Изобретение относится к усовершенствованному способу получения соединений 23-(С1-С6 алкилоксимов)-LL-F28249. Обозначение LL -F28249 используют для описания ряда соединений, продуцируемых ферментативным бульоном подвида noncyanoyenus вида Streptomyces cyane- ogrisens, депонированного в коллекции NRRL под по номером 15773.
Целью данного изобретения является создание способа получения соединений 23-(С1-С6 алкилоксим)-LL-F28249, и более конкретно - 23-метилоксим)-LL-F 28249 α (моксидектина) сильнодействующего эндектоцидного агента.
Данное изобретение относится к способу получения соединений 23-(С1-С6 алкилоксим)-LL-F 28249, включающему защиту гидроксильной группы в положении 5 соединений LL-F 28249 n-нитробензоилхлоридом с образованием соот- ветствующего соединения 5-0-(n-нитробензоил)-LL-F 28249, окисление указанного соединения с образованием прои- зводного 5-0-(n-нитробензоил)-23-оксо-LL-F 28249 в кристаллическом состоянии; реакцию указанного производства с С1-С6 алкоксиламином или его солью с образованием интермедиата 23-(С1-С6 алкилоксим) - 5-0-(n-нитробензоил)-LL-F 28249 в кристаллическом состоянии; и снятие защиты с указанного интермедиата в присутствии основания с образованием целевого соединения 23-(С1-С6 алкилоксим)-LL-F 28249. Возможно снятие защиты с кристаллических производных 5-0-(n-нитробензоил) - 23-оксо-LL-F 28249 в присутствии основания с образованием соответствующих соединений 23-оксо-LL-F 28249, и реакция указанных соединений с С1-С6алкоксиламином или его солью с образованием целевого соединения 23-(С1-С6 алкилоксим)-LL-F 28249. Известен способ получения соединения 23-(С1-С6 алкилоксимов)-LL-F 28249 [1] , их использование в качестве противогельминтных, инсектицидных, противонематодных, противоэктопаразитарных и акарицидных агентов описаны в [2].
Данное изобретение относится к упрощенному за счет более простой очистки и выделения промежуточного соединения, способу получения 23-(С1-С6 алкоксимных) производных соединений LL-F 28249. Соединения LL-F 28249 изображаются нижеследующей общей структурной формулой: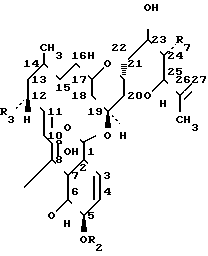

Компонент R1 R2 R3 R7
LL-F 28249α CH(CH3)2 H CH3 CH3
LL-D 28249β CH3 H CH3 CH3
LL-F 28249C CH3 CH3 CH3 CH3
LL-F 28249е CH(CH3)2 H H CH3
LL-F 28249f CH2CH3 H CH3 CH3
LL-D 28249h CH(CH3)2 H CH3 CH2CH3
LL-F 28249i CH(CH3)2 H CH2CH3 CH3
LL-F 28249k CH(CH3)2 CH3 CH3 CH3
Далее, предлагается способ получения соединений 23-(С1-С6алкилоксим)-LL-F 28249, в котором с кристаллических производных 5-0-(n-нитробензоил) - 23-оксо-LL-F 28249 в присутствии основания снимают защиту, получая соответствующие соединения 23-оксо LL-F 28249, и приводят реакцию указанных соединений с С1-С6 алкоксиламином или его солью с получением целевого соединения 23-(С1-С6 алкилоксим) LL-F 28249.
При использовании в качестве исходного соединения LL-F 28249 и солянокислого метоксиламина в качестве С1-С6 алкоксиламина предлагаемый способ можно проиллюстрировать диаграммой, в которой ПНБ обозначает функциональную группу - n-нитробензоил (см. чертеж).
Защита гидроксильной группы в положении 5 соединения LL-F 28249 α осуществляется реакцией между LL-F 28249 α и n-нитробензоилхлоридом в присутствии органического растворителя, такого как, например, толуол, хлористый метилен, этил- ацетат, ацетонитрил и им подобные, предпочтительно в присутствии толуола, и органического основания, такого, как, например пиридин, триэтиламин, N-метилпирролидон, и им подобные, предпочтительно в присутствии триэтиламина.
Неожиданно было найдено, что окисление соединений 5-0-(n-нитробензоил) LL-F 28249 дает кристаллический продукт - соответствующее соединение 5-0-(n-нитробензоил)-23-оксо-LL-F 28249. Кристаллическое состояние интермедиата позволяет осуществлять его простую и эффективную очистку путем перекристаллизации их подходящего органического растворителя и устранить сложную, длительную процедуру очистки с помощью таких методов, как колоночная хроматография. Окисление соединения 5-0-(n-нитробензоил)-LL-F 28249 успешно осуществляется с использованием окисля- ющей системы, выбранной из группы, состоящей из пиридиндихромата и уксусного ангидрида; пиридиндихромата и диметилформамида; трет-бутоксиалюминия и ортобензохинона; пятиокиси фосфора и диметилсульфоксида; дициклогексилкарбодиимида и диметилсульфоксида; двуокиси марганца; и уксусного ангидрида и диметилсульфоксида.
Предпочтительной окисляющей системой для окисления соединений 5-0-(n-нитробензоил)-LL-F 28249 является двуокись марганца в присутствии растворителя, такого как, например, хлористый метилен, ацетонитрил, этилацетат или им подобные, пред- почтительно - в присутствии этилацетата.
Более предпочтительной окисляющей системой для окисления соединений 5-0-(n-нитробензоил)-LL-F 28249 является уксусный ангидрид и диметилсульфоксид в присутствии пиридина и кислоты, такой, как, например уксусная кислота, трифторуксусная кислота, дихлоруксусная кислота, монохлоруксусная кислота и им подобные, предпочтительно - монохлоруксусная кислота. Неожиданно было найдено, что использование уксусного ангидрида и диметилсульфоксида в присутствии пиридина и кислоты сильно увеличивает выход производных 5-0-(n-нитробензоил)-23-оксо- LL-F 28249 по сравнению с использованием только уксусного ангидрида и диметилсульфоксида. Например, если окисление уксусным ангидридом и диметилсульфоксидом проводят в присутствии пиридина и монохлоруксусной кислоты, то выход 5-0-(n-нитробензоил)-23-оксо-LL-F 28249 α составляет 76%; если то же окисление проводят в отсутствие пиридина и монохлоруксусной кислоты, выход составляет 2-4%.
Кристаллическое производное 5-0-(n-нитробензоил)-23-оксо-LL-F 28249 можно очистить перекристаллизацией из подходящего растворителя (предпочтительно из н-пропанола) перед снятием защиты (удалением n-нитробензоильной группы) или реакцией с солянокислым С1-С6алкоксиламином.
Предпочтительно проводят реакцию раствора неочищенного продукта реакции - 5-0-(n-нитробензоил)-23-оксо-LL-F 28249 - в органическом растворителе, таком как толуол, с водным раствором солянокислого С1-С6алкоксиламина и ацетата натрия при перемешивании до тех пор, пока не завершится образование оксима. Полученный таким образом интермедиат 23-(С1-С6 алкилоксим)-5-0-(n-нитробензоил) - 28249 выделяют и очищают перекристаллизацией из подходящего растворителя, предпочтительно из н-бутанола.
В перекристаллизованном соединении 23-(С1-С6алкилоксим)-5-0-(n-нитробензоил)-LL-F 28249 снимают защитную группу реакцией с гидроокисью натрия при 0оС-25оС, получая целевой продукт - 23 - (С1-С6 алкилоксим)-LL-F 28249. Снятие защиты осуществляют реакцией между раствором соединения 23-(С1-С6 алкилоксим) - 5-0-(n-нитробензоил)-LL-F 28249 в органическом растворителе, таком как толуол, диоксан, н-бутанол и т.п., предпочтительно в диоксане, с водным раствором гидроокиси натрия при 0оС - 25оС и выделением полученного соединения 23-(С1-С6 алкилоксим) LL-F 28249 из органической фазы с использованием обычных методик, таких как концентрирование и фильтрование или удаление растворителя.
Таким образом, принципиальное отличие настоящего изобретения заключается в следующем: а) использование новой n-нитробензоильной группы для получения кристаллического производного 5-(n-нитробен- зоил)-23-кетона или 5-(n-нитробензоил) - 23-метоксима, каждый из которых может быть перекристаллизован, и в) окисление 5-замещенного производного LL-F 28249 α уксусным ангидридом, ДМСО, пиридином и кислотой с образованием 5-замещенного 23-кетона; n-нитробензоильная группа является не только хорошей защитной группой, но также позволяет провести простую очистку промежуточных соединений кристаллизацией, а не дорогостоящую очистку колоночной хроматографией. Новизна окисления уксусным ангидридом - ДМСО заключается в тех специфических условиях реакции, которые необходимы в данном случае для того, чтобы произошло окисление. Экспериментальные условия, приводимые ранее в химической литературе для уксусного ангидрида - ДМСО, не дают продукта.
Уникальность 5-(n-нитробензоил) производного обнаруживается при рассмотрении работы Sutherland и др. [3]; [1]. В примере 1, а окисление 5-кето- производного дихромата натрия приводит к 5,23-дикето-фактору А - промежуточному соедине- нию, охарактеризованному в виде смолы. Даже после очистки смолы колоночной хроматографией целевой продукт выделен в виде пены. Кроме того, необходимость хроматографической очистки ясно показана также и последующим промежуточным соединением в этой последовательности - 5-кето-23-метоксиимино-Фактора А, получен- ным метоксииминированием 5, 23-дикето-Фактора А (пример 1, в). Несмотря на применение хроматографической очистки промежуточное 5-кето-23-метоксииминопроизводное выделено в виде твердой пены. Другие примеры производных Фактора А, требующих очистки хроматографированием, приводятся в патенте Великобритании 2176182 в примерах 2-4, 6, 8, 11, 14, 18, 21, 22, 28, 30, 40, 41, 44, 45-52, 56, 57, 60, 69, 70, 76, 88, 103, 105, 109, 111, 112 и 123-127. То, что 5-(n-нитробензоил) - 23-кетон или 5-(n-нитробензоил)-23-метоксим, а не 5,23-дикето- и 5-кето-23 (Е)-метоксимопроизводное могут быть очищены простой кристаллизацией с получением чистых кристаллических продуктов, является положительным преимуществом предложенного способа перед способом [1]. 0-ацилирование n-нитробензоилхлоридом существенно отличается от примера, в котором n-нитробензойную кислоту используют вместе с трифенилфосфином-диэтилазодикарбоксилатом. В [4] 0-алкилирование 5-трет-бутилдиметилсилилокси-фактора А n-нитробензойной кислотой и трифенилфосфином происходит с обращением конфигурации 23-гидроксила, что не имеет места в случае использования в качестве ацилирующего средства n-нитробензоилхлорида. Важный вывод при этом заключается в том, что промежуточное соединение имеет n-нитробензоатную группу в 23-положении с обращенной конфигурацией и продукт охарактеризован в виде пены даже после двойной хроматографи- ческой очистки. Напротив 5-(n-нитробензоил)-23-кетон и 5-(n-нитробензоил)-23-метоксим, относящиеся к настоящей заявке, не имеют обращенной конфигурации в положении, содержащем n-нитробензоил, и оба являются кристаллическими продуктами.
Многие известные окислители дают неудовлетворительные результаты при окислении 5-замещенного F-28249 α. Даже приведенная таблица 11 показывает, что несколько окислителей, известных способностью с хорошими выходами превращать вторичные спирты в кетоны, не пригодны для данной системы. Хотя окислители на основе хрома до некоторой степени успешно окисляют 23-гидроксил, тем не менее, однако, часто комплексообразование ионов металлического хрома с конечным продуктом мешает его очистке и в результате приводит к снижению выходов и чистоты. Дихромат натрия и серная кислота, описанные для окисления 23-гидроксила 5-кето-фактора А в [4] вряд ли приемлемы для аналогичного превращения 5-0-алкилированного производного. В предложенном способе показано, что 0-замещение в 5-положении делает такое производное уязвимым к действию кислот. Длительное действие кислоты на 5-0-замещенное производное приводит к ароматизации низшего шестичлен- ного цикла в результате дегидратации с участием 7-гидроксила и 1,2-элиминирования при участии 5-n-нитротензоатной группы.
Условия окисления в присутствии уксусного ангидрида - ДМСО, приведенные в настоящем изобретении, также являются уникальными. Хорошо известная экспериментальная система уксусной ангидрид - ДМСО [5]. Дает менее 2% 23-кетона из 5-(n-нитробензоил)-F-28249 α.
П р и м е р 1. Получение 5-0-(n-нитробензоил)-LL-28249 α.
Перемешиваемый раствор LL-F 28249 α (6,36 г, 10,4 ммоля) в хлористом метилене обрабатывают пиридином (1,98 г, 25,0 ммоля) и n-нитробензоилхлоридом (2,45 г, 13,2 ммоля) при 20о-25оС. Через 4 ч реакционную смесь при 20о - 25оС обрабатывают насыщенным раствором бикарбоната натрия и хлористым метиленом и перемешивают до полного растворения. Фазы разделяют, органическую фазу промывают последовательно насыщенным раствором бикарбоната натрия, 5% -ной соляной кислотой и насыщенным раствором хлористого натрия, и концентрируют в вакууме, получая целевое соединение в виде твердой пены в количестве 7,9 г (количественный выход); идентифицируют жидкостной хроматографией, Н1ЯМР-анализом и масс-спектроскопическим анализом.
П р и м е р 2. Получение 5-0-(n-нитробензоил)-LL-F 28249 α.
Перемешиваемый раствор LL-28249 α (6,13 г, 10,0 ммолей) в толуоле обрабатывают триэтиламином (2,53 г, 25 ммолей), охлаждают до 15оС, обрабатывают несколькими порциями n-нитробензоилхлорида (2,60 г, 14 ммолей) при температуре от 15о до 22оС, и перемешивают в течение 6 ч при 20о-24оС. Реакционную смесь обрабатывают водой, перемешивают в течение 10 мин, и фильтруют. Фильтрат отделяют, органическую фазу промывают последовательно насыщенным раствором бикарбоната натрия, 3 соляной кислотой и водой, и концентрируют в вакууме, получая целевой продукт в виде твердой пены в количестве 7,55 г.
П р и м е р 3. Получение 5-0-(n-нитробензоил)-23-оксо LL-F 28249 α с использованием пиридиндихромата и диметилфор- мамида.
Перемешиваемый раствор 5-0-(n-нитробензоил) LL-F 28249α (3,12 г, 4,10 ммоля) в диметилформамиде обрабатывают пиридиндихроматом (18,8 г, 50 ммолей), взятым в виде одной порции, перемешивают при 20о-25оС в течение 6 ч и выливают в воду. Реакционную смесь перемешивают в течение 15 мин и фильтруют. Отфильтрованный осадок промывают водой, сушат на воздухе и переносят в этилацетат. Полученную смесь кипятят с обратным холодильником в течение 15 мин, обрабатывают диатомитом и фильтруют. Фильтрат концентрируют в вакууме с образованием красно-коричневого твердого вещества, которое перекристаллизовывают из н-пропанола, получая целевой продукт в виде кристаллов белого цвета в количестве 3,33 г (выход - 52% в расчете на LL-F 28249 α); темп. пл. 217-221оС, идентифицирован Н1ЯМР-анализом и масс-спектроскопическим анализом.
П р и м е р 4. Получение 5-0-(n-нитробензоил)-23-оксо-LL-F 28249 α с использованием пиридиндихромата и уксусного ангидрида.
Раствор 5-0-(n-нитробензоил) LL-F 28249 (0,38 г, 0,5 ммоля) в хлористом метилене добавляют к свежеприготовленной смеси пиридиндихромата (0,19 г, 0,5 ммоль) и уксусного ангидрида (0,3 г, 3,0 ммоля) при интенсивном перемешивании. Реакционную смесь перемешивают при комнатной температуре в течение 15 мин, кипятят с обратным холодильником в течение 6-8 ч, охлаждают до комнатной температуры и обрабатывают водой. После интенсивного перемешивания фазы разделяют и органическую фазу промывают насыщенным раствором бикарбоната натрия и концентрируют в вакууме, получая остаток. Этот остаток переносят в этилацетат и хроматографируют, используя силикагель и этилацетат в качестве элюента, получая бледное желто-серое вещество. Это твердое вещество перемешивают со смесью этилацетат : гексан (объемн. отношение 55:45), фильтруют, и фильтрат концентрируют в вакууме, получая целевое соединение и в виде бледно-желтого твердого вещества в количестве 0,26 г, которое идентифицируют Н1ЯМР - и ЖХВД - анализами.
П р и м е р 5. Получение 5-0-(n-нитробензоил)-23-оксо LL-F 28249 α с использованием трет-бутоксиалюминия и орто- бензохинона.
Перемешиваемую смесь 5-0-(n-нитробензоил) LL-F 28249 (0,38 г, 0,50 ммоля), трет-бутоксиалюминия (0,184 г, 0,75 ммоля и ортобензохинона (0,216 г, 2,0 ммоля) в толуоле кипятят с обратным холодильником в течение 2 ч, охлаждают до комнатной температуры, обрабатывают толуолом и разбавленной серной кислотой (16%), перемешивают 5 мин и фильтруют. Фильтрат отделяют и промывают органическую фазу водой и концентрируют в вакууме, получая стекловидный твердый остаток. Остаток переносят в этилацетат и фильтруют через нейтральную окись алюминия. Фильтрат концентрируют в вакууме, получая целевой продукт в виде твердого белого вещества в количестве 0,324 г (выход 71%, согласно данным ЖХВД - анализа).
П р и м е р 6. Получение 5-0-(n-нитробензоил)-23-оксо-LL-F 28249 α с использованием пятиокиси фосфора и диметил- сульфоксида.
Раствор 5-0-(n-нитробензоил)-LL-F 28249 (0,38 г, 0,50 ммоля) и диметилсульфоксида (0,75 г, 9,6 ммолей) в хлористом метилене обрабатывают порошкообразной пятиокисью фосфора (0,107 г, 0,75 ммоля), взятой в виде одной порции, перемешивают при 20о-25оС в течение 19 ч, обрабатывают добавляемым по каплям триэтиламином (0,30 г, 3,0 ммоля), перемешивают в течение 30 мин, затем обрабатывают хлористым метиленом и водой и перемешивают в течение 5 мин. Фазы разделяют, и органическую фазу промывают разбавленной соляной кислотой (7%) и концентрируют в вакууме, получая целевой продукт в виде белого твердого вещества в количестве 0,28 г (чистота 47%, по данным ЖХВД-анализа).
П р и м е р 7. Получение 5-0-(n-нитробензоил)-23-оксо-LL-F 28249 α с использованием двуокиси марганца и хлористого метилена в качестве растворителя.
Раствор 5-0-(n-нитробензоил)-LL-F 28249 α (0,19 г, 0,25 ммоля) в хлористом метилене обрабатывают двуокисью марганца (8,0 г, 92 ммоля), перемешивают при 20о - 25оС в течение 2 ч, обрабатывают затем хлористым метиленом, перемешивают в течение 5 мин и фильтруют. Фильтрат концентрируют в вакууме, получая целевое соединение в виде белого твердого вещества в количестве 0,08 г, чистота 51% по данным ЖХВД - анализа.
П р и м е р 8. Получение 5-0-(n-нитробензоил)-23-оксо-LL-F 28249 α с использованием двуокиси марганца и ацетонитрила в качестве растворителя.
Раствор 5-0-(n-нитробензоил)-LL-F 28249 α (0,19 г, 0,25 ммоля) в ацетонитриле обрабатывают двуокисью марганца (8,0 г, 92 ммоля), перемешивают в течение 3 часов при комнатной температуре, затем обрабатывают ацетонитрилом, перемешивают в течение 5 мин и фильтруют. Отфильтрованный осадок суспендируют в хлористом метилене и фильтруют. Ацетонитрильный и метиленхлоридный фильтраты совмещают, промывают водой и концентрируют в вакууме, получая целевое соединение в виде белого твердого вещества в количестве 0,14 г, чистота 69%, по данным ЖХВД - анализа.
П р и м е р 9. Получение 5-0-(n-нитробензоил)-23-оксо-LL-F 28249 α с использованием двуокиси марганца и этилацетата в качестве растворителя.
Раствор 5-0-(n-нитробензоил-LL-F 28249 α (1,0 г, 1,3 ммоля) в этилацетате обрабатывают двуокисью марганца (20,0 г, 230 ммолей), перемешивают при 20о - 25оС в течение 3 ч и фильтруют. Отфильтрованный осадок промывают этилацетатом. Фильтраты собирают вместе, обрабатывают двуокисью марганца (8,0 г, 92 ммоля), перемешивают при 20о - 25оС в течение 3 ч и фильтруют. Отфильтрованный осадок промывают этилацетатом; фильтраты собирают вместе и концентрируют в вакууме, получая целевой продукт в виде белого твердого вещества в количестве 0,8 г (чистота 70% по данным ЖХВД - анализа). Твердый продукт перекристаллизовывают из н-пропанола, получая белые кристаллы с температурой плавления 218 - 222оС.
П р и м е р 10. Получение 5-0-(n-нитробензоил)-23-оксо-LL-F 28249 α с использованием дициклогексилкарбодиимида и диметилсульфоксида.
Раствор 5-0-(n-нитробензоил)-LL-F 28249 α (0,33 г, 0,50 ммоля) в бензоле обрабатывают последовательно диметилсульфоксидом (0,78 г, 10 ммолей), пиридином (0,04 г, 0,5 ммоля), трифторуксусной кислотой (0,03 г, 0,25 ммоля) и дициклогексилкарбодиимидом (0,31 г, 1,5 ммоля), перемеши- вают при 20о-25оС в течение 21 ч, затем обрабатывают бензолом и фильтруют. Отфильтрованный осадок промывают бензолом. Собранные вместе фильтраты промывают водой и концентрируют в вакууме, получая целевое соединение в виде твердого вещества оранжево-коричневого цвета в количестве 0,34 г, выход 75% по данным ЖХВД - анализа.
П р и м е р 11. Получение 5-0-(n-нитробензоил)-23-оксо-LL-F 28249 α с использованием уксусного ангидрида и диметил- сульфоксида в присутствии пиридинтрифторацетата.
Смесь 5-0-(n-нитробензоил)-LL-F 28249 α (0,38 г, 0,5 ммоля), диметилсульфоксида (0,78 г, 10 ммолей) и пиридинтрифторацетата (0,97 г, 0,5 ммоля) в этилацетате обрабатывают добавляемым по каплям уксусным ангидридом (0,26 г, 2,5 ммоля), перемешивают в течение 24 ч при 20о - 25оС и обрабатывают этилацетатом и водой. Фазы разделяют: органическую фазу промывают водой и концентрируют в вакууме, получая остаток в виде вязкого масла. Остаток переносят в хлористый метилен и концентрируют в вакууме, получая целевой продукт в виде желтого твердого вещества в количестве 0,36 г; идентифицирован ЖХВД - анализом.
П р и м е р 12. Получение 5-0-(n-нитробензоил)-23-оксо-LL-F 28249 α с использованием уксусного ангидрида и диме- тилсульфоксида в присутствии пиридина и дихлоруксусной кислоты.
Смесь 5-0-(n-нитробензоил)-LL-F 28249α (7,26 г, 10 ммолей) и пиридина (31,6 г, 400 ммолей) обрабатывают диметилсульфоксидом (15,6 г, 200 ммолей) и дихлоруксусной кислоты (1,29 г, 10 ммолей), охлаждают до 2о-3оС, обрабатывают добавляемым по каплям уксусным ангидридом (5,1 г, 50 ммолей) при 3о-7оС и обрабатывают хлористым метиленом и водой. Реакционную смесь перемешивают при температуре окружающей среды в течение 15-30 мин, и разделяют фазы. Органическую фазу промывают холодной разбавленной соляной кислотой (5% ) и 5%-ным раствором хлористого натрия и концентрируют в вакууме, получая целевой продукт в виде твердой пены желтого цвета в количестве 7,47 г, чистота 73% по данным ЖХВД - анализа.
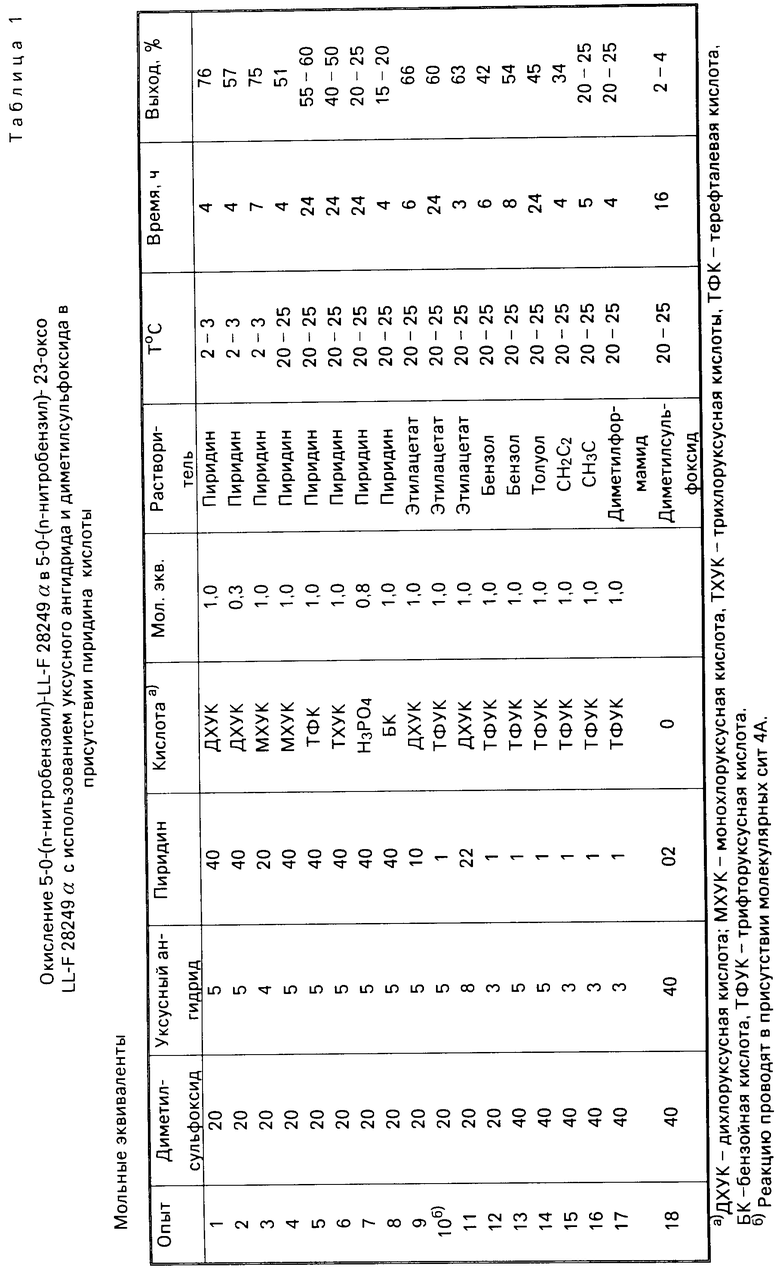
Используя по существу тот же способ, но меняя используемый кислотный реагент, получают следующие выходы продукта, приведенные в табл. 1.
П р и м е р 13. Получение 5-0-(n-нитробензоил)-23-оксо-LL-F 28249 α c использованием уксусного ангидрида и диметил- сульфоксида в присутствии пиридина и монохлоруксусной кислоты.
Смесь 5-0-(n-нитробензоил)-LL-F 28249 α (1,52 г, 2,0 ммоля) и пиридина (3,16 г, 40 ммолей) в толуоле обрабатывают диметилсульфоксидом (3,12 г, 40 ммолей) и монохлоруксусной кислотой (0,19 г, 2,0 ммоля), охлаждают до 2о-3оС, обрабатывают добавляемым по каплям уксусным ангидридом (0,82 г, 8,0 ммоля) при 3о-5оС, перемешивают при 2о-3оС в течение 7 ч и далее обрабатывают толуолом и водой. После перемешивания реакционной смеси при 15о-20оС в течение 10 мин фазы разделяют. Органическую фазу промывают последовательно холодной 2,4 N соляной кислотой и водой при 15-20оС и концентрируют в вакууме, получая целевой продукт в виде твердой пены желтого цвета в количестве 1,4 г, чистота 71% по данным ЖХВД-анализа.
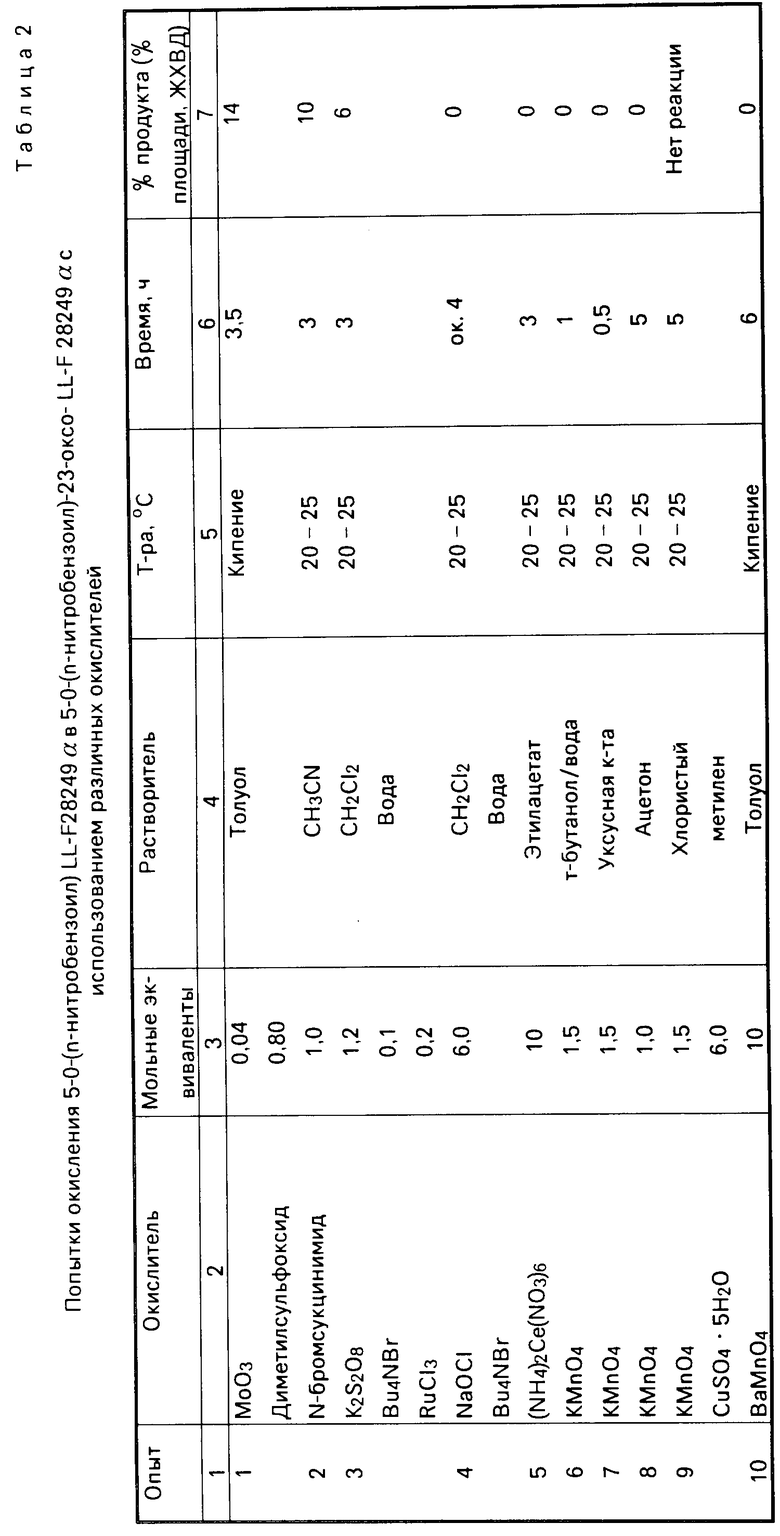
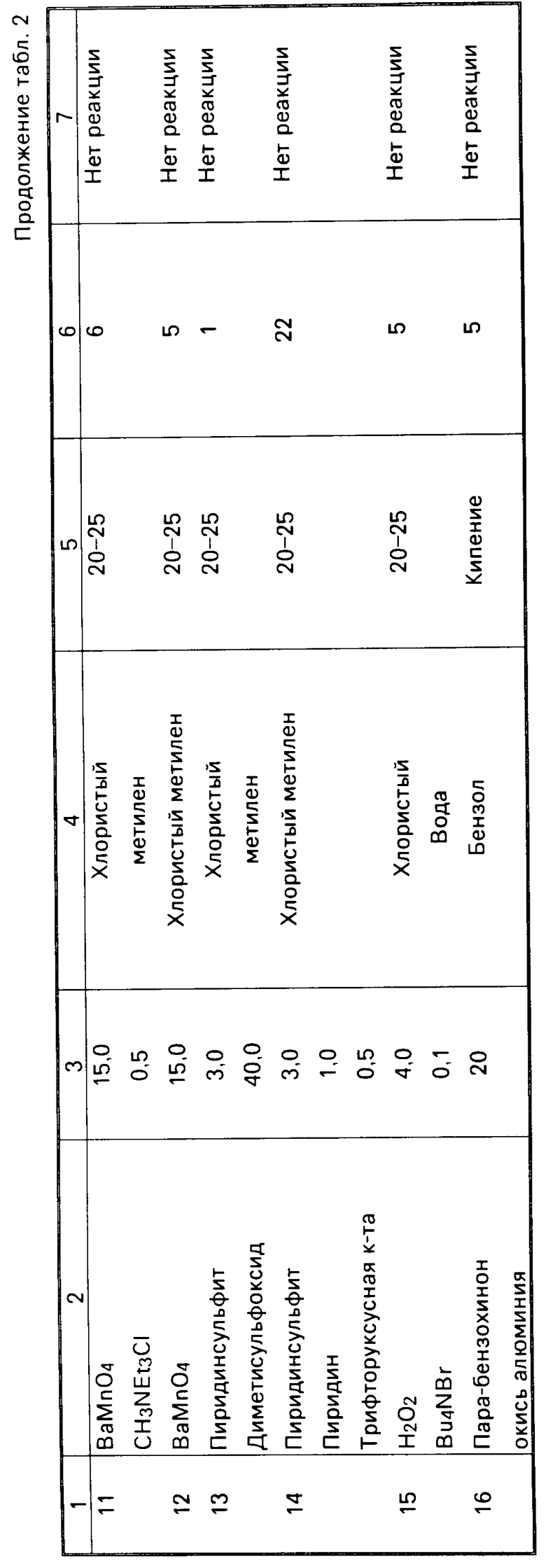
П р и м е р 14. Оценка окисления 5-0-(n-нитробензоил)-LL-F 28249 α с образованием 5-0-(n-нитробензоил)-23-оксо-LL-F 28249 α с использованием различных окислителей.
Была проведена оценка различных систем, окисляющих 5-0-(n-нитробензоил)-LL-F 28249 α в 5-0-(n-нитробензоил)-23-оксо-LL-F 28249 α. Реагенты, условия проведения реакции и процент полученного 5-0-(n-нитробензоил)-23-оксо-LL-F 28249 α определен- ного ЖХВД-анализом, приведены в табл. 2.
П р и м е р 15. Получение 5-0-(n-нитробензоил)-23-(метилоксим)-LL-F 28249 α.
Раствор 5-0-(n-нитробензоил)-23-оксо-LL-F 28249 α, (10,67 г, 14,0 ммолей) в н-бутаноле обрабатывают раствором солянокис- лого метоксиламина (2,34 г, 28,1 ммоля) и безводного ацетона натрия (2,30 г, 28,1 г ммоля) в воде при 20-22оС, перемешивают в течение 2 ч при 20о-22оС и фильтруют. Отфильтрованный осадок сушат на воздухе и перекристаллизовывают из н-бутанола (горячее фильтрование), получая целевое соединение в виде бесцветного твердого вещества в количестве 3,6 г, чистота 91% по данным ЖХВД.
П р и м е р 16. Получение 5-0-(n-нитробензоил)-23-(метилоксим)-LL-F 28249 α.
Раствор 5-0-(n-нитробензоил)-23-оксо-LL-F 28249 α (1,5 г, 2,0 ммоля) в толуоле обрабатывают раствором солянокислого метонсиламина (0,25 г, 3,0 ммоля) и безводного ацетата натрия (0,25 г, 3,0 ммоля) в воде и перемешивают при 20о-25оС в течение 10 ч. Толуольную фазу отделяют, промывают водой и концентрируют в вакууме, получая твердый остаток. Этот остаток перекристаллизовывают из н-бутанола, получая целевой продукт, 0,65 г, идентифицирован ЖХВД - анализом.
П р и м е р 17. Получение 23-(метилоксим)-LL-F 28249 α.
Раствор 5-0-(n-нитробензоил)-23-(метилоксим)-LL-F 28249 α (1,58 г, 2,0 ммоля) в диоксане обрабатывают добавляемым по каплям 4%-ным раствором гидроокиси натрия (3,0 г, 3,0 ммолей NaOH) при 8-12оС, перемешивают в течение 3 ч при 8-12оС, обрабатывают толуолом и водой, и перемешивают в течение 5 мин при температуре окружающей среды. Фазы разделяют, и органическую фазу промывают 10%-ным хлористым натрием и концентрируют в вакууме, получая целевое соединение в виде твердой пены белого цвета в количестве 1,15 г, чистота 89%, по данным ЖХВД-анализа.
П р и м е р 18. Получение 23-оксо-LL-F 28249 α.
Смесь 5-0-(n-нитробензоил)-23-оксо-LL-F 28249 α (1,52 г, 2,0 ммоля) и 4% -ной гидроокиси натрия (3,3 г, 3,3 ммоля NaOH) в диоксане перемешивают при 23оС в течение 2 ч, обрабатывают толуолом и водой и встряхивают. Фазы разделяют, и органическую фазу промывают водой и концентрируют в вакууме, получая целевое соединение в виде твердой пены в количестве 0,90 г, идентифицировано Н1ЯМР.
П р и м е р 19. Получение 23-(метилоксим)-LL-F 28249 α.
Смесь 23-оксо-LL-F 28249 α (0,90 г, 1,5 ммоля), солянокислого метоксиламина (0,42 г, 5,0 ммоля), безводного ацетата натрия (0,41 г, 5,0 ммолей), уксусной кислоты и диоксана перемешивают при 20о-25оС в течение 22 ч, обрабатывают толуолом и водой и перемешивают в течение 5 мин. Фазы разделяют, и органическую фазу промывают водой и концентрируют в вакууме, получая целевое соединение в виде твердой пены в количестве 0,84 г, чистота 71%, по данным ЖХВД-анализа.
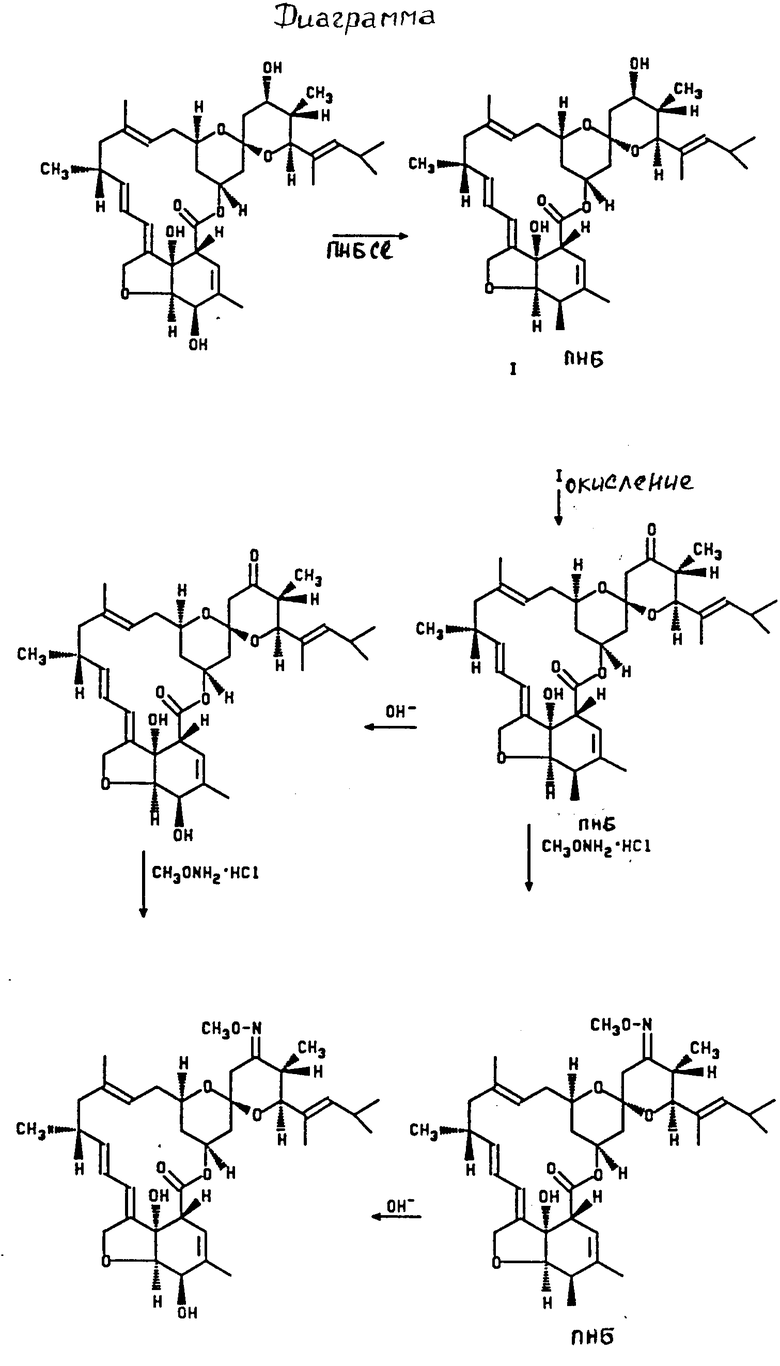
Использование: в медицине в качестве сильнодействующего эндектоцидного агента. Сущность изобретения: продукт-23-C1-C6 алкилоксимы) - L, L - F - 28249. Реагент 1: L, L - F - 28249. Реагент 2:(n-нитробензоилхлорид. Реагент 3: 5 - 0 - n-нитробензоил) - L, L - F - 28249. Реагент 4: уксусный ангидрид и диметилсульфаксид. Условия процесса окисления: в присутствии пиридина и кислоты и полученное производное 5 - о - (n-нитробензоил) - 23 - оксо - L, L - F - 28249 в кристаллическом состоянии подвергают в любом порядке оксилированию действием C1-C6-алкоксиамина или его соли и снятию защиты. 2 табл.
СПОСОБ ПОЛУЧЕНИЯ 23-(C1- C6-АЛКИЛОКСИМОВ)-LL-F-28249, отличающийся тем, что LL-F-28249 подвергают алкилированию п-нитробензоилхлоридом, полученное 5-О-(п-нитробензоил)-LL-F-28249 окисляют уксусным ангидридом и диметилсульфоксидом в присутствии пиридина и кислоты и полученное производное 5-О-(п-нитробензоил)-23-оксо-LL-F-28249 в кристаллическом состоянии подвергают в любом порядке оксилированию действием C1-C6-алкоксиамина или его соли и снятию защиты.
| Кипятильник для воды | 1921 |
|
SU5A1 |
| J.D.Albright et al, JACS, 1967,89, с.2416. | |||
