Область изобретения
Настоящее изобретение в целом относится к областям органической химии и антипролиферативных и проапоптотических соединений. Более конкретно, данное изобретение относится к соединениям на основе хромана и их производным и их применениям в качестве агентов пролиферации клеток, агентов проапоптоза, иммуномодулирующих и противовирусных агентов.
Предпосылки изобретения
Биология пролиферации клеток и гибели клеток (апоптоза) является чрезвычайно сложной, включающей в себя многочисленные внутриклеточные пути передачи сигналов и многочисленные взаимодействующие продукты генов. У раковых клеток могут проявляться множественные дефекты в нормальных механизмах регуляции пролиферации клеток, позволяющие им увеличиваться в числе. Кроме того, у раковых клеток проявляются дефекты в механизмах, участвующих в элиминации отклоняющихся от нормы клеток посредством многостадийных процессов, называемых запрограммированной смертью клеток или апоптозом. Таким образом, комбинации нерегулируемой пролиферации клеток и супрессии путей передачи сигналов, индуцирующих смерть клеток, дают раковым клеткам преимущества как в росте, так и в выживании.
Увеличиваются ли клетки в числе или нет, зависит от баланса экспрессии негативно действующих и позитивно действующих на рост продуктов регуляторных генов и присутствия или отсутствия функциональных путей передачи сигналов гибели клеток. Негативно действующие на рост регуляторные гены способствуют блокированию клеток в клеточном цикле. Позитивно действующие на рост регуляторные гены стимулируют клетки к прогрессированию в течение всего клеточного цикла. Гены, участвующие в апоптозе, могут быть либо проапоптотическими, либо антиапопототическими, и динамический баланс между ними определяет, живет ли клетка или умирает.
Раковые клетки, для того чтобы выживать и увеличиваться в числе, с течением времени подвергаются серии мутационных событий, которые удаляют регуляторные механизмы управления, что дает им возможность неконтролируемо расти и выживать даже в присутствии проапопототических сигналов и развивать свойства, которые позволяют им избежать обнаружения и удаления защитной системой иммунного ответа. Раковые опухоли могут вызывать смерть индивидуумов, если они не удаляются хирургически или не подвергаются эффективному лечению лекарственными средствами.
Существует большое разнообразие патологических клеточно-пролиферативных состояний, в случае которых для обеспечения терапевтических преимуществ требуются новые терапевтические стратегии и агенты. Эти патологические состояния могут иметь место в почти во всех типах клеток, способных к ненормальной пролиферации клеток или отклоняющейся от нормы отвечаемости на сигналы гибели клеток. Среди типов клеток, которые проявляют патологические или отклоняющиеся от нормы характеристики роста и гибели, находятся (1) фибробласты, (2) сосудистые эндотелиальные клетки и (3) эпителиальные клетки. Таким образом, необходимы новые способы для лечения локальных или рассеянных патологических состояний во всех или почти во всех системах органов и тканей индивидуумов.
Большинство видов рака, являются ли они специфически мужскими, такими как рак предстательной железы или рак яичка, или специфически женскими, такими как рак молочной железы, яичника или шейки матки, или же поражают в равной степени мужчин и женщин, такие как рак печени, кожи или легкого, подвергаются со временем усиленным генетическим повреждениям и эпигенетическим событиям и в конечном счете становятся высокометастатическими и трудными для лечения. Хирургическое удаление локализованных случаев рака оказалось эффективным только в том случае, когда рак не распространился за пределы первичного повреждения. Как только рак распространился на другие ткани и органы, хирургические процедуры должны быть дополнены другими, более специфическими процедурами для уничтожения (ликвидации) больных или злокачественных клеток. Большинство обычно используемых дополнительных процедур для лечения пораженных или злокачественных клеток, таких как химиотерапия или радиобиологическое воздействие, не являются локализованными на опухолевых клетках и, хотя они обладают пропорционально более высоким разрушающим действием на злокачественных клетках, часто повреждают до некоторой степени и нормальные клетки.
Некоторые производные токоферолов, токотриенолов и витамина Е использовали в качестве проапоптотических и ингибирующих синтез ДНК агентов. Структура витамина Е состоит из хроманола в качестве головной части и алкильной боковой цепи. Существуют восемь главных встречающихся в природе форм витамина Е: альфа (α), бета (β), гамма (γ) и дельта (δ) токоферолы и α, β, γ и δ токотриенолы. Токоферолы отличаются от токотриенолов тем, что они имеют насыщенную фитильную боковую цепь, а не ненасыщенную изопренильную боковую цепь. Четыре формы токоферолов и токотриенолов различаются по числу метильных групп на хромановой головной части (α имеет три, β и γ имеют две и δ имеет одну).
R,R,R-α-токоферилсукцинат является производным R,R,R-α-токоферола, которое структурно модифицировано посредством сложноэфирной связи, так что оно содержит сукцинильную часть вместо гидроксильной части в положении 6 хромановой головной части. Эта присоединенная эфирной связью сукцинатная часть молекулы R,R,R-α-токоферола оказалась наиболее сильнодействующей формой витамина Е, влияющей на биологические действия запуска апоптоза и ингибирования синтез ДНК. Эта форма витамина Е индуцирует апоптоз опухолевых клеток, не оказывая индуцирующего влияния апоптоз эффектов на нормальные клетки. Главное преимущество этой формы витамина Е в качестве противоракового агента состоит в том, что многие раковые клетки либо экспрессируют низкие уровни эстераз, либо не экспрессируют эстеразы, которые могут отщеплять сукцинатную часть молекулы с превращением тем самым сукцинатной формы R,R,R-α-токоферола в свободный R,R,R-α-токоферол. R,R,R-α-токоферол не проявляет ни сильнодействующей антипролиферативной, ни запускающей апоптоз биологической активности. Однако связанный эфирной связью сукцинат витамина Е является неэффективным in vivo, поскольку природные эстеразы в хозяине отщепляют сукцинатную часть молекулы, образуя неэффективный противораковый агент, R,R,R-α-токоферол.
В существующем уровне техники отсутствуют эффективные средства ингибирования нежелательной или неконтролируемой пролиферации клеток в большом разнообразии патофизиологических состояний, одновременно не влияющих или мало влияющих на нормальные клетки. Данное изобретение удовлетворяет этой давнишней потребности и пожеланию в данной области.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
В одном варианте данного изобретения предлагается соединение, имеющее структурную формулу
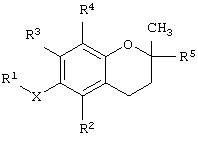
где Х обозначает кислород, азот или серу; R1 выбран из группы, состоящей из алкила, алкенила, алкинила, арила, гетероарила, карбоновой кислоты, карбоксилата, карбоксамида, сложного эфира, тиоамида, сложного тиолового эфира, тиоловой кислоты, сахарида, алкоксисвязанного сахарида, амина, сульфоната, сульфата, фосфата, спирта, простого эфира и нитрила;
R2 выбран из группы, состоящей из водорода, метила, бензилкарбоновой кислоты, бензилкарбоксилата, бензилкарбоксамида, бензилового сложного эфира, сахарида и амина; R3 выбран из группы, состоящей из водорода, метила, бензилкарбоновой кислоты, бензилкарбоксилата, бензилкарбоксамида, бензилового сложного эфира, сахарида и амина; R4 выбран из группы, состоящей из метила, бензилкарбоновой кислоты, бензилкарбоксилата, бензилкарбоксамида, бензилового эфира, сахарида и амина; и R5 выбран из группы, состоящей из алкила, алкенила, алкинила, арила, гетероарила, карбоксила, амида и сложного эфира; причем, когда Х представляет собой кислород, R2 представляет собой метил, R3 представляет собой метил, R4 представляет собой метил и R5 представляет собой фитил, R1 не является масляной кислотой.
В другом варианте данного изобретения предлагается способ лечения клеточно-пролиферативного заболевания, предусматривающий введение животному фармакологически эффективной дозы соединения, имеющего структурную формулу
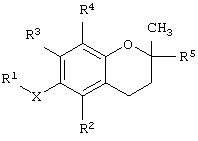
где Х обозначает кислород, азот или серу; R1 выбран из группы, состоящей из алкила, алкенила, алкинила, арила, гетероарила, карбоновой кислоты, карбоксилата, карбоксамида, сложного эфира, тиоамида, сложного тиолового эфира, тиоловой кислоты, сахарида, алкоксисвязанного сахарида, амина, сульфоната, сульфата, фосфата, спирта, простого эфира и нитрила;
R2 выбран из группы, состоящей из водорода, метила, бензилкарбоновой кислоты, бензилкарбоксилата, бензилкарбоксамида, бензилового сложного эфира, сахарида и амина; R3 выбран из группы, состоящей из водорода, метила, бензилкарбоновой кислоты, бензилкарбоксилата, бензилкарбоксамида, бензилового сложного эфира, сахарида и амина; R4 выбран из группы, состоящей из метила, бензилкарбоновой кислоты, бензилкарбоксилата, бензилкарбоксамида, бензилового сложного эфира, сахарида и амина; и R5 выбран из группы, состоящей из алкила, алкенила, алкинила, арила, гетероарила, карбоксила, амида и сложного эфира.
Еще в одном варианте данного изобретения предлагается фармацевтическая композиция, содержащая описанное здесь соединение и фармацевтически приемлемый носитель.
Еще в одном варианте данного изобретения предлагается способ индукции апоптоза клетки, предусматривающий стадию контактирования указанной клетки с фармакологически эффективной дозой соединения данного изобретения.
Другие и дополнительные аспекты, признаки, эффекты и преимущества данного изобретения будут очевидными из следующего описания предпочтительных в настоящее время вариантов данного изобретения, приведенных с целью раскрытия изобретения.
Краткое описание чертежей
Для того чтобы предмет настоящей заявки, в рамках которого достигаются вышеуказанные признаки, преимущества и цели данного изобретения, а также другие цели, которые станут очевидными, мог быть понятым в деталях, более конкретные описания данного изобретения, вкратце суммированного выше, могут быть даны со ссылкой на определенные варианты изобретения, которые иллюстрируются в прилагаемых чертежах. Эти чертежи образуют часть данного описания. Однако следует отметить, что прилагаемые чертежи иллюстрируют предпочтительные варианты данного изобретения и, следовательно, не должны рассматриваться в их объеме как ограничивающие.
Фиг.1 показывает общую структуру токоферола, токотриенола и других соединений на основе хромана.
Фиг.2 показывает соединения 1-29 на основе токоферола, синтезированные и испытанные в настоящее время.
Фиг.3А, 3В и 3С показывают общие синтетические органические подходы для химического варьирования соединений хроманола в положении R1.
Фиг.4 показывает общие синтетические органические подходы для химического варьирования соединений хроманола в положении R2.
Фиг.5 показывает общие синтетические органические подходы для химического варьирования соединений хроманола в положениях R3 и R4.
Фиг.6 показывает общие синтетические органические подходы для химического варьирования соединений хроманола в положении R5.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Как он применяется в настоящем описании, термин "индивидуум" будет относиться к животным и человеку.
Как он применяется в настоящем описании, термин "биологически ингибирующий" или "ингибирование" роста пролиферирующих клеток включает частичное или полное ингибирование роста и, как подразумевается, включает уменьшение в скорости пролиферации или роста этих клеток. Биологически ингибирующая доза композиции по данному изобретению может быть определена оценкой воздействия тестируемого элемента на рост злокачественных или ненормально пролиферирующих клеток-мишеней в культуре ткани, на рост опухолей в животных и культуре клеток или любым другим способом, известным лицам с обычной квалификацией в данной области.
Как он применяется в настоящем описании, термин "индукция запрограммированной гибели клеток или апоптоза" включает частичную или полную гибель клеток для клеток, проявляющих установленные морфологические и биохимические апоптотические характеристики. Доза композиции по данному изобретению, которая индуцирует апоптоз, может быть определена оценкой воздействия тестируемого элемента на рост злокачественных или ненормально пролиферирующих клеток-мишеней в культуре ткани, на рост опухолей в животных и культуре клеток или любым другим способом, известным лицам с обычной квалификацией в данной области.
Как он применяется в настоящем описании, термин "индукция остановки (задержки) клеточного цикла" включает остановку (задержку) роста вследствие блокирования обработанных клеток в фазе клеточного цикла GO/G1 или G2/M. Доза композиции по данному изобретению, которая индуцирует остановку клеточного цикла, может быть определена оценкой эффектов тестируемого элемента на рост злокачественных или ненормально пролиферирующих клеток-мишеней в культуре ткани, на опухолевый рост в животных и культуре клеток или любым другим способом, известным лицам с обычной квалификацией в данной области.
Как он применяется в настоящем описании, термин “индукция клеточной дифференцировки” включает остановку роста вследствие индукции обработанных клеток к прохождению клеточной дифференцировки стадии, в которой клеточная пролиферация не происходит. Доза композиции по данному изобретению, которая индуцирует клеточную дифференцировку, может быть определена оценкой воздействия тестируемого элемента на рост злокачественных или ненормально пролиферирующих клеток-мишеней в культуре ткани, на рост опухолей в животных и культуре клеток или любым другим способом, известным лицам с обычной квалификацией в данной области.
В данном изобретении предлагаются токоферолы, токотриенолы и другие производные хромана с производными или без производных насыщенной фитильной или ненасыщенной изопренильной боковых цепей и их аналоги. С использованием простых эфиров и некоторых других химических связей для присоединения различных остатков к токоферолу, токотриенолу и другим производным хромана получают новые противораковые соединения для применения in vivo. Общие структуры новых соединений данного изобретения показаны на фиг.1, а возможные пути их синтеза предложены на фиг.3-6. Новые признаки этих молекул включают в себя химическую функционализацию (присоединение функциональных групп) положений R1-R5 структуры хромана и химическую функционализацию (присоединение функциональных групп) фитильной и изопренильной боковых цепей, в частности, соединений на основе токоферолов и токотриенолов (фиг.1). Особенно предпочтительные соединения включают 2,5,7,8-тетраметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)уксусную кислоту (1), 2,5,7,8-тетраметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)пропионовую кислоту (2), 2,5,8-триметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)уксусную кислоту (7), 2,7,8-триметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)уксусную кислоту (8), 2,8-диметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)уксусную кислоту (9), 2-(N,N-(карбоксиметил)-2-(2,5,7,8-тетраметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)уксусную кислоту (12), 2,5,7,8-тетраметил-(2RS-(4RS,8RS,12-триметилтридецил)хроман-6-илокси)уксусную кислоту (15), 2,5,7,8-тетраметил-2R-(2RS,6RS,10-триметилеундецил)-хроман-6-илокси)уксусную кислоту (17), 3-(2,5,7,8-тетраметил-(2R-(4R,8,12-триметилтридецил)хроман-6-илокси)-пропил-1-аммонийхлорид (19), 2,5,7,8-тетраметил-(2R-(4R,8R,12-триметилтридецил)хроман-3-ен-6-илокси)уксусную кислоту (20), 2-(2,5,7,8-тетраметил-(2R-(4R,8,12-триметилтридецил)хроман-6-илокси)этилтриэтиламмонийсульфат (21), 6-(2,5,7,8-тетраметил-(2R-(4R,8,12-триметилтридецил)хроман)-уксусную кислоту (22), 2,5,7,8-тетраметил-(2R-(гептадецил)-хроман-6-илокси)уксусную кислоту (25), (2,5,7,8-тетраметил-2R-(4,8-диметил-1,3,7-Е,Z-нонотриен)хроман-6-илокси)уксусную кислоту (26) и Е,Z,RS,RS-(фитилтриметилбензолтиол-6-илокси)уксусную кислоту (27).
Разработанные с учетом фармакодинамики соединения данного изобретения имеют улучшенный терапевтический индекс и являются сильными ингибиторами роста раковых клеток; т.е. они демонстрируют высокую противоопухолевую активность с минимальными побочными эффектами. Эти соединения, которые не могут быть легко деградированы, так как в организме млекопитающих нет известных расщепляющих простую эфирную связь ферментов (этеразы), могут быть использованы в лечении разновидностей рака и нарушений, включающих в себя избыточную пролиферацию клеток, а также для клеток, которые во множестве накапливаются вследствие угнетенных механизмов убивания клеток, с минимальными побочными эффектами. Соединения согласно изобретению ингибируют рост раковых клеток путем индукции апоптоза и остановки синтеза ДНК. Индукция апоптоза этими соединениями опосредована активацией TGF-P, стресс-киназы, и путей передачи сигналов Fas/Fas-лиганда. Индукция апоптоза посредством других путей, например, через получение керамидов, также не исключена. Эти ингибирующие рост свойства позволяют использовать эти соединения в лечении пролиферативных заболеваний, в том числе рака различных типов клеток и линий дифференцировки, не-неопластических гиперпролиферативных заболеваний и расстройств в связи с дефектами в путях трансдукции апоптотических сигналов. Некоторые из соединений данного изобретения являются как сильными индукторами апоптоза, так и сильными ингибиторами синтеза ДНК опухолевых клеток, представляющих различные линии клеточной дифференцировки.
Иллюстрируется терапевтическое применение соединений данного изобретения в лечении видов рака и других заболеваний и расстройств, в которых участвует избыточная пролиферация клеток или неспособность клеток умирать. Показано, что эти новые производные (таблицы 1 и 2 в конце описания) при концентрациях ЕС50 индуцируют апоптоз раковых клеток молочной железы человека (клетки рака молочной железы MDA MB 435, MDA MB 231 и MCF-7), раковых клеток предстательной железы человека (PC-3, DU-145 и LnCaP), опухолевых клеток яичника человека (С-170), опухолевых клеток шейки матки человека (ME-180), клеток эндометрия человека (RL-95-2) лимфоидных клеток человека (миеломы, Raji, Ramos, Jurkat и HL-60), раковых клеток ободочной кишки (НТ-29 и DLD-1) и раковых клеток легкого (А-549). Показано, что эти новые производные не индуцируют апоптоз нормальных эпителиальных клеток молочной железы человека (HMECs) и иммортализованных, но неонкогенных клеток молочной железы MCF-10A.
Эти новые соединения и способы данного изобретения могут быть использованы для лечения неопластических заболеваний и не-неопластических заболеваний. Характерными примерами неопластических заболеваний являются рак яичника, рак шейки матки, рак эндометрия, рак мочевого пузыря, рак легкого, рак молочной железы, рак предстательной железы, рак яичка, глиомы, фибросаркомы, ретинобластомы, меланомы, саркомы мягких тканей, остеосаркомы, рак ободочной кишки, рак почки, рак поджелудочной железы, базально-клеточный рак (базалиома) и плоскоклеточный рак. Характерные примеры не-неопластических заболеваний выбраны из группы, состоящей из псориаза, доброкачественных пролиферативных заболеваний кожи, ихтиоза (диффузной кератомы), папилломы, рестеноза, склеродермии и гемангиомы.
Соединения и способы данного изобретения могут быть использованы для лечения не-неопластических заболеваний, которые развиваются вследствие неспособности выбранных клеток подвергаться нормальной запрограммированной гибели клеток или апоптозу. Характерными примерами заболеваний и расстройств, которые имеют место вследствие неспособности клеток отмирать, являются аутоиммунные заболевания. Аутоиммунные заболевания характеризуются деструкцией иммунокомпетентными клетками (иммуноцитами) собственных клеток, тканей и органов. Характерная группа аутоиммунных заболеваний включает в себя аутоиммунный тиреоидит, множественный склероз, тяжелую псевдопаралитическую миастению, системную красную волчанку, герпетиформный дерматит, глютеновую болезнь (чувствительность к недостаточности глютена) и ревматоидный артрит. Данное изобретение не ограничивается аутоиммунитетом, но включает все расстройства, имеющие иммунный компонент, такие как воспалительный процесс, участвующий в образовании сердечно-сосудистых бляшек, или индуцированное ультрафиолетовым излучением повреждение кожи.
Соединения и способы данного изобретения могут быть использованы для лечения расстройств и заболеваний, которые развиваются вследствие вирусных инфекций. Характерными примерами заболеваний и нарушений, которые имеют место вследствие вирусных инфекций, являются вирусы иммунодефицита человека (ВИЧ). Поскольку эти соединения работают на внутриклеточных сетях передачи сигналов, они обладают способностью воздействовать на любой тип наружного сигнала, такого как цитокины, вирусы, бактерии, токсины, тяжелые металлы и т.д.
Способы данного изобретения могут быть использованы для лечения любого животного. Наиболее предпочтительно способы данного изобретения применимы для человека.
Обычно для достижения фармакологически эффективного убивания клеток и антипролиферативных эффектов эти соединения и аналоги могут вводиться в любой терапевтически эффективной дозе. Предпочтительно структурно модифицированные токоферолы и токотриенолы и их аналоги вводят в дозе от приблизительно 0,1 мг/кг до приблизительно 100 мг/кг. Более предпочтительно структурно модифицированные токоферолы и токотриенолы и их аналоги вводят в дозе от приблизительно 1 мг/кг до приблизительно 10 мг/кг.
Введение композиций согласно данному изобретению может быть местным, внутриглазным, парентеральным, пероральным, интраназальным, внутривенным, внутримышечным, подкожным или выполняемым любым другим подходящим способом. Вводимая доза зависит от возраста, клинической стадии и степени нарушения или генетического предрасположения индивидуума, местоположения, веса, типа сопутствующего лечения, если оно проводится, и характера патологического или злокачественного состояния. Эффективная система доставки, используемая в способе данного изобретения, может применяться в таких формах, как капсулы, таблетки, жидкие растворы, суспензии или эликсиры для перорального введения или стерильные жидкие формы, такие как растворы, суспензии или эмульсии. Для местного применения она может использоваться в такой форме, как мази, кремы или спреи. Любой инертный носитель предпочтительно используют в комбинации с подходящими солюбилизирующими агентами, такими как солевой раствор или забуференный фосфатом солевой раствор или любой такой носитель, в котором соединения, используемые в способе, имеют подходящие характеристики растворимости, такой как этанол, ацетон или ДМСО.
Существует большое разнообразие патологических раковых и нераковых клеточно-пролиферативных состояний и случаев накопления клеток вследствие отсутствия нормальной гибели клеток, для которых композиции и способы данного изобретения будут обеспечивать терапевтическую пользу. Эти патологические состояния могут встречаться почти во всех типах клеток, способных к ненормальной пролиферации клеток или дефектных в механизмах запрограммированной гибели клеток. Среди типов клеток, у которых проявляется патологический или ненормальный рост или ненормальная гибель, находятся (1) фибробласты, (2) сосудистые эндотелиальные клетки и (3) эпителиальные клетки. Из вышеописанного можно видеть, что способы данного изобретения применимы в лечении местных или рассеянных состояний во всех или почти во всех системах органов и тканей индивидуумов.
Специально предполагается, что фармацевтические композиции могут быть приготовлены с использованием новых соединений на основе хромана и их производных данного изобретения. В этом случае, фармацевтическая композиция содержит новые соединения данного изобретения и фармацевтически приемлемый носитель. Лицо с обычной квалификацией в данной области смогло бы легко определить, без чрезмерного экспериментирования, подходящие дозы и способы введения соединений и аналогов данного изобретения.
Таким образом, данное изобретение относится к созданию и эффективному применению новых агентов, которые могут специфически поражать раковые клетки и либо отрицательно регулировать (даун-регулировать) сигналы стимуляции роста, и/или положительно регулировать сигналы ингибирования роста, и/или отрицательно регулировать сигналы выживания, и/или положительно регулировать сигналы гибели. Более конкретно, в данном изобретении создаются и характеризуются новые агенты, которые активируют факторы ингибирования роста, запускают пути передачи сигналов гибели клеток и ингибируют синтез ДНК.
Следующие ниже примеры приведены с целью иллюстрации различных вариантов данного изобретения и не предназначены для какого-либо ограничения данного изобретения.
ПРИМЕР 1
Методология органического синтеза
Синтез различных токоферолов, токотриенолов или других производных хромана с производными насыщенной фитильной или ненасыщенной изопренильной боковых цепей или без них возможен посредством структурной модификации циклической системы хромана (фиг.3-8). Структурные переменные R1, R2, R3, R4, R5 и Х иллюстрируют группы на группе хромана, которые модифицируют. С использованием химии (способа) алкилирования может быть синтезировано большое число соединений, содержащих различные группы R1, в частности, когда Х является кислородом. После алкилирования дополнительная химическая модификация групп R1 позволяет синтезировать широкий спектр новых соединений. Бромирование метильных групп бензила на группе хромана обеспечивает промежуточные продукты, которые позволяют варьирование групп R2, R3 и R4. Варьирование группы R5 также является возможным, в частности, когда исходным соединением является коммерчески доступная 6-гидрокси-2,5,7,8-тетраметилхроман-2-карбоновая кислота. Варьирование Х до групп, иных, чем кислород, который представляет Х в токоферолах и токотриенолах, может быть выполнено с использованием способа с палладием (для Х=СН2) и нуклеофильного ароматического замещения (для Х=N или S). Другие возможные модификации в отношении структуры хромана включают в себя ненасыщенность в положениях 3-4, сокращение цикла (уменьшение числа членов в цикле) с получением пятичленного фуранильного кольца и замещение гетероатомами (N или S) кислорода кольца хромана.
Используемые реагенты были либо коммерчески доступны, либо получены в соответствии с известной методикой. Безводные СН2Сl2 и ТГФ получали путем дистилляции. Все другие растворители имели реактивную чистоту. Условия безводной реакции поддерживали в атмосфере аргона с небольшим избыточным давлением в высушенной в термостате стеклянной посуде. Хроматографию на силикагеле проводили с использованием 230-400 меш диоксида кремния, купленного у фирмы ЕМ Science. Стандартные 1H- и 13С-ЯМР-спектры получали на спектрометре Varian Unity при частотах 300, 132 МГц и 75,033 МГц соответственно. ЯМР-спектры относили к TMS (0 м.д.) или к пику изотопной примеси CDCl3 (7,26 и 77,0 м.д. для 1H и 13С соответственно). Масс-спектроскопию высокого разрешения с ионизацией электронным ударом выполняли в Центре масс-спектроскопии при Университете Техаса в Austin.
ПРИМЕР 2
Синтез и характеристика новых соединений токоферола
2,5,7,8-тетраметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)уксусная кислота (1)

Раствор R,R,R-α-токоферола (0,5 г, 1,16 ммоль) в N,N-диметилформамиде (20 мл) обрабатывали метилбромацетатом (3,4 г, 8,3 ммоль) и избытком порошкообразного NaOH (1,2 г, 30 ммоль). Полученную желтую суспензию интенсивно перемешивали в течение 24 часов при комнатной температуре. Реакцию подкисляли 5 н. НСl и экстрагировали диэтиловым эфиром (3×30 мл). Объединенные эфирные слои промывали H2O (3×30 мл) и солевым раствором (1×30 мл) и затем сушили над Na2SO4. Эфирный раствор концентрировали до желтого масла, которое очищали хроматографией на силикагеле с элюированием 19% (об/об) EtOAc и 2%-ной уксусной кислотой в гексанах. Полученную желтую жидкость растворяли в диэтиловом эфире (30 мл), промывали H2O (3×20 мл) и солевым раствором (1×20 мл) и затем сушили над Na2SO4. Полученный раствор концентрировали до светло-желтого масла и сушили в вакууме в течение 48 часов. Это давало соединение 1 в виде воскообразного твердого вещества не чисто белого цвета (0,50 г, 88%). 1H-ЯМР (CDCl3/TMS, м.д.): 0,87 (м, 12Н, 4а'-, 8а'-, 12а'-, 13'-СН3), 1,0-1,6 (м, 24Н, 4'-, 8'-, 12'-СН, 1'-, 2'-, 3'-, 5'-, 6'-, 7'-, 9'-, 10'-, 11'-CH2, 2а-СН3), 1,81 (м, 2Н, 3-СН2), 2,07, 2,14, 2,16 (3×с, 9Н, 5а-, 7а-, 8а-СН3), 2,59 (т, J=6,6 Гц, 2Н, 4-СН2), 4,34 (с, 2Н, ОСН2); 13С-ЯМР (СDСl3, м.д.): 11,7, 11,8, 12,7 (5а-, 7а-, 8а-СН3), 19,6, 19,7 (СН3), 20,6, 21,0 (СН2), 22,6, 22,7 (СН3), 23,8 (2а-СН3), 24,4, 24,8 (СН2), 28,0 (СН), 31,2 (3-CH2), 32,7, 32,8, (СН), 37,3, 37,4, 37,5, 39,4, 40,0 (СН2), 69,2 (ОСН2), 75,0 (2-С), 117,8, 123,2, 125,4, 127,3 (арил С), 147,0, 148,5 (арил С-O), 173,7 (СООН); HRMS (CI, m/z): 489, 394374 (М+Н+, Рассчитано для С31Н53O4 489,394386). Все отнесения подтверждали с использованием HMQC, DEPT-135 и 1H-NOSEY.
2,5,7,8-тетраметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)пропионовая кислота (2)
Соединения 2-6 синтезируют способом, идентичным синтезу 1, с использованием подходящих бромалкановых кислот.

(Выход 89%). 1H-ЯМР (CDCl3/TMS, м.д.): 0,87 (м, 12Н, 4а'-, 8а'-, 12а'-, 13'-СН3), 1,0-1,6 (м, 24Н, 4'-, 8'-, 12'-СН, 1'-, 2'-, 3'-, 5'-, 6'-, 7'-, 9'-, 10'-, 11'-СН2, 2а-СН3), 1,81 (м, 2Н, 3-СН2), 2,09, 2,14, 2,19 (3×с, 9Н, 5а-, 7а-, 8а-СН3), 2,59 (т, J=6,6 Гц, 2Н, 4-СН2), 2,85 (т, J=6,4 Гц, 2Н, СН2СООН), 3,96 (т, J=6,4 Гц, 2Н, ОСН2); 13С-ЯМР (СDСl3, м.д.): 11,7, 11,8, 12,7 (5а-, 7а-, 8а-СН3), 19,6, 19,7 (СН3), 20,6, 21,0 (СН2), 22,6, 22,7 (СН3), 23,8 (2а-СН3), 24,4, 24,8 (СН2), 28,0 (СН), 31,2 (3-СН2), 32,7, 32,8, (СН), 37,3, 37,4, 37,5, 39,4, 40,0 (СН2), 67,5 (ОСН2), 74,8 (2-C), 117,8, 122,2, 125,8, 127,8 (арил С), 147,6, 148,5 (арил С-O), 177,1 (СООН); HRMS (CI, m/z): 503,408610 (М+H+, Рассчитано для С32Н55O4 503,410036).
2,5,7,8-тетраметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)масляная кислота (3)

(Выход 85%). 1H-ЯМР (CDCl3/TMS, м.д.): 0,87 (м, 12Н, 4а'-, 8а'-, 12а'-, 13'-СН3), 1,0-1,6 (м, 26Н, 4'-,8'-,12'-СН, 1'-, 2'-, 3'-, 5'-, 6'-, 1'-, 9'-, 10'-, 11'-СН2, 2а-СН3), 1,81 (м, 2Н, 3-СН2), 2,14, 2,17, 2,21 (3×с, 9Н, 5а-, 7а-, 8а-СН3), 2,62 (т, J=6,6 Гц, 2Н, 4-СН2), 2,72 (т, J=7,2 Гц, 2Н, СН2СООН), 3,74 (т, J=6,1 Гц, 2Н, ОСН2); 13С-ЯМР (CDCl3, м.д.): 11,7, 11,8, 12,7 (5а-, 7а-, 8а-СН3), 19,6, 19,7 (СН3), 20,6, 21,0 (СН2), 22,6, 22,7 (СН3), 23,9 (2а-СН3), 24,4, 24,8 (СН2), 28,0 (СН), 30,09, 31,2 (3-СН2), 32,7, 32,8, (СН), 37,3, 37,4, 37,5, 39,4, 40,0 (CH2), 71,3 (ОСН2), 74,8 (2-С), 117,5, 122,7, 125,8, 127,7 (арил С), 147,8, 147,9 (арил С-O), 178,9 (СООН); HRMS (CI, m/z): 516,424374 (M+H+, Рассчитано для С33Н57O4 516,424386).
2,5,7,8-тетраметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)валериановая кислота (4)

(Выход 90%). 1H-ЯМР (CDCl3/TMS, м.д.): 0,87 (м, 12Н, 4а'-, 8а'-, 12а'-, 13'-СН3), 1,0-1,6 (м, 28Н, 4'-,8'-,12'-СН, 1'-, 2'-, 3'-, 5'-, 6'-, 7'-, 9'-, 10'-, 11'-СН2, 2а-СН3), 1,81 (м, 2Н, 3-СН2), 2,09, 2,14, 2,18 (3×с, 9Н, 5а-, 7а-, 8а-СН3), 2,49 (т, J=6,8 Гц, 2Н, СН2СООН), 2,59 (т, J=6,6 Гц, 2Н, 4-СН2), 3,68 (т, J=5,5 Гц, 2Н, ОСН2); 13С-ЯМР (CDCl3, м.д.): 11,7, 11,8, 12,7 (5а-, 7а-, 8а-СН3), 19,6, 19,7 (СН3), 20,6, 21,0, 21,4 (СН2), 22,6, 22,7 (СН3), 23,8 (2а-СН3), 24,4, 24,8 (СН2), 28,0 (СН), 30,0 (СН2), 31,2 (3-СН2), 32,7, 32,8, (СН), 35,8, 37,3, 37,4, 37,5, 39,4, 40,0 (СН2), 72,2 (ОСН2), 74,9 (2-С), 117,8, 123,2, 125,4, 127,3 (арил С), 147,6, 148,3 (арил С-O), 178,7 (СООН); HRMS (CI, m/z): 530,433514 (M+H+, Рассчитано для С34Н59O4 530,433516).
2,5,7,8-тетраметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)гексановая кислота (5)
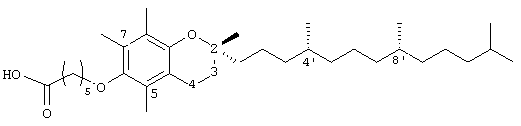
(Выход 77%). 1H-ЯМР (СDСl3/ТМS, м.д.): 0,87 (м, 12Н, 4а'-, 8а'-, 12а'-, 13-СН3), 1,0-1,6 (м, 30Н, 4'-, 8'-, 12'-СН, 1'-, 2'-, 3'-, 5'-, 6'-, 7'-, 9'-, 10'-, 11'-CH2, 2а-СН3), 1,81 (м, 2Н, 3-СН2), 2,08, 2,12, 2,16 (3×с, 9Н, 5а-, 7а-, 8а-СН3), 2,32 (т, J=6,5 Гц, 2Н, СН2СООН), 2,57 (т, J=6,6 Гц, 2Н, 4-СН2), 3,64 (т, J=5,5 Гц, 2Н, ОСН2); 13С-ЯМР (СDСl3, м.д.): 11,8, 11,9, 12,7 (5а-, 7а-, 8а-СН3), 19,6, 19,7 (СН3), 20,6, 21,0 (СН2), 22,6, 22,7 (СН3), 23,8 (2а-СН3), 24,4, 24,6, 24,8, 25,7 (СН2), 28,0 (СН), 30,0 (СН2), 31,3 (3-СН2), 32,7, 32,8, (СН), 34,08, 37,3, 37,3, 37,4, 39,3, 40,0 (СН2), 72,6 (ОСН2), 74,7 (2-С), 117,4, 122,7, 125,4, 127,8 (арил С), 147,6, 148,2 (арил С-O), 179,6 (СООН); HRMS (CI, m/z): 545,457026 (М+Н+, Рассчитано для С35Н61O4 545,456986).
2,5,7,8-тетраметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)октановая кислота (6)

(Выход 91%). 1Н-ЯМР (CDCl3/TMS, м.д.): 0,87 (м, 12Н, 4а'-, 8а'-, 12а'-, 13'-СН3), 1,0-1,6 (м, 34Н, 4'-, 8'-, 12'-СН, 1'-, 2'-, 3'-, 5'-, 6'-, 7'-, 9'-, 10'-, 11'-CH2, 2а-СН3), 1,81 (м, 2Н, 3-СН2), 2,08, 2,11, 2,16 (3×с, 9Н, 5а-, 7а-, 8а-СН3), 2,36 (м, 2Н, СН2СООН), 2,58 (т, J=6,6 Гц, 2Н, 4-СН2), 3,62 (т, J=5,5 Гц, 2Н, ОСН2); 13С-ЯМР (СDСl3, м.д.): 11,7, 11,8, 12,7 (5а-, 7а-, 8а-СН3), 19,6, 19,7 (СН3), 20,6, 21,0 (СН2), 22,6, 22,7 (СН3), 23,8 (2а-СН3), 24,4, 24,6, 24,8, 25,1, 25,7, 26,6 (СН2), 28,0 (СН), 30,0 (СН2), 31,3 (3-СН2), 32,7, 32,8, (СН), 34,08, 37,3, 37,3, 37,4, 39,3, 40,0 (СН2), 72,7 (ОСН2), 74,6 (2-С), 117,6, 122,8, 125,4, 127,6 (арил С), 147,5, 148,2 (арил С-O), 179,4 (СООН); HRMS (CI, m/z): 573,484396 (М+Н+, Рассчитано для С37Н65O4 573,488286).
2,5,8-триметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)уксусная кислота (7)
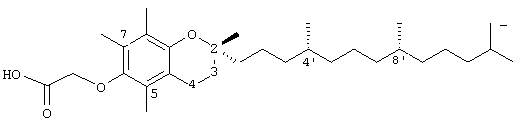
Раствор R,R,R-α-токоферола (75 мг, 0,18 ммоль) в N,N-диметилформамиде (2 мл) обрабатывали метилбромацетатом (0,4 г, 2,8 ммоль) и избытком порошкообразного NaOH (0,5 г, 12,5 ммоль). Полученную желтую суспензию интенсивно перемешивали в течение 24 часов при комнатной температуре. Реакцию подкисляли 5 н. НСl и экстрагировали диэтиловым эфиром (3×10 мл). Объединенные эфирные слои промывали Н2О (3×10 мл) и солевым раствором (1×10 мл) и затем сушили над Na2SO4. Эфирный раствор концентрировали до желтого масла, которое очищали хроматографией на силикагеле с элюированием 19% (об/об) EtOAc и 2%-ной уксусной кислотой в гексанах. Полученную желтую жидкость растворяли в диэтиловом эфире (30 мл), промывали Н2О (3×10 мл) и солевым раствором (1×20 мл) и затем сушили над Na2SO4. Полученный раствор концентрировали до светло-желтого масла и сушили в вакууме в течение 48 часов. Это давало соединение 7 в виде воскообразного твердого вещества не чисто белого цвета (80 мг, 97%). 1H-ЯМР (CDCl3/TMS, м.д.): 0,87 (м, 12Н, 4а'-, 8а'-, 12а'-, 13'-СН3), 1,0-1,6 (м, 24Н, 4'-, 8'-, 12'-CH, 1'-, 2'-, 3'-, 5'-, 6'-, 7'-, 9'-, 10'-, 11'-СН2, 2а-СН3), 1,81 (м, 2Н, 3-СН2), 2,08, 2,12, 2,16 (2×с, 6Н, 5а-, 8а-СН3), 2,61 (т, J=6,6 Гц, 2Н, 4-СН2), 4,59 (с, 2Н, ОСН2), 6,53 (с, 1Н, арил СН); 13С-ЯМР (CDCl3, м.д.): 11,2, 16,1 (5а-, 8а-СН3), 19,6, 19,7 (СН3), 20,7, 21,0 (СН2), 22,6, 22,7 (СН3), 23,8 (2а-СН3), 24,4, 24,8 (СН2), 27,9 (СН), 31,3 (3-СН2), 32,7, 32,8, (СН), 37,2, 37,4, 37,5, 39,4, 40,0 (СН2), 66,8 (ОСН2), 74,8 (2-С), 113,8, 120,7, 123,1, 127,3 (арил С), 147,1, 148,2 (арил С-O), 175,3 (СООН); HRMS (CI, m/z): 475,377840 (M+H+, Рассчитано для С30Н51O4 475,378736).
2,7,8-триметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)уксусная кислота (8)

Раствор R,R,R-α-токоферола (100 мг, 0,24 ммоль) в N,N-диметилформамиде (5 мл) обрабатывали метилбромацетатом (1,1 г, 7,4 ммоль) и избытком порошкообразного NaOH (1,0 г, 25 ммоль). Полученную желтую суспензию интенсивно перемешивали в течение 24 часов при комнатной температуре. Реакцию подкисляли 5 н. НСl и экстрагировали диэтиловым эфиром (3×10 мл). Объединенные эфирные слои промывали Н2О (3×10 мл) и солевым раствором (1×10 мл) и затем сушили над Na2SO4. Эфирный раствор концентрировали до желтого масла, которое очищали хроматографией на силикагеле с элюированием 19% (об/об) EtOAc и 2%-ной уксусной кислотой в гексанах. Полученную желтую жидкость растворяли в диэтиловом эфире (30 мл), промывали Н2О (3×10 мл) и солевым раствором (1×10 мл) и затем сушили над Na2SO4. Полученный раствор концентрировали до светло-желтого масла и сушили в вакууме в течение 48 часов. Это давало соединение 8 в виде воскообразного твердого вещества не чисто белого цвета (110 мг, 97%). 1H-ЯМР (CDCl3/TMS, м.д.): 0,87 (м, 12Н, 4а'-, 8а'-, 12а'-, 13'-СН3), 1,0-1,6 (м, 24Н, 4'-, 8'-, 12'-СН, 1'-, 2'-, 3'-, 5'-, 6'-, 7'-, 9'-, 10'-, 11'-СН2, 2а-СН3), 1,81 (м, 2Н, 3-СН2), 2,12, 2,19 (2×с, 6Н, 7а-, 8а-СН3), 2,61 (т, J=6,6 Гц, 2Н, 4-СН2), 4,59 (с, 2Н, ОСН2), 6,39 (с, 1Н, арил СН); 13С-ЯМР (СDСl3, м.д.): 11,9, 12,0 (7а-, 8а-СН3), 19,6, 19,7 (СН3), 20,7, 21,0 (CH2), 22,6, 22,7 (СН3), 23,8 (2а-СН3), 24,4, 24,8 (СН2), 27,9 (СН), 31,2 (3-СН2), 32,7, 32,8, (СН), 37,2, 37,4, 37,5, 39,4, 40,0 (СН2), 66,6 (ОСН2), 75,7 (2-С), 110,68, 117,7, 125,0, 126,3 (арил С), 146,9, 148,7 (арил С-O), 175,0 (СООН); HRMS (CI, m/z): 475,377962 (M+H+, Рассчитано для С30Н51O4 475,378736).
2,8-диметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)уксусная кислота (9)
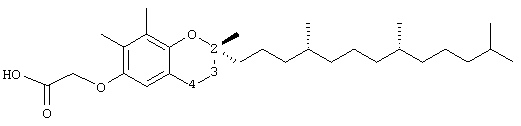
Раствор R,R,R-α-токоферола (100 мг, 0,25 ммоль) в N,N-диметилформамиде (5 мл) обрабатывали метилбромацетатом (1,1 г, 7,4 ммоль) и избытком порошкообразного NaOH (1,0 г, 25 ммоль). Полученную желтую суспензию интенсивно перемешивали в течение 24 часов при комнатной температуре. Реакцию подкисляли 5 н. НСl и экстрагировали диэтиловым эфиром (3×10 мл). Объединенные эфирные слои промывали Н2О (3×10 мл) и солевым раствором (1×10 мл) и затем сушили над Na2SO4. Эфирный раствор концентрировали до желтого масла, которое очищали хроматографией на силикагеле с элюированием 19% (об/об) EtOAc и 2%-ной уксусной кислотой в гексанах. Полученную желтую жидкость растворяли в диэтиловом эфире (30 мл), промывали Н2О (3×10 мл) и солевым раствором (1×10 мл) и затем сушили над Na2SO4. Полученный раствор концентрировали до светло-желтого масла и сушили в вакууме в течение 48 часов. Это давало соединение 9 в виде воскообразного твердого вещества не чисто белого цвета (111 мг, 98%). 1H-ЯМР (CDCl3/TMS, м.д.): 0,87 (м, 12Н, 4а'-, 8а'-, 12а'-, 13'-СН3), 1,0-1,6 (м, 24Н, 4'-, 8'-, 12'-СН, 1'-, 2'-, 3'-, 5'-, 6'-, 7'-, 9'-, 10'-, 11'-CH2, 2а-СН3), 1,81 (м, 2Н, 3-СН2), 2,15 (с, 3Н, 7а-, 8а-СН3), 2,71 (т, J=6,6 Гц, 2Н, 4-СН2), 4,59 (с, 2Н, ОСН2), 6,48 (д, J=3,0 Гц, 1Н, арил СН), 6,61 (в, 1Н, J=3,0 Гц, 1Н, арил СН); 13С-ЯМР (CDCl3, м.д.): 16,2 (8а-СН3), 19,6, 19,7 (СН3), 21,0 (СН2), 22,6, 22,7 (СН3), 24,0 (2а-СН3), 24,4, 24,8 (СН2), 27,9 (СН), 31,2 (3-CH2), 32,7, 32,8, (СН), 37,2, 37,4, 37,5, 39,4, 40,0 (СН2), 66,7 (OCH2), 75,8 (2-С), 112,3, 115,6, 121,3, 127,5 (арил С), 149,9 (арил С-O), 174,8 (СООН); HRMS (CI, m/z): 460,355022 (M+H+, Рассчитано для С30Н51O4 460, 355262).
2,5,7,8-тетраметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)ацетамид (10)

Раствор соединения 1 (0,1 г, 0,2 ммоль) в СН2Сl2 (5 мл) обрабатывали N-гидроксисукцинимидом (26 мг, 0,23 ммоль) и дициклогексилкарбодиимидом (46 мг, 0,23 ммоль). Спустя 2 минуты образовался белый осадок. Полученную суспензию перемешивали в течение 2 часов. Реакцию перемешивали в течение еще 6 часов. Реакционную смесь охлаждали до -30°С и фильтровали. Фильтрат концентрировали и полученное бесцветное масло очищали хроматографией на силикагеле с элюированием EtOAc (35%, об/об) в гексанах. Это давало белое твердое вещество (75 мг, 76%). 1Н-ЯМР (CDCl3/TMS, м.д.): 0,87 (м, 12Н, 4а'-, 8а'-, 12а'-, 13'-СН3), 1,0-1,6 (м, 24Н, 4'-, 8'-, 12'-СН, 1'-, 2'-, 3'-, 5'-, 6'-, 7'-, 9'-, 10'-, 11'-CH2, 2а-СН3), 1,81 (м, 2Н, 3-СН2), 2,10, 2,12, 2,16 (3×с, 9Н, 5а-, 7а-, 8а-СН3), 2,59 (т, J=6,6 Гц, 2Н, 4-СН2), 4,19 (с, 2Н, OCH2), 6,36-6,92 (2×шир., 2Н, NH); 13С-ЯМР (СDСl3, м.д.): 11,7, 11,8, 12,7 (5а-, 7а-, 8а-СН3), 19,6, 19,7 (СН3), 20,6, 21,0 (СН2), 22,6, 22,7 (СН3), 23,8 (2а-СН3), 24,4, 24,8 (СН2), 28,0 (СН), 31,2 (3-СН2), 32,7, 32,8, (СН), 37,3, 37,4, 37,5, 39,4, 40,0 (СН2), 70,9 (ОСН2), 74,9 (2-С), 117,8, 123,3, 125,4, 127,3 (арил С), 146,5, 148,4 (арил С-O), 172,1 (СООН); HRMS (CI, m/z): 488,409341 (М+H+, Рассчитано для С31Н54NО3 488,410370).
Метил-2,5,7,8-тетраметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)ацетат (11)

Раствор соединения 1 (0,1 г, 0,2 ммоль) в СН2Сl2 (5 мл) обрабатывали N,N-диметиламинопиридином (26 мг, 0,23 ммоль), метанолом (1 мл) и дициклогексилкарбодиимидом (46 мг, 0,23 ммоль). Спустя 2 минуты образовался белый осадок. Полученную суспензию перемешивали в течение 6 часов. Реакционную смесь охлаждали до -30°С и фильтровали. Фильтрат концентрировали и полученное бесцветное масло очищали хроматографией на силикагеле с элюированием EtOAc (40%, об/об) в гексанах. Это давало белое твердое вещество (82 мг, 80%). 1H-ЯМР (CDCl3/TMS, м.д.): 0,87 (м, 12Н, 4а'-, 8а'-, 12а'-, 13'-СН3), 1,0-1,6 (м, 24Н, 4'-, 8'-, 12'-СН, 1'-, 2'-, 3'-, 5'-, 6'-, 7'-, 9'-, 10'-, 11'-CH2, 2а-СН3), 1,81 (м, 2Н, 3-СН2), 2,10, 2,16, 2,20 (3×с, 9Н, 5а-, 7а-, 8а-СН3), 2,59 (т, J=6,6 Гц, 2Н, 4-СН2), 3,85 (с, 3Н, ОСН3), 4,32 (с, 2Н, ОСН2); 13С-ЯМР (CDCl3, м.д.): 11,7, 11,8, 12,7 (5а-, 7а-, 8а-СН3), 19,6, 19,7 (СН3), 20,6, 21,0 (СН2), 22,6, 22,7 (СН3), 23,8 (2а-СН3), 24,4, 24,8 (СН2), 28,0 (СН), 31,2 (3-СН2), 32,7, 32,8, (СН), 37,3, 37,4, 37,5, 39,4, 40,0 (СН2), 50,2 (ОСН2), 69,8 (ОСН2), 74,9 (2-С), 117,6, 123,0, 125,6, 127,5 (арил С), 147,6, 148,2 (арил С-O), 172,1 (СООН); HRMS (CI, m/z): 503,408411 (М+Н+, Рассчитано для С32Н55O4 503,410036).
2-(N,N-(карбоксиметил)-2-(2,5,7,8-тетраметил-(2R(4R,8R,12-триметилтридецил)хроман-6-илокси)уксусная кислота

Раствор соединения 1 (0,2 г, 0,4 ммоль) в СН2Сl2 (5 мл) обрабатывали диэтилиминодиацетатом (77 мг, 0,4 ммоль) и 0-7-азабензотриазол-1-ил-N,N,N',N'-тетраметилуронийгексафторфосфатом (HATU) (46 мг, 0,23 ммоль). Спустя 12 часов реакционную смесь концентрировали до пасты и затем очищали хроматографией на силикагеле с элюированием EtOAc (30%, об/об) в гексанах. Это давало целевой промежуточный диэфирный продукт в виде бесцветного масла (150 мг, 55%). 1H-ЯМР (CDCl3/TMS, м.д.): 0,87 (м, 12Н, 4а'-, 8а'-, 12а'-, 13'-СН3), 1,0-1,6 (м, 30Н, 4'-, 8'-, 12'-СН, 1'-, 2'-, 3'-, 5'-, 6'-, 7'-, 9'-, 10'-, 11'-CH2, 2а-СН3), 1,78 (м, 2Н, 3-СН2), 2,08, 2,13, 2,17 (3×с, 9Н, 5а-, 7а-, 8а-СН3), 2,58 (т, J=6,8 Гц, 2Н, 4-СН2), 4,19 (к, J=7,4 Гц, 4Н, ОСН2), 4,30 (3×с, 6Н, ОСН2); 13С-ЯМР (СDСl3, м.д.): 11,7, 11,8, 12,7 (5а-, 7а-, 8а-СН3), 14,0 (СН3), 19,6, 19,7 (СН3), 20,6, 21,0 (СН2), 22,6, 22,7 (СН3), 23,8 (2а-СН3), 24,4, 24,8 (СН2), 28,0 (СН), 31,2 (3-СН2), 32,7, 32,8, (СН), 37,3, 37,4, 37,5, 39,4, 40,0 (СН2), 48,1, 49,4 (NCH2), 61,2, 61,5 (ОСН2), 71,8 (ОСН2), 74,8 (2-С), 117,5, 122,9, 125,6, 127,4 (арил С), 148,0, 148,1 (арил С-O), 168,8, 169,0 (CO); MS (CI, m/z): 660 (М+Н+, Рассчитано для С39Н65NO7 659,47610).
Раствор диэфирного промежуточного продукта (0,15 г, 0,23 ммоль) в этаноле (4 мл) обрабатывали 1 н. NaOH (1 мл). Полученную мутную смесь перемешивали при 70°С в течение 15 часов. Реакционную смесь подкисляли 1 н. НСl и этанол удаляли в вакууме. Полученный водный раствор экстрагировали СНСl3 (5×20 мл) и объединенные органические слои сушили над Nа2SO4. Это давало соединение 12 (0,13 г, 52%) в виде белого твердого вещества. 1H-ЯМР (СDСl3/ТМS, м.д.): 0,87 (м, 12Н, 4а'-, 8а'-, 12а'-, 13'-СН3), 1,0-1,6 (м, 24Н, 4'-, 8'-, 12'-СН, 1'-, 2'-, 3'-, 5'-, 6'-, 7'-, 9'-, 10'-, 11'-CH2, 2а-СН3), 1,70 (м, 2Н, 3-СН2), 2,01, 2,05, 2,08 (3×с, 9Н, 5а-, 7а-, 8а-СН3), 2,47 (м, 2Н, 4-СН2), 4,18 (м, 4Н, 2×NCH2), 4,31 (м, 2Н, ОСН2); 13С-ЯМР (СDСl3, м.д.): 11,5, 11,6, 12,4 (5а-, 7а-, 8а-СН3), 19,4, 19,5 (СН3), 20,6, 21,0 (СН2), 22,6, 22,7 (СН3), 23,8 (2а-СН3), 24,4, 24,8 (СН2), 28,0 (СН), 31,2 (3-CH2), 32,4, 32,5, (СН), 37,0, 37,2, 37,5, 39,1, 40,0 (СН2), 48,1, 49,4 (NCH2), 71,1 (ОСН2), 71,1 (ОСН2), 74,8 (2-С), 117,5, 122,9, 125,4, 127,2 (арил С), 147,8, 148,1 (арил С-O), 168,8, 169,0 (CO); HRMS (CI, m/z): 604,420882 (М+Н+, Рассчитано для С35H58NO7 604,421329).
2-(2,5,7,8-тетраметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)этан-1-ол (13)

Раствор R,R,R-α-токоферола (0,5 г, 1,16 ммоль) в N,N-диметилформамиде (20 мл) обрабатывали иодэтанолом (1,7 г, 10 ммоль) и избытком порошкообразного NaOH (1,0 г, 25 ммоль). Полученную желтую суспензию интенсивно перемешивали в течение 24 часов при комнатной температуре. Реакцию подкисляли 5 н. НСl и экстрагировали диэтиловым эфиром (3×30 мл). Объединенные эфирные слои промывали Н2О (3×30 мл) и солевым раствором (1×30 мл) и затем сушили над Na2SO4. Эфирный раствор концентрировали до желтого масла, которое очищали хроматографией на силикагеле с элюированием 19% (об/об) EtOAc и 2%-ной уксусной кислотой в гексанах. Полученную желтую жидкость растворяли в диэтиловом эфире (30 мл), промывали Н2O (3×20 мл) и солевым раствором (1×20 мл) и затем сушили над Na2SO4. Полученный раствор концентрировали до светло-желтого масла и сушили в вакууме в течение 48 часов. Это давало соединение 13 в виде желтого масла (0,40 г, 73%). 1H-ЯМР (CDCl3/TMS, м.д.): 0,87 (м, 12Н, 4а'-, 8а'-, 12а'-, 13'-СН3), 1,0-1,6 (м, 24Н, 4'-, 8'-, 12'-СН, 1'-, 2'-, 3'-, 5'-, 6'-,7'-, 9'-, 10'-, 11'-CH2, 2а-СН3), 1,81 (м, 2Н, 3-CH2), 2,07, 2,14, 2,16 (3×с, 9Н, 5а-, 7а-, 8а-СН3), 2,59 (т, J=6,6 Гц, 2Н, 4-СН2), 3,79 (м, 2Н, ОСН2), 3,94 (м, 2Н, ОСН2); 13С-ЯМР (СDСl3, м.д.): 11,7, 11,8, 12,7 (5а-, 7а-, 8а-СН3), 19,6, 19,7 (СН3), 20,6, 21,0 (СН2), 22,6, 22,7 (СН3), 23,8 (2а-СН3), 24,4, 24,8 (CH2), 28,0 (СН), 31,2 (3-СН2), 32,7, 32,8, (СН), 37,3, 37,4, 37,5, 39,4, 40,0 (СН2), 63,1, 69,2 (OCH2), 75,0 (2-С), 117,8, 123,4, 128,3 (арил С), 149,2, 149,5 (арил С-O); MS (CI, m/z): 475 (М+H+, Рассчитано для С31Н54O3 474,40729).
2-(2,5,7,8-пентаметил-хроман-6-илокси)уксусная кислота (14)

Раствор 2-(2,5,7,8-пентаметил-6-хроманола (0,3 г, 1,36 ммоль) в N,N-диметилформамиде (20 мл) обрабатывали метилбромацетатом (0,8 г, 5,3 ммоль) и избытком порошкообразного NaOH (0,7 г, 18 ммоль). Полученную желтую суспензию интенсивно перемешивали в течение 24 часов при комнатной температуре. Реакцию подкисляли 5 н. НСl и экстрагировали диэтиловым эфиром (3×30 мл). Объединенные эфирные слои промывали Н2O (3×30 мл) и солевым раствором (1×30 мл) и затем сушили над Na2SO4. Эфирный раствор концентрировали до желтого масла, которое очищали хроматографией на силикагеле с элюированием 30% (об/об) EtOAc и 2%-ной уксусной кислотой в гексанах. Полученную желтую жидкость растворяли в диэтиловом эфире (30 мл), промывали Н2О (3×20 мл) и солевым раствором (1×20 мл) и затем сушили над Na2SO4. Полученный раствор концентрировали до светло-желтого масла и сушили в вакууме в течение 48 часов. Это давало соединение 14 в виде белого твердого вещества (0,31 г, 82%). 1H-ЯМР (CDCl3/TMS, м.д.): 1,31 (с, 6Н, СН3), 1,81 (т, J=7,8 Гц, 3-СН2), 2,10, 2,16, 2,19 (3×с, 9Н, 5а-, 7а-, 8а-СН3), 2,61 (т, J=7,8 Гц, 2Н, 4-СН2), 4,39 (с, 2Н, ОСН2); 13С-ЯМР (CDCl3, м.д.): 11,7, 11,8, 12,7 (5а-, 7а-, 8а-СН3), 20,9, 26,8, 32,7 (алкил), 69,1 (ОСН2), 72,9 (2-С), 117,5, 123,2, 125,5, 127,3 (арил), 147,0, 148,6 (O-арил); HRMS (CI, m/z): 279,159238 (М+H+, Рассчитано для С16Н23О4 279,159634).
2,5,7,8-тетраметил-(2RS-(4RS,8RS,12-триметилтридецил)хроман-6-илокси)уксусная кислота (15)
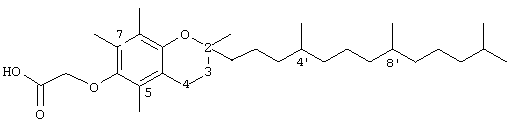
Раствор полностью рацемического -α-токоферола (0,5 г, 1,16 ммоль) в N,N-диметилформамиде (20 мл) обрабатывали метилбромацетатом (3,4 г, 8,3 ммоль) и избытком порошкообразного NaOH (1,2 г, 30 ммоль). Полученную желтую суспензию интенсивно перемешивали в течение 24 часов при комнатной температуре. Реакцию подкисляли 5 н. НСl и экстрагировали диэтиловым эфиром (3×30 мл). Объединенные эфирные слои промывали Н2O (3×30 мл) и солевым раствором (1×30 мл) и затем сушили над Na2SO4. Эфирный раствор концентрировали до желтого масла, которое очищали хроматографией на силикагеле с элюированием 19% (об/об) EtOAc и 2%-ной уксусной кислотой в гексанах. Полученную желтую жидкость растворяли в диэтиловом эфире (30 мл), промывали Н2О (3×20 мл) и солевым раствором (1×20 мл) и затем сушили над Nа2SO4. Полученный раствор концентрировали до светло-желтого масла и сушили в вакууме в течение 48 часов. Это давало соединение 15 в виде воскообразного твердого вещества не чисто белого цвета (80%). 1H-ЯМР (CDCl3/TMS, м.д.): 0,88 (м, 12Н, 4а'-, 8а'-, 12а'-, 13'-СН3), 1,0-1,6 (м, 24Н, 4'-, 8'-, 12'-СН, 1'-, 2'-, 3'-, 5'-, 6'-, 7'-, 9'-, 10'-, 11'-СН2, 2а-СН3), 1,84 (м, 2Н, 3-СН2), 2,07, 2,14, 2,16 (3×с, 9Н, 5а-, 7а-, 8а-СН3), 2,61 (т, J=6,6 Гц, 2Н, 4-СН2), 4,34 (с, 2Н, ОСН2); 13С-ЯМР (СDСl3, м.д.): 11,5, 11,7, 12,6 (5а-, 7а-, 8а-СН3), 19,6, 19,7 (СН3), 20,6, 21,3 (CH2), 22,6, 22,8 (СН3), 23,8 (2а-СН3), 24,5, 24,9 (СН2), 29,0 (СН), 31,6 (3-СН2), 32,7, 32,8, (СН), 37,5, 37,8, 37,9, 39,5, 41,0 (СН2), 69,3 (OCH2), 75,1 (2-С), 117,9, 123,3, 125,5, 127,3 (арил С), 147,0, 148,0 (арил С-O), 173,9 (СООН); HRMS (CI, m/z): 489, 394375 (М+Н+, Рассчитано для С31Н53O4 489,394383).
2,5,7,8-тетраметил-(2R-(карбокси)хроман-6-ил))уксусная кислота (16)

Раствор (-)-(R)-6-гидрокси-2,5,7,8-тетраметилхроман-2-карбоновой кислоты (0,34 г, 1,36 ммоль) в N,N-диметилформамиде (20 мл) обрабатывали метилбромацетатом (0,8 г, 5,3 ммоль) и избытком порошкообразного NaOH (0,7 г, 18 ммоль). Полученную желтую суспензию интенсивно перемешивали в течение 24 часов при комнатной температуре. Реакцию подкисляли 5 н. НСl и экстрагировали диэтиловым эфиром (3×30 мл). Объединенные эфирные слои промывали Н2O (3×30 мл) и солевым раствором (1×30 мл) и затем сушили над Na2SO4. Эфирный раствор концентрировали до желтого масла, которое очищали хроматографией на силикагеле с элюированием 30% (об/об) EtOAc и 2%-ной уксусной кислотой в гексанах. Полученную желтую жидкость растворяли в диэтиловом эфире (30 мл), промывали Н2О (3×20 мл) и солевым раствором (1×20 мл) и затем сушили над Na2SO4. Полученный раствор концентрировали до светло-желтого масла и сушили в вакууме в течение 48 часов. Это давало соединение 16 в виде белого твердого вещества 0,33 г, 80%). 1H-ЯМР (CDCl3/TMS, м.д.): 1,52 (с, 3Н, 2а-СН3), 2,10 (м, 2Н, 3-СН2), 2,12, 2,16, 2,19 (3×с, 9Н, 5а-, 7а-, 8а-СН3), 2,56 (т, J=6,5 Гц, 2Н, 4-СН2), 4,36 (с, 2Н, ОСН2).
2,5,7,8-тетраметил-2В-(2RS,6RS,10-триметилундецил)хроман-6-илокси)уксусная кислота (17)
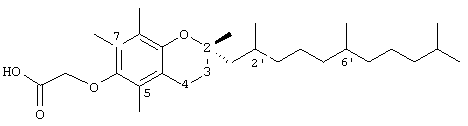
Раствор 10 г (40 ммоль)) (-)-(S)-6-гидрокси-2,5,7,8-тетраметилхроман-2-карбоновой кислоты и моногидрата 0,5 г п-толуолсульфоновой кислоты в 200 мл метанола перемешивали и нагревали с обратным холодильником при дефлегмации в течение 4 часов. После охлаждения раствор разбавляли водой и экстрагировали диэтиловым эфиром. Объединенные эфирные слои промывали насыщенным водным раствором бикарбоната натрия, Н2O и солевым раствором (1×30 мл) и затем сушили над Na2SO4. Полученный раствор концентрировали и сушили в вакууме в течение 48 часов. Это давало 10 г (95%) метил-(-)-(S)-6-гидрокси-2,5,7,8-тетраметилхроман-2-карбоксилата в виде бесцветного твердого вещества, которое использовали без дополнительной очистки. 1Н-ЯМР (CDCl2/TMS, м.д.): 1,52 (с, 3Н, 2а-СН3), 2,10 (м, 2Н, 3-СН2), 2,12, 2,16, 2,19 (3×с, 9Н, 5а-, 7а-, 8а-СН3), 2,56 (т, J=6,5 Гц, 2Н, 4-СН2), 3,55 (с, 3Н, ОСН3); MS (CI, m/z): 264,422 (М+H+, Рассчитано для С15Н20O4 265,3224).
К раствору 2 г (7,58 ммоль) этого эфира в 7,5 мл N,N-диметилформамида (ДМФ) добавляли 2,6 г (18,8 ммоль) безводного гранулярного карбоната калия с последующим добавлением 2,3 мл (20 ммоль) бензилхлорида. Полученную суспензию перемешивали при комнатной температуре в течение 41 часа, затем выливали в 50 мл воды и обрабатывали эфиром обычным образом. Продукт освобождали от избытка бензилхлорида при 50°С под высоким вакуумом. Получали 2,69 г (100%) чистого (ТСХ) метилового эфира (-)-(S)-бензилокси-2,5,7,8-тетраметилхроман-2-карбоновой кислоты в виде желтого твердого вещества, т.пл. 102-106°С. Аналитический образец этого соединения готовили в виде бесцветного твердого вещества с т.пл. 108-109°С (из смеси эфир/метанол). 1H-ЯМР (CDCl2/TMS, м.д.): 1,54 (с, 3Н, 2а-СН3), 2,01 (м, 2Н, 3-СН2), 2,14, 2,17, 2,19 (3×с, 9Н, 5а-, 7а-, 8а-СН3), 2,51 (т, J=6,7 Гц, 2Н, 4-СН2), 3,64 (с, 3Н, ОСН3), 5,12 (с, 2Н, 6-ОСН2), 7,15 (м, 5Н, ArH); MS (CI, m/z): 355,232 М+Н+, Рассчитано для С22H25О4 354,448.
Раствор 3,54 г (10 ммоль) вышеуказанного алкоксиэфира (ether ester) в 20 мл толуола и 10 мл СН2Сl2 перемешивали с охлаждением на бане со смесью сухой лед/ацетон с одновременным добавлением по каплям 12 мл (18 ммоль) 25% диизобутилалюминийгидрида в толуоле (Texas Alkyls) на протяжении 10 минут. После перемешивания при приблизительно -70°С в течение 30 минут реакционную смесь осторожно разлагали (-70°С) 10 мл МеОН. После добавления 50 мл воды и 50 мл 1 н. водного раствора Н2SО4, смесь нагревали до комнатной температуры и обрабатывали эфиром обычным образом с получением 3,2 г (100%) неочищенного альдегида [(+)-S-6-бензилокси-2,5,7,8-тетраметилхроман-2-карбальдегида] в виде вязкого масла, которое очищали хроматографией на силикагеле с элюированием 19% EtOAc в гексане. 1H-ЯМР (CDCl2/TMS, м.д.): 1,53 (с, 3Н, 2а-СН3), 2,11 (м, 2Н, 3-СН2), 2,24, 2,27, 2,29 (3×с, 9Н, 5а-, 7а-, 8а-СН3), 2,481 (т, J=6,7 Гц, 2Н, 4-СН2), 5,19 (с, 2Н, 6-ОСН2), 7,20 (м, 5Н, АrН), 9,6 (с, 1Н, СНО); MS (CI, m/z): 325,332 М+Н+, Рассчитано для С21H24O3 324,422.
Раствор 9,6 г псевдоионона растворяли в 100 мл 95% этанола; после добавления 0,68 г бор гидрида натрия в этаноле при комнатной температуре эту смесь перемешивали в течение 2 часов и затем оставляли стоять в течение ночи. Смесь добавляли к раствору 2 г гидроксида натрия в 500 мл воды. Смесь экстрагировали эфиром и эфирный экстракт промывали водой, сушили и концентрировали. Дистилляция остаточного масла в вакууме давало бесцветное масло (псевдоионол); т. кип. 112-120°С/5 мм рт.ст. 7,7 г (80%).
К раствору 2,97 г псевдоионола в 10 мл ацетонитрила добавляли при перемешивании и поддержании температуры ниже 30°С 4,53 г гидрохлорида трифенилфосфина, который получали пропусканием сухого хлористого водорода в раствор трифенилфосфина в безводном эфире. После выдерживания этой смеси в течение ночи при комнатной температуре ацетонитрил удаляли при пониженном давлении при температуре ниже 50°С. К остатку добавляли 4,47 г (+)-S-6-бензилокси-2,5,7,8-тетраметилхроман-2-карбальдегида в 15 мл диметилформамида и эту смесь перемешивали. После получения прозрачного раствора к нему добавляли по каплям при перемешивании метилат натрия, полученный из 0,352 г натрия и 7 мл безводного метанола, при температуре ниже 15°С. Реакционная смесь становилась красной в результате образования илида. По завершении добавления перемешивание продолжали в течение 30 минут при 10°С; затем эту смесь постепенно нагревали до 80°С, когда красная окраска исчезала. Продукт выливали в 200 мл 50%-ного водного метанола, сушили и концентрировали в вакууме. Остаточное масло растворяли в 20 мл эфира и добавляли эфирный раствор хлорида ртути до прекращения дальнейшего образования осадка. После фильтрования осадка и промывания фильтрата водой, сушки и концентрирования получали 4,7 г желтого масла. Неочищенную смесь цис- и транс-алкена (MS (CI, m/z): 485,22, М+Н+, Рассчитано для С34Н44O2 484,7255) растворяли в 30 мл этилацетата и добавляли 0,80 г 5% палладия на угле, и эту смесь встряхивали под давлением 40 psi (275,8 кПа) Н2 в течение 30 часов и затем фильтровали через целит и хорошо промывали этилацетатом. Фильтрат концентрировали и очищали хроматографией на силикагеле с элюированием EtOAc в гексане (1:9) с получением 2,5,7,8-тетраметил-(2R-(2RS,6RS,10-триметилундецил))-6-хроманола (выход 60%); 1H-ЯМР (CDCl2/TMS, м.д.): 0,97 (м, 12Н, 2а'-, 6а'-, 10а'-, 11а'-СН3), 1,1-1,7 (м, 20Н, 2а'-, 6а'-, lOa'-CH, 1'-, 3',4'-,5'-,7'-,8'-,9'-СН2 2а-СН3), 1,88 (м, 2Н, 3-СН2), 2,17, 2,19, 2,20 (3×с, 9Н, 5а-, 7а-, 8а-СН3), 2,63 (т, J=6,7 Гц, 2Н, 4-СН2); (MS (CI, m/z): 403,27, М+Н+, Рассчитано для С27Н46O2 402,6632.
Раствор 2,5,7,8-тетраметил-(2R-(2RS,6RS,10-триметилундецил))-6-хроманола (0,466 г, 1,16 ммоль) в N,N-диметил-формамиде (20 мл) обрабатывали метилбромацетатом (3,4 г, 8,3 ммоль) и избытком порошкообразного NaOH (1,2 г, 30 ммоль). Полученную желтую суспензию интенсивно перемешивали в течение 24 часов при комнатной температуре. Реакцию подкисляли 5 н. НСl и экстрагировали диэтиловым эфиром (3×30 мл). Объединенные эфирные слои промывали Н2O (3×30 мл) и солевым раствором (1×30 мл) и затем сушили над Na2SO4. Эфирный раствор концентрировали до желтого масла, которое очищали хроматографией на силикагеле с элюированием 19% (об/об) EtOAc и 2%-ной уксусной кислотой в гексанах. Это давало соединение 17 с выходом 76%. 1H-ЯМР (CDCl2/TMS, м.д.): 0,97 (м, 12Н, 2а'-, 6а'-, 10а'-, 11а'-СН3), 1,2-1,7 (м, 20Н, 2'-, 6'-, 10'-CH, 1'-, 3'-, 4'-, 5'-, 7'-, 8'-, 9'-СН2 2а-СН3), 1,92 (м, 2Н, 3-СН2), 2,18, 2,20, 2,23 (3×с, 9Н, 5а-, 7а-, 8а-СН3), 2,68 (т, J=6,8 Гц, 2Н, 4-СН2), 4,48 (с, 2Н, ОСН2); (MS (CI, m/z): 461,44 М+Н+, Рассчитано для С29Н48O4 460,700.
2,5,7,8-тетраметил-2R-(2,6,10-триметил)-1,3,5,9-E:Z-декатетраен)хроман-6-илокси)уксусная кислота (18)
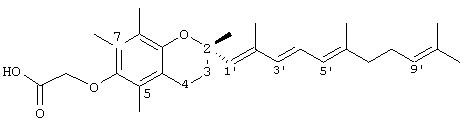
К раствору метил-(-)-(S)-6-гидрокси-2,5,7,8-тетраметилхроман-2-карбоксилата (20 г, 0,075 моль) в 50 мл безводного ДМФ добавляли имидазол (13 г, 0,1911 моль) и третбутилдиметилсилилхлорид (14 г, 0,0933 моль). Смесь перемешивали при 23°С в течение 24 часов и затем обрабатывали эфиром и выливали в 1 н. НСl. Органические экстракты сушили (солевой раствор, Na2SO4) и концентрировали в вакууме. Неочищенный продукт очищали флеш-хроматографией (9:1 гексан:этилацетат) с получением 6-[диметил-(1,1-диметилэтил)-силил]-2,5,7,8-тетраметилхроман-2-карбоксилата (ТВS-защищенный метиловый эфир). 1H-ЯMP (CDCl2/TMS, м.д.): 0,12 (с, 6Н), 1,102 (с, 3Н), 1,18 (с, 3Н), 1,48 (с, 3Н), 1, 645 (с, 3Н), 2,07 (с, 3Н), 2,2 (т, J=6,6 Гц, 2Н), 2,48-2,7 (м, 2Н), и 3,72 (с, 3Н, ОСН3); (MS (CI, m/z): 379,32, М+Н+, Рассчитано для С21Н34O4 378,586.
Раствор 3,78 г (10 ммоль) вышеуказанного сложного алкоксиэфира (ether ester) в 20 мл толуола и 10 мл CH2Cl2 перемешивали при охлаждении на бане со смесью сухой лед/ацетон при добавлении по каплям 12 мл (18 ммоль) 25% диизобутилалюминийгидрида в толуоле (Texas Alkyls) на протяжении 10 минут. После перемешивания при приблизительно -70°С в течение 30 минут реакционную смесь осторожно разлагали (-70°С) 10 мл МеОН. После добавления 50 мл воды и 50 мл 1 н водного раствора H2SO4 смесь нагревали до комнатной температуры и обрабатывали эфиром обычным образом с получением 3,2 г (100%) неочищенного альдегида [(+)-S-6-[диметил-(1,1-диметилэтил)силил]-2,5,7,8-тетраметилхроман-2-карбальдегида] в виде вязкого масла, которое очищали хроматографией на силикагеле с элюированием 19% EtOAc в гексане. Концентрирование этого раствоpa с последующим высушиванием под вакуумом в течение 48 часов давало TBDS-альдегид (78%) в виде твердого вещества с т.пл. 66-68°С. 1H-ЯМР (CDCl2/TMS, м.д.): 0,12 (с, 6Н), 1,1 (с, 9Н), 1,38 (с, 3Н), 1,64 (с, 3Н), 2,12 (с, 3Н), 2,16 (с, 3Н), 2,3-2,2 (м, 2Н), 2,53 (м, 2Н), и 9,82 (д, J=1,4 Гц, 1Н); MS (CI, m/z): 349,40 М+Н+, Рассчитано для С20Н32SiO3 348,560.
К раствору 2,97 г псевдоионола в 10 мл ацетонитрила добавляли при перемешивании и поддержании температуры ниже 30°С 4,53 г гидрохлорида трифенилфосфина, который получали пропусканием сухого хлористого водорода в раствор трифенилфосфина в безводном эфире. После выдерживания этой смеси в течение ночи при комнатной температуре, ацетонитрил удаляли при пониженном давлении при температуре ниже 50°С. К остатку добавляли 4,80 г [(+)-S-6-[диметил-(1,1-диметилэтил)силил]-2,5,7,8-тетраметилхроман-2-карбальдегида] в 15 мл диметилформамида и эту смесь перемешивали. После получения прозрачного раствора к нему добавляли по каплям при перемешивании метилат натрия, полученный из 0,352 г натрия и 7 мл безводного метанола, при температуре ниже 15°С. Реакционная смесь становилась красной в результате образования илида. По завершении добавления перемешивание продолжали в течение 30 минут при 10°С; затем эту смесь постепенно нагревали до 80°С, причем красная окраска исчезала. Продукт выливали в 200 мл 50% водного метанола, сушили и концентрировали в вакууме. Оставшееся масло растворяли в 20 мл эфира и добавляли эфирный раствор хлорида ртути до прекращения дальнейшего образования осадка. После фильтрования осадка и промывания фильтрата водой, сушки и концентрирования получали 4,7 г желтого масла. Неочищенную содержащую силиловый эфир смесь цис- и транс-алкена растворяли в ТГФ и добавляли тетра-н-бутиламмонийфторид (0,031 моль). После перемешивания при 23°С в течение 40 минут эту смесь выливали в воду и экстрагировали в эфир. Эфирный экстракт сушили, концентрировали и очищали хроматографией на силикагеле с элюированием EtOAc в гексане (1:9) с получением 2,5,7,8-тетраметил-2R-(2,6,10-триметил-1,3,5,9-Е:Z-декатетраен)-6-хроманола (выход 68%). 1H-ЯМР (CDCl2/TMS, м.д.): 1,28 (с, 3Н, 2аСН3), 1,65 (с, 3Н), 1,70 (с, 6Н), 1,72 (с, 3Н), 1,9 (м, 6Н), 2,18 (с, 3Н), 2,35 (с, 6Н), 2,53 (т, J=6,6 Гц, 2Н, 4СН2), 5,13-5,27 (м, 3Н) и 6,44 (м, 2Н); MS (CI, m/z): 395,17 М+Н+, Рассчитано для С37Н38O2 394,60.
Раствор 2,5,7,8-тетраметил-2R-(2,6,10-триметил-1,3,5,9-Е:Z-декатетраен)-6-хроманола (0,457 г, 1,16 ммоль) в N,N-диметилформамиде (20 мл) обрабатывали метилбромацетатом (3,4 г, 8,3 ммоль) и избытком порошкообразного NaOH (1,2 г, 30 ммоль). Полученную желтую суспензию интенсивно перемешивали в течение 24 часов при комнатной температуре. Реакцию подкисляли 5 н. НСl и экстрагировали диэтиловым эфиром (3×30 мл). Объединенные эфирные слои промывали Н2O (3×30 мл) и солевым раствором (1×30 мл) и затем сушили над Na2SO4. Эфирный раствор концентрировали до желтого масла, которое очищали хроматографией на силикагеле с элюированием 19% (об/об) EtOAc и 2%-ной уксусной кислотой в гексанах. Полученную жидкость растворяли в диэтиловом эфире (30 мл), промывали Н2O (3×20 мл) и солевым раствором (1×20 мл) и затем сушили над Na2SO4. Полученный раствор концентрировали и сушили в вакууме в течение 48 часов. Это давало соединение 18 с выходом 67%. 1Н-ЯМР (CDCl3/TMS, м.д.): 1,24 (с, 3Н, 2аСН3), 1,63 (с, 3Н), 1,72 (с, 6Н), 1,74 (с, 3Н), 1,92 (м, 6Н), 2,18 (с, 3Н), 2,29 (с, 6Н), 2,43 (т, J=6,6 Гц, 2Н, 4СН2), 4,68 (с, 2Н, ОСН2), 5,10-5,27 (м, 3Н) и 6,34 (м, 2Н); MS (CI, m/z): 452,24 М-Н+, Рассчитано для C27H38O2 452,63.
3-(2,5,7,8-тетраметил-(2R-(4R,8,12-триметилтридецил)хроман-6-илокси)пропил-1-аммонийхлорид (19)

Раствор гидробромида 3-бромпропиламина (1,0 г, 4,6 ммоль) в смеси 2:1 диоксан/Н2O (45 мл) охлаждали до 0°С и обрабатывали К2СО3 (6,22 г, 45 ммоль) и ди-третбутилдикарбонатом (1,5 г, 6,9 ммоль). Реакцию перемешивали в течение 15 часов при нагревании до комнатной температуры. Диоксан удаляли в вакууме и оставшуюся водную смесь подкисляли 5 н. НСl и экстрагировали этилацетатом (5×25 мл). Объединенные органические слои сушили с МgSO4 и получали 3-бром-N-(трет-бутоксикарбонил)пропиламин в виде бесцветного масла (0,93 г, 93%). 1H-ЯМР (СDСl3/ТМS, м.д.): 1,41 (с, 9Н, СН3), 2,02 (квинтет, J=6,4 Гц, 2Н, СН2), 3,23 (м, 2Н, NCH2), 3,41 (т, J=6,6 Гц, СН2Вr), 4,8 (шир, 1Н, NH); 13С-ЯМР (CDCl3, м.д.): 28,3 (СН3), 30,7, 32,6, 38,9 (СН2), 79,3 (четвертичный С), 155,9 (СО); MS (CI, m/z): 239,241 (М+Н+, Рассчитано для C8H16BrNO2 237,03644).
Раствор R,R,R-α-токоферола (0,5 г, 1,16 ммоль) в N,N-диметилформамиде (15 мл) обрабатывали 3-бром-N-(трет-бутоксикарбонил)пропиламином (0,9 г, 3,8 ммоль) и избытком порошкообразного NaOH (0,32 г, 8 ммоль). Полученную желтую суспензию интенсивно перемешивали в течение 24 часов при комнатной температуре. Реакцию подкисляли 5 н. НСl и экстрагировали диэтиловым эфиром (3×30 мл). Объединенные эфирные слои промывали Н2О (3×30 мл) и солевым раствором (1×30 мл) и затем сушили над Na2SO4. Эфирный раствор концентрировали до желтого масла, которое очищали хроматографией на силикагеле с элюированием EtOAc (10% об/об) в гексанах. Это давало целевой эфир в виде бесцветного масла (0,45 г, выход 66%). 1H-ЯМР (CDCl3/TMS, м.д.): 0,87 (м, 12Н, 4а'-, 8а'-, 12а'-, 13'-СН3), 1,0-1,6 (м, 33Н, 4'-, 8'-, 12'-СН, 1'-, 2'-, 3'-, 5'-, 6'-, 7'-, 9'-, 10'-, 11'-СН2, 2а-СН3), 1,81 (м, 2Н, 3-CH2), 1,99 (квинтет, J=6,2 Гц, 2Н, СН2), 2,07, 2,14, 2,16 (3×с, 9Н, 5а-, 7а-, 8а-СН3), 2,59 (т, J=6,6 Гц, 2Н, 4-СН2), 3,43 (м, 2Н, NCH2), 3,73 (т, J=5,7 Гц, 2Н, ОСН2), 4,34 (с, 2Н, ОСН2); 13С-ЯМР (CDCl3, м.д.): 11,7, 12,08, 12,9 (5а-, 7а-, 8а-СН3), 19,6, 19,7 (СН3), 20,6, 21,0 (СН2), 22,6, 22,7 (СН3), 23,7 (2а-СН3), 24,4, 24,8 (СН2), 27,9 (СН), 31,2 (3-СН2), 32,7, 32,8, (СН), 37,2, 37,4, 37,5, 39,3, 40,1 (СН2), 70,2 (ОСН2), 74,8 (2-С), 117,5, 122,9, 125,5, 127,5 (арил С), 147,5, 148,0 (арил С-O), 156,0 (CO); MS (CI, m/z): 589 М+Н+, Рассчитано для С37Н65NO4 587,49136.
Вышеуказанный N-защищенный простой эфир (0,1 г, 0,17 ммоль) растворяли в 4 н. НСl в диоксане (1 мл, 4 ммоль) и перемешивали в течение 4 часов. Диоксан удаляли продуванием потока аргона над реакционной смесью. Полученный материал сушили в вакууме в течение 8 часов с получением соединения 19 в виде белого твердого вещества (82 мг, 99%). 1Н-ЯМР (CDCl3/TMS, м.д.): 0,87 (м, 12Н, 4а'-, 8а'-, 12а'-, 13'-СН3), 1,0-1,6 (м, 33Н, 4'-, 8'-, 12'-CH, 1'-, 2'-, 3'-, 5'-, 6'-, 7'-, 9'-, 10'-, 11'-CH2, 2а-СН3), 1,81 (м, 2Н, 3-СН2), 1,99 (квинтет, J=6,2 Гц, 2Н, СН2), 2,07, 2,11, 2,15 (3×с, 9Н, 5а-, 7а-, 8а-СН3), 2,29 (м, 2Н, СН2), 2,59 (т, J=6,6 Гц, 2Н, 4-СН2), 3,43 (м, 2Н, NCH2), 3,79 (т, J=5,7 Гц, 2Н, ОСН2); 13С-ЯМР (СDСl3, м.д.): 11,8, 11,9, 12,7 (5а-, 7а-, 8а-СН3), 19,6, 19,7 (СН3), 20,6, 21,0 (СН2), 22,6, 22,7 (СН3), 23,9 (2а-СН3), 24,4, 24,8 (СН2), 28,0 (СН), 28,4 (СН3), 31,2 (3-СН2), 32,7, 32,8, (СН), 37,3, 37,4, 37,4, 39,4, 40,0 (СН2), 74,8 (ОСН2), 75,0 (2-С), 117,5, 122,9, 126,0, 127,3 (арил С), 147,8, 148,0 (арил С-О); HRMS (CI, m/z): 487,438887 (М+Н+, Рассчитано для С32Н57NО2 487,438935).
2,5,7,8-тетраметил-(2R-(4R,8R,12-триметилтридецил)хроман-3-ен-6-илокси)уксусная кислота (20)

Раствор ацетата R,R,R-α-токоферола (2 г, 4,2 ммоль) в безводном толуоле (150 мл) нагревали до кипения с обратным холодильником и затем обрабатывали 2,3-дихлор-5,6-дициано-1,4-бензохиноном (0,96 г, 4,2 ммоль) в виде 4 порций с интервалами 1 час. Реакцию нагревали с обратным холодильником в течение 24 часов. Во время этого периода времени реакционная смесь становилась темно-красной и затем она давала осадок светлого окрашенного твердого вещества. Реакцию охлаждали до комнатной температуры, фильтровали и фильтрат концентрировали. Полученное темное окрашенное масло очищали хроматографией на силикагеле с элюированием этилацетатом (10%, об/об) в гексанах. Это давало целевой ацетат хромена в виде бесцветного масла (1,74 г, 88%). 1H-ЯМР (CDCl3/TMS, м.д.): 0,87 (м, 12Н, 4а'-, 8а'-, 12а'-, 13'-СН3), 1,0-1,6 (м, 24Н, 4'-, 8'-, 12'-СН, 1'-, 2'-, 3'-, 5'-, 6'-, 7'-, 9'-, 10'-, 11'-CH2, 2а-СН3), 2,07, 2,13, 2,18 (3×с, 9Н, 5а-, 7а-, 8а-СН3), 2,35 (с, 3Н, СН3СО-), 5,61, 6,52 (2×д, J=10,0 Гц, 2Н, СН); 13С-ЯМР (СDСl3, м.д.): 11,5, 11,6, 13,1 (5а-, 7а-, 8а-СН3), 14,1, (СН3), 19,6, 19,7 (СН3), 20,4, 21,4 (СН2) 22,6, 22,7 (СН3), 24,4, 24,8 (СН2), 25,8 (2а-СН3), 27,9 (СН), 30,8 (3-СН3), 32,7, 32,8, (СН), 37,2, 37,4, 39,4, 40,0 (СН2), 60,3 (2-С), 117,6, 119,7, 122,3, 122,6 128,9, 129,6 (арил и винил С), 141,2, 148,4 (арил С-O), 169,4 (СО); HRMS (CI, m/z): 471,375799 (M+H+, Рассчитано для С31H50О3 470,375996).
Раствор хроменацетата (1,0 г, 2,13 ммоль) в этаноле (20 мл) обрабатывали 2 н. NaOH (20 мл) и перемешивали при 60°С в течение 90 мин. Реакционную смесь охлаждали, подкисляли 5 н. НСl и этанол удаляли в вакууме. Полученный водный раствор экстрагировали эфиром и концентрировали до светло-желтого масла, которое очищали гель-хроматографией на силикагеле с элюированием этилацетатом (15%, об/об) в гексанах. Это давало целевой промежуточный продукт хромен-6-ол в виде бесцветного масла (0,92 г, 98%). 1H-ЯМР (CDCl3/TMS, м.д.): 0,87 (м, 12Н, 4a'-, 8a'-, 12a'-, 13'-СН3), 1,0-1,6 (м, 24Н, 4'-, 8'-, 12'-СН, 1'-, 2'-, 3'-, 5'-, 6'-, 7'-, 9'-, 10'-, 11'-CH2, 2а-СН3), 2,14, 2,18, 2,19 (3×с, 9Н, 5а-, 7а-, 8а-СН3), 5,63, 6,55 (2×д, J=10,0 Гц, 2Н, СН); 13С-ЯМР (CDCl3, м.д.): 10,8, 11,6, 12,4 (5а-, 7а-, 8а-СН3), 19,6, 19,7 (СН3), 21,3 (CH2) 22,6, 22,7 (СН3), 24,4, 24,8 (CH2), 25,2 (2а-СН3), 27,9 (СН), 30,9 (3-СН2), 32,7, 32,8, (СН), 37,2, 37,4, 37,5, 39,3, 40,5 (СН2), 50,8 (2-С), 116,2, 117,8, 120,1, 122,3 123,0 130,0 (арил и винил С), 144,6, 145,3 (арил С-O), 169,4 (СО); HRMS (CI, m/z): 428,365275 (M+H+, Рассчитано для C29H48O2 428,365431).
Раствор промежуточного продукта хромен-6-ола (0,9 г, 2,1 ммоль) в N,N-диметилформамиде (20 мл) обрабатывали метилбромацетатом (3,4 г, 8,3 ммоль) и избытком порошкообразного NaOH (1,2 г, 30 ммоль). Полученную желтую суспензию интенсивно перемешивали в течение 24 часов при комнатной температуре. Реакцию подкисляли 5 н. НСl и экстрагировали диэтиловым эфиром (3×30 мл). Объединенные эфирные слои промывали Н2O (3×30 мл) и солевым раствором (1×30 мл) и затем сушили над Na2SO4. Эфирный раствор концентрировали до желтого масла, которое очищали хроматографией на силикагеле с элюированием 19% (об/об) EtOAc и 2%-ной уксусной кислотой в гексанах. Полученную желтую жидкость растворяли в диэтиловом эфире (30 мл), промывали Н2O (3×20 мл) и солевым раствором (1×20 мл) и затем сушили над Na2SO4. Полученный раствор концентрировали до светло-желтого масла и сушили в вакууме в течение 48 часов. Это давало соединение 19 в виде бесцветного масла (0,90 г, 88%). 1H-ЯМР (СDСl3/ТМS, м.д.): 0,87 (м, 12Н, 4а'-, 8а'-, 12а'-, 13'-СН3), 1,0-1,6 (м, 24Н, 4'-, 8'-, 12'-СН, 1'-, 2'-, 3'-, 5'-, 6'-, 7'-, 9'-, 10'-, 11'-CH2, 2а-СН3), 2,07, 2,10, 2,19 (3×с, 9Н, 5а-, 7а-, 8а-СН3), 4,37 (с, 2Н, ОСН2), 5,62, 6,50 (2×д, J=10,0 Гц, 2Н, СН); 13С-ЯМР (CDCl3, м.д.): 11,3, 11,5, 12,9 (5а-, 7а-, 8а-СН3), 19,6, 19,7 (СН3), 21,3 (СН2) 22,6, 22,7 (СН3), 24,4, 24,8 (СН2), 25,6 (2а-СН3), 27,9 (СН), 30,9 (3-СН2), 32,7, 32,8, (СН), 37,2, 37,4, 37,5, 39,3, 40,9 (СН2), 60,5 (ОСН2), 69,1 (2-С), 118,0, 119,8, 122,8, 122,9, 129,6, 19,8 (арил и винил С), 147,5, 147,8 (арил С-O), 173,4 (СО); HRMS (CI, m/z): 487,378731 (M+H+, Рассчитано для C31H51O4 487,378736).
2-(2,5,7,8-тетраметил-(2R-(4R,8R,12-триметилтридецил)-хроман-6-илокси)этилтриэтиламмонийсульфат (21)
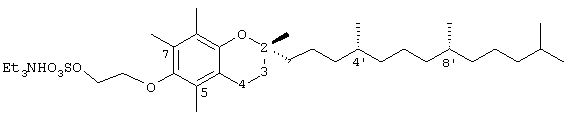
Раствор 2-(2,5,7,8-тетраметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)) этан-1-ола (13) (0,1 г, 0,21 ммоль) в безводном ДМФА (2 мл) и пиридине (0,6 мл) обрабатывали комплексом триоксид серы-N,N-диметилформамид (0,16 г, 1,0 ммоль) и полученный раствор перемешивали в течение 24 часов. Реакционную смесь гасили 1 н. NaOH и затем экстрагировали СН2Сl2 (5×5 мл). Газообразный аммиак барботировали через раствор СН2Сl2 в течение 10 мин. Полученный раствор концентрировали до желтой пасты и очищали хроматографией на силикагеле с элюированием МеОН (10%, об/об) и триэтиламином (2%) в СН2Сl2. Это давало соединение 21 в виде желтого полутвердого вещества (92 мг, 77%). 1H-ЯМР (CDCl3/TMS, м.д.): 0,87 (м, 12Н, 4а'-, 8а'-, 12а'-, 13'-СН3), 1,0-1,6 (м, 33Н, 4'-, 8'-, 12'-СН, 1'-, 2'-, 3'-, 5'-, 6'-, 7'-, 9'-, 10'-, 11'-СН2, 2а-СН3), 1,81 (м, 2Н, 3-СН2), 1,95, 2,01, 2,05 (3×с, 9Н, 5а-, 7а-, 8а-СН3), 2,45 (т, J=6,6 Гц, 2Н, 4-СН2), 3,05 (м, 6Н, NCH2), 3,79 (м, 2Н, ОСН2), 4,21 (м, 2Н, ОСН2); 13С-ЯМР (СDСl3, м.д.): 9,46 (СН3), 12,4, 12,6, 13,5 (5а-, 7а-, 8а-СН3), 20,3, 20,4 (СН3), 21,3, 21,7 (СН2), 23,3, 23,4 (СН3), 24,5 (2а-СН3), 25,1, 25,5 (СН2), 28,6, (СН), 31,9 (3-СН2), 33,3, 33,4 (СН), 37,9, 38,1 40,0 40,8 (СН2), 46,9 (NCH2), 67,4, 71,9 (OCH2), 75,5 (2-С), 118,3, 123,5, 126,5, 128,3 (арил С), 148,5 (арил С-O); HRMS (CI, m/z): 554,364102 (M-NН3, Рассчитано для С31Н54O6S 554,364119).
6-(2,5,7,8-тетраметил-(2R-(4R,8R,12-триметилтридецил)хроман) уксусная кислота (22)
Раствор R,R,R-α-токоферола (1,0 г, 2,3 ммоль) в безводном СН2Сl2 (25 мл) охлаждали до 0°С. Добавляли диизопропилэтиламин (2 мл, 11,6 ммоль) с последующим добавлением по каплям трифторметилсульфонового ангидрида (5,0 г, 17,7 ммоль). Раствор сразу же становился темным и ему давали нагреваться до комнатной температуры при перемешивании в течение 24 часов. Реакцию гасили Н2O и затем экстрагировали диэтиловым эфиром (2×100 мл). Объединенные эфирные слои промывали 1 н. НСl (50 мл), Н2O (50 мл), солевым раствором (50 мл) и затем сушили MgSO4. Эфирный раствор концентрировали до желтого масла и очищали хроматографией на силикагеле с элюированием этилацетатом (3%, об/об) в гексане. Это давало целевой промежуточный продукт трифлат в виде желтого масла (1,3 г, выход количественный). 1H-ЯМР (CDCl3/TMS, м.д.): 0,87 (м, 12Н, 4а'-, 8а'-, 12а'-, 13'-СН3), 1,0-1,6 (м, 24Н, 4'-, 8'-, 12'-СН, 1'-, 2'-, 3'-,5'-, 6'-, 7'-, 9'-, 10'-, 11'-СН2, 2а-СН3), 1,81 (м, 2Н, 3-СН2), 2,07, 2,13, 2,21 (3×с, 9Н, 5а-, 7а-, 8а-СН3), 2,62 (т, J=6,6 Гц, 2Н, 4-СН2), 13С-ЯМР (СDСl3, м.д.): 11,9, 13,2, 14,0 (5а-, 7а-, 8а-СН3), 19,6, 19,7 (СН3), 20,6, 21,0 (CH2), 22,6 22,7 (СН3), 23,8 (2а-СН3), 24,4, 24,8 (СН2), 28,0 (СН), 31,2 (3-СН2), 32,7, 32,8 (СН), 37,3, 37,4, 37,5, 39,4, 40,0 (СН2), 75,6 (2-С), 118,4, 124,4, 126,7, 128,1 (арил С), 139,6, 150,9 (арил С-O); 19F-ЯМR (СDСl3, м.д.): -73,52; HRMS (CI, m/z): 563,337803 (M+H+, Рассчитано для С30Н50O4F3S 563,338192).
Раствор трифлата (1,3 г, 2,31 ммоль) в безводном ДМФА (23 мл) обрабатывали LiCl (0,98 г, 4,52 ммоль), трифенилфосфином (0,37 г, 1,4 ммоль), 2,6-ди-трет-бутил-4-метилфенолом (2-3 кристалла), трибутил(винил)оловом (0,73 г, 2,31 ммоль) и дихлорбис(трифенилфосфин)палладием(II) (0,24 г, 0,35 ммоль). Эту смесь нагревали до 120°С и перемешивали. Спустя 2 часа добавляли дополнительное количество трибутил (винил) олова (0,73 г, 2,31 ммоль). Спустя 8 часов реакцию охлаждали до комнатной температуры и добавляли к смеси Н2О (50 мл) и диэтилового эфира (50 мл). Эфирный слой промывали 1 н. HCl (6×30 мл) и насыщенным раствором KF (6×30 мл). Эфирный раствор сушили над Na2SO4 и затем концентрировали до темного масла. Этот материал очищали хроматографией на силикагеле с элюированием этилацетатом (3%, об/об) в гексане с получением промежуточного продукта 6-винилхромана в виде прозрачного масла (0,38 г, 38%). 1H-ЯМР (СDСl3/ТМS, м.д.): 0,87 (м, 12Н, 4а'-, 8а'-, 12а'-, 13'-СН3), 1,0-1,6 (м, 24Н, 4'-, 8'-, 12'-СН, 1'-, 2'-, 3'-, 5'-, 6'-, 7'-, 9'-, 10'-, 11'-СН2, 2а-СН3), 1,86 (м, 2Н, 3-СН2), 2,20, 2,24, 2,28 (3×с, 9Н, 5а-, 7а-, 8а-СН3), 2,62 (т, J=6,8 Гц, 2Н, 4-СН2), 5,18, 5,56 (2×дд, Jgem=2,3 Гц, Jcis=11,2 Гц, Jtrans=18,7 Гц, 2Н, =СН2), 6,77 (дд, J=18,7, 11,2 Гц, 1Н, СН); 13С-ЯМР (CDCl3, м.д.): 11,9, 16,3, 17,2 (5а-, 7а-, 8а-СН3), 19,7, 19,8 (СН3), 20,8, 21,1 (СН2), 22,6, 22,7 (СН3), 23,8 (2а-СН3), 24,5, 24,8 (СН2), 28,0 (СН), 31,2 (3-СН2), 32,7, 32,8 (СН), 37,3, 37,5 37,5 39,4, 40,1 (СН2), 74,9 (2-С), 116,7, 119,0, 122,0, 129,8, 131,2 132,8, 136,8 (арил/винил С), 150,9 (арил С-O); HRMS (CI, m/z): 440,401602 (M+H+, Рассчитано для С31H52О 440,401812).
Раствор промежуточного продукта 6-винилхромана (0,12 г, 0,27 ммоль) в безводном ТГФ (1 мл) охлаждали до 0°С и обрабатывали 9-борабицикло [3.3.1] нонаном (0,60 мл, 0,5 М в ТГФ, 0,3 ммоль). Реакционную смесь нагревали с обратным холодильником в течение 8 часов. Реакцию гасили водой (1,5 мл) и обрабатывали NаВО3·4Н2O и полученную суспензию перемешивали в течение ночи. Диэтиловый эфир (4 мл) и реакционную смесь экстрагировали СН2Сl2 (2×20 мл). Органические слои концентрировали до прозрачного масла, которое очищали хроматографией на силикагеле с элюированием этилацетатом (50%, об/об) в гексане. Это давало целевой промежуточный продукт 6-(2-гидроксиэтил)хроман в виде бесцветного масла (30 мг, 24%). 1H-ЯМР (CDCl3/TMS, м.д.): 0,87 (м, 12Н, 4а'-, 8a'-, 12а'-, 13'-СН3), 1,0-1,6 (м, 24Н, 4'-, 8'-, 12'-СН, 1'-, 2'-, 3'-, 5'-, 6'-, 7'-, 9'-, 10'-, 11'-СН2, 2а-СН3), 1,81 (м, 2Н, 3-СН2), 2,17, 2,24, 2,28 (3×с, 9Н, 5а-, 7а-, 8а-СН3), 2,68 (т, J=6,8 Гц, 2Н, 4-СН2), 3,01 (т, J=7,5 Гц, 2Н, Аr-СН2), 3,74 (т, J=7,5 Гц, 2Н, ОСН2); 13С-ЯМР (СDСl3, м.д.): 12,0, 15,1, 16,0 (5а-, 7а-, 8а-СН3), 19,6, 19,7 (СН3), 20,6, 21,0 (СН2), 22,6, 22,7 (СН3), 23,8 (2а-СН3), 24,4, 24,8 (СН2), 28,0 (СН), 31,2 (3-СН2), 32,7, 32,8 (СН), 37,3, 37,4, 37,5, 39,4, 40,0 (СН2), 62,2 (ОСН2), 72,6 (2-С), 116,8, 122,3, 124,9, 132,4, 133,9 (арил С), 150,1 (арил С-O); HRMS (CI, m/z): 458,412154 (M+H+, Рассчитано для С31H52О2 458,412384).
Раствор хлорхромата пиридиния (32 мг, 0,1 ммоль) в безводном СН2С12 (0,5 мл) обрабатывали раствором промежуточного продукта 6-(2-гидроксиэтил)хромана (32 мг, 0,07 ммоль) в CH2Cl2 (0,5 мл). Реакцию перемешивали в течение 2 часов, причем после этого периода времени тонкослойная хроматография не обнаруживала видимого исходного материала. Добавляли диэтиловый эфир (2 мл) и полученный раствор фильтровали через тонкий слой целита. Фильтрат концентрировали и получали желтое масло (20 мг). Масло растворяли в трет-ВuОН (0,5 мл) и обрабатывали фосфатным буфером (0,5 мл, 1 н., рН=4,0), 2-метил-2-бутеном (0,1 мл) и NaClO2 (5,4 мг, 0,05 ммоль). После перемешивания в течение 40 минут реакционную смесь экстрагировали СНСl3 (6×10 мл) и объединенные органические слои сушили Na2SO4. СНСl3-раствор концентрировали до желтого масла, которое очищали препаративной тонкослойной хроматографией с элюированием этилацетатом (30%, об/об) и уксусной кислотой (1%) в гексанах. Это давало соединение 22 в виде бесцветного масла (20 мг, 63%). 1H-ЯМР (CDCl3/TMS, м.д.): 0,87 (м, 12Н, 4а'-, 8а'-, 12а'-, 13'-СН3), 1,0-1,6 (м, 24Н, 4'-, 8'-, 12'-СН, 1'-, 2'-, 3'-, 5'-, 6'-, 7'-, 9'-, 10'-, 11'-CH2, 2а-СН3), 1,81 (м, 2Н, 3-СН2), 2,17, 2,24, 2,28 (3×с, 9Н, 5а-, 7а-, 8а-СН3), 2,66 (т, J=6, 8 Гц, 2Н, 4-СН2), 3,71 (с, 2Н, СН2СООН); 13С-ЯМР (СDСl3, м.д.): 12,0, 15,3, 16,2 (5а-, 7а-, 8а-СН3), 19,6, 19,7 (СН3), 20,6, 21,0 (СН2), 22,6, 22,7 (СН3), 23,8 (2а-СН3), 24,4, 24,8 (СН2), 28,0 (СН), 28,9, 31,2 (3-СН2), 32,7, 32,8 (СН), 37,3, 37,4 37,5 39,4, 40,0 (СН2), 72,6 (2-С), 117,1, 122,2, 124,9, 132,4, 132,7 (арил С), 150,2 (арил С-O), 179,2 (СООН); HRMS (CI, m/z): 472,391583 (M+H+, Рассчитано для С31Н52O3 472,391644).
2,5,7,8-тетраметил-(2R-(гептил)хроман-6-илокси)уксусная кислота (23)
Раствор гексилтрифенилфосфонийбромида (0,880 г, 2,05 ммоль) в 11,2 мл безводного ДМЭ перемешивали при комнатной температуре с добавлением 0,86 мл (2,06 ммоль) 2,4 М н-бутиллития в гексане. Полученный красный раствор перемешивали в течение 2 часов при комнатной температуре, затем добавляли раствор [(+)S-6-бензилокси-2,5,7,8-тетраметилхроман-2-карбальдегида (306 мг, 0,944 ммоль) в 3 мл безводного ДМЭ и перемешивание продолжали в течение 3 часов при 65-75°С. После охлаждения реакционную смесь выливали в холодную разведенную Н2SO4 и обрабатывали эфиром обычным образом. Эфир упаривали в вакууме с получением маслянистого материала.
Продукт выделяли колоночной хроматографией и элюировали хлороформом с получением 46% продукта. Смесь цис- и транс- алкена растворяли в 30 мл этилацетата и добавляли 50 мг 5% палладия на угле и эту смесь встряхивали под давлением 40 psi (275,8 кПа) Н2 в течение 10 часов и затем фильтровали через целит и хорошо промывали этилацетатом. Фильтрат концентрировали и очищали хроматографией на силикагеле с элюированием EtOAc в гексане (1:9) с получением (2R)-2,5,7,8-тетраметил-2-(гептил)-6-хроманола (выход 60%). 1H-ЯМР (CDCl3/TMS, м.д.): 0,89 (с, 3Н), 1,3-1,5 (м, 15Н), 1,89 (м, 2Н), 2,2 (с, 3Н), 2,08 (с, 3Н), 2,23 (с, 3Н), и 2,48 (т, J=6,5 Гц, 2Н); MS (CI, m/z): 305,35 М+Н+, Рассчитано для С20Н32O2 304,4746).
Раствор 2,5,7,8-тетраметил-2-(гептил)-6-хроманола (0,353 г, 1,16 ммоль) в N,N-диметилформамиде (20 мл) обрабатывали метилбромацетатом (3,4 г, 8,3 ммоль) и избытком порошкообразного NaOH (1,2 г, 30 ммоль). Полученную желтую суспензию интенсивно перемешивали в течение 24 часов при комнатной температуре. Реакцию подкисляли 5 н. НСl и экстрагировали диэтиловым эфиром (3×30 мл). Объединенные эфирные слои промывали Н2O (3×30 мл) и солевым раствором (1×30 мл) и затем сушили над Na2SO4. Эфирный раствор концентрировали до желтого масла, которое очищали хроматографией на силикагеле с элюированием 19% (об/об) EtOAc и 2%-ной уксусной кислотой в гексанах. Полученную жидкость растворяли в диэтиловом эфире (30 мл), промывали Н2О (3×20 мл) и солевым раствором (1×20 мл) и затем сушили над Na2SO4. Полученный раствор концентрировали и сушили в вакууме в течение 48 часов.
Это давало соединение 23 с выходом 36%. 1Н-ЯМР (CDCl3/TMS, м.д.): 0,88 (с, 3Н), 1,2-1,5 (м, 15Н), 1,88 (м, 2Н), 2,1 (с, 3Н), 2,18 (с, 3Н), 2,2 (с, 3Н), 2,55 (т, J=6,5 Гц, 2Н) и 4,78 (с, 2Н); HRMS (CI, m/z): 363,2535 (M+H+, Рассчитано для С22Н35O4 363,2541).
2,5,7,8-тетраметил-(2R-(тридецил)хроман-6-илокси)уксусная кислота (24)
Соединения 24 и 25 синтезировали по способу, идентичному синтезу соединения 23, с использованием бромида фосфония.
1H-ЯМР (CDCl3/TMS, м.д.): 0,83 (с, 3Н), 1,25-1,57 (м, 27Н), 1,88 (м, 2Н), 2,1 (с, 3Н), 2,18 (с, 3Н), 2,20 (с, 3Н), 2,55 (т, J=6,6 Гц, 2Н) и 4,48 (с, 2Н); MS (CI, m/z): 447,14 М+Н+, Рассчитано для С28Н46O4 446,6732.
2,5,7,8-тетраметил-(2R-(гептадецил)хроман-6-илокси)уксусная кислота (25)

1Н-ЯМР (CDCl3/TMS, м.д.): 0,86 (с, 3Н), 1,15-1,67 (м, 35Н), 1,88 (м, 2Н), 2,16 (с, 3Н), 2,20 (с, 3Н), 2,23 (с, 3Н), 2,55 (т, J=6,4 Гц, 2Н) и 4,78 (с, 2Н); MS (CI, m/z): 503,45 М+Н+, Рассчитано для С32Н54O4 502,781.
2,5,7,8-тетраметил-2R-(4,8-диметил-1,3,7-нонотриен)хроман-6-илокси)уксусная кислота (26)

Соединение 26 синтезировали по способу, идентичному синтезу соединения 18, с использованием нерола вместо псевдоионола. 1H-ЯМР (СDСl3/ТМS, м.д.): 1,24 (с, 3Н, 2аСН3), 1,63 (м, 1Н), 1,68 (с, 3Н), 1,74 (с, 6Н), 1,92 (м, 6Н), 2,18 (с, 3Н), 2,29 (с, 6Н), 2,43 (т, J=6,6 Гц, 2Н, 4CH2), 4,68 (с, 2Н, ОСН2), 5,64 (м, 2Н) и 5,27 (м, 1Н); MS (CI, m/z): 413,24 М+Н+, Рассчитано для С26Н36O4 412,0115.
EZ,RS,RS,RS-(фитилтриметилбензолтиол-6-илокси)-уксусная кислота (27)
2,3,6-триметилфенол (1,6 г, 11,8 ммоль) растворяли в 50 мл безводного метанола, который дезоксигенировали барботированием азотом. К этому раствору добавляли тиоцианат аммония (2,2 г, 28,9 ммоль), и затем раствор охлаждали до 0°С и барботировали газообразным хлором. Сначала бесцветный гомогенный раствор становится розовым и затем зеленым с образованием затем белого цвета. Раствор перемешивали в течение 1 часа при 0°С и затем еще в течение часа при 20°С. Растворенный хлор удаляли барботированием азотом и осадок удаляли фильтрованием. Упаривание фильтрата при пониженном давлении с последующей сушкой под высоким вакуумом (0,1 тор) давало 2,20 г (97%) 2,3,5-триметил-4-гидроксифенилтиоцианата в форме достаточной чистой для следующей стадии в этом синтезе. Аналитическую пробу перекристаллизовывали из гексанов с получением белых кристаллов, т.пл. 100,3°С. 1H-ЯМР (CDCl2): δ 7,2 (с, 1Н), 5,0 (с, 1Н), 2,4 (с, 3Н), 2,2 (с, 6Н).
2,3,5-триметил-4-гидроксифенилтиоцианат (2 г, 10,35 ммоль) растворяли в 100 мл безводного эфира, содержащего 25 мл безводного тетрагидрофурана. Этот раствор добавляли по каплям на протяжении 1 часа к 100 мл безводного эфира, содержащего LiAlH4 (0,9 г, 24 ммоль) при комнатной температуре. Спустя еще один час при 20°С непрореагировавший LiAlH4 разрушали охлаждением гетерогенной смеси до 0°С и добавлением увлажненного эфира (50 мл), Н2О (50 мл) и 1 н. НСl (50 мл). Добавляли дополнительные 50 мл воды и органическую фазу отделяли и промывали водой (2×50 мл), раствором NаНСО3 (2×50 мл), водой (2×50 мл) и насыщенным NaCl (50 мл). Органическую фазу сушили над безводным МgSO4 и фильтровали и растворитель удаляли при пониженном давлении. Колоночная хроматография на силикагеле с 5%-ным этилацетатом в гексане давала 1,8 г (90%) 2,3,5-триметил-4-гидроксибензолтиола в виде белого порошка, т.пл. 86°C [Лит. 1т.пл. 86°С].
Раствор 2,3,5-триметил-4-гидроксибензолтиола (3 г, 17,83 ммоль), изофитола (4,8 г, 16,19 ммоль), безводного хлорида цинка (1,2 г, 8,8 ммоль) и 0,2 мл ледяной уксусной кислоты в 30 мл абсолютного эфира нагревали с обратным холодильником в течение 1 часа. Затем растворитель удаляли в вакууме при 50°С и полученное красное масло растворяли в смеси 50 мл петролейного эфира и 20 мл 70%-ного водного метанола. Эфирный слой сушили (Na2SO4) и упаривали в вакууме с получением красного масла, которое очищали хроматографией на силикагеле с элюированием смесью гексан:эфир (9:1) с получением 3 г (38%) EZ, RS, RS, RS-фитилтриметилгидроксибензолтиола в виде желтого масла. 1H-ЯМР (CDCl2): δ 7,11 (с, 1Н, Аr-Н), 5,23 (т, 1Н, винильный Н), 4,62 (с, 1Н, ОН), 3,34 (д, 2Н, Ar-S-CH2-), 2,41 (с, 3Н, Аr-СН3), 2,19 (с, 3Н, Аr-СН3), 2,18 (с, 3Н, Аr-СН3), 0,83-1,92 (м, 39Н, цепь фитола).
Раствор фитилтриметилгидроксибензолтиола (3 г, 6,7 ммоль) в N,N-диметилформамиде (80 мл) обрабатывали метилбромацетатом (7,4 г, 48,3 ммоль) и избытком порошкообразного NaOH (7 г, 175 ммоль). Полученное розовое масло перемешивали при комнатной температуре в течение 24 часов. Реакционную смесь подкисляли 5 н. НСl и экстрагировали эфиром (3×150 мл). Объединенные эфирные слои промывали Н2О (3×150 мл) и солевым раствором (1×150 мл) и затем сушили (Na2SO4). Эфирный раствор концентрировали до желтого масла, которое очищали хроматографией на силикагеле с элюированием 20% EtOAc в гексане с получением 3 г (88%) EZ, RS, RS, RS(фитилтриметилбензолтиол-6-илокси)уксусной кислоты в виде желтого масла. 1H-ЯМР (CDCl2): δ 10,90 (с, 1Н, СООН), 8,08 (с, 1Н, Аr-Н), 5,30 (т, 1Н, винильный Н), 4,35 (с, 2Н, СН2СООН), 3,42 (д, 2Н, Ar-S-CH2-), 2,34 (с, 3Н, Аr-СН3), 2,25 (с, 3Н, Ar-СН3), 2,22 (с, 3Н, Аr-СН3), 0,83-1,94 (м, 39Н, цепь фитила). HRMS (CI, m/z): 504,362821 (М+Н+, Рассчитано для С31Н53О3S 504, 363718).
(R)-2-[(2,5,7,8-тетраметил-2-(3-пропенметиловый сложный эфир)хроман-6-илокси]уксусная кислота (28)

К суспензии бромида (карбометоксиметил)трифенилфосфония (1,8 г, 4,32 ммоль) в 12 мл ТГФ добавляли по каплям 1,66 мл н-BuLi (2,5 М в гексане). Полученный раствор удаляли при комнатной температуре в течение 2 часов и затем добавляли через канюлю раствор (+)S-6-[диметил-(1,1-диметилэтил)силил]-2,5,7,8-тетраметилхроман-2-карбальдегида (1,31 г, 3,76 ммоль) в 7 мл ТГФ. Раствор перемешивали при комнатной температуре в течение 44 часов и затем добавляли 10 мл 1 н. водной НСl. Слои разделяли и затем водную фазу экстрагировали эфиром (3×15 мл). Объединенный органический слой промывали солевым раствором, сушили над Na2SO4 и фильтровали. После концентрирования фильтрата неочищенный алкен очищали флеш-хроматографией с элюированием дихлорметаном с получением смеси цис- и транс-алкена с выходом 93%. Содержащую силиловый эфир смесь цис- и транс- алкена (3,76 ммоль) растворяли в ТГФ и добавляли тетра-н-бутиламмонийфторид (0,041 моль). После перемешивания при 23°С в течение 1,5 часа смесь выливали в воду и экстрагировали в эфир. Эфирный экстракт сушили, концентрировали и очищали хроматографией на силикагеле с элюированием EtOAc в гексане (3:7) и как цис-, так и транс- изомер 2,5,7,8-тетраметил-2R-(3'-пропенметиловый сложный эфир)-6-хроманола выделяли и характеризовали (выход 68%). 1H-ЯМР (CDCl2/TMS, м.д.): 1,65 (с, 3Н, 2а СН3), 2,12 (м, 2Н, 3СН2), 2,39 (с, 9Н, СН3), 2,48 (м, 2Н, 4 СН2), 3,78 (с, 3Н, ОСН3), 6,11 (д, 1Н, СН=) и 7,13 (д, 1Н, СН=).
Раствор 2,5,7,8-тетраметил-2R-(3'-пропенметиловый сложный эфир)-6-хроманола (0,353 г, 1,16 ммоль) в N,N-диметилформамиде (20 мл) обрабатывали метилбромацетатом (3,4 г, 8,3 ммоль) и избытком порошкообразного NaOH (1,2 г, 30 ммоль). Полученную желтую суспензию интенсивно перемешивали в течение 24 часов при комнатной температуре. Реакцию подкисляли 5 н. НСl и экстрагировали диэтиловым эфиром (3×30 мл). Объединенные эфирные слои промывали Н2О (3×30 мл) и солевым раствором (1×30 мл) и затем сушили над Na2SO4. Эфирный раствор концентрировали до желтого масла, которое очищали хроматографией на силикагеле с элюированием 19% (об/об) EtOAc и 2%-ной уксусной кислотой в гексанах. Полученную жидкость растворяли в диэтиловом эфире (30 мл), промывали Н2О (3×20 мл) и солевым раствором (1×20 мл) и затем сушили над Na2SO4. Полученный раствор концентрировали и сушили в вакууме в течение 48 часов. Это давало соединение 28 с выходом 40%). 1H-ЯМР (CDCl3/TMS, м.д.): 1,68 (с, 3Н, 2а СН3), 2,11 (м, 2Н, 3СН2), 2,36 (с, 9Н, СН3), 2,56 (м, 2Н, 4 СН2), 3,70 (с, 3Н, ОСН3), 4,78 (с, 2Н, ОСН2), 6,03 (д, 1Н, СН=) и 7,03 (д, 1Н, СН=). MS (CI, m/z): 337,24 М+Н+, Рассчитано для C18H24O6 336,3867.
2,5,7,8-тетраметил-(2R-(пропионат)хроман-6-илокси)уксусная кислота (29)

Смесь цис- и транс-алкенового изомера 2,5,7,8-тетраметил-2R-(3-пропенметиловый эфир)-6-хроманола растворяли в 30 мл этилацетата и добавляли 50 мг 5% палладия на угле и смесь встряхивали под давлением 40 psi (275,8 кПа) H2 в течение 24 часов и затем фильтровали через целит и хорошо промывали этилацетатом. Фильтрат концентрировали и очищали хроматографией на силикагеле с элюированием EtOAc в гексане (1:9) с получением соединения 29. 1H-ЯМР (CDCl3/TMS, м.д.): 1,62 (с, 3Н, 2а СН3), 2,0-2,3 (м, 6Н, CH2), 2,41 (с, 9Н, СН3), 2,53 (м, 2Н, 4 CH2), 3,67 (с, 3Н, ОСН3) и 4,88 (с, 2Н, OCH2); MS (CI, m/z): 339,34 М+Н+, Рассчитано для С18H26О6 338,4025.
ПРИМЕР 3
Условия культуры клеток
Все испытуемые клеточные линии культивировали при 37°С в 5% СO2 в стандартных средах, дополненных фетальной телячьей сывороткой, с использованием установленных стандартных условий. Прикрепившиеся к пластику клетки диссоциировали трипсином, промывали, считали и использовали непосредственно в экспериментах. Все клетки подвергали стандартным испытаниям, чтобы проверить отсутствие загрязнения микоплазмой.
ПРИМЕР 4
Растворимость и разведение новых соединений токоферола и токотриенола
Со всеми соединениями обращались таким образом, как если бы они были светочувствительными (фотодеградируемыми). Все соединения сначала растворяли в абсолютном этаноле, а затем разводили до конечной концентрации 0,5% этанола с помощью подходящих сред.
ПРИМЕР 5
Определение эффективной концентрации (EC50) для индукции апоптоза
В то время как исходные структурно не модифицированные формы токоферолов не проявляют эффективных апоптотических свойств против набора (“батареи”) опухолевых клеток, пятнадцать из двадцати девяти соединений R,R,R-α-токоферола, структурно модифицированных посредством связанных простой эфирной связью частей молекул различного состава и размера, были чрезвычайно эффективными в индуцировании апоптоза опухолевых клеток, не обнаруживая индуцирующих апоптоз свойств на нормальных клетках. Соединения 1, 2, 3, 7, 8, 9, 12, 15, 17, 19, 20, 21, 22, 25, 26 и 27 проявляют эффективные ингибирующие рост (индуцирующие апоптоз) свойства, специфические в отношении раковых клеток человека из большого разнообразия линий дифференцировки, в том числе (i) молочной железы (эстроген-чувствительной линии клеток 7 рака молочной железы человека M.D. Anderson, MDA-MB-435, MCF-7 McGuire, рака молочной железы человека Michigan Cancer Foundation; эстроген-нечувствительной клеточной линии метастатического; и эстроген-нечувствительной клеточной линии метастатического рака молочной железы человека M.D. Anderson, MDA-MB-231); (ii) предстательной железы (андроген-чувствительной клеточной линии рака предстательной железы человека, LnCaP и андроген-нечувствительной клеточной линии рака предстательной железы человека, РС-3, и клеточной линии DU-145); (iii) промиелоцитарных лейкозных клеток (линии промиелоцитарных лейкозных клеток человека, HL-60), линий лимфоидных клеток Jurkat и HL-60; (iv) шейки матки (клеточной линии рака шейки матки человека, ME-180); (v) яичника (клеточной линии рака яичника человека, клеток С-170); (vi) эндометрия (клеточной линии рака эндометрия человека, клеток RL-95-2); (vii) клеточных линий DLD-1 ободочной кишки и (viii) линии клеток легкого А-549. Нормальные первичные клетки молочной железы (нормальные первичные эпителиальные клетки молочной железы человека раннего пассажа, НМЕС) и иммортализованные, неонкогенные клетки молочной железы человека (иммортализованные, но неонкогенные клетки молочной железы человека, культура Michigan Cancer Foundation 10A, MCF-10A) не подвергаются апоптозу при культивировании с вышеуказанными разработанными с учетом фармакодинамики формами токоферола.
Эффективной терапевтической дозой новых реагентов для борьбы с ростом рака называют ингибирующую рост концентрацию (IС50) или эффективную концентрацию (EC50), которая блокирует на 50% раковый рост через ингибирование синтеза ДНК, блокирование клеточного цикла и/или гибель клеток. Апоптотические ЕС50 для набора ("батареи") испытуемых раковых клеток для пятнадцати новых соединений данного изобретения представлены в таблицах 1 и 2.
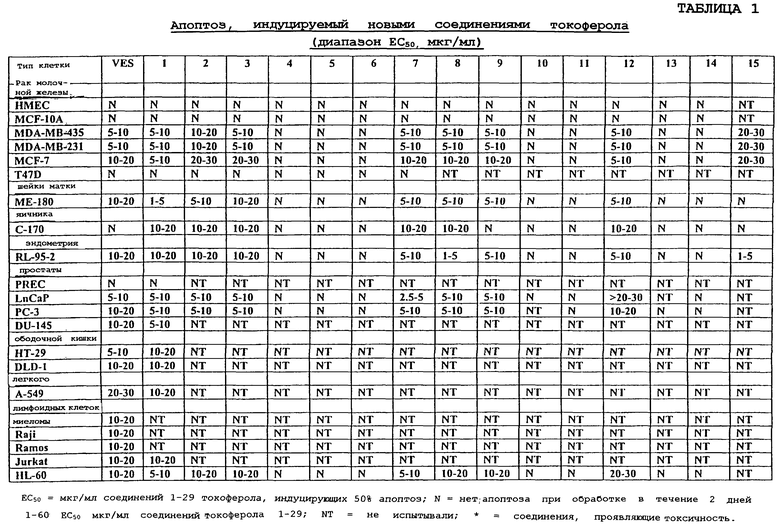
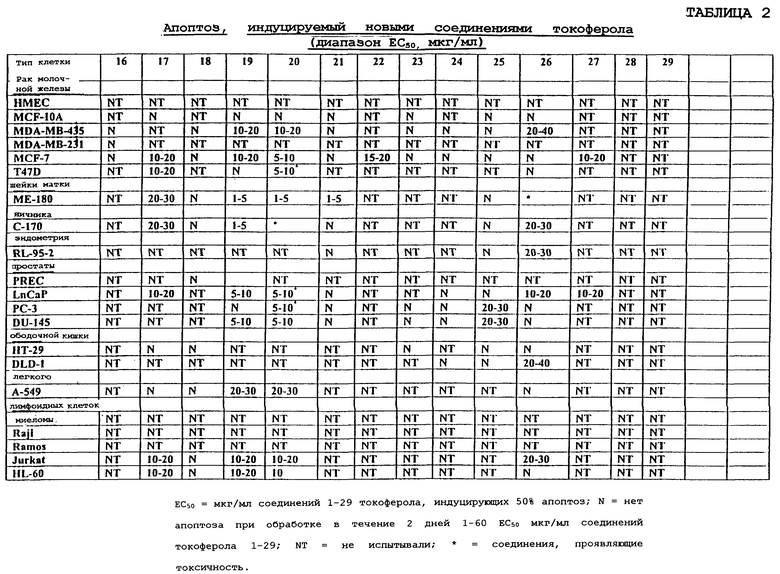
ПРИМЕР 6
Биотест на апоптоз
Клетки культивировали в количестве 1,5×105 клеток на лунку в 12-луночных планшетах. Клеткам давали прикрепляться в течение ночи, затем инкубировали с новыми тестируемыми соединениями при 0,01, 0,1, 1, 5, 10 и 20 г/мл в течение 1, 2 и 3 дней. После обработки клетки (плавающие + освобожденные трипсином прикрепленные клетки) осаждали, промывали и окрашивали 2 г/мл DAPI (дигидрохлорид 4',6-диамидин-2'-фенилиндола) в 100% метаноле в течение 15 минут при 37°С и/или окрашивали с помощью TUNEL, затем наблюдали с использованием микроскопа Zeiss ICM 405. Клетки, ядро которых содержало явно конденсированный или фрагментированный хроматин, оценивались как апоптотические. Данные представлены в виде процента клеток, подвергнутых апоптозу.
ПРИМЕР 7
Биотест на остановку синтеза ДНК
Для анализа синтеза ДНК все клетки использовали при 2,5×105 на мл. Клетки обрабатывали каждым из соединений 1-29 (таблицы 1 и 2) при концентрациях 0,01, 0,1, 1, 5, 10 и 20 мкг/мл и 200 мкл каждой группы обработки высевали в четырех повторностях в 96-луночные культуральные планшеты (Corning, Corning NY). Эксперименты проводили в двух повторностях, один планшет использовали для испытания жизнеспособности, а другой планшет для испытания поглощения 3H-TdR для мониторинга синтеза ДНК. Планшеты культивировали в течение 48 часов при 37°С, 5% СO2. За восемь часов перед окончанием инкубации 3H-TdR добавляли к одному из двух повторных планшетов и инкубацию продолжали в течение 8 часов. Затем клетки собирали (для сбора прикрепленных клеток требовалась трипсинизация) и поглощение изотопа определяли в виде импульсов в минуту (имп/мин). Для исследований жизнеспособности в конце инкубации клетки удаляли из лунок и жизнеспособность проверяли способом исключения с трипановым синим. Процент жизнеспособности и процент синтеза ДНК рассчитывали в сравнении с необработанными или обработанными носителем клетками каждой группы обработки.
ПРИМЕР 8
Биотест на остановку клеточного цикла
Клетки культивировали с новыми испытуемыми агентами в течение 2-3 дней, фиксировали в 95% этаноле и окрашивали пропидийиодидом в течение ночи. Содержание ДНК определяли с использованием проточного цитометра Coulter Epics Elite с аргоновым лазером, установленным на 488 нм. Размер клеток измеряли одновременно и результаты анализировали как процент клеток в каждой фазе клеточного цикла с использованием программы Coulter Multicycle Program.
ПРИМЕР 9
Биотест на дифференцировку клеток
Для определения, индуцируют ли новые соединения клеточную дифференцировку, клетки культивировали на покровных стеклах, фиксировали в 95% этаноле и окрашивали специфическим для липидов красителем для детектирования липидов молока. Кроме того, клетки исследовали посредством иммуногистологии и Вестерн-анализами на присутствие молочного белка казеина с использованием поликлональных антител, полученных в лаборатории.
ПРИМЕР 10
Эффекты остановки синтеза ДНК
Клетки культивировали в течение 48 часов, импульсно метили 8 часов тритированным тимидином, собирали и считали. Результаты представлены в виде импульсов в минуту. Проверку остановки синтеза ДНК определяют по понижению поглощения тритированного тимидина клетками, обработанными тест-соединениями. Дополнительное подтверждение остановки синтеза ДНК получают окрашиванием пропидийиодидом и стандартными анализами клеточного цикла.
ПРИМЕР 11
Механизмы индукции апоптоза
Механизм индукции апоптоза этими соединениями, по-видимому, включает в себя три различающихся пути передачи сигналов апоптоза; а именно, активацию латентного трансформирующего фактора роста бета (TGF-P), активацию путей передачи сигналов Fas/Fas-лиганда и путь передачи сигнала стресс-киназы (c-Jun N-концевой киназы).
TGF-β представляют собой сильные ингибирующие рост молекулы, которые, как известно, ингибируют рост клеток посредством ингибирования синтеза ДНК или индукцией апоптоза. TGF-β участвуют только в индукции апоптотического пути, т.е. в настоящее время нет доказательства того, что TGF-β осуществляют остановку синтеза ДНК; однако, эта возможность не была полностью исключена. TGF-β образуются и секретируются клетками в латентной неактивной форме. Чтобы быть эффективными в качестве ингибиторов роста клеток, латентные TGF-β должны быть активированы индукцией поверхностных белков клеток, которые обеспечивают правильную структуру для процессинга и активирования протеаз, которые разрезают латентный белок и высвобождают активный TGF-β.
Показано, что соединения данного изобретения активируют протеазы, такие как протеазы семейства катепсина D, и положительно регулируют маннозо-6-фосфатный рецептор, который связывает неактивный TGF-β и делает возможной активацию через протеазы. Активный TGF-β передает сигнал через рецепторы TGF-β I и II клеточной мембраны для активации находящихся ниже по ходу трансдукции сигналов киназ, называемых стресс-киназами или c-Jun N-концевыми киназами (JNK), которые фосфорилируют и активируют факторы транскрипции cJun, ATF-2 и Elk-1. Пролонгированная активация фактора транскрипции c-Jun заставляет опухолевые клетки подвергаться апоптозу. Эти факторы транскрипции, действующие в виде гомодимеров или гетеродимеров со множеством партнеров факторов транскрипции, активируют проапоптотические гены и/или отрицательно регулируют антиапоптотические гены, приводя к фрагментации ДНК. Соединения данного изобретения не генерируют антипролиферативного результата в отношении трансдукции сигналов TGF-β в нормальных неопухолевых клетках.
Второй индуцирующий апоптоз механизм, называемый путем передачи сигналов Fas/Fas-лиганда, активируется новыми соединениями данного изобретения. Активированная передача сигналов Fas/Fas-лиганда может приводить к быстрой гибели клеток в результате апоптоза. Таким образом, для того чтобы опухолевые клетки могли избежать гибели под действием Fas/Fas-лиганда, они должны инактивировать этот наиболее важный путь апоптоза. Механизм инактивации пути передачи сигналов Fas/Fas-лиганда опухолевыми клетками варьируется; однако многие опухолевые клетки отрицательно регулируют (снижают) экспрессию Fas-рецептора и Fas-лиганда на их мембранах.
Наиболее важно то, что, как было показано, R,R,R-2-(2,5,7,8-тетраметил-2-(4,8,12-триметилтридецил)хроман-6-илокси)уксусная кислота (1) индуцирует устойчивые к Fas/Fas-лиганду опухолевые клетки к их превращению в чувствительные к Fas/Fas-лиганду клетки. Соединение 1 обладает также способностью усиливать экспрессию Fas-лиганда на мембране клеток предстательной железы LNCaP. Исследования показывают, что резистентные к передаче сигнала Fas раковые клетки молочной железы человека удерживают Fas-рецептор в их цитоплазме, но при культивировании с соединением 1 Fas-рецептор транспортируется из цитоплазмы в мембрану; посредством этого он делает эти клетки чувствительными к передаче сигнала Fas. Кроме того, это соединение является синергическим в случае aнти-Fas-инициируемого апоптоза, поскольку достигают уничтожения большого количества клеток как для клеток рака молочной железы, так и для клеток рака предстательной железы человека при совместном введении по сравнению с обработками по отдельности. Способность соединения 1 превращать резистентные к передаче сигнала Fas опухолевые клетки в чувствительные к передаче сигнала Fas опухолевые клетки и проявлять синергические эффекты в их уничтожении обеспечивает чрезвычайно важный механизм для деструкции опухолевых клеток как иммуной системой защиты, так и фармацевтическим вмешательством. Соединения данного изобретения не активируют путь передачи сигнала Fas нормальных неопухолевых клеток.
Данные соединения активируют путь трансдукции сигналов JNK-киназы, возможно, посредством трансдукции сигналов TGF-β и Fas/Fas-лиганда. Пролонгированная активация JNK приводит к пролонгированной активации факторов транскрипции c-Jun и ATF-2, которые, как постулируется, играют роль в экспрессии или репрессии проапоптотических и антиапоптотических генов, соответственно.
ПРИМЕР 12
Механизм индукции остановки синтеза ДНК, остановки клеточного цикла и клеточной дифференцировки.
Механизмы ингибирования роста посредством остановки синтеза ДНК, остановки клеточного цикла и индукцией клеточной дифференцировки не были охарактеризованы так полно, как механизм ингибирования роста посредством апоптозам. Исследования показывают, что соединения данного изобретения глубоко влияют на клеточный цикл, индуцируя остановку синтеза ДНК приблизительно 95% опухолевых клеток в пределах 24 часов обработки. Опухолевые клетки, культивируемые с описанными здесь соединениями, имеют ингибированный рост в фазе G1 клеточного цикла, подвергаются морфологическим изменениям и экспрессируют липиды молока, что указывает на то, что клетки с блокированным клеточным циклом подверглись дифференцировке. Р21, ген, известный как ингибитор вступления клеток из фазы клеточного цикла G1 в фазу S клеточного цикла, и мРНК, а также белок гена Р21 положительно регулируют обработкой раковых клеток молочной железы человека MDA-MB-435 соединением 1.
ПРИМЕР 13
Возможности in vivo в отношении для раковых клеток человека
Данное изобретение имеет возможности для применения в качестве терапевтических агентов. Проводятся исследования in vivo опухолевого роста и метастазирования опухолевых клеток человека, эктопически или ортотопически трансплантированных в животных с ослабленным иммунитетом, таких как “голые” мыши, или в исследованиях in vivo с использованием хорошо известных животных моделей. Ингибирование роста опухолевых клеток человека, трансплантированных в мышей с ослабленным иммунитетом, обеспечивает данные для клинических испытаний. Исследования in vivo включают в себя две модели опухолевых клеток человека: модель метастатического нечувствительного к эстрогену рака молочной железы MDA-MB-435 и модель нечувствительного к андрогену рака предстательной железы РС-3 человека.
Модель рака молочной железы MDA-MB-435
Не содержащие патогенов клетки рака молочной железы человека MDA-MB-435, стабильно трансфицированные маркерным белком (флуоресцентным зеленым белком), выращивают в виде твердой опухоли в “голых” мышах или SCID мышах с ослабленным иммунитетом. Опухоли извлекают и срезы 1 мм равного размера ортотопически трансплантируют в жировое тело молочной железы или эктопически трансплантируют в заднюю часть бока самок “голых” мышей. Определяют рост опухолей, метастазирование и смерть животных. Рост опухолей измеряют оценками при помощи штангенциркуля размера опухоли. Во время умерщвления опухоли извлекают, измеряют их размер и используют для гистохимического исследования. Органы, такие как селезенка, лимфатические узлы, легкие и костный мозг исследуют на метастатические клетки MDA-MB-435 гистохимическим окрашиванием срезов тканей на экспрессию маркерного флуоресцентного зеленого белка.
Модель рака предстательной железы РС-3
Не содержащие патогенов клетки рака предстательной железы человека РС-3, стабильно трансфицированные маркерным белком (флуоресцентным зеленым белком), выращивают в виде твердой опухоли в “голых” мышах. Опухоли извлекают и срезы 1 мм равного размера эктопически трансплантируют в заднюю часть бока самцов “голых” мышей. Определяют рост опухолей, метастазирование и смерть животных. Во время умерщвления опухоли извлекают, измеряющих размер и используют для гистохимического исследования. Органы, такие как селезенка, лимфатические узлы, легкие и костный мозг исследуют на метастатические клетки РС-3 гистохимическим окрашиванием тканей на экспрессию маркерного флуоресцентного зеленого белка.
Модель животного с раком кожи
Рак кожи индуцируют в бесшерстных мытах SENCAR и SKH-1 ультрафиолетовым излучением и химическими обработками (DMBA). Кроме того, используют мышей, специфически экспрессирующих онкоген Her-2/neu в базальных клетках кожи, которые самопроизвольно развивают рак кожи. Описанные здесь соединения наносят местно на кожу один раз в день до и после инициации рака кожи и оценивают развитие образования кожных папиллом. Контрольных мышей обрабатывают идентично, за исключением того, что они получают обработку носителем, наносимым местно на их кожу. Мониторингу подвергают эффективность данных соединений в лечении папилломы, а также их способность влиять на злокачественное перерождение при нанесении перед предраковым прогрессированием.
ПРИМЕР 14
Применение новых соединений
Перед началом экспериментов in vivo соединения данного изобретения, для которых выявлено наивысшее количество уничтоженных опухолевых клеток, вводят “голым” мышам, SCID, трансгенным и другим мытам при изменяющихся уровнях для определения наивысшего уровня соединения, который может быть введен безопасно без неблагоприятных эффектов. Соединения вводят подходящим для модели образом; например, перорально, инъекциями, в том числе инъекциями непосредственно в орган-мишень, или локально (местно). После определения наивысшего уровня данных соединений, который может ими переноситься, и эффективных путей введения эти новые соединения вводят мышам один раз в день и определяют рост и прогрессирование опухолей, как описано выше.
ПРИМЕР 15
Определение предельно допустимой (максимальной переносимой) дозы (ПДД)
Для определения предельно допустимой (максимальной переносимой) дозы (ПДД) соединения 1 мышей штамма 25 Balb/c делят на следующие 5 групп:
Группа 1. Необработанные
Группа 2. Обработанные носителем (этиловый спирт + арахисовое масло)/0,1 мл зондового питания/мышь/сутки
Группа 3. Соединение №1 при 20 мг/0,1 мл зондового питания/мышь/сутки
Группа 4. Соединение №1 при 10 мг/0,1 мл зондового питания/мышь /сутки
Группа 5. Соединение №1 при 5 мг/0,1 мл зондового питания/мышь/сутки
Соединение 1 растворяют в 100% этаноле и разводят до соответствующего уровня в лишенном витамина Е арахисовом масле для доставки 20, 10 и 5 мг/0,1 мл объема через желудочный зонд. Контроль-носитель состоит из 100% этанола плюс лишенное витамина Е арахисовое масло. Мышей обрабатывают один раз в день в течение 30 дней. Массы всего тела определяют один раз в неделю после начала обработок. Среди групп нет различий в массах мышей. Мыши остаются активными и не обнаруживают признаков токсичности.
Анализы методом жидкостной хроматографии высокого разрешения (ВЖХ) проводят на пробах сыворотки и ткани с интервалами в одну неделю во время 30-дневной обработки. Соединение 1 детектируют в сыворотке и ткани из всех трех тест-групп.
ПРИМЕР 16
Получение исходного раствора, носителя и разведении исходного раствора соединения 1
Исходный раствор соединения 1
Растворяют 2 грамма соединения №1 в 5 мл 100% этанола (EtOH) и проводят интенсивное перемешивание при 37°С.
Соединение 1 при 20 мг/0,1 мл зондового питания/мышь:
Объединяют 1 мл исходного раствора соединения 1, 3 мл лишенного витамина Е арахисового масла и 400 мг соединения №1 (сухого) и проводят интенсивное перемешивание при 37°С.
Соединение 1 при 10 мг/0,1 мл зондового питания/мышь:
Объединяют 1 мл исходного раствора соединения 1 и 3 мл лишенного витамина Е арахисового масла.
Соединение 1 при 5 мг/0,1 мл зондового питания/мышь:
Объединяют 0,5 мл исходного раствора соединения 1 и 3 мл лишенного витамина Е арахисового масла.
Носитель:
Объединяют 1 мл EtOH и 3 мл лишенного витамина Е арахисового масла.
ПРИМЕР 17
Химиопрофилактические свойства соединения 1 в модели рака крыс ACI
Соединение 1 используют in vivo для лечения трансплантированных опухолей молочной железы, предстательной железы и ободочной кишки человека, трансплантированных в "голых" мышей с ослабленным иммунитетом. Химиопрофилактическая эффективность соединения 1 in vivo против рака молочной железы человека показана на модели рака молочной железы крыс ACI, с раком, инициированным эстрогеном. Приблизительно у 90% крыс, имплантированных гранулами эстрогена, развивается рак молочной железы в пределах 6 месяцев после имплантации эстрогена.
Соединение 1 растворяют в 100% этаноле и разводят до соответствующей дозы с использованием не содержащего витамина Е арахисового масла. Предельно допустимую дозу (ПДД, максимальную дозу соединения, которая может быть введена без неблагоприятных воздействий) определяют, как описано в примерах 14 и 15. Соединение 1 вводят при ПДД и S ПДД. Крыс ACI в 4-недельном возрасте субпанникулярно имплантируют гранулами эстрогена в области плеча. Соединение 1 при ПДД и 1/2 ПДД вводят через желудочный зонд. Опухоли молочной железы детектируют в контрольной группе приблизительно через 100 дней после имплантации эстрогена. У девяноста процентов контрольных крыс развивается рак молочной железы в пределах 6 месяцев после имплантации эстрогена. Несущих опухоли животных из контрольной группы и из обработанной группы умерщвляют через различные интервалы времени после начала обработки и ткань молочной железы исследуют на видимые опухоли и дополнительно исследуют гистологическими анализами.
Квалифицированному специалисту в данной области будет вполне понятно, что данное изобретение хорошо приспособлено для достижения целей и получения упомянутых результатов и преимуществ, и они присущи этому изобретению. Данные примеры вместе со способами, методиками, обработками, молекулами и конкретными соединениями, описанными здесь, являются в данном случае типичными предпочтительными вариантами и не предназначены для ограничения объема данного изобретения. Изменения в них и другие возможности применения, которые возникнут у специалистов в данной области, находятся в сфере сущности данного изобретения, определенной объемом прилагаемой формулы изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ТОКОФЕРОЛЫ, ТОКОТРИЕНОЛЫ, ДРУГИЕ ХРОМАНЫ И ПРОИЗВОДНЫЕ ПО БОКОВЫМ ЦЕПЯМ И ИХ ПРИМЕНЕНИЕ | 2001 |
|
RU2263672C2 |
| СПОСОБ ПОЛУЧЕНИЯ (2RS)-2,5,7,8-ТЕТРАМЕТИЛ-2-[(4RS,8RS)-4,8,12-ТРИМЕТИЛТРИДЕЦИЛ]-ХРОМАН-6-ИЛ-N-[3-ОКСОЛУП-20(29)-ЕН-28-ОИЛ]-ГЛИЦИНАТА | 2008 |
|
RU2440366C2 |
| ПРОИЗВОДНЫЕ БОРОНОВОЙ КИСЛОТЫ | 2018 |
|
RU2793315C2 |
| СПОСОБ ПОЛУЧЕНИЯ (S)-(-)-6-БЕНЗИЛОКСИ-3,4-ДИГИДРО-2,5,7,8-ТЕТРАМЕТИЛ-2Н-1-БЕНЗОПИРАН-2-ИЛМЕТАНОЛА | 2010 |
|
RU2443695C1 |
| ЛИПОСОМАЛЬНАЯ ДОСТАВКА СОЕДИНЕНИЙ, ОСНОВАННЫХ НА ВИТАМИНЕ Е | 2002 |
|
RU2328273C2 |
| ЛИГАНДЫ, ТРОПНЫЕ К ПРОСТАТИЧЕСКОМУ СПЕЦИФИЧЕСКОМУ МЕМБРАННОМУ АНТИГЕНУ И ИХ ПРИМЕНЕНИЕ ДЛЯ ПОЛУЧЕНИЯ ДВОЙНЫХ КОНЪЮГАТОВ С ТЕРАПЕВТИЧЕСКИМИ АГЕНТАМИ НА ИХ ОСНОВЕ ДЛЯ КОМБИНИРОВАННОЙ ТЕРАПИИ ПСМА ЭКСПРЕССИРУЮЩИХ ОПУХОЛЕЙ | 2023 |
|
RU2841078C1 |
| Способ получения хиральных S-монотерпенилцистеинов | 2018 |
|
RU2675238C1 |
| ХАЛКОНЫ, ОБЛАДАЮЩИЕ АНТИПРОЛИФЕРАТИВНОЙ АКТИВНОСТЬЮ | 1998 |
|
RU2203883C2 |
| НОВЫЕ НАФТО[2,1-b]КАРБАЗОЛПРОИЗВОДНЫЕ ФУЗИДОВОЙ КИСЛОТЫ, СПОСОБ ПОЛУЧЕНИЯ И АНТИМИКРОБНЫЕ СВОЙСТВА | 2019 |
|
RU2746947C2 |
| Хиральные S-монотерпенилцистеины | 2018 |
|
RU2689381C1 |
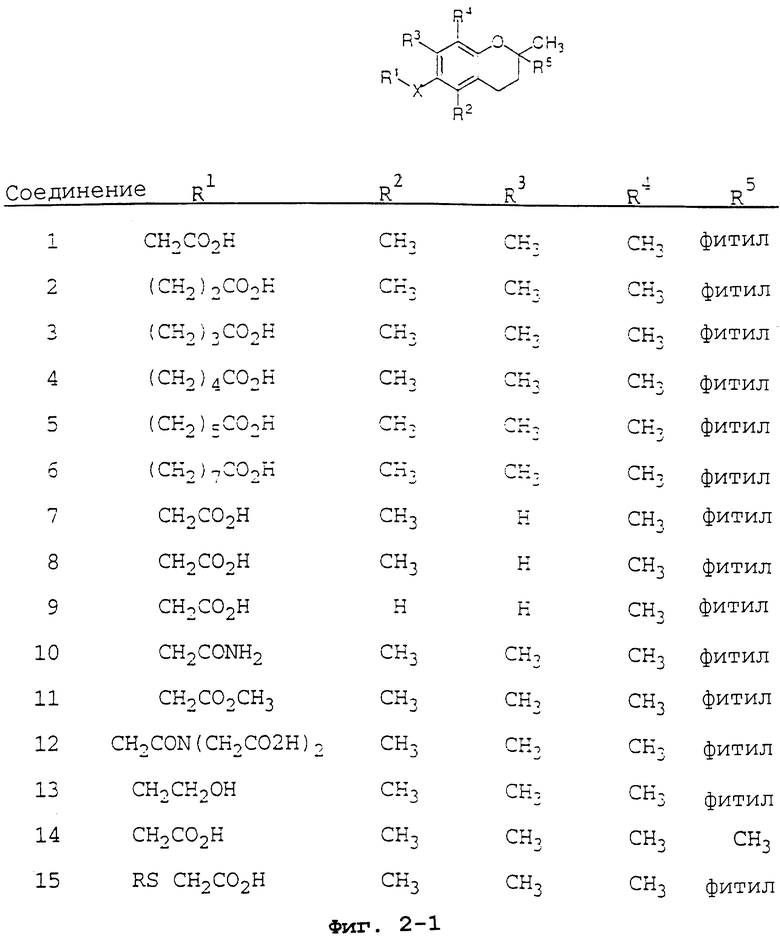
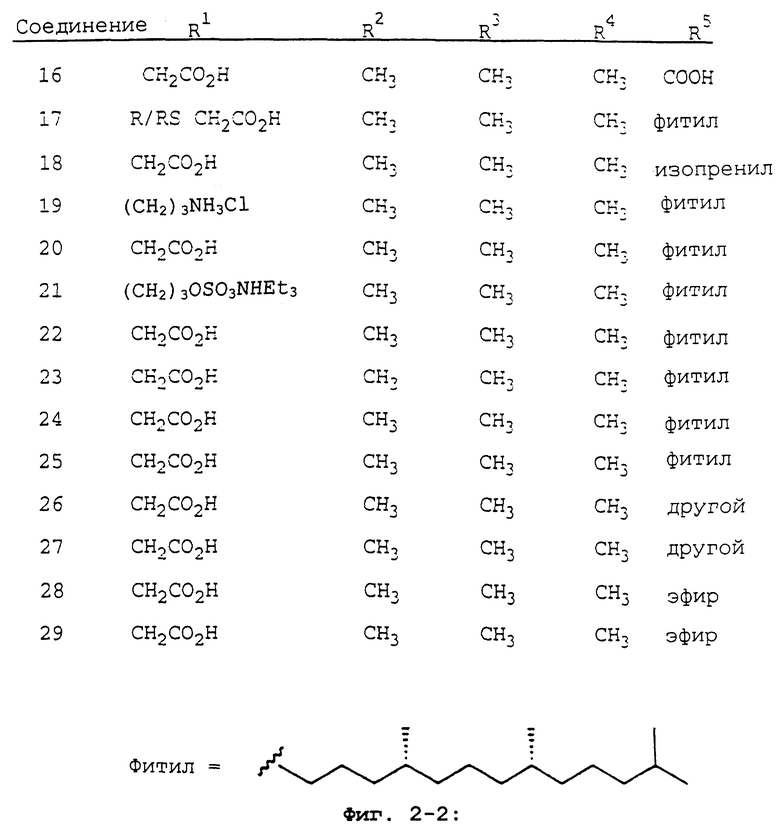
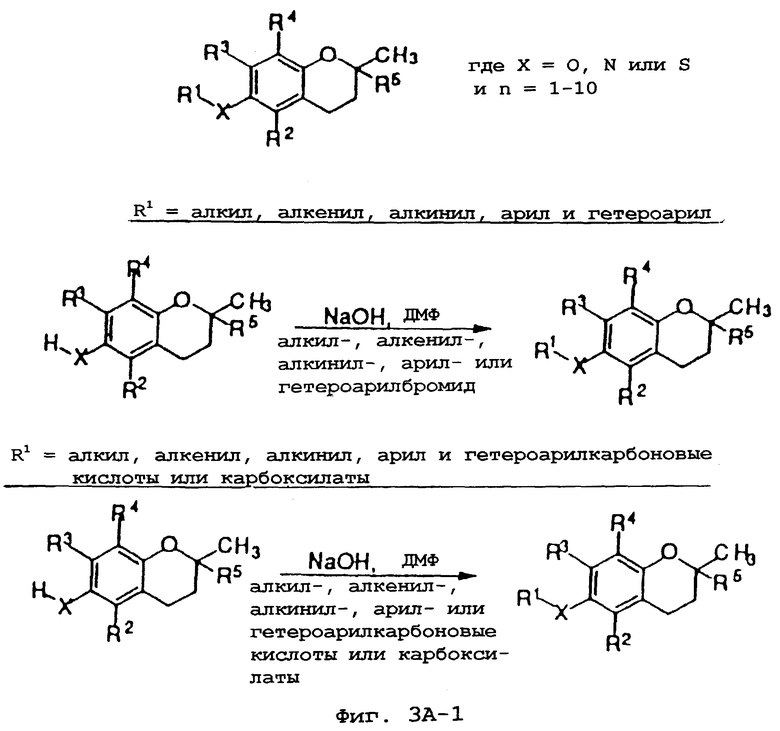



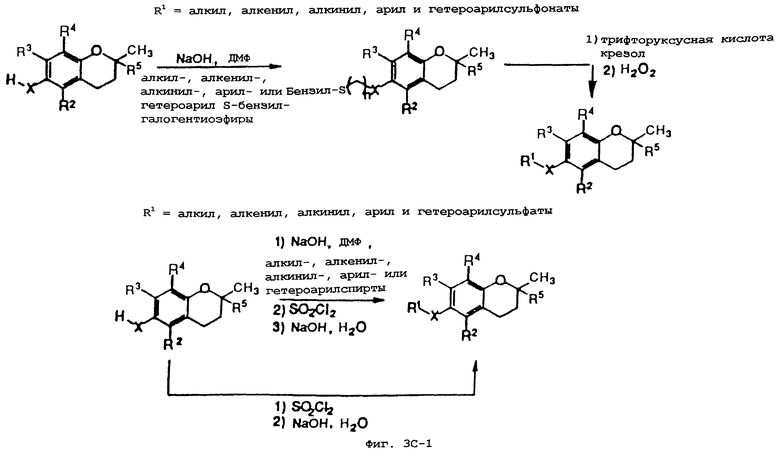
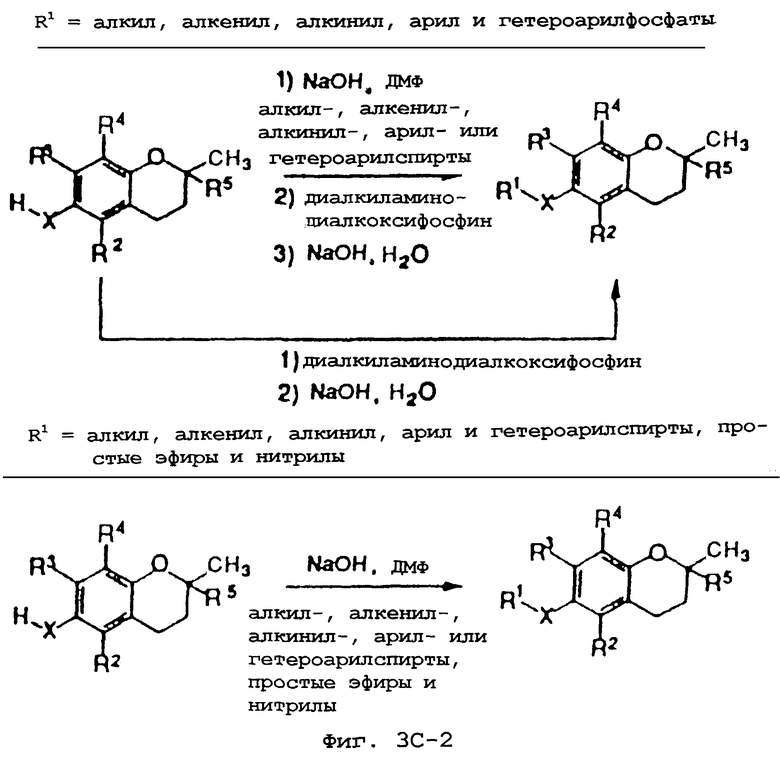

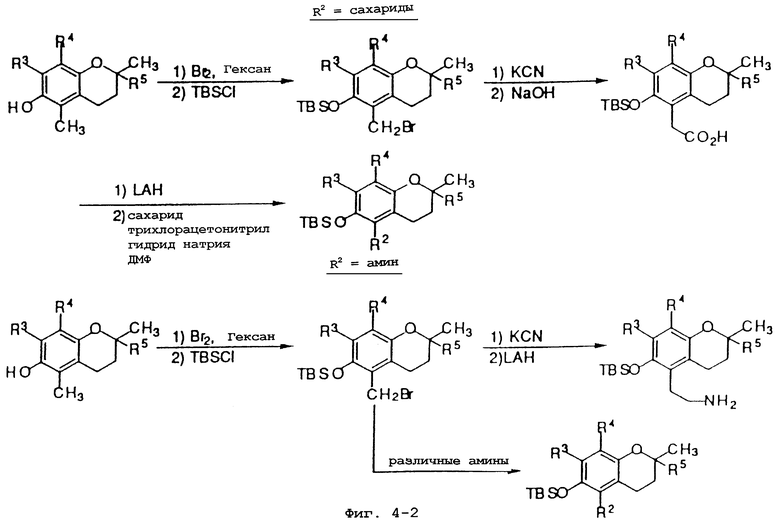

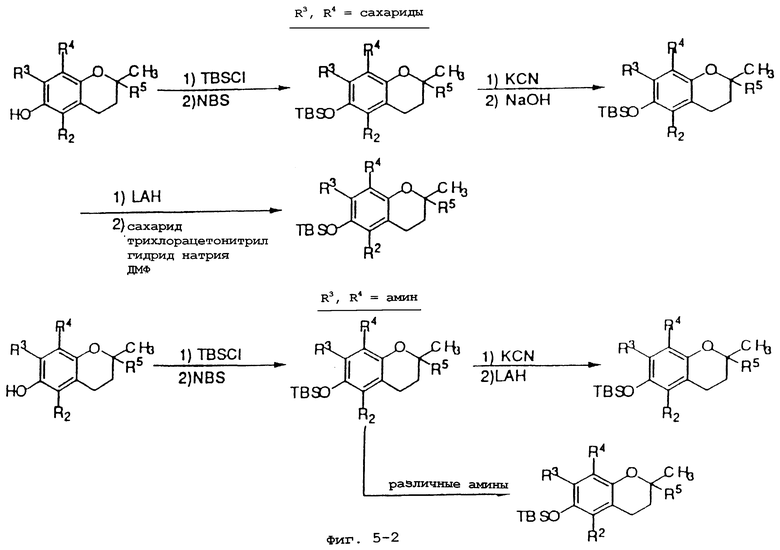
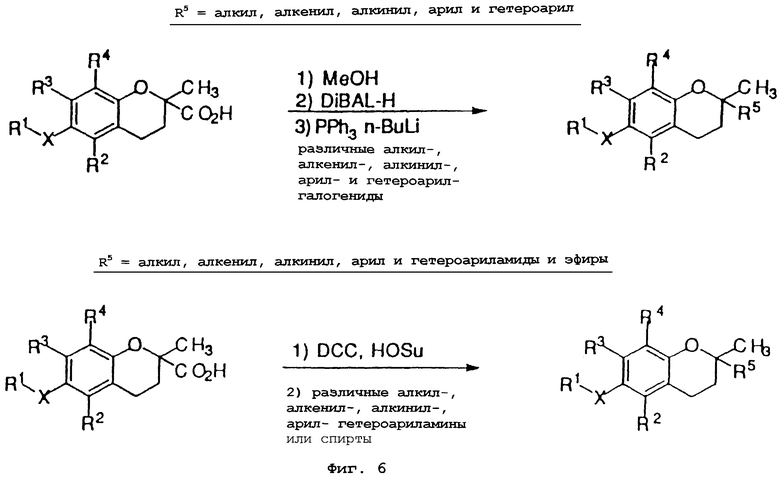
Изобретение относится к новым производным токоферола, токотриенола и другим производным хромана и боковых цепей формулы 1:  ,
,
где Х представляет собой кислород; R1 представляет собой группу -С1-10алкилен-СООН, -С1-4алкилен-CONH2, -C1-4 алкилен-СОО-С1-4алкил, -С1-4алкилен-CON(С1-4алкилен-COOH)2, -С1-4алкилен-ОН, -СН2(СН2)2-NH3-CI или -С1-4алкилен-OSO3NH(С1-4алкил)3; R2 и R3 представляют собой водород или метил; R4 представляет собой метил; и R5 представляет собой группу -С7-17 алкил, -СООН, -С7-16-олефиновую группу, содержащую от 3 до 5 этиленовых связей, -С=С-СОО-С1-4алкил или -С1-4алкилен-СОО-С1-4алкил; при условии, что R1 не может являться ни группой -С2-4алкилен-СООН, ни -C1-4 алкилен-CONH2, ни -С1-4алкилен-ОН, когда каждый из R2, R3 и R4 представляет собой метил, а R5 представляет собой -С16алкил, а также к способу лечения клеточно-пролиферативных заболеваний и способу индукции апоптоза клетки. Технический результат - получение новых производных токоферола, токотриенола и других производных хромана и боковых цепей, обладающих ингибирующим действием в отношении нежелательной или неконтролируемой пролифекации клеток. 3 н. и 15 з.п. ф-лы., 2 табл., 14 ил.
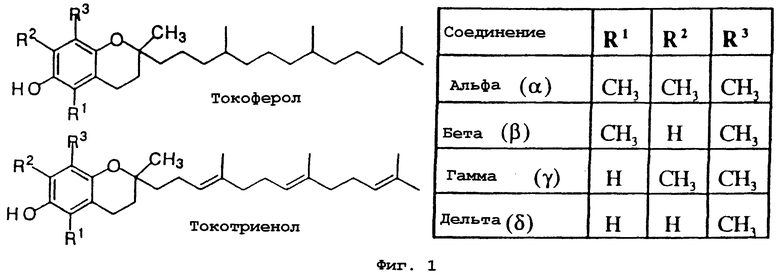

где Х представляет собой кислород;
R1 представляет собой группу -С1-10алкилен-СООН, -C1-4алкилен-CONH2, -C1-4 алкилен-СОО-С1-4алкил, -С1-4алкилен-СОN(С1-4алкилен-СООН)2, -С1-4алкилен-ОН, -CH2(CH2)2-NH3-Cl или -С1-4алкилен-OSO3NH(С1-4алкил)3;
R2 и R3 представляют собой водород или метил;
R4 представляет собой метил;
R5 представляет собой группу -С7-17алкил, -СООН, -С7-16-олефиновую группу, содержащую от 3 до 5 этиленовых связей, -С=C-СОО-С1-4алкил или -С1-4алкилен-СОО-С1-4алкил;
при условии, что R1 не может являться ни группой -С2-4алкилен-СООН, ни -C1-4 алкилен-СОNН2, ни -С1-4алкилен-ОН, когда каждый из R2, R3 и R4 представляет собой метил, а R5 представляет собой -С16алкил.
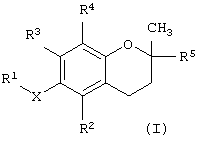
где X представляет собой кислород;
R1 представляет собой группу -С1-10алкилен-СООН, -С1-4алкилен-СONН2, -C1-4 алкилен-СОО-С1-4алкил, -С1-4алкилен-СОN(С1-4алкилен-СООН)2, -С1-4алкилен-ОН, -CH2(CH2)2-NH3-Cl или -С1-4алкилен-OSO3NН(С1-4алкил)3;
R2 и R3 представляют собой водород или метил;
R4 представляет собой метил;
R5 представляет собой группу -С7-17алкил, -СООН, -С7-16-олефиновую группу, содержащую от 3 до 5 этиленовых связей, -С=С-СОО-С1-4алкил или -С1-4алкилен-СОО-С1-4алкил,
или содержащей их фармацевтической композиции.
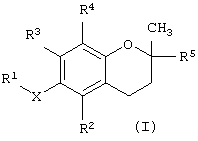
где X представляет собой кислород;
R1 представляет собой группу -С1-10алкилен-СООН, -С1-4алкилен-СОNН2, -C1-4 алкилен-СОО-С1-4алкил, -С1-4алкилен-СОN(С1-4алкилен-СООН)2, -С1-4алкилен-ОН, -СН2(CH2)2-NH3-Cl или -С1-4алкилен-OSO3NН(С1-4алкил)3;
R2 и R3 представляют собой водород или метил;
R4 представляет собой метил;
R5 представляет собой группу -С7-17алкил, -СООН, -С7-16-олефиновую группу, содержащую от 3 до 5 этиленовых связей, -С=С-СОО-С1-4алкил или -С1-4алкилен-СОО-С1-4алкил.
| RU 96104250 А, 10.06.1998.WO 98104250 А1, 30.04.1998.US 5114957 А, 19.05.1992. |

