Изобретение касается новых соединений, которые промотируют продуцирование и секретирование человеческого фактора роста нервов.
Открытие фактора роста нервов (ФРН) было описано Леви-Монталейни и др. в 1954 г. ФРГ является одним из факторов питания и факторов роста, необходимых для поддержания роста и функционирования нервной системы. Недавно было установлено, что ФРН может ускорять восстановление нарушений периферийной нервной системы и восстанавливать нарушения в функционировании центральной нервной системы, в частности, при заболевании Элзхаймера и ишемии головного мозга. Однако применение самого ФРН нежелательно ввиду того, что он является высокомолекулярным протеином с мол. м. 13000 для мономерной формы и 26000 для димерной формы, а в больше части случаев по соображениям безопасности.
Известно, что непротрансмиттеры на основе пирокатехина такие, что адреналин или норадреналин, и соединения, аналогичные пирокатехину, могут промотировать ФРН-продуцирование.
Целью изобретения является разработка доступного способа получения соединений формулы I
(R1O)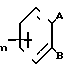 где А - водород;
где А - водород;
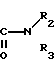
В - группа -(СН2)m-C(O)-N(R2)(R3); или А и В вместе образуют группу формулы IV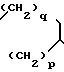
 n=2 или 3; каждый из R1 может принимать одинаковые или различные значения и означают: атом водорода; С1-С8-алканоил группу, возможно замещенную карбоксильной группой или ее С1-С3-алкиловым эфиром; аминоацетильную или карбамоильную группу, замещенную по атому азота С1-С3-алкилом; бензоил или,
n=2 или 3; каждый из R1 может принимать одинаковые или различные значения и означают: атом водорода; С1-С8-алканоил группу, возможно замещенную карбоксильной группой или ее С1-С3-алкиловым эфиром; аминоацетильную или карбамоильную группу, замещенную по атому азота С1-С3-алкилом; бензоил или,
две группы R1 могут образовывать С1-С3-алкилен связующую группу;
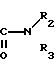
R2 означает С1-C16-алкил, моно-, ди-, или трициклоалкил, необязательно замещенный фенилом, ароматическая гетероциклическая группа, содержащая 5 или 6 атомов в кольце, из которых по крайней мере один является серой, и/или из которых 1 или 2 являются азотом, причем этот гетероцикл может быть сконденсирован с циклогексилом или фенилом и необязательно замещен 1-3 заместителями, выбранными из группы: С1-С3-алкил карбоксильная группа или ее С1-С4-алкиловый эфир; фенил, возможно замещенный 1-3 заместителями, выбранными из группы, включающей галоген, С1-С6-алканоил, С1-С6-алкил, С1-С3-алкоксигруппу, аминогруппу и бензоильную группу, возможно замещенную атомом галогена; инданильную группу;
R3 - атом водорода или фенил;
m - целое число от 1 до 4; р и q независимо означают целое число 1, а связь, обозначенная прерывистой линией, может быть одинарной или двойной;
или их фармакологически приемлемых кислотно-аддитивных, который заключается в том, что осуществляют взаимодействие галоизоангидрида формулы II
(R1O)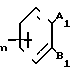 где каждый R1 имеет указанные значения, кроме водорода, и А1 и В1 имеют указанные значения, но где фрагмент -N(R2) (R3) заменен атомом галогена с амином формулы (III)
где каждый R1 имеет указанные значения, кроме водорода, и А1 и В1 имеют указанные значения, но где фрагмент -N(R2) (R3) заменен атомом галогена с амином формулы (III)
HN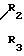
Соединения I проявляют активность при промотировании продуцирования и секретирования ФРН. Кроме того, они имеют низкую токсичность. Поэтому соединения настоящего изобретения могут быть использованы для лечения слабоумия, ишемии головного мозга и, более обще, для лечения различных нарушений нервной системы.
Приводимые ниже примеры иллюстрируют получение соединений настоящего изобретения из известных исходных соединений или из исходных соединений, которые могут быть получены с использованием процедур, аналогичных тем, что используют для известных соединений. Когда приводят Rf, то его получают при помощи тонкослойной хроматографии на силикагеле Мерк Арт 5715 толщиной 0,25 мм, проявляемом 1: 1 смесью гексана или этилацетата. Примеры для ссылок включены для получения некоторых исходных соединений.
Пример для ссылок 1. 6,7-Диметокси-2-метоксикарбонил-1-тетралон.
2,78 г (0,0135 моля) 6,7-диметокси-1-тетралона растворяли в 100 мл диметилкарбоната и при подаче потока азота добавляли 0,26 г метилата натрия. Смесь дефлегмировали в течение 5 ч. После охлаждения реакционную смесь нейтрализовали 2 н. раствором уксусной кислоты, затем разбавляли этилацетатом. Смесь последовательно промывали водным раствором кислого карбоната натрия и 0,5 н. раствором хлористоводородной кислоты. Органический слой сушили над безводным сульфатом натрия и концентрировали отгонкой. Остаток подвергали очистке при помощи хроматографической колонны через силикагель, чтобы получить 2,8 г (0,0106 моля, 74,5%) соединения из заголовка примера в виде белых кристаллов.
Пример для ссылок 2. Метил 6,7-диметокси-1,2,3,4-тетрагидро-2-нафтоат.
1 г (0,038 моля) соединения, полученного в примере для ссылок 1, растворяли в смеси 50 мл уксусной кислоты и 30 мл метанола и подвергали гидрогенизации в присутствии 10% палладия на древесном угле в качестве катализатора. После того, как абсорбировалось примерно 170 мл водорода, катализатор удаляли фильтрацией, а фильтрат концентрировали отгонкой, чтобы получить 0,91 г (3,64 ммоля, 96,3%) соединения из заголовка примера в виде белых кристаллов.
Пример для ссылок 3. 6,7-Диокси-1,2,3,4-тетрагидро-2-нафтойная кислота.
Раствор 0,9 г соединения, полученного в примере для ссылок 2, растворенного в 30 мл 47% бромистоводородной кислоты, дефлегмировали в течение 4. После концентрирования остаток смешивали с водой при охлаждении льдом и перемешивали, чтобы осадить кристаллы, которые собирали фильтрацией, чтобы получить 0,7 г (3,36 ммоля, 98,8%) соединения из заголовка примера в виде бледно-желтых кристаллов.
Пример для ссылок 4. 6,7-Диацетокси-1,2,3,4-тетрагидро-2-нафтойная кислота.
В раствор 0,7 г соединения, полученного в примере для ссылок 3, растворенного в 30 мл уксусного ангидрида, добавляли одну каплю серной кислоты и смесь перемешивали в течение 2 ч. В смесь добавляли 30 мл воды, затем перемешивали 2 ч. Реакционную смесь разбавляли простым эфиром и промывали последовательно водой и водным насыщенным раствором хлорида натрия. После осушки раствора над безводным сульфатом магния растворитель отгоняли дистилляцией. Остаток смешивали с толуолом и концентрировали при помощи азеотропной дистилляции, чтобы получить 0,96 г (3,28 ммоля, 97,8%) соединения из заголовка примера.
Пример для ссылок 5. Метил 1-окси-6,7-диметокси-1,2,3,4-тетрагидро-2-нафтоат.
В раствор 1,9 г (7,19 ммоля) соединения, полученного в примере для ссылок 1, растворенного в 30 мл метанола, добавляли 4 г борогидрида натрия при температуре 45оС в течение 20 мин. После перемешивания в течение 40 мин смесь концентрировали до примерно половины первоначального объема и разбавляли этил ацетатом. Смесь последовательно промывали водным раствором кислого карбоната натрия, 0,5 н. раствором хлористоводородной кислоты и водным насыщенным раствором хлорида натрия. После сушки раствора над безводным сульфатом натрия растворитель отгоняли, чтобы получить 1,9 г (7,14 ммоля, 99,3%) соединения из заголовка примера.
Пример для ссылок 6. Метил 6,7-диметокси-3,4-дигидро-2-нафтоат.
В раствор 1,9 г соединения, полученного в примере для ссылок 5, растворенного в 70 мл дихлорметана, последовательно добавляли 2,98 мл триэтиламина и 0,78 мл тионил хлорида при температуре 0оС, и смеси давали возможность нагреться до комнатной температуры, а затем перемешивали в течение 1 ч. Далее в реакционную смесь при 0оС добавляли воду. После разбавления водой и этилацетатом смесь промывали водным раствором кислого карбоната натрия, 0,5 н. раствором хлористоводородной кислоты и водным насыщенным раствором хлорида натрия. Органический слой сушили над безводным сульфатом натрия и растворитель удаляли отгонкой, чтобы получить соединение из заголовка примера.
Пример для ссылок 7. 6,7-Диокси-3,4-дигидро-2-нафтойная кислота.
В раствор соединения, полученного в примере для ссылок 6, растворенного в 5 мл дихлорметана, по каплям добавляли 30 мл 1М дихлорметанового раствора трибромида бора при температуре -78оС в атмосфере азота. Реакционной смеси давали возможность нагреться до комнатной температуры, а затем перемешивали 3 ч. После охлаждения его сливали в ледяную воду и перемешивали в течение 1 ч, а затем экстрагировали этилацетатом, соединенные экстракты промывали водным насыщенным раствором хлорида натрия и сушили над безводным сульфатом натрия. После обработки раствора активированным древесным углем растворитель удаляли дистилляцией. Остаток подвергали очистке при помощи хроматографической колонны через силикагель, чтобы получить 0,63 г (3,06 ммоля, 42,9%)соединения из заголовка примера в виде белых кристаллов.
Пример для ссылок 8. 6,7-Диацетокси-3,4-дигидро-2-нафтойная кислота.
В раствор 0,7 г соединения, полученного в примере для ссылок 7, растворенного в 30 мл уксусного ангидрида, добавляли одну каплю серной кислоты и смесь перемешивали в течение 2 ч, затем добавляли 30 мл воды. После перемешивания в течение 2 ч реакционную смесь разбавляли простым эфиром и промывали последовательно водой и водным насыщенным раствором хлорида натрия. После сушки раствора над безводным сульфатом магния растворитель удаляли дистилляцией. Остаток смешивали с толуолом, а оставшуюся уксусную кислоту удаляли при помощи азеотропной дистилляции, чтобы получить 0,96 г (3,28 ммоля, 97,8%) соединения из заголовка примера.
П р и м е р 1. N-(2,5-дихлорфенил)-3,4-диацетоксифенилпропионамид.
2,5-Дихлоранилин (1,62 г, 10 мМ) и пиридин (1 мл) растворяли в 20 мл дихлорметана. В смесь добавляли 3,4-диацетоксифенилпропионил хлорид (2,62 г) при охлаждении льдом при перемешивании, затем перемешивали 15 мин при той же температуре. После перемешивания при комнатной температуре в течение 30 мин реакционную смесь разбавляли водой и органический слой отделяли. Водный слой экстрагировали дихлорметаном (2х20 мл). Соединенные экстракты промывали водным раствором кислого карбоната натрия, а затем разбавленной хлористоводородной кислотой. После сушки промытого экстракта над безводным сульфатом натрия растворитель выпаривали, чтобы получить 3,2 г соединения из заголовка примера, температура точки плавления (ттп) 96-97оС.
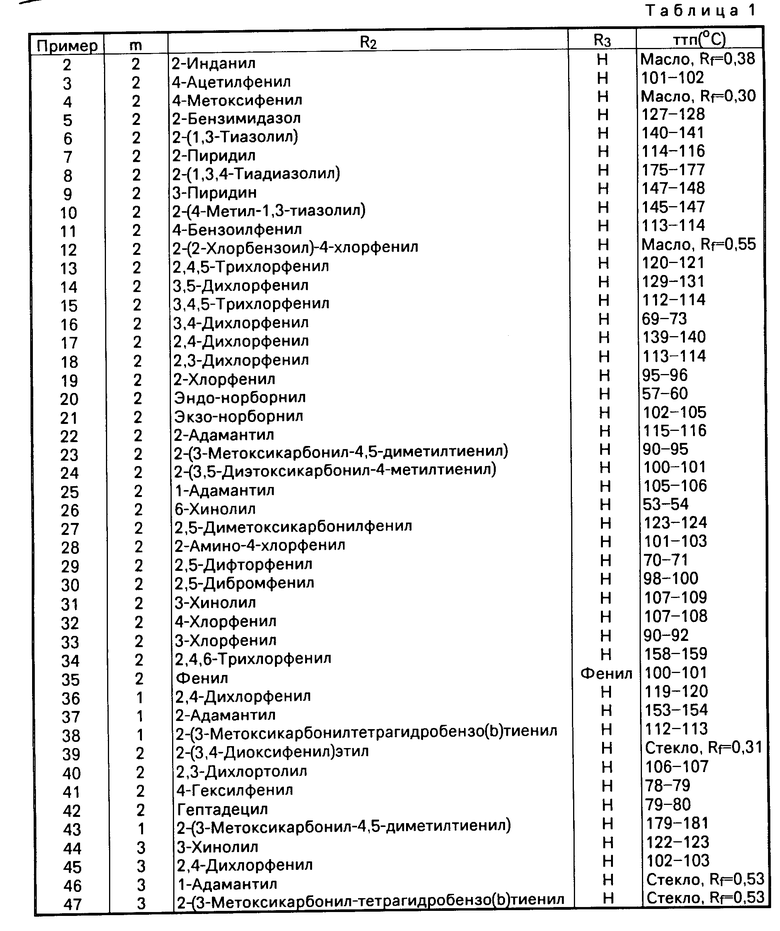
П р и м е р ы 2-47. При помощи процедуры, аналогичной той, что описана в примере 1, синтезировали приводимые в табл. 1 соединения формулы I с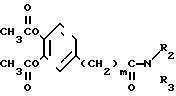
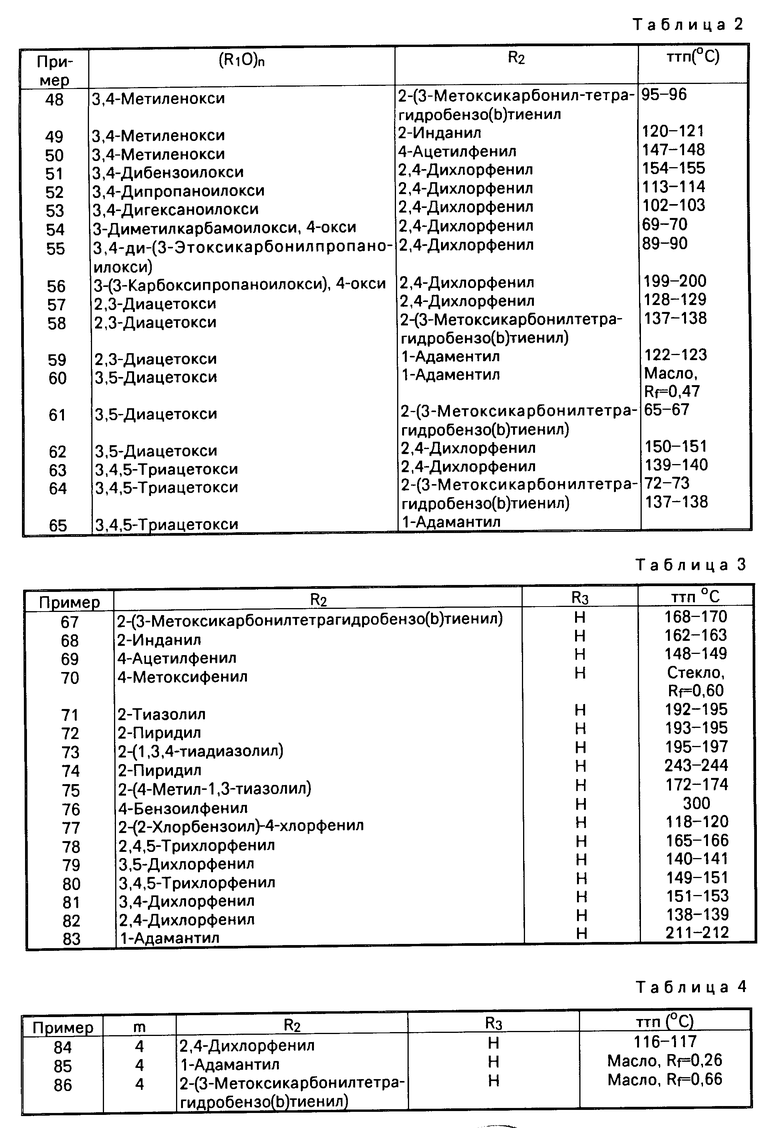
П р и м е р ы 48-66. При помощи процедуры, аналогичной описанной в примере 1, синтезировали следующие соединения формулы (Id) (табл. 2). В примерах 54 и 56 заместители (R1О)n содержат 4-оксигруппу и указанную группу в 3-позиции. В каждом из остальных примеров соответствующие заместители R1О одинаковы.
(R1O) (Id)
(Id)
П р и м е р 66. N-(2,5-дихлорфенил)-3,4-диоксифенилпропионамид.
Амид (1,0 г), полученный в примере 1, суспендировали в 20 мл метанола и в суспензию добавляли 10 мг металлического натрия, затем перемешивали в течение 2 ч до тех пор, пока суспензия не станет прозрачной. Реакционную смесь разбавляли 200 мл воды и подкисляли разбавленной хлористоводородной кислотой. Полученный в результате светло-желтый осадок собирали фильтрацией и подвергали рекристаллизации из этанола, чтобы получить 0,5 г соединения из заголовка примера, температура точки плавления 130-131оС.
П р и м е р ы 67-83. При помощи процедуры, аналогичной той, что была описана в примере 1, синтезировали следующие соединения формулы I е (табл. 3):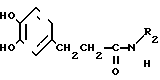
П р и м е р ы 84-86. При помощи процедуры, аналогичной той, что была использована в примере 1, синтезировали следующие соединения формулы I с (табл. 4):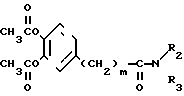
П р и м е р 87. Смесь 1,63 г (6 мМ) оксисоединения из примера 66, 1,75 г (10 мМ) трет-бутоксигликокола и 2,06 г (10 мМ) ДСС в 30 мл тетрагидрофурана перемешивали при комнатной температуре в течение 3 ч. В конце этого времени растворитель удаляли выпариванием при пониженном давлении. Остаток подвергали хроматографии на силикагеле и элюировали объемной 1:1 смесью гексана и этилацетата, чтобы получить сложный эфир трет-бутоксигликокола оксисоединения. Весь продукт растворяли в 10 мл диоксана. При охлаждении льдом добавляли 20 мл 4 н. диоксана (НСl), затем смеси давали возможность отстояться при охлаждении льдом в течение 1 ч. Растворитель удаляли выпариванием при пониженном давлении, а остаток тщательно промывали по очереди диэтиловым простым эфиром и тетрагидрофураном, чтобы получить 2,1 г соединения из заголовка примера с температурой точки плавления выше 220оС.
П р и м е р ы 88-90. При помощи процедуры, аналогичной той, что была использована в примере 87, синтезировали следующие соединения формулы If (табл. 5):
2HCl
П р и м е р 91. N-(2,4-дихлорфенил)-6,7-диацетокси-1,2,3,4-тетрагидро-2-нафтамид.
В раствор 0,17 г (0,58 ммоля) соединения (полученного в примере для ссылок 4, растворенного в 6 мл дихлорметана, добавляли одну каплю диметилфермамида и смесь охлаждения до 0оС. После добавления 0,4 г оксалилхлорида температуру смеси поднимали до комнатной и смесь перемешивали 2 ч. Затем растворитель полностью удаляли дистилляцией. Остаток растворяли в 6 мл дихлорметана и в раствор добавляли 0,113 г (0,7 ммоля) 2,4-дихлоранилина и 0,071 мг (0,7 ммоля) триэтиламина. После перемешивания в течение 3 ч смесь разбавляли этилацетатом и промывали последовательно водой, разбавленным водным раствором кислого карбоната калия и водным насыщенным раствором хлорида натрия. Органический слой сушили над безводным сульфатом натрия и растворитель отгоняли дистилляцией. Остаток подвергали очистке при помощи хроматографической колонны на силикагеле, чтобы получить в результате 0,201 г (0,461 ммоля, 79,2% ) соединения из заголовка примера, температура точки плавления 166-168оС.
П р и м е р 92. N-(3-хинолил)-6,7-диацетокси-1,2,3,4-тетрагидро- 2-нафтамид.
В раствор 0,17 г (0,58 ммоля) соединения, полученного в примере для ссылок 4, растворенного в 6 мл дихлорметана, добавляли одну каплю диметилформамида и смесь охлаждали до 0оС. После добавления 0,4 г оксалилхлорида температуру смеси поднимали до комнатной, затем перемешивали в течение 2 ч. Реакционную смесь полностью освобождали от растворителя, остаток растворяли в 6 мл дихлорметана и в раствор добавляли 0,084 г (0,58 ммоля) 3-аминохинолина и 0,088 мг (0,82 ммоля) триэтиламина. После перемешивания смеси в течение 3 ч ее разбавляли этилацетатом и последовательно промывали водой, разбавленным водным раствором кислого карбоната натрия и водным насыщенным раствором хлорида натрия. Органическую фазу сушили над безводным сульфатом натрия, и растворитель удаляли дистилляцией. Остаток подвергали очистке на хроматографической колонне через силикагель, чтобы получить 0,207 г (0,495 ммоля, 84,1%) соединения из заголовка примера, температура точки плавления 194-196оС.
П р и м е р 93. N-(1-Адамантил)-6,7-диацетокси-1,2,3,4-тетрагидро-2-нафтамид.
В раствор 0,13 г (0,45 ммоля) соединения, полученного в примере для ссылок 4, растворенного в 6 мл дихлорметана, добавляли одну каплю диметилформамида и смесь охлаждали до 0оС. После добавления 0,4 г оксалилхлорида температуру поднимали до комнатной и смесь перемешивали 2 ч. Реакционную смесь полностью освобождали от растворителя. Остаток растворяли в 6 мл дихлорметана и в раствор добавляли 0,067 г (0,58 ммоля) 1-адаментиламина и 0,068 мг (0,67 ммоля) триэтиламина. После перемешивания смеси в течение 3 ч ее разбавляли этилацетатом и последовательно промывали водой, разбавленным водным раствором кислого карбоната натрия и водным насыщенным раствором хлорида натрия. Органическую фазу сушили над безводным сульфатом натрия и растворитель удаляли дистилляцией. Остаток подвергали очистке на хроматографической колонне через силикагель, чтобы получить 0,114 г (0,268 ммоля, 60,3% ) соединения из заголовка примера, температура точки плавления 234-235оС.
П р и м е р 94. N-(2,4-дихлорфенил)-6,7-диацетокси-3,4-дигидро-2-нафтамид.
В раствор 0,075 г (0,26 ммоля) соединения, полученного в примере для ссылок 8, растворенного в 5 мл дихлорметана, добавляли одну каплю диметилформамида и смесь охлаждали до 0оС. После добавления 0,3 г оксалилхлорида температуру поднимали до комнатной, затем смесь перемешивали 2 ч. Реакционную смесь полностью освобождали от растворителя. Остаток растворяли в 5 мл дихлорметана и в раствор добавляли 0,050 г (0,31 ммоля) 2,4-дихлоранилина и 0,089 мг (0,39 ммоля) триэтиламина. После того, как смесь перемешивали 3 ч, ее разбавляли этилацетатом и промывали последовательно водой, разбавленным водным раствором кислого карбоната натрия и водным насыщенным раствором хлорида натрия. Органическую фазу сушили над безводным сульфатом натрия, а растворитель удаляли дистилляцией. Остаток подвергали очистке при помощи хроматографической колонны через силикагель, чтобы получить 0,100 г (0,23 ммоля, 89,4%) соединения из заголовка примера, температура точки плавления 180-182оС.
П р и м е р 95. N-(3-хинолил)-6,7-диацетокси-3,4-дигидро-2-нафтамид.
В раствор 0,082 г (0,28 ммоля) соединения, полученного в примере для ссылок 8, растворенного в 5 мл дихлорметана, добавляли одну каплю диметилформамида, и смесь охлаждали до 0оС. После добавления 0,3 г оксалилхлорида температуру поднимали до комнатной, затем смесь перемешивали 2 ч. Реакционную смесь полностью освобождали от растворителя. Остаток растворяли в 5 мл дихлорметана, и в раствор добавляли 0,049 г (0,34 ммоля) 3-аминохинолина и 0,043 мг (0,42 ммоля) триэтиламина. После того, как смесь перемешивали 3 ч, ее разбавляли этилацетатом и промывали последовательно водой, разбавленным водным раствором кислого карбоната натрия и водным насыщенным раствором хлорида натрия. Органическую фазу сушили над безводным сульфатом натрия, и растворитель удаляли дистилляцией. Остаток подвергали очистке при помощи хроматографической колонны через силикагель, чтобы получить 0,096 г (0,231 ммоля, 81,6%) соединения из заголовка примера, температура точки плавления 214-216оС.
П р и м е р 96. N-(1-адамантил)-6,7-диацетокси-3,4-дигидро-2-нафтамид.
В раствор 0,072 г (0,25 ммоля) соединения, полученного в примере для ссылок 8, растворенного в 5 мл дихлорметана, добавляли одну каплю диметилформамида, и смесь охлаждали до 0оС. После добавления 0,3 г оксалилхлорида температуру поднимали до комнатной, затем перемешивали 2 ч. Реакционную смесь полностью освобождали от растворителя. Остаток растворяли в 5 мл дихлорметана, и в раствор добавляли 0,045 г (0,30 ммоля) 1-адамантиламина и 0,038 мг (0,37 ммоля) триэтиламина. После того, как смесь перемешивали 3 ч, ее разбавляли этилацетатом и промывали последовательно водой, разбавленным водным раствором кислого карбоната натрия и водным насыщенным раствором хлорида натрия. Органическую фазу сушили над безводным сульфатом натрия, и растворитель удаляли дистилляцией. Остаток подвергали очистке на хроматографической колонне через силикагель, чтобы получить 0,082 г (0,194 ммоля, 78,4% ) соединения из заголовка примера, температура точки плавления 224-226оС.
Примеры изучения активности по промотированию ФРН.
Фурукава и др. установили, что L-М-клетки линии клеток фибробласта соединительной ткани мыши могут продуцировать и секретировать относительно большие количества ФРН, причем пирокатехинамины ускоряют такое продуцирование и секретирование ФРН (I.Biol. Chem, т. 261, с. 6039-6047, 1986). Следуя процедуре испытания, описанной в статье Фурукавы, но используя соединения настоящего изобретения и известные промоторы ФРН (эпинофрин, изопротеренол, Л-ДОПА и кофеиновую кислоту), анализировали активности по продуцированию и секретированию ФРН. Испытуемые соединения настоящего изобретения использовали в концентрации 10 γ/мл, а известные соединения использовали в концентрации 20 γ/мл.
Культуральную среду 199, содержащую 0,5% пептона, использовали для культивирования L-М-клеток. Примерно 5х104 L-М-клеток помещали в каждое углубление культурной пластины с 24 углублениями и выращивали в СО2-инкубаторе (37оС, 5% СО2) до слияния. После удаления культурной среды культивированные клетки промывали один раз культурной средой 199, содержащей 0,5% альбумина бычьей сыворотки (Франция V, Сигма). Испытываемые соединения добавляли до необходимой концентрации в культурную среду 199, содержащую 0,5% альбумина бычьей сыворотки, и обрабатывали 0,5 мл L-М-клеток. После культивирования L-М-клеток в инкубаторе СО2 в течение 24 ч среду извлекали и определяли уровень ФРН.
ФРН определяли с использованием ферментного иммуноанализа. Раствор (каждый 75 мл) антитела антимышиного β -ФРН (0,3 мк/г/мл, рН 9,6; Боерингер Маннгейм) пипеткой наносили в каждое углубление полистироловой пластины с 96 углублениями. После выдерживания пластины при комнатной температуре в течение 1 ч антитела удаляли при помощи трехкратного промывания моющим раствором. Раствор (50 мл) стандартного β -ФРН (фирма Уако Пью Кемикал Индастриз Лтд. ) или извлеченную среду (50 мл) для каждого из испытуемых соединений наносили пипеткой в каждое углубление. После выдерживания пластины при комнатной температуре в течение 6-8 ч стандартный β -ФРН или испытываемый раствор удаляли и каждое углубление промывали три раза. В каждое углубление пипеткой наносили раствор (50 мл) моноклонального антитела β -ФРН (100 мг/мл, рН 7,0; Боерингер Маннгейм), меченного β -галактозидазой. После выдерживания пластины при температуре 4оС в течение 15-18 ч меченные ферментом антитела удаляли, а углубления промывали три раза, затем наносили пипеткой раствор (100 мл) хлорфенол красного β -D-галактопиранозида (1 мг/мл, рН 7,3; Боерингер Маннгейм) в каждое углубление. Окраске давали возможность проявиться (2-3 ч при комнатной температуре), а поглощение определяли в диапазоне 570 нм. Количество ФРН вычисляли из стандартной кривой. Результаты выражали в виде относительного значения (%), которое вычисляли относительно количества ФРН, продуцированного и секретированного клетками, которые не обрабатывали испытываемыми соединениями. Числовые значения (% от контрольного) выражали в виде среднего значения для 3 углублений от контрольного (без добавления испытуемых соединений). Результаты приведены в табл. 6.
Отсюда следует, что соединения настоящего изобретения проявляют исключительно высокую активность в качестве промотора продуцирования и секретирования ФРН.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ БЕНЗОЛА И ИХ СОЛИ | 1992 |
|
RU2042663C1 |
| ТРИЦИКЛИЧЕСКИЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1992 |
|
RU2072997C1 |
| МОДИФИЦИРОВАННЫЕ ОЛИГОДЕЗОКСИРИБОНУКЛЕОТИДЫ, КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ | 1994 |
|
RU2111971C1 |
| СПОСОБ ПОЛУЧЕНИЯ ИЗОКСАЗОЛОВЫХ ПРОИЗВОДНЫХ | 1991 |
|
RU2017737C1 |
| ПЕПТИДНОЕ СОЕДИНЕНИЕ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 1993 |
|
RU2106357C1 |
| ПРОИЗВОДНЫЕ БЕНЗОПИРАНА ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1991 |
|
RU2038354C1 |
| АНАЛОГИ ЛИПИДА ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ | 1992 |
|
RU2076107C1 |
| ТИАЗОЛИДИНОВЫЕ СОЕДИНЕНИЯ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, СПОСОБ СНИЖЕНИЯ СОДЕРЖАНИЯ САХАРА В КРОВИ У МЛЕКОПИТАЮЩИХ | 1992 |
|
RU2095354C1 |
| ПИПЕРИДИЛОКСИ- ИЛИ ХИНУКЛИДИНИЛОКСИ-ИЗОКСАЗОЛЬНЫЕ ПРОИЗВОДНЫЕ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ СВОЙСТВАМИ УМЕНЬШАТЬ НАРУШЕНИЯ ПОЗНАВАТЕЛЬНОЙ СПОСОБНОСТИ | 1993 |
|
RU2079496C1 |
| Способ получения производных тианафтена или их фармацевтически приемлемых солей | 1987 |
|
SU1739848A3 |

Сущность изобретения: продукт - производное фенола ф-лы 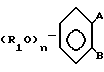 где A - H, B - (CH2)m-C(O)-N(R2)(R3) или A и B вместе образуют группу
где A - H, B - (CH2)m-C(O)-N(R2)(R3) или A и B вместе образуют группу 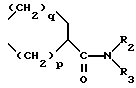 или его фармакологически приемлемые кислотно-аддитивные соли. Реагент 1: галоидангидрид ф-лы
или его фармакологически приемлемые кислотно-аддитивные соли. Реагент 1: галоидангидрид ф-лы 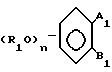 Реагент 2: амин HNR2R3, с последующим переводом полученного соединения в соль и в случае необходимости снятием гидроксилзащитной группы. 6 табл.
Реагент 2: амин HNR2R3, с последующим переводом полученного соединения в соль и в случае необходимости снятием гидроксилзащитной группы. 6 табл.
Способ получения производных фенола общей формулой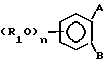
где A - водород;
B - группа - (CH2)m - C(O) - N(R2)(R3), или A и B - вместе образуют группу общей формулы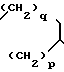
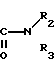
n = 2 или 3;
каждый R1 одинаковый или различный водород, (C1 - C8)-алканоилгруппа, возможно замещенная карбоксильной группой или ее (C1- C3)-алкиловым эфиром, аминоацетиальная или карбамоильная группа, замещенная по атому азота C1 - C3-алкилом;
бензоил или две группы R1 могут образовывать C1 - C3-алкилен свзующую группу;
R2 - C1 - C16-алкил, моно-, ди- или трициклоалкил, необязательно замещенный фенилом, ароматическая гетероциклическая группа, содержащая 5 или 6 атомов в кольце, из которых по крайней мере один является серой и/или из которых один или два являются азотом, причем этот гетероцикл может быть сконденсирован с циклогексилом или фенилом и необязательно замещен 1 - 3-мя заместителями, выбранными из группы C1 - C3-алкил, карбоксильная группа или ее C1 - C4-алкиловый эфир, фенил, возможно замещенный 1 - 3-мя заместителями, выбранным из группы включающей галоген, C1 - C6-алканоил, C1 - C6-алкил, C1 - C3-алкоксигруппу, аминогруппу и бензоильную группу, возможно замещенную атомом галогена, инданильную группу;
R3 - водород или фенил;
m = 1 - 4 - целое число
p и q = 1
связь, обозначенная прерывистой линией, может быть одинарной или двойной;
или их фармакологически приемлемых кислотно-аддитивных солей, отличающийся тем, что осуществляют взаимодействие галоидоангидрида общей формулы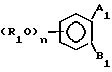
где R1 имеет указанные значения, кроме водорода;
A1 и B1 имеют значения, как и A и B но фрагмент -N(R2)(R3) заменен атомом галогена с амином общей формулы
HN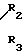
переводом полученного соединения в соль и, в случае необходимости, снятием гидроксилзащитной группы.
Приоритет по признакам:
23.05.89 при А - водород, В - (CH2)mC(O)N(R2)(R3);
07.08.89 при А и В вместе образующими группу общей формулы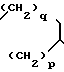
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
