Изобретение относится к новым производным бензопирана, которые имеют противогипотензивную активность и могут быть использованы при лечении и профилактике сердечно-сосудистых заболеваний.
Соединениями настоящего изобретения являются 3,4-дигидро-2,2-диметил-4(1-оксоизоиндолин-2-ил)-6- перфторалкил- сульфонил-2Н-1-бензопиран-3-ола, соединения 3,4-дигидро-4-(1,2-дигидро-2-оксо-1-пиридил)-2,2-диметил-6- трифторметил- сульфонил-2Н-1-бензопиран-3-ола, нитраты этих соединений, аналоги эти соединений, имеющие углерод углеродную ненасыщенную связь 3,4-положении пиранового цикла, и производные этих соединений и их нитраты, в которых изоиндолинильный или пиридильный цикл является замещенным.
Известны соединения этого типа и с таким типом активности [1-5] например, 6-циано-3,4-дигидро-2,2-диметил-4-(2-оксо-1- пирролидинил)-2Н- 1-бензопиран-3-ол, формулы А, 3,4-дигидро-2,2-диметил-6-трифторметилсульфонил-4-(2-оксо-1- пирроли- динил)-2Н-1-бензопиран-3-ол формулы В и 3,4-дигидро-2,2-диметил-4-(1-оксоизоиндо- лин-2-ил)-6- трифторметилсульфонил-2Н-1-бензопиран-3-ол формулы С:
 (A)
(A)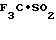
 (B)
(B)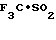
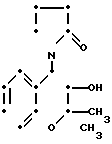
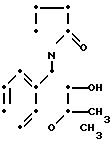
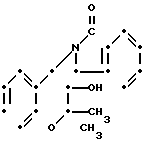 (C)
(C)
Изомер соединения А [(±)-6-циано-3,4-дигидро-2,2-диметилтранс-4-(2-оксо-1-пир- ролидинил)-2Н- 1-бензопиран-3-ол] известен как Кромакалим находится в стадии исследования в качестве возможного коммерческого сосудорасширяющегося средства.
Хотя эти известные соединения имеют приемлемые и полезные активности, их активность продолжительность действия являются меньшими, чем желательно.
В предлагаемом изобретении описан ограниченный ряд соединений, которые имеют одинаковый класс активности, сочетание 1-оксоизоиндолин-2-ильной или 1,2-дигидро-2-оксо-1-пиридильной группы в положении 4 и перфторалкилсульфональной группы в положении 6 и которые в результате являются более мощными (сильно действующими) и имеют большую продолжительность действия.
Существом настоящего изобретения являются производные бензопирана, обладающие сосудорасширяющей и гипотен- зивной активностью.
Целью изобретения является создание соединений, обладающих улучшенной активностью.
Соединениями согласно настоящему изобретению являются соединения формулы
R1SO2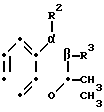 (I) где R1 перфторалкильная группа, имеющая 1-3 атома углерода;
(I) где R1 перфторалкильная группа, имеющая 1-3 атома углерода;
R2 группа формулы
-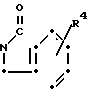 (II) где R4 атом водорода или галогена, или нитрогруппа, или группу формулы
(II) где R4 атом водорода или галогена, или нитрогруппа, или группу формулы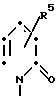 (III) где R5 атом водорода, алкильная группа, имеющая 1-4 атома углерода, алкоксигруппа, имеющая 1-4 атома углерода;
(III) где R5 атом водорода, алкильная группа, имеющая 1-4 атома углерода, алкоксигруппа, имеющая 1-4 атома углерода;
α-β простая углерод-углеродная связь (СН-СН) или углерод-углеродная двойная связь (С=С),
когда α-β представляет простую углерод-углеродную связь, R3 гидроксигруппа или нитрооксигруппа (-O-NO2), или когда α-β представляет двойную углерод-углеродную связь, R3 атом водорода, или их фармацевтически приемлемые соли при условии, что R1 не является трифторметильной группой, когда R2 представляет группу формулы (II), где α-β простая связь; R3 гидроксигруппа; R4 атом водорода.
Изобретение предлагает также способ получения соединений по настоящему изобретению.
В соединениях формулы (I), где R1 перфторалкильная группа, алкильная часть имеет 1-3 атома углерода и может быть линейной или разветвленно-цепной группой, хотя является предпочтительно линейно-цепной группой. Такими группами являются трифторметильная, пентафторэтильная, гептафторпропильная и гептафторизопропильная группы, из которых трифторметильная и пентафторэтильная группы являются предпочтительными, а трифторметильная группа являются более предпочтительной.
R2 может представлять указанную группу формулы (II) или (III), из которой группа формулы (II) является предпочтительной.
Когда R5 представляет алкильную группу, она может быть линейно- или разветвленно-цепной алкильной группой, имеющей 1-4 атома углерода. Примеры включают метильную, этильную, пропильную, изопропильную, бутильную, изобутильную, вторбутильную и третбутильную группы, предпочтительно метильную, этильную, пропильную, изопропильную или бутильную группу, наиболее предпочтительно метильную группу.
Когда R5 представляет алкоксигруппу, она может быть линейно- или разветвленно-цепной алкоксигруппой, имеющей 1-4 атома углерода. Примеры включают метокси, этокси, пропокси-, изопропокси-, бутокси-, изобутокси-, вторбутокси и третбутоксигруппы, предпочтительно метокси-, этокси-, пропокси-, изопропокси- или бутоксигруппу, наиболее предпочтительно метоксигруппу.
Предпочтительно R5 представляет собой атом водорода, метильную группу, метоксигруппу.
Когда R2 представляет группу формулы (II), предпочтительно чтобы α-β представляла бы простую углерод-углеродную связь и чтобы R3представляла бы гидроксигруппу или нитрооксигруппу. В этом случае, предпочтительно чтобы оксоизоиндолинильная группа была бы замещенной, т.е. R4 не представляла бы атом водорода. Когда R2 представляет группу формулы (III) лучше, чтобы α-β представляла бы углерод-углеродную двойную связь.
В соединениях формулы (I), где R2 группа формулы (II), R4гидроксигруппа, соединение является кислотным, поэтому оно может образовывать соль. Отсутствует определенное ограничение на природу этих солей при условии, что, если они предназначаются для терапевтического использования, они должны быть фармацевтически приемлемыми. Если же они предназначаются не для терапевтических использований, например в качестве интермедиатов при получении других и возможно более действующих соединений, то не применяется даже этого ограничение. Примеры таких солей: соли с щелочным металлом, таким как натрий, калий или литий, соли с щелочно-земельными металлом, таким как барий или кальций, соли с другим металлом, таким как магний или алюминий, соли органических оснований, такие как соль с дициклогексиламином, и соли с основной аминокислотой, такой как лизин или аргинин.
Когда α-β представляет простую углерод-углеродную связь, соединения настоящего изобретения могут образовывать различные стереоизомеры и оптические изомеры. Хотя все из этих стереоизомеров и оптических изомеров, включая рацематы, представленные здесь общей формулой (I), настоящее изобретение рассматривает как индивидуальные изомеры, так и смеси их. Предпочтительными соединениями являются соединения, содержащие 3,4-трансконфигурацию, в частности соединения, содержащие (3S, 4P)-конфигурацию. Когда применяются методики стереоспецифического синтеза или используются оптически активные соединения в качестве исходных веществ, индивидуальные изомеры могут быть получены непосредственно; с другой стороны, если получается смесь изомеров, то индивидуальные изомеры могут быть получены с помощью обычных методик разделения.
Предпочтительными классами соединений настоящего изобретения являются те соединения формулы (I), в которых
1) R1 трифторметильная группа или пентафторэтильная группа;
2) R2 группа формулы (II), где R4 атом водорода, или нитрогруппа, или группа формулы (III), где R5 атом водорода, метильная группа, или метоксигруппа.
Более предпочтительными соединениями настоящего изобретения являются те соединения формулы (I), в которой:
3) R2 группа формулы (II), где R4 атом водорода;
α-β простая углерод-углеродная связь;
R3 нитрооксигруппа,
либо α-β углерод углеродная двойная связь;
R3 атом водорода;
4) R2 группа формулы (II), где R4 нитрогруппа;
α-β углерод-углеродная простая связь;
R3 гидрооксигруппа или нитрооксигруппа,
либо α-β углерод-углеродная двойная связь;
R3 атом водорода;
5) R2 группа формулы (III), где R5 атом водорода;
α-β углерод-углеродная простая связь;
R3 нитрооксигруппа.
В особенности те соединения, в которых R1 определено выше в п. (1), а R2, R3 и α-β определены в пп. (3), (4) или (5).
Более предпочтительными соединениями настоящего изобретения являются те соединения формулы (I), в которой:
6) R1 трифторметильная группа,
в особенности те соединения, где R1 является таким, как определено в п. (6), а R2, R3 и α-β являются такими, как определено в п. (4).
Примеры некоторых соединений настоящего изобретения представлены следующими формулами (I-1) и (I-2), в которых символы являются такими, как определено в табл. 1 и 2, т.е. табл. 1 относится к формуле (I-1), а табл. 2 к формуле (I-2).
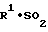
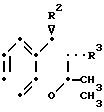 (I-1)
(I-1)
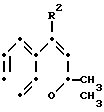 (I-2)
(I-2)
Из перечисленных соединений предпочтительными являются соединения 1-2, 1-5, 1-6, 1-9, 1-13, 1-15, 1-16, 1-20, 1-21, 1-22, 1-23, 2-1, 2-2 и 2-19. Наиболее предпочтительными соединениями являются соединения под номерами:
1-5. Нитрат 3,4-дигидро-2,2-диметил-4-(1-оксоизоиндолин-2-ил)-6 -трифторметилсульфонил-2Н-1-бензопиран-3-ила;
1-9. 3,4-Дигидро-2,2-диметил-4-(6-фтор-1-оксоизоиндолин-2-ил)-6 -трифторметилсульфонил-2Н-1-бензопиран-3-ол;
1-13. 3,4-Дигидро-2,2-диметил-4-(4-нитро-1-оксоизоиндолин-2-ил)-6- трифторметилсульфонил-2Н-1-бензопиран-3-ол;
1-15. 3,4-Дигидро-2,2-диметил-4-(6-нитро-1-оксоизоиндолин-2-ил)-6- трифторметилсульфонил-2Н-1-бензопиран-3-ол;
1-16. 2,4-Дигидро-2,2-диметил-4-(4-фтор-1-оксоизоиндолин-2-ил)-6- трифторметилсульфонил-2Н-1-бензопиран-3-ол;
2-1. 2,2-Диметил-4-(1-оксоизоиндолин-2-ил)-6-трифторметилсульфонил -2Н-1-бензопиран;
и их фармацевтически приемлемые соли.
Соединения настоящего изобретения могут быть получены целым рядом методов, широко известных для получения соединений этого типа. Например, они могут быть получены путем взаимодействия соединения формулы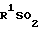
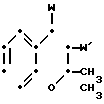 (IV) где R1 определено выше;
(IV) где R1 определено выше;
a) W аминогруппа;
W' гидроксилгруппа;
б) W и W' вместе представляют атом кислорода, чтобы образовать эпоксигруппу;
в случае а) с соединением формулы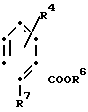 (V) где R4 определено выше;
(V) где R4 определено выше;
R6 алкильная группа, имеющая 1-4 атома углерода;
R7 формильная группа или галоидметильная группа, предпочтительно бромометильная, хлорметильная или йодометильная группа, или,
в случае б) с соединением формул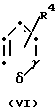
 где R4 и R5 определены выше;
где R4 и R5 определены выше;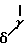 группа формулы
группа формулы
N OS
OS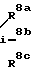
или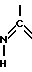 O где R8a, R8b, R8c являются одинаковыми или различными и каждый представляет алкильную группу, имеющую 1-4 атома углерода; чтобы получить соединение формулы
O где R8a, R8b, R8c являются одинаковыми или различными и каждый представляет алкильную группу, имеющую 1-4 атома углерода; чтобы получить соединение формулы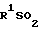
 (Ia) где R1 и R2 определены выше.
(Ia) где R1 и R2 определены выше.
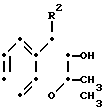
Если это необходимо, соединение формулы (Ia) может быть далее образовано основанием, чтобы получить соединение формулы
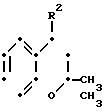 (Ib) где R1 и R2 определены выше.
(Ib) где R1 и R2 определены выше.
Кроме того, соединение формулы (Ia) может быть подвергнуто реакции с нитрирующим реагентом, чтобы получить соединение формулы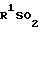
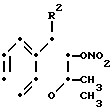 (Ic)
(Ic)
Если это необходимо, продукт реакции может быть далее превращен в соль обычными способами.
Соединения настоящего изобретения могут быть получены так, как показано в следующих схемах реакции А и В.
Схема реакции А (см. фиг. 1) показывает получение соединения формулы (I), где R2 группа формулы (II);
α-β представляет простую связь;
R3 гидроксигруппа, т.е. соединения формулы (Id),
и соответствующего соединения, в котором α-β представляет двойную связь, т.е. соединения формулы (Ie).
В приведенных формулах R1, R4 и R6 определены выше, а R7a атом галогена, предпочтительно атом брома, хлора или йода.
В стадии А1 этой схемы реакции аминоспиртовое соединение формулы (IIa) подвергается реакции либо с формилза- мещенным сложным эфиром (формулы Va), либо с галоидзамещенным сложным эфиром формулы (Vb), чтобы получить соединение формулы (Id).
Реакция аминоспиртового соединения формулы (IIa) с формилзамещенным сложным эфиром формулы (Va) обычно и предпочтительно проходит в инертном растворителе; она предпочтительно осуществляется также в присутствии восстановителя.
Отсутствует определенное ограничение на природу применяемого растворителя при условии, что отсутствует неблагоприятное воздействие на реакцию или на вовлеченные в реакцию реагенты и что он может растворять реагенты, по меньшей мере до некоторой степени. Примеры подходящих растворителей: спирты, такие как метанол, этанол или пропанол, карбоновые кислоты, такие как уксусная кислота или пропионовая кислота, сложные эфиры кислот, такие как этилацетат, кетоны, такие как ацетон или метилэтилкетон, вода, и смеси любых двух или более этих растворителей, из них спирты, в особенности метанол и пропанол, являются предпочтительными, а пропанол является наиболее предпочтительным.
Примеры восстановителей, которые могут быть использованы в этой реакции, включают борогидриды и цианоборогидриды, такие как цианоборогидрид натрия, борогидрид натрия, цианоборогидрид цинка и борогидрид цинка, из которых цианоборогидрид натрия и цианоборогидрид цинка являются наиболее предпочтительными. Если требуется, соединения цинка могут быть получены из соответствующих натриевых соединений и цинковой соли, например цианоборогидрид цинка может быть получен из цианоборогидрида натрия и хлористого цинка во время реакции.
Реакция может происходить в широком пределе температур, при этом определенная температура реакции не является критической в настоящем изобретении. Определено, что удобно проводить реакцию при 0-150оС, более предпочтительно от комнатной температуры до 100оС. Время, необходимое для реакции, также может варьировать широко в зависимости от многих факторов, особенно от температуры реакции и природы реагентов. Однако при условии, что реакция осуществляется при перечисленных условиях, период времени устанавливается от 1 до 72 ч, более предпочтительно от 1 до 36 ч.
Аминоспиртовое соединение формулы (IIa) может быть подвергнуто реакции в стадии А1 с галоидзамещенным сложноэфирным соединением формулы (Vb). Эта реакция предпочтительно проводится в инертном растворителе, предпочтительно в присутствии основания.
Примеры оснований, которые могут быть использованы для этой реакции, включают карбонаты щелочных металлов, такие как карбонат калия, карбонаты натрия или лития, гидрокарбонаты щелочных металлов, такие как гидрокарбонат натрия или гидрокарбонат калия, гидриды щелочных металлов, такие как гидрид натрия, гидроокиси щелочных металлов, такие как гидроокись калия, третичные амины, такие как триэтиламин или диизопропилэтиламин. Из них предпочтительны третичные амины, карбонаты щелочных металлов и гидрокарбонаты щелочных металлов.
Отсутствует ограничение на природу применяемого растворителя при условии, что он не имеет неблагоприятного воздействия на реакцию или на вовлеченные в реакцию реагенты и что он может растворять реагенты по меньшей мере до некоторой степени. Примеры подходящих растворителей включают нитрилы, такие как ацетонитрил, ароматические углеводороды, такие как бензол, толуол или ксилол, простые эфиры, такие как диэтиловый эфир, третрагидрофуран или диоксан, галоидированные углеводороды, особенно галоидированные алифатические углеводороды, такие как хлористый метилен или дихлорэтан, амиды, такие как диметилформамид или диметилацетамид; из них нитрилы и амиды являются предпочтительными.
Реакция может происходить в широком пределе температур, при этом определенная температура реакции не является критической (решающей) в настоящем изобретении.
Удобно проводить реакцию при температуре от 0 до 200оС, более предпочтительно от комнатной температуры до 170оС. Время, требуемое для реакции, также можно широко варьировать в зависимости от многих факторов, особенно температуры реакции и природы реагентов. Однако при условии, что реакция осуществляется при перечисленных условиях, обычно достаточен период времени от 1 до 48 ч, более предпочтительно от 1 до 10 ч.
После завершения реакции целевое соединение изобретения может быть извлечено из реакционной смеси обычными методами. Например, по одной подходящей последовательности извлечения реакционная смесь освобождается от растворителя перегонкой или выливается в воду и экстрагируется с помощью несмешивающегося с водой органического растворителя. Экстракт затем высушивают и растворитель удаляют перегонкой. Если это необходимо, целевое соединение может быть очищено в дальнейшем обычным методом, например путем перекристаллизации, или различными методами хроматографии, особенно колоночной хроматографии.
В стадии А2 схемы реакции А соединение формулы (I), где связь формулы α-β является двойной связью, а R3 атом водорода, т.е. соединение формулы (Ie), может быть получено обработкой спиртового соединения формулы (Id) с помощью основания в присутствии инертного растворителя.
Примеры оснований, которые могут быть использованы в этой реакции, включают гидроокиси щелочных металлов, такие как гидроокись натрия или гидроокись калия, содовый тальк (каталог Мерка N 1567) гидриды щелочных металлов, такие как гидрид натрия или гидрид калия; из них содовый тальк является предпочтительным.
Отсутствует определенное ограничение на природу применяемого растворителя при условии, что он не имеет неблагоприятного воздействия на реакцию или на вовлеченные в реакцию реагенты и что он может растворять реагенты по меньшей мере до некоторой степени. Примеры подходящих растворителей включают простые эфиры, такие как диэтиловый эфир, тетрагидрофуран и диоксан, сульфоксиды, такие как диметилсульфоксид, амиды, особенно амиды жирных кислот, такие как диметилформамид или диметилацетамид; из них предпочтительными являются простые эфиры.
Реакция может протекать в широком пределе температур, при этом определенная температура реакции не является решающей в настоящем изобретении. Обнаружено, что реакцию удобно проводить при температуре примерно от комнатной температуры до 180оС, более предпочтительно от 50 до 150оС. Время, необходимое для реакции, также можно широко варьировать в зависимости от многих факторов, особенно температуры реакции и природы реагентов. Однако при условии, что реакция осуществляется при перечисленных условиях, обычно достаточен период времени от 5 мин до 2 ч, более предпочтительно от 10 мин до 1 ч.
После завершения реакции целевое соединение изобретения может быть извлечено из реакционной смеси обычным методом. Например, по одной подходящей методике извлечения реакционная смесь освобождается от растворителя перегонкой или выливается в воду и экстрагируется с помощью несмешивающегося с водой органического растворителя. Затем экстракт высушивается, а растворитель удаляется перегонкой. Если это необходимо, целевое соединение может быть дополнительно очищено обычным методом, например путем перекристаллизации, или различными методами хроматографии, особенно колоночной хроматографией.
В схеме реакции В показано, как получить соединение формулы (I), где R2 группа формулы (III), связь, представленная знаком α-β, является углерод-углеродной простой связью, R3 гидроксигруппа, т.е. соединение формулы (If), или соединение формулы (I), где R2 группа формулы (III), и связь, представленная знаком α-β, является углерод-углеродной двойной связью, R3 атом водорода, т.е. соединение формулы (Ij).
В стадии В1 схема реакции В (см. фиг. 2) соединение формулы (If) может быть получено реакцией эпоксисоединения формулы (IVa) с силильным эфирным соединением формулы (VIa) или с пиридоновыми соединением формулы (VIb).
Реакция соединения формулы (IVa) с силильным простым эфирным соединением формулы (VIa) предпочтительно осуществляется в присутствии десилилирующего реагента, а также предпочтительно в инертном растворителе.
Примеры десилилирующих реагентов, которые могут быть использованы для этой реакции, включают фтористый тетрабутиламмоний и трехфтористый бор, из которых предпочтителен фтористый тетрабутиламмоний.
Отсутствует определенное ограничение на природу применяемого растворителя при условии, что он не имеет неблагоприятного воздействия на реакцию или на реагенты, вовлекаемые в реакцию, и что он может растворять реагенты по меньшей мере до некоторой степени. Примеры подходящих растворителей включают простые эфиры, такие как диэтиловый эфир, тетрагидрофуран или диоксан, углеводороды, в особенности ароматические углеводороды, такие как бензол, толуол или ксилол, и алифатические углеводороды, такие как гексан или пентан; из них предпочтительными являются простые эфиры.
Реакция может протекать в широком пределе температур, и определенная температура реакции не является решающей в настоящем изобретении. Обнаружено, что реакцию удобно проводить при температуре от 0 до 50оС (предпочтительно примерно при комнатной температуре). Время, необходимое для реакции можно широко варьировать в зависимости от многих факторов, особенно температуры реакции и природы реагентов. Однако при условии, что реакция осуществляется при перечисленных условиях, обычно достаточен период времени от 10 до 96 ч, более предпочтительно от 12 до 80 ч.
Реакция соединения формулы (IVa) с пиридоновым соединением формулы (VIb) предпочтительно проводится в инертном растворителе в присутствии основания.
Отсутствует определенное ограничение на природу основания, применяемого в этой реакции, а примеры оснований, которые могут быть использованы, включают амины, такие как пиридин, триэтиламин или 4-диметиламинопиридин, гидриды щелочных металлов, такие как гидрид натрия или гидрид калия, алкоксиды щелочных металлов, такие как метоксид натрия, этоксид натрия, этоксид калия или третбутоксид калия, гидроксиды четвертичного аммония, такие как гидроокись бензилтриметиламмония, гидроокись тетрабутиламмония или гидроокись тетраэтиламмония, карбонаты щелочных металлов, такие как карбонат калия, карбонат натрия или карбонат лития; из них предпочтительны гидриды щелочных металлов, в частности гидрид натрия, гидроокиси четвертичного аммония, в частности гидроокись бензилтриметиламмония, и амины, в частности пиридин.
Отсутствует определенное ограничение на природу применяемого растворителя при условии, что он не имеет неблагоприятного воздействия на реакцию или на реагенты, вовлеченные в реакцию, и что он может растворять реагенты по меньшей мере до некоторой степени. Когда гидрид щелочного металла используется в реакции в качестве основания, примеры подходящих растворителей включают простые эфиры, такие как диэтиловый эфир, тетрагидрофуран или диоксан, амиды, в особенности амиды жирных кислот, такие как диметилформамид или диметилацетамид, и сульфоксиды, такие как диметилсульфоксид. Когда алкоксиды щелочных металлов, амины или гидроокиси четвертичного аммония используются в реакции в качестве основания, предпочтительно использовать спирты, такие как метанол, этанол или пропанол, в качестве растворителя. Когда карбонаты используются для реакции в качестве основания, предпочтительно использовать кетоны, такие как ацетон или метилэтилкетон, или спирты, такие как метанол или этанол, в качестве растворителя. В частности, когда гидрид щелочного металла используется в качестве основания, предпочтительно использовать амид, в частности диметилформамид, или сульфоксид, в частности диметилсульфоксид, в качестве растворителя. Когда другие основания используются для реакции, предпочтительно использовать спирт, в частности метанол или этанол, в качестве растворителя.
Реакция может протекать в широком пределе температур, при этом определенная температура реакции не является решающей в настоящем изобретении. Обнаружено, что реакцию удобно проводить при температуре от 0 до 150оС, более предпочтительно от 0 до 50оС. Время, требуемое для реакции, также можно варьировать в зависимости от многих факторов, особенно температуры реакции и природы реагентов. Однако при условии, что реакция осуществляется при перечисленных условиях, обычно достаточен период времени от 30 мин до 48 ч, более предпочтительно от 1 до 30 ч.
После завершения реакции целевое соединение изобретения может быть извлечено из реакционной смеси обычным методом. Например, по одной подходящей последовательности извлечения реакционная смесь освобождается от растворителя перегонкой или выливается в воду и экстрагируется с помощью несмешивающегося с водой органического растворителя. Затем экстракт высушивают и растворитель удаляют перегонкой. Если это необходимо, целевое соединение может быть дополнительно очищено обычным методом, например путем перекристаллизации, или различными методами хроматографии, особенно колоночной хроматографией.
В стадии В2 схемы реакции В, те соединения формулы (I), где R3 атом водорода, а связь, представленная знаком α-β, является углерод-углеродной двойной связью, т. е. соединения формулы (Ig), могут быть получены, если это необходимо. Эта реакция является такой же, как и реакция, описанная в стадии А2 схемы реакции А, и может быть проведена, используя те же реагенты и при тех же условиях.
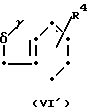
Соединение формулы (Id) или (Ie) (схема реакции А) может быть получено по методике, описанной в схеме реакции В, но заменяя пиридильное соединение формулы (VIa) или (VIb) на соответствующее изоиндолиновое соединение формул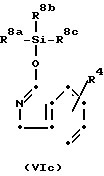
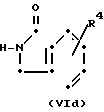 где R4, R8a, R8b и R8c определены выше.
где R4, R8a, R8b и R8c определены выше.
Соединения формулы (I), в которой связь, представленная знаком α-β, является углерод-углеродной простой связью, а R3 представляет нитрооксигруппу (-ONO2), т.е. соединения формулы
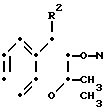 O2 (Ih) где R1 и R2 определены выше, могут быть получены реакцией соответствующего соединения формулы (I). где R3 представляет гидроксигруппу, которые можно получить по схемам реакций А и В, например соединения формулы (Id) или (If), с нитрующим реагентом.
O2 (Ih) где R1 и R2 определены выше, могут быть получены реакцией соответствующего соединения формулы (I). где R3 представляет гидроксигруппу, которые можно получить по схемам реакций А и В, например соединения формулы (Id) или (If), с нитрующим реагентом.
Реакция с нитрующим реагентом обычно и предпочтительно проводится в присутствии инертного растворителя.
Отсутствует определенное ограничение на природу применяемого растворителя при условии, что он не имеет неблагоприятного воздействия на реакцию или на реагенты, вовлеченные в реакцию, и что он может растворять реагенты по меньшей мере до некоторой степени. Примеры подходящих растворителей включают галоидированные углеводороды, в особенности галоидированные алифатические углеводороды, такие как хлористый метилен, дихлорэтан, четыреххлористый углерод из хлороформ, простые эфиры, такие как диэтиловый эфир, диметоксиэтан или тетрагидрофуран, углеводороды, в особенности алифатические углеводороды, такие как гексан или гептан. Из них предпочтительными являются галоидированные алифатические углеводороды.
Примеры нитрующих реагентов, которые могут быть использованы для этой реакции, включают соли нитрония, такие как тетрафторборат нитрония (NO2BF4), гексафторфосфат нитрония (NO2PF6) или трифторметансульфонат нитрония (NO2CF3SO3), и саму азотную кислоту. Из них предпочтительными являются соли нитрония.
Реакция может протекать в широком пределе температур, определенная температура реакции не является решающей при температуре от 0 до 100оС, более предпочтительно от 10 до 50оС. Время, требуемое для реакции, можно варьировать широко в зависимости от многих факторов, особенно температуры реакции и природы реагентов. Однако при условии, что реакция осуществляется при перечисленных условиях, обычно достаточно период времени от 10 мин до 10 ч, более предпочтительно от 20 мин до 3 ч.
После завершения реакции целевое соединение изобретения может быть извлечено из реакционной смеси обычным методом. Например, по одной методике извлечения реакционная смесь освобождается от растворителя перегонкой или она выливается в воду и экстрагируется с помощью несмешивающегося с водой органического растворителя. Затем экстракт высушивают, а растворитель удаляют перегонкой. Если это необходимо, получающийся остаток может быть в дальнейшем очищен обычным методом, например путем перекристаллизации, или различными методами хроматографии, особенно колоночной хроматографией.
В соединениях формулы (I), где связь, представленная знаком α-β, является углерод-углеродной простой связью, соединение может образовывать оптически активные изомеры. В этом случае оптически активный изомер может быть получен путем использования оптически активного соединения формулы (IIa). Оптически активное соединение формулы (IIa) может быть получено из рацемата с помощью обычного метода, например обработкой рецемата соединений формулы (II) оптически активной карбоновой кислотой [например, (+)- или (-)-винной кислотой, (+)- или (-)-дибензоилвинной кислотой, (-)-яблочной кислотой или (-)-миндальной кислотой] или оптически активной сульфоновой кислотой (например, камфорсульфоновой кислотой), после чего следует перекристаллизация получающихся солей.
Если это необходимо, фармацевтически приемлемая соль соединения формулы (I) может быть получена обработкой свободного кислотного соединения подходящим основанием, например гидроокисью щелочного металла, такой как гидроокись натрия или гидроокись калия, или органическим амином, таким как триэтиламин или пиридин, в присутствии инертного растворителя, например диоксана, тетрагидрофурана или диэтилового эфира в подходящем растворителе, а затем отгонкой растворителя.
Исходные вещества формул (IIa) и (IVa), используемые в схемах реакций А и В, легко могут быть получены обычным методом, например, как показано в схемах реакций D, Е и F (см. фиг. 3-5).
В приведенных формулах R1 является таким, как определено выше, а R9представляет аралкильную группу, в которой алкильная часть имеет 1-4 атома углерода и является замещенной одной или более арильными группами, предпочтительно от 1 до 3 таких групп.
Арильные группы являются карбоциклическими арильными группами, имеющими 6-10, предпочтительно 6-10, наиболее предпочтительно 6, атомов углерода замещенными или незамещенными.
Аралкильная группа, представленная R9, является предпочтительно замещенной или незамещенной бензильной, дифенилметильной (бензгидрильной), трифенилметильной (тритильной), фенетильной, 1-фенилэтильной, 2-фенилпропильной или 3-фенилпропильной группой, более предпочтительно бензольной, метоксибензильной, дифенилметильной или трифенилме- тильной группой.
Схема реакции D.
В стадии D1 схемы реакции D аралкилтиофенол формулы (VIII) может быть получен обработкой 4-меркаптофенола, который имеет формулу (VII), гидридом щелочного металла, например гидридом натрия, в инертном растворителе, например амиде, таком как диметилформамид, или простом эфире, таком как тетрагидрофуран, а затем проводят реакцию получающейся соли щелочного металла с соединением формулы
R9-X', (XIX) где R9 определено выше; X' атом галогена. Реакция предпочтительно протекает при температуре от 0оС до комнатной температуры и требует периода времени от 30 мин до 5 ч.
В стадии D2 схемы реакции D соединение формулы (IX) может быть получено реакцией соединения формулы (VIII) с соединением формулы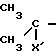 C ≡ CH (XX) где X' определено выше, в инертном растворителе, например кетоне, таком как ацетон или метилэтилкетон, спирте, таком как метанол или этанол, или смеси любых двух или более этих растворителей, и в присутствии основания, например карбоната щелочного металла, такого как карбонат натрия или карбонат калия. Реакция предпочтительно протекает при 50-100оС и обычно требует периода времени от 10 до 60 ч. Если это необходимо, соединения йода, например йодистый калий, йодистый натрий или т.п. могут присутствовать в реакционной смеси.
C ≡ CH (XX) где X' определено выше, в инертном растворителе, например кетоне, таком как ацетон или метилэтилкетон, спирте, таком как метанол или этанол, или смеси любых двух или более этих растворителей, и в присутствии основания, например карбоната щелочного металла, такого как карбонат натрия или карбонат калия. Реакция предпочтительно протекает при 50-100оС и обычно требует периода времени от 10 до 60 ч. Если это необходимо, соединения йода, например йодистый калий, йодистый натрий или т.п. могут присутствовать в реакционной смеси.
В стадии D3 схемы реакции D пирановое соединение формулы (X) может быть получено обработкой соединения формулы (IX) при 100-200оС в течение 1-5 ч в инертном растворителе, например ароматическом углеводороде, таком как толуол, хлорбензол, дихлорбензол или ксилол.
В стадии D4 схемы реакции D меркаптосоединение формулы (XI) может быть получено реакцией соединения формулы (X) с солью ртути, например трихлорацетатом ртути или ацетатом ртути, в инертном растворителе, например водной органической карбоновой кислоте, такой как водная уксусная кислота. Реакция предпочтительно протекает при 0-50оС и обычно требует периода времени от 30 мин до 2 ч. Получающаяся соль ртути подвергается затем реакции с сероводородом или с предшест- венником сероводорода, например сернистый натрий/соляная кислота. Реакция предпочтительно протекает при 0-50оС в течение от 10 мин до 1 ч. В первой из этих стадий, если это необходимо, в реакционной смеси могут также присутствовать катионные стабилизаторы, такие как анизол.
В стадии D5 схемы реакции D соединение формулы (XII) может быть получено реакцией соединения формулы (XI) с соединением формулы
R1-X', (XXI) где R1 и X' определены выше, в инертном растворителе, например амиде, таком как диметилформамид, или простом эфире, таком как тетрагидрофуран, в присутствии основания, например гидрида щелочного металла, такого как гидрид натрия, алксида щелочного металла, такого как третбутоксид калия, или органического амина, такого как триэтиламин. Реакция предпочтительно протекает при 0-50оС в течение 2-10 ч. С другой стороны, реакция с соединением формулы (XXI), в которой R1 трифторметильная группа, может быть достигнута облучением ультрафиолетовым излучением, например ультрафиолетовым излучением, генерируемым ртутной лампой, в присутствии органического амина, такого как аммиак или триэтиламин, и в инертном растворителе, например жидком аммиаке или амиде, таком как диметилформамид, при относительно низкой температуре, предпочтительно при температуре от -78оС до комнатной температуры, и предпочтительно в течение 1-10 ч.
В стадии D6 схемы реакции D соединение формулы (IVa), которое может быть одним из целевых исходных веществ для использования при получении соединений настоящего изобретения, может быть получено реакцией соединения формулы (XII) с окислителем, например перекись, такая как 3-хлорнадбензойная кислота, надуксусная кислота или перекись водорода, в инертном растворителе, например галоидированном углеводороде, таком как хлористый метилен или дихлорэтан. Реакция предпочтительно протекает при 0-50оС в течение от 30 мин до 3 ч. В этом реакции целевое соединение формулы (IVa) предпочтительно получается, используя примерно 3 моль или более окислителя на моль соединения формулы (XII).
В стадии D7 схемы реакции D соединение формулы (IIa) может быть получено реакцией соединений формулы (IVa) c водным аммиаком или с этанольным раствором, содержащим аммиак, предпочтительно при температуре от 0 до 50оС и предпочтительно в течение периода времени от 10 ч до 5 дн.
Схема реакции E.
Схема реакции Е показывает альтернативную методику получения соединения формулы (XII), получаемого в стадии D5 схемы реакции D.
В стадии Е1 схема реакции Е тиофенол формулы (VII) подвергается реакции с соединением формулы (XXI) по методу, описанному в стадии D5 схемы реакции D, чтобы получить соединение формулы (XIII).
В стадии Е2 схемы реакции Е соединение формулы (XIV) может быть получено реакцией соединения формулы (XIII) с соединением формулы (XX) по методу, описанному в стадии D2 схемы реакции D.
В стадии Е3 схемы реакции Е соединение формулы (XII) может быть получено реакцией соединения формулы (XIV) по методу, описанному в стадии D3 схемы реакции D.
Схема реакции F.
Схема реакции F показывает альтернативную методику получения соединения формулы (IVa), которое является одним из исходных веществ, которые могут быть использованы в способе настоящего изобретения, чтобы получить предлагаемые соединения.
В стадии F1 схемы реакции F соединение формулы (XIV) может быть получено реакцией соединения формулы (XV) с соединением формулы
R1SO2-X' (XXII) где R1 и X' определены выше, в инертном растворителе, например галоидированном углеводороде, таком как хлористый метилен, или нитрованном углеводороде, таком как нитрометан или нитробензол, и в присутствии кислоты Льюиса, например окись алюминия или хлорное железо (3). Реакция предпочтительно протекает при 0-50оС в течение от 30 мин до 24 ч.
В стадии F2 схемы реакции F соединение формулы (XVII) может быть получено реакцией соединения формулы (XVI) с восстановителем, например борогидридом натрия или литийалюминийгидридом, в инертном растворителе, например метаноле, водном этаноле, диэтиловом эфире или тетрагидрофуране. Реакция предпочтительно протекает при 0-40оС в течение 0,5-3 ч.
В стадии F3 схемы реакции F соединение формулы (XVIII) может быть получено реакцией соединения формулы (XVII) с дегидратирующим реагентом, например пиридин/хлорокись фосфора, пиридин/хло- ристый тионил или паратолуолсульфоновая кислота, предпочтительно в инертном растворителе, например пиридине или бензоле. Реакция предпочтительно протекает при температуре от -10 до 150оС в течение 0,5-3 ч.
В стадии F4 схемы реакции F соединение формулы (IVa) может быть получено реакцией соединения формулы (XVIII) по методу, описанному в стадии D6 схемы реакции D, за которым следует реакция окисления.
После завершения любой из этих реакций целевые соединения, получаемые по реакции, могут быть извлечены из реакционной смеси обычным методом. Например, по одной подходящей последовательности извлечения растворитель отгоняется из реакционной смеси или реакционная смесь выливается в воду, экстрагируется с помощью несмешивающегося с водой органического растворителя и высушивается, после чего растворитель отгоняется. Получающийся остаток, если необходимо, может быть дополнительно очищен обычным способом, например перекристаллизацией, или различными методами хроматографии, особенно колоночной хроматографией.
Соединения настоящего изобретения имеют превосходную сосудорасширяющую активность, как это показано в следующем примере испытания. Таким образом, соединения изобретения, являются весьма полезными для профилактики и лечения целого ряда расстройств, для которых такая активность обычно показывается, включая следующие заболевания и расстройства:
гипертензия, застойная сердечная недостаточность, стенокардия (грудная жаба);
обратимая обструкция дыхательных путей, астма;
пептическая язва;
локальная алопеция (облысение);
инконтиненция (недержание),
в частности для лечения сердечно-сосудистых заболеваний.
П р и м е р испытания. Сосудорасширяющая активность.
Крыса была умерщвлена кровопусканием. Грудная аорта была немедленно вырезана. После этого удаляют соединительную ткань и жировую ткань и получают препарат спиральных узких полосок, в котором гладкие кровеносные сосуды вырезают, чтобы образовать спиральные узкие полоски. Препарат суспендируют в трубку Магнуса и, после того как препарат становится стабильным, добавляют фенилэфрин (1 х 10-6 М), чтобы вызвать сокращение кровеносных сосудов. Когда препарат достигнет стабильного состояния, образец, содержащий испытуемое соединение, применяют совокупно, чтобы наблюдать ответную релаксационную (расслабляющую) реакцию. Когда ответная реакция достигнет максимума, добавляют 10-4 М папаверина и получающуюся индуцированную ответную реакцию принимают за 100% Эксперимент проводят, используя каждое испытуемое соединение при нескольких различных дозах. Величины релаксации, достигаемые при каждой из доз (моль/л), измеряют и концентрацию испытуемого соединения, показывающую 30%-ную релаксацию (IC30 моль/л) вычисляют.
В каждом испытании в качестве контрольных соединений используют Кромакалим и 3,3-дигидро-2,2-диметил-6-трифтор- метилсульфонил-4- (2-оксо-1-пирролидинид)-2Н-1-бензопиран-3-ол. Относительная активность представлена в табл. 3, где сое- динения изобретения идентифицируются по номерам примеров, которые демонстрируются ниже.
Соединения настоящего изобретения проявляют сильнодействующий гипотензивный эффект. Они также показывают превосходную продолжительность срока действия при гипотензивном испытании путем перорального ввода (приема) ин виво спонтанно (самопроизвольно) гипертензивным крысам.
Соединения настоящего изобретения могут быть использованы для профилактики и лечения сердечно-сосудистых и других нарушений, включая гипертензию. С этой целью они могут быть использованы, если это требуется, в смеси с другими действующими соединениями и/или с обычными носителями, разбавителями, адъювантами (вспомогательными средствами при приготовлении лекарственной формы) и/или наполнителями, чтобы образовать фармацевтический препарат. Кроме того, они могут быть назначены для приема без добавок. Форма фармацевтического препарата зависит от пути приема (ввода), например, для перорального приема соединения могут быть составлены в форме порошков, гранул, сиропов, таблеток или капсул, для парентерального введения они могут быть включены в состав в виде инъекций или ингаляций. Хотя дозирование можно варьировать в зависимости от симптомов пациента, природы и остроты заболевания или нарушения, а также пути и способа введения, в случае перорального приема соединения обычно могут быть назначены в суточной дозе 0,1-500 мг, в частности 0,2-100 мг, а в случае внутривенного введения в суточной дозе 0,02-100 мг, в частности 0,1-30 мг. Соединения могут быть назначены в однократной дозе или в небольших дозах, например дважды в день.
Получение соединений настоящего изобретения иллюстрируется следующими примерами.
П р и м е р 1. Транс-3,4-дигидро-4-(1,2-дигидро-2-оксо-1-пиридил)-2,2диметил- 6-трифторметилсульфонил-2Н-1-бензопиран-3-ол (соединение 1-24).
К раствору 1 г 3,4-эпокси-3,4-дигидро-2,2-диметил-6-трифторметилсульфонил- 2Н-1-бензопирана (полученного так, как описано в примере получения 8) в 1,2 мл безводного тетрагидрофурана добавляют 1,63 г 2-триметилсилилоксипиридина, раствор (2,54 г) фтористого тетрабутиламмония в безводном тетрагидрофуране добавляют к получающейся смеси с помощью шприца, охлаждая при этом льдом и в атмосфере азота. Реакционную смесь перемешивают при комнатной температуре в течение 72 ч, после чего ее выливают в воду и экстрагируют этилацетатом. Затем экстракт промывают водой, 5%-ным водным раствором соляной кислоты, водным раствором гидрокарбоната натрия и насыщенным водным раствором хлористого натрия, после чего его высушивают над безводным сульфатом магния. Далее растворитель удаляют перегонкой при пониженном давлении. Получающийся остаток очищают с помощью колоночной хроматографии через силикагель, используя 2: 1 по объему смесь этилацетата и циклогексана в качестве элюента, чтобы получить 503 мг указанного соединения в виде примерно 1:1 смеси вращательных изомеров, плавящийся при 219-220оС.
Спектр ядерного магнитного резонанса (гексадейтерированный диметилсульфоксид) δ м.д.
1,24 (3/2Н, синглет);
1,32 (3/2Н, синглет);
1,47 (3/2H, синглет);
1,52 (3/2Н, синглет);
4,02-4,13 (1/2Н, мультиплет);
4,38-4,47 (1/2Н, мультиплет);
5,07 (1/2H, дублет, I=10 Гц);
5,95 (1Н, дублет, I=6 Гц);
6,55 /1/2H, дублет, I=10 Гц);
6,1-6,4 /2H, мультиплет);
7,0-7,9 (5Н, мультиплет).
Инфракрасный спектр поглощения (KBr) νмакс˙ см-1:
3270, 1663, 1580.
П р и м е р 2. Транс-3,4-дигидро-4-(1,2-дигидро-2-оксо-1-пиридил) -2,2-диметил-6-трифторметилсульфонил-2Н-1-бензопиран-3-ол (соединение 1-24).
К суспензии 48 мг гидрида натрия (в виде 55 мас. дисперсии в нефти) в безводном диметилсульфоксиде добавляют 105 мг 2-гидроксипиридина; получившуюся смесь перемешивают при комнатной температуре в течение 15 мин. К концу этого времени к смеси добавляют в атмосфере азота и при комнатной температуре 309 мг 3,4-эпокси-3,4-дигидро-2,2-диметил-6-трифторметил-сульфонил- 2Н-1-бензопирана (полученного так, как описано в примере получения 8). Смесь перемешивают затем при комнатной температуре в течение 24 ч, после чего ее выливают в воду и экстрагируют этилацетатом. Экстракт промывают водой и насыщенным водным раствором хлористого натрия, затем его высушивают над безводным сульфатом натрия. После этого растворителя удаляют перегонкой при пониженном давлении и получающийся остаток очищают с помощью колоночной хроматографии через силикагель, используя 3:1 по объему смесь этилацетата и циклогексана в качестве элюента, чтобы получить 95 мг указанного соединения.
Спектры ядерного магнитного резонанса и инфракрасного поглощения полученного таким образом соединения идентичны примеру 1.
П р и м е р 3. 2,2-Диметил-4-(1-оксоизоиндолин-2-ил)-6- трифторметилсульфонил-2Н-1-бензопиран (соединение 2-1).
F3C·SO2 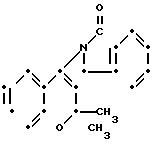
К раствору 200 г транс-3,4-дигидро-2,2-диметил-4-(1-оксоизоиндолин-2-ил)-6- трифторметилсульфонил-2Н-1-бензопиран-3- ола (полученного так, как описано в примере получения 13) в 6 мл диоксана добавляют 200 мг содового талька (каталог Мерка N 1567). Получившуюся смесь перемешивают в течение 30 мин, нагревая при этом ее на масляной бане, поддерживаемой при 140оС. Реакционную смесь охлаждают, а затем освобождают от нерастворимых веществ путем фильтрации. Далее растворитель удаляют перегонкой при пониженном давлении. Остаток растворяют в 20 мл хлористого метилена, а получающийся раствор промывают водой и насыщенным водным раствором хлористого натрия. Раствор высушивают над безводным сульфатом магния, а затем при пониженном давлении перегонкой удаляют растворитель. Маслянистый остаток, полученный таким образом, перекристаллизовывают из этилацетата, чтобы получить 81 мг указанного соединения, плавящегося при 110-115оС.
Спектр ядерного магнитного резонанса (CDCl3) δ м.д.
1,61 (6Н: синглет);
4,65 (2Н, синглет);
5,89 (1Н, синглет);
7,07 (1Н, дублет, I=8 Гц);
7,5-7,7 (4Н, мультиплет);
7,82 (1Н, дублет, дублетов, I=2 и 8 Гц);
7,94 (1Н, дублет, I=8 Гц).
Инфракрасный спектр поглощения (KBr) νмакс см-1:
1690.
Масс-спектр (м/е): 432 (M+).
П р и м е р 4. Транс-6-гептафторпропилсульфонил-3,4-лигидро-2,2-диметил-4-(1 -оксоизоиндолин-2-ил)-2Н-1-бензопиран-3-ол (соединение 1-3).
F7C3·SO2 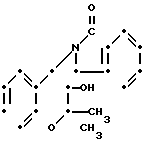
Раствор 0,142 г хлористого цинка и 0,065 г цианоборогидрида натрия в 3 мл метаноле добавляют к раствору 0,40 транс-4-амино-6-гептафторпропилсульфонил-3,4- дигидро-2,2-диметил- 2Н-1-бензопиран-3-ола (полученного так, как описано в примере получения 4) и 0,156 г метилового эфира 2-формилбензойной кислоты в 6 мл метанола. Получившуюся смесь перемешивают при комнатной температуре в течение 1 ч, а затем при 50оС в течение 24 ч. К концу этого времени смесь охлаждают с помощью льда, а затем смешивают с насыщенным водным раствором гидрокарбоната натрия. Затем удаляют перегонкой метанол при пониженном давлении, а получающийся остаток разбавляют водой и экстрагируют хлористым метиленом. Экстракт промывают водой и насыщенным водным раствором хлористого натрия, высушивают над безводным сульфатом магния, затем растворитель удаляют перегонкой при пониженном давлении. Получающийся остаток перекристаллизовывают из этилацетата, чтобы получить 0,356 г указанного соединения, плавящегося при 241-249оС. Спектр ЯМР (гексадейтерированный диметилсульфоксид) δ м.д.
1,32 (3Н, синглет);
1,54 (3Н, синглет);
3,9-4,2 (2Н, мультиплет);
4,59 (1Н, дублет, I=17 Гц);
5,32 (1н, уширенный синглет);
5,98 (1Н, дублет, I=6 Гц);
7,24 (1Н, дублет, I=9 Гц);
7,37 (1Н, уширенный синглет);
7,5-7,7 (3Н, мультиплет);
7,80 (1Н, дублет, I=7 Гц);
7,89 (1Н, дублет, дублетов, I=2 и 9 Гц).
ИК-спектр поглощения (KBr) νмакс см-1: 3478, 1667.
Масс-спектр (м/е): 541 (М+).
П р и м е р 5. 6-Гептафторпропилсульфонил-2,2-диметил-4-(1-оксоизоиндолин-2-ил) -2Н-1-бензопиран (соединение 2-3).
F7C3·SO2 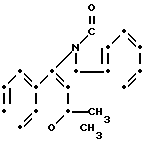
Следуя примеру 3, но используя 100 мг транс-6-гептафторпропилсульфонил-3,4-дигидро-2,2-диметил-4-(1 -оксоизоиндолин-2-ил)-2Н-1-бензопиран-3-ола (полученного так, как описано в примере получения 4) и 100 мг содового талька, получают 30 мг указанного соединения, плавящегося при 164-166оС. Спектр ЯМР (CDCl3) δ м.д.
1,61 (6Н, синглет);
4,64 (2Н, синглет);
5,89 (1Н, синглет);
7,07 (1Н, дублет, I=9 Гц);
7,5-7,7 (4Н, мультиплет);
7,82 (1Н, дублет дублетов, I=2 и 9 Гц);
7,95 (1Н, дублет, I=7 Гц).
ИК-спектр поглощения (KBr) νмакс см-1: 1686.
Масс-спектр (м/е): 523 (М+).
П р и м е р 6. Транс-4-(1,2-дигидро-2-ок-со-1-пиридил)-2,2-диметил-6- трифторметилсульфонил-2Н-1-бензопиран (соединение 2-11).
F3C·SO2 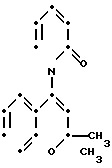
Следуя примеру 3, но используя 406 мг транс-3,4-дигидро-4-(1,2-дигидро-2-оксо-1-пиридил)-2,2-диметил -6-трифторметилсульфонил-2Н-1-бензопиран-3-ола (полученного так, как описано в примере 1) и 406 мг содового талька (каталог Мерка N 1657), получают 124 мг указанного соединения, плавящегося при 214-215оС.
Спектр ЯМР (CDCl3) δ м.д.
1,60 (3Н, синглет);
1,67 (3Н, синглет);
5,86 (1Н, синглет);
6,28 (1Н, дублет, триплетов, I=1 и 7 Гц);
6,66 (1Н, дублет, I=9 Гц);
7,06 (1Н, дублет, I=9 Гц);
7,15 (1Н, дублет дублетов, I=2 и 7 Гц);
7,27 (1Н, дублет, I=2 Гц);
7,46 (1Н, мультиплет);
7,82 (1Н, дублет дублетов, I= 2 и 9 Гц).
Масс-спектр (м/е): 385 (М+).
П р и м е р 7. Транс-3,4-дигидро-2,2-диметил-4-(1-оксоизоиндолин-2-ил)- 6-трифторметилсульфонил-2Н-1-бензопиран-3-ил нитрат (соединение 1-5).
F3C·SO2 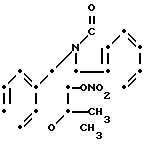
К раствору 300 мг транс-3,4-дигидро-2,2-диметил-4-(1-оксоизоиндолин-2-ил)-6- трифторметилсульфонил-2Н-1-бензопиран- 3-ола (полученного так, как описано в примере получения 13) в 5 мл хлористого метилена добавляют 116 мг тетрафторбората нитрония и получившуюся смесь перемешивают при комнатной температуре в течение 1,5 ч. К концу этого времени реакционную смесь разбавляют 20 мл воды и экстрагируют с 15 мл хлористого метилена. Экстракт промывают водой и насыщенным водным раствором хлористого натрия, после чего его высушивают над безводным сульфатом магния. Затем растворитель удаляют перегонкой при пониженном давлении, а получающийся остаток очищают с помощью колоночной хроматографии через силикагель, используя 1:4 по объему смесь этилацетата и циклогексана в качестве элюента, чтобы получить 88 мг указанного соединения в виде кристаллов, плавящихся при 189,5-190оС.
Спектр-ЯМР (CDCl3) δ м.д.
1,48 (3Н, синглет);
1,61 (3Н, синглет);
4,06 (1Н, дублет, I=17 Гц);
4,33 (1Н, дублет, I=17 Гц);
5,54 (1Н, дублет, I=10 Гц);
5,92 (1Н, дублет, I=10 Гц);
7,17 (1Н, дублет, I=9 Гц);
7,4-7,6 (4Н, мультиплет);
7,8-8,0 (2Н, мультиплет).
Масс-спектр (м/е): 440 (М+ NO2), 423 (M+ HNO3).
П р и м е р 8. Транс-3,4-дигидро-4-(1,2-дигидро-3-метил-2-оксо-1-пиридил)- 2,2-ди- метил-6-трифторметилсульфонил-2Н-1-бензопиран-3-ол (соединение 1-41).
F3C·SO2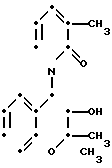
К раствору 200 мг 3,4-эпокси-3,4-дигидро-2,2-диметил-6- трифторметилсульфонил-2Н-1-бензопирана (полученного так, как описано в примере получения 8) в 0,2 мл безводного тетрагидрофурана добавляют 441 мг 3-метил-2-триметилсилилоксипиридина. К получающейся смеси добавляют затем раствор 509 мг фтористого третрабутиламмония в безводном тетрагидрофуране, охлаждая при этом льдом и в атмосфере азота. Затем реакционную смесь перемешивают при комнатной температуре в течение 5 дн, после чего ее обрабатывают таким же путем, как и в примере 1. Далее продукт реакции очищают с помощью колоночной хроматографии через силикагель, используя 1:2 по объему смесь этилацетата и циклогексана в качестве элюента, чтобы получить 135 мг указанного соединения, плавящегося при 215-216оС. Спектр ЯМР (CDCl3) δ м.д.
1,41 (3Н, синглет);
1,58 (3Н, синглет);
1,0-2,2 (1Н, уширенный синглет);
2,24 (3Н, синглет);
3,88 (1Н, дублет, I=10 Гц);
6,19 (1Н, триплет, I=7 Гц);
6,48 (1Н, дублет, I=10 Гц);
6,71 (1Н, дублет, I=7 Гц);
7,14 (1Н, дублет, I=9 Гц);
7,28 (1Н, дублет, I=7 Гц);
7,46 (1Н, уширенный синглет);
7,87 (1Н, дублет дублетов, I=2 и 9 Гц).
ИК-спектр поглощения (KBr) νмакс см-1:
3324, 1652, 1590, 1361.
Масс-спектр (м/е): 418 (М++1).
П р и м е р 9. Транс-3,4-дигидро-4-(1,2-дигидро-3-метокси-2-оксо-1-пиридил)- 2,2-диметил-6-трифторметилсульфонил-2Н-1-бензопиран-3-ол (соединение 1-42).
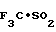
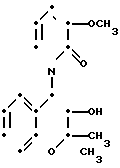
К суспензии 38 мг гидрида натрия (в виде 55%-ной дисперсии в нефти) в 3 мл диметилсульфоксида добавляют 98 мг 3-метокси-2(1Н)-пиридона. Получившуюся смесь перемешивают при комнатной температуре в течение 30 мин. После этого добавляют раствор 200 мг 3,4-эпокси-3,4-дигидро-2,2-диметил-6- трифторметилсульфонил-2Н-1-бензопирана (полученного так, как описано в примере получения 8) в 1,5 мл диметилсульфоксина при той же температуре и в атмосфере азота. Реакционную смесь перемешивают при комнатной температуре в течение 4 дн, после чего ее обрабатывают таким же образом, как описано в примере 2. Затем продукт реакции очищают с помощью колоночной хроматографии через силикагель, используя 1:2 по объему смесь этилацетата и циклогексана в качестве элюента, чтобы получить 16 мг указанного соединения, плавящегося при 223-225оС.
ЯМР-спектр (CDCl3) δ м.д.
1,42 (3Н, синглет);
1,58 (3Н, синглет);
3,75 (1Н, дублет, I=5 Гц);
3,88 (1Н, дублет дублетов, I=5 и 10 Гц);
3,90 (3Н, синглет);
6,20 (1Н, триплет, I=7 Гц);
6,43 (1Н, дублет дублетов, I=1,5 и 7 Гц);
6,50 (1Н, дублет, I=10 Гц);
6,69 (1Н, дублет дублетов, I=1,5 и 7 Гц);
7,13 (1Н, дублет, I=9 Гц);
7,45 (1Н, уширенный синглет);
7,86 (1Н, дублет дублетов, I=2 и 9 Гц).
ИК-спектр поглощения (KBr), νмакс см-1:
3312, 1655, 1603, 1371.
П р и м е р 10. Транс-3,4-дигидро-2,2-диметил-4-(6-нитро-1-оксоизоиндолин-2-ил) -6-трифторметилсульфонил-2Н-1-бензопи-ран-3-ол (соединение 1-15).
Раствор 0,164 г хлористого цинка и 0,075 г цианоборогидрида натрия в 3,3 мл пропанола добавляют к раствору 0,326 г транс-4-амино-3,4-дигидро-2,2-диметил-6-трифторметилсульфонил- 2Н-1-бензопиран-3-ола, полученного из его хлоргидрата так, как описано в примере получения 9, и 0,224 г метилового эфира 2-формил-5-нитробензойной кислоты в 3,3 мл пропанола. Получившуюся смесь нагревают при кипячении с обратным стеканием флегмы в масляной бане, поддерживаемой при 140оС в течение 1,5 ч. После этого реакционную смесь разбавляют со 150 мл этилацетата, получающийся раствор промывают 1 н. водным раствором соляной кислоты, водой, насыщенным раствором гидрокарбоната натрия и насыщенным водным раствором хлористого натрия. Затем смесь высушивают над безводным сульфатом натрия, после чего растворитель удаляют перегонкой при пониженном давлении. Получившийся остаток перекристаллизовывают из смеси этилацетата и гексана, чтобы получить 0,424 г указанного соединения, плавящегося при 264-266оС. ЯМР-спектр (гексадейтерированный диметилсульфоксид), δ м.д.
1,34 (3Н, синглет);
1,54 (3Н, синглет);
3,86-4,24 (1Н, мультиплет);
4,26 (1Н, дублет, I=20 Гц);
4,84 (1Н, дублет, I=20 Гц);
5,38 (1Н, дублет, I=10 Гц);
5,86-6,16 (1Н, мультиплет);
7,14 (1Н, дублет, I=10 Гц);
7,46-7,60 (2Н, мультиплет);
7,80-8,14 (2Н, мультиплет);
8,44-8,74 (2Н, мультиплет).
ИК-спектр поглощения (KBr), νмакс см-1:
3300, 1680.
Масс-спектр (м/е): 487 (М++1).
Элементарный анализ. C20H17F3N2O7S:
Вычислено, С 49,38, Н 3,52, N 5,76, F 11,72, S 6,59.
Найдено, C 49,78, H 3,84, N 5,38, F 11,69, S 6,74.
П р и м е р 11. Транс-3,4-дигидро-2,2-диметил-4-(7-нитро-1-оксоизоиндолин-2ил) -6-трифторметилсульфонил-2Н-1-бензопиран-3-ол (соединение 1-14).
Следуя примеру 10, но используя 0,326 г транс-4-амино-3,4-дигидро-2,2-диметил-6-трифторметилсульфонил- 2Н-1-бензопиран-3-ола, полученного из его хлоргидрата по примеру получения 9, 0,244 г метилового эфира 2-формил-6-нитробензойной кислоты, 0,164 г хлористого цинка и 0,075 г цианоборогидрида натрия, получают 0,391 г указанного соединения, плавящегося при 283-284оС.
ЯМР-спектр (гексадейтерированный диметилсульфоксид), δ м.д.
1,34 (3Н, синглет);
1,54 (3Н, синглет);
3,46 (1Н, уширенный синглет);
4,02 (1Н, дублет, I=10 Гц);
4,22 (1Н, дублет, I=18 Гц);
4,76 (1Н, дублет, I=18 Гц);
5,34 (1Н, дублет, I=10 Гц);
7,29 (1Н, дублет, I=8 Гц);
7,46-7,62 (1Н, мультиплет);
7,76-8,02 (4Н, мультиплет).
ИК-спектр поглощения (KBr), νмакс см-1:
3440, 1670.
Масс-спектр (м/е): 486 (М+).
Элементарный анализ. C20H17F3N2O7S:
Вычислено, C 49,39, H 3,52, N 5,76, F 11,72, S 6,59.
Найдено, C 49,43, H 3,50, N 5,58, F 11,59, S 6,58.
П р и м е р 12. Транс-3,4-дигидро-2,2-диметил-4-(4-нитро-1-оксоизоиндолин-2-ил)-6- трифторметилсульфонил-2Н-1-бензопиран-3-ол (соединение 1-13).
К раствору 0,367 г этилового эфира 2-бромометил-3-нитробензойной кислоты в 7,4 мл диметилформамида добавляют 0,362 г транс-4-амино-3,4-дигидро-2,2-диметил-6-трифторметилсульфонил-2Н- 1-бензопиран-3-ола, полученного из его хлоргидрата по примеру получения 9, и 0,30 мл триэтиламина. Получившуюся смесь перемешивают при 100оС в течение 1 ч в атмосфере азота. Затем реакционную смесь перемешивают при кипячении с обратным стеканием флегмы растворителя (при 152оС) в течение дополнительно 1 ч, после чего смесь выливают в разбавленную водную соляную кислоту и экстрагируют этилацетатом. Затем экстракт промывают водой, насыщенным водным раствором гидрокарбоната натрия и насыщенным водным раствором хлористого натрия, после чего его высушивают над безводным сульфатом натрия. Далее растворитель удаляют перегонкой при пониженном давлении и остаток очищают с помощью колоночной хроматографии через силикагель, используя 1:1:1 по объему смесь этилацетата, циклогексана и тетрагидрофурана в качестве элюента. Продукт реакции, полученный из элюента, перекристаллизовывают из смеси тетрагидрофурана и гексана, чтобы получить 0,295 г указанного соединения, плавящегося при 292-295оС.
ЯМР-спектр (гексадейтерированный диметилсульфоксид), δ м.д.
1,34 (3Н, синглет);
1,54 (3Н, синглет);
4,14 (1Н, дублет дублетов, I=6 и 10 Гц);
4,58 (1Н, дублет, I=18 Гц);
5,18 (1Н, дублет, I=18 Гц);
5,38 (1Н, дублет, I=10 Гц;
5,92 (1Н, дублет, I=6 Гц);
7,22 (1Н, дублет, I=8 Гц);
7,48-7,64 (1Н, мультиплет);
7,74-8,06 (2Н, мультиплет);
8,28 (1Н, дублет, I=8 Гц);
8,54 (1Н, дублет дублетов, I=2 и 8 Гц).
ИК-спектр поглощения (KBr), νмакс см-1:
3460, 1676.
Масс-спектр (м/е): 468 (М+-18).
Элементарный анализ. C20H17F3N2O7S:
Вычислено, C 49,39, H 3,52, N 5,76, F 11,72, S 6,59.
Найдено, C 49,49, H 3,69, N 5,72, F 11,69, S 6,58.
П р и м е р 13. Транс-4-(4-фтор-1-оксоизоиндолин-2-ил)-3,4-дигидро-2,2-диметил-6- трифторметансульфонил-2Н-1-бензопиран-3-ол (соединение 1-16).
Следуя примеру 12, но используя 0,272 г метилового эфира 2-бромометил-3-фторбензойной кислоты, 0,326 г транс-4-амино-3,4-дигидро-2,2-диметил-6-трифторметил-сульфонил-2Н-1- бензопиран-3-ола и 0,15 мл триэтиламина, получают после очистки с помощью колоночной хроматографии через силикагель, используя 1:1 по объему смесь циклогексана и этилацетата в качестве элюента, 0,294 г указанного соединения, плавящегося при 259-261оС после перекристал- лизации из смеси этилацетата и гексана.
ЯМР-спектр (гексадейтерированный диметилсульфоксид), δ м.д.
1,34 (3Н, синглет);
1,54 (3Н, синглет);
4,08 (1Н, дублет, I=10 Гц);
4,16 (1Н, дублет, I=18 Гц);
4,76 (1Н, дублет, I=18 Гц);
5,36 (1Н, дублет, I=10 Гц);
5,90 (1Н, уширенный синглет);
7,20 (1Н, дублет, I=8 Гц);
7,36-8,02 (5Н, мультиплет).
ИК-спектр поглощения (KBr), νмакс см-1:
3460, 1670.
Масс-спектр (м/е): 460 (М++1).
Элементарный анализ. C20H17F4NO5S:
Вычислено, C 52,29, H 3,73, N 3,05, F 16,54, S 6,98.
Найдено, C 52,29, H 3,88, N 3,05, F 16,81, S 7,30.
П р и м е р получения 1. 4-(Гептафторпропил)-тиофенол.
К суспензии 2,4 г гидрида натрия (в виде 55%-ной дисперсии в нефти) в 64 мл диметилформамида в атмосфере азота при охлаждении со льдом добавляют 3,16 г 4-гидрокситиофенола и получающуюся смесь перемешивают в течение 15 мин. Затем к смеси добавляют 3,6 мл гептафторпропил-1-йодистого и эту смесь перемешивают при комнатной температуре в течение 5 ч. К концу этого времени реакционную смесь выливают в разбавленную водную соляную кислоту и экстрагируют этилацетатом. Экстракт промывают водой, насыщенным водным раствором гидрокарбоната натрия и водным раствором хлористого натрия, после чего растворитель удаляют при пониженном давлении. Получающийся остаток очищает с помощью колоночной хроматографии через силикагель, используя 1:1 по объему смесь циклогексана и этилацетата в качестве элюента, чтобы получить 3,4 г указанного соединения.
ЯМР-спектр (CDCl3) δ м.д.
5,87 (1Н, уширенный синглет);
6,85 (2Н, дублет, I=9 Гц);
7,52 (2Н, дублет, I=9 Гц).
Масс-спектр (м/е): 294 (М+).
П р и м е р получения 2. 6-(Гептафторпропилтио)-2,2-диметил-2Н-1-бензопиран.
К 10,8 мл водного раствора, содержащего 0,72 г гидроокиси натрия, добавляют 11 мл диоксанового раствора из 3,4 г 4-(гептафторпропил)-тиофенола, полученного так, как описано в примере получения 1), 1,85 г 3-хлор-3-метил-1-бутина и 2,50 г 40 мас. водного раствора гидроокиси триметил- бензиламмония. Получившуюся смесь нагревают при кипячении с обратным стеканием флегмы в течение 15 ч. После этого реакционную смесь выливают в воду и экстрагируют этилацетатом. Экстракт промывают разбавленным водным раствором гидроокиси натрия, водой, разбавленной водной соляной кислотой, и насыщенным водным раствором хлористого натрия, после чего его высушивают над безводным сульфатом натрия. Затем растворитель удаляют перегонкой при пониженном давлении, чтобы получить 3,81 г маслянистого вещества в качестве остатка. Все это растворяют в 16 мл дихлорбензола и получающийся раствор нагревают при кипячении с обратным стеканием флегмы в течение 2 ч в атмосфере азота. Затем растворитель отгоняют из реакционной смеси перегонкой при пониженном давлении и получающийся остаток очищают с помощью колоночной хроматографии через силикагель, используя циклогексан в качестве элюента, чтобы получить 1,67 г указанного соединения.
ЯМР-спектр (CDCl3) δ м.д.
1,42 (6Н, синглет);
5,64 (1Н, дублет, I=10 Гц);
6,30 (1Н, дублет, I=10 Гц);
7,20-7,46 (2Н, мультиплет);
6,78 (1Н, дублет, I=9 Гц).
П р и м е р получения 3. 3,4-Эпокси-6-гептафторпропилсульфонил-3,4-дигидро-2, 2-диметил-2Н- 1-бензопиран.
К раствору 1,65 г 6-гептафторпропилтио-2,2-диметил-2Н-1-бензопирана, полученного так, как описано в примере получения 2, в 33 мл хлористого метилена добавляют 2,79 г 3-хлорнадбензойной кислоты (85%-ной чистоты). Получившуюся смесь перемешивают при комнатной температуре в течение 1,5 ч. После этого реакционную смесь разбавляют этилацетатом, а затем промывают 1 н. водным раствором гидроокиси натрия, водой и насыщенным водным раствором хлористого натрия. Рас- твор высушивают над безводным сульфатом натрия, а затем остаток получаемый перегонкой растворителя при пониженном давлении, очищают с помощью колоночной хроматографии через силикагель, используя 5:1 по объему смесь циклогексана и этилацетата в качестве элюента, чтобы получить 1,21 г указанного соединения.
ЯМР-спектр (CDCl3) δ м.д.
1,32 (3Н, синглет);
1,60 (3Н, синглет);
3,66 (1Н, дублет, I=4 Гц);
3,98 (1Н, дублет, I=4 Гц);
7,00 (1Н, дублет, I=8 Гц);
7,90 (1Н, дублет дублетов, I=8 и 2 Гц);
8,05 (1Н, дублет, I=2 Гц).
ИК-спектр поглощения (жидкая пленка) νмакс см-1:
1330, 1100.
Масс-спектр (м/е): 408 (М+).
П р и м е р получения 4. Транс-4-амино-6-гептафторпропилсульфонил-3,4-дигидро-2,2-диметил- 2Н-1-бензопиран-3-ол.
К раствору 1,086 г 3,4-эпокси-6-гептафторпропилсульфонил-3,4-дигидро-2,2-диметил-2Н-1- бензопирана, полученного так, как описано в примере получения 3, в 30 мл этанола добавляют 30 мл (28 об.) водного аммиака и получающуюся смесь оставляют стоять при комнатной температуре в течение 3 дн. К концу этого времени реакционную смесь выливают в воду и экстрагируют хлористым метиленом. Экстракт промывают водой и высушивают над безводным сульфатом натрия. Растворитель удаляют перегонкой при пониженном давлении, а затем получающийся остаток очищают с помощью колоночной хроматографии через силикагель, используя 1: 1 по объему смесь циклогексана и этилацетата в качестве элюента, Фракции, содержащие целевое соединение, перекристаллизовывают затем из смеси этилацетата и гексана, чтобы получить 0,796 г указанного соединения, плавящегося при 186-177оС.
ЯМР-спектр (CDCl3) δ м.д.
1,26 (3H, cинглет);
1,54 (3Н, синглет);
1,80-2,50 (3Н, мультиплет);
3,34 (1Н, дублет, I=10 Гц);
3,74 (1Н, дублет, I=10 Гц);
6,98 (1Н, дублет, I=8 Гц);
7,80 (1Н, дублет дублетов, I=8 и 2 Гц);
8,12 (1Н, дублет, I=2 Гц).
ИК-спектр поглощения (KBr) νмакс см-1: 3140.
Масс-спектр (м/е): 426 (M++1).
Элементарный анализ. C14H14F7NO4S:
Вычислено, C 39, 54, H 3,32, N 3,29, F 31,27, S 7,54.
Найдено, C 39,85, H 3,51, N 3,26, F 31,23, S 7,33.
П р и м е р получения 5. 6-(4-Метоксибензилтио)-2,2-диметил-2Н-1-бензопиран.
5 а) 4-(4-Метоксибензилтио)-фенол.
Раствор 50,47 г парагидрокситиофенола в 250 мл тетрагидрофурана добавляют к суспензии 38,4 г гидрида натрия (в виде 55%-ной дисперсии в нефти) в 380 мл диметилформамида, охлаждая при этом льдом и добавляя в атмосфере азота. Получившуюся смесь перемешивают в течение 0,5 ч. После этого раствор 54,2 мл хлористого параметоксибензила в 120 мл тетрагидрофурана добавляют по каплям к получающейся смеси, которую затем перемешивают в течение 1 ч. Затем реакционную смесь нейтрализуют добавлением уксусной кислоты, охлаждая при этом льдом, после чего ее выливают в воду. Далее смесь экстрагируют этилацетатом и экстракт промывают водой и насыщенным водным раствором хлористого натрия. Затем получающийся раствор высушивают, удаляют растворитель перегонкой при пониженном давлении, а остаток очищают с помощью колоночной хроматографии через силикагель, используя 1:3 по объему смесь циклогексана и тетрагидрофурана в качестве элюента. Элюированный продукт реакции перекристаллизовывают из смеси тетрагидрофурана и гексана, чтобы получить 88,99 г указанного соединения.
ИК-спектр поглощения (Нуйол, торговое название), νмакс см-1:
3380, 1450, 1250.
5 b) 6-(4-Метоксибезилтио)-2,2-диметил-2Н-1-бензопиран.
Раствор 73,84 г 3-хлор-3-метил-1-бутина в 225 мл метанола, 11,95 г йодистого калия и 99,50 г карбоната калия добавляют к всему количеству 4-(4-меотксибензилтио)-фенола, полученного так, как описано в стадии а), в 0,9 л метилэтилкетона. Полученную смесь нагревают при кипячении с обратным стеканием флегмы в течение 40 ч в атмосфере азота. К концу этого времени реакционную смесь экстрагируют этилацетатом и экстракт промывают водой и насыщенным водным раствором хлористого натрия. Затем его высушивают над безводным сульфатом натрия, после чего растворитель удаляют перегонкой при пониженном давлении. Остаток очищают с помощью колоночной хроматографии через силикагель, используя 5: 1 смесь по объему циклогексана и этилацетата в качестве элюента, чтобы получить 67,68 г простого эфира в виде масла. Раствор всего содержимого этого простого эфира в 340 мл ортодихлорбензола нагревают затем при кипячении с обратным стеканием флегмы в течение 2 ч в атмосфере азота. После этого реакционную смесь освобождают от растворителя перегонкой при пониженном давлении и остаток очищают с помощью колоночной хроматографии через силикагель, используя 20:1 по объему смесь циклогексана и этилацетата в качестве элюента. Элюированный продукт реакции перекристаллизовывают из гексана, чтобы получить 48,74 г указанного соединения, плавящегося при 58-59оС.
ЯМР-спектр (CDCl3) δ м.д.
1,38 (6Н, синглет);
3,72 (3Н, синглет);
3,90 (2Н, синглет);
5,56 (1Н, дублет, I=10 Гц);
6,22 (1Н, дублет, I=10 Гц);
6,58 -7,28 (7Н, мультиплет).
Масс-спектр (м/е): 312 (М+).
Элементарный анализ. C19H20O2S:
Вычислено, C 73,04; H 6,45, S 10,26.
Найдено, С 73,33, H 6,49, S 10,39.
П р и м е р получения 6. 6-Меркапто-2,2-диметил-2Н-1-бензопиран.
В 12 мл 80%-ной уксусной кислоты растворяют с нагреванием 0,313 г 6-(4-метоксибензилтио)-2,2-диметил-2Н-1-бензопирана и к получающемуся раствору добавляют 0,12 мл анизола и 0,512 г трифторацетата ртути. Полученную смесь перемешивают при 40оС в течение 1 ч, после чего реакционную смесь экстрагируют этилацетатом. Экстракт промывают водой, водным раствором гидрокарбоната натрия и насыщенным водным раствором хлористого натрия, после чего его высушивают над безводным сульфатом натрия. Затем удаляют растворитель перегонкой при пониженном давлении и остаток очищают с помощью колоночной хроматографии через силикагель, используя 5:1 по объему смесь циклогексана и этилацетата в качестве элюента. Элюи- рованный продукт реакции переосаждают из смеси гексана и этилацетета, чтобы получить 0,283 г ртутной соли указанного соединения в виде кристаллов.
ЯМР-спектр (CDCl3) δ м.д.
1,40 (6Н, синглет);
5,58 (1Н, дублет, I=10 Гц);
6,20 (1Н, дублет, I=10 Гц);
6,60 (1Н, дублет, I=8 Гц);
7,02-7,23 (2Н, мультиплет).
Масс-спектр (м/е): 584 (М++1).
Затем к раствору 2,33 г этой ртутной соли в смеси 46 мл тетрагидрофурана и 46 мл метанола добавляют по каплям 48 мл водного раствора (4,80 г) нонагидрата сернистого натрия. К полученной смеси при охлаждении льдом добавляют 4 мл концентрированной соляной кислоты. Затем смесь перемешивают при комнатной температуре в течение 0,5 ч, после чего ее освобождают от нерастворяющихся веществ фильтрацией с помощью активного древесного угля. Фильтрат экстрагируют этилацетатом. Экстракт промывают водой, насыщенным раствором гидрокарбоната натрия и насыщенным раствором хлористого натрия в воде, после чего его высушивают над безводным сульфатом натрия. Затем растворитель удаляют перегонкой при пониженном давлении, чтобы получить 1,47 г указанного соединения в виде масла.
ЯМР-спектр (CDCl3) δ м.д.
1,42 (6Н, синглет);
3,28 (1Н, синглет);
5,58 (1Н, дублет, I=10 Гц);
6,22 (1Н, дублет, I=10 Гц);
6,64 (1Н, дублет дублетов, I=8 и 2 Гц);
6,92-7,30 (2Н, мультиплет).
Масс-спектр (м/е): 192 (М+).
П р и м е р получения 7. 2,2-Диметил-6-трифторметилтио-2Н-1-бензопиран.
В 30 мл жидкого аммиака суспендируют 1,45 г 6-меркапто-2,2-диметил-2Н-1-бензопирана, полученного так, как описано в примере получения 6, и к суспензии при -60оС в атмосфере азота добавляют 8,23 г йодистого трифторметила. Полученную смесь облучают затем при -65оС в течение 2 ч, а затем при 25оС в течение 2 ч, используя ртутную лампу низкого давления. После этого реакционную смесь разбавляют этилацетатом и промывают разбавленной водой соляной кислотой, водой, водным раствором гидрокарбоната натрия и насыщенным водным раствором хлористого натрия. Органический слой высушивают затем над безводным сульфатом натрия и концентрируют перегонкой при пониженном давлении. Полученный остаток очищают с помощью колоночной хроматографии через силикагель, используя 20:2:1 по объему смесь циклогексана, хлороформа и этилацетата в качестве элюента, чтобы получить 1,17 г указанного соединения в виде масла.
ЯМР-спектр (CDCl3) δ м.д.
1,46 (6Н, синглет);
5,62 (1Н, дублет, I=10 Гц);
6,28 (1Н, дублет, I=10 Гц);
6,76 (1Н, дублет, I=8 Гц);
7,20-7,46 (2Н, мультиплет).
Масс-спектр (м/е): 260 (М+).
П р и м е р получения 8. 3,4-Эпокси-3,4-дигидро-2,2-диметил-6-трифторметилсуль- фонил-2Н-1 -бензопиран.
Следуя методике, подобной описанной в примере получения 3, но используя 1,15 г 2,2-диметил-6-трифторметилтио-2Н-1-бензопирана, полученного так, как описано в примере получения 7, и 4,59 г 3-хлорнадбензойной кислоты (70%-ной чистоты), получают 0,74 г указанного соединения в виде масла.
ЯМР-спектр (CDCl3) δ м.д.
1,32 (3Н, синглет);
1,60 (3Н, синглет);
3,56 (1Н, дублет, I=4 Гц);
4,00 (1Н, дублет, I=4 Гц);
7,02 (1Н, дублет, I=8 Гц);
7,92 (1Н, дублет дублетов, I=8 и 2 Гц);
8,06 (1Н, дублет, I=2 Гц).
Масс-спектр (м/е): 308 (М+).
П р и м е р получения 9. Транс-4-амино-3,4-дигидро-2,2-диметил-6-трифторметил-сульфонил-2Н- 1-бензопиран-3-ола хлоргидрат.
Следуя примеру получения 4, но используя 0,68 г 3,4-эпокси-3,4-дигидро-2,2-диметил-6-трифторметилсульфонил-2Н-1- бензопирана, полученного так, как описано в примере получения 8, и 28 об. водного раствора аммиака, получают 0,55 г указанного соединения в виде кристаллов, плавящегося при 256-259оС.
ЯМР-спектр (гексадейтерированный диметилсульфоксид) δ м.д.
1,22 (3Н, синглет);
1,50 (3Н, синглет);
3,78 (1Н, дублет, I=10 Гц); 4,16-5,26 (2Н, мультиплет);
7,20 (1Н, дублет, I=8 Гц);
7,98 (1Н, дублет дублетов, I=8 и 2 Гц);
8,62 (1Н, дублет, I=2 Гц);
9,04 (3Н, уширенный синглет).
ИК-спектр поглощения (KBr) νмакс см-1:
3330, 1360.
Масс-спектр (м/е): 326 (М++1).
Элементарный анализ.
C12H15ClF3 NO4S:
Вычислено, C 39,84, H 4,18, N 3,87, Cl 9,80, F 15,75, S 8,86.
Найдено, C 40,05, H 4,41, N 3,89, Cl 9,98, F 15,76, S 8,79.
П р и м е р получения 10. 2,2-Диметил-6-пентафторэтилтио-2Н-1- бензопиран.
Раствор 1,463 г 6-меркапто-2,2-диметил-2Н-1-бензопирана, полученного так, как описано в примере получения 6, в 15 мл диметилформамида добавляют по каплям, охлаждая при этом со льдом, к суспензии 0,366 г гидрида натрия (в виде 55%-ной дисперсии в нефти) в 15 мл диметилформамида в атмосфере азота. После перемешивания реакционной смеси в течение 15 мин добавляют 5,53 г йодистого перфторэтила. Далее реакционную смесь перемешивают, охлаждая ее со льдом в течение 1 ч, а затем при комнатной температуре в течение 2 ч. После этого реакционную смесь выливают в 150 мл разбавленной водной соляной кислоты и экстрагируют этилацетатом. Экстракт промывают водой, водным раствором гидрокарбоната натрия и насыщенным водным раствором хлористого натрия, после чего его высушивают над безводным сульфатом натрия. Растворитель удаляют перегонкой при пониженном давлении и полученный остаток очищают с помощью колоночной хроматографии через силикагель, используя 20:1 по объему смесь циклогексана и этилацетата в качестве элюента, чтобы получить 1,935 г указанного соединения в виде масла.
ЯМР-спектр (СDCl3) δ м.д.
1,40 (6Н, синглет);
5,64 (1Н, дублет, I=10 Гц);
6,30 (1Н, дублет, I=10 Гц);
6,78 (1Н, дублет, I=8 Гц);
7,24 (1Н, дублет, I=2 Гц);
7,38 (1Н, дублет дублетов, I=8 и 2 Гц).
Масс-спектр (м/е): 310 (M+).
П р и м е р получения 11. 3,4-Эпокси-3,4-дигидро-2,2-диметил-6-пентафторэтилсуль- фонил-2Н- 1-бензопиран.
Следуя методике, описанной в примере получения 3, но используя 1,87 г 2,2-диметил-6-пентафторэтилтио-2Н-1-бензопира-на, полученного так, как описано в примере получения 10 и 5,35 г 3-хлорнадбензойной кислоты, получают 1,70 г указанного соединения в виде масла.
ЯМР-спектр (CDCl3) δ м.д.
1,34 (3Н, синглет);
1,62 (3Н, синглет);
3,56 (1Н, сигнлет, I=4 Гц);
4,00 (1Н, дублет, I=4 Гц);
7,02 (1Н, дублет, I=8 Гц);
8,92 (1Н, дублет дублетов, I=8 и 2 Гц);
9,06 (1Н, дублет, I=2 Гц).
Масс-спектр (м/е): 358 (М+).
П р и м е р получения 12. Транс-4-амино-3,4-дигидро-2,2-диметил-6-пентафтор- этил-2Н-1- бензопиран-3-ола хлоргидрат.
Следуя методике, описанной в примере получения 4, но используя 0,247 г 3,4-эпокси-3,4-дигидро-2,2-диметил-6-пентафтор- этилсульфонил-2Н-1- бензопиран-3-ола, полученного так, как описано в примере получения 11 и водный раствор аммиака, получают 0,245 г указанного соединения в виде кристаллов.
Т.пл. 250-252оС.
ЯМР-спектр (гексадейтерированный диметилсульфоксид), δ м.д.
1,28 (3Н, синглет);
1,48 (3Н, синглет);
3,62-3,92 (1Н, мультиплет);
4,36 (1Н, дублет, I=10 Гц);
6,46-6,70 (1Н, мультиплет);
7,20 (1Н, дублет, I=8 Гц);
7,94 (1Н, дублет дублетов, I=8 и 2 Гц);
8,58 (1Н, дублет, I=2 Гц);
8,98 (3Н, уширенный синглет).
ИК-спектр поглощения (KBr) νмакс см-1:
3230, 3020, 2940, 1220, 1120.
Масс-спектр (м/е): 374 (М++1).
П р и м е р получения 13. Транс-3,4-дигидро-2,2-диметил-4-(1-оксоизоиндолин-2-ил)-6- трифторметилсульфонил-2Н-1-бензопиран-3-ол.
F3C·SO2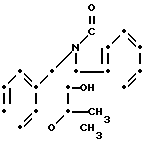
Раствор 0,18 г хлористого цинка и 0,083 г цианоборогидрида натрия в 3 мл метанола добавляют к раствору 0,40 транс-4-амино-3,4-дигидро-2,2-диметил-6-трифторметил-сульфонил-2Н- 1-бензопиран-3-ола, полученного из его хлоргидрата, как описано в примере получения 9, 0,20 г метилового эфира и 2-формилбензойной кислоты в 6 мл метанола. Полученную смесь перемешивают при комнатной температуре в течение 1 ч, а затем при 50оС в течение 24 ч. После этого смесь охлаждают со льдом, а затем смешивают с насыщенным водным раствором гидрокаpбоната натрия. Затем удаляют метанол перегонкой при пониженном давлении, получающийся остаток разбавляют водой и экстрагируют хлористым метиленом. Экстракт промывают водой и насыщенным водным раствором хлористого натрия, высушивают над безводным сульфатом магния и растворитель удаляют перегонкой при пониженном давлении. Получающийся остаток перекристаллизовывают из этилацетата, чтобы получить 0,34 г указанного соединения, плавящегося при 279-281оС.
ЯМР-спектр (гексадейтерированный диметилсульфоксид) δ м.д.
1,35 (3Н, синглет);
1,56 (3Н, синглет);
3,98 (1Н, дублет дублетов, I=6 и 11 гц);
4,08 (1Н, дублет, I=17 Гц);
4,60 (1Н, дублет, I=17 Гц);
5,43 (1Н, дублет, I=11 Гц);
5,89 (1Н, дублет, I=6 Гц);
7,17 (1Н, дублет, I=9 Гц);
7,4-8,1 (6Н, мультиплет).
ИК-спектр поглощения (KBr) νмакс см-1:
3473, 1671.
Масс-спектр (м/е): 4411 (М+).
Элементарный анализ. C20H18F3NO5S:
Вычислено, C 54,42, H 4,11, N 3,17.
Найдено, C 54,50, H 4,31, N 2,88.
| название | год | авторы | номер документа |
|---|---|---|---|
| ФУНГИЦИДНЫЕ ПРОИЗВОДНЫЕ ОКСЕТАНА И ИХ СОЛИ | 1992 |
|
RU2044736C1 |
| КРЕМНИЙСОДЕРЖАЩИЕ СОЕДИНЕНИЯ ИЛИ ИХ СОЛИ, ОБЛАДАЮЩИЕ ФУНГИЦИДНОЙ АКТИВНОСТЬЮ, И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1992 |
|
RU2069213C1 |
| α,ω ДИАРИЛАЛКАНЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1993 |
|
RU2105752C1 |
| СТЕРОИДНЫЕ СОЕДИНЕНИЯ, СПОСОБ ПОЛУЧЕНИЯ СОЕДИНЕНИЙ | 1993 |
|
RU2097387C1 |
| ЧЕТЫРЕХКООРДИНАЦИОННЫЕ КОМПЛЕКСЫ ДВУХВАЛЕНТНОЙ ПЛАТИНЫ | 1992 |
|
RU2039064C1 |
| АЗАСТЕРОИДНЫЕ СОЕДИНЕНИЯ | 1991 |
|
RU2070204C1 |
| ГЕКСАГИДРОНАФТАЛИНОВЫЕ СЛОЖНОЭФИРНЫЕ СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1993 |
|
RU2104997C1 |
| ПРОИЗВОДНЫЕ ТИАЗОЛИДИН-2,4-ДИОНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1992 |
|
RU2103265C1 |
| КРИСТАЛЛИЧЕСКИЙ ПИВАЛОИЛОКСИМЕТИЛ (IR, 5S, 6S)-[(4R)-2-ОКСО-4-ПИРРОЛИДИНИЛТИО]-6-[(IR)-1-ГИДРОКСИЭТИЛ]-1-МЕТИЛ-1-КАРБАПЕН-2-ЕМ-3-КАРБОКСИЛАТ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 1993 |
|
RU2090567C1 |
| ТИАЗОЛИДИНОВЫЕ СОЕДИНЕНИЯ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, СПОСОБ СНИЖЕНИЯ СОДЕРЖАНИЯ САХАРА В КРОВИ У МЛЕКОПИТАЮЩИХ | 1992 |
|
RU2095354C1 |

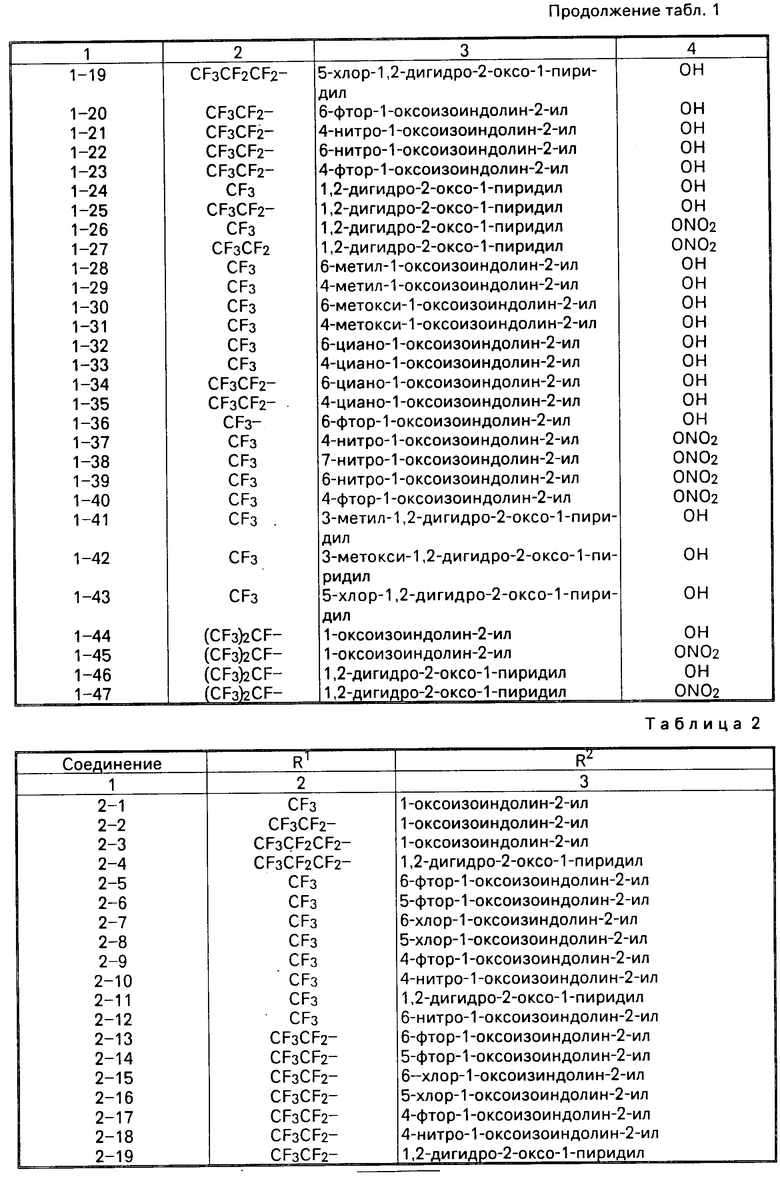
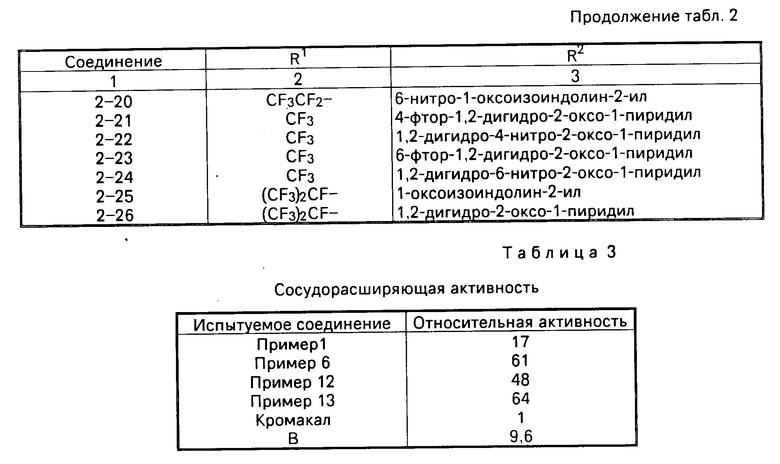
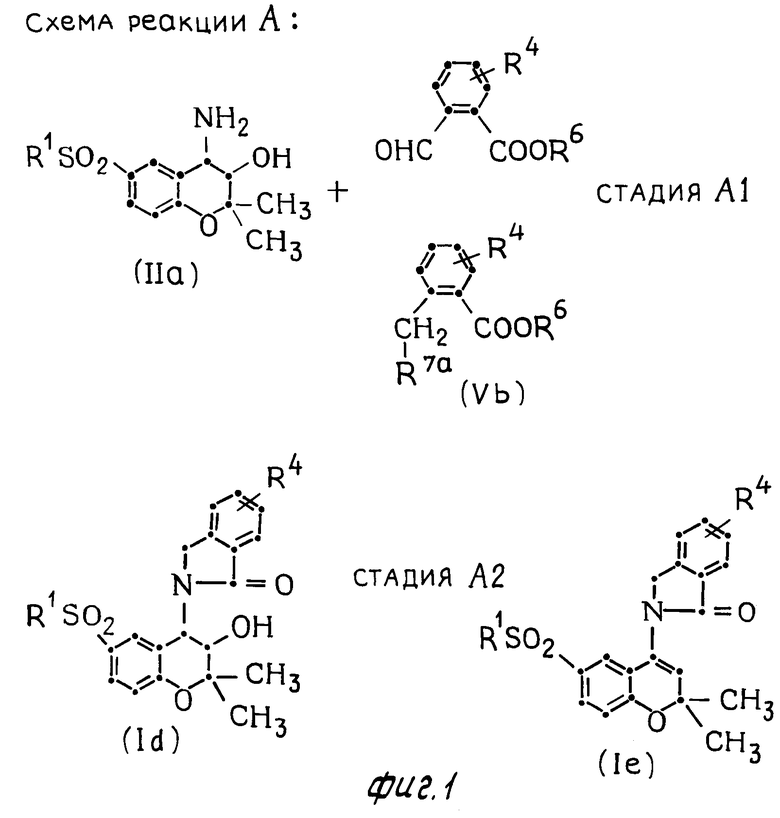
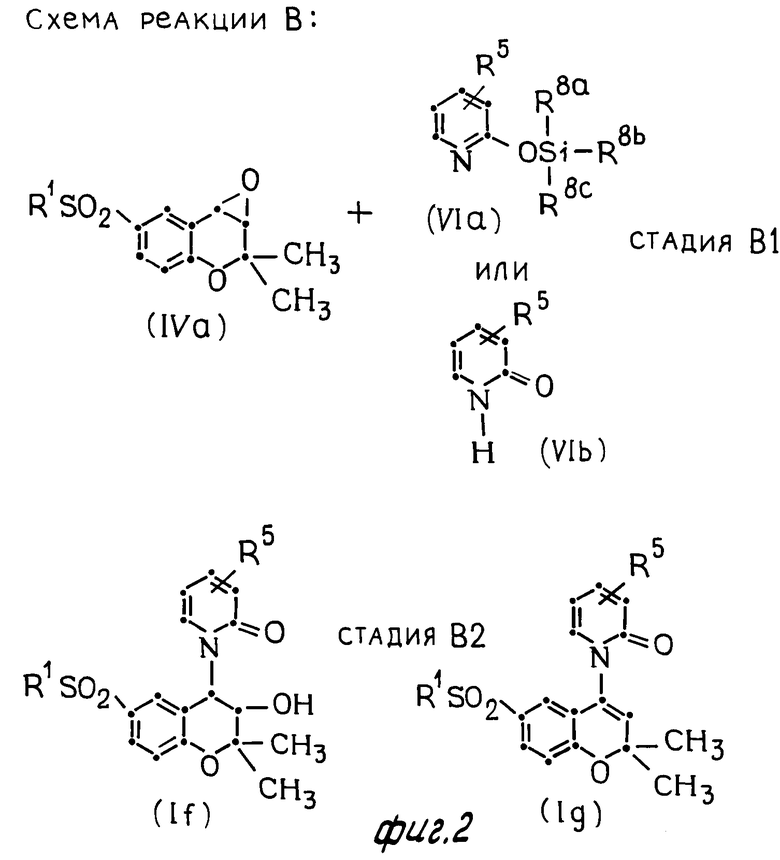
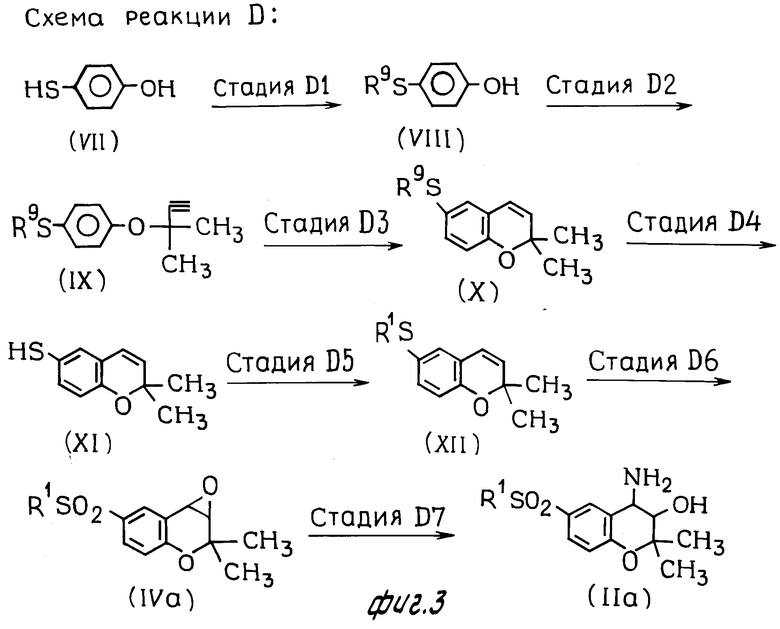
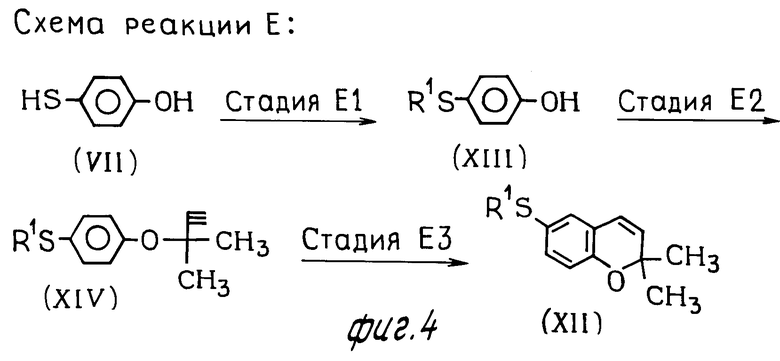
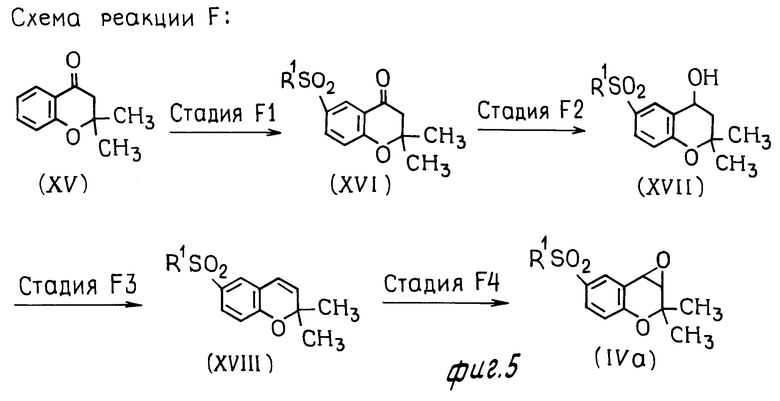
Использование: в медицине в качестве противогипотензивного средства. Сущность изобретения: продукт - производные бензопирана ф-лы I, где R1, R2, α-β, R3 и R4 имеют соответствующие значения. Реагент 1: соединение ф-лы IV, где а) W-NH2- группа; W′-OH- группа или б) W и W′ вместе атом кислорода и образуют этоксигруппу. Реагент 2: в случае а) соединение ф-лы V, где R4 указано выше; R6 - C1-C4 -алкил; R7 - формил или галоидметил, или в случае б) соединение ф-лы VI или VIа, где R4 и R5 являются такими, как определено выше; а -α-γ- представляет группу ф-лы VII или VIII, где R8a, R8b и R8c - C1-C4- алкил, и получают соединение ф-лы Iа, где R1 и R2 указаны выше, и, если необходимо, обрабатывают соединение ф-лы Iа основанием и получают соединение ф-лы Iв, где R1 и R2 - указано выше, или, если необходимо соединение ф-лы Iа подвергают взаимодействию с нитрующим реагентом и получают соединение ф-лы Iс, где R1 и R2 указано выше, или, если необходимо, выделяют соединение ф-лы Ia, Ib и Ic в виде фармацевтически приемлемой соли. Относительная активность по сравнению с кромакалимом составляет 17 - 64%. Структура соединений ф-л: 2 с. и 1 з.п. ф-лы, 5 ил., 3 табл.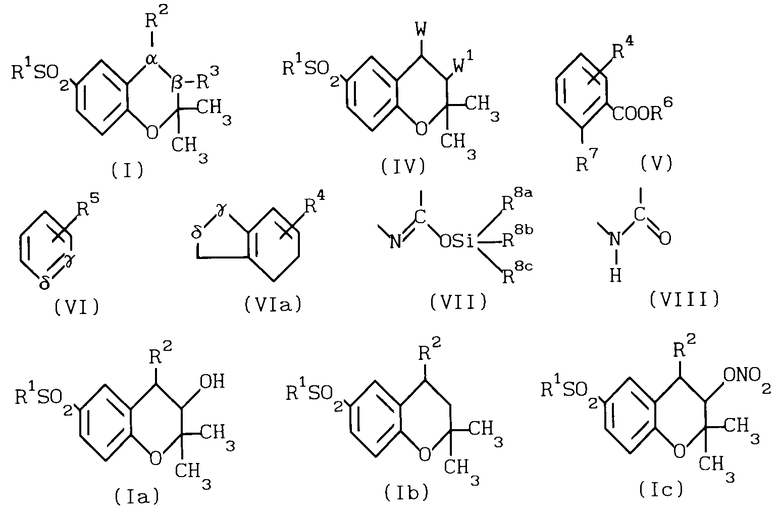
где R1 C1 C3-перфторалкильная группа;
R2 группа общей формулы II
где R4 водород, нитрогруппа или группа общей формулы III
где R5 водород, C1 C4-алкильная группа, C1 - C4-алкоксигруппа;
α-β углерод-углеродная простая связь или углерод-углеродная двойная связь;
при a-β углерод-углеродная простая связь R3 гидроксигруппа или нитроксигруппа, или при a-β углерод-углеродная двойная связь R3 водород;
или их фармацевтически приемлемые соли, при условиичто R1 не представлет трифторметильную группу, когда R2 группа формулы II, где R4 водород, a-β простая связь, а R3 гидроксигруппа.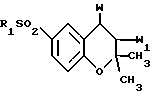
где R1 имеет указанные в п.1 значения,
(а) W аминогруппа, W1 гидроксигруппа;
(b) W и W1 вместе кислород, чтобы образовать эпоксигруппу;
(c) в случае (а), с соединением формулы V
где R4 имеет указанные в п.1 значения;
R6 C1 C4-алкильная группа;
R7 формальная группа или галоидметильная группа,
или, в случае (в), с соединением общей формулы VI или VIа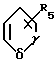

где R4 и R5 имеют указанные в п.1 значения;
группа общей формулы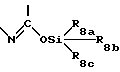
или формулы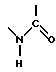
где R8a,R8b и R8c/ одинаковые или различные, каждый C1 - C4-алкильная группа;
и получают соединение общей формулы Iа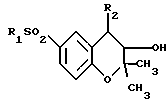
где R1 и R2 имеют указанные в п.1 значения,
и, если необходимо, обрабатывают соединения общей формулы Iа основанием и получают соединение общей формулы Iв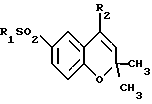
где R1 и R2 имеют указанные в п.1 значения,
или, если необходимо, соединения общей формулы Iа подвергают взаимодействию с нитрующим реагентом и получают соединение общей формулы Iс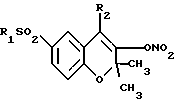
где R1 и R2 имеют указанные в п.1 значения,
или, если необходимо, соединения общих формул Iа, Iв и Iс выделяют в виде фармацевтически приемлемой соли.
| Кипятильник для воды | 1921 |
|
SU5A1 |
| Патент США N 4908378, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
