соединения ЕР-489, заявитель синтезировал следующее соединение примера 19 ЕР-489:
CH
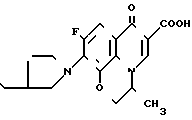 а затем измерил противомикробную активность полученного соединения, использовав метод, описанный в примере испытания настоящей заявки.
а затем измерил противомикробную активность полученного соединения, использовав метод, описанный в примере испытания настоящей заявки.
Результаты приведены в табл. 4, в сравнении с данными по противомикробной активности соединения изобретения.
Из результатов табл. 4 видно, что соединения по изобретению проявляют сильную противомикробную активность и широкий спектр действия против грамотрицательных и грамположительных бактерий. Антимикробная активность соединений по изобретению против грамотрицательных бактерий более, чем в 8 раз, превышают активность соединения ЕР-489, а активность против грамположительных бактерий превышает активность соединения ЕР-489 в 2-8 раз.
Таким образом, соединение настоящего изобретения обладает очень высокой противомикробной активностью, которую нельзя ожидать от соединения, описанного в ЕР-489.
Данные по противомикробной активности соединения по примеру 31, описанному в ЕР N 208210 (далее ЕР-210)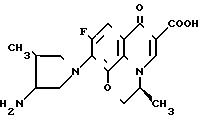 и соединения по настоящему изобретению формулы:
и соединения по настоящему изобретению формулы: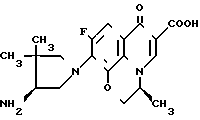 приведены в табл. 5 и 6 ниже. (В тех случаях, когда того же штамма не было, сравнение проводили с использованием одного и того же вида).
приведены в табл. 5 и 6 ниже. (В тех случаях, когда того же штамма не было, сравнение проводили с использованием одного и того же вида).
Определение противомикробной активности соединения ЕР-210 проводили так же, как описано в примере испытаний настоящего описания.
Как описано в табл. 5 и 6, противомикробная активность соединения настоящего изобретения находится почти на том же уровне, что и активность соединения ЕР-210, но активность настоящего соединения против грамположительных бактерий в 4-8 раз выше, чем активность соединения ЕР-210.
Таким образом, соединение настоящего изобретения значительно превосходит по противомикробной активности соединение ЕР-210. Активность такого высокого уровня нельзя ожидать от соединений, описанных в ЕР-210.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПИРОСОЕДИНЕНИЕ ИЛИ ЕГО СОЛИ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ, ОБЛАДАЮЩАЯ ПРОТИВОМИКРОБНОЙ АКТИВНОСТЬЮ | 1989 |
|
RU2094432C1 |
| ПРОИЗВОДНЫЕ ПИРИДОНКАРБОНОВОЙ КИСЛОТЫ И КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1993 |
|
RU2100351C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО 8-ХЛОРХИНОЛОНА | 1991 |
|
RU2049778C1 |
| ПРОИЗВОДНЫЕ АМИНОМЕТИЛПИРРОЛИДИНА, ИМЕЮЩИЕ АРОМАТИЧЕСКИЕ ЗАМЕСТИТЕЛИ | 2000 |
|
RU2255938C2 |
| БИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ 1,4-ДИГИДРО-4-ОКСОХИНОЛИН-3-КАРБОНОВОЙ КИСЛОТЫ ИЛИ ИХ СОЛИ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ АНТИМИКРОБНОЙ АКТИВНОСТЬЮ | 1993 |
|
RU2125046C1 |
| ПРОИЗВОДНЫЕ ПИРИМИДИНИЛПИРАЗОЛА И ПРОТИВООПУХОЛЕВОЕ СРЕДСТВО | 1995 |
|
RU2146675C1 |
| ЦИКЛОАЛКИЛЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ АМИНОМЕТИЛПИРРОЛИДИНА И АНТИБАКТЕРИАЛЬНЫЙ АГЕНТ НА ИХ ОСНОВЕ | 1999 |
|
RU2248970C2 |
| ПРОИЗВОДНЫЕ ДИАЗАБИЦИКЛОАЛКЕНОВ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1993 |
|
RU2126008C1 |
| ГЕКСАЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ | 1992 |
|
RU2071476C1 |
| СРЕДСТВО ПРОТИВ КИСЛОТОУСТОЙЧИВЫХ БАКТЕРИЙ, СОДЕРЖАЩЕЕ ПИРИДОНКАРБОНОВЫЕ КИСЛОТЫ В КАЧЕСТВЕ АКТИВНОГО КОМПОНЕНТА | 2001 |
|
RU2299205C2 |
Использование: в медицине, в частности в качестве антимикробных веществ. Сущность изобретения: продукт - оптически активные производные пиридобензоксазина формулы 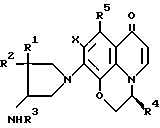 , где R1 и R2 - одинаковые или разные, C1-C4-алкил; R3 - водород; R4 - метил с β -конфигурацией; R5 - водород или метил; X - фтор или хлор, или их соли. 3 з.п. ф-лы, 6 табл.
, где R1 и R2 - одинаковые или разные, C1-C4-алкил; R3 - водород; R4 - метил с β -конфигурацией; R5 - водород или метил; X - фтор или хлор, или их соли. 3 з.п. ф-лы, 6 табл.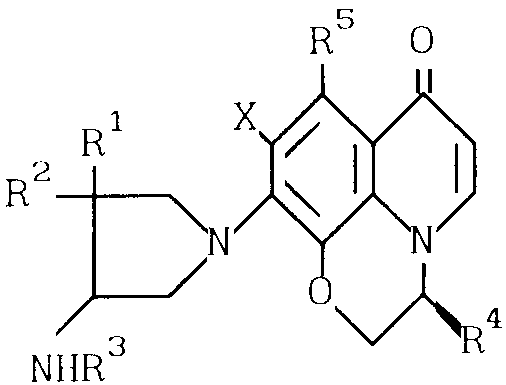
ОПТИЧЕСКИ АКТИВНЫЕ ПРОИЗВОДНЫЕ ПИРИДОБЕНЗОКСАЗИНА ИЛИ ИХ СОЛИ.
| СПОСОБ НАНЕСЕНИЯ ПОЛИМЕРНОГО ЗАЩИТНОГО ПОКРЫТИЯ НА ВНУТРЕННЮЮ ПОВЕРХНОСТЬ ТРУБОПРОВОДА | 1993 |
|
RU2028210C1 |
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |

