Изобретение относится к усовершенствованному способу получения 6,7-дихлор-1,5-дигидроимидазо-[2,1-b]-хиназолин-2(3H)-она, к новым промежуточным продуктам, используемым для получения его, а также к способу получения указанных н овых промежуточных продуктов.
Таким образом предложен усовершенствованный способ получения 6,7-дихлор-1,5-дигидроимидазо[2,1-b]хиназолин-2(3Н)-она формулы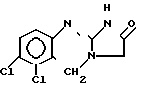 (I) (далее именуемого, как "анагрелид").
(I) (далее именуемого, как "анагрелид").
Известно, что анагрелид это ценное соединение, препятствующее агрегации тромбоцитов крови, которое является сильнодействующим ингибитором циклической аденазин монофосфат фосфодиэстеразы.
Известно несколько способов получния анагрелида.
Известен способ получения гидробромида анагрелида, который заключается в том, что диамин общей формулы NH-CH2-R (II) где R сложная эфирная группа, подвергается кипячению с бромцианом в этиловом спирте. Реакция протекает довольно долго, она продолжается 22 ч. Усовершенствованный способ раскрывается в патенте США N 4146718. Указанным способом диамин общей формулы (II) подвергается кипячению с хлорцианом, бромцианом или иодцианом в течение длительного времени, т.е. 12-18 ч, приводя к получению в качестве промежуточного продукта соли 2-иминохиназолина общей формулы
NH-CH2-R (II) где R сложная эфирная группа, подвергается кипячению с бромцианом в этиловом спирте. Реакция протекает довольно долго, она продолжается 22 ч. Усовершенствованный способ раскрывается в патенте США N 4146718. Указанным способом диамин общей формулы (II) подвергается кипячению с хлорцианом, бромцианом или иодцианом в течение длительного времени, т.е. 12-18 ч, приводя к получению в качестве промежуточного продукта соли 2-иминохиназолина общей формулы (IV) где R обозначает сложную эфирную группу, а Наl представляет собой галоген. Затем основание, выделенное в свободном состоянии из этого промежуточного продукта, подвергается кипячению в течение 4 ч в этиловом спирте с образованием анагрелида.
(IV) где R обозначает сложную эфирную группу, а Наl представляет собой галоген. Затем основание, выделенное в свободном состоянии из этого промежуточного продукта, подвергается кипячению в течение 4 ч в этиловом спирте с образованием анагрелида.
Недостатки обоих способов состоят в применении чрезвычайно токсичных галогенидов дициана, требующих особой технологии и специального оборудования для производства указанного соединения в промышленном масштабе.
Согласно способу, описанному в патенте США N 3932407, диамин общей формулы (II) подвергается реакции циклизации с 1,1-карбонилдиимидазолом, полученный при этом соответствующий сложный эфир хиназолин-2-она общей формулы (V) где R это сложная эфирная группа, реагирует с хлорангидридом фосфорной кислоты с образованием соответствующего эфира хиназолина общей формулы
(V) где R это сложная эфирная группа, реагирует с хлорангидридом фосфорной кислоты с образованием соответствующего эфира хиназолина общей формулы где R это сложная эфирная группа. Последнее из указанных соединений подвергается затем кипячению вместе с раствором аммиака в этиловом спирте, давая анагрелид. Недостатки этого способа заключаются в том, что 1,1-карбонилдиимидазол- вещество малораспространненное, и, кроме того, при реакции сложного эфира хиназолин-2-она общей формулы (V) с хлорангидридом фосфорной кислоты чувствительная к действию кислот сложная эфирная группа распадается, вызывая образование значительного количества побочных продуктов. Заключительная ступень этого синтеза также неблагоприятная, так как реакция с раствором аммиака в этиловом спирте должна проводиться при повышенном давлении и протекает она довольно длительное время, т.е. 16 ч.
где R это сложная эфирная группа. Последнее из указанных соединений подвергается затем кипячению вместе с раствором аммиака в этиловом спирте, давая анагрелид. Недостатки этого способа заключаются в том, что 1,1-карбонилдиимидазол- вещество малораспространненное, и, кроме того, при реакции сложного эфира хиназолин-2-она общей формулы (V) с хлорангидридом фосфорной кислоты чувствительная к действию кислот сложная эфирная группа распадается, вызывая образование значительного количества побочных продуктов. Заключительная ступень этого синтеза также неблагоприятная, так как реакция с раствором аммиака в этиловом спирте должна проводиться при повышенном давлении и протекает она довольно длительное время, т.е. 16 ч.
Известна реакция циклизации соединения общей формулы (VI), где R -% этоксикарбонил, с раствором аммиака в этиловом спирте, но эта реакция также осуществляется при повышенном давлении в закрытом сосуде высокого давления при 120оС в течение 16 ч, поэтому этот способ также невыгоден для производства в промышленном масштабе.
Известен еще один способ (патент США N 3.932.467) получения соединения формулы (I). В соответствии с этим способом диамин общей формулы (VIII) где R сложная эфирная группа, реагирует с галоиддианом с образованием хиназолина общей формулы
(VIII) где R сложная эфирная группа, реагирует с галоиддианом с образованием хиназолина общей формулы (IX) где R сложная эфирная группа, который затем посредством реакции циклизации превращется в производное имидазохиназолинона общей формулы
(IX) где R сложная эфирная группа, который затем посредством реакции циклизации превращется в производное имидазохиназолинона общей формулы (X)
(X)
Последнее соединение хлорируется при повышенном давлении в присутствии безводного хлорида железа, давая анагрелид. Серьезный недостаток этого способа, в дополнении к вышеупомянутым, заключается в том, что в ходе процесса хлорирования получаются не только нужное 6,7-дихлорпроизводное, но и другие дихлоризомеры, хлорированные по другим положениям ароматического кольца, и их отделение от cоединения формулы (I) довольно затруднительно. В патенте ничего не говорится о выделении, идентификации и даже о температуре плавления соединения формулы (I), лишь описывается 6-хлор-7-бром-аналог этого соединения, а данные, касающиеся выхода, приведены только для неочищенного продукта. Кроме того, в патенте не сообщается ни о потерях, связанных с очисткой, ни о разделении изомеров.
Согласно способу, описанному в патенте США N 4208521, по которому, диамин общей формулы (II), где R обозначает сложную эфирную группу, реагирует с амидин- производным общей формулы C-x (VII) где Х обозначает отщепляющуюcя группу, с прямым получением анагрелида. В качестве отщепляющихся групп указываются амино-, алкилтио-, арилтио-группы, а также азотсодержащие гетероциклические группы, которые могут иметь в своем составе заместителей. В патенте, однако, описывается только наиболее реакционноспособный 1-амидино-3,5-диметилпиразол, который подвергается кипячению в течение 24 ч для получения анагрелида с выходом в 40% Очень длительное время реакции и низкий выход делают этот способ неэкономичным для производства в промышленном масштабе.
C-x (VII) где Х обозначает отщепляющуюcя группу, с прямым получением анагрелида. В качестве отщепляющихся групп указываются амино-, алкилтио-, арилтио-группы, а также азотсодержащие гетероциклические группы, которые могут иметь в своем составе заместителей. В патенте, однако, описывается только наиболее реакционноспособный 1-амидино-3,5-диметилпиразол, который подвергается кипячению в течение 24 ч для получения анагрелида с выходом в 40% Очень длительное время реакции и низкий выход делают этот способ неэкономичным для производства в промышленном масштабе.
Цель изобретения создание способа, который устраняет недостатки известных способов и позволяет также осуществить подходящим образом получение соединения формулы (I) в промышленном масштабе.
Цель достигается тем, что предложен способ получения 6,7-дихло-1,5-дигидроимидазо[2,1-Ь] хиназолин-2(3Н)-она формулы (I) и его фармацевтически пригодных солей, который заключается в том, что производное 2-цианоиминохиназолина общей формулы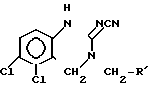 (III) где R' обозначает цианогруппу или группу формулы COORI, причем RIпредсталяет собой низший алкил, который в случае необходимости может иметь в своем составе фенильный заместитель, подвергается реакции термической циклизации в кислой среде.
(III) где R' обозначает цианогруппу или группу формулы COORI, причем RIпредсталяет собой низший алкил, который в случае необходимости может иметь в своем составе фенильный заместитель, подвергается реакции термической циклизации в кислой среде.
Термин "низший алкил" относится к линейным или разветвленным алкильным группам, имеющим 1-6, предпочтительно 1-4 атома углерода, таким как метильная, этильная, пропильная, изопропильная, бутильная, трет-бутильная, пентильная, гексильная и т.д. группы. Эти группы в случае необходимости могут иметь в качестве заместителя одну фенильную группу у любого атома углерода.
Реакция циклизации осуществляется в смеси инертного растворителя и минеральной кислоты при кипячении в течение 20-180 мин. В реакции могут быть использованы растворители, имеющие температуру кипения выше 80оС, предпочтительно этиленгликоль, диметиловый эфир диэтиленгликоля, диэтиловый эфир диэтиленгликоля, 2-метоксиэтиловый спирт, N-метилпирролидон, диметилформамид, ацетонитрил или диоксан. В качестве минеральной кислоты предпочтительно используется хлористоводородная кислота.
Реакция также может быть осуществлена при использовании в качестве растворителя низшей органической киcлоты, предпочтительно уксусной. В таких случаях нет необходимости использовать минеральную кислоту.
Реакция термической циклизации предпочтительно проводится при 80-130оС, при этом реакция предпочтительно заканчивается через 20-180 мин. После окончания реакции реакционную смесь охлаждают и продукт отфильтровывают либо в виде соли, или после нейтрализации в виде свободного основания.
Исходные вещества общей формулы (III) новые соединения.
Согласно изобретению получены новые производные 2-цианоиминохиназолина общей формулы (III), где R представляет собой цианогруппу или группу формулы COOR1, при этом в последней группе R1обозначает низший алкил, который в случае необходимости может иметь в своем составе фенильный заместитель.
Кроме того, предложен также способ получения соединений общей формулы (III), который заключается в том, что диамин общей формулы (II), где R представляет собой цианогруппу или группу формулы COOR1, при этом в последней группе R1 обозначает низший алкил, который в случае необходимости может иметь в своем составе фенильный заместитель, реагирует с цианопроизводным общей формулы (XI) где L отщепляющаяся группа, предпочтительно группа формулы XR2, при этом в последней группе Х обозначает кислород или серу, а R2 низшую алкильную группу, которая в случае необходимости может иметь в своем составе фенильный заместитель.
(XI) где L отщепляющаяся группа, предпочтительно группа формулы XR2, при этом в последней группе Х обозначает кислород или серу, а R2 низшую алкильную группу, которая в случае необходимости может иметь в своем составе фенильный заместитель.
Реакция веществ, имеющих общие формулы (II) и (XI), проводится в инертном по отношению к реагентам растворителе, предпочтительно в спирте или же неполярном, или полярном апротонном растворителе. В качестве наиболее предпочтительных растворителей можно указать следующие: изопропиловый спирт, бензол, диметилформамид или ацетонитрил. Реакция осуществляется при 0-100оС, предпочтительно 20-80оС, наиболее предпочтительно при комнатной температуре.
Вещества общей формулы (III) могут быть выделены из реакционной смеси известными методами, вообще стандартным фильтрованием, за которым следует перекристаллизация из соответствующего растворителя.
Вещества общей формулы (II), использующиеся в качестве исходных, могут быть получены, например, посредством восстановления нитро- и цианогрупп 2,3-дихлор-6-нитробензонитрила с последующей реакцией образовавшегося при этом соединения формулы (XII) с эфиром галогенуксусной кислоты или нитрилом галогенуксусной кислоты общей формулы
(XII) с эфиром галогенуксусной кислоты или нитрилом галогенуксусной кислоты общей формулы
Х СН2 R (XIII) где Х обозначает галоген, а R группа, указанная выше.
Изобретение касается способа получения анагрелида формулы (I), который обладает преимуществами по сравнению с известными способами благодаря устранению токсичных реагентов. простым ступеням реакции, непродолжительному времени реакции, высокому выходу и чистоте продукта.
П р и м е р 1. 6,7-Дихлор-1,5-дигидроимидазо[2.1-Ь]хиназолин-2(3Н)-он гидрохлорид полугидрат.
К раствору, состоящему из 150 мл этиленгликоля и 15 мл (0,15 моля) концентрированной хлористоводородной кислоты малыми порциями в течение 10 мин при 115оС добавляют 24,5 г (0,075 моля) этил-(2-цианоимино-5,6-дихлор-1,2,3,4-тетрагидро- хиназолин-3-ил) ацетата, затем смесь выдерживают при той же температуре еще в течение 20 мин. Затем ее охлаждают до комнатной температуры и нейтрализуют, добавляя по каплям 10%-ный раствор карбоната калия. Выделившиеся кристаллы отфильтровывают. Таким образом получают 17,75 г 93,0% ) 6,7-дихлор-1,5-дигидроимидазол[2,1-Ь] хиназолин-2(3Н)-она основания (температура плавления >300оС, содержание указанного вещества, определенное методом жидкостной хроматографии высокого разрешения 99,5%), которое растворяют в горячей смеси, состоящей из 150 мл метилового спирта и 150 мл концентрированного раствора хлорводорода в этиловом спирте (содержание хлорводорода 0,75г моля/1000 мл). Затем смесь охлаждают, выделившийся продукт отфильтровывают и промывают небольшим количеством этилового спирта.
Выход 17,4 г (82.8%).
Температура плавления: 300оС, содержание продукта, определенное методом жидкостной хроматографии высокого разрешения 99,9%
П р и м е р 2. 6.7-Дихлор-1,5-дигидроимидазо[2,1-Ь]-хиназолин-2(3Н)-он гидрохлорид полугидрат.
К раствору, состоящему из 10 мл этиленгликоля и 0,5 мл (0,005 моля) концентрированной хлористоводородной кислоты, нагретому до 120оС, добавляют 0,28 г (0,001 моля) (2-цианимино-5,6-дихлор-1,2,3,4-тетрагидрохина- золин-3-ил)ацетонитрила, раствор перемешивают при той же температуре в течение 20 мин. Затем его охлаждают до комнатной температуры и нейтрализуют 10%-ным раствором карбоната калия. Выделившиеся кристаллы отфильтровывают. Таким образом получают 0,27 г (96,4% ) неочищенного 6,7-дихлор-1,5-дигидроимидазо[2,1-Ь] -хиназо-лин-2(3Н)-она основания (температура плавления >300оС, содержание указанного вещества, определенное методом жидкостной хроматографии высокого разрешения 96,3%), которое растворяют в горячей смеси, состоящей из 7 мл метилового спирта и 6 мл концентрированного раствора хлорводорода в этиловом спирте (содержание хлорводорода 0,76 моля/1000 мл) и оставляют кристаллизоваться. Затем смесь охлаждают, выделившийся продукт отфильтровывают и промывают небольшим количством этилового спирта.
Выход 0,25 г (78,2%).
Температура плавления >300оС, содержание продукта, определенное методом жидкостной хроматографии высокого разрешения 99,9%
П р и м е р 3. 6.7-Дихлор-1,5-дигидроимидазо[2,1-Ь]хиназолин-2(3Н)-он гидрохлорид полугидрат.
К смеси, состоящей из 15 мл этиленгликоля и 2 мл (0,02 моля) концентрированной хлористоводородной кислоты, нагретой до 120оС, добавляют 2,50 г (0,01 моля) (2-цианимино-5,6-дихлор-1,2,3,4-тетрагидрохин- азолин-3-ил)ацетонитрила, раствор перемешивают при той же температуре в течение 20 мин. Затем его охлаждают до комнатной температуры и нейтрализуют 10%-ным раствором карбоната калия. Выделившиеся кристаллы отфильтровывают. Таким образом получают 2,72 г (97,1%) неочищенного 6,7-дихлор-1,5-дигидроимидазо[2,1-Ь]хиназолин-2(3Н)-она в виде основания (температура плавления >300оС, содержание указанного соединения, определенное методом жидкостной хроматографии высокого разрешения 97,2% ), которое растворяют в горячей смеси, состоящей из 15 мл метилового спирта и 16 мл концентрированного раствора хлороводорода в этиловом спирте (одержание хлороводорода 0,76 моля/1000 мл) и оставляют кристаллизоваться. Затем смесь охлаждают, выделившийся продукт отфильтровывают и промывают небольшим количеством этилового спирта.
Выход 2,56 г (84,5%).
Температура плавления >300оС, содержание продукта, определенное методом жидкостной хроматографии высокого разрешения 99,2%
П р и м е р 4. 6.7-Дихлор-1,5-дигидроимидазо[2,1-Ь]хиназолин-2(3Н)-он.
0,163 г (0,0005 моля) этил (2-цианимино-5,6-дихлор-1,2,3,4-тетрагидрохиназолин-3-ил) ацетата добавляют к 2 мл уксусной кислоты при комнатной температуре, полученную при этом суспензию нагревают до 100оС при перемешивании и выдерживают при той же температуре в течение 30 мин. Затем ее охлаждают, выделившиеся кристаллы отфильтровывают и промывают ацетонитрилом.
Выход 0,116 г (91%).
Температура плавления >300оС, содержание продукта, определенное методом жидкостной хроматографии высокого разрешения 99,15%
П р и м е р 5. 6,7-Дихлор-1,5-дигидроимидазо[2,1-Ь]-хиназолин-2-(3Н)-он гидрохлорид полугидрат.
0,2 мл концентрированной хлористоводородной кислоты и 0,163 г (0,0005 моля) этил (2-цианимино-5,6-дихлор-1,2,3,4-тетрагидрохиназолин-3-ил) ацетата добавляют к 2 мл диметилового эфира диэтиленгликоля при комнатной температуре. Реакционную смесь нагревают до 110оС и выдерживают при той же температуре в течение 60 мин. Затем раствор охлаждают,выделившиеся кристаллы отфильтровывают и промывают этиловым спиртом.
Выход 0,121 г (80%).
Содержание продукта, определенное методом жидкостной хроматографии высокого разрешения 86%
Маточный раствор подщелачивают до рН 8, добавляя триэтиламин, полученное при этом свбодное основание отфильтровывают и промывают этиловым спиртом.
Выход 0,010 г (7,8%).
Содержание продукта, определенное методом жидкостной хроматографии высокого разрешения 90%
Суммарный выход 87,8%
П р и м е р 6. 6.7-Дихлор-1,5-дигидроимидазо[2,1-Ь]хиназолин-2(3Н)-он гидрохлорид полугидрат.
Реакцию проводят согласно примеру 5 за исключением того, что вместо диметолового эфира диэтиленгликоля используют диэтиловый эфир диэтиленгликоля.
Выход 0,123 г (81%).
Содержание продукта, определенное методом жидкостной хроматографии высокого разрешения 87,8%
Выход свободного основания, полученного из маточного раствора 0,011 г (8,2% ), содержание продукта, определенное методом жидкостной хроматографии высокого разрешения 91,3%
Суммарный выход 89,2%
П р и м е р 7. 6,7-Дихлор-1,5-дигидроимидазо[2,1-Ь] хиназолин-2[3Н]-он гидрохлорид полугидрат.
Поступают по примеру 5 за исключением того, что вместо диметилового эфира диэтиленгликоля используют диметиловый эфир этиленгликоля, а реакция проводится при 90оС в течение 100 мин.
Выход 0,124 г (81,8%).
Содержание продукта, определенное методом жидкостной хроматографии высокого разрешения 88,2%
Выход свободного основания, полученного из маточного раствора 0,014 г (11% ), содержание продукта, определенное методом жидкостной хроматографии высокого разрешения 93%
Суммарный выход 92,8%
П р и м е р 8. 6,7-Дихлор-1,5-дигидроимидазо[2,1-Ь]хиназолин-2(3Н)-он гидрохлорид полугидрат.
Поступают по примеру 5 за исключением того, что вместо диметилового эфир диэтиленгликоля используют 2-метоксиэтиловый спирт, а реакционную смесь оставляют кристаллизоваться при 0оС вместо комнатной температуры.
Выход 0,100 г (66%).
Содержание продукта, определенное методом жидкостной хроматографии высокого разрешения, 79%
Выход свободного основания, полученного из маточного раствора 0,01 г (7,8%). Суммарный выход 73,8%
П р и м е р 9. 6,7-Дихлор-1,5-дигидроимидазо[2,1-хиназолин-2(3Н)-она гидрохлорид полугидрат.
Проводят реакцю по примеру 8 за исключением того, что вместо 2-метоксиэтилового спирта используется 2-этоксиэтиловый спирт, а реакционную смесь оставляют реагировать в течение 785 мин вместо 60 мин.
Выход 0,106 г (70%).
Маточный раствор подщелачивают, добавляют концентрированный раствор аммиака, и получают 0,013 г (10,3%) свободного основания. Содержание продукта, определенное методом жидкостной хроматографии высокого разрешения 87,3% Суммарный выход 80,3%
П р и м е р 10. 6,7-Дихлор-1,5-дигидроимидазо[2,1-Ь] хиназолин-2(3Н)-он.
0,163 г (0,0005 моля) этил (2-цианимино-5,6-дихлор-1,2,3,4-тетрагидрохиназо- лин -3-ил) ацетата и 0,2 мл концентрированной хлористоводородной кислоты добавляют к 0,2 мл диметилформамида, полученную при этом суспензию нагревают до 120оС при перемешивании и выдерживают при той же температуре в течение 180 мин. В ходе реакции к смеси каждые 60 мин добавляют дополнительные порции хлористоводородной кислоты в общем количестве 0,2 мл. Полученный при этой раствор охлаждают, раствор подщелачивают (рН 8), добавляя триэтиламин, и оставляют его стоять на ночь в холодильнике. Выделившиеся кристаллы отфильтровывают и промывают ацетонитрилом.
Выход 0,070 г (54,7%), содержание продукта, определегнное методом жидкостной хроматографии высокого разрешения 87,3%
П р и м е р 11. 6,7-Дихлор-1,5-дигидроимидазо[2,1-Ь]хиназолин-2(3Н)-он.
Реакцию ведут по примеру 10 за исключением того, что вмеасто диметилформамида используют N-метилпирролидон, а реакция проводится при 130оС в течение 120 мин.
Выход 0,076 г (59,4%), содержание продукта, определенное методом жидкостной хроматографии высокого разрешения 87,8%
П р и м е р 12. 6,7-Дихлор-1,5-дигидроимидазо[2,1-b]хиназолин-2(3Н)-он гидрохлорид полугидрат.
0,140 г (0,0005 моля) (2-цианимино-5,6-дихлор-1,2,3,4-тетрагидрохиназолин-3-ил)-ацетонитрила и 0,2 мл концентрированной хлористоводородной кислоты добавляют к 2 мл ацетонитрила. Реакционную смесь нагревают до 80оС при перемешивании и выдерживают при той же температуре в течение 50 мин. Затем ее охлаждают. выделившиеся кристаллы отфильтровывают и промывают ацетонитрилом.
Выход 0,138 г (91,1%), содержание продукта, определенное методом жидкостной хроматографии высокого разрешения 99,0%
П р и м е р 13. 6,7-Дихлор-1,5-дигидроимидазо[2,1-b]хиназолин-2(3Н)-он.
Поступают по примеру 12 за исключением того, что вместо ацетонитрила используют диоксан, а реакционную смесь перемешивают при 100оС в течение 30 мин.
Выход 0,139 г (91,7%), содержание продукта, определенное методом жидкостной хроматографии высокого разрешения 99,2%
П р и м е р 14. 6,7-Дихлор-1,5-дигидроимидазо[2,1-b]хиназолин-2(3Н)-он.
Реакцию проводят по примеру 12 за исключением того, что вместо ацетонитрила использдуют диметилформамид, а реакционную смесь перемешивают при 100оС в течение 100 мин. В ходе реакции добавляют дополнительные порции концентрированной хлористоводородной кислоты (2х0,2 мл) через 30 и 60 мин соответственно. Затем смесь охлаждают, делают реакцию среды щелочной (рН 8), добавляя 10%-ный раствор карбоната калия, выделившиеся кристаллы отфильтровывают и промывают небольшим количеством воды и ацетонитрила.
Выход 0,080 г (62,5%) содержание продукта, определенное методом жидкостной хроматографии высокого разрешения 88%
П р и м е р 15. 6,7-Дихлор-1,5-дигидроимидазо[2,1-b]хиназолин-2(3Н)-он.
Поступают по примеру 14 за исключением того, что вместо диметилформамида используют N-метилпирролидон.
Выход 0,078 г (61,0%), содержание продукта, определенное методом жидкостной хроматографии высокого разрешения 87,3%
П р и м е р 16. 6,7-Дихлор-1,5-дигидроимидазо[2,1-b]хиназолин-2(3Н)-он гидрохлорид полугидрат.
0,140 г (0,0005 моля) (2-цианимино-5,6-дихлор-1,2,3,4-тетрагидрохиназолин-3-ил)-ацетонитрила и 0,2 мл концентрированной хлористоводородной кислоты добавляют к 2 мл 2-метоксиэтилового спирта, реакция в смеси идет при 100оС в течение 50 мин. Затем смесь охлаждают и оставляют стоять на ночь в холодильнике. После этого выделившиеся кристаллы отфильтровывают и промывают ацетонитрилом.
Выход 0,110 г (72,6%), содержание продукта, определенное методом жидкостной хроматографии высокого разрешения 82%
Реакцию среды фильтрата делают щелочной (рН 8), добавляя концентрированный раствор аммиака, а выделившиеся кристаллы фильтруют и промывают водой и ацетонитрилом.
Выход свободного основания, полученного из маточного раствора 0,011 г (8,6% ), содержание продукта, определнное методом жидкостной хроматографии высокого разрешения 87,2%
Суммарный выход 81,2%
П р и м е р 17. 6,7-Дихлор-1,5-дигидроимидазо[2,1-b]хиназолин-2(3Н)-он гидрохлорид полугидрат.
Реакцию проводят по примеру 16 за исключением того, что вместо 2-метоксиэтилового спирта используют 2-этоксиэтиловый спирт.
Выход 0,115 г (76г,0%), содержание продукта, определенное методом жидкостной хроматографии высокого разрешения 84,5%
Выход свободного основания, полученного из маточного раствора 0,011 г (8,6% ), содержание продукта, определенное методом жидкостной хроматографии высокого разрешения 88,3%
Суммарный выход 84,6%
П р и м е р 18. 6,7-Дихлор-1,5-дигидроимидазо[2,1-b]хиназолин-2(3Н)-он гидрохлорид полугидрат.
Реакцию проводят по примеру 16 за исключением того, что вместо 2-метоксиэтилового спирта используют диметиловый эфир этиленгликоля, а реакционную смесь выдерживают при 90оС в течение 70 мин.
Выход 0,128 г (84,5%), содержание продукта, определенное методом жидкостной хроматографии высокого разрешения 92,5%
При добавлении к маточному раствору триэтиламина получается 0,011 г (8.6%) 6,7-дихлор-1,5-дигидроимидазо[2,1-b]хиназолин-2(3Н)он в виде основания. Содержание продукта, определенное методом жидкостной хроматографии высокого разрешения: 95,7% суммарный выход: 93,1%
П р и м е р 19. 6,7-Дихлор-1,5-дигидроимидазо[2,1-b]хиназолин-2(3Н)-он гидрохлорид полугидрат.
Поступают так, как описано в примере 18 за исключением того, что вместо диметилового эфира этиленгликоля используют диметиловый эфир диэтиленгликоля.
Выход 0,127 г (83,8%), содержание продукта, определенное методом жидкостной хроматографии высокого разрешения 92,3%
Выход свободного основания, полученного из маточного раствора 0,011 г (8,6% ), содержание продукта, определенное методом жидкостной хроматографии высокого разрешения 95,8%
Суммарный выход 92,4%
П р и м е р 20. 6,7-Дихлор-1,5-дигидроимидазо[2,1-b]хиназолин-2(3Н)-он гидрохлорид полугидрат.
Поступают так, как описано в примере 18 за исключением того, что вместо 2-метоксиэтилового спирта используют диэтиловый эфир диэтиленгликоля.
Выход 0,125 г (82,5%), содержание продукта, определенное методом жидкостной хроматографии высокого разрешения 92,0%
Выход свободного основания, полученного из маточного раствора 0,011 г (8,6% ), содержание продукта, определенное методом жидкостной хроматографии высокого разрешения 94,9%
Суммарный выход 91,1%
П р и м е р 21. 6,7-Дихлор-1,5-дигидроимидазо[2,1-b]хиназолин-2(3Н)-он гидрохлорид полугидрат.
0,140 г (0,0005 моля) 2-цианоимино-5,6-дихлор-1,2,3,4-тетрагидрохиназолин-3-ил)- ацетонитрила и 0,2 мл воды добаляют к 2 мл уксусной кислоты. Реакционную смесь нагревают до 80оС при перемешивании и выдерживают при той же температуре в течение 30 мин. Затем ее охлаждают, выделившиеся кристаллы фильтруют и промывают ацетонитрилом.
Выход 0,118 г (92,2%).
Содержание продукта, определенное методом жидкостной хроматографии высокого разрешения 99,3%
Получение исходных веществ.
П р и м е р 22. Этил(2-цианимино-5,6-дихлор-1,1,3,4-тетрагидрохиназолин-3-ил)ацетат.
К суспензии, состоящей из 2,38 г (0,01 моля) дифенил-N-цианоимидокарбоната и 20 мл ацетонитрила при 20-30оС в течение 30 мин при перемешивании по каплям добавляют раствор 2,76 г (0,01 моля) этил (2-амино-5,6-дихлорбензил)-аминоацетата в 20 мл ацетонитрила. Реакционную смесь перемешивают далее в течение 1 ч при комнатной температуре, выделившийся продукт отфильтровывают и промывают небольшим количеством ацетонитрила.
Выход 1,87 г (57,4%).
Температура плавления 274-278оС (содержание продукта, определенное методом жидкостной хроматографии высокого разрешения 99,2%).
Маточный раствор в горячем состоянии обрабатывают активированным углем и выпаривают досуха в вакууме. Осадок растворяют в 10 мл этилацетата, полученный раствор сначала экстрагируют 3 раза, используя порции по 50 мл 10%-ного раствора карбоната калия каждый раз, затем дважды, используя по 5 мл воды каждый раз, затем сушат, выпаривают в вакууме и оставляют кристаллизоваться. Выделившийся продукт отфильтровывают с получением еще 0,66 г (20,2%) вещества, указанного в заголовке примера 22.
Температура плавления 275-278оС (содержание продукта, определенное методом жидкостной хроматографии высокого разрешения, 99,5%).
Суммарный выход 77,6%
П р и м е р 23. Этил (2-цианимино-5,6-дихлор-1,2,3,4-тетрагидрохиназолин-3-ил)ацетат.
К раствору, состоящему из 0,28 г (0,001 моля) этил (2-амино-5,6-дихлорбензол) аминоацетата (содержание продукта, определенное методом газовой хроматографии 82%) и 2 мл бензола, при комнатной температуре и при перемешивании добавляют 0,24 г (0,001 моля) дифенил-N-цианимидкарбоната, реакционную смесь перемешивают при комнатной температуре в течение 20 мин. Выделившийся продукт отфильтровывают и промывают небольшим количеством бензола.
Выход 0,21 г (64,4%).
Температура плавления 263-270оС (содержание продукта, определенное методом жидкостной хроматографии высокого разрешения, 98,2%).
Продукт подвергается перекристаллизации из 4 мл диметилформамида с получением 0,17 г (52%) чистого вещества, указанного в заголовке примера 23.
Температура плавления 268-275оС (содержание продукта, определенное методом жидкостной хроматографии высокого разрешения, 99,9%).
П р и м е р 24. Этил (2-цианимино-5,6-дихлор-1,2,3,4-тетрагидрохиназолин-3-ил)ацетат.
Поступают так, как описано в примере 23, за исключением того, что вместо бензола в качестве растворителя используют 2 мл диметилформамида.
Выход 0,19 г (58,3%).
Температура плавления 271-278оС (содержание продукта, определенное методом жидкостной хроматографии высокого разрешения, 99,7%).
П р и м е р 25. Этил (2-цианимино-5,6-дихлор-1,2,3,4-тетрагидрохиназолин-3-ил)ацетат.
Поступают так, как описано в примере 23 за исключением того, что вместо бензола в качестве растворителя используют 2 мл изопропилового спирта.
Выход 0,23 г (70,6%).
Температура плавления 268-274оС (содержание продукта, определенное методом жидкостной хроматографии высокого разрешения, 99,5%).
П р и м е р 26. Этил (2-цианимино-5,6-дихлор-1,2,3,4-тетрагидрохиназолин-3-ил)ацетат.
К раствору, cостоящему из 7,68 г (0,023 моля) этил(2-амино-5,6-дихлорбензил)аминоацетата (содержание указанного вещества, определенное методом газовой хроматографии 82% ) и 50 мл изопропилового спирта, добавляют 3,8 г (0,026 моля) диметил-N-цианимидкарбоната, реакционную смесь кипятят в течение 3 ч при перемешивании. Затем ее охлаждают, выделившийся продукт отфильтровывают и промывают небольшим количеством холодного изопропилового спирта.
Выход 2,8 г (37,3%).
Температура плавления 272-278оС (содержание продукта, определенное методом жидкостной хроматографии высокого разрешения, 99,5%).
П р и м е р 27. (2-Цианимино-5,6-дихлор-1,2,3,4-тетрагидрохиназолин-3-ил)ацетони- трил.
К суспензии, состоящей из 5,0 г (0,0208 моля) дифенил-N-цианимидкарбоната и 100 мл ацетонитрила, добавляют 4,61 г (0,02 моля) (2-амино-5,6-дихлорбензиламино)ацетонит- рила, реакционную смесь нагревают до 60оС в течение 20 мин при перемешивании и оставляют реагировать при той же температуре в течение еще 1 ч. Затем ее охлаждают, выделившийся продукт отфильтровывают и промывают небольшим количеством ацетонитрила.
Выход 3,53 г (60,9%).
Температура плавления >280оС (содержание продукта, определенное методом жидкостной хроматографии высокого разрешения, 99,5%).
Фильтрат выпаривают приблизительно до одной трети от его первоначального объема с получением 1,2 г (20,7%) вещества, указанного в заголовке примера 27.
Температура плавления >280оС (содержание продукта, определенное методом жидкостной хроматографии высокого разрешения 98,7%).
Суммарный выход 81,6%
П р и м е р 28. Бензил (2-цианимино-5,6-дихлор-1,2,3,4-тетрагидрохиназолин-3-ил)ацетат.
К суспензии, состоящей из 7,2 г (0,030 моля) дифенил-N-цианимидкарбоната и 120 мл л ацетонитрила добавляют 10,20 г (0,030 моля) бензил (2-амин-5,6-дихлорбензил) аминацетата, реакционную смесь нагревают до 60о в течение 20 мин при перемешивании и оставляют реагировать при той же температуре еще в течение 1 ч. Затем ее охлаждают, выделившийся продукт отфильтровывают и промывают небольшим количеством ацетонитрила и ацетона.
Выход 6,94 г (60,0%).
Температура плавления 278-285оС (содержание продукта, определенное методом жидкостной хроматографии высокого разрешения, 98,9%).
Фильтрат выпаривают приблизительно до одной трети от его первоначального объема с получением еще 2,0 г (17,3%) продукта.
Температура плавления 279-284оС (содержание продукта, определенное методом жидкостной хроматографии высокого разрешения 99,4%).
Суммарный выход 77,3%
П р и м е р 29. Этил (3,4-дигидр-5,6-дихлор-2(1Н)-цианоиминохиназолин-3-ил)ацетат.
К раствору, состоящему из 19,8 г (0,1 М) дибензил (N-цианимиддитиокарбоната) в 100 мл диметилформамида, добавляют 27,7 г (0,1 М) 6-амино-2,3-дихлорбензилглицин этилового эфира, реакционную смесь перемешивают при 100оС в течение 5 ч. Затем ее охлаждают, добавляют к ней 100 мл воды и оставляют кристаллизоваться. Выделившийся продукт отфильтровывают и промывают небольшим количеством изопропилового спирта.
Выход 10,1 г (31%).
Температура плавления 274-277оС.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ 5-(ЗАМЕЩЕННЫЙ АМИНО)-1,2,4-ТРИАЗОЛ-[1,5А]-ПИРИМИДИНА, ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ КИСЛОТНО-АДДИТИВНЫЕ СОЛИ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ КАРДИОТОНИЧЕСКИМ И КОРОНАРОРАСШИРЯЮЩИМ ДЕЙСТВИЕМ, И СПОСОБ КАРДИОТОНИЧЕСКОГО И/ИЛИ КОРОНАРОРАСШИРЯЮЩЕГОСЯ ВОЗДЕЙСТВИЯ НА СЕРДЦЕ | 1992 |
|
RU2097382C1 |
| ПРОИЗВОДНЫЕ ДИГИДРОПИРИМИДОТИАЗИНА ИЛИ ИХ ТЕРАПЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ ПРИСОЕДИНЕНИЯ КИСЛОТЫ, ОБЛАДАЮЩИЕ ПРОТИВОАНГИННОЙ И АНТИВОСПАЛИТЕЛЬНОЙ АКТИВНОСТЬЮ | 1990 |
|
RU2022965C1 |
| СПОСОБ ПОЛУЧЕНИЯ 1-(3-ХЛОРФЕНИЛ)-4-МЕТИЛ-7,8-ДИМЕТОКСИ-5Н-2,3-БЕНЗОДИАЗЕПИНА | 1992 |
|
RU2065852C1 |
| ПРОИЗВОДНЫЕ 3-(2-ЗАМЕЩЕННЫЙ ФЕНИЛ)-2-(ЗАМЕЩЕННЫЙ ИМИНО)-ТИАЗОЛИДИНОВ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ АНТИАНГИНАЛЬНЫМ И/ИЛИ АНТИНЕВРАЛГИЧЕСКИМ ЭФФЕКТОМ | 1992 |
|
RU2065855C1 |
| ПРОИЗВОДНЫЕ ТРИАЗОЛИЛГИДРАЗИДА И ИХ ФАРМАЦЕВТИЧЕСКИ ПРИГОДНЫЕ СОЛИ | 1990 |
|
RU2039051C1 |
| ПРОИЗВОДНЫЕ 1,2,4-ТРИАЗОЛИЛТИОАМИДОВ | 1990 |
|
RU2045521C1 |
| СПОСОБ ПОЛУЧЕНИЯ СОЛИ 3-ЭТИЛ-5-МЕТИЛ-2-(2-АМИНОЭТОКСИМЕТИЛ)-4-(2-ХЛОРФЕНИЛ)-6-МЕТИЛ-1,4-ДИГИДРО-3,5-ПИРИДИНДИКАРБОКСИЛАТА И БЕНЗОЛСУЛЬФОКИСЛОТЫ (БЕЗИЛАТА АМЛОДИПИНА) | 1998 |
|
RU2163597C2 |
| ПРОИЗВОДНОЕ 2-(1,2,4-ТРИАЗОЛ-1-ИЛ)-1,3,4-ТИАДИАЗОЛА, СПОСОБ ЕГО ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ | 1998 |
|
RU2180903C2 |
| СПОСОБ ПОЛУЧЕНИЯ ФЛУОКСЕТИНА | 1997 |
|
RU2173679C2 |
| ТРИЦИКЛИЧЕСКИЕ ДИКАРБОНИЛЬНЫЕ ПРОИЗВОДНЫЕ И ЛЕКАРСТВЕННЫЙ ПРЕПАРАТ НА ИХ ОСНОВЕ | 1995 |
|
RU2145606C1 |
Сущность изобретения: продукт -6,7-дихлор- 1,5-дигидроимидазо /2,1-b/ хиназолин-2/3H/-она, БФ C10H7C12N3O т.пл. > 300°С. Реагент: этил- (2-цианоимино-5,6- дихлор-1,2,3,4 -тетрагидрохиназолин -3-ил)ацетат. Условия реакции: циклизация реагента при температуре выше 80°С в среде растворителя. 3 с. и 8 з.п. ф-лы.

где R1 цианогруппа или группа СООR2, где R2 низший алкил, который в случае необходимости может иметь в своем составе фенильный заместитель,
подвергают реакции термической циклизации в кислой среде, в среде органического растворителя с выделением целевого продукта в виде свободного основания или в виде его фармацевтически приемлемой соли.
4. Способ по п.2, отличающийся тем, что в качестве минеральной кислоты используют хлористо-водородную кислоту.
где R1 цианогруппа или группа СООR2, где R2 низший алкил, который в случае необходимости может иметь в своем составе фенильный заместитель.
где R1 цианогруппа или группа СООR2, где R2 - низший алкил, который в случае необходимости может иметь в своем составе фенильный заместитель,
отличающийся тем, что диамин общей формулы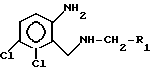
где R1 имеет указанные значения,
подвергают взаимодействию с производным общей формулы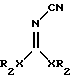
где R2 низший алкил, который может иметь в своем составе фенильный заместитель;
Х кислород или сера.
| US N 4208521, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| Способ получения фтористых солей | 1914 |
|
SU1980A1 |
