Изобретение относится к новым производным бензола, промотирующим выработку или секрецию фактора роста нервной ткани (ФРН) и в то же время оказывающим слабое побочное действие.
Сообщение об открытии ФРН было сделано в 1954 г. Леви-Монталейни и др. ФРН является питательным веществом и фактором роста, необходимым для роста и поддержания функционирования нервных тканей. Недавние исследования, проведенные на животных, позволили установить, что ФРН ускоряет восстановление поврежденной периферической нервной ткани и является эффективным терапевтическим средством при лечении дисфункций центральной нервной системы, особенно болезни Альцгеймера и церебральной ишемии.
Вместе с тем, ФРН является белком с большой молекулярной массой (мол.масса мономерной формы равна 13000, димерной формы 26000), и поэтому имеются проблемы, связанные с его введением в организм в виде лекарства, и с общими требованиями к безопасности.
Известно также, что катехиновые неротрансмиттеры, такие как адреналин и норадреналин, и аналоги катехина могут промотировать образование ФРН. Эти соединения обладают побочным действием, в особенности связанным с нервным возбуждением.
В Европейском патенте N 399814, опубликованном 28 ноября 1990 г. раскрываются производные фенола, которые промотируют выработку и секрецию фактора нервной ткани человека. Родственные соединения, имеющие такое же применение, раскрываются в Японской патентной заявке N 1-217211, поданной 25 августа 1989 г. и опубликованной в форме Японского патента Кокаи N 3-83921 9 апреля 1991 г.
Целью изобретения является разработка производных бензола, представляющих собой эффективные лекарственные средства, промотирующие фактор роста нервной ткани или использующихся в качестве интермедиатов при получении таких лекарств. Более конкретной целью изобретения является разработка вышеуказанных лекарственных средств, обладающих слабым побочным действием, в особенности низким уровнем нервного возбуждения. Другие цели изобретения включают разработку лекарственных композиций для терапевтического лечения повреждений периферической нервной системы и лечения нарушений в работе центральной нервной системы, в особенности болезни Альцгеймера и мозговой ишемии.
Согласно предлагаемому изобретению разработаны новые производные бензола, отвечающие общей формуле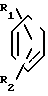 _
_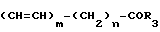 (I) в которой R1 аминогруппа или нитрогруппа;
(I) в которой R1 аминогруппа или нитрогруппа;
R2 аминогруппа, ацетоксигруппа, бензилоксигруппа, гидроксильная группа или трифторацетамидо; группа, выбранная из числа членов, образующих группу А заместителей, или защищенная гидроксильная группа;
R3 аминогруппа или замещенная аминогруппа, содержащая в качестве заместителей 1 или 2 группы из числа заместителей группы А;
m целое число от 0 до 1,
n целое число от 0 до 2.
Группа А заместителей состоит из следующих членов: С1-С4-алкильных групп, содержащих в качестве заместителей от 1 до 3 групп, выбранных из заместителей группы В, гетероциклильных групп и метоксикарбонилзамещенных гетероциклильных групп.
Группа В заместителей состоит из следующих членов: атомов галогена, арилкарбонильных и С1-С4-алкильных групп; гетероциклильные группы представляют собой 5-6-членные ароматические или карбоциклические кольца, содержащие от 1 до 2 гетероатомов, выбранных из атомов азота, кислорода и серы, необязательно сконденсированные с одним арильным кольцом, при условии, что когда m 0, n представляет собой целое число 2, а также их соли.
Соединение формулы I, в которых R1 нитрогруппа, являются промежуточными для соединений формулы I, в которых R1 аминогруппа.
Соединение формулы I предлагаемого изобретения могут существовать в виде солей. Примеры предпочтительных солей включают: соли неорганических кислот, таких как галогеноводородные кислоты, например гидрохлориды, гидрофториды, гидробромиды и гидроиодиды, а также соли других неорганических кислот, например нитраты перхлораты, сульфаты или фосфаты, соли органических кислот, таких как алкансульфокислоты, более конкретно могущих быть галогенозамещенными алкансульфокислот с числом атомов углерода в алкильной группе от 1 до 3 и с числом атомов галогена от 1 до 5, например метансульфонаты, трифторметансульфонаты, этансульфонаты, трифторэтансульфонаты или пентафторэтансульфонаты, таких как арилсульфокислоты, более конкретно могущих быть алкилзамещенными арилсульфокислот с числом атомов углерода в алкильной группе от 1 до 3 и с числом атомов галогена от 1 до 5, например метансульфонаты, трифторметансульфонаты, этансульфонаты, трифторэтансульфонаты или пентафторэтансульфонаты, таких как арилсульфокислоты, более конкретно могущих быть алкилзамещенными арилсульфокислот с числом алкильных заместителей, каждый из которых содержит от 1 до 3 атомов углерода, например бензолсульфонаты или n-толуолсульфонаты, а также других органических кислот, например фумараты, сукцинаты, цитраты, тартраты, оксалаты или малеаты, а также соли аминокислот, например глютаматы или аспараты. Примеры предпочтительных солей включают также ониевые соли, такие как соли, образующие, когда член группы А заместителей является заместителем при третичном атоме азота, например, когда член группы А заместителей является четвертой группой при насыщенном атоме азота, через который гетероциклическая группа R3 связана с остальной частью соединения. Соли, являющиеся фармацевтически приемлемыми, составляют одну из отличительных особенностей описываемого изобретения.
Соединения предлагаемого изобретения могут существовать в оптически активных формах. При наличии в молекуле асимметрического атома углерода возможно существование стереоизомеров с R конфигурацией и S-конфигурацией. Если m равно 1, то возможно существование геометрических изомеров. Изобретение охватывает все эти индивидуальные изомеры и любые их смеси.
Активность в промотировании ФРН
Фурукава и др. сделали сообщение о том, что образующие фибробласты LM-клетки из соединительной ткани мышей могут вырабатывать и секретировать относительно большое количество ФРН и что катехинамины ускоряют выработку и секрецию ФРН (I. Biol. Chem. 261, 6039-6047, 1986). По методике испытаний, описанной в статье Фурукавы, были определены уровни активности в выработке и секреции ФРН при использовании соединений согласно предлагаемому изобретению и хорошо известных промоторов ФРН-эпинефрина, изопротеренола, L-DOPA и кофеиновой кислоты. Соединения согласно изобретению испытывали при концентрации 10 γ/мл, а известные соединения при концентрации 20 γ/мл.
Для выращивания LМ-клеток использовали питательную среду 199, содержащую 0,5% пептона [о питательной среде 199 см. например: Морган и др. Proc. Soc. Exp. Brol. Med. 73,1 (1950) или Морган и др. I.Natl. Cancer Inct. 16,557 (1955)] В каждую ячейку имеющей 24 ячейки чашки для выращивания клеточных культур помещали около 5 х 104/М-клеток и выращивали их до слияния, используя инкубатор с СО2 (37оС, 5% СО2). После удаления питательной среды выращенные клетки промывали один раз промывным раствором, который представлял собой среду 199, содержащую 0,5% бычьего сывороточного альбумина (Фракция V, Сигма). Испытуемые соединения добавляли до достижения вышеуказанной концентрации к среде 199, содержащей 0,5% бычьего сывороточного альбумина, и полученным составом обрабатывали 0,5 мл LМ-клеток. После выращивания LM-клеток в инкубаторе со СО2 в течение 24 ч среду отделяли и определяли содержание ФРН.
Количественное определение ФРН осуществляли с помощью метода иммуноферментного анализа 3 (Коршинг, Тоенен и др. Рroc.Natl.Acad.Sci. USA, 80, 3513-3516, 1983). С помощью пипетки в каждую ячейку чашки из полистирола, имеющей 96 ячеек, помещали 75 мкл раствора антитела на β-ФРН мыши (0,3 мкг/мл, рН 9,6, Берингер Маннгейм). Чашку оставляли на 1 ч при комнатной температуре, затем антитело удаляли троекратной промывкой промывочным раствором. Затем в ячейки с помощью попитки вносили по 5 мкл стандартного раствора β-ФРН (Вако Пьюре Кемикал Индастриз Лтд) или отделенной от культуры клеток среды. Чашку оставляли на 68 ч при комнатной температуре, после чего стандартный раствор β-ФРН или испытуемый раствор удаляли, и каждую ячейку промывали три раза. Затем в каждую ячейку вносили по 50 мкл раствора моноклонального антитела на β-ФРН (100 миллиединиц) мл, рН 7, Берингер Маннгейм) меченного β-галактозидазой. Чашку оставляли стоять при 4оС на 15-18 ч, после чего меченное ферментом антитело удаляли, ячейки трижды промывали и пипеткой вносили в каждую из них 100 мкл раствора хлорфенолового красного β-D-галактопиранозида (1 мг/мл, рН 7,3, Берингер Маннгейм). Давали развиться окраске (по истечении 2-3 ч при комнатной температуре) и определяли поглощение при 570 нм.
Количество ФРН рассчитывали согласно стандартной кривой. Результаты представлены в виде относительных величин (в), которые даны по отношению к количеству ФРН, вырабатываемому и секретируемому клетками, которые не были обработаны испытуемыми соединениями. Численные величины (% от контроля) представляют собой средние значения для трех ячеек с контролем без добавления испытуемых соединений. Известное от контроля соединение Эпинефрин 140±24 Изопротеренол 168±22 L-DOPA 117±7 Кофеиновая кислота 123±14 Соединение примера от контроля 22 380 34 606 40 276 43 491 53 315
Нетрудно видеть, что новые производные фенила согласно описываемому изобретению включают соединения, обладающие очень высокой активностью в промотировании выработки и секреции ФРН. Они, кроме того, являются низкотоксичными. Новые активные производные можно использовать при терапевтическом лечении слабоумия, церебральной ишемии и различных типов нервных дисфункций.
Таким образом, предлагаемое изобретение обеспечивает лекарственные композиции, которые содержат соединение общей формулы I, исключая соединения-интермедиаты, в которых R1 представляет собой нитрогруппу, и фармацевтически приемлемый носитель.
Примеры способов введения активных соединений I изобретения в организм включают: оральное введение в виде таких лекарственных форм, как таблетки, капсулы, гранулы или сиропы, и парэнтеральное введение в виде составов для инъекций и суппозиториев. Лекарственная композиция может быть приготовлена с использованием соответствующих добавок, таких как носители, связующие, дезинтеграторы, смазывающие вещества, стабилизаторы или корригенты, в соответствии с известными методиками. Дозировка варьируется в зависимости от характера симптомов и возраста пациента, но обычно составляет от 0,1 до 1000 мг/кг в день, предпочтительно от 1 до 100 мг/кг в день, эта дневная дозировка может быть введена взрослому пациенту в один прием или разделена на несколько меньших доз.
Соединения изобретения, отвечающие общей формуле I, могут быть получены способом, который также обеспечивается изобретением, способ включает образование амида по реакции между реакционноспособным производным карбоновой кислоты общей формулы II с соединением общей формулы III с получением соединения I изобретения в соответствии с нижеследующей схемой реакции: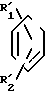 _
_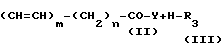
________________→ 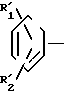 (CH=CH)m-(CH2)n-
(CH=CH)m-(CH2)n-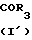 +H-Y где R11 представляет собой нитрогруппу, замещенную аминогруппу, содержащую в качестве заместителей 1 или 2 группы, выбранные из числа членов группы А заместителей согласно ее определению в п. I формулы изобретения, или защищенную аминогруппу; R2' представляет собой замещенную аминогруппу, содержащую в качестве заместителей 1 или 2 группы, выбранные из числа членов группы А заместителей согласно ее определению в п. I формулы изобретения, защищенную аминогруппу, замещенную гидроксильную группу, содержащую в качестве заместителя группу, выбранную из числа членов группы А заместителей согласно ее определению в п. 1 формулы изобретения, или защищенную гидроксильную группу; Y представляет собой уходящую группу, а R3, m и n имеют вышеприведенные значения, после чего этот продукт превращают, если это необходимо или желательно, в другое соединение согласно предлагаемому изобретению, например, следующим образом:
+H-Y где R11 представляет собой нитрогруппу, замещенную аминогруппу, содержащую в качестве заместителей 1 или 2 группы, выбранные из числа членов группы А заместителей согласно ее определению в п. I формулы изобретения, или защищенную аминогруппу; R2' представляет собой замещенную аминогруппу, содержащую в качестве заместителей 1 или 2 группы, выбранные из числа членов группы А заместителей согласно ее определению в п. I формулы изобретения, защищенную аминогруппу, замещенную гидроксильную группу, содержащую в качестве заместителя группу, выбранную из числа членов группы А заместителей согласно ее определению в п. 1 формулы изобретения, или защищенную гидроксильную группу; Y представляет собой уходящую группу, а R3, m и n имеют вышеприведенные значения, после чего этот продукт превращают, если это необходимо или желательно, в другое соединение согласно предлагаемому изобретению, например, следующим образом:
1. Если R1' представляет собой нитрогруппу, то осуществляют восстановительное превращение в соединение общей формулы I, в котором R1представляет собой аминогруппу.
2. Если R1' представляет собой защищенную аминогруппу, то удаляют защитную группу, и/или
3. Если R2' представляет собой защитную аминогруппу или защищенную гидроксильную группу, то удаляют защитную группу.
Способ включает реакцию между реакционноспособным производным карбоновой кислоты, имеющим общую формулу II и соединением, имеющим общую формулу III. Предпочтительно эту реакцию проводят в среде инертного растворителя в присутствии основания, получая в результате соединение формулы I' и HY.
Природа уходящей группы Y не является существенной, предпочтительно Y представляет собой уходящую группу из числа тех групп, которые обычно используются в подобных нуклеофильных реакциях получения амидов. Как правило, уходящая группа Y представляет собой атом галогена, такого как хлор, бром или иод, алкансульфонилоксигруппу с числом атомов углерода в алкильной группе от 1 до 6, такую как метансульфонилокси или этансульфонилоксигрупп, галогеноалкансульфонилоксигруппу с числом атомов углерода в алкильной группе от 1 до 3 и с числом галогеновых заместителей от 1 до 6, такую как трифторметансульфонилокси- или пентафторэтансульфонилоксигруппа, или арилсульфонилоксигруппу, могущую иметь от 1 до 3 алкильных заместителей, каждый из которых содержит от 1 до 3 атомов углерода, такую как бензолсульфонилокси- или n-толуолсульфонилоксигруппа. Более предпочтительно группа Y представляет собой атом галогена.
Круг инертных растворителей не ограничивается указанием каких-либо конкретных типов этих растворителей, при условии, что растворитель не оказывает влияния на протекание реакции и может растворять какие-то количества исходных реагентов. Примеры предпочтительных растворителей включают ароматические углеводороды, такие как бензол, толуол или ксилол, галогеноуглеводороды, такие как хлористый метилен или хлороформ, или простые эфиры, такие как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран или диоксан.
Круг оснований также не ограничивается указанием каких-либо конкретных типов, при условии, что основание является эффективным для реакции такого рода. Примеры предпочтительных оснований включают органические основания, такие как триэтиламин, трибутиламин, диизопропилэтиламин, N-метилморфолин, пиридин, 4-(N,N-диметиламино) пиридин, N,N-диметиланилин, N,N-диэтиланилин, 1,5-диазабицикло 4.3.0 нона-5-ен, 1,4-диазабицикло [2.2.2] октан (ДАБЦО) или 1,8-диазабицикло [5.4.0]ундек-7-ен(ДБУ).
Протекание реакции образования амида можно ускорить добавлением четвертичной аммонийной соли, такой как бензилтриэтиламмонийхлорид или тетрабутиламмоний хлорид, или краун-эфира, такого как дибензил-18-краун-6.
Предпочтительно реакцию проводят при температуре от -10 до 50оС, более предпочтительно от 0 до 30оС, время реакции обычно составляет от 1 до 3 ч, хотя оно и меняется в зависимости от таких факторов, как температура реакции, конкретный тип исходных соединений, вспомогательные реагенты и инертный растворитель.
По окончании реакции получения амида целевое соединение общей формулы I' изобретения можно выделить из реакционной смеси с помощью обычных методов выделения. Например, реакционную смесь нейтрализуют и после отфильтровывания всех нерастворимых веществ добавляют не смешивающийся с водой органический растворитель. Затем органический экстракт отделяют, промывают водой и перегоняют для удаления растворителя, получая целевое соединение. При необходимости целевое соединение можно подвергнуть дальнейшей очистке с помощью таких стандартных методов, как перекристаллизация, переосаждение и/или хроматография.
Далее полученное соединение может быть подвергнуто одному или нескольким возможным превращениям. Порядок их следования не является существенным и разные реакции депротектирования можно проводить одновременно. Выделение продукта реакции получения амида перед осуществлением превращений может быть необязательным.
Возможное превращение (1), при котором нитрогруппу превращают в аминогруппу, можно осуществить хорошо известными методами восстановления нитрогрупп в аминогруппы. Предпочтительные методы восстановления включают:
а) реакцию с использованием металла, например амальгамы натрия, или переходного металла, такого как олово, цинк, железо, трихлорид титана или дихлорид олова. Предпочтительная система растворителей включает водный метанол, водный ацетон, водный тетрагидрофуран, обычно с добавлением соляной кислоты и с возможным добавлением хлорида аммония. Примеры растворителей включают системы хлорид аммония/вода метанол и вода-соляная кислота-ацетон;
б) реакцию с использованием гидрида, такого как боргидрид щелочного металла, например боргидрид натрия или боргидрид лития, такого как гидрид алюминия, например литийалюминийгидрид или литийтриэтоксиалюминийгидрид, или другого гидридного реагента, например натрийтеллургидрида, в среде простого эфира, такого как диэтиловый эфир или тетрагидрофуран, или в их смеси. Кроме того, в случае использования боргидрида натрия или натрийтеллургидрида растворитель может представлять собой спирт, такой как метанол или этанол;
в) каталитическое восстановление при комнатной температуре с использованием катализатора, такого как палладий на угле, платина или никель Ренея, в среде спирта, такого как метанол или этанол, простого эфира, такого как тетрагидрофуран или диоксан, жирной кислоты, такой как уксусная кислота, или их смесей с водой,
г) реакцию с использованием кислоты Льюиса, такой как хлорид алюминия, тетрахлорид олова или тетрахлорид титана, и гидрогенизованных синильных соединений, таких как гидротриэтилсилан или гидротрифенилсилан; или
д) восстановление радикальным восстановителем, таким как гидротрибутилолово, гидротрифенилолово или гидродибутилолово в присутствии радикального инициатора, такого как азобисизобутиронитрил, или трифенилбора в качестве катализатора.
Из этих методов более предпочтительным является каталитическое восстановление по методу в.
Возможное превращение (2), при котором осуществляют удаление защитной группы из защищенной аминогруппы, а также ту часть возможного превращения (3), при котором также осуществляют удаление защитной группы из защищенной аминогруппы, можно выполнить хорошо известными методами, которые варьируются в зависимости от природы наличествующей защитной группы.
Если защитная группа для аминогруппы представляет собой силильную группу, ее можно удалить обработкой соединением, способным генерировать фторид-анион, таким как тетрабутиламмонийфторид. Обработку, как правило, проводят в среде растворителя. Специальные ограничения на растворитель не вводятся при условии, что он не оказывает отрицательного воздействия на реакцию. Предпочтительно использовать простой эфир, такой как тетрагидрофуран или диоксан. В отношении температуры и времени реакции специальные ограничения также не вводятся, обычно реакцию удаления силильной защитной группы проводят при комнатной температуре в течение 10-18 ч.
Если защитная группа для аминогруппы представляет собой алифатическую ацильную группу, ароматическую ацильную группу, алкоксикарбонильную группу или замещенную метиленовую группу, образующую основание Шиффа, депротектирование можно осуществить обработкой кислотой или основанием в присутствии водного растворителя. При использовании кислоты какие-либо конкретные ограничения в отношении ее природы отсутствуют, предпочтительно она представляет собой неорганическую кислоту, такую как соляная кислота, серная кислота, фосфорная кислота или бромистоводородная кислота. При использовании основания какие-либо конкретные ограничения в отношении его природы также отсутствуют, при условии, что оно не оказывает воздействия на остальную часть соединения, предпочтительно основание представляет собой алкоксид металла, такой как метоксид натрия, карбонат щелочного металла, такой как карбонат натрия или карбонат калия, гидроокись щелочного металла, такую как гидроокись натрия или гидроокись калия, или аммиак, например, в виде водного раствора или в виде смеси концентрированного аммиака с метанолом. На выбор растворителя также не накладываются конкретные ограничения, и, как правило, используют какой-либо из растворителей, обычно применяемых для реакций гидролиза, предпочтительно воду, органический растворитель, например спирт, такой как метанол, этанол или пропанол, или простой эфир, такой как тетрагидрофуран или диоксан, или смешанный растворитель, содержащий воду и органический растворитель. Температура и время реакции варьируются в зависимости от исходных соединений и используемых для гидролиза кислоты или основания, и поэтому не требуют введения специальных ограничений. С целью сведения к минимуму побочных реакций депротектирование обычно проводят при температуре от 0 до 150оС в течение 1-10 ч.
Если защитная группа для аминогруппы представляет собой аралкильную группу или аралкилоксикарбонильную группу, то может быть использован целый ряд методов депротектирования. В общем случае эта группа может быть удалена взаимодействием с восстановителем в среде растворителя, предпочтительно каталитическим восстановлением при комнатной температуре в присутствии катализатора, взаимодействием с окислителем, обработкой щелочным металлом, или обработкой галогенидом.
Выбор растворителя для депротектирования (снятия защиты) каталитическим восстановлением не имеет специальных ограничений при условии, что этот растворитель не принимает участия в реакции, предпочтительно он представляет собой спирт, такой как метанол, этанол или изопропанол, простой эфир, такой как диэтиловый эфир, тетрагидрофуран или диоксан, ароматический углеводород, такой как толуол бензол или ксилол, алифатический углеводород, такой как гексан или циклогексан, сложный эфир, такой как этилацетат или пропилацетат, жирную кислоту, такую как уксусная кислота, или смешанный растворитель, содержащий вышеуказанные растворители и воду. На выбор катализатора специальные ограничения также не накладываются, может быть использован один из тех катализаторов, которые обычно применяют для каталитического восстановления, например, палладий на угле, никель Ренея, оксид платины, платиновую чернь, родий на окиси алюминия, трифенилфосфин хлорид родия, или палладий на сульфате бария. Давление реакции особо не ограничивается, обычно реакцию проводят при давлении от 1 до 10 атм. Температура и время реакции могут варьироваться в зависимости от природы исходных веществ и типа используемого катализатора, но обычно реакцию проводят при температуре от 0 до 100оС в течение 5 мин 24 ч.
В случае окислительного депротектирования выбор растворителя не подвержен специальным ограничениям при условии, что растворитель не принимает участия в реакции. Предпочтительным растворителем является водно-органический растворитель, в котором органическая часть может представлять собой кетон, такой как ацетон, галогеноуглеводород, такой как хлористый метилен, хлороформ или четыреххлористый углерод, нитрил, такой как ацетонитрил, простой эфир, такой как диэтиловый эфир, тетрагидрофуран или диоксан, амид, такой как диметилформамид, диметилацетамид, или гексаметилфосфоротриамид, или сульфоксид, такой как диметилсульфоксид. Окислителем, как правило, является один из тех окислителей, которые обычно используются для реакции этого типа, и поэтому его выбор не является специально ограничиваемым, примеры включают персульфат калия, персульфат натрия, церийаммонийнитрат (ЦАН) и 2,3-дихлор-5,6-дициано-n-бензохинон (ДДХ). Температура и время реакции могут варьироваться в зависимости от исходных веществ и типа используемого окислителя, но, как правило, реакцию проводят при температуре от 0 до 150оС в течение 10 мин 24 ч.
В случае депротектирования с использованием щелочного металла реакцию предпочтительно проводят, применяя такой щелочной металл, как металлический литий или металлический натрий, в среде спирта, такого как метанол или этанол, предпочтительно при температуре от -78 до -20оС.
В случае депротектирования путем обработки галогенидом предпочтительные реагенты включают комплекс хлорида алюминия с иодидом натрия или алкилсилилгалогенид, такой как триметилсилилиодид, в среде растворителя. На выбор растворителя специальные ограничения не накладываются при условии, что растворитель не принимает участия в реакции. Предпочтительным растворителем является нитрил, такой как ацетонитрил, галогеноуглеводород, такой как хлористый метилен или хлороформ, или их смеси. Температура и время реакции могут варьироваться в зависимости от исходных веществ, но как правило, реакцию проводят при температуре от 0 до 50оС в течение от 5 мин до 3 дней.
Если защитная группа для аминогруппы представляет собой алкенилоксикарбонильную группу, то ее, как правило, можно удалить путем обработки основанием в условиях, аналогичных описанным выше для депротектирования в случае, когда защитная группа представляет собой алифатическую ацильную группу, ароматическую ацильную группу, алкоксикарбонильную группу или замещенную метиленовую группу, образующую основание Шиффа.
Если защитная группа для аминогруппы представляет собой аллилоксикарбонильную группу, то ее можно легко удалить, используя палладий вместе с трифенилкарбонилом или с тетракарбонилом никеля, вклад побочных реакций при этом минимален.
В зависимости от выбранных защитных групп и выбранных условий реакции депротектирования, можно провести реакцию таким образом, чтобы депротектирование защищенной аминогруппы одновременно приводило и к депротектированию защищенной гидроксильной группы, которое предусмотрено как часть возможного превращения (3).
Та часть возможного превращения (3), которая относится к удалению защитной группы из защитной гидроксильной группы, может быть осуществлена хорошо известными методами, варьирующимися в зависимости от природы наличествующей защитной группы.
Если защитная группа для гидроксильной группы представляет собой силильную группу, аралкильную группу, аралкилоксикарбонильную группу, алифатическую ацильную группу, ароматическую ацильную группу, алкоксикарбонильную группу или алкенилоксикарбонильную группу, то ее можно удалить с помощью соответствующих методов, описанных для удаления этой группы при ее использования в качестве защитной группы для аминогруппы.
Если защитная группа для гидроксильной группы представляет собой алкоксиметильную, тетрагидропиранильную, тетрагидротиопиранильную, тетрагидрофуранильную или замещенную этильную группу, то ее в общем случае можно удалить путем обработки кислотой в среде растворителя. Хотя выбор кислоты специально не ограничивается, предпочтительно кислота представляет собой кислоту Бренстеда, неорганическую кислоту, такую как соляная кислота или серная кислота, органическую кислоту, такую как уксусная кислота или n-толуолсульфокислота, или сильно кислую катионообменную смолу, такую как Dowex 50 W. На выбор растворителя специальных ограничений также не накладывается при условии, что растворитель не принимает участия в реакции, предпочтительно растворитель представляет собой спирт, такой как метанол или этанол, простой эфир, такой как тетрагидрофуран или диоксан, или смесь какого-либо из этих растворителей с водой. Температура и время реакции могут варьироваться в зависимости от исходных веществ и типа используемой кислоты, но обычно реакцию проводят при температуре от 0 до 50оС в течение 10 мин 18 ч.
В зависимости от выбранных защитных групп и выбранных условий реакции депротектирования, можно провести реакцию таким образом, чтобы депротектирование защищенной гидроксильной группы одновременно приводило и к депротектированию защищенной аминогруппы, которое предусмотрено как возможное превращение (2), и как часть возможного превращения (3).
После завершения одного или нескольких возможных превращений целевое соединение общей формулы I предлагаемого изобретения может быть выделено из реакционной среды с помощью известных методов выделения. Например, к реакционной смеси добавляют не смешивающийся с водой органический растворитель, образующий органический экстракт, который можно промыть водой, после чего, удалив растворитель перегонкой, получить целевое соединение. При необходимости целевое соединение можно подвергнуть дальнейшей очистке с помощью стандартных методик, таких как перекристаллизация, переосаждение и/или хроматография.
Реакционноспособное производное карбоновой кислоты общей формулы II, являющееся исходным реагентом в способе согласно изобретению, можно приготовить из соответствующей "родительской" карбоновой кислоты, например, с помощью стандартных методов галоидирования. Это галоидирование обычно осуществляют обработкой стандартным галоидирующим агентом. Предпочтительно галоидирующим агентом является: тионилгалогенид, такой как тионилхлорид, тионилбромид или тионилиодид, сульфурилгалогенид, такой как сульфурилхлорид, сульфурилбромид, или сульфурилиодид, тригалогенид фосфора, такой как трихлорид фосфора, трибромид фосфора, или трииодид фосфора, пентагалогенид фосфора, такой как пентахлорид фосфора, пентабромид фосфора или пентаиодид фосфора, или оксигалогенид фосфора, такой как оксихлоридфосфора, оксибромид фосфора или оксииодид фосфора. Особенно предпочтительным галоидирующим агентом является оксигалогенид фосфора.
Сама "родительская" кислота должна представлять собой известное соединение или такое соединение, которое можно легко получить известными способами. Такую карбоновую кислоту, как 3-нитро-4-аминокоричная кислота, содержащую и нитрозаместитель, и аминный заместитель, можно синтезировать, например, способом, описанным в Annalen Chimica, 48, 958-991 (1958) или в Chem. Ber. 16, 2042. Такую карбоновую кислоту, как 3-нитро-4-гидроксикоричная кислота, содержащую и нитрозаместитель, и гидроксильный заместитель, можно синтезировать, например, способом, описанным в I.Chem. Soc. 3072 (1952) или в I.Am.Chem.Soc. 79, 4114 (1957). Карбоновую кислоту, имеющую два аминных заместителя, или карбоновую кислоту, имеющую и аминный заместитель, и гидроксильный заместитель, можно получить восстановлением соответствующего нитросоединения.
Карбоновые кислоты, имеющие большее число атомов углерода, можно получить или аналогичными способами, или способами, основанными на наращивании углеродной цепи. Например, известные аминокоричную кислоту или гидроксикоричную кислоту можно защитить соответственно по аминогруппе или по гидроксильной группе, подвергнуть реакции наращивания углеродной цепи, пронитровать и депротектировать, после чего, возможно, восстановить нитрозаместитель.
Для увеличения числа атомов углерода в карбоновой кислоте можно использовать целый ряд хорошо известных реакций. Как правило, исходную карбоновую кислоту восстанавливают до соответствующего спирта, затем активируют гидроксильную группу, получая уходящую группу, такую как атом галогена, например атом хлора, брома или иода, такую как алкансульфонилоксигруппа, например метансульфонилокси- или этансульфонилоксигруппа, такую как галогеноалкансульфонилоксигруппа, например трифторметансульфонилоксигруппа или пентафторэтансульфонилоксигруппа, или такую как арилсульфонилоксигруппа, например бензолсульфонилокси- или n-толуолсульфонилоксигруппа, после чего удлиняют углеродную цепь активированного таким образом соединения на один или два атома углерода, используя один из ниже следующих способов, обычно осуществляемых при температуре от -78 до 50оС.
В качестве способов удлинения углеродной цепи активированного соединения на один атом углерода могут быть упомянуты следующие:
1. Использование 1,3-дициана.
2. Использование цианида металла.
3. Реакция с двуокисью углерода после приготовления реактива Гриньяра.
В качестве способов удлинения углеродной цепи активированного соединения на два атома углерода могут быть упомянуты следующие:
1. Использование производного малоновой кислоты.
2. Ацидолиз с использованием производного ацетоуксусной кислоты.
Способ с использованием 1,3-дициана может быть осуществлен путем реакции 1,3-дициана с органическим или неорганическим основанием, таким как гидрид натрия, метоксид калия, гидроокись калия или диизопропилнитрид лития, в среде простого эфира, такого как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран или диоксан, в атмосфере инертного газа, такого как азот, с образованием соли 1,3-дициана с металлом, которая затем реагирует с активированным соединением, с последующим гидролизом сильной кислотой, такой как соляная кислота.
Способ с использованием цианида металла может быть осуществлен путем реакции цианида металла с активированным соединением с получением соответствующего цианосоединения, и с последующим гидролизом этого соединения известными способами.
Способ, в котором используется реакция с двуокисью углерода после приготовления реактива Гриньяра, может быть осуществлен путем приготовления из активированного соединения реактива Гриньяра с использованием известных методик, с последующей реакцией этого реактива с двуокисью углерода по стандартным методикам.
Способ с использованием производного малоновой кислоты может быть осуществлен путем реакции производного малоновой кислоты с основанием металла в среде простого эфира, такого как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран или диоксан, с получением соли металла, с последующей реакцией этой соли с активированным соединением по стандартным методикам, и с декарбонилированием и/или гидролизом полученного продукта. Выбор основания металла может зависеть от рКа производного малоновой кислоты, но предпочтительно основание представляет собой неорганическое основание, например карбонат щелочного металла, такой как карбонат натрия или карбонат калия, гидрид щелочного металла, такой как гидрид лития, гидрид натрия или гидрид калия, или гидроокись щелочного металла, такую как гидроокись натрия, гидроокись калия или гидроокись бария, органическое основание металла, например алкоксид щелочного металла, такой как метоксид натрия или этоксид натрия, бутиллития или диизопропиламид лития.
Способ ацидолиза с использованием производного ацетоуксусной кислоты может быть осуществлен путем реакции производного ацетоуксусной кислоты с основанием металла, таким как одно из оснований, названных выше для способа с малоновой кислотой, в среде простого эфира, такого как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран или диоксан, с получением соли металла на метиленовом остатке производного ацетоуксусной кислоты с последующей реакцией активированного соединения с этой солью по известной методике, и гидролизом полученного продукта кислотой.
Может быть также использована реакция Виттига, которая представляет собой общий метод увеличения длины углеродной цепи карбоновой кислоты на любое желаемое число атомов углерода. При использовании этого метода может быть получена карбоновая кислота, имеющая в нужном положении двойную связь, или же эта двойная связь может быть удалена восстановлением. Реакцию Виттига обычно проводят между реагентом Виттига и соответствующем альдегидом, с последующим возможным восстановлением двойной связи в полученном продукте с помощью стандартных методов.
Подбирая нужное сочетание этих реакций, можно синтезировать карбоновую кислоту, имеющую нужную длину углеродной цепи и нужную степень ненасыщенности.
Нитрование карбоновой кислоты с целью введения в нее нитрогруппы обычно осуществляют стандартными методами. Например, это можно сделать, используя нитратное производное, способное вводить в соединение нитрогруппу, например дымящую азотную кислоту, при температуре от комнатной до 50оС в среде кислотного растворителя, например, смеси уксусной кислоты и уксусного ангидрида.
Нижеследующие примеры иллюстрируют получение соединений согласно изобретению из известных исходных соединений или из таких исходных соединений, которые можно приготовить, используя методики, аналогичные используемым для известных соединений. Для справки приведены также Примеры, иллюстрирующие приготовление некоторых исходных соединений. Приведен также Пример на приготовление лекарственной формы.
П р и м е р 1. N-(1-адамантил)амид 4-ацетокси-3-нитрокоричной кислоты.
1 г 4-ацетокси-3-нитрокоричной кислоты (приготовление описано в примере на приготовление) 2 растворяют в 30 мл хлористого метилена. К полученному раствору добавляют одну каплю диметилформамида и охлаждают полученную смесь до 0оС. Добавляют к смеси 3 г оксалилхлорида, нагревают ее до комнатной температуры, после чего перемешивают 2 ч. По истечении этого времени перегонкой при пониженном давлении полностью удаляют растворитель. Полученный остаток растворяют в 20 мл хлористого метилена, добавляют 0,1 г 1-адамантиламина и 0,4 г триэтиламина, и перемешивают полученную смесь 3 ч. Затем к раствору добавляют этилацетат, который промывают водой, разбавленным водным раствором бикарбоната натрия и насыщенным водным раствором хлорида натрия (в указанном порядке). Органический слой сушат над безводным сульфатом натрия и перегонкой при пониженном давлении удаляют растворитель. Полученный остаток очищают колоночной хроматографией на силикагеле, используя в качестве элюента смесь этилацета: н-гексан состава 1:1, получают 1,2 г целевого соединения, т.пл. 179-180оС.
П р и м е р 2. N-(1-адамантил)амид 4-гидрокси-3-нитрокоричной кислоты.
1 г N-(1-адамантил)амида 4-ацетокси-3-нитрокоричной кислоты (полученного, как описано в примере 1) растворяют в 30 мл метанола, затем добавляют 30 мл 4 водного раствора гидроокиси натрия, и полученную смесь перемешивают 3 ч при комнатной температуре. По окончании этого времени метанол удаляют перегонкой при пониженном давлении. Добавление к остатку 3N водного раствора соляной кислоты дает кристаллы целевого соединения с т.пл. 162-163оС.
П р и м е р 3. N-(1-адамантил)амид 3-(3-амино-4-гидроксифенил)пропионовой кислоты.
Кристаллы N-(1-адамантил)амида 4-гоидрокси-3-нитрокоричной кислоты, полученные, как описано в примере 2, собирают фильтрованием и растворяют в 30 мл метанола. К раствору добавляют 300 мг 10% палладия на угле и проводят каталитическое восстановление, пробулькивая через раствор водород при атмосферном давлении в течение 60 мин. По окончании этого времени катализатор отфильтровывают и перегонкой при пониженном давлении удаляют метанол. Полученный остаток очищают колоночной хроматографией на силикагеле, используя в качестве элюента этиацетат, получают 0,78 г целевого соединения в виде кристаллов, т.пл. 79-80оС.
П р и м е р 4. N,N-диэтиламид 4-ацетокси-3-нитрокоричной кислоты.
По методике примера 1, используя 400 мг диэтиламина, получают 950 мг целевого соединения в виде маслообразного вещества.
Rf: 0,85 (подвижная фаза: этилацетат).
П р и м е р 5. N,N-диэтиламид 4-гидрокси-3-нитрокоричной кислоты.
По методике примера 2, используя 900 мг N,N-диэтиламида 4-ацетокси-3-нитрокоричной кислоты (получен, как описано в примере 4), получают 750 мг целевого соединения в виде маслообразного вещества.
Rf: 0,5 (подвижная фаза: смесь этилацетат:гексан, объемное отношение 1: 1).
П р и м е р 6. N,N-диэтиламид 3(4-гидрокси-3-аминофенил)пропионовой кислоты.
По методике примера 3, используя 700 мг N,N-диэтиламида 4-гидрокси-3-нитрокоричной кислоты (получен, как описано в примере 5), получают 550 мг целевого соединения, т.пл. 92-93оС).
П р и м е р 7. N,N-дифениламид 4-ацетокси-3-нитрокоричной кислоты. По методике примера 1, используя 600 мг дифениламина, получают 1000 мг целевого соединения, т.пл. 170, 5-171,5оС.
П р и м е р 8. N,N-дифениламид 4-гидрокси-3-нитрокоричной кислоты.
По методике примера 2, используя 950 мг N,N-дифениламида 4-ацетокси-3-нитрокоричной кислоты (получен, как описано в примере 7), получают 700 мг целевого соединения, т.пл. 197-198оС.
П р и м е р 9. N,N-дифениламид 3-(4-гидрокси-3-аминофенил)пропионовой кислоты.
По методике примера 3, используя 650 мг N,N-дифениламида 4-гидрокси-3-нитрокоричной кислоты (получен, как описано в примере 8), получают 450 мг целевого соединения, т.пл. 170,5-171,5оС.
П р и м е р 10. N-[2-(3-метоксикарбонилтетрагидробензотиенил)] амид 4-ацетокси-3-нитрокоричной кислоты.
По методике примера 1, используя 800 мг 2-амино-3-метоксикарбонилтетрагидробензотиофена, получают 1000 мг целевого соединения, т.пл. 184-185оС.
П р и м е р 11. N-[2-(3-метоксикарбонилтетрагидробензотиенил)]амид-4-гидрокси-3-нитрокорично й кислоты.
По методике примера 2, используя 950 мг N-[2-(3-метоксикарбонилтетрагидробензотиенил)] амида 4-ацетокси-3-нитрокоричной кислоты (получен, как описано в примере 10), получают 600 мг целевого соединения, т.пл. 223-224оС.
П р и м е р 12. N-(4-бензилокси-3-нитроциннамоил)пиперидин.
По методике примера 1, используя 1 г 4-бензилокси-3-нитрокоричной кислоты и 400 мг пиперидина, получают 0,8 г целевого соединения, т.пл. 155-156оС.
П р и м е р 13. N-[3-(4-гидрокси-3-аминофенид)пропионил]пиперидин.
0,5 г N-(4-бензилокси-3-нитроциннамоил)пиперидина (получен, как описано в примере 12) растворяют в 30 мл метанола. К полученному раствору добавляют 300 мг 10% палладия на угле и проводят каталитическое восстановление, пробулькивая через раствор водород, при атмосферном давлении в течение 60 мин. Затем катализатор удаляют фильтрованием и при пониженном давлении отгоняют метанол. Полученный остаток очищают колоночной хроматографией на силикагеле, используя в качестве элюента этилацетат, получают 0,35 г целевого соединения в виде кристаллов, т.пл. 149-150оС.
П р и м е р 14. N-изобетиламид 4-бензилокси-3-нитрокоричной кислоты.
По методике примера 1, используя 400 мг изобутиламина, получают 850 мг целевого соединения, т.пл. 166-167оС.
П р и м е р 15. N-изобутиламид 3-(4-гидрокси-3-аминофенил)пропионовой кислоты.
По методике примера 13, используя 800 мг N-изобутиламида 4-бензилокси-3-нитрокоричной кислоты (получен, как описано в примере 14), получают 600 мг целевого соединения в виде маслообразного вещества.
Rf: 0,19 (подвижная фаза: этилацетат).
П р и м е р 16. N-(2,4-дихлорфенил)амид 4-бензилокси-3-нитрокоричной кислоты.
По методике примера 12, используя 600 мг 2,4-дихлоранилина, получают 900 мг целевого соединения, т.пл. 152-153оС.
П р и м е р 17. N-(2,4-дихлорфенил)амид 3-(4-гидрокси-3-аминофенил)пропионовой кислоты.
По методике примера 13, используя 850 мг N-(2,4- дихлорфенил)амида 4-бензилокси-3-нитрокоричной кислоты (получен, как описано в примере 16), получают 650 мг целевого соединения, т.пл. 130-131оС.
П р и м е р 18. N-(2,5-диметилфенил)амид 4-бензилокси-3-нитрокоричной кислоты.
По методике примера 12, используя 600 мг 2,5-диметиланилина, получают 850 мг целевого соединения в виде маслообразного вещества.
Rf: 0,90 (подвижная фаза: этилацетат).
П р и м е р 19. N-(2,5-диметилфенил)амид 3-(4-гидрокси-3-аминофенил)пропионовой кислоты.
По методике примера 13, используя 800 мг N-(2,5-диметилфенил)амида 4-бензилокси-3-нитрокоричной кислоты (получен, как описано в примере 18), получают 600 мг целевого соединения, т. пл. 162-163оС.
П р и м е р 20. N-(1-адамантил)амид 3-3-нитро-4-трифторацетамидофенил)про-пионовой кислоты 1 г 3-(3-нитро-4-трифторацетамидофенил)пропионовой кислоты (приготовление описано в примере 5 на приготовление) растворяют в 300 мл хлористого метилена. К полученному раствору добавляют одну каплю диметилформамида и охлаждают его до 0оС. Затем к раствору добавляют 3 г оксалилхлорида и, после того как температура полученной смеси достигает комнатной, перемешивают ее 2 ч. Затем перегонкой при пониженном давлении полностью отгоняют растворитель и остаток растворяют в 20 мл хлористого метилена. К полученному раствору добавляют 0,6 г 1-адамантиламина и 0,4 г триэтиламина и перемешивают полученную смесь 3 ч. По окончании этого времени к раствору добавляют этилацетат, который затем промывают водой, разбавленным водным раствором бикарбоната натрия и насыщенным водным раствором хлорида натрия (в указанном порядке). Органический слой отделяют и сушат над безводным сульфатом натрия. Затем перегонкой при пониженном давлении удаляют растворитель. Полученный остаток очищают колоночной хроматографией на силикагеле, используя в качестве элюента этилацетат, получают 1,0 г целевого соединения в виде маслообразного вещества.
Rf: 0,71 (подвижная фаза: этилацетат).
П р и м е р 21. N-(1-адамантил)амид 3-(3-нитро-4-аминофенил)пропионовой кислоты.
0,5 г N-(1-адамантил)амида 3-(3-нитро-4-трифторацетамидо-фенил)пропионовой кислоты (получен, как описано в примере 20) растворяют в 10 мл метанола. Затем к раствору добавляют 10 мл 2N водного раствора гидроокиси натрия и перемешивают 3 ч при комнатной температуре. По окончании этого времени метанол удаляют перегонкой при пониженном давлении. Экстрагирование остатка этилацетатом дает целевое соединение в виде маслообразного вещества.
Rf: 0,38 (подвижная фаза: смесь этилацетат: гексан, объемное соотношение 1:1).
П р и м е р 22. N-(1-адамантил)амид 3-(3,4-диаминофенил)пропионовой кислоты.
500 мл N-(1-адамантил)амида 3-(3-нитро-4-аминофенил)пропионовой кислоты (получен, как описано в примере 21) растворяют в 20 мл метанола. К раствору добавляют 200 мг 10% палладия на угле и проводят каталитическое восстановление, пробулькивая через раствор водород, при атмосферном давлении в течение 120 минут. Затем катализатор удаляют фильтрованием, и при пониженном давлении отгоняют метанол. Полученный остаток очищают колоночной хроматографией на силикагеле, используя в качестве элюента смесь этилацетат: метанол состава 19: 1, получают 0,21 г целевого соединения в виде кристаллов, т.пл. 151-152оС.
П р и м е р 23. N-(2,4-дихлорфенил)амид 3-(3-нитро-4-трифторацетамидофенил)про- пионовой кислоты.
По методике примера 20, используя 80 мг 2,4-дихлоранилина, получают 800 мг целевого соединения, т.пл. 193-194оС.
П р и м е р 24. N-(2,4-дихлорфенил)амид 3-(3-нитро-4-аминофенил)пропионовой кислоты.
По методике примера 21, используя 700 мг N-(2,4-дихлорфенил)амида 3-(3-нитро-4-трифторацетамидофенил)-пропионовой кислоты (получен, как описано в примере 23), получают 350 мг целевого соединения, т.пл. 164-165оС.
П р и м е р 25. N-(2,4-дихлорфенил)амид 3-(3,4-диаминофенил)пропионовой кислоты.
По методике примера 22, используя 300 мг N-(2,4-дихлорфенил)амид 3-(3-нитро-4-аминофенил)пропионовой кислоты (получен, как описано в примере 24), получают 150 мг целевого соединения, т.пл. 149-150оС.
П р и м е р 26. N-[3-(3-нитро-4-трифторацетамидофенил)пропионил]-морфолин.
По методике примера 20, используя 400 мг морфолина, получают 900 мг целевого соединения в виде маслообразного вещества.
Rf: 0,50 (подвижная фаза: этилацетат).
П р и м е р 27. N[3-(3-нитро-4-аминофенил)пропионил]морфолин.
По методике примера 21, используя 800 мг N-[3-(3-нитро-4-трифторацетамидофенил)пропионил] морфолина (получен, как описано в примере 26), получают 600 мг целевого соединения в виде маслообразного вещества.
Rf: 0,50 (подвижная фаза: смесь этилацетат гексан, объемное соотношение 1:1).
П р и м е р 28. N-[3-(3,4-диаминофенил)пропионил]морфолин.
По методике примера 22, используя 500 мг N-[3-(3-нитро-4-аминофенил)пропионил]морфолина (получен, как описано в примере 27), получают 300 мг целевого соединения в виде маслообразного вещества.
Rf: 0,37 (подвижная фаза: смесь этилацетат метанол, объемное отношение 4:1).
П р и м е р 29. N, N-диэтиламид 3-(3-нитро-4-трифторацетамидофенил)пропионовой кислоты.
По методике примера 20, используя 400 мг диэтиламина, получают 1000 мг целевого соединения в виде маслообразного вещества.
Rf: 0,70 (подвижная фаза: этилацетат).
П р и м е р 30. N,N-диэтиламил 3-(3-нитро-4-аминофенил)пропионовой кислоты.
По методике примера 21, используя 900 мг N,N-диэтиламина 3-(3-нитро-4-трифторацетамидофенил)пропионовой кислоты (получен, как описано в примере 29), получают 600 мг целевого соединения, т.пл. 98-99оС.
П р и м е р 31. N,N-диэтиламид 3-(3,4-диаминофенил)пропионовой кислоты.
По методике примера 22, используя 550 мг N,N-диэтиламида 3-(3-нитро-4-аминофенил)пропионовой кислоты (получен, как описано в примере 30), получают 300 мг целевого соединения в виде кристаллов.
Rf 0,53 (подвижная фаза смесь этилацетат метанол, объемное соотношение 4:1).
П р и м е р 32. N-(3-хинолил)амид 3-(3-нитро-4-трифторацетамидофенил)пропио-новой кислоты.
По методике примера 20, используя 700 мг 3-аминохинолина, получают 900 мг целевого соединения в виде маслообразного вещества.
Rf 0,50 (подвижная фаза этилацетат).
П р и м е р 33. N -(3-хинолил)амид 3-(3-нитро-4-аминофенил)пропионовой кислоты.
По методике примера 21, используя 850 мг N-(3-хинолил)амида 3-(3-нитро-4-трифторацетамидофенил)пропионовой кислоты (получен, как описано в примере 32), получают 600 мг целевого соединения, т.пл. 219,5-220,5оС.
П р и м е р 34. N-(3-хинолил)амид 3-(3,4-диаминофенил)пропионовой кислоты.
По методике примера 22, используя 500 мг N-(3-хинолил)амида 3-(3-нитро-4-аминофенил)пропионовой кислоты (получен, как описано в примере 33), получают 300 мг целевого соединения в виде кристаллов.
Rf 0,42 (подвижная фаза: смесь этилацетат: метанол, объемное соотношение 4:1).
П р и м е р 35. N-(2,5-диметилфенил)амид 3-(3-нитро-4-трифторацетамидофенил)пропионовой кислоты.
По методике примера 20, используя 600 мг 2,5-диметиланилина, получают 900 мг целевого соединения в виде маслообразного вещества.
Rf 0,45 (подвижная фаза: смесь этилацетат: гексан, объемное соотношение 1:1).
П р и м е р 36. N-(2,5-диметилфенил)амид 3-(4-амино-3-нитрофенил)-пропионовой кислоты.
По методике примера 21, используя 800 мг N-(2,5-диметилфенил)амида 3-(3-нитро-4-трифторацетамидофенил)пропионовой кислоты (получен, как описано в примере 35), получают 600 мг целевого соединения, т.пл. 187-188оС.
П р и м е р 37. N-(2,5-диметилфенил)амид-3-(3,4-диаминофенил)пропионовой кислоты.
По методике примера 22, используя 500 мг N-(2,5-диметилфенил)амида 3-(4-амино-3-нитрофенил)пропионовой кислоты (получен, как описано в примере 36), получают 300 мг целевого соединения, т.пл. 120-121оС.
П р и м е р 38. N, N-дифениламид 3-(3-нитро-4-трифторацетамидофенил)пропио-новой кислоты.
По методике примера 20, используя 600 мг дифениламина, получают 950 мг целевого соединения в виде маслообразного вещества.
Rf 0,56 (подвижная фаза: смесь этилацетат: гексан, объемное соотношение 1:1).
П р и м е р 39. N,N-дифениламил 3-(4-амино-3-нитрофенил)пропионовой кислоты.
По методике примера 21, используя 900 мг N,N-дифениламида 3-(3-нитро-4-трифторацетамидофенил)пропионовой кислоты (получен, как описано в примере 38), получают 750 мг целевого соединения, т.пл. 189-190оС.
П р и м е р 40. N,N-дифениламил 3-(3,4-диаминофенил)пропионовой кислоты.
По методике примера 22, используя 600 мг N,N-дифениламида 3-(4-амино-3-нитрофенил)пропионовой кислоты (получен, как описано в примере 39), получают целевое соединение, т.пл. 122-123оС, в количестве 300 мг.
П р и м е р 41. N-(4-бензоилфенил)амид 3-(3-нитро-4-трифторацетамидофенил)про- пионовой кислоты.
По методике примера 20, используя 800 мг 4-бензоиланилина, получают 1,1 г целевого соединения в виде маслообразного вещества.
Rf 0,63 (подвижная фаза смесь этилацетат гексан, объемное соотношение 3: 2).
П р и м е р 42. N-(4-бензоилфенил)амид 3-(4-амино-3-нитрофенил)пропионовой кислоты.
По методике примера 21, используя 1,0 г N-(4-бензоилфенил)амида 3-(3-нитро-4-трифторацетамидофенил)пропионовой кислоты (получен, как описано в примере 41), получают 800 мг целевого соединения, т.пл. 72-73оС.
П р и м е р 43. N-(4-бензоилфенил)амид 3-(3,4-диаминофенил)пропионовой кислоты.
По методике примера 22, используя 700 мг N-(4-бензоилфенил)амида 3-(4-амино-3-нитрофенил)пропионовой кислоты (получен, как описано в примере 42), получают 500 мг целевого соединения, т.пл. 156-157оС.
П р и м е р 44. N-[3-(3-нитро-4-трифторацетамидофенил)пропионио]пиперидин.
По методике примера 20, используя 400 мг пиперидина, получают 800 мг целевого соединения, т.пл. 102-103оС.
П р и м е р 45. N-[3-(4-амино-3-нитрофенил)пропионил]пиперидин.
По методике примера 21, используя 700 мг N-[3-(3-нитро-4трифторацетамидофенил)пропионил] пиперидина (получен, как описано в примере 44), получают 500 мг целевого соединения, т.пл. 140-141оС.
П р и м е р 46. N-[3-(3,4-диаминофенил)пропионил]пиперидин.
По методике примера 22, используя 400 мг N-[3-(4-амино-3-нитрофенил)пропионил] пиперидина (получен, как описано в примере 45), получают 200 мг целевого соединения в виде маслообразного вещества.
Rf 0,37 (подвижная фаза: смесь этилацетат: метанол, объемное соотношение 9:1).
П р и м е р 47. N-метил-N-фениламид 3-(3-нитро-4-трифторацетамидофенил)про- пионовой кислоты.
По методике примера 20, используя 500 мг N-метилаланилина, получают 1,1 г целевого соединения в виде маслообразного вещества.
Rf: 0,40 (подвижная фаза: смесь этилацетат: гексан, 1:1).
П р и м е р 48. N-метил-N-фениламид 3-(4-амино-3-нитрофенил)пропионовой кислоты.
По методике примера 21, используя 900 мг N-метил-N-фениламида 3-(3-нитро-4-трифторацетамидофенил)пропионовой кислоты (получен, как описано в примере 47), получают 700 мг целевого соединения в виде маслообразного вещества.
Rf 0,33 (подвижная фаза: смесь этилацетат: гексан, объемное соотношение 1:1).
П р и м е р 49. N-метил-N-фениламид 3-(3,4-диаминофенил)пропионовой кислоты.
По методике примера 22, используя 600 мг N-метил-N-фениламида 3-(4-амино-3-нитрофенил)пропионовой кислоты (получен, как описано в примере 48), получают 400 мг целевого соединения в виде маслообразного вещества.
Rf 0,47 (подвижная фаза: смесь этилацетат: метанол, объемное отношение 9:1).
П р и м е р 50. Гидрохлорид N-(3-хинолил)амида 3-(3,4-диаминофенил)пропионовой кислоты.
К раствору 1 г N-(3-хинолил)амида 3-(3,4-диаминофенил)пропионовой кислоты (получен, как описано в примере 34) в 20 мл метанола при охлаждении льдом добавляют 5 мл 4N раствора хлористого водорода в диоксане, после чего отгоняют растворитель. Остаток перекристаллизовывают из смеси метанола и диэтилового эфира, получая 0,7 г целевого соединения, т.пл. 246-247оС.
П р и м е р 51. N-(1-инданил)амид 4-ацетокси-3-нитрокоричной кислоты.
По методике примера 1, используя 600 мг 1-инданиламина, получают 1,0 г целевого соединения, т.пл. 177-178оС.
П р и м е р 52. N-(1-инданил)амид 4-гидрокси-3-нитрокоричной кислоты.
По методике примера 2, используя 900 мг N-(1-инданил)амида 4-ацетокси-3-нитрокоричной кислоты (получен, как описано в примере 51), получают 700 мг целевого соединения, т.пл. 203-205оС.
П р и м е р 53. N-(1-инданил)амид 3-(4-гидрокси-3-аминофенил)пропионовой кислоты.
По методике примера 3, используя 600 мг N-(1-инданил)амида 4-гидрокси-3-нитрокоричной кислоты (получен как описано в примере 52), получают 400 мг целевого соединения, т.пл. 130-131оС.
П р и м е р 54. N-(4-ацетокси-3-нитроциннамоил)морфолин.
По методике примера 1, используя 400 мг морфолина, получают 900 мг целевого соединения в виде маслообразного вещества.
Rf 0,24 (подвижная фаза: смесь этилацетат: гексан, объемное соотношение 1:1).
П р и м е р 55. N-(4-гидрокси-3-нитроциннамоил)морфолин.
По методике примера 2, используя 800 мг N-(4-ацетокси 3-нитроциннамоил)морфолина (получен, как описано в примере 54), получают 600 мг целевого соединения в виде маслообразного вещества.
Rf 0,24 (подвижная фаза: смесь этилацетат: гексан, объемное соотношение 1:1).
П р и м е р 56. N-[3-(4-гидрокси-3-аминофенил)пропионил]морфолин.
По методике примера 3, используя 500 мг N-[4-гидрокси-3-нитроциннамоил)морфолина (получен, как описано в примере 55), получают 400 мг целевого соединения в виде маслообразного вещества.
Rf 0,27 (подвижная фаза этилацетат).
Пример на приготовление N 1.
4-гидрокси-3-нитрокоричная кислота
16,7 г 4-гидрокси-3-нитробензальдегида и 33,5 г метилтрифенилфосфоранилиденацетата растворяют в 100 мл хлористого метилена и полученный раствор перемешивают 2 ч при комнатной температуре. По окончании этого времени перегонкой при пониженном давлении удаляют растворитель. К полученному остатку добавляют 300 мл метанола и 18 г гидроокиси натрия и полученный раствор 2 ч кипятят с обратным холодильником. Затем перегонкой при пониженном давлении удаляют метанол. Остаток смешивают с 1 л воды и отфильтровывают нерастворившиеся вещества. Фильтрат подкисляют добавлением 3N водного раствора соляной кислоты, выпавшие кристаллы собирают фильтрованием и растворяют их в 500 мл тетрагидрофурана. Полученный раствор смешивают с активированным углем, 5 мин кипятят с обратным холодильником и фильтруют в горячем состоянии. Раствор концентрируют до объема примерно 100 мл, отгоняя растворитель при пониженном давлении, и оставляют стоять при комнатной температуре, получая 15,0 г целевого соединения в виде твердого вещества желтого цвета.
Пример на приготовление N 2.
4-ацетокси-3-нитрокоричная кислота
10 г 4-гидрокси-3-нитрокоричной кислоты (полученной, как описано в примере на приготовление N 1) растворяют в 30 мл уксусного ангидрида. К раствору добавляют одну каплю серной кислоты и перемешивают его 2 ч при комнатной температуре. К раствору добавляют 50 мл воды и перемешивают его еще 2 ч. По окончании этого времени добавляют этилацетат, полученную смесь промывают водой и насыщенным водным раствором хлористого натрия (в указанном порядке) и сушат над безводным сульфатом натрия. Растворитель удаляют отгонкой при пониженном давлении и смешивают остаток с толуолом, оставшуюся уксусную кислоту удаляют азеотропной перегонкой, получая 10,2 г целевого соединения в виде кристаллов.
Пример на приготовление N 3.
Этиловый эфир 4-ацетоаминофенилпропионовой кислоты
10 г этилового эфира 4-нитрокоричной кислоты растворяют в 100 мл уксусной кислоты, добавляют к раствору 1 г 10% палладия на угле и проводят каталитическое восстановление, пробулькивая через раствор водород при атмосферном давлении в течение 60 мин. Затем катализатор удаляют фильтрованием и при пониженном давлении отгоняют уксусную кислоту. Полученный таким образом остаток смешивают с 50 мл уксусного ангидрида и дают полученному раствору стоять при комнатной температуре до утра. Затем при пониженном давлении отгоняют растворитель. После очистки хроматографированием на колонке с силикагелем получают 7,8 г целевого соединения в виде кристаллов.
Пример на приготовление N 4.
3-(4-амино-3-нитрофенил)пропионовая кислота
5 г этилового эфира 4-ацетоаминофенилпропионовой кислоты (полученного, как описано в примере на приготовление N 3) растворяют в 30 мл уксусной кислоты и 30 мл уксусного ангидрида. К этому раствору при 40оС по каплям в течение 1 ч добавляют 10 мл дымящей азотной кислоты (плотность 1,5). Затем реакционную смесь по каплям выливают в 300 мл ледяной воды. Выпавшие кристаллы собирают фильтрованием, добавляют к ним 30 мл концентрированной соляной кислоты и 30 мл уксусной кислоты и 8 кипятят с обратным холодильником. Затем отгонкой при пониженном давлении удаляют растворитель. После очистки хроматографированием на колонке с силикагелем получают 2,2 г целевого соединения в виде масла.
Значение Rf продукта, определенное методом тонкослойной хроматографии на силикагеле с использованием этилацетата в качестве подвижной фазы, равно 0,62.
Пример на приготовление N 5.
3-(3-нитро-4-трифторацетамидофенил)пропионовая кислота
2 г 3-(4-амино-3-нитрофенил)пропионовой кислоты (полученной, как описано в примере на приготовление N 4) растворяют в 10 мл трифторуксусного ангидрида. Полученный раствор перемешивают 2 ч при комнатной температуре, после чего смешивают с этилацетатом, промывают водой и насыщенным водным раствором хлористого натрия и сушат над безводным сульфатом натрия.
Растворитель удаляют отгонкой при пониженном давлении, остаток смешивают с толуолом и удаляют оставшуюся трифторуксусную кислоту азеотропной перегонкой. Получают 1,5 г целевого соединения в виде кристаллов.
Приготовление лекарственной формы Приготовление капсул
Порошки нижеследующих ингредиентов тщательно смешивают и просеивают через сито 60 меш (стандарт Тайлера):
N-(1-адамантил)амид
3-(3-амино-4-гидрок-
сифенил)пропионо-
вой кислоты (полу-
чен, как описано в примере 3) 25,0 мг Лактоза 153,6 мг Кукурузный крахмал 100,0 мг Стеарат магния 1,4 мг Всего 280,0 мг 280 мг полученного порошка помещают в желатиновую капсулу N 3.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНОЕ АМИНОКИСЛОТЫ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 1992 |
|
RU2104265C1 |
| ПРОИЗВОДНЫЕ БЕНЗОПИРАНА ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1991 |
|
RU2038354C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ФЕНОЛА ИЛИ ИХ ФАРМАКОЛОГИЧЕСКИ ПРИЕМЛЕМЫХ КИСЛОТНО-АДДИТИВНЫХ СОЛЕЙ | 1990 |
|
RU2022961C1 |
| ЗАМЕЩЕННОЕ КОНДЕНСИРОВАННОЕ ГЕТЕРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ, СПОСОБ ФАРМАКОЛОГИЧЕСКОГО ВОЗДЕЙСТВИЯ, СПОСОБ ИНГИБИРОВАНИЯ 5-ЛИПОКСИГЕНАЗЫ, ИНГИБИРОВАНИЯ ПРОДУКЦИИ ЛИПИДНЫХ ПЕРОКСИДОВ ИЛИ СНИЖЕНИЯ УРОВНЯ САХАРА В КРОВИ | 1998 |
|
RU2196141C2 |
| ПРОИЗВОДНЫЕ БИФЕНИЛА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1993 |
|
RU2109736C1 |
| ПРОИЗВОДНЫЕ ТЕТРАГИДРОТИЕНО(3,2-С)ПИРИДИНА ИЛИ ИХ ФУРО-АНАЛОГИ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1992 |
|
RU2089553C1 |
| ПРОИЗВОДНЫЕ НИТРОКСИАЛКИЛАМИДА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1994 |
|
RU2098406C1 |
| СПОСОБ ПОЛУЧЕНИЯ ОПТИЧЕСКИ АКТИВНОГО ПРОИЗВОДНОГО ИНДОЛОБЕНЗОХИНОЛИНА ИЛИ ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫХ СОЛЕЙ | 1990 |
|
RU2045528C1 |
| ПРОИЗВОДНЫЕ ПИПЕРАЗИНКАРБОНОВОЙ КИСЛОТЫ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1992 |
|
RU2091373C1 |
| ПРОИЗВОДНЫЕ НУКЛЕОЗИДОВ ПИРИМИДИНА ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1992 |
|
RU2085557C1 |
Использование: в медицине, в частности в качестве веществ, способных промотировать выработку или секрецию фактора роста нервной ткани. Сущность изобретения: продукт производное бензола ф-лы 1 R1R2Ph-(CH=CH)m-(CH2)n-C(O)R3, где m 0 или 1, n 0 -2; R1 амино или нитрогруппа; R2 амино, ацетокси, бензилокси, трифторацетамидо- или ОН -группа; R3 аминогруппа, которая может быть моно- или ди-замещена заместителями из группы "А". Реагент 1: реакционноспособное соединение ф-лы 2: R1R2Ph-(CH=CH)mC(O)Y. Реагент ф-лы 3: H-R3 Условия реакции: лучше в среде инертного растворителя в присутствии основания. 1 табл.
Производные бензола общей формулы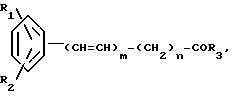
где R1 аминогруппа или нитрогруппа;
R2 аминогруппа, ацетоксигруппа, бензилоксигруппа, гидроксильная группа или трифторацетамидо;
R3 аминогруппа или замещенная аминогруппа, содержащая в качестве заместителей 1 или 2 группы, выбранные из числа членов заместителей группы А;
m целое число от 0 до 1;
n целое число от 0 до 2;
группа А заместителей состоит из следующих членов: C1 - C4-алкильных групп, адамантильной группы, фенильных групп, замещенных фенильных групп, содержащих в качестве заместителей от 1 до 3 групп, выбранных из числа членов заместителей группы В, гетероциклильных групп и метоксикарбонилзамещенных гетероциклильных групп;
группа В заместителей состоит из следующих членов: арилкарбонила, атомов галогена и C1 C4-алкильных групп;
гетероциклильные группы представляют собой 5 6-членные ароматические или карбоциклические кольца, содержащие от 1 до 2 гетероатомов, выбранных из атомов азота, кислорода и серы, необязательно сконденсированные с одним арильным кольцом, при условии, что, когда m 0, n целое число 2,
и их соли.
| СКВАЖИННЫЙ ПРИБОР ДЛЯ АКУСТИЧЕСКОГО ВИДЕОКАРОТАЖА | 0 |
|
SU399814A1 |
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
