Некоторые имидазолинил бензойные и нафтойные кислоты, сложные эфиры и соли, их применение в качестве гербицидных агентов предложены в патентах США NN 4 188 487; 4 297 128 и 4 554 013, и в патентной заявке Великобритании N 2 172 866 A, и Европейской патентной заявке 86200304.3. Однако, имидазолинил бензазолы настоящего изобретения не описаны и не предложены в вышеупомянутых патентах и патентных заявках. Соединения с конденсированным гетеропиридином и их гербицидное использование описано в патентах США N 4 650 514 и 4 752 323. Хотя известно большое количество гербицидно активных имидазолиниловых соединений, гораздо более эффективные имидазолиниловые соединения были бы полезны для фермеров, агрономов, предпринимателей и для борьбы с нежелательными растительными видами.
Цель изобретения состоит в том, чтобы предложить эффективные гербицидные имидазолинил орто-карбокси-2-бензогетероциклические соединения и индологетероциклические дионы для уничтожения разнообразных однодольных и двудольных растительных видов таких, как те виды, которые в общем случае очень трудно поддаются уничтожению в сельскохозяйственной практике.
Изобретение относится к 2-/2-имидазолин-2-ил/бензогетероциклическим соединениям, которые имеют следующую структуру: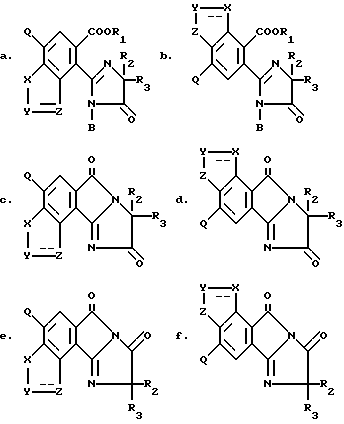
в которых: R1 является водородом, ди/C1-C4/ алкилимино,
C1-C12 алкилом, может быть замещенным одним-тремя заместителями: C1-C4 алкокси, C1-C4 алкилтио, галогеном, окси, C1-C4 циклоалкилом, бензоилокси, фурилом, фенилом, возможно замещенным нитро, одним-тремя галогенами, C1-C4 алкильными группами или C1-C4 алкокси группами, карбокси, C1-C4 алкоксикарбонилом, циано или три/C1-C4/ алкиламмоний галидом;
C3-C12 алкенилом, может быть замещенным одним-тремя заместителями: C1-C4 алкокси, фенилом, галогеном или C1-C4 алкоксикарбонилом;
C3-C6 циклоалкилом, может быть замещенным одной-тремя C1-C4 алкильными группами;
C3-C16 алкинилом, может быть замещенным одним-тремя галогенами или катионом;
R2 является C1-C4 алкилом;
R3 является C1-C4 алкилом или C3-C6 циклоалкилом, а когда R2 и R2 взяты вместе с углеродом, к которому они присоединены, они могут представлять C1-C4 циклоалкил, возможно замещенный метилом;
B является водородом, COR4 или SO2R5 с той оговоркой, что когда B является COR4 или SO2R5, R1 отличен от водорода или катиона, а R9 отличен от водорода;
R4 является C1-C11 алкилом, хлорметилом или фенилом, возможно замещенным галогеном, нитро- или C1-C4 алкилом;
R5 является C1-C4 алкилом или фенилом, возможно замещенным C1-C4 алкилом;
X, Y и Z каждый независимо является CR6, CR7R8, N или NR9 с той оговоркой, что, по крайней мере, один из X, Y и Z должен быть N или NR9; конфигурация представляет либо простую связь, либо двойную связь с той оговоркой, что когда любой из X, Y или Z является CR7R8 или NR9, тогда === конфигурация, присоединенная к нему, является простой связью, и с еще одной оговоркой, что,по крайней мере, одна из === конфигураций представляет простую связь;
конфигурация представляет либо простую связь, либо двойную связь с той оговоркой, что когда любой из X, Y или Z является CR7R8 или NR9, тогда === конфигурация, присоединенная к нему, является простой связью, и с еще одной оговоркой, что,по крайней мере, одна из === конфигураций представляет простую связь;
R6, R7 и R8 являются независимо водородом, галогеном, C1-C4 алкокси или C1-C4 алкилом, может быть замещенным одним окси или одним-тремя галогенами, C1-C4 алкокси группами или C1-C4 алкилтио группами;
R9 является водородом или C1-C4 алкилом, возможно замещенным окси или одним-тремя галогенами, C1-C4 алкокси группами или C1-C4 алкилтио-группами;
Q является водородом, галогеном, C1-C4 алкокси или C1-C4 алкилом, возможно замещенным одним-тремя следующими заместителями: галогеном, C1-C4 алкокси, C1-C4 алкилтио или C2-C4 алкенилом;
их оптическим изомерам, когда R2 и R3 не- одинаковы или когда R7 и R8 неодинаковы;
их таутомерам и геометрическим изомерам, и их присоединенным солям кислот, за тем исключением, когда R1 является сольобразующим катионом.
Кроме того, в соответствии с настоящим изобретением предлагаются способы получения вышеупомянутых соединений и способы уничтожения нежелательных однодольных и двудольных растительных видов при помощи их применения.
Изобретение относится к 2-/2-имидазолин-2-ил/бензогетероциклическим соединениям, имеющим структуру: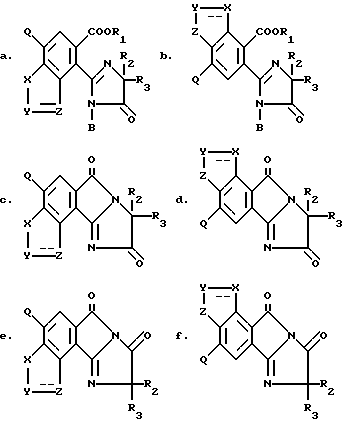
в которых R1 является водородом, ди/C1-C4/алкиламино,
C1-C12алкилом, возможно замещенным одним-тремя заместителями: C1-C4 алкокси, C1-C4 алкилтио, галогеном, окси, C3-C6 циклоалкилом, бензилокси, фурилом, фенилом, возможно замещенным одним нитро, одним-тремя галогенами, C1-C4 алкильными группами или C1-C4 алкокси группами, карбокси, C1-C4 алкоксикарбонилом, циано или три/C1-C4/алкиламмоний галидом;
C3-C12 алкенилом, возможно замещенным одним-тремя заместителями: C1-C4 алкокси, фенилом, галогеном или C1-C4 алкоксикарбонилом;
C3-C6 циклоалкилом, возможно замещенным одним-тремя C1-C4 алкильными группами;
C3-C16 алкинилом, возможно замещенным одним-тремя галогенами или катионом;
R2 является C1-C4 алкилом;
R3 является C1-C4 алкилом или C3-C6 циклоалкилом, а когда R2 и R3 взяты вместе с атомом углерода, к которому они присоединены, они могут представлять C3-C6 циклоалкил, возможно замещенный метилом;
B является водородом, COR4 или SO2R5 с той оговоркой, что когда B является COR4 или SO2R5 отличен от водорода или катиона, а R9 отличен от водорода;
R4 является C1-C11 алкилом, хлорметилом или фенилом, возможно замещенным галогеном, нитро или C1-C4 алкилом;
R5 является C1-C4 алкилом или фенилом, возможно замещенным C1-C4 алкилом;
X, Y и Z каждый независимо является CR6, CR7R8, NR9 или N с той оговоркой, что по крайней мере один из X, Y или Z является N или NR9; конфигурация представляет либо простую связь, либо двойную связь с той оговоркой, что когда любой из X, Y или Z является CR7R8 или NR9, тогда === конфигурация, присоединенная к нему, представляет простую связь, и о еще одной оговоркой, что по крайней мере одна из === конфигураций представляет простую связь;
конфигурация представляет либо простую связь, либо двойную связь с той оговоркой, что когда любой из X, Y или Z является CR7R8 или NR9, тогда === конфигурация, присоединенная к нему, представляет простую связь, и о еще одной оговоркой, что по крайней мере одна из === конфигураций представляет простую связь;
R6, R7 и R8 независимо являются водородом, галогеном, C1-C4 алкокси или C1-C4 алкилом, возможно замещенным одним окси или одним-тремя галогенами, C1-C4 алкокси группами или C1-C4 алкилтио группами;
R9 является водородом или C1-C4 алкилом, возможно замещенным одним окси или одним-тремя галогенами, C1-C4 алкокси группами или C1-C4 алкилтио группами;
Q является водородом, галогеном, C1-C4 алкокси или C1-C4 алкилом, возможно замещенным одним-тремя следующими заместителями: галогеном, C1-C4 алкокси, C1-C4 алкилтио или C2-C4 алкенилом;
их оптическим изомерам, когда R2 и R3 не- одинаковы или когда R7 и R8 неодинаковы;
таутомерам и их геометрическим изомерам, и их присоединенным солям кислот за тем исключением, когда R1 является соль-образующим катионом.
Термин "галоген" обозначает F, Cl, Br или I. Термин катион, как он используется в настоящем патентном описании и формуле изобретения, обозначает щелочные металлы, щелочно-земельные металлы, марганец, медь, железо, цинк, кобальт, свинец, серебро, никель, аммоний или органический аммоний. К щелочным металлам относятся натрий, калий и литий. Среди катионов органического аммония пригодными для использования в соответствии с настоящим изобретением являются моноалкиламмоний, диалкиламмоний, триалкиламмоний, тетраалкиламмоний, моноалкениламмоний, диалкениламмоний, триалкениламмоний, моноалкиниламмоний, диалкиниламмоний, моноалканоламмоний, диалканоламмоний, C5-C6 циклоалкиламмоний, пиперидиний, морфолиний, пирролидиний, бензил аммоний и т.п.
Среди орто-карбокси-/5-оксо-2-имидазолин-2-ил/бензогетероциклов, описанных в соответствии с настоящим изобретением, используют орто-/2-имидазолин-2-ил/индолкарбоксилаты, орто-/2-имидазолин-2-ил/индазолкарбоксилаты, орто-/2-имидазолин-2-ил/бензимидазолкарбоксилаты, орто-/2-имидазолин-2-ил/бензотриазол карбоксилаты и т.п.
Уже давно известна необходимость в агрономической практике в более эффективных гербицидных агентах и, в частности, в эффективных гербицидных агентах, которые можно использовать в присутствии важных сельскохозяйственных культур так, чтобы не причинить ущерба вышеупомянутым растениям. Без адекватного контроля нежелательные растительные виды могут погубить или снизить урожайность культур, снизить качество и ценность урожая, и снизить эффективное получение и сбор урожая. Гербицидные имидазолиниловые бензогетероциклы, являющиеся предметом настоящего изобретения, обладают свойствами эффективного контроля самых разнообразных нежелательных однодольных и двудольных растительных видов и, кроме того, демонстрируют хорошую селективность относительно ценных широколиственных культур таких, как соевые бобы и сахарная свекла.
Гербицидно активные имидазолинил бензогетероциклические соединения, имеющие структуру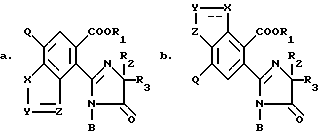
в которых B является водородом, а R1, R2, R3, X, Y, Z уже были определены выше, могут быть получены из их предшествующих имиднитриловых соединений, имеющих структуру формулы I: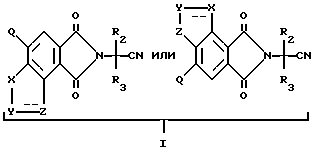
Нитриловые группы в соединениях формулы I могут быть гидролизованы в присутствии серной кислоты, чтобы получить соответствующие амиды, а в полученных в результате имидамидах кольцо может быть разомкнуто в присутствии соответствующего нуклеофила такого, как алкоголят щелочного металла, чтобы получить промежуточные соединения диамида сложного эфира формулы II и их региоизомеры. Диамиды сложного эфира формулы II могут быть превращены в целевые соединения, имеющие структуру a или b, в результате взаимодействия пентахлорида фосфора в присутствии растворителя. В случае, когда R6, R7, R8 или R9 содержат одну или несколько оксигрупп, эти оксигруппы превращают в хлор-группы при помощи этой реакции. Эта реакционная последовательность иллюстрируется на технологической схеме I.
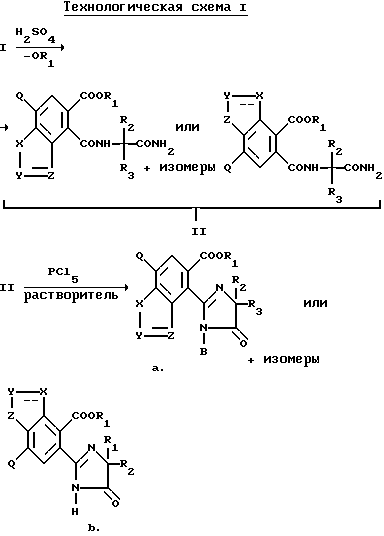
Региоизомеры могут быть отделены при помощи стандартных хроматографических приемов таких, как обращенно-фазовая жидкостная хроматография.
В качестве альтернативы, соединения, имеющие структуру a и b, которые приведены выше, и в которых R1 является водородом, могут быть получены в две стадии в результате взаимодействия соответствующего фталевого ангидрида с аминоамидом формулы III в присутствии основания такого, как триэтиламин, и не обязательно в присутствии растворителя, чтобы получить промежуточные соединения диамида кислоты и их региоизомеры, и замыкания кольца вышеупомянутых промежуточных соединений в водном основании щелочного металла с последующим подкислением, чтобы получить целевые орто-2-/имидазолин-2-ил/бензогетероциклические карбоновые кислоты, имеющие структуру a или b, и их региоизомеры, как это показано на технологической схеме II.
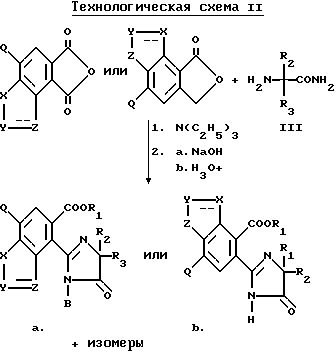
Региоизомеры могут быть отделены с использованием стандартных хроматографических приемов таких, как обращенно-фазовая жидкостная хроматография.
Еще один способ получения соединений структуры a или b, которые описаны выше, в которых R1 и B являются водородом, аналогичен способу, предложенному в патенте США N 4 758 667, в соответствии с которым сложный диэфир формулы IV обрабатывают алкоголятом щелочного металла таким, как третичн.-бутилат калия и аминоамидом формулы III в присутствии инертного растворителя такого, как ксилол, и затем обрабатывают водной кислотой, чтобы получить целевые имидазолинил бензогетероциклы, как это показано на схеме III, на которой R10 является C1-C8 алкилом.

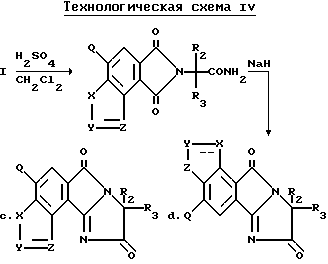
Соединения, имеющие структуру c и d, могут быть получены из соответствующих имиднитрилов формулы I при помощи кислотного гидролиза нитриловых групп, чтобы получить соответствующие имидамиды, и их циклизации в присутствии гидрида щелочного металла такого, как гидрид натрия, чтобы получить целевые индологетероциклические дионы, как это показано на технологической схеме IV.
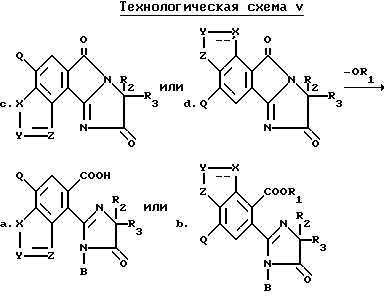
Соединения, имеющие структуру a или b, в которых R10 отличен от водорода, а B является водородом, могут быть получены из соединений, имеющих структуру c или d в результате взаимодействия вышеупомянутых соединений с соответствующим нуклеофилом таким, как алкоголят щелочного металла, как это показано на технологической схеме V.
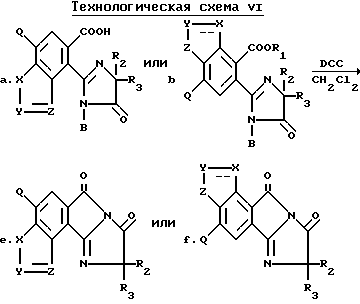
Соединения, имеющие структуру e или f, могут быть получены в результате взаимодействия орто-/2-имидазолин-2-ил/бензогетероциклических карбоновых кислот, имеющих структуру a или b, в которых B является водородом, с дициклогексилкарбодиамидом (ДЦК) в присутствии непротического растворителя, как это показано на технологической схеме VI.
Соединения структуры c и d, в которой Z является NR9 или N, X и Y являются CH и Q является водородом, могут быть получены из соответствующего 3-винилпиррола при помощи образования соответствующего имиднитрилового промежуточного соединения формулы I в результате реакции Дилза-Адлера с соответствующим образом замещенным малеимидом нитрила формулы V, и последующего окисления, используя окисляющий реагент, известный в этой области техники, такой, как двуокись марганца, чтобы обеспечить необходимое окислительное состояние. Образование 3-винилпирролового исходного материала осуществляют через реакцию Виттига. Полученное таким образом имидовое нитриловое промежуточное соединение формулы I может быть превращено в целевые индологетероциклические дионы формулы VI в результате последовательности реакции, описанной выше, и проиллюстрированной на технологической схеме IV. Схема реакций приведена на помещенной ниже технологической схеме VII.
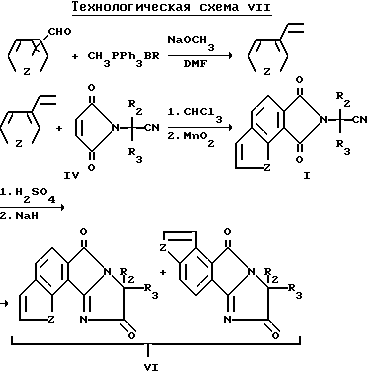
И как уже было указано выше, дионы формулы VI могут быть превращены в соответствующие имидазолинил бензогетероциклы структур a и b, как это показано в технологической схеме V, в которой Z является NR9 или N, X и Y являются CH2, а B и Q являются водородом.
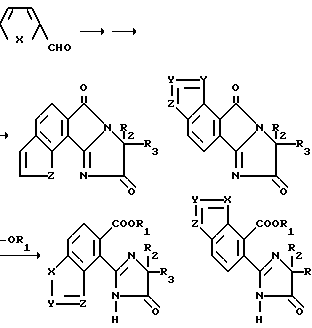
Аналогично соединения, имеющие структуры a, b, c и d, в которых X является NR9 или N, Y и Z являются CH2, а B и Q являются водородом, могут быть получены из 2-пиррол-карбоксальдегида при помощи повторения последовательности реакций, приведенной на технологических схемах VII и V соответственно, как это показано ниже.
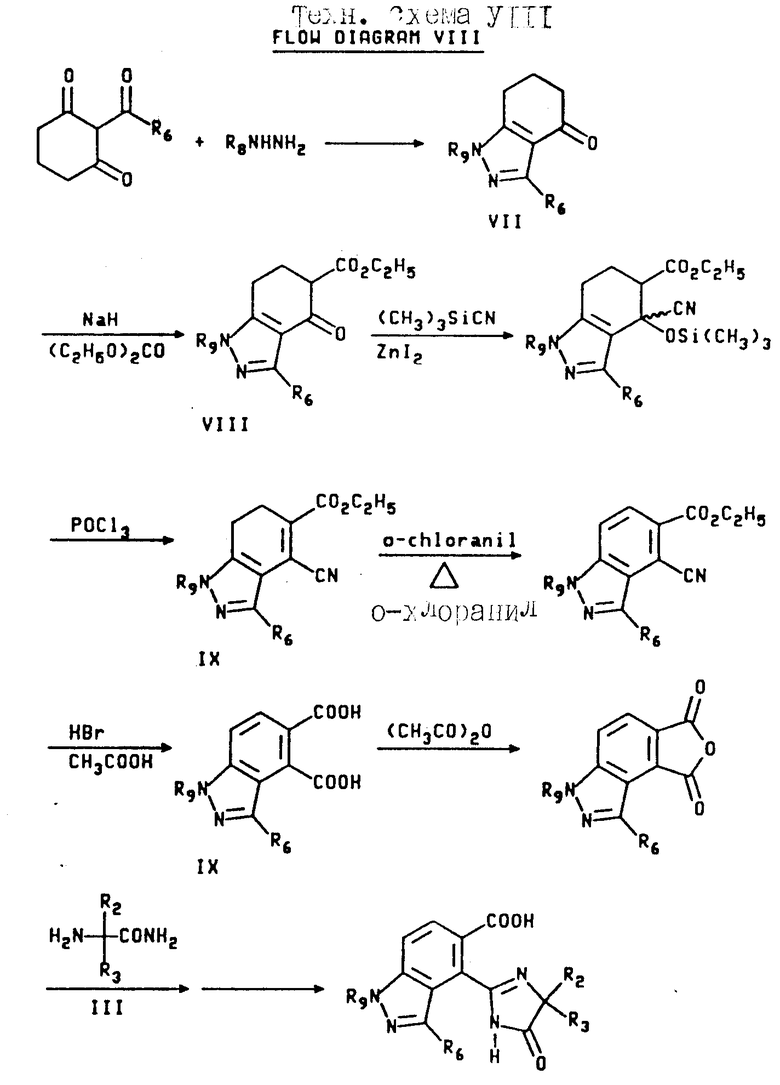
Соединения, имеющие структуру a, в которой X и Y являются N или NR9, Z является CR6, а B и Q являются водородом, получают из 2-кето-1,3-циклогександиона следующим образом: конденсация вышеупомянутого диона с соответствующим образом замещенным гидразином обеспечивает 1,3-двузамещенный дигидроиндазолон формулы VII; обработка промежуточного соединения формулы VII гидридом натрия и этилкарбонатом дает тетрагидроиндазол-5-карбоксилат формулы VIII; обработка кетона формулы VIII триметилсилилцианидом и иодидом цинка с последующим снятием защиты/дегидрированием дает дигидро-4-цианоиндазол-5-карбоксилат формулы IX; дегидрогенизация соединения формулы IX и последующая обработка реакционного продукта бромистым водородом и уксусной кислотой дает 1,3-диметил-1Н-индазол-4,5-дикарбоновую кислоту; обработка уксусным ангидридом обеспечивает соответствующий ангидрид, который может региоспецифически подвергнут разрыву кольца с использованием соответствующего аминоамида формулы III в целевой имидазолинил бензогетероцикл, имеющий структуру a, как это подтверждается в технологической схеме II. Реакционная последовательность иллюстрируется в технологической схеме VIII (см. в конце описания).
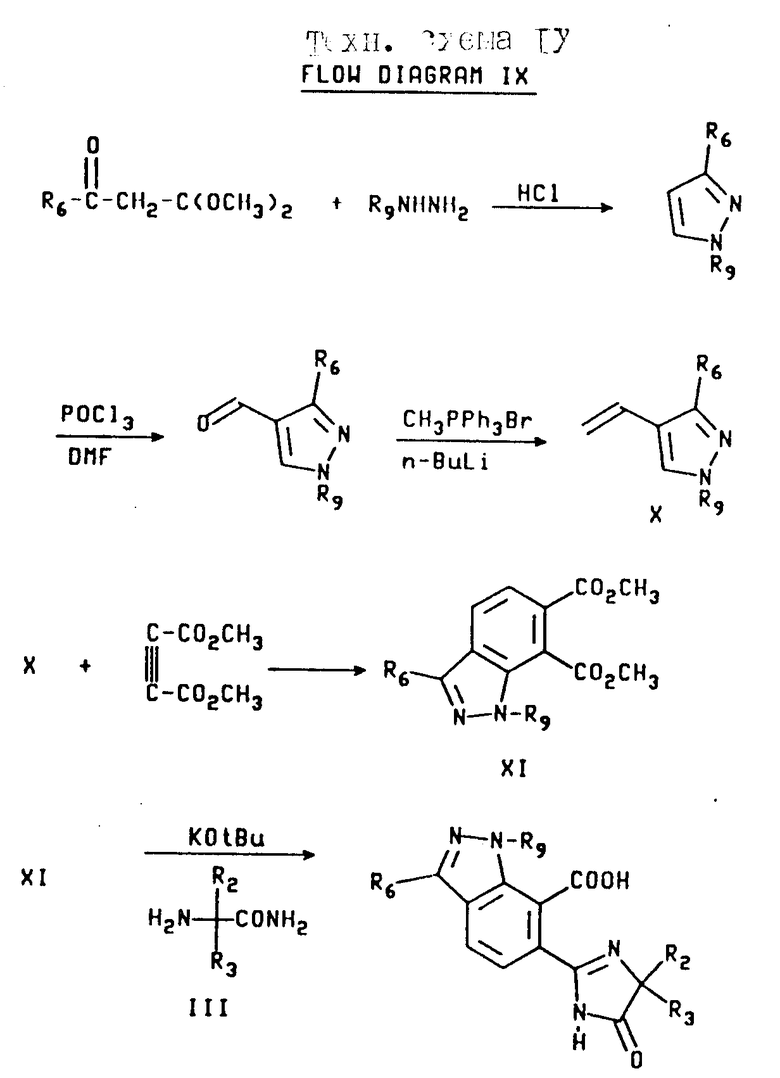
Имидазолинил индазолы, имеющие структуру b, в которой X является CR6, а Y и Z являются N или NR9, могут быть получены из дикетала из соответствующим образом замещенного кетоацетальдегида, как это показано на технологической схеме IX (см.в конце описания).
Конденсация диметилацетала, соответствующим образом замещенного кетоацетальдегида с соответствующим гидразином с последующей обработкой кислотой, дает 1,5-двухзамещенный пиразол, как это показано. Обработка вышеупомянутого пиразола оксихлоридом фосфора и диметилформамидом (ДМФ) и после реакции Виттига получают винилпиразол формулы X. В результате реакции Дилза-Адлера пиразола формулы X с диметил ацетилендикарбоксилатом получают сложный диэфир формулы XI, который затем превращают в продукт имидазолинил индазола, используя процедуру, проиллюстрированную на технологической схеме III.
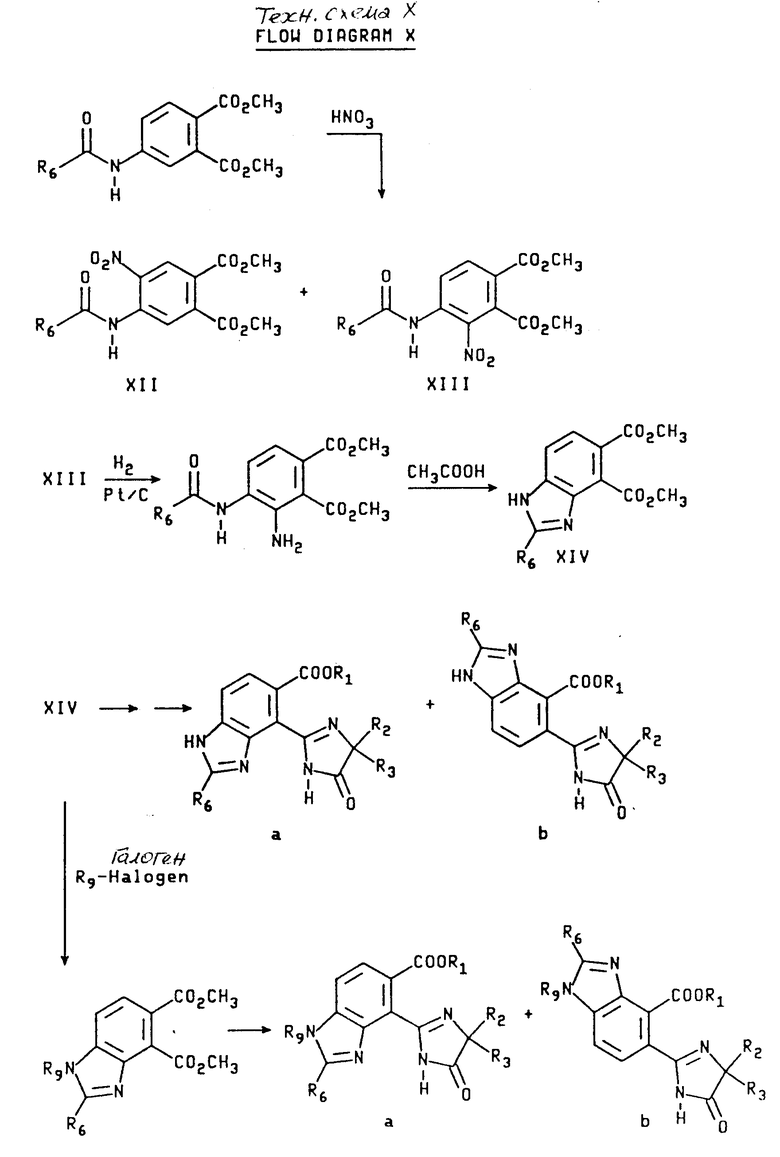
Орто-имидазолинил бензимидазолкарбоксилаты, имеющие структуру a и b, могут быть получены из их общего предшествующего соединения, 4-карбоксамидофталата, которое получают из ацилирования 4-аминофталата. Используя процедуру нитрования, описанную Р.Л. Уилльямсом и С.У. Шалаби в Journal of Heterocyche Chemistry, 1973, /10/, 891 получают промежуточные соединения формулы XII и XIII. Разделение соединений XII и XIII осуществляют при помощи фракционной рекристаллизации. Гидрогенизация соединения XIII с последующей циклизацией полученного в результате амидоанилина дает дикарбоксилат бензимидазола формулы XIV. Сложный диэфир формулы XIV может быть непосредственно превращен в имидазолинил бензимидазолы структуры a и b, используя приемы, описанные выше, и технологические схемы II и III. В качестве альтернативы сложный диэфир формулы XIV можно подвергнуть алкилированию с подходящим алкилгалидом, чтобы получить бензимидазолдикарбоксилат формулы XV, который превращают в целевой имидазолинил бензимидазол, имеющий структуру a и b при помощи процедуры, показанной на технологических схемах II и III. Реакционная последовательность, начинающаяся с подходящего 4-карбоксамидофталата, приведена на технологической схеме X (cм.в конце описания).
Аналогичным образом промежуточное соединение формулы XII может быть превращено в орто-имидазолинилбензимидазол карбоксилат формул XVI и XVII, как это показано на технологической схеме XI (см.в конце опмсания).
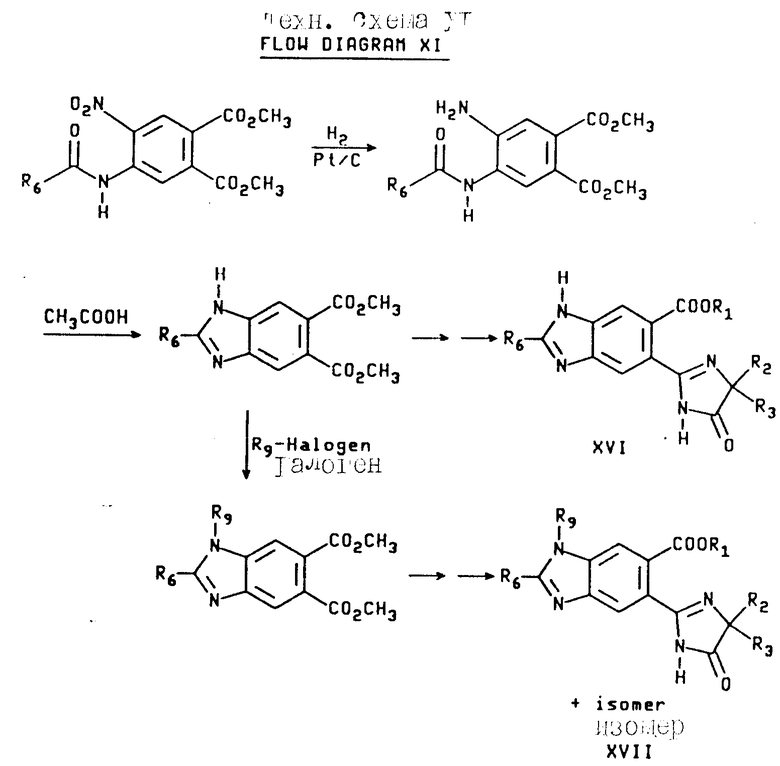
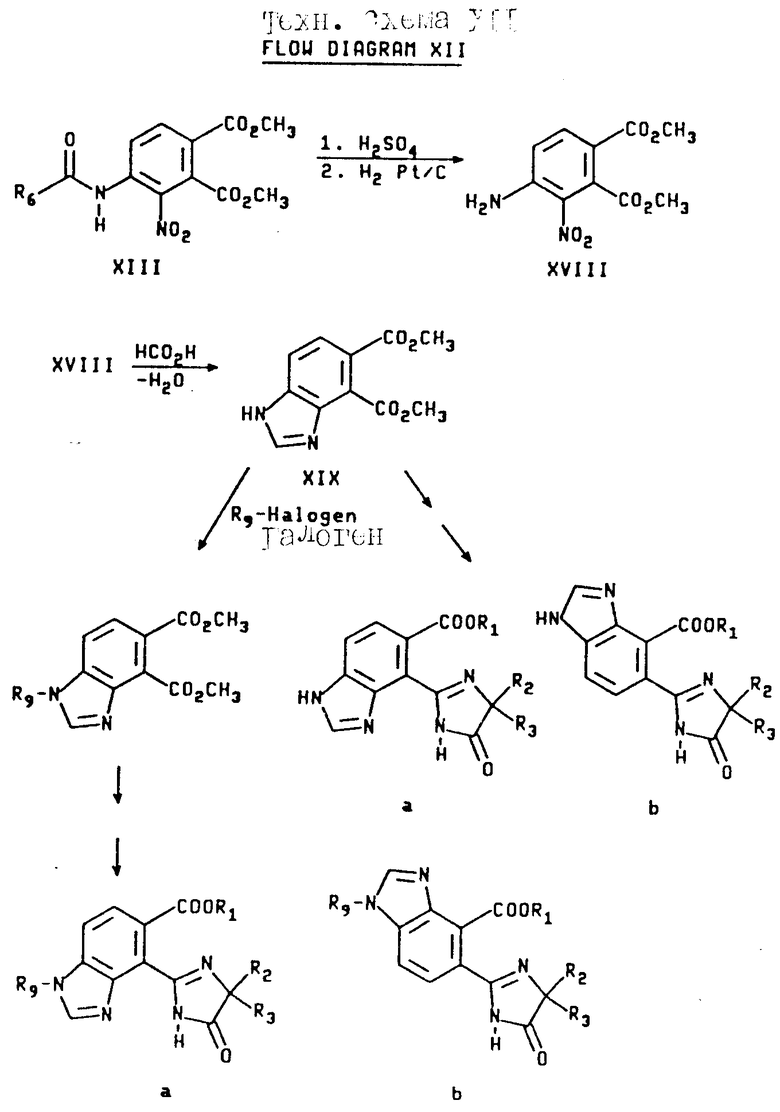
Соединения имидазолинил бензимидазола, в которых R6 является водородом, могут быть получены из соединений формулы XIII при помощи последовательного кислотного гидролиза амидной группы и восстановления нитрогруппы через каталитическую гидрогенизацию, чтобы получить диамин формулы XVIII. В результате обработки диамина муравьиной кислотой и азеотропного удаления воды получают бензимидазол дикарбоксилат формулы XIX, который затем непосредственно превращают в целевые соединения, имеющие структуру a или b, в которой X и Y являются N и Y является CH или сложный диэфир формулы XVIII подвергают алкилированию, как это показано выше, а замещенный бензимидазол дикарбоксилат превращают в целевые соединения, имеющие структуру a или b, в которых X и Z являются N или NR9, а Y является CH. Превращения дикарбоксилатов в финальные имидазолинил бензимидазолы показаны на технологических схемах II и III, а реакционная последовательность, начинающаяся с соединений формулы XIII, показана ниже на технологической схеме XII (см.в конце опсания).
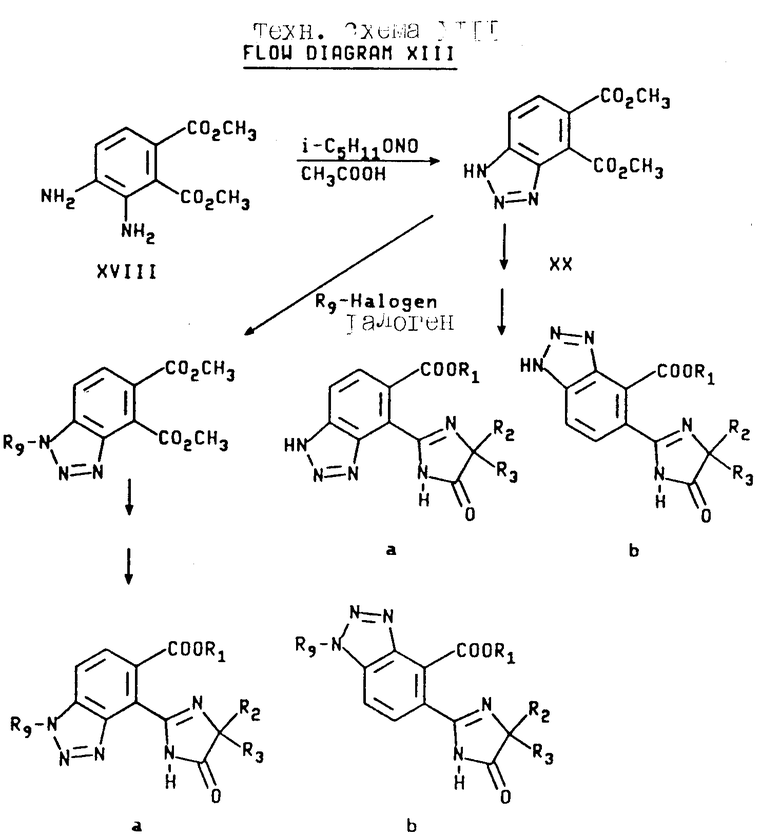
Соединения настоящего изобретения, имеющие структуры a, и b, в которых X, Y и Z независимо являются N или NR9,получают с использованием диамина формулы XVIII. Реакция вышеупомянутого диамина с изоамилнитритом в присутствии уксусной кислоты дает сложный диэфир бензотриазола формулы XX, который затем превращают в целевые соединения, имеющие структуру a и b, в которых X, Y и Z являются N или NR9 при помощи процедуры, описанной выше для сложных диэфиров бензимидазола формулы XIV и XIX. Эта реакционная схема иллюстрируется на технологической схеме XIII (см. в конце описания).
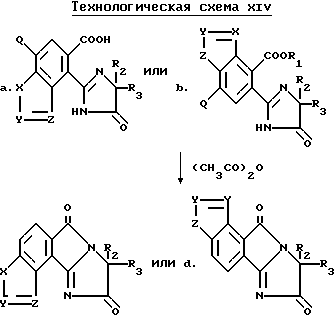
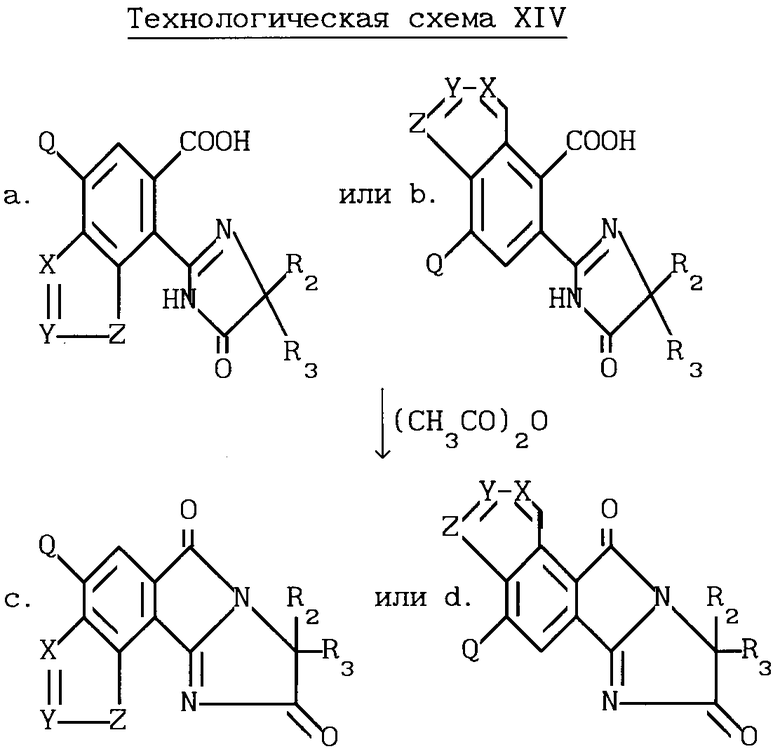
Разумеется, все соединения, описанные выше, имеющие структуру a или b, в которых R1 и B являются водородом, могут быть превращены в соответствующие имидазолинил индологетероциклические дионы, имеющие структуру e и f, при помощи повторения процедуры, проиллюстрированной на технологической схеме VI. Соответствующие имидазолинил индологетероциклические дионы, имеющие структуру c и d, могут быть получены в результате взаимодействия вышеупомянутых имидазолинил бензогетероциклов с ангидридом кислоты, не обязательно в присутствии растворителя, как это показано на технологической схеме XIV.
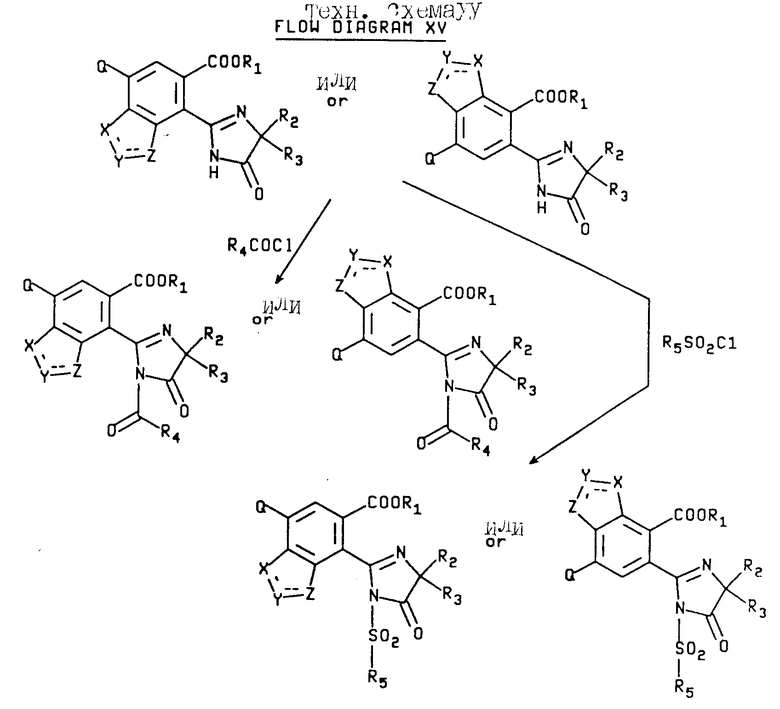
Соединения, имеющие структуру a или b, в которых R1 отличен от водорода или катиона, R9 отличен от водорода, а B является COR4 или SO2R5, могут быть получены при помощи взаимодействия соединений, имеющих структуру a или b, в которых R1 отличен от водорода или катиона, R9 отличен от водорода, а B является водородом, с ацилгалидом таким, как ацилхлорид или сульфонилгалидом таким, как сульфонил хлорид, чтобы получить целевые продукты, в которых B является COR4 или SO2R5. Реакция приведена на технологической схеме XV (см.в конце описания).
В качестве альтернативы соединения, имеющие структуру a, или b, в которой R1 отличен от водорода или катиона, R9 отличен от водорода, а B является COR4, могут быть получены в результате взаимодействия соединений, имеющих структуру a или b, в которых R1 отличен от водорода или катиона, R9 отличен от водорода, а B является водородом, с ангидридом кислоты формулы /R4CO/2O.
Имидазолинил бензогетероциклические соединения настоящего изобретения являются в высшей степени эффективными при уничтожении разнообразных нежелательных однодольных растительных видов таких, как просто петушье, лисохвост, нимфалина, осока, овсюг, свинорой и т.п. и двудольных растительных видов таких, как вьюнок полевой, матрикарий, ипомея, горчица полевая, амброзия высокая, лимнохарис и т.п. Уничтожение вышеупомянутых растительных видов может быть осуществлено при помощи применения соединений настоящего изобретения к листве вышеупомянутых растений или к почве или воде, содержащей семена, или другие его средства размножения в дозе от примерно 0,016 до 4,0 кг/га.
Неожиданным образом было установлено, что некоторые соединения настоящего изобретения хорошо переносятся широколиственными культурными растениями такими, как соевые бобы и сахарная свекла, когда вышеупомянутые соединения применяют к листве вышеупомянутых культур или к почве, содержащей семена или другие их средства размножения в дозе от примерно 0,016 до 1,000 кг/га.
Соевые бобы играют все более возрастающую роль как источник высококачественного протеина и являются наиболее важными пищевыми бобами, получаемыми в настоящее время. Сахарная свекла является основным источником сахара в Северной Америке, приблизительно одну треть сахара, потребляемого здесь, получают из сахарной свеклы. В Европе сахарная свекла является главным источником рафинированного сахара. При помощи комбинации уничтожения сорняков и толерантности культурных растений применение соединений настоящего изобретения дает возможность улучшить управление урожаем, временем сбора урожая, качеством и количеством урожая.
Имидазолинил бензогетероциклические соединения могут быть применены в форме жидких брызг таких, как водные концентраты, эмульгируемые концентраты и т. п. или в форме твердых форм таких, как смачиваемые порошки, диспергируемые гранулы, гранулированные формы и т.п.
Если гербицидно-активные соединения растворимы в воде, их можно просто растворить в воде и применить в виде водной струи. Вышеупомянутые соединения могут быть также скомпонованы в форме эмульгируемых концентратов и разбавлены водой непосредственно перед применением в форме струи. Общая композиция эмульгируемого концентрата может быть получена при помощи растворения примерно 5-25 мас. активного соединения в примерно 65-90 мас. N-метил-пирролидона, изофорона, бутилцеллозольва, метилацетата и т.п. и диспергирования в нем примерно 5-10 мас. неионного поверхностно-активного агента такого, как алкилфеноксиполиэтокси спирт.
Композиции смачиваемых порошков могут быть получены при помощи измельчения вместе примерно 20-45 мас. тонко измельченного носителя такого, как каолин, бентонит, диатомовая земля, аттапульгит и т.п. с примерно 40-80 мас. гербицидно-активного соединения и примерно 2-5 мас. неионного поверхностно-активного агента такого, как алкилфеноксиполиэтокси спирт.
Общие гранулированные продукты могут быть получены при помощи растворения активного соединения в растворителе таком, как метилен хлорид, N - метилпирролидон и т.п. и разбрызгивания полученного в результате раствора на носитель-глину такой, как аттапульгит или каолин,таким образом, чтобы получить примерно 3-20 мас. активного соединения и примерно 80-97 мас. носителя.
Чтобы упростить последующее понимание настоящего изобретения, следующие примеры представлены в первую очередь для целей иллюстрации некоторых его дополнительных подробностей. Настоящее изобретение не ограничено ничем, кроме пунктов формулы изобретения.
Сокращение кг/га обозначает килограммы/гектар. Сокращения NOE, 1H-ЯМР и ИК обозначают ядерный эффект Оверхауза, протонный ядерный магнитный резонанс и инфракрасный спектр, соответственно. Термин ЖХВД обозначает жидкостную хроматографию высокого давления. Все части являются массовыми частями, если не указано противное.
Пример 1. Получение диметил 4-ацетамидо-5-нитрофталата /I/ и диметил 4-ацетамидо-3-нитрофталата /II/.
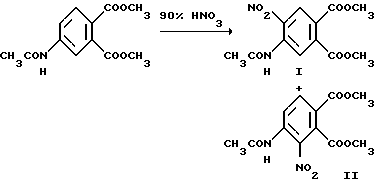
Диметил 4-ацетамидофталат /100,4 г, 0,400 моль/ добавляли в дымящуюся азотную кислоту /90% 600 мл/ при температуре от 0 до 5oC. Когда добавление закончено /0,5 ч/, смесь перемешивали 2,5 ч при 0-10oC, соединяли с холодным метиленхлоридом и встряхивали с измельченным льдом. Водный слой отделяли и затем экстрагировали метиленхлоридом. Соединенные органические слои промывали ледяной водой, раствором бикарбоната натрия и холодной водой, сушили /сульфат магния/ и концентрировали под вакуумом, чтобы получить твердый остаток. Остаток подвергали рекристаллизации дважды из метанола, чтобы получить продукт I из заголовка примера в виде оранжево-коричневых игл, температура точки плавления 119-120oC. Первоначальную маточную жидкость концентрировали под вакуумом, а остаток подвергали рекристаллизации несколько раз из четыреххлористого углерода, чтобы получить соединение II из заголовка примера в виде ярко-желтых игл, температура точки плавления 124-125oC. Выход соединения I равен 38,3 г (32,9%), а выход соединения II равен 33,1 г (28,1%).
Пример 2. Получение диметил 2-метил-4,5-бензимидазолдикарбоксилата.
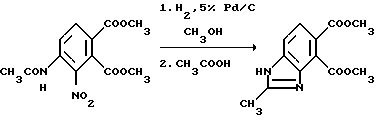
Смесь диметил 4-ацетамидо-3-нитрофталата (142,1 г, 0,480 моль) в метаноле с 5% платины на углероде (катализатор) подвергали гидрогенизации в гидрогенизаторе Парра при комнатной температуре. Реакционные смеси фильтровали через диатомовую землю и концентрировали под вакуумом, чтобы получить твердый остаток. Это твердое вещество диспергировали в ледяной уксусной кислоте и толуоле и нагревали с перемешиванием при температуре дефлегмирования в течение 4 ч при азеотропном удалении воды. Полученную в результате горячую реакционную смесь медленно добавляли в насыщенный раствор бикарбоната натрия при энергичном перемешивании, фильтровали и фильтровальную лепешку сушили, чтобы получить продукт из заголовка примера в виде белого твердого вещества (71,6 г, 60,1%), температура точки плавления 97-103oC.
Пример 3. Получение 2-метил-4,5-бензимидазолдикарбоновой кислоты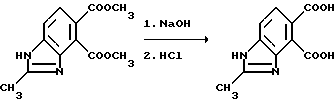
Смесь диметил 2-метил-4,5-бензимидазолдикарбоксилата и 8 эквивалентов 10N раствора гидрата окиси натрия перемешивали 4 ч при температуре 70oC, охлаждали и подкисляли хлористоводородной кислотой, чтобы получить продукт из заголовка примера в виде кристаллов кремового цвета, температура точки плавления 270oC (разлож.).
Пример 4. Получение 4-/4-изопропил-4-метил-5-оксо-2-имидазолин-2-ил/-2-метил-4-бензимидазол карбоновой кислоты (I) и 5-/4-изопропил-4-метил-5-оксо-2-имидазолин-2-ил/-2-метил-4- бензимидазолкарбоновой кислоты /II/, 4:1-смесь.
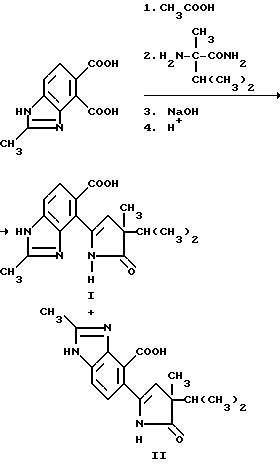
Смесь 2-метил-4,5-бензимидазолдикарбоксилата /5,00 г, 22,7 ммоль/ и уксусного ангидрида перемешивали в течение 6 ч при температуре дефлегмирования, охлаждали и концентрировали под вакуумом, чтобы получить остаток. Остаток переносили в ацетонитрил и обрабатывали a-метил-валирамидом /5,90 г, 45,3 ммоль/. Полученную в результате смесь перемешивали 15 ч при температуре дефлегмирования, охлаждали и выдерживали в течение ночи. Отвержденную реакционную смесь переносили в 5N раствор гидрата окиси натрия, нагревали до температуры дефлегмирования на 10 ч при перемешивании и фильтровали горячей. Фильтрат подкисляли до pH 4 концентрированной HCl, чтобы получить коричневое твердое вещество в осадке, который удаляли фильтрацией. Коричневое твердое вещество нагревали в метаноле, фильтровали горячим, а фильтрат концентрировали под вакуумом, чтобы получить продукт из заголовка примера в отношении 4: 1 для I:II, соответственно, в виде желтого порошка (0,93 г, 13,0%), температура точки плавления 256oC (разлож.).
Пример 5. Получение диметил 1,2-диметил-4,5-бензимидазолдикарбоксилата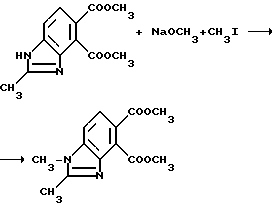
Метилат натрия (1,98 г, 36,6 ммоль) добавляли в раствор диметил 2-метил-4,5-бензимидазолдикарбоксилата (7,44 г, 33,3 ммоль) в метаноле и тетрагидрофуране. Спустя 5 мин добавляли йодметан (2,18 мл, 35,0 ммоль) и смесь перемешивали в течение 24 ч при комнатной температуре. Добавляли дополнительно метилат натрия (1,80 г, 33,3 ммоль) и йодметан (2,07 мл, 33,3 ммоль) и перемешивание продолжали еще в течение 15 ч. Реакционную смесь концентрировали под вакуумом, чтобы получить остаток, который диспергировали в разбавленной хлористоводородной кислоте и обрабатывали бикарбонатом натрия до pH 8 и экстрагировали хлороформом. Соединенные органические экстракты сушили (сульфат магния) и концентрировали под вакуумом, чтобы получить остаток, который подвергали рекристаллизации из этилацетата, чтобы получить продукт из заголовка примера в виде оранжевого твердого вещества, температура точки плавления 205-208oC.
Пример 6. Получение 1,2-диметил-4,5-бензимидазол-дикарбоновой кислоты
Смесь диметил 1,2-диметил-4,5-бензимидазолдикарбоксилата (1,70 г, 6,49 ммоль) и 2N раствора гидрата окиси натрия (25 мл, 12,5 ммоль) перемешивали 5 ч при 100oC. Смесь охлаждали, подкисляли до pH 4 при помощи хлористоводородной кислоты и фильтровали, чтобы получить продукт из заголовка примера в виде белого порошка (1,32 г, 86,8%) температура точки плавления 305-308oC (разлож.).
Пример 7. Получение 1,2-диметил-4,5-бензимидазолдикарбонового ангидрида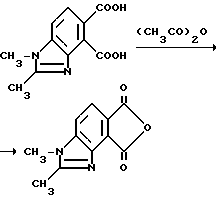
Смесь 1,2-диметил-4,5-бензимидазолдикарбоновой кислоты (1,00 г, 4,27 ммоль) и уксусного ангидрида (10 мл) перемешивали 4 ч при температуре дефлегмации, выдерживали в течение ночи при комнатной температуре и фильтровали, чтобы получить продукт из заголовка примера в виде желтых кристаллов (0,830 г, 89,8%), температура точки плавления 295oC (разлож.).
Пример 8. Получение 4-[/1-карбамоил-1,2-диметилпропил/карбамоил]-1,2- диметил-6-бензидимазолкарбоновой кислоты /I/, 5-[/1-карбамоил-1,2-диметилпропил/карбамоил] -1,2-диметил-4- бензимидазолкарбоновой кислоты /II/, 4: 1-смесь
Смесь 1,2-диметил-4,5-бензимидазолдикарбонового ангидрида (0,830 г, 3,85 ммоль), а-метил-валирамида (0,550 г, 4,22 ммоль) и ацетонитрила перемешивали 4 ч при температуре дефлегмирования, охлаждали в течение ночи и фильтровали, чтобы получить продукт из заголовка примера в виде белого твердого вещества (1,24 г, 93,2% ), температура точки плавления 246-248oC, которое идентифицировали при помощи 1Н-ЯМР, как 4:1-смесь соединения I и соединения II, соответственно.
Пример 9. Получение 4-/4-изопропил-4-метил-5-оксо-2-имидазолин-2-ил/-1,2- диметил-5-бензимидазолкарбоновой кислоты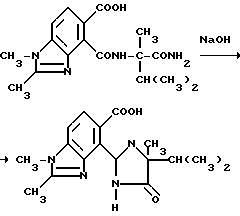
Раствор 4-[/1-Kaрбамоил-1,2-диметилпропил/-кaрбамоил] -1,2- диметил-6-бензимидазолкарбоновой кислоты (5,70 г, 16,5 ммоль) и 10N раствора гидрата окиси натрия (9,88 мл, 98,8 ммоль) перемешивали 3 часа при температуре дефлегмации, охлаждали до 0oC и подкисляли до pH 4 хлористоводородной кислотой и фильтровали. Фильтровальную лепешку сушили, чтобы получить продукт из заголовка примера в виде белого твердого вещества (4,11 г, 75,8%), температура точки плавления 280-285oC (разложение).
Пример 10. Получение 9-изопропил-2,3,9-триметилимидазол [1',2':1,2] пирроло [3,4-e] бензимидазол-6,8/3H,9H/-диона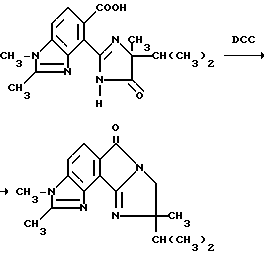
Смесь 4-/4-изопропил-4-метил-5-оксо-2-имидазолин-2-ил/-1,2- диметил-5-бензимидазолкарбоновой кислоты (0,942 г, 2,87 ммоль), дициклогексилкарбодиимида (0,590 г, 2,87 ммоль) и тетрагидрофурана перемешивали в течение 3 ч при температуре дефлегмации, охлаждали и концентрировали под вакуумом. Остаток подвергали хроматографии (силикагель, этилацетатэлюент), чтобы получить продукт из заголовка примера в виде белого порошка (0,210 г, 23,6%), температура точки плавления 258-263oC.
Пример 11. Получение метил 4-/4-изопропил-4-метил-5-оксо-2-имидазолин-2-ил/- 1,2-диметил-5-бензимидазолкарбоксилата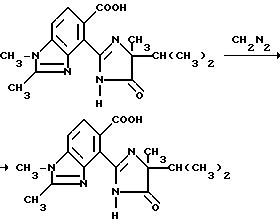
Раствор диазометана в простом эфире по каплям добавляли в раствор 4-/4-изопропил-4-метил-5-оксо-2-имидазолин-2-ил/-1,2- диметил-5-бензимидазолкарбоновой кислоты (2,50 г, 7,61 ммоль) в метаноле до тех пор, пока не установится желтая окраска. Реакционную смесь нейтрализовали уксусной кислотой и концентрировали под вакуумом. После препаративной ЖХВД (силикагель, этил ацетат -элюэнт) получали продукт из заголовка примера в виде не совсем белых кристаллов (0,750 г, 28,9%), температура точки плавления 240-242oC.
Пример 12. Получение диметил 1-бензил-2-метил-4,5-бензимидазолдикарбоксилата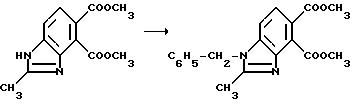
Гидрид натрия (0,750 г, 80% масляная дисперсия, 25,0 ммоль) добавляли порциями в раствор диметил 2-метил-4,5-бензимидазолдикарбоксилата (6,07 г, 24,6 ммоль) в сухом диметилформамиде при 0oC при перемешивании. После того, как выделение водорода прекращается, бензилбромид (2,93 мл, 24,6 ммоль) добавляли в реакционную смесь и перемешивание продолжали в течение 16 ч. Реакционную смесь концентрировали под вакуумом и полученный в результате остаток диспергировали в метиленхлорид и воду. Фазы разделяли и водную фазу экстрагировали метиленхлоридом. Органические фазы соединяли, сушили и концентрировали под вакуумом, чтобы получить твердый остаток. После рекристаллизации твердого вещества из этилацетата получали продукт из заголовка примера в виде не совсем белого порошка (3,81 г, 45,0%), температура точки плавления 192-195oC.
Пример 13. Получение 1-бензил-2-метил-4,5-бензимидазолдикарбоновой кислоты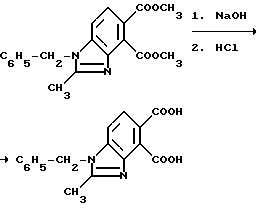
Смесь диметил 1-бензил-2-метил-4,5-бензимидазолдикарбоксилата (5,50 г, 16,2 ммоль), метанола, 10N раствора гидрата окиси натрия (13 мл, 130 ммоль) и воды перемешивали в течение 4 ч при 70oC, охлаждали, подкисляли до pH 4 хлористоводородной кислотой и фильтровали, чтобы получить продукт из заголовка примера в виде не совсем белого твердого вещества (4,94 г, 98,4%), температура точки плавления 224-226oC.
Пример 14. Получение 1-бензил-2-метил-4,5-бензимидазолкарбонового ангидрида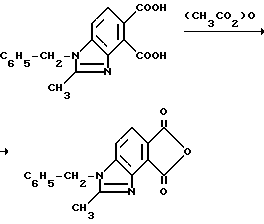
Смесь 1-бензил-2-метил-4,5-бензимидазолдикарбоновой кислоты (4,40 г, 14,2 ммоль) и уксусного ангидрида перемешивали 5 ч при температуре дефлегмирования, охлаждали до 0oC и фильтровали, чтобы получить продукт из заголовка примера в виде бледно-желтого твердого вещества (3,78 г, 91,1%), который идентифицировали при помощи 1H-ЯМР-спектроскопии.
Пример 15. Получение 1-бензил-4-[/1-карбамоил-1,2-диметилпропил/карбамоил] 2-метил-5-бензимидазолкарбоновой кислоты /I/ и 1-бензил-5- [/1-карбамоил-1,2-диметилпропил/-карбамоил]-2-метил-4-бензимидазолка- рбоновой кислоты (II), 4:1-смесь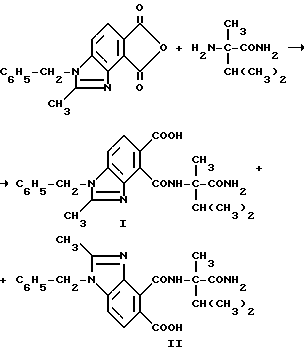
Смесь 1-бензил-2-метил-4,5-бензимидазолдикарбонового ангидрида /3,70 г, 12,7 ммоль/, а-метилвалерамида /1,70 г, 13,0 ммоль/ и ацетонитрила перемешивали в течение 6 ч при температуре дефлегмирования, охлаждали до 0oC и фильтровали, чтобы получить продукт из заголовка примера, 4:1-смесь соединения I и соединения II соответственно, в виде не совсем белого порошка (4,08 г, 76,1% ), температура точки плавления 194-196oC. Отношение продуктов определяли при помощи 1H-ЯМР-спектрального анализа.
Пример 16. Получение 1-бензил-4-/4-изопропил-4-метил-5-оксо-2-имидазолин-2-ил/-2-метил-5- бензимидазолкарбоновой кислоты.
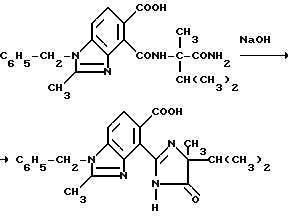
Смесь 1-бензил-4-[/1-карбамоил-1,2-диметилпропил/карбамоил] -2-метил-5- бензимидазолкарбоновой кислоты (3:58 г, 8,47 ммоль), 10 N раствора гидрата окиси натрия (5,06 мл, 50,8 ммоль) и воды перемешивали в течение 4 ч при температуре дефлегмирования, охлаждали, подкисляли до pH 4 хлористоводородной кислоты и фильтровали. Фильтровальную лепешку подвергали рекристаллизации из ацетонитрила, чтобы получить продукт из заголовка примера в виде не совсем белого порошка (1,06 г, 31,0%), температура точки плавления 198-208oC (разложение).
Пример 17. Получение метил 1-бензил-4-/4-изопропил-4-метил-5-оксо-2-имидазолин-2-ил/-2-метил-5- бензимидазолкарбоксилата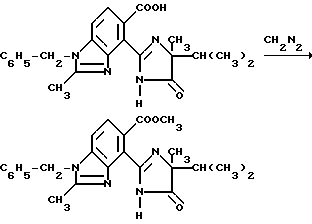
Раствор диазометана в простом эфире добавляли в суспензию 1-бензил-4-/4-изопропил-4-метил-5-оксо-2-имидазолин-2-ил/-2- метил-5-бензимидазолкарбоновой кислоты (1,15 г, 2,84 ммоль) в метаноле до тех пор, пока не установится желтая окраска. Реакцию перемешивали 5 мин, прекращали добавлением 2 капель уксусной кислоты и концентрировали под вакуумом. Полученный в результате остаток подвергали очистке при помощи ЖХВД (силикагель, этилацетат элюент), чтобы получить продукт из заголовка примера в виде белого порошка (0,480 г, 40,3%), температура точки плавления 194-196oC.
Пример 18. Получение диметил 1-этил-2-метил-4,5-бензимидазолдикарбоксилата
Гидрид натрия (1,38 г, 46,0 ммоль, 80% масляной дисперсии) добавляли порциями в раствор диметил 1-бензил-2-метил-4,5-бензимидазолдикарбоксилата (10,9 г, 43,8 ммоль) в сухом диметилформамиде при 0oC. После того, как выделение водорода прекращается, этил иодид (3,68 мл, 46,1 ммоль) добавляли. Реакционную смесь перемешивали при комнатной температуре в течение ночи, обрабатывали этилацетатом и фильтровали. Фильтрат концентрировали под вакуумом; полученный в результате остаток подвергали рекристаллизации из смеси 50% этилацетата:гексанов, чтобы получить продукт из заголовка примера в виде ярко-желтого порошка (3,84 г, 31,7%), температура точки плавления 126,5-128oC.
Пример 19. Получение 1-этил-2-метил-4,5-бензимидазолдикарбоновой кислоты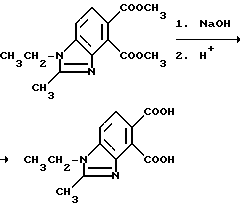
Смесь диметил 1-этил-2-метил-4,5-бензимидазолдикарбоксилата (5,30 г, 19,2 ммоль), метанола и 5N раствора гидрата окиси натрия (30 мл, 150 ммоль) перемешивали в течение 4 ч при 100oC, охлаждали, подкисляли хлористоводородной кислотой и фильтровали, чтобы получить продукт из заголовка примера в виде лимонно-желтого порошка (4,07 г, 85,5%), который идентифицировали при помощи 1H-ЯМР-спектрального анализа.
Пример 20. Получение 1-этил-2-метил-4,5-бензимидазолдикарбонового ангидрида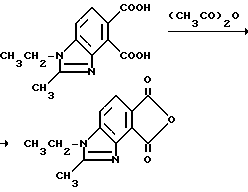
Смесь 1-этил-2-метил-4,5-бензимидазолдикарбоновой кислоты (4,00 г, 16,1 ммоль) и уксусного ангидрида (50 мл) перемешивали 5 ч при температуре дефлегмации, охлаждали и фильтровали. Фильтровальную лепешку промывали простым эфиром и сушили, чтобы получить продукт из заголовка примера в виде золотистых пластинок (3,37 г, 90,8%), который идентифицировали при помощи 1H-ЯМР-спектрального анализа.
Пример 21. Получение 4-[/1-карбамоил-1,2-диметилпропил/карбамоил]-1-этил-2-метил-5- бензимидазолкарбоновой кислоты (I) и 5-[/1-карбамоил-1,2-диметилпропил/карбамоил] -1-этил-2-метил-4- бензимидазолкарбоновой кислоты (II), 4:1 смесь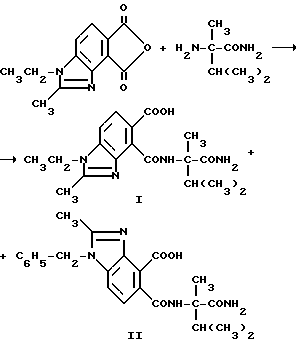
Смесь 1-этил-2-метил-4,5-бензимидазолдикарбонового ангидрида (3,30 г, 14,3 ммоль), а-метилвалерамида (1,90 г, 14,5 ммоль) и ацетонитрила перемешивали 2 ч при температуре дефлегмации в течение ночи при комнатной температуре и 6 ч при температуре дефлегмации. Смесь охлаждали, концентрировали под вакуумом до 50% первоначального объема и фильтровали, чтобы получить продукт из заголовка примера в виде 4:1-смеси соединения I и соединения II ярко-желтого цвета (порошок) (4,59 г, 88,8%), температура точки плавления 240-243oC.
Пример 22. Получение 1-этил-4-/4-изопропил-4-метил-5-оксо-2-имидазолин-2-ил/- 2-метил-5-бензимидазолкарбоновой кислоты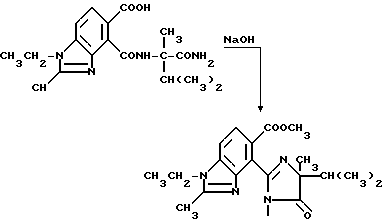
Смесь 4-[/1-карбамоил-1,2-диметилпропил/карбамоил] -1-этил- 2-метил-5-бензимидазолкарбоновой кислоты (4,09 г, 11,4 ммоль), 10 N раствора гидрата окиси натрия (6,8 мл, 68,0 ммоль) и воды перемешивали в течение 2 ч при температуре дефлегмации, охлаждали, подкисляли до pH 4 хлористоводородной кислотой и фильтровали. Фильтровальную лепешку подвергали рекристаллизации из ацетонитрила, чтобы получить продукт из заголовка примера в виде белого порошка (1,10 г, 28,2%), температура точки плавления 250-256oC (разлож).
Пример 23. Получение метил 1-этил-4-/4-изопропил-4-метил-5-оксо-2-имидазолин- 2-ил/-2-метил-5-бензимидазолкарбоксилата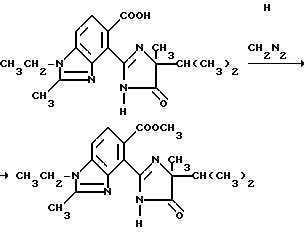
Раствор диазометана в простом эфире добавляли по каплям в смесь исходной карбоновой кислоты (2,10 г, 6,13 ммоль) в метаноле до тех пор, пока желтый цвет не стабилизировался. Спустя 5 мин реакцию прекращали уксусной кислотой и концентрировали под вакуумом. Полученный в результате остаток подвергали хроматографии (силикагель, ЖВХД, этилацетат элюент), чтобы получить продукт из заголовка примера в виде не совсем белого порошка (1,03 г, 47,2%), температура точки плавления 189-191oC.
Пример 24. Получение диметил 2-метил-5,6-бензимидазолкарбоксилата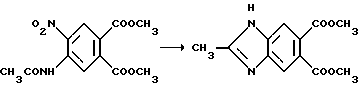
Смесь диметил 4-ацетамидо-5-нитрофталата, метанола и 5% платины на углероде подвергали гидрогенизации в гидрогенизаторе Парра. Реакционную смесь фильтровали через диатомовую землю и фильтрат концентрировали под вакуумом. Полученный таким образом сырой диамин (промежуточное соединение, 48,09 г, 0,180 ммоль) смешивали с пара-толуол/моно/сульфокислотой (51,4 г, 0,270 моль) и толуолом (400 мл) и перемешивали в течение 2 ч при температуре дефлегмации при азеотропном удалении воды, охлаждали и концентрировали под вакуумом. Полученный в результате остаток подвергали рекристаллизации из метанола, чтобы получить пара-толуол сульфонатную соль продукта из заголовка примера. Эту соль растворяли в горячей воде, превращали в щелочную бикарбонатом натрия и экстрагировали метиленхлоридом. Соединенные экстракты промывали соляным раствором, сушили (сульфат магния) и концентрировали под вакуумом, чтобы получить красное твердое вещество. После рекристаллизации порции красного твердого вещества получали продукт из заголовка примера в виде белых кристаллов, температура точки плавления 151-152oC.
Пример 25. Получение диметил 1,2-диметил-5,6-бензимидазолдикарбоксилата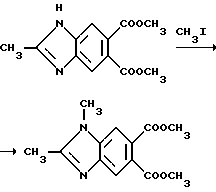
Метилат натрия (1,96 г, 36,3 ммоль) добавляли в раствор диметил 2-метил-5,6-бензимидазолдикарбоксилата (8,50 г, 34,2 ммоль) в метаноле. Реакционную смесь перемешивали 0,5 ч при комнатной температуре, обрабатывали иодметаном (2,15 мл, 34,5 ммоль), перемешивали в течение ночи, подкисляли до pH 6 уксусной кислотой, обрабатывали бикарбонатом натрия до pH 8 и экстрагировали хлороформом. Экстракты соединяли, сушили (сульфат магния) и концентрировали под вакуумом, чтобы получить продукт из заголовка примера в виде розового твердого вещества (6,25 г, 63,7%). Небольшую порцию подвергали рекристаллизации из этилацетата, чтобы получить продукт из заголовка примера в виде розовых кристаллов, температура точки плавления 147-148oC.
Пример 26. Получение 1,2-диметил-5,6-бензимидазолдикарбоновой кислоты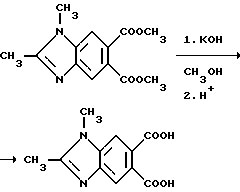
Смесь диметил 1,2-диметил-5,6-бензимидазолдикарбоксилата (2,80 г, 10,7 ммоль), гидрата окиси калия (1,49 г, 26,7 ммоль), воды и метанола перемешивали при комнатной температуре в течение 16 ч, а затем концентрировали под вакуумом. Полученный в результате остаток переносили в минимальное количество воды, охлаждали, подкисляли до pH 3 хлористоводородной кислотой и фильтровали, чтобы получить продукт из заголовка примера в виде розовых кристаллов (2,28 г, 91,2%), температура точки плавления 308-312oC.
Пример 27. Получение 1,2-диметил-5,6-бензимидазолдикарбонового ангидрида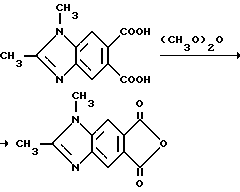
Смесь 1,2-диметил-5,6-бензимидазолдикарбоновой кислоты (1,25 г, 5,34 ммоль) и уксусного ангидрида перемешивали в течение 2 ч при температуре дефлегмации, охлаждали до 5oC и фильтровали. Фильтровальную лепешку сушили, чтобы получить продукт из заголовка примера в виде коричневых игл (1,04 г, 90,1%), температура точки плавления 310-315oC).
Пример 28. Получение 6-[/1-карбамоил-1,2-диметил-пропил/-карбамоил]-1,2- диметил-5-бензимидазолкарбоновой кислоты (I) и 5-[/1-карбамоил-1,2-диметилпропил/карбамоил]-1,2-диметил-6- бензимидазол карбоновой кислоты (II)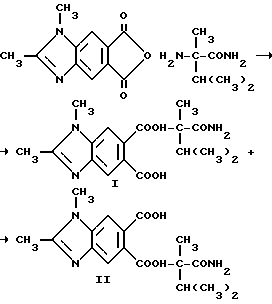
Смесь 1,2-диметил-5,6-бензимидазолдикарбонового ангидрида (0,500 г, 2,31 ммоль), а-метилвалирамида (0,300 г, 2,31 ммоль) и ацетонитрила перемешивали в течение 3 ч при температуре дефлегмации, охлаждали и фильтровали. В результате рекристаллизации фильтровальной лепешки из метанола получали соединение I в виде белого порошка (0,230 г, 28,8), температура точки плавления 169-171oC. Маточную жидкость концентрировали под вакуумом в желтое твердое вещество, которое подвергали рекристаллизации из метанола, чтобы получить соединение из заголовка примера II в виде белой пены (0,240 г, 30,0%), температура точки плавления 145-150oC.
Пример 29. Получение хлордигидрата 6-/4-изопропил-4-метил-5-оксо-2-имидазолин-2-ил/-1,2-диметил-5- бонзимидазолкарбоновой кислоты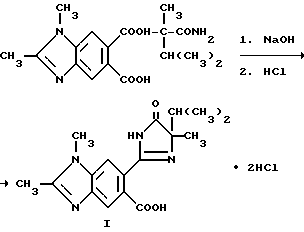
Смесь 6-[/1-карбамоил-1,2-диметилпропил/карбамоил] -1,2-диметил-5- бензимидазолкарбоновой кислоты (0,330 г, 0,910 ммоль) и 2M раствора гидрата окиси натрия (3,0 мл, 6,0 ммоль) перемешивали в течение 2,5 часов при температуре дефлегмации, охлаждали, подкисляли до pH 3 хлористоводородной кислотой и фильтровали. Фильтровальную лепешку подвергали рекристаллизации из этанола, чтобы получить продукт из заголовка примера в виде белого твердого вещества (0,120 г, 31,5%), температура точки плавления 256-258oC.
Пример 30. Получение метил 5-/4-изопропил-4-метил-5-оксо-2-имидазолин-2-ил/-1,2-диметил- бензимидазолкарбоксилата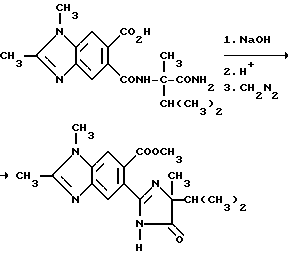
Смесь исходного диамина (0,510 г, 1,47 ммоль) и 2M раствора гидрата окиси натрия (5,0 мл, 10,0 ммоль) перемешивали в течение 2,5 ч при температуре дефлегмации, охлаждали, подкисляли до pH 3 хлористоводородной кислотой и фильтровали. Фильтрат концентрировали под вакуумом, чтобы получить белое твердое вещество, которое растворяли в метаноле и обрабатывали раствором диазометана в простом эфире до тех пор, пока не установится желтая окраска. Реакцию быстро прекращали уксусной кислотой и концентрировали под вакуумом. Полученный в результате остаток подвергали хроматографии (силикагель, 10% метанол:этилацетат элюент), чтобы получить продукт из заголовка примера в виде белого порошка (0,140 г, 27,8%), температура точки плавления 138-140oC.
Пример 31.
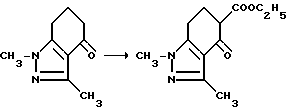
Суспензию гидрида натрия (14,6 г, 0,365 моль, 60% масляная дисперсия) в сухом бензоле при 5-10oC обрабатывали 1,5,6,7-тетрагидро-1,3-диметил-4H-индазол-4-оном (30,0 г, 0,183 моль). Реакционную смесь обрабатывали по каплям этил карбонатом (45,0 мл, 0,365 моль) и этанолом (1,0 мл) при охлаждении, перемешивали в течение 11 ч при температуре дефлегмации, охлаждали до комнатной температуры и обрабатывали уксусной кислотой (24,1 г, 0,400 моль) и водой. Фазы разделяли, органическую фазу удаляли, а водную фазу экстрагировали простым эфиром. Органические фазы соединяли, промывали водой, сушили (сульфат натрия) и концентрировали под вакуумом. Полученный в результате остаток дважды вытесняли толуолом, подвергали рекристаллизации из (2: 1) метилена хлорида/гептана, а второй раз - из 50% тетрагидрофурана/гептана, чтобы получить продукт из заголовка примера (27,6 г, 63,9%), температура точки плавления 76,5-79oC.
Пример 32. Получение этил 4-циано-4,5,6,7-тетрагидро-1,3-диметил-4-/триметил- силиокси/-1H-индазол-5-карбоксилата
Раствор этил 4,5,6,7-тетрагидро-1,3-диметил-4-оксо-1H-индазол-5-карбоксилата (41,0 г, 0,174 моль) в бензоле перемешивали при температуре дефлегмации с азеотропным удалением воды в течение нескольких часов, охлаждали до 65oC, обрабатывали триметилсилил цианидом (58,0 мл, 0,440 моль), перемешивали 15 мин при 65oC, обрабатывали иодидом цинка (2,00 г, 0,00688 моль), перемешивали 6 ч при 65oC, выдерживали в течение ночи при комнатной температуре и концентрировали под вакуумом, чтобы получить продукт из заголовка примера в виде масла, который идентифицировали при помощи ИК- и масс-спектроскопии.
Пример 33. Получение этил 4-циано-6,7-дигидро-1,3-диметил-1H-индазол-5-карбоксилата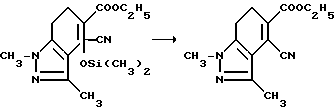
Раствор этил 4-циано-4,5,6,7-тетрагидро-1,3-диметил-4-/три- метилсилилокси/-1H-индазол-5-карбоксилата (28,1 г, 0,119 моль) в пиридине смешивали с оксихлоридом фосфора (43,0 мл, 0,461 моль), перемешивали в течение 5 ч при температуре дефлегмации и концентрировали под вакуумом. Полученный в результате черный остаток разбавляли этилацетатом и водой, и обрабатывали бикарбонатом натрия до pH 6. Фазы разделяли, органическую фазу оставляли, а водную фазу экстрагировали этил ацетатом. Органические фазы соединяли, промывали водным раствором бикарбоната натрия и водой, сушили (сульфат магния) и концентрировали под вакуумом, чтобы получить черный остаток. Остаток растирали с этилацетатом, чтобы получить продукт из заголовка примера в виде рыжевато-коричневого твердого вещества (15,5 г, 52,9%), которое идентифицировали при помощи 1H-ЯМР- и ИК-спектроскопии.
Пример 34. Получение этил 4-циано-1,3-диметил-1H-индазол-5-карбоксилата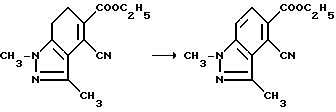
Смесь этил 4-циано-6,7-дигидро-1,3-диметил-1H-индазол-5- карбоксилата (13,5 г, 55,0 ммоль), орто-хлоранила (16,3 г, 66,0 ммоль) и сухого бензола перемешивали в течение 1,5 ч при температуре дефлегмирования, охлаждали и фильтровали. Фильтрат концентрировали под вакуумом, фильтровали через две подушки из нейтральной окиси алюминия и выпаривали до сухого состояния, чтобы получить желтое твердое вещество. В результате рекристаллизации из этилацетата получали продукт из заголовка примера (6,33 г, 47,2%), температура точки плавления 170-175,5oC.
Пример 35. Получение 1,3-диметил-1H-индазол-4,5-дикарбоновой кислоты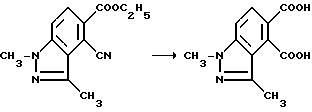
Смесь этил 4-циано-1,3-диметил-1H-индазол-5-карбоксилата (7,03 г, 28,7 ммоль), концентрированной бромистоводородной кислоты (35 мл) и уксусной кислоты (35 мл) перемешивали в течение 2 ч при температуре дефлегмирования, охлаждали, разбавляли водой (200 мл) и перемешивали 2 ч. После фильтрации реакционной смеси получали продукт из заголовка примера в виде бледно-голубого твердого вещества (6,57 г, 97,8% ), температура точки плавления 228,5-232,5oC.
Пример 36. Получение 1,3-диметил-1Н-индазол-4,5-дикарбонового ангидрида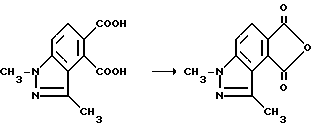
Смесь 1,3-диметил-1H-индазол-4,5-дикарбоновой кислоты (6,30 г, 26,9 ммоль) и уксусного ангидрида перемешивали при температуре дефлегмирования 3 ч, охлаждали до 5oC и фильтровали. Фильтровальную лепешку сушили воздухом, чтобы получить продукт из заголовка примера в виде игл цвета шартреза (4,80 г, 82,5%), температура точки плавления 214-215,5oC.
Пример 37. Получение 4-[/1-карбамоил-1,2-диметилпропил/карбамоил]1,3-диметил- 1H-индазол-5-карбоновой кислоты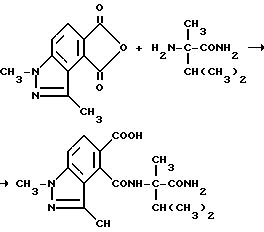
Смесь 1,3-диметил-1H-индазол-4,5-дикарбонового ангидрида (1,60 г, 7,40 ммоль, триэтиламина/1,12 мл, 8,10 ммоль), а-метилваринамида (1,06 г, 8,14 ммоль), диметоксиэтана и диметилформамида перемешивали в течение 16 ч при комнатной температуре и концентрировали под вакуумом. Полученный в результате остаток разбавляли водой и фильтровали, чтобы получить продукт из заголовка примера в виде белого твердого вещества. После подкисления фильтрата до pH получали дополнительный продукт, который идентифицировали при помощи 1H-ЯМР, ИК- и масс-спектроскопии. Общий выход 2,00 г (78,1%).
Пример 38. Получение метил 4-[/1-карбамоил-1,2-диметилпропил/карбамоил]- 1,3-диметил-1H-индазол-5-карбоксилата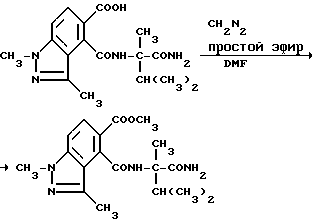
Раствор 4-[/1-карбамоил-1,2-диметилпропил/карбамоил] 1,3- диметил-1H-индазол-5-карбоновой кислоты (0,720 г, 2,07 ммоль) в сухом диметилформамиде обрабатывали достаточным количеством диазометана в простом эфире, чтобы получить постоянную желтую окраску. Избыток диазометана удаляли добавлением уксусной кислоты, реакционную смесь концентрировали под вакуумом. Полученный в результате остаток вытесняли 2 раза ксилолом, подвергали рекристаллизации из 50%-ного водного метанола, чтобы получить продукт из заголовка примера в виде белого твердого вещества, температура точки плавления 221-222oC.
Пример 39. Получение метил 4-/4-изопропил-4-метил-5-оксо-2-имидазолин-2-ил/-1,3-диметил- 1H-индазол-5-карбоксилата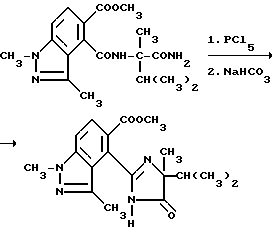
Смесь метил 4-[/1-карбамоил-1,2-диметилпропил/карбамоил]- 1,3-диметил-1H-индазол-5-карбоксилата (0,600 г, 1,67 ммоль), пентахлорида фосфора (1,04 г, 5,00 ммоль) и сухого толуола (7,0 мл) перемешивали в течение 3,5 ч при 90oC, охлаждали и фильтровали. Фильтровальную лепешку промывали толуолом, из нее приготавливали шлам в воде, обрабатывали бикарбонатом натрия (0,370 г, 4,40 ммоль) до pH 8,5 и фильтровали, чтобы получить продукт из заголовка примера в виде белого твердого вещества (0,490 г, 85,7%), температура точки плавления 130-150oC.
Пример 40. Получение 4-/4-изопропил-4-метил-5-оксо-2-имидазолин-2-ил/-1,3- диметил-1H-индазол-5-карбоновой кислоты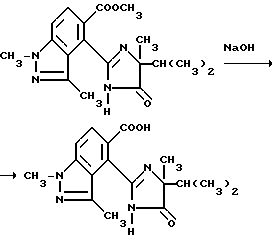
Смесь метил 4-/4-изопропил-4-метил-5-оксо-2-имидазолин-2-ил/- 1,3-диметил-1H-индазол-5-карбоксилата (0,310 г, 0,905 ммоль), 2N раствора гидрата окиси натрия (0,91 мл, 1,82 ммоль) и тетрагидрофурана (11 мл) перемешивали в течение 1,5 ч при 42-52oC, охлаждали в ледяной ванне, подкисляли до pH 3-3,5 при помощи 5N серной кислоты и экстрагировали хлороформом. Органические экстракты сушили (сульфат натрия) и концентрировали под вакуумом, чтобы получить остаток, который подвергали рекристаллизации из ацетонитрила, чтобы получить продукт из заголовка примера в виде белого твердого вещества (0,160 г, 53,2%), температура точки плавления 156,5-166oC.
Пример 41. Получение 1-метилпиррол-3-карбоксальдегида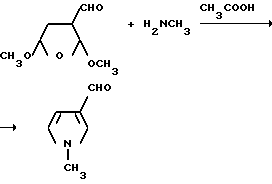
Монометиламин (20 г, 0,64 моля) добавляли через конденсатор с сухим льдом в перемешиваемый раствор 2,5-диметокси-3-тетрагидрофуранкарбоксальдегида (48,1 г, 0,300 моль) в ледяной уксусной кислоте (500 мл) при 10oC. Конденсатор сухого льда заменяли водяным конденсатором и реакционную смесь перемешивали 2 ч при 110oC, охлаждали до комнатной температуры и подвергали дистилляции при 25-30oC/4,0 мм рт. ст. чтобы удалить уксусную кислоту. Остаток разбавляли ледяной водой, промывали простым эфиром, охлаждали, обрабатывали гидратом окиси натрия до pH 7 и экстрагировали метиленхлоридом. Органические экстракты соединяли, сушили (сульфат магния) и концентрировали под вакуумом, чтобы получить продукты из заголовка примера в виде красной жидкости (11,0 г, 33,6%), который идентифицировали при помощи 1H-ЯМР-спектроскопии.
Пример 42. Получение 1-метил-3-винилпиррода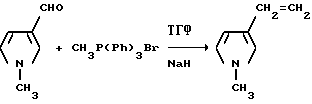
В перемешиваемый шлам гидрида натрия (7,00 г, 0,174 моль, 60%-ная масляная дисперсия) в сухом тетрагидрофуране в атмосфере азота добавляли бромид метилтрифенилфосфония (51,8 г, 0,145 моль). Смесь перемешивали 1 ч при температуре дефлегмирования, охлаждали до 45oC и обрабатывали по каплям раствором 1-метилпиррол3-карбоксальдегида (15,8 г, 0,145 моль) в тетрагидрофуране. Полученную в результате смесь перемешивали 6 дней при комнатной температуре и фильтровали через нейтральную окись алюминия с петролейным эфиром, в качестве дополнительного элюата. Фильтрат концентрировали под вакуумом в желто-белое полутвердое вещество, которое разбавляли петролейным эфиром и снова фильтровали через нейтральную окись алюминия. Бесцветный фильтрат концентрировали под вакуумом, чтобы получить продукт из заголовка примера в виде бледно-желтого масла (10,1 г, 65,2%), которое идентифицировали при помощи 1H-ЯМР-спектроскопии.
Пример 43. Получение гексагидро-а-изопропил-а,1-диметил-6,8- диоксобензо[2,1-b:3,4-c']дипиррол-7/1H/-ацетонитрила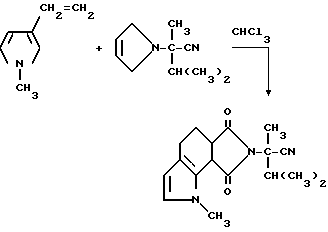
Смесь 1-метил-3-винилпиррола (9,50 г, 0,089 моль), а-изопропил-а-метил-2,5-диоксо-3-пирролин-1-ацетонитрила (17,0 г, 0,089 моль) и хлороформа перемешивали в течение ночи при комнатной температуре и концентрировали под вакуумом в янтарный масляный остаток. Этот остаток подвергали испарительной хроматографии (силикагель, элюирование градиентом: 50% гексаны: метиленхлорид до простого эфира), чтобы получить продукт из заголовка примера в виде оранжевого стекла (13,0 г, 48,8%), которое идентифицировали при помощи 1H-ЯМР-спектроскопии.
Пример 44 Получение 6,8-дигидро-а-изопропил-а, 1-диметил-6,8- диоксобензо[2,1-b:3,4-c']дипиррол-7/1H/-ацетонитрила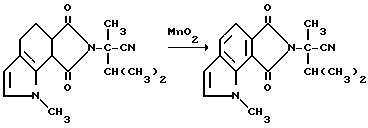
Смесь гексагидро-2-изопропил-а,1-диметил-6,8- диоксобензо[2,1-b:3,4-c'] дипиррол-7/1H/-ацетонитрила (9,30 г, 30,0 ммоль), двуокиси марганца (7,00 г, 80,5 ммоль) и хлорбензола перемешивали при температуре дефлегмирования в течение ночи, обрабатывали дополнительным количеством двуокиси марганца (3,50 г, 40,2 ммоль) и нагревали еще на 16 ч до температуры дефлегмирования. Добавляли третью порцию двуокиси марганца (3,50 г, 40,2 ммоль) и реакционную смесь перемешивали 3 дня при температуре дефлегмирования, охлаждали до 25oC и концентрировали под вакуумом, чтобы получить черный остаток. Остаток подвергали испарительной хроматографии (силикагель, 50% простой эфир:гексан), чтобы получить продукт из заголовка примера в виде желтого твердого вещества (4,30 г, 48,5%), температура точки плавления 134-138oC.
Пример 45. Получение 6,8-дигидро-а-изопропил-а,1-диметил-6,8- диоксобензо[2,1-b:3,4-c']дипиррол-7/1H/-ацетамида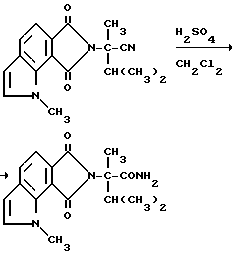
Раствор 6,8-дигидро-в-изопропил-а, 1-диметил-6,8- диоксобензо[2,1-b: 3,4-c'] дипиррол-7/1H/-ацетонитрила (5,30 г, 18,0 ммоль) в метиленхлориде по каплям добавляли в концентрированную серную кислоту при температуре 5-10oC при быстром перемешивании. Ледяную ванну снимали и реакционную смесь перемешивали в течение ночи при окружающей температуре, сливали на измельченный лед, разбавляли метиленхлоридом, обрабатывали 50%-ным раствором гидрата окиси натрия при охлаждении ледяной ванной до pH 3-4 и разделяли. Водный слой экстрагировали метиленхлоридом. Соединенные органические слои сушили (сульфат магния) и концентрировали под вакуумом, чтобы получить остаток в виде оранжевой пены, который подвергали рекристаллизации из метиленхлорида, чтобы получить продукт из заголовка примера в виде желтых кристаллов (1,30 г, 23,2%), температура точки плавления 184-189oC.
Пример 46. Получение 8-изопропил-1,8-диметил-1H-имидазо[1',2':1,2]пирроло[3,4-g]индол-6,9- -диона и 8-изопропил-1,8-диметил-1H-имидазо[2',1':5,1] пирроло[3,4-g]-индол- 7,10-диона (1:1 смесь)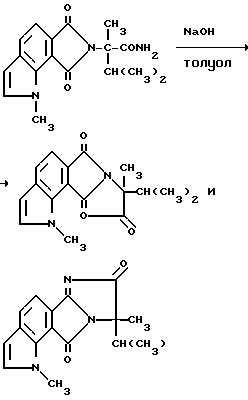
Гидрид натрия (0,240 г, 6,00 ммоль, 60%-ная масляная дисперсия) порциями добавляли в смесь 6,8-дигидро-а-изопропил-а-1- диметил-6,8-диоксобензо[2,1-b: 3,4-c']дипиррол-7/1H/-ацетамида (0,900 г, 2,87 ммоль) и сухого толуола при температуре дефлегмирования. Спустя 1 ч при температуре дефлегмирования смесь охлаждали до комнатной температуры и фильтровали через диатомовую землю. Фильтрат концентрировали под вакуумом, чтобы получить продукт из заголовка примера в виде оранжевого твердого вещества (0,900 г, 100%), который идентифицировали при помощи 1H-ЯМРспектрального анализа.
Пример 47. Получение метил [7-/4-изопропил-4-метил-6-оксо-2-имидазолин-2-ил/-1- метил] индол-6-карбоксидата и метил [6-/4-изопропил-4- метид-5-оксо-2-имидазолин-2-ил/-1-метил]индол-7-карбоксилата (1:1-смесь)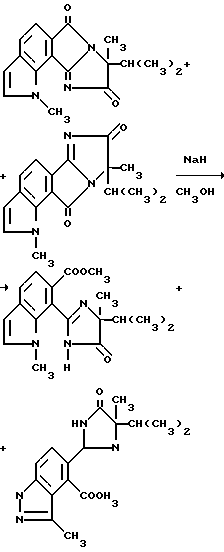
Каталитическое количество гидрида натрия (60%-ная масляная дисперсия (добавляли в раствор 1:1-смеси 8-изопропил-1,8-диметил1H- имидазо[1',2':1,2] пирроло[3,4-g] индол-6,9-диона и 8-изопропил-1,8-диметил-1H-имидазо[2', 1': 5,1] пирроло[3,4-g] -индол -7,10 диона (0,900 г, 3,00 ммоль) в метаноле при комнатной температуре до pH 10. Спустя 5 дней добавляли еще гидрид натрия до pH 10-11 и реакционную смесь нагревали при помощи паровой ванны 1 ч. После охлаждения до комнатной температуры добавляли уксусную кислоту до pH 6 и смесь концентрировали под вакуумом. Остаток разбавляли метиленхлоридом и водой; органический слой сушили (сульфат магния) и концентрировали под вакуумом, чтобы получить продукт из заголовка примера в виде желтой пены (0,350 г, 37,7%). Идентификацию осуществляли при помощи 1H-ЯМР-спектрального анализа.
Пример 48. Получение 7-/4-изопропил-4-метил-5-оксо-2-имидазолин-2-ил/-1- метилиндол-6-карбоновой кислоты и 6-/4-изопропил-4-метил-5-оксо-2-имидазолин-2-ил/-1-метилиндол-7- карбоновой кислоты (1:1-смесь)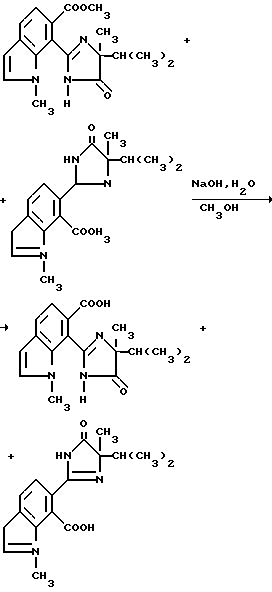
I-I-смесь метил[7-/4-изопропил-4-метил-5-оксо-2-имидазолин- 2-ил/-1-метил] индол-6-карбоксилата и метил[6-/4-изопропил-4-метил-5-оксо-2-имидазолин-2-ил/-1-метил]индол- 7-карбоксилата /0,350 г, 1,07 ммоль/ в метаноле и 1,93N раствора гидрата окиси натрия (0,60 мл, 1,16 ммоль) перемешивали в течение ночи при комнатной температуре. Реакционную смесь концентрировали под вакуумом и остаток разбавляли водой, обрабатывали концентрированной хлористоводородной кислотой до pH 3 и фильтровали, чтобы получить продукт из заголовка примера в виде ярко-желтого твердого вещества (0,140 г), температура точки плавления 122-140oC.
Пример 49. Получение 1-метил-2-винилпиррола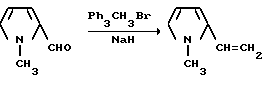
Бромид метилтрифенилфосфония (35,7 г, 0,100 моль) добавляли в шлам гидрида натрия (4,80 г, 0,120 моль, 60%-ная масляная дисперсия) в сухом тетрагидрофуране при 10oC в атмосфере азота. Смесь перемешивали при температуре дефлегмирования 1 ч, охлаждали до 35oC, обрабатывали по каплям раствором 1-метилпиррол-2-карбоксальдегида (10,9 г, 0,100 моль) в тетрагидрофуране, перемешивали 3 дня при окружающей температуре, 2 ч при дефлегмировании и 16 ч при комнатной температуре. Реакционную смесь фильтровали через нейтральную окись алюминия с петролейным эфиром. Прозрачный желтый фильтрат концентрировали под вакуумом, чтобы получить ярко-желтое полутвердое вещество, которое переносили в петролейный эфир и фильтровали через лепешку из диатомовой земли на нейтральной окиси алюминия. Полученный в результате бесцветный фильтрат концентрировали под вакуумом, чтобы получить продукт из заголовка примера в виде прозрачного бесцветного масла /7,80 г, 72,5%/, которое идентифицировали при помощи 1H-ЯМР-спектроскопии.
Пример 50. Получение 3,3а, 4,5,6,8в-гексагидро-а-изопропил-а, 6-диметил1,3- диоксобензо/1,2-b:3,4-c'/дипиррол-2/1H/-ацетонитрила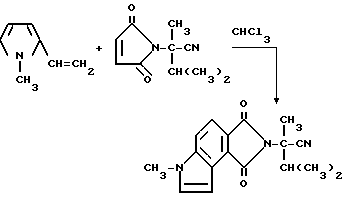
Смесь 1-метил-2-винилпиррола (3,90 г, 36,0 ммоль), а-изопропил-а-метил-2,5-диоксо-3-пирролин-1-ацетонитрила (7,00 г, 36,0 ммоль) и хлороформа перемешивали в течение ночи при комнатной температуре и концентрировали под вакуумом. Полученный в результате остаток подвергали испарительной хроматографии (силикагель, 50%-ный простой эфир: гексаны - элюент), чтобы получить продукт из заголовка примера в виде прозрачного желтого стекла (63,5 г, 58,8%).
Пример 51. Получение 3,6-дигидро-а-изопропил-а,6-диметил-1,3- диоксобензо[1,2-b:3,4-c']дипиррол-2/1H/-ацетонитрила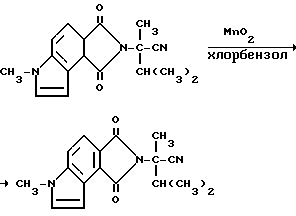
Активированную двуокись марганца (27,0 г, 0,310 моль) добавляли в раствор 3,3а, 4,5,6,8в-гексагидро-а-изопропил-а, 6-диметил-1,3- диоксобензо[1,2--b: 3,4-c']дипиррол-2/1H/-ацетонитрила (51,3 г, 0,167 моль) в хлорбензоле. Реакционную смесь перемешивали в течение ночи при температуре дефлегмирования, охлаждали и фильтровали дважды через диатомовую землю с метиленхлоридом. Фильтрат концентрировали под вакуумом в черное масло, которое подвергали испарительной хроматографии дважды метиленхлоридом, затем смесью гексанов: метиленхлорида, чтобы получить желтое твердое вещество (5,50 г, 11,6% ). В результате рекристаллизации из гексанов: метиленхлорида получали продукт из заголовка примера, температура точки плавления 123-128oC.
Пример 52. Получение 3,6-дигидро-а-изопропил-а,6-диметил-1,3- диоксобензо[1,2-b:3,4-c']дипиррол-2/1H/-ацетамида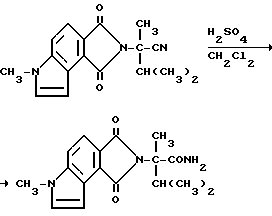
Концентрированную серную кислоту (5,50 мл) добавляли медленно в раствор 3,6-дигидро-а-изопропил-а, 6-диметил-1,3- диоксобензо[1,2-b: 3,4-c'] дипиррол-2/1H/-ацетонитрила (5,50 г, 19,0 ммоль) в метиленхлориде при 10oC. Добавляли измельченный лед. Смесь перемешивали 24 ч при окружающей температуре и сливали на измельченный лед, обрабатывали 6N раствором гидрата окиси натрия до pH 2 и экстрагировали метиленхлоридом. Органический слой отделяли, сушили (сульфат магния) и концентрировали под вакуумом, чтобы получить остаток в виде оранжевой пены. После испарительной хроматографии (силикагель, простой эфир, затем этилацетат элюенты) получали продукт из заголовка примера в виде желтой пены (1,53 г, 25,7%), температура точки плавления 159-168oC.
Пример 53. Получение 8-изопропил-3,8-диметил-1H- имидазо[1', 2':1,2] пирроло[3,4-e] индол-6,9-диона и 8-изопропил-3,8-диметил-1H-имидазо[2',1': 5,1]пирроло[3,4-e]индол- 7,10-диона (1:1-смесь)
3,6-дигидро-а-изопропил-а, 6-диметил-1,3- диоксобензо[1,2-b; 3,4-c']дипиррол-2/1H/-ацетамид (2,60 г, 8,30 моль) порциями добавляли в суспензию гидрида натрия (0,800 г, 16,6 моль, 50%-ная масляная дисперсия) в толуоле при температуре дефлегмирования в атмосфере азота. После перемешивания в течение 20 ч при температуре дефлегмирования смесь фильтровали горячей через диатомовую землю, желтый фильтрат концентрировали под вакуумом в темно-желтый масляный остаток, который растирали с простым эфиром, чтобы получить продукт из заголовка примера в виде желтого твердого вещества (1,60 г, 65%), который идентифицировался при помощи 1H-ЯМР-спектроскопии.
Пример 54. Получение метил [5-/4-изопропил-4-метил-5-оксо-2- имидазолин-2-ил/-1-метил] индол-4-карбоксилата (I) и метил [4-/4-изопропил-4-метил-5-оксо-2-имидазолин-2-ил/-1-метил]индол- 5-карбоксилата (II)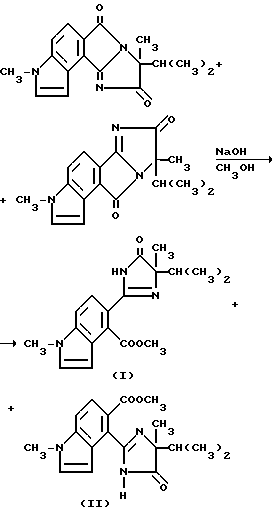
Каталитическое количество гидрида натрия (60%-ная масляная дисперсия) добавляли в раствор 1:1-смеси 8-изопропил-3,8-диметил-1H- имидазо[1',2':1,2] пирроло-[3,4-e] индол-6,9-диона и 8-изопропил-3,8-диметил-1H-имидазо[2',1': 5,1] пирроло[3,4-e] индол-7,1- 0-диона (1,00 г, 3,39 ммоль) в метаноле до pH 10. Реакционную смесь перемешивали в течение 1 ч при комнатной температуре, обрабатывали ледяной уксусной кислотой /2 капли/ и концентрировали под вакуумом. Полученный в результате остаток разбавляли метиленхлоридом и водой. Фазы разделяли и органическую фазу сушили (сульфат магния) и концентрировали под вакуумом, чтобы получить оранжевую пену, которую подвергали испарительной хроматографии (силикагель), чтобы получить 3 фракции, которые находились в порядке уменьшения Rf:
1. Соединение I из заголовка примера, бледно-желтое твердое вещество (0,100 г);
2. Смесь соединений I и II из заголовка примера (0,60 г);
3. Соединение II из заголовка примера, бледно-желтое твердое вещество (0,080 г).
Эти фракции идентифицировали при помощи 1H-ЯМР-спектроскопии.
Пример 55. Получение 5-/4-изопропил-4-метил-5-оксо-2- имидазолин-2-ил/-1-метилиндол-4-карбоновой кислоты и 4-/4-изопропил-4-метил-5-оксо-2-имидазолин-2-ил/-1-метилиндол-5- карбоновой кислоты (1:1-смесь)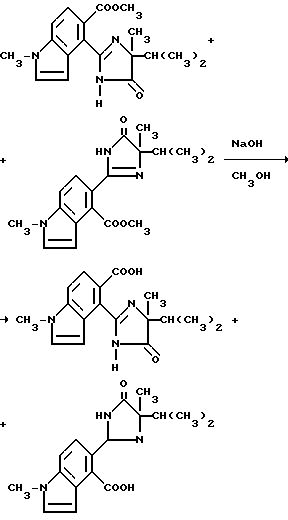
Смесь 1,93N раствора гидрата окиси натрия (0,70 мл, 1,34 ммоль), 1:1-смеси метил[5-/4-изопропил-4-метил-5-оксо-2-имидазолин-2-ил/-1- метил] индол-4-карбоксилата и метил 4-/4-изопропил-4-метил-5-оксо-2- имидазолин-2-ил/-1-метил/индол-5-карбоксилата (0,400 г, 1,22 ммоль) и метанола перемешивали 4 дня при комнатной температуре и концентрировали под вакуумом. Полученный в результате остаток разбавляли водой, охлаждали, подкисляли до pH 3 концентрированной хлористоводородной кислотой и экстрагировали этилацетатом. Органические экстракты сушили (сульфат магния) и концентрировали под вакуумом, чтобы получить смесь продуктов из заголовка примера в виде твердого вещества (0,160 г, 41,9% ), температура точки плавления 266-280oC, которое идентифицировали при помощи 1H-ЯМР-анализа.
Пример 56. Получение диметил 1H-бензотриазол-4,5-дикарбоксилата 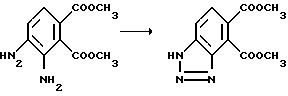
Перемешиваемый раствор метил 3,4-диаминофталата (2,24 г, 10 ммоль) в уксусной кислоте и метиленхлориде обрабатывали одной порцией охлажденным льдом раствором изоамилнитрила (1,6 мл) в 1 мл метиленхлорида при 5oC. Охлаждающую ванну снимали и реакционной смеси давали возможность нагреться в результате экзотермии до 40oC, затем нагревали до 80oC на 30 мин, охлаждали и концентрировали под вакуумом, чтобы получить коричневое масло. Этот остаток подвергали хроматографии, используя окись алюминия E и 2-5% метанол в хлороформе в качестве элюента, чтобы получить продукт из заголовка примера в виде твердого вещества цвета буйволовой кожи (0,73 г, 31% выход), температура точки плавления 147-150oC.
Пример 57. Получение диметил 1-метил-1H-бензотриазол-4,5- дикарбоксилата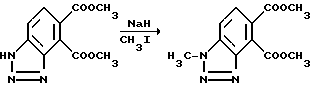
Перемешиваемый раствор диметил 1H-бензотриазол-4,5-дикарбоксилата (10,1 г, 43 ммоль) в диметилформамиде обрабатывали порцией при охлаждении гидридом натрия. Когда выделение газа прекращалось, реакционную смесь обрабатывали по каплям иодметаном (6,7 г, 46 ммоль), перемешивали при комнатной температуре в течение 2 ч, концентрировали под вакуумом и разбавляли смесью хлороформа и воды. Слои разделяли, органический слой промывали соляным раствором, сушили над сульфатом магния и концентрировали под вакуумом, чтобы получить остаток. Остаток подвергали хроматографии, используя силикагель и 35%-ный этилацетат в гексанах, чтобы получить продукт из заголовка примера в виде белого твердого вещества, 3,15 г, температура точки плавления 146-147oC. Структуру определяли при помощи ЯМР-, ЯЭО-экспериментов.
Пример 58. Получение 4-/4-изопропил-4-метил-5-оксо-2-имидазолин- 2-ил/-1-метил-1H-бензотриазол-5-карбоновой кислоты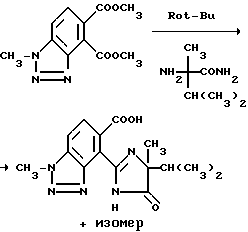
Смесь диметил 1-метил-1H-бензотриазол-4,5-дикарбоксилата (0,73 г, 2,9 ммоль) и а-метилвалерамида (0,40 г, 3,1 ммоль) в толуоле обрабатывали порциями третичн.-бутилатом калия (0,68 г, 6,1 ммоль) в течение 30 минут, нагревали до температуры дефлегмирования на 5 часов, давали возможность охладиться до комнатной температуры в течение 26 часов, обрабатывали 5 мл 2N раствора гидрата окиси натрия и перемешивали в течение 1 часа. Фазы разделяли и органическую фазу экстрагировали водой. Видные фазы соединяли, подкисляли до pH 3 концентрированной HCl, концентрировали под вакуумом до 1/2 первоначального объема, охлаждали до 5-10oC и фильтровали, чтобы получить 3:2-смесь продукта из заголовка примера и его региоизомера в виде белого твердого вещества, температура точки плавления 140-194oC.
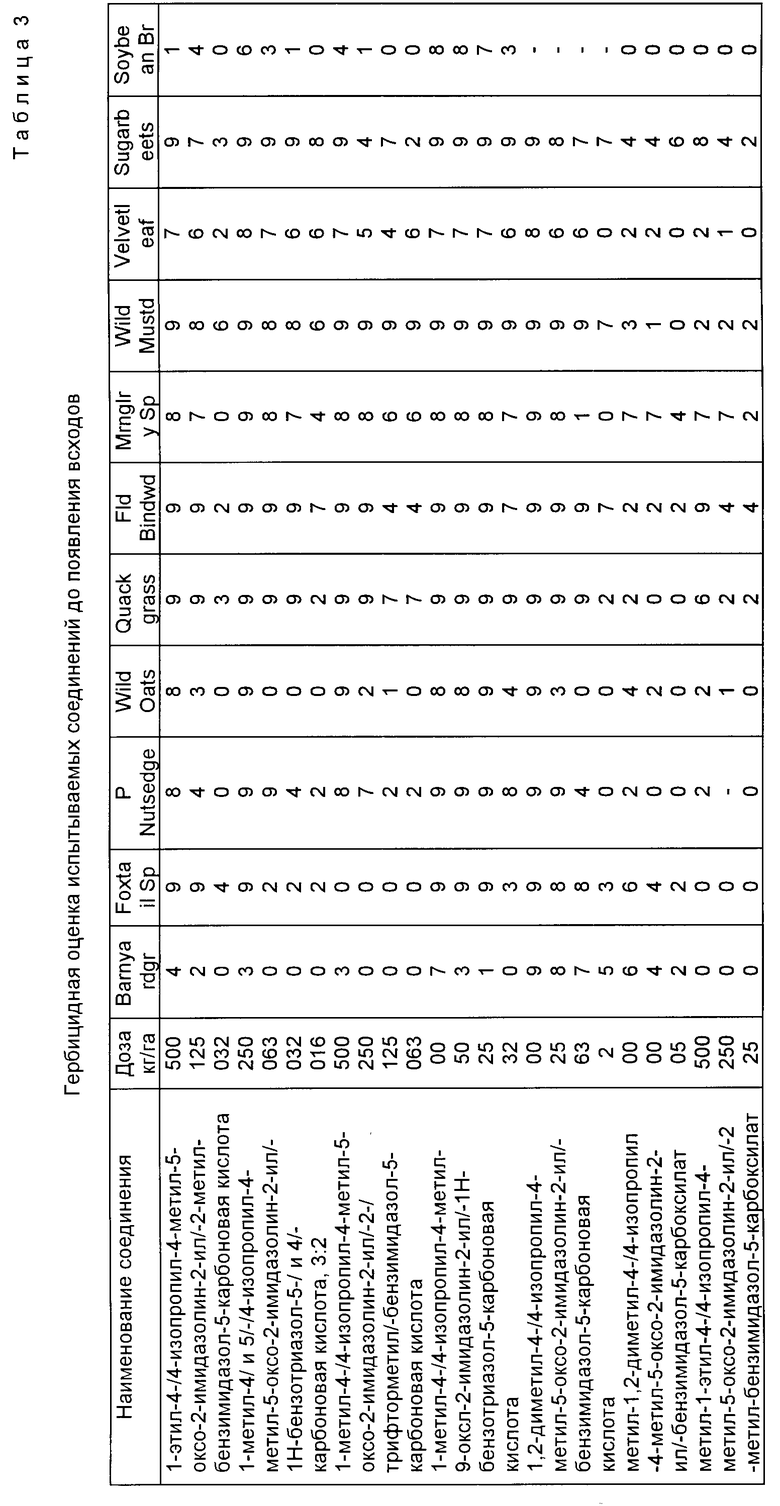
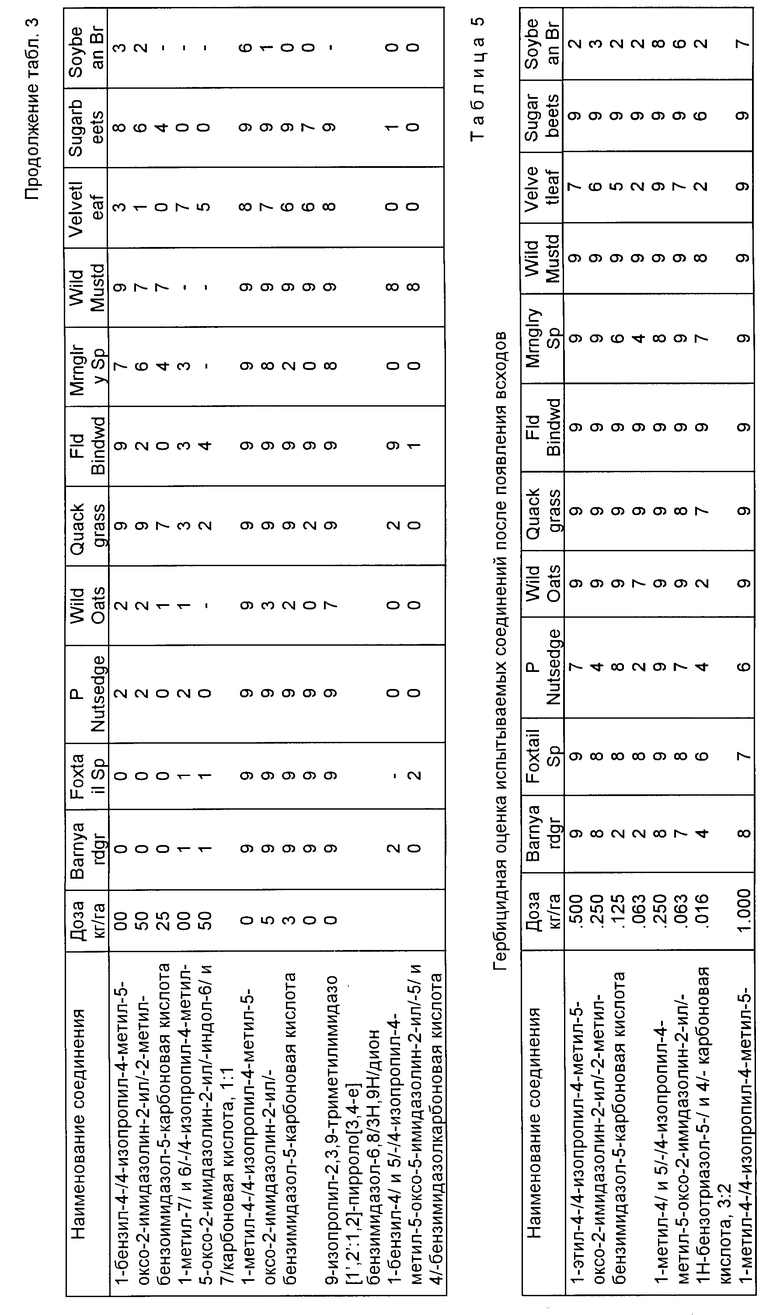
Пример 59. Гербицидная оценка испытываемых соединений до появления всходов и используемые растительные виды представлены в табл. 1, 2 соответственно.
Гербицидную активность до появления всходов имидазолинил бензогетероциклических соединений, являющихся предметом настоящего изобретения, подтверждали при помощи следующих испытаний, в соответствии с которыми семена разнообразных однодольных и двудольных растительных видов индивидуально смешивали с почвой, используемой для выращивания рассады, и высаживали сверху на глубину приблизительно один дюйм (25,4 мм) в чашки емкостью одна пинта (0,568 л). После посадки чашки опрыскивали водным раствором ацетона, содержащим испытываемое соединение. Вышеупомянутый испытываемый раствор состоял из 50/50 смеси ацетон/вода и испытываемого соединения в достаточном количестве, чтобы гарантировать эквивалент примерно 0,016 кг/га-4,0 кг/га активного соединения при применении к почве через распылительный наконечник, работающий при давлении 40 фунтов на кв. дюйм (2,8 кг/см2) в течение заранее определенного времени. Обработанные чашки затем помещали на полки в теплицу и ухаживали в соответствии с известными процедурами.
Через 4-5 недель после обработки испытываемые чашки оценивали в соответствии со шкалой, которая приведена ниже. Результаты гербицидных оценок выражаются по шкале 0-9. Эта шкала основана на визуальном наблюдении за состоянием растения, пороками развития, размерами, хлорозом и общим внешним видом растений по сравнению с контрольными.
Полученные данные собраны в табл. 3. Когда осуществляли больше, чем один эксперимент для данного соединения, приведено среднее значение.
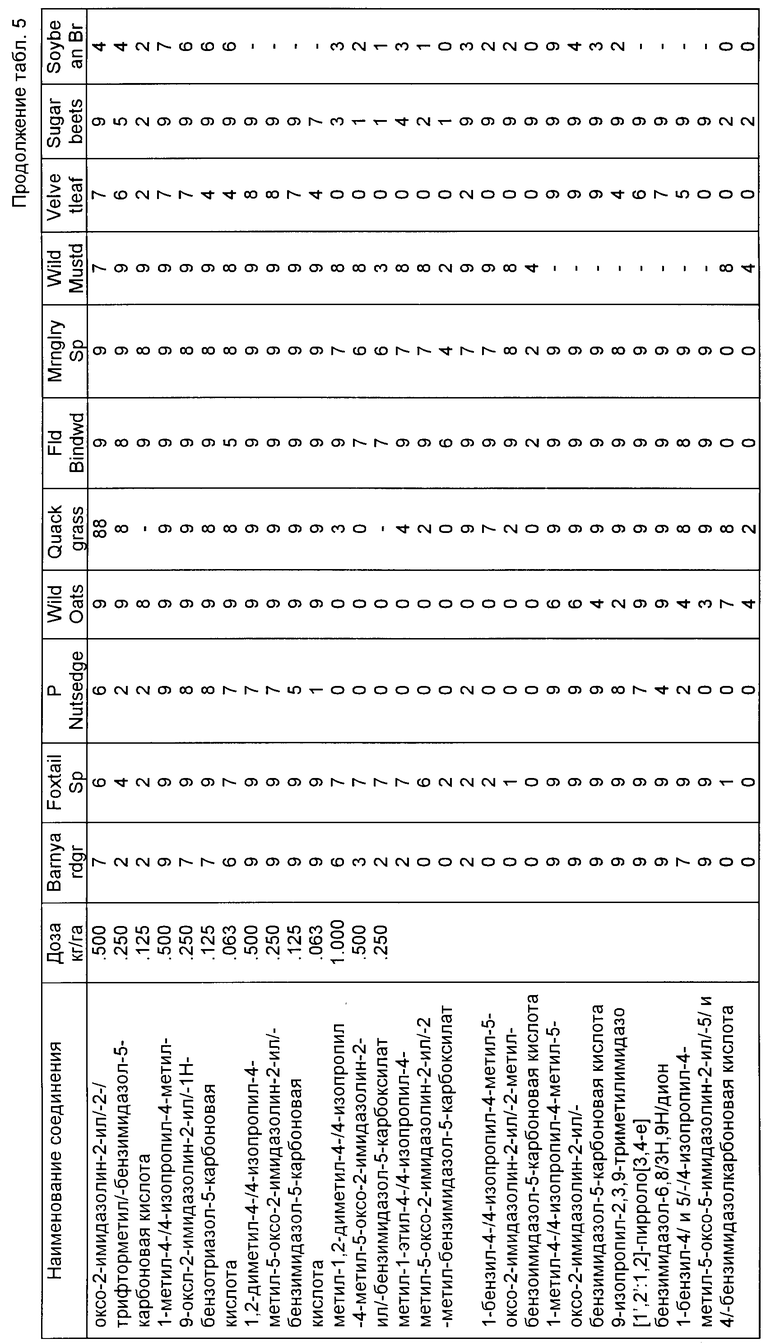
Пример 60. Гербицидная оценка испытываемых соединений после появления всходов.
Используемые растительные виды представлены в табл. 4.
Гербицидную активность после появления всходов имидазолинил бензогетероциклических соединений, являющихся предметом настоящего изобретения, подтверждали при помощи следующих испытаний, в которых самые разнообразные однодольные и двудольные растения обрабатывали растворами испытываемых соединений в водном ацетоне. Вышеупомянутые испытываемые растворы состояли из 50/50 смеси ацетона/воды, содержащей 0,50% NDBYF® 20, монолаурата полиоксиэтилен сорбитана, поверхностно-активного агента, производимого фирмой Атлас Кемикэл Индастриз, и испытываемых соединений в достаточном количестве, чтобы гарантировать эквивалент примерно 0,016 кг/га-1,00 кг/га активного соединения при применении к растениям через распылающий наконечник, работающий под давлением 40 фунтов на кв. дюйм /2,8 кг/см пер./ в течение заранее определенного времени. В этих испытаниях всходы растений выращивали на временных участках в течение примерно двух недель. На растения разбрызгивали испытываемый раствор, помещали в теплицу и ухаживали в соответствии с известными процедурами для теплиц.
Спустя 4-5 недель после обработки растения анализировали и оценивали в соответствии с системой оценок, которая была приведена выше. Гербицидная активность соединений, являющихся предметом настоящего изобретения, очевидна из данных, которые приведены в табл. 5, помещенной ниже.
Если для данного соединения осуществляют более одного испытания, то данные усредняются.

Предлагаются орто-карбокси-/5-оксо-2-имидазолин-2- ил/бензогетероциклические соединения, в которых конденсированная гетероциклическая кольцевая система представляет собой 5-элементное кольцо, содержащее один, два или три атома азота, производные вышеупомянутых гетероциклических соединений и способ их применения для уничтожения однодольных и двудольных растительных видов. 2 с.п. ф-лы, 5 табл.
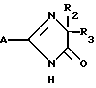
где A означает группу формулы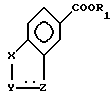
где R1 водород, алкил C1 C4,
X, Y и Z каждый независимо CR или N при условии, что по крайней мере один из X, Y и Z должен быть N,
R водород или алкил C1 C4,
R2 и R3 алкил C1 C4.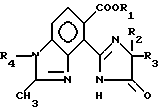
где R1 водород, алкил C1 C4,
R2 и R3 алкил C1 C4,
R4 алкил C1 C4, отличающийся тем, что осуществляют взаимодействие соединения формулы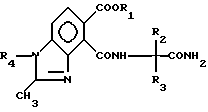
где R1 R4 имеют вышеуказанные значения, с водным основанием с последующим добавлением минеральной кислоты.
| Патент США N 4188487, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
