Изобретение относится к ряду пиразоло (4.3-d) пиримидин-7-онов, которые представляют собой сильнодействующие и селективные ингибиторы циклической гуанозин-3'-5'-монофосфатфосфодиэстеразы (цГМФ ФДЭ), которая полезна в самых различных областях терапии, включая лечение разных сердечно-сосудистых заболеваний, в частности ангины, гипертонии, сердечной недостаточности, атеросклероза.
Соединения настоящего изобретения проявляют селективность в отношении ингибирования скорее цГМФ ФДЭ, как следствие этого селективного ингибирования ФДЭ повышается содержание цГМФ, что, в свою очередь, вызывает улучшение антиагрегационных условий для кровяных пластинок, проявляются антисосудоспазматическое и сосудорасширяющее действия, а также потенциация эффектов релаксационного фактора, вызванного эндотелием (РФВЭ), и нитрососудорасширительных средств. Таким образом, такие соединения могут быть использованы при лечении ряда заболеваний, включая сюда стабильную, нестабильную и вариантную (Prinzmetal) ангину, гипертонию, гиперемическую сердечную недостаточность, атеросклероз, пониженную пропускную способность кровеносных сосудов, например постподкожную транслуминальную коронарную ангиопластику (пост-ПТКА), заболевания периферийных сосудов, удар, бронхиты, хроническую астму, аллергическую астму, аллергические риниты, глаукому и те заболевания, которые характеризуются расстройствами подвижности кишечника, например синдром кишечного раздражения (СКР).
В описании к заявке на европейский патент EP-A-0201188 предлагаются некоторые пиразоло(4,3-d)пиримидин-7-оны в качестве аденозиновых рецепторных антагонистов и ингибиторов ФДЭ, которые могут быть использованы для лечения сердечно-сосудистых заболеваний, в частности сердечной недостаточности или кардиальной недостаточности. Тем не менее, такие соединения не являются ни особенно сильнодействующими ингибиторами ФДЭ, ни соединениями, о которых сказано, что они селективно ингибируют цГМФ ФДЭ.
Соединения настоящего изобретения отвечают формуле (I):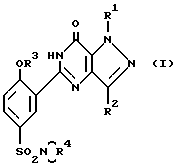
где
R1 - водородный атом, метил или этил; R2 - метил, CH2 OH, CH2OCH3 или н-пропил; R3 - этил, н-пропил или аллил; R4, взятый совместно с азотным атомом, с которым он связан, образует 4-N(R5)-пиперидино или 4-N-(R6)-пиперазинильную группу; R5 - водородный атом, N(CH3)2 или CONH2; R6 - водородный атом, метил, 2-пропил, 2-гидроксиэтил, CSNH2, C(NH)NHCH3 или C(NH)SCH3, предлагаются также их фармацевтически приемлемые соли.
Соединения формулы (I) могут содержать по одному или более центру ассиметрии, благодаря чему они могут существовать в форме энантиомеров или диастереоизомеров. Рамками настоящего изобретения охватываются как смеси таких изомеров, так и индивидуальные отдельные изомеры.
Соединения формулы (I) могут также существовать в таутометрических формах, причем рамками настоящего изобретения охватываются как смеси, так и отдельные индивидуальные таутомеры.
Кроме того, рамками настоящего изобретения охватываются радиоактивные меченые производные соединений формулы (I), которые приемлемы для использования в ходе биологических исследований.
Фармацевтически приемлемые соли соединений формулы (I), которые содержат основные центры, представляют собой кислые аддукты, полученные с использованием фармацевтически приемлемых кислот. Примеры таких аддуктов охватывают гидрохлоридные, гидробромидные, сульфатные или бисульфатные, фосфатные или кислые фосфатные, ацетатные, цитратные, фумаратные, глюконатные, лактатные, малеитные, сукцинатные и тартратные соли. Соединения формулы (I) способны с основаниями также образовывать фармацевтически приемлемые соли металлов. К их примерам относятся натриевые и калиевые соли.
Соединения предпочтительного ряда отвечают формуле (I), где R1 - метил; R2- н-пропил; R3 - этил, н-пропил или аллил; R4, взятый вместе с азотным атомом, с которым он связан, образует 4-N-(R6)-пиперазиниловую группу; R5 - водородный атом; а R6 - водородный атом, алкил C1-C3- или 2-оксиэтил.
Класс особенно предпочтительных индивидуальных соединений настоящего изобретения охватывает 5-[2-аллилокси-5-/4-метил- пиперазинилсульфонил)-фенил] -1-метил-3-н-пропил-1,6-дигидро-7Н-пиразоло(4,3-d)пиримидин-6-он; 5-[2-этокси-5-/пиперазинилсульфонил)-фенил]-1-метил-3-н-пропил-1,6- дигидро-7Н-пиразоло(4,3-d)пиримидин-7-он; 5-[2-этокси-5-/4-метилпиперазинилсульфонил)-фенил] -1-метил-3-н-пропил-1,6 дигидро-7Н- пиразоло(4,3-d)пиримидин-7-он; 5-{ 2-этокси-5-[4-/2-пропил/-пиперазинил-сульфонил] -фенил]-1-метил-3-н-пропил-1,6-дигидро-7Н- пиразоло(4,3-d)пиримидин-7-он; 5-{2-этокси-5-[4-/2-оксиэтил/-пиперазинилсульфони] -фенил} -1-метил-3-н- пропил-1,6-дигидро-7Н-пиразоло/4,3-d/пиримидин-7-он; 1-метил-5-[5-пиперазинилсульфонил/-2-н-пропоксифенил] -3-н-пропил-1,6-дигидро-7Н -пиразоло(4,3-d)пиримидин-7-он и 5 {5-[4-(2-оксиэтил)-пиперазинилсульфонилсульфонил] -2-н-пропоксифенил} -1-метил-3-н-пропил-1,6-дигидро-7Н- пиразоло/4,3-d/пиримидин-7-он.
Соединения общей формулы (I) могут быть получены реакцией соединения общей формулы (II):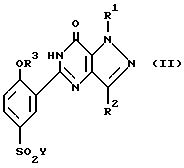
где значения символов R1 - R3 определены выше, а Y обозначает атом галогена, предпочтительнее атом хлора, с соединением общей формулы (III).
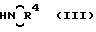
где значения символа R4 определены выше. Эту реакцию обычно проводят при комнатной температуре, предпочтительнее в присутствии растворителя, например алканола, содержащего от одного до трех углеродных атомов, с использованием избытка соединения (III) с целью удалить кислый побочный продукт (НУ).
Соединения общей формулы (II) могут быть получены из соединений общей формулы (IV).

(где значения символов R1-R3 определены выше) в соответствии с известными методами введения группы SO2Y (где значения символа Y определены выше) в ароматическое кольцо, например, когда символом Y обозначен хлорный атом, путем воздействия хлорсульфокислотой при температуре 0oC или близкой к ней.
В том случае, когда R3 - группа, чувствительная к удалению в условиях хлорсульфонилирования, например аллил, указанную группу можно вводить на заключительной стадии синтеза. Таким образом, фенол общей формулы (IV), где R3 - водородный атом, а значения символов R1 и R2 определены выше, который можно получить удалением защитной группы О-аллилового аналога через посредство палладия, как это проиллюстрировано в примере 25, хлор сульфируют с получением соединения общей формулы (II), где Y - атом хлора, R3 - водородный атом, а значения символов R1 и R3 определены выше. Затем проводят реакцию этого последнего с соответствующим амином (III), в результате чего образуется соединение общей формулы (I), где R3 - водородный атом, а значения символов R1, R2, R4 и R5 определены выше, которое наконец О-алкилируют с получением соединения общей формулы (I), где значения символов R1 - R5 определены выше для формулы (I). Эту реакцию алкилирования можно проводить в стандартных условиях с использованием соответствующего алкилгалогенида, например аллилбромида, в присутствии основания, в частности карбоната калия, в среде приемлемого растворителя, например 2-бутанона, при температуре кипения реакционной смеси с обратным холодильником. По другому варианту реакцию алкилирования можно проводить в реакционных условиях, предложенных Мицунобу.
В случае других соединений формулы (IV), которые могут оказаться не совместимыми с условиями реакции хлорсульфонирования, например, в таких, у которых R2-оксиалкил C1-C6, гидроксильная группа может быть защищена ацильной группой, в частности ацетильной или бензоильной группой. В дальнейшем упомянутую защитную группу удаляют на заключительной стадии синтеза в стандартных условиях основного гидролиза, в результате чего образуются соединения общей формулы (I), где R2 - оксиалкил C1-C6, а значения символов R1, R3-R5 определены выше для формулы (I). Эти последние соединения могут быть также получены в данном случае в качестве побочных продуктов хлорсульфонирования соответствующих алкоксианалогов, то есть соединений общей формулы (IV), где R2 - алкокси (C1-C3) алкил (C1-C6), с последующей реакцией сырого продукта с требуемым амином (III), как это проиллюстрировано в примере 48.
Соединения общей формулы (IV) могут быть получены из соединений общей формулы (V):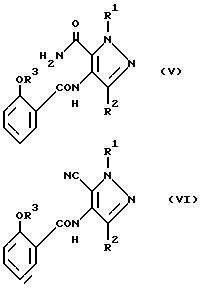
(где значения символов R1-R3определены выше) путем осуществления известных методов циклизации с получением пимидинового кольца. Так, например, циклизацию можно проводить путем обработки соединения (V) основанием, в частности гидратом окиси натрия (или карбонатом калия), возможно в присутствии перекиси водорода, в водно-этанольной среде при температуре кипения с обратным холодильником в течение 2 - 40 ч. В этих условиях в качестве предшественника соединения (IV) можно также использовать связанный нитрил общей формулы (VI), где значения символов R1-R3 определены выше.
В соответствии с другой процедурой циклизации соединения общей формулы (IV) могут быть получены путем обработки соединения (V) фосфорной кислотой при температуре 140oC или близкой к ней в течение 6 - 18 ч.
Соединения общих формул (V) и (VI) могут быть получены из соединений общих формул соответственно (VII) и (VIII).
(где значения символов R1 и R2 определены выше) реакцией с соединением общей формулы (IX) (где значения символов R3и Y определены выше).
Эту реакцию обычно проводят с использованием избытка (IX) в присутствии избытка алифатического третичного амина, в частности триэтиламина, который выполняет функцию поглотителя кислого побочного продукта (НУ), возможно в присутствии катализатора, в частности 4-диметиламинопиридина, в среде инертного растворителя, в частности дихлорметана, при температуре от 0 до 25oC в течение 2 - 6 ч.
В том случае, когда амины формулы (III), аминопиразолы формул (VII) и (VIII) и ацилгалогениды формулы (IX) не являются технически доступными, их можно получать согласно обычным процедурам синтеза, которые описаны в известной литературе, из легкодоступных исходных материалов с использованием стандартных реагентов и в стандартных реакционных условиях.
Некоторые соединения общей формулы (I), где R4 совместно с азотным атомом, с которым он связан, образует 4-N-(R6)-пиперазиниловую группу, а значения символа R6 определены выше, но он не обозначает водородного атома, могут быть получены непосредственно из соответствующего 4-N-незамещенного пиперазинового аналога, то есть из соединения общей формулы (I), где R6 - водородный атом, с использованием соответствующих стандартных процедур синтеза.
Все вышеуказанные реакции совершенно обычны, а соответствующие реагенты и условия их проведения могут быть легко определены, если воспользоваться стандартными учебниками и примерами, которые приведены ниже. Их альтернативы и варианты совершенно очевидны любому специалисту в данной области в отношении возможности получения всех соединений, отвечающих формуле (I).
Биологическую активность соединений настоящего изобретения определяют согласно нижеследующим методам испытаний.
Фосфодиэстеразная активность
Сродство соединения с цГМФ и цАМФ ФДЭ оценивают путем определения его величины IC50 (концентрация ингибиторов, которая требуется для 50%-го ингибирования энзимного действия), ФДЭ-энзимы выделяют из кровяных пластин кролика и почки крысы, по существу, по методу У.Дж.Томпсона и др.(Biochem, 1971, 10, 311). Независимый от кальция/кольмодулина [Ca/CAM] цГМФ ФДЭ-энзим с GMP PDE и цГМФ-ингибированный цАМФ ФДЭ-энзим получают из кровяных пластинок кролика, тогда как из четырех основных ФДЭ-энзимов крысиной почки выделяют Ca/CAM-зависимую цГМФ ФДЭ (фракция I). Испытания проводят в соответствии с модифицированным "периодическим" методом У. Дж. Томпсона и М.М.Эпплмана (Biochem, 1979, 18, 5228). Результаты таких испытаний показывают, что соединения настоящего изобретения являются сильнодействующими и селективными ингибиторами обеих цГМФ ФДЭ.
Действие, препятствующее агрегированию кровяных пластинок
Это действие оценивают путем определения способности соединения ингибировать агрегирование кровяных пластинок в лабораторных условиях, которое инициировано активирующим кровяные пластинки фактором (АПФ), и потенциировать антиагрегативное действие на кровяные тельца в лабораторных условиях активаторов гуанилатциклазы, в частности нитропруссида и РФВЭ. Промытые кровяные пластины готовят по существу по методу Дж.Ф.Мустарда и др. (Methoas in Enzymol., 1989, 169, 3), а агрегирование определяют в соответствии со стандартной турбидиметрической технологией, которая описана Г.В.Р.Борном в J. Physiol (Лондон), 1962, 162, 67P.
Антигипертоническое действие.
Это действие оценивают после внутривенного или перорального введения соединения в организм крыс, страдающих спонтанной гипертонией. С помощью канюли, имплантированной в сонную артерию находящихся в сознании либо анастезированных животных.
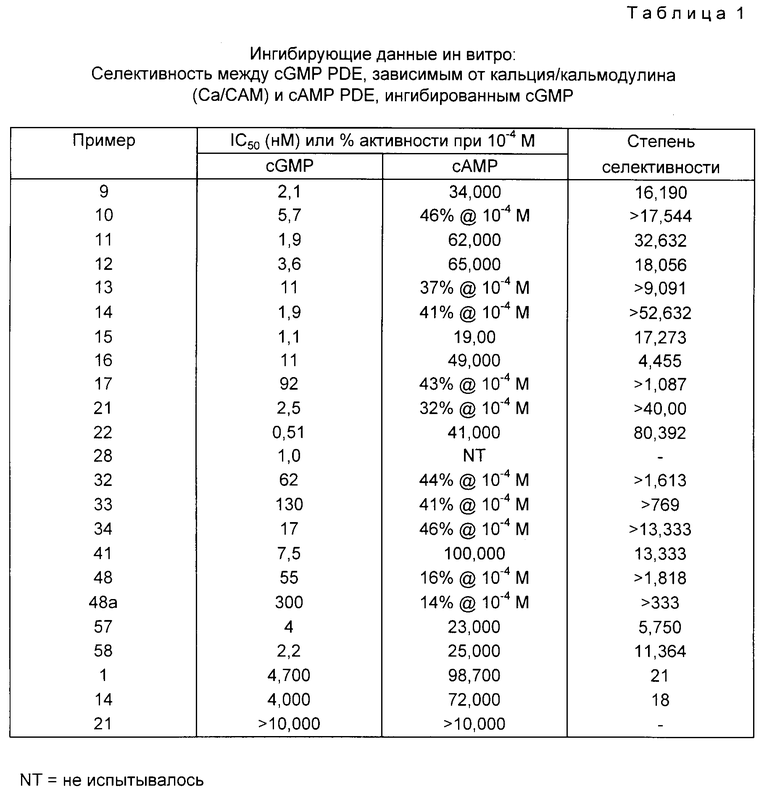
Ингибирующая активность соединений настоящего изобретения в отношении cGMP PDE и cAMP PDE ин витро определялась как описано выше и сравнивалась с показателями активности некоторых соединений, описанных в EP-A-0201188. Эти данные приведены в прилагаемой таблице, в которой ингибирование зависимого от кальция/кальмодулина (Ca/CAM) cGMP PDE сравнивается с ингибированием CAMP PDE, ингибированным cGMP, при этом показатель селективности представляет отношение последних величин к первым величинам. Фермент-ингибирующие активности выражаются или в виде величин IC50 (ингибирующая концентрация) в наномолях, или в виде активностей при 10-4 молярной конц. Следует отметить, что, чем ниже числовая величина, тем более сильной является ингибирующая активность соединения. В противоположность этому, в колонке степень селективности, чем выше числовая величина, тем более селективным является соединение по его PDE ингибирующему профилю.
Двадцать примеров в верхней части табл. 1 (примеры 9 - 58) представляют соединения настоящего изобретения, причем пример 48a относится к побочному продукту, полученному в примере 48. Три примера в нижней части табл. 1 (примеры 1, 14 и 21) взяты из EP-A-0201188, в котором они имеют те же самые номера. Результаты демонстрируют, что все двадцать соединений настоящей заявки являются значительно более сильными и селективными, чем три соединения, выбранные из EP-A-0201188.
Соединения изобретения испытывались при терапевтических дозах до 3 мг/кг внутривенно и орально на крысах и собаках, при этом не обнаружено никаких чрезмерных признаков токсичности. Прежде, чем будет установлена безопасность соединений для людей, потребуется провести обширные испытания, однако по имеющимся сведениям можно сказать, что соединения изобретения могут быть отнесены к соединениям с низкой токсичностью.
Для введения в организмы людей с целью лечения или профилактики ангины, гипертонии или гипертонической сердечной недостаточности пероральная доза соединений обычно находится в интервале 4-800 мг ежедневно для взрослого пациента среднего веса (70 кг). Так, например, для типичного взрослого пациента индивидуальные таблетки или капсулы содержат по 2-400 мг активнодействующего вещества в среде подходящей и фармацевтически приемлемой основы для приготовления лекарств или носителя для введения в организм в виде одинарных или многократных доз по одному или несколько раз в день. Дозы для внутривенного, буккального или подъязычного введения в организм обычно находятся в интервале 1-400 мг/одинарную дозу в зависимости от потребности. На практике фактический режим дозирования подбирается врачом таким образом, чтобы он оказался наиболее приемлемым для данного индивидуального пациента, поскольку такой режим изменяется в зависимости от возраста, веса и реакции конкретного пациента. Вышеприведенные дозы являются примерами для среднего случая, однако в отдельных случаях конкретные дозы могут превосходить максимальные или быть ниже минимальных пределов диапазона дозировок, причем все возможные дозировки охватываются рамками настоящего изобретения.
Для использования людьми соединения формулы (I) могут быть введены в организм индивидуально, но обычно их вводят в смеси с фармацевтическим носителем, выбираемым в зависимости от намечаемого пути введения в организм и стандартной фармацевтической практики. Так, например, их можно вводить перорально, буквально или подъязычно в форме таблеток, содержащих основу для приготовления лекарств, в частности крахмал или лактозу, или капсул, или шариков как индивидуально, так и в смеси с основой для приготовления, или же в форме эликсиров или суспензий, содержащих вкусовые, ароматизирующие или окрашивающие добавки. Такие соединения можно также вводить в организм парентерально, например внутривенно, внутримышечно, подкожно или интракоронарно. Для парантерального введения в организм их лучше всего использовать в форме стерильного водного раствора, который может содержать другие вещества, например соли и глюкозу, в количестве, достаточном для того, чтобы сделать раствор изотоническим относительно крови.
Таким образом, в соответствии с еще одним аспектом настоящего изобретения предлагается фармацевтическая композиция, которая включает в себя соединение формулы (I) или его соль, фармацевтически приемлемую для использования в медицине, в частности для лечения ангины, гипертонии или гиперемической сердечной недостаточности человека.
Рамками настоящего изобретения охватывается также использование соединений формулы (I) или их фармацевтически приемлемых солей для приготовления медикаментов, применяемых при лечении стабильной, нестабильной и вариантной (Prinzmetal) ангины, гипертонии, гипертонической сердечной недостаточности, атеросклероза, удара, заболевания периферийных сосудов, пониженной пропускной способности, например пост-ПТКА, хронической астмы, бронхитов, аллергической астмы, аллергического ринита, глаукомы или тех заболеваний, которые характеризуются расстройствами подвижности кишечника, в частности СКР.
В дальнейшем процессы получения соединений настоящего изобретения более конкретно проиллюстрированы со ссылками на нижеследующие примеры экспериментов. Степень чистоты соединений при этом проверяли обычным путем, тонкослойным хроматографическим анализом (ТСХ) с использованием пластин Nerck Kieselgel 60 F254. IH-ядерномагнитную резонансную спектрограмму записывали с помощью спектрометра Nicolet GE-300, причем во всех случаях она соответствовала предложенному строению.
Пример 1. Этиловый эфир 1-метил-3-н-пропилпиразол-5-карбоновой кислоты.
24,1 г (0,132 моль) этилового эфира 3-н-пропилпиразол-5-карбоновой кислоты (полученной по методу, описанному в Chem Pharm. Bull., 1984, 32, 1568) в смеси с 16,8 г (0,133 моль) диметилсульфата выдерживали при температуре 90oC в течение 2,5 ч. Эту смесь растворили в дихлорметане и раствор промыли раствором карбоната натрия. Органическую фазу отделили, высушили над сульфатом магния и выпарили в вакууме, получив твердый продукт. В результате хроматографической обработки на 300 г силикагеля, элюируя дихлорметаном, продукт получили в виде 20,4 г (79%-ный выход) бесцветного масла. Rf - 0,8 (двуокись кремния, дихлорметан, метанол, уксусная кислота в соотношении 80:20:1).
Пример 2. 1-метил-3-н-пропилпиразол-5-карбоновая кислота. 20,2 г (0,10 моль) этилового эфира 1-метил-3-пропилпиразол-5-карбоновой кислоты суспендировали в 50 мл (0,30 моль) 6н. водного раствора гидрата окиси натрия. Эту смесь выдерживали при температуре 80oC в течение 2 ч, а затем разбавили 50 мл воды и подкислили добавлением 25 мл концентрированной соляной кислоты. В результате фильтрования в виде бледно-коричневых кристаллов получили 12,3 г (71%-ный выход) карбоновой кислоты с температурой плавления 150-154oC. Для C8H12N2O2
Найдено %: C 56,99: H 7,25: N 16,90
Вычислено %: C 57,13: H 7,19: N 16,66.
Пример 3. 1-метил-4-нитро-3-н-пропилпиразол-5-карбоновая кислота.
12,1 г (0,072 моль) 1-метил-3-н-пропилпиразол-5-карбоновой кислоты порциями добавили в смесь 13 мл олеума и 11 мл дымящей азотной кислоты, поддерживая температуру на уровне ниже 60oC. После завершения операции добавления смесь выдерживали в течение ночи при температуре 60oC, а затем охладили до комнатной температуры с последующим выливанием на лед. В результате отфильтровывания осадка в виде белого твердого вещества получили 11,5 г (75%-ный выход) нитропиразола с температурой плавления 124 - 127oC.
Найдено %: C 45,43, H 5,22; N 19,42.
Для C8H11N3O4
Вычислено, %: C 45,57; H 5,20; N 19,71.
Пример 4. 1-метил-4-нитро-3-н-пропилпиразол-5-карбоксамид.
11,3 г (0,053 г моль) 1-метил-4/нитро-3-н-пропилпиразол-5-карбоновой кислоты добавили в 50 мл хлористого топлива, а образовавшуюся смесь выдержали при температуре кипения с обратным холодильником в течение 3 ч. Затем реакционную смесь охладили и выпариванием в вакууме удалили избыток хлористого тионила. Маслоподобный остаток растворили в 50 мл ацетона и раствор осторожно добавили в смесь 50 г льда с 50 мл концентрированного водного раствора гидрата окиси аммония. Остаток собрали фильтрованием, в результате чего в виде бледно-желтого твердого материала получили 8,77 г (78%-ный выход) пиразолкарбоксамида с температурой плавления 141-143oC.
Найдено, %: C 45,22, H 5,71, N 26,12;
Для C8H12N4O3
Вычислено, %: C 45,28; H 5,70; N 16,40.
Пример 5. 4-амино-1-метил-3-н-пропилпиразол-5-карбоксамид.
3,45 г (16,2 ммоль) 1-метил-4-нитро--3-н-пропилпиразол-5-карбоксамида и 18,4 г (81 ммоль) дигидрата хлорида двухвалентного олова суспендировали в этаноле и смесь выдерживали при температуре кипения с обратным холодильником в течение 2 ч. Образовавшийся раствор охладили до комнатной температуры, подкисляли до величины pH, равной 9, добавлением 2н. водного раствора гидрата окиси натрия с последующей экстракцией обработкой 3 порциями по 150 мл дихлорметана. Органические экстракты собрали, высушили над сульфатом магния и выпарили в вакууме. В результате растирания остатка в диэтиловом эфире получили в виде не совсем белого твердого продукта 2,77 г (94%-ный выход) аминопиразол с температурой плавления 98 - 101oC.
Найдено, %: C 52,84; H 7,81; N 30,38.
Для C8H14N4O
Вычислено %: C 52,73; H 7,74; N 30,75.
Пример 6. 4-/2-этоксибензамидо/-1-метил-3-н-пропилпиразол-5-карбоксиамид
Раствор 6,1 г (33,0 ммоль) 2-этоксибензоилхлорида в 50 мл дихлорэтана добавили в перемешиваемый раствор 3,0 г (16,4 ммоль) 4-амино-1-метил-3-н-пропилпиразол-5-карбоксиамида, 0,02 г/0,164 ммоль/ 4-диметиламинопиридина и 3,34 г (33,0 ммоль) триэтиламина в 50 мл дихлорметана при температуре 0oC. Образовавшейся смеси позволили нагреться до комнатной температуры и ее перемешали в течение дополнительных 2 ч. Растворитель выпарили в вакууме, остаток растворили в 250 мл смеси дихлорметана с метанолом в соотношении 19 : 1, а затем раствор промыли 100 мл 1 н. соляной кислоты, высушили над сульфатом магния и выпарили в вакууме. Сырой материал подвергли хроматографической обработке на 200 г силикагеля, элюируя смесью дихлорметана с метанолом в соотношении 97 : 3, в результате чего получили розовое твердое вещество, кристаллизация которого из смеси этилацетата с гексаном позволила получить в виде бледно-розового твердого продукта 2,2 г (40%-ный выход) пиразол-5-карбоксамида с температурой плавления 153 - 155oC.
Найдено, %: C 61,66; H 6,77; N 16,9.
Для C17H22N4O3
Вычислено, %: C 61,80; H 6,71; N 16,96.
Пример 7. 5-/2-этоксифенил/-1-метил-3-н-пропил-1,6-дигидро-7H- пиразоло/4,3-d/пиримидин-7-он
223 г (0,676 моль) 4-/2-этоксибензамидо/-1-метил-3-н- пропилпиразол-5-карбоксамида порциями добавили в раствор 54 г (1,35 моль) гидрата окиси натрия с 224 мл 30%-ной перекиси водорода в 2000 мл воды. Затем добавили 700 мл этанола и образовавшуюся смесь выдержали при температуре кипения с обратным холодильником в течение 2,5 ч, охладили, после чего выпарили в вакууме. Образовавшийся твердый продукт обработали 380 мл 2н. соляной кислоты с использованием внешнего охлаждения и смесь подвергли экстракционной обработке 700 мл и 3 порциями по 200 мл дихлорметана. Объединенные органические экстракты промыли последовательно 3 порциями по 0400 мл насыщенного водного раствора карбоната натрия и 30 мл рассола, после чего высушили над сульфатом натрия и выпарили в вакууме.
В результате хроматографической обработки остатка на 1000 г силикагеля с использованием градиента элюирования метанолом в дихлорметане (0 - 1%) с последующим растиранием сырого продукта в 300 мл диэтилового эфира получили в виде бесцветного твердого материала 152,2 г (72%-ный выход) соединения, указанного в заголовке, с температурой плавления 143 - 146oC.
Найдено, %: C 65,56; H 6,44; N 18,14.
Для C17H20N4O2
Вычислено, %: C 65,36; H 6,45; N 17,94%.
Пример 8. 5-/5-хлорсульфонил-2-этоксифенил/-1-метил-3-н-пропил- 1,6-дигидро-7H-пиразоло/4,3-d/ пиримидин-7-он.
10,0 г (32,1 ммоль) 5-/2-этоксифенил/-1-метил-3-н-пропил-1,6-дигидро-7H-пиразол/4,3-d/ пиримидин-7-она порциями добавили в 20 мл хлорсульфоновой кислоты при температуре 0oC в азотной атмосфере. После перемешивания в течение ночи реакционный раствор осторожно добавили в 150 мл смеси льда с водой и водную смесь подвергли экстракционной обработке 4 порциями по 100 мл смеси дихлорметана с метанолом в соотношении 9 : 1. Объединенные экстракты высушили над сульфатом натрия и выпарили в вакууме, в результате чего получили в виде белого твердого вещества 12,8 г (97%-ный выход) требуемого хлористого сульфонила с температурой плавления 179 - 181oC.
Найдено, %: C 50,07; H 4,71; N 13,29.
Для C17H19ClN4O4S
Вычислено, %: C 49,70; H 4,66; N 13,64.
Пример 9. 5-[2-этокси-5-/4-карбамоилпиперидинилсульфонил/-фенил/- фенил] -1-метил-3-н-пропил-1,6-дигидро-7H-пиразоло(4,3-d) пиримидин-7-он.
703 мг (5,50 ммоль) 4-карбамоилпиперидина добавили в перемешиваемую суспензию 750 г (1,80 ммоль) 5-/5-хлорсульфонил-2-этоксифенил)-1-метил-3-н-пропил-1,6-дигидро-7H- пиразоло/4,3 - d)пиримидин-7-она в 50 мл этанола при комнатной температуре. Образовавшуюся смесь перемешивали в течение 4 дн. с последующим удалением растворителя в вакууме. Остаток растворили в 100 мл смеси дихлорметана с метанолом в соотношении 9 : 1 и раствор промыли 100 мл насыщенного водного раствора карбоната натрия. Затем водную фазу подвергли дополнительной экстракционной обработке 3 порциями по 100 мл смесями дихлорметана с метанолом, все органические фракции собрали, высушили над сульфатом магния и выпарили в вакууме с получением твердого продукта. В результате кристаллизации из смеси метанола с диметилформамидом в виде не совсем белого твердого продукта получали 446 мг (49%-ный выход) указанного в заголовке сульфонамида с температурой плавления 274 - 276oC.
Найдено, %: C 55,36; H 6,01; N 16,65
Для C23H29N6O5S
Вычислено, %: C 55,08; H 5,83; N 16,75.
Примеры 10 - 14.
В соответствии с процедурой примера 9 с использованием соответствующего амина получили нижеследующие соединения (см. табл. 2):
Пример 16. 5-[2-Этокси-5-[4-/метилтиоимидоил/-пиперазинилсульфонил]-фенил] -1-метил-3-н-пропил-1,6-дигидро-7H-пиразоло(4,3-d) пиримидин-7-онгидроиодид
Смесь 0,78 г (1,5 ммоль) 5-[2-этокси-5-/4- тиокарбамоилпиперазинилсульфонил/-фенил] -1-метил-3-н-пропил-1,6- дигидро-7H-пиразоло/4,3-d)пиримидин-7-она с 426 мг (3,0 ммоль)иодистого метила и 20 мл метанола перемешивали при температуре кипения с обратным холодильником в течение 2 ч, после чего ей дали остыть. Образовавшееся белое твердое вещество удалили фильтрованием и кристаллизовали из смеси этилацетата с метанолом, получив в виде бесцветных кристаллов 0,70 г (71%-ный выход) соединения, указанного в заголовке с температурой плавления 227-228oC.
Найдено,%: C 41,43; H 4,79; N 14,42
Для C23H31N7O4S2 • HI
Вычислено,%: C 41,75; H 4,88; N 14,82.
Пример 17. 5-{2-этокси-5-[4-/метиламидино/- пиперазинилсульфонил]-фенил} -1-метил-3-н-пропил-1,6-дигидро-7H-пиразоло/4,3-d)-7-онгидроиодид
0,5 г(0,75 ммоль) 5-{2-этокси-5-[4-метилтиоимидоил)-пиперазинилсульфонил] -фенил}-1-метил-3-н-пропил-1,6-дигидро-7H- пиразоло/4,3-d)пиримидин-7-онгидроиодида добавляли в 33%-ный раствор метиламина в 20 мл этанола и смесь перемешивали при комнатной температуре в течение 18 ч. Раствор выпарили в вакууме и остаток растерли в диэтиловом эфире. В результате хроматографической обработки образовавшегося твердого продукта на 10 г силикагеля с использованием метанола в дихлорметане в качестве элюента с градиентом (0-4%), после чего сырой продукт растерли в диэтиловом эфире, получили светло-коричневый порошок. Кристаллизация из смеси этилацетата с метанолом позволила получить в виде бесцветных кристаллов 112 мг соединения, указанного в заголовке примера (23%-ный выход) с температурой плавления 253-255oC.
Найдено,%: C 42,90; H 5,09; N 17,41.
Для C23H32N8O4 • HI
Вычислено,%: C 42,86; H 5,16; N 17,39.
Пример 18. 1-метил-4-/2-н-пропоксибензамидо/-3-н-пропилпиразол-5-карбоксамид
Этот амид получили из 2-н-пропоксибензоилхлорида в соответствии с процедурой, изложенной в примере 6, в результате чего получили твердый продукт (63%-ный выход) розового цвета с температурой плавления 148-149oC.
Найдено,%: C 62,97; H 7,00; N 16,29.
Для C18H24N4O3
Вычислено,%: C 62,77; H 7,02; N 16,27.
Пример 19. 1-метил-5-/2-н-пропоксифенил/3-н-пропил-1,6-дигидро-7H-пиразоло/4,3-d)пиримидин-7-он
0,34 г (0,99 ммоль) 1-метил-4-/2-р-пропоксибензамидо/3-н-пропилпиразол-5-карбоксамида добавляли в перемешиваемую смесь 1,0 мл 30%-ной перекиси водорода, 0,54 г (3,92 ммоль) карбоната калия, 10 мл воды и 5 мл этанола. Эту смесь выдерживали при температуре кипения с обратным холодильником в течение 38 ч, а затем выпаривали в вакууме. Остаток суспендировали в 20 мл воды, а затем смесь подкислили добавлением 2 н. соляной кислоты и подвергли экстракционной обработке 3 порциями по 20 мл дихлорметана. Экстракты объединили, высушили под сульфатом натрия и выпарили в вакууме. Полученный остаток подвергли хроматографической обработке на 6 г силикагеля с использованием метанола в дихлорметане с градиентом элюирования 0,01-1,0%, в результате чего получили масло, последующее растирание которого в диэтиловом эфире позволило получить в виде белого твердого вещества 0,19 г (59%-ный выход) целевого продукта с температурой плавления 111-114oC.
Найдено,%: C 66,26; H 6,92; N 17,15
Для C18H22N4O2
Вычислено,%: C 66,23; H 6,80; N 17,17
Пример 20. 5-/5-хлорсульфонил-2-н-пропоксифенил/-1-метил- 3-н-пропил-1,6-дигидро-7H-пиразоло(4,3-d)-пиримидин-7-он
Этот сульфонилхлорид получили из 5-/2-н-пропоксифенил)-1- метил-3-н-пропил-1,6-дигидро-7H-пиразоло(4,3-d)пиримидин-7-она в соответствии с процедурой примера 8 в виде белого твердого вещества (выход - 92%).
Найдено,%: C 51,26; H 5,02; N 12,90
Для C18H21ClN4O4S
Вычислено,%: C 50,88; H 4,98; N 13,19.
Пример 21. 1-метил-5-[5-/пиперазинилсульфонил)-2-н-пропоксифенил]-3-н-пропил-1,6-дигидро-7H-пиразоло/4,3-d)пиримидин-7-он
Этот сульфонамид получили из пиперазина и 5-/5-хлорсульфонил-2- н-пропоксифенил/1-метил-3-н-пропил-1,6-дигидро- 7H-пиразоло /4,3-d)пиримидин-7-она в соответствии с процедурой примера 9 в виде белого твердого продукта (70%-ный выход) с температурой плавления 185-186oC.
Найдено, %: C 56,17; H 6,38; N 17,65
Для C22H30N6C4S
Вычислено, %: C 55,67; H 6,37; N 16,61.
Пример 22. 5-{5-[4-/2-оксиэтил/-пиперазинилсульфонил]-2-н-пропоксифенил} -1-метил-3-н-пропил-1,6-дигидро-7H-пиразоло /4,3-d)-пиримидин-7-он
Этот сульфонамид получили из N-/2-оксиэтил/-пиперазина и 5-/5-хлорсульфонил-2-н-пропоксифенил/-1-метил-3-н-пропил-1,6- дигидро-7H-пиразоло/4,3-d/пиримидин-7-она в соответствии с процедурой примера 9 в форме бесцветных иглоподобных кристаллов (66%-ный выход) с температурой плавления 158-159oC.
Найдено, %: C 55,83; H 6,58; N 16,13
Для C24H34N6O5S
Вычислено, %: C 55,58; H 6,61; N 16,20.
Пример 23. 4-/2-аллилоксибензамидо)-1-метил-3-н-пропилпиразол-5-карбоксамид
Раствор 3,93 г (0,02 моль) 2-аллилоксибензоилхлорида в 20 мл дихлорметана по каплям добавляли в перемешиваемый частичный раствор 3,64 г (0,02 моль) 4-амино-1-метил-3-н-пропилпиразол-5-карбоксамида в 50 мл пиридина и образовавшуюся смесь перемешивали при комнатной температуре в течение ночи в сухой атмосфере. Растворитель выпарили в вакууме и остаток разделили между 50 мл дихлорметана и 50 мл насыщенного водного раствора карбоната натрия. Органический слой отделили, а водный слой подвергли исчерпывающей экстракционной обработке дополнительным количеством дихлорметана. Объединенные органические растворы промыли 3 порциями по 30 мл 2М соляной кислоты, а затем 30 мл рассола и высушили над сульфатом натрия. После фильтрования и выпаривания в вакууме фильтрата сырой продукт кристаллизовали из этилацетата, получил 4,525 г (66%-ный выход) соединения, указанного в заголовке примера, с температурой плавления 132 - 134oC.
Найдено, %: C 63,49, H 6,42, N 16,33.
Для C16H22N4O3
Вычислено, %: C 63,14, H 6,48; N 16,36.
Пример 24. 5-(2-аллилоксифенил/-1-метил-3-н-пропил-1,6-дигидро-7H-пиразоло (4,3-d)пиримидин-7-он
Смесь 1,2 г (0,0035 моль) 4-/2-аллилоксибензамидо/-1-метил-3-н-пропилпиразол-5/карбоксамида с 0,70 г (0,018 моль) гидрата окиси натрия, 34 мл воды и 8 мл этанола прокипятили с обратным холодильником в течение 5 ч. После охлаждения раствор подвергли исчерпывающей экстракционной обработке этилацетатом. Объединенные экстракты промыли 30 мл рассола, высушили над сульфатом натрия, профильтровали и выпарили растворитель в вакууме, в результате чего получили сырой продукт, который кристаллизовали из смеси этилацетата с гексаном, выделив 0,476 г (37%-ный выход) соединения, указанного в заголовке примера, с температурой плавления 116 - 119oC.
Найдено, %: C 67,00; H 6,21; N 17,23
Для C18H20N4O2
Вычислено, %: C 66,65; H 6,21; N 17,27.
Пример 25. 5-/2-оксифенил/-1-метил-3-н-пропил-1,6-дигидро-7H-пиразоло/4,3-d/пиримидин-7-он
Смесь 0,25 г (0,0008 моль)5-/2-аллилоксифенил)-1-метил-3-н-пропил-1,6-дигидро-7H-пиразоло (4,3-d)пиримидин-7-она с 0,145 г (0,0015 моль) фенола, 0,031 г (0,0015 моль) пиперидина и 0,046 г (0,00004 моль) тетракис-(трифенилфосфин)-палладия (0) в 5 мл абсолютированного этанола прокипятили в течение ночи с обратным холодильником в атмосфере азота. Смеси дали охладиться, растворитель выпарили в вакууме и остаток растворяли в 40 мл этилацетата. Этот раствор промыли 3 порциями по 10 мл воды, 3 порциями по 10 мл 1М соляной кислоты и 10 мл рассола. После сушки над сульфатом натрия и фильтрования фильтрат выпарили в вакууме, получив сырой продукт. После растирания в диэтиловом эфире и кристаллизации из смеси этилацетата с пентаном получили 0,021 г (10%-ный выход) фенола, указанного в заголовке, с температурой плавления 233 - 238oC.
Найдено, %: C 63,17; H 5,65; N 19,52
Для C15H16N4O2
Вычислено, %: C 63,36; H 5,67; N 19,71.
Пример 26. 5-/5-хлороульфонил-2-оксифенил/-1-метил-3-н-пропил-1,6-дигидро-7H- пиразоло/4,3-d/пиримидин-7-он
5-/2-оксифенил/-1-метил-3-н-пропил-1,6-дигидро-7H-пиразоло(4,3-d)пиримидин-7-он (0,239 г, 0,00084 моль) порциями добавили в 3 мл перемешиваемой хлорсульфоновой кислоты, охлажденной до температуры 0oC, в атмосфере азота, а образовавшийся темнокрасный раствор перемешивали при комнатной температуре в течение 18 ч. Затем реакционную смесь осторожно, по каплям добавили в перемешиваемую смесь воды со льдом, получив коричневый твердый продукт. Эту последнюю смесь подвергли экстракционной обработке 3 порциями по 30 мл дихлорметана, объединенные экстракты высушили над сульфатом натрия и профильтровали, фильтрат выпарили в вакууме, получив 0,24 г (75%-ный выход) коричневого твердого вещества, использованного на следующей стадии без дополнительной очистки; Rf-0,3(двуокись кремния; дихлорэтан, метанол в соотношении 95:5).
Пример 27. 5-[2-окси-5-/4-метилпиперазинилсульфонил/-фенил]-1-метил-3-н-пропил-1,6-дигидро-7H-пиразол (4,3-d)пиримидин-7-он
Раствор 0,235 г (0,0006 моль) 5-/5-хлорсульфонил-2-окси-фенил)-1-метил-3-н-пропил-1,6-дигидро-7H-пиразоло (4,3-d)пиримидин-7-она и 0,5 мл (0,0045 моль) N-метилпиперазина в 40 мл этанола перемешивали при комнатной температуре в течение 18 ч. Этот раствор выпарили в вакууме и остаток разделили между 40 мл этилацетата и 40 мл воды. Тонкодисперсный осадок отфильтровали, промыли водой, затем этилацетатом и кристаллизовали из смеси этилацетата с ДМФ, в результате чего в форме не совсем белого порошка получили 0,260 г (49%-ный выход) соединения, указанного в заголовке с температурой плавления 283-284oC.
Найдено, %: C 53,53; H 5,89; N 18,40.
Для C20H26N6O4S
Вычислено, %: C 53,80; H 5,87; N 18,82.
Пример 28. 5-[2-аллилокси-5-/4-метилпиперазинил ульфонил/-фенил]-1-метил-3-н-пропил-1,6-дигидро-7H-пиразоло(4,3-d) пиримидин-7-он
0,02 мл (0,00023 моль) аллилбромида добавили в перемешиваемую суспензию 0,103 г (0,00023 моль) 5-[2-окси-5-/4-метил-пиперазинилсульфонил/-фенил]-1-метил-3-н-пропил-1,6-дигидро-7H-пиразоло/4,3-d/пиримидин-7-она и 0,032 г (0,00023 моль) карбоната калия в 10 мл 2-бутанона и смесь выдержали при температуре кипения с обратным холодильником в течение 8 ч. После охлаждения реакционную смесь выпарили в вакууме и остаток суспендировали в 20 мл воды. Водную суспензию подвергли экстракционной обработке 3 порциями по 20 мл этилацетата, объединенные экстракты высушили над сульфатом натрия и после фильтрования выпарили в вакууме с получением масла. В результате хроматографической обработки в колонке с 2 г силикагеля с использованием метанола в дихлорметане с градиентом элюирования 0-3%, после чего выпарили в вакууме соответствующие фракции, получили полутвердый продукт, который растворили в ацетоне, раствор выпарили в вакууме, выделив 0,011 г (10%-ный выход) соединения, указанного в заголовке, с температурой плавления 151 - 153oC. RF=0,5 (двуокись кремния, дихлорметан, метанол, 95:5), m/е 487 (М++1).
Пример 29. 4-(этоксибензамидо/-1,3-диметилпиразол-5-карбоксамид
Этот амид получили из 4-амино-1,3-диметилпиразол-5-карбоксамида (в свою очередь, полученного по методу из журнала J.Med. Chem., 1987, 30, 91) в соответствии с процедурой примера 6 в форме белого твердого продукта (81%-ный выход) с температурой плавления 178-181oC.
Найдено, %: C 59,89; H 6,05; N 18,44.
Для C15H18N4O3
Вычислено, %: C 59,59, H 6,00; N 18,53.
Пример 30. 5-(2-этоксифенил)-1,3-диметил-1,6-дигидро-7H-пиразоло(4,3-d)пиримидин-7-он
1,6 г (5,29 моль) 4-/2-этоксибензамидо)-1,3-диметил-пиразил-5-карбоксамида добавили в 50 г полифосфорной кислоты и смесь выдержали при температуре 140oC в течение 6 ч. Затем раствор охладили, вылили в 100 мл смеси воды со льдом, после чего подщелачили добавлением 10%-го раствора гидрата окиси натрия в воде и подвергли экстракционной обработке 3 порциями по 100 мл дихлорметана. Органические экстракты объединили, высушили над сульфатом магния и выпарили в вакууме. Остаток подвергли хроматографической обработке на силикагеле, элюируя смесью дихлорметана с метанолом. В результате кристаллизации сырого продукта из водного этанола в виде бесцветного твердого вещества получили соединение, указанное в заголовке, с температурой плавления 201-204oC.
Найдено, %: C 63,43; H 5,57; N 19,35.
Вычислено для C15H16N4O2, %:
C 63,36; H 5,67; N 19,71.
Пример 31. 5-(5-хлорсульфонил-2-этоксифенил)-1,3-диметил-1,6-дигидро-7H-пиразоло (4,3-d)пиримидин-7-он
Этот сульфонилхлорид получили из 5-/2-этоксифенил)-1,3-диметил-1,6-дигидро-7H-пиразоло(4,3-d)пиримидин-7-она в соответствии с процедурой примера 8 с достижением количественного выхода в виде белого твердого вещества, RF - 0,3 (двуокись кремния; диэтиловый эфир). Это соединение в дальнейшем использовали без дополнительной очистки.
Примеры 32-34.
В соответствии с процедурой примера 9 с использованием 5-/5-хлорсульфонил-2-этоксифенил)-1,3-диметил-1,6-дигидро-7H-пиразоло (4,3-d)пиримидин-7-она и соответствующего амина получили нижеследующие соединения (см. табл. 3):
Пример 35. 4-Нитро-3-н-пропилпиразол-5-карбоновая кислота 3-н-пропилпиразол- 5-карбоновую кислоту (полученную по методу из журнала Chem.Pharm. Bull. , 1984, 32, 1568) нитровали в соответствии с процедурой примера 3, в результате чего в виде бесцветного твердого вещества с 75%-ным выходом получили соединение, указанное в заголовке, с температурой плавления 169-173oC.
Найдено, %: C 42,35; H 4,56; N 21,07.
Для C7H9N3O4
Вычислено, %: C 42,21; H 4,55; N 21,10.
Пример 36. 4-нитро-3-н-пропилпиразол-5-карбоксамид
Смесь 7,8 г (39,2 моль) 4-нитро-3-н-пропилпиразол-5-карбоновой кислоты с 35 мл хлористого тионила прокипятили с обратным холодильным в течение 3 ч. Растворитель удалили выпариванием в вакууме и твердый остаток порциями добавили при температуре 0oC в 40 мл водного раствора гидрата окиси аммония. Смесь разбавили 60 мл воды и подвергли экстракционной обработке смесью в соотношении 9: 1 дихлорметана с метанолом в виде 3 порций по 100 мл. Органические фракции объединили, высушили над сульфатом магния и выпарили в вакууме, а остаток кристаллизовали из этанола, в результате чего в виде бесцветного твердого вещества получили 1,0 г (13%-ный выход) карбоксамида с температурой плавления 202-206oC.
Найдено, %: C 42,35; H 5,01; N 28,38
Для C7H10N4O3
Вычислено, %: C 42,42; H 5,09; N 28,27.
Пример 37. 4-амино-3-н-пропилпиразол-5-карбоксамид
Раствор 198 мг (1,0 ммоль) 4-нитро-3-н-пропилпиразол-5-карбоксамида в 5 мл метанола по каплям добавили в смесь 113 мг (2,97 ммоль) боргидрида натрия с 5 мг 10%-го палладия на угле и 3 мл воды. Эту смесь перемешивали при комнатной температуре в течение 3 ч, профильтровали и выпариванием в вакууме удалили из нее растворитель. В результате кристаллизации остатка из смеси этилацетата с метанолом в виде не совсем белого твердого вещества получили 61 мг (36%-ный выход) соединения, указанного в заголовке, с температурой плавления 196-201oC. Rf -0,4 (двуокись кремния; дихлорметан, метанол, гидрат окиси аммония в соотношении 90:10:1).
Найдено, %: C 48,96; H 6,98; N 32,08.
Для C7H12N4O
Вычислено, %: C 49,98; H 7,19; N 33,31.
Пример 38. 4-/2-Этоксибензамидо/-3-н-пропилпиразол-5-карбоксамид
Амид, указанный в заголовке, получили из 4-амино-3-н-пропилпиразол-5-карбоксамида в соответствии с процедурой примера 6 в виде белого твердого вещества (64%-ный выход) с температурой плавления 209-211oC.
Найдено, %: C 60,73; H 6,41; N 17,80.
Для C16H20N4O3
Вычислено, %: C 60,74; H 6,37; N 17,71.
Пример 39. 5-/2-этоксифенил/-3-н-пропил-1,6-дигидро-7H-пиразоло (4,3-d)пиримидин-7-он
Указанное в заголовке соединение получили из 4-/2-этоксибензамидо)-3-н-пропилпиразол-5-карбоксамида в соответствии с процедурой примера 30 в виде белого твердого вещества (16%-ный выход) с температурой плавления 199-201oC.
Найдено, %: C 64,44; H 6,19; N 18,44.
Для C16H18N4O2
Вычислено, %: C 64,41; H 6,08; N 18,78.
Пример 40. 5-5/-хлорсульфонил-2-этоксифенил/-3-н-пропил-1,6-дигидро-7H-пиразоло /4,3-d/-пиримидин-7-он
Указанный в заголовке сульфонилхлорид получили из 5-/2-этоксифенил/-3-н-пропил-1,6-дигидро-7-H-пиразоло (4,3-d)пиримидин-7-она в соответствии с процедурой примера 8 в виде белого твердого вещества (78%-ный выход). Rf - 0,25 (двуокись кремния; диэтиловый эфир).
Его использовали без предварительной очистки.
Пример 41. 5-[2-этокси-5-/4-метилпиперазинил/-сульфонилфенил] - -3-н-пропил-1,6-дигидро-7H-пиразоло /4,3-d/пиримидин-7-он
Указанный в заголовке сульфонамид получили из 5-/5-хлорсульфонил-2-этоксифенил/-3-н-пропил-1,6-дигидро-7H-пиразоло /4,3-d/пиримидин-7-она в соответствии с процедурой примера 9 в виде белого твердого вещества (70%-ный выход) с температурой плавления 236-239oC.
Найдено, %: C 54,84; H 6,27; N 18,10.
Для C21H28N6O4S
Вычислено, %: C 54,78; H 6/13; N 18,25.
Пример 42. 3-бромметил-5-хлор-1-метил-4-нитропиразол
10,7 г (60,0 ммоль) N-бромсукцинимида добавили в раствор 8,78 г (50,0 ммоль) 5-хлор-1,3-диметил-4-нитропиразола в 100 мл четыреххлористого углерода и раствор прокипятили с обратным холодильником с одновременным облучением видимым светом (вольфрамовая лампа на 150 Вт) в течение 3 дн. Через определенные интервалы по ходу всей реакции добавили 6 порциями по 50 кг перекиси бензоила. Затем выпариванием в вакууме удалили растворитель и остаток подвергли хроматографической обработке на силикагеле, элюируя смесью в соотношении 1 : 1 дихлорметана с гексаном, в результате чего в виде не совсем белого твердого вещества получили 8,0 г (63%-ный выход) бромида с температурой плавления 80 - 82oC.
Найдено,%: С 23, 95; H 2,05; N 16,31
Для C5H5BrClN3O2
Вычислено,%: C 23,60; H 1,98; N 16,51.
Пример 43. 5-хлор-3-метоксиметил-1-метил-4-нитропиразол
5,0 г (19,6 моль) раствора 3 бромметил-5-хлор-1-метил-4-нитропиразола в 50 мл метанола обработали 5,75 г (33,8 ммоль) нитрата серебра и смесь прокипятили с обратным холодильником в течение 2 ч. Охлажденную реакционную смесь профильтровали и фильтрат выпаривали в вакууме. Остаток разделили между 100 мл этилацетата и 50 мл воды, подвергнув затем водную фазу экстрактной обработке дополнительными 50 мл этилацетата. Органические экстракты объединили, высушили над сульфатом магния и выпаривали в вакууме. В результате хроматографической обработки на силикагеле, элюируя смесью дихлорметана с метанолом в соотношении 97:3, получили в виде твердого белого вещества 1,6 г (40%-ный выход) пиразола, указанного в заголовке, с температурой плавления 59-63oC.
Найдено,%: C 34,65; H 3,83; N 20,05
Для C6H8ClN3O3
Вычислено,%: C 35,05; H 3,92; N 20,44.
Пример 44. 5-циано-3-метоксиметил-1-метил-4-нитропиразол
Раствор 205 мг (1,0 ммоль) 5-хлор-3-метоксиметил-1-метил-4-нитропиразола. 130 мг (2,0 ммоль) цианистого калия и 10 мг 18-крон-6 в 2 мл ацетона прокипятили с обратным холодильником в течение ночи. Растворитель выпаривали в вакууме и остаток разделили между 20 мл этилацетата с 20 мл воды. Органическую фазу отделили, высушивали над сульфатом магния и выпаривали в вакууме, после чего остаток подвергли хроматографической обработке на силикагеле, элюируя смесью в соотношении 1:1 этилацетата с пентаном. В результате растирания сырого продукта в диэтиловом эфире получили 38 мг (19%-ный выход) желтого твердого вещества с температурой плавления 48 - 50oC.
Найдено,%: C 42,89; H 4,15; N 28,78
Для C7H8N4O3
Вычислено,%: C 42,86; H 4,11; N 28,56
Пример 45. 4-амино-5-циано-3-метоксиметил-1-метилпиразол
Указанное в заголовке соединения получили из 5-циано-3-метоксиметил-1-метил-4-нитропиразола в соответствии с процедурой примера 5 в виде на совсем белого твердого продукта (68%-ный выход) с температурой плавления 82-84oC.
Найдено,%: C 50,81; H 6,313; N 33,94.
Для C7H10N4
Вычислено,%: C 50,59; H 6,07; N 33,72.
Пример 46. 5-циано-4-/2-этоксибензамидо/-3-метоксиметил-1-метилпиразол
Указанное в заголовке соединение получили из 4-амино-5-циано-3-метоксиметил-1-метилпиразола в соответствии с процедурой примера 6 в виде не совсем белого твердого вещества (61%-ный выход) с температурой плавления 103 - 105oC.
Найдено,%: C 61,21; H 5,98; N 17,80.
Для C16H18N4O3
Вычислено,%: C 61,13; H 5,77; N 17,83.
Пример 47. 5-/2-этоксифенил)-3-метоксиметил-1-метил-1,6-дигидро-7Н-пиразоло (4,3-d)пиримидин-7-он
Указанное в заголовке соединения получили из 5-циано-4-/2-этоксибензамидо/-3-метоксиметил-1-метилпиразола в соответствии с процедурой примера 7 посредством получения по месту использования 5-первичного амидного производного в виде белого твердого вещества (38%-ный выход) с температурой плавления 160-161oC.
Найдено,%: C 61,35; H 5,75; N 17,98
Для C16H18N4O3
Вычислено,%: C 61,13; H 5,77; N 17,83.
Пример 48. 3-метоксиметил-1-метил-5-[5-/4-метилпиперазинилсульфонил-2-этоксифенил] -1,6-дигидро-7H-пиразоло(4,3-d) пиримидин-7-он
470 мг (1,50 ммоль) 5-/2-этоксифенил/-3-метоксиметил-1-метил-1,6-дигидро-7H-пиразоло/4,3-d) пиримидин-7-она растворили при температуре 0oC в хлорсульфоновой кислоте. Этот раствор перемешивали при комнатной температуре в течение 2 ч, а затем осторожно добавили в 50 мл смеси воды со льдом. Образовавшийся раствор нейтрализовали насыщенным раствором карбоната натрия, затем подвергли экстракционной обработке смесью в соотношении 20:1 дихлорметана с метанолом в виде 2 порций по 50 мл. Объединенные органические экстракты выпаривали в вакууме и остаток растворили в 5 мл этанола, после чего раствор обработали 450 мг (4,5 ммоль) N-метилпиперазина. По истечении 1 ч выдержки при комнатной температуре растворитель выпаривали в вакууме и остаток подвергли хроматографической обработке на силикагеле, элюируя смесью дихлорметана с метанолом и водным раствором гидрата окиси аммония в объемном соотношении 90:10:1.
В результате растирания в этилацетате сырого продукта в виде белого твердого вещества получили 49 мг (7%-ный выход) соединения, указанного в заголовке примера, с температурой плавления 198-199oC.
Найдено,%: C 52,94; H 6,04; N 17,67.
Для C21H28N6O5S
Вычислено,%: C 52,93; H 5,92; N 17,64.
Из смеси этилацетата с метанолом в соответствии с процедурой хроматографии и кристаллизации в виде белого твердого вещества выделили 51 мг (7%-ный выход) 3-оксиметил-1-метил-5-[5-/4-метилпиперазинилсульфонил/-2-этоксифенил] -1,6-дигидро-7H-пиразоло/4,3-d)пиримидин-7-она с температурой плавления 209-210oC.
Найдено,%: C 51,94; H 5,77; N 18,05.
Для C20H26N6O5S
Вычислено,%: C 51,94; 5,67; N 18,17.
Пример 49. 1-этил-3-н-пропилпиразол-5-карбоновая кислота, этиловый эфир.
Этот пиразол получили из этилового эфира 3-н-пропилпиразол-5-карбоновой кислоты и диэтилсульфата в соответствии с процедурой, изложенной в примере 1, в виде бесцветного малоподобного продукта (72%-ный выход), RF - 0,5 (двуокись кремния, этилацетат, гексан в соотношении 1:1).
Пример 50. 1-этил-3-н-пропилпиразол-5-карбоновая кислота
Эту карбоновую кислоту получили из этилового эфира 1-этил-3-н-пропилпиразол-5-карбоновой кислоты в соответствии с процедурой, которая описана в примере 2, в виде бледно-коричневого твердого вещества (89%-ный выход) с температурой плавления 73-77oC.
Найдено,%: C 58,62; H 7,69; N 15,23.
Для C9H14N2O2
Вычислено,%: C 59,32; H 7,74; N 15,37.
Пример 51. 1-этил-4-нитро-3-н-пропилпиразол-5-карбоновая кислота
Указанное в заголовке соединение получили из 1-этил-3-н-пропилпиразол-5-карбоновой кислоты в соответствии с процедурой, которая описана в примере 3, в виде бесцветного твердого вещества (96%-ный выход) с температурой плавления 120-123oC.
Найдено,%: C 47,61; H 5,81; N 18,54.
Для C9H13N3O4
Вычислено,%: C 47,57; H 5,77; N 18,49.
Пример 52. 1-этил-4-нитро-3-н-пропилпиразол-5-карбоксамид
Указанный в заголовке амид получили из 1-этил-3-н-пропилпиразол-5-карбоновой кислоты в соответствии с процедурой, которая описана в примере 4, в виде не совсем белого твердого вещества (86%-ный выход) с температурой плавления 119-120oC.
Найдено,%: C 47,83; H 6,18; N 24,34.
Для C9H14N4O3
Вычислено,%: C 47,78; H 6,24; N 24,77.
Пример 53. 4-амино-1-этил-3-н-пропилпиразол-5-карбоксамид
Указанное в заголовке соединение получили из 1-этил-4-нитро-3-н-пропилпиразол-5-карбоксамида в соответствии с процедурой, которая описана в примере 5, в виде не совсем белого твердого вещества (100%-ный выход) с температурой плавления 93-97oC.
Найдено,%: C 55,17; H 8,34; N 28,93.
Для C9H16N4O
Вычислено,%: C 55,08; H 8,22; N 28,55.
Пример 54. 4-/2-этоксибензамидо/-1-этил-3-н-пропилпиразол-5-карбоксамид
Указанный в заголовке амид получили из 4-амино-1-этил-3-н-пропилпиразол-5-карбоксамида и 2-этоксибензоилхлорида в соответствии с процедурой, которая описана в примере 6 в виде бесцветного твердого вещества (73%-ный выход) с температурой плавления 139-141oC.
Найдено,%: C 63,03; H 7,15; N 16,50.
Для C18H24N4O3
Вычислено,%: C 62,77; H 7,02; N 16,27.
Пример 55. 5-/2-этоксифенил/-1-этил-3-н-пропил-1,6-дигидро-7H-пиразоло(4,3-d)пиримидин-7-он
Указанное в заголовке соединение получили из 4-/2-этоксибензамино/-1-этил-3-н-пропилпиразоло-5-карбоксамида в соответствии с процедурой примера 7, в виде бесцветного твердого вещества (46%-ный выход) с температурой плавления 112-114oC.
Найдено,%: C 66,59; H 6,85; N 17,26
Для C18H22N4O2
Вычислено,%: C 66,23; H 6,79; N 17,17/
Пример 56. 5-/5-хлорсульфонил-2-этоксифенил/-1-этил-3-н-пропил-1,6-дигидро-7H-пиразоло/4,3-d/пиримидин-7-он
Указанное в заголовке примера соединение получили из 5-/2-этоксифенил/-1-этил-3-н-пропил-1,6-дигидро-7H-пиразоло /4,3-d/пиримидин-7-она в соответствии с процедурой примера 8 в виде метиленхлоридного сольвата (86%-ный выход) с температурой плавления 170-172oC.
Найдено,%: C 49,82; H 4,84; N 12,77.
Для C18H21ClN4S1/6CH2Cl2
Вычислено,%: C 49,70; H 4,90; N 12,77.
Пример 57. 5-[2-этокси-5-/4-метилпиперазинилсульфонил/-фенил]-1-этил-3-н-пропил-1,6-дигидро-7H-пиразоло/4,3-d/пиримидин-7- он
Указанный в заголовке сульфонамид получили из 5-/5-хлорсульфонил-2-этоксифенил/-1-этил-3-н-пропил-1,6-дигидро-7H-пиразоло(4,3-d)пиримидин-7-она и N-метилпиперазина в соответствии с процедурой примера 9 в виде бесцветного твердого вещества (43%-ный выход) с температурой плавления 160-162oC.
Найдено,%: C 57,24; H 6,17; N 16,83
Для C23H32N6O4S
Вычислено,%: C 56,54; H 6,60; N 17,20.
Rf-0,35% (двуокись кремния; дихлорметан, метан в соотношении 9:1).
Пример 58. 5-{2-этокси-5-[4-/2-оксиэтил/-пиперазинилсульфонил]-фенил}-1-этил-3-н-пропил-1,6-дигидро-7H-пиразоло(4,3-d)пиримидин-7-он
Указанный в заголовке сульфонамид получили из 5-/5-хлорсульфонил-2-этоксифенил)-1-этил-3-н-пропил-1,6-дигидро-7H-пиразоло/4,3-d)пиримидин-7-она и N-/2-оксиэтил/-пиперазина в соответствии с процедурой примера 9 в виде бесцветного твердого вещества (88%-ный выход) с температурой плавления 191-193oC.
Найдено,%: C 55,74; H 6,55; N 15,78.
Для C24H34N6O5S
Вычислено,%: C 55,58; H 6,61; H 16,20.

Изобретение относится к новым производным пиразоло/4,3-d/пиримидин-7-она формулы I, где R1 - H, CH3, C2H5, R2 - CH3, CH2OH, CH2OCH3 или н - C3H7, R3 - C2H5, CH2 = CH - CH2, R4 вместе с атомом азота, к которому он присоединен, составляет 4-(R5)-пиперидино- или 4-N (R6)-пиперазильную группу, R5 - H, N(CH3)2, CONH2, R6 - H, CH3, i - C3H7, CH2CH2OH, CSNH2, C(NH)NHCH3 или C(NH)S CH3, и их фармацевтически приемлемые соли фармацевтической композиции, проявляющей ингибирующую активность в отношении циклической гуанозин-31,51-монофосфатфосфодиэстеразы (ЦГМФ), которая содержит 1-400 мг на разовую дозу соединения формулы (I) в смеси с фармацевтически приемлемым разбавителем или носителем; способу лечения или профилактики состояний, обусловленных активностью ЦГМФ, сущность которого состоит в назначении человеку эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли или вышеуказанной композиции. Изобретение также относится к промежуточным соединениям формул II и IY, в которых R1, R2, R3 имеют значения, определенные выше, в соединениях формулы II R3 может быть атомом H, Y - атом Cl. Формулы соединений I, II и IY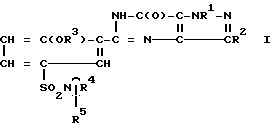

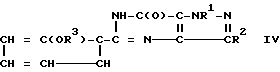 5 с. и 4 з.п.ф-лы, 3 табл.
5 с. и 4 з.п.ф-лы, 3 табл.
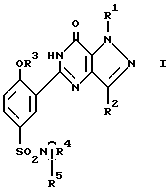
где R1 - атом водорода, метил или этил;
R2 - метил, CH2OH, CH2, OCH3 или н-пропил;
R3 - этил, н-пропил или аллил;
R4 вместе с атомом азота, к которому он присоединен, составляет 4- (R5) - пиперидино- или 4-N-(R6-пиперазинильную группу;
R5 - атом водорода, N(CH3)2- или CONH2-группа;
R6 - атом водорода, метил, 2-пропил, 2-гидроксиэтил, CSNH2-, C(NH)NHCH3- или C(NH)SCH3-группы,
и их фармацевтически приемлемые соли.
5-[2-аллилокси-5-(4-метилпиперазинилсульфонил)фенил] -1- метил-3-н-пропил-1,6-дигидро-7H-пиразоло[4,3-d]пиримидин-7-она;
5[2-этокси-5-(пиперазинилсульфонил)фенил] -1-метил-3-н-пропил- 1,6-дигидро-7H-пиразоло[4,3-d]пиримидин-7-она;
5-[2-этокси-5-(4-метилпиперазинилсульфонил)фенил] -1-метил-3- н-пропил-1,6-дигидро-7H-пиразоло[4,3-d]пиримидин-7-она;
5-{ 2-этокси-5-[4-(2-пропил)-пиперазинилсульфонил] фенил}-1- метил-3-н-пропил-1,6-дигидро-7H-пиразоло[4,3-d]пиримидин-7-она;
5-{ 2-этокси-5-[4-(2-гидроксиэтил)пиперазинилсульфонил] - фенил} -1-метил-3-н-пропил-1,6-дигидро-7H-пиразоло[4,3-d]пиримидин-7-она;
1-метил-5-[5-(пиперазинилсульфонил)-2-н-пропоксифенил] -3- н-пропил-1,6-дигидро-7H-пиразоло[4,3-d] пиримидин-7-она и 5-{ 5-[4-(2-гидроксиэтил)пиперазинсульфонил] -2-н-пропоксифенил} -1- метил-3-н-пропил-1,6-дигидро-7H-пиразоло[4,3-d]пиримидин-7-она,
и его фармацевтически приемлемой соли.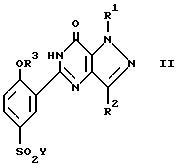
где R1 и R2 имеют значения, определенные в п. 1;
R3 имеет значения, определенные в п. 1, а также может быть атомом водорода;
Y - атом хлора.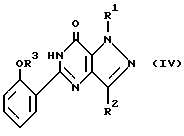 ,
,
где R1 и R2 имеют значения, определенные в п. 1;
R3 имеет значение, определенное в п. 1, а также может быть атомом водорода.
| EP, A, 0201188, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
