Изобретение относится к усовершенствованному способу получения N-фосфонометилглицина, известного гербицида и регулятора роста растений.
В области усовершенствования способов получения этого важного для сельского хозяйства соединения проводятся непрерывные исследования.
Так наиболее близкими по сути и достигаемому результату являются способы получения N-фосфонометилглицина, основанные на реакции трихлорида, фосфора с триазином в среде уксусной и соляной кислоты.
Способ по изобретению заключается в том, что N-замещенный азанорборнен общей формулы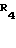

где R2-R7 каждый независимо друг от друга означает водород или С1-С4 алкил, С6Н5 или NO2,
Y группа COOR, CON(R1)2 или CN, где R и R1 каждый независимо друг от друга означает водород или С1-С4 алкил, подвергают взаимодействию с 1,0-5,0 мол. эквивалентами соединения трехвалентного фосфора формулы II или III
 - (OR2)2 где Х атом хлора или брома
- (OR2)2 где Х атом хлора или брома
R2 водород или С1-С4 алкил, в среде растворителя, в частности ароматического углерода, галоидированного ароматического углеводорода, галоидированного углеводорода, низшего алкилового спирта или ацетонитрила, или же карбоновой кислоты С1-С4, предпочтительнее уксусной кислоты, при температуре, при которой реакция протекает с удобной скоростью. Эта скорость зависит от температуры, при которой протонированное N-замещенное азадициклоалкеновое соединение подвергается обратной реакции Дильса-Альдера в конкретном используемом растворителе. В том случае, когда соединения формулы II используют в присутствии негидроксилированного растворителя, необходимо стехиометрическое количество гидроксилированного соединения. Ключевой аспект данной реакции состоит в том, что азотный атом в соединении формулы I является частично тетракоординированным. Типичные температуры находятся в интервале приблизительно 20-120оС, предпочтительнее примерно 35-80оС, причем при указанных температурах продолжительность реакции составляет от 2 до 24 ч. В случае использования соединения трехвалентного фосфора формулы III продуктом реакции является сложный эфир, который можно гидролизовать в соответствии со стандартными процедурами, в частности, удалением растворителя в вакууме, а затем обработкой остатка либо водным раствором минеральной кислоты, либо водным раствором щелочного основания и выдержкой при температуре приблизительно 30-100оС до полного завершения гидролиза и образования N-фосфонометилглицина. В случае использования соединения трехвалентного фосфора формулы II в присутствии уксусной кислоты основным продуктом такой реакции является N-ацетилированное соединение, которое гидрализуют совмещением реакционной смеси приблизительно с 1-5 об.ч. воды и выдержкой при температуре кипения с обратным холодильником. Гидролиз завершают в течение 3-6 ч при 100оС.
Операцию добавления соединения РХ3 проводят с охлаждением до температуры ниже 30оС. В случае использования соединений трехвалентного фосфора либо формулы II, либо формулы III процесс выделения получаемого N-фосфонометилглицина из гидролизной реакционной смеси проводят путем удаления побочных продуктов фильтрованием, концентрированием фильтрата, доведением величины рН до 1,3-1,5 с целью осаждения целевого продукта и отфильтровыванием N-фосфонометилглицинового целевого продукта.
П р и м е р 1. Получение N-фосфонометилглицина через N-замещенный азанорборненовый продукт и трихлорид фосфона в уксусной кислоте.
Перемешиваемую смесь 4,7 г (0,030 мол.) N-карбоксиметилазанорборнена в 40 мл уксусной кислоты обрабатывают 8,2 г (0,060 мол.) трихлорида фосфора в течение 10 мин при 20-25оС. Реакционную смесь выдерживают далее при 35-40оС в течение 6 ч, охлаждают до 25оС в течение 16-часового периода времени, а затем совмещают со 100 мл воды. После фильтрования фильтрат кипятят с обратным холодильником в течение 6 ч, а затем концентрируют до конечного объема 40 мл путем перегонки. После охлаждения до 20оС и фильтрования фильтрат обрабатывают добавлением в него 50%-ного раствора гидроокиси натрия до величины рН 1,5. Далее эту смесь концентрируют и охлаждают с получением твердого осадка. Устанавливают, что этот твердый продукт является соединением, указанным в заголовке примера, его выход, как показывает ЖХВД анализ, составляет 46% а степень чистоты 87%
П р и м е р 2. Получение N-фосфонометилглицина через N-замещенный азанорборненовый продукт и трихлорид фосфора в пропионовой кислоте.
Перемешиваемую смесь N-карбоксиметилазанорборнена (4,6 г, 0,030 мол.) с 40 мл пропионовой кислоты в течение 10-минутного периода времени обрабатывают при 20-25оС 8,2 г (0,060 мол.) трихлорида фосфора. Затем реакционную смесь выдерживают при 45-50оС в течение 4 ч с последующей выдержкой при 74-80оС в течение 1 ч. После выпаривания основной части пропионовой кислоты в вакууме остаток диспергируют в 75 мл воды и выдерживают при температуре кипения с обратным холодильником в течение 16 ч. В результате охлаждения до 25оС и фильтрования реакционной смеси получают фильтрат, который содержит продукт, указанный в заголовке примера, с достижением 55-60%-ного выхода, как это устанавливают ЖХВЛ-анализом.
П р и м е р 3. Получение N-фосфонометилглицина через N-карбоксиметилазанорборненовый продукт и диметилфосфит.
Перемешиваемую смесь 25% -ного водного раствора N-карбоксиметилазанорборнена (5,9 г, 0,010 мол.) с 15 мл уксусной кислоты обрабатывают 1,65 г (0,015 мол. ) диметилфосфита и выдерживают при 80оС в течение 3 ч. Далее реакционную смесь охлаждают до 25оС и концентрируют в вакууме. Остаток диспергируют в 10 мл концентрированной соляной кислоты и 4 мл воды и кипятят с обратным холодильником в течение 3 ч.
П р и м е р 4. Получение N-фосфонометилглицина через N-карбоэтоксиметилазанорборненовый промежуточный продукт и трихлорид фосфора в уксусной кислоте.
Перемешиваемую смесь 9,05 г (0,050 мол.) N-карбоэтоксиметилазанорборнена с 25 мл уксусной кислоты обрабатывают 10,3 г (0,075 мол.) трихлорида фосфора при температуре 15-25оС, а затем выдерживают при 37-43оС в течение 6 ч. После охлаждения до 25оС реакционную смесь по каплям добавляют в 100 мл воды, приготовив шлам. Твердый материал удаляют фильтрованием, а фильтрат концентрируют приблизительно до 50 мл остаточного объема перегонкой при 100оС, причем за этот промежуток времени N-ацетиловое соединение гидролизуется. Смесь охлаждают до 25оС и фильтруют. Величину рН фильтрата доводят до 1,4 добавлением 50%-ной гидроокиси натрия и охлаждают до 10оС. Образующийся осадок отфильтровывают, получив 6,0 г N-фосфонометилглицина с достижением 66% -ного выхода при 93%-ной степени чистоты, как это устанавливают ЖХВД-анализом.
П р и м е р 5. Получение N-фосфонометилглицина через N-карбоэтоксиметилазанорборненовый продукт и диметилфос- фит.
Перемешиваемый раствор 1,8 г (0,010 мол.) N-карбоэтоксиметилазанорборнена в 10 мл ацетонитрила обрабатывают 2,2 г (0,020 мол.) диметилфосфита и кипятят с обратным холодильником в течение 2 ч. Реакционную смесь охлаждают до комнатной температуры и концентрируют в вакууме. Остаток диспергируют в 10 мл 3 н. соляной кислоты и кипятят с обратным холодильником в течение приблизительно 8 ч. ЖХВД-анализ показывает 50%-ный выход N-фосфонометилглицина.
П р и м е р 6. Получение N-фосфонометилглицина через N-карбоэтоксиметилазанорборненовый промежуточный продукт и 1,0 эквивалент диэтилфосфита.
Перемешиваемый раствор 1,8 г (0,010 мол.) N-карбоэтоксиметилазанорборнена в 10 мл ацетонитрила обрабатывают 1,4 г (0,010 мол.) диэтилфосфита и кипятят с обратным холодильником в течение 2 ч. Реакционную смесь охлаждают до комнатной температуры и концентрируют в вакууме. Остаток диспергируют в 15 мл 3,4 н. раствора гидрата окиси натрия и кипятят с обратным холодильником в течение 3 ч, а затем охлаждают до 30оС, обрабатывают концентрированной соляной кислотой, добавляя ее до величины рН, равной 1,5, и фильтруют. Анализ фильтрата ЖХВД указывает на 40%-ный выход N-фосфонометилглицина.
П р и м е р 7. Получение N-фосфонометилглицина через N-карбоэтоксиметилазанорборнена и 2,0 эквивалента диэтилфос- фита.
Перемешиваемый раствор 1,8 г (0,010 мол.) N-карбоксиметилазанорборнена в 10 мл этанола обрабатывают 2,7 г (0,20 мол.) диэтилфосфита и кипятят с обратным холодильником в течение 2 ч. Затем реакционную смесь охлаждают до 25оС и концентрируют в вакууме. Остаток диспергируют в концентрированной и кипятят с обратным холодильником до полного завершения гидролиза, как показывает ЖХВД-анализ. Затем реакционную смесь охлаждают до 25оС и обрабатывают добавлением 50%-ной гидроокиси натрия до величины рН 1,5 и фильтруют. Фильтрат концентрируют в вакууме и остаток суспендируют в воде, а затем фильтруют, получая с достижением 40%-ного выхода продукт, указанный в заголовке примера, в виде твердого вещества, идентифицированного ЖХВД- и ЯМР-анализами.
П р и м е р 8. Получение N-фосфонометилглицина через N-метоксиметил-5-метилазанорборнен.
Раствор 0,91 г (5,0 ммол.) N-карбометоксиметил-5-метилазанорборнена в уксусной кислоте обрабатывают добавлением отдельными порциями 1,03 г (7,5 ммол. ) трихлорида фосфора, выдерживают при температуре 45-50оС в течение 5 ч, обрабатывают водой и кипятят с обратным холодильником в течение 5 ч. Далее реакционную смесь охлаждают до комнатной температуры, фильтруют и фильтрат концентрируют в вакууме, получая продукт, указанный в заголовке данного примера с достижением 61%-ного выхода, как определяют ЖХВД-анализом.
П р и м е р 9. Получение N-фосфонометилглицина через N-цианометилазанорборнен.
Раствор 9,80 г (0,073 мол.) N-цианометилазанорборнена в уксусной кислоте обрабатывают 11,6 г (0,085 мол.) трихлорида фосфора в течение 5-минутного периода при температуре 25-37оС, выдерживают при температуре 50оС в течение 3 ч, обрабатывают водой и прокипятили с обратным холодильником до полного завершения гидро- лиза, как это устанавливают ЖХВД-анализом. Далее реакционную смесь охлаждают до комнатной температуры и фильтруют. ЖХВД-анализ фильтрата и фильтровального пирога показывает достижение 24%-ного выхода продукта, указанного в заголовке примера.
П р и м е р 10. Получение N-фосфонометилглицина через N-карбоксамидометилазанорборнена.
Раствор 5,0 г (0,033 мол.) N-карбоксамидометилазанорборнена в уксусной кислоте обрабатывают 5,3 г (0,038 мол.) трихлорида фосфора в течение 2-минутного периода времени, выдерживают при температуре 50оС в течение 3,5 ч, обрабатывают водой и кипятят с обратным холодильником до завершения гидролиза, как это определяют ЖХВД-анализом. Реакционную смесь охлаждают и фильтруют. ЖХВД-анализ фильтрата и фильтровального пирога доказывает 42%-ный выход продукта, указанного в заголовке данного примера.
П р и м е р 11. Получение N-фосфонометилглицина через N-карбометоксиметилазанорборненовый продукт в среде различных кислороднесодержащих растворителей.
Перемешиваемый раствор N-карбометоксиметилазанорборнена (15,9 г, 0,095 мол. ) в кислороднесодержащем растворителе обрабатывают 4,3 г (0,239 мол.) воды с последующим добавлением в течение 10-минутного периода времени трихлорида фосфора. Затем реакционную смесь выдерживают при 50оС в течение 4 ч, охлаждают, обрабатывают дополнительным количеством воды и кипятят с обратным холодильником до завершения гидролиза, как это показывает ЖХВД-анализ. Реакционную смесь охлаждают и фильтруют. Фильтрат разделяют и водную фазу анализируют ЖХВД в присутствии продукта, указанного в заголовке данного примера.
В ходе проведения каждого эксперимента порцию конечной водной фазы концентрируют в вакууме и продукт, указанный в заголовке примера, идентифицируют 1Н- и 13С-ЯМР-спектральными анализами. Данные приведены в таблице.
Данный способ отличается от описанных в патентах США способов получения N-фосфонометилглицина тем, что используются легкодоступные и недорогостоящие материалы и достигается более высокий выход (до 80 против 40%).
Использование: в качестве гербицида и регулятора роста растений. Сущность изобретения: продукт N-фосфонометилглицин. Реагент 1: азабициклоалкан ф-лы 1, где R2-R7 - независимо друг от друга водород или C1-C4 -алкил, C6H5 или NO2, Y - означает группу COOR, CON(R1)2 или CN, где R и R1 - независимо означают водород или C1-C4 алкил. Реагент 2: соединение трехвалентного фосфора ф-лы 2 или 3. Условия реакции: в присутствии кислоты и растворителя при 25 - 200°С. Структура соединений ф-л I, II, III.  PX3 (II)
PX3 (II)  1 табл.
1 табл.
СПОСОБ ПОЛУЧЕНИЯ N-ФОСФОНОМЕТИЛГЛИЦИНА взаимодействием азотсодержащего гетероциклического соединения с соединением трехвалентного фосфора в присутствии кислоты и растворителя, отличающийся тем, что в качестве азотсодержащего гетероциклического соединения используют азабициклоалкан общей формулы I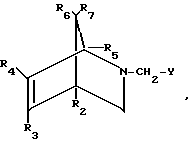
где R2 R7, каждый независимо друг от друга, водород или С1 С4-алкил, С6Н5 или NO2;
Y группа COOR, CON(R1)2 или CN, где R и R1, каждый независимо друг от друга, водород или С1 С4-алкил,
подвергают взаимодействию с приблизительно 1,0 5,0 моль соединения трехвалентного фосфора общих формул II или III
PX3;
где X хлор или бром;
R2 водород или С1 С4-алкил,
при 25 200oС, затем в случае соединения формулы III, удаляют растворитель из реакционной смеси и обрабатывают последнюю водным раствором минеральной кислоты или водным раствором основания щелочного металла, либо, в случае соединения формулы II, охлаждают реакционную смесь до 15 25oС, смешивают с водой, фильтруют и гидролизуют.
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
