Настоящее изобретение относится к новому способу получения 3-этил-5-метил-2-(2-аминоэтокси-метил)-4-(2-хлор-фенил)-6-метил-1,4- дигидро-3,5-пиридин-дикарбоксилата бензолсульфокислоты. Это соединение является известным фармакологически активным ингредиентом, имеющим Международное непатентованное название (МНН) безилат амлодипина.
Безилат амлодипина является кальциевым антагонистом дигидропиридиндикарбоксилатного типа и обладает выраженными антигипертензивными и антиангинальными свойствами.
В соответствии с известными методиками, дигидропиридиновый фрагмент амлодипина получают, используя синтез Ганча, предложенный ранее для реакций данного типа. Главной особенностью метода, описанного в патентных публикациях ЕР 89, 167 и HU 186,868, является то, что первичная аминогруппа образуется на последней стадии синтеза, либо в результате удаления защитной группировки из защищенной аминогруппы, либо в результате восстановления соответствующего азида. Защищенная аминогруппа либо азидогруппа вводятся в молекулу путем взаимодействия с ацетоацетатной компонентой синтеза Ганча.
В соответствии с первым вышеуказанным способом в синтезе Ганча взаимодействуют 4-(2-аминоэтокси)-ацетоуксусный эфир, 2- хлор-бензальдегид и эфир аминокротоновой кислоты, либо в частном случае данного способа, 4-(2-аминоэтокси)-ацетоуксусный эфир сначала конденсируется с 2-хлорбензальдегидом, и далее таким образом полученное "илидное" производное взаимодействует с аминокротоновым эфиром. Амлодипин получают путем удаления защитной группировки из производного дигидропиридина, содержащего защищенную первичную аминогруппу, полученного в ходе синтеза Ганча.
В соответствии с другим вышеуказанным способом в синтезе Ганча взаимодействуют 4-(2-азидоэтокси)-ацетоуксусный эфир, 2- хлорбензальдегид и эфир аминокротоновой кислоты. Первичная аминогруппа амлодипина образуется при восстановлении азидогруппы.
В отмеченной выше публикации ЕР описываются соли амлодипина и подходящих кислот. Указанные соли получают из амлодипина путем реакции солеобразования. Наиболее важной из них является соль малеиновой кислоты.
Недостатком вышеуказанных методов является относительно низкий выход продукта на каждой стадии реакции (выход в синтезе Ганча даже не описывается). Более того, известно, что азиды являются взрывоопасными веществами (см. ссылку С.А. 105. 11321t в отношении азидосоединений).
В патентной публикации DE 3,710,457 описываются соль амлодипина и бензолсульфокислоты и способ ее получения. Согласно данному патенту указанная соль обладает значительными преимуществами перед другими известными солями амлодипина, в особенности при введении этой соли в лекарственные композиции. Безилат амлодипина получают путем реакции амлодипинового основания или аммонийной соли амлодипииа с раствором бензолсульфокислоты в инертном растворителе и последующим выделением таким образом полученного безилата амлодипина из реакционной смеси.
В патентной публикации ЕР 599,220 описывается новый способ получения безилата амлодипина. Производную амлодипина, содержащую тритильную защитную группировку на первичной аминогруппе, получают путем стандартного синтеза Ганча. Тритильная защитная группировка удаляется в процессе гидролиза, проводимом в присутствии бензолсульфокислоты. Таким образом, безилат амлодипина получают без выделения амлодипинового основания. Существенным недостатком данного способа является низкий выход продукта. Другим значительным недостатком является то, что чистый конечный продукт можно получить только с использованием довольно сложных методов. По отношению к 2-хлорбензальдегиду полный выход составляет 7%.
Целью данного изобретения было преодолеть указанные недостатки известных методов и разработать способ, позволяющий получить безилат амлодинина с высоким выходом, простой в исполнении и исключающий выделение амлодипинового основания.
Указанная цель была достигнута и был получен способ, описанный в данном изобретении.
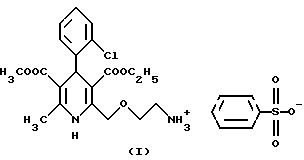
Согласно данному изобретению был разработан способ получения соли 3-этил-5-метил-2-(2-аминоэтокси-метил)-4-(2-хлор-фенил)-6- метил-1,4-дигидро-3,5-пиридин-дикарбоксилата и бензолсульфокислоты формулы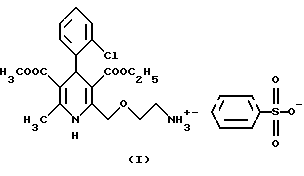
включающий
a1) гидролиз соединения формулы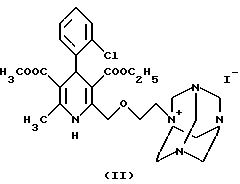
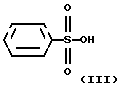
бензолсульфокислотой формулы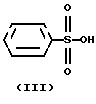
в смеси воды и органического растворителя. Безилат амлодипина также может быть получен:
а2) реакцией соединения формулы.
с гексаметилентетрамином формулы
и реакцию таким образом полученного соединения формулы II с бензолсульфокислотой формулы III, или
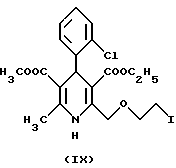
a3) замещением в соединении
хлора на иод, реакцию таким образом полученного соединения формулы IX с гексаметилентетрамином формулы X и реакцию таким образом полученного соединения формулы II с бензолсульфокислотой формулы III; или реакций
a4) соединения формулы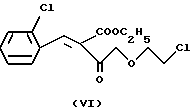
с метил-3-аминокротонатом формулы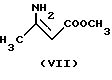
замещением в полученном таким образом соединении формулы VIII хлора на иод, реакцию таким образом полученного соединения формулы IX с гексаметилентетрамином формулы X и реакцию таким образом полученного соединения формулы II с бензолсульфокислотой формулы III; или
a5) реакций соединения формулы
с 2-хлорбензальдегидом формулы
реакцией таким образом полученного соединения формулы VI с метил-3-аминокротонатом формулы VII, замещением в полученном таким образом соединении формулы VIII хлора на иод, реакцию таким образом полученного соединения формулы IX с гексаметилентетрамином формулы X и реакцию таким образом полученного соединения формулы XI с бензолсульфокислотой формулы XII; или
а6) реакцией этил-4-бром-ацетоацетата формулы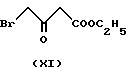
с этиленхлоргидрином формулы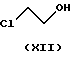
реакцией таким образом полученного соединения формулы IV с 2-хлорбензальдегидом формулы V, реакцию таким образом полученного соединения формулы VI с метил-3-аминокротонатом формулы VII, замещением в полученном таким образом соединении формулы VIII хлора на иод, реакцию таким образом полученного соединения формулы IX с гексаметилентетрамином формулы X и реакцию таким образом полученного соединения формулы II с бензолсульфокислотой формулы III; или
а7) замещением в соединении формулы IV хлора на иод, реакций таким образом полученного соединения формулы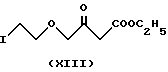
с 2-хлорбензальдегидом формулы V, реакций таким образом полученного соединения формулы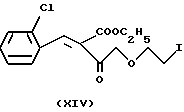
с метил-3-аминокротонатом формулы VII, реакцию таким образом полученного соединения формулы IX с гексаметилентетрамином формулы X и реакцию таким образом полученного соединения формулы II с бензолсульфокислотой формулы III.
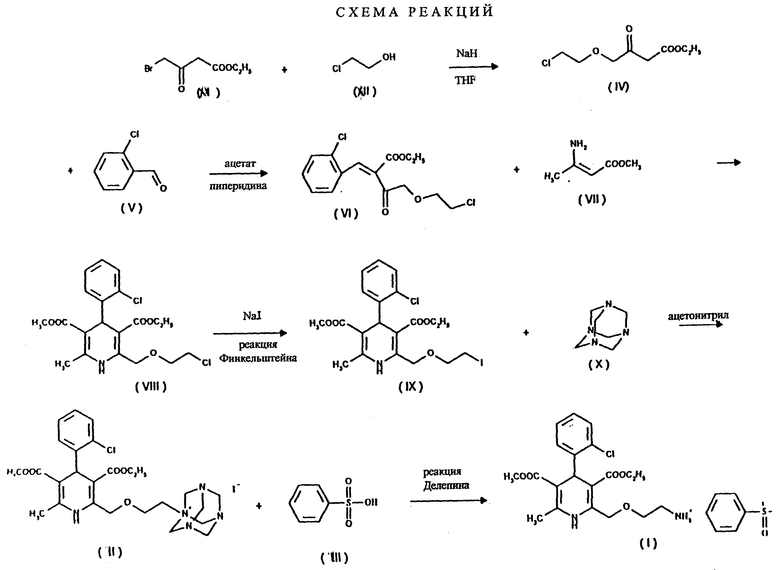
Отличительной чертой данного изобретения является то, что в синтезе Ганча бензилиденовое производное формулы VI, полученное из этил-4-(2-хлорэтокси)-ацетоацетата формулы IV и 2-хлор- бензальдегида формулы V, взаимодействует с эфиром аминокротоновой кислоты формулы VII, в результате чего получают с хорошим выходом новое производное 2-хлорэтокси-дигидропиридина формулы VIII и после замещения хлора на иод новое производное 2-иодэтокси- дигидропиридина формулы IX.
Соединения формул VIII и IX являются новыми, ранее не описанными в литературе. Безилат амлодипина получают с помощью нового, обладающего рядом достоинств метода аммонолиза галогенопроизводного путем так называемой реакции Делепина. Галоген замещается на аминогруппу, причем в качестве источника аммиака используется гексаметилентетрамин. Полученная соль уротропина разлагается бензолсульфокислотой, безилат амлодипина выделяют прямо из реакционной смеси. Данный процесс является новым, ранее не описанным в литературе.
Способ по данному изобретению отличается от описанных ранее тем, что первичная аминогруппа вводится в молекулу не в составе эфира ацетоуксусной кислоты в защищенной форме и последующим снятием защитной группировки, но посредством реакции замещения галогена на аминогруппу, проводимой после синтеза Ганча, с использованием соответствующего соединения галогена. Отличительной чертой данного изобретения является то, что указанная реакция проводится способом, ранее не описанным в литературе, в результате которого образуется не амлодипиновое основание, а требуемая соль - безилат амлодипина.
Данная реакция представлена на схеме. Первая стадия синтеза по данному изобретению представляет собой реакцию алкилирования этиленхлоргидрина формулы XII (2-хлорэтанола) этил-4- бромацетоацетатом формулы XI. Таким образом полученное соединение формулы IV является новым. Данная реакция проводится известным способом: применяется подходящий инертный растворитель, предпочтительно алифатический или алициклический эфир, например тетрагидрофуран, температура около - 10oC или ниже. Образующийся бромистый водород связывается с веществом основного характера, предпочтительно с гидридом натрия, который используется в форме суспензии в масле, либо в форме, предварительно очищенной от парафинов. Реакционная смесь обрабатывается обычным способом кислотой, нейтрализуется и экстрагируется. Чистый продукт получают фракционной перегонкой в вакууме.
Следующая стадия синтеза представляет собой реакцию альдольной конденсации эфира хлорэтоксиацетоуксусной кислоты формулы IV с 2-хлорбензальдегидом формулы V. Продуктом реакции является "илидное" производное формулы VI. Реакцию между альдегидом и производным ацетоуксусной кислоты предпочтительно проводить в присутствии катализатора ацетата пиперидина, в результате чего получают 2-хлорбензилиден-4-хлорэтоксиацетоацетат формулы VI с довольно высоком выходом. Используемое количество катализатора ацетата пиперидина - 0,01 - 0,1 моля в расчете на 1 моль соединения формулы IV. Для проведения реакции могут быть использованы полярные протонные растворители, предпочтительно спирты, особенно изопропанол. Температура реакции - 10-60oC, хотя можно проводить реакцию и при комнатной температуре. Время проведения реакции от 5 до 10 часов, предпочтительно - 10 часов. Затем из реакционной смеси отгоняют растворитель и промывают ее водой. Таким образом полученный продукт используется для дальнейших химических превращений.
Также можно использовать соединение формулы VI для последующей химической реакции без отгонки растворителя.
Следующая стадия синтеза заключается в получении соединения формулы VIII с помощью синтеза Ганча. Реакцию проводят при кипячении смеси производного бензилидена формулы VI и аминокротоната формулы VII в подходящем органическом растворителе. В качестве растворителя предпочтительно использовать C1-4 - спирт (в частности, изопропанол, метанол или этанол), либо полярный апротонный растворитель (напр. ацетонитрил), либо смесь указанных растворителей.
Также реакцию можно проводить между 2-хлорбензальдегидом формулы V, кетоэфиром формулы IV и аминокротонатом формулы VII без выделения производного бензилидена формулы VI. Время реакции - не более 15-20 часов. После завершения реакции реакционную смесь обрабатывают обычным способом, (охлаждение, фильтрация).
На следующей стадии синтеза соединение формулы VIII превращают в соединение формулы IX. Хлор замещают на иод с помощью известной реакции Финкельштейна. Реакцию проводят с иодидом щелочного металла, например иодидом натрия. Согласно литературным данным, с точки зрения растворимости взаимодействующих веществ, в качестве растворителя лучше использовать ацетон. По настоящему изобретению реакцию можно проводить в ацетоне. Однако использование высококипящих спиртов (например, изопропанола) дает ряд преимуществ. Реакцию можно проводить при нагревании, предпочтительно при кипении реакционной смеси. Время реакции составляет не более 20-25 часов. Далее реакционную смесь обрабатывают обычным способом (фильтрация после интенсивного охлаждения). Производное дигидропиридина формулы IX получают с высоким выходом.
Альтернативным способом по данному изобретению является замещение хлора на иод в хлорэтоксиацетоацетате формулы IV. В результате реакции получают иодацетоацетат формулы XIII. Реакцию проводят в ацетоне при температуре кипения реакционной смеси. Конечный продукт очищают фракционной перегонкой. Таким образом полученное соединение формулы XIII превращают в производное бензидилена формулы XIV путем реакции с 2-хлорбензальдегидом формулы V. Данную реакцию проводят как описано выше (см. реакцию между соединениями Формул IV и V). Таким образом полученное соединение формулы XIV реагирует с соединением формулы VII с получением производного иодэтокси-дигидропиридина формулы IX. Данная реакция проводится аналогично описанной ранее реакции Ганча.
Следующая стадия синтеза по данному изобретению заключается в получении четвертичной соли путем реакции иод-производного формулы IX - как более реакционноспособного, чем хлорэтоксидигидропиридин формулы VIII - с гексаметилентетрамином (уротропином) формулы X. По литературным данным соли такого типа обычно получают в неполярных апротонных растворителях. Однако было обнаружено, что применение в качестве растворителей низших спиртов (например, метанола, этанола, изопропанола) или ацетонитрила дает лучшие результаты для реакции между соединениями формул IX и X. Реагенты можно использовать в эквимолярных количествах, однако уротропин формулы X лучше взять в избытке 10- 15%. Реакцию следует проводить при температуре в диапазоне от комнатной до точки кипения, оптимальная температура 40-55oC. Реагенты можно одновременно добавлять в растворитель, либо, что предпочтительнее, постепенно добавлять небольшими порциями иодпроизводное формулы IX в раствор гексаметилентетрамина. Время реакции составляет 20-50 часов, как правило, 30-40 часов. Четвертичная соль дигидропиридин-уротропина формулы II осаждается в виде твердого осадка и может быть легко отфильтрована при комнатной температуре. Степень чистоты полученного продукта является достаточной для получения конечного продукта и не требует дополнительной очистки.
Следующая стадия синтеза представляет собой гидролиз четвертичной соли формулы II с бензолсульфокислотой формулы III, в результате которого получают безилат амлодипина формулы I.
Гидролиз с бензолсульфокислотой проводят в смеси воды и органического растворителя. Для этой цели можно использовать смешиваемые с водой, частично смешиваемые с водой или не смешиваемые с водой органические растворители. Из смешиваемых с водой растворителей лучше применять спирты с неразветвленной и разветвленной цепью, имеющие 1-3 атома углерода (метанол, этанол, изопропанол). Из частично смешиваемых и не смешиваемых с водой растворителей лучше использовать спирты с 4-8 атомами углерода (например, n-бутанол) или этилцетат. Реакцию можно проводить в диапазоне температур от комнатной до точки кипения растворителя, либо при кипячении. Бензолсульфокислоту следует брать, по крайней мере, в 4-мольном избытке по отношению к соединению формулы II. Для практических целей избыток бензолсульфокислоты не должен превышать 10 молярных эквивалентов. Для создания наиболее благоприятной среды реакцию проводят с использованием 5 молярных эквивалентов бензолсульфокислоты. Далее реакционную смесь обрабатывают обычным способом.
Исходные реагенты формул XII, V, X и III имеются в продаже. Бромацетоацетат формулы XI - известное соединение и может быть получено методами, описанными в патентных публикациях US 3,786,082 и ЕР 102,893. Аминокротонат формулы VI также хорошо известен (HU 202,474).
Промежуточные соединения Формул IV, VI, VIII, IX, II, XIII и XIV являются новыми, ранее не описанными в литературе.
Новизна по настоящему изобретению состоит в том, что получены новые соединения формул IV, VI, VIII, IX, XIII и XIV и разработаны методы их синтеза.
Соединения дигидропиридин-дикарбоксилатной структуры являются смешанными эфирами и содержат асимметрический центр. Такие соединения могут существовать в виде пары энантиомеров, которые можно разделить методами, известными из литературных источников. Настоящее изобретение включает индивидуальные изомеры (право- и левовращающие изомеры) и их смеси (включая рацемические смеси).
Согласно способу по настоящему изобретению, конечный продукт формулы I получают из новых промежуточных продуктов, ранее не описанные в литературе. Гидролиз четвертичной соли формулы II бензолсульфокислотой является новым процессом.
Достоинство способа по настоящему изобретению состоит в высоком выходе продуктов на каждой стадии синтеза. Выход на стадии циклизации Ганча по данному изобретению выше, чем в других известных реакциях циклизации при получении амлодипина. Другое достоинство способа по настоящему изобретению состоит в том, что нет необходимости выделять основание амлодипина, так как четвертичная соль получается за одну стадию при осаждении. Данный способ подходит для промышленного производства, причем не требует специального оборудования.
Другие детали по настоящему изобретению описаны в следующих примерах без ограничения объема охраны указанных примеров.
Пример 1 Этил-4-(2-хлорэтокси)-ацетоацетат (IV)
10,38 г (0,25 моля) 57,8 % гидрида натрия добавляют к 110 мл тетрагидрофурана. Смесь охлаждают до температуры между -10oC и -20oC, затем при этой температуре добавляют 10,08 г (0,125 моля) этиленхлоргидрина (XII), продувая смесь азотом. Смесь перемешивают 20 минут, после чего добавляют раствор 26,18 г (0,125 моля) этил-4- бромацетоацетата (XI) в 35 мл тетрагидрофурана. Реакционную смесь перемешивают 20 минут, допуская нагревание до комнатной температуры, выдерживают при этой температуре 6 часов и вливают в 270 мл 1 N соляной кислоты при охлаждении, затем экстрагируют дихлорметаном. Органический слой высушивают, растворитель отгоняют. Кубовый остаток очищают от парафинов обработкой смесью ацетонитрила и бензола (1:1). Продукт получают фракционной перегонкой в вакууме. Таким образом получают 17,47 г требуемого вещества с выходом 67%, с температурой кипения 110oC при 2 мм рт.ст.
Элементарный анализ соединения формулы C8H13ClO4 (208,41):
рассчитанный: C 46,05%, H 6,28%, Cl 16,99%
определенный: C 46,45%; H 6,11%; Cl 16,52%.
Пример 2
Этнил-4-(2-иодэтокси)-ацетоацетат (XIII)
К раствору 19 г (91 милимоль) этил-4-(2-хлорэтокси)- ацетоацетата (IV) в 380 мл ацетона добавляют 134,4 г (91 милимоль) иодида натрия. Реакционную смесь кипятят 13 часов. Неорганическое вещество отфильтровывают, фильтрат отгоняют в вакууме. Кубовый остаток растворяют в дихлорметане, раствор промывают водой, высушивают и отгоняют. Продукт получают фракционной перегонкой в вакууме. Точка кипения 170oC при 0,1 мм рт. ст. Таким образом получают 18,9 г требуемого вещества с выходом 67%.
Элементарный анализ соединения формулы C8H13JO4 (300,091):
рассчитанный C 32,02%; H 4,37%;
определенный C 31,86%; H 4,36%.
Пример 3
Этил-4(2-хлорэтокси)-2-(2-хлорбензилиден)-ацетоацетат (VI)
16,64 г (0,118 милимоля) 2-хлорбензальдегида (V) и 24,7 г (0,118 милимоля) этил-4-(2-хлорэтокси)-ацетоацетата (IV), растворенные в 365 мл изопропанола, в присутствии ацетата пиперидина, в качестве катализатора [10 г (11,8 милимоля) пиперидина +0,7 г (11,8 милимоля) уксусной кислоты] реагируют при комнатной температуре в течение 10 часов. Затем растворитель отгоняют, кубовый остаток растворяют в дихлорметане, промывают водой и высушивают. Органическую фазу отгоняют в вакууме. Таким образом получают 37,9 г требуемого продукта в виде масла желтого цвета, с выходом 97%.
Элементарный анализ соединения формулы C15H16Cl2O4 (331,203):
рассчитанный: C 54,39%; H 4,87%; Cl 21,41%;
определенный: C 53,69%; H 5,03%; Cl 20,98%.
Пример 4
Этил-4-(2-иодэтокси)-2-(2-хлорбензилиден)-ацетоацетат (XIV)
10 г (33 милимоля) этил-4-(2-иодэтокси)-ацетоацетата (XIII) и 4,64 г (33 милимоля) 2-хлорбензальдегида (V), растворенные в 100 мл изопропанола, реагируют в присутствии ацетата пиперидина в качестве катализатора [0,28 г (3,3 милимоля) пиперидина +1,198 г (3,3 милимоля) уксусной кислоты] при комнатной температуре в течение 10 часов. Затем растворитель отгоняют, кубовый остаток растворяют в дихлорметане, промывают водой и высушивают. Органическую фазу отгоняют в вакууме. Таким образом получают 11,55 г требуемого продукта в виде масла красно-бурого цвета, с выходом 83%.
Элементарный анализ соединения формулы C15H16ClJO4 (422,643):
рассчитанный: C 42,63%; H 3,82%; Cl 8,39%;
определенный C 43,00%; H 4,12%; Cl 8,13%.
Пример 5
3-этил-5-метил-2-(2-хлорэтоксиметил)-4-2-хлорфенил)-6-метил-1,4- дигидро-3,5-пиридиндикарбоксилат (VIII)
36 г (0,1087 моля) этил-4-(2-хлорэтокси)-2-(2-хлорбензилиден)- ацетоацетата (VI) и 12,5 г (0,1087 моля) метил-3-аминокротоната (VII), растворенные в 355 мл изопропанола реагируют при температуре кипения реакционной смеси в течение 20 часов. Затем смесь охлаждают до температуры от 0oC до -5oC и оставляют на ночь в холодильнике. На следующее утро осадок отфильтровывают, хорошо промывают холодным изопропанолом и диизопропиловым эфиром. Далее продукт перекристаллизовывают из диизопропилового эфира или водного раствора уксусной кислоты (если необходимо). Таким образом получают 21,88 г требуемого вещества с температурой плавления 152-154oC, выход составляет 47%.
Элементарный анализ соединения формулы C20H23Cl2NO5 (428,32)
рассчитанный: C 56,08%; H 5,41%; N 3,27%; Cl 16,56%;
определенный: C 56,10%; H 5,42%; N 3,37%; Cl 16,18%.
Пример 6
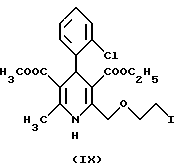
3-этил-5-метил-2-(2-иодэтокси)-метил-4-(2-хлорфенил)-6-метил-1,4- дигидро-3,5-пиридиндикарбоксилат (IX)
Метод а)
Смесь из 16 г (37 милимолей) 3-этил-5-метил-2-(2- хлорэтоксиметил)-4-(2-хлорфенил)-6-метил-1,4-дигидро-3,5- пиридиндикарбоксилата (VIII), 55,46 г (370 милимолей) иодида натрия и 183 мл изопропанола перемешивают при кипячении в течение 20 часов. Затем реакционную смесь охлаждают до температуры между 0oC и -5oC и выдерживают в холодильнике в течение ночи. На следующее утро осадок отфильтровывают и промывают холодным изопропанолом. Продукт перекристаллизовывают из изопропанола. Таким образом получают 16,35 г требуемого вещества, температура плавления которого 152-154oC, с выходом 85%.
Элементарный анализ соединения формулы C20H23ClJNO5 (428,32):
рассчитанный: C 46,22%; H 4,46%; N 2,69%; Cl 6,82%;
определенный: C 45,92%; H 4,45%; N 2,73%; Cl 6,77%.
Метод b)
Смесь из 11 г (26 милимолей) этил-4-(2-иодэтокси)- 2-(2-хлорбензилиден)-ацетоацетата (XIV), 2,99 г (26 милимолей) метил-3-аминокротоната (Vll) и 110 мл изопропанола кипятят в течение 8 часов. Затем растворитель отгоняют, кубовый остаток перекристаллизовывают из холодного изопропанола, фильтруют и промывают холодным изопропанолом. Продукт перекристаллизовывают из изопропанола. Таким образом получают 2,97 г требуемого вещества, температура плавления которого 152-155oC, с выходом 22%.
Пример 7
Иодид-3-этил-5-метил-2-(2-ил-этоксиметил)-4-(2- хлорфенил)-6-метил-1,4-дигидро-3,5-пиридинкарбоксилат-гексамина (II)
1,77 г (12,7 милимоля) гексаметилентетрамина (X) добавляют к 15 мл ацетонитрила. Смесь перемешивают при комнатной температуре в течение 10 минут, нагревают до 45-50oC, затем 6,0 г (115 милимолей) 3-этил-5-метил-2-(2-иодэтокси)-метил-4-(2-хлорфенил)-6- метил-1-, 4-дигидро-3,5-пиридиндикарбо-ксилата (IX) добавляют небольшими порциями в течение 2 часов. Реакционную смесь перемешивают при этой температуре 40 часов, охлаждают до комнатной температуры, фильтруют и хорошо промывают ацетонитрилом и дихлорметаном. Таким образом получают 6,83 г требуемого вещества в виде белого порошка, температура плавления которого 177-179oC, с выходом 90%.
Элементарный анализ соединения формулы C26H35ClJNO5O5 (659,957):
рассчитанный: C 47,32%; H 3,35%; N 10,61%;
определенный: C 46,84%; H 5,42%; N 10,40%.
Пример 8.
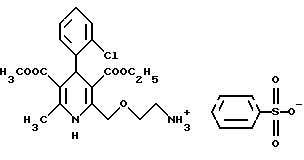
Соль 3-этил-5-метил-2-(2-аминоэтоксиметил)-4(2-хлорфенил)-6-метил-1,4- дигидро-3,5-пиридиндикарбоксилата и бензолсульфокислоты (безилат амлодипина) (I)
Метод а)
Смесь 3.3 г (5 милимолей) иoдидa 3-этил-5-метил-2-(2-ил-этoкcимeтил)-4-(2-хлорфенил)-6-метил-1,4-дигидро- 3,5-пиридиндикарбоксилат-гексамина (II), 3,95 г (25 милимолей) бензолсульфокислоты (III), 350 мл n-бутанола и 350 мл воды нагревают до кипения и кипятят в течение 45 минут при интенсивном перемешивании. Затем реакционную смесь охлаждают до комнатной температуры, разделяют водный и органический слои. Органический слой промывают водой, высушивают и отгоняют растворитель.
Кубовый остаток кристаллизуют при выдерживании в холодном этилацетате в холодильнике в течение ночи. На следующее утро кристаллический продукт отфильтровывают, промывают холодным этилацетатом и высушивают. Высушенный продукт тщательно промывают водой, сушат и перекристаллизовывают из ацетонитрила. Таким образом получают 1,5 г безилата амлодипина. Выход 52,9 %, температура плавления 202-203oC.
Метод b)
Смесь 8,9 г (0,013 моля) иодида 3-этил-5-метил-2-(2- ил-этоксиметил)-4-(2-хлорфенил)-6-метил-1,4-дигидро-3,5- пиридиндикарбоксилат-гексамина (II), 10,54 г (0,066 моля) бензолсульфокислоты (III), 535 мл метанола и 535 мл воды кипятят в течение 1 часа. Затем реакционную смесь охлаждают до комнатной температуры, промывают водой, высушивают и отгоняют растворитель. Кубовый остаток кристаллизуют при выдерживании в ацетонитриле в холодильнике в течение ночи. Таким образом получают 4,18 г безилата амлодипина. Выход 55,4%, температура плавления 205-206oC (ацетонитрил).
Элементарный анализ соединения формулы C26H31ClN2O8S (567,055):
рассчитанный: C 55,07%; H 5,51%; N 4,94%; Cl 6,25%; S 5,65%;
определенный: C 54,71%; H 5,55%; N 4,95%; Cl 6,05%, S 5,57%.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ 3-ЭТИЛ-5-МЕТИЛ (*01+)2[(2-АМИНОЭТОКСИ)-МЕТИЛ]-4-(2-ХЛОРФЕНИЛ)-1,4-ДИГИДРО-6-МЕТИЛ-3,5-ПИРИДИНДИКАРБОКСИЛАТМОНОБЕНЗОЛСУЛЬФОНАТА И ПРОМЕЖУТОЧНОЕ ДЛЯ НЕГО | 1993 |
|
RU2105759C1 |
| СПОСОБ ПОЛУЧЕНИЯ 3-ЭТИЛ-5-МЕТИЛ(±)2[2-АМИНОЭТОКСИ)-МЕТИЛ]-4-(2-ХЛОРФЕНИЛ)-1,4-ДИГИДРО- 6-МЕТИЛ-3,5-ПИРИДИНДИКАРБОКСИЛАТ МОНОБЕНЗОЛСУЛЬФОНАТА | 1999 |
|
RU2146672C1 |
| ПРОИЗВОДНЫЕ ПИПЕРАЗИНИЛАЛКИЛТИОПИРИМИДИНА, СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ И СПОСОБ ПОЛУЧЕНИЯ | 2000 |
|
RU2245334C2 |
| СПОСОБ ПОЛУЧЕНИЯ ФЛУОКСЕТИНА | 1997 |
|
RU2173679C2 |
| ПРОИЗВОДНОЕ 2-(1,2,4-ТРИАЗОЛ-1-ИЛ)-1,3,4-ТИАДИАЗОЛА, СПОСОБ ЕГО ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ | 1998 |
|
RU2180903C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ 2,3-БЕНЗОДИАЗЕПИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ЛЕЧЕНИЯ | 2000 |
|
RU2243228C2 |
| СПОСОБ ПОЛУЧЕНИЯ КВЕТИАПИНА И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ | 2001 |
|
RU2258067C2 |
| СПОСОБ ПОЛУЧЕНИЯ ФАРМАЦЕВТИЧЕСКОГО ПРОДУКТА - КАРВЕДИЛОЛА И ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ | 1998 |
|
RU2216539C2 |
| СПОСОБ ПОЛУЧЕНИЯ МОНОБЕНЗОЛСУЛЬФОНАТА 3-ЭТИЛ-5-МЕТИЛОВОГО ЭФИРА 2-[(2-АМИНОЭТОКСИ)МЕТИЛ]-4-(2-ХЛОРФЕНИЛ)-1,4-ДИГИДРО-6-МЕТИЛ-3,5-ПИРИДИНДИ КАРБОНОВОЙ КИСЛОТЫ | 1999 |
|
RU2142942C1 |
| ПРОИЗВОДНЫЕ ДИГИДРОПИРИМИДОТИАЗИНА ИЛИ ИХ ТЕРАПЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ ПРИСОЕДИНЕНИЯ КИСЛОТЫ, ОБЛАДАЮЩИЕ ПРОТИВОАНГИННОЙ И АНТИВОСПАЛИТЕЛЬНОЙ АКТИВНОСТЬЮ | 1990 |
|
RU2022965C1 |
Изобретение относится к новому способу получения безилата амлодипина формулы I, обладающему выраженными антигипертензивными и антиангинальными свойствами. Способ заключается в том, что соединение II подвергают гидролизу бензолсульфокислотой III в смеси воды и органического растворителя, как смешиваемого, так и не смешиваемого с водой, например н.бутанола, этилацетата, одноатомного спирта с 1 - 3 атомами углерода. Исходное соединение формулы II получают взаимодействием соединения формулы IX с гексаметилентетрамином X. Достоинство способа по настоящему изобретению заключается в том, что он прост в исполнении, обеспечивает высокий выход продуктов реакций и нет необходимости выделять основание амплодипина. 5 з.п. ф-лы.
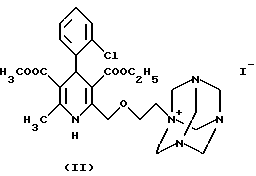

отличающийся тем, что соединение формулы II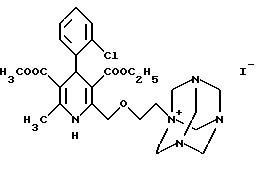
подвергают гидролизу бензолсульфокислотой формулы III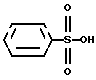
в смеси воды и органического растворителя.
| Преобразователь действующего значения напряжения переменного тока в напряжение постоянного тока | 1976 |
|
SU599220A1 |
| ИЗЛОЖНИЦА ДЛЯ СЛИТКОВ СТАЛИ | 1972 |
|
SU419297A1 |
| ЬЭЗ.ЧАЯ IМШТ1Ш'Т?} | 0 |
|
SU352853A1 |
| Способ получения бензолсульфонатной соли 3-этил-5-метилового эфира 2- (2-аминоэтоксиметил )-4-(2-хлорфенил)-6-метил-1,4-дигидропиридин-3,5-дикарбоновой кислоты | 1987 |
|
SU1498388A3 |
| 0 |
|
SU168841A1 |

