Изобретение относится к ряду пирозоло[4,3-d]пиримидин-7-онов, которые представляют собой сильнодействующие и селективные ингибиторы циклической гуанозин-3',5'-монофосфатфосфодиэстеразы (цГМФ ФДЭ), которая полезна в самых различных областях терапии, включая лечение разных сердечно-сосудистых заболеваний, в частности гипертонии, сердечной недостаточности и атеросклероза.
Соединения изобретения проявляют селективность в отношении ингибирования скорее цГМФ ФДЭ, чем циклических аденозин-3',5'-монофосфатфосфодиэстеразы (цАМФ ФДЭ), и как следствие этого селективного ингибирования ФДЭ повышается содержание цГМФ, что в свою очередь вызывает улучшение антиагрегационных условий для кровяных пластинок, проявляются антисосудоспазматическое и сосудорасширяющее действие, а также потенциация эффектов релаксационного фактора, вызванного эндотелием (РФВЭ) и нитрососудорасширительных средств. Таким образом, такие соединения могут быть использованы при лечении ряда заболеваний, включая сюда стабильную, нестабильную и вариантную (Prinzmetal) ангину, гипертонию, гиперемическую сердечную недостаточность, атеросклероз, пониженную пропускную способность кровеносных сосудов, например постподкожную транслуминальную коронарную ангиопластику (пост-ПТКА), заболевания периферийных сосудов, удар, бронхиты, хроническую астму, аллергическую астму, аллергические риниты, глаукому и те заболевания, которые характеризуются расстройствами подвижности кишечника, например синдромом кишечного раздражения (СКР).
В описании к заявке на европейский патент ЕР-А-0201188 предлагаются некоторые пиразоло/4,3-d/пиримидин-7-оны в качестве аденозиновых рецепторных антагонистов и ингибиторов ФДЭ, которые могут быть использованы для лечения сердечно-сосудистых заболеваний, в частности сердечной недостаточности или кардиальной недостаточности. Тем не менее такие соединения не являются ни особенно сильнодействующими ингибиторами ФДЭ, ни соединениями, о которых сказано, что они селективно ингибируют цГМФ ФДЭ.
Согласно изобретению предложен способ получения пиразолопиримидиновых соединений I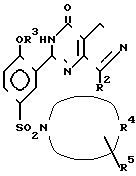 где R1 атом водорода, метил или этил;
где R1 атом водорода, метил или этил;
R2 метил, СН2ОН, СН2ОСН3 или н-пропил;
R3 н-пропил или аллил;
R4, взятый вместе с азотным атомом, в котором он связан, образует пиперидино или 4-N-(R6) пиперазинильную группу;
R5 атом водорода, N(CH3)2 или CONH2;
R6 атом водорода, метил, 2-пропил, 2-гидроксил этил, CSNH2 или C(NH)NHCH3; или их фармацевтически приемлемых солей, отличающийся тем, что соединение формулы II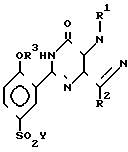 где R1, R2 и R3 имеют значения, определенные выше, или R3 Н, R2 CH2OCOCH3, CH2OCOPh, а
где R1, R2 и R3 имеют значения, определенные выше, или R3 Н, R2 CH2OCOCH3, CH2OCOPh, а
Y атом хлора, брома или фтора, подвергают взаимодействию с соединением формулы III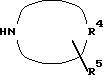 где R4 и R5 имеют приведенные значения, полученный продукт выделяют и при необходимости переводят в его фармацевтически приемлемую соль, или соль R3 атом водорода, осуществляют последующее О-алкилирование фенольной группы и необязательное превращение полученного продукта в его фармацевтически приемлемую соль или, если группа R2 является гидроксигруппой, защищенной ацетильной или бензольной группой, указанную защитную группу с помощью основного гидролиза удаляют и затем при необходимости переводят в его фармацевтически приемлемую соль.
где R4 и R5 имеют приведенные значения, полученный продукт выделяют и при необходимости переводят в его фармацевтически приемлемую соль, или соль R3 атом водорода, осуществляют последующее О-алкилирование фенольной группы и необязательное превращение полученного продукта в его фармацевтически приемлемую соль или, если группа R2 является гидроксигруппой, защищенной ацетильной или бензольной группой, указанную защитную группу с помощью основного гидролиза удаляют и затем при необходимости переводят в его фармацевтически приемлемую соль.
Предпочтительные соединения формулы I, где R1 метил; R2-п-пропил; R3 этил, н-пропил, или аллил; R4 взятый вместе с атомом азота с которым он связан, образует 4-(N)-(R6)-пинеразинильную группу; R5 атом водорода, R6 атом водорода, метил, 2-пропил или 2-гидроксиэтил.
Соединения формулы I могут содержать по одному или более центра асимметрии, благодаря чему они могут существовать в форме энантиомеров или диастереоизомеров. Рамками изобретения охватываются как смеси таких изомеров, так и индивидуальные отдельные изомеры.
Соединения формулы I могут также существовать в таутометрических формах, причем рамками изобретения охватываются как смеси, так и отдельные индивидуальные таутомеры.
Кроме того, изобретением охватываются радиоактивные меченые производные соединений формулы I, которые приемлемы для использования в ходе биологических исследований.
Фармацевтически приемлемые соли соединений формулы I, которые содержат основные центры, представляют собой кислые аддукты, полученные с использованием фармацевтически приемлемых кислот. Примеры таких аддуктов охватывают гидрохлоридные, гидробромидные, сульфатные или бисульфатные, фосфатные или кислые фосфатные, ацетатные, цитратные, фумаратные, глюконатные, лактатные, малеатные, сукцинатные и тартратные соли. Соединения формулы I способны с основаниями также образовывать фармацевтически приемлемые соли металлов. К их примерам относятся натриевые и калиевые соли.
Соединения особенно предпочтительного ряда отвечают формуле I, где R1 метил, R2 н-пропил; R3 этил, н-пропил или аллил; R4 взятый вместе с азотным атомом, с которым он связан, образует 4-N-(R6)-пиперазиниловую группу; R5 водородный атом; R6 водородный атом, алкил С1-С3 или 2-оксиэтил.
Класс особенно предпочтительных индивидуальных соединений изобретения охватывает 5-[2-аллилокси-5-/4-метилпиперазинилсульфонил/-фенил] -1-метил- 3-н-пропил-1,6-дигидро-7Н-пирозоло(4,3-d)пи- римидин-7-он; 5-[2-этокси-5-/пиперазинилсульфонил/фенил] -1-метил-3-н-пропил- 1,6-дигидро-7Н-пиразоло(4,3-d)пиримидин-7-он; 5-[2-этокси-5-/4-метилпиперазинилсульфонил/-фенил] -1-метил-3-н- пропил-1,6-дигидро-7Н-пиразоло(4,3-d)пиримидин-7-он; 5-{2-этокси-5-[4-/2-пропил/-пиперазинилсульфонил] -фенил} -1-метил- 3-н-пропил-1,6-дигидро-7Н-пиразоло/4,3-d/пири- мидин-7-он; 5-{2-этокси-5-[4-/2-оксиэтил/пиперазинилсульфонил] -фенил} -1-ме- тил- 3-н-пропил-1,6-дигидро-7Н-пиразоло/4,3-d/пиримидин-7-он; 1-метил-5-[5-пиперазинилсульфонил/-2-н-пропоксифенил] -3-н-пропил-1,6- дигидро-7Н-пиразоло(4,3-d)пиримидин-7-он и 5-{ 5-[4-/2-оксиэтил/пиперазинилсульфонилсульфонил/-2-н-про- поксифенил} 1-метил-3-н-пропил-1,6-дигидро-7Н-пиразоло/4,3-d/пиримидин-7-он.
Соединения общей формулы I могут быть получены реакцией соединения общей формулы II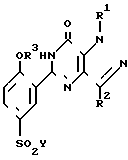 где значения символов R1-R3 определены выше, а Y обозначает атом галогена, предпочтительнее атом хлора с соединением общей формулы III
где значения символов R1-R3 определены выше, а Y обозначает атом галогена, предпочтительнее атом хлора с соединением общей формулы III
HN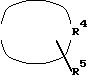 где значения R4 и R5 определены выше. Эту реакцию обычно проводят при комнатной температуре, предпочтительнее в присутствии растворителя, например алканола, содержащего от одного до трех углеродных атомов, с использованием избытка соединения III с целью удалить кислый побочный продукт (НУ).
где значения R4 и R5 определены выше. Эту реакцию обычно проводят при комнатной температуре, предпочтительнее в присутствии растворителя, например алканола, содержащего от одного до трех углеродных атомов, с использованием избытка соединения III с целью удалить кислый побочный продукт (НУ).
Соединения общей формулы II могут быть получены из соединений общей формулы IV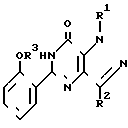 где значения символов R1-R3 определены выше, в соответствии с известными методами введения группы SO2, где значения Y определены выше в ароматическое кольцо, например, когда символом Y обозначен хлорный атом, путем воздействия хлорсульфокислотой при температуре 0оС или близкой к ней.
где значения символов R1-R3 определены выше, в соответствии с известными методами введения группы SO2, где значения Y определены выше в ароматическое кольцо, например, когда символом Y обозначен хлорный атом, путем воздействия хлорсульфокислотой при температуре 0оС или близкой к ней.
В том случае, когда R3 группа, чувствительная к удалению в условиях хлорсульфонилирования, например аллил, причем указанную группу можно вводить на заключительной стадии синтеза. Таким образом, фенол общей формулы IV, где R3 водородный атом; R1 и R2 определены выше, который можно получить удалением защитной группы О-аллилового аналога через посредство палладия, как это проиллюстрировано в примере 25, хлорсульфируют с получением соединения общей формулы II, где Y атом хлора; R3 водородный атом; R1 и R3 определены выше. Затем проводят реакцию этого последнего с соответствующим амином III, в результате чего образуется соединение общей формулы I, где R3 водородный атом; R1, R2, R4 и R5 определены выше, которое О-алкилируют с получением соединения общей формулы I, где R1-R5 определены выше для формулы I. Эту реакцию алкилирования можно проводить в стандартных условиях с использованием соответствующего алкилгалогенида, например аллилбромида, в присутствии основания, в частности карбоната калия, в среде приемлемого растворителя, например 2-бутанона, при температуре кипения реакционной смеси с обратным холодильником. По другому варианту реакцию алкилирования можно проводить в реакционных условиях, предложенных Мицунобу.
В случае других соединений формулы IV, которые могут оказаться не совместимыми с условиями реакции хлорсульфонирования, например таких, у которых R2 оксиалкил С1-С6, гидроксильная группа может быть защищена ацильной группой, в частности ацетильной или бензоильной группой. В дальнейшем упомянутую защитную группу удаляют на заключительной стадии синтеза в стандартных условиях основного гидролиза, в результате чего образуются соединения общей формулы I, где R2 оксиалкил С1-С6; R1, R3-R5 определены выше для формулы I. Эти соединения могут быть также получены в данном случае в качестве побочных продуктов хлорсульфонирования соответствующих алкоксианалогов, то есть соединений общей формулы IV, где R2 алкокси (С1-С3) алкил (С1-С6), с последующей реакцией сырого продукта с требуемым амином III, как это проиллюстрировано в примере 48.
Соединения общей формулы IV могут быть получены из соединений общей формулы V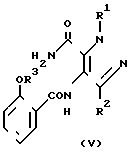
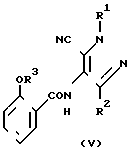 где R1-R3 определены выше, путем осуществления известных методов циклизации с получением пимидинового кольца. Так, например, циклизацию можно проводить путем обработки соединения Y основанием, в частности гидратом окиси натрия или карбонатом калия, возможно в присутствии перекиси водорода, в водно-этанольной среде при температуре кипения с обратным холодильником в течение 2-40 ч. В этих условиях в качестве предшественника соединения IV можно также использовать связанный нитрил общей формулы VI, где R1-R3 определены выше.
где R1-R3 определены выше, путем осуществления известных методов циклизации с получением пимидинового кольца. Так, например, циклизацию можно проводить путем обработки соединения Y основанием, в частности гидратом окиси натрия или карбонатом калия, возможно в присутствии перекиси водорода, в водно-этанольной среде при температуре кипения с обратным холодильником в течение 2-40 ч. В этих условиях в качестве предшественника соединения IV можно также использовать связанный нитрил общей формулы VI, где R1-R3 определены выше.
В соответствии с другой процедурой циклизации соединения общей формулы IV могут быть получены путем обработки соединения V фосфорной кислотой при 140оС или близкой к ней в течение 6-18 ч.
Соединения общих формул V и VI могут быть получены из соединений общих формул соответственно VII и VIII
H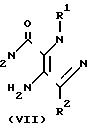
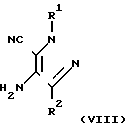 где R1 и R2 определены выше, реакцией с соединением общей формулы IX
где R1 и R2 определены выше, реакцией с соединением общей формулы IX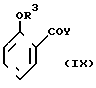 где R3 и Y определены выше.
где R3 и Y определены выше.
Эту реакцию обычно проводят с использованием избытка (IX) в присутствии избытка алифатического третичного амина, в частности триэтиламина, который выполняет функцию поглотителя кислого побочного продукта (НУ), возможно в присутствии катализатора, в частности 4-диметиламинопиридина, в среде инертного растворителя, в частности дихлорметана, при 0 25оС в течение 2-6 ч.
В том случае, когда амины формулы III, аминопиразолы формул VII и VIII и ацилгалогениды формулы IX не являются технически доступными, их можно получать согласно обычными процедурам синтеза, которые описаны в известной литературе, из легкодоступных исходных материалов с использованием стандартных реагентов и в стандартных реакционных условиях.
Некоторые соединения общей формулы I, где R4 совместно с азотным атомом, с которым он связан, образует 4-N-(R6)-пиперазиниловую группу, значения R6 определены выше, но он не обозначает водородного атома, могут быть получены непосредственно из соответствующего 4-N-незамещенного пиперазинового аналога, то есть из соединения общей формулы I, где R6 водородный атом, с использованием соответствующих стандартных процедур синтеза.
Все указанные реакции совершенно обычны, а соответствующие реагенты и условия их проведения могут быть легко определены, если воспользоваться стандартными учебниками и примерами, которые приведены ниже. Их альтернативы и варианты совершенно очевидны любому специалисту в данной области в отношении возможности получения всех соединений, отвечающих формуле I.
Биологическую активность соединений изобретения определяют согласно следующим методам испытаний.
Фосфодиэстеразная активность.
Сродство соединения с цГМФ и цАМФ ФДЭ оценивают путем определения его величины IC50 (концентрация ингибитора, которая требуется для 50%-ного ингибирования энзимного действия). ФДЭ-энзимы выделяют из кровяных пластин кролика и почки крысы, по существу по методу У.Дж.Томпсона и др. (Biochem. 1971, 10, 311). Независимый от кальция) кольмодулина [Ca/CAM] цГМФ ФДЭ-энзим и цГМФ-ингибированный цАМФ ФДЭ-энзим получают из кровяных пластинок кролика, тогда как из четырех основных ФДЭ-энзимов крысиной почки выделяют Са/САМ-зависимую цГМФ ФДЭ (фракция I). Испытания проводят в соответствии с модифицированным "периодическим" методом У. Дж.Томпсона и М.М.Эпплмана (Biochem. 1979, 18, 5228). Результаты таких испытаний показывают, что соединения настоящего изобретения являются сильнодействующими и селективными ингибиторами обеих цГМФ ФДЭ.
Действие, препятствующее агрегированию кровяных пластинок.
Это действие оценивают путем определения способности соединения ингибировать агрегирование кровяных пластинок в лабораторных условиях, которое инициировано активирующим кровяные пластинки фактором (АПФ), и потенциировать антиагрегативное действие на кровяные тельца в лабораторных условиях активаторов гуанилатциклазы, в частности нитропруссида и рФВЭ. Промытые кровяные пластины готовят по существу по методу Дж.Ф.Мустарда и др. (Methods in Enzymol. 1989, 169, 3), а агрегирование определяют в соответствии со стандартной турбидиметрической технологией, которая описана Г.В.Р.Борном в J. Physiol (Лондонэ, 1962, 162, 67Р.).
Антигипертоническое действие.
Это действие оценивают после внутривенного или перорального введения соединения в организм крыс, страдающих спонтанной гипертонией. С помощью канюли, имплантированной в сонную артерию либо находящихся в сознании, либо анастезированных животных.
Для введения в организм людей с целью лечения или профилактики ангины, гипертонии или гиперемической сердечной недостаточности пероральная доза соединений обычно находится в интервале 4-800 мг ежедневно для взрослого пациента средней массы 70 кг. Так, например, для типичного взрослого пациента индивидуальные таблетки или капсулы содержат по 2-400 мг активнодействующего вещества в среде подходящей и фармацевтически приемлемой основе для приготовления лекарств или носителя для введения в организм в виде одинарных или многократных доз по одному или несколько раз в день. Дозы для внутривенного, буккального или подъязычного введения в организм обычно находятся в интервале 1-400 мг/одинарную дозу в зависимости от потребности. На практике фактический режим дозирования подбирается врачом таким образом, чтобы он оказался наиболее приемлемым для данного индивидуального пациента, поскольку такой режим изменяется в зависимости от возраста, массы и реакции конкретного пациента. Приведенные дозы являются примерами для среднего случая, однако в отдельных случаях конкретные дозы могут превосходить максимальные или быть ниже минимальных пределов диапазона дозировок, причем все возможные дозировки охватываются рамками изобретения.
Для использования людьми соединения формулы I могут быть введены в организм индивидуально, но обычно их вводят в смеси с фармацевтическим носителем, выбираемым в зависимости от намечаемого пути введения в организм и стандартной фармацевтической практики. Так, например, их можно вводить перорально, буккально или подъязычно в форме таблеток, содержащих основу для приготовления лекарств, в частности крахмал или лактозу, или капсул или шариков как индивидуально, так и в смеси с основной для приготовления лекарств, или же в форме эликсиров или суспензий, содержащих вкусовые, ароматизирующие или окрашивающие добавки. Такие соединения можно также вводить в организм парентерально, например внутривенно, внутримышечно, подкожно или интракоронарно. Для парентерального введения в организм их лучше всего использовать в форме стерильного водного раствора, который может содержать другие вещества, например соли и глюкозу в количестве, достаточном для того, чтобы сделать раствор изотоническим относительно крови.
Таким образом, в соответствии с еще одним аспектом изобретения предлагается фармацевтическая композиция, которая включает в себя соединение формулы I или его соль, фармацевтически приемлемую для использования в медицине, в частности для лечения ангины, гипертонии или гиперемическую сердечную недостаточность человека.
Рамками изобретения охватывается также использование соединений формулы I или их фармацевтически приемлемых солей для приготовления медикаментов, применяемых при лечении стабильной, нестабильной и вариантной (Prinzmetal) ангины, гипертонии, гиперемической сердечной недостаточности, атеросклероза, удара, заболевания периферийных сосудов, пониженной пропускной способности, например, пост-ПТКА, хронической астмы, бронхитов, аллергической астмы, аллергического ринита, глаукомы или тех заболеваний, которые характеризуются расстройствами подвижности кишечника, в частности СКР.
В дальнейшем процессы получения соединений изобретения более конкретно проиллюстрированы со ссылками на следующие примеры экспериментов. Степень чистоты соединений при этом проверяли обычным путем, тонкослойным хроматографическим анализом (ТСХ) с использованием пластин Merek Kieselgel 60 F254. 1Н-ядерномагнитную резонансную спектрограмму записывали с помощью спектрометра Nicolet QE-300, причем во всех случаях она соответствовала предложенному строению.
П р и м е р 1. Этиловый эфир 1-метил-3-н-пропилпиразол-5-карбоновой кислоты.
24,1 г (0,132 моля) этилового эфира 3-н-пропилпиразол-5-карбоновой кислоты (полученной по методу, описанному в Chem. Pharm. Bull. 1984, 32, 1568) в смеси с 16,8 г (0,133 моля) диметилсульфата выдержали при 90оС в течение 2,5 ч. Эту смесь растворили в дихлорметане и раствор промыли раствором карбоната натрия. Органическую фазу отделили, высушили над сульфатом магния и выпарили в вакууме, получив твердый продукт. В результате хроматографической обработки на 300 г силикагеля, элюируя дихлорметаном, продукт получили в виде 20,4 г (79% -ный выход) бесцветного масла. Rf 0,8 (двуокись кремния; дихлорметан, метанол, уксусная кислота в соотношении 80:20:1).
П р и м е р 2. 1-Метил-3-н-пропилпиразол-5-карбоновая кислота.
20,2 г (0,10 моля) этилового эфира 1-метил-3-н-пропилпиразол-5-карбоновой кислоты суспендировали в 50 мл (0,30 моля) 6 н.водного раствора гидрата окиси натрия. Эту смесь выдержали при 80оС в течение 2 ч, а затем разбавили 50 мл воды и подкислили добавлением 25 мл концентрированной соляной кислоты. В результате фильтрования в виде бледно-коричневых кристаллов получили 12,3 г (71% -ный выход) карбоновой кислоты с температурой плавления 150-154оС.
Найдено, C 56,99; H 7,25; N 16,90.
C8H12N2O2.
Вычислено, C 57,13; H 7,19; N 16,6.
П р и м е р 3. 1-Метил-4-нитро-3-н-пропилпиразол-5-карбоновая кислота.
12,1 г (0,072 моля) 1-метил-3-н-пропилпиразол-5-карбоновой кислоты порциями добавили в смесь 13 мл олеума и 11 мл дымящей азотной кислоты, поддерживая температуру на уровне ниже 60оС. После завершения операции добавления смесь выдержали в течение ночи при 60оС, а затем охладили до комнатной температуры с последующим выливанием на лед. В результате отфильтровывания осадка в виде белого твердого вещества получили 11,5 г (75%-ный выход) нитропиразола с температурой плавления 124-127оС.
Найдено, C 45,43; H 5,22; N 19,42.
C8H11N3O4.
Вычислено, C 45,57; H 5,20; N 19,71.
П р и м е р 4. 1-Метил-4-нитро-3-н-пропилпиразол-5-карбоксамид.
11,3 г (0,053 моля) 1-метил-4-нитро-3-н-пропилпиразол-5-карбоновой кислоты добавили в 50 мл хлористого тионила, и образовавшуюся смесь выдержали при температуре кипения с обратным холодильником в течение 3 ч. Затем реакционную смесь охладили и выпариванием в вакууме удалили избыток хлористого тионила. Маслоподобный остаток растворили в 50 мл ацетона и раствор осторожно добавили в смесь 50 г льда с 50 мл концентрированного водного раствора гидрата окиси аммония. Остаток собрали фильтрованием, в результате чего в виде бледно-желтого твердого материала получили 8,77 г (78%-ный выход) пиразолкарбоксамида с температурой плавления 141-143оС.
Найдено, C 45,22; H 5,71; N 26,12.
C8H12N4O3.
Вычислено, C 45,28; H 5,70; N 26,40.
П р и м е р 5. 4-Амино-1-метил-3-н-пропилпиразол-5-карбоксамид.
3,45 г (16,2 ммоля) 1-метил-4-нитро-3-н-пропилпиразол-5-карбоксамида и 18,4 г (81 ммоля) дигидрата хлорида двухвалентного олова суспендировали в этаноле и смесь выдержали при температуре кипения с обратным холодильником в течение 2 ч. Образовавшийся раствор охладили до комнатной температуры, подкислили до величины рН, равной 9, добавлением 2 н.водного раствора гидрата окиси натрия с последующей экстракционной обработкой 3 порциями по 150 мл дихлорметана. Органические экстракты собрали, высушили над сульфатом магния и выпарили в вакууме. В результате растирания остатка в диэтиловом эфире получили в виде не совсем белого твердого продукта 2,77 г (94%-ный выход) аминопиразола с температурой плавления 98-101оС.
Найдено, C 52,84; H 7,81; N 30,38.
C8H14N4O.
Вычислено, C 52,73; H 7,74; N 30,75.
П р и м е р 6. 4-(2-Этоксибензамидо)-1-метил-3-н-пропилпиразол-5-карбоксамид.
Раствор 6,1 г (33,0 моля) 2-этоксибензоилхлорида в 50 мл дихлорэтана добавили в перемешиваемый раствор 3,0 г (16,4 ммоля) 4-амино-1-метил-3-н-пропилпиразол-5-карбоксиамида, 0,02 г (0,164 ммоля) 4-диметиламинопиридина и 3,34 г (33,0 ммоля) триэтиламина в 50 мл дихлорметана при 0оС. Образовавшейся смеси позволили нагреться до комнатной температуры и ее перемешали в течение 2 ч. Растворитель выпарили в вакууме, остаток растворили в 250 мл смеси дихлорметана с метанолом в соотношении 19:1, а затем раствор промыли 100 мл 1 н.соляной кислоты, высушили над сульфатом магния и выпарили в вакууме. Сырой материал подвергали хроматографической обработке на 200 г силикагеля, элюируя смесью дихлорметана с метанолом в соотношении 97:3, в результате чего получили розовое твердое вещество, кристаллизация которого из смеси этилацетата с гексаном позволила получить в виде бледно-розового твердого продукта 2,2 г (40%-ный выход) пиразол-5-карбоксамида с температурой плавления 153-155оС.
Найдено, C 61,66; H 6,77; N 16,95.
C17H22N4O3.
Вычислено, C 61,80; H 6,71; N 16,96.
П р и м е р 7. 5-(2-Этоксифенил)-1-метил-3-н-пропил-1,6-дигидро-7Н-пиразоло/4,3-d/ пиримидин-7-он.
223 г (0,676 моля) 4-(2-этоксибензамидо)-1-метил-3-н-пропилпиразол-5-карбоксамида порциями добавили в раствор 54 г (1,35 моля) гидрата окиси натрия с 224 мл 30% -ной перекиси водорода в 2000 мл воды. Затем добавили 700 мл этанола образовавшуюся смесь выдержали при температуре кипения с обратным холодильником в течение 2,5 ч, охладили, после чего выпарили в вакууме. Образовавшийся твердый продукт обработали 380 мл 2 н.соляной кислоты с использованием внешнего охлаждения и смесь подвергали экстракционной обработке 700 мл и 3 порциями по 200 мл дихлорметана. Объединенные органические экстракты промыли последовательно 3 порциями по 400 мл насыщенного водного раствора карбоната натрия и 30 мл рассола, после чего высушили над сульфатом натрия и выпарили в вакууме.
В результате хроматографической обработки остатка на 1000 г силикагеля с использованием градиента элюирования метанолом в дихлорметане (0-1%) с последующим растиранием сырого продукта в 300 мл диэтилового эфира получили в виде бесцветного твердого материала 152,2 г (72%-ный выход) соединения, указанного в заголовке, с температурой плавления 143-146оС.
Найдено, C 65,56; H 6,44; N 18,14.
C17H20N4O2.
Вычислено, C 65,36; H 6,45; N 17,94.
П р и м е р 8. 5-(5-Хлорсульфонил-2-этоксифенил)-1-метил-3-н-пропил-1,6-диги- дро- 7Н-пиразоло(4,3-d)пиримидин-7-она.
10,0 г (32,1 ммоля) 5-(2-этоксифенил)-1-метил-3-н-пропил-1,6-дигидро-7Н-пиразол(4,3-d) пиримидин-7-она порциями добавили в 20 мл хлорсульфоновой кислоты при 0оС в азотной атмосфере. После перемешивания в течение ночи реакционный раствор осторожно добавили в 150 мл смеси льда с водой и водную смесь подвергали экстракционной обработке 4 порциями по 100 мл смеси дихлорметана с метанолом в соотношении 9:1. Объединенные экстракты высушили над сульфатом натрия и выпарили в вакууме, в результате чего получили в виде белого твердого вещества 12,8 г (97%-ный выход) требуемого хлористого сульфонила с температурой плавления 179-181оС.
Найдено, C 50,07; H 4,71; N 13,29.
C17H19ClN4O4S.
Вычислено, C 49,70; H 4,66; N 13,64.
П р и м е р 9. 5-[2-Этокси-5-(4-карбамоилпиперидинилульфонил)-фенил]-1-метил-3-н- пропил-1,6-дигидро-7Н-пиразоло(4,3-d)пиримидин-7-он.
703 мг (5,50 ммоля) 4-карбамоилпиперидина добавили в перемешиваемую суспензию 750 г (1,80 г (1,80 ммоля) 5-(5-хлорсульфонил-2-этоксифенил)-1-метил-3-н-пропил-1,6-дигидро- 7Н-пиразоло(4,3-d)пиримидин-7-она в 50 мл этанола при комнатной температуре. Образовавшуюся смесь перемешивали в течение 4 дней с последующим удалением растворителя в вакууме. Остаток растворили в 100 мл смеси дихлорметана с метанолом в соотношении 9:1 и раствор промыли 100 мл насыщенного водного раствора карбоната натрия. Затем водную фазу подвергали дополнительной экстракционной обработке 3 порциями по 100 мл смесями дихлорметана с метанолом, все органические фракции собрали, высушили над сульфатом магния и выпарили в вакууме с получением твердого продукта. В результате кристаллизации из смеси метанола с диметилформамидом в виде не совсем белого твердого продукта получили 446 мг (49%-ный выход) указанного в заголовке сульфонамида с температурой плавления 274-276оС.
Найдено, C 55,36; H 6,01; N 16,65.
C23H29N6O5S.
Вычислено, C 55,08; H 5,83; N 16,75.
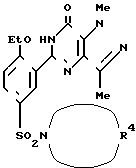
П р и м е р ы 10-14. В соответствии с процедурой примера 9 с использованием соответствующего амина получили нижеследующие соединения
Данные приведены в табл. 1.
П р и м е р 16. 5-{2-Этокси-5-[4-(метилтиоимидоил)-пиперазинилсульфонил] -фе- нил} -1- метил-3-н-пропил-1,6-дигидро-7Н-пиразоло(4,3-d)пиримидин-7- ангидроиодид.
Смесь 0,78 г (1,5 ммоля) 5-[2-этокси-5-(4-тиокарбамоилпиперазинилсульфонил)-фе- нил]-1-метил- 3-н-пропил-1,6-дигидро-7Н-пиразоло(4,3-d)пиримидин-7-она с 426 мг (3,0 ммоля) иодистого метила и 20 мл метанола перемешивали при температуре кипения с обратным холодильником в течение 2 ч, после чего ей дали остыть. Образовавшееся белое твердое вещество удалили фильтрованием и кристаллизовали из смеси этилацетата с метанолом, получив в виде бесцветных кристаллов 0,70 г (71%-ный выход) соединения, указанного в заголовке, с температурой плавления 227-228оС.
Найдено, C 41,43; H 4,79; N 14,42.
C23H31N7O4S2.
Вычислено, C 41,75; H 4,88; N 14,82.
П р и м е р 17. 5-{2-Этокси-5-[4-(метиламидино)-пиперазинилсульфонил]-фенил}-1- метил-3-н-пропил-1,6-дигидро-7Н-пиразоло (4,3-d)-7-онгидроиодид.
0,5 г (0,75 ммоля) 5-{2-этокси-5-[4-метилтиоимидоил)-пиперазинилсульфонил]-фе- нил} -1-метил-3-н-пропил-1,6-дигидро-7Н-пиразоло(4,3-d)пиримидин-7- онгидроиодида добавили в 33%-ный раствор метиламина в 20 мл этанола и смесь перемешивали при комнатной температуре в течение 18 ч. Раствор выпарили в вакууме и остаток растерли в диэтиловом эфире. В результате хроматографической обработки образовавшегося твердого продукта на 10 г силикагеля с использованием метанола в дихлорметане в качестве элюента с градиентом (0-4%), после чего сырой продукт растерли в диэтиловом эфире, получили светло-коричневый порошок. Кристаллизация из смеси этилацетата с метанолом позволила получить в виде бесцветных кристаллов 112 мг соединения, указанного в заголовке примера (23%-ный выход) с температурой плавления 253-255оС.
Найдено, C 42,90; H 5,09; N 17,41.
C23H32N8O4S; HI.
Вычислено, C 42,86; H 5,16; N 17,39.
П р и м е р 18. 1-Метил-4-(2-н-пропоксибензамидо)-3-н-пропилпиразол-5-карбокса- мид.
Этот амид получили из 2-н-пропоксибензоилхлорида в соответствии с процедурой, изложенной в примере 6, в результате чего получили твердый продукт (63%-ный выход) розового цвета с температурой плавления 148-149оС.
Найдено, C 62,97; H 7,00; N 16,29.
C18H24N4O3.
Вычислено, C 62,77; H 7,02; N 16,27.
П р и м е р 19. 1-Метил-5-(2-н-пропоксифенил)-3-н-пропил-1,6-дигидро-7Н-пиразо- ло (4,3-d)пиримидин-7-он.
0,34 г (0,99 ммоля) 1-метил-4-(2-н-пропоксибензамидо)-3-н-пропилпиразол-5-кар- боксамида добавили в перемешиваемую смесь 1,0 мл 30%-ной перекиси водорода, 0,54 г (3,92 ммоля) карбоната калия, 10 мл воды и 5 мл этанола. Эту смесь выдержали при температуре кипения с обратным холодильником в течение 38 ч, а затем выпарили в вакууме. Остаток суспендировали в 20 мл воды, а затем смесь подкисляли добавлением 20 н.соляной кислоты и подвергли экстракционной обработке 3 порциями по 20 мл дихлорметана. Экстракты объединили, высушили над сульфатом натрия и выпарили в вакууме. Полученный остаток подвергли хроматографической обработке на 6 г силикагеля с использованием метанола в дихлорметане с градиентом элюирования 0,0-1, в результате чего получили масло, последующее растирание которого в диэтиловом эфире позволило получить в виде белого твердого вещества 0,19 г (59%-ный выход) целевого продукта с температурой плавления 111-114оС.
Найдено, C 66,26; H 6,92; N 17,15.
C18H22N4O2.
Вычислено, C 66,23; H 6,80; N 17,17.
П р и м е р 20. 5-(5-Хлорсульфонил-2-н-пропоксифенил)-1-метил-3-н-пропил-1,6- дигидро-7Н-пиразоло(4,3-d)пиримидин-7-он.
Этот сульфонилхлорид получили из 5-(2-н-пропоксифенил)-1-метил-3-н-пропил-1,6- дигидро-7Н-пиразоло (4,3-d)пиримидин-7-она в соответствии с процедурой примера 8 в виде белого твердого вещества (выход 92%).
Найдено, C 51,26; H 5,02; N 12,90.
C18H21ClN4O4S.
Вычислено, C 50,88; H 4,98; N 13,19.
П р и м е р 21. 1-Метил-5-[5-(пиперазинилсульфонил)-2-н-пропоксифенил] -3-н- пропил- 1,6-дигидро-7Н-пиразоло(4,3-d)пиримидин-7-он.
Этот сульфонамид получили из пиперазина и 5-(5-хлорсульфонил-2-н-пропоксифенил)-1-метил-3-н-пропил-1,6- дигидро-7Н-пиразоло(4,3-d)пирими- дин-7-она в соответствии с процедурой примера 9 в виде белого твердого продукта (70%-ный выход) с температурой плавления 185-186оС.
Найдено, C 56,17; H 6,38; N 17,65.
C22H30N6O4S.
Вычислено, C 55,67; H 6,37; N 16,61.
П р и м е р 22. 5-{5-[4-(2-Оксиэтил)-пиперазинилсульфонил]-2-н-пропоксифенил}-1- метил-3-н-пропил-1,6-дигидро-7Н-пиразоло (4,3-d)пиримидин-7-он.
Этот сульфонамид получили из N-(2-оксиэтил)-пиперазина и 5-(5-хлорсульфонил-2-н-пропоксифенил)-1-метил-3-н-пропил-1,6- дигидро-7Н-пиразоло(4,3-d)пиримидин-7-она в соответствии с процедурой примера 9 в форме бесцветных иглоподобных кристаллов (66%-ный выход) с температурой плавления 158-159оС.
Найдено, C 55,83; H 6,58; N 16,13.
C24H34N6O5S.
Вычислено, C 55,58; H 6,61; N 16,20.
П р и м е р 23. 4-(2-Аллилоксибензамидо)-1-метил-3-н-пропилпиразол-5-карбокса- мид.
Раствор 3,93 г (0,02 моля) 2-аллилоксибензоилхлорида в 20 мл дихлорметана по каплям добавили в перемешиваемый частичный раствор 3,64 г (0,02 моля) 4-амино-1-метил-3-н-пропилпиразол-5-карбоксамида в 50 мл пиридина и образовавшуюся смесь перемешивали при комнатной температуре в течение ночи в сухой атмосфере. Растворитель выпарили в вакууме и остаток разделили между 50 мл дихлорметана и 50 мл насыщенного водного раствора карбоната натрия. Органический слой отделили, а водный слой подвергли исчерпывающей экстракционной обработке дополнительным количеством дихлорметана. Объединенные органические растворы промыли 3 порциями по 30 мл 2М соляной кислоты, а затем 30 мл рассола и высушили над сульфатом натрия. После фильтрования и выпаривания в вакууме фильтрата сырой продукт кристаллизовали из этилацетата, получив 4,525 г (66%-ный выход) соединения, указанного в заголовке примера, с температурой плавления 132-134оС.
Найдено, C 63,49; H 6,42; N 16,33.
C18H22N4O3.
Вычислено, C 63,14; H 6,48; N 16,36.
П р и м е р 24. 5-(2-Аллилоксифенил)-1-метил-3-н-пропил-1,6-дигидро-7Н-пиразоло (4,3-d)пиримидин-7-он.
Смесь 1,2 г (0,0035 моля) 4-(2-аллилоксибензамидо)-1-метил-3-н-пропилпиразол-5-карбоксамида с 0,70 г (0,018 моля) гидрата окиси натрия, 34 мл воды и 8 мл этанола прокипятили с обратным холодильником в течение 5 ч. После охлаждения раствор подвергли исчерпывающей экстракционной обработке этилацетатом. Объединенные экстракты промыли 30 мл рассола, высушили над сульфатом натрия, профильтровали и выпарили растворитель в вакууме, в результате чего получили сырой продукт, который кристаллизовали их смеси этилацетата с гексаном, выделив 0,476 г (37%-ный выход) соединения, указанного в заголовке примера, с температурой плавления 116-119оС.
Найдено, C 67,00; H 6,21; N 17,23.
C18H20N4O2.
Вычислено, C 66,65; H 6,21; N 17,27.
П р и м е р 25. 5-(2-Оксифенил)-1-метил-3-н-пропил-1,6-дигидро-7Н-пиразоло(4,3-d) пиримидин-7-он.
Смесь 0,25 г (0,0008 моля) 5-(2-аллилоксифенил)-1-метил-3-н-пропил-1,6-дигидро -7Н-пиразоло (4,3-d)пиримидин-7-она с 0,145 г (0,0015 моля) фенола, 0,131 г (0,0015 моля) пиперидина и 0,046 г (0,00004 моля) тетракис-(трифенилфосфин)-палладия в 5 мл абсолютированного этанола прокипятили в течение ночи с обратным холодильником в атмосфере азота. Смеси дали охладиться, растворитель выпарили в вакууме и остаток растворили в 40 мл этилацетата. Этот раствор промыли 3 порциями по 10 мл воды, 3 порциями по 10 мл 1М соляной кислоты и 10 мл рассола. После сушки над сульфатом натрия и фильтрования фильтрат выпарили в вакууме, получив сырой продукт. После растирания в диэтиловом эфире и кристаллизации из смеси этилацетата с пентаном получили 0,021 г (10%-ный выход) фенола, указанного в заголовке, с температурой плавления 233-238оС.
Найдено, C 63,17; H 5,65; N 19,52.
C15H16N4O2.
Вычислено, C 63,36; H 5,67; N 19,71.
П р и м е р 26. 5-(5-Хлорсульфонил-2-оксифенил)-1-метил-3-н-пропил-1,6-дигидро-7Н- пиразоло(4,3-d)пиримидин-7-он.
5-(2-Оксифенил)-1-метил-3-н-пропил-1, 6-дигидро-7Н-пиразоло(4,3-d) пиримидин-7-он (0,239 г, 0,00084 моля) порциями добавили в 3 мл перемешиваемой хлорсульфоновой кислоты, охлажденной до 0оС, в атмосфере азота, а образовавшийся темно-красный раствор перемешивали при комнатной температуре в течение 18 ч. Затем реакционную смесь осторожно по каплям добавили в перемешиваемую смесь воды со льдом, получив коричневый твердый продукт. Эту последнюю смесь подвергли экстракционной обработке 3 порциями по 30 мл дихлорметана, объединенные экстракты высушили над сульфатом натрия и профильтровали, фильтрат выпарили в вакууме, получив 0,24 г (75%-ный выход) коричневого твердого вещества, использованного на следующей стадии без дополнительной очистки: Rf 0,3 (двуокись кремния; дихлорэтан, метанол в соотношении 95:5).
П р и м е р 27. 5-[2-Окси-5-(4-метилпиперазинилсульфонил)-фенил]-1-метил-3-н- пропил- 1,6-дигидро-7Н-пиразоло(4,3-d)пиримидин-7-он.
Раствор 0,235 г (0,0006 моля) 5-(5-хлорсульфонил-2-оксифенил)-1-метил-3-н-про- пил-1,6-дигидро-7Н- пиразоло(4,3-d)пиримидин-7-она и 0,5 мл (0,0045 моля) N-метилпиперазина в 40 мл этанола перемешивали при комнатной температуре в течение 18 ч. Этот раствор выпарили в вакууме и остаток разделили между 40 мл этилацетата и 40 мл воды. Тонкодисперсный осадок отфильтровывали, промыли водой, затем этилацетатом и кристаллизовали из смеси этилацетата с ДМФ, в результате чего в форме не совсем белого порошка получили 0,260 г (49% -ный выход) соединения, указанного в заголовке с температурой плавления 283-284оС.
Найдено, C 53,53; H 5,89; N 18,40.
C20H26N6O4S.
Вычислено, C 53,80; H 5,87; N 18,82.
П р и м е р 28. 5-[2-Аллилокси-5-(4-метилпиперазинилсульфонил)-фенил]-1-ме- тил-3- н-пропил-1,6-дигидро-7Н-пиразоло(4,3-d)пиримидин-7-он.
0,02 мл (0,00023 моля) аллилбромида добавили в перемешиваемую суспензию 0,103 г (0,00023 моля) 5-[2-окси-5-(4-метилпиперазинилсульфонил)-фенил]-1-метил-3-н- пропил-1,6-дигидро-7Н-пиразоло(4,3-d)пи- римидин-7-она и 0,032 г (0,00023 моля) карбоната калия в 10 мл 2-бутанона и смесь выдержали при температуре кипения с обратным холодильником в течение 8 ч. После охлаждения реакционную смесь выпарили в вакууме и остаток суспендировали в 20 мл воды. Водную суспензию подвергли экстракционной обработке 3 порциями по 20 мл этилацетата, объединенные экстракты высушили над сульфатом натрия и после фильтрования выпарили в вакууме с получением масла. В результате хроматографической обработки в колонке с 2 г силикагеля с использованием метанола в дихлорметане с градиентом элюирования 0-3% после чего выпарили в вакууме соответствующие фракции, получили полутвердый продукт, который растворили в ацетоне, раствор выпарили в вакууме, выделив 0,011 г (10%-ный выход) соединения, указанного в заголовке, с температурой плавления 151-153оС, Rf 0,5 (двуокись кремния, дихлорметан, метанол, 95:5), м/е 487 (М+1).
П р и м е р 29. 4-(Этоксибензамидо)-1,3-диметилпиразол-5-карбоксамид.
Этот амид получили из 4-амино-1,3-диметилпиразол-5-карбоксамида (в свою очередь полученного по методу из журнала J.Med.Chem. 1987, 30, 91) в соответствии с процедурой примера 6 в форме белого твердого продукта (81%-ный выход) с температурой плавления 178-181оС.
Найдено, C 59,89; H 6,05; N 18,44.
C15H18N4O3.
Вычислено, C 59,59; H 6,00; N 18,53.
П р и м е р 30. 5-(2-Этоксифенил)-1,3-диметил-1,6-дигидро-7Н-пиразоло(4,3-d) пиримидин-7-он.
1,6 г (5,29 моля) 4-(2-этоксибензамидо)-1,3-диметилпиразол-5-карбоксамида добавили в 50 г полифосфорной кислоты и смесь выдержали при 140оС в течение 6 ч. Затем раствор охладили, вылили в 100 мл смеси воды со льдом, после чего подщелачили добавлением 10%-ного раствора гидрата окиси натрия в воде и подвергли экстракционной обработке 3 порциями по 100 мл дихлорметана. Органические экстракты объединили, высушили над сульфатом магния и выпарили в вакууме. Остаток подвергли хроматографической обработке на силикагеле, элюируя смесью дихлорметана с метанолом. В результате кристаллизации сырого продукта из водного этанола в виде бесцветного твердого вещества получили соединение, указанное в заголовке, с температурой плавления 201-204оС.
Найдено, C 63,43; H 5,57; N 19,35.
C15H16N4O2.
Вычислено, C 63,36; H 5,67; N 19,71.
П р и м е р 31. 5-(5-Хлорсульфонил-2-этоксифенил)-1,3-диметил-1,6-дигидро-7Н- пиразоло(4,3-d)пиримидин-7-он.
Этот сульфонилхлорид получили из 5-(2-этоксифенил)-1,3-диметил-1,6-дигидро-7Н-пиразоло(4,3-d) пиримидин-7-она в соответствии с процедурой примера 8 с достижением количественного выхода в виде белого твердого вещества. Rf 0,3 (двуокись кремния диэтиловый эфир). Это соединение в дальнейшем использовали без дополнительной очистки.
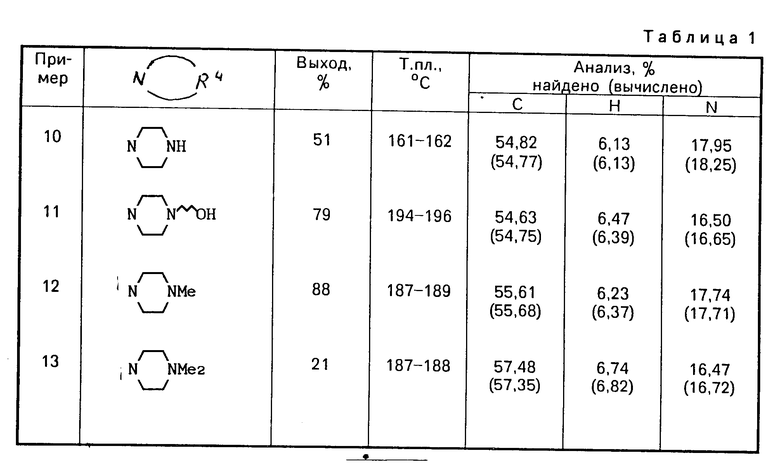
П р и м е р ы 32-34. В соответствии с процедурой примера 9 с использованием 5-(5-хлосульфонил-2-этоксифенил)-1,3-ди- метил-1,6-дигидро-7Н- пиразоло(4,3-d)пиримидин-7-она и соответствующего амина получили следующие соединения. Данные приведены в табл. 2.
П р и м е р 35. 4-Нитро-3-н-пропилпиразол-5-карбоновая кислота.
3-н-пропилпиразол-5-карбоновую кислоту (полученную по методу из журнала Chem. Pharm. Bull. 1984, 32, 1568) нитровали в соответствии с процедурой примера 3, в результате чего в виде бесцветного твердого вещества с 75%-ным выходом получили соединение, указанное в заголовке, с температурой плавления 169-173оС.
Найдено, C 42,35; H 4,56; N 21,07.
C7H9N3O4.
Вычислено, C 42,21; H 4,55; N 21,10.
П р и м е р 36. 4-Нитро-3-н-пропилпиразол-5-карбоксамид.
Смесь 7,8 г (39,2 ммоля) 4-нитро-3-н-пропилпиразол-5-карбоновой кислоты с 35 мл хлористого тионила прокипятили с обратным холодильником в течение 3 ч. Растворитель удалили выпариванием в вакууме и твердый остаток порциями добавили при 0оС в 40 мл водного раствора гидрата окиси аммония. Смесь разбавили 60 мл воды и подвергли экстракционной обработке смесью в соотношении 9: 1 дихлорметана с метанолом в виде 3 порций по 100 мл. Органические фракции объединили, высушили над сульфатом магния и выпарили в вакууме, а остаток кристаллизовали из этанола, в результате чего в виде бесцветного твердого вещества получили 1,0 г (13%-ный выход) карбоксамида с температурой плавления 202-206оС.
Найдено, C 42,35; H 5,01; N 28,38.
C7H10N4O3.
Вычислено, C 42,42; H 5,09; N 28,27.
П р и м е р 37. 4-Амино-3-н-пропилпиразол-5-карбоксамид.
Раствор 198 мг (1,0 ммоля) 4-нитро-3-н-пропилпиразол-5-карбоксамида в 5 мл метанола по каплям добавили в смесь 113 мг (2,97 ммоля) боргидрида натрия с 5 мг 10%-ного палладия на угле и 3 мл воды. Эту смесь перемешивали при комнатной температуре в течение 3 ч профильтровали и выпариванием в вакууме удалили из нее растворитель. В результате кристаллизации остатка из смеси этилацетата с метанолом в виде не совсем белого твердого вещества получили 61 мг (36% -ный выход) соединения, указанного в заголовке, с температурой плавления 196-201оС. Rf 0,4 (двуокись кремния; дихлорметан, метанол, гидрат окиси аммония в соотношении 90:10:1).
Найдено, C 48,96; H 6,98, N 32,08.
C7H12N4O.
Вычислено, C 49,98; H 7,19; N 33,31.
П р и м е р 38. 4-(2-Этоксибензамидо)-3-н-пропилпиразол-5-карбоксамид.
Амид, указанный в заголовке, получили из 4-амино-3-н-пропилпиразол-5-карбоксамида в соответствии с процедурой примера 6 в виде белого твердого вещества (64%-ный выход) с температурой плавления 209-211оС.
Найдено, C 60,73; H 6,41; N 17,80.
C16H20N4O3.
Вычислено, C 60,74; H 6,37; N 17,71.
П р и м е р 39. 5-(2-Этоксифенил)-3-н-пропил-1,6-дигидро-7Н-пиразоло(4,3-d) пиримидин-7-он.
Указанное в заголовке соединение получили из 4-(2-этоксибензамино)-3-н-пропилпиразол-5-карбоксамида в соответствии с процедурой примера 30 в виде белого твердого вещества (16%-ный выход) с температурой плавления 199-201оС.
Найдено, C 64,44; H 6,19; N 18,44.
C16H18N4O2.
Вычислено, C 64,41; H 6,08; N 18,78.
П р и м е р 40. 5-(5-Хлорсульфонил-2-этоксифенил)-3-н-пропил-1,6-дигидро-7Н- пиразоло(4,3-d)пиримидин-7-он.
Указанный в заголовке сульфонилхлорид получили из 5-(2-этоксифенил)-3-н-пропил-1,6-дигидро-7Н-пиразоло(4,3-d) пиримидин-7-она в соответствии с процедурой примера 8 в виде белого твердого вещества (78%-ный выход). Rf 0,25 (двуокись кремния диэтиловый эфир).
Его использовали без предварительной очистки.
П р и м е р 41. 5-[2-Этокси-5-(4-метилпиперазинил)-сульфонилфенил]-3-н-пропил-1, 6- дигидро-7Н-пиразоло(4,3-d)пиримидин-7-он.
Указанный в заголовке сульфонамид получили из 5-(5-хлорсульфонил-2-этоксифенил)-3-н-пропил-1,6-дигидро-7Н- пиразоло(4,3-d)пиримидин-7-она в соответствии с процедурой примера 9 в виде белого твердого вещества (70%-ный выход) с температурой плавления 236-239оС.
Найдено, C 54,84; H 6,27; N 18,10.
C21H28N6O4S.
Вычислено, C 54,76; H 6,13; N 18,25.
П р и м е р 42. 3-Бромметил-5-хлор-1-метил-4-нитропиразол.
10,7 г (60,0 ммоля) N-бромсукцинимида добавили в раствор 8,78 г (50,0 ммоля) 5-хлор-1,3-диметил-4-нитропиразола в 100 мл четыреххлористого углерода и раствор прокипятили с обратным холодильником с одновременным облучением видимым светом (вольфрамовая лампа на 150 Вт) в течение 3 дней. Через определенные интервалы по ходу всей реакции добавили 6 порциями по 50 мл перекиси бензоила. Затем выпариванием в вакууме удалили растворитель и остаток подвергли хроматографической обработке на силикагеле, элюируя смесью в соотношении 1:1 дихлорметана с гексаном, в результате его в виде не совсем белого твердого вещества получили 8,0 г (63%-ный выход) бромида с температурой плавления 80-82оС.
Найдено, C 23,95; H 2,05; N 16,31.
C5H5BrClN3O2.
Вычислено, C 23,60; H 1,98; N 16,51.
П р и м е р 43. 5-Хлор-3-метоксиметил-1-метил-4-нитропиразол.
5,0 г (19,6 моля) раствора 3-бромметил-5-хлор-1-метил-4-нитропиразола в 50 мл метанола обработали 5,75 г (33,8 ммоля) нитрата серебра и смесь прокипятили с обратным холодильником в течение 2 ч. Охлажденную реакционную смесь профильтровали и фильтрат выпарили в вакууме. Остаток разделили между 100 мл этилацетата и 50 мл воды, подвергнув затем водную фазу экстрактной обработке дополнительными 50 мл этилацетата. Органические экстракты объединили, высушили над сульфатом магния и выпарили в вакууме. В результате хроматографической обработки на силикагеле, элюируя смесью дихлорметана с метанолом в соотношении 97: 3, получили в виде твердого белого вещества 1,6 г (40% -ный выход) пиразола, указанного в заголовке, с температурой плавления 59-63оС.
Найдено, C 34,65; H 3,83; N 20,05.
C6H8ClN3O3.
Вычислено, C 35,05; H 3,92; N 20,44.
П р и м е р 44. 5-Циано-3-метоксиметил-1-метил-4-нитропиразол.
Раствор 205 мг (1,0 ммоля) 5-хлор-3-метоксиметил-1-метил-4-нитропиразола, 130 мг (2,0 ммоля) цианистого калия и 10 мл 18-крон 6 в 2 мл ацетона прокипятили с обратным холодильником в течение ночи. Растворитель выпарили в вакууме и остаток разделили между 20 мл этилацетата с 20 мл воды. Органическую фазу отделили, высушили над сульфатом магния и выпарили в вакууме, после чего остаток подвергали хроматографической обработке на силикагеле, элюируя смесью в соотношении 1:1 этилацетата с пентаном. В результате растирания сырого продукта в диэтиловом эфире получили 38 мг (19%-ный выход) желтого твердого вещества с температурой плавления 48-50оС.
Найдено, C 42,89; H 4,15; N 28,78.
C7H8N4O3.
Вычислено, C 42,86; H 4,11; N 28,56.
П р и м е р 45. 4-Амино-5-циано-3-метоксиметил-1-метилпиразол.
Указанное в заголовке соединение получили из 5-циано-3-метоксиметил-1-метил-4-нитропиразола в соответствии с процедурой примера 5 в виде не совсем белого твердого продукта (68%-ный выход) с температурой плавления 82-84оС.
Найдено, C 50,81; H 6,13; N 38,94.
C7H10N4.
Вычислено, C 50,59; H 6,07; N 33,72.
П р и м е р 46. 5-Циано-4-(2-этоксибензамидо)-3-метоксиметил-1-метилпиразол.
Указанное в заголовке соединение получили из 4-амино-5-циано-3-метоксиметил-1-метилпиразола в соответствии с процедурой примера 6 в виде не совсем белого твердого вещества (61%-ный выход) с температурой плавления 103-105оС.
Найдено, C 61,21; H 5,98; N 17,80.
C16H18N4O3.
Вычислено, C 61,13; H 5,77; N 17,83.
П р и м е р 47. 5-(2-Этоксифенил)-3-метоксиметил-1-метил-1,6-дигидро-7Н-пира- золо (4,3-d)пиримидин-7-он.
Указанное в заголовке соединение получили из 5-циано-4-(2-этоксибензамидо)-3-метоксиметил-1-метилпиразола в соответствии с процедурой примера 7 посредством получения по месту использования 5-первичного амидного производного в виде белого твердого вещества (38%-ный выход) с температурой плавления 160-161оС.
Найдено, C 61,35; H 5,75; N 17,98.
C16H18N4O3.
Вычислено, C 61,13; H 5,77; N 17,83.
П р и м е р 48. 3-Метоксиметил-1-метил-5-[5-(4-метилпиперазинилсульфонил)-2- этоксифенил]-1,6-дигидро-7Н-пиразоло(4,3-d) пиримидин-7-он.
470 мл (1,50 ммоля) 5-(2-этосифенил)-3-метоксиметил-1-метил-1,6-дигидро-7Н-пи- разоло (4,3-d)пиримидин-7-она растворили при температуре 0оС в хлорсульфоновой кислоте. Этот раствор перемешивали при комнатной темпертуре в течение 2 ч, а затем осторожно добавили в 50 мл смеси воды со льдом. Образовавшийся раствор нейтрализовали насыщенным раствором карбоната натрия, затем подвергли экстракционной обработке смесью в соотношении 20:1 дихлорметана с метанолом в виде 2 порций по 50 мл. Объединенные органические экстракты выпарили в вакууме и остаток растворили в 5 мл этанола, после чего раствор обработали 450 мг (4,5 ммоля) N-метилпиперазина. По истечении 1 ч выдержки при комнатной температуре растворитель выпарили в вакууме и остаток подвергли хроматографической обработке на силикагеле, элюируя смесью дихлорметана с метанолом и водным раствором гидрата окиси аммония в объемном соотношении 90:10:1. В результате растирания в этилацетате сырого продукта в виде белого твердого вещества получили 49 мг (7%-ный выход) соединения, указанного в заголовке примера, с температурой плавления 198-199оС.
Найдено, C 52,94; H 6,04; N 17,67.
C21H28N6O5S.
Вычислено, C 52,93; H 5,92; N 17,64.
Из смеси этилацетата с метанолом в соответствии с процедурой хроматографии и кристаллизации в виде белого твердого вещества выделили 5 мг (7%-ный выход) 3-оксиметил-1-метил-5-[5-(4-метилпиперази- нилсульфонил)-2- этоксифенил] -1,6-дигидро-7Н-пиразоло(4,3-d)пиримидин-7-она с температурой плавления 209-210оС.
Найдено, C 51,94; H 5,77; N 18,05.
C20H26N6O5S.
Вычислено, C 51,94; H 5,67; N 18,17.
П р и м е р 49. 1-Этил-3-н-пропилпиразол-5-карбоновая кислота, этиловый эфир.
Этот пиразол получили из этилового эфира 3-н-пропилпиразол-5-карбоновой кислоты и диэтилсульфата в соответствии с процедурой, изложенной в примере 1, в виде бесцветного маслоподобного продукта (72%-ный выход).
Rf 0,5 (двуокись кремния; этилацетат, гексан в соотношении 1:1).
П р и м е р 50. 1-Этил-3-н-пропилпиразол-5-карбоновая кислота.
Эту карбоновую кислоту получили из этилового эфира 1-этил-3-н-пропилпиразол-5-карбоновой кислоты в соответствии с процедурой, которая описана в примере 2, в виде бледно-коричневого твердого вещества (89%-ный выход) с температурой плавления 73-77оС.
Найдено, C 58,62; H 7,69; N 15,23.
C9H14N2O2.
Вычислено, C 59,32; H 7,74; N 15,37.
П р и м е р 51. 1-Этил-4-нитро-3-н-пропилпиразол-5-карбоновая кислота.
Указанное в заголовке соединение получили из 1-этил-3-н-пропилпиразол-5-карбоновой кислоты в соответствии с процедурой, которая описана в примере 3, в виде бесцветного твердого вещества (96%-ный выход) с температурой плавления 120-123оС.
Найдено, C 47,61; H 5,81; N 18,54.
C9H13N3O4.
Вычислено, C 47,57; H 5,77; N 18,49.
П р и м е р 52. 1-Этил-4-нитро-3-н-пропилпиразол-5-карбоксамид.
Укаанный в заголовке амид получили из 1-этил-3-н-пропилпиразол-5-карбоновой кислоты в соответствии с процедурой, которая описана в примере 4, в виде не совсем белого твердого вещества (86%-ный выход) с температурой плавления 119-120оС.
Найдено, C 47,38; H 6,18; N 24,34.
C9H14N4O3.
Вычислено, C 47,78; H 6,24; N 24,77.
П р и м е р 53. 4-Амино-1-этил-3-н-пропилпиразол-5-карбоксамид.
Указанное в заголовке соединение получили из 1-этил-4-нитро-3-н-пропилпиразол-5-карбоксамида в соответствии с процедурой, которая описана в примере 5, в виде не совсем белого твердого вещества (100%-ный выход) с температурой плавления 93-97оС.
Найдено, C 55,17; H 8,34; N 28,93.
C9H16N4O.
Вычислено, C 55,08; H 8,22; N 28,55.
П р и м е р 54. 4-(2-Этоксибензамидо)-1-этил-3-н-пропилпиразол-5-карбоксамид.
Указанный в заголовке амид получили из 4-амино-1-этил-3-н-пропилпиразол-5-карбоксамида и 2-этоксибензоилхлорида в соответствии с процедурой, которая описана в примере 6, в виде бесцветного твердого вещества (73%-ный выход) с температурой плавления 139-141оС.
Найдено, C 63,03; H 7,15; N 16,50.
C18H24N4O3.
Вычислено, C 62,77; H 7,02; N 16,27.
П р и м е р 55. 5-(2-Этоксифенил)-1-этил-3-н-пропил-1,6-дигидро-7Н-пиразоло (4,3-d)пиримидин-7-он.
Указанное в заголовке соединение получили из 4-(2-этоксибензамидо)-1-этил-3-н-пропилпиразоло-5-карбоксамида в соответствии с процедурой примера 7, в виде бесцветного твердого вещества (46%-ный выход) с температурой плавления 112-114оС.
Найдено, C 66,59; H 6,85; N 17,26.
C18H22N4O2.
Вычислено, C 66,23; H 6,79; N 17,17.
П р и м е р 56. 5-(5-Хлорсульфонил-2-этоксифенил)-1-этил-3-н-пропил-1,6-дигид- ро- 7Н-пиразоло(4,3-d)пиримидин-7-он.
Указанное в заголовке примера соединение получили из 5-(2-этоксифенил)-1-этил-3-н-пропил-1,6-дигидро-7Н-пиразоло(4,3-d) пиримидин-7-она в соответствии с процедурой примера 8 в виде метиленхлоридного сольвата (86% -ный выход) с температурой плавления 170-172оС.
Найдено, C 49,82; H 4,84; N 12,77.
C18H21ClN4O4S.
Вычислено, C 49,70; H 4,90; N 12,77.
П р и м е р 57. 5-[2-Этокси-5-(4-метилпиперазинилсульфонил)-фенил]-1-этил-3-н- пропил- 1,6-дигидро-7Н-пиразоло(4,3-d)пиримидин-7-он.
Указанный в заголовке сульфонамид получили из 5-(5-хлорсульфонил)-2-этоксифенил)-1-этил-3-н-пропил-1,6-дигидро-7Н- пиразоло(4,3-d)пиримидин-7-она и N-метилпиперазина в соответствии с процедурой примера 9 в виде бесцветного твердого вещества (43%-ный выход) с температурой плавления 160-162оС.
Найдено, C 57,24; H 6,17; N 16,83.
C23H32N6O4S.
Вычислено, C 56,54; H 6,60; N 17,20.
Rf 0,35 (двуокись кремния; дихлорметан, метан в соотношении 9:1).
П р и м е р 58. 5-{2-Этокси-5-[4-(2-оксиэтил)-пиперазинилсульфонил]-фенил}-1-этил- 3-н-пропил-1,6-дигидро-7Н-пиразоло(4,3- d)пиримидин-7-он.
Указанный в заголовке сульфонамид получили из 5-(5-хлорсульфонил-2-этоксифенил)-1-этил-3-н-пропил-1,6-дигидро-7Н- пиразоло(4,3-d)пиримидин-7-она и N-(2-оксиэтил)-пиперазина в соответствии с процедурой примера 9 в виде бесцветного твердого вещества (88% -ный выход) с температурой плавления 191-193оС.
Найдено, C 55,74; H 6,55; N 15,78.
C24H34N6O5S.
Вычислено, C 55,58; H 6,61; N 16,20.
Использование: в фармакологии, в частности в способе получения замещенных пиразолопиримидиновых соединений. Сущность изобретения: продукт соединения ф-лы I, где R1 водород, метил или этил; R2 - метил; CH2OH, CH2-OCH3 или н-пропил; R3 этил, пропил, аллил; R4 совместно с азотом, с которым он связан, образует 4-N-(R6) пиперазинильную группу; R5 водород и др. R6 - водород и др. или их фармацевтически приемлемых солей. Реагент I: соединения ф-лы II, где Y хлор, бром и др. Реагент 2: соединения ф-лы III. Условия реакции: после взаимодействия реагентов I и II целевой продукт выделяют или переводят в формацевтически приемлемую соль, или О-алкилируют фенольную группу, или если R2 гидроксигруппа, то защищают ацетильной или бензоильной группой с последующим ее удалением с помощью гидролиза. 2 табл. Структура соединений ф-лы I, II и III см.ниже. 2 з. п. ф-лы.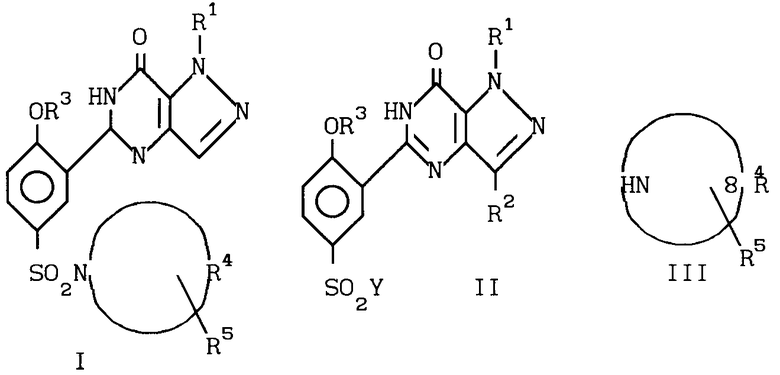
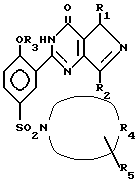
где R1- водород, метил или этил,
R2- метил, CH2OH, CH2OCH3 или н-пропил;
R3-этил, н-пропил или аллил;
R4 вместе с атомом азота с которым от связан, образует пиперидино или 4-N-(R6)-пиперазинильную группу;
R5- водород, N((СН3)2 или CONH2;
R6- водород, метил, 2-пропил, 2-гидроксиэтил, CSNH2 или C(NH)NHCH3,
или их фармацевтически приемлемых солей, отличающийся тем, что соединение общей формулы II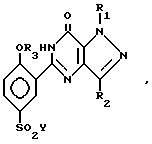
где R1, R2 и R3 имеют указанные значения или R3 - водород, R2 CH2OCOCH3, CH2OCOPh;
Y- хлор, бром, фтор,
подвергают взаимодействию с соединением общей формулы III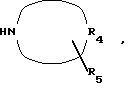
где R4 и R5 имеют указанные значения,
полученный продукт выделяют и при необходимости переводят в его фармацевтически приемлемую соль, или, если R3- водород, осуществляют последующее О-алкилирование фенольной группы и необязательное превращение полученного продукта в его фармацевтически приемлемую соль, или, если R2 гидроксигруппа, защищенная ацетильной или бензольной группой, указанную защитную группу с помощью основного гидролиза удаляют и затем при необходимости переводят в его фармацевтически приемлемую ось.
| Устройство для замыкания, по выбору, на расстоянии электрических цепей | 1930 |
|
SU20188A1 |
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
