Изобретение относится к производным хинона, проявляющим превосходную активность в качестве медикаментов.
Среди переносчиков воспалений, недавно были отмечены лейкотриены и тромбоксаны. Каждый из них вызывает воспаление как таковой или во взаимодействии с другим, чтобы вызвать, или участвовать в распространении воспаления. Однако было найдено немного соединений, которые проявляют активность, как при ингибировании образования лейкотриенов, так и при ингибировании производства тромбоксанов.
Изобретатели изобретения выполнили интенсивные исследования в течение многих лет для того, чтобы получить вещество, проявляющее обе упомянутые выше активности, и было найдено, что производная хинона, которая будет описана ниже, проявляет как хорошо сбалансированное соотношение активностей, так и является превосходным так называемым двойным ингибитором. Изобретение было выполнено на основе этого наблюдения.
Настоящее изобретение обеспечивает производную хинона, представленную следующей общей формулой (I), или его фармакологически приемлемую соль
A__CH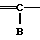 R1 в которой А является группой, представленной формулой
R1 в которой А является группой, представленной формулой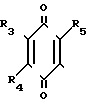 в которой R3, R4 и R5 являются одинаковыми или отличаются друг от друга, и каждый представляет собой атом водорода, гидроксильную группу, низшую алкильную группу, низшую алкокси-группу, алкоксиалкильную группу, алкоксиалкокси-группу, циклоалкилалкокси-группу, тиоловую группу или тиоалкильную группу, при условии, что исключается случай, когда и R3, и R4 каждый является низшей алкокси-группой одновременно) или группой, представленной формулой
в которой R3, R4 и R5 являются одинаковыми или отличаются друг от друга, и каждый представляет собой атом водорода, гидроксильную группу, низшую алкильную группу, низшую алкокси-группу, алкоксиалкильную группу, алкоксиалкокси-группу, циклоалкилалкокси-группу, тиоловую группу или тиоалкильную группу, при условии, что исключается случай, когда и R3, и R4 каждый является низшей алкокси-группой одновременно) или группой, представленной формулой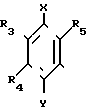 (в которой R3, R4 и R5 являются одинаковыми или отличаются друг от друга, и каждый представляет собой атом водорода, гидроксильную группу, низшую алкильную группу, низшую алкокси-группу, алкокси- алкильную группу, алкоксиалкоксигруппу, циклоалкилалкокси-группу, тиоловую группу или тиоалкильную группу, при условии, что исключается случай, когда и R3и R4 каждый является низшей алкокси-группой одновременно, Х и Y являются одинаковыми или отличаются друг от друга, и каждый представляет собой гидроксильную группу, или защищенную гидроксильную группу);
(в которой R3, R4 и R5 являются одинаковыми или отличаются друг от друга, и каждый представляет собой атом водорода, гидроксильную группу, низшую алкильную группу, низшую алкокси-группу, алкокси- алкильную группу, алкоксиалкоксигруппу, циклоалкилалкокси-группу, тиоловую группу или тиоалкильную группу, при условии, что исключается случай, когда и R3и R4 каждый является низшей алкокси-группой одновременно, Х и Y являются одинаковыми или отличаются друг от друга, и каждый представляет собой гидроксильную группу, или защищенную гидроксильную группу);
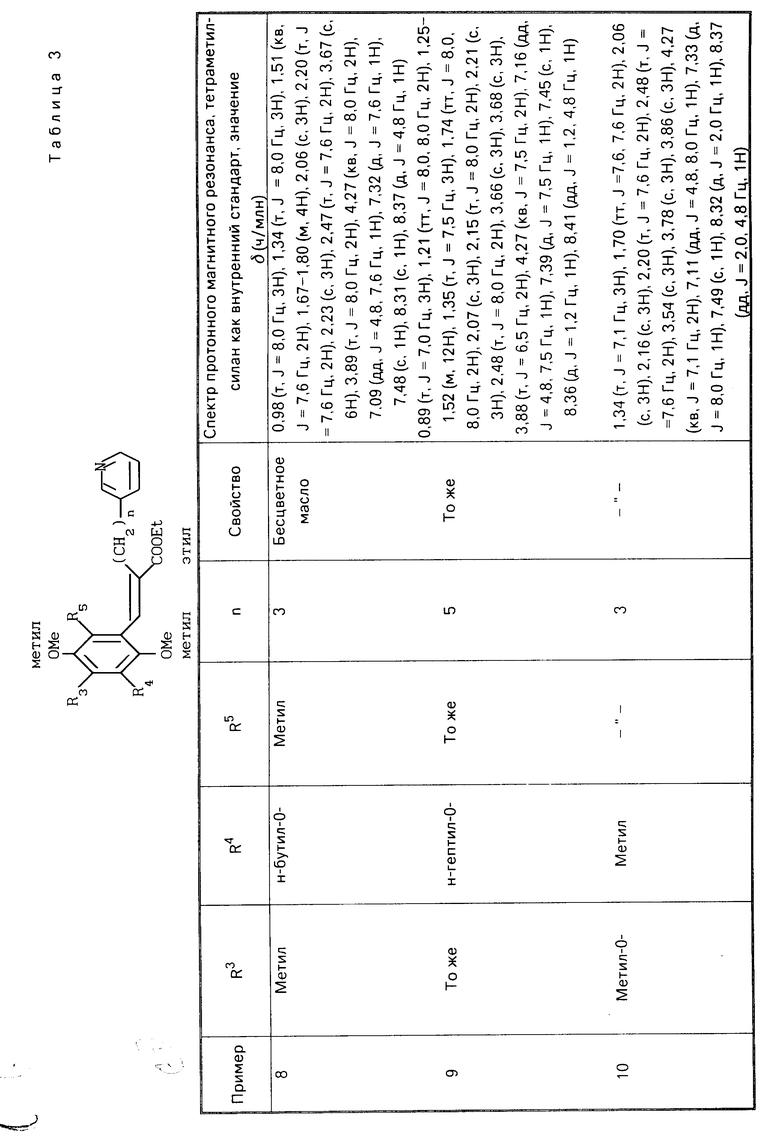
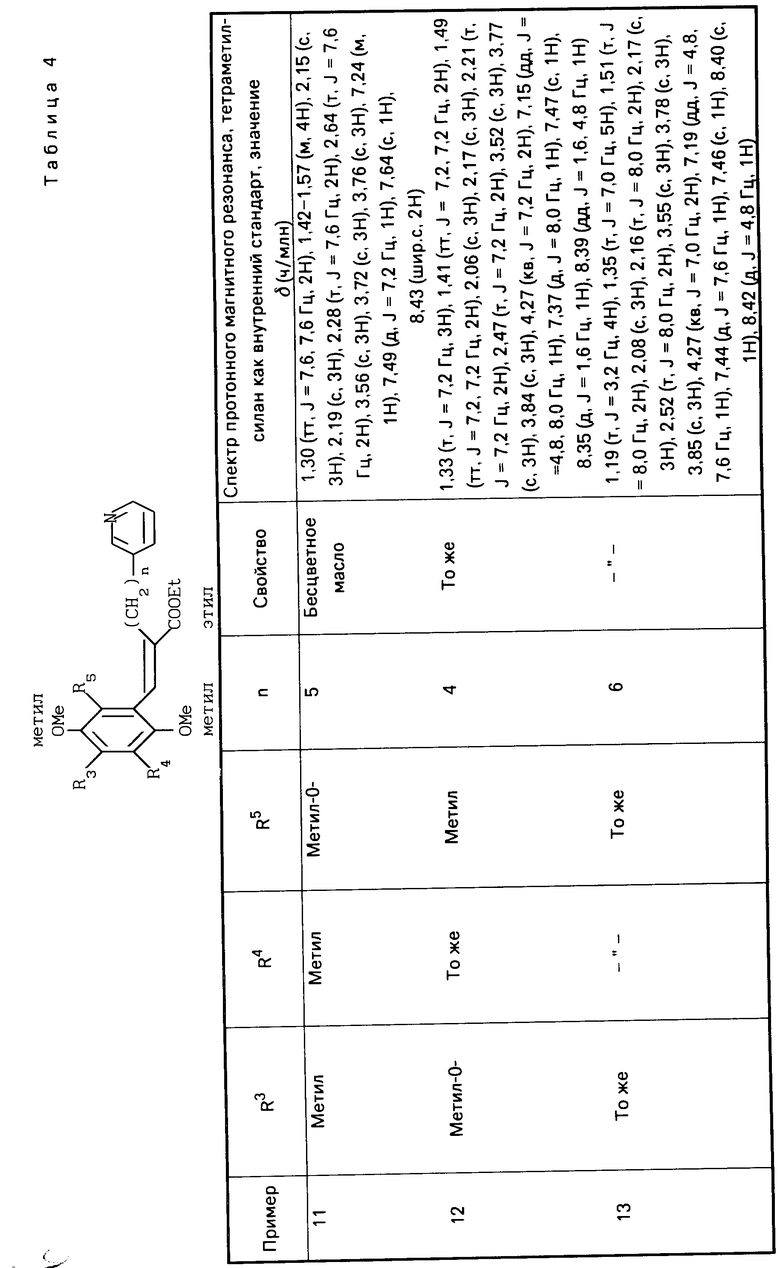
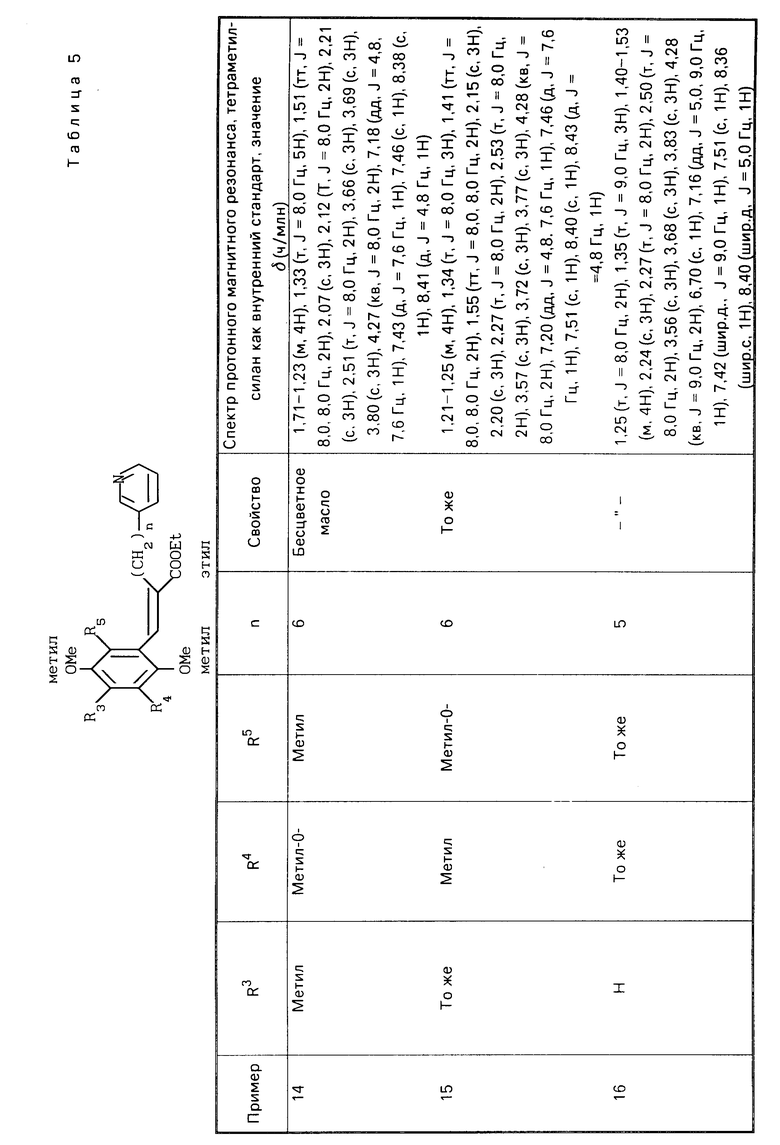
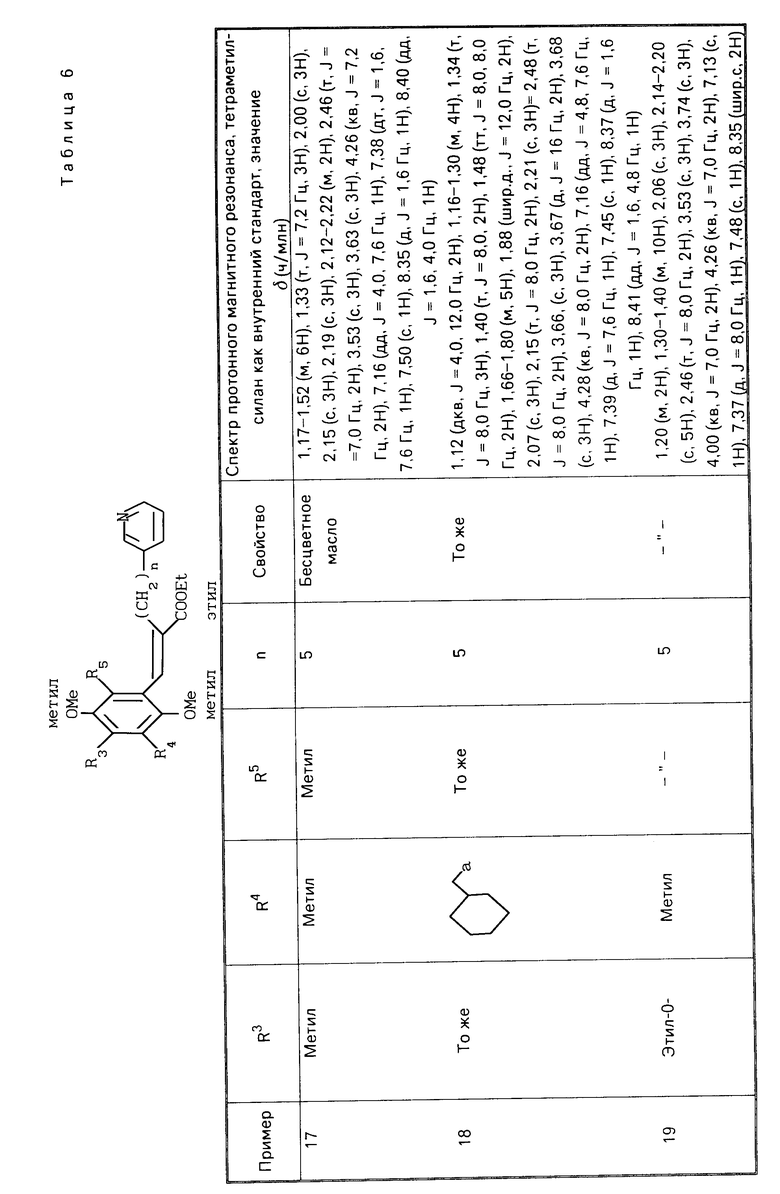
R1 является гетероарилалкильной группой, и В представляет собой карбоксильную группу или защищенную карбоксильную группу.
Предпочтительно производные хинона или их фармакологически приемлемые соли настоящего изобретения включают такие, в которых в общей формуле (I) R1 является гетероарилалкильной группой, R3 является низшей алкокси-группой, R4 является низшей алкильной группой, R5является низшей алкильной группой, Х является гидроксильной или алкокси-группой, Y является гидроксильной или алкокси-группой, В является карбоксильной группой.
Более предпочтительные производные хинона или их фармакологически приемлемые соли включают те, в которых в общей формуле (I) R1 является пиридилгексильной или пиридилпентильной группой, R3 является метокси- или метильной группой, R4 является метильной или метокси-группой, R5является метильной или метокси-группой, Х является гидроксильной или метокси-группой, Y является гидроксильной или метокси-группой.
Хинонную часть производной хинона или его фармакологически приемлемой соли настоящего изобретения преимущественно выбирают из группы, состоящей из перечисленных производных хинона,
(Е)-3-(2-метокси-3,5-диметил-1,4-бензо- хинон-6-ил)-2-[5-(3-пиридил)пентил]-2-про- пеновая кислота,
(Е)-3-(2-метокси-5,6-диметил-1,4-бензо- хинон-3-ил-2-[5-(3-пиридил)пентил]-2-про- пеновая кислота,
(Е)-3-(2-метокси-5,6-диметил-1,4-бензо- хинон-3-ил)-2-[6-(3-пиридил)пентил]-2-про- пеновая кислота,
(Е)-3-(2,4,5-триметокси-3,6-диметилфе- нил)-2-[5-(3-пиридил)пентил]-2-пропеновая кислота,
(Е)-3-(2,5-дигидрокси-4-метокси-3,6-ди- метилфенил)-2-[5-(3-пиридил)пентил]-2-про- пеновая кислота.
(Е)-3-(2,3,5-триметокси-4,6-диметилфе- нил)-2-[5-(3-пиридил)пентил] -2-пропеновая кислота.
Хинонная часть производной хинона или его фармакологически приемлемой соли настоящего изобретения более предпочтительно является (Е)-3-(2-метокси-3,6-диметил-1,4-бензохинон-5-ил)-2-[5-(3-пиридил)- пентил]-2-пропеновой кислотой.
Изобретение также представляет ингибитор образования лейкотриенов и/или тромбоксанов, включающий в качестве активного компонента производную хинона или его фармакологически приемлемую соль изобретения.
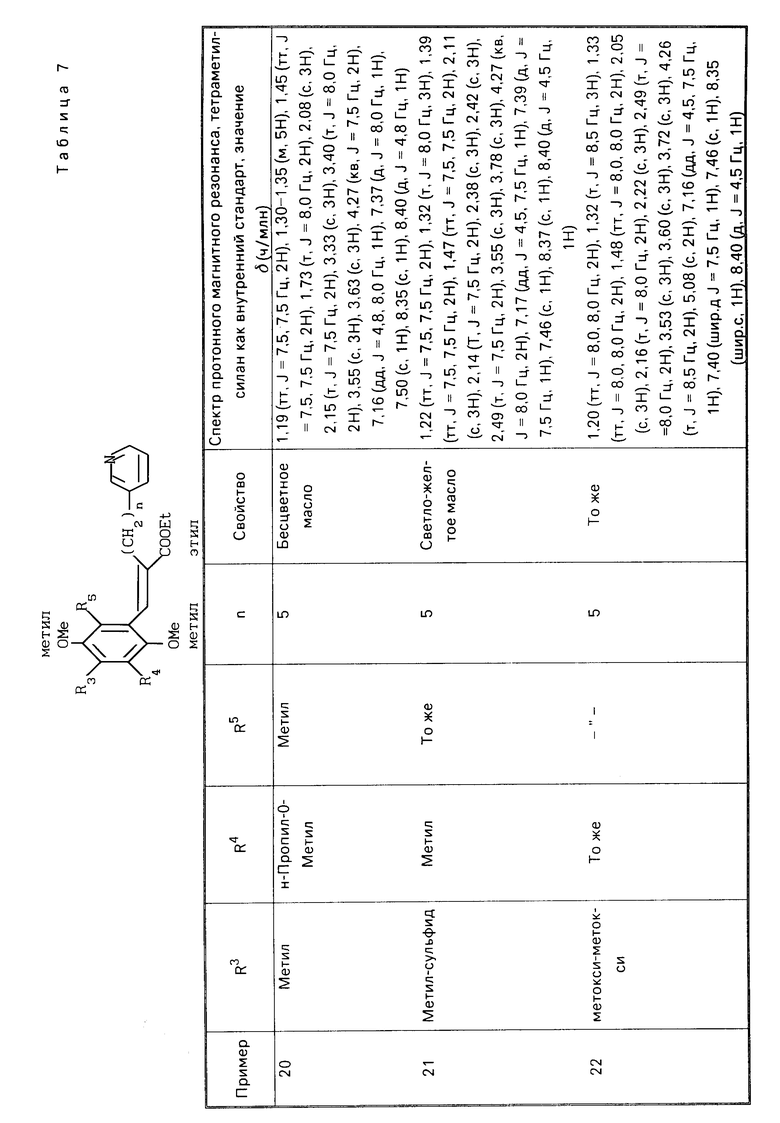
Изобретение также представляет терапевтический и предотвращающий агент для заболеваний, в которых эффективными являются ингибиторы образования лейкотриенов и/или тромбоксанов, который включает в качестве активного компонента производную хинона или его фармакологически приемлемую соль настоящего изобретения.
Изобретение предоставляет фармакологическую композицию, которая включает терапевтически эффективное количество производной хинона или его фармакологически приемлемой соли настоящего изобретения, и фармакологически приемлемый носитель.
Изобретение также предоставляет применение производной хинона или его фармакологически приемлемой соли настоящего изобретения для получения лекарства для лечения заболевания, которое обусловлено образование лейкотриена и/или тромбоксана А2.
Изобретение также обеспечивает применение производной хинона или его фармакологически приемлемой соли настоящего изобретения для получения лекарства для лечения заболевания, выбранного из группы, состоящей из астмы, хронического гепатита, острого гепатита, гепатита, вызванного медикаментом, вирусного гепатита, алкогольного гепатита, желтухи, цирроза, инфаркта миокарда, грудной жабы, церебрального эмболизма, церебрального тромбоза, почечной недостаточности, нефроза и нефрита.
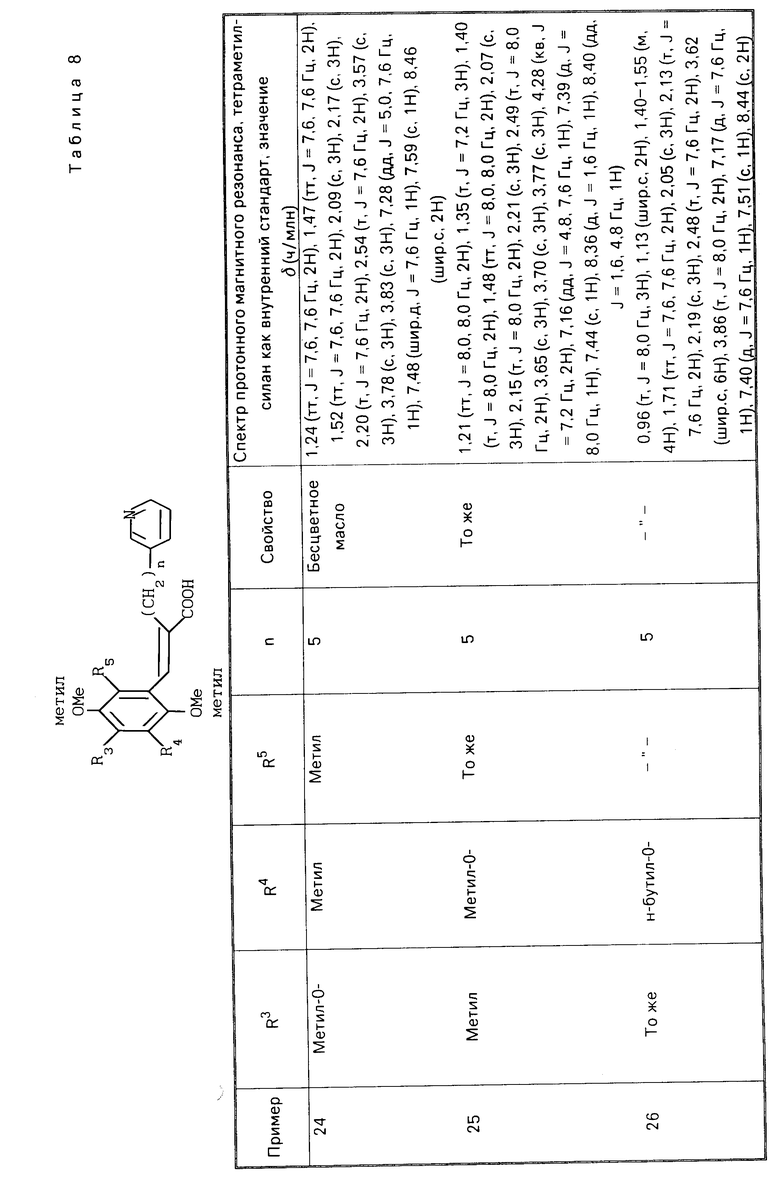
Изобретение представляет собой способ лечения заболевания, который включает назначение фармацевтически эффективного количества производной хинона или его фармакологически приемлемой соли настоящего изобретения пациенту, который страдает от заболевания, вызванного образованием лейкотриена и/или тромбоксана А2.
Настоящее изобpетение также представляет способ лечения заболевания, который включает назначение фармацев- тически эффективного количества производной хинона или его фармакологически приемлемой соли настоящего изобретения пациенту, который страдает от заболевания, выбранного из группы, состоящей из астмы, хронического гепатита, острого гепатита, гепатита, вызванного медикаментом, вирусного гепатита, алкогольного гепатита, желтухи, цирроза, инфаркта миокарда, грудной жабы, церебрального эмболизма, церебрального тромбоза, почечной недостаточности, нефроза и нефрита.
Вещество изобретения является производной хинона, представленной следующей общей формулой (I), или его фармакологически приемлемой солью
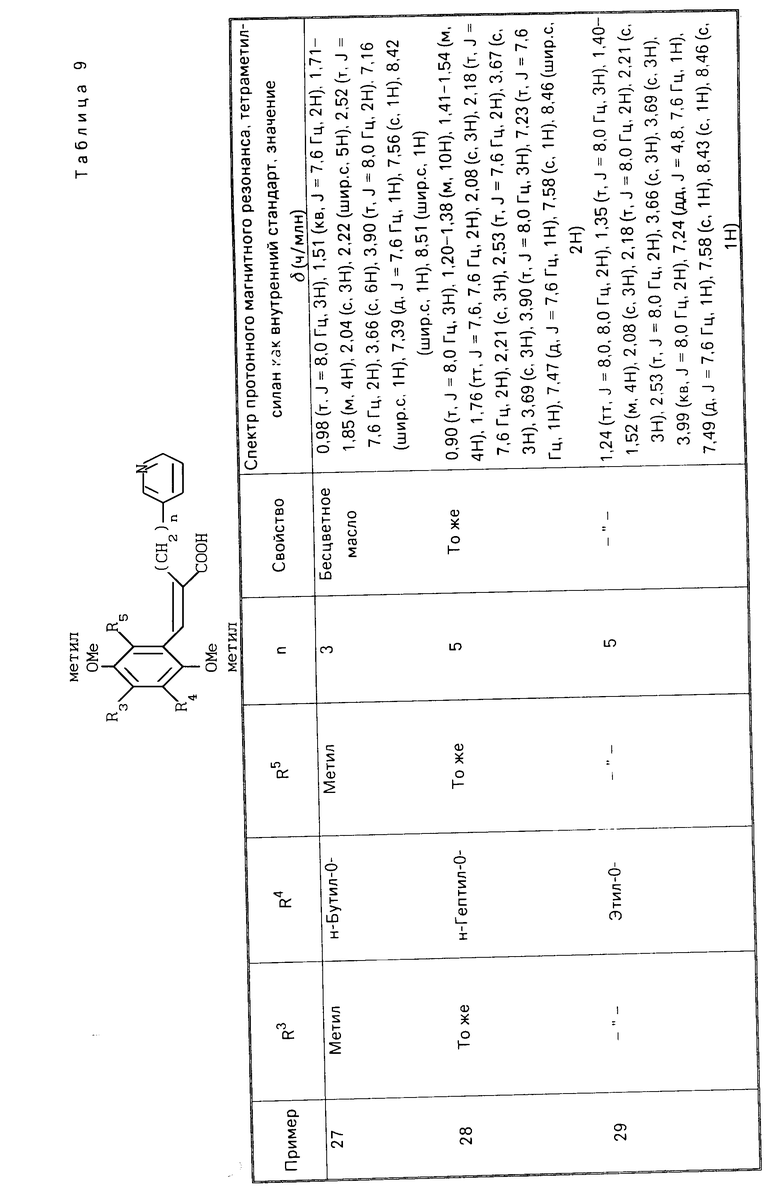
A__CH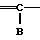 R1 в которой А является группой, представленной формулой
R1 в которой А является группой, представленной формулой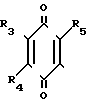 (в котоpой R3, R4 и R5 являются одинаковыми или отличаются друг от друга, и каждый представляет собой атом водорода, гидроксильную группу, низшую алкильную группу, низшую алкокси-группу, алкоксиалкильную группу, алкоксиалкокси-группу, циклоалкилалкокси-группу, тиоловую группу или тиоалкильную группу, при условии, что исключается случай, когда и R3 и R4 каждый является низшей алкокси-группой одновременно) или группой, представленной формулой
(в котоpой R3, R4 и R5 являются одинаковыми или отличаются друг от друга, и каждый представляет собой атом водорода, гидроксильную группу, низшую алкильную группу, низшую алкокси-группу, алкоксиалкильную группу, алкоксиалкокси-группу, циклоалкилалкокси-группу, тиоловую группу или тиоалкильную группу, при условии, что исключается случай, когда и R3 и R4 каждый является низшей алкокси-группой одновременно) или группой, представленной формулой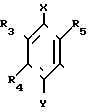 (в которой R3, R4 и R5 являются одинаковыми или отличаются друг от друга, и каждый представляет собой атом водорода, гидроксильную группу, низшую алкильную группу, низшую алкокси-группу, алкоксиалкильную группу, алкоксиалкокси-группу, циклоалкилалкокси-группу, тиоловую группу или тиоалкильную группу, при условии, что исключается случай, когда и R3 и R4 каждый является низшей алкокси-группой одновременно, Х и Y являются одинаковыми или отличаются друг от друга, и каждый представляет собой гидроксильную группу, или защищенную гидроксильную группу); R1 является гетероарилалкильной группой; и В представляет собой карбоксильную группу или защищенную карбоксильную группу.
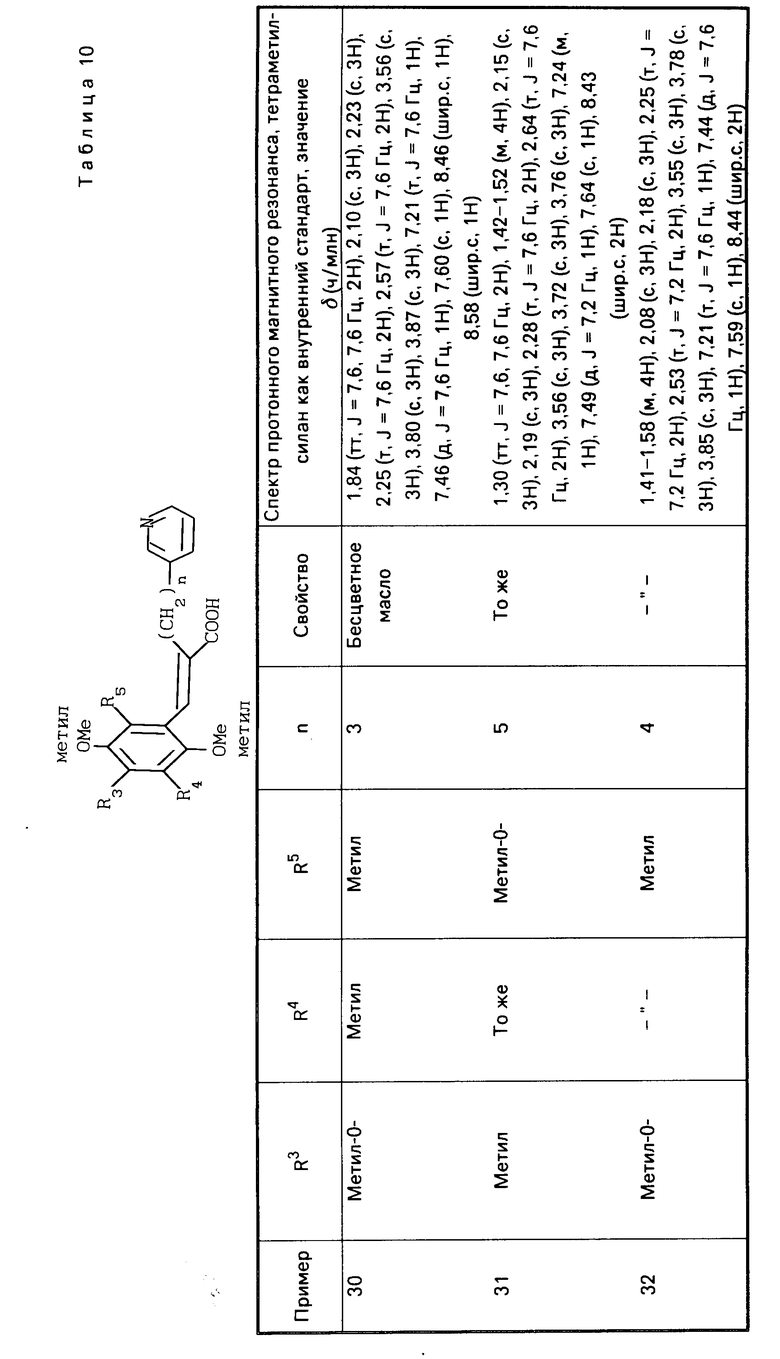
(в которой R3, R4 и R5 являются одинаковыми или отличаются друг от друга, и каждый представляет собой атом водорода, гидроксильную группу, низшую алкильную группу, низшую алкокси-группу, алкоксиалкильную группу, алкоксиалкокси-группу, циклоалкилалкокси-группу, тиоловую группу или тиоалкильную группу, при условии, что исключается случай, когда и R3 и R4 каждый является низшей алкокси-группой одновременно, Х и Y являются одинаковыми или отличаются друг от друга, и каждый представляет собой гидроксильную группу, или защищенную гидроксильную группу); R1 является гетероарилалкильной группой; и В представляет собой карбоксильную группу или защищенную карбоксильную группу.
В приведенном определении вещества (I), согласно изобретению, низшая алкильная группа, определенная в отношении радикалов R3, R4 и R5, является неразветвленной или разветвленной алкильной группой, имеющей 1-8 атомов углерода. Примеры такой группы включают: метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил (амил), изопентил, неопентил, трет-пентил, 1-метил-бутил, 2-метилбутил, 3-метилбутил, 1,2-диметилпропил, н-гексил, изогексил, 1-метилпентил, 2-метилпентил, 3-метилпентил, 1,1-диметилбутил, 1,2-диметилбутил, 2,2-диметилбутил, 1,3-диметилбутил, 2,3-диметилбутил, 3,3-диметилбутил, 1-этилбутил, 2-этилбутил, 1,1,2-триметилпропил, 1,2,2-триметилпропил, 1-этил-1-метилпропил, 1-этил-2-метилпропил, гептил и октил. Среди этих групп желательными являются метильная, этильная, н-пропильная и изопропильная группы.
Низшая алкокси-группа, определенная в отношении заместителей R3, R4и R5, представляет собой одну из упомянутых выше низших алкокси-групп, причем их примеры включают метокси-, этокси и н-пропокси-группы, среди которых наиболее желательной является метокси-группа.
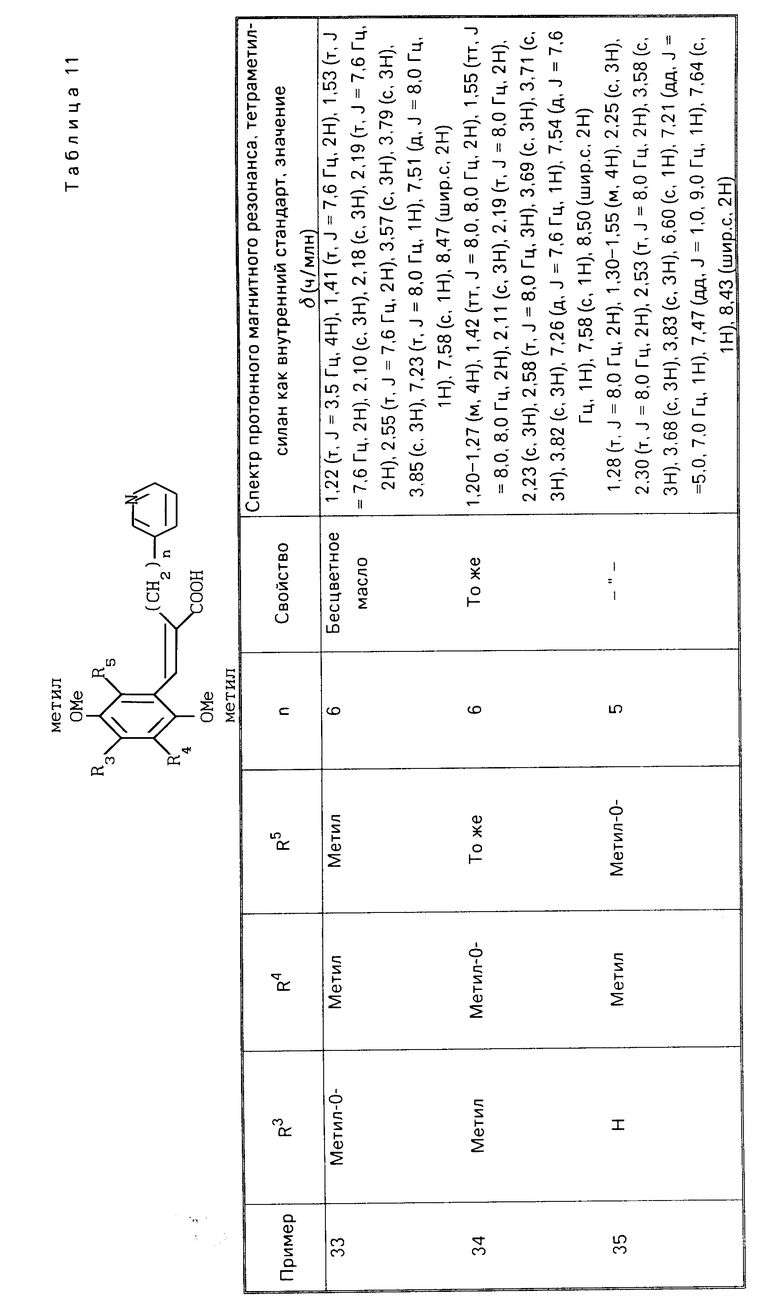
Алкоксильная группа, составляющая алкоксиалкильную группу, определенную в отношении заместителей R3, R4 и R5, имеет 1-8 атомов углерода, предпочтительно 1 или 2 атома, в то время как алкильная группа, например, алкиленовая цепочка, составляющая ее, имеет 1-10 атомов углерода.
Алкоксиалкоксильная группа, определенная в связи с заместителями R3, R4 и R5, представляет собой одну, произведенную из вышеупомянутых низших алкокси-групп, и их примеры включают метоксиметокси- метоксиэтокси-, этоксиэтокси- и метоксипропокси-группы.
Циклоалкильная группа, составляющая циклоалкилалкокси-группу, определенную в связи с заместителями R3, R4 и R5, имеет 3-7 атомов, предпочтительно 5 или 6 атомов углерода, в то время как входящая в ее состав алкокси-группа является такой же, как определено выше в связи с низшей алкоксильной группой.
Алкильная группа, входящая в состав тиоалкильной группы, определенной в связи с заместителями, R3, R4 и R5 имеет 1-8 атомов углерода, предпочтительно 1 или 2 атома углерода.
В настоящем изобретении оба заместителя R3 и R4 не могут быть одновременно низшей алкоксильной группой, такой как метокси-группа.
Наиболее желательным сочетанием заместителей R3, R4 и R5 является случай, в котором один из заместителей представляет собой низшую алкокси-группу, такую как метокси-группа, а другие (каждый из них) являются низшей алкильной группой, такой как метильная и этильная группы, которые могут быть одинаковыми или различными друг от друга.
Гетероалкильная группа, входящая в состав гетероарилалкильной группы, определенной в связи с заместителем R1, предпочтительно является пяти- или шестичленным азотсодержащим гетероарильным циклом, причем конкретные приметы такой группы включают пиридильную группу и имидазолильную группу, которые могут быть либо незамещенными, либо каждая может быть замещена низшей алкильной группой, такой как метильная и этильная, низшей алкокси-группой, такой как метокси-группа и этокси-группа, или атомом галогена, таким как атом хлора и брома.
Алкильная группа, входящая в состав гетероарилалкильной группы, т.е. алкиленовая цепочка, имеет 1-10 атомов углерода, предпочтительно 2-8 атомов углерода, более предпочтительно 4-6 атомов углерода. Кроме того, алкиленовая цепочка может иметь замещенную низшую алкильную группу, такую как метильная и этильная группа, при любом атоме углерода.
Защищенная гидроксильная группа, определенная в связи с заместителями Х и Y, может быть, например, гидроксильной группой, защищенной вышеупомянутой низшей алкильной группой, такой как метильная и этильная группы, т.е. алкоксигруппой, или гидроксильной группой, защищенной ацильной группой, такой как ацетильная, пропионильная, бутироильная, пивалоильная и никотиноильная группы, т. е. группы, имеющие сложноэфирную связь. Этой группой может быть любая, которая может быть отщеплена некоторым образом в живом организме, чтобы регенерировалась гидроксильная группа.
Защищающая группа, входящая в состав защищенной карбоксильной группы, определенной в связи с группой В, включает низшие алкильные группы, такие как метильная, этильная и трет-бутильная группы, низшие алкильные группы, замещенные фенильной группой, которая может быть замещенной, такие как пара-метоксибензильная группа, пара-нитробензильная группа, 3,4-диметоксибензильная, дифенилметильная, тритильная (трифенилметильная) и фенетильная группы, галогенированные низшие алкильные группы, такие как 2,2,2-трихлорэтильная и 2-иодэтильная группы, низшие алканоилокси (низший алкил)-группы, такие как пивалоилоксиметильная группа, ацетоксиметильная, пропионилоксиметильная, бутирилоксиметильная, валерилоксиме- тильная, 1-ацетоксиэтильная, 2-ацетоксиэтильная, 1-пивалоилоксиэтильная и 2-пивалоилоксиэтильная группы, высшие алканоилокси (низший алкил) группы, такие как пальмитоилоксиэтильная группа, гептадеканоилоксиметильная и 1-пальмитоилоксиэтильная группы; низшие алк- оксикарбонилокси (низший алкил) группы, такие как метоксикарбонилоксиметильная группа, 1-бутоксикарбонилоксиэтильная группа, 1-трет-бутоксикарбонилоксиэтильная, 1-этоксикарбонилоксиэтильная и 1-(изопропоксикарбонилокси) этильная группы; карбокси (низший алкил)-группы, такие как карбоксиметильная и 2-карбоксиэтильная группы; гетероциклические группы, такие как 3-фталидильная группа, бензоилокси(низший алкил)-группы, которые могут быть замещены, такие как 4-глицилоксибензоилоксиметильная группа и 4-(N-(трет-бутоксикарбонил)-глицилокси)- бензоилоксиметильная группа; (замещенный диоксолен) низший алкилгруппы, такие как (5-метил-2-оксо-1,3-диоксолен-4-ил) метильная группа, циклоалкил-замещенные (низший алканоилокси) (низший алкил)-группы, такие как 1-циклогексил-ацетилоксиэтильная группа; и циклоалкил- оксикарбонилокси (низший алкил)-группы, такие как 1-циклогексилоксикарбонилоксиэтильная группа.
Кроме того, защищенная карбоксильная группа может быть амидом кислоты.
Защищенная карбоксильная группа может быть любой такой, из которой может быть отщеплена защищающая группа некоторым образом в живом организме, для того, чтобы регенерировать карбоксильную группу.
Фармакологически приемлемая соль, в соответствии с настоящим изобретением, включает соли неорганических кислот, такие как гидрохлорид, гидробромид, сульфат и фосфат; соли органических кислот, такие как ацетат, малеат, тартрат, метансульфонат, бензолсульфонат и толуолсульфонат; и соли аминокислот, такие как аргининат, аспаргат и глутамат.
Кроме того, производная хинона настоящего изобретения может образовывать металлические соли, такие как соли натрия, калия, кальция и магния. Фармакологически приемлемая соль настоящего изобретения включает эти соли металлов.
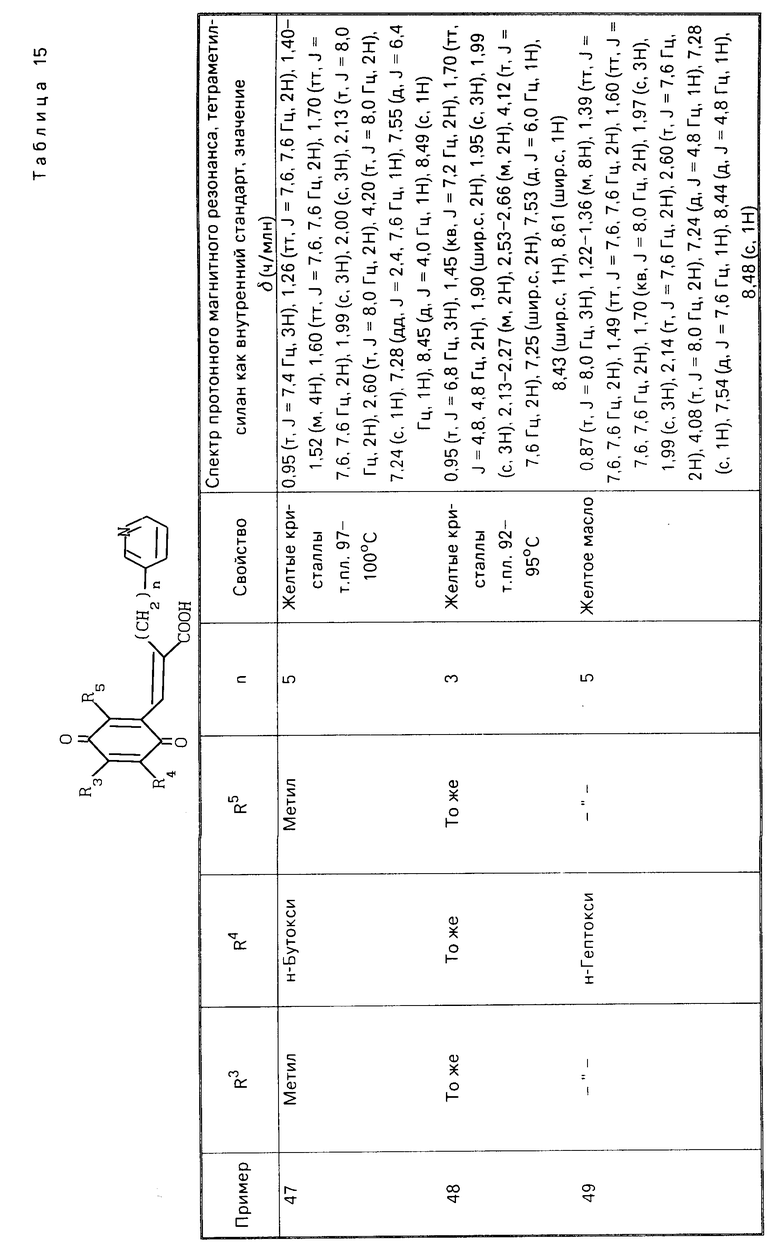
Хотя вещество настоящего изобретения может быть представлено в виде геометрических размеров (включающих цис- и транс-изомеры) вследствие наличия двойной связи в его молекуле, нет необходимости упоминать, что настоящее изобретение включает все эти изомеры.
Теперь будут описаны характерные способы получения веществ в соответствии с настоящим изобретением.
Способ приготовления I.
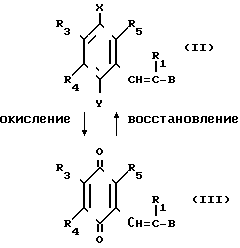 (в приведенных формулах значения Х, Y, R1, R4, R3, R5 и В определено).
(в приведенных формулах значения Х, Y, R1, R4, R3, R5 и В определено).
В приведенной выше схеме реакции вещества (II) и (III) являются веществами настоящего изобретения. Как видно из схемы реакции, производная бензохинона, представленная формулой (III), может быть получена путем взаимодействия производной гидрохинона, представленной формулой (II) c окисляющим агентом, в то время как производная гидрохинона (II) может быть получена посредством восстановления производной бензохинона (III).
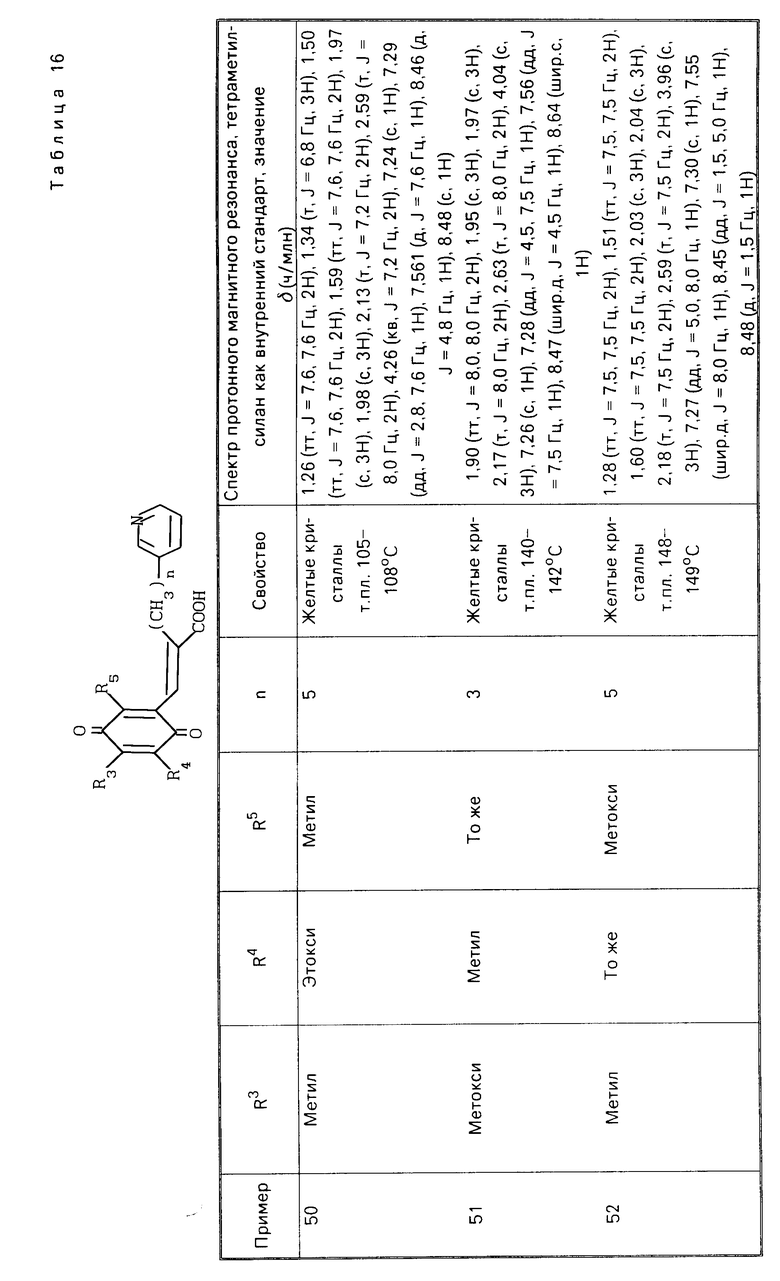
При окислении производной гидрохинона, представленной формулой (II), в качестве окисляющего агента применяется, например, церий (IV) аммоний-нитрат, гексагидрат хлорного железа или оксид свинца. Количество используемого окисляющего агента предпочтительно составляет от 2 до 10 молей на моль гидрохинона. Предпочтительный растворитель, который используется при окислении, включает ацетонитрил, бензол, этилацетат, диоксан, этанол, 1,2-диметоксиэтан и их смеси с водой. Окисление проводится при 0-90оС, предпочтительно 0-20оС. Время реакции обычно составляет примерно 1-12 ч.
Напротив, при восстановлении производной хинона в производную гидрохинона, которая является одним из целевых веществ, в качестве восстанавливающего агента предпочтительно используется боргидрид натрия или гидросульфит натрия. Предпочтительный растворитель, который применяется при восстановлении, включает этанол, тетрагидрофуран, этилацетат, 1,2-диметоксиэтан и их смеси с водой. Температура реакции предпочтительно составляет 0-40оС, более предпочтительно 0-20оС.
Способ приготовления 2. Производная гидрохинона (II), которая является одним из целевых веществ в соответствии с настоящим изобретением, также может быть получена следующим способом: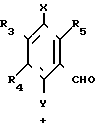 (IV) (IV)
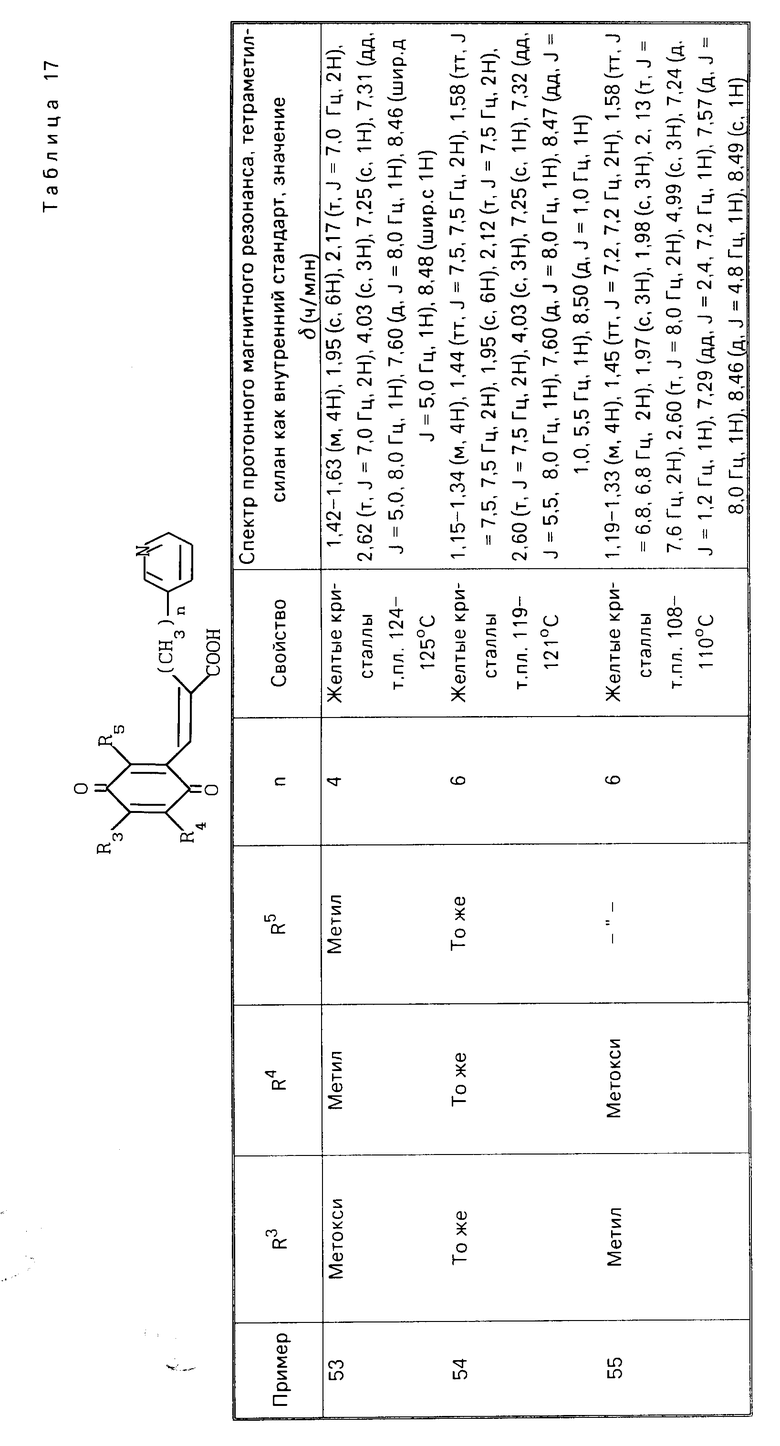
(IV) (IV)
(C2H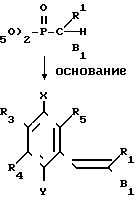 (
( (в приведенной выше схеме реакции, R1, R3, R4, R5, X и Y такие как определено выше, и В1 является группой, выбранной среди тех, которые определены в отношении В, за исключением карбоксильной группы).
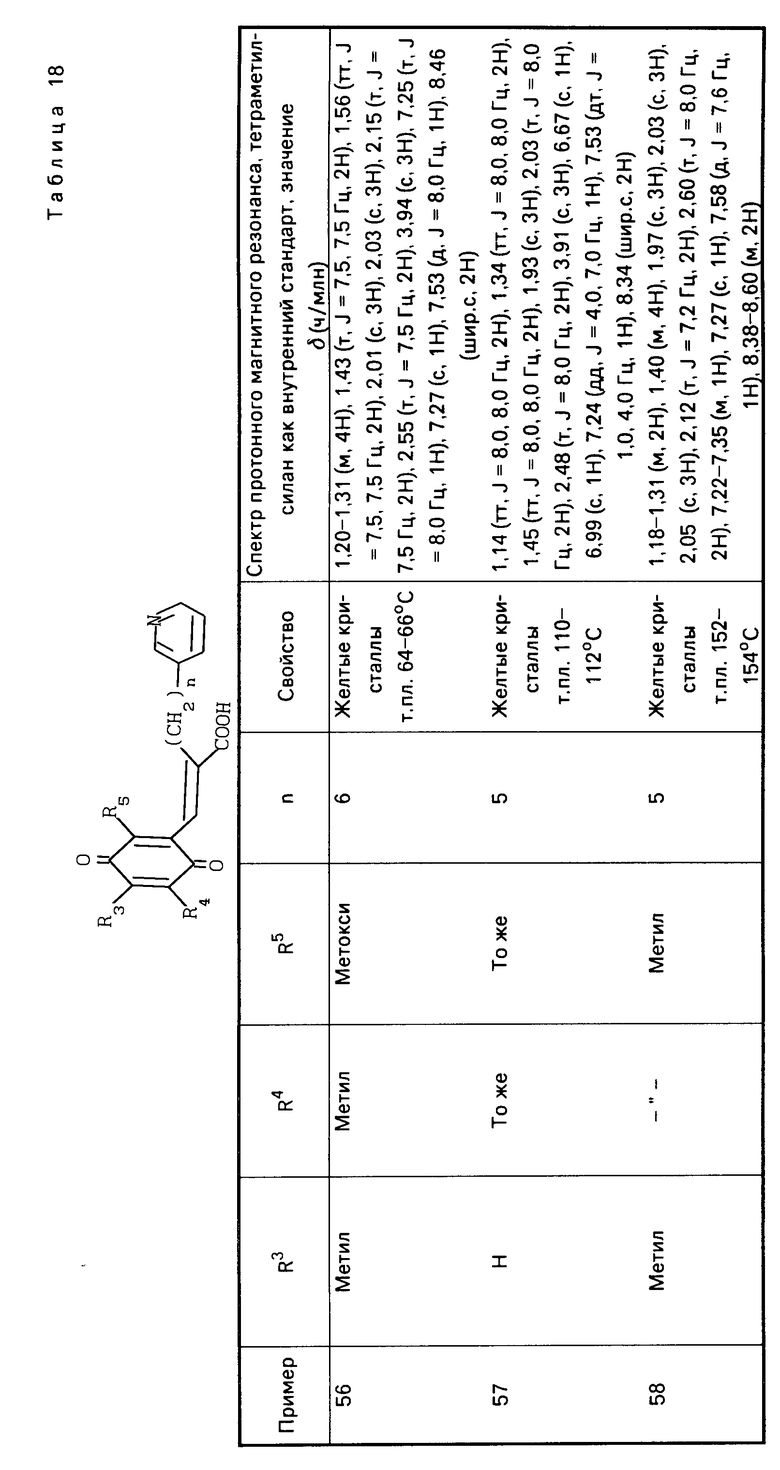
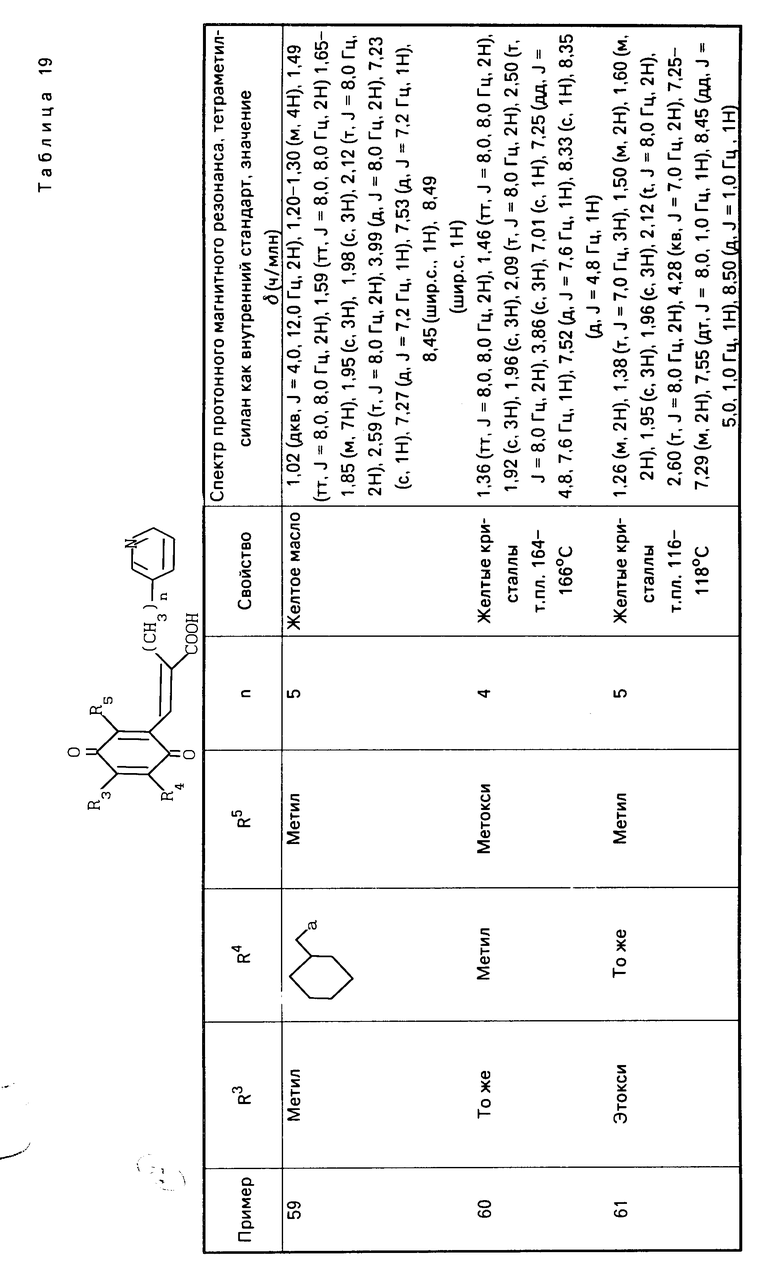
(в приведенной выше схеме реакции, R1, R3, R4, R5, X и Y такие как определено выше, и В1 является группой, выбранной среди тех, которые определены в отношении В, за исключением карбоксильной группы).
Более точно, целевое вещество, представленное общей формулой (II'), может быть приготовлено путем взаимодействия альдегидной производной, представленной общей формулой (IV) с фосфонатом, представленным общей формулой (V), в присутствии основания по реакции Виттига (см. например, журнал I.A.C.S, 1961, том 83, с. 1733).
Основание, которое используется в этой реакции, включает гидриды щелочных металлов, такие как гидрид натрия и гидрид калия, и алкоголяты щелочных металлов, такие как метилат натрия, этилат натрия и трет-бутоксид калия. Предпочтительные примеры растворителя, подлежащего использованию в этой реакции, включают бензол, толуол, дихлорметан, тетрагидрофуран, диоксан, диметоксиэтан и диметилформамид. Температура реакции составляет 0-100оС, предпочтительно 20-80оС.
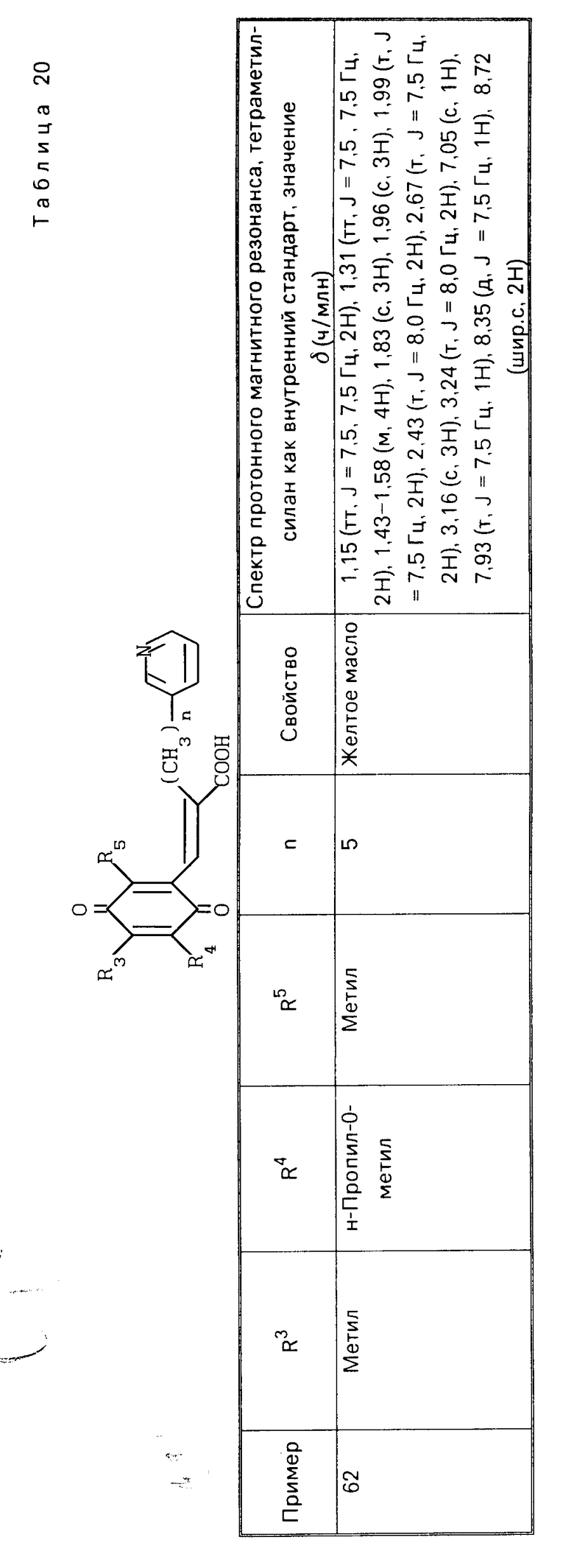
Способ приготовления 3. Производная гидрохинона, представленная общей формулой (I), в которой В является карбоксильной группой, может быть получена с помощью следующего процесса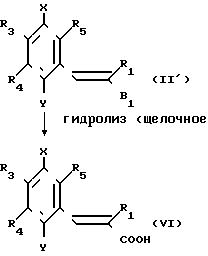
 (в приведенной выше схеме реакции R1, R3, R4, R5, X и Y и В1 такие, как определено выше).
(в приведенной выше схеме реакции R1, R3, R4, R5, X и Y и В1 такие, как определено выше).
Более конкретно, вещество, представленное общей формулой (VI), которое представляет собой одно из целевых веществ, может быть получено посредством омыления соединения, представленного общей формулой (II'), щелочью в соответствии с традиционным процессом.
Это омыление проводится с использованием традиционной щелочи, такой как щелочная каустическая сода или поташ.
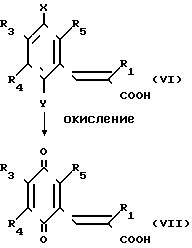
Вещество (VI), полученное таким способом, может быть легко окислено в соединение, представленное общей формулой (VII), которое является одним из целевых веществ в соответствии с настоящим изобретением, таким же образом, как описано в препаративном способе I.
 (в приведенной выше схеме реакции R1, R3, R4, R5, Х и Y такие, как определено выше).
(в приведенной выше схеме реакции R1, R3, R4, R5, Х и Y такие, как определено выше).
С п о с о б п р и г о т о в л е н и я 4. Вещество, представленное общей формулой (I), в которой В является защищенной карбоксильной группой, может быть получено с помощью следующего способа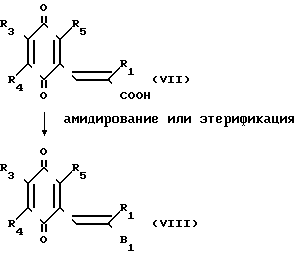 (в приведенной выше схеме реакции R1, R3, R4, R5 и В такие, как определено выше).
(в приведенной выше схеме реакции R1, R3, R4, R5 и В такие, как определено выше).
Когда целевое вещество (II') или (VIII) является сложным эфиром, оно может быть приготовлено путем этерификации карбоновой кислоты, представленной общей формулой (VII), с помощью традиционного процесса.
Растворитель, который подлежит использованию в этой реакции омыления, может быть любым, который является инертным по отношению к этерификации. Температура реакции конкретно не ограничивается, но изменяется в зависимости от характера реакционно-способной производной.
Когда целевое вещество (II') или (VIII) является амидом, оно может быть приготовлено, путем превращения карбоксильной кислоты, представленной общей формулой (VII), или ее реакционноспособной производной, в соответствующий амид с помощью традиционного процесса.
Реакционноспособная производная вещества (VII) включает галогениды кислот, такие как хлорангидрид и бромангидрид кислоты, азид кислоты, активные сложные эфиры этих кислот с N-гидроксибензотриазолом или N-гидроксисукцинимидом, симметричные ангидриды, и смешанные ангидриды кислот с алкилкарбоновыми кислотами или пара-толуолсульфокислотой.
Когда вещество (VII) является свободной карбоксильной кислотой, является предпочтительным проведение амидирования вещества (VII) в присутствии конденсирующего агента, такого как дициклогексилкарбодимид и 1,1'-карбонилдиимидазол.
Реакция амидирования проводится в органическом растворителе, который инертен в отношении амидирования, например, в пиридине, тетрагидрофуране, диоксане, диэтиловом эфире, бензоле, толуоле, ксилоле, хлористом метилене, дихлорэтане, хлороформе, диметилформамиде, этилацетате или ацетонитриле.
Температура реакции конкретно не ограничивается, но изменяется в зависимости от характера реакционноспособной производной.
Способы приготовления веществ в соответствии с настоящим изобретением могут быть проиллюстрированы следующей схемой реакции: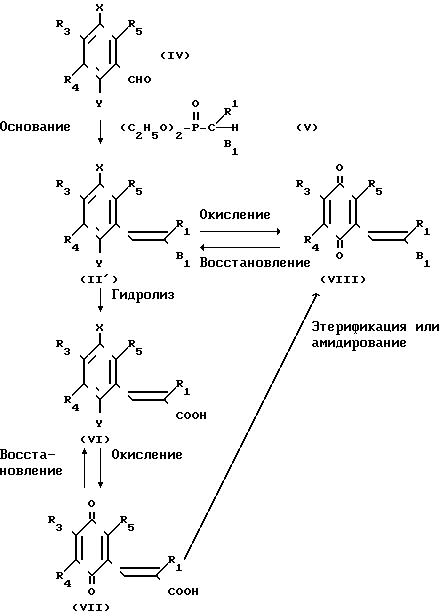
Теперь будут описаны экспериментальные примеры для того, чтобы проиллюстрировать действие вещества в соответствии с изобретением.
Экспериментальный пример. Ингибирующая активность против образования лейкотриена В4 (LTB4) и тромбоксана В2 (ТхВ2) из брюшной инфильтрационной клетки крысы.
Методика эксперимента.
10 мл 6% -ного (мас./об.) раствора гликогена (тип II из Ойстера, фирма Сигма) в физиологическом рассоле вводят инъекцией в брюшную полость самок крыс Фишера, имеющих массу 150-200 г. Через 20 ч удаляют из нее брюшные эксудативные клетки, промывают их и суспендируют в сбалансированном солевом растворе Хэнкса (НВSS) в концентрации 5 млн/мл. Эту суспензию выливают на культуральную пластину с 96 ячейками (Костар, зарегистрированный торговый знак), в которой расположен испытуемый медикамент, разбавленный до заданной концентрации, в количестве 10 мкл/ячейка, в количестве 100 мкл/ячейка. Полученную пластинку культивируют при 37оС в течение 5 мин. Добавляют ионофор кальция А-23187 (Кальбиохем, зарегистрированный торговый знак) в окончательной концентрации 2 мкг/мл. После взаимодействия при 37оС еще в течение 10 мин пластинку помещают на лед и добавляют раствор BW 755 С в окончательной концентрации 100 мкмоль/л. Полученную пластинку подвергают центрифугированию со скоростью 15000 об/мин в течение 10 мин. Выделяют жидкость над осадком и определяют количества лейкотриена и тромбоксана в ней с помощью ферментного иммуноанализа с забором ЕIA, производство ф. Кайман.
Результаты следующие.
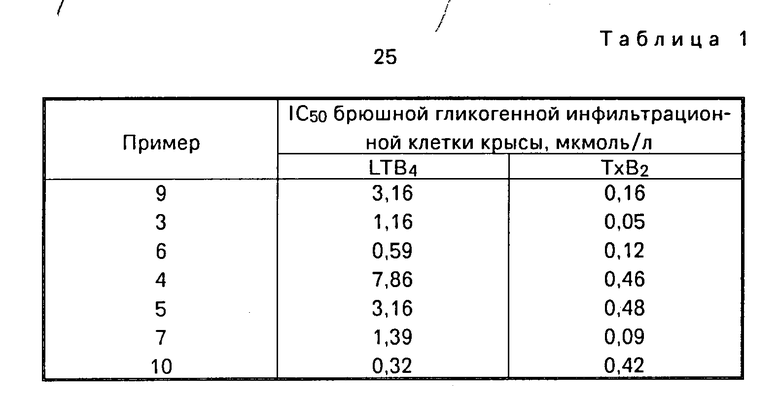
Ингибирующие активности (IC50) каждого вещества (показаны номером примера, которые будут описаны ниже) против образования лейкотриена В и тромбоксана В приведены в табл. 1.
Из результатов приведенных выше экспериментальных Примеров можно понять, что вещества настоящего изобретения обладают ингибирующей активностью против образования лейкотриенов, а также тромбоксана.
Соответственно, производные хинона согласно настоящему изобретению являются эффективными для заболеваний, в которых действенной является ингибирующая активность образования лейкотриена и/или образования тромбоксана А2. Примеры таких заболеваний включают астму, различные болезни печени (такие как хронический гепатит, острый гепатит, медикаментозный гепатит, вирусный гепатит, алкогольный гепатит, желтуха и цирроз), ишемические болезни сердца (такие как инфаркт миокарда и грудная жаба), церебральные ишемические заболевания (такие как церебральный эмболизм и церебральный тромбоз) и различные болезни почек (такие как почечная недостаточность, нефроз и нефрит).
Кроме того, вещества настоящего изобретения являются весьма безопасными и поэтому являются ценными в этом отношении.
При использовании веществ настоящего изобретения в качестве ингибиторов против образования лейкотриена и/или тромбогексана, для того, чтобы излечить или предотвратить различные заболевания, они могут назначаться перорально в виде порошка, гранулы, капсулы или сиропа, или парентерально в виде свечей, инъекций, наружных препаратов или капель. Хотя дозировка этих веществ изменяется значительно в зависимости от симптомов, возраста пациента и рода заболевания, обычно она составляет 0,1-2000 мг, предпочтительно примерно 2-500 мг, более предпочтительно примерно 5-150 мг/сутки для взрослого пациента, причем доза может назначаться от 1 до нескольких порций в сутки.
Вещества настоящего изобретения могут быть превращены в фармацевтические препараты посредством использования традиционных усиливающих лекарства носителей в соответствии с общепринятыми способами.
Твердый препарат для перорального назначения в соответствии с настоящим изобретением готовится посредством добавления носителя и, в случае необходимости, связующего, измельчителя, смазывающего вещества, красителя и/или корригента к активному компоненту, и формования полученной смеси в таблетки, покрытые таблетки, гранулы, порошок или капсулы.
Примеры носителей включают лактозу, кукурузный крахмал, сахарозу, глюкозу, сорбитол, кристаллическую целлюлозу и диоксид кремния. Примеры связующих включают поливиниловый спирт, поливиниловый эфир, этилцеллюлозу, метилцеллюлозу, арабскую камедь, трагакант, желатин, шеллак, гидроксипропилцеллюлозу, гидроксипропилметилцеллюлозу, цитрат кальция, декстрин и пектин. Примеры смазывающих веществ включают стеарат магния, тальк, полиэтиленгликоль, диоксид кремния и отвержденное растительное масло. Примеры красителя включают те, которые одобрены как фармацевтические добавки и примеры корригентов включают порошок какао, траву мяты (ментол), ароматический порошок, мятное масло (масло перечной мяты), борнеол и измельченную кору коричного дерева. Конечно, таблетки и гранулы могут быть соответственно покрыты сахаром, желатином и т.п. если это необходимо.
Инъекции согласно настоящему изобретению готовятся посредством добавления модификатора рН, буфера, стабилизатора и/или солюбилизирующего агента к активному компоненту при необходимости, и превращения смеси в инъекции для подкожного, внутримышечного или внутривенного назначения.
П р и м е р. Примеры изобретения описаны ниже, хотя не обходимо сказать, что изобретение не ограничивается ими.
Приготовление исходных соединений для получения веществ настоящего изобретения описаны в следующих препаративных примерах.
Символы в химических формулах, которые будут даны ниже, имеют следующие соответствующие значения: Ме-метильная группа, Et-этильная группа, Н-Вu н-бутильная группа, н-Нер-н-гептильная группа, МОМО-метоксиметокси-группа.
П р е п а р а т и в н ы й п р и м е р 1. Этиловый эфир 2-диэтилфосфоно-7-(3-пиридил)гептановой кислоты
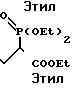
Растворяют в 2 л хлористого метилена 221 г 5-(3-пиридил)пентанола и затем добавляют 142 г триэтиламина. В полученную смесь прикапывают 161 г хлористого мезила при охлаждении льдом. По завершении прикапывания полученную смесь перемешивают 1 ч при охлаждении льдом. Органический слой промывают дважды водой, сушат над сульфатом магния и перегоняют в вакууме, чтобы удалить растворитель. В остатке получают светло-красное масло.
Отдельно суспендируют 59 г гидрида натрия (55%-ная суспензия в масле) в 500 мл диметилформамида и затем по каплям добавляют 300 г триэтилового эфира фосфоноуксусной кислоты. Полученную смесь перемешивают 1 ч при 50-60оС, затем добавляют раствор указанного выше остатка (светло-красное масло) в 500 мл диметилформамида. Полученную смесь перемешивают 18 ч при 50-60оС. После завершения реакции к реакционной смеси добавляют 3 л этилацетата, и смесь промывают дважды насыщенным водным раствором хлорида натрия. Органический слой сушат над безводным сульфатом магния и перегоняют в вакууме, чтобы удалить растворитель. Остаток очищают на хроматографической колонке с силикагелем (элюенты н-гексан/этилацетат (30-50%), этилацетат/метанол (5%)), получая 226 г указанного в заголовке соединения в виде светло-красного масла.
П р и м е р 1. Этиловый эфир (Е)-3-(2,4,5-триметокси-3,6-диметилфенил)-2-( 5-(3-пиридил)пентил)-2-пропеновой кислоты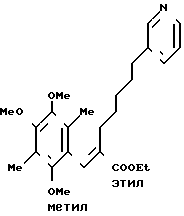 Суспендируют 24 г (0,6 моля) 60%-ного гидрида натрия в 200 мл диметилформамида, и полученную суспензию перемешивают при комнатной температуре.
Суспендируют 24 г (0,6 моля) 60%-ного гидрида натрия в 200 мл диметилформамида, и полученную суспензию перемешивают при комнатной температуре.
В указанную выше суспензию постепенно прикапывают раствор 222 г (0,6 моля) этилового эфира 2-диэтилфосфоно-7-пиридил)-гептановой кислоты (реактив Виттига), приготовленного в препаративном примере 1, в 300 мл диметилформамида. После завершения прикапывания полученную смесь перемешивают 1 ч при комнатной температуре, чтобы получить прозрачный раствор. К нему добавляют по каплям раствор 122 г (0,54 моль) 2,4,5-триметокси-3,6-диметилбензальдегида в 200 мл диметилформамида. Полученную смесь перемешивают в течение ночи при нагревании до 50оС.
Реакционную смесь выливают на 1 л воды с льдом. Полученную смесь экстрагируют 1 литром этилацетата (2 раза). Органический слой сушат над сульфатом магния и перегоняют, чтобы удалить растворитель. Остаток очищают на хроматографической колонке с силикагелем (элюент гексан/этил ацетат (10-30% н-гексан).
Получают 177 г указанного в заголовке вещества в виде светло-желтого масла.
ПМР-спектр (400 МГц, в дейтерохлороформе), δ (ч/млн): 1,21 (тт. I 7,5, 7,5 Гц, 2Н), 1,35 (т. I 7,1 Гц, 3Н), 1,38 (тт. I 7,5, 7,5 Гц, 2Н) 1,46 (тт. I 7,5 и 7,5 Гц, 2Н), 2,07 (с. 3Н), 2,16 (Т. I 7,5 Гц, 2Н), 2,16 (С, 3Н), 2,48 (т. I 7,5 Гц, 2 Н), 3,54 (с. 3Н), 3,76 (с. 3Н), 3,82 (с. 3Н), 4,13 (кв, I 7,1 Гц, 2Н), 7,16 (дд. I 5,5 7,8 Гц) 7,39 (дт. I 1,5 и 5,5 Гц, 1Н). 7,45 (с. 1Н) 8,36 (д. I 1,5 Гц, 1Н), 8,39 (дд. I 1,5 и 5,5 Гц, 1Н).
П р и м е р 2. (Е)-3-(2,4,5-триметокси-3,6-диметилфенил) -2-[5-(3-пиридил)пентил]-2-пропеновая кислота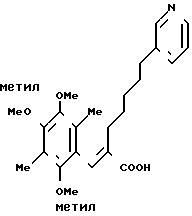
177 г (0,39 моль) сложного эфира, приготовленного в примере 1, растворяют в 500 мл этанола и затем добавляют 100 мл водного раствора 78 г гидроксида натрия. Образовавшуюся смесь нейтрализуют 6-нормальной соляной кислотой. Полученную смесь дважды экстрагируют 1 л этилацетата. Органический слой промывают водным раствором хлорида натрия, сушат над безводным сульфатом магния и перегоняют, чтобы удалить растворитель. Получают 159 г указанного в заголовке вещества в виде безцветного масла. ПМР-спектр (400 МГц, в дейтерохлороформе), δ (ч/мин) 1,24 (тт. I 7,6, 7,6 Гц, 2Н), 1,47 (тт. I 7,6, 7,6 Гц, 2Н), 1,52 (тт. I 7,6, 7,6 Гц, 2Н), 2,09 (с. 3Н), 2,17 (с. 3Н) 2,20 (т. I 7,6 Гц, 2Н), 2,54 (т. I 7,6 Гц, 2Н) 3,57 (с. 3Н) 3,78 (с. 3Н), 7,23 (дд, I 5,0 Гц 7,5 Гц, 1Н) 7,48 (шир. д, I 7,6 Гц, 1Н) 7,59 (с. 1Н) 8,46 (шир. с 2Н).
П р и м е р 3. (1) (Е)-3-(2-метокси-3,6-диметил-1,4-бензохинон-5 -ил)-2-[5-(3-пиридил)пентил]-2-пропеновая кислота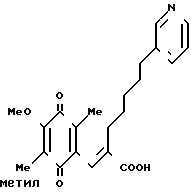
159 г (0,39 моль) карбоновой кислоты, приготовленной в Примере 2, растворяют в смесь 800 мл ацетонитрила и 400 мл воды. Полученный раствор охлаждают в ледяной бане, и затем по каплям постепенно добавляют 700 мл водного раствора 527 г (0,96 моль) церий (IV)-аммоний нитрата. Образовавшуюся смесь перемешивают 30 мин и устанавливают значение рН смеси при 5 насыщенным раствором гидрокарбоната натрия, затем добавляют 3 л воды. Полученную смесь дважды экстрагируют 6 л этилацетата. Органический слой промывают насыщенным водным раствором хлорида натрия, сушат над безводным сульфатом магния и перегоняют, чтобы удалить растворитель. Полученное масло подвергают кристаллизации из небольшого количества этилацетата, образуются 114 г желтых кристаллов. Эти кристаллы подвергают перекристаллизации из смеси этанол/вода, получая 90 г указанного в заголовке вещества. Т.пл. 134-135оС. ПМР-спектр (400 МГц, в дейтерохлороформе), δ (ч/млн) 1,26 (тт. I 7,0, 7,0 Гц, 2Н), 1,50 (тт. I 7,0, 7,0 Гц 2Н), 1,95 (с, 3Н), 1,96 (с. 3Н)= 2,12 (т. I 7,0 Гц, 2Н) 2,60 (т. I 7,0 Гц, 2Н), 4,01 (с. 3Н), 7,26), с. 1Н), 7,27 (дд, I 5,0, 8,5 Гц, 1Н), 7,55 (шир. д, I 8,5 Гц, 1Н) 8,44 (шир. д, I 5,0 Гц, 1Н), 8,50 (шир. с. 1Н).
(2) (Е)-3-(2-Метокси-3,6-диметил-1,4-бензохинон-5-ил)-2 -[5-(3-пиридил)пентил]-2-пропеновая кислота, гидрохлорид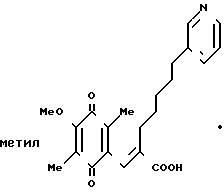 HCl
HCl
Гидрохлорид описанного выше вещества (I) был получен по традиционной методике. Т. пл. 138-139оС. ПМР-спектр (400 МГц, в пердейтеродиметилформамиде), δ (ч/млн) 1,18 (тт. I 7,2, 7,2 Гц, 2Н), 1,37 (тт. I 7,2, 7,2 Гц, 2Н) 1,54 (тт. I 7,2, 7,2 Гц, 2Н) 1,82 (с. 3Н) 1,84 (с. 2Н) 2,04 (т. I 7,2 Гц, 2Н), 2,71 (т. I 7,2 Гц, 2Н), 3,92 (с. 3Н), 7,04 (д, I 1,2 Гц, 1Н) 7,97 (дд, I 2,4, 8,0 Гц, 1Н), 8,41 (д, I 8,0 Гц, 1Н), 8,7644 (д, I 5,60 Гц, 1Н) 8,79 (с. 1Н).
П р и м е р 4. (Е)-3-(2,5-Дигидрокси-4-метокси-3,6-диметил-фенил) -2-[5-(3-пиридил)пентил]-2-пропеновая кислота
метил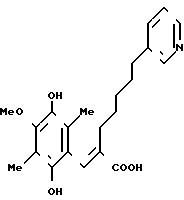
1,0 г хинона, приготовленного в примере 3, суспендируют в 150 мл этилацетата и образовавшуюся суспензию полностью смешивают с раствором 2 г гидросульфита натрия в 50 мл воды. Органический слой отделяют, сушат над безводным сульфатом магния и концентрируют в вакууме, чтобы получить 660 мг указанного в заголовке вещества в виде белого аморфного порошка. ПМР-спектр (400 МГц, в дейтерохлороформе), δ (ч/млн) 1,21 (тт. I 7,0, 7,0 Гц, 2Н), 1,44 (тт. I 7,0, 7,0 Гц, 2Н), 1,51 (тт. I 7,0, 7,0 Гц, 2Н), 2,06 (с. 3Н), 2,17 (с. 3Н), 2,23 (т. I 7,0 Гц, 2Н), 2,54 (т. I 7,0 Гц, 2Н), 3,76 (с. 3Н), 5,22 (шир. с. 3Н) 7,26 (дд, I 5,5; 7,0 Гц, 1Н), 7,43 (с. 1Н), 7,51 (дд, I 1,5, 7,0 Гц, 1Н), 8,4-8,47 (м. 2Н).
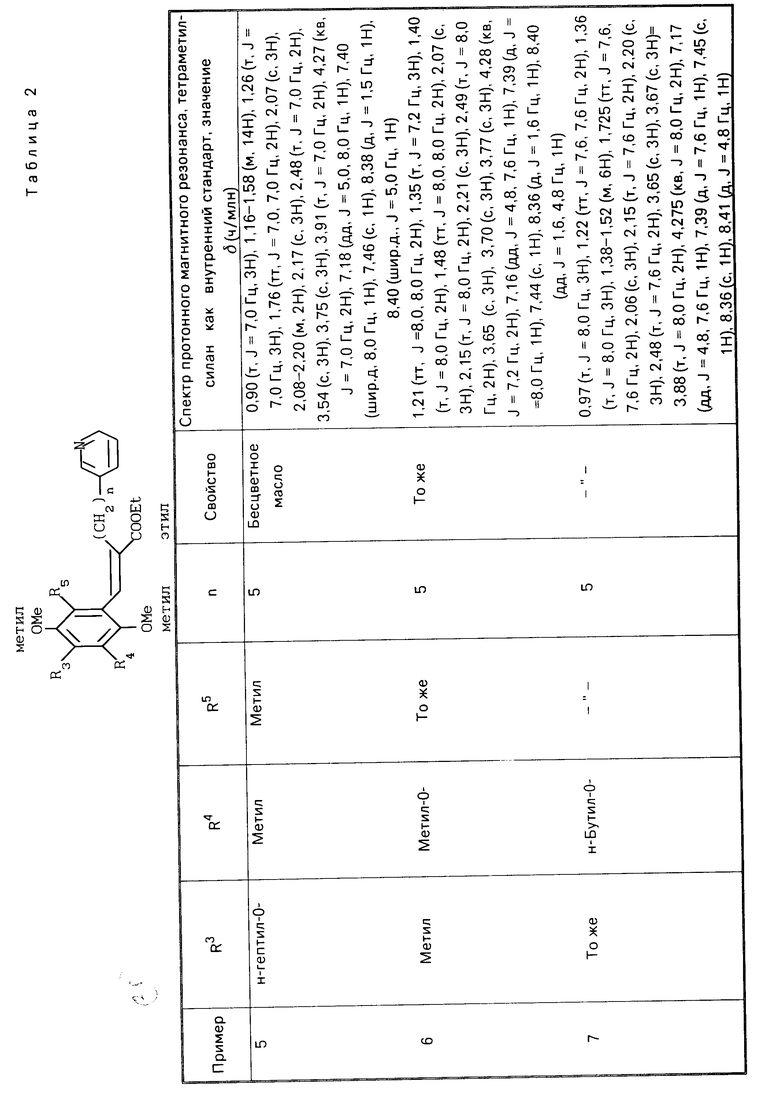
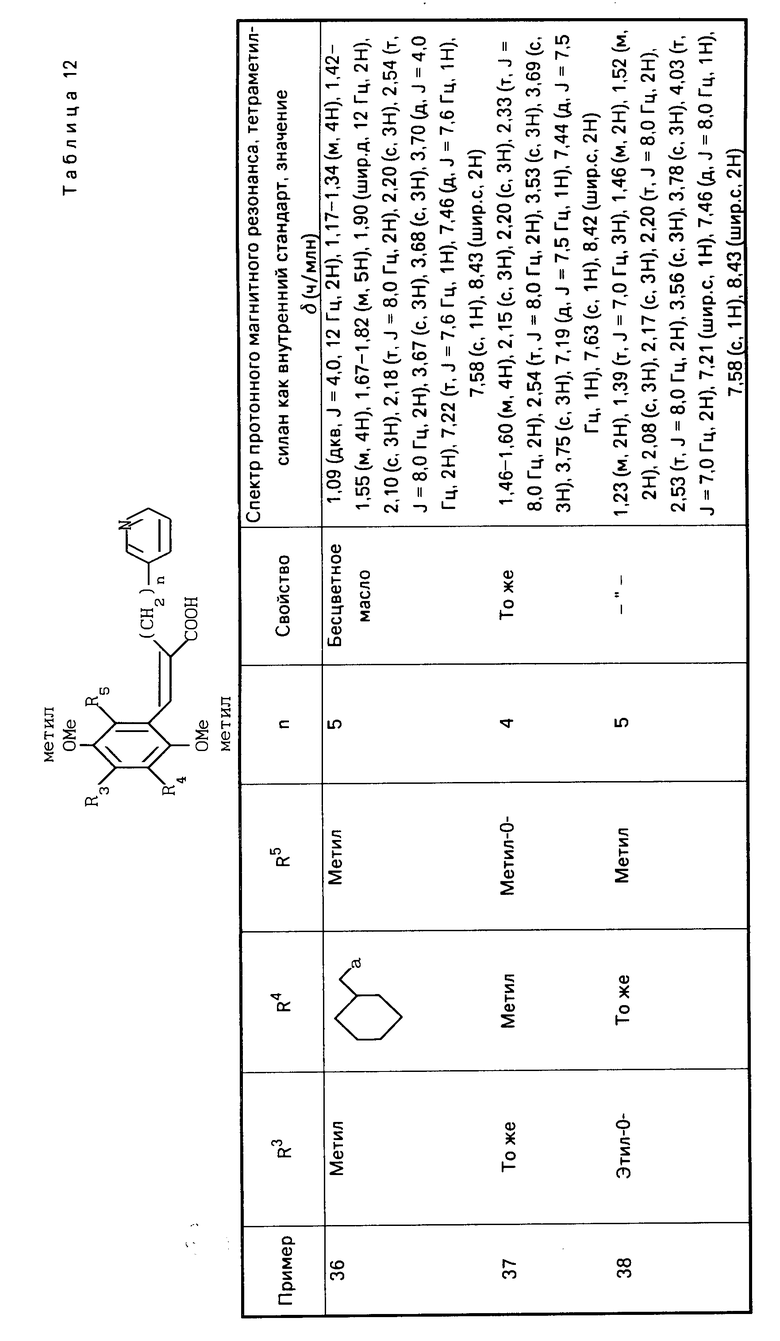
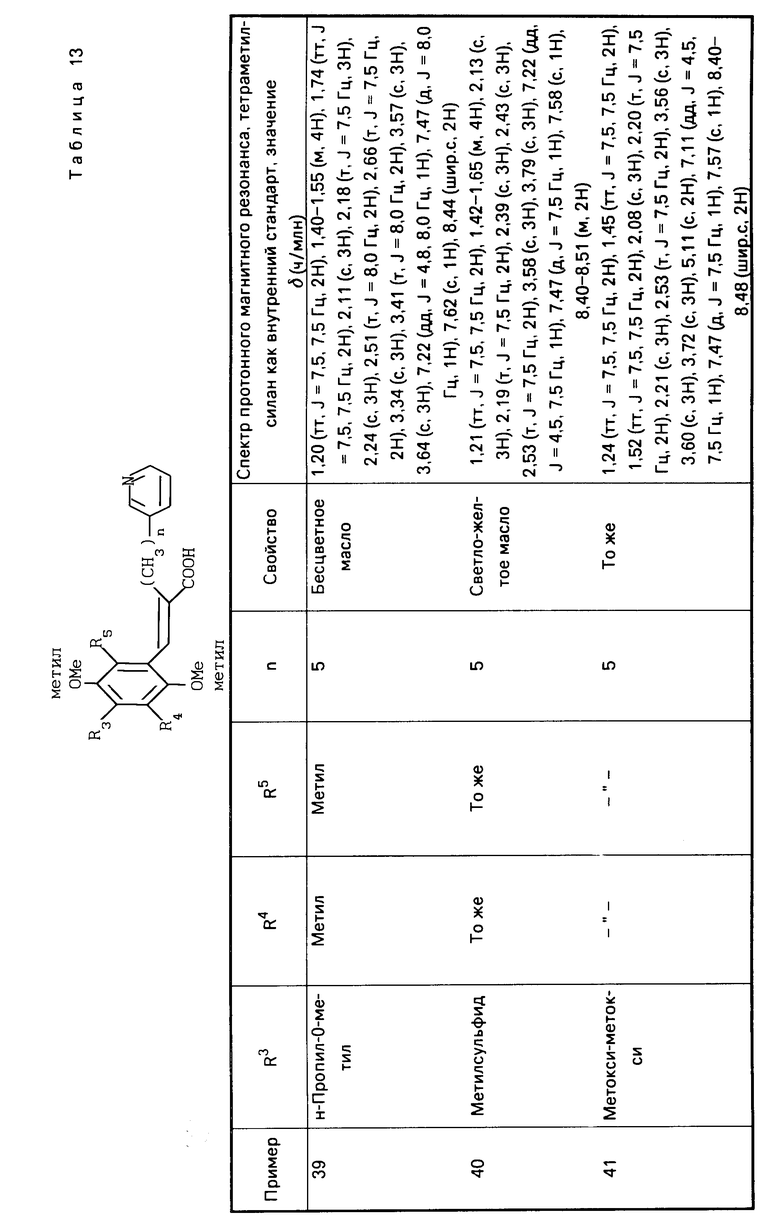
П р и м е р ы 5-22. Вещества, перечисленные в следующих табл. 2-7, каждое было получено таким же образом, как в примере 1.
П р и м е р 23. Этиловый эфир (Е)-3-(2-метокси-3,6-диметил-1,4-бензохинон-5-ил )-2-[6-(3-пиридил)гексил]-2-прокислоты, гидрохлорид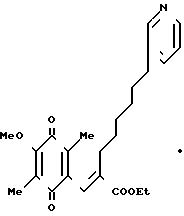 HCl
HCl
0,60 г этилового эфира (Е)-3-(2,4,5-триметокси-3,6-диметилфенил)-2-[6- (3-пиридил)гексил]-2-пропеновой кислоты, полученного в примере13, растворяют в смеси 20 мл ацетонитрила и 10 мл воды. Полученный раствор перемешивают при охлаждении льдом и затем добавляют по частям 1,66 г церий (IV)-аммоний нитрата. Полученную смесь перемешивают 3 ч при охлаждении льдом и затем добавляют 100 мл этилацетата. Полученную смесь промывают 2 раза насыщенным водным раствором бикарбоната натрия и затем насыщенным водным раствором хлорида натрия, сушат над безводным сульфатом магния и фильтруют. Фильтрат концентрируют до объема 10 мл адсорбируют на хроматографической колонке с силикагелем и элюируют смесью этилацетат/гексан (I/I). Фракции, содержащие целевое вещество, объединяют и пробулькивают через них газообразный хлористый водород в течение 1 мин. Растворитель отгоняют в вакууме, получая указанное в заголовке вещество (0,40 г) в виде желтого масла.
ПМР-спектр (400 МГц, в пердейтеродиметилформамиде), δ (ч/млн) 1,07-1,33 (м. 6Н), 1,23 (т. I 7,0 Гц, 3Н), 1,45-1,57 (т. I8,0 Гц, 2Н), 1,80 (с. 3Н), 1,81 (с. 3Н), 2,03 (т. I 8,0 Гц, 2Н), 2,49 (т. I 8,0 Гц, 2Н), 3,91 (с. 3Н), 4,18 (кв. I 7,0 Гц, 2Н), 7,03 (с. 1Н) 7,93 (шир. т. I 8,0 Гц, 1Н) 8,37 (шир. I 8,0 Гц, 1Н), 8,73 (д, I 8,0 Гц, 1Н), 8,76 (с. 1Н).
П р и м е р ы 24-41. Вещества, перечисленные в следующих табл. 8-13, каждое было получено таким же образом, как в примере 2.
П р и м е р 42. (Е)-3-(4-гидрокси-2,5-диметокси-3,6-диметилфенил) -2-[5-(3-пиридил)пентил]-2-пропеновая кислота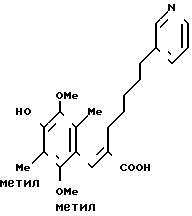
1,85 г вещества, приготовленного в примере 41, растворяют в 15 мл ацетона и затем добавляют 2,5 мл концентрированной соляной кислоты. Образовавшуюся смесь перемешивают при комнатной температуре 10 ч. Нейтрализуют насыщенным водным раствором гидрокарбоната натрия и экстрагируют этилацетатом. Органический слой промывают водным раствором хлорида натрия, сушат над безводным сульфатом магния и фильтруют. Фильтрат перегоняют, чтобы удалить растворитель. Получают 7,59 г указанного в заголовке вещества в виде белого стеклообразного вещества. ПМР-спектр (в дейтерохлороформе), δ (ч/млн) 1,21 (тт. I 7,5, 7,5 Гц 2Н), 1,49 (тт. I 7,5, 7,5 Гц, 2Н), 2,02 (т. I 7,5 Гц 2Н), 2,12 (с. 3Н), 2,17 (с. 3Н), 2,50 (т. I 7,5 Гц, 2Н) 3,56 (с. 3Н), 3,74 (с. 3Н), 7,18-7,28 (м. 1Н), 7,24 (д, I 5,5 Гц, 1Н), 7,39 (с. 1Н), 8,35-8,50 (м. 2Н).
П р и м е р 43. (Е)-3-(2-Гидрокси-3,6-диметил-1,4-бензохинон-5-ил) -2-[5-(3-пиридил)пентил]-2-пропеновая кислота, гидрохлорид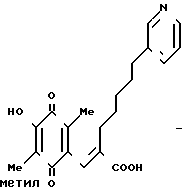 HCl
HCl
1,26 г (Е)-3-(4-гидрокси-2,5-диметокси-3,6-диметилфенил)-2-[5-(3-пиридил) гексил] -2-пропеновой кислоты, полученной в примере 42, растворяют в смеси 60 мл ацетонитрила и 30 мл воды. Полученный раствор охлаждают до температуры льда и затем постепенно добавляют по каплям 20 мл водного раствора, 3,63 г церий (IV)-аммоний нитрата. К полученной смеси добавляют 5% -ный водный раствор бикарбоната натрия, доводя значение рН до 6. Образовавшуюся смесь экстрагируют этилацетатом. Органический слой промывают водой, сушат над безводным сульфатом магния и фильтруют. К фильтрату добавляют 1,6 мл 6-нормальной соляной кислоты и 100 мл этанола, и удаляют растворитель перегонкой в вакууме. Остаток подвергают перекристаллизации из этанола, получая 0,8 г указанного в заголовке вещества в виде желтых кристаллов. ПМР-спектр (в пердейтеродиметилформамиде) δ (ч/млн) 1,05-1,18 (м. 2Н), 1,20-1,38 (м. 2Н), 1,40-1,54 (м. 2Н), 1,73 (с. 3Н), 1,80 (с. 3Н), 2,00 (шир.т. I 7,0 Гц, 2Н), 2,66 (т. I 7,0 Гц, 2Н) 7,04 (шир. с. 1Н), 7,91 (дд, J 5,0 Гц и 8,0 Гц, 1H) 8,35 (шир. д. J 8,0 Гц, 1H) 8,65-8,80 (м. 1Н), 8,75 (с. 1Н).
Масс-спектр: МН+ 370.
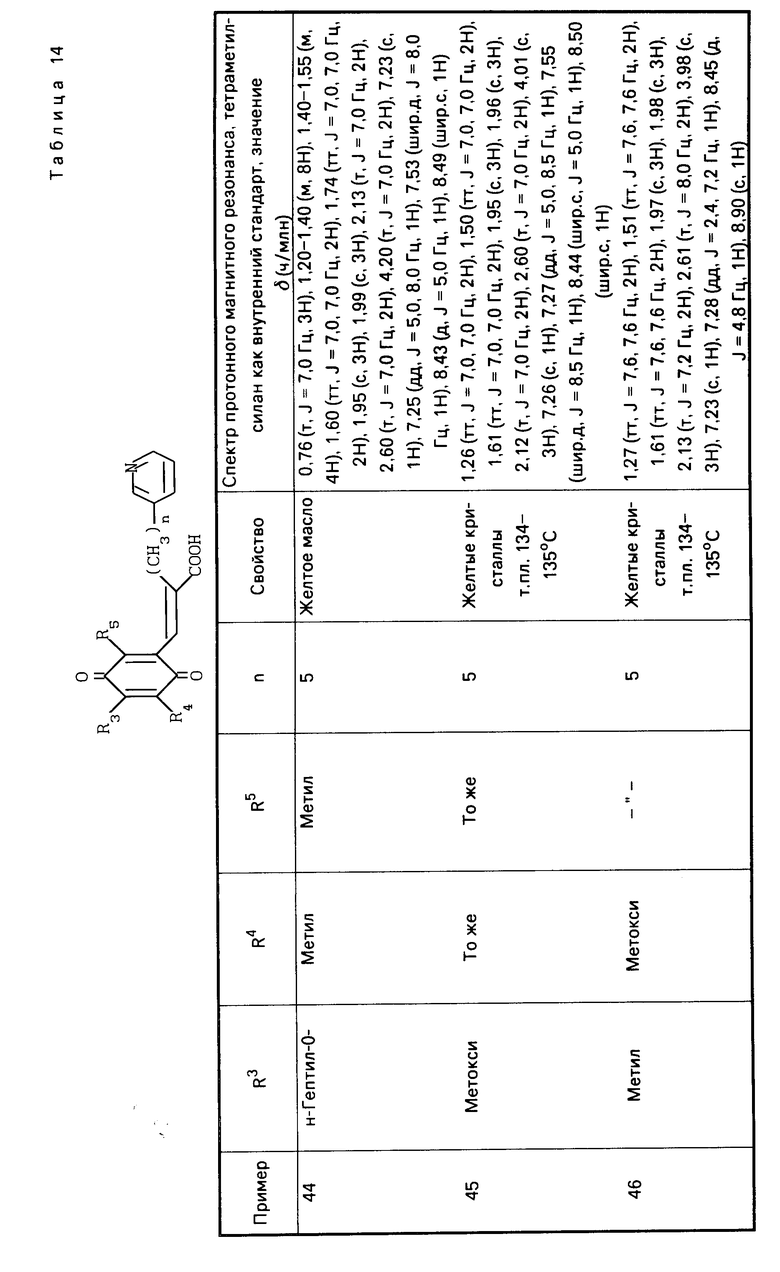
П р и м е р ы 44-62. Вещества, перечисленные в следующих табл. 14-20 каждое, было получено таким же образом, как в примере 3.
П р и м е р 63. Этиловый эфир 3-(2-метокси-3,5-диметил-1,4-бензохинон-6-ил)-2-[5- (3-пиридил)пентил]-2-пропеновой кислоты, гидрохлорид.
ме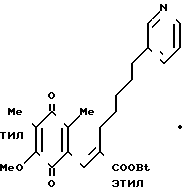 HCl
HCl
1,0 г вещества, полученного в примере 46, растворяют в 10 мл этанола и затем добавляют 0,7 мл концентрированной серной кислоты. Полученную смесь нагревают с обратным холодильником в течение 12 ч, охлаждают, нейтрализуют и экстрагируют этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, сушат над безводным сульфатом магния и концентрируют в вакууме. Остаток очищают на хроматографической колонке с силикагелем, которую элюируют смесью этилацетат (0-30%/гексан. Остаток растворяют в небольшом количестве этанола и затем добавляют 0,25 мл концентрированной серной кислоты. Полученную смесь концентрируют в вакууме, получая указанное в заголовке вещество в виде желтого масла. ПМР-спектр (400 мГц, в пердейтеродиметилформамиде), δ (ч./млн) 1,17 (тт. I 8,0 Гц 8,0 Гц, 6Н), 1,27 (т. I 6,4 Гц, 3Н), 1,36 (тт. I 8,0, 8,0 Гц, 2Н), 1,55 (тт. I 8,0, 8,0 Гц, 2Н), 1,85 (с. 3Н), 1,88 (с. 3Н), 2,07 (т. I 8,0 Гц, 2Н), 2,73 (т. I 8,0 Гц, 2Н), 3,88 (с. 3Н), 4,20 (кв. I 6,4 Гц, 2Н), 7,06 (с. 1Н), 8,00 (дд, I 1,6 и 8,0 Гц, 1Н), 8,45 (д, I 8,0 Гц, 1Н), 8,78 (д, I 5,2 Гц, 1Н), 8,81 (с. 1Н).
П р и м е р 64. 3-(2-Метокси-3,5-диметил-1,4-бензохинон-6-ил)-2-[5-(3-пиридил)- пентил]-1-оксо-1-морфолин-2-пропен.
1,4 г вещества, приготовленного в примере 46, растворяют в 200 мл хлористого метилена. Образовавшийся раствор дважды промывают 100 мл 10%-ного раствора гидросульфита натрия в воде. Органический слой отделяют, сушат над безводным сульфатом магния и концентрируют в вакууме. К остатку добавляют 20 мл диметилформамида и 1 г гидросульфита натрия. Полученную смесь охлаждают до 0оС и затем по каплям добавляют 1,1 г дифенилфосфорилазида, 0,32 г морфолина и 0,38 г триэтиламина в указанной последовательности. Полученную смесь перемешивают при комнатной температуре в течение суток, затем добавляют воду. Полученную смесь экстрагируют этилацетатом. Органический слой перемешивают, пробулькивая через него воздух в течение 2 ч, затем промывают насыщенным водным раствором хлорида натрия, сушат над сульфатом магния и концентрируют в вакууме. Остаток очищают на хроматографической колонке с силикагелем (элюент-этилацетат), получая 710 мг указанного в заголовке вещества в виде оранжевого масла. ПМР-спектр (400 МГц, в дейтерохлороформе), δ (ч/млн), 1,27 (тт. I 8,0 Гц, 2Н), 1,36 (тт. I 8,0 8,0 Гц, 2Н), 1,55 (тт. I 8,0 Гц, 8,0 Гц, 2Н), 1,98 (с. 6Н), 2,10 (т. I 8,0 Гц, 2Н) 2,55 (т. I 8,0 Гц, 2Н), 3,65-3,80 (м. 3Н), 3,96 (с. 3Н), 5,97 (д, I 1,2 Гц, 1Н), 7,18 (дд, I 3, 6, 8,0 Гц, 1Н), 7,44 (дт, I 1,2 и 8,0 Гц, 1Н), 8,39 (д, I 1,5 Гц, 1Н), 8,42 (дд, I 1,5 и 3,6 Гц, 1Н).
Испытание токсичности (I). Испытание на повторяющееся в течение 4 недель пероральное назначение крысам.
М е т о д и к а. Полученное в примере 3 вещество перорально вводилось в дозировке 30 и 100 мг/кг на протяжении 4 недель самкам крысы Slc SD в возрасте 7 недель (каждая группа содержала 5 животных). Это вещество суспендировали в водном 0,5%-ном (по массе) растворе метилцеллюлозы и вводили 1 раз в сутки.
В течение периода назначения наблюдали за общим состоянием животных, а также измеряли вес тела и потребление пищи. По завершении последнего введения были проведены гематологические, гемобиохимические исследования, анализы мочи, после вскрытия были взвешены внутренние органы. Кроме того, на оптическом микроскопе было проведено патогистологическое исследование печени и почек крыс.
Р е з у л ь т а т ы. По обеих дозировках 30 и 100 мг/кг не было замечено никаких изменений.
Испытание токсичности (2). Испытание на повторяющееся в течение 4 недель пероральное назначение собакам.
М е т о д и к а. Полученное в примере 3 вещество перорально вводилось в дозировке 30 и 100 мг/кг в течение 4 недель самкам гончих в возрасте 8 месяцев (в каждой группе было по 2 животных). Это вещество растиралось с лактозой и вводилось 1 раз в сутки.
В течение периода назначения наблюдали за общим состоянием животных, а также измеряли вес тела и потребление пищи. После завершения первого, седьмого и последнего введения было осуществлено гемо-биохимическое исследование. После окончания седьмого и последнего введения, кроме того, было проведены гематологические исследования и анализы мочи. Более того, было осуществлено вскрытие и после последнего назначения определяли вес внутренних органов. На оптическом микроскопе было проведено пато-гистологическое исследование печени, почек и надпочечников собак.
Р е з у л ь т а т ы. При обеих дозировках 30 и 100 мг/мг не наблюдали никаких изменений. Таким образом, для вещества, полученного в примере 3, было установлено, что токсилогически не воздействующая доза составляет 100 мг/кг.
Т Т а б л и ц а 19
Т а б л и ц а 20
Т а б л и ц а 16
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНОЕ БЕНЗОТИАЗОЛА ИЛИ ЕГО ФАРМАКОЛОГИЧЕСКИ ПРИЕМЛЕМАЯ СОЛЬ | 1992 |
|
RU2041216C1 |
| ПРОИЗВОДНЫЕ ПЕПТИДОВ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ, ПОСРЕДНИКОМ КОТОРЫХ ЯВЛЯЕТСЯ ТАХИКИНИН | 1991 |
|
RU2073683C1 |
| ДИАМИНОВЫЕ СОЕДИНЕНИЯ ИЛИ ИХ КИСЛОТНО-АДДИТИВНЫЕ СОЛИ | 1990 |
|
RU2045522C1 |
| ХИНОНОВЫЕ ПРОИЗВОДНЫЕ И ИХ ФАРМАКОЛОГИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ | 1992 |
|
RU2049771C1 |
| ПРОИЗВОДНОЕ ОКСАСПИРО [2,5] ОКТАНА И СПОСОБ ЕГО ПОЛУЧЕНИЯ (ВАРИАНТЫ) | 1989 |
|
RU2068412C1 |
| ПРОИЗВОДНЫЕ БУТЕНОВОЙ ИЛИ ПРОПЕНОВОЙ КИСЛОТЫ | 1992 |
|
RU2041871C1 |
| ОПТИЧЕСКИ АКТИВНЫЕ ПРОИЗВОДНЫЕ ПИРИДОБЕНЗОКСАЗИНА ИЛИ ИХ СОЛИ | 1991 |
|
RU2029771C1 |
| АМИДЫ КИСЛОТ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2000 |
|
RU2208608C2 |
| ПРОИЗВОДНЫЕ ПЕПТИДОВ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ | 1992 |
|
RU2043365C1 |
| ПРОИЗВОДНЫЕ 3(2Н)-ПИРИДАЗИНОНА ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1991 |
|
RU2054004C1 |
Изобретение относится к производным хинона ф-лы I 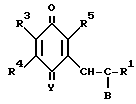 или его фармакологически приемлемым солям, где R1 гетероалкильная группа, в которой гетероалкил это пяти- или шестичленный цикл, содержащий в качестве гетероатома азот, а алкильная часть содержит 1-10 атомов углерода, B-карбоксильная или защищенная карбоксильная группа R3,R4,R5 - одинаковые или отличающиеся друг от друга и каждый представляет собой атом водорода, гидроксильную группу C1-C8 алкил, C1-C8 алкокси, C1-C8 алкокси C1-C10 алкил, C1-C8 алкокси C1-C8 алкокси, C3-C7 циклоалкил C1-C8 алкокси, тио C1-C8 алкил, тио C1-C8 алкил, за исключением случая, когда R3 и R4 каждый одновременно является низшей C1-C8 алкоксигруппой, или группой, представляемой формулой II
или его фармакологически приемлемым солям, где R1 гетероалкильная группа, в которой гетероалкил это пяти- или шестичленный цикл, содержащий в качестве гетероатома азот, а алкильная часть содержит 1-10 атомов углерода, B-карбоксильная или защищенная карбоксильная группа R3,R4,R5 - одинаковые или отличающиеся друг от друга и каждый представляет собой атом водорода, гидроксильную группу C1-C8 алкил, C1-C8 алкокси, C1-C8 алкокси C1-C10 алкил, C1-C8 алкокси C1-C8 алкокси, C3-C7 циклоалкил C1-C8 алкокси, тио C1-C8 алкил, тио C1-C8 алкил, за исключением случая, когда R3 и R4 каждый одновременно является низшей C1-C8 алкоксигруппой, или группой, представляемой формулой II 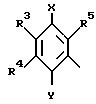 , где R3,R4,R5 имеют значения, указанные выше, X и Y одинаковые или различные и каждый представляет собой гидроксил или C1-C8 алкокси. Соединение I или его фармакологически премлемые соли обладают активностью в ингибировании образования лейкотриенов и тромбоксанов и может быть использовано в медицине в качестве терапевтического средства. 2 с. и 8 з. п. ф-лы, 20 табл.
, где R3,R4,R5 имеют значения, указанные выше, X и Y одинаковые или различные и каждый представляет собой гидроксил или C1-C8 алкокси. Соединение I или его фармакологически премлемые соли обладают активностью в ингибировании образования лейкотриенов и тромбоксанов и может быть использовано в медицине в качестве терапевтического средства. 2 с. и 8 з. п. ф-лы, 20 табл.
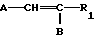
где A группа общей формулы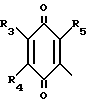
где R3, R4 и R5 одинаковые или различные и каждый - водород, гидроксильная группа, C1-C8-алкил, С1-С8-алкокси, С1-С8-алкокси-С1-С10-алкил,
С1-С8-алкокси-С1-С8-алкокси, С3-С7-циклоалкил-С1-С8-алкокси, тио-С1-С8-алкил, за исключением случая, когда R3 и R4 каждый одновременно являются низшей С1-С8-алкоксигруппой, или группа общей формулы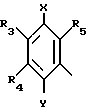
где R3, R4 и R5 имеют указанные значения;
X и Y, одинаковые или различные и каждый гидроксил или С1-С8-алкокси;
R1 гетероарилалкильная группа, в которой гетероалкил это 5- или 6-членный цикл, содержащий в качестве гетероатома атом азота, а алкильная часть содержит 1-10 атомов углерода;
B карбоксильная или защищенная карбоксильная группа.
Приоритет по признакам:
11.03.91 при R3, R4 и R5, одинаковые или различные и каждый водород, С1-С8-алкил, С1-С8-алкокси.
| УСТРОЙСТВО для АВТОМАТИЧЕСКОГО ОБЪЕМНОГОТИТРОВАНИЯ | 0 |
|
SU234729A1 |
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
