Изобретение относится к новым диаминовым соединениям и их кислотно-аддитивным солям и церебральным защитным лекарственным препаратам, содержащим эти соединения или их соли, более конкретно к диаминовым соединениям и их кислотно-аддитивным солям, которые характеризуются прекрасным церебральным защитным действием и используются в качестве лекарственных средств при лечении расстройств церебральных функций или для предотвращения развития таких расстройств, и к церебральным защитным лекарственным средствам, содержащим диаминовые соединения или их кислотно-аддитивные соли.
Современное увеличение численности пожилых городских жителей привело к увеличению числа больных, страдающих различными функциональными расстройствами мозга, характеризующимися тем, что они сопровождаются внутримозговым кровоизлиянием, церебральным инфарктом, субарахноидным кровоизлиянием, преходящим нарушением мозгового кровообращения, церебрососудистыми расстройствами и т. п. Эти болезни следует рассматривать в основном, как следствие снижения кровообращения мозга, кислородной недостаточности крови, расстройств обмена веществ, таких, как гипогликемия.
Обратили внимание на причины болезней, которые подлежат лечению, и сосредоточились на лечении последствий церебральной ишемии, церебрососудистого слабоумия и т.д. Такие стандартные лекарственные средства включают лекарственные препараты для улучшения мозгового кровообращения, лекарственные препараты для улучшения церебрального обмена веществ, лекарственные препараты для улучшения мозговых функций, лекарственные препараты для предотвращения тромбоцитной коагуляции, которые уже использовались в клинической практике. В последнее время, независимо от причин заболеваний, предложены церебральные защитные лекарственные средства, которые привлекли внимание в качестве лекарства для лечения церебральных функциональных расстройств. Они предназначены для устранения церебральных функциональных расстройств или для предотвращения развития заболеваний посредством защиты мозга от ишемии или гипоксии.
Однако такие лекарства, предназначенные для лечения церебральных функциональных заболеваний, характеризуются только недостаточным клиническим действием и не выявляют четкого церебрального защитного воздействия при испытании на животных в условиях единичного дозированного орального приема, хотя большинство из них предназначены для благоприятного использования при оральном приеме.
Таким образом, до сих пор остается актуальным создание церебральных защитных лекарственных средств, характеризующихся прекрасным клиническим эффектом, и благоприятных для орального приема.
При названных обстоятельствах было проведено тщательные исследования для решения упомянутых вопросов и найдено, что новые динаминовые соединения формулы (I) и их кислотно-аддитивные соли обнаруживают прекрасное церебральное защитное действие, и это действие является надежным даже в случае орального способа приема. Это изобретение было выполнено, основываясь на вышеприведенных результатах.
В соответствии с изобретением предлагаются диаминовые соединения, представленные формулой I и их кислотно-аддитивные соли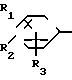 A
A Y
Y
 Y
Y A
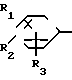
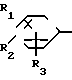
A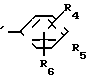 (I) где R1, R2, R3, R4, R5 и R6 являются одинаковыми или отличаются друг от друга и представляют собой индивидуально гидроксильную группу, низшую алкильную группу, низшую алкокси группу, низшую ацилокси группу, среди которых низшая алкильная группа и низшая алкокси группа могут быть замещены галогена; R7 и R8 связанные друг с другом, представляющие алкиленовую группу, имеющую от 1 до 4 углеродных атомов; A и A' являются одинаковыми или отличаются друг от друга и индивидуально представляют собой простую связь, -NH-, -NHCO -> -CONH -> -SO2NH- (здесь знак ->> означает присоединение к Y или Y'), Y и Y' являются одинаковыми или отличаются друг от друга и представляют собой низшую алкиленовую группу или низшую алкениленовую группу.
(I) где R1, R2, R3, R4, R5 и R6 являются одинаковыми или отличаются друг от друга и представляют собой индивидуально гидроксильную группу, низшую алкильную группу, низшую алкокси группу, низшую ацилокси группу, среди которых низшая алкильная группа и низшая алкокси группа могут быть замещены галогена; R7 и R8 связанные друг с другом, представляющие алкиленовую группу, имеющую от 1 до 4 углеродных атомов; A и A' являются одинаковыми или отличаются друг от друга и индивидуально представляют собой простую связь, -NH-, -NHCO -> -CONH -> -SO2NH- (здесь знак ->> означает присоединение к Y или Y'), Y и Y' являются одинаковыми или отличаются друг от друга и представляют собой низшую алкиленовую группу или низшую алкениленовую группу.
Соединение I, соответствующие изобретению, характеризуются прекрасным церебральным защитным действием, совершенно безопасны и проявляют свое сильное воздействие даже в условиях орального способа приема, являясь, таким образом, полезным в качестве церебрального защитного лекарственного средства.
Примерами наиболее предпочтительных соединений I являются соединения, представленные формулой I':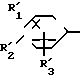 (CH2)
(CH2) (CH2)
(CH2)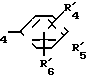 (I')
(I')
в которой R1-R6 отдельно представляют низшую алкоксильную группу.
Соединения I, соответствующие изобретению, могут быть получены посредством любого из следующих способов (1)-(9):
(1)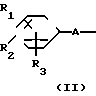 Y
Y X + H
X + H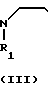
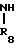
__→  A
A Y
Y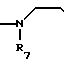
 Y
Y A
A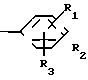 (IV)
(IV)
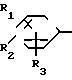
где Х представляет собой атом галогена, метилсульфонилокси группу или толилсульфонилокси группу, а обозначения R1-R3, R7, R8, A и Y имеют те же значения, что были определены выше.
В соответствии с этим способом, соединения IV данного изобретения могут быть получены взаимодействием соединений II с этиленовыми диаминовыми производными III.
Этот способ I применяется для получения соединений IV, которые являются соединениями I изобретения, в которых R1, R2, R3, A и Y являются соответственно идентичными R4, R5, R6, A' и Y'. На разновидности A(A') и Y(Y') не налагаются какие-либо ограничения.
Реакция протекает в присутствии подходящего растворителя, предпочтительно в присутствии основания, при температуре от комнатной до 200оС в интервале от нескольких минут до 10 ч. Примерами растворителей, используемых в этом процессе, являются эфиры, такие, как диэтиловый эфир, диоксан, тетрагидрофуран; углеводороды, такие, как хлороформ, дихлорметан, этанол и н-пропанол; пиридин; диметилформамид; диметилсульфоксид; и вода.
Примерами оснований могут служить такие неорганические основания, как карбонат калия, гидроокись натрия и гидроокись калия; и органические основания, такие, как триэтиламин, диизопропилэтиламин, ди-т-бутиламин, ди- метиламинопиридин и пирролидинопиридин.
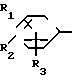
(2)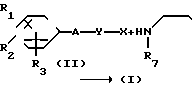
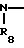 Y
Y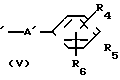
где обозначения от R1 до R8, A, A', Y, Y' и Х отдельно имеют те же значения, которые были определены ранее.
В соответствии с этим способом (2), соединения (I) данного изобретения могут быть получены реакцией между соединениями (II) и диаминовыми производными (V).
Этот способ (2) применим для получения запланированных соединений (I) независимо от их ввода. Реакция проводится в условиях, аналогичных тем, которые описаны в способе (I) выше.
(3)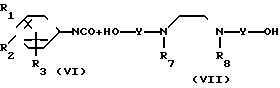
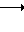
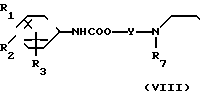
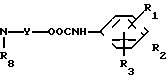
в которой обозначения от R1 до R3, R7, R8 и Y имеют те же значения, которые были определены здесь ранее.
В соответствии с этим способом (3) соединения (VIII) данного изобретения, которые имеют уретановую связь, могут быть получены реакцией между фенилизоцианатными производными VI и дигидроксиаминами VII. Этот способ (3) применим для получения соединений VIII, которые представляют собой соединения (I), в которых R1, R2, R3 и V являются идентичными R4, R5, R6 и Y' соответственно, а A и A' представляют собой -NHCOO ->>
Реакция между соединениями (VI) и соединениями VII проводится в растворителях, аналогичных тем, которые описаны в способе (I), предпочтительно в углеводородах, при перемешивании в условиях нагревания в интервале от 50 до 200оС в течение времени от 10 мин до 5 ч. Исходное вещество, фенилизоцианатные производные (VI) могут быть получены, например, взаимодействием соответствующих галоидных бензоилов с азидом натрия.
(4)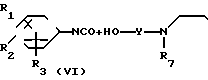
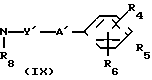 __→
__→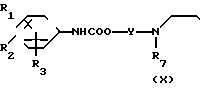
где обозначения от R1 до R8, A', Y и Y' имеют те же значения, что были определены ранее.
В соответствии со способом (4) соединения (Х) данного изобретения могут быть получены реакцией между фенилизоцианатными производными (VI) и спиртами (IX). Этот способ (4) применим для получения соединений (X), которые представляют собой соединения (I) данного изобретения, в которых А является -NHCOO ->> Реакция протекает в условиях, аналогичных тем, что описаны в способе (3).
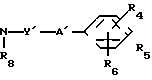
(5)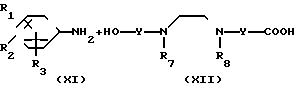 __→
__→  NHCO
NHCO Y
Y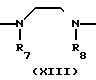 Y
Y COOHNH
COOHNH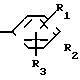
где обозначения от R1 до R3, R7, R8 и Y имеют те же значения, что были определены ранее.
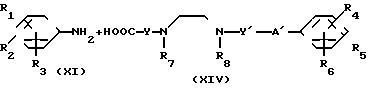
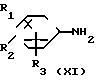
В соответствии со способом (5), соединения XIII данного изобретения могут быть получены реакцией между аминами (XI) и карбоксильными кислотами (XII). Этот способ (5) применим для получения соединений (XIII), которые представляют собой соединения (I) данного изобретения, в которых R1, R2, R3 и Y являются идентичными R4, R5, R6 и Y' соответственно, а A и A' представляют собой -NHCO ->>
Для осуществления этой реакции можно следовать реакции для получения амида кислоты, которая является обычно применимой в области пептидного синтеза.
Подробно упоминается несколько реакций, которые включают (а): реакцию между свободным амином (XI) и свободной карбоксильной кислотой (XII) в присутствии конденсирующего агента, (b): реакцию между свободным амином XI и реакционноспособным производным карбоксильной кислоты XII и (с): реакцию между реакционноспособным производным амина XI и свободный карбоксильной кислотой XII. Примерами конденсирующих агентов, применяемых в реакции (а)1 являются дициклогексилкарбодиимид, N, N'-дисукцинимидилкарбамат, N,N'-карбонилдиимидазол и дифенилфосфорилазид. Условия реакции могут быть различными в зависимости от применяемых конденсирующих агентов и в случае использования дициклогексилкарбодиимида реакция заканчивается прежде всего взаимодействием карбоксильной кислоты XII и дициклогексилкарбодиимида в растворителе, после чего следует добавление амина XI, а затем перемешивание смеси при от -30 до 100оС в течение интервала времени от нескольких часов до нескольких часов до нескольких дней. Может быть использован любой растворитель, который упоминается в способе (I), приведенном ранее. Примеры реакционноспособных производных карбоксильных кислот (XII) в способе (b) включают галоидангидриды кислот, ангидриды кислот, смесь ангидридов кислот, активные эфиры и азиды кислот. Примеры реакционноспособных производных аминов (XI) в способе (с) включают изоцианаты и фосфазо соединения.
(6)
__→  NHCO
NHCO Y
Y
 Y
Y A
A
где R1-R8, Y, Y' и A' имеют те же значения, которые были определены ранее.
В соответствии с данным способом (6), соединения (XV) данного изобретения могут быть получены реакцией между аминами XI и карбоксильными кислотами XIV. Этот способ (6) применим для получения соединений XV, которые представляют собой соединения (I) данного изобретения, в которых А имеет значение NHCO ->> Реакция может проводиться в условиях, аналогичных описанным в способе (5).
(7)

 где обозначения от R1 до R3, R7, R8, Y и Х имеют те же значения, что определены здесь ранее.
где обозначения от R1 до R3, R7, R8, Y и Х имеют те же значения, что определены здесь ранее.
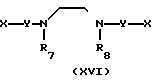
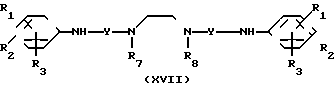
В соответствии с этим способом (7), соединения (XVII) данного изобретения могут быть получены реакцией между аминами (XI) и соединениями (XVI). Этот способ (7) применим для получения соединений (XVII), в которых R1, R2, R3 и Y идентичны R4, R5, R6 и Y' соответственно, а A и A' представляют собой -NH.
Это взаимодействие осуществляется реакцией аминов (XI) с соединениями (XVI) в растворителе, аналогичном тому, что описан в способе (I) при перемешивании при температурах от -30 до 200оС в интервале от 1 до 20 ч.
(8)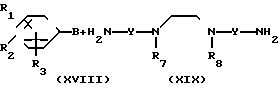
__→  A
A Y
Y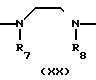 Y
Y A
A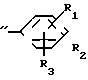 где B представляет собой СОХ', SO2X' (здесь Х' является атомом галогена) или NCO, A" представляет собой -CONH ->> SO2NH ->> или -NHCONH-, a R1-R3, R7, R8 и Y отдельно имеют те же значения, что были определены ранее.
где B представляет собой СОХ', SO2X' (здесь Х' является атомом галогена) или NCO, A" представляет собой -CONH ->> SO2NH ->> или -NHCONH-, a R1-R3, R7, R8 и Y отдельно имеют те же значения, что были определены ранее.
В соответствии с этим способом (8) соединения (XX) данного изобретения могут быть получены реакцией между соединениями (XVIII) и аминами (XIX). Этот способ (8) применим для получения соединений (XX), которые представляют собой соединения (I), в которых R1, R2, R3 и Y идентичны R4, R5, R6 и Y' соответственно, а A и A' представляют собой -CONH ->> SO2NH ->> или -NHCONH-.
Эта реакция осуществляется взаимодействием соединений (VIII) и аминов (XIX) в растворителе, аналогичном описанному в способе (I), при перемешивании при температуре от -30 до 100оС в интервале времени от 10 мин до 10 ч.
(9)
__→  A
A Y
Y Y
Y A
A
где обозначения от R1 до R8, A', A", Y, Y' и B имеют те же значения, что определены выше.
В соответствии с этим способом (9) соединения (XXII) данного изобретения могут быть получены реакцией между соединениями (XVIII) и аминами (XXI). Этот способ (9) применим для получения соединений (XXII), которые представляют собой соединения (I), в которых A представляет собой -CONH ->> SO2NH ->> или -NHCONH-. Реакция проводится в условиях, аналогичных условиям в способе (8).
Когда соединение (I) получаются в соответствии с любым из способов от (1) до (9) и если радикал от R1 до R6 содержит амино группу, соответствующие нитро соединения могут подвергаться реакции, за которой следует восстановление нитро группы, осуществляемое стандартным методом. Кроме того, группы от R1 до R6, являющиеся гидроксильными группами, низшими алкоксильными группами и низшими алкоксикарбонилокси группами, могут быть подвергнуты взаимному превращению одной в другую стандартным методом.
Полученные соединения (I), соответствующие изобретению, могут быть выделены и очищены стандартным методом, предпочтительно, при желании могут комбинироваться солевой обмен, экстракция растворителем и хроматография.
Соединения (I) изобретения, которые получены с использованием приведенных способов, при желании могут быть превращены в кислотно-аддитивные соли с использованием по существу известных методов. Примеры используемых кислот включают неорганические кислоты, такие, как серная кислота, хлористоводородная кислота, азотная кислота, фосфорная кислота и бромистоводородная кислота, и органические кислоты, такие, как уксусная кислота, молочная кислота, янтарная кислота, винная кислота, яблочная кислота, лимонная кислота, фумаровая кислота, метансульфоновая кислота и толуолсульфоновая кислота.
Полученные таким образом соединения (I) или их кислотно-аддитивные соли изобретения выявляют прекрасное церебральное защитное действие и обладают низкой токсичностью.
Для использования соединений (I) изобретения в качестве лекарственных средств они могут быть изготовлены в виде таблеток, капсул, гранул, порошка, инъекций, суппозиториев и т.д. с применением подходящих наполнителей, носителей, разбавителей и т. п. могут приниматься оральным или неоральным способом, но особенно предпочтительным является оральный способ приема. Получение лекарственного препарата осуществляется с использованием по сути своей известных методов. Например, препараты для орального приема могут быть получены смешением соединений (I) изобретения с наполнителями, такими, как крахмал, маннитол и лактоза; связующими, такими, как натрий карбоксиметилцеллюлоза и гидроксипропилцеллюлоза; дерегуляторами, такими, как кристаллическая целлюлоза и карбоксиметилцеллюлоза; смазывающим веществом, таким, как тальк, стеарат магния; регуляторами текучести, такими, как ангидрид кремниевой кислоты в соответствующей комбинации.
Соединения (I) данного изобретения предпочтительно принимаются оральным способом с дозировкой от 10 до 3,000 мг в день, которая принимается от 1 до 3 раз в день, хотя могут вноситься и изменения в зависимости от возраста и симптомов заболевания пациентов.
П р и м е р ы. Теперь изобретение описывается детально посредством примеров.
П р и м е р 1. Получение N,N'-бис-[4-(3,4,5-триметоксифенил)бутил] гомопиперазин. 2HCl:
7,5 г 1-хлор-4-(3,4,5-триметоксифенил)бутана, 1,3 г гомопиперазина, 4,5 г карбоната натрия и 5,3 г йодида калия добавлялись к 42 мл диметилформамида и перемешивались в течение 1 ч при 100оС. Реакционная смесь добавлялась к водному раствору NaCl, после чего следовала экстракция этилацетатом. Этилацетатный слой экстрагировался разбавленной хлористоводородной кислотой, а водный слой промывался этилацетатом. Затем к этому раствору добавлялся NaOH для создания основного значения рН и осуществлялась экстракция эфиром. Эфирный слой промывался водным раствором NaCl, высушивался и растворитель испарялся. Остаток очищался на силикагелевой хроматографической колонке с образованием 4,7 г свободного основания.
Полученное свободное основание превращалось в гидрохлорид с использование стандартного метода и перекристаллизовывалось из смеси метанол-эфир, образуя 2,2 г нужного соединения, имеющего точку плавления от 191 до 194оС (разложение). 1Н-ЯМР (CDCl3): δ
2,60 (4Н, уш.т. I=8 Гц)
3,82 (6Н, с)
3,86 (12Н, С)
6,37 (4Н, с)
ИКС (KBr); см-1
1587,1238, 1122
П р и м е р 2. Получение N,N'-бис-[(E)-4-(3,4,5-триметоксифенил)-3-бутенил]гомопи- перазин 2HCl:
Была использована методика примера 1 с применением 1,37 г (Е)-4-(3,4,5-триметоксифенил)-3-бутенилбромида с получением 619 мг свободного основания. Полученное свободное основание превращалось в гидрохлорид с использованием стандартного способа с последующим высаждением смесью диоксан-эфир с образованием 534 мг нужного соединения в виде палевого желтого аморфного вещества.
1Н-ЯМР (CDCl3) δ:
3,80 (6Н, с)
3,88 (12Н, с)
6,53 (2Н, д, I=15,6 Гц).
6,76 (4Н, с)
ИКС (KBr); см-1
1580, 1502, 1451, 1416
Примеры 3-8.
Используя любую из методик примера 1 или примера 2 были получены следующие соединения.
П р и м е р 3. N,N'-диметил-N,N'-бис-[3-(3,4,5-триметоксифенилкарбамоил)пропил] этилендиамин. 2 малеиновая кислота:
Точка плавления: от 159 до 161оС
1Н-ЯМР (CDCl3+DNSO-d6): δ
2,68 (6Н, с)
3,76 (6Н, с)
3,80 (12Н, с)
6,27 (4Н, с)
6,96 (4Н, с)
ИКС (KBr); см-1
1690, 1615, 1412, 1123
П р и м е р 4. N,N'-бис-[4-(3,4,5-триметоксифенил)пропил] гомопиперазин 2 малеиновая кислота:
Точка плавления: от 137 до 139оС
1Н-ЯМР (CDCl3); δ
3,81 (3Н, с)
3,84 (6Н, с)
6,26 (4Н, с)
6,39 (4Н, с)
ИКС (KBr); см-1
1587, 1497, 1124, 861
П р и м е р 5. N,N']-бис-[(Z)-4-(3,4,5-триметоксифенил)-3-бутенил] гомопиперазин 2HCl;
Аморфный порошок,
1Н-ЯМР (CD3OD); δ
3,78 (6Н, с)
3,95 (12Н, с)
6,49 (2Н, д, I=10 Гц)
6,55 (4Н, с)
ИКС (KBr); см-1
1577, 1502, 1456
П р и м е р 6. N,N'-бис-[3-(3,4,5-триметоксибензоилтио)пропил]-гомопиперазин 2HCl
Точка плавления: от 214 до 216оС (разложение)
1Н-ЯМР (CDCl3);δ
2,61 (2Н, м)
3,95 (18Н, с)
7,24 (4Н, с)
ИКС (KBr); см-1
1650, 1583, 1449, 1410
П р и м е р 7. N,N'-диметил-N,N'-бис-[3-(3,4,5-триметоксибензоилтио)-пропил]этил- ендиамин 2HCl:
Точка плавления: от 193 до 195оС
1Н-ЯМР (DMSO-d6);δ
2,87 (6Н, с)
3,79 (6Н, с)
3,88 (12Н, с)
7,18 (4Н, с)
ИКС (KBr); см-1
1661, 1584, 1231, 1124
П р и м е р 8. N,N'-бис-[5-(3,4,5-триметоксифенил)-N-пентил]-гомопиперазин 2HCl
Аморфный порошок
1Н-ЯМР (CD3OD);δ
2,60 (4Н, т, I=7 Гц)
3,70 (6Н, с)
3,80 (12Н, с)
6,49 (4Н, с)
ИКС (KBr); см-1
1585, 1503, 1455, 1420
П р и м е р 9. Получение N,N'-бис-[3-(3,4,5-триметоксифенилкарбамоил)-пропил] гомопиперазин 2 малеиновая кислота:
7,2 г N-[3-(3,4,5-триметоксифенилкарбамоил)пропил] гомопиперазина, 2,8 г карбоната калия и 3,4 г йодида калия добавляли к 10 мл диметилформамида и к смеси было добавлено 5,7 г 1-хлор-3-(3,4,5-триметоксифенилкарбамоил)пропана при перемешивании при 70оС, за счет следовало перемешивание в течение 30 мин при той же температуре и последующее перемешивание в течение 30 мин при 80оС. К этому было добавлено 5,9 г названных хлористых соединений и 2,8 г карбоната калия и полученная смесь перемешивалась в течение 30 мин при 80оС. Реакционная смесь растворялась в этилацетате, промывалась водой и экстрагировалась разбавленной хлористоводородной кислотой, рН доводилось до щелочного значения добавлением NaOH и проводилось экстрагирование хлороформом. Хлороформовый слой промывался водой, сушился, после чего проводилось удаление растворителя с получение 20,1 г неочищенного продукта. Этот продукт очищался с использованием хроматографической колонки с силикагелем, превращался в малеинат стандартным способом и перекристаллизовывался из смеси метано-эфир, образуя 5,5 г целевого продукта.
Точка плавления: от 181 до 184 оС.
1Н-ЯМР (CDCl3+DMSO-d6); δ
3,77 (6Н, с)
3,83 (12Н, с)
6,27 (4Н, с)
6,99 (4Н, с)
ИКС (KBr); см-1
1644, 1505, 1229, 1124.
П р и м е р 10. Получение N-[5-(3,4,5-триметоксифенил)пентил]-N'-[3-(2,6-диметил-4- гидроксифенилкарбамоил)пропил] гомопиперазина:
Осуществляли взаимодействие 1,0 г 1-[(3,4,5-триметоксифенил)-пентил] гомопипер- азина и 1,4 г 1-хлор-3-(2,6-диметил-4-гидроксифенилкарбамоил)пропана, проводя процесс аналогично тому, как описано в примере 9, с получением 0,6 г нужного соединения.
Маслянистое вещество
1Н-ЯМР (CDCl3); δ
2,08 (6Н, с)
2,84 (9Н, с)
6,35 (2Н, с)
6,42 (2Н, с)
ИКС (CHCl3); см-1
3320, 1652, 1588, 1401.
П р и м е р 11. Получение N-[5-(3,4,5-триметоксифенил)пентил]-N'- [3-(4-ацетилокси-2,6-диметилфенилкарбамоил)пропил]-гомо- пиперазин 2 малеиновая кислота:
Соединение, полученное в примере 10, превращалось в оксалат с использованием стандартного способа, а затем подвергалось ацетилированию уксусным ангидридом и пиридином. Ацетилированный продукт обычным способом превращался в малеинат и перекристаллизовывался из смеси метанол-эфир, образуя 4,8 г нужного соединения.
Точка плавления: от 85 до 89оС (разложение)
1Н-ЯМР (CDCl3+DMSO-d6); δ
2,19 (6Н, с)
2,27 (3Н, с)
3,80 (3Н, с)
3,85 (6Н, с)
6,27 (4Н, с)
6,79, 6,39 (2Н, с) для каждого значения
ИКС (KBr); см-1
1750, 1207, 1121, 872
П р и м е р 12. Получение N,N'-бис-[4-(3,4,5-триметоксифенил)оксибутил] гомопиперазин 2 малеиновая кислота:
Осуществлялось взаимодействие 711 мг 1-[4-(3,4,5-триметоксифенил)оксибутил] -гомо- пиперазина и 818 мг 1-бром-4-(3,4,5-триметоксифенил)оксибутана, проводя процесс аналогично тому, как описано в примере 9, с получением 785 мг свободного основания. Полученное свободное основание превращалось в малеинат с использованием стандартного способа и перекристаллизовывалось из смеси метанол-эфир с образование 959 мг нужного соединения.
Точка плавления: от 124 до 126оС
1Н-ЯМР (СDCl3);δ
3,78, 3,83 (9Н, с) для каждого значения
6,13 (4Н, с)
6,25 (4Н, с)
Примеры с 13 по 36.
Следующие соединения были получены согласно методикам примеров 9, 10, 11 или 12, описанным выше.
П р и м е р 13. N-[3-(3,4,5-триметоксифенилкарбамоил)пропил]-N'-[3-(4-ацетилокси-2,6 -диметоксифенилкарбамоил)пропил] -гомопиперазин 2 малеиновая кислота:
Аморфный порошок
1Н-ЯМР (DMSO-d6);δ
2,25 (3Н, с)
3,60 (3Н, с)
3,70, 3,72 (3Н, с) для каждого
6,10 (4Н, с)
6,48, 6,97 (2Н, с) для каждого
ИКС (KBr); см-1
1754, 1651, 1123, 997
П р и м е р 14. N-[3-(3,4,5-триметоксифенилкарбамоил)пропил]-N'-[3-(4-гидрокси- 2,6- диметилфенилкарбамоил)пропил]-гомопиперазин:
Маслянистое вещество
1H-ЯМР (CDCl3);δ
2,05 (6Н, с)
3,79 (6Н, с)
3,80 (3Н, с)
6,36, 6,89 (2Н, с) для каждого
П р и м е р 15. N-[3-(3,4,5-триметоксифенилкарбамоил)пропил]-N'-[3-(4-ацетилкси- 2,6- диметилфенилкарбамоил)пропил]-гомопиперазин:
Маслянистое вещество
1Е-ЯМР (CDCl3); δ
2,19 (6Н, с)
2,27 (3Н, с)
3,80 (3Н, с)
3,83 (6Н, с)
6,78, 6,92, (2Н, с) для каждого
ИКС (CHCl3); см-1
1749, 1609, 1223, 1128
П р и м е р 16. N-[3-(3,4,5-триметоксифенилкарбамоил)пропил)-N'-[3-[4-(2-трифторэто- кси) -2,6-диметилфенилкарбамоил]-пропил]гомопиперазин 2 малеиновая кислота:
Точка плавления: от 125 до 129оС (разложение)
1H-ЯМР (DMSO-d6);δ
2,11 (6Н, с)
3,61 (3Н, с)
3,73 (6Н, с)
4,69 (2Н, кв, I=9 Гц)
6,10 (4Н, с)
6,78, 6,98 (2Н, с) для каждого ИКС (KBr); см-1
3476, 1662, 1509, 1124
П р и м е р 17. N-[3-(3,4,5-триметоксифенилкарбамоилокси)пропил]-N'-[3-(4-(2-трифторэтокси) -2,6-диметилфенилкарбамоил] -пропил] гомопиперазин 2HCl:
Аморфный порошок
1Н-ЯМР (DMSO-d6); δ
2,11 (6Н, с)
3,60 (3Н, с)
3,72 (6Н, с)
4,70 (2Н, кв, I=9 Гц)
6,79, 6,85 (2Н, с) для каждого
ИКС (KBr); см-1
3398, 1716, 1645, 1605
П р и м е р 18. N,N'-бис-[3-[4-(2-трифторэтокси)-2,6-диметилфенилкарбамоил]пропил] гомопиперазин 2HCl:
Точка плавления: 231-233оС
1H-ЯМР (DMSO-d6); δ
2,11 (12Н, с)
4,69 (4Н, кв, I=9 Гц)
6,78 (4Н, с)
ИКС (KBr); см-1
3397, 1645, 1277, 1160
П р и м е р 19. N,N'-бис-[4-[3,4,5-триметоксифенилкарбамоил)бутил) гомопиперазин 2 малеиновая кислота:
Точка плавления: от 138 до 141оС
1Е-ЯМР (CDCl3+DMSO-d6); δ
3,77 (6Н, с)
3,84 (12Н, с)
6,26 (4Н, с)
7,01 (4Н, с)
ИКС (KBr); см-1
3321, 1684, 1610, 1124
П р и м е р 20. N-[3-(2,4,6-триметоксифенилкарбамоил)пропил]-N'-[3-(3,4,5- триметоксифенилкарбамоил)пропил]-гомопипера- зин:
Маслянистое вещество
1Н-ЯМР (CDCl3); δ
3,77, 3,80, 3,82 (6Н, с) для каждого
6,09, 6,95 (2Н, с) для каждого
ИКС (CHCl3); см-1
1671, 1604, 1506, 1129
П р и м е р 21. N,N'-бис-[3-(2,4,6-триметоксифенилкарбамоил)пропил]-гомопипер- азин:
Маслянистое вещество
1Н-ЯМР (CDCl3); δ
3,78 (12Н, с)
3,80 (6Н, с)
6,14 (4Н, с)
ИКС (CHCl3); см-1
1668, 1608, 1507, 1130
П р и м е р 22. N-[3-(4-ацетилокси-2,6-диметилфенилкарбамоил-пропил] N'-[3-(2,4,6-триметоксифенилкарбамоил)пропил] гомопиперазин 2 малеиновая кислота:
Точка плавления: 60oC (разложение)
1H-ЯМР (CDCl3+DMSO-d6); δ
2,18 (6Н, с)
2,27 (3Н, с)
3,76 (6Н, с)
3,79 (3Н, с)
6,31 (4Н, с)
6,77, 6,12 (2Н, с) для каждого
ИКС (KBr); см-1
3647, 1511, 1361, 1203
П р и м е р 23. N-[3-(4-ацетилокси-2,6-диметилфенилкарбамоил)пропил]-N'-[3-(4-ме- токси-2,6 -диметилфенилкарбамоил)пропил)-гомопиперазин 2 малеиновая кислота:
Точка плавления: 90oC (разложение)
1Н-ЯМР (CDCl3+DMSO-d6); δ
2,16, 2,18 (6Н, с) для каждого
2,28 (3Н, с)
3,76 (3Н, с)
6,30 (4Н, с)
6,59, 6,78 (2Н, с) для каждого
ИКС (KBr); см-1
3232, 1750, 650, 1576
П р и м е р 24. N,N'-бис-[3-(4-ацетитлокси-2,6-диметилфенилкарбамоил)-пропил]го- мопиперазин 2 малеиновая кислота:
Точка плавления: 80oC (разложение)
1Н-ЯМР (CDCl3+DMS-d6); δ
2,20 (12H, c)
2,29 (6H, c)
6,29 (4H, с)
6,79 (4Н, с)
ИКС (KBr); см-1
3474, 1750, 1655, 1207
П р и м е р 25. N-[3-2,4,6-триметоксифенилкарбамоил)пропил]-N'-[3-(4-метокси-2,6- диметилфенилкарбамоил)пропил] -гомопиперазин 2 малеиновая кислота:
Точка плавления: 85оС (разложение)
1Н-ЯМР (CDCl3+DMSO-d6); δ
2,16 (6H, с)
3,76 (6Н, с)
3,77, 3,80 (3Н, с) для каждого
6,28 (4Н, с)
6,14, 6,59 (2Н, с) для каждого
ИКС (KBr); см-1
3338, 1652, 1591, 1203
П р и м е р 26. N,N'-бис [3-(4-метокси-2,6-диметилфенилкарбамоил)-пропил]гомопи- перазин:
Точка плавления: от 158 до 161оС
1Н-ЯМР (CDCl3+DMSO-d6); δ
2,20 (12Н, с)
3,77 (6Н, с)
6,60 (4Н, с)
ИКС (KBr); см-1
3268, 1647,1603, 1149
П р и м е р 27. N-[3-(4-метокси-2,6-диметилфенилкарбамоил)пропил)-N'-[2-(3,4,5- триметоксифенил)этил] гомопиперазин:
Точка плавления: от 94 до 96оС
1Н-ЯМР (CDCl3); δ
2,24 (6Н, с)
3,80 (3Н, с)
3,90 (3Н, с)
6,48, 6,71 (2Н, с) для каждого
ИКС (KBr): см-1
3302, 1654, 1587, 1127
П р и м е р 28. N-[3-(4-метокси-2,6-диметилфенилкарбамоил)-пропил] N'-[3-(3,4-5-триметоксифенил)пропил] гомопиперазин:
Маслянистое вещество
1Н-ЯМР (CDCl3); δ
2,19 (6Н, с)
3,78 (3Н, с)
3,86 (9Н, с)
6,45, 6,66 (2Н, с) для каждого
ИКС (CHCl3); см-1
1650, 1589, 1232, 1124
П р и м е р 29. N-[3-(4-гидрокси-2,6-диметилфенилкарбамоил)пропил] N'-[3-(3,4,5-триметоксифенилкарбамоил)пропил]-гомопиперазин:
Маслянистое вещество
1Н-ЯМР (CDCl3); δ
2,08 (6H, c)
3,82 (3Н, с)
3,83 (6Н, с)
6,34, 6,38 (2Н, с) для каждого
ИКС (Пленка); см-1
3235, 1648, 1235, 1124
П р и м е р 30. N-[3-(4-гидрокси-2,6-диметоксифенилкарбамоил)пропил] N'-[3-(2,4,6-триметоксифенилкарбамоил)пропил] гомопиперазин:
Аморфный порошок
1Н-ЯМР (CDCl3); δ
3,64, 3,77 (6Н, с) для каждого
3,79 (3Н, с)
5,88, 6,13 (2Н, с) для каждого
ТКС (CHCl3); см-1
1656, 1597, 1153, 1130
П р и м е р 31. N-[3-(4-ацетилокси-2,6-диметоксифенилкарбамоил)-пропил] N'-[3-(2,4,6-триметоксифенилкарабамоил)про- пил]-гомопиперазин 2 малеиновая кислота:
Аморфный порошок
1Н-ЯМР (CDCl3+DMSO-d6); δ
2,26 (3Н, с)
3,71 (12Н, с)
3,78 (3Н, с)
6,10 (4Н, с)
6,24, 6,51 (2Н, с) для каждого
ИКС (KBr); cм-1
3374, 1755, 1646, 1124
П р и м е р 32. N-[3-(4-бензилокси-2,6-диметилфенилкарбамоил)-пропил]-N'-[2-(3,4,5- триметоксифенил)этил]-гомопиперазин:
Маслянистое вещество
1Е-ЯМР (CDCl3); δ
2,22 (6H, c)
3,86 (3H, c)
3,88 (6Н, с)
5,06 (2Н, с)
6,46, 6,78 (2Н, с) для каждого
ИКС (CHCl3); см-1
1661, 1589, 1486, 1461
П р и м е р 33. N-[3-(4-бензилокси-2,6-диметилфенилкарбамоил)-пропил]-N'-[5-(3,4,5- триметоксифенил)-N-пентил] гомопиперазин:
Маслянистое вещество
1Н-ЯМР (CDCl3); δ
2,22 (6Н, с)
3,88 (9Н, с)
5,06 (2Н, с)
6,46, 6,78 (2Н, с) для каждого
ИКС (CHCl3); cм-1
1661, 1589, 1486, 1461
П р и м е р 34. N-[3-(4-метилсульфонилокси-2,6-диметилфенилкарбамоил)пропил]-N' -[5- (3,4,5-триметоксифенил)-N-пентил] гомопиперазин:
Маслянистое вещество
1Н-ЯМР (CDCl3); δ
2,26 (6Н, с)
3,36 (3Н, с)
3,86 (3Н, с)
3,90 (6Н, с)
6,46, 7,08 (2Н, с) для каждого ИКС (CHCl3); см-1
2924, 1662, 1588, 1476
П р и м е р 35. N-метил-N-[3-(3,4,5-триметоксифенилкарбамоил)-пропил]-N-метил-N' -[3- (3,4,5-триметоксибензоилтио)-пропил]этилендиамин 2 малеиновая кислота
Точка плавления: от 137 до 139оС
1Н-ЯМР (CDCl3+DMSO-d6); δ
2,65, 2,76 (3Н, с) для каждого
3,78, 3,90 (3Н, с) для каждого
3,83, 3,91 (6Н, с) для каждого
6,28 (4Н, с)
6,98, 7,19 (2Н, с) для каждого
ИКС (KBr); см-1
1660, 1582, 1355, 1126
П р и м е р 36. N-[3-(3,4,5-триметоксифенилкарбамоил) пропил]-N'-[3-(3,4,5-триметоксифенил)пропил] гомопиперазин 2 малеиновая кислота:
Точка плавления: от 89 до 91оС
1Н-ЯМР (СDCL3+DMSO-d6) δ
2,23 (3Н, с)
2,16 (6Н, с)
3,78 (3Н, с)
6,24 (4Н, с)
ИКС (KBr); см-1
1754, 1653, 1616, 1123
П р и м е р 37. Получение N,N'-бис-[3-(4-аминофенилкарбамоилокси)пропил] гомопиперазина:
1 г п-нитробензоилохлорида растворяли в 22 мл ацетона и к этому раствору при перемешивании, на ледяной бане добавляли раствор азида натрия, после чего следовало перемешивание при той же температуре в течение 1 ч. Из реакционной смеси отгоняли ацетон, а к остатку добавляли для растворения хлороформ, промывали водой, сушили и отгоняли хлороформ с получением неочищенного соединения азида кислоты. Это соединение разбавляли 20 мл толуола и нагревали в течение 3 мин при 100оС, затем добавляли 520 мл N,N'-бис-(3-гидроксипропил) гомопиперазина и перемешивали в течение 1 часа при 100оС. Полученные нитросоединения восстанавливали дихлоридом олова и HCl стандартным способом с получением 507 мг нужного соединения.
Аморфный порошок
1H-ЯМР (CDCl3); δ
4,13 (4Н, т, I=6 Гц)
6,63. 7,14 (4Н, д, I=9 Гц) для каждого
ИКС (CHCl3); см-1
3423, 1710, 1516, 825
П р и м е р ы с 38 по 45.
Следующие соединения были получены способом, аналогичным тому, что описан в примере 37.
П р и м е р 38. N,N'-бис[2-(3,4,5-триметоксифенилкарбамоилокси)-этил] гомопиперазин 2 малеиновая кислота:
Точка плавления: от 140 до 143оС
1Н-ЯМР (CDCl3+CD3OD); δ
3,80 (6Н, с)
3,84 (12Н, с)
6,28 (4Н, с)
6,80 (4Н, с)
ИКС (KBr); см-1
3472, 1726, 1607, 1123
П р и м е р 39. N,N'-бис-[3-(3,4,5-триметоксифенилкарбамоилокси)-пропил] гомопиперазин:
Маслянистое вещество
1Н-ЯМР (CDCl3); δ
3,82 (18H, с)
4,24 (4Н, т, I=6 Гц)
6,72 (4Н, с)
ИКС (СHCl3); см-1
3422, 1725, 1604, 1128
П р и м е р 40. N,N'-бис [3-(4-гидрокси-3,5-диметоксифенилкарбамоилокси)про- пил] гомопиперазин:
Аморфный порошок
1Н-ЯМР (CDCl3); δ
3,84 (12Н, с)
4,20 (4Н, т, I=6 Гц)
6,72 (4Н, с)
ИКС (СНСl3); см-1
3518, 3424, 1720, 1623
П р и м е р 41. N,N'-бис-[3-(4-этоксикарбонилокси-3,5-диметоксифенилкарбамоил- окси)пропил] гомопиперазин:
Аморфный порошок:
1Н-ЯМР (CDCl3); δ
1,36 (6H, т, I=8 Гц)
3,82 (12Н, с)
6,74 (4Н, с)
ИКС (СНСl3); см-1
3425, 1757, 11720, 1616.
П р и м е р 42. N,N'-бис-[3-(4-(2-трифторэтокси)-3,5-диметоксифенилкарбамоило- кси]пропил] гомопиперазин 2HCl:
Точка плавления: от 211 до 214оС (разложение)
1Н-ЯМР (DMSO-d6); δ
3,74 (12H, с)
4,36 (4Н, кв, I=10 Гц)
6,88 (4Н, с)
ИКС (KBr); см-1
3383, 1728, 1608, 1127
П р и м е р 43. N,N'-бис-[3-(4-фторфенилкарбамоилокси)пропил]-гомопиперазин 2HCl:
Точка плавления от 204 до 207оС (разложение)
1Н-ЯМР (DMSO-d6); δ
4,15 (4Н, уш. т, I=6Гц)
7,10 ≈7,30 (6Н, м)
7,60 ≈7,80 (2Н, м)
ИКС (KBr); см-1
3295, 1720, 1534, 1230
П р и м е р 44. N,N'-[3-(3-ацетилокси-4,5-диметоксифенилкарбамоилокси)пропил] гомопиперазин:
Маслянистое вещество
1Н-ЯМР (CDCl3); δ
2,30 (6Н, с)
3,78, 3,84 (6Н, с) для каждого
6,66, 7,26 (2Н, д, I=2 Гц) для каждого ИКС (CHCl3); см-1
1761, 1726, 1507, 1193
П р и м е р 45. N,N'-[3-(4-ацетилокси-3,5-диметоксифенилкарбамоилокси)пропил] гомопиперазин:
Аморфный порошок
1Н-ЯМР (CDCl3); δ
2,32 (6Н, с)
3,80 (12Н, с)
4,24 (4Н, т, I=6 Гц)
6,76 (4Н, с)
ИКС (СНСl3); см-1
3425, 1760, 1727, 1615
П р и м е р 46. Получение N-[2-(3,4,5-триметоксифенилкарбамоилокси)этил] -N'-[2-(4- ацетилокси -3,5-диметоксифенилкарбамоилокси)этил] гомопиперазин 2 малеиновая кислота:
N-[2-(3,4,5-триметоксифенилкарбамоилокси)этил] -N'-[2-гидроксиэтил) гомопиперазин и 4-ацетилокси-3,5-диметоксибензоилхлорид взаимодействовали в соответствии с методикой, описанной в примере 37. 307 мг азида кислоты, полученного в результате этой реакции, добавлялось к 10 мл толуола и перемешивалось в течение 1 ч при 100оС. Из реакционной смеси выпаривали растворителя, затем следовала очистка с использованием силикагелевой хроматографической колонки (элюат: смесь хлороформ-метанол (30:1-20:1)). Было получено 519 мг свободного основания. Полученное вещество превращалось в малеинат с применением стандартного способа с получением в результате 489 мг нужного соединения в виде бесцветных порошкообразных кристаллов.
Точка плавления: от 103 до 107оС (разложение)
1Н-ЯМР (DMSO-d6); δ
3,60 (3Н, с)
3,70, 3,72 (6Н, с) для каждого
6,14 (4Н, с)
6,85, 6,91 (2Н, с) для каждого ИКС (KBr); см-1
1728, 1760 (передержка), 1607, 1222
П р и м е р ы 47, 48. Была использована методика примера 46 для получения следующих соединений.
П р и м е р 47. N-[3-(3,4,5-триметоксифенилкарбамоилокси)пропил]-N'-[3-(3-ацетил- окси-4,5- диметоксифенилкарбамоилокси)-пропил] гомопиперазин:
Аморфный порошок
1Н-ЯМР (CDCl3); δ
2,31 (3Н, с)
3,78, 3,80, 3,85 (3Н, с) для каждого
3,84 (6Н, с)
3,69 (4Н, с)
ИКС (CHCl3); см-1
1760, 1725, 1510, 1196
П р и м е р 48. N-[3-(3,4,5-триметоксифенилкарбамоилокси)пропил]-N'-[3-(3-гидрокси-4, 5-диметоксифенилкарбамоилокси)пропил]-гомопиперазин:
Аморфный порошок
1Н-ЯМР (CDCl3); δ
3,80 (3Н, с)
3,83, 3,84 (6Н, с) для каждого
6,48 (1Н, д, I=2 Гц)
6,69 (3Н, уш. с)
ИКС (СHCl3); см-1
1723, 1606, 1508, 1128
П р и м е р 49. Получение N,N'-бис-[2-(3,4,5-триметоксифенилкарбамоил)этил]го- мопиперазина:
852 мг N, N'-бис-(2-карбоксиэтил)гомопиперазина суспендировали в 25 мл пиридина и полученную суспензию разбавляли в 1,75 мл диоксанового раствора HCl (4 ммоль/мл) при перемешивании и охлаждении льдом, а затем добавляли 1,73 г дициклогексилкарбодиимида при 0оС и перемешивании в течение еще 2 ч. Затем к этому добавляли 1,92 г 3,4,5-триметоксианилина и осуществляли перемешивание в течение ночи при комнатной температуре. После окончания реакции осадки отфильтровывали, а фильтрат подвергли конденсированию. Конденсированный фильтрат растворяли разбавленной хлористоводородной кислотой и промывали этилацетатом. Водный слой нейтрализовали и экстрагировали хлороформом. Полученный экстракт промывали водой и сушили для удаления растворителя. Образующийся сырой продукт очищали с использованием препаративной тонкослойной хроматографии с применением силикагеля и перекристаллизовывали из смеси этилацетат-эфир, получая 216 г нужного продукта в виде палево-желтых призм.
Точка плавления: от 135 до 137оС
1Н-ЯМР (СDCl3); δ
2,52 (4Н, т, I=6 Гц)
6,91 (4Н, с)
ИКС (KBr); cм-1.
1677, 1603, 1506, 1234.
П р и м е р 50. Получение N,N'-бис-[3-(3,4,5-триметоксианилино)пропил] пипера- зин 4 HCl:
1,83 г 3,4,5-триметоксианилина и 2,45 г N,N'-(3-бромпропил)пиперазина растворялись в смеси из 20 мл метанола, 3 мл диметилсульфоксида и 0,5 мл воды и перемешивались с обратным холодильником в течение 7 ч. Полученный в результате реакции раствор конденсировался, подщелачивался NaOH, затем экстрагировался хлороформом. Слой хлороформа промывался водным раствором NaCl, после чего следовала сушка и выпаривание растворителя. Остаток подвергался очистке на силикагелевой хроматографической колонке, а полученное свободное основание превращалось в гидрохлорид с использованием стандартного способа. Перекристаллизация из смеси метанол-этанол дала 350 мг целевого соединения в виде палево-коричневых призм.
Точка плавления: от 240 до 242оС (разложение)
1Н-ЯМР (DMSO-d6); δ
3,63 (3H, c)
3,78 (6H, c)
6,64 (2Н, уш. с)
ИКС (KBr); см-1
3389, 1613, 1506, 1243
П р и м е р 51. Методика примера 50 была использована для получения следующего соединения:
N,N'-бис-[3-(3,4,5-триметоксианилино)пропил]-гомопиперазин 4HCl:
Точка плавления: от 214 до 216оС (разложение)
1Н-ЯМР (CDCl3+DMSO-d6); δ
3,79 (34Н, уш. с)
7,08 (2Н, уш. с)
ИКС (KBr); см-1
3390, 1612, 1504, 1243.
П р и м е р 52. Получение N,N'-бис-[3-(2,4,6-триметоксибензоиламино)пропил] гомопиперазин 2 малеиновая кислота:
13,1 г N, N'-бис-(3-т-бутоксикарбониламинопропил)-гомопиперазина растворялось в 136 мл этанола и к раствору добавлялось 26,8 мл концентрированной HCl, после чего следовало кипячение с обратным холодильником в течение 30 мин. После окончания реакции образующееся вещество высушивалось при пониженном давлении. Остаток растворялся в хлороформе, сушился, и растворитель испаряли. Оставшееся вещество растворяли в тетрагидрофуране и фильтровали с помощью Celite, после чего растворитель удаляли. Остаток растворяли в 299 мл безводного пиридина и к раствору добавляли заранее подготовленный раствор, полученный растворением хлорида кислоты из 13,4 г 2,4,6-триметоксибензойной кислоты стандартным способом в 44 мл бензола при охлаждении на ледяной бане. Смесь перемешивали в течение 3 ч при комнатной температуре. После окончания реакции, растворитель удаляли, а остаток растворяли в хлороформе, после чего осуществляли экстракцию водным раствором 5% уксусной кислоты. Водный слой промывали хлороформом, подщелачивали карбонатом натрия и экстрагировали хлороформом. Хлороформовый слой промывали водой и сушили, а полученный неочищенный продукт после удаления растворителя превращали в малеинат с использованием стандартного метода.
Перекристаллизация из смеси этанол-ацетон дала 6,7 г нужного соединения.
Точка плавления: от 105 до 107оС (разделение)
1Н-ЯМР (CDCl3+DMSO-d6); δ
3,78 (12Н, с)
3,83 (6Н, с)
6,23 (4Н, с)
6,10 (4Н, с)
ИКС (KBr); см-1
3343, 1605, 1123, 861
П р и м е р 53. Получение N,N'-бис-[3-(3,4,5-триметоксибензолсульфониламино)пропил] гомопиперазин 2HCl:
Была осуществлена методика примера 52 с использованием 650 мг 3,4,5,-триметоксибензолсульфонил хлорида. Было получено 323 мг свободного основания. Это свободное основание было превращено в гидрохлорид и перекристаллизовано из смеси метанол-эфир с образованием 320 мг нужного соединения в виде бесцветных порошкообразных кристаллов.
Точка плавления: от 233 до 227оС (разложение)
1Н-ЯМР (DMSO-d6); δ
3,73 (6Н, с)
3,85 (12Н, с)
7,09 (4Н, с)
ИКС (KBr); см-1
1591, 1411, 1315, 607.
П р и м е р 54. Получение N,N'-бис-[3-[3-(3,4,5-триметоксифенил) уреидо] пропил]гомопиперазин.
1,35 г соединения азида кислоты, полученного из 3,4,5-триметоксибензоилхлорида по методу, аналогичному описанному в примере 36, растворяли в 16 мол диоксана и перемешивали на горячей бане температурой 100оС в течение 3 ч. После охлаждения смеси до комнатной температуры, добавляли раствор, полученный растворением 550 мг N,N'-бис-(3-аминопропил)гомопиперазина в 5 мл диоксана, и перемешивали в течение 2 ч при комнатной температуре. Растворитель удаляли из реакционной смеси, а остаток очищали с использованием окисноалюминиевой хроматографической колонки, получая 810 мг целевого соединения в виде стекловидного вещества.
1Н-ЯМР (СDCl3); δ
3,809 (18Н, уш.с)
6,76 (4Н, с)
ИКС (CHCl3) cм-1
3326, 1655, 1546, 1127.
П р и м е р ы 55-58. Следующие соединения были получены в соответствии с методом, описанным в примерах 52, 53 или 54.
П р и м е р 55. N,N'-бис-[3-(3,4,5-триметоксибензоиламино)пропил]-гомопипера-зин:
Точка плавления от 139 до 140оС
1Н-ЯМР (CDCl3); δ
3,88 (18Н, с)
7,06 (4Н, с)
ИКС (KBr); см-1
3255, 1627, 1581, 1120
П р и м е р 56. N,N'-бис-[3-(4-гидрокси-3,5-диметоксибензоиламино)пропил]гомо-пиперазин:
Точка плавления: от 205 до 207оС (разложение)
1Н-ЯМР (DMSO-d6); δ
3,80 (12Н, с)
7,20 (4Н, с)
ИКС (KBR); см-1
33760, 1628, 1589, 1116
П р и м е р 57. N,N'-бис-[3-(п-аминобензоиламино)пропил]-гомопиперазин:
Аморфный порошок
1Н-ЯМР (CDCl3); δ
3,51 (4Н, кв, I=5 Гц)
6,64, 7,62 (4Н, д, I=9 Гц)
ИКС (CHCl3); см-1
1620, 1497, 1283, 837
П р и м е р 58. N,N'-бис-[3-(2,4,6-триметоксибензилсульфониламино)пропил]-гомо- пиперазин:
Аморфный порошок
1Н-ЯМР (СDCl3); δ
3,02 (4Н, т, I=7 Гц)
3,88, 3,92 (общий 20Н, с)
6,21 (4Н, с)
П р и м е р 59. Получение N-[3-(3,5-диметокси-4-гидроксибензоиламино)пропил]-N'-[3-(3,4,5- триметоксибензоиламино)пропил] гомопиперазин.
510 мг N-(3-аминопропил)-N'-[3-(3,4,5-триметоксибензоиламино)пропил] гомопиперазина взаимодействовали с 378 мг 3,4-диметокси-4-этоксикарбонилоксибен-зоил хлоридом с использованием стандартного метода, образуя 800 мг амидного соединения, имеющего гидроксильную группу.
Этот продукт растворялся в 11 мл метанола, к нему добавлялось 1,5 мл 2н. NaOH и осуществлялось перемешивание в течение 30 мин при 60оС. После окончания реакции 580 мг неочищенного продукта, полученного стандартным методом, перекристаллизовывалось из смеси метанол-эфир с образованием 500 мг целевого продукта в виде палево-желтых призм.
Точка плавления: от 147 до 150оС
1Н-ЯМР (CDCl3); δ
3,90 (15Н, с)
7,10 (2Н, с)
7,12 (2Н, с)
ИКС (KBr); см-1
3377, 3267, 1629, 1119
П р и м е р ы 60-62. Следующие примеры были осуществлены в соответствии с методикой, описанной в примере 59.
П р и м е р 60. N-[3-(2-трифторметилбензоиламино)пропил] -N'-[3- (3,4,5-триметоксибензоиламино)пропил] гомопиперазин:
Маслянистое вещество
1Н-ЯМР (CDCl3); δ
3,84 (3Н, с)
3,90 (6Н, с)
7,00 (2Н, с)
ИКС (Пленка); см-1
3272, 1639, 1545, 1123
П р и м е р 61. N-[3-(2,6-диметокси-4-гидроксибензоиламино)пропил] -N'-[3-(2,4,6-триметоксибензоиламино)пропил] гомопиперазин:
Аморфный порошок
1Н-ЯМР (CDCl3); δ
3,74, 3,78 (6Н, с) для каждого
3,81 (3Н, с)
6,22, 6,08 (2Н, с) для каждого
ИКС (KBr); см-1
3400, 1605, 1125
П р и м е р 62. N-[3-(4-ацетилокси-2,6-диметоксибензоиламино)пропил]-N'-[3-(2,4, 6- триметоксибензоиламино)пропил] гомопиперазин:
Аморфный порошок
1Н-ЯМР (CDCl3); δ
2,26 (3Н, с)
3,77, 3,78 (6Н, с) для каждого
3,80 (3Н, с)
6,09, 6,30 (2Н, с) для каждого
ИКС (CHCl3); см-1
1750, 1645, 1605, 1460.
П р и м е р 63. Получение N-[3-(3,4,5-триметоксифенилкарбамоил)пропил] -N'- [5-(3,4,5-триметоксифенил) пентил] гомопиперазин.
515 мг N-(3-карбоксипропил)-N'-[5-(3,4,5-триметоксифенил)пентил] гомопиперазина растворяли в 10 мл безводного тетрагидрофурана и к этому раствору добавляли 268 мг 3,4,5-триметоксианилина и 301 мл дициклогексилкарбодиимида при перемешивании на ледяной бане, за чем следовало дальнейшее перемешивание при комнатной температуре в течение ночи. После завершения реакции проводили выделение и очистку в соответствии с общепринятым методом с получением 551 мг целевого соединения в виде маслянистого вещества.
1Н-ЯМР (CDCl3); δ
3,86, 3,88 (общее 18Н, с)
6,45, 6,95 (2Н, с) для каждого
ИКС (СHCl3); см-1
1670, 1602, 1501, 1430.
П р и м е р ы 64-65. Следующие соединения были получены в соответствии с методикой, описанной в примере 63.
П р и м е р 64. N-[3-(3,4,5-триметоксифенилкарбамоил)пропил]-N'-[2-(3,4,5-тримето- ксифенил) этил] гомопиперазин.
Маслянистое вещество
1Н-ЯМР (CDCl3); δ
3,94 (9Н, с)
6,44 (2Н, с)
6,96 (2Н, с)
ИКС (Пленка); см-1
1674, 1602, 1541, 1504.
П р и м е р 65. N-[3-(4-ацетилокси-2,6-диметилфенилкарбамоил)-пропил]-N'-[2-(3,4,5- триметоксифенил)этил] гомопиперазин.
Маслянистое вещество
1Н-ЯМР (CDCl3); δ
2,24, 2,30 (общее 9Н, с)
3,92 (9Н, с)
6,50, 6,90 (2Н, с) для каждого
ИКС (CHCl3); см-1
1749, 1665, 1588, 1418
Примеры испытаний.
Среди соединений I изобретения типичные соединения подвергались испытаниям на церебральное защитное действие и токсичность.
(а) Действие против кислородной недостаточности.
1) Действие соединений в течение продолжительного времени в условиях газообразного азота.
Испытаниям подвергались группы мужских особей мышей ddY (в каждой группе по 10 мышей) массой 20-27 г. Испытуемые соединения вводились в суспензию 0,5% метилцеллюлозы и давались мышам оральным способом (обозначенным как п. о. ) или внутрибрюшинным способом (обозначенным как в.б.), подкожным (п.к.). Спустя 60 мин после орального приема или 30 мин после внутрибрюшинного приема к животным применялось кислородное голодание и измерялась продолжительность времени выживания.
Антигипоксические испытания осуществлялись помещением единичной особи мыши в 300-мл-вый прозрачный пластиковый контейнер, через который пропускалась смесь газа, содержащего 95% азота и 5% кислорода, при скорости прохождения 80 л/ч. Этот газ вытекал из контейнера через отверстие, в боковой стенке. Определялся промежуток времени от начальной точки пропускания смеси газов до окончания дыхательных движений мыши. Результаты выражались в виде разности продолжительности времени по отношению к группе мышей, не подвергнутых воздействию, которая приведена в табл. 1.
Статическая обработка проводилась в соответствии с методом Mean Whithey U.
2) Действие соединений на продолжительность выживания при приеме цианата калия:
Испытанию подвергались группы мужских особей ddY мышей массой 23-31 г и (в группе 10 мышей), Испытуемые соединения вводились в суспензию 0,5% метилцеллюлозы и вводились мышам оральным или внутрибрюшинным способом. Спустя 1 ч после орального приема или 15 мин после внутрибрюшинного приема было проведено впрыскивание 3 мг/кг цианата калия через каудальную вену в течение 20 ч. Измерялся отрезок времени от начала инъекции цианата калия до точки прекращения дыхательных движений. Результаты выражались в виде разницы в продолжительности времени по сравнению с мышами не подвергнутой группы испытаний, и приведены в табл. 2.
(b) Действие против ишемических церебральных поражений:
Испытания проводились на группах крыс Scl:Wistar возраста около 10 недель (в каждой группе от 4 до 5 крыс). Под действием эфирного наркоза крысам было проведено вскрытие в передней части шеи по средней линии и были выделены для воздействия общие каротидные артерии и лигированы хирургическим швом. Спустя 3,5 ч после лигирования была без анестезии проведена рециркуляция крови.
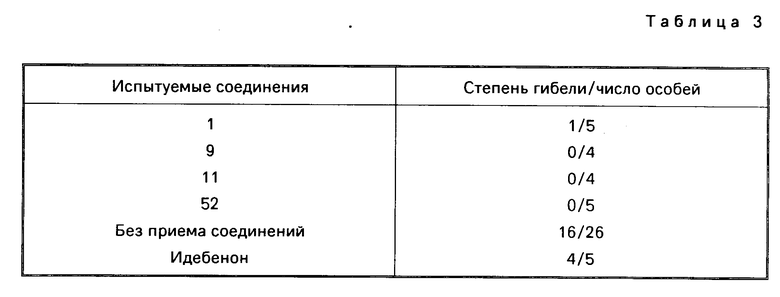
Испытуемые соединения вводились крысам подкожно, каждое соединение в количестве 30 мг/кг за 60 и 30 мин перед лигированием. Идебенон, который представлял собой контрольное соединение в этом исследовании, также вводился крысам в аналогичных условиях в количестве 150 мг/кг. Гибель крыс через 72 ч после рециркуляции показана в табл. 3.
(с) Испытание острой токсичности.
Группа мужских особей крыс Slc: Wistar возраста около 10 недель подвергались испытаниям. Испытуемые соединения были суспендированы в водном 5%-ном гуммиарабике и вводились крысам в количестве от 300 до 1000 мг/кг. Поведение крыс наблюдалось в точках, соответствующих 0,5, 1, 2 и 4 ч после орального приема, крыс кормили и наблюдали в течение дальнейших 3 дней.
Было установлено, что соединения примеров 1, 9, 11 и 52 не вызывали каких-либо отклонений в поведении крыс или гибели крыс при оральной дозировке 1000 мг/кг.
Промышленная применимость.
Соединения I, соответствующие изобретению, характеризуются прекрасными церебральным защитным действием, являются очень безопасными и проявляют сильное действие при оральном приеме и, следовательно, являются полезными в качестве церебральных защитных лекарственных средств. Соответственно, медицинские препараты, содержащие эти соединения, успешно применяются для лечения болезней, вызванных внутримозговыми кровоизлияниями, церебральным инфарктом, субарахноидным кровоизлиянием, временным ишемическим приступом, расстройствами мозгового кровообращения и т.п. или для предотвращения развития таких болезней.
| название | год | авторы | номер документа |
|---|---|---|---|
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ ИЛИ ИХ КИСЛОТНО-АДДИТИВНЫЕ СОЛИ | 1991 |
|
RU2057754C1 |
| ПРОИЗВОДНЫЕ ИНДОЛИЛПИПЕРИДИНА ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ | 1991 |
|
RU2039056C1 |
| ПРОИЗВОДНЫЕ БУТЕНОВОЙ ИЛИ ПРОПЕНОВОЙ КИСЛОТЫ | 1992 |
|
RU2041871C1 |
| ПРОИЗВОДНЫЕ ИНДОЛА ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1991 |
|
RU2045518C1 |
| ПРОИЗВОДНЫЕ ГЛИЦЕРИНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1989 |
|
RU2040521C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ БЕНЗИМИДАЗОЛА ИЛИ ИХ СОЛЕЙ | 1990 |
|
RU2023713C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ N-АКРИЛОИЛПИПЕРАЗИНА | 1990 |
|
RU2024513C1 |
| ПРОИЗВОДНЫЕ АРОИЛПИПЕРАЗИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ЛЕЧЕНИЯ | 1998 |
|
RU2258702C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО ИМИДАЗОЛА | 1990 |
|
RU2026293C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПИРАЗОЛПИРИДИНОВОГО ПРОИЗВОДНОГО ИЛИ ЕГО СОЛИ | 1990 |
|
RU2007403C1 |
Использование: в медицине. Сущность изобретения: новые диаиминовые соединения общей формулы I, где R1-R6 одинаковые или различные и представляют собой низший алкил, гидрокси, низший ацилокси-, низший алкокси-радикал, возможно замешенный атомами галогена, или их кислотно-аддитивные соли; Y,Y′ одинаковые или различные и представляют собой низший алкилен или низший алкенилен; A,A′ одинаковые или различные и означают простую связь, -NH, -NHCO, -CONH, SO2NH, R7 и R8 вместе образуют C1-C4 алкиленовую группу. 3 табл. Структура формулы I. 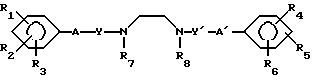
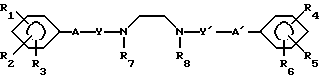
где R1 R6 одинаковые или различные, низший алкил, гидрокси, низший ацилокси, низший алкоксирадикал, возможно замещенный атомами галогена;
Y и Y', одинаковые или различные, низший алкилен или низший алкенилен;
A, A', одинаковые или различные, простая связь, NH, NHCO, CONH, SO2NH;
R7 и R8 вместе образуют С1 С4-алкиленовую группу
или их кислотно-аддитивные соли.
| Merk Indem, p.777. |

