Изобретение относится к новому и усовершенствованному способу получения флуоксетина и его фармацевтически приемлемым солям присоединения кислот.
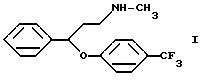
Известно, что N-метил-{3-фенил-3-[4-(трифторметил)-фенокси]-пропил}-амин формулы I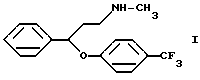
(далее "флуоксетин") является ценным антидепрессантом, который проявляет свое действие в виде избирательной замедляющей поглощение серотонина активностью (патент Венгрии N 172723). В патенте США N 4018895 описывается применение флуоксетина для лечения депрессий.
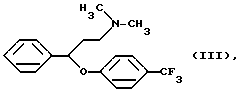
Согласно патенту Венгрии N 173723 флуоксетин получают следующим образом. 3-Диметиламинпропиофенон восстанавливают дибораном в тетрагидрофуране, таким образом полученный N,N-диметил-(3-гидрокси-3-фенилпропил)-амин обрабатывают соляной кислотой и хлористым тионилом. Гидрохлорид N,N-диметил-(3-фенил-3-хлорпропил)-амина кипятят с 4-трифторметилфенолом в щелочной среде в течение 5 дней. Полученный таким образом N,N-диметил-{3-фенил-3-[4-трифторметил)-фенокси]-пропил}-амин формулы III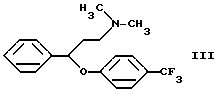
деметилируют в две стадии. В первой стадии производное диметиламина формулы III реагирует с бромцианом, после чего N-метил-N-циан-{3-фенил-3-[4-(трифторметил)-фенокси] -пропил}-амин кипятят с этиленгликолем в растворе гидроксида калия при 130oC в течение 2 ч. Основание флуоксетина формулы I очищают с помощью соли оксалата и переводят в гидрохлорид флуоксетина путем реакции с газообразным хлористым водородом в эфире. Этот способ имеет ряд недостатков. С одной стороны, наносится вред окружающей среде в результате выброса ядовитых материалов (например, бромциана), с другой - синтез включает много стадий с длительным временем реакции, что делает весь процесс неэкономичным. Общий выход продукта реакции составляет менее 20%. Еще один недостаток этого способа заключается в том, что 4-трифторметилфенол является дорогостоящим веществом, которое не всегда имеется в распоряжении.
Согласно патенту Венгрии N 204769 флуоксетин формулы I получают из N-бензил-N-метил-(2-бензоилэтил)-амина. Это исходное вещество гидрируют в присутствии платино-палладиевого катализатора на угле в этилацетате в автоклаве в атмосфере водорода при 50oC и давлении 5•105 Па. Таким образом полученный N-метил-(3-гидрокси-3-фенилпропил)-амин O-арилируют 4-хлортрифторметилбензолом в N-метилпирролидоне в присутствии третичного бутилата калия и иодида калия. Полученный таким образом флуоксетин формулы I переводят в гидрохлорид. Недостаток этого способа заключается в том, что используемый в качестве исходного реагента N-метилбензиламин является труднодоступным дорогостоящим веществом. Другой недостаток заключается в том, что способ, требующий использования специального катализатора и очистки продукта, является сложным. В цитируемом патенте не указаны характеристики и доступность катализатора.
Согласно патенту Венгрии N 207035 N-метил-(3-гидрокси-3-фенилпропил)-амин реагирует с небольшим избытком амида натрия в диметилсульфоксиде, затем к гомогенной реакционной смеси добавляется небольшой избыток 4-хлортрифторметилбензола или его раствор в диметилсульфоксиде при 60-80oC. После завершения реакции весь растворитель диметилсульфоксид или часть его отгоняют в вакууме. Затем реакционную смесь вливают в воду. Полученный таким образом флуоксетин формулы I экстрагируют, переводят в гидрохлорид и соль перекристаллизуют.
Этот способ имеет ряд недостатков. Реакцию необходимо проводить в безводной среде. Более того, очень сложна работа с диметилсульфоксидом, который подвержен термическому разложению и отгоняется в вакууме. Еще одним недостатком является то, что конечный продукт получается недостаточной чистоты и поэтому требует перекристаллизации.
Предметом выложенной венгерской заявки на патент N P 9202128 было устранить указанные недостатки. Согласно этой заявке N-метил-(3-гидрокси-3-фенилпропил)-амин реагирует с 30%-ным избытком гидроксида натрия в диметилсульфоксиде при 100oC в течение 1 ч. Затем к реакционной смеси добавляют по каплям 4-хлортрифторметилбензол, проводят реакцию при 100-120oC в течение 10-20 ч. Серьезный недостаток данного способа заключается в довольно продолжительном времени реакции. Другим недостатком является то, что используется диметилсульфоксид, который подвержен разложению при высоких температурах, что усложняет работу с этой реакционной смесью.
Согласно патенту Испании N 556009 флуоксетин формулы I получают с помощью реакции 4-трифторметилфенола с молярным эквивалентом N-этоксикарбонил-N-метил-(3-фенил-3-метансульфонилоксипропил)-амина или N-ацетил-N-метил-(3-фенил-3-метансульфонилоксипропил)-амина в щелочной спиртовой среде при 60-100oC. Удаляют защитную группу обработкой 3-6 N раствором неорганической кислоты (например, соляной, серной, бромистоводородной) в этаноле или воде, или в смеси воды с этанолом. Реакцию проводят при кипячении реакционной смеси. Таким образом полученное маслянистое основание флуоксетина формулы I очищают на силикагельной колонке. Этот способ имеет ряд недостатков. Используемый в качестве исходного реагента 4-трифторметилфенол является труднодоступным, дорогостоящим и вредным для здоровья веществом. Другим недостатком является то, что в производное пропиламина вводятся две защитные группы. Конечный продукт формулы I получается в виде масла, которое необходимо очищать на силикагельной колонке. При воспроизведении способа согласно патенту Испании N 556009 было обнаружено, что в результате гидролиза N-метил-N-этоксикарбонил-{3-фенил-3-[4-(трифторметил)-фенокси]-пропил}-амина 4 N соляной кислотой получают смесь небольшого количества сильно загрязненного соединения формулы I и непрореагировавшего исходного материала как основного компонента смеси. Этот способ практически непригоден для промышленного производства.
Согласно патенту EP-A 380924 соединение формулы I получают путем восстановления этилбензоилацетата борогидридом натрия в среде метанола, реакцией этил-3-гидрокси-3-фенилпропионата с метиламином в спиртовой среде и O-арилированием N-метил-(3-гидрокси-3-фенилпропионат)-амида в тетрагидрофуране в присутствии трифенилфосфина и азидокарбоновой кислоты 4-трифторметилхлорбензолом. После завершения реакции полученный продукт восстанавливают в основание флуоксетина формулы I литий-алюминийгидридом в тетрагидрофуране и переводят в соль способом, известным из уровня техники. Этот способ требует использования токсических легковоспламеняющихся и вредных для окружающей среды материалов, поэтому он не подходит для промышленного производства. Другим недостатком данного способа является то, что полученный продукт имеет недостаточную чистоту, и, следовательно, требует сложной дорогостоящей очистки, которая приводит к потерям вещества.
Согласно выложенной венгерской заявке N T/63144 флуоксетин формулы I и его соли присоединения кислот получают с помощью новых промежуточных соединений. Согласно этому способу N,N-диметил-(3-гидрокси-3-фенилпропил)-амин формулы IV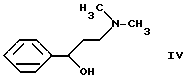
получают известным способом путем реакции с этилхлорформиатом в инертном растворителе (например, толуоле) при нагревании с обратным холодильником. В качестве связывающего кислоту агента используют, например, карбонат натрия, гидрокарбонат натрия. Таким образом получают соединение формулы VI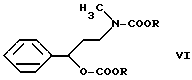
Если реакцию проводят в отсутствии агента, связывающего кислоту, нужное производное карбамата формулы VI, то получают из промежуточного продукта N, N-диметил-{3-фенил-3-(этоксикарбонилокси)-пропил}-амина формулы VIII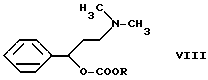
в две стадии. Производное карбамата формулы VI кипятят в щелочном растворе этанола с получением N-метил-(3-гидрокси-3-фенилпропил)-амина формулы VII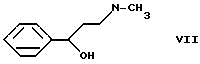
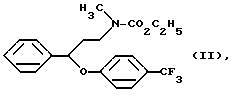
Из этого соединения получают натриевую соль в N,N-диметилацетамиде реакцией с гидридом натрия известным способом, далее эту соль O-арилируют 4-хлортрифторметилбензолом. Гидрохлорид получают реакцией с газообразным хлористым водородом в толуоле. Полученный гидрохлорид флуоксетина очищают перекристаллизацией из горячей воды, а горячий водный раствор дополнительно обрабатывают активированным углем. Необходимо отметить, что в этой заявке особо указывается на то, что N-метил-N-этоксикарбонил-{ 3-фенил-3-[4- (трифторметил)-фенокси]-пропил}-амин формулы II
стабилен до 130oC в используемых условиях реакции щелочного гидролиза, и соединение формулы II устойчиво при обработке щелочью. Описанный способ имеет ряд недостатков. Алкоксикарбонильную группу вводят в гидроксигруппу, а затем эту группу необходимо удалить. Способ включает несколько стадий. Выход конечного продукта мал. Более того, полученный продукт требует очистки.
Следует отметить, что в противоположность соединению формулы VI N-метил-N-этоксикарбонил-{ 3-фенил-3-[4-(трифторметил)-фенокси]-пропил}-амин формулы II устойчив в условиях реакции щелочного гидролиза и стабилен до 130oC.
Согласно EP-A 617006 N-бензил-N-метил-(3-гидрокси-3-фенилпропил)-амин реагирует с 4-хлортрифторметилбензолом в диметилацетамиде. Полученный N-бензил-N-метил-{ 3-фенил-3-[4-(трифторметил)-фенокси]-пропил}-амин реагирует с метилхлорформиатом, затем полученный таким образом N-метил-N-метоксикарбонил-{3-фенил-3-[4-(трифторметил)-фенокси]-пропил}-амин гидролизуют с получением флуоксетина формулы I гидроксидом натрия в метаноле. Основание флуоксетина переводят в оксалат известным способом. Недостатком этого способа является использование двух защитных групп. Удаление двух защитных групп и гидролиз в щелочной метанольной среде приводит к получению сильно загрязненного основания формулы I, которое очищают через соль оксалата, затем соль оксалата переводят в основание, а из этого свободного основания получают гидрохлорид.
Согласно способу, описанному Робертсоном и др. (J. Labelled Comp. Radiopharm. 24, 1937 (1987)) N,N-диметил-(3-гидрокси-3-фенилпропил)-амин этерифицируют 4-хлортрифторметилбензолом, затем таким образом полученное производное диметила деметилируют менее токсичным фенилхлорформиатом вместо ядовитого бромциана. Однако в результате получают сильно загрязненный продукт, который необходимо очищать ВЭЖХ. Этот способ является дорогостоящим и непригоден для промышленного производства.
Целью настоящего изобретения было преодолеть указанные недостатки известных способов и создать способ получения флуоксетина формулы I высокой чистоты и отвечающего строгим требованиям Фармакопеи, который является приемлемым для промышленного использования.
Указанная цель была достигнута благодаря способу по настоящему изобретению.
В данном изобретении был разработан способ получения N-метил-{3-фенил-3-[4-(трифторметил)-фенокси] -пропил} -амина формулы I и его фармацевтически приемлемых солей присоединения кислот путем реакции N,N-диметил-{3-фенил-3-[4-трифторметил)-фенокси] -пропил} -амина формулы III и этилхлорформиата, гидролиза и декарбоксилирования N-метил-N-этоксикарбонил-{ 3-фенил-3-[4-(трифторметил)-фенокси] -пропил}-амина формулы II, и, если необходимо, получения соли, который включает проведение реакции соединения формулы III с этилхлорформиатом в толуоле или ксилоле или их смеси при температуре ниже 90oC; удаление примесей и побочных продуктов путем обработки разбавленной кислотой; отделение органической фракции, содержащей производное уретана формулы II, и реакцию указанной органической фракции без выделения производного уретана формулы II с гидроксидом щелочного металла при температуре кипения реакционной смеси в присутствии воды и, необязательно, н-бутанола; удаление неорганических соединений; и, если необходимо, превращение таким образом полученного основания формулы I в его фармацевтически приемлемую соль присоединения кислоты.
Настоящее изобретение основано на том факте, что в процессе получения соединения формулы I большинство примесей образуется, с одной стороны, при деметилировании соединения формулы III, с другой стороны, при щелочном гидролизе и декарбоксилировании соединения формулы II.
Существенные признаки изобретения можно суммировать следующим образом:
- ацилирование проводят в среде толуола или ксилола:
- промежуточное соединение формулы II не выделяют;
- большую часть примесей и побочных продуктов удаляют экстракцией разбавленной кислотой;
- гидролиз производного уретана формулы II проводят в присутствии воды и необязательно н-бутанола в той же реакционной смеси при температуре кипения реакционной смеси.
Согласно способу по настоящему изобретению подавляются побочные реакции, ведущие к образованию примесей.
Было обнаружено, что при проведении реакции соединения формулы III и этилхлорформиата в толуоле или ксилоле вместо ранее используемого бензола при температуре ниже 90oC и последующей обработке реакционной смеси разбавленной кислотой примеси переходят в водно-кислотную фазу, а органическая фаза, содержащая соединение формулы II, получается свободной от примесей. Соединение формулы II не выделяют, а сразу гидролизуют в растворе толуола или ксилола.
Реакцию соединения формулы III и этилхлорформиата проводят в толуоле или ксилоле при 80-90oC. Также можно работать предпочтительно при температуре 80-85oC. В качестве реакционной среды также можно использовать промышленный ксилол, состоящий из орто-, мета- и параксилола. Изомеры ксилола не влияют на реакцию, поэтому вместо чистых изомеров ксилола можно успешно использовать промышленный ксилол, состоящий из смеси изомеров. Реакцию проводят до тех пор, пока не прекратится выделение газообразного хлористого метила.
После окончания реакции реакционную смесь экстрагируют водным раствором кислоты. Для этой цели предпочтительно используют разбавленную соляную кислоту. Предпочтительным является использование 0,75-0,85 моль (особенно 0,8 моль) разбавленной неорганической кислоты, в частности разбавленной соляной кислоты на 1 моль исходного соединения формулы III. Примеси остаются в водно-кислотной фазе, которую можно легко отделить от верхнего органического слоя. Органический слой представляет собой чистый, не содержащий примесей раствор соединения формулы II, который можно прямо использовать для щелочного гидролиза без выделения соединения формулы II.
Настоящее изобретение основано на том факте, что соединение формулы II можно гидролизовать гидроксидом щелочного металла в толуоле или ксилоле в присутствии небольшого количества воды в течение очень короткого времени реакции для получения соединения формулы I. Этот факт тем более удивителен, что из уровня техники известно, что трифторметильная группа гидролизуется в щелочной среде с образованием карбоксигруппы. Согласно J. Org. Cherm. 26, 2707 (1961) при нагревании 2-трифторметил-4-нитрохлорбензола с гидроксидом калия образуется 5-нитросалициловая кислота как единственный продукт реакции. В J. Am. Chem. Soc. 79, 1745 (1957) описано, что при нагревании 6-трифторметил-2-индолкарбоновой кислоты или ее метилового эфира с раствором гидроксида натрия образуется 2,6-индолдикарбоновая кислота. При реакции соответствующего изомера 4-трифторметила с гидроксидом натрия получается 2,4-индолдикарбоновая кислота. Согласно J. Am. Chem. Soc. 69, 2346 (1947) реакция паратрифторметилфенола и гидроксида натрия дает парагидроксибензойную кислоту. В резюме к этой статье авторы утверждают, что орто- и паратрифторметилфенолы разрушаются под воздействием водных растворов гидроксидов щелочных металлов. Исходя из вышеизложенного не является очевидным тот факт, что в результате реакции соединения формулы II с гидроксидом щелочного металла в толуоле или ксилоле трифторметильная группа не вступит в реакцию, а будет получено соединение формулы I высокой чистоты, не содержащее примесей.
Реакция гидролиза соединения формулы II в метаноле или этаноле требует много времени - около 30 ч. Время реакции можно сократить примерно до 12-15 ч путем проведения реакции в этиленгликоле, но в этом случае качество продукта не удовлетворяет строгим требованиям Фармакопеи даже после повторной очистки. Длительное время реакции и жесткие условия реакции вызывают образование продуктов вторичного и третичного разрушения, имеющих очень близкие химические свойства соединения формулы I, поэтому их очень трудно удалить.
Было обнаружено, что при проведении реакции щелочного гидролиза соединения формулы II в толуоле или ксилоле в присутствии небольшого количества воды, реакция проходит за 2-10 ч, причем побочные продукты и продукты разрушения либо не образуются вообще, либо образуются в минимальных количествах. Реакцию проводят в присутствии 20-40 мл - предпочтительно 30 мл - воды в расчете на 1 моль производного уретана формулы II. Проведение реакции можно облегчить путем проведения гидролиза в присутствии н-бутанола, добавленного в таком же количестве, что и вода. В этом случае реакционная смесь легче фильтруется, и при экстрагировании водой не образуется трудно разлагаемой эмульсии.
Предпочтительнеe реакцию органической фазы, полученной в результате реакции соединения формулы III, проводить с этилхлорформиатом в толуоле или ксилоле и экстракции реакционной смеси разбавленным раствором кислоты и отделения водно-кислотной фазы, можно проводить с гидроксидом щелочного металла в присутствии воды и н-бутанола. В качестве гидроксида щелочного металла можно использовать гидроксид натрия, гидроксид калия или их смесь. Реакцию проводят при температуре кипения реакционной смеси. Время реакции - 2-4 ч.
Неорганические соединения (смесь гидроксидов щелочных металлов и солей) можно легко удалить из реакционной смеси путем фильтрации или центрифугирования. Органическую фазу, содержащую соединение формулы I, промывают водой, сушат и выделяют основание формулы I путем выпаривания раствора. Также из раствора толуола или ксилола можно проводить прямое осаждение фармацевтически приемлемой соли присоединения кислоты путем добавления раствора нужной неорганической кислоты с органическим растворителем. Согласно особо предпочтительной реализации гидрохлорид соединения формулы I осаждают прямо из толуола или ксилола этилацетатом, содержащим соляную кислоту. Этот способ дает без дополнительной очистки соединение формулы I со степенью чистоты для Фармакопеи.
Фармацевтически приемлемые соли присоединения кислот соединений формулы I можно получить с неорганическими или органическими кислотами (например, галогениды водорода, карбонат, гидрокарбонат, сульфат, ацетат, фумарат, малеат, цитрат, аскорбинат и т. д.). Для фармацевтических целей особенно предпочтительным является гидрохлорид соединения формулы I.
Преимущества способа настоящего изобретения можно суммировать следующим образом:
- реакцию проводят без выделения уретана формулы II (одностадийный процесс);
- при деметилировании соединения формулы III образуется минимальное количество побочных продуктов;
- образующиеся в небольшом количестве побочные продукты можно легко удалить;
- в используемых условиях реакции гидролиз и декарбоксилирование проходят очень быстро - примерно на один порядок быстрее - следовательно, время реакции намного короче, и подавляются побочные реакции;
- образующиеся в процессе реакции неорганические соли можно легко удалить из реакционной смеси с помощью фильтрации и центрифугирования;
- требуемое соединение формулы I осаждается в чистом виде из не смешивающихся с водой органических растворителей, предпочтительно в виде гидрохлорида, причем его можно сразу фильтровать даже при промышленном производстве;
- ограничено применение вредных для здоровья и окружающей среды исходных веществ;
- в результате получается с большим выходом продукт с высокой степенью чистоты, который удовлетворяет требованиям Фармакопеи.
Дополнительные подробности настоящего изобретения описаны в следующих примерах, причем объем охраны не ограничивается только указанными примерами.
Пример 1
N-метил-{3-фенил-3-[4-(трифторметил)-фенокси]-пропил}-амин
22,1 г (0,0683 моль) N,N-диметил-{3-фенил-3-[4-трифторметил)-фенокси]-пропил}-амина растворяли в 75 мл ксилола. Для этой цели использовали имеющуюся в продаже смесь орто-, мета- и параксилола, содержащую около 20 вес.% ортоксилола, около 60 вес.% метаксилола и около 20 вес.% параксилола. К этому раствору при 80-85oC добавляли по каплям при перемешивании раствор 22,4 г (0,203 моль) этилхлорформиата в 20 мл ксилола. Реакционную смесь перемешивали при этой температуре до прекращения выделения газообразного хлористого метила.
Реакционную смесь разбавляли 25 мл ксилола, затем экстрагировали смесью 5 мл концентрированной соляной кислоты и 20 мл воды. Нижний водно-кислотный слой отделяли, органический слой промывали 10 мл N раствора гидроксида натрия. Фазу ксилола отделяли, разбавляли свежей порцией ксилола до объема 140 мл. Анализ газовой хроматографией показал, что раствор содержал 22,4 г N-метил-N-этоксикарбонил-{ 3-фенил-3-[4-(трифторметил)-фенокси] -пропил} -амина (уретана).
К этому раствору добавляли 27,32 г (0,683 моль) гидроксида натрия, 1,85 мл воды и 1,85 мл н-бутанола. Реакционную смесь кипятили при интенсивном перемешивании. Время реакции - 3,5 ч. Момент окончания реакции определяли тонкослойной хроматографией (ТСХ) (элюент: смесь метанол:дихлорметан:гидроксид аммония в соотношении 9:1:1; сорбент: силикагель; детектирование: УФ-излучение, иод, реактив Драгендорфа).
После завершения реакции реакционную смесь охлаждали до 25oC, фильтровали в вакууме через фильтр из пористого стекла, осажденный продукт промывали ксилолом. Органическую фазу промывали трижды по 40 мл воды до pH 4 и сушили над безводным сульфатом магния. Раствор ксилола подкисляли рассчитанным количеством хлористого водорода в этилацетате (содержание хлористого водорода примерно 10-20 вес.%), осажденные кристаллы фильтровали и сушили до постоянного веса. Таким образом было получено 17,06 г гидрохлорида N-метил-{3-фенил-3-[4-(трифторметил)-фенокси]-пропил}-амина в виде белых кристаллов.
Выход 83,95%.
Чистота (ТСХ): максимум одно неизвестное пятно (максимум 1% примесей).
Температура плавления: 155-158oС.
Содержание (основания): 98,5-101,5%
Содержание (по ВЭЖХ): 98,5-101,5%
Примеси (по ВЭЖХ):
- общее максимальное количество 1,5%;
- примеси неизвестной природы, максимальное количество каждой 0,1%;
- примеси известной природы (промежуточные продукты, продукты разложения), максимальное количество каждой 0,3%.
Продукт отвечает требованиям Фармакопеи Великобритании и США.
Пример 2
N-метил-{3-фенил-3-[4-(трифторметил)-фенокси]-пропил}-амин
87,4 г (0,27 моль) N,N-диметил-{3-фенил-3-[4-трифторметил)-фенокси]-пропил}-амина и 300 мл толуола помещали в сосуд с мешалкой. К этому раствору при 80oC добавляли по каплям при перемешивании раствор 89,7 г (0,81 моль) этилхлорформиата в 68 мл ксилола. Температура реакционной смеси имела значение 80-82oC. Нагревание и перемешивание продолжали до прекращения выделения газообразного хлористого метила. Реакционную смесь разбавляли 100 мл толуола, затем добавляли по каплям смесь 20,6 мл концентрированной соляной кислоты и 80 мл воды при перемешивании в течение 5 мин. Слои разделяли. Органическая фаза содержала 92,7 г (90%) N-метил-N-этоксикарбонил-{3-фенил-3-[4-(трифторметил)-фенокси]-пропил}-амина.
Это соединение гидролизовали раствором 45 г гидроксида калия и 65,2 г гидроксида натрия в 7,66 мл воды и 7,66 мл н-бутанола. Реакционную смесь кипятили при перемешивании в течение 8 ч, причем температура кипения уменьшилась со 110 до 106oC (наружная температура 134-136oC). Реакционную смесь обрабатывали, как описано в примере 1. Таким образом было получено 65,6 г нужного соединения в форме гидрохлорида с выходом 78%. Чистота этого продукта была такой же, как чистота продукта, полученного в примере 1.
Пример 3
Синтез проводили, как описано в примере 2, за исключением того, что гидролиз N-метил-N-этоксикарбонил-{3-фенил-3-[4-(трифторметил)-фенокси]-пропил} -амина проводили в растворе 97,2 г гидроксида натрия в 7,66 мл воды и 7,66 мл н-бутанола. Время реакции - 8 ч.
Таким образом было получено 74 г гидрохлорида N-метил-{3-фенил-3-[4-(трифторметил)-фенокси] -пропил}-амина с выходом 88%. Чистота этого продукта была такой же, как чистота продукта, полученного в примере 2.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ СОЛИ 3-ЭТИЛ-5-МЕТИЛ-2-(2-АМИНОЭТОКСИМЕТИЛ)-4-(2-ХЛОРФЕНИЛ)-6-МЕТИЛ-1,4-ДИГИДРО-3,5-ПИРИДИНДИКАРБОКСИЛАТА И БЕНЗОЛСУЛЬФОКИСЛОТЫ (БЕЗИЛАТА АМЛОДИПИНА) | 1998 |
|
RU2163597C2 |
| ПРОИЗВОДНОЕ 2-(1,2,4-ТРИАЗОЛ-1-ИЛ)-1,3,4-ТИАДИАЗОЛА, СПОСОБ ЕГО ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ | 1998 |
|
RU2180903C2 |
| СПОСОБ ПОЛУЧЕНИЯ {2-[4-(АЛЬФА-ФЕНИЛ-П-ХЛОРБЕНЗИЛ)ПИПЕРАЗИН-1-ИЛ]-ЭТОКСИ}-УКСУСНОЙ КИСЛОТЫ И НОВЫЕ ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ | 2000 |
|
RU2248974C2 |
| СПОСОБ ПОЛУЧЕНИЯ 1-(3-МЕРКАПТО-(2S-МЕТИЛПРОПИОНИЛ)-ПИРРОЛИДИН-(2S)-КАРБОНОВОЙ КИСЛОТЫ | 1991 |
|
RU2026286C1 |
| ПРОИЗВОДНЫЕ 1,2,4-ТРИАЗОЛИЛТИОАМИДОВ | 1990 |
|
RU2045521C1 |
| СПОСОБ ПОЛУЧЕНИЯ 6-МЕТИЛ-2-(4-МЕТИЛФЕНИЛ)-ИМИДАЗОЛО[1,2-А]-ПИРИДИН-3-(N, N-ДИМЕТИЛАЦЕТАМИДА), ЭФИРЫ, КРИСТАЛЛИЧЕСКИЕ ЭФИРЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ | 2000 |
|
RU2254334C2 |
| ПРОИЗВОДНЫЕ 3(2Н)-ПИРИДАЗИНОНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ЛЕЧЕНИЯ | 1992 |
|
RU2130019C1 |
| ПРОИЗВОДНЫЕ ДИГИДРОПИРИМИДОТИАЗИНА ИЛИ ИХ ТЕРАПЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ ПРИСОЕДИНЕНИЯ КИСЛОТЫ, ОБЛАДАЮЩИЕ ПРОТИВОАНГИННОЙ И АНТИВОСПАЛИТЕЛЬНОЙ АКТИВНОСТЬЮ | 1990 |
|
RU2022965C1 |
| 2-ФЕНИЛ-2-МЕТИЛАМИНОЭТОКСИ-1,7,7-ТРИМЕТИЛБИЦИКЛО[2,2,1]ГЕПТАН, ЕГО ОПТИЧЕСКИ АКТИВНЫЕ ИЗОМЕРЫ И ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ ДЛЯ ФАРМАЦЕВТИЧЕСКИХ КОМПОЗИЦИЙ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ), ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 1997 |
|
RU2199523C2 |
| СПОСОБ ПОЛУЧЕНИЯ N-МЕТИЛ-3-ФЕНИЛ-3-(4-ТРИФТОРМЕТИЛФЕНОКСИ)-ПРОПИЛАМИНА ГИДРОХЛОРИДА | 2007 |
|
RU2336264C1 |
Описывается способ получения флуоксетина, а именно N-метил-{3-фенил-3-[4-(трифторметил)-фенокси] -пропил} -амина формулы I и его фармацевтически приемлемых солей присоединения кислот путем реакции N,N-диметил-{3-фенил-3-[4-(трифторметил)-фенокси] -пропил} -амина формулы III и этилхлорформиата, гидролиза и декарбоксилирования N-метил-N-этоксикарбонил-{ 3-фенил-3-[4-(трифторметил)-фенокси] -пропил} амина формулы II, и, если необходимо, получения соли, который включает произведение реакции соединения формулы III с этилхлорформиатом в толуоле, или ксилоле, или их смеси при температуре ниже 90oC; удаление из реакционной смеси примесей и побочных продуктов путем обработки разбавленной кислотой; отделение органической фазы, содержащей производное уретана формулы II, и реакцию указанной органической фазы, без выделения производного уретана формулы II, с гидроксидом щелочного металла при температуре кипения реакционной смеси в присутствии воды и необязательно н-бутанола; удаление неорганических соединений, и, если необходимо, превращение таким образом полученного основания формулы I в его фармацевтически приемлемую соль присоединения кислоты. Соединения формулы I известны как ценные антидепрессанты. Технический результат - упрощение процесса. 7 з.п. ф-лы. 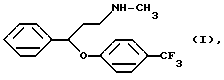
и его фармацевтически приемлемых солей присоединения кислот путем реакции N,N-диметил-{3-фенил-3[4-трифторметил)-фенокси]-пропил}-амина формулы III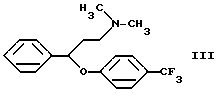
и этилхлорформиата, гидролиза и декарбоксилирования N-метил-N-этокси-карбонил-{3-фенил-3-[4-(трифторметил)-фенокси]-пропил}-амина формулы II
и, если необходимо, получения соли, который включает проведение реакции соединения формулы III с этилхлорформиатом в толуоле, или ксилоле, или их смеси при температуре ниже 90oC, удаление из реакционной смеси примесей и побочных продуктов путем обработки разбавленной кислотой, отделение органической фазы, содержащей производное уретана формулы II, и реакцию указанной органической фазы, без выделения производного уретана формулы II, с гидроксидом щелочного металла при температуре кипения реакционной смеси в присутствии воды и, необязательно н-бутанола, удаление неорганических соединений, и, если необходимо, превращение таким образом полученного основания формулы I в его фармацевтически приемлемую соль присоединения кислоты.
| Установка для приготовления проб сыпучего материала | 1974 |
|
SU529842A1 |
| Способ получения малеинового ангидрида | 1973 |
|
SU617006A3 |
| US 4018895 A, 19.04.1977 | |||
| RU 94037967 A, 27.08.1996. | |||
