Изобретение касается новых пептидных производных и их фармацевтически приемлемых солей, которые обладают фармакологической активностью.
Более конкретно оно касается новых пептидных производных и их фармацевтически приемлемых солей, которые обладают фармакологическими активностями, такими как антагонизм к веществу Р, антагонизм к нейрокинину А (вещество К) или аналогичным.
Таким образом, изобретение предусматривает пептидные производные и их фармацевтически приемлемые соли, полезные для профилактики и лечения астмы и т.д.
Пептидные производные согласно изобретению могут быть представлены следующей формулой I:
CH
где R1 водород, фенил (низший)алкоксикарбонил, низший алканоил, С15-С20-алканоил, бензоил, тиенил (низший)алканоил, фенил(низший)алкеноил, замещенный низшей алкенильной группой, или фенил (низший)алканоил, замещенный низшей алкильной группой;
R2 гидроксигруппа;
R3 карбокси или низший алкоксикарбонил, или
R2 и R3 соединены вместе, образуя группу формулы -O- -,
-,
R4 гидрокси-, низший алканоилокси или бензилокси;
R5 гидрокси-, низший алканоилокси или бензилокси;
R6 гидрокси-, низший алканоилокси, бензилокси или низший алкокси, и представляет одинарную или двойную связь.
представляет одинарную или двойную связь.








где R1 низшая алкоксигруппа.
Исходные соединения II и III являются новыми и могут быть получены следующими способами.




Способы получения целевых и исходных соединений согласно изобретению поясняются следующим образом.
Процесс 1 (способ 1). Соединение Ia или его соль можно получить, если подвергнуть соединение II или его соль реакции циклизации. Эту реакцию проводят традиционным методом синтеза циклических пептидов, таким как метод с использованием смешанных ангидридов кислот, активированных сложных эфиров, карбодиимидный метод или тому подобные. Реакцию обычно проводят в традиционном растворителе, таком как спирт, тетрагидрофуран, этилацетат, N,N-диметилформамид, дихлорметан, хлороформ или любой другой растворитель, который не оказывает отрицательного влияния на течение реакции. Температура реакции не является критической, и реакцию обычно проводят при охлаждении и до нагревания.
Способ 2. Соединение Ia или его соль можно получить, подвергая соединение III или его соль реакции циклизации. Эту реакцию проводят традиционным методом синтеза циклических пептидов, таким как метод с использованием смешанных ангидридов кислот, активированных сложных эфиров, карбодиимидный метод и тому подобные. Реакцию обычно проводят в традиционном растворителе, таком как спирт, тетрагидрофуран, этилацетат, N,N-диметилформамид, дихлорметан, хлороформ или любой другой растворитель, который не влияет отрицательно на реакцию. Температура реакции не является критической и реакцию обычно проводят при температурах от охлаждения до нагревания.
Способ 3. Соединение Ic или его соль можно получить, подвергая соединение Ib или его соль реакции деацилирования. Подходящий метод такой реакции может включать обычный метод, такой как гидролиз, восстановление и тому подобные.
i) Гидролиз.
Гидролиз предпочтительно проводят в присутствии основания или кислоты, включая кислоту Льюиса. Подходящее основание может включать неорганическое основание и органическое основание, такое как гидроокись, или карбонат, или бикарбонат щелочного металла (натрия, калия и так далее), щелочноземельного металла (т. е. магния, кальция, и так далее), триалкиламин (т,е, триметиламин, триэтиламин и так далее), пиколин, 1,5-диазабицикло(4.3.0)-оно- 5-ен, 1,4-диазабицикло(2,2,2)-октан, 1,8-диазабицикло-(5.4.0)-ундец-7-ен или тому подобные.
Подходящая кислота может включать органическую кислоту (т.е. муравьиную кислоту, уксусную кислоту, пропионовую кислоту, трихлоруксусную кислоту, трифторуксусную кислоту и так далее) и неорганическую кислоту (т.е. хлористоводородную кислоту, бромистоводородную кислоту, серную кислоту, хлористый водород, бромистый водород и так далее). Отщепление с использованием кислоты Льюиса, такой как тригалоидуксусная кислота (т.е. трихлоуксусная кислота, трифторуксусная кислота и так далее) или тому подобной предпочтительно проводят в присутствии улавливающих катионы агентов (т.е. анизол, фенол и так далее).
Реакцию обычно проводят в растворителе, таком как вода, спирт (т.е. метанол, этанол и так далее), хлористый метилен, тетрагидрофуран, их смесь или любой другой растворитель, который не оказывает вредного воздействия на реакцию. Жидкое основание или кислота может быть также использовано в качестве растворителя. Температура реакции не является критической и реакцию обычно проводят при температурах от охлаждения до нагревания.
i) Восстановление.
Восстановление проводят традиционным методом, включающим химическое восстановление и каталитическое восстановление. Подходящими катализаторами для использования при химическом восстановлении являются сочетание металла (т.е. цинка, железа и так далее) или металлического соединения (т.е. хлорида хрома, ацетата хрома и так далее) и органической или неорганической кислоты (т. е. муравьиной кислоты, уксусной кислоты, пропионовой кислоты, трифторуксусной кислоты, пара-толуолсульфокислоты, хлористоводородной кислоты, бромистоводородной кислоты и так далее).
Подходящими катализаторами, используемыми при каталитическом восстановлении, являются традиционные катализаторы, такие как платиновые катализаторы (т. е. платиновая пластинка, губчатая платина, платиновая чернь, коллоидная платина, окись платины, платиновая проволока и так далее), палладиевые катализаторы (т.е. губчатый палладий, палладиевая чернь, окись палладия, палладий на угле, коллоидный палладий, палладий на сульфате бария, палладий на карбонате бария и так далее), никелевые катализаторы (т.е. измельченный никель, окись никеля, никель Ренея и так далее), кобальтовые катализаторы (т.е. измельченный кобальт, кобальт Ренея и так далее), железные катализаторы (т. е. измельченное железо, железо Ренея и так далее), медные катализаторы (т.е. измельченная медь, медь Ренея, медь Ульмана и так далее) и тому подобные. Восстановление обычно проводят в традиционном растворителе, который не влияет отрицательно на реакцию, таком как вода, метанол, этанол, пропанол, N, N-диметилформамид, тетрагидрофуран или их смесь. В дополнение в случае использования упомянутой выше кислоты в жидком виде, применяемой при химическом восстановлении, она может быть также использована в качестве растворителя.
Температура реакции этого метода восстановления не является критической и реакцию обычно проводят при температурах от охлаждения до нагревания.
Способ 4. Соединение Ib или его соль можно получить, подвергая реакции ацилирования соединение Ic или его реакционноспособное производное при аминогруппе или его соль. Подходящее реакционноспособное производное при аминогруппе соединения Ic может включать основание Шиффа иминного типа или его таутомерный изомер енаминового типа, полученное при реакции соединения Ic с карбонильным соединением, таким как альдегид, кетон и тому подобное; силильное производное, полученное взаимодействием соединения Ic с силильным соединением, таким как бис-(триметилсилил)-ацетамид, моно-(триметилсилил)-ацетамид, бис-(триметилсилил)-мочевина или тому подобными; производное, образованное при взаимодействии соединения Ic с треххлористым фосфором или фосгеном и тому подобными.
Подходящее ацилирующее средство, используемое в настоящей реакции ацилирования, может включать традиционное ацилирующее средство и может быть выражено формулой
Ra1-OH (XIV) (где Ra1 имеет значение, определенное ранее) или его реакционноспособным производным или его солью.
Подходящее реакционноспособное производное соединения формулы XIV может включать галоидангидрид, ангидрид кислоты, активированный амид, активированный сложный эфир и тому подобное. Подходящим примером может быть хлорангидрид; азид кислоты, смешанный ангидрид кислоты с кислотой, такой как замещенная фосфорная кислота (т.е. диалкилфосфорная кислота, фенилфосфорная кислота, дифенилфосфорная кислота, дибензилфосфорная кислота, галогенизированная фосфорная кислота и так далее), диалкилфосфористая кислота, сернистая кислота, тиосерная кислота, сульфокислота (т.е. метансульфокислота, и так далее), алкилуглекислота, алифатическая карбоновая кислота (т.е. пивалиновая кислота, валериановая кислота, изовалериановая кислота, 2-этилмасляная кислота или трихлоруксусная кислота и так далее) или ароматическая карбоновая кислота (т.е. бензойная кислота и так далее); симметрический ангидрид кислоты, активированный амид с имидазолом, 4-замещенный имидазол, диметилпиразол, триазол или тетразол; или активированный сложный эфир (т.е. цианометиловый сложный эфир, метоксиметиловый эфир, диметилиминометиловый эфир ((CH3)2N+= CH-), виниловый эфир, пропаргиловый эфир, паранитрофениловый эфир, 2,4-динитрофениловый эфир, трихлорфениловый эфир, пентахлорфениловый эфир, метилсульфонилфениловый эфир, фенилазофениловый эфир, фениловый тиоэфир, паранитрофениловый тиоэфир, паракрезиловый тиоэфир, карбоксиметиловый тиоэфир, пираниловый эфир, пиридиловый эфир, пиперидиловый эфир, 8-хинолиловый тиоэфир, и так далее) или сложный эфир с N-гидроксисоединением (т.е. N,N-диметилгидроксиламин, 1-гидрокси-2-(1Н)-пиридон, N-гидроксисукцини- мид, N-гидроксифталимид, 1-гидрокси-6-хлор-1Н-бензотиазол и так далее) и тому подобные. Эти реакционноспособные произ- водные могут быть необязательно выбраны из них в соответствии с видом используемого соединения формулы XIV.
Реакцию обычно проводят в традиционном растворителе, таком как спирт (т. е. метанол, этанол и так далее), ацетон, диоксан, ацетонитрил, хлористый метилен, тетрагидрофуран, N,N-диметилформамид, пиридин или любой другой растворитель, который не влияет отрицательно на реакцию. Этот традиционный растворитель может быть также использован в смеси с водой.
Если соединение XIV используют в форме свободной кислоты или в форме ее соли в этой реакции, то реакцию предпочтительно проводят в присутствии традиционного конденсирующего средства, такого как N,N'-дициклогексилкарбодиимид, N-циклогексил- N'-морфолиноэтилкарбодиимид, N-циклогексил-N'-(4- диэтиламиноциклогексил)-карбодиимид, N,N'-диэтилкарбодиимид, N,N'-диизо- пропилкарбодиимид, N-этил-N'-(3-диметиламинопропил)- карбодиимид, N,N-карбонилбис-(2-метилимидазол), пентаметилен- кетен-N-циклогексиламин, дифенилкетен-N- циклогексилимин, этоксиацетилен, 1-алкокси-1-хлорэтилен, триалкилфосфит, этил- полифосфат, изопропиловый эфир полифосфорной кислоты, оксихлорид фосфора (хлористый фосфорил), треххлористый фосфор, хлористый оксалил, трифенилфосфин, хлористый тионил, соль 2-этил-7-оксибензизоксазолия, гидроокись 2-этил-5-(мета-сульфофенил)- изоксазолия, внутримолекулярная соль, 1-(парахлорбензолсульфонилокси)-6-лхлор-1Н-бен- зотриазол, так называемый реактив Вильсмайера, полученный взаимодействием N,N-диметилформамида с хлористым тионилом, фосгеном, трихлорметиловым эфиром хлормуравьиной кислоты, оксихлоридом фосфора и так далее.
Реакция может быть также проведена в присутствии неорганического или органического основания, такого как бикарбонат щелочного металла, три(низший)алкиламин, пиридин, N-(низший) алкилморфолин, N,N-ди(низший)алкилбензиламин или тому подобного. Температура реакции не является критической и реакцию обычно проводят при температурах от охлаждения до комнатной температуры.
Способ 5. Соединение Ie или его соль можно получить, подвергая соединение Id или его соль реакции ацилирования. На эту реакцию имеется ссылка в примерах 2, 4, 5, 7, 8, 17 и 18, описанных ниже.
Способ 6. Соединение If или его соль можно получить, подвергая гидролизу соединение Ia или его соль. Эту реакцию гидролиза можно провести со ссылкой на указанный выше способ 3.
Способ 7. Соединение Ig или его соль можно получить, подвергая соединение If или его соль реакции этерификации. Этерифицирующее средство, используемое в этой реакции, может включать одно из традиционных средств, такое как спирт или его реакционноспособный эквивалент (т.е. галогенид, сульфонат, сульфат, диазосоединение и так далее) или тому подобное.
Реакцию обычно проводят в традиционном растворителе, таком как ацетон, диоксан, спирт, хлористый метилен, хлористый этилен, н-гексан, тетрагидрофуран, этилацетат, N,N-диметилформамид или любой другой растворитель, который не оказывает отрицательного воздействия на реакцию. Температура реакции не является критической и реакцию обычно проводят при температурах от охлаждения до нагревания.
Способ 8. Соединение Ii или его соль можно получить, подвергая восстановлению соединение Ih или его соль. Подходящими катализаторами, используемыми при каталитическом восстановлении, являются традиционные восстановители, такие как платиновые катализаторы (т.е. платиновая пластинка, губчатая платина, платиновая чернь, коллоидная платина, окись платины, платиновая проволока и так далее), палладиевые катализаторы (т.е. губчатый палладий, палладиевая чернь, окись палладия, палладий на угле, коллоидный палладий, палладий на сульфате бария, палладий на карбонате бария и так далее), никелевые катализаторы (т. е. измельченный никель, окись никеля, никель Ренея и так далее), кобальтовые катализаторы (т.е. измельченный кобальт, кобальт Ренея и так далее), железные катализаторы (т.е. измельченное железо, железо Ренея и так далее), медные катализаторы (т.е. измельченная медь, медь Ренея, медь Ульмана, и так далее) и тому подобные.
Реакцию обычно проводят в традиционном растворителе, таком как ацетон, диоксан, спирт, тетрагидрофуран, этилацетат, диметилформамид, диметилсульфоксид или любой другой растворитель, который не влияет отрицательно на реакцию. Температура реакции не является критической и реакцию обычно проводят при температурах от охлаждения до нагревания.
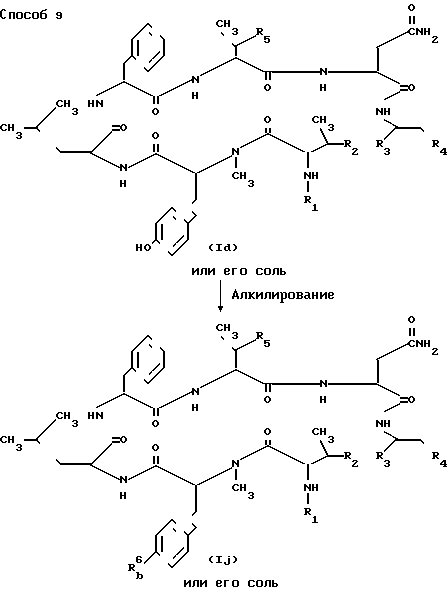
Способ 9. Соединение Ij или его соль можно получить, подвергая реакции алкилирования соединение Id или его соль. Эта реакция описана ниже со ссылкой на пример 19.
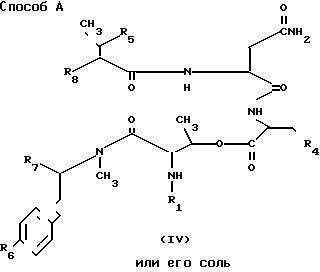
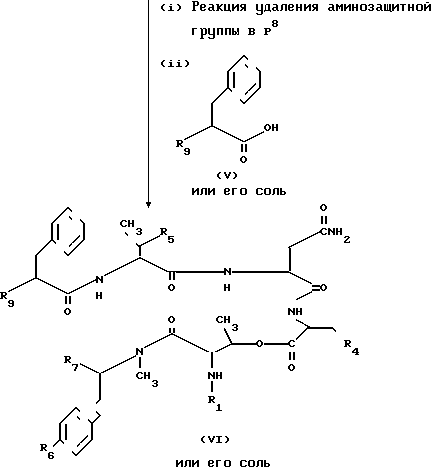
Способ А. Соединение II или его соль можно получить взаимодействием соединения IV или его соли согласно схеме реакции, показанной в способе А. Каждая реакция в этой схеме может быть проведена традиционным методом, принятым в синтезе пептидов. Исходное соединение IV или его соль можно получить методами, описанными ниже.
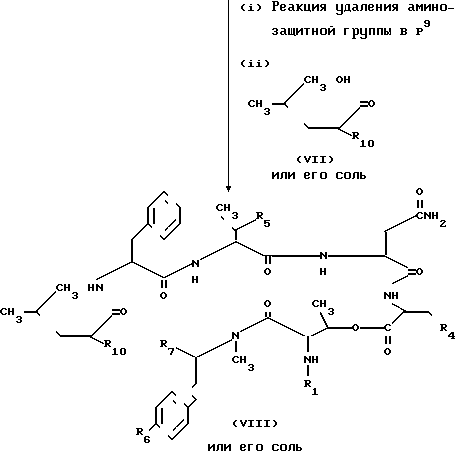
Способ В. Соединение III или его соль можно получить реакцией соединения IX или его соли по схеме реакции, показанной в способе В. Каждая реакция в этой схеме может быть проведена традиционным методом для пептидного синтеза. Исходное соединение IX или его соль можно получить методами, описанными ниже.
Получение ферментацией.
WS-9326В и WS-9326А согласно изобретению можно получить ферментацией WS-9326А и/или WS-9326В продуцирующих штаммов, принадлежащих к роду Streptomyces, такому как Streptomyces Violaceoniger N 9326 в питательной среде. Специфические микроорганизмы, использованные для получения WS-9326А и WS-9326В, будут описаны ниже.
Микроорганизмы.
Микроорганизм, который может быть использован для продуцирования WS-9326А и WS-9326В, является штаммом-продуцентом WS-9326А и/или WS-9326В, принадлежащим к роду Streptomyces, из которого Streptomyces violaceoniger N 9326 недавно был выделен из образца почвы, взятого в городе Суа префектура Нагано, Япония.
Лиофилизированный образец вновь выделенного Streptomyces violaceoniger N 9326 депонирован в научно-исследовательском институте агентства промышленных знаний и технологии (1-3, Higashi-1-chome, Tsukuba-shi, Ibaraki-ken, 305, Japan) под номером FEPM BP-1667 (дата депонирования 20 января 1988 года).
Понятно, что продуцирование новых WS-9326А и WS-9326В не ограничивается описываемым здесь конкретным организмом, который приведен лишь в целях иллюстрации. Это изобретение включает также использование любых мутантов, которые способны продуцировать WS-9326А и WS-9326В, включая природные мутанты, а также искусственные мутанты, которые могут быть получены от описанного здесь организма традиционными средствами, такими как облучение рентгеновскими лучами, ультрафиолетовым излучением, обработка N-метил-N'-нитро-N-нитрозогуанидином, 2-аминопурином и тому подобными. Streptomyces violaceoniger N 9326 имеет следующие морфологические, культуральные, биологические и физиологические характеристики.
Морфологические характеристики.
Были использованы методы, описанные Ширлингом и Готлибом (Shirling, E.B. and D.Sottlieb, Методы для характеристики видов Streptomyces, International Journal of Systematic Bacteriology, 16, 313-340, 1966) для этого таксономического исследования. Морфологические наблюдения были выполнены на оптическом и электронном микроскопах на культуре, выращенной при 30оС в течение 14 дней на агаре с дрожжево-солодовым экстрактом, на агаре из овсяной муки на агаре из крахмала и неорганических солей.
Вегетативный мицелий развит хорошо без фрагментации. Воздушный мицелий разветвлен монополиально и образует спиральные цепи спор с 10-30 спорами на цепь. Споры имеют гладкую поверхность и овальную форму с размерами 0,6-0,8 х 0,8-1,3 мкм. Отвердевших гранул, спорангия и зооспор не наблюдали.
Культуральные характеристики.
Культуральные характеристики наблюдали на десяти видах сред, описанных Ширлингом и Готлибом в указанном выше источнике и Ваксманом (Waksman, S.A. Actinomycetes, vol.2; Classification, identification and description of genera and species. The Williams and Wilkins Co. Baltimore, 1961). Выращивание проводили при 30оС в течение 21 дня. Название цвета, использованные в этом исследовании, взяты из колориметрического руководства Метуэна (kornerup, A. and J.H.Wanscher: Handbook of Colour, Methuen, London, 1978).
Культуральные характеристики штамма 9326 приведены ниже.
Даны следующие сокращения: G рост, А воздушный мицелий, R цвет обратной стороны, S растворимый пигмент.
Среда Культуральные характеристики л
л


 Агар из овсяной муки
Агар из овсяной муки 




 -
-






 зиновый
зиновый 






 тельный
тельный 

 Агар Беннета
Агар Беннета 




Воздушный мицелий имел окраску от серого до коричневато-серого цвета. Часть колонии становилась черной и влажной и показывала гигроскопические свойства на большинстве агаровых сред. Обратная сторона культуры была желтовато-коричневая, коричневая и темно-коричневая. Пигмент мицелия обратной стороны не чувствителен к рН среды. Меланоидные пигменты и другие растворимые пигменты не продуцирует.
Анализ клеточных стенок был выполнен по методам Беккера и соавторов (Becker B. M.P.Lechevalier, R.E.Jordon and H.A.Lechevalier "Быстрая дифференциация между Nocardia и Streptomyces хроматографией на бумаге цельного клеточного гидролизата". Appl. Microbiol. 12, 421-423, 1964) и Ямагуши (Jamagushi, T. "Сравнение состава клеточных мембран морфологически различных актиномицетов. J. Bacteriol. 89, 444-453, 1965). Анализ цельных клеточных гидролизатов штамма N 9326 показал присутствие LL-диаминопимелиновой кислоты. Следовательно, клеточная стенка этого штамма, по-видимому, типа I.
Биологические и физиологические свойства.
Физиологические свойства и усвоение источников углерода показаны ниже.
Использование источников углерода определяли методами Придхема и Готлиба (Pridham T.J. and D.Gottlieb "Использование углеродных соединений некоторыми актиномицетами, как вспомогательное средство определения видов". J.Bacteroil. 56, 107-114, 1948).
Физиологические свойства штамма N 9326
Условия Характеристики Температурный интервал для роста 11 47оС Оптимальный интервал температур для роста 29 31оС Разжижение желатина Положительно Свертывание молока Отрицательно Пептонизация молока Положительно Гидролиз крахмала Положительно Продуцирование меланоидного пигмента Отрицательно Разложение целлюлозы Отрицательно
Усвоение углерода штаммом N 9326
(+ означает усвоение).
Соединение Рост
D-глюкоза +
Сахароза +
D-ксилоза +
D-фруктоза +
L-рамноза +
Рафиноза +
L-арбиноза +
Инозит +
Маннит +
Морфология и химические характеристики штамма N 9326 позволяют четкое отнесение организма к роду Streptomyces.
Было установлено, что штамм N 9326 имеет близкое сходство со Streptomyces violaceoniger. Поэтому штамм N 9326 был идентифицирован как Streptomyces violaceoniger и назван Streptomyces violaceoniger N 9326.
Получение WS-9326А и WS-9326В.
Новые WS-9326А и WS-9326В согласно изобретению могут быть получены выращиванием штамма-продуцента WS-9326А и/или WS-9326В, принадлежащего к роду Streptomyces (т.е. Streptomyces violaceoniger N 9326, FEPM ВР-1667) в питательной среде. В общем WS-9326А и WS-9326В могут быть получены выращиванием штамма-продуцента WS-9326А и/или WS-9326В в водной питательной среде, содержащей источник усваиваемого углерода и азота, предпочтительно в аэробных условиях (т.е. встряхиваемая культура, погруженная культура и так далее).
Предпочтительными источниками углерода в питательной среде являются углеводы, такие как глюкоза, ксилоза, галактоза, глицерин, крахмал, декстрин и тому подобные. Другими источниками, которые могут быть использованы, являются мальтоза, рамноза, рафиноза, арабиноза, манноза, салицин, сукцинат натрия и тому подобные. Предпочтительными источниками азота являются дрожжевой экстракт, пептон, клейковина, хлопковая мука, соевая мука, экстракт от замачивания кукурузы, пшеничные проростки, высушенные дрожжи, молотое перо, порошок земляного ореха и так далее, а также неорганические и органические соединения азота, такие как соли аммония (т.е. нитрат аммония, сульфат аммония, фосфат аммония и так далее), мочевина, аминокислоты и тому подобные.
Источники углерода и азота, хотя и преимущественно используемые в сочетании, не обязательно используются в чистой форме, поскольку менее чистые материалы, которые содержат следы ростовых факторов и значительные количества минеральных питательных веществ, также пригодны для использования. Если желают, то они могут быть добавлены к минеральным солям среды, таким как карбонат натрия или кальция, фосфат натрия или калия, хлористый натрий или калий, иодистый натрий или калий, соли магния, соли меди, соли кобальта и тому подобные. Если необходимо, особенно когда культуральная среда сильно вспенивается, то могут быть добавлены антивспениватели, такие как жидкий парафин, жировое масло, растительное масло, минеральное масло или силикон.
Как условия для получения WS-9326А и WS-9326В в больших количествах предпочтительны аэробные условия погруженной культуры. Для получения небольших количеств используют встряхиваемые или поверхностные культуры в колбах или бутылях. Кроме того, если выращивание проводят в больших резервуарах, то предпочтительно использование вегетативной формы организма для инокулирования в ферментационные емкости, чтобы избежать латентный период развития культуры в процессе получения WS-9326А и WS-9326В. Таким образом, желательно сначала получить посевную культуру организма инокулированием относительно малого количества культуральной среды спорами или мицеллами микроорганизма и выращиванием этой инокулированной среды и затем перенести выращенный вегетативный инокулят в асептических условиях в большие резервуары. Среда, в которой получен вегетативный инокулят, является аналогичной или отличается от среды, используемой для получения WS-9326А и WS-9326В.
Перемешивание и аэрация культуральной смеси могут быть осуществлены различными путями. Перемешивание может быть проведено лопастями винта или аналогичными механическими мешалками, вращением или встряхиванием бродильного чана, различными нагнетающими устройствами или пропусканием стерильного воздуха через среду. Аэрация может быть осуществлена путем пропускания стерильного воздуха через ферментационную среду.
Брожение обычно проводят при температуре 20-40оС, предпочтительно при 25-35оС, в течение примерно 50-150 ч, которые могут варьировать в соответствии с условиями брожения и масштабами производства. Полученные WS-9326А и WS-9326В могут быть затем выделены из культуральной среды традиционными средствами, которые обычно используются для выделения других известных биологически активных веществ. Полученные WS-9326А и WS-9326В были найдены в культуральном фильтрате и мицелии, и таким образом WS-9326А и WS-9326В могут быть выделены и очищены из фильтрата и мицелия, которые получены при фильтрации или центрифугировании культуральной жидкости традиционным методом, таким как концентрирование при пониженном давлении, лиофилизация, экстракция традиционным растворителем, регулирование рН, обработка обычной смолой (т.е. анионо- или катионообменная смола, неионогенная абсорбирующая смола и так далее), обработка традиционным абсорбентом (т.е. активированным углем, кремневой кислотой, силикагельцеллюлозой, глиноземом и тому подобными), кристаллизация, перекристаллизация и тому подобные.
WS-9326А, полученный описанным выше способом, обладает следующими физическими и химическими свойствами.
Форма и цвет: бесцветный порошок.
Цветная реакция: положительная на сульфат церия, реакция на пары иода, реакция на хлористое железо (3) железо (3) цианистый калий; отрицательная реакция на нингидрин, реакция Молиша, реакция на хлористое железо (3), реакция Эрлиха, реакция Паули.
Растворимость: растворим метанол, этанол; слабо растворим ацетон, этилацетат; нерастворим вода, хлороформ.
Точка плавления 187-190оС.
Удельное вращение: (α)D23 -84o (С 1,0, метанол).
Спектр ультрафиолетового поглощения (метанол): λмакс= 280 нм (ε= 34,700).
Спектр поглощения инфракрасного излучения: (KBr) νмакс= 3300, 3050, 2950, 2920, 2860, 1730, 1650, 1610, 1560, 1540, 1530, 1510, 1440, 1380, 1340, 1280, 1240, 1170, 1110, 1080, 1060, 1040, 970, 880, 860, 830 см-1.
Вычислено, C 60,43; H 6,76; N 10,44
C54H68N8O13 2H2O
Найдено, C 60,18; H 6,61; N 10,32
Данные тонкослойной хроматографии приведены в табл.1.
Молекулярная формула C54H68N8O13.
Молекулярная масса (FAB-масс-спектр) m/z 1037 (М+Н)+.
Свойства вещества: кислое вещество.
Спектр ядерного магнитного резонанса на ядрах 13С: (100 МГц, СD3ОD), δ: 175,69 (с), 174,70 (с), 173,73 (с), 173,38 (с), 172,89 (с), 171,04 (с), 170,45 (с), 167,79 (с), 167,15 (с), 159,20 (с), 140,05 (д), 139,12 (с), 138,71 (с), 135,27 (д), 135,85 (с), 132,11 (д), 132,03 (с), 131,69 (д)х2, 130,70 (д), 129,90 (д), 130,70 (д), 129,90 (д), 129,61 (д)х2, 128,55 (д), 128,04 (д), 129,99 (д), 127,38 (д), 126,09 (с), 123,70 (д), 115,63 (д), 73,46 (д), 71,34 (д), 62,80 (т), 59,53 (д), 56,91 (д), 56,76 (д), 55,55 (д), 53,64 (д), 52,10 (д), 39,85 (т), 37,18 (т), 37,09 (т), 34,58 (кв), 31,37 (т), 24,56 (д), 23,63 (т), 22,71 (кв), 22,52 (кв), 21,17 (кв), 17,19 (кв), 14,13 (кв).
Спектр ядерного магнитного резонанса на ядрах 1Н, (400 МГц, СД3ОД) δ: 7,80 (1Н, д, I 8 Гц), 7,67 (1Н, д, I 16 Гц), 7,45-7,14 (9Н, м), 7,06 (2Н, д, I 8 Гц), 6,83 (1Н, с), 6,65 (2Н, д, I 8 Гц), 6,59 (1Н, д, I 12 Гц), 5,88 (1Н, дт, I 12 и 7 Гц), 5,55 (1Н, м), 5,35 (1Н, широкий сигнал), 5,10 (1Н, дд, I 3 и 9,5 Гц), 4,68 (1Н, д, I 10 Гц), 4,55 (1Н, т, I 6 Гц), 4,48 (1Н, дд, I 3 и 12 Гц), 3,92 (2Н, д, I 6 Гц), 3,70 (1Н, т, I 7,5 Гц), 3,62 (1Н, м), 3,46 (1Н, дд, I 3 и 14 Гц), 2,94 (1Н, дд, I 3 и 16 Гц), 2,89 (3Н, с), 2,74 (1Н, дд, I 9,5 и 16 Гц), 2,69 (1Н, дд, I 12 и 14 Гц), 2,14 (2Н, м), 1,5-1,4 (2Н, м), 1,20 (3Н, д, I 6 Гц), 1,08 (3Н, д, I 6 Гц), 1,0-0,8 (2Н, м), 0,91 (3Н, т, I 7 Гц), 0,6 (1Н, м), 0,53 (3Н, д, I 6 Гц), 0,51 (3Н, д, I 6 Гц).
Аминокислотный анализ.
WS-9326 (5 мг) гидролизовали при 110оС в течение 20 ч с хлористоводородной кислотой (2 мл) в запаянной пробирке. Смесь выпаривали досуха и получали продукты гидролиза, которые затем анализировали на автоматическом анализаторе аминокислот Хиташи 835.
Результаты аминокислотного анализа. Треонин (2), лейцин (1), фенилаланин (1), аспарагиновая кислота (1), серин (1), метиламин (1) и аммиак (1).
Что касается WS-9326А, то следует заметить, что спектры ядерного магнитного резонанса на ядрах 13С и 1Н, показали, что WS-9326А существует по меньшей мере в двух стабильных конформациях в растворе СД3ОД, и химические сдвиги являются сдвигами основного конформера WS-9326А. WS-9326В, полученный в соответствии с описанным выше способом, обладает следующими физическими и химическими свойствами.
Форма и цвет: бесцветный аморфный порошок
Цветная реакция: положительная реакция на сульфат церия, реакция на пары иода; отрицательная реакция на нингидрин.
Растворимость: растворим в метаноле, слабо растворим в этаноле и не растворим в воде, ацетоне, этилацетате и хлороформе.
Точка плавления 165-170оС (разлагается).
Удельное вращение: (α)D23 -64о (С 1,0, метанол)
Спектр ультрафиолетового поглощения (метанол): λмакс= 283 нм (ε= 27000).
Молекулярная формула C54H70N8O13
Вычислено, C 60,32; H 6,94; N 10,42
C54H70N8O13 2H2O
Найдено, C 59,97; H 6,87; N 10,29
Молекулярная масса (FAB-масс-спектр) m/z 1061,6 (М + а)+.
Данные тонкослойной хроматографии приведены в табл.2.
Спектр инфракрасного поглощения (KBr): νмакс 3300, 3050, 2950, 1735, 1660, 1530, 1510, 1450, 1400, 1380, 1340, 1260, 1220, 1080, 980, 920 см-1.
Спектр ядерного магнитного резонанса на ядрах 13С (100 МГц, СD3ОD) δ: 174,99 (с), 174,54 (с), 173,60 (с), 173,41 (с), 173,30 (с), 171,27 (с), 170,74 (с), 168,69 (с), 157,59 (с), 140,53 (д), 139,35 (с), 139,18 (с), 135,76 (д), 134,17 (с), 131,15 (д)х2, 130,93 (д), 130,35 (д), 130,93 (л), 130,35 (д), 129,88 (д)х2, 129,39 (д)х2, 128,70 (д), 129,58 (с), 128,13 (д), 127,64 (д), 127,53 (д), 121,99 (д), 116,45 (д)х2, 72,76 (д), 70,82 (д), 62,73 (т), 62,67 (д), 56,33 (д)х2, 56,19 (д), 53,36 (д), 52,24 (д), 40,24 (т), 37,55 (т), 37,08 (т), 33,69 (т), 31,57 (т), 29,93 (кв), 24,61 (д), 23,70 (кв), 23,59 (т), 22,16 (кв), 21,36 (кв), 17,12 (кв), 14,23 (кв).
Спектр ядерного магнитного резонанса на ядрах 1Н (400 МГц, СD3ОD) δ: 7,86 (1Н, д, I 16 Гц), 7,80 (1Н, широкий, д, I 8 Гц), 7,12-7,42 (11Н, м), 6,77 (2Н, д, I 8,5 Гц), 6,61 (1Н, д, I 11,5 Гц), 5,88 (1Н, дт, I 7,5 и 11,5 Гц), 5,08 (1Н, дд, I 3,5 и 10 Гц), 5,04 (1Н, кв, I 6,5 Гц), 4,66 (1Н, дд, I 3,5 и 13 Гц), 4,65 (1Н, д, I 11 Гц), 4,56 (1Н, дд, I 2,5 и 7 Гц), 4,48 (1Н, дд, I 4,5 и 11 Гц), 4,46 (1Н, с), 3,88 (2Н, м), 3,64 (2Н, м), 3,51 (1Н, дд, I 3,5 и 14 Гц), 3,17 (1Н, дд, I 4,5 и 14 Гц), 3,01 (1Н, дд, I 11 и 14 Гц), 2,94 (1Н, дд, I 3,5 и 16 Гц), 2,71 (3Н, с), 2,71 (1Н, дд, I 10 и 16 Гц), 2,64 (1Н, дд, I 13 и 14 Гц), 2,04 (2Н, м), 1,43 (2Н, м), 1,28 (2Н, м), 1,20 (3Н, д, I 6 Гц), 0,95 (3Н, д, I 6,5 Гц), 0,87 (3Н, т, I 7,5 Гц), 0,53 (1Н, м), 0,52 (6Н, д, I 10,5 Гц).
Что касается WS-9326В, то следует заметить, что спектры ядерного магнитного резонанса на ядрах 13С и 1Н, показывают, что WS-9326В существует по меньшей мере в двух стабильных конформациях в растворе СD3ОD и химические сдвиги являются сдвигами основного конформера WS-9326В.
Из анализа представленных выше физических и химических свойств и по результатам дальнейших исследований по идентификации химической структуры химическое строение WS-9326А и WS-9326В идентифицировано и интерпретировано следующим образом: WS-9326A
R-
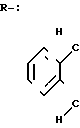

 где R /(E)-3-/2-((Z)-1-пентенил)-фенил/- пропеноил/ WS-9326B
где R /(E)-3-/2-((Z)-1-пентенил)-фенил/- пропеноил/ WS-9326B
R-
R (E)-3-/2-((Z)-1-пентенил)-фенил/-пропеноил
Подходящие фармацевтически приемлемые соли целевого соединения I являются традиционными нетоксичными солями и могут включать соль с основанием или кислотно-аддитивную соль, такую как соль с неорганичвеским основанием, например соль щелочного металла (т.е. соль лития, соль натрия, соль калия и так далее), соль щелочноземельного металла (т.е. соль кальция, соль магния и так далее), соль аммония; соль с органическим основанием, например, соль органического амина (т.е. соль триэтиламина, соль пиридина, соль николина, соль этаноламина, соль триэтаноламина, соль дициклогексиламина соль N,N'-дибензилэтилендиамина и так далее) и тому подобные; аддитивная соль неорганической кислоты (т. е. гидрохлорид, гидробромид, сульфат, фосфат, и так далее); аддитивная соль органической карбоновой или сульфокислоты (т.е. формиат, ацетат, трифторацетат, малеат, тартрат, метансульфонат, бензолсульфонат, паратолуолсульфонат и так далее); соль с основной или кислой аминокислотой (т.е. аргинин, аспарагиновая кислота, глутаминовая кислота и так далее) и тому подобные. Подходящие соли соединений (Ia-Ij), II и III могут быть названы соли, приведенные в качестве примера для соединения I.
Термин "низший" означает от 1 до 6 углеродных атомов, если не оговорено особо. Термин "высший" означает от 7 до 20 углеродных атомов, если не указано специально. Подходящий "ацил" или ацильная часть молекулы в термине "ацилокси"может включать карбамоил, алифатическую ацильную группу и ацильную группу, соедржащую ароматическое кольцо, которое относится к ароматическому ацилу, или гетероциклическое кольцо, которое относится к гетероциклическому ацилу. Подходящий пример упомянутого ацила может быть проиллюстрирован следующим образом.
Алифатический ацил, такой как низший или высший алканоил (т.е. формил, ацетил, пропаноил, бутаноил, 2-метилпропаноил, пентаноил, 2,2-диметилпропаноил, гексаноил, гептаноил, октаноил, наноил, деканоил, ундеканоил, додеканоил, тридеканоил, тетрадеканоил, пентадеканоил, гексадеканоил, гептадеканоил, октадеканоил, нонадеканоил, икозаноил и так далее); низший или высший алкоксисульфонил (т. е. метоксисульфонил, этоксисульфонил и так далее) или тому подобные.
Ароматический ацил, такой как ароил (т.е. бензоил, толуоил, нафтоил и так далее); ар (низший)алканоил, т.е. фенил (низший) алканоил (т.е. фенилацетил, фенилпропаноил, фенилбутаноил, фенилизобутаноил, фенилпентаноил, фенилгексаноил и так далее), нафтил (низший) алканоил (т.е. нафтилацетил, нафтилпропаноил, нафтилбутаноил и так далее) и тому подобные; ар(низший)алкеноил, т. е. фенил(низший)алкеноил, (т.е. фенилпропеноил, фенилбутеноил, фенилметакрилоил, фенилпентеноил, фенилгексеноил и так далее), нафтил(низший) алкеноил (т.е. нафтилпропеноил, нафтилбутеноил, нафтилпентеноил и так далее) и тому подобные; ар(низший) алкоксикарбонил, т.е. фенил(низший)алкоксикарбонил (т. е. бензилоксикарбонил, и так далее) и тому подобные арилоксикарбонил (т. е. феноксикарбонил, нафтилоксикарбонил и так далее); арил окси(низший)алканоил (т. е. феноксиацетил, феноксипропионил, и так далее); арилглиоксилоил (т. е. фенилглиоксилоил, нафтилглиоксилоил и так далее); аренсульфонил (т. е. бензолсульфонил, паратолуолсульфонил и так далее) или тому подобные.
Гетероциклический ацил, такой как гетероциклический карбонил (т.е. теноил, фуроил, никотиноил и так далее); гетероциклический (низший)алканоил (т.е. тиенилацетил, тиенилпропаноил, тиенилбутаноил, тиенилпентаноил, тиенилгексаноил, тиазолилацетил, тиадиазолилацетил, тетразолилацетил и так далее); гетероциклический глиоксилоил (т.е. тиазолилглиоксилоил, тиенилглиоксилоил и так далее) или тому подобные, в которых подходящая гетероциклическая часть молекулы в терминах "гетероциклический карбонил", "гетероциклический (низший)алканоил" и "гетероциклический глиоксилоил" как упоминалось выше означает более детально насыщенный или ненасыщенный, моноциклический или полициклический радикал, содержащий по меньшей мере один гетероатом, такой как кислород, серу, азот и тому подобные. И особенно предпочтительной группой может быть гетероциклическая группа, такая как ненасыщенная 3-8-членная, более предпочтительно 5-6-членная гетероциклическая группа, содержащая от 1 до 4 атомов азота, например пирроил, пиролинил, имидазолил, пиразолил, пиридил и его N-оксид, дигидропиридил, пиримидил, пиразинил, пиридазинил, триазолил (т.е. 4Н-1,2,4-триазолил, 1Н-1,2,3-триазолил, 2Н-1,2,3-триазолил и так далее), тетразолил (т.е. 1Н-тетразолил, 2Н-тетразолил, и так далее) и тому подобные; насыщенная 3-8-членная (более предпочтительно 5-6-членная) гетероциклическая группа, содержащая 1-4 атома азота, например пирролидинил, имидазолидинил, пиперидино, пиперазинил и так далее; ненасыщенная гетероциклическая конденсированная группа, содержащая от 1 до 4 атомов азота, например индолил, изоиндолил, индолизинил, бензимидазолил, хинолил, изохинолил, индазолил, бензотриазолил, и так далее; ненасыщенная 3-8-членная (более предпочтительна 5-6-членная гетеромоноциклическая группа, содержащая от 1 до 2 атомов кислорода и 1-3 атома азота, например, оксазолил, изоксазолил, оксадиазолил (т.е. 1,2,4-оксадиазолил, 1,3,4-оксадиазолил, 1,2,5-оксадиазолил и так далее) и тому подобные; насыщенная 3-8-членная (более предпочтительна 5-6-членная) гетеромоноциклическая группа, содержащая 1-2 атома кислорода и 1-3 атома азота, например морфолинил, зиднонил и так далее; ненасыщенная конденсированная гетероциклическая группа, содержащая 1-2 атома кислорода и 1-3 атома азота, например бензоксазолил, бензоксадиазолил и так далее; ненасыщенная 3-8-членная (более предпочтительная 5-6-членная) гетеромоноциклическая группа, содержащая 1-2 атома серы и 1-3 атома азота, например тиазолил, изотиазолил, тиадиазолил (т. е. 1,2,3-тиадиазолил, 1,2,4-тиадиазолил 1,3,4-тиадиазолил, 1,2,5-тиадиазолил и так далее), дигидротиазинил и так далее; насыщенная 3-8-члденная (более предпочтительна 5-6-членная) гетеромоноциклическая группа, содержащая один-два атома серы, например тиенил, дигидродитиинил, дигидродитионил и так далее; ненасыщенная конденсированная гетероциклическая группа, содержащая один-два атома серы и один-три атома азота, например бензотиазолил, бензотиадиазолил и так далее: ненасыщенная 3-8-членная (более предпочтительно 5-6-членная гетеромоноциклическая группа, содержащая атом кислорода, например фурил и так далее; ненасыщенная 3-8-членная (более предпочтительна 5-6-членная) гетеромоноциклическая группа, содержащая атом кислорода и один-два атома серы, например дигидрооксатиинил и так далее); ненасыщенная конденсированная гетероциклическая группа, содержащая 1-2 атома серы, например бензотиенил, бензодитиинил и так далее; ненасыщенная конденсированная гетероциклическая группа, содержащая атом кислорода и один-два атома серы, например бензоксатиинил и так далее и тому подобные. Ацильная часть молекулы, как утверждалось выше, может иметь от одного до десяти одинаковых или различных подходящих заместителей, таких как низших алкил (т.е. метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, пентил, гексил и так далее); низший алкенил (т.е. винил, аллил, 1-пропенил, 1- или 2- или 3-бутенил, 1- или 2- или 3- или 4-пентенил, 1- или 2- или 3- или 4- или 5-гексенил и так далее); низший алкокси (т.е. метокси, этокси, пропокси и так далее); низший алкилтио (т.е. метилтио, этилтио и так далее); низший алкиламино (т.е. метиламино и так далее); цикло(низший)алкил (т.е. циклопентил, циклогексил и так далее); цикло(низший)алкенил (т.е. циклогексенил, и так далее); галоген; амино; защищенное амино; окси; защищенная оксигруппа; циано; нитро; карбокси; защищенная карбоксигруппа; сульфо; сульфамоил, имино, оксо; амино(низший)алкил (т.е. аминометил, аминоэтил и так далее); карбомоилокси: гидрокси(низший) алкил (т.е. гидроксиметил, 1- или 2-гидроксиэтил, 1- или 2- или 3-гидроксипропил и так далее); циано(низший)алкенилтио (т.е. циановинилтио и так далее) или тому подобные.
Подходящая гидроксизащитная группа в термине "защищенная гидроксигруппа" может включать фенил(низший) алкил (т.е. бензил и так далее), ацил, как упомянуто выше, и тому подобные. Подходящая "защищенная карбоксигруппа" может в ключать этерифицированную карбоксигруппу.
Подходящим примером сложного эфирного фрагмента в этерифицированной карбоксигруппе может быть фрагмент низшего алкилового эфира (т.е. метилового эфира, этилового эфира, пропилового эфира, изопропилового эфира, бутилового эфира, изобутилового эфира, трет-бутилового эфира, пентилового эфира, гексилового эфира, 1-циклопропилэтилового эфира и так далее), который может иметь по меньшей мере один подходящий заместитель, например, низший алканоилокси (низший)-алкиловый сложный эфир (например ацетоксиметиловый эфир, пропионилоксиметиловый эфир, бутирилоксиметиловый эфир, валерилоксиметиловый эфир, пивалоилоксиметиловый эфир, гексаноилоксиметиловый эфир, 1- или 2-ацетоксиэтиловый эфир, 1- или 2- или 3-ацетоксипропиловый эфир, 1- или 2-или 3- или 4-ацетоксибутиловый эфир, 1- или 2-пропионилоксиэтиловый эфир, 1- или 2- или 3-пропионилоксипропиловый эфир, 1- или 2-бутирилоксипропиловый эфир, 1-или 2-изобутирилоксиэтиловый эфир, 1- или 2-пивалоилоксиэтиловый эфир, 1- или 2-гекса- ноилоксиэтиловый эфир, изобутирилоксиметиловый эфир, 2-этилбутирилоксиметиловый эфир, 3,3-диметилбутирилоксиметиловый эфир, 1- или 2-пентаноилоксиэтиловый эфир и так далее, низший алкансульфонил (низший)алкиловый сложный эфир (т. е. 2-метилсульфониловый эфир и так далее), моно-, ди- или три-галоидо (низший)алкиловый сложный эфир (т. е. 2-иодоэтиловый эфир, 2,2,2-трихлорэтиловый эфир и так далее); низший алкоксикарбонилокси (низший)алуиловый сложный эфир (т. е. метоксикарбонилоксиметиловый эфир, этоксикарбонилоксиметиловый эфир, 2-метоксикарбонилоксиэтиловый эфир, 1-этоксикарбонилоксиэтиловый эфир, 1-изопропоксикарбонилоксиэтиловый эфир и так далее), фталидилиден (низший)алкиловый сложный эфир или (5-низший алкил-2-оксо-1,3-диоксол-4-ил) (низший)алкиловый сложный эфир, т.е. (5-метил-2-оксо-1,3-диоксол -4-ил)-метиловый эфир, (5-этил-2-оксо-1,3-диоксол-4-ил)-метиловый эфир, (5-пропил-2-оксо- 1,4-диоксол-4-ил)-этиловый эфир и так далее; низший алкениловый сложный эфир (т.е. виниловый эфир, алкиловый эфир и так далее); низший алкиниловый эфир (т.е. этиниловый эфир, пропиниловый эфир и так далее); ар(низший)алкиловый сложный эфир, который может иметь по меньшей мере один подходящий заместитель, такой как моно- (или ди- или три-) фенил(низший)алкиловый сложный эфир, который может иметь по меньшей мере один подходящий заместитель (т.е. бензиловый эфир, 4-метоксибензиловый эфир, 4-нитробензиловый эфир, фенетиловый эфир, тритиловый эфир, бензгидриловый эфир, бис-(метоксифенил)-метиловый эфир, 3,4-диметоксибензиловый эфир, 4-окси-3,5-ди-трет-бутилбензиловый эфир и так далее); ариловый сложный эфир, который может иметь по меньшей мере один подходящий заместитель (т.е. фениловый эфир, 4-хлорфениловый эфир, толиловый эфир, трет-бутилфениловый эфир, ксилиловый эфир, мезитиловый эфир, кумениловый эфир и так далее); фталидиловый эфир и тому подобные.
Подходящий "низший алкокси" может включать метокси, этокси, пропокси, изопропокси, бутокси, изобутокси, трет-бутокси, пентилокси, гексилокси и тому подобные.
Подходящая "аминозащитная группа" в термине "защищенная аминогруппа" может включать ацил, как упоминалось выше, и тому подобное. Подходящий "ар(низший)алкеноил" в термине "ар(низший) алкеноил, замещенный низшей алкенильной группой" может включать фенил(низший)алкеноил (т.е. фенилпропеноил, фенилбутеноил, фенилметакрилоил, фенилпентеноил, фенилгексеноил и так далее), нафтил(низший)алкеноил (т.е. нафтилпропеноил, нафтилбутеноил, нафтилпентеноил, и так далее) и тому подобные.
Подходящим "низшим алкенилом" в термине "ар(низший)алкеноил, замещенный низшей алкенильной группой" может быть винил, аллил, 1-пропенил, 1- или 2- или 3-бутенил, 1- или 2- или 3- или 4-пентенил, 1- или 2- или 3- или 4- или 5-гексенил и тому подобные.
Подходящий "ар(низший)алканоил" в термине "ар(низший)алканоил, замещенный низшей алкильной группой" может включать фенил (низший)алканоил (т.е. фенилацетил, фенилпропаноил, фенилбутаноил, фенилизобутилил, фенилпентаноил, фенилгексаноил и так далее), нафтил(низший)алканоил (т.е. нафтилацетил, нафтилпропаноил, нафтилбутаноил и так далее) и тому подобные. Подходящий "низший алкил" в термине "ар(низший)алкил, замещенный низшей алкильной группой" может включать метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, пентил, гексил и тому подобные.
Предпочтительными осуществлениями целевого соединения (1) являются следующие
R1 представляет собой водород, ар(низший)алкоксикарбонил, (более предпочтителен фенил(низший)алкоксикарбонил), низший алканоил, высший алканоил (более предпочтителен алканоил с 15-20 углеродными атомами), ароил (более предпочтителен бензоил), гетероциклический (низший)алканоил (более предпочтителен тиенил (низший)алканоил), ар(низший)алкеноил, замещенный низшей (алкенильной группой (более предпочтителен фенил(низший)алкеноил, замещенный низшей алкенильной группой) или ар(низший)алканоил, замещенный низшей алкильной группой (более предпочтителен фенил(низший)алканоил, замещенный низшей алкильной группой);
R2 является гидроксигруппой и
R3 означает карбоксигруппу или этерифицированную карбоксигруппу (более предпочтителен низший алкоксикарбонил) или
R2 и R3, соединенные вместе, образуют группу формулы -O- -;
-;
R4 представляет собой гидроксигруппу, ар(низшую)алкоксигруппу (более предпочтительно фенил(низшую)алкоксигруппу) или ацилоксигруппу (более предпочтительно низшую алканоилоксигруппу);
R5 представляет собой гидроксигруппу, ар(низшую)алкоксигруппу (более предпочтительно фенил(низший)алкокси) или ацилоксигруппу (более предпочтительно низшую алканоилоксигруппу);
R6 представляет собой гидроксигруппу, низшую алкоксигруппу, ар(низшую)алкоксигруппу (более предпочтительно фенил(низшую) алкоксигруппу) или ацилоксигруппу (более предпочтительно низший алканоилокси) и простая связь или двойная связь.
простая связь или двойная связь.
Биологические свойства пептидных производных.
Пептидные производные формулы (1) и их фармацевтически приемлемые соли обладают фармакологическими активностями, такими как антагонизм к веществу Р, антагонизм к нейтрокинину А (веществу К) или тому подобным и поэтому они полезны для профилактики и лечения астмы и тому подобного.
В качестве примера демонстрации такой фармакологической активности приводятся ниже данные фармакологических исследований.
(1) анализ на связывание меченого лиганда
а) получение неочищенной мембраны.
Мозг (головной).
Использовали самцов крыс линий Вистар (средний вес 200 г) и все реактивы были закуплены у Sigma Chemical Company. Весь мозг (4 г) измельчали на мелкие кусочки и гомогенизировали в 8 объемах охлажденной льдом среды 1 (50 ммолей трис-HCl, рН 7,5 5 ммолей хлористого марганца, 0,02% альбумина бычьей сыворотки, 2 мкг/мл химостатина, 4 мкг/мл лейпептина и 40 мкг/мл бацитрацина) на ультра-диспергаторе (Jamato модель LK-21). Гомогенат либо хранили при -20оС, либо использовали немедленно в экспериментах на связывание.
Легкие.
Самцов альбиносов морских свинок линии Хартлей (вес 600 г) умерщвляли обезглавливанием. Удаляли трахеи и легкие и хранили при -80оС до использования. Эти ткани (150 г) оттаивали и гомогенизировали в 500 мл буфера (0,25 М сахарозы, 50 ммолей трис-HCl с рН 7,5, 0,1 ммоля этилендиаминтетрауксусной кислоты) на компактном смесителе (Matsuden MI-761). Ткань гомогенизировали в ультра-дисперсере (Jamato модель LK-21) с установкой максимального интервала в 10 с при 10 с интервалах с охлаждением между гомогенизациями (общее время гомогенизации составляло 60 с). Гомогенат центрифугировали (при 900 х g в течение 10 мин) для удаления сгустков ткани и надосадочный слой центрифугировали при 14000 х g в течение 20 мин, чтобы получить шарики, которые расценивались как сырые мембранные фракции. Шарики повторно суспендировали в среде 1, гомогенизировали на тефлонном гомогенизаторе и центрифугировали при 14000 х g в течение 20 мин. Шарики хранили при -20оС.
в) связывание меченого тритием вещества Р с препаративными мембранами.
Меченое тритием вещество Р (1 нмоль, New England Nuclaar) инкубировали с 50 мл мембранного препарата в среде 1 при 4оС в течение 30 мин в конечном объеме 250 мл. В конце инкубационного периода его содержимое быстро фильтровали через фильтр Whatman GF/B из стекловолокна (предварительно обработанный 0,1% -ным полиэтиленимином в течение 3 ч до использования), используя собиратель клеток (Brandel M-24S). Фильтры затем промывали десять раз общим количеством 3 мл буфера (50 ммолей трис-HCl с рН 7,5) при 0оС. Радиоактивность считали в 3 мл аквазола-2 в сцинтилляционном счетчике Паккарда (Packard FRJ-CARB 4530).
(2) Действие WS-9326А или тетрагидро-WS-9326В на трахею морских свинок.
Спиральные полоски трахеи получали от взрослых самцов белых морских свинок линии Хартлей (600 г) стандартным методом и помещали в заключенную в футляр 30 мл стеклянную тканевую ванну. Растяжение трахеальных полос измеряли изометрически посредством преобразования усилия сдвига, соединенного с полиграфом (биопсиограф 180 система San-Ei Instrument). Трахеальные полосы (1 мм шириной и 50 мм длиной) суспендировали при остаточном растягивающем напряжении 500 мг в 30 мл банях, содержащих теплый (37оС) насыщенный кислородом (95% кислорода, 5% углекислого газа) раствор Тирода следующего состава: NaCl 137 ммолей (8 г/л), KCl 2,7 ммоля (0,2 г/л), CaCl2 ˙ 2H2O 1,8 ммоля (0,264 г/л), MgCl2 ˙ 6H2O 1,02 ммоля (0,208 г/л), NaHCO3 11,9 ммоля (1г/л), NaH2PO4 ˙ 2H2O 0,42 ммоля (0,0666 г/л) и глюкозы 5,5 ммоля (1 г/л). Ткани приводили в состояние равновесия в течение 90 мин и затем испытывали WS-9326А или тетрагидро-WS-9326А против различных бронхоконстрикторов (вещество Р 10-8 М и нейрокинин А 10-9 М). Растяжение регистрировали записывающим устройством (San-Ei Rectigraph-8S, San-Ei Instrument).
3) Действие WS-9326А или тетрагидро-WS-9326А на сужение бронхов, вызванное нейрокинином А и капсаицином.
Самцов морских свинок линии Хартлей, весивших 300-500 гл, обездвиживали пентобарбиталом натрия (10 мг/животное, вводимым внутрибрюшинно). Яремную вену канюлировали для введения нейрокинина А (или капсаицина) и лекарства. Катетер был также интубирован в трахею для искусственной вентиляции легких. Животное получало вдыхаемый воздух посредством миниатюрного дыхательного насоса (Harvard B-34, производительность 5 мл/толчок, 60 толчков в минуту). Устойчивость к раздуванию легких измеряли модифицированным методом обильного излияния Концетта-Ресслера.
Агонист вводили внутривенно и антагонистическое лекарство вводили согласно схеме, приведенной на фиг.1 (приготовлено в смеси 0,1% метилцеллюлозы и физиологического раствора).
(4) Влияние внутритрахеального введения WS-9326А или тетрагидро-WS-9326А на индуцированное нейрокинином А сужение бронхов у морских свинок.
Для испытания действия ингаляции WS-9326А или тетрагидро-WS-9326А на сужение бронхов WS-9326А или тетрагидро-WS-9326А растворяли в диметилсульфоксиде и вводили внутритрахеально. Метод почти не отличался от описанного выше.
Как показано в табл.7 и 8, WS-9326А и тетрагидро-WS-9326А оказались сильнодействующими средствами.
5) Острая токсичность.
Острую токсичность WS-9326А определяли на мышах ddI (в возрасте 5 недель, самцы) однократной инъекцией возрастающей дозы испытуемого соединения внутрибрюшинно 5 мышам. Значение летальной дозы ЛД50 WS-9326А было выше 250 мг/кг, не ниже 500 мг/кг (500 мг/кг > ЛД50 > 250 мг/кг).
Фармацевтическая композиция согласно изобретению может быть использована в форме фармацевтического препарата, например, в твердой, полутвердой или жидкой форме, которая содержит пептидные производные формулы (1) или их фармацевтически приемлемые соли в качестве активного ингредиента в смеси с органическим или неорганическим носителем или наполнителем, пригодным для внешнего, внутреннего или парентерального применения. Активный ингредиент может быть смешан, например, с обычным нетоксичным фармацевтически приемлемым носителем (ями) для изготовления таблеток, гранул, капсул, растворов, эмульсий, суспензий и других форм, пригодных для использования. Носители, которые могут быть использованы, включают воду, глюкозу, лактозу, аравийскую камедь, желатин, маннит, крахмальную пасту, трисиликат магния, тальк, кукурузный крахмал, кератин, коллоидную двуокись кремния, картофельный крахмал, мочевину и другие носители, пригодные для использования при производстве препаратов в твердом, полутвердом или жидком состоянии, и в дополнение могут быть использованы вспомогательные средства, стабилизаторы, осушители, красители и ароматические вещества. Активное целевое соединение включают в фармацевтическую композицию в количестве, достаточном для получения желаемого действия в зависимости от течения процесса и условий заболеваний.
Поскольку дозирование терапевтически эффективного количества пептидных производных формулы (1) или их фармацевтически приемлемых солей варьируется в зависимости от возраста и состояния каждого отдельного больного, подлежащего лечению, то в общем вводят для лечения болезни дневную дозу 0,01-1000 мг, предпочтительно 0,1-500 мг и наиболее предпочтительно 0,5-100 мг активного ингредиента и однократную дозу примерно 0,5 мг, 1 мг, 5 мг, 10 мг, 50 мг, 100 мг, 250 мг и 500 мг.
В этом описании аминокислоты, пептиды, защитные группы и так далее указаны сокращениями, принятыми 1UPAC-1UB (комисией по биологической номенклатуре), которые являются обычными в данной области. Кроме того, в последующих примерах и получениях использованы и другие сокращения в дополнение к сокращениям, принятым Комиссией по биологической номенклатуре.
В описании использованы следующие сокращения:
Thr L-треонин
Ser L-серин
Tyr L-тирозин
Asn L-аспарагин
allo-Thr L-аллотреонин
D-Phe D-фенилаланин
Leu L-лейцин
n-Hex н-гексан
Et этил
Z бензилоксикарбонил
Рас фенацил
Bзl бензил
Вос трет-бутоксикарбонил
Me метил
Tce 2,2,2-трихлорэтил
Mmp 4-метоксиметоксифенил
Sit трет-бутилметилсилил
Ас ацетил
П р и м е р 1. Ферментация. Водная посевная среда (160 мл), содержащая растворимый крахмал (1%), сахарозу (1%), глюкозу (1%), хлопковую муку (1%), пептон (0,5%), соевую муку (0,5%) и карбонат кальция (0,2%) (рН устанавливали 7,0 посредством 6н гидроокиси натрия), была налита в каждую из двадцати 500 мл колб Эрленмейера и стерилизована при 120оС в течение 30 мин. Петлю культуры Streptomyces violaceoniger N 9326 на скошенном агаре инокулировали в каждую из сред и выращивали на роторном вибраторе (220 оборотов в минуту, дуга качания 5,1 см) при 30оС в течение трех дней. Полученная посевная культура была внесена в 160 л стерильной ферментационной среды, содержащей глицерин (3%), соевую муку (0,5%), порошок сои (1,5%), карбонат кальция (0,2%) и иодистый натрий (NaI) (0,001%) в 200-литровом встряхиваемом бродильном баке из нержавеющей стали. Брожение проводили при 30оС три дня при аэрации 160 л/мин и перемешивании при 200 оборотах в минуту. Количество WS-9326А в бродильном бульоне оценивали количественно методом высокопроизводительной жидкостной хроматографии с использованием насоса Hitachi модель 655. Использовали колонку из стали (4,6 мм внутренний диаметр, высота 250 мм), упакованную R-ODS-5 (УМС упакованная колонка) при скорости тока 1,0 мл/мин. В качестве подвижной фазы использовали смесь метанола и воды в отношении 8:2. Пробу для высокопроизводительной жидкостной хроматографии готовили следующим образом. Равный объем ацетона добавляли к бульону при энергичном перемешивании и выдерживали один час и затем центрифугировали. 5 мл надосадочного слоя инъецировали в инжектор проб Hitachi модель 655.
Выделение и очистка. Равное количество ацетона добавляли к культуральному бульону (150 л) при перемешивании. Смесь выдерживали при комнатной температуре 1 ч и затем фильтровали. Фильтрат концентрировали до 80 л при пониженном давлении, устанавливали рН 7,0 хлористоводородной кислотой и затем экстрагировали 80 л этилацетата. Экстракт концентрировали досуха при пониженном давлении и помещали на колонку силикагеля (кизельгель 60, 70-230 меш, 3 л). Колонку промывали н-гексаном (10 л), смесью н-гексанэтилацетат в отношении 1:1 (10 л), этилацетатом (20 л) и активное вещество элюировали из колонки ацетоном (6 л). Активные фракции сушили при пониженном давлении и подвергали колоночной хромато- графии на силикагеле (кизельгель 60, 70-230 меш, Merck 1,2 л). Колонку промывали смесью хлороформ-метанол в отношении 20: 1 (5 л) и целевое соединение элюировали раствором хлороформ-метанол в отношении 10: 1 (6 л). Фракции сушили при пониженном давлении и получали порошок. Порошок растворяли в небольшом количестве метанола и вносили на колонку NS-геля (Nihon Seimitsu 500 мл). Целевое соединение элюировали смесью метанол-вода в отношении 8:2 (2 л) и концентрировали до 300 мл при пониженном давлении и затем экстрагировали 500 мл этилацетата. Экстракт концентрировали досуха при пониженном давлении и получали порошок (5 г). Порошок (5 г) растворяли в 10 мл метанола (500 мг/мл) и подвергали высокопроизводительной жидкостной хроматографии с использованием стальной колонки (20 мм внутренний диаметр, 250 мм длиной), упакованной D-ODS-5 (УМС-упакованная колонка) и элюировали смесью метанол-вода в отношении 8:2 при скорости потока 9,9 мл/мин. Полученная таким образом активная фракция концентрировалась при пониженном давлении и затем экстрагировалась этилацетатом. Экстракт концентрировали досуха при пониженном давлении и получали чисто белый порошок (150 мг) WS-9326А.
П р и м е р 2. К раствору WS-9326А (300 мг) в пиридине (4,5 мл) добавляли уксусный ангидрид (1,5 мл) и 4-диметилалминопиридин (1 мг) и реакционную смесь оставляли на ночь при комнатной температуре. Реакционную смесь выпаривали досуха и получали масло, которое очищали препаративной тонкослойной хроматографией (смесь хлороформ-метанол в отношении 10:1). Полученный продукт растирали с диэтиловым эфиром и получали триацетил-WS-9326А (332 мг) в виде бесцветного порошка. Физические и химические свойства триацетил-WS-9326А следующие:
Форма и цвет: бесцветный порошок.
Цветная реакция: реакция положительная на сульфат церия, на серную кислоту и на пары иода, отрицательная: реакция на нингидрин.
Растворимость: растворяется в метаноле, диметилсульфоксиде; слабо растворим в хлороформе, диэтиловом эфире; нерастворим в н-гексане.
Точка плавления 141-143оС.
Удельное вращение: (α)D23-122о (с 1,0, метанол).
Спектр ультрафиолетового поглощения (метанол); λмакс= 283 нм (ε= 132000).
Молекулярная формула C60H74N8O16.
Вычислено, C 60,09; H 6,56; N 9,34
C60H74N8O16 ˙ 2H2O
Найдено, C 60,19; H 6,42; N 9,27
Молекулярный вес: FAB-масс-спектр: m/z 1163,6 (М+Н)+.
Данные тонкослойной хроматографии приведены в табл.9.
11) спектр инфракрасного поглощения:
νмаксКВг 3350, 3020, 2950, 2920, 2850, 1730, 1650, 1520, 1440, 1360, 1230, 1200, 1160, 1100, 1060, 1040, 910 см-1;
12) свойства вещества: нейтральное вещество;
13) спектр ядерного магнитного резонанса на ядрах 13С: (100 МГц, СDCl3-CD3OH в отношении 10:1) δ: 174,20 (с), 173,23 (с), 173,06 (с), 171,32 (с), 171,02 (с), 170,84 (с), 169,79 (с), 169,59 (с), 169,55 (с), 168,52 (с), 167,03 (с), 166,36 (с), 151,02 (с), 140,74 (д), 138,82 (с), 138,74 (с), 137,12 (с), 135,23 (д), 133,75 (с), 131,31 (с), 130,20 (д), 129,96 (д)х2, 129,34 (д), 129,21 (д)х2, 128,56 (д)х2, 127,24 (д), 126,95 (д), 126,74 (д), 126,63 (д), 126,50 (д), 122,10 (д)х2, 121,29 (д), 70,99 (д), 69,22 (д), 63,73 (т), 58,13 (д), 56,10 (д), 53,22 (д), 52,66 (д), 52,18 (д), 49,93 (д), 39,75 (т), 39,39 (кв), 39,06 (т), 35,75 (т), 30,65 (т), 24,26 (д), 23,15 (кв), 22,79 (т), 21,42 (кв), 21,21 (кв), 20,99 (кв), 20,83 (кв), 17,05 (кв), 16,18 (кв), 13,82 (кв).
14) спектр ядерного магнитного резонанса на ядрах 1Н: (400 МГц, CDCl3-CD3OH в отношении 10:1): 8,25 (1Н, д, I 8 Гц), 8,02 (1Н, д, I 8 Гц), 7,88 (1Н, д, I 16 Гц), 7,86 (1Н, д, I 8 Гц), 7,70 (1Н, д, I 6 Гц), 7,61 (1Н, д, I 8 Гц), 7,45 (1Н, д, I 7 Гц), 7,32-7,15 (6Н, м), 7,03 (2Н, д, I 8 Гц), 7,00-6,94 (3Н, м), 6,88-6,79 (4Н, м), 6,70 (1Н, с), 6,49 (1Н, д, I 12 Гц), 5,76 (1Н, дт, I 12 и 7,5 Гц), 5,54 (1Н, широкий синглет), 5,50-5,45 (2Н, м), 4,93 (1Н, м), 4,75 (1Н, м), 4,65-4,56 (2Н, м), 4,46 (1Н, дд, I 6 и 11 Гц), 4,41 (1Н, т, I 6 Гц), 4,22 (1Н, м), 4,18 (1Н, дд, I 8 и 11 Гц), 3,56 (3Н, с), 2,90 (1Н, дд, I 6 и 16 Гц), 2,85-2,80 (2Н, м), 2,56 (1Н, дд, I 4 и 16 Гц), 2,26 (3Н, с), 2,00 (3Н, с), 1,96-1,89 (2Н, м), 1,85 (3Н, с), 1,58 (1Н, м), 1,35 (3Н, д, I 6 Гц), 1,32-1,20 (3Н, м), 1,07 (3Н, д, I 6 Гц), 0,84 (1Н, м), 0,72 (3Н, д, I 6 Гц), 0,71 (3Н, т, I 7,5 Гц), 0,65 (3Н, д, I 6 Гц).
П р и м е р 3. Ферментация. Водная посевная среда (160 мл), содержащая растворимый крахмал (1%), сахарозу (1%), хлопковую муку (1%), пептон (0,5%), соевую муку (0,5%) и карбонат кальция (0,2%), разливали в каждую из десяти 500 мл колб Эрленмейера и стерилизовали при 120оС в течение 30 мин. Петлю культуры Streptomyces violaceoniger N 9326 на скошенном агаре инокулировали в каждую из сред и выращивали на роторном вибраторе (220 оборотов в минуту, дуга качания 5,1 см) при 30оС три дня. Полученную посевную культуру инокулировали в водную посевную среду (160 л), содержащую растворимый крахмал (1%), сахарозу (1%), глюкозу (1%), хлопковую муку (1%), пептон (0,5%), соевую муку (0,5% ), карбонат кальция (0,2%), адеканол-LG-109 (деформирующее средство, товарный знак Asahi Denka Co.) (0,07%) и силикон КМ-70 (деформирующее средство, товарный знак Shin-etsu Chemical Co.) (0,05%) в 500-литровом встряхиваемом ферменторе из нержавеющей стали, который стерилизовали предварительно при 120оС в течение 30 мин. Ферментацию проводили при 30оС один день при аэрации 160 л в минуту и перемешивали при 200 оборотах в минуту.
Полученный посевной культуральный бульон (60 л) инокулировали в стерилизованную питательную среду, содержащую глицерин (3,0%), соевую муку (1,0%), костную муку цыпленка (1,0% ), карбонат кальция (0,2%), иодистый натрий (0,001% ), адеканол-LG-109 (0,07%) и силикон КМ-70 (0,05%) в 4000-литровом встряхиваемом ферменторе из нержавеющей стали, который предварительно стерилизовали при 120оС в течение 30 мин, и выращивали при 30оС четыре дня при аэрации 3000 л в минуту и перемешивании при 100 оборотах в минуту.
Процесс ферментации контролировали высокопроизводительной жидкостной хроматографией с использованием насоса Hitachi модель 655. Стальную колонку, упакованную силикагелем с обратной фазой, "УМС-упакованная колонка R-ODS-5" (торговое название Yamamura Chemical Institute) использовали при скорости потока 1,0 мл/мин. Использованной подвижной фазой являлся водный раствор 45% ацетонитрила. Образцы для анализа методом высокопроизводительной хроматографии готовили следующим образом. К пробе бульона добавляли равный объем ацетона при энергичном перемешивании, смесь выдерживали 1 ч и затем центрифугировали. 5 мл надосадочной жидкости набирали в инжектор Hitachi модель 655 для проведения высокопроизводительной жидкостной хроматографии.
Выделение и очистка.
Полученный таким образом культуральный бульон фильтровали с помощью диатомовой земли (перлит топко 34, товарный знак Сева Кемикал Индастри Лтд) (15 кг). Мицелиальный остаток на фильтре экстрагировали этилацетатом (1600 л) и экстракт фильтровали. Фильтрат (1400 л) вводили в колонку с активированным углем (Сирасаги, товарный знак, Такеда Фармасьютикал Ко, Лтд (200 л). Колонку промывали этилацетатом (1200 л) и затем проводили элюирование смесью этилацетат-метанол в отношении 5:1. Активные фракции (фракции от 50 до 1030 л) объединяли и концентрировали до 45 л при пониженном давлении. н-Гексан (120 л) добавляли к полученному раствору при перемешивании. Смесь выдерживали при комнатной температуре 1 ч и затем фильтровали с помощью кремнезема Силика 600 (Чуо Силика Ко. Лтд.) (3 кг) и целевое вещество элюировали метанолом (20 л). Элюат концентрировали досуха при пониженном давлении. Остаток (500 г) растворяли в смеси метанол уксусная кислота дихлорметан в отношении 1: 1: 2 (4 л) и вводили в колонку силикагеля (Кизельгель 60, 70-230 меш, 70 л). Колонку проявляли смесью метанол-уксусная кислота-дихлорметан в отношении 1:1:2 (0,5 л) и дихлорметаном (25 л). Целевые вещества элюировали смесью дихлорметан-метанол в отношении 10:1 и дихлорметан-метанол в отношении 8:1. Активные фракции объединяли и концентрировали при пониженном давлении. Остаток растворяли в метаноле (1 л). К полученному раствору при помешивании добавляли ацетонитрил (9 л). Смесь выдерживали при комнатной температуре 1 ч и выпавший осадок собирали фильтрацией. Операцию по сбору преципитата повторяли три раза. Полученный таким образом преципитат промывали ацетонитрилом (1 л), сушили и получали белый порошок (190 г) WS-9326А. Полученные фильтраты на операциях сбора преципитата объединяли и концентрировали досуха при пониженном давлении. Остаток (11,7 г) растворяли 80%-ным водным метанолом и полученный раствор пропускали через колонку с активированным углем (300 мл). Колонку промывали 80%-ным водным метанолом (1 л) и элюирование проводили метанолом (6 л). Активные фракции объединяли и концентрировали досуха при пониженном давлении. Остаток (3,4 г) растворяли метанолом (12 мл). Полученный раствор вводили в колонку с обратной фазой силикагеля (УМС упакованная колонка Р-354 S-15/30 (ODS), диаметром 50 мм, длиной 300 мм х 2; изготовитель Yamamura Chemical Institute), уравновешенную 50%-ным водным ацетонитрилом. Колонку проявляли 50%-ным водным ацетонитрилом, используя воду для высокопроизводительной жидкостной хроматографии (система 500). Элюаты, содержащие WS-9326В (фракции от 3 до 3,5 л), объединяли, концентрировали досуха и получали белый порошок (790 мг) WS-9326В.
П р и м е р 4. К раствору WS-9326А (100 мг) в пиридине (1 мл) добавляли уксусный ангидрид (0,01 мл) и смесь оставляли на ночь при комнатной температуре. Смесь выпаривали досуха, чтобы получить масло, которое очищали препаративной тонкослойной хроматографией (смесь хлороформ-метанол, в отношении 9: 1). Полученный продукт растирали с диэтиловым эфиром и получали моноацетил-WS-9326А (55 мг) в виде бесцветного порошка. Физические и химические свойства моноацетил-WS-9326А следующие.
Форма и цвет: бесцветный порошок;
Молекулярная формула C56H70N8O14;
Молекулярный вес: FAB-масс-спектр: m/z 1079,4 (М+Н)+;
Данные тонкослойной хроматографии приведены ниже.
Стационарная фаза: пластинка силикагеля (Merck Art 5715).
Проявляющий растворитель: смесь хлороформ-метанол в отношении 10:1 по объему.
Значение Rf 0,17.
Спектр инфракрасного поглощения: νмаксКВг= 3300, 2920, 1730, 1650, 1500, 1360, 1190, 1170, 910 см-1;
Свойства вещества: нейтральное вещество.
Спектр ядерного магнитного резонанса на 1Н (400 МГц, CDCl3-CD3OD в отношении 5:1).
П р и м е р 5. К раствору WS-9326A (100 мг) в пиридине (1 мл) добавляли уксусный ангидрид (0,03 мл) и смесь оставляли на ночь при комнатной температуре. Смесь выпаривали досуха и получали масло, которое очищали тонкослойной хроматографией (смесь хлороформ:метанол в отношении 9:1), что давало 72 мг диацетил-WS-9326А в виде бесцветного порошка. Физические и химические свойства диацетил-WS-9326А следующие.
Форма и цвет: бесцветный порошок.
Молекулярная формула C58H72N8O15.
Молекулярный вес: FAB-масс-спектр: m/z 1121,4у (М+Н)+.
Данные тонкослойной хроматографии приведены ниже.
Стационарная фаза: пластинка силикагеля (Merck Art 5715).
Проявляющий растворитель: смесь хлороформ-метанол в отношении 10:1 по объему.
Значение Rf 0,35
Спектр инфракрасного поглощения: νмаксКВг= 3300, 3020, 2950, 1730, 1650, 1520, 1500, 1360, 1200, 1170, 1100, 1040, 960, 910 см-1;
Свойства вещества: нейтральное вещество.
Спектр ядерного магнитного резонанса на 1Н (400 МГц, CDCl3-CD3OD в отношении 5:1).
П р и м е р 6. WS-9326А (100 мг) растворяли в метаноле (2 мл) и раствор гидрогенизировали над палладиевой чернью (25 мг) под давлением водорода в 1 атм при комнатной температуре в течение 4 ч. Смесь фильтровали и фильтрат выпаривали досуха при пониженном давлении. Образовавшийся продукт растирали с диэтиловым эфиром и получали тетрагидро-WS-9326А (92 мг) в виде бесцветного порошка. Физические и химические свойства тетрагидро-WS-9326А следующие.
Форма и цвет: бесцветный порошок.
Спектр ультрафиолетового поглощения (метанол): λмакс= 287 нм (ε= 13000).
Молекулярная формула C54H72N8O13.
Молекулярный вес: FAB-масс-спектр: m/z 1041,6 (М+Н)+.
Спектр ядерного магнитного резонанса на 13С (100 МГц, CD3OD).
Спектр ядерного магнитного резонанса на 1Н (400 МГц, CD3OD).
П р и м е р 7. К раствору тетрагидро-WS-9326А (1100 мг) в пиридине (10 мл) добавляли уксусный ангидрид (3 мл) и 4-диметиламинопиридин (3 мг) и реакционную смесь оставляли на ночь при комнатной температуре. Раствор выпаривали досуха, чтобы получить масло, которое очищали хроматографией на колонке силикагеля (смесь хлороформ-метанол в отношении 20:1). Полученный чистый продукт растирали с диэтиловым эфиром и получали тетрагидротриацетил-WS-9326А (998 мг) в виде бесцветного порошка. Физические и химические свойства тетрагидротриацетил-WS-9326А следующие.
Форма и цвет: бесцветный порошок.
Спектр ультрафиолетового поглощения (метанол): λмакс= 280 нм (ε= 13000).
Молекулярная формула C60H78N8O16
Вычислено, C 60,80; H 6,80; N 9,45
C60H78N8O16H2O
Найдено, C 61,03; H 6,70; N 9,41
Молекулярный вес: FAB-масс-спектр: m/z 1167,6 (М+Н)+;
Спектр ядерного магнитного резонанса на ядрах 13С: (100 МГц, CDCl3) δ: 173,3 (с), 129,23 (д), 172,96 (с), 128,95 (д)х2, 172,90 (с), 129,61 (д)х2, 172,81 (с), 170,87 (с), 170,56 (с), 170,50 (с), 169,46 (с), 169,16 (с), 168,48 (с), 167,99 (с), 165,52 (с), 150,70 (с), 140,84 (с), 138,93 (с), 138,58 (с), 136,83 (с), 131,04 (с), 129,71 (д)х2, 128,61 (д)х2, 128,52 (д), 126,80 (д), 126,07 (д), 126,01 (д), 125,85 (д), 121,87 (д)х2, 70,49 (д), 60,10 (д), 63,32 (т), 58,28 (д), 56,19 (д), 52,63 (д), 52,07 (д), 51,63 (д), 49,23 (д), 39,30 (т), 39,17 (кв), 38,31 (т), 36,52 (т), 35,22 (т), 32,60 (т), 31,77 (т), 30,74 (т), 27,66 (т), 24,08 (д), 22,82 (кв), 22,50 (т), 21,46 (кв), 20,74 (кв), 20,63 (кв), 16,77 (кв), 16,22 (кв), 13,97 (кв);
7) Спектр ядерного магнитного резонанса на ядрах 1Н (400 МГц, CDCl3), δ: 8,18 (1Н, д, I 8 Гц), 7,66 (1Н, д, I 8 Гц), 7,65 (1Н, д, I 8 Гц), 7,29 (2Н, д, I 8 Гц), 7,22 (1Н, д, I 8 Гц), 7,15-7,02 (12Н, м), 6,96 (1Н, д, I 7 Гц), 6,67 (1Н, с), 6,21 (1Н, широкий сигнал), 5,51 (1Н, широкий сигнал), 5,43 (1Н, м), 5,36 (1Н, широкий дублет, I 8 Гц), 4,85-4,75 (2Н, м), 4,66-4,58 (2Н, м), 4,40 (1Н, дд, I 11 и 6 Гц), 4,34 (1Н, м), 4,23 (1Н, дд, I 11 и 9 Гц), 4,07 (1Н, м), 3,53 (3Н, с), 3,04-2,84 (5Н, м), 2,75-2,50 (4Н, м), 2,46 (1Н, дд, I 16 и 5 Гц), 2,28 (3Н, с), 1,99 (3Н, с), 1,85 (3Н, с), 1,66-1,50 (3Н, м), 1,37-1,27 (4Н, м), 1,27 (3Н, д, I 7 Гц), 1,19 (1Н, м), 1,03 (3Н, д, I 7 Гц), 0,88 (1Н, м), 0,86 (3Н, т, I 6 Гц), 0,72 (3Н, д, I 6 Гц), 0,65 (3Н, д, I 6 Гц).
П р и м е р 8. Триацетил-WS-9326А (100 мг) растворяли в метаноле (3 мл) и раствор гидрогенизировали над палладиевой чернью (35 мг) под давлением водорода в одну атмосферу при комнатной температуре в течение 3 ч. Смесь фильтровали и фильтрат выпаривали досуха при пониженном давлении. Остаток растирали с диэтиловым эфиром и получали соединение (90 мг) в виде бесцветного порошка. Это соединение было идентично во всех отношениях тетрагидротриацетил- WS-9326А, полученному в примере 7.
Из анализа указанных выше физических и химических свойств и в результате дальнейших исследований по идентификации химической структуры химические структуры триацетил-WS-9326А, моноацетил- и диацетил-WS-9326А, тетрагидро-WS-9326А и тетрагидротриацетил- WS-9326А были идентифицированы согласно фиг. 2-4.
П р и м е р 9.
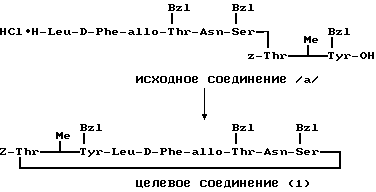
К раствору исходного соединения (а) в дихлорметане (1000 мл) добавляли триэтиламин (350 мл) и 1-этоксикарбонил-2-этокси-1,2- дигидрохинолин (6,17 г) при комнатной температуре. После перемешивания смеси в течение 24 ч при комнатной температуре растворитель выпаривали. К остатку добавляли хлороформ и смесь промывали водой, 1н. хлористоводородной кислотой, водой, насыщенным водным раствором бикарбоната натрия и водой. После осушения смеси над сульфатом магния и фильтрации растворитель выпаривали. Остаток подвергали колоночной хроматографии ("Lober column", размер С) и элюировали 3%-ным метанолом в хлороформе. Фракции, содержащие целевое соединение, выпаривали и получали целевое соединение формулы (1) (1,06 г).
(α)D18-95,3о (с 0,33, хлороформ);
ИК-спектр (CHCl3): 1660, 1600, 1510 см-1;
ЯМР-спектр (CDCl3, δ): 0,88 (3Н, д, I 6 Гц), 0,93 (3Н, д, I 6 Гц), 1,09 (3Н, д, I 6,5 Гц), 1,37 (3Н, д, I 6,5 Гц), 2,82 (3Н, с), 4,87 (2Н, с), 6,93 (2Н, д, I 8 Гц).
П р и м е р 10.

К раствору исходного соединения (А) (42 мг) в дихлорметане (4 мл) и N, N-диметилформамиде (0,1 мл) добавляли N-оксисукцинимид (20,4 мг) и водорастворимый солянокислый карбодиимид (8,2 мг). После перемешивания в течение 15 ч при комнатной температуре к смеси добавляли водорастворимый гидрохлорид карбодиимида (4 мг) с интервалами 1,5 ч до исчезновения исходного соединения (А). Растворитель удаляли в вакууме, остаток растворяли в этилацетате (10 мл) и промывали разбавленной хлористоводородной кислотой и водой. После просушивания над сульфатом магния растворитель удаляли в вакууме и остаток растворяли в трифторуксусной кислоте (1 мл) и анизоле (0,1 мл). После перемешивания в течение 30 мин при комнатной температуре растворитель удаляли в вакууме. Остаток растворяли в N,N-диметилформамиде (2 мл) и смесь добавляли к пиридину (40 мл). После перемешивания в течение 16 ч при комнатной температуре растворитель удаляли в вакууме. Остаток подвергали препаративной тонкослойной хроматографии (Merck 5744) и проявляли смесью хлороформ-метанол в отношении 10:1 и получали целевое соединение (1) (15,2 мг).
ИК-спектр (KBr): 1636, 1510 см-1;
ЯМР-спектр (CD3OD) δ: 6,24 (1Н, с)
(α)D20+18,0о (с 0,1, метанол).
П р и м е р 11.
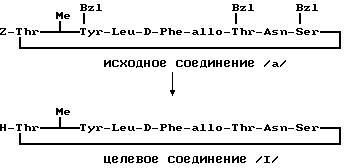
Исходное соединение (а) (240 мг) гидрогенизировали при давлении примерно 0,3 кг/см2 с палладием (200 мг) в смеси муравьиной кислоты и метанола в отношении 1: 24 (10 мл) в течение 7 ч. Затем смесь фильтровали, фильтрат выпаривали и получали целевое соединение (1) (140 мг).
ИК-спектр (KBr): 1730, 1650, 1510 см-1;
(αD21-21,04о (с 0,1, метанол).
П р и м е р 12.
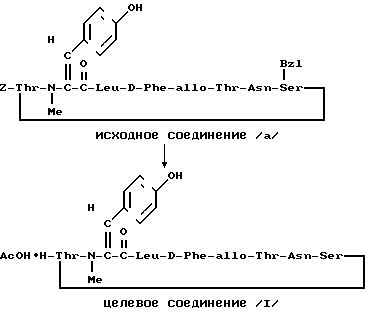
Исходное соединение (а) (22 мг) растворяли в растворе фтористый водород-пиридин (0,8 мл) и в анизоле (0,2 мл) в наполненном азотном мешке. После перемешивания в течение 1 ч при комнатной температуре к смеси добавляли несколько кусочков льда и устанавливали рН раствора 8,0 водным раствором бикарбоната натрия. Смесь помещали в ионообменную колонку с диамином НР-20 (10 мл) и промывали водой. Продукт элюировали метанолом и очищали тонкослойной хроматографией (Merck 5715), смесь хлороформ-метанол-вода в отношении 3:1: 0,1 (по объему) и получали целевое соединение (1) (13,0 мг).
ИК-спектр (KBr): 1635, 1510 см-1;
ЯМР-спектр (CD3OD, δ): 7,05 (1Н, с);
(α)D20-90,6о (с 0,1, метанол).
Тонкослойная хроматография: Rf 0,35 (Merck Art 5715, смесь хлороформ-метанол-вода в отношении 3:1:0,1).
П р и м е р 13.
 ие /b/
ие /b/
R(E)-3-/2-((Z)-1-пентенил)-фенил/-пропеноил.
К раствору исходного соединения (а) (6,0 мг) в дихлорметане (1,5 мл), бис-(диметилсилил)-ацетамиде (30 мкл) и N,N-диметилформамиде (0,3 мл) добавляли 0,02 М раствор исходного соединения (b) (0,4 мл). После перемешивания в течение 1 ч при комнатной температуре к смеси добавляли 4-диметил- аминопиридин (0,1 мг). Исходное соединение (b) добавляли к смеси с 30-минутными интервалами до исчезновения исходного соединения (а). К смеси добавляли разбавленную хлористоводородную кислоту и органический слой промывали водой. После выпаривания в вакууме остаток подвергали препаративной тонкослойной хроматографии (Merck 5715) и проявляли смесью хлороформ-метанол-вода в отношении 65:25:4 по объему. Получали целевое соединение (1) (0,2 мг).
Это соединение было идентично соединению WS-9326А, полученному в примере 1.
П р и м е р 14.

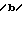
К раствору исходного соединения (а) (11 мг) в пиридине (1 мл) добавляли 0,02 М раствор исходного соединения (b) в дихлорметане (0,6 мл). После перемешивания в течение 1 ч при комнатной температуре исходное соединение (b) добавляли к смеси с интервалами в 1 ч до исчезновения исходного соединения (а). К смеси добавляли метанол (2 мл) и растворитель удаляли в вакууме. Остаток растворяли в этилацетате (10 мл) и промывали разбавленной хлористоводородной кислотой и водой. После просушивания над сульфатом магния растворитель удаляли в вакууме. Остаток подвергали препаративной тонкослойной хроматографии (Merck 5715) и проявляли смесью хлороформ-метанол-вода в отношении 3:1:0,1 по объему. Получали целевое соединение (а) (2 мг).
ИК-спектр (KBr): 1640, 1510 см-1.
П р и м е р 15.
 (b)
(b)
R(E)-3-/2-((Z)-1-пентенил)фенил/-пропеноил.
К раствору исходного соединения (а) (49,7 мг) в пиридине (1 мл) добавляли 0,1 М раствора исходного соединения (b) в дихлорметане (1,2 мл) в атмосфере азота и смесь перемешивали 3,5 ч при комнатной температуре. К реакционной смеси добавляли этилацетат и смесь промывали водой, 7%-ной уксусной кислотой, водой и насыщенным водным раствором хлористого натрия. После просушивания над сульфатом магния и фильтрации растворитель выпаривали, остаток подвергали препаративной тонкослойной хроматографии (0,5 мм х 2) и проявляли 20% -ным метанолом в хлороформе и поучали целевое соединение (1) (20,6 мг).
Это соединение было идентично соединению WS-9326В, полученному в примере 3.
П р и м е р 16. Следующие соединения были получены способом, аналогичным описанному в примере 15.
R-
(1) R-:бензоил
(α)D21-45,8о (с 0,74, метанол); точка плавления 176-178оС.
Тонкослойная хроматография: Rf 0,48 (Merck Art 5715), смесь хлороформ-метанол в отношении 5:1).
ИК-спектр (KBr): 1720 (изгиб пика), 1655, 1640 см-1
(2) R-:2-(2-тиенил)-ацетил
(α)D23-16,8о (с 0,73, метанол); точка плавления 160-163оС.
Тонкослойная хроматография: Rf 0,24 (Merck Art 5715, смесь хлороформ-метанол в отношении 5:1).
ИК-спектр (KBr): 1720 (изгиб пика), 1650 см-1
(3) R-:ацетил
(α)D21-37,4о (с 0,72, метанол); точка плавления 231-233оС.
Тонкослойная хроматография: Rf 0,41 (Merck Art 5715, смесь хлороформ-метанол в отношении 5:1).
ИК-спектр (KBr): 1720 (изгиб пика), 1650 см-1.
П р и м е р 17.
R-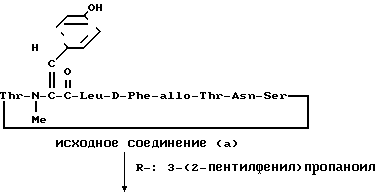
R-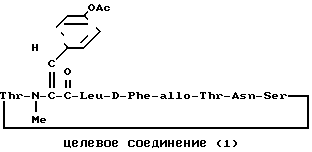
К раствору исходного соединения (а) (100 мг) в пиридине (1 мл) добавляли уксусный ангидрид (11 мл) и смесь оставляли при комнатной температуре на ночь. Смесь выпаривали досуха и получали масло, которое очищали препаративной тонкослойной хроматографией (смесь хлороформ-метанол в отношении 9: 1). Получали целевое соединение (1) (52 мг).
Тонкослойная хроматография: Rf 0,17 (Merck Art 5715, смесь хлороформ-метанол в отношении 10:1).
ИК-спектр (нуйол): 3300, 1760, 1730, 1650, 1530, 1510, 1200, 1160, 1070, 910 см-1.
П р и м е р 18.
R-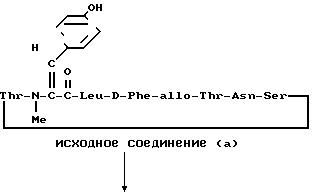
R-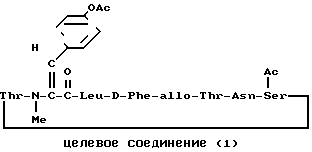
R-:3-(2-пентилфенил)-пропаноил.
К раствору исходного соединения (а) (100 мг) в пиридине (1 мл) добавляли уксусный ангидрид (25 мл) и смесь оставляли на ночь при комнатной температуре. Смесь выпаривали досуха, а масляный остаток очищали препаративной тонкослойной хроматографией (смесь хлороформ-метанол в отношении 9:1) и получали целевое соединение (1) (78 мг).
Тонкослойная хроматография: Rf 0,36 (Merck Art 5715, смесь хлороформ-метанол в отношении 10:1).
ИК-спектр (нуйол): 3300, 1760, 1740, 1650, 1540, 1510, 1300, 1220, 1200, 1170, 1050, 920 см-1.
П р и м е р 19.

R-:3-(2-пентилфенил)-пропаноил.
К раствору исходного соединения (а) (0,21 г) в метаноле (3 мл) добавляли раствор (3 мл) диазометана в диэтиловом эфире. После перемешивания в течение 5 мин растворитель удаляли в вакууме. Остаток подвергали препаративной тонкослойной хроматографии (Merck 5744) и проявляли 20%-ным метанолом в хлороформе. Получали целевое соединение (1) (45 мг).
ИК-спектр (нуйол): 3300, 1645, 1530, 1510 см-1.
П р и м е р 20.
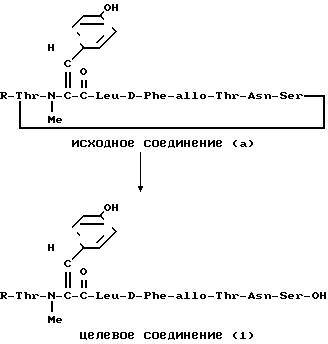
R-:3-(2-пентилфенил)пропаноил.
К раствору исходного соединения (а) (1,0 г) в метаноле (15 мл) добавляли 1н. гидроокись натрия (5 мл) при 0оС. После перемешивания в течение 1 ч в раствор добавляли 1н. хлористоводородную кислоту (1 мл). Растворитель удаляли в вакууме, а остаток растворяли в смеси этилацетата (20 мл) и разбавленной хлористоводородной кислоты (30 мл). Органический слой промывали рассолом, сушили над сульфатом магния и выпаривали в вакууме. Полученное твердое вещество промывали этилацетатом и получали целевое соединение (1) (0,95 г).
ИК-спектр (нуйол): 3300, 1710 (изгиб пика), 1645, 1510 см-1.
П р и м е р 21.
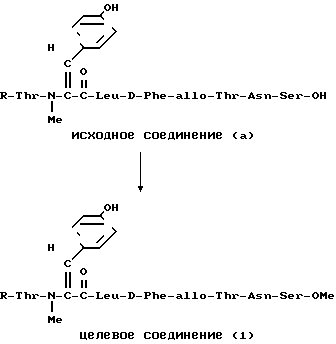
R-:3-(2-пентилфенил)-пропаноил.
К раствору исходного соединения (а) (1,0 г) в метаноле (2 мл) и этилацетате (20 мл) добавляли раствор 92 мл) 10% триметилсилилдиазометана в н-гексане. После перемешивания в течение 5 мин растворитель удаляли в вакууме. Остаток вводили в колонку силикагеля (Merck 7734) и элюировали смесью хлороформ-метанол в отношении 10:1 по объему, получали целевое соединение (1) в виде твердого вещества (0,55 г).
ИК-спектр (нуйол): 3300, 1735, 1645, 1530, 1510 см-1.
Получение 1.
 Z
Z
 -
-
К раствору исходного соединения (а) (2,53 г) в метаноле (10 мл) и воде (3 мл) добавляли карбонат цезия (1,63 г). После удаления растворителя остаток растворяли в N,N-диметилформамиде и смесь добавляли к бромистому фенацилу (1,92 г). Смесь перемешивали 30 мин при комнатной температуре. Растворитель отфильтровывали, остаток растворяли в этилацетате и промывали водой. После просушивания смеси над сульфатом магния и фильтрации, растворитель выпаривали и получали кристаллы целевого соединения (1) (3,7 г).
(α)D20-20,3о (с 1,0, хлороформ);
ИК-спектр (нуйол): 1745, 1690, 1545 см-1
Получение 2.
исход ение (a)
ение (a)  исход
исход динение (b)
динение (b) B
B
К раствору исходного соединения (а) (1,48 г) и исходного соединения (b) (2,5 г) в дихлорметане (80 мл) добавляли гидрохлорид 1-этил-(3-диметиламинопропил)-карбодиимида (0,764 г) и 4-диметиламинопиридин (0,488 г) при 0оС. После перемешивания в течение 6 ч растворитель выпаривали. Остаток растворяли в этилацетате и смесь промывали водой, 1н. хлористоводородной кислотой, насыщенным водным раствором бикарбоната натрия и водой. После просушивания над сульфатом магния и фильтрации растворитель выпаривали и получали целевое соединение (1) (2,58 г).
(α)D20+9,76о (с 0,3, хлороформ);
ИК-спектр (CHCl3): 1755, 1720, 1705, 1505 см-1
ЯМР-спектр (CDCl3, ): 1,37 (3Н, д, I 6 Гц), 1,45 (9Н, с), 3,70 (1Н, м), 3,88 (1Н, м), 4,52 (2Н, м), 4,23 (1Н, м), 5,17 (2Н, с), 5,37 (2Н, с).
Получение 3.




К раствору исходного соединения (а) в 90%-ной водной уксусной кислоте (100 мл) добавляли цинковый порошок (11 г) при перемешивании и смесь перемешивали 2 ч при охлаждении льдом и 1 ч при комнатной температуре. После фильтрации смеси фильтрат концентрировали, устанавливали рН 2,0 лимонной кислотой и экстрагировали этилацетатом. После просушивания экстракта над сульфатом магния и фильфтрации растворитель выпаривали. Остаток промывали петролейным эфиром и получали целевое соединение (1) (5,4 г).
ЯМР-спектр (CDCl3, δ): 1,30 (3Н, д, I 6 Гц), 1,40 (9Н, с), 3,60 (1Н, м), 3,82 (1Н, м).
Получение 4.
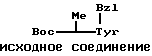
 _____→
_____→ 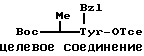
К раствору исходного соединения (а) (7,7 г) в дихлорметане (60 мл) добавляли 2,2,2-трихлорэтанол (2,105 мл), гидрохлорид 1-этил-3-(3-диметиламинопиридин) (244 мг) при перемешивании при 0оС и смесь перемешивали 1 ч при 0оС и выпаривали. Остаток растворяли в этилацетате и промывали водой, 1н. хлористоводородной кислотой, водой и насыщенным водным раствором бикарбоната натрия. После осушивания смеси над сульфатом магния и фильтрации фильтрат выпаривали и получали целевое соединение (1) (9,37 г).
(α)D19-29,84о (с 0,4, хлороформ);
ИК-спектр (хлороформ): 1755, 1690, 1610, 1510 см-1.
Получение 5.
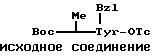 e
e _____→
_____→ 
Исходное соединение (а) (9,3 г) охлаждали до 0оС и добавляли к трифторуксусной кислоте (15 мл). Смесь перемешивали 30 мин при 0оС и выпаривали. Остаток растворяли в этилацетате и промывали водой, насыщенным водным раствором бикарбоната натрия и водой. После сушки над сульфатом магния и фильтрации растворитель отгоняли и получали целевое соединение 1) (6 г).
ЯМР-спектр (CDCl3) δ: 2,45 (3Н, с), 3,03 (2Н, м), 3,62 (1Н, т, I 7 Гц), 4,75 (2Н, м), 5,06 (2Н, с), 6,94 (2Н, д, I 8 Гц), 7,15 (2Н, д, I 8 Гц), 7,3-7,5 (5Н, м).
Получение 6.
 (b)
(b)
К раствору исходного соединения (а) (4,07 г) и исходного соединения (b) (4,70 г) в дихлорметане (40 мл) добавляли 1-этоксикарбонил-2-этокси-1,2-дигидрохинолин (2,8 г) и смесь перемешивали 24 ч при комнатной температуре. Растворитель выпаривали, а остаток растворяли в этилацетате и промывали 1н. хлористоводородной кислотой, водой, насыщенным водным раствором бикарбоната натрия и водой. После просушивания над сульфатом магния и фильтрования растворитель выпаривали и получали целевое соединение (1) (4,35 г)
(α)D20-27,88о (с 0,12, хлороформ);
ИК-спектр (CHCl3): 1750, 1710, 1655, 1510 см-1; ЯМР-спектр (CDCl3, δ): 1,18 (3Н, д, I 6 Гц), 1,36 (9Н, с), 2,92 (3Н, с), 4,69 (2Н, с), 4,91 (2Н, с), 5,01 (2Н, с), 6,80 (2Н, д, I 8 Гц), 7,02 (2Н, д, I 8 Гц).
Получение 7.
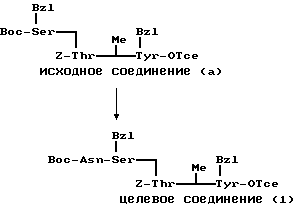
Исходное соединение (а) (4,35 г) охлаждали до 0оС и добавляли к трифторуксусной кислоте (20 мл). После перемешивания смеси в течение 45 мин при 0оС растворитель выпаривали. Остаток растворяли в этилацетате и промывали насыщенным водным раствором бикарбоната натрия и водой. После просушивания раствора над сульфатом магния и фильтрования растворитель выпаривали. Остаток растворяли в дихлорметане (100 мл). К раствору добавляли Boc-Asn (1,2 г), хлоргидрат 1-этил-3-(3-диметиламинопропил)-карбодиимида (990 мг) и 1-гидроксибензотриазол (700 мг). Смесь перемешивали 4 ч при 0оС и промывали 1н. хлористоводородной кислотой, водой, насыщенным водным раствором бикарбоната натрия и водой, и растворитель выпаривали. Получали целевое соединение (1) (4,74 г).
(α)D20-15,7о (с 0,1, хлороформ);
ИК-спектр (KBr): 1740, 1690, 1645, 1510 см-1;
ЯМР-спектр (CDCl3, δ): 1,25 (3Н, д, I 6 Гц), 1,43 (9Н, с), 3,00 (3Н, с), 2,55 (1Н, м), 2,80 (1Н, м), 3,05 (1Н, м), 3,37 (1Н, м), 3,62 (1Н, м), 3,82 (1Н, м), 6,88 (2Н, д, I 8,5 Гц), 7,09 (2Н, д, I 8,5 Гц).
Получение 8.

Целевое соединение (1) получали взаимодействием исходного соединения (а) способом, аналогичным описанному в получении 7.
(α)D22-13,04о (с 0,11, хлороформ);
ИК-спектр (KBr): 1740, 1700, 1655, 1510 см-1;
ЯМР-спектр (CDCl3, δ): 1,18, 1,27 (каждый 3Н, д, I 6 Гц), 1,43 (9Н, с), 2,52 (1Н, м), 2,80 (1Н, м), 3,00 (3Н, с), 3,36 (1Н, м), 4,99 (2Н, с), 6,87 (2Н, д, I 8 Гц), 7,10 (2Н, д, I 8 Гц).
Получение 9.
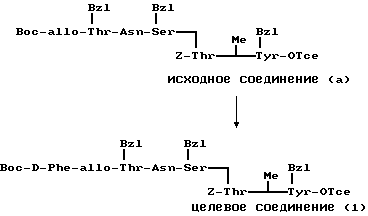
Целевое соединение (1) получали реакцией исходного соединения (а) способом, аналогичным описанному в получении 7.
(α)D21-15,19о (с 0,1, хлороформ);
ИК-спектр (CDCl3, KBr): 1740, 1650, 1635, 1510 см-1;
ЯМР-спектр (CDCl3, δ): 1,11 (3Н, д, I 6 Гц), 1,27 (3Н, д, I 6 Гц), 1,33 (9Н, с), 3,01 (3Н, с), 4,72 (2Н, с), 4,98 (2Н, с), 6,87 (2Н, д, I 8 Гц), 7,10 (2Н, д, I 8 Гц).
Получение 10.
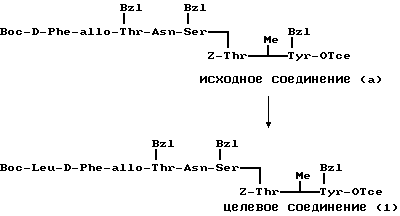
Целевое соединение (1) получали реакцией исходного соединения (а) способом, аналогичным описанному в получении 7.
(α)D21-19,07о (с 0,1, CHCl3);
ИК-спектр (KBr): 1740, 1635, 1510 см-1;
ЯМР-спектр (CDCl3) δ: 0,81 (6Н, м), 1,13, 1,28 (каждый 3Н, д, I 6 Гц), 1,40 (9Н, с), 3,02 (3Н, с), 4,96 (2Н, с), 7,09, 6,87 (каждый 2Н, д, I 8 Гц).
Получение 11.
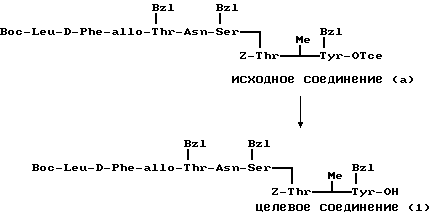
К раствору исходного соединения (А) (4,2 г) в 90%-ной водной уксусной кислоте (80 мл) добавляли порошок цинка (9 г) при 0оС и смесь перемешивали при 0оС 2 ч и при комнатной температуре 1 ч. После фильтрования смеси фильтрат концентрировали. К остатку добавляли хлороформ и смесь промывали 1н. хлористоводородной кислотой и водой. После просушивания смеси над сульфатом магния и фильтрования растворитель выпаривали. Остаток подвергали хроматографии на колонке силикагеля (150 г) и элюировали 2% метанолом в хлороформе и затем 8% метанолом в хлороформе. Фракции, содержащие целевое соединение, выпаривали и получали целевое соединение (1) (3,4 г).
(α)D21-23,42о (с 0,1, метанол)
ИК-спектр (KBr): 1635, 1510 см-1
Получение 12
Исходное соединение (а) (3,4 г) охлаждали до 0оС и добавляли трифторуксусную кислоту (20 мл). После перемешивания смеси 1 ч при 0оС растворитель выпаривали. Остаток растворяли в растворе хлористого водорода в диоксане и затем выпаривали. Остаток растворяли в хлороформе и промывали водой. После осушения раствора над сульфатом магния и фильтрования растворитель выпаривали и получали целевое соединение (1) (3,27 г).
(α)D21-18,87о (с 0,12, метанол);
ИК-спектр (KBr): 1650, 1510 см-1.
Получение 13. К раствору гидроокиси калия (26,8 г) в этаноле (500 мл) добавляли глицин (14,6 г) и 4-метоксиметоксибензальдегид (48,5 г) при комнатной температуре. После перемешивания при комнатной температуре в течение 19 ч растворитель удаляли в вакууме. Остаток растворяли в воде и подкисляли хлористоводородной кислотой. Раствор промывали этилацетатом и устанавливали рН 6,0 бикарбонатом натрия. В осадок выпадало белое твердое вещество, которое собирали и получали 0-метоксиметил-β-окситирозин (9,2 г). Точка плавления 164-166оС.
ИК-спектр (KBr): 1610 см-1.
Получение 14. К раствору 0-метоксиметил-β-окситирозина (21,0 г) в 1н. гидроокиси натрия (250 мл) добавляли диметилсульфат (16,5 г). После перемешивания в течение 20 мин при 90оС раствор подкисляли разбавленной хлористоводородной кислотой на ледяной бане. Кислый раствор промывали диэтиловым эфиром и устанавливали рН 6,0 1н. гидроокисью натрия. После выпаривания твердое вещество собирали фильтрацией и получали 0-метоксиметил-N-метил- β-окситирозин (5,2 г). Точка плавления 59-60оС.
ИК-спектр (KBr): 3400, 1700 см-1.
Получение 15. К раствору 0-метоксиметил-N-метил-β-окситирозина (15,1 г) и бис-(триметилсилил)-ацетамида (25 мл) в дихлорметане (150 мл) добавляли раствор 2-нитрофенилсульфонилхлорида (11,2 г) в дихлорметане (50 мл). После перемешивания в течение 2 ч при 0оС к раствору добавляли бис(триметилсилил)ацетамид (10 мл) и хлористый 2-нитрофенилсульфонил (5,6 г). Смесь перемешивали 4 ч при комнатной температуре и добавляли 1н. гидроокись натрия (200 мл). Органический слой промывали водой (300 мл) и водные растворы объединяли. После подкисления водного раствора разбавленной хлористоводородной кислотой продукт экстрагировали этилацетатом (300 мл) и экстракт промывали водой (по 100 мл три раза). После просушивания раствора над сульфатом магния растворитель удаляли в вакууме и получали 0-метоксиметил-N-метил-N-(2-нитрофенилтио)- β-окситирозин (20,5 г). Точка плавления 59-60оС.
ИК-спектр (KBr): 3400, 1700 см-1.
Получение 16. К раствору 0-метоксиметил-N-метил-N-(2-нитрофенилтио)-β-гидро- кситирозина (20,0 г) в этилацетате (100 мл) добавляли диазометан в диэтиловом эфире (80 мл). После перемешивания в течение 10 мин растворитель удаляли в вакууме. Остаток вводили в колонку силикагеля (Merck 7734), 500 г и элюировали хлороформом. Получали метиловый эфир 0-метоксиметил-N- (2-нитрофенилтио)-β-окситирозина (трео-изомер 8,82 г) (эритро-изомер 6,63 г).
Трео-изомер: ИК-спектр (пленка): 3500, 2950, 1735 см-1.
Тонкослойная хроматография: Rf 0,31 (Merck Art 5715, смесь этилацетат-н-гексан в отношении 1:1).
Эритро-изомер: ИК-спектр (пленка): 3500, 2950, 1735 см-1.
Тонкослойная хроматография: Rf 0,40 (Merck Art 5715, этилацетат-н-гексан в отношении 1:1).
Получение 17. К раствору метилового эфира 0-метоксиметил-N-метил- N-(2-нитрофенилтио)-β-гидрокситирозина (эритро-изомер) (3,85 г) в дихлорметане (30 мл) добавляли триэтиламин (1,38 г), 4-диметиламинопропи ламин (0,45 г) и хлористый бензоил (1,92 г). После перемешивания в течение 16 ч при комнатной температуре к смеси добавляли 3-диметиламинопропиламин (3,3 г) и растворитель удаляли в вакууме. Остаток растворяли в этилацетате (30 мл) и промывали разбавленной хлористоводородной кислотой, водным раствором бикарбоната натрия и водой. После выпаривания остаток помещали в колонку силикагеля (Merck 7734, 150 г) и элюировали смесью н-гексан-этилацетат в отношении 5:2 по объему и получали метиловый эфир 0-метоксиметил-N-метил-N-(2-нитрофенилтио)- β-бензоилокситирозина (эритро-изомер, 4,49 г).
ИК-спектр (пленка): 2950, 1740 см-1.
Тонкослойная хроматография: Rf 0,23 (Merck Art 5715, смесь этилацетат-н-гексан в отношении 1:2).
Получение 18. Следующее соединение было получено способом, аналогичным описанному в получении 17: метиловый эфир 0-метоксиметил-N-метил-N-(2-нитрофенилтио)-β-бензоилокситирозина (трео-изомер); точка плавления 114-115оС.
ИК-спектр (CHCl3): 2950, 1740 см-1.
Тонкослойная хроматография: Rf 0,26 (Merck Art 5715, смесь этилацетат-н-гексан в отношении 1:2).
Получение 19. К раствору метилового эфира 0-метоксиметил-N- метил-N-(2-нитрофенилтио)-β-бензоилокситирозина (4,94 г) в дихлорметане (50 мл) добавляли тиофенол (4,8 мл) и трифторуксусную кислоту при 0оС (2,5 мл). После перемешивания в течение 30 мин к смеси добавляли водный раствор бикарбоната натрия. Органический слой промывали водным раствором бикарбоната натрия и рассолом. После выпаривания остаток помещали на колонку с силикагелем (Merck 7734) (100 г) и элюировали 5%-ным метанолом в хлороформе. Получали метиловый эфир 0-метоксиметил-N-метил-β-бензоилокситирозина (трео-изомер, 0,32 г).
Тонкослойная хроматография: Rf 0,31 (Merck Art 5715, смесь этилацетат-н-гексан в отношении 1:1).
Получение 20. Следующее соединение получали способом, аналогичным описанному в получении 19: метиловый эфир 0-метоксиметил-N-метил-β-бензоилокситирозина (эритро-изомер).
Тонкослойная хроматография: Rf 0,25 (Merck Art 5715, смесь этилацетат-н-гексан в отношении 1:1).
Получение 21. К раствору N-бензилоксикарбонил-α-треонина (3,7 г) и метилового эфира 0-метоксиметил-N-метил-β-бензоил- окситирозина (трео-изомер, 3,11 г) в дихлорметане (50 мл) добавляли этиловый эфир 1,2-дигидро-2-этокси-1-хинолинкарбоновой кислоты (2,9 г). После перемешивания в течение 20 мин при комнатной температуре растворитель удаляли в вакууме. Остаток растворяли в этилацетате (50 мл) и промывали дистиллированной хлористоводородной кислотой, водным раствором бикарбоната натрия и водой. После выпаривания остаток вводили в колонку силикагеля (Merck 7734, 100 г) и элюировали смесью н-гексан-этилацетат в отношении 1:1 (по объему) и получали метиловый эфир N-(N-бензилоксикарбонил-α-треонил)-0- метоксиметил-N-метил-α-бензоилокситирози- на (трео-изомер, 2,04 г).
ИК-спектр (пленка): 3400, 2950, 1740 (изгиб пика), 1720 см-1.
Тонкослойная хроматография: Rf 0,36 (Merck Art 5715, смесь метанол-хлороформ в отношении 3:97).
Получение 22. Следующее соединение получали способом, аналогичным описанному в получении 21: метиловый эфир N-(N-бензилоксикарбонил-α-треонил)-0-метокси- метил-N-метил-β-бензоилокситирозина (эритро-изомер).
ИК-спектр (пленка): 2950, 1740, 1730 (изгиб пика) см-1.
Тонкослойная хроматография: Rf 0,23 (Merck Art 5715, смесь этилацетат-н-гексан в отношении 1:2).
Получение 23.
Z-Th
 -OMe
-OMe
Целевое соединение (1)
К раствору метилового эфира β-бензоилокси-N-(N-бензилоксикарбонил-α-треон- ил)-0-метоксиметил-N-метилтирозина (трео-изомер, 1,20 г) в толуоле (20 мл) добавляли 1,8-диазабицикло(5.4.0)-ундец-7-ен (0,30 г). После перемешивания в течение получаса при комнатной температуре к смеси добавляли 7% -ную хлористоводородную кислоту (10 мл). Органический слой промывали водой и водным раствором бикарбоната натрия. После сушки органического слоя над сульфатом магния растворитель удаляли в вакууме и получали целевое соединение (1) (0,95 г).
ИК-спектр (пленка): 3400, 2950, 1720 см-1.
(α)D22-7,7о (с 0,64, метанол).
Целевое соединение (1) было также получено взаимодействием метилового эфира β-бензоилокси-N-(N-бензилоксикарбонил-α -треонил)-0-метоксиметил-N-метилтирозина (эритроизомер) вместо метилового эфира β-бензоилокси-N-(N-бензилоксикарбонил-α-треонил)-0-метоксиметил-N-мети(трео- изомер).
Получение 24.
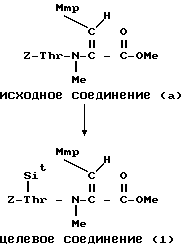
К раствору исходного соединения (а) (1,0 г) в N,N-диметилформамиде (10 мл) добавляли трет-бутилдиметилсилилхлорид (0,75 г) и имидазол (0,34 г). После перемешивания в течение 16 ч при комнатной температуре к смеси добавляли этилацетат (30 мл) и лед (50 г). Органический слой промывали разбавленной хлористоводородной кислотой, водным раствором бикарбоната натрия и водой. Растворитель удаляли в вакууме. Остаток вводили в колонку силикагеля (Merck 7734, 30 г), элюировали хлороформом и получали целевое соединение (1) (1,21 г).
ИК-спектр (пленка): 2950, 1720 см-1,
(α)D22-55,9о (с 0,56, метанол).
Получение 25.
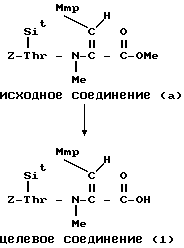
К раствору исходного соединения (а) (0,95 г) добавляли 1н. водный раствор гидроокиси натрия (4,8 мл). После перемешивания в течение двух дней при 30оС растворитель удаляли в вакууме. Остаток растворяли в этилацетате (20 мл) и промывали разбавленной хлористоводородной кислотой и водой. После выпаривания получали целевое соединение (1) (0,81 г).
ИК-спектр (пленка): 3300, 2950, 1720, 1700 (изгиб пика) см-1
(α)D2282,9о (с 1,06, метанол).
Получение 26.
 b)
b)
К смеси исходного соединения (а) (1,60 г) и исходного соединения (b) (5,50 г) в дихлорметане (50 мл) добавляли триэтиламин (1,25 г) и этиловый эфир 1,2-дигидро-2-этокси-1-хинолинкарбоновой кислоты (3,04 г). После перемешивания в течение 15 ч при комнатной температуре белое твердое вещество отфильтровывали и к фильтрату добавляли исходное соединение (b) (2,23 г), триэтиламин (0,50 г) и этиловый эфир 1,2-дигидро-2-этокси-1-хинолинкарбоновой кислоты (1,24 г). Смесь перемешивали 18 ч и растворитель удаляли в вакууме. Остаток растворяли в этилацетате (50 мл) и промывали разбавленной хлористоводородной кислотой, водным раствором бикарбоната натрия и водой. После выпаривания в вакууме остаток помещали в колонку силикагеля (Merck 7734, 100 г), элюировали смесью н-гексан-этилацетат в отношении 2:7 (по объему) и получали целевое соединение (1) (0,87 г).
ИК-спектр (пленка): 2950, 1760, 1740 (изгиб пика), 1720, 1660 см-1:
(α)D20-31,5о (с 1,07, метанол).
Получение 27.
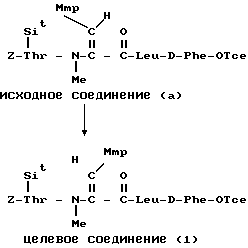
Раствор исходного соединения (а) (0,85 г) в толуоле (100 мл) и ацетоне (10 мл) облучали ультрафиолетовой лампой (100 В) в течение полутора часов при 0оС. После выпаривания остаток помещали в колонку силикагеля (Merck 7734, 50 г), элюировали смесью н-гексан-этилацетат в отношении 2:1 (по объему) и получали целевое соединение (1) (0,18 г).
Тонкослойная хроматография: Rf 0,22 (Merck Art 5715, смесь н-гексан-этилацетат в отношении 2:1).
ИК-спектр (KBr): 3300, 1740, (изгиб пика), 1640 см-1.
Получение 28.

Исходное соединение (а) (0,17 г) растворяли в 67%-ном водном растворе уксусной кислоты (10 мл). После перемешивания в течение 28 ч при 25оС растворитель удаляли в вакууме. Остаток растворяли в этилацетате (20 мл) и промывали водным раствором бикарбоната натрия и водой. После концентрирования остаток промывали н-гексаном и растворитель удаляли в вакууме. Получали целевое соединение (1) (0,15 г).
ИК-спектр (KBr): 3250, 1740 (изгиб пика), 1635 см-1.
Тонкослойная хроматография: Rf 0,18 (Merck Art 5715, смесь н-гексан-этилацетат в отношении 1:1).
Получение 29.


К раствору исходного соединения (а) (0,14 г) в дихлорметане (5 мл) добавляли исходное соединение (b) (0,10 г), водорастворимый гидрохлорид карбодиимида (65 мг) и 4-диметиламинопиридина (4 мг). Затем после 12-часового перемешивания при комнатной температуре к смеси добавляли N,N-диметиламинопропиламин (50 мг) и растворитель выпаривали в вакууме. Остаток растворяли в этилацетате (20 мл) и промывали разбавленной хлористоводородной кислотой и водой. После выпаривания остаток вводили в колонку силикагеля (Merck 7734, 10 г), элюировали смесью н-гексан-этилацетат в отношении 1:1 (по объему) и получали целевое соединение (1) (0,16 г).
ИК-спектр (KBr): 3300, 1700, 1640, 1495 см-1.
Тонкослойная хроматография: Rf 0,38 (Merck Art 5715, смесь н-гексан-этилацетат в отношении 1:1).
Получение 30.
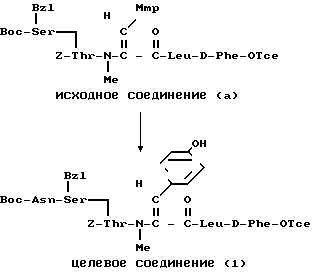
Исходное соединение (а) (145 мг) растворяли в смеси 4н. хлористого водорода в диоксане (3 мл) и анизоле (0,1 мл). После 30-минутного перемешивания при комнатной температуре растворитель удаляли в вакууме. Остаток растворяли в дихлорметане (3 мл). К раствору добавляли N-трет-бутоксикарбонил-α-аспарагин (35 мг), триэтиламин (13 мг), 1-оксибензотриазол (18 мг) и растворимый в воде гидрохлорид карбодиимида (29 мг). После перемешивания в течение 1 ч при комнатной температуре к смеси добавляли 7%-ную хлористоводородную кислоту (5 мл). Органический слой промывали водой. После выпаривания остаток подвергали препаративной тонкослойной хроматографии (Merk 5744) и проявляли 6% -ным метанолом в хлороформе и получали целевое соединение (1) (110 мг).
ИК-спектр (KBr): 3300, 1650, 1505 см-1.
Тонкослойная хроматография: Rf 0,44 (Merck Art 5715, смесь хлороформ-метанол в отношении 10:1).
Получение 31.

Исходное соединение (а) (105 мг) растворяли в смеси 4н. хлористого водорода в диоксане (3 мл) и анизоле (0,1 мл). После перемешивания в течение получаса при комнатной температуре растворитель удаляли в вакууме. Остаток растворяли в дихлорметане (3 мл). К раствору добавляли N-трет-бутоксикарбонил-α-аллотреонин (22 мг), триэтиламин (9 мг), 1-оксибензотриазол (12 мг) и растворимый в воде гидрохлорид карбодиимида (19 мг). После перемешивания в течение 8 ч при комнатной температуре к смеси добавляли 7%-ную хлористоводородную кислоту (5 мл). Органический слой промывали водой. После выпаривания остаток подвергали препаративной тонкослойной хроматографии (Merck 5744) и проявляли 6%-ным метанолом в хлороформе и получали целевое соединение (1).
Тонкослойная хроматография: Rf 0,73 (Merck Art 5715, смесь хлороформ-метанол в отношении 5:1).
ИК-спектр (KBr): 3300, 1740, (плечо), 1650, 1500 см-1.
Получение 32.
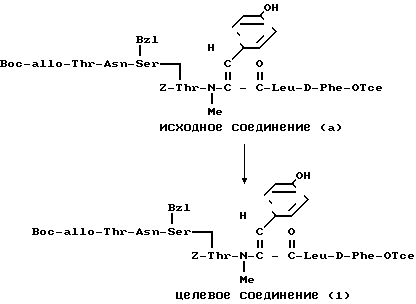
К раствору исходного соединения (а) (58,5 мг) в 90%-ном водном растворе уксусной кислоты (1 мл) добавляли порошок цинка (30 мг). После перемешивания в течение 9 ч при комнатной температуре порошок цинка (30 мг) добавляли к смеси с интервалами в 1 ч до исчезновения исходного соединения (а). После фильтрации растворитель удаляли в вакууме. Остаток растворяли в этилацетате (10 мл), промывали водой и выпаривали в вакууме. Остаток подвергали препаративной тонкослойной хроматографии (Merck 5744), проявляли смесью этилацетат-ацетон-уксусная кислота-вода в отношении 6:3:1:1 (по объему) и получали целевое соединение (1) (43,5 мг).
ИК-спектр (KBr): 3300, 1650, 1505 см-1.
Тонкослойная хроматография: Rf 0,16 (Merck Art 5715, смесь хлороформ-метанол-ацетол в отношении 10:1:0,1).
Получение 33. К раствору фталальдегида (6,7 г) в дихлорметане (30 мл) добавляли этоксикарбонилметилентрифенилфосфоран (17,42 г) и смесь перемешивали 30 мин при комнатной температуре. Растворитель выпаривали и остаток растворяли в диэтиловом эфире. После фильтрования смеси фильтрат выпаривали. Остаток отгоняли под вакуумом (125оС, 0,6 мм Н) и получали этиловый эфир (К)-3-(2-фтормилфенил)-актиловой кислоты (6 г).
Спектр ЯМР (CDCl3, δ): 1,24 (3Н, т, I 6,5 Гц), 4,19 (2Н, кв, I 6,5 Гц), 6,28 (1Н, д, I 15 Гц), 7,5 (3Н, м), 7,77 (1Н, м), 8,43 (1Н, д, I 15 Гц), 10,18 (1Н, с).
Получение 34. К раствору бромистого бутилтрифенилфосфония (3,2 г) в тетрагидрофуране (50 мл) добавляли трет-бутилат калия (900 мг) в атмосфере азота и смесь перемешивали 30 мин при комнатной температуре. К смеси добавляли раствор этилового эфира (Е)-3-(2-формилфенил)-акриловой кислоты (30 мл). Смесь перемешивали 1 ч. После выпаривания растворителя остаток растворяли в диэтиловом эфире и промывали рассолом и водой. Раствор сушили над сульфатом магния, фильтровали и выпаривали. Остаток подвергали хроматографии на колонке силикагеля (100 г) и элюировали смесью н-гексан-этилацетат в отношении 3: 1. Фракции, содержащие целевое соединение, выпаривали и получали этиловый эфир (Е)-3-/2-((Z)-1-пентенил)-фенил/-акриловой кислоты (2,00 г).
ЯМР-спектр (CDCl3, δ): 0,38 (3Н, т, I 7 Гц), 1,34 (3Н, т, I 6,5 Гц), 1,42 (2Н, м), 2,05 (2Н, м), 4,27 (2Н, кв, I 6,5 Гц), 5,85 (1Н, дт, I 7 и 11 Гц), 6,39 (1Н, д, I 16 Гц), 6,56 (1Н, д, I 11 Гц), 7,3 (3Н, м), 7,61 (1Н, м), 7,92 (1Н, д, I 16 Гц).
Получение 35. К раствору этилового эфира (Е)-3-/2-((Z)-1-пентенил)-фенил/-акриловой кислоты (2,0 г) в 20%-ном водном метаноле добавляли гидроокись калия (2,3 г). Смесь перемешивали 2 ч при 60оС, устанавливали рН 1,0 хлористоводородной кислотой и экстрагировали этилацетатом. После просушивания экстракта над сульфатом магния и фильтрации растворитель удаляли выпариванием. Остаток растворяли в смеси н-гексан-этилацетат в отношении 4:1. Этот раствор добавляли к дициклогексиамину (1,63 мл) и получали кристаллы. Кристаллы растворяли в этилацетате и промывали 1н. серной кислотой. Раствор сушили над сульфатом магния, фильтровали, выпаривали и получали (Е)-3/2-((Z)-1-пентенил)-фенил/-акриловую кислоту (0,92 г).
ИК-спектр (нуйол): 1690, 1680, 1620 см-1.
Получение 36. (E)-3-/2-((Z)-1-пентенил)-фенил/-акриловую кислоту (1,08 г) растворяли в смеси дихлорметана (10 мл), хлористого оксалила (0,5 мл) и N, N-диметилформамида (0,05 мл). После перемешивания в течение 1 ч при комнатной температуре в атмосфере азота растворитель выпаривали. Остаток растворяли в н-гексане и смесь фильтровали. Фильтрат выпаривали и получали хлористый (Е)-3-/2-((Z)-1-пентенил)-фенил/-пропеонил (1,15 г).
ИК-спектр (чистый): 1750, 1730, 1605, 1585 см-1.
ЯМР-спектр (CDCl3, δ): 0,88 (3Н, т, I 6,5 Гц), 1,45 (2Н, м), 2,06 (2Н, м), 5,95 (1Н, дт, I 11 и 7 Гц), 6,58 (1Н, д, I 11 Гц), 6,66 (1Н, д, I 16 Гц), 7,4 (3Н, м), 7,69 (1Н, м), 8,12 (1Н, д, I 16 Гц).
П р и м е р 22. Следующее соединение получали способом, аналогичным описанному в примере 14.
R-
R=3-(2-пентилфенил)-пропаноил.
Молекулярный вес: FAB-масс-спектр: m/z 1041,6 (М+Н)+.
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения производных пептидов WS-9326 А | 1989 |
|
SU1826970A3 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ТИАЗОЛА ИЛИ ИХ СОЛЕЙ С ГАЛОИДВОДОРОДНОЙ КИСЛОТОЙ | 1990 |
|
RU2010026C1 |
| WS-7622А ДИ-СУЛЬФАТЫ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1991 |
|
RU2087482C1 |
| ПРОИЗВОДНЫЕ ПЕПТИДОВ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ, ПОСРЕДНИКОМ КОТОРЫХ ЯВЛЯЕТСЯ ТАХИКИНИН | 1991 |
|
RU2073683C1 |
| ТРИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ | 1989 |
|
RU2007406C1 |
| АМИДЫ КИСЛОТ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2000 |
|
RU2208608C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ СОЕДИНЕНИЯ WS7622A | 1990 |
|
RU2021280C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПИРАЗОЛПИРИДИНОВОГО ПРОИЗВОДНОГО ИЛИ ЕГО СОЛИ | 1990 |
|
RU2007403C1 |
| ПРОИЗВОДНЫЕ ТИАЗОЛА | 1991 |
|
RU2048468C1 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛО [1,5-А] ПИРИДИНА, ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1992 |
|
RU2069662C1 |

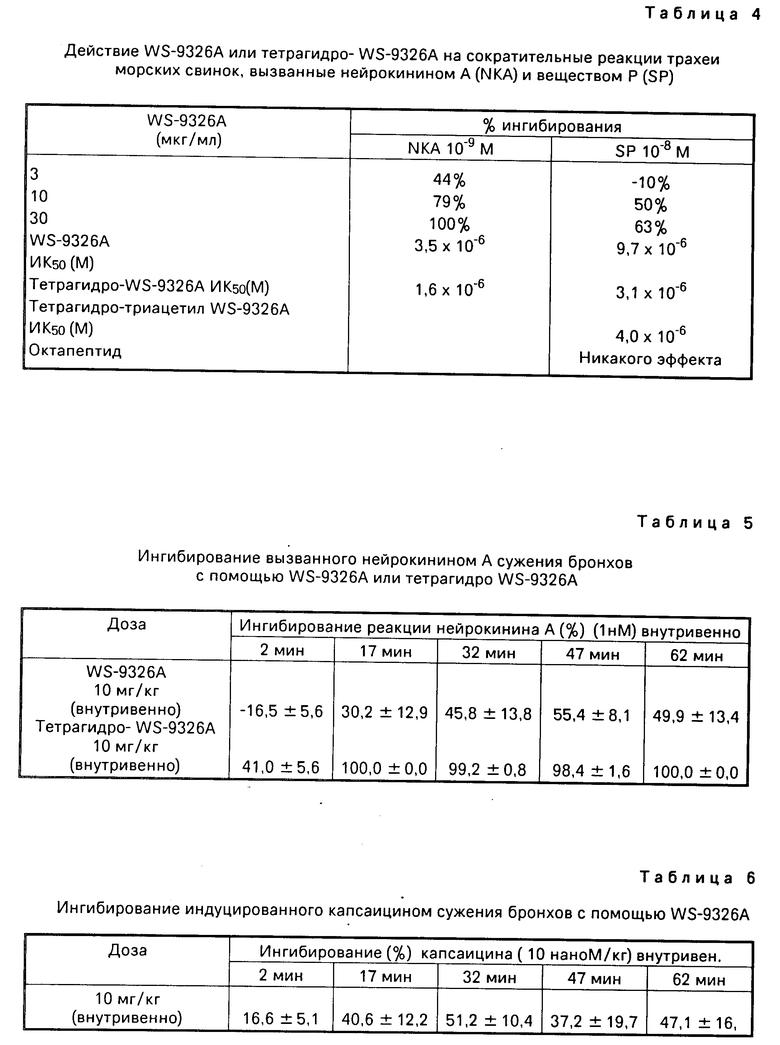



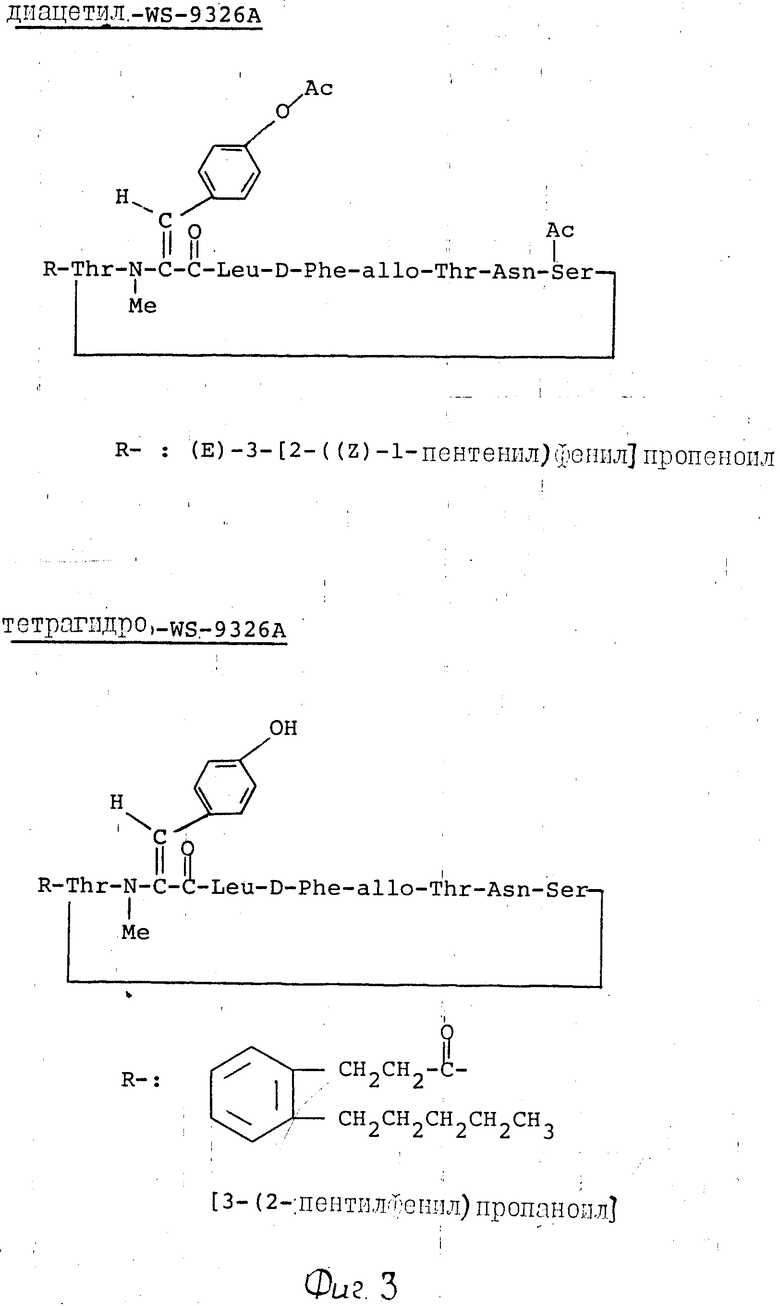
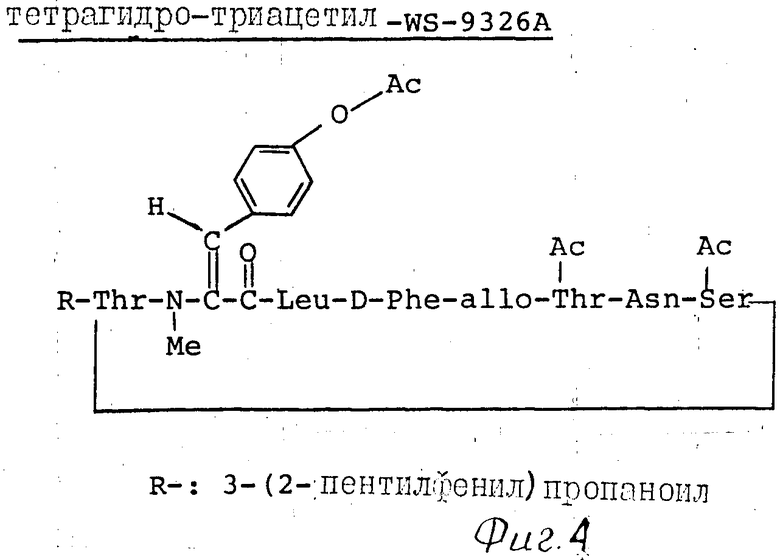
Использование: в биохимии и медицине в качестве ингибиторов реакции нейроксинина А. Сущность изобретения: производные пептидов ф-лы I (см.ниже), где R1 - H, фенил (низший) алкоксикарбонил, низший алканоил, C15-C20- алканоил, бензоил, тиенил (низший) алканоил, фенил (низший) алкеноил, замещенный низшей алкенильной группой, фенил (низший) аланоил, замещенный низшей алкильной группой; R2 -гидроксигруппа; R3 - карбокси или низший алкоксикарбонил; или R2 и R3 соединены вместе, образуя группу ф-лы -О-С(О)-; R4 -гидрокси, низший алканоилокси, бензилокси; R5 гидрокси, низший алканоилокси или бензилокси; R6 -гидрокси, низший алканоилокси или бензоилокси или низший алкокси, и одинарная или двойная связь. 4 ил. 8 табл.
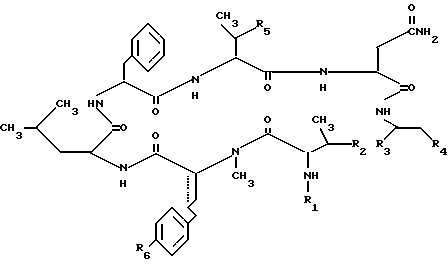
где R1 водород, фенил(низший)алкоксикарбонил, низший алканоил, С15 С20-алканоил, бензоил, тиенил(низший)алканоил, фенил(низший)алкеноил, замещенный низшей алкенильной группой, фенил(низший)алканоил, замещенный низшей алкильной группой;
R2 гидроксигруппа;
R3 карбокси или низший алкоксикарбонил,
или R2 и R3, соединенные вместе, представляют собой группу формулы
R4 гидрокси-, низший алканоилокси или бензилокси;
R5 гидрокси-, низший алканоилокси или бензилокси;
R6 гидрокси-, низший алканоилокси, бензилокси или низший алкокси; одинарная или двойная связь,
одинарная или двойная связь,
или их фармацевтически приемлемые соли.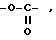
R4 гидроксигруппа, R5 гидроксигруппа, R6 - гидроксигруппа,  двойная связь.
двойная связь.
где R1 3-(2-пентилфенил)-пропаноил.
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Brady stephen F | |||
| и др | |||
| J.Org | |||
| Cheeu | |||
| Кузнечная нефтяная печь с форсункой | 1917 |
|
SU1987A1 |
