Изобретение относится к новому способу получения делмопинола recINN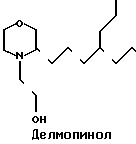 (I) а также новых промежуточных продуктов, использованных в процессе синтеза.
(I) а также новых промежуточных продуктов, использованных в процессе синтеза.
Делмопинол является соединением, которое показало многообещающие результаты как ингибитор пятен сыпи, поэтому его можно использовать в качестве компонента, например, полосканий для рта или зубных паст. Делмопинол является морфолиновым соединением. Известны некоторые промышленные методы, которые можно использовать для приготовления морфолиновых соединений такого типа. Вплоть до настоящего времени делмопинол приготовляли в больших количествах и с желаемыми выходами, согласно процессу, содержащему 16 стадий. Очевидно, что такой промышленный процесс поглощает большое количество времени и усилий.
Цель изобретения создание промышленного процесса, требующего меньше времени и усилий, дающего желаемые выходы и большие количества соединений.
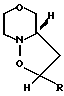
Цель достигается тем, что делмопинол получают в результате реакции моно- или полиненасыщенного соединения общей формулы (II) СН2-СН-R, где R представляет собой 2-пропилпентил, необязательно содержащий одну внутреннюю двойную связь, или 2-замещенный-2-пропилпентил, где заместитель во 2 положении является удаляемой группой, подвергают взаимодействию с морфолиннитроном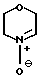 (III) c получением соединения общей формулы
(III) c получением соединения общей формулы (IV) где R имеет вышеуказанные значения, в виде смеси син- и антиизомеров, с последующим восстановительным размыканием кольца в полученном соединении с образованием смеси соединений (Va), (Vb) и (Vc), имеющих формулы
(IV) где R имеет вышеуказанные значения, в виде смеси син- и антиизомеров, с последующим восстановительным размыканием кольца в полученном соединении с образованием смеси соединений (Va), (Vb) и (Vc), имеющих формулы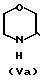


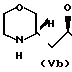 H
H
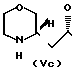 H
H где R представляет 2-пропилпентил, обработкой полученной смеси соединений хлорирующим агентом с получением смеси соединения общей формулы (Va) и хлораналогов соединений общей формулы (Vb) и (Vc), дехлорированием этой смеси и N-алкиллированием полученного при этом соединения общей формулы (Va) с последующим выделением целого продукта.
где R представляет 2-пропилпентил, обработкой полученной смеси соединений хлорирующим агентом с получением смеси соединения общей формулы (Va) и хлораналогов соединений общей формулы (Vb) и (Vc), дехлорированием этой смеси и N-алкиллированием полученного при этом соединения общей формулы (Va) с последующим выделением целого продукта.
Моно- и полиненасыщенные 4-пропилгептиловые соединения (II) получаются согласно примерам 1-5.
Уходящими группами на стадии а) могут быть любыми из обычных уходящих групп и обычно выбираются из следующего набора: гидрокси, алкокси, ацетокси или тетрагидропиранилокси.
Морфолиннитрон (III), использованный в качестве исходного продукта может быть получен из N-гидроксилморфолина путем окисления, например, желтым оксидом ртути, палладием или другими окислителями, либо из того же предшественника путем фотохимического или электрохимического окисления. Он может быть также получен непосредственно из морфолина путем окисления 2-(фенилсульфонил)-3-фенилоксахиридином или путем катализатора, например, диоксида селения или вольфрамата натрия.
Морфолиннитрон недостаточно стабилен для того, чтобы его можно было выделить, и поэтому используется непосредственно для реакции с ненасыщенными соединениями (II).
Соединения IV-анти и IV-син (как рецематы) получаются согласно примерам 6-12 с желаемыми выходами, а непрореагировавшее исходное соединение легко может быть выделено и снова запущено в реакцию. Образующиеся соединения являются диастереомерами с соотношением 90-98% IV-анти и 2-10 IV-син. Стереохимия аддуктов основана на аналогии. Стереохимия соединений IV, равное как и степень ненасыщенности, не являются решающими факторами для синтеза в целом. Все соединения IV приводят к одному и тому же конечному продукту через следующие стадии.
Восстановительное раскрытие кольца можно провести путем обработки соединения (IV) и (V), желательнее всего, кислотой, например, пара-толуолсульфоновой кислотой, в низшем спирте, например (предпочтительно) в изопропаноле, в восстановитель.
Последняя состоит из катализатора, желательнее всего Pd-С6 при давлении водорода лучше всего 3-7 атм.
Получение соответствующих хлораналогов (Vb) и (Vc) проводят путем реагирования реакционной смеси с хлорирующим агентом, желательнее всего кипячением с тионил хлоридом.
Стадию дехлорирования проводят путем гидрирования, используя при этом в качестве катализатора лучше всего никель Ренея.
Стадию алкилирования лучше всего проводить обработкой хлорэтанолом и иодидом калия и, в промежутках, гидроксидом калия, получая желаемый 3-(4-пропилгепти)-4-морфолин-этанол.
Наиболее важным в изобретении является получение промежуточных продуктов (IV) и (V), поскольку они являются ключевыми промежуточными продуктами в процессе получения делмопинола.
Изобретение иллюстрируется примерами, из которых 1-5 касаются получения конечных алкенов/алкинов. Примеры 6-12 относятся к получению изоксазолидинов (IV) и изоксазолинов (V), а примеры 13-15 иллюстрируют конечное получение делмопинола.
П р и м е р 1. Получение 4-пропил-1-гептена (табл.1а).
К 100 г 4-пропилгептил бромида в 400 мл бензола добавляли 90 г трет-ВиОК в 300 мл диметилсульфоксида. В процессе добавления температуру поддерживали ниже 50оС. Смесь перемешивали в течение 2 ч, после чего добавляли 600 мл воды. Органическую фазу отделяли, а водную фазу экстрагировали петролейным эфиром (т.пл. 40-60оС). Сложные органические фазы промывали водой и солевым раствором.
После осушивания с Na2SO4 и выпаривания остаток перегоняли.
Выход: 23,2 г (т.пл. 56-90оС/75 мм рт.ст.), IH-ЯСР (СD Cl3): δ0,9 (6Н, СН3), 1,2 (9Н, СН2СН), 2,0 (2Н, СН2С-С), 4,8-5,1 (2Н, СН2=С), 5,5-6,0 (1H, CH=C).
П р и м е р 2. Получение 4-пропил-1,3-гептадиена (табл.1в) и цис/транс-4-пропил-1,4-гептадиена (табл.1с).
К 80 г РВr3 в 250 мл сухого диэтилового эфира медленно добавляли 46 г 4-гидрокси-4-пропил-1-гептена при (-30) (-20)оС. После добавления температуру поддерживали в интервале (-25) (-10)оС в течение 2 ч, а затем +5оС в течение 15 ч. Реакционную смесь выливали на лед (500 г) и добавляли диэтиловый эфир (500 мл). Эфирную фазу отделяли и промывали раствором NaHCO3 (2х250 мл), осушали в МgSO4 и выпаривали. Остаток 60,0 г добавляли к 250 мл бензола и 94 г 1,8-диазо-бицикло[5.4.0]ундек-7-она (1,5-5) и нагревали с обратным холодильником в течение 2 ч. После охлаждения добавляли 1000 мл диэтилового эфира и эфирный раствор промывали с 5 М НСl (2х300 мл) и водой (3х250 мл), осушали с МgSO4 и выпаривали. Остаток (38,2 г) перегоняли и получали 30,6 г фракции, кипящей при 48-56оС (8 мм рт.ст.). ГХ показала, что она состоит из 47% цис/транс-4-пропил-1,4-гептадиена (не выделяли) и 46% 4-пропил-1,3-гептадиена, 1,4- и 1,3-изомеры разделяли методом препаративной газожидкостной хроматографии (Perkin Elmer F 21) на колонке 12м х 8 мм с 20% карбоваксом 20 М, 180оС и при давлении азота 1,9 атм.
IЯМР (СD Cl3):
1 δ 0,9 (6Н, СН3), 1,3-1,5 (4Н, СН2ОС=С), 1,9-2,2 (4Н, СН2С=С), 4,9-5,1 (2Н, СН2=С), 5,8-5,9 (IH, C=CHO-C), 6,5-6,7 (IH, C=CCH=C)
IC: δ 0,8-0,9 (6Н, СН3), 1,3-1,5 (2Н, СН2СО=C), 1,9-2,1(4Н, СН2С=С), 2,6-2,8 (2Н, С= ССН2С= С), 4,9-5,1 (2Н, СН2=С), 5,1-5,3 (1Н, СН=С), 5,6-5,9 (IH, CH=C).
П р и м е р 3. Получение 4-гидрокси-4-пропил-1-гептена (табл.1d).
113 г 4-гептанола в 1000 мл сухого диэтилового эфира медленно добавляли к раствору аллилмагния бромида, приготовленного из 38,5 г Mg и 178 г аллилбромида в 500 мл сухого диэтилового эфира. После добавления смесь кипятили с обратным холодильником в течение 10 ч. Реакционную смесь выливали на смесь 150 г льда, 450 мл 20% NH4Сl и 350 мл 5М НСl. Эфирную фазу отделяли, а водную фазу экстрагировали диэтиловым эфиром (3х300 мл). После этого составные органические фазы промывали раствором Na2CO3 и водой, осушали Na2SO4 и выпаривали. Остаток перегоняли.
Выход: 142 г (т.пл. 38-40оС, 0,1 мм рт.ст.).
IH-ЯМР (СDCl3): δ0,9 (6Н, СН3), 1,3-1,6 (9Н, СН2ОН), 2,1-2,3 (СН2С=С), 5,0-5,2 (СН2=С), 5,6-6,1 (СН=С).
П р и м е р 4. Получение 2-пропилпентил тозилата.
К смеси 52 г 2-пропилпентанола и 36 г пара-толуолсульфоновой кислоты в 175 мл хлороформа добавляли при 0-3оС и при атмосферном давлении паров азота 48 г пиридина. Смесь выдерживали в течение 30 мин при 0оС, а затем в течение 19 ч при комнатной температуре. После охлаждения реакционной смеси добавляли 3М НСl (300 мл). Органическую фазу отделяли и промывали водой и солевым раствором. Осушка с Na2SO4 и выпаривание дали 110 г 2-пропилпентил тозилата. IH-ЯМР (СDCl3):δ0,8 6Н, СН3), 1,1-1,8 (9Н, СН2, СН), 2,4-(3Н, АrСН), 3,9(2Н, ОСН2), 7,2-7,9 (4Н, ArH).
П р и м е р 5. Получение 4-пропил-1-гептина (табл. IIа).
18,4 г комплекса ацетилида лития с этилендиамином были заряжены в наполненной аргоном колбе. После этого добавляли диметилсульфоксид (100 мл) и смесь охлаждали до 15оС. Медленно добавляли 50 г 2-пропилпентил-паратолуолсульфоната. После добавления смесь перемешивали при комнатной температуре в течение 1 ч, а затем добавляли 50 мл воды, осторожно и энергично перемешивали, при этом поддерживали температуру ниже 35оС. Смесь выливали в 600 мл воды и экстрагировали гексаном (3х100 мл). Сложные гексановые фазы промывали солевым раствором и осушали с Na2SO4. Гексан отгоняли, а остаток перегоняли при пониженном давлении.
Выход: 13,1 г (т. пл. 75-80оС, 85 мм рт.ст. IH-ЯМР (СDCl3):δ0,9 (6Н, СН3), 1,3 (9Н, СН2, СН), 1,9 (1Н, СН-С), 2,2 (2Н, СН2С=С).
П р и м е р 6. Общая процедура получения изоксазолидинов IV и изоксазолина V (метод А).
К смеси концевых алкенов/алкинов (10 г), морфолина (19 г) и Na2WO4, 2Н2О (2,7 г) в метаноле (50 г) добавляли 35% Н2О2 (43 г) с такой скоростью, чтобы температура кипения была 50-60оС. К смеси, кипевшей в течение 18 ч при 50-60оС, добавляли еще порцию этанола (100 мл). Большую часть метанола/этанола испаряли в вакууме, после чего добавляли воду (300 мл) и смеси экстрагировали диэтиловым эфиром (4х50 мл). Органическую фазу промывали водой и селевым раствором. Осушка с Na2SO4 и выпаривание давали изоксазолидины IV/изоксазолин V. Возможны и другие комбинации растворителей, например, СНCl3, толуол и СН3ССl3.
П р и м е р 7. Получение изоксазолидина IV.
70 г 35% Н2О2 добавляли к смеси 31 г морфолина, 125 мл метанола, 125 мл этанола, 19 г 4-гидрокси-4-пропил-1-гептена и 4,8 г Na2WO4, 2Н2О с такой скоростью, чтобы поддерживать температуру кипения 50-80оС. Дополнительно добавляли 200 мл этанола и выдерживали смесь в течение 18 ч при 50-60оС. Большую часть метанола/этанола испаряли в хорошем вакууме, после чего добавляли 600 мл воды и смесь экстрагировали эфиром (4х200 мл). Эфирную фазу обрабатывали 5М НСl (4х100 мл) и получали 13,5 г исходного вещества. Кислую водную фазу подщелачивали и экстрагировали эфиром. Осушка с Na2SO4 выпаривали дали 5,9 г IVd (90% анти + 10% син).

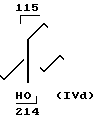
MS (70 еV): m/Z 257 (M+), 214, 196 (214-H2O), 115- 102, 86 IH-ЯМR (СDCl3):
IVd:δ0,9 (6Н, СН3), 1,2-2,0 (12Н, СН2), 2,2-3,2 (3Н, СН2N, CHN), 3,2-4,0 (5Н, СН2О, СН-О), 4,6 (IH, OH).
П р и м е р 8. Получение изоксазолидина IV (метод Б).
735 г 30% Н2О2 добавляли в 330 г морфолина и 52 г NaWO4, 2Н2О в 400 мл воды, медленно и при охлаждении. Температуру реакционной смеси поддерживали ниже 20оС. Половину этой натриевой смеси добавляли затем к кипящей с обратным холодильником смеси 100 г 4-гидрокси-4-пропил-1-гептена и 900 мл метанола. После добавления кипячение с обратным холодильником продолжали еще 2,5 ч, после чего вторую половину смеси нитронов добавляли в кипящую смесь и кипятили затем с обратным холодильником еще 2,5 ч. После охлаждения смесь экстрагировали толуолом (750 мл). Толуольную смесь экстрагировали 5% НСl (650 мл). Из органической фазы было выделено 57 г исходного вещества 4-гидрокси-4-пропил-1-гептена. Водную фазу доводили до рН 8,8 с помощью 5М NaOH и экстрагировали толуолом (500 мл). После осушки с Na2SO4 и выпаривания было вновь получено 37 г IVd в виде смеси син-анти.
MS (70 eV): m/Z 257 (М+), 214, 196 (214-Н2О), 115, 102, 86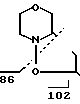
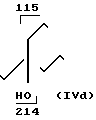
IH-ЯМP (CD, Cl3):
IVd:δ0,9 (6Н, СН3), 1,2-2,0 (12Н, СН2), 2,2-3,2 (3Н, СН2N, CHN), 3,2-4,0 (5Н, СН2О, СН-0), 4,6 (1Н, ОН).
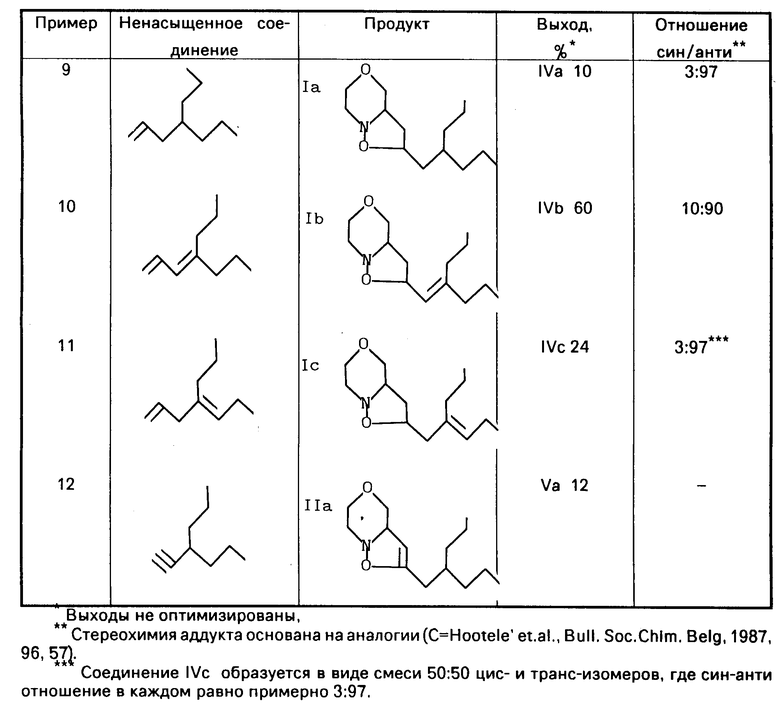
П р и м е р ы 9-12 использованы для подтверждения процессов, описанных в примере 6, представлены в таблице.
Данные спектрального анализа для соединений примеров 9-11 приведены на отдельных страницах.
В примере 12 продукт (Va) не был выделен в чистом виде. Выход был определен методом IH-ЯМР (СD Cl3): δ0,9 (6Н, СН3), 1,3 (9Н, СН2СН), 4,5 (1Н, СН= С). Продукт был использован в качестве промежуточного продукта в последующих реакциях без образования каких-либо побочных продуктов.
П р и м е р 9. Соединение IVа
MS (70 eV): m/z 241 (M+), 128, 102, 86
П р и м е р 10. Соединение IVB.
MS (70 eV): m/z 239 (M+), 210, 138, 102, 86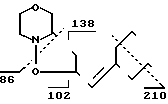
1H-ЯMP (CDCl3):
IVb: δ0,9 (6H, CH3), 1,1-2.2 (10 H, CH2), 2,4-4,2 (7H, CH2N, CHN, CH2O, CH-O), 5.1-5.3 (2H, CH=C, O-CHC=O).
П р и м е р 11. Соединение IVс.
MS (70 eV): m/z 239 (M+), 196, 128, 102, 86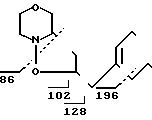

П р и м е р 13. Восстановительное размыкание кольца изоксазолидина IV.
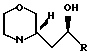
Смесь 10 г изоксазолидина IVd 27 г пара-толуолсульфоновой кислоты и 1,5 г 10% Pd в 100 мл изопропанола встряхивали в склянке Парра при 70-80оС и 3-7 атм давления водорода в течение 15 ч. После охлаждения реакционную смесь отфильтровывали и изопропанол испаряли в хорошем вакууме. Добавляли избыток 5 М NaOH и смесь экстрагировали диэтиловым эфиром. После осушки и выпаривания былa вновь получено смесь соединений Va, Vb и Vс (R=2-пропилпентил) в количестве 8,8 г.
П р и м е р 14. Хлорирование гидроксиалкилморфолинов Vb и Vc (R=2-пропилфенил) и последующее дехлорирование.
15 мл тионилхлорида добавляли и 5,0 г смеси соединений Vd, Vb и Vc (R= 2-пропилпентил) в 7 мл хлороформа и затем смесь перемешивали при 20оС в течение 3 ч и нагревали с обратным холодильником в течение 1 ч. После выпаривания добавляли 5 М NaOH (25 мл) и смесь экстрагировали диэтиловым эфиром (3х15 мл). Сложные эфирные фазы промывали водой и солевым раствором. Осушка и выпаривание дали 4,8 г хлораналогов и Va.
Эту смесь, вместе с 5 г катализатора никель Ренея, 5 г триэтиламина и 250 л диоксана, гидролизовали при 100оС и давлении водорода 120 атм в течение 24 ч. Реакционную смесь отфильтровывали через целит и выпаривали. Добавляли 30 мл 5М NaOH и смесь экстрагировали диэтиловым эфиром (3х15 мл). После осушки и выпаривания было снова выделено 4,3 г чистого 3-(4-пропилгептил)-морфолина.
MS (70 eV) m/Z 227 (М+), 154, 86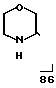

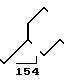
IН-ЯМР (СDCl3):
δ0,9(6Н, СН3), 1,2 (15Н, СН2, СН), 1,6 (1Н, NH), 2,6-3,9 (7Н, СН2О, СН2N, CHN).
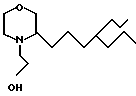
П р и м е р 15. Получение 3-(4-пропилгептил)-4-морфолинэтанола.
Смесь 2,5 г 3-(4-пропилгептил)морфолина, 3,5 г хлорэтанола, 1,1 г иодистого калия и 7 мл этанола нагревали с обратным холодильником в течение 5 ч. Затем добавляли 0,3 г КОП в 1,5 мл этанола и кипятили с обратным холодильником продолжали еще 2 ч, после чего добавляли, еще 0,2 г КОН в 1,0 мл этанола. Кипятили еще 7 ч с обратным холодильником, добавляли третью порцию 0,1 г КОН в 0,5 мл этанола. Затем кипятили еще 2 ч, растворитель испаряли и добавляли 10 мл воды. Смесь экстрагировали диэтиловым эфиром (3х10 мл) и сложные органически фазы промывали водой и солевым раствором. После осушки и выпаривания было выделено 2,5 г 3-(4-пропилгептил)-4-морфолин-этанола.
MS(70 eV): m/Z 271 (M+), 240, 130, 100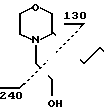
 m/Z100
m/Z100
1H-ЯМР (СD Cl3):δ0,9 (6Н, СН3), 1,1-1,6 (15Н, СН2, СН), 2,3-2,5 и 2,7-3,0 (6Н, ОН, СН2N, CHN), 3,4-3,8 (6Н, СН2О).
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ДЕЛМОПИНОЛА И ЕГО ПРОИЗВОДНЫХ | 2006 |
|
RU2404169C2 |
| EMM-28 - новый синтетический кристаллический материал, его получение и применение | 2016 |
|
RU2721569C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПИРАЗОЛОВ | 2015 |
|
RU2712192C2 |
| АЛКИЛЗАМЕЩЕННЫЕ ЦИКЛИЧЕСКИЕ АМИНЫ В КАЧЕСТВЕ СЕЛЕКТИВНЫХ ЛИГАНДОВ D3-ДОПАМИНА | 1997 |
|
RU2185372C2 |
| ПИРИДИЛ- ИЛИ ПИРИМИДИЛСОДЕРЖАЩИЕ ПРОИЗВОДНЫЕ ПИПЕРАЗИНА ИЛИ 1,4-ДИАЗАЦИКЛОГЕПТАНА ИЛИ ИХ ФАРМАКОЛОГИЧЕСКИ АКТИВНЫЕ КИСЛОТНО-АДДИТИВНЫЕ СОЛИ, ОБЛАДАЮЩИЕ ПСИХОТРОПНЫМ ДЕЙСТВИЕМ | 1989 |
|
RU2021269C1 |
| ПРОИЗВОДНЫЕ ХИНОЛИНА | 1999 |
|
RU2197481C2 |
| ПРОИЗВОДНЫЕ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ N-АДАМАНТИЛМЕТИЛА В КАЧЕСТВЕ ФАРМАЦЕВТИЧЕСКИХ КОМПОЗИЦИЙ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 2002 |
|
RU2300525C2 |
| ТОКОФЕРОЛЫ, ТОКОТРИЕНОЛЫ И ДРУГИЕ ПРОИЗВОДНЫЕ ХРОМАНА И БОКОВЫХ ЦЕПЕЙ И СПОСОБЫ ЛЕЧЕНИЯ С ИХ ИСПОЛЬЗОВАНИЕМ | 1999 |
|
RU2232758C2 |
| САМОЭМУЛЬГИРУЮЩАЯСЯ КОМПОЗИЦИЯ ДЛЯ ЛИПОФИЛЬНЫХ СОЕДИНЕНИЙ | 1998 |
|
RU2203648C2 |
| ТИЕНОПИРИМИДИНДИОНЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ) И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2002 |
|
RU2294937C9 |
Сущность изобретения: моно- или полиненасыщенное соединение ф-лы (II) CH2= CH-R, где R 2-пропиллентил, необязательно содержащий одну внутреннюю двойную связь, или 2-замещенный-2-пропилпентил, где заместитель в положении 2 является уходящей группой, подвергают взаимодействию с морфолиннитроном ф-лы (III) с получением соединения ф-лы (IY), где R указано выше, в виде смеси син- и антиизомеров и последующим восстановительным раскрытием кольца в полученном соединении. Полученную при этом смесь соединений ф-лы (Va), (Vb) и (Vc), где R 2-пропилпентил обрабатывают хлорирующим агентом. Полученную в результате хлорирования смеси соединения ф-лы (Va) с хлоранологами соединений (Vb) и (Vc) подвергают дехлорированию. Полученное соединение ф-лы (Va)N алкилируют и выделяют целевой продукт. 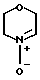

 2 с. и 4 з.п. ф-лы, 1 табл.
2 с. и 4 з.п. ф-лы, 1 табл.
отличающийся тем, что, с целью упрощения способа, моно- или полиненасыщенное соединение общей формулы II
СН2=СН-R,
где R 2-пропилпентил, необязательно содержащий одну внутреннюю двойную связь, или 2-замещенный-2-пропилпентил, где заместитель в положении 2 является уходящей группой,
подвергают взаимодействию с морфолиннитроном формулы III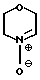
с получением соединения общей формулы IV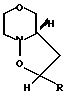
где R имеет указанные значения,
в виде смеси син- и антиизомеров с последующим восстановительным раскрытием кольца в полученном соединении с образованием смеси соединений общих формул Va, Vb и Vc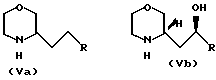

где R 2-пропилпентил,
обработкой полученной смеси соединений хлорирующим агентом с получением смеси соединения формулы Va и хлораналогов соединений общей формулы Vb и Vc, дехлорированием этой смеси и N-алкилированием полученного при этом соединения общей формулы Va с последующим выделением целевого продукта.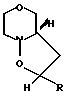
где R 2-пропилпентил, необязательно имеющий одну внутреннюю двойную связь, или 2-замещенный 2-пропилпентил, где заместитель в положении 2 является уходящей группой.
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| C.Hootele'et al., Bull.Soc.Chim.Belg., 1987, 96, 57. | |||
