Изобретение относится к 2-сахаринилметил- и 4,5,6,7- тетрагидро-2-сахаринилметилфосфатам, -фосфонатам и -фосфинатам, ингибирующим ферментную активность; к композициям, содержащим указанные соединения, к способу их использования и лечении деструктивных нарушений, и к способам их получения.
Ингибирование протеолитических ферментов с помощью нетоксичных реагентов используется при лечении деструктивных нарушений, таких, как эмфизема, ревматоидный артрит, и панкреатит, в основе которых лежит протеолиз. В биомедицинских исследованиях широко используются протеазные ингибиторы. Наиболее распространенным классом протеолитических ферментов являются сериновые протеазы. На основании их субстратной специфичности, некоторые сериновые протеазы относят к группе химотрипсинов или эластаз.
Химотрипсин и химотрипсин подобные ферменты расщепляют пептидные связи в белках в тех участках, где со стороны карбокси конца имеется обычно Tip, Tyr, Phe, Met, Leu или другой аминокислотный остаток, который содержит ароматические или большие алкильные боковые цепи. Эластаза и эластаза-подобные ферменты обычно расщепляют пептидные связи в тех участках, где с карбоксильной стороны связи имеются Ala, Val, Ser, Leu, или другие аналогичные, более мелкие аминокислоты. Химотрипсин- и эластаза подобные ферменты были обнаружены в лейкоцитах, тучных клетках, и панкреотическом соке у высших организмов, и секретируются многими типами бактерий, дрожжей и паразитов.
В публикации японского патента 72/00419 раскрывается ряд 2-сахаринилметилбензоатов, включая 2-сахаринилметилбензоат per Se и 2-сахаринилметил 2,4-дихлоробензоат, и 4-нитробензоат. Указанные соединения обладают сильной активностью против перикуляриоза риса, ризоктониоза риса, гельминтоспориоза риса и бактериоза листьев риса.
Sunkel и др. J. Med. Chem. 31, 1886-1890 (1988) раскрывают ряд 2-сахаринил-низший алкил-1,4-дигидропиридин -3-карбоксилатов, обладающих способностью ингибировать агрегацию тромбоцитов, а также антитромбиновой активностью.
В патенте США (Chen) раскрывают различные 2-ароил-метилсахарины, используемые в качестве фотографических реагентов и пленочных компонентов.
В патенте США 4195023 (Mulvey и др.) раскрываются R1-2-R2 CO-1,2-бензизотиазол-3-оны, где R1 является галогеном, алкокси, алкиламино, диалкиламино, алкокси-карбонилом, амино, нитро, или водородом в бензоидном кольце, а R2 является водородом, алкилом, алкенилом, алкинилом, циклоалкилом, галогенфенилом, гетероарилом, или замещенным гетероарилом; и R1-2-A-CO-сахарины, где R1 имеет значения, аналогичные значениям заместителей бензоидного кольца в 1,2-бензизотиазол-3-онах, а A является алкилом, алкенилом, алкинилом, циклоалкилом, фторофенилом, гетероарилом или замещенным гетероарилом. Указанные соединения обладают способностью к ингибированию эластазы, и используются для лечения эмфиземы. Zimmerman и др. (J. Biol Chem. 255 (20), 9848-9851 (1980)) раскрывают N-ацилсахарины, где ацильной группой является фуроил, феноил, бензоил, циклопропаноил, этилбутирил и акрилоид, которые обладают способностью к ингибированию сериновых протеаз.
В Chemical Abstracts 81, 22249 (1974) раскрывается 4-метилфенил-2-сахаринилкарбоксилат, обладающий бактерицидной и фунгицидной активностью.
Известно несколько классов соединений, являющихся ингибиторами сериновой протеазы. Например, в патенте США 4659855 (Powers) раскрываются производные арилсульфонилфторида, используемые в качестве ингибиторов эластазы. В патентах США 4547371, 4623645, (Doherty и др.) раскрывают цефалоспоринсульфоны и сульфоксиды, соответственно, которые, как было установлено, являются сильными ингибиторами эластазы и используются для лечения воспалительных заболеваний, в частности, артритов и эафиземы.
В работе Jeshima и др. J. Biol. Chem. 257(9). 5085-5091 (1982) представлены результаты исследований сериновых протеаз (эластазы лейкоцитов человека, панкреатической эластазы свиньи, катепсина G, и бычьего химотрипсина Aα)) с использованием 4-нитрофенилэфиров и тиоэфиров N-трифторацетилантранилатов, 2-замещенных-4Н-3,1-бензоксазин-4-онов, 2-замещенных-4-хиназолинонов и 2-замещенных-4-хлорохиназолинов.
В работе Cha (Biochem Pharmacol, 24, 2177-2185 (1975)) обсуждаются кинетические подходы к изучению связывания ингибиторов с макромолекулами, такими, как ферменты; и методы определения таких параметров, как константы ингибирования, скорости реакции, и концентрации связанных и несвязанных ферментов.
В патенте США 4276298 (Jones и др.) раскрываются 2-R-1,2-бензизотиазолинон-1,1-диоксиды, где R является фенилом, замещенным фторо, динитро, трифторметилом, циано, алкоксикарбонилом, алкилкарбонилом, карбоксидом, карбамоилом, алкилациламино, алкилсульфонилом, N, N-диалкилсульфамоилом, трифторометокси, трифторометилтио, трифторметилсульфонилом и трифторометилсульфинилом, или пиридилом, замещенным так же, как R, в том случае, когда R является фенилом за исключением того, что пиридил может быть также моно-нитрозамещенным. Указанные соединения обладают способностью ингибировать протеолитический фермент, в частности, эластазу, и используются для лечения эмфиземы, ревматоидного артрита и других воспалительных заболеваний.
Powers и др. (Biochem. 24, 2048 2058 (1985)) описывают исследования ингибирования четырех химотрипсин-подобных ферментов таких как, катепсин G, протеазы I и II тучных клеток крыс, химаза кожи человека, и химотрипсин Aα с использованием N-фуроилсахарина и N-(2,4-дицианофенил)сахарина.
Svoboda и др. (Coll. Czech. Chem. Commun. 51, 1133 1139 (1986)) раскрывают получение 4-гидрокси-2Н-1,2- бензотиазин-3-карбоксилатов путем внутримолекулярной конденсации Дикмана сложных эфиров 2Н-1,2-бензизотиазол-3-он-2-ацетат-1,1-диоксида.
Патенты США 4350752 и 4363865 (Reczek и др.) и патент США 4410618 (Vanmeter и др.) относятся к фотореагентам (Reczek 4350752 и Vanmeter и др.) и фотографическим красителям (Reczek 4363865) и раскрывают различные 2-замещенные-сахарины, используемые в области фотографии, например, в качестве фотореагентов, связанных посредством гетероатома с имидометильной защитной группой (Reczek 4350752), и диффундируемых в носителе фотографических красителей, связанных с атомом азота имида посредством 1,1-алкиленовой группы (Reczer 4363865); и N-ацилметилимиды, которые раскрываются как блокированные фотореагенты, имеющие группу органического фотореагента, содержащего гетероатом, посредством которого он связывается с защитной группой (Vanmeter).
В патенте США 3314960 (Freed и др.) раскрываются 2-(1,1,3-триоксо-1,2-бензизотиазол-2-ил)глутаримиды, которые, как указано, могут быть использованы в качестве селативных средств.
В патенте Франции 1451417, 2-хлорометилсахарин раскрывается в качестве промежуточного соединения для получения N-метилсахарин d1-транс-хризантемата, используемого в качестве инсектицида, а в патенте США 3002884 (L0) раскрываются 2-хлоро-2-бромо, и 2-иодометилсахарины, используемые в качестве фунгицидных средств.
В PCT-заявке WO 90/13549 (Dunlap и др.) раскрывается серия производных 2-замещенных сахаринов, используемых в качестве ингибиторов протеолитических ферментов.
Изобретение относится к 4-R1-R2-R3-2-сахарин-илметил-, 4-R4-4-R5-6-R6-4,5,6,7-тетрагидро-2-сахаринилметил-, 4,7-C-4,5,6,7-тетрагидро-2-сахаринилметилфосфатам, -фосфонатам, и фосфинатам формул I, II и IIA, соответственно (см. ниже), которые обладают ингибирующей активностью по отношению к протеолитическому ферменту, и которые могут быть использованы для лечения деструктивных нарушений.
Изобретение также относится к композициям для лечения дегенеративных расстройств, которые включают в себя фармацевтический носитель и эффективное количество соединения формул I, II или IIA, ингибирующее протеолический фермент.
Кроме того, изобретение относится к способу использования соединений формул I, II или IIA для лечения дегенеративных нарушений, который заключается в том, что пациенту, нуждающемуся в таком лечении, вводят препарат, содержащий эффективное количество соединения формул I, II или IIA, необходимое для ингибирования протеолитического фермента.
Изобретение также относится к способу получения соединения формул I, II или IIA, который заключается в том, что 4-R1-R2-R3-2-галогенметилсахарин, 4-R4-4-R5-6-R6-4,5,6,7-тетрагидро-2-галогенметилсахарин или 4,7-C-4,5,6,7-тетрагидро-2-галогенметил сахарин подвергают реакции с фосфатом, фосфонатом или фосфиновой кислотой формулы III (см. ниже) в присутствии акцептора кислоты.
В частности, изобретение относится к 4-R1-R2-R3-2-сахаринилметил- и 4-R4-4-R5-6-R6-4,5,6,7-тетрагидро-2- сахаринилметилфосфатам, -фосфонатам, и -фосфинатам формул: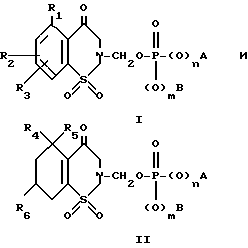 ,
,
где R1 представляет собой водород, галоген, низший алкил, перфторо-низший алкил, перхлоро-низший алкил, низший алкенил, низший алкинил, циано, амино, низший алкиламино, ди-низший алкиламино, карбоксамидо, низший алкокси, бензилокси, гидрокси, низший алкоксикарбонил или фенил;
R2 и R3 независимо представляют собой водород или заместитель в любом из подходящих 5-, 6- или 7-положений, выбранный из группы, включающей в себя галоген, циано, нитро, N B, 1-низший алкил -2-пирролил, низший алкилсульфониламино, полифторо-низший алкилсульфониламино, полихлоро-низший алкилсульфониламино, аминосульфонил, низший алкил, полифторо-низший алкил, полихлоро-низший алкил, циклоалкил, низший алкокси, гидрокси, карбокси, карбоксамидо, гидрокси-низший алкил, формил, аминонометил, полифторо-низший алкилсульфонил, полихлоро-низший алкилсульфонил, низший алкилсульфониламиносульфонил, низший алкоксикарбонил-низший алкиламино, низший алкилкарбониламино, низший алкокси-поли-низший алкиленокси, циклоалкилокси, гидрокси-низший алкокси, полигидроксиалкокси, или его ацеталь или кеталь, полиалкоксиалкокси, (низший алкокси)2P(O)-O-, -SR, -SOR, -SO2R, -OCOR, -O-(C1-10-алкилен)-COOR, -O-(C1-10-алкилен)-COOH и -O-(C2-10алкилен)-N B, где R представляет собой низший алкил, фенил, бензил или нафтил, или фенил или нафтил, замещенный 1 2 заместителями, выбранными из низшего алкила, низшего алкокси, или галогена, а N B, в любом случае, представляет собой амино, низший алкиламино, ди-низший алкиламино, 1-азеотидинил, 1-пирролидинил, 1-пиперидинил, 4-морфолинил, 1-пиперазинил, 4-низший-алкил-1-пиперазинил, 4-бензил-1-пиперазинил, 1-имидазолил, или (карбокси-низший алкил)амино;
либо R2 и R3 вместе представляют собой 3-атомный или 4-атомный незамещенный или метилированный насыщенный мостик, соединяющий атомы углерода в 5,6- или 6,7-положениях, причем, атомы мостика состоят из одного из двух атомов углерода и двух одинаковых или различных гетероатомов, выбранных из атомов кислорода, серы и азота;
R4 является водородом, низшим алкилом или фенилом;
R5 является водородом или первичным низшим алкилом; или R4 и R5, взятые вместе, являются этиленом;
R6 является водородом или низшим алкокси;
m и n независимо являются 0 или 1;
если m и n являются 1, то A и B независимо представляют собой водород, низший алкил, фенил, низший алкоксифенил или бензил; или взятые вместе, они представляют собой: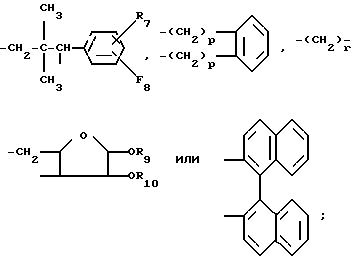
где R7 и R8 независимо представляют собой водород или хлор, а каждый из R9 и R10 является водородом, либо вместе взятые, они представляют собой изопропилиден, p 0 или 1, а r 2, 3 или 4;
если m 1, а n 0, то A и B независимо представляют собой низший алкил, фенил, бензил или 2-пиридинил; и
если m и n равны 0, то A и B независимо представляют собой низший алкил, фенил или низший алкокифенил.
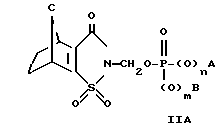
где C представляет собой метилен, этилен, или диметилметилен, а предпочтительно метилен, а A, B, m и n являются такими, как они были определены для формул I и II.
Предпочтительными соединениями формулы I являются соединения, в которых:
R1 представляет собой водород, низший алкил, предпочтительно C1-4-алкил, а более предпочтительно пропил, изопропил или втор-бутил; или низший алкокси, предпочтительно метокси или этокси;
R2 представляет собой низший алкокси, в частности, C1-3-низший алкокси, а более предпочтительно метокси или изопропокси; полиалкоксиалкокси, в частности 2,3-диметоксипропокси; низший алкокси-поли-низший алкиленокси, в частности, метокси-низший алкиленоксиэтокси; или полигидроалкокси, либо его кеталь или ацеталь, а предпочтительно 2,3-дигидроксипропокси, или его диметилкеталь;
R3 представляет собой водород или низший алкокси, в частности, метокси;
m и n, оба равны 0 или 1;
если m и n равны 1, то A и B независимо представляют собой водород, низший алкил, фенил, низший алкоксифенил, или бензил;
если m и n равны 0, то A и В независимо представляют собой низший алкоксифенил.
Другими предпочтительными соединениями формулы I являются соединения в которых:
R1 представляет собой водород; низший алкил, в частности, C1-4-алкил, а предпочтительно изопропил или втор-бутил; или низший алкокси, в частности метокси или этокси;
R2 представляет собой водород, гидрокси или низший алкокси, в частности, метокси или этокси; или полигидроксиалкокси или его кеталь или ацеталь, в частности, дигидроксиалкокси или его кеталь или ацеталь, а предпочтительно 2,3-дигидроксипропокси или его диметилкеталь;
R3 является водородом;
m и n оба являются 0 или 1;
если m и n являются 1, то A и B являются независимо, предпочтительно оба, водородом, низшим алкилом, в частности, C1-4-низшим алкилом, а более предпочтительно, метилом, этилом, изопропилом или бутилом, фенилом, низшим алкоксифенилом, или бензилом; и
если m и n являются 0, то A и B независимо является, предпочтительно оба, низшим алкилом, в частности, C1-4-низшим алкилом, а более предпочтительно, бутилом, фенилом, или низшим алкоксифенилом, в частности метоксифенилом, а более предпочтительно 4-метоксифенилом.
Предпочтительными соединениями формулы II являются соединения, в которых:
R4 представляет собой низший алкил, в частности, метил, этил или изопропил, предпочтительно метил;
R5 представляет собой водород или метил;
R6 представляет собой водород или низший алкокси;
m и n оба являются 0 или 1;
если m и n равны 1, то A и B независимо являются водородом, низшим алкилом, фенилом или бензилом; и
если m и n равны 0, то A и B независимо являются низшим алкилом, фенилом или низшим алкоксифенилом.
Другими предпочтительными соединениями формулы II являются соединения, в которых:
R4 представляет собой низший алкил, предпочтительно метил;
R5 представляет собой первичный низший алкил, предпочтительно, метил;
R6 представляет собой водород или низший алкокси, а предпочтительно водород;
m и n оба являются 0 или 1, предпочтительно 1;
A и B независимо являются, предпочтительно оба, низшим алкилом, в частности, C1-4-низшим алкилом.
Особенно предпочтительными соединениями формулы II являются соединения, в которых:
R4 представляет собой водород или низший алкил; и
R5 представляет собой водород или первичный низший алкил, либо R4 и R5, взятые вместе, представляют собой этилен.
Следует отметить, что в химической литературе, соединения, имеющие общие структурные формулы I и II обозначаются как 1,2-бензизотиазол-3(2H)-он, 1,1-диоксиды. Однако, для краткости, указанные соединения часто называют производными сахарина, и это название будет далее использоваться при описании соединений изобретения и их биологических свойств.
Используемые в описании термины "низший алкил", "низший алкокси" и "низший алкан" означают моновалентные алифатические радикалы, включая радикалы разветвленной цепи, состоящие и 1-10 атомов углерода, Так, например, низшим алкилом (или низшим алканом) указанных групп могут быть метил, этил, изопропил, н-бутил, втор-бутил, трет-бутил, н-пентил, 2-метил-3-бутил, 1-метилбутил, 2-метилбутил, неопентил, н-гексил, 1-метилпентил, 3-метилпентил, 1-этилбутил, 2-этилбутил, 2-гексил, 3-гексил, 1,1,3,3-тетраметилпентил, 1,1-диметилоксил и т.д.
Используемые в описании термины "циклоалкил" и "циклоалкокси" означают радикалы, имеющие от 3 до 7 атомов углерода, примерами которых могут служить циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклопропилокси, циклобутилокси, циклопентилокси и циклогептилокси.
Используемый в описании термин "галоген" означает фтор, хлор, бром или йод.
Используемые в описании термины "низший алкенил" или "низший алкинил" означают моновалентные незамещенные радикалы, включая радикалы разветвленной цепи, содержащие 2-10 атомов углерода, например, такие, как 1-этинил, 1-(2-пропенил), 1-(2-бутенил), 1-(1-метил-2-пропенил), 1-(4-метил-2-пентинил), 4,4,6-триметил-2-гептенил, 1-этинил, 1-(2-пропинил), 1-(2-бутинил), 1-(1-метил-2-пропинил), 1-(4-метил-2-пентинил) и т.п.
Используемый в описании термин C2-10 алкилен означает двухвалентные насыщенные радикалы, включая радикалы разветвленной цепи, содержащие 2-10 атомов углерода, которые имеют свободные валентности по различным атомам углерода; а термин C1-10 означает двухвалентные насыщенные радикалы, включая радикалы разветвленной цепи, содержащее 1-10 атомов углерода, которые имеют свободные валентности по одинаковым или свободным атомами углерода. Примерами таких радикалов могут служит 1,2-этилен, 1,3-пропилен, 1,4-бутилен, 1-метил-1,2-этилен, 1,8-октилен и т.п. а в случае лишь C1-10, то также метилен, этилен, пропилиден и т.п.
Используемый в описании термин низший алкокси-поли-низший алкиленокси означает такие радикалы, в которых низший алкокси имеет значения приведенные выше, поли означает 2-4, а низший алкилен в низшем-алкиленокси означает двухвалентные радикалы, включая разветвленные радикалы, содержащие от 2 до 5 атомов углерода. Так, например, этот термин включает в себя: CH3(OCH2CH2)pO-
CH3CH2[OCH2CH(CH3]pO-, где p 2 4 и т.п.
Используемый в описании термин гидрокси-низший алкокси означает низший алкокси как он был определен выше, замещенный гидрокси-группой, но не на C-1 атома углерода, и может быть, например, представлен 2-гидроксиэтокси т.п.
Используемый в описании термин полиалкоксиалкокси означает моновалентные алифатические алкокси радикалы с 3 5 атомами углерода, замещенные 2 4 метокси или этокси-группами, ни одна из которых не является связанной с тем же или C-1-атомом углерода.
Соединения изобретения ингибируют активность сериновых протезов, в частности, эластазы лейкоцита человека и химотрипсин-подобных ферментов, и поэтому могут быть использованы в лечении заболеваний, связанных с дегенеративными состояниями, например, таких, как эмфизема, ревматоидный артрит, панкреатит, фиброзно-кистозная дегенерация, хронический бронхит, распираторный дистресс-синдром у взрослого человека, воспалительные заболевания кишечника, псориаз, пузырчатка, и дефицит ингибитора α- -трипсина 1.
Соединения формул I, II и IIA получают с помощью реакции 4-R1-R2-R3-2-галогенметилсахарина, 4-R4-4-R5-6-R6-4,5,6,7- тетрагидро-2-галогенметилсахарина или 4,7-C-4,5,6,7-тетрагидро-2-галогенметилсахарина соответственно с сложным диэфиром фосфорной кислоты, сложным моноэфиром фосфорной кислоты, или фосфиновой кислоты формулы: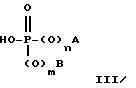
где A, B, m и n имеют значения, определенные выше, за исключением того, что если m и n равны 1, то A и B не являются водородом. Эта реакция может быть проведена в присутствии акцептора кислоты, такого, как карбонат щелочного металла, тринизший алкиламин, или 1,8-диазабицикло[5.4.0]ундек-7-ен (обозначаемый далее ДБУ). Эту реакцию также проводят в органическом растворителе, который является инертным в отношении условий реакции, например, таком, как ацетон, метилэтилкетон (МЭК), ацетонитрил, тетрагидрофуран (ТГФ), диэтилэфир, диметилформамид (ДМФ), N-метилпирролидон, метилендихлорид (МДХ), ксилол, толуол, или низшие алканолы, при температуре в пределах от комнатной температуры до температуры кипения используемого растворителя.
Соединения формул I, II и IIA, где m n равны 1, а A и B являются водородом, получают путем гидрогенолиза соответствующих соединений, в которых m и n равны 1, а A и B являются бензилом.
4-R1-R2-R3-2-галогенметилсахарины, необходимые для получения соединений формулы 1, получают способами, описанными D'Alelio и др. J. Macromol. Sci-Chem. A3(5), 941 (1969) и Saari и др. I. Hct. chem. 23. 1253 (1986). В способа, описанном Saari сложный метиловый эфир соответствующей антранилиновой кислоты получают традиционным методом из замещенной антраниловой кислоты и диазотированного сложного эфира. Затем диазониевую роль подвергают реакции с диоксидом серы и хлоридом меди и получают сульфонилхлорид, который, в свою очередь, подвергают реакции с концентрированным гидроксидом аммония, и получают замещенные производные сахарина формулы IV. После реакции этих производных с формальдегидом в низшеалканоловом растворителе получают 4-1-R2-R3-2-гидрокси-метилсахарины формулы V, которые затем подвергают реакции с тионилгалидом или тригалидом фосфора, в результате чего получают соответствующие производные 4-R1-R2-R3-2-галогенметилсахарина формулы VI.
4-R1-R2-R3-2-галогенметилсахарины формулы VI, где R1, R2, R3, имеют значения, приведенные выше, а X является хлором или бромом, могут быть получены с помощью реакции соответствующего 4-R1-R2-R3-2-фенилтиометилсахарина с сульфурилгалидом в инертном органическом растворителе, например, МДС, этилендихлориде (ЭДХ) или тетрахлорметана, при температуре от около 0oC до около 30oC. 4-R1-R2-R3-2-фенилтиометилсахарины, в свою очередь, получают с помощью реакции 4-R1-R2-R3-сахарина формулы IV с галогенметилфенилсульфидом в инертном органическом растворителе, таком, как толуол, ксилол, ДМФ, или МДХ, при температуре в диапазоне от комнатной температуры до точки кипения используемого растворителе. Эта реакция может быть осуществлена с помощью реакции галогенметилфенилсульфида либо с таллиевой солью производного сахарина формулы IV (полученного путем реакции производного сахарина с низшим алкоксидом таллия в низшем алканоле), либо с ди-низшей алкиламмониевой солью производного сахарина (полученного как описано ниже) в присутствии галида тетра-низший алкиламмония, такого, как бромид тетрабутиламмония (БТБА); либо с производным сахарина формулы VI per se в присутствии галида тетра-низший алкиламмония, либо с производным сахарина формулы IV per se в присутствии галида тетра-низший-алкиламмония и низшего алкоксида щелочного металла, такого, как т-бутоксид калия.
Сахарины формулы IV могут быть также превращены в хлорметилсахарины формулы VI, где X является хлором с помощью одностадийной реакции с избытком формальдегида или с эквивалентным количеством формальдегида, таким, как параформальдегид или 1,3,5-триоксан, и хлоросиланом, предпочтительно хлоротриметилсиланом, в присутствии кислоты Льюиса, предпочтительно каталитического количества хлорида олова, и в инертном растворителе, предпочтительно 1,2-дихлороэтане (этилендихлориде, ЭДХ).
Описанные методы проиллюстрированы ниже, при этом R1, R2 и R3 имеют значения, приведенные выше, Alk является низшим алкилом, X является галогеном, а pH является фенилом.
Соединения формулы IV могут быть также получены посредством реакции 2-R1-R2-R3-N,N-ди-низший алкилбензамида формулы VII с одним молярным эквивалентом низший-алкил щелочного металла, такого, как литий, необязательно в присутствии тетра-низший-алкилэтилендиамина, и в инертном органическом растворителе, например в ТГФ; и реакции полученной соли щелочного металла либо с диоксидом серы при температуре в интервале от -50 до -80oC с последующей реакцией полученного сульфината щелочного металла с гидроксиламин-O-сульфоновой кислотой в присутствии водного основания, либо с сульфорилгалидом, в затем с аммиаком. Если выбирают метод с использованием диоксид-гидроксиламид-O-сульфоновой кислоты, то перед добавлением сульфината щелочного металла, гидроксиламин-O-сульфоновую кислоту желательно нейтрализовать основанием, предпочтительно одним эквивалентом водного гидроксида натрия. Полученный 2-R1-R2-R3-6-амино-сульфонил-N, N-ди-низший-алкилбензамид затем нагревают в кислой среде для осуществления его циклизации с получением ди-низший-алкиламмониевой соли нужного 2-R1-R2-R3-сахарина формулы IV, который может быть непосредственно использован в последующей реакции, или если необходимо, может быть гидролизован в разбавленной кислоте; и выделяют свободный сахарин. Циклизацию предпочтительно проводить в ледяной уксусной кислоте при нагревании с обратным холодильником. Описанный метод проиллюстрирован ниже, где R1, R2, R3 и Alk имеют значения, определенные выше, а щелочным металлом является литий.
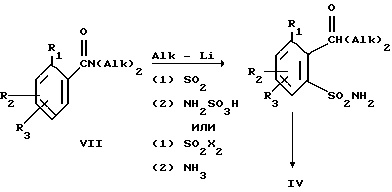
Соединения формулы IV, где R1 является либо первичным, любо вторичным низшим алкилом, которые могут быть использованы в качестве промежуточных соединений для получения соединений формулы I, описанных выше, получают одним из следующих способов.
Соединения формулы IV, где R1 является первичным низшим алкилом, получают посредством реакции 4-метил-R2- R3-сахарина (формула IV, где R1 является CH3) с двумя молярными эквивалентами низший-алкиллития в инертном органическом растворителе, например, ТГФ, и реакции полученной литиевой соли с одним молярным эквивалентом низший-алкилгалидом; причем, обе реакции проводят при температуре в диапазоне от около -50 до -80oC.
Соединения формулы IV, где R1 является первичным низшим алкилом, а R2 и R3 не являются водородом, или R1 является вторичным низшим алкилом, а R2 и R3 являются такими, как они были определены для формулы I, получают посредством реакции 2-первичный-низший-алкил- R2-R3-N,N-ди-низший-алкилбензамида (формулы VII, где R1 не является первичным низшим алкилом) с одним молярным эквивалентом либо низший-алкиллития в присутствии тетра-низший-алкилэтилендиамина, либо с ди-низшим-алкиламином лития, необязательно в присутствии тетра-низший-алкилэтилендиамина, в инертном органическом растворителе, например, ТГФ; и реакции полученной литиевой соли с одним молярным эквивалентом низшего алкилгалида при температуре в диапазоне от около -50 до -80oC. Полученный 2-первичный или вторичный низший алкил -R2-R3-N,N-ди-низший алкил-бензамид затем превращают в соединения формулы IV, где R1 является первичным или вторичным низшим алкилом, посредством той же последовательности реакций, описанных выше, то есть посредством реакции 2-первичный- или вторичный-низший-алкил-R2-R3- N,N-ди-низший алкилбензамида с одним молярным эквивалентом низший алкиллития; реакции полученной литиевой соли либо с диоксидом серы, а затем с гидроксиламин-O-сульфоновой кислотой в присутствии основания, либо с сульфурилгалидом, а затем с аммиаком; и циклизации целевого 4-первичный- или вторичный-низший-алкил-R2-R3- сахарина формулы IV.
Если в исходном материале 2-низший алкил-R2-R3-N,N-низший-алкилбензамиде, 2-низшая алкильная группа является метилом, то после алкилирования получают соединения, в которых 2-низшая алкильная группа является либо прямой, либо разветвленной, в зависимости от того, использовался ли для алкилирования низший алкилгалид с прямой или разветвленной цепью. С другой стороны, если в исходном материале 2-низшая алкильная группа содержит более, чем один атом углерода, то алкилирование происходит на атоме углерода, смежном с бензольным кольцом, и дает продукты, имеющие вторичную низшую алкильную группу во 2-положении.
Предпочтительный способ получения соединений IV, где R1 является н-низшим алкилом, а R2 и R3 являются водородом, включает в себя реакцию блокирования бензильных протонов исходного материала VII триалкилсилильной группой, что позволяет ввести литий в 6-положение, и получить сульфонамид, описанный выше. Этот способ (где R11-CH2 является н-низшим алкилом) иллюстрируется ниже.
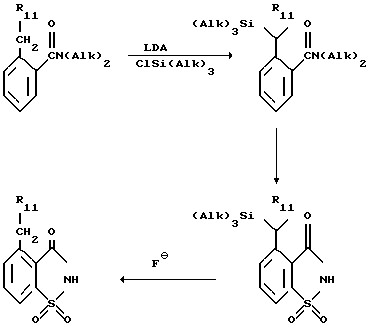
2-н-низший-алкилбензамид подвергают силированию с образованием бензильного аниона, используя алкиллитий или предпочтительно диалкиламид лития (ДАЛ), в инертном растворителе, предпочтительно ТГФ, и обрабатывают соответствующим хлоротриалкилсиланом, предпочтительно хлоротриметилсиланом. Сахарин синтезировали как описано выше, а силильную группу удаляют путем обработки источником аниона фторида, предпочтительно фторидом цезия в ДМФ, или фторидом тетра-н-бутиламмония в инертном растворителе.
Для получения некоторых из целевых сахаринов и промежуточных соединений тетрагидросахарина, в некоторых случаях требуется образование двух колец, составляющих сахариновое ядро. Для получения сахаринов формулы IV, где R1 является водородом, 3,3-дитиобис-пропионовую кислоту превращают в бис-хлоран-гидрид с помощью реакции кислоты с тионилхлоридом, после чего хлорангидрид подвергают реакции с двумя молярными эквивалентными бензиламина и получают бис-N-бинзиламид. После реакции полученного соединения с сульфурилхлоридом в органическом растворителе, таком, как МДХ, ЭДХ или тетрахлорметан, получают 5-хлоро-2-бензил-3Н-изотианол, который затем окисляют с помощью одного молярного эквивалента перкислоты, такой, как пербензойная кислота или 3-хлорпербензойная кислота, с получением 5-хлоро-2-бензил-3(2Н)-изотиазолон-1-оксида. Полученное соединение затем нагревают под давлением с 2-низшим-алкоксифураном в органическом растворителе, таком, как бензол, толуол или ксилол, в результате чего получают 4-низший алкокси-7-гидрокси-2-бензил-1,2-бензизотиазол-3(2Н)-он 1-оксид. По желанию, 7-гидроксигруппа может быть затем подвергнута реакции с низшим алкилгалидом или низшим алкил-(O-низшим-алкилен)p-галидом, где галидом является бромид, хлорид или иодид, в результате чего может быть получен соответствующий 4,7-ди-низший алкокси или 4-низший алкокси -7-низший-алкил-(O-низший-алкилен)p-O] -2-бензил1,2-бензизотиазол-3(2H)-он 1-оксид. После окисления полученного продукта одним молярным эквивалентом перкислоты, как описано выше, с последующем каталитическим дебензилированием, получают соответствующие 4-низший алкокси-7-гидрокси-сахарины. Описанный способ проиллюстрирован ниже (BZ бензил).
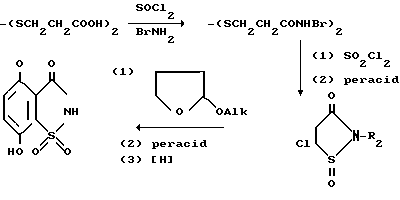
Если необходимо получить 4,5,6,7-тетрагидросахарин формулы VIII, то может быть использована следующая модификация (фиг. 2).
5-хлоро-2-бензил-2Н-изотиазол-3-он 1-оксид может быть окислен с использованием соответствующего окисляющего агента, предпочтительно перекисью водорода в уксусной кислоте, с получением 1,1-диоксида, который затем подвергают реакции в типичных условиях Diels Alder с соответствующим диеном и восстанавливают до получения 2-бензилтетрагидросахарина, который затем подвергают гидрогенолизу, как описано выше, с получением тетрагидросахарина VIII, который, в свою очередь может быть конвертирован в промежуточное соединение, 2-галогенметиловое производное, способом, описанным выше для получения соединения VI из соединения IV.
Альтернативно, соединения формулы I, где R1 является низшим алкилом или фенилом, а R2 и R3 являются водородом, могут быть синтезированы из 2-циклогексенона (фиг.3)
2-Циклогексенон подвергают реакции с купратом (R1)2CuZ, где Z является литием или Mg(X1)2, где X1 является бромидом, хлоридом или иодидом, а затем с метилцианоформатом и гексаметилфосфорамидом (ГМФА) в соответствии с методом Winkler и др. [Tet. Lett. 1987, 1051 и J. Org. Chem, 54, 4491 (1989)] Полученный сложный β--кетоэфир подвергают реакции с бензилмеркаптаном в присутствии кислой монтмориллонитовой глины KSF, в результате чего получают смесь региоизомеров бензилтиоэнолового эфира. Эту смесь ароматизируют путем обработки дихлородицианобензохиноном (ДДХ) и окисляют хлорным газом в водной кислоте, в результате чего получают сульфонилхлоридэфир, который может быть затем конвертирован в соответствующее промежуточное соединение VI, как было показано ранее.
4,5,6,7-тетрагидросахарины, которые являются исходными материалами для получения соединений формулы II, где R6 представляет собой водород, синтезируют способом, аналогичным предыдущему способу (фиг. 4).
3-низший алкил-2-циклогексенон подвергают реакции с соответствующим ди-(низший алкил)литийкупратом в эфирном растворителе, предпочтительно в диэтилэфире, при -50 +20oC, предпочтительно при около 0oC, и полученный аддукт обрабатывают in situ метилцианоформатом и гексаметилфосфорамидом. Полученный таким образом 6,6-ди-(низший алкил)-2-оксоциклогексана карбоксилат подвергают реакции с бензилмеркаптаном, как описано выше, а смесь 2-(бензилтио)циклогексенкарбоксилатов подвергают восстановительному хлорированию, как описано выше, и получают в результате смесь сложных хлоросульфониловых эфиров, которую затем обрабатывают аммиаком, как описано выше, и получают целевой 4,4-ди(низший алкил)-4,5,6,7-тетрагидросахарин VIIIA, который может быть затем конвертирован в промежуточное 2-галогенметиловое производное, как было описано ранее.
Следует отметить, что описанное выше каждое из превращений сахарина IV в 2-галогенметилсахарин VI может быть в равной степени применимо к превращению тетрагидросахаринов VIII и VIIIA в соответствующие 2-галогенметилтетрагидросахарины.
Фосфонаты, фосфаты и фосфиновая кислота формулы III принадлежат к хорошо известному классу фосфорных соединений. Этим классам фосфорных соединений, а также способам их получения посвящено множество работ, например, M. Regitz, Organische Phospoh Verbindungen I и II, Hauben-Weyl, Methoden Der Organischen Chemie, Vilrte Auflage, Erweiterungs-Und-Folge-Bande, Bande E1 и E2, Georg Fhieme Verlag, Штуттгарт-Нью-Йорк, 1982; Robert Engel, Ph. D. Synthesis of Carlon-Phosphorus Bonds, CRC Press, Inc. Boca Raton, Флорида, 1988; J. Jankowska и др. Synthesis (1984); 408; K. Nagasawa, Chem. and Pharm. Bull. 7, 397 (1959); и J. G. Moffatt и др. J. Am. Chem. 79, 1194 (1957).
Для изменений функциональных групп в соединениях изобретения могут быть использованы простые химические преобразования, которые являются традиционными и хорошо известны специалистам. Например, могут быть осуществлены следующие реакции: каталитическое восстановление нитро-групп с получением соответствующих амино-замещенных соединений; окисление сульфидов или сульфоксидов с получением соответствующих сульфоксидов или сульфонов; омыление сложных эфиров с получением соответствующих карбоновых кислот; каталитическое дебензилирование феноловых простых эфиров, бензиламинов или бензилфосфатов с получением соответствующих фенолов, дебензилированных аминов и дебензилированных фосфатов; или реакция фенолов с алкилирующим агентом в присутствии основания или спирта в присутствии связывающего агента с получением нужных простых эфиров.
Стандартные процедуры биологических тестов, которым были подвергнуты характерные соединения изобретения, показали, что указанные соединения обладают активностью, ингибирующей эластазу лейкоцитов человека, и поэтому могут быть использованы для лечения дегенеративных нарушений, таких, как эмфизема, ревматоидный артрит, панкреатит, фибро-кистозная дегенерация, хронический бронхит, респираторный дистресс-синдром у взрослого человека, воспалительные заболевания кишечника, псориаз, пузырчатка и дефицит ингибитора α--трипсина 1.
Соединения изобретения, имеющие основные функции, могут быть превращены в аддитивную кислую соль с помощью реакции основания с кислотой. Аналогичным образом, свободное основание может быть получено из кислой аддитивной соли путем обработки этой соли холодным слабым водным основанием, например, таким, как карбонат щелочного металла и бикарбонат щелочного металла. Полученные таким образом, основания могут быть затем подвергнуты взаимодействию с той же самой или другой кислотой с получением той же самой или другой кислотой аддитивной соли. Таким образом, основания и все их кислые аддитивные соли являются легко взаимопревращаемыми.
Аналогично, соединения изобретения, обладающие кислотными функциями (т. е. карбоновая кислота и фосфат), могут быть преобразованы в их солевые формы с помощью реакции кислоты или фосфата с основанием, таким, как гидроксиды щелочного металла или аммония, или с органическими основаниями, такими, как алкил-, диалкил- или триалкиламины, и наоборот, кислоты и фосфаты могут быть получены из их солей путем обработки этих солей водными кислотами.
Соединения изобретения и их соли обладают присущей им фармакологической активностью, тип которой будет более полно описан ниже. Эта свойственная им активность может быть использована в фармацевтических целях посредством применения свободных оснований или свободных кислот указанных соединений, либо солей, образованных из их фармацевтически приемлемых кислот и оснований (то есть, кислот или оснований, анионы или катионы которых являются безвредными для организма животного), в эффективных дозах, таких, чтобы преимущественные свойства, присущие основной структурной молекуле, представленной свободным основанием или свободной кислотой, не подвергались неблагоприятному воздействию побочных эффектов, приписываемых анионам или катионам.
При использовании указанной фармакологической активности соли, очевидно, что предпочтительно использовать фармацевтически приемлемые соли. Хотя нерастворимость в воде, высокая токсичность или недостаток кристаллических свойств могут сделать некоторые конкретные виды солей неприемлемыми или менее желательными для использования их в данной фармацевтической композиции, однако, эти водонерастворимые или токсичные соли могут быть превращены в их соответствующие фармацевтически приемлемые основания путем разложения указанных солей с помощью водного основания или водной кислоты, как описано выше; или, альтернативно, они могут быть превращены в любую нужную фармацевтически приемлемую соль с помощью двойной реакции разложения с использованием аниона или катиона, например, с помощью ионообменных процедур.
Кроме того, независимо от их фармацевтической ценности, эти соли могут быть использованы как характеризующие или идентифицирующие производные свободных оснований или свободных кислот, либо они могут быть использованы в процедурах выделения или очистки. Подобно всем солям, указанные производные соли, применяемые для характеризации или очистки, могут быть, если это необходимо, использованы для восстановления фармацевтически приемлемых свободных оснований или свободных кислот с помощью реакции этих солей с водным основанием или водной кислотой, либо, альтернативно, они могут быть превращены в любую фармацевтически приемлемую соль, например, с помощью ионообменных реакций.
Новая отличительная особенность соединений изобретения, в данном случае, заключается в идее получения свободных оснований и кислот, а также катионных и анионных форм указанных соединений, имеющих основные и/или кислотные функции, а не какой-либо конкретной кислотной или основной части, либо аниона кислоты или катиона основания, ассоциированных с солевыми формами соединений; скорее можно сказать, что кислотные или основные части, либо анионы или катионы, которые могут быть ассоциированными с солевыми формами, не являются сами по себе ни новыми ни критическими, и поэтому они могут быть любыми анионами кислоты или катионами основания, способными образовывать соли с основаниями или кислотами.
Соединения изобретения могут быть получены для фармацевтического использования путем их введения в виде разовых лекарственных форм, таких, как таблетки или капсулы для перорального введения, содержащие активное соединение либо в чистом виде, либо в сочетании с соответствующими адъювантами, такими, как карбонат кальция, крахмал, лактоза, тальк, стеарат магния, аравийская камедь и т.п. Кроме того, соединение настоящего изобретения могут быть использованы в качестве препаратов для перорального и парентерального введения, или для аэрозольных ингаляций, которые могут быть изготовлены в виде водных растворов водорастворимых солей соединений настоящего изобретения или в виде водных растворов спиртов, гликолей или маслянистых растворов, или эмульсий типа "масло-вода", содержащих указанные соединения; причем, указанные препараты могут быть изготовлены с использованием традиционной техники получения лекарственных средств.
Процентное содержание активного компонента в таких препаратах может варьироваться в соответствии с той дозой, которую необходимо получить. Доза, необходимая для введения пациенту, будет зависеть от клинической оценки, включающей в себя следующие факторы: способ введения, продолжительность лечения, физические данные и состояние пациента, реакционная способность активного компонента и восприимчивость пациента к данному лекарственному средству. Эффективное количество дозы активного компонента может быть легко определено лечащим врачом после рассмотрения всех указанных факторов и использования наилучшей клинической оценки с точки зрения интересов пациента.
Молекулярные структуры соединений изобретения были установлены на основании исследования их ИК- и ЯМР-спектров. Эти структуры были подтверждены рассчитанными и найденными значениями, полученными для элементного анализа. Все точки плавления приводятся без поправок.
Получение исходных материалов.
Приготовление I. Измельченный гидроксид калия (7,4 г, 0,132 М) смешивали с диметилсульфоксидом (ДМСО) (100 мл) и смесь перемешивали в течение 5 мин. Затем к смеси добавляли 6-метилантранилиновую кислоту (10,0 г, 0,066 М) и добавляли по капле иодометан (4,52 мл, 0,073 М). Реакционную смесь перемешивали в течение 30 мин при комнатной температуре, затем разбавляли 250 мл простого эфира, промывали водой (3х100 мл), высушивали сульфатом магния и концентрировали. Сырой продукт фильтровали через прокладку силикагеля ["флеш-сорта (32 63)] элюировали смесью простого эфира и гексана (1:9), в результате чего получали 4,23 г (39%) метил 6-метилантранилата в виде маслянистого вещества.
Полученный таким образом метил 6-метилантранилат (4,23 г, 0,026 М) растворяли в 25 мл уксусной кислоты и раствор охлаждали до 0oC. Затем добавляли концентрированную соляную кислоту 45 мл и получали суспензию коричнево-желтого цвета. После этого, размешивая, по капле добавляли раствор 1,89 г (0,027 М) нитрита натрия в 8 мл воды, и полученный оранжевый раствор размешивали 1 ч при 0oC, а затем добавляли 6 порциями к смеси 2,18 г (0,013 М) дигидрата хлорида меди и диоксида серы (6,3 г), в 33 мл уксусной кислоты и 6 мл воды при 0oC. Темно-зеленый раствор перемешивали при комнатной температуре в течение ночи, выливали в 300 мл ледяной воды, и отделенное твердое вещество собирали и высушивали путем отсасывания, в результате чего получали 1,11 г метил 2-хлоросульфонил-6-метилбензоата, который тотчас же добавляли к 100 мл ледяного гидроксида аммония и перемешивали при комнатной температуре в течение ночи. Затем раствор подкисляли до pH 1 концентрированной соляной кислотой, и полученный осадок собирали и высушивали воздухом, в результате чего получали 729 мг (12%) 4-метилсахарина, т. пл. 224 226oC.
Смесь 1,0 г (0,005 М) 4-метилсахарина, 0,33 г (0,001 М) БТБА и 1,2 г (0,0075 М) хлорометилфенилсульфида в 25 мл толуола нагревали с обратным холодильником приблизительно 16 ч, а затем охлаждали, разбавляли этилацетатом и раствор промывали водным бикарбонатом и водой. Органический слой осушали досуха и получали 0,74 г 2-фенилтиометил-4-метилсахарина.
Полученный раствор (0,74 г, 0,002 М) растворяли в 25 мл МДХ, и раствор, перемешивая, обрабатывали по капле в течение около 2 ч. раствором 0,47 г (0,003 М) сульфорилхлорида в МДХ и реакционную смесь осушали досуха. Твердый желтый остаток перетирали с гексаном, фильтровали и высушивали, в результате чего получали 0,46 г 2-хлорометил-4-метилсахарина в виде бледно-желтого твердого вещества.
Приготовление 2. В соответствии с процедурой, описанной в приготовлении 1, 5,0 г (0,029 М) 6-хлороантранилиновой кислоты и 2,75 мл (0,044 М) иодометана подвергали реакции в присутствии 4,08 г (0,073 М) порошка гидроксида калия, в результате чего получали 4,22 г (78%) метил 6-хлороантранилата в виде маслянистого вещества.
4-Хлоросахарин получали тем же методом, что и 4-метилсахарин, с использованием 4,22 г (0,023 М) метил 6-хлороантранилата в 22 мл уксусной кислоты и 40 мл концентрированной соляной кислоты и 1,68 г (0,024 М) нитрита натрия в 7 мл воды, в результате чего получали диазониевую соль, которую добавляли к 1,93 (0,011 М) дигидрата хлорида меди и 6,5 г диоксида серы в 30 мл уксусной кислоты и 5 мл воды. Полученный метил 2-хлоро-сульфонил-6-хлоробензоат обрабатывали 150 мл гидроксида аммония, как описано выше, в результате чего получали 3,07 г (62%) 4-хлоросахарина в виде бледно-желтого твердого вещества, т.пл. 245-246oC.
Гидроксиметил-4-хлоросахарин получали путем нагревания раствора 1,00 г (0,0046 М) 4-хлоросахарина и 3,22 мл водного 37%-го формалина в этаноле. Все попытки кристаллизовать вязкий маслянистый продукт привели к разложению до исходного материала, и полученный таким образом продукт использовали в следующей стадии без характеризации.
Полученный таким образом сырой 2-гидроксиметил-4-хлоросахарин (609 мг, 0,0025 М) перемешивали с 5 мл диэтилового эфира, и добавляли 3 мл тионилхлорида. Полученную смесь нагревали до полного растворения, затем перемешивали в течение ночи при комнатной температуре, разбавляли 20 мл простого эфира, фильтровали через прокладку из цеолита с нанесенным поверх песком и элюировали простым эфиром. Растворитель удаляли, в результате чего получали 430 мг сырого производного хлорметила. Часть полученного продукта (225 мг) удаляли для следующих реакций. Остаток (205 мг) подвергали флеш-хроматографии на силикагеле и элюировали 40%-ым эфиром/пентаном, в результате чего получали 137 мг 2-хлорметил-4-хлоросахарина, т.пл. 135-136oC.
Приготовление 3. К суспензии 6,0 г (0,03 М) иодида меди в 100 мл ТРФ добавляли 25 мл диметилсульфида, и полученный желтый раствор охлаждали до -78oC и по капле обрабатывали раствором 23 мл (0,06М) 3,0 М раствора фенилбромида магния в диэтиловом эфире. Полученный бледно-желто-оранжевый раствор перемешивали при -78oC в атмосфере азота в течение 1 ч, а затем обрабатывали 3,02 г (0,03 М) 2-циклогексенона в 10 мл ТГФ. Полученную смесь нагревали до 0oC в течение 2 ч, снова охлаждали до -78oC, обрабатывали 15 мл гексаметилфосфорамида, затем перемешивали 30 мин, обрабатывали 8,0 г (0,09 М) метилцианоформата и нагревали при комнатной температуре в течение ночи. Реакционную смесь выливали в 2 н. соляную кислоту (100 мл), и органическую фазу отделяли, водную фазу подвергали обратной экстракции с МДХ. Объединенные органические экстракты высушивали досуха в вакууме и остаток перетирали с насыщенным хлоридом аммония, затем водой, солевым раствором и снова высушивали досуха, в результате чего получали 3,2 г метил 2-фенилциклогексан-6-он карбоксилата в виде масляного вещества.
Полученное соединение (3,0 г, 0,013 М), 4,8 г (0,039 М) бензилмеркаптана и 1,0 г смолы Amberlyst®-15 (Rohm и Haas.) в хлороформе нагревал с обратным холодильником 20 ч, затем смесь обрабатывали еще 1,5 г смолы и нагревали еще 4 ч. Затем смесь охлаждали до комнатной температуры, фильтровали, фильтрат высушивали досуха в вакууме, остаток перетирали с гексаном, а твердое вещество собирали путем фильтрации, в результате чего получали 0,85 г (19%) смеси метил 2-бензилтио-6-фенилциклогекс-2-ен-карбоксилата и метил 2-бензилтио-6-фенилциклогекс-1-ен карбоксилата, 0,6 г (0,0018 М) которых нагревали с 2,0 г 2,3-дихлоро-5,6-дицианобензохинона в 25 мл толуола, перемешивали в атмосфере азота в течение 24 ч. Смесь фильтровали через слой силикагеля, элюируя смесью МДХ и гексана (2:1), и элюат высушивали досуха, в результате чего получали 0,3 г (67%) метил 2-бензилтио-6-фенилбензоата.
Полученное соединение (0,52 г, 0,0016 М), растворенное в 10 мл МДХ, разбавляли 20 мл уксусной кислоты и 5 мл воды, затем смесь охлаждали до -10oC, и через эту смесь барботировали газообразный хлор до тех пор, пока экзотермическая реакция не начнет ослабевать. После этого смесь перемешивали 10 мин и высушивали досуха в вакууме, в результате чего получали 0,41 г (85%) метил-2-хлоро-сульфонил-6-фенилбензоата, который растворяли в 10 мл ТГФ и добавляли к 25 мл концентрированного раствора гидроксида аммония, охлаждая при этом в бане из льда/ацетона. Реакционную смесь экстрагировали МДХ, органическую фазу отбрасывали, а водный слой подкисляли до pH 1 концентрированной соляной кислотой и экстрагировали МДХ. Органические экстракты промывали солевым раствором, высушивали и выпаривали досуха, в результате чего получали 0,33 г (97%) 4-фенилсахарина.
В соответствии с процедурой, описанной в приготовлении 1, 4-фенилсахарин (0,33 г, 0,0012 М) подвергали реакции с 0,3 г (0,0019 М) хлорметилфенилсульфида в 15 мл толуола в присутствии 0,08 г (0,0025 М) ТБАБ, и продукт, 2-фенилтиометил-6-фенилсахарин (0,48 г, 100%), обрабатывали сульфурилхлоридом в МДХ, в результате чего получали 0,36 г (95%) 2-хлорметил-4-фенилсахарина.
Приготовление 3B. К суспензии безводного CuCN (2,16 г, 0,025 М) в безводном простом эфире (100 мл) при -78oC добавляли трет-бутиллитий (29,0 мл 1,7 М раствора в пентане, 0,05 М). После перемешивания в течение 1 ч при -78oC м 30 мин при -45oC, реакционную смесь снова охлаждали до -78oC. Затем добавляли раствор циклогексенона (2,4 г, 0,025 М) в простом эфире (25 мл) и продолжали перемешивание 15 мин при -78oC и 30 мин при -45oC. Полученную смесь снова охлаждали до -78oC, и добавляли ГМФА (10 мл) в простом эфире (25 мл). Через 5 мин добавляли метилцианоформат (2,55 г, 0,03 М) в простом эфире (25 мл), и реакционную смесь нагревали до 0oC в течение 2 ч. Полученную смесь гасили 2 н. HCl (100 мл), слои отделяли, а органическую фазу промывали насыщенным раствором NH4Cl (3х50 мл), водой (2х50 мл), солевым раствором (1х50 мл) и высушивали (Na2SO4). Растворитель удаляли в вакууме и очищали путем дистилляции Kygelrohr (температура бани 100 115oC при 0,6 мм), в результате чего получали 4,7 г (88%) метил 2-(1,1-диметилэтил)-циклогексан-6-он карбоксилата.
Циклогексанон (4,6, 0,022 М) смешивали с бензилмеркаптаном (2,95 г, 0,024 М), кислой монтмориллонитовой глиной, (7,5 г в безводном толуоле (7,5 г). Смесь нагревали с обратным холодильником в атмосфере азота с азеотропным удалением воды в течение 6 ч, охлаждали до комнатной температуры и оставляли в течение ночи. Твердые вещества отфильтровывали и промывали простым эфиром. Объединенный фильтрат промывали 10%-ым Na2CO3, водой, солевым раствором и высушивали. Растворитель удаляли в вакууме, остаток очищали с помощью флеш-хроматографии на силикагеле (10% эфира в гексанах), и получали смесь 4,4 г (66%) метил 2-бензилтио-6-(1,1-диметилэтил)-циклогекс-2-ен-карбоксилата и 2-бензилтио-6-(1,1-диметилэтил)циклогекс-1-ен-карброксилата, которую перемешивали с ДДХ (17,5 г, 0,077 М) в толуоле (50 мл) в течение 16 ч. Красную реакционную смесь фильтровали через 15 см слой силикагеля, элюируя смесью гексанов; МДХ: простого эфира (6:3:1) (1000 мл). Элюаты промывали 10% -ым раствором NaOH, водой, солевым раствором, и высушивали. Растворитель удаляли в вакууме и очищали с помощью хроматографии на силикагеле (5% простого эфира в гексанах), в результате чего получали 1,6 г (40%) метил 2-бензилтио-6-(1,1-диметилбензоата).
Бензилтиобензоат (1,3 г, 0,004 М), растворенный в МДХ (5 мл) разбавляли уксусной кислотой (25 мл) и водой (2 мл), смесь охлаждали до -10oC, и обрабатывали газообразным хлором до тех пор, пока экзотермическая реакция не начнет ослабевать. Затем смесь перемешивали 10 мин осушали досуха в вакууме. Остаток очищали с помощью флеш-хроматографии на силикагеле (1:1 гексана: МДХ), и получали 0,8 г (67% ) метил 2-хлоросульфонил-6-(1,1-диметилэтил)бензоата, который растворяли в ТГФ (5 мл) и добавляли к раствору концентрированного гидроксида аммония (25 мл), охлаждая в бане ил льда/ацетона. После перемешивания в течение 16 ч при комнатной температуре, реакционную смесь концентрировали в вакууме и подкисляли до pH 2 н. HCl. Разделенные твердые вещества собирали путем фильтрации и кристаллизовали из простого эфира, в результате чего получали 0, 64 г (95%) 4-(1,1-диметилэтил)-сахарина, т. пл. 185-187oC.
4-(1,1-диметилэтил)сахарин (0,025 г, 1,0 мМ) смешивали с хлорометилфенилсульфидом (0,25 г, 1,5 мМ) и тетрабутилбромидом аммония (0,2 г, 0,6 мМ) в толуоле (25 мл) и нагревали с обратным холодильником в атмосфере азота в течение 16 ч. Полученную смесь охлаждали до комнатной температуры, выпаривали досуха и очищали с помощью хроматографии на силикагеле (80%) МДХ в гексане, в результате чего получали 0,35 г (98%) 2-фенилтиометил-4-(1,1-диметилэтил)сахарина, который обрабатывали сульфурилхлоридом (0,25 г, 1,8 мМ) в МДХ и получали 0,21 г (75%) 2-хлорометил-4-(1,1-диметилэтил)сахарина.
Приготовление 4. Смесь 3,22 г (0,012 М) 4-бромосахарина [Japanese Pat. Desclosure 58/79, 0,34: C.A. 100, 7773 W (1984)] 1,63 г (0,015 М) т-бутоксида калия 0,39 г (0,0012 М) ТБАБ и 3,0 мл (0,022 М) хлорометилфенилсульфида в 100 мл толуола нагревали с обратным холодильником в атмосфере азота в течение 8 ч и затем перемешивали при комнатной температуре около 16 ч. Реакционную смесь разбавляли этилацетатом, органический слой промывали водой, разбавленным карбонатом калия и солевым раствором, осушали сульфатом магния и высушивали досуха в вакууме. Полученное твердое вещество перекристаллизовали из толуолгексана, в результате чего получали 3,86 г (84%) 4-бромо-2-фенилтиометилсахарина, т. пл. 174,5-178oC.
К полученному раствору (3,27 г, 0,0085 М) в 85 мл МДХ добавляли по капле при перемешивании, 1,02 мл (0,0127 М) сульфурилхлорида. Эту смесь перемешивали при комнатной температуре в течение полутора часов, концентрировали в вакууме и остаток перетирали с гексаном, в результате чего получали 2,61 г сырого продукта, который перекристаллизовали из толуола-гексана, и получали 2,24 г (85%) 2-хлорометил-4-бромосахарина, т. пл. 157-159oC.
Приготовление 5. К раствору 8,0 мл (0,053 М) тетраметилэтилендиамина (ТМЭДА) в 350 мл ТГФ при -70oC добавляли 42 мл (0,055 М), 1,3 М раствора втор-бутиллития в циклогексане и эту смесь перемешивали в течение 15 мин. К этому раствору добавляли по капле при перемешивании раствор 10,36 г (0,050 М) 2-метокси-N,N-диэтилбензамида в 150 мл ТГФ, поддерживая, при этом температуру при 60oC или ниже. После перемешивания в течение 20 мин, в реакционную смесь барботировали диоксид серы, поддерживая при этом температуру реакции ниже -50oC, до тех пор, пока реакционная смесь не даст кислую реакцию при смачивании лакмусовой бумаги. Эту смесь перемешивали при комнатной температуре в течение 2 ч, разбавляли 450 мл гексана, а выделенный твердый материал собирали, растворяли в 200 мл воды, и полученную смесь, размешивая, порциями обрабатывали 65 г ацетата натрия и 21,5 г (0,19 М) гидроксиламин-О-сульфоновой кислоты. Выделенное белое твердое вещество собирали и высушивали, в результате чего получали 7,04 г (49%) 2-аминосульфонил-6-метокси-N, N-диэтилбензамида, т. пл. 190-194,5oC.
Смесь полученного продукта (4,3 г, 0,015 М) в 75 мл диоксана и 25 мл концентрированной соляной кислоты нагревали в паровой бане в течение 70 ч, затем охлаждали, концентрировали в вакууме, разбавляли водой и льдом, и полученную смесь делали сильно основной с помощью концентрированного гидроксида натрия. После этого, смесь промывали МДХ, водный слой подкисляли разбавленной соляной кислотой и экстрагировали МДХ. Экстракты осушали сульфатом магния и высушивали досуха, в результате чего получали 1,29 г (40%) 4-метоксисахарина. Альтернативно и предпочтительно, процедуру циклизации 2-аминосульфонил-6-метокси-N, N-диэтилбензамида в 4-метоксисахарин с 65%-ным выходом осуществляли при нагревании в ледяной уксусной кислоте в течение 6,5 ч.
Повторяли процедуру, описанную в приготовлении 4 (см. выше), 1,14 г (0,0053 М) 4-метоксисахарина подвергали реакции с 1,31 мл (0,0097 М) хлорометилфенилсульфида в толуоле в присутствии 0,72 г (0,0064 М) т-бутоксида калия и 174 мг (0,00054 М) бромида тетрабутиламмония, и получали 1,23 г (69% ) 4-метокси-2-фенилтиометилсахарина, т. пл. 152,5-154,5oC (из этилацетата-гексана), 1,02 г (0,003 М) которого обрабатывали 0,36 мл (0,0045 М) сульфурилхлорида в МДХ, и получали 282 мг (36%) 2-хлорометил-4-метоксисахарина, т. пл. 169-174oC.
Приготовление 6А. К раствору 4,74 мл (0,031 М) тетраметилэтилендиамина в 300 мл ТГФ (который до использования был пропущен через окись алюминия) добавляли 5,8 г (0,03 М) 2-этил-N,N-диэтилбензамида. Этот раствор охлаждали до -78oC и обрабатывали раствором 34,9 мл (0,031 М) 0,9 М раствора в-бутиллития в циклогексане. После добавления вышеуказанного раствора, смесь перемешивали 20 мин и затем обрабатывали раствором 3,2 мл (0,04 М) этилиодида, поддерживая при этом температуру при -78oC. Затем эту температуру повышали до комнатной, смесь перемешивали около 16 ч и выливали в воду. Полученный маслянистый продукт отделяли и хроматографировали на силикагеле, элюируя смесью 10%-го этилацетата и гексана, в результате чего получали 2,86 г (43%) 2-втор-бутил-N,N-диэтилбензамида в виде желтого маслянистого продукта.
В соответствии с процедурой, описанной в приготовлении 5 (см. выше), 2-втор-бутил-N, N-диэтилбензамид (10,45 г, 0,045 М), растворенный в 70 мл ТГФ, добавляли к раствору 39,2 мл (0,047 М) 1,2 М раствора втор-бутиллития в циклогексане и 7,1 мл (0,047 М) тетраметилендиамина в 250 мл ТГФ, поддерживая температуру при -78oC. После добавления, смесь перемешивали еще 0,5 ч при -78oC и затем обрабатывали диоксидом серы при -70oC, а затем нагревали до комнатной температуры. После этого смесь высушивали досуха в вакууме, а остаток растворяли в воде и, перемешивая, добавляли к холодному раствору 15,2 г (0,134 М) гидроксиламин-О-сульфоновой кислоты и 15,4 мл (0,134 М) 35% гидроксида натрия, и получали 10,1 г (72%) 2-аминосульфонил-6-втор-бутил-N,N-диэтилбензамида.
Полученное соединение (6,83 г 0,22 М) растворяли в 100 мл ледяной уксусной кислоты, и раствор нагревали с обратным холодильником в течение 13 ч, а затем высушивали досуха. Остаток перетирали с диэтиловым эфиром, и собирали путем фильтрации, в результате чего получали 5,7 г (83%) диэтиламмониевой соли 4-втор-бутилсахарина.
Полученное соединение (3,0 г, 0,0096 М) подвергали реакции с 1,13 мл (0,012 М) хлорометилфенилсульфида в толуоле, в результате чего получали 3,47 г (100%) 2-фенилтиометил-4-втор-бутилсахарина.
Полученное соотношение (3,2 г, 0,0097 М) подвергали реакции с 2,3 мл (0,029 М) сульфурилхлорида в 20 мл МДХ и получали 2,4 г (87%) 2-хлорометил-4-втор-бутилсахарина.
Приготовление 6B. 2 С помощью процедуры, аналогичной описанной в приготовлении 6A, 9,2 г, (32,9 мМ) 3,4-диметокси-2-пропил-N,N-диэтилбензамида подвергали реакции с диоксидом серы и 5,6 г (49,4 мМ) гидроксиламин-О-сульфоновой кислоты, и получали 7,4 г (63%) 2-аминосульфонил-4,5-диметокси-6-пропил-N,N-диэтилбензамида, который был циклизован в уксусную кислоту с количественным выходом и фенилтиометилирован с использованием 1,42 мл (15 мМ) хлоромутилфенилсульфида, и получали 4,07 г 5,6-диметокси-2-фенилтио-4-пропилсахарина. 3,59 г (8,8 мМ) фенилтиоэфира подвергали реакции с 2,12 мл (36,4 мМ) сульфурилхлорида и получали 2,84 г (97%) 2-хлорометил-5,6-диметокси-4-пропилсахарина.
3,4-диметокси-2-пропил-N, N-диэтилбензамид получали с помощью следующей процедуры:
К раствору 0,216 М н-бутиллития в 250 мл простого эфира при комнатной температуре добавляли по капле 138,2 г (0,216 М) вератрола в 100 мл простого эфира и 32,6 мл (0,216 М) ТМЭДА. Реакционную смесь перемешивали при комнатной температуре в течение 14 ч и 21,9 мл (0,225 М) н-пропила в течение 1 ч при комнатной температуре и обрабатывали водным раствором 1 н. HCl, в результате чего получали 14 г (36% ) 2,3-диметокси-бензолпропана, который бромировали с использованием 14,52 г (81,6 мМ) N-бромосуцинимида на 36 г Кизельгеля в 400 мл CCl4 в соответствии с методом Hisatoshi и др. [Bull. Chem. Soc. Jap 32, 591-593 (1989)] в результате чего получали 19,6 г (98%) 6-бромо-2,3-диметоксибензолпропана.
Бромобензол (14,2 г, 54,8 мМ) растворяли в 200 мл простого эфира, охлаждали до -78oC, и добавляли 25,2 мл (63 мМ) 2,5N н-бутиллития в гексане. Реакционную смесь нагревали до 0oC, выдерживали в течение 1 ч, охлаждали до -70oC и добавляли 9 мл (71,2 мМ) диэтилкарбамилхлорида. Реакцию доводили до комнатной температуры и гасили насыщенным хлоридом аммония. После экстракции и осушки, продукт кристаллизовали из гексана, и получали 9,5 г (62%) 3,4-диметокси-2- пропил-N,N-диэтилбензамида, т.пл. 65-67oC.
Приготовление 6C. В соответствии с процедурой, описанной в приготовлении 6B, 10,75 г (30 мМ) 2-аминосульфонил-4,5-диметокси-6- изопропил-N,N-диэтилбензамида циклизовали, и получали 6,43 г 5,6-диметокси-4-изопропилсахарина (т. пл. 186-188oC из простого эфира-гексана), 5 г (17,5 мМ) которого подвергли фенилтиометилированию с использование 2,48 мл (26,3 мМ) фенилтиометилхлорида в соответствии с процедурой, описанной в получении 5, и хлорированию 3-мя эквивалентами сульфурилхлорида с получением 2-хлорометил-5,6-диметокси-4- изопропилсахарина, с выходом 85% т. пл. 117-119oC после кристаллизации и этилацетата-гексана.
Требуемый бензамид получали из 2,3-диметокси- α--метилбензолэтана путем бромирования с последующим карбамилированием, как описано в приготовлении 6B, в результате чего получали промежуточное соединение 3,4-диметокси-2-изопропил-N, N-диэтилбензамил. Раствор 66 мл 0,96 М втор-бутиллития добавляли к 16,1 г (57,6 мМ) бензамида в 400 ТГФ при -78oC в атмосфере азота. После перемешивания в течение 1 ч, оранжевый анион канюлировали в избыточное количество диоксида серы при -60oC. Реакцию доводили до комнатной температуры и перемешивали 18 ч доля удаления SO2. Затем добавляли 10 мл сульфурилхлорида при 0oC и реакционную смесь выпаривали. Сульфонилхлорид экстрагировали в EtOAc-простой эфир, промывали водой, высушивали и выпаривали. Остаток растворяли в 80 мл ТГФ, и добавляли 17 мл конц. NH4OH при 0oC. Реакционную смесь быстро перемешивали при комнатной температуре, выпаривали, и перетирали в простом эфире-гексане (2:1), в результате чего получали 12,89 г (62%) 2-аминосульфонил-4,5-диметокси-6-изопропил-N, N-диэтилбензамина, т. пл. 138-140oC.2 Приготовление 7. К раствору 9,3 мл (0,058 М) тетраметилэтилендиамина в 340 мл ТГФ при -78oC добавляли 52 мл (1,1 М) раствор (0,067 М) втор-бутиллития в циклогексане. Затем этот раствор обрабатывали раствором 11,37 г (0,052 М) 2-пропил-N,N-диэтилбензамида в 75 мл ТГФ при -78oC, и полученный раствор перемешивали 15 мин, а затем обрабатывали раствором 8,3 мл (0,104 М) этилиодида в ТГФ. После этого раствор перемешивали 1,5 ч при -78oC, а затем гасили путем добавления по капле насыщенного хлорида аммония при -78oC. После этого полученную смесь нагревали до комнатной температуры, разбавляли диэтиловым эфиром, последовательно промывали разбавленной соляной кислотой, водой, насыщенным бикарбонатом натрия, и солевым раствором, а затем осушали досуха, в результате чего получали 12,91 г сырого продукта, который подвергали хроматографии на силикагеле, элюируя 10%-ой смесью этилацетата и гексана, и получали 3,23 г (25%) 2-(3-пентил)-N,N-диэтилбензамида в виде желтого маслянистого вещества.
В соответствии с процедурой, описанной в приготовлении 5 (см. выше), 2-(3-пентил)-N, N-диэтилбензамид (3,05 г, 0,0115 М) в ТГФ подвергали реакции с 10,5 мл (0,126 М) 1,2 М раствора втор-бутиллития в циклогексане в присутствии 2,1 мл (0,014 М) тетраметилендиамина. Затем полученную соль лития подвергали реакции сначала с диоксидом серы, а затем с гидроксиламин-О-сульфонатом натрия, в результате чего получали 1,97 г (52%) 2-аминосульфонил-6-(3-пентил)-N, N-диэтилбензамида в виде бледно-желтых кристаллов, т. пл. 118-120oC (размягчение при 102oC), 1,84 г (0,0056 M) которого циклизовали в 22 мл ледяной уксусной кислоты, при нагревании с обратным холодильником, в результате чего получали 1,28 г (70% ) диэтиламмониевой соли 4-(3-центил)сахарина, т. пл. 107,5-109,5oC.
Полученное соединение 0,0037 М подвергали реакции с 0,74 мл (0,0055 М) хлорометилфенилсульфида в присутствии 166 мг (0,0004 М) БТБА в 45 мл толуола, и получали 1,93 г 2-фенилтиометил-4-(3-пентил)сахарина в виде бледно-желтого маслянистого вещества, 1,93 г (0,0037 М) которого подвергали реакции с 0,59 мл (0,0073 М) сульфурилхлорида в 37 мл МДХ, в результате чего получали 1,2 г 2-хлорметил-4-(3-пентил)сахарина в виде бледно-желтого маслянистого вещества.
Приготовление 8. Раствор 50,0 г (0,27 М) 2,4-диметоксибензойной кислоты в 60 мл (98,0 г, 0,82 М) тионилхлорида нагревали с обратным холодильником в течение 3 ч, затем охлаждали, а избыток тионилхлорида отгоняли. Полученный 2,4-диметоксибензоилхлорид растворяли в 150 мл МДХ и полученный раствор обрабатывали раствором 68 мл (48 г, 0,66 М) диэтиламина в 500 мл МДХ, а затем охлаждали до 0oC. После завершения добавления, смесь перемешивали в течение 15 ч при комнатной температуре, затем промывали насыщенным бикарбонатом натрия, водой, солевым раствором и высушивали досуха, а остаток дистиллировали в вакууме, в результате чего получали 44,78 г (69%) 2,4-диметокси-N,N-диэтилбензамида, т. кип. 155-163 C/0,4 мм.
В соответствии с процедурой, описанной выше в приготовлении 5, 10,0 г (0,042 М) продукта в 250 мл ТГФ подвергали реакции с 40,57 мл 1,1 М раствора (0,044 М) втор-бутиллития в циклогексане и 6,35 мл (0,042 М) тетраметилэтилендиамина в ТГФ. Полученную соль лития сначала подвергали реакции примерно с 40 мл диоксида серы, а затем с водным раствором (0,13 М) гидроксиламин-О-сульфоната натрия, в результате чего получали 8,26 г 2-аминосульфонил-4,6-диметокси-N,N-диэтилбензамида, 7,0 г (0,022 М) которого циклизовали в 80 мл ледяной уксусной кислоты, нагретой с обратным холодильником, в результате чего получали 6,6 г (94%) диэтиламмониевой соли 4,6-диметоксисахарина, которая была использована как таковая в следующей стадии без дополнительной очистки.
Полученное соединение (6,0 г, 0,019 М) подвергали реакции с 3,82 мл (0,028 М) хлорометилфенилсульфида в присутствии 0,611 г (0,0019 М) БТБА в 200 мл толуола, в результате чего получали 6,2 г (89%) 2-фенилтиометил-4,6-диметоксисахарина, 5,82 г (0,016 М), которого подвергали реакции с 3,23 граммами (0,0019 М) сульфурилхлорида в 100 мл МДХ, и получали 4,63 г (100%) 2-хлорометил-4,6-диметоксисахарина, т. пл. 185-187oC.
Приготовление 9A-9C. В соответствии с процедурой, описанной выше в приготовлении 5, заменяя используемый в этой процедуре 2-метокси-N,N-диэтилбензамид соответствующим 2-R1-R2-R3- замещенным-N,N-диэтилбензамидом, в результате чего с помощью промежуточных соединений, 2-фенилтиометилсахаринов, получали 4-R1-R2-R3-2-галогенометилсахарины, представленные в таблице A, где в каждом случае R3 является водородом. В указанной таблице для каждого 2-незамещенных сахаридов, 2-фенилтиометил-сахаринов и 2-хлорометилсахаринов, приводятся, везде, где это возможно, температура плавления (198>C), растворитель для перекристаллизации и выход в колонках, озаглавленных "т. пл. /раств. " и "Выход". Во всех случаях, промежуточные соединения, а именно 2-фенилтиометилсахарины были использованы непосредственно, в последующих стадиях без дополнительной характеризации или очистки.
Приготовление 10. В соответствии с процедурой, описанной в приготовлении 2 18,3 г (0,1 M) сахарина подвергали реакции с 70 мл 37% формалина в этаноле, и получали 3,58 г (70%) 2-гидроксиметилсахарина. Полученное соединение (25 г, 0,117 M) подвергали реакции с 63,3 г (0,234 M) трибромида фосфора в диэтилэфире, в результате чего получали 29,8 г (92%) 2-бромометилсахарина, т. пл. 155 157oC.
Приготовление 11. К раствору 4 г (0,0175 M) 6-нитросахарина в 240 мл этанола, добавляли 4,4 г (0,175 M) этоксида таллия, и полученную смесь оставляли при комнатной температуре в течение 1 ч, затем охлаждали около 16 ч, и полученное твердое вещество собирали и высушивали, в результате чего получали 7,6 г (100%) таллиевой соли 6-нитросахарина. Полученный продукт суспендировали в 50 мл ДМФ, смесь обрабатывали 3,07 граммами (0,0194 M) хлорметилфенилсульфида, а затем эту смесь нагревали при около 63oC в течение 5 ч оставляли при комнатной температуре около 16 ч, после чего ее выливали в ледяную воду. Сырой продукт, полученный путем фильтрации, перемешивали в МДХ и фильтровали для удаления таллиевой соли. Этот фильтрат удаляли из растворителя, а полученное бледно-желтое твердое вещество обрабатывали ультразвуковом с использованием теплового этанола, и снова собирали и высушивали, в результате чего получали 4,6 г (75%) 6-нитро-2-фенилтиометилсахарина, т. пл. 161-163oC. Полученное соединение подвергали реакции с сульфурилхлоридом в МДХ, с использованием процедуры, описанной выше в приготовлении 4, в результате чего получали 3,7 г 2-хлорометил-6-нитросахарина.
Приготовление 12. Раствор 49,8 г (0,199 M) 2-гидрокси-5-(1,1,3,3-тетраметилбутил)бензойной кислоты в 200 мл метанола нагревали до 50oC, а затем обрабатывали по капле примерно 80 г серной кислоты, по крайней мере, для поддержания реакции при температуре перегонки. Затем эту реакционную смесь нагревали с обратным холодильником еще 11 ч, после чего ее охлаждали и распределяли между водой и этилацетатом. Органический слой промывали насыщенным бикарбонатом натрия, солевым раствором, высушивали сульфатом натрия, и осушали досуха, в результате чего получали 48,6 г (92%) метил 2-гидрокси-5-(1,1,3,3-тетраметилбутил)-бензоата.
Полученное соединение, растворенное в 250 мл ДМФ, обрабатывали сначала 40,4 г (0,36 M) 1,4-диазабицикло[2,2,2]-октана, а затем 33,4 граммами (0,27 M) N, N-диметилхлоротиокарбамата в 100 мл ДМФ. Эту реакционную смесь нагревали при 45oC около 8 ч, охлаждали, выливали в ледяную воду, концентрировали соляной кислотой, а затем экстрагировали этилацетатом. Объединенные органические экстракты промывали разбавленной соляной кислотой, бикарбонатом натрия, а затем солевым раствором, после чего высушивали досуха и получали 48,2 г (76%) метил 2-(N,N-диметилтиокарбамилокси)-5-(1,1,3,3-тетраметилбутил)бензоата, который нагревали при 220oC в течение 15 ч, растворяли в толуоле и подвергали хроматографии на силикагеле, элюируя смесью этилацетата и толуола (1:9), в результате чего получали 3,6 г (14%) метил 2-(N,N-диметилкарбамилтио)-5-(1,1,3,3-тетраметилбутил)бензоата.
Раствор полученного соединения (0,025 M) в 40 мл МДХ обрабатывали, перемешивая, 80 м ледяной уксусной кислоты, а затем добавляя 16 мл воды. Реакционную смесь охлаждали до 0oC, и через реакционную смесь барботировали хлор примерно в течение 5 мин, поддерживая при этом температуру между 5 и 24oC. Реакционную смесь перемешивали еще 30 мин, концентрировали в вакууме, а оставшийся раствор выливали в ледяную воду. После экстракции смеси этилацетатом и выделения продукта из объединенных органических экстрактов, получали 6,8 г (78%) метил 2-хлорсульфонил-5-(1,1,3,3-тетраметилбутил)-бензоата.
Полученный продукт 9,0 г (0,026 M) растворяли в ТГФ и добавляли к 100 мл концентрированного гидроксида аммония с охлаждением в ледяной бане. Полученный раствор перемешивали около 16 часов, затем концентрировали в вакууме, и концентрированный раствор подкисляли до pH 3 концентрированной соляной кислотой. Полученную смесь перемешивали несколько ч, а отделенное твердое вещество собирали, промывали водой и высушивали, в результате чего получали 9,0 г 5-(1,1,3,3-тетраметилбутил)сахарина, т. п. 213 215oC.
В соответствии с процедурой, описанной в приготовлении 11, (9,0 г, 0,30 M) продукта подвергали реакции с этоксидом таллия в этаноле, и полученную таллиевую соль подвергали реакции с 3,33 г (9021 M) хлорометилфенилсульфида в ДМФ, в результате чего получали 5,76 г (66%) 2-фенилтиометил-5-(1,1,3,3-тетраметилбутил)сахарина, 3,3 г (0,007 M) которого обрабатывали 0,944 г сульфокрилхлорида в МДХ, и получали 1 г (41%) 2-хлорметил-5-(1,1,3,3-тетраметил)бутилсахарина.
Приготовление 13. В соответствии с процедурой, описанной выше в приготовлении 12, 15,5 г (0,086 M) этил 2-гидрокси-6-метилбензоата подвергали реакции с 15,9 г (0,129 M) N,N-диметилхлоротиокарбамата в присутствии 19,3 г (0,172 M) 1,4-диазабицикло [2,2,2]октана в ДМФ, и получали 22,1 г (96%) этил 2-(N,N-диметилтиокарбамилокси)-6-метил-бензоата, который нагревали при 220oC около 10 ч. Полученный продукт очищали с помощью хроматографии на силикагеле в МДХ, в результате чего получали этил 2-(N,N-диметилкарбамилтио)-6-метилбензоата в виде красно-коричневого маслянистого вещества.
Раствор полученного соединения (22,6 г, 0,0844 M) в 170 мл МДХ обрабатывали 340 м ледяной уксусной кислоты и 68 мл воды, охлаждая в бане из льда и ацетона, и через реакционную смесь барботировали хлор в течение 10 15 мин. Из реакционного сосуда удаляли избыток хлора и МДХ, а смесь выливали в воду и распределяли между МДХ и водой. Органический слой высушивали и выпаривали досуха, в результате чего получали 19 г этил 2-хлоросульфонил-6-метилбензоата, 5 г (0,019 M) которого подвергли реакции с концентрированным гидроксидом аммония в ТГФ, и получали 6,1 г (67%) 4-метилсахарина.
В соответствии с процедурой, описанной выше в приготовлении 11, полученный продукт (10,1 г, 0,0512 M) превращали в литиевую соль с помощью реакции с 12,8 г (0,0512 M) этоксида таллия в этаноле и полученную таллиевую соль подвергали реакции с 6,7 г (0,0427 M) хлорометилфенилсульфида в ДМФ, в результате чего получали 6,85 г (50%) 2-фенилтиометил-4-метилсахарина.
Полученное соединение (6,7 г 0,021 M) подвергали реакции с сульфурилхлоридом в МДХ, и получали 4,9 г (95%) 2-хлорометил-4-метилсахарина.
Приготовление 14A. Смесь 75 г (0,36 M), 3,3-дитиобиспропионовой кислоты, 102 мл тионилхлорида и каталитическое количество пиридина перемешивали около 24 ч и затем выпаривали досуха в вакууме. Остаток обрабатывали с использованием МДХ и снова выпаривали досуха, для удаления остатка тионилхлорида и ангидрида, в результате чего получали 87 г (98%) соответствующего бис-хлорангидрида, 44,8 г (0,18 M) которого растворяли в ТГФ и добавляли по капле к раствору 77,16 г (0,72 M) бензиламина в ТГФ. Полученную смесь перемешивали 2 ч при 40 45oC, охлаждали, а осажденное твердое вещество собирали, промывали водой и высушивали, в результате чего получали 59 г (84%) 3,3 дитиобиспропионовой кислоты N,N'-дибензилкарбоксамида, т. пл. 162 165oC.
Полученное соединение 7,0 г (0,018 M) подвергали реакции с 10,25 (0,076 M) сульфурилхлорида в МДХ и получали смесь 2-бензил-2H-изотиазол-3-она и 5-хлоро-2-бензил-22-изотиазол-3-она, которые были отделены друг от друга, в основном, с использованием ультразвука в МДХ (который солюбилизировал, по большей части, первое соединение). Нерастворенный материал собирали путем фильтрации и хроматографировали на силикагеле с использованием МДХ. Таким образом получали 5-хлоро-2-бензил-2H-изотиазол-3-он, т. пл. 58 68oC.
Раствор 10 г (0,044 M) полученного соединения в МДХ охлаждали до 0oC и раствор обрабатывали 7,6 (0,044 M) 3-хлоропербензойной кислоты, полученную смесь перемешивали 10 минут, а затем обрабатывали второй частью 7,6 г пербензойной кислоты. Затем эту реакционную смесь фильтровали, фильтр промывали МДХ, фильтрат промывали насыщенным бикарбонатом натрия, солевым раствором, высушивали сульфатом натрия, а затем осушали досуха, после чего остаток хроматографировали в МДХ на силикагеле, а полученный продукт элюировали смесью гексана и МДХ (50:50), в результате чего получали 7,15 г (46%) 5-хлоро-2-бензил-2H-изотиазол-3-он-оксида.
Раствор 1,1 г (0,045 M) полученного соединения в 8 мл бензола обрабатывали 0,55 г (0,0051 M) 2-метоксифурана, нагревали в сосуде под давлением при 70oC в течение полутора ч, а затем охлаждали, и полученное твердое вещество собирали, промывали бензолом и высушивали, в результате чего получали 2-бензил-7-гидрокси-4-метоксибензизотиазол-3-он-1-оксида, т. пл. 235 237oC.
Смесь полученного продукта (1,85 г, 0,006 M), 2,48 г (0,018 M) карбоната калия и 1,70 г (0,012 M) метилиодида в ацетоне нагревали с обратным холодильником в течение 1,5 ч, а затем охлаждали и выливали в воду. Выделенное твердое вещество собирали путем фильтрации, промывали водой, и высушивали, в результате чего получали 1,70 г (89%) 2-бензил-4,7-диметоксибензизотиазол-3-он-1-оксида, 1,13 г (0,0035 M) которого оксидировали 1,20 граммами (0,007 M) 3-хлоропербензойной кислоты в МДХ с использованием процедуры, описанном выше, и получали 1,03 г (88%) 2-бензил-4,7-диметоксисахарина.
Смесь 2,07 г (0,0062 M) полученного продукта, 1,37 г (0,02 M) формата аммония и 1,5 г 10% палладированного угля в 80 мл метанола нагревали с обратным холодильником в течение 1 ч, затем охлаждали и фильтровали, а фильтрат высушивали досуха, и получали 0,92 г (57%) аммониевой соли 4,7-диметоксисахарина.
Раствор 1,11 г (0,0042 M) аммониевой соли растворяли в ДМФ, добавляли 0,67 г (0,0042 M) хлорометилфенилсульфида, а затем нагревали с обратным холодильником в течение 8 ч охлаждали, и выливали в ледяную воду. Выделенное твердое вещество собирали, промывали водой и высушивали, в результате чего получали 0,50 г (33%) 2-фенилтиометил-4,7-диметоксисахарина.
Полученное соединение (0,5 г, 0,0013 M) подвергали реакции в сульфурилхлоридом в МДХ, используя процедуру, описанную выше в приготовлении 4, в результате чего получали 0,22 г (58%) 2-хлорометил-4,7-диметоксисахарина.
Приготовления 14B и 14C.
В соответствии с процедурой, описанной в приготовлении 14A, другие производные 2-хлорометилсахарина получали следующим образом:
Приготовление 14B. 5,8 г (0,024 M) 5-хлоро-2-бензил-2H-изотиазол-3-он-1-оксида подвергали реакции с 3,76 г (0,0335 M) 2-этоксифурана, и получали 3,05 г (40%) 2-бензил-4-этокси-7-гидроксибензизотиазолил-3-он-1-оксида, 5,7 г которого подвергали реакции с 3,6 г (0,0197 M) 2-(2-метоксиэтокси)этилбромида в присутствии 4,95 г (0,0358 M) карбоната калия в 125 мл метилкетона и 25 мл ДМФ, в результате чего получали 7,0 г (93%) 2-бензил-4-этокси-7-[2-(2-метоксиэтокси)этокси] бензизотиазол-3-он 1-оксида, который оксидировали, как указано ранее, с использованием 3-хлоропербензойной кислоты в МДХ, и получали 2-бензил-4-этокси-7(2-(2-метоксиэтокси)этокси]сахарина. 6,6 г (0,015 M) полученного соединения дебензилировали 3,34 г (0,053 M) формата аммония в присутствии 6,4 г 10% палладированного угля в метаноле, и получали аммониевую соль 4-этокси-7-[2-(2-метоксиэтокси)этокси] сахарина, которую подвергали реакции с 2,38 г (0,015 M) хлорометилфенилсульфида в 100 мл ДМФ, в результате чего получали 1,46 г (21%) 2-фенилтиометил-4-этокси7-[2-(2-метоксиэтокс)этокси] сахарина, т.пл. 73-75oC (из изопропанола). 1,4 г (0,0029 M) полученного продукта обрабатывали 0,4 г (0,0029 M) сульфурилхлорида в МДХ, и получали 1,16 г 100% 2-хлорометил-4-этокси-7-[2-(2-метоксиэтокси)-этокси]сахарина.
Приготовление 14C. 3,03 г (0,01 M) 2-бензил-7-гидрокси-4-метоксибензизотиазол-3-он-1-оксида приготовление 14A подвергали реакции с 2,01 г (0,011 M) 2-(2-метоксиэтокси)этилбромида в метилэтилкетоне в присутствии 2 г (0,015 M) карбоната калия, и получали 2,58 г (64%) 2-бензил-4-метокси-7-[2-(2-метоксиэтокси)этокси] бензизотиазол-3-он-1-оксида, который оксидировали с использованием 1,1 г (0,0063 M) 3-хлоропербензойной кислоты в МДХ, и получали 2-бензил-4-метокси-7-[2-(2-метоксиэтокси)этокси] сахарин. 0,25 г (0,0006 M) полученного продукта дебензилировали с использованием 0,13 г (0,0021 M) формата аммония в метаноле в присутствии 0,25 г 10% паллидированного угля, в результате чего получали 0,21 г (100%) аммониевую соль 4-метокси-7-[2-(2метоксиэтокси)-этоксисахарина. 1,4 г (0,004 M) аммониевой соли подвергали реакции с 0,63 г (0,004 M) хлорометилфенилсульфида в ДМФ, и получали 2-фенилтиометил-4-метокси-7-[2-(2-метокси-этокси)этокси] сахарин, который подвергали реакции с сульфурилхлоридом в МДХ в результате чего получали 0,53 г (35%) 2-хлорометил-4-метокси-7-[2-(2-метоксиэтокси)этокси]сахарина.
Приготовление 15. Раствор 1,89 г (0,011 M) диэтиламинотрифторида серы (ДАТС) в 20 мл МДХ добавляли к суспензии 2,13 г (0,01 M) 2-гидроксиметилсахарина в 25 мл МДХ, поддерживали реакционную температуру при -78oC.
Эту реакционную смесь перемешивали при -78oC в течение 1 ч температуру медленно доводили до комнатной, а полученную смесь перемешивали 16 ч и затем выливали в ледяную воду. Органический слой отделяли и промывали водой, высушивали сульфатом магния и осушали досуха, в результате чего получали 2,2 г продукта, который перекристаллизовали из этилацетата, в результате чего получали 1,6 г (74%) 2-фторометилсахарина, т.пл. 96-98oC.
Приготовление 16A. К раствору 0,5 г (0,0025 M) 4-метилсахарина в ТГФ, охлажденном до -78oC в сухой бане из льда и ацетона, добавляли по капле, перемешивали, раствор 5,2 мл 1,3 M раствора втор-бутил-лития в циклогексане. Полученную смесь перемешивали еще 1 ч при -78oС, а затем обрабатывали 0,16 мл (0,025 M) метилиодида в течение 1,5 ч. Полученную смесь перемешивали в течение 1 ч и 45 мин, гасили в 25 мл 1 н. соляной кислоты, после чего реакционная смесь делали сильно основной, в водную смесь экстрагировали хлороформом, подкисляли, а затем экстрагировали этилацетатом. Объединенные органические экстракты промывали 10%-ым тиосульфатом натрия, затем солевым раствором и высушивали досуха сульфатом натрия, в результате чего получали продукт, ПРМ-спектр которого обнаруживал смесь, состоящую из 74% 4-этилсахарина и 21% 4,7-диметилсахарина.
В соответствии с процедурой, описанной выше, в приготовлении 4, сырой материал (0,47 г, 0,0022 M) подвергали реакции с 0,24 мл (0,0028 М) хлорометилфенилсульфида в толуоле, в присутствии бромида тетрабутиламмония, и полученный продукт хроматографировали на силикагеле, элюируя МДХ, и 5 мл фракции собирали. Первые 420 мл элюата отбрасывали. После выпаривания следующих 20 фракций, получали 0,07 г материала, преимущественно 2-фенилтиометил-4,7-диметилсахарина, который отбрасывали. Следующие 25 фракций дали 0,37 г 2-фенилтиометил-4-этилсахарина, который подвергали реакции с сульфурилхлоридом в МДХ, в результате чего получали 0,19 г (66%) 2-хлорометил-4-этилсахарина.
Приготовление 16В. В соответствии с процедурой, описанной в приготовлении 16А, 10 г (0,051 М) 4-метилсахарина в ТГФ подвергали реакции с 86 мл (0,10 М) 1,18 М раствора втор-бутиллития в циклогексане, и полученный раствор обрабатывали 4,5 мл (0,050 М) этилиодида, в результате чего получали 10,15 г (89%) 4-пропилсахарина, который подвергали реакции с 5,32 мл (0,056 М) хлорометилфенилсульфида в толуоле, в присутствии бромида тетрабутиламмония, и получали сырую смесь, из которой с помощью флеш-хроматографии на силикагеле выделяли 2-фенилтиометил -4- пропилсахарина в виде маслянистого вещества, 1,8 г (0,0052 М) которого подвергали реакции с 1,25 миллилитрами (0,016 М) сульфурилхлорида в МДХ, и получали 0,94 г (66%) 2-хлоро-метил-4-пропилсахарина.
Приготовление 16С. Предпочтительная альтернатива для приготовления 16А заключается в следующем:
К раствору 5,13 г (25 мМ) N,N,2-триэтилбензамида в ТГФ (50 мл) при -78oC добавляли LDA (Aldrich 2,0 М, 15,63 мл, 31,25 мМ). Полученный раствор нагревали до -10oC в ледяной воде в течение 1 ч, затем охлаждали до -78oC в сухой бане из льда и ацетона. ТМ SCl (6,34 мл, 50 мМ) добавляли примерно при -78oC, а затем реакционную смесь доводили до комнатной температуры в течение 1 ч. Эту реакционную смесь гасили насыщенным NH4Cl, экстрагировали простым эфиром (2х100 мл), высушивали MgSO4, выпаривали, а остаток дистиллировали в Kugelrohr (130 - 140oC, 0,65 мм), в результате чего получали 6,51 г (94%) N, N-диэтил-2-(1-(триметилсилил)этил]бензамида.
К раствору втор-бутиллития (0,97 М, 5,10 мл, 4,96 мМ), ТМЭДА (0,75 мл, 4,96 мМ) в ТГФ при -78oC добавляли избыточное количество SO2, и нагревали до комнатной температуры. ТГФ удаляли в вакууме, а затем подвергали реакции при 0oC с двумя эквивалентами раствора гидроксида натрия (1:1) (0,36 г, 9,0 мМ) и гидроксиламин-О-сульфоновой кислоты (1,0 г, 9,0 мМ) в H2O. Полученную реакционную смесь перемешивали при комнатной температуре в течение 4 ч, экстрагировали EtOAc и подвергали флеш-хроматографии на силикагеле с использованием 20% смеси этилацетата и гексана, в результате чего получали 0,62 г (47%) 2-аминосульфрнил-N,N-диэтил-6-[1-(триметилсилил)этил]-бензамида. Бензамид (0,95 г, 2,66 М) нагревали с обратным холодильником в ледяной уксусной кислоте (20 мл) в течение 18 ч, выпаривали досуха, перетирали с горячим циклогексаном (30 мл) и со следовым количеством EtOAc (3 мл), и охлаждали путем разрыхления и фильтрования. Таким образом получали 0,81 г (85%) 4-[1-(триметилсилил)этил]сахарин, т.пл. 123 125oC.
К триметилсилилэтилсахарину (0,25 г, 0,70 мМ) в ДМФ (9 мл) при комнатной температуре добавляли H2O (1 мл) и фторид цезия (0,75 г, 4,94 мМ, 7 эквивалентов). Через 7 ч реакционную смесь выливали в 5% NaOH и экстрагировали с использованием EtOAc. Водный слой подкисляли 12h•HCl, экстрагировали Et2O EtOAc (1:1), высушивали Na2SO4, фильтровали и выпаривали, в результате чего получали бесцветное твердое вещество с количественным выходом. Это вещество перекристаллизовывали из 5% Et2O-гексанов, в результате чего получали 0,091 г (64%) 4-этилсахарина, т.пл. 183-185oC.
Приготовление 17. 0,07 г образца соединения, полученного в ранних фракциях при хроматографическом разделении, описанном выше в приготовлении 16А, и состоящего преимущественно из 2-фенилтиометил-4,7-диметилсахарина, подвергали реакции с 0,05 мл сульфурилхлорида в МДХ, а продукт перекристаллизовывали из циклогексанаэтилацетата, в результате чего получали 20 мг (51%) 2-хлорометил-4,7-диметилсахарина, т.пл. 107 108oC.
Приготовление 18А. К раствору 40,0 г (0,174 М) 2-изопропил-4-метоксибромобензола в 600 мл диэтилэфира при 0oC добавляли 103,68 мл (0,175 М) 1,69 М раствора бутиллития в диэтилэфире. После завершения добавления, полученный раствор охлаждали до 0oC в течение 1 ч и перемешивали еще 5 ч при комнатной температуре, а затем снова охлаждали до -78oC и обрабатывали раствором 23,68 г (0,175 М) N, N-диэтилкарбамилхлорида в 80 мл диэтилэфира. Полученный раствор перемешивали около 12 ч, реакционную температуру затем повышали, а реакцию гасили с использованием раствора насыщенного хлорида аммония. Водный и органический слой разделяли, водный слой подвергали обратному экстрагированию этилацетатом, и полученные объединенные органические экстракты промывали один раз солевым раствором, а затем высушивали досуха, в результате чего получали сырой продукт, который подвергали флеш-хроматографии на силикагеле, элюируя 30% этилацетатом/гексаном, и получали 34,4 г (79%) 2-изопропил-4-метокси-N,N-диэтилбензамида в виде маслянистого вещества, которое использовали как таковое в последующей стадии без дополнительной очистки. Маслянистое вещество может быть, если это необходимо, подвергнуто дистилляции, т.кип. при 123-129oC/0,2 0,3 мм.
В соответствии с процедурой, описанной выше в приготовлении 5, полученное соединение (15,0 г, 0,060 М) в 100 мл диэтилэфира, подвергали реакции с 77,8 мл (0,784 М) 1,2 М раствора втор-бутиллития в циклогексане в присутствии 6,98 г (0,06 М) тетраметилэтилендиамина. Полученную литиевую соль подвергали реакции сначала с 50 мл диоксида серы, а затем с 0,181 М гидроксиламин-O-сульфонатом натрия, и получали 11,6 г (59%) 2-аминосульфонил-6-изопропил-4-метокси-N, N-диэтилбензамида, т. пл. 103-105oC (из этилацетата/гексана), 11,0 г (0,034 М) которого циклизовали в 200 мл ледяной уксусной кислоты, нагретой с обратным холодильником, в результате чего получали 10,3 г диэтиламмониевой соли 4-изопропил-6-метоксисахарина, т.пл. 132-135oC.
Полученное соединение (0,030 М) подвергали реакции с 6,14 мл (7,25 г, 0,046 М) хлорометилфенилсульфида в присутствии 0,98 г (0,003 М) БТБА в 250 мл толуола, и получали 10,1 г (88%) 2-фенилтиометил-4-изопропил-6-метоксисахарина, в виде маслянистого вещества, 9,7 г (0,026 М) которого подвергали реакции с 3,1 мл (5,21 г, 0,039 М) сульфурилхлорида в МДХ, в результате чего получали 6,9 г (88% ) 2-хлорометил-4-изопропил-6-метоксисахарина, т.пл. 151-152oC.
Приготовление 18В. Альтернативная процедура также заключается в следующем:
К раствору 300 мл N,N,N',N'-тетраметилэтилендиамина (ТМЭДА) (1,99 М) в 4 л безводного простого эфира, добавляли 1550 мл втор-бутиллития (1,3 М) и систему охлаждали до -70oC в атмосфере азота. Затем в течение 30 мин, по капле добавляли раствор 454,2 г 2-изопропил-4-метокси-N,N-диэтилбензамида (1,82 М) в 300 мл безводного простого эфира. Во время добавления, температуру поддерживали при -60oC или ниже. После завершения добавления, реакционную смесь перемешивали пи -70oC в течение 1 ч и нагревали до -50oC. После поддержания температуры при -50oC в течение 30 мин смесь снова охлаждали до -70oC. К этому перемешанному раствору добавляли через канюлю раствор 200 г SO2 200 мл сухого простого эфира, предварительно охлажденного до -40oC, при положительном давлении азота в течение 20 мин. Температуру реакционной смеси в течение добавления поддерживали ниже -40oC. Белый порошкообразный осадок сульфината ариллития отделялся почти тотчас же. После добавления, ледяную баню удаляли, а реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Затем ее охлаждали до -5oC и к этому перемешанному раствору добавляли 190 мл сульфурилхлорида (2,36 М) по капле, в течение 15 мин, поддерживая температуру в течение добавления ниже 10oC. После дополнительного 30-ти минутного размешивания при 0-5oC, белый нерастворимый осадок отфильтровывали и промывали 2-литрами безводного простого эфира. Растворитель удаляли при атмосферном давлении и получали сульфонилхлорид в виде сырого темного маслянистого вещества. Этот сырой сульфонилхлорид растворяли в 1,4 л ТГФ, охлаждали до -10oC, и добавляли порциями, в течение 15 мин, 540 мл концентрированного NH4CH (28%) (в течение добавления поддерживали температуру при 15oC или ниже). После перемешивания в течение 15 мин при комнатной температуре. ТГФ и избыток аммиака удаляли в вакууме, в результате получали темное маслянистое вещество, которое разбавляли 6,0 л воды и подкисляли 3 н. HCl до значения pH 1. Полученное светло-желтое вещество собирали путем фильтрации и промывали 800 мл воды. Полученное твердое вещество высушивали при 60oC в вакууме в течение 18 ч, и перекристаллизовывали из смеси 800 мл этилацетата и 3 л гексана, в результате чего получали 429 г 2-аминосульфонил-6-изопропил-4-метокси-N,N-диэтилбензами, а т. пл. 122-125oC.
Раствор 429,6 г диэтилбензамида (1,31 М) в 1,5 л уксусной кислоты нагревали с обратным холодильником в течение 20 ч. Затем его охлаждали до комнатной температуры, а растворитель удаляли в вакууме. Маслянистый остаток растворяли в 6 литрах воды и доводили до значения pH 1 6 н.соляной кислоты. Полученный сырой продукт собирали путем фильтрации и промывали 2 л воды. Полученное твердое вещество высушивали в вакууме при 60oC в течение 18 ч и перекристаллизовали из этилацетата и гексана, в результате чего получали 303 г (91%) 4-изопропил-6-метоксисахарина, т. пл. 188oC.
К суспензии 24 г параформальдегида (0,8 М) и 86,4 г хлоротриметилсилана (1,6 М) в 200 мл 1,2-дихлорэтана добавляли 0,8 мл безводного хлорида олова (IV), и полученный раствор перемешивали на паровой бане в течение 1 ч. По окончании этого периода, к прозрачному раствору добавляли 51 г (0,2 М) 4-изопропил-6-метоксисахарина, а реакционную смесь нагревали с обратным холодильником еще 18 ч. Затем ее охлаждали до комнатной температуры, выливали в воду, а полученный органический слой отделяли и промывали 50 мл 2 н. раствора гидроксида натрия. Этот органический слой высушивали безводным сульфатом магния и концентрировали в вакууме, в результате чего получали сырой продукт. После этого, его очищали путем кристаллизации из этилацетата и гексана, и получали 57 г (87%) 2-хлорометил-4-изопропил-6-метоксисахарина, т. пл. 151oC.
Приготовление 19. К раствору 1,0 г (0,0039 М) 4-изопропил-6-метокси-сахарина в 15 мл МДХ, добавляли при комнатной температуре 1,28 г (5,12 мл) 1 М раствора трибромида бора в МДХ. После завершения добавления, реакционную смесь нагревали с обратным холодильником в течение 5 ч, затем охлаждали и высушивали досуха, а остаток обрабатывали льдом и насыщенным бикарбонатом натрия. Полученный водный раствор сначала экстрагировали этилацетатом, а затем подкисляли до pH 1 с использованием концентрированной соляной кислоты. После экстрагирования смеси этилацетатом/диэтиловым эфиром (8:2), высушивания органических экстрактов и удаления растворителя в вакууме, получали 0,9 г (96%) 6-гидрокси-4-изопропилсахарина в виде белого кристаллического твердого вещества, которое было использовано как таковое в последующей стадии.
Также использовали альтернативную процедуру. К перемешанной суспензии 62,74 г (0,47 М) AlCl3 в 500 мл хлороформа при 0oC, добавляли 43,9 г (0,7 М) этанэтиола. В течение нескольких минут образовывался прозрачный раствор. К этому раствору в течение 30 мин добавляли 20,0 г (0,078 М) 4-изопропил-6-метоксисахарина в 550 мл хлороформа. Этот раствор нагревали до комнатной температуры и перемешивали 3-4 ч при 60oC. После охлаждения, полученную смесь выливали в ледяную воду и подкисляли разбавленной HCl. Выделенное твердое вещество собирали путем фильтрации, промывали водой и высушивали, в результате чего получали 18,4 г (97%) 6-гидрокси-4-изопропилсахарина.
В соответствии с процедурой, описанной выше в приготовлении 4, 6-гидрокси-4-изопропилсахарин (0,004 М) подвергали реакции с 0,61 мл (0,00416 М) хлорофенилметилсульфида в толуоле в присутствии 0,133 г (0,004 М) БТБА, в результате чего получали 0,32 г (21%) 6-гидрокси-4-изопропил-2-фенилтиометилсахарина, т. пл. 127-129,5oC, 1,78 г которого подвергали реакции с 0,43 мл (0,73 г) сульфурилхлорида в МДХ, и получали 1,2 г (84%) 2-хлорометил-6-гидрокси-4-изопропил-сахарина, т.пл. 149-150oC.
Приготовление 19A. В соответствии с процедурой, описанной выше в приготовлении 19, 4-метоксисахарин может быть превращен последовательно в 4-гидроксисахарин, 4-гидрокси-2-фенилтиометилсахарин и 2-хлорометил-4-гидроксисахарин.
Приготовление 20. 2 5 г (0,0207 М) 6-гидрокси-4-изопропилсахарина растворяли в 150 мл метанола, после чего добавляли 3,4 г (0,0104 М) CS2CO3. Полученную смесь размешивали при комнатной температуре в течение 3-4 ч. Избыточное количество метанола удаляли при пониженном давлении, а полученный остаток осушали в условиях высокого вакуума в течение 2-х ч. После этого, остаток растворяли в 110 мл ДМФ, и добавляли 0,32 г (0,0209 М) хлорометилфунилсульфида. Размешанную смесь нагревали при 70-75oC в течение 12 ч, а затем охлаждали, обрабатывали ледяной водой и экстрагировали 600 мл смеси этилацетата и эфира (4:1). Органический слой промывали водой и насыщенным NaCl, а затем осушали. Растворитель удаляли при пониженном давлении. Полученный остаток очищали с помощью флеш-хроматографии с использованием 20% этилацетата в МДХ. В результате этой процедуры получали 4,5 (60%) 6-гидрокси-4- изопропил-2-фенил-тиометилсахарина (т. пл. 150-151,5oC), который затем подвергали реакции с сульфурилхлоридом в соответствии с процедурой, описанной в приготовлении 19, и получали 2-хлорометил-6-гидрокси-4-изопропилсахарин, как в указанном приготовлении 19.
Приготовление 21. К раствору 5-хлоро-2-бензил-4-изотиазолин-3-она (J. Hlt. Chem. 8, 571, 1971) (9,4 г, 0,04 М) в МДХ (100 мл) добавляли одной порцией 80-85% 3-хлоропероксибензойную кислоту (10,8 г, 0,06 М), и полученную смесь в течение ночи перемешивали в атмосфере азота. Осажденное твердое вещество отфильтровывали и промывали МДХ (50 мл). Объединенный фильтрат выпаривали почти досуха, и остаток распределяли между этилацетатом (300 мл) и насыщенным NaHCO3 (100 мл). Образовавшиеся слои отделяли, а органическую фазу промывали насыщенным NaHCO3 (2х100 мл), солевым раствором (1х100 мл) и осушали. После удаления растворителя в вакууме, получали 10,0 г (99%) 5-хлоро-2-бензил-4-изотиазолин-3(2H)-он-1-оксида в виде светло-желтого маслянистого вещества.
1-оксид (10,0 г, 0,04 М) в ледяной уксусной кислоте (200 мл) обрабатывали 30% H2O2 (100 мл, 0,88 М) и нагревали в паровой бане 2 ч, в течение которых добавляли еще 30 мл (0,26 М) 30% H2O2. После дополнительного 1 ч нагревания на паровой бане, реакционную смесь охлаждали до комнатной температуры, и выливали в холодную воду (1 л) и размешивали. Осажденное твердое вещество собирали путем фильтрации, промывали водой (2х100) и затем гексанами, и осушали воздухом, в результате чего получали 4,8 г (45%) 5-хлоро-2-бензил-4-изотиазолин-3(2H)-она 1,1-диоксида в виде бесцветного твердого продукта.
Диоксид (1,2 г, 4,7 мМ) смешивали с 2,02 г (11 мМ) 2-триметилсилокси-5-метилгекса-1,3-диена (полученного из 5-метилгекс-3-ена в соответствии с методом E.J.Cozey и др. Tet. Lett. 495, 1984) в толуоле (50 мл) и нагревали с обратным холодильником в течение 20 ч в атмосфере азота. Полученную смесь охлаждали до комнатной температуры и концентрировали в вакууме. Остаток растворяли в ТГФ (25 мл) и обрабатывали 2 н. HCl (10 мл). После 10- минутного перемешивания при комнатной температуре в атмосфере азота, добавляли простой эфир (100 мл), и слои разделяли. Органическую фазу промывали водой, солевым раствором, затем высушивали и выпаривали досуха, в результате чего получали бледно-желтую пену. Эту пену растворяли в толуоле (30 мл), добавляли ДБН (1,5 мл), и размешивали 2 ч при комнатной температуре. После этого добавляли МДХ (100 мл) и 2 н. HCl (50 мл) и продолжали размешивать еще 5 мин. Слои отделяли, а органическую фазу промывали водой и солевым раствором, и осушали. После удаления растворителя в вакууме, и очистки остатка с помощью флеш-хроматографии на силикагеле (Гексаны:МДХ:простой эфир, 5:4:1), получали 0,6 г (39%) 2-бензил-4- изопропил-6-оксотетрагидросахарина в виде бледно-желтой пены.
Этот тетрагидросахарин (0,59 г, 1,7 мМ) растворяли в толуоле (50 мл), а затем добавляли диметилгидрохлорид (1,5 г, 18,0 мМ) и 4А сита (2,0 г). Полученную смесь нагревали с обратным холодильником с азеотропным удалением воды в течение 96 ч. В течение этого 96-часового периода, через каждые 12 ч добавляли дополнительное количество диметиламингидрохлорида (0,8 г, 10,0 мМ) и 4A сита, и по истечении этого времени, реакционную смесь охлаждали до комнатной температуры и фильтровали. Остаток на фильтре промывали диэтилэфиром (100 мл), и объединенные фильтраты концентрировали в вакууме, в результате чего получали 0,63 г (99%) 2-бензил-4-изопропил-6-диметиламино (4,5-дигидросахарина в виде бледно-желтого твердого вещества.
К раствору дигидросахарина (0,63 г, 1,7 мМ) в кипящем с обратным холодильником хлороформе 850 мл), частями, в течение 4 ч добавляют активированный диоксид марганца (4,3 г, 59,5 мМ 59,5 мМ). После добавления последней части диоксида марганца, реакцию нагревали с обратным холодильником еще 1 ч, затем охлаждали до комнатной температуры, и фильтровали через прокладку Super-Cel, элюируя этилацетатом. Объединенные элюаты концентрировали в вакууме, а остаток очищали с помощью флеш-хроматографии на силикагеле (элюируя смесью гексанов, МДХ и простого эфира, 5:4:1), в результате чего получали 0,32 г (50%) 2-бензил-4-изопропил-6-диметиламино-сахарина в виде бесцветного твердого вещества.
2-бензилсахарин (0,32 г, 0,9 мМ) в метане (20 мл) обрабатывали форматом аммония (0,24 г, 3,8 мМ) и 10% палладированного угля (0,25), и в течение 1 ч нагревали с обратным холодильником, после чего смесь охлаждали до комнатной температуры и фильтровали через прокладку Super-Cel, элюируя метанолом (100 мл). Объединенные элюаты концентрировали в вакууме. Остаток растворяли в МДХ (10 мл), затем к нему добавляли ледяную уксусную кислоту (0,25 мл), размешивали 5 мин, и выпаривали досуха в вакууме, в результате чего получали 0,25 г (100%) 4-изопропил-6-диметиламиносахарина в виде бесцветной пены.
В соответствии с процедурой, описанной в приготовлении 1, смесь 4-изопропил-6-диметиламиносахарина (0,27 г, 1,0 мМ), хлорометилфенилсульфида (0,32 г, 2,0 мМ) и бромида тетрабутиламмония (0,1 г, 0,2 мМ) в толуоле конвертировали в 0,22 г (56%) 2-фенилтиометил-4- изопропил-6-диметиламиносахарина, который обрабатывали сульфурилхлоридом (1,86 мл, 0,31 мл, 0,31 М раствор, 0,6 мМ), в результате чего получали 0,15 г желтой камеди, которая содержала 25% (как было определено с помощью ЯМР) 2-хлорометил-4-изопропил-6-диметиламино-7-хлоросахарина.
Приготовление 22. 31 г 4-изопропил-1,2-диметоксибензола обрабатывали N-бромосукцинимидом, а затем бутиллитием и диэтилкарбамулхлоридом, как в получении 6B, в результате чего получали 15,2 г 2-изопропил-4,5-диметокси-N, N-диэтилбензамил в виде вязкого маслянистого вещества. Полученный бензамид обрабатывали, как и в получении 18B, бутиллитием и диоксидом серы, затем сульфурилхлоридом, а затем аммиаком, в результате чего получали 4,5 г сульфонамида с т.пл. 181-182oС (после кристаллизации из эфира). Полученное соединение циклизовали в уксусной кислоте в соответствии с процедурой приготовления 18B, и получали в результате 2,86 г, 6,7-диметокси-4-изопропилсахарина, т.пл. 210-212oC (из этилацетатагексана).
К раствору 0,5 г 4-изопропил-6,7-диметоксисахарина в 3 мл ДМФ добавляли 0,5 мл диизопропилэтиламина при комнатной температуре. Через 15 мин добавляли 0,35 г хлорометилфенилсульфида, и полученную смесь нагревали при 80oC в течение 16 ч. Реакционную смесь выливали в EtOA, и промывали водным раствором Na2CO3, водным раствором 2 н. HCl, и насыщенным водным раствором NaCl. Органический слой осушали сульфатом натрия, а растворитель удаляли. В результате хроматографии с использованием МДХ, получали 0,35 г целевого продукта, который затем немедленно использовали. 0,35 г образец фенилтиометилсахарина в 3 мл МДХ обрабатывали 0,1 мл сульфурилхлорида в течение 30 мин при 20oC, с последующим удалением растворителя и растиранием с гексаном, и получали 0,3 г 2-хлоро-метил-6,7-диметокси-4- изопропилсахарина.
Приготовление 23. К раствору 5,7 г метилпиперонилата в 20 мл сухого эфира добавляли 30 мл 3,0 метилмагнийбромида в простом эфире в течение 20 м при 0oC. Полученную смесь размешивали 20 ч, а затем разбавляли 200 мл простого эфира и промывали водой. Органический слой осушали сульфатом натрия, растворителя удаляли, и получали 5,6 г сырого 3,4-диметокси-(1'-гидрокси-1'-метилэтил/бензола. Этот продукт тотчас же обрабатывали в 50 мл уксусной кислоты с 1 г 10% Pd/c в течение 20 ч при добавлении водорода 50 фунт/кв. дюйм (344,7 кПа). После удаления катализатора путем фильтрации и удаления растворителя, получали 4,5 г 5-изопропил-1,3-бензодиоксола. Полученный изопропиодиоксол подвергали бромированию, амидированию, сульфонирования и циклизации в соответствии с процедурой, описанной в приготовлении 22, в результате чего получали 700 мг 4-изопропил-6,7-метилендиокситсахарина с т. пл. 226-228oC (из этилацетата/гексана). В соответствии с процедурой приготовления 22, 500 мг сахарина подвергали хлорометилированию, и получали в результате 300 мг 2-хлорометил-4-изопропил-6,7-метилендиоксисахарина, т. пл. 174-176oC.
Приготовление 24. В соответствии с процедурой, описанной в приготовлении 18А, 5 г 2-бромо-N,N-диметиламина конвертировали в 3,5 г N,N-диэтил-2-диметиламинобензамида. Полученный амид подвергали реакции методом, описанным в приготовлении 18B, и получали 65 мг 4-диметиламиносахарина, т. п. 228-229oC (из эфира-гексана).
В результате реакции 4-диметиламиносахарина с хлорометилфенилсульфидом в присутствии т-бутоксида калия и бромида тетрабутиламмония получали 4-диметиламино-2-фенилтиометилсахарин. А в результате реакции полученного соединения с сульфурилхлоридом в МДХ получали 4-диметиламино-2-хлоро-метилсахарин. Альтернативно, в результате реакции 4-диметиламиносахарина с параформальдегидом и хлоротриметилсиланом в присутствии каталитического количества хлорида олова в этилендихлориде получали 4-диметиламино-2-хлорометилсахарин.
Приготовление 25. К раствору 1,0 г (2,75 мм) 6-гидрокси-4-изопропил-2-фенилтиометилсахарина в ТГФ добавляли 0,73 г (2,78 мм) трифенилфосфина, 0,14 г (3,04 мм) этанола и 0,48 г (2,76 мм) диэтилазодикарбоксилата при комнатной температуре. Полученную смесь перемешивали в течение 10-12 ч. Реакцию повторяли, исходя из 3,73 г (10,28 мм) 6-гидрокси-соединения. Реакционные смеси объединяли, и подвергали флеш-хроматографии на силикагеле, элюируя этилацетатом в гексане (10% а затем 15%), в результате чего получали 4,37 г (85%) 6-этокси-4-изопропил-2-фенилтиометилсахарина, т. пл. 111,5-112,5oC, который затем конвертировали в 2-хлорометил-6-этокси-4-изопропилсахарин с выходом 91% (т. пл. 127-128oC), в соответствии с процедурой, описанной в приготовлении 18А.
Другие 4-R1-R2-R3-сахарины формулы IV, используемые в качестве промежуточных соединений для получения соединений формулы I, могут быть получены следующим образом.
Осуществляют реакцию 2-трифторометилбензойной кислоты с тионилхлоридом, в результате чего получают 2-трифторметилбензоилхлорид, после реакции которого с диэтиламином получают 2-трифторометил-N,N-диэтилбензамид. В соответствии с процедурой, описанной в приготовлении 5, в результате реакции полученного соединения с втор-бутиллитием, и реакции полученной литиевой соли с диоксидом серы, а затем с гидроксиламил-О-сульфонатом натрия, получали 2-трифторметил-6-аминосульфонил-N, N-диэтилбензамид, после нагревания которого в ледяной уксусной кислоте, получали 4-трифторметилсахарин.
Аналогичным образом, в результате реакции 2-трихлорметилбензойной кислоты с тионилхлоридом получали 2-трихлорометилбензоилхлорил, который затем подвергали реакции с диэтиламином и получали 2-трихлорметил-N,N-диэтилбензамид. В соответствии с процедурой, описанной в получении 5, полученный амид подвергали реакции с втор-бутиллитием, а полученную в результате литиевую соль подвергали реакции с диоксидом серы, а затем с гидроксиламин-О-гидрофонатом натрия, в результате чего получали 2-трихлорметил-6-аминосульфонил-N, N-диэтилбензамид, и после его нагревания в ледяной уксусной кислоте, получали 4-трихлорометилсахарин.
В результате реакции 4-циклогексилбензойной кислоты с тионилхлоридом получали 4-циклогексилбензоилхлорид, после реакции которого с диэтиламином получали 4-циклогексил-N, N-диэтилбензамид. Затем, в соответствии с процедурой, описанной в приготовлении 5, осуществляли реакцию полученного соединения с втор-бутиллитием, с последующей реакцией полученной литиевой соли с диоксидом серы, а затем с гидроксиламин-О-сульфонатом натрия, в результате чего получали 4-циклогексил-2-аминосульфонил-N,N- диэтилбензамид, после нагревания которого в ледяной уксусной кислоте, получали 6-циклогексилсахарин.
В результате бензилирования 6-гитросахарина получали 2-бензил-6-нитросахарин, который затем подвергали восстановительной реакции с использованием хлорида олова и водного хлорводорода, и получали 2-бензил-6-аминосахарин. После реакции этого соединения с метансульфонилхлоридом, трифторметилсульфонилхлоридом или трихлорметилсульфонилхлоридом в МДХ в присутствии пиридина с последующим гидрогенолизом 2-бензил-защитной группы получали, соответственно, 6-метилсульфониламиносахарин, 6-трифторметилсульфониламиносахарин, или 6-трихлорметилсульфониламиносахарин.
В результате диазотирования 6-аминосахарина с использованием азотистой кислоты в кислой среде и разложения полученной диазониевой соли в присутствии цианида меди или хлорида меди и диоксида серы, или хлорида меди и щелочно-металлической соли метилмеркаптана или трифторметилмеркаптана, получали, соответственно, 6-цианосахарин, 6-хлоросульфонилсахарин, 6-метилтиосахарин или 6-трифторометилтиосахарин. После реакции 6-хлоросульфонилсахарин in situ с аммиаком или метансульфониламидом получали, соответственно, 6-аминосульфонилсахарин и 6-метансульфониламиносульфонилсахарин. В результате окисления 6-метилтиосахарина и 6-трифторометилтиосахарина с использованием двух молярных эквивалентов 3-хлоропербензойной кислоты получали 6-метилсульфонилсахарин и 6-трифторметилсульфонилсахарин, соответственно.
В результате гидролиза 6-цианосахарина при нагревании с водным раствором гидроксида натрия получали сахарин-6-карбоновую кислоту. После N-бензилирования 6-цианосахарина получали 2-бензил-6-цианосахарин. После щелочного гидролиза этого соединения получали 2-бензилсахарин-6-карбоновую кислоту, которую затем превращали в хлорид 2-бензилсахарин-6-карабоновой кислоты с помощью реакции с тионилхлоридом с последующей исчерпывающей гидрогенизацией в присутствии палладированного угля, получали 6-гидроксиметилсахарин. После окисления этого соединения с использованием "пиридин-трихлорид хрома, 2:1" (Реагент Коллинза) в МДХ получали 6-формилсахарин, который затем подвергали восстановительному аминированию с использованием аммиака, в результате чего получали 6-аминометилсахарин.
После реакции 4-трифторометилбензойной кислоты с тионилхлоридом получали хлорид 4-трифторометилбензоилхлорид, который затем подвергали реакции с диэтиламином в результате 4-трифторметил-N,N-диэтилбензамил. В соответствии с процедурой, описанной в приготовлении 5, осуществляли реакцию полученного соединения с втор-бутиллитием, и реакцию полученной в результате литиевой соли с диоксидом серы, а затем с гидроксиламин-N,N-сульфонатом натрия, в результате чего получали 4-трифторометил-2-аминосульфонил-N,N-диэтилбензамид, который затем нагревали в ледяной уксусной кислоте и получали 6-трифторметилсахарин.
В результате реакции 4-трихлорометилбензойной кислоты с тионилхлоридом получали 4-трихлорометилбензоилхлорид, который затем подвергали реакции с диэтиламином и получали 4-трихлорометил-N,N-диэтилбензамид. В соответствии с процедурой, описанной в приготовлении 5, полученное соединение подвергали реакции в втор-бутиллитием, а полученную соль лития подвергали реакции с диоксидом серы, а затем гидроксиамин-О-сульфонатом натрия, в результате чего получали 4-трихлорометил-2-аминосульфонил-N,N-диэтилбензамид, после нагревания которого в ледяной уксусной кислоте, получали 6-трихлорметилсахарин.
В результате реакции 2-этенилбензойной кислоты с тионилхлоридом получали 2-этенилбензоилхлорид, а после реакции этого соединения с диэтиламином получали 2-этинил-N,N-диэтилбензамин. Полученное соединение подвергали реакции с втор-бутиллитием, а полученную литиевую соль подвергали реакции с диоксидом серы, а затем с гидроксиламин-О-сульфонатом натрия, в результате чего получали 2-этенил-6-аминосульфонил-N, N-диэтилбензамид, после нагревания которого в ледяной уксусной кислоте получали 4-этенилсахарин.
В результате реакции 2-этенил-6-аминосульфонил-N, N-диэтилбензамида с бромом получали 2-(1,2-дибромэтил)-6-аминосульфонил-N, N-диэтилбензамид, который подвергали реакции с амидом натрия в аммиаке, и получали в результате 2-этинил-6-аминосульфонил-N,N-диэтилбензамид, после нагревания которого в ледяной уксусной кислоте получали 4-этинилсахарин.
Этил 2-аминобензоат подвергали реакции с двумя молярными эквивалентами бензилхлорида в ацетоне карбоната калия и получали этил 2-(N,N-дибензиламино)-бензоат, который затем подвергали омыленную в водном этаноловом растворе гидроксида калия и полученный продукт выделяли из нейтральной среды, в результате чего получали 2-(N,N-дибензиламино)-бензойную кислоту.
Полученную кислоту подвергали реакции с тионилхлоридом, и получали 2-(N, N-дибензиламино)бензоилхлорид, после реакции которого с диэтиламином получали 2-N,N-диэтиламино-N,N-диэтилбензамид. Это соединение подвергали реакции с втор-бутиллитием, а полученную литиевую соль подвергали реакции с диоксидом серы, а затем с гидроксиламин-О-сульфонатом натрия, в результате чего получали 2-(N, N-дибензил)-6-аминосульфонил-(N, N-диэтилбензамид), после нагревания которого в ледяной уксусной кислоте получали 4-N,N-дибензиламино сахарин, который затем подвергали каталитическому дибензилированию в атмосфере водорода и в присутствии палладированного угля, в результате чего получали 4-аминосахарин. После восстановительного алкилирования этого соединения, которое осуществляли с использованием одного молярного эквивалента формальдегида в муравьиной кислоте, получали 4-метиламиносахарин.
Осуществляли селективное N-бензилирование цезиевой соли 6-гидрокси-4-изопропилсахарина (приготовление 19) с использованием бензилбромида, и реакцию 2-бензил-6-гидрокси-4-изопропилсахарина с N,N-диэтилтиокарбамилхлоридом в ДМФ в соответствии с процедурой, описанной в приготовлении 12, в результате чего получали 2-бензил-4-изопропил-6-(N, N-диэтилтиокарбамилокси)сахарин, который затем подвергали нагреванию и перегруппировке с получением 2-бензил-4-изопропил-6-(N, N-диэтилкарбамилтио)сахарина. После гидролиза этого соединения с использованием щелочи получали 2-бензил-4-изопропил-6-меркаптосахарин, который подвергали реакции с метилиодидом и обменному гидрогенолизу, в результате чего получали 4-изопропил-6-метилтиосахарин. Это соединение окисляли с использованием одного или двух молярных эквивалентов 3-хлорбензойной кислоты и получали в результате 4-изопропил-6-метилсульфинилсахарин и 4-изопропил-6-метилсульфонилсахарин.
В результате реакции 2-изопропил-4-фторобензойной кислоты с тионилхлоридом получали 2-изопропил-4-фторобензоилхлорид, который затем подвергали реакции с диэтиламином, и получали в результате 2-изопропил-4-фторо-N,N-диэтилбензамид. После реакции этого соединения с втор-бутиллитием, и реакции полученной литиевой соли с диоксидом серы, а затем с гидроксиламин-О-сульфонатом натрия, получали 2-изопропил-4-фторо-2-аминосульфонил-N,N-диэтилбензамид, после нагревания которого в ледяной уксусной кислоте получали 4-изопропил-6-фторосахарин.
Полученное соединение подвергали реакции с тиофенолом, 4-метилфенилтиофенолом, 4-метаксифенилтиофенолом, 4-хлорофенилтиофенолом, 1-меркапто-4-метилнафталином или 1-меркаптонафталином с последующим нагреванием реагентов в ДМФ, и получили в результате 4-изопропил-6-фенилтиосахарин, 4-изопропил-6-(4-метилфенилтио)сахарин, 4-изопропил-6-(4-метокси-фенилтио)сахарин, 4-изопропил-6-(4-хлорофенилтио)-сахарин, 4-изопропил-6-(4-метил-1-нафталтио)сахарин и 4-изопропил-6-(1-нафталтио)сахарин, соответственно, после чего эти соединения подвергали реакции окисления в каждом случае с использованием одного или двух молярных эквивалентов 3-хлорбензойной кислоты, и получали в результате 4-изопропил-6-фенилсульфинил-сахарин, 4-изопропил-6-фенилсульфонилсахарин, 4-изопропил-6-(4-метилфенилсульфинил) сахарин, 4-изопропил-6-(4-метилфенилсульфонил)сахарин, 4-изопропил-6-(4-метоксифенилсульфинил)сахарин, 4-изопропил-6-(4-метоксифенилсульфонил)сахарин, 4-изопропил-6-(4-хлорфенилсульфинил)сахарин, 4-изопропил-6-(4-хлорфенилсульфонил)сахарин, 4-изопропил-6-(4-метил-1-нафтилсульфинил)сахарин, 4-изопропил-6-(4-метил-1-нафтилсульфонил)сахарин, 4-изопропил-6-(1-нафтилсульфинил)сахарин, и 4-изопропил-6-(1-нафтилсульфонил)сахарин.
2-бензил-6-гидрокси-4-изопропилсахарин подвергали реакции с одним молярным эквивалентом уксусного ангидрида, бензоилхлорида, или хлорида 1-нафтилкарбоновой кислоты, с последующим в каждом случае, обменным гидрогенолизом, в результате чего получали, соответственно, 4-изопропил-6- ацетоксисахарин, 4-изопропил-6-бензоилокси-сахарин и 4-изопропил-6-(1-нафтил-карбонил-окси)сахарин.
После нагревания 4-изопропил-6-фторосахарина в ДМФ с азетидином, пирролидином, пиперидином, морфолином, 1-бензилпиперазином, 1-метилпиперазином, имидазолом, т-бутил-альфа-амино-ацетатом или аммиаком получали, соответственно, 4-изопропил-6-1-азетидинил сахарин, 4-изопропил-6- (1-пирролидинил)сахарин; 4-изопропил-6-(1-пиперидинил)сахарин, 4-изопропил-6-(5-морфолин)сахарин, 4-изопропил-6-(4-бензил-1-пиперазинил)сахарин, 4-изопропил-6-(4- метил-1-пиперазинил)сахарин, 4-изопропил-6-(1-1Н-имидазолил)сахарин, 4-изопропил-6-(т-бутоксикарбонил-метиламино)сахарин, и 4-изопропил-6-аминосахарин.
4-Иизопропил-6-(4-бензил-1-пиперазинил)сахарин подвергали каталитическому дебензилированию в атмосфере водорода и в присутствии палладированного угля, в результате чего получали 4-изопропил-6-(1-пиперазинил)сахарин.
4-Изопропил-6-(т-бутоксикарбонилметиламино)сахарин подвергали гидролизу с использованием разбавленной соляной кислоты, после чего продукт выделяли из нейтральной среды и получали 4-изопропил-6- карбоксиметиламиносахарин.
После реакции 4-изопропил-6-аминосахарина с одним молярным эквивалентом ацетилхлорида получали 4-изопропил-6-ацетиламиносахарин.
С помощью щелочного гидролиза проводили реакцию омыления 4-карбометоксахарина (приготовление 9D) с получением соответствующей сахарин-4-карбоновой кислоты, которую с помощью реакции с тионилхлоридом превращали в хлорангидрид, и после реакции указанного хлорангидрида с аммиаком получали сахарин-4-карбоксамид.
После диазотирования аминосахарина с азотистой кислотой в кислой среде и разложения полученной джазониевой соли в присутствии цианида меди получали 4-цианасахарин.
4-R1-R2-R3-2-хлорометилсахарины формулы VI, представленные в табл. 2, где, в каждом случае, R3 является водородом, а X является Cl, могут быть получены с помощью реакции 4-R1-R2-R3-сахаринов, которые, в свою очередь, получают с использованием хлорометилфенилсульфида в присутствии т-бутоксида калия и бромида тетрабутиламмония, с последующей реакцией полученных 4-R1-R2-R3-2-фенилтиометилсахаринов с сульфурилхлоридом в МДХ; и/или с помощью реакции 4-R1-R2-R3-сахаринов, которые получают с использованием параформальдегида и хлоротриметилсилана в присутствии каталитического количества хлорида олова в этилендихлориде.
Приготовление 87. Изотиазол-6-карбоксальдегид подвергали реакции с 3-(трифенилфосфоранилиден)пропаноатом лития в стандартных условиях Виттига, и получали 4-(5-изотиазолил)-3-бутеновую кислоту, которую затем восстанавливали и циклизовали с использованием хлорида алюминия, в результате чего получали 4-оксо-4,5,6,7- тетрагидро-1,2-бензизитиазол, 4-оксо-соединение подвергали реакции с метилентрифенилфосфораном в стандартных условиях Виттига, и в полученное 4-метиленовое соединение посредством реакции Симмонса-Смита вводили метилен, в результате чего получали 6,7-дигидроспиро[1,2-бензизотиазол-4(5Н),1'-циклопропан] после окисления которого перекисью водорода в уксусной кислоте получали 6,7-дигидроспиро[3-оксо-1,2-бензизотиазол-4(5H), 1'-циклопропан 1,1-диоксил] 4-спироциклопропил-4,5,6,7-тетрагидросахарин). Это соединение подвергали хлорометилированию в соответствии с процедурой, описанной в приготовлении 1, и получали 2-хлорометил-4-спироциклопропил-4,5,6,7-тетрагидросахарин.
Приготовление 88. 2-Бензил-4-изопропил-6-оксотетрагидросахарин приготовления 21 подвергали восстановлению с использованием борогидрида натрия и метилированию с использованию метилиодида в присутствии гидрида натрия, в результате чего получали 2-бензил-4-изопропил-6- метокситетрагидросахарин. Это соединение дебензилировали и хлорометилировали в соответствии с описанием в приготовлении 21, и получали 2-хлорометил-4-изопропил-6-метокси-4,5,6,7-тетрагидросахарин.
Приготовление 89. К свежедистиллированному циклопентадиену 25 мл при 0oC добавляли 4-бромо-2-(трет-бутил)изотиазол-3(2H)-он 1,1-диоксид (Helu. Chim. Acba. 72, 1416, 1989) (7,9 г, 0,03 M). Полученную смесь размешивали 16 ч при 0oC, а затем концентрировали в вакууме. Остаток очищали путем фильтрации через силикагель, элюируя гексанами (500 мл), а затем 20%-ым этилацетатом в гексане (500 мл) и снова 20%-ым этилацетатом в гексане (500 мл). Элюаты концентрировали в вакууме, и получали 9,8 г (100%) норбореновский аддукт, 3а-бромо-2-т-бутил-3а, 4,7,7а-тетрагидро4,7-метано-1,2-бензизотиазол-3(2H)-он 1,1-диоксид, в виде белого твердого вещества.
Этот аддукт (0,4 г, 1,2 мМ) в 25 мл этилацетата, содержащий 5% Pd на CaCO3 (0,2 г), размешивали в атмосфере водорода в течение 4 ч, и полученную реакционную смесь фильтровали через прокладку силикагеля, элюируя этилацетатом (100 мл). Элюаты концентрировали в вакууме, а остаток кристаллизовали из гексанов, в результате чего получали 0,4 г (100%) бромонорборнан, 3а-бромо-2-т-бутил-3а, 4,5,6,7,7а-гексагидро-4,7-метано-1,2-бензизотиазол-3(2H)-он 1,1-диоксид, в виде белого кристаллического вещества.
К раствору бромоборбонана (3,7 г, 0,011 М) в толуоле 825 мл) при 0oC добавляли диазабициклононен (1,37 г, 0,011 М) в толуоле (10 мл). После 20-ти минутного размешивания при 0oC, к реакционной смеси добавляли силикагель (25 г). Полученную суспензию загружали поверх 15-ти сантиметровой прокладки из силикагеля и элюировали 20% -ым этилацетатом в гексанах (800 мл). Элюаты концентрировали в вакууме, и получали 2,8 г (100%) дегидробромированного соединения, 2-т-бутил-4,5,6,7-тетрагидро- 4,7-метано-1,2-бензизотиазол-3(2H)-он-1,1-диоксида, в виде белого твердого вещества.
Это дегидробромированное соединение (2,8 г, 0,011 М) в трифторуксусной кислоте (30 мл) нагревали с обратным холодильником в течение 48 ч и оставляли при комнатной температуре на 4 дн. Полученную смесь концентрировали в вакууме, обрабатывали метанолом (20 мл) и выпаривали досуха. Остаток растворяли в простом эфире (100 мл) и промывали насыщенным NaHCO3 (1х50 мл). Слои отделяли, а водную фазу подкисляли до pH 1 с помощью 2 н. HCl экстрагировали МДХ (2х100 мл). Объединенные органические экстракты осушали и концентрировали в вакууме, в результате чего получали 0,9 г (42%) бицикло[2.2.1]сахариновое производное, 4,5,6,7-тетрагидро-4,7-метано-1,2-бензизотиазол-3(2H)-он 1,1-диоксид (4,5,6,7-тетрагидро-4,7-метано-сахарин), в виде белого твердого вещества.
Смесь производного бицикло(2.2.1)сахарина (0,9 г, 5 мМ), хлорометилфенилсульфида (0,07 г, 7 мМ) и бромид тетрабутиламмония (0,36 г, 0,16 мМ) в толуоле (50 мл) нагревали с обратным холодильником в атмосфере азота в течение 16 ч, охлаждали до комнатной температуры, и выпаривали досуха в вакууме. Остаток очищали с помощью флеш-хроматографии на силикагеле (100 г) с использованием 100% МДХ в качестве элюента, и получали 1,05 г (72%) соответствующего 2-фенилтиометилового производного в виде вязкого маслянистого продукта.
Этот продукт (1,05 г, 3 мМ) в дихлорметане (100 мл) обрабатывали сульфурилхлоридом (0,66 г, 5 мМ) и размешивали в течение 2 ч. Полученный желтый раствор разбавляли МДХ (100 мл), промывали насыщенным раствором NaHCO3 и концентрировали в вакууме. Остаток очищали с помощью флеш-хроматографии на силикагеле (33% ) МДХ в гексане, и получали в результате 0,66 г (81%) 2-хлорометил-4,5,6,7-тетрагидро-4,7-метано-1,2-бензизотиазол-3(2H)-он 1,1-диоксид (2-хлорометил-4,5,6,7-тетрагидро-4,7-метаносахарин).
Приготовления 90 и 91. В соответствии с процедурами, описанными в приготовлении 89, очевидно, что циклогексадиен или 1,1-диметил-циклопентадиен может быть подвергнут реакции с 4-бромо-2-(т-бутил)изотиазол-3(2H)-он 1,1-диоксидом с получением, соответственно, 3а-бромо-2-т-бутил-3а,4,7,7а-тетрагидро-4,7-этано или 4,7-диметилметано-1,2-бензизотиазол-3(2H)-он 1,1-диоксида, который может быть гидрогенизован с получением 3а-бромо-2-т-бутил-3а, 4,5,6,7,7а-гексагидро-4,7-этано (или 4,7-диметилметано)-1,2-бензизотиазол-3(2H)-он 1,1-диоксида, который, в свою очередь, может быть дегидробромирован с получением 2-т-бутил-4,5,6,7-тетрагидро-4,7-этано (или 4,7-диметилметано)-1,2-бензизотиазол-3(2H)-он 1,1-диоксида, который может быть затем деалкирован с получением 4,5,6,7-тетрагидро-4,7- этано (или 4,7-диметилметано)-1,2-бензизотиазол-3(2H)-он 1,1-диоксида, который, в свою очередь, может быть подвергнут реакции с хлорометилфенилсульфидом с получением 2-фенилтиометил-4,5,6,7-тетрагидро-4,7-этано (или 4,7-диметилметано)-1,2-бензоизотиазол-3(2H)он 1,1-диоксида, который может быть затем подвергнут реакции с сульфурилхлоридом с получением 2-хлорометил-4,5,6,7-тетрагидро-4,7-этано (или 4,7-диметилметано)-1,2-бензоизотиазол-3(2H)-она, т. е. 2-хлорометил-4,5,6,7-тетрагидро-4,7-этаносахарина (приготовление 90) или 2-хлорометил-4,5,6,7-тетрагидро-4,7-диметилметаносахарина (получение 91).
Приготовления 92E-94E. Общий способ получения метил 2-алкилциклогексан-6-онкарбоксилата.
К суспензии безводного Cul (10 мМ) в безводном ТГФ (100 мл) добавляли Me2S (10 мМ), и полученный раствор охлаждали до -78oC. Затем, в течение 15 мин, добавляли реагент Гриньяра (алкилбромид магния) (20 мМ). После 1-часового размешивания при -78oC, добавляли раствор циклогексенона (10 мМ) в ТГФ, и продолжали размешивать еще в течение 15 мин. К полученной смеси добавляли ГМФА (5 мл), а через 15 мин добавляли метилциклоформат (30 мМ) в ТГФ (20 мл), после чего реакционную смесь нагревали до комнатной температуры и размешивали в течение ночи. Затем, реакционную смесь гасили с помощью 2 н. HCl (50 мл). Слои отделяли, а водную фазу экстрагировали Et2O (1х100 мл). Объединенные органические экстракты промывали насыщенным раствором NH4Cl (3х50 мл), водой (2х50 мл), солевым раствором (1х50 мл), и осушали сульфатом натрия (Na2SO4). После удаления растворителя в вакууме и очистки либо с помощью дистилляции в Kugelrohr, либо с помощью флеш-хроматографии, получали целевой метил 2-алкилциклогексан-6-онкарбоксилат (табл. C).
Общий способ получения метил 2-бензилтио-6- алкилциклогекс-2-енкарборксилата и метил 2-бензилтио-6-алкилциклогекс-1-енкарбоксилата.
Смесь метил 2-алкилциклогексан-6-он-карбоксилата (1 экв.), бензилмеркаптана (1,1 экв. ) и кислой монтмориллонитовой глины KSF (масса которой в 1,5 раза превышает массу метил 2-алкилциклогексан-6-онкарбоксилата) в безводном толуоле (50-100 мл) нагревали с обратным холодильником в атмосфере азота с азеотропным удалением воды в течение 12-14 ч, а затем охлаждали до комнатной температуры. Полученное твердое вещество отфильтровывали и промывали простым эфиром. Объединенный фильтрат промывали 10%-ым Na2CO3, водой, солевым раствором, и осушали. После удаления растворителя в вакууме и очистки остатка с помощью флеш-хроматографии на силикагеле (10% эфир в гексане), получали смесь метил 2-бензилтио-6-алкилциклогекс-2-енкарбоксилата и метил 2-бензилтио-6-алкилциклогекс-1-енкарбоксилата (табл. D), которые затем использовали в следующей стадии в виде смеси.
Общий способ получения 4-алкилтетрагидросахаринов.
Раствор метил 2-бензилтио-6-алкилциклогекс-2-енкарбоксилата и метил 2-бензилтио-6-алкилциклогекс-1-енкарбоксилата (1-10 мМ смеси) в 10 мл МДХ разводили 20-50 мл ледяной уксусной кислоты и 1-5 мл воды, полученную смесь охлаждали до -10oC, и через эту смесь барботировали газообразный хлор до тех пор, пока экзотермическая реакция не начнет ослабевать. Затем смесь размешивали в течение 10 мин и осушали досуха, в результате чего получали смесь метил 2-хлоросульфонил-6-алкилциклогекс-2-енкарбоксилата и метил 2-хлоросульфонил-6-алкилциклогекс-1-енкарбоксилата, которую растворяли в 10 мл ТГФ и добавляли к 25 мл раствора концентрированного гидроксида аммония, охлаждая, при этом, в бане со льдом и ацетоном. После 2-х часового размешивания, реакционную смесь концентрировали в вакууме, а остаток растворяли в воде, подкисляли до pH 1 с помощью 2 н. HCl, и экстрагировали МДХ. Органическую фазу осушали и концентрировали в вакууме, в результате чего получали смесь метил 2-аминосульфонил-6-алкил-циклогекс-2-енкарбоксилата и метил 2-хлоросульфонил-6-алкилциклогекс-1-енкарбоксилата, которую растворяли в 10 мл ТГФ и добавляли к 25 мл раствора концентрированного гидроксида аммония, охлаждая, при этом, в бане со льдом и ацетоном. После 2-х часового размешивания, реакционную смесь концентрировали в вакууме, а остаток растворяли в воде, подкисляли до pH 1 с помощью 2 н. HCl, и экстрагировали МДХ. Органическую фазу осушали и концентрировали в вакууме, в результате чего получали смесь метил 2-аминосульфонил-6-алкилциклогекс-2-енкарбоксилата и метил 2-аминосульфонил-6-алкилциклогекс-1-екнкарбоксилата. Полученную смесь растворяли в метаноле и добавляли к свежеприготовленному раствору метоксида натрия (10-50 мМ), после чего размешивали при комнатной температуре в течение 12 ч. Реакционную смесь концентрировали в вакууме, разбавляли водой и экстрагировали простым эфиром. Органическую фазу отбрасывали, а водную фазу подкисляли до pH 1 с помощью концентрированной HCl и экстрагировали МДХ. Органические экстракты промывали солевым раствором, осушали им выпаривали досуха, в результате чего получали 4-алкил-4,5,6,7-терагидро-1,2-бензизотиазол-3(2H)-он-1,1-диоксид (4-алкил-4,5,6,7-тетрагидросахарин) (табл. E).
Смесь 4-алкил-4,5,6,7-тетрагидро-1,2-бензизотиазол-3(2H)-он 1,1-диоксида (4-алкил-4,5,6,7-тетрагидросахарина) (1,0 экв. ), хлорометилфенилсульфида (1,5 экв.), и бромида тетрабутиламмония (0,2 экв.) в толуоле (25 мл/г сахарина) нагревали с обратным холодильником в атмосфере азота в течение 16-24 ч, а затем охлаждали до комнатной температуры. Полученную смесь выпаривали досуха, а остаток хроматографировали на силикагеле, элюируя смесью гексана и МДХ (1:1 1:3), в результате чего получали соответствующий 2-фенилтиометил-4-алкил-4,5,6,7-тетрагидро-1,2-бензизотиазол-3(2H)-он 1,1-диоксид (2-фенилтиометил-4-алкил-4,5,6,7-тетрагидросахарин) (табл. F).
Раствор 2-фенилтиометил-4-алкил-4,5,6,7-тетрагидросахарина (1,0 экв.) обрабатывали сульфурилхлоридом (1,5 экв.), и размешивали в течение 2 ч. Полученный желтый раствор осушали досуха, и получали 2-хлорометил-4-алкил-4,5,6,7-тетрагидросахарин. В полученных соединениях алкилом является метилом (92E), этилом (93E) и изопропилом (94E).
Приготовление 95. В соответствии с общими процедурами, описанными в приготовлениях 92В-94В 92Е-94Е, и используя в качестве исходного материала метилциклогексан-6-онкарбоксилат, последовательно получали: смесь метил 2-бензилтиоциклогекс-1 (и 2)-ен-карбоксилата (выход 40%); смесь метил 2-хлоросульфонил-циклогекс-1-(и 2)-енкарбоксилата; смесь метил 2-аминосульфонилциклогекс-1-(и 2)-енкарбоксилата, 4,5,6,7-тетрагидро-1,2-бензизотиазол-3(2H)-он 1,1-диоксида 4,5,6,7-тетрагидросахарин (выход 50% ), 2-фенилтиометил-4,5,6,7-тетрагидро-1,2-бензизотиазол-3(2H)-он 1,1-диоксида-2-фенилтиометил-4,5,6,7-тетрагидросахарин (выход 40%) и 2-хлорометил-4,5,6,7-тетрагидро-1,2-бензизотиазол-3(2H)-он 1,1-диоксида (2-хлорометил-4,5,6,7-тетрагидросахарин).
Приготовление 96. Метил 2,2-диметилциклогексан-6-он-карбоксилат.
К суспензии безводного Cul (70,0 г, 0,37 М) в безводном эфире (500 мл) при 0oC добавляли метиллития в свободной форме (520 мл или 1,4-М раствор в эфире, 0,73 М). После 15-ти минутного размешивания при 0oC, добавляли раствор 3-метил-2-циклогексенона (20,0 г, 0,18 М) в эфире (50 мл), и продолжали размешивать еще 1 ч. К полученной смеси добавляли ТГФ (50 мл) и ГМФА (25 мл), а через 15 мин добавляли метил-цианоформат (45,0 г, 0,53 М) в ТГФ (20 мл), после чего реакционную смесь нагревали до комнатной температуры и размешивали в течение 3 ч. Реакционную смесь гасили с помощью 2 н. HCl (50 мл). Слои разделяли и водную фазу экстрагировали Et2O (1х500 мл). Объединенные экстракты промывали насыщенным раствором NH4Cl (3х50 мл), водой (2х50 мл), солевым раствором (1х50 мл) и осушали сульфатом натрия. После удаления растворителя в вакууме и очистки путем дистилляции в Kugelrohr, получали 34,0 г (99%) метил-2,2-ди-метилциклогексан-6-он карбоксилата, т.кип. 80-84oC/0,6 мм который затем превращали в 2-хлорометил-4,4-диметил-4,5,6,7-тетрагидросахарин в соответствии с общими способами, описанными в приготовлениях 92В-94В 92Е-94Е.
Получение конечных продуктов.
Пример 1. Раствор 2-бромометилсахарина (2,0 г, 7,2 мМ), дибутилфосфата (2,29 г, 10,9 мМ) и N,N-диизопропиленэтиламина (1,41 г, 10,9 мМ) в 40 мл метилендихлорида размешивали при комнатной температуре в течение 48 ч. Реакционную смесь концентрировали, а остаток подвергали флеш-хроматографии на силикагеле, элюируя 30%-ым этилацетатом в гексанах, в результате чего получили 2,35 г (80%) дибутил 2-сахаринилметилфосфата в виде бесцветного маслянистого продукта.
Пример 2. Раствор 2-хлорометил-4-этоксисахарина (2,0 г, 7,3 мМ), диэтилфосфата (1,68 г, 10,9 мМ), и триэтиламина (1,53 мл, 10,9 мМ) в 25 мл метилендихлорида нагревали с обратным холодильником в течение 58 ч. После охлаждения, реакционную смесь концентрировали, а остаток подвергали флеш-хроматографии на силикагеле, элюируя смесь этилацетата и гексана, в результате чего получали 2,0 г (73%) диэтил 4-этокси-2- сахаринилметилфосфата в виде бесцветного маслянистого продукта.
Пример 3. К раствору дибензилфосфата (0,69 г, 2,48 мМ) в 30 мл метанола при комнатной температуре добавляли карбонат цезия (0,403 г, 1,24 мМ). После 2-х часового размешивания, растворитель выпаривали, а остаток осушали в высоком вакууме и суспендировали в 10 мл N,N-диметилформамида. К суспензии добавляли 2-хлорометил-4-изопропил-6-метоксисахарин (0,5 г, 1,6 мМ), и полученную смесь размешивали 24 ч в масляной бане при 50oC. После охлаждения, смесь разводили ледяной водой и экстрагировали 200 миллилитрами смеси простого эфира и этилацетата (4:1). Органический слой отделяли и последовательно промывали водой, а затем насыщенным солевым раствором. Экстракт осушали сульфатом магния, фильтровали, концентрировали, и остаток подвергали флеш-хроматографии на силикагеле, элюируя смесью этилацетата и гексана, в результате чего получали 0,46 г (52%) дибензил 4-изопропил-2-сахаринилметилфосфата в виде маслянистого вещества, который кристаллизовался после выдерживания, т.пл. 75,5-76,5oC.
В соответствии с процедурами, описанными выше в примерах 1 3 (обозначаемыми далее методами 1 3), получали соединения формулы I, представленные ниже в табл. 1. В каждом из примеров 4 9, для получения продуктов использовали 2 бромометилсахарин. В примерах 10 21, для получения продуктов использовали соответствующий 2-хлорометилсахарин.
В примерах 1 17 и 20, для получения соединений, в качестве исходных материалов использовали фосфат и фосфиновую кислоту, которые представляли собой коммерческие материалы. Диизопропилфосфат, используемый в примерах 18, 19 и 21, получали следующим образом:
Смесь диизопропилхлорофосфата (10 г, 50 мМ) (см. R.A. Melvor и др. Can. J. Chem. 34, 1819 (1956)) в 10 мл дистиллированной воды размешивали в масляной бане при 80oC в течение 2 ч. Полученную смесь концентрировали в вакууме, а оставшуюся воду удаляли путем азеотропной дистилляции с бензолом (3х100 мл). После осушки в высоком вакууме получали 8,8 г (97%) диизопропилфосфата в виде маслянистого вещества, которое затем использовали без очистки.
Пример 22. Диизопропил 6-(2,2-диметил-1,3-диоксан-4-ил)метокси-4-изопропил-2-сахаринилметилфосфат.
Диэтилазодикарбоксилат (0,96 г, 5,55 мМ) добавляли к смеси диизопропил 6-гидрокси-4-изопропил-2-сахаринилметилфосфата (2,37 г, 5,44 мМ), трифенилфосфина (1,44 г, 5,5 мМ) и глицериндиметилкеталя (2,2-диметил-1,3-диоксолан-4-метанол) (0,79 г, 5,98 мМ) в 40 мл ТГФ, и полученную смесь размешивали 15 ч при комнатной температуре. Избыточное количество растворителя удаляли при пониженном давлении, а остаток подвергали флеш-хроматографии (SiO2; этилацетат-МДХ), в результате чего получали первую фракцию с выходом 0,49 г (19,7% ) целевого соединения в виде густого маслянистого продукта, и вторую фракцию с выходом 2,0 г смеси, содержащей около 85% целевого соединения.
Пример 23. 2-Сахаринилметилфосфат.
Дибензил 2-сахаринилметилфосфат (1,1 г), растворенный в 50 мл метанола, подвергали гидрогенизации в присутствии 10% палладированного угля (0,3 г) в течение приблизительно 6 ч при нормальном плавлении. Раствор концентрировали при пониженном давлении, и получали 2-сахаринилметилфосфат в виде густого маслянистого остатка, который затем растворяли в метаноле и обрабатывали циклогексаламином (0,59 мл). После выдерживания при комнатной температуре выделялись кристаллы соли, которые собирали путем фильтрации, промывали смесью метанола и эфира и осушали, в результате чего получали 0,503 г (43,9% ) дициклогексиламиновой соли целевого соединения, т. пл. (усадка при 214-215oC).
Пример 24. a) Метил 2,2-диметилциклогексан-6-онкарбоксилат.
К суспензии безводного Cul (50,0 г, 0,37 М) в безводном эфире (500 мл) при 0oC добавляли свободный метиллитий (520 мл 1,4 М-раствор в эфире, 0,73 М). После 15-ти минутного размешивания при 0oC, добавляли раствор 3-метил-2-циклогексен-1-она (20,0 г, 0,18 М) в эфире (50 мл), и продолжали размешивать в течение 1 ч. К полученной смеси добавляли ТГФ (50 мл) и ГМФА (25 мл), а через 15 мин добавляли метилцианоформат (45,0 г, 0,53 М) и ТГФ (20 ил), после чего смесь нагревали до комнатной температуры и размешивали 3 ч. Затем реакцию гасили с помощью 2 н. HCl (50 мл). Слои отделяли, а водную фазу экстрагировали Et2O (1х500 мл). Объединенные органические экстракты промывали насыщенным распором NH4Cl (3х50 мл), H2O (2х50 мл), солевым раствором (1х50 мл), и осушали сульфатом натрия. После удаления растворителя в вакууме и очистки путем дистилляции (Kugelrohr), получали 34,0 г (99%) метил 2,2-диметилциклогексан-6-онкарбоксилата; т. кип. 0,6 80-84oC.
b) Метил-2-бензилтио-6,6-диметилциклогекс-2-не-карбоксилат и метил-2-бензилтио-6,6-диметилциклогекс-1-ен-карбоксилат.
Кислую смолу Амберлист-15 (Amberlyst-15) (25,0 г) смешивали с полифосфорной кислотой (3,0 г) и фосфорной кислотой (3,0 г) и нагревали в вакууме при 60oC в течение 2 ч. Полученную смолу смешивали с метил 2,2-диметилциклогексан-6-он-карбоксилатом (34,0 г, 0,18 М), бензилмеркаптаном (50,0 г, 0,40 М) и измельченным ситом 4 А (20,0 г) в безводном дихлорэтане (700 мл) нагревали с обратным холодильником в атмосфере азота в течение 18 ч, и охлаждали до комнатной температуры. Твердое вещество отфильтровывали, фильтрат концентрировали в вакууме, а избыток бензилмеркаптана отгоняли. Остаток очищали с помощью хроматографии (МДХ:гексаны 2:1), и получали 11,3 г (21%) смеси метил 2-бензилтио-6,6-диметилциклогексен-2-ен-карбоксилата и метил 2-бензилтио-6,6-диметилциклогекс-1-ен-карбоксилата.
c) Полученную смесь (11,3 г, 0,04 М) растворяли в МДХ (25 мл), разводили ледяной уксусной кислотой (65 мл), а затем водой (10 мл) охлаждали до -10oC, и через смесь барботировали газообразный хлор до тех пор, пока экзотермическая реакция не прекратится. После этого смесь размешивали 10 мин и осушали досуха, в результате чего получали смесь метил 2-хлоросульфонил-6,6-диметилциклогексен-2-енкарбоксилата и метил 2-хлоросульфонил-6,6-диметилциклогекс-1-енкарбоксилата, которую затем растворяли в 10 мл ТГФ и добавляли к 25 мл раствора конц. гидрокиси аммония, охлаждая при этом в бане из льда и ацетона. После 2-х часового размешивания, реакционную смесь концентрировали в вакууме, остаток растворяли в воде, подкисляли до pH 1 с использованием 2 н. HCl, и экстрагировали МДХ. Органическую фазу осушали и концентрировали в вакууме, в результате чего получали смесь метил 2-аминосульфонил-6,6-диметилциклогекс-2-енкарбоксилата и метил 2-аминосульфонил-6,6-диметилциклогекс-1-енкаробоксилата. Полученную смесь растворяли в метаноле (25 мл) и добавляли к свежеприготовленному раствору метоксида натрия (0,20 М), после чего размешивали 12 ч при комнатной температуре. Реакционную смесь концентрировали в вакууме, разбавляли водой и экстрагировали простым эфиром. Органическую фазу отбрасывали, а водную фазу подкисляли до pH 1 концентрированной HCl и экстрагировали МДХ. Органических экстракты промывали солевым раствором, осушали и выпаривали досуха, в результате чего получали 3,5 г (42%) 4,4-диметил-4,5,6,7-тетрагидро-1,2-бензизотиазол-3(2H)-он 1,1-диоксида (4,4-диметил-4,5,6,7-тетрагидросахарина).
d) Смесь 4,4-диметил-4,5,6,7-тетрагидро-1,2-бензизотиазол-3(2H)-он 1,1-диоксида (1,0 г, 4,7 мМ), хлорометилфенилсульфида (1,1 г, 7,0 мМ) и тетрабутилбромида аммония (0,3 г, 0,93 мМ) в толуоле (25 мл) нагревали с обратным холодильником в атмосфере азота в течение 16-24 ч, а затем охлаждали до комнатной температуры. Полученную смесь выпаривали досуха, а остаток хроматографировали на силикагеле, элюируя смесью гексана и МДХ (1:1 1:3), в результате чего получали 1,0 г (67%) 2-фенилтиометил-4,4-диметил-4,5,6,7-тетрагидро-1,2-бензизотиазол-3(2H)-он 1,1-диоксида (2-фенилтиометил-4,4-диметил-4,5,6,7-тетрагидросахарин).
e) Раствор 2-фенилтиометил-4,4-лиметил- 4,5,6,7-тетрагидро-1,2-бензизотиазол-3(2H)-она, 1,1-диоксида (0,8 г, 2,4 мМ) обрабатывали сульфурилхлоридом (0,48 г, 3,5 мМ), и размешивали 2 ч. Полученный желтый раствор осушали досуха, разводили простым эфиром (100 мл) и промывали насыщенным раствором NaHCO3 и солевым раствором. Органическую фазу осушали и концентрировали в вакууме, в результате чего получали 0,6 г (95%) 2-хлорометил-4,4-диметил-4,5,6,7-тетрагидро-4,4-бензизотиазол-3(2H)-она, 1,1-диоксида (2-хлорометил-4,4-диметил-4,5,6,7-тетрагидросахарин), который обрабатывали диэтиофосфатом (1,05 г, 6,8 мМ) и триэтиламином (0,7 г, 6,9 мМ) в дихлороэтане (15 мл) при 50oC в течение 16 ч, а затем охлаждали до комнатной температуры. Полученную смесь осушали досуха и очищали с помощью флеш-хроматографии на силикагеле (40% гексан в этилацетате), в результате чего получали 0,18 г (21%) диэтил 4,4-диметил-4,5,6,7-тетрагидро-3-оксобензизотиазолин-2-илметил 1,1-ди-оксидфосфата (диэтил 4,4-диметил-4,5,6,7-тетрагидро-2-сахаринилметилфосфат) с виде бесцветного маслянистого продукта.
Пример 25. Диизопропил 6-этокси-4-изопропил-2-сахаринил-метофосфат (пример 19) может быть также получен способом, описанным в примере 22, то есть, с помощью реакции диизопропил 6-гидрокси-4- изопропил-2-сахаринилметилфосфата с трифенилфосфином, этанолом и диэтилазодикарбоксилатом.
Если повторить процедуру примера 22, но вместо глицериндиметилкеталя использовать соответствующий спирт, то можно получить диизопропил 4-R1-R2-R3-2-сахаринилметилфосфаты (табл. 2).
2,3-диметокси-1-пропанол, который использовали в примере 29, синтезировали следующим образом.
Раствор 10,0 г (0,055 М) DL--α--O-бензил глицерина в небольшом количестве ТГФ добавляли к суспензии 15,38 г (0,137 М) трет-бутоксида калия в 300 мл ТГФ. Смесь размешивали в течение 1 ч при комнатной температуре, и добавляли 18,72 г (0,132 М) иодометана. В результате тотчас же выделялось более твердое вещество. Реакцию размешивали 10 ч при комнатной температуре, затем охлаждали, осторожно разбавляли раствором хлорида натрия и экстрагировали эфиром. Органический слой промывали водой, затем 5%-ым HCl, снова водой, и насыщенный NaCl, после чего осушали. Растворитель удаляли, а остаток очищали с помощью флеш-хроматографии, в результате чего получали 1-бензилокси-2,3-диметоксипропан 9,16 г 79% в виде маслянистого продукта.
Раствор 8,8 г (0,042 М) полученного продукта в 200 мл MeOH подвергали гидрогенизации с использованием 1,1 г 10% Pd/C при давлении 50 фунт/кв. дюйм (344,7 кПа). Затем катализатор удаляли путем фильтрации, а растворитель выпаривали при пониженном давлении, в результате чего получали 4,4 г (87%) 2,3-диметокси-1-пропанола.
Пример 33. Посредством реакции диизопропил 6-гидрокси-4-изопропил-2-сахаринметилфосфата (1 М) в МДХ с ангидридом трифторметансульфоновой кислоты (1,3 М) в присутствии триэтиламина (3 М) при 0oC может быть получен, после стандартной обработки и очистки, диизопропил 4-изопропил-6-трифторметансульфонилокси-2-сахаринметилфосфат.
Пример 34. Посредством реакции продукта примера 33 (1 М) в атмосфере азота и в п-диоксане с 1-метил-2-триметилстаннил-пирролом (1,6 М) в присутствии тетракис (трифенилфосфин) палладия(O) (0,02 М), хлорида лития (3,1 М) и 2,6-ди-трет-бутил-4-метилфенола (9,1 М) при нагревании с обратным холодильником и после стандартной обработки и очистки, может быть получен диизопропил 4-изопропил-6-[2-(1-метилпропирролил)]-2-сахаринметилфосфат.
Пример 35. Посредством реакции продукта примера 33 (1 М) в ТГФ с 40%-ым водным диметиламином (4,4 М) при комнатной температуре и после стандартной обработки и очистки, может быть получен диизопропил-6-диметилоамино-4-изопропил-2-сахаринметилфосфат.
Пример 36. Посредством реакции диизопропил 6-гидрокси-4-изопропил-2-сахаринметилфосфата в толуоле с ди-(фтор-бутоксиметил)метиламином при 80oC и после стандартной обработки и очистки может быть получен сложный эфир диизопропил 4-изопропил-8-метил-2,3,7,8-тетрагидро-9H-1,3-оксазино-[6,5-g]-3-оксибензизотиазол-2-илметил 1,1-диоксида фосфорной кислоты.
Пример 37. Посредством обработки диизопропил 6-(2,2-диметил-1,3-диоксилан-4-ил)метокси-4-изопропил-2-сахаринилметилфосфата (пример 22) (1 М) моногидрата п-толуолсульфоновой кислоты (0,8 М) в метаноле - хлороформе при комнатной температуре, после стандартной обработки и очистки, может быть получен диизопропил 6-(2,3-дигидроксипропокси)-4-изопропил-2-сазаринилметилфосфат.
Пример 38. Посредством реакции диизопропил 6-гидрокси-4-изопропил-2-сахаринилметилфосфата (1 М) в ацетоне с трет-бутилбромоацетатом (3,4 М) в присутствии безводного карбоната калия (1,96 М) при комнатной температуре, и после стандартной обработки и очистки, может быть получен диизопропил 6-(2-третбутокси-2-оксоэтокси)-4- изопропил-2-сахаринилметилфосфат.
Пример 39. В соответствии с процедурой примера 38, но используя бензил бромоацетат вместо трет-бутилбромацетата, может быть получен диизопропил 6-(2-бензилокси-2-оксоэтокси)-4-изопропил-2-сахаринилметилфосфат, который может быть затем превращен в диизопропил 6-карбоксиметокси-4-изопропил-2-сахаринилметилфосфат путем гидрогенизации в присутствии палладированного угля.
Пример 40. В соответствии с процедурой, описанной в примере 3, но используя 2-хлорметил-4-гидроксисахарин вместо 2-хлорометил-4-изопропил-6-метоксисахарин, может быть получен дибензил 4-гидрокси-2-сахаринилметилфосфат.
Пример 41. В соответствии с процедурой, описанной в примере 22, дибензил 4-гидрокси-2-сахаринилметилфосфат может быть подвергнут реакции с диэтилдиазокарбоксилатом, трифенилфосфином и бензиловым спиртом с получением дибензил 4-бензилокси-2-сахаринилметилфосфата.
В соответствии с процедурой, описанной в примере 3, каждый из 4-R1-R2-R3-2-хлорметилсахаринов приготовлений 1-86; 4-R4-4-R5-6-R6-хлорметил-4,5,6,7-тетрагидросахаринов приготовлений 87, 88, 92Е-94Е, 95 и 96; и 4,7-метано-, 4,7-диметилметано- и 4,7-этано-4,5,6,7-тетрагидросахаринов приготовлений 89-91, соответственно, может быть подвергнут реакции каждого из сложных ди-эфиров фосфорной кислоты, сложных моно-эфиров фосфорной кислоты и фосфиновых кислот формулы III, представленных в табл. 3 и на фиг. 5, с получением соответствующих соединений формул I, II и IIA, соответственно.
Сложный моно(2-метилпропил)-эфир метилсульфоновой кислоты и сложный монофениловый эфир метилфосфоновой кислоты в табл. 3 могут быть получены путем гидролиза из (2-метилпропил) эфира метилфосфонохлорангидрида и фенилового эфира метилфосфонохлорангидрида, соответственно.
Этилфенилфосфиновая кислота и изопропилфосфиновая кислота в табл. 3 могут быть получены путем традиционного O-алкильного расщепления из соответствующих метиловых эфиров, например, посредством реакции с триметилсилилбромидом [(CH3)3SiBr] и гидролиза образовавшегося таким образом сложного триметилового эфира.
Традиционный кислотный гидролиз соединения, полученного в результате каждой из реакций хлорметиловых соединений приготовлений 1-96, с соединением формулы III, где A и B, вместе взятые представляют собой:
где R9 и R10 вместе представляют собой изопропилиден (фиг. 5) дает соответствующие соединения, где R9 и R10, каждый является водородом.
Изменение константы ингибирования, Ki, ЭЛЧ (эластазы лейкоцитов человека)-ингибирующего комплекса было описано для констант истинно обратимого ингибирования, относящихся к конкурентным ингибиторам [Cha, Biochem. Pharmacol. 24, 2177-2185 (1975)] Однако, соединения настоящего изобретения не образуют истинно обратимых ингибирующих комплексов, и до некоторой степени поглощаются ферментом. Поэтому, в настоящем эксперименте, вместо 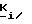 определения
определения 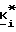 , которое вычисляли как отношение
, которое вычисляли как отношение 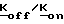 , то есть, отношение скорости реактивации фермента в скорости инактивации фермента. Сначала измеряли величины
, то есть, отношение скорости реактивации фермента в скорости инактивации фермента. Сначала измеряли величины 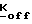 и
и 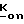 , а затем вычисляли
, а затем вычисляли 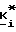 .
.
Скорость инактивации,  ферментной активности для испытуемых соединений определяли путем измерения ферментной активности соответствующего фермента как функции времени после добавления испытуемого соединения. Путем построения кривой зависимости логарифма ферментной активности от времени, получали наблюдаемую скорость инактивации,
ферментной активности для испытуемых соединений определяли путем измерения ферментной активности соответствующего фермента как функции времени после добавления испытуемого соединения. Путем построения кривой зависимости логарифма ферментной активности от времени, получали наблюдаемую скорость инактивации, 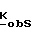 , которая может быть представлена в виде
, которая может быть представлена в виде 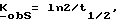 , где t 1/2 представляет собой время, необходимое для того, чтобы ферментная активность снизилась на 50% Таким образом, скорость инактивации равна:
, где t 1/2 представляет собой время, необходимое для того, чтобы ферментная активность снизилась на 50% Таким образом, скорость инактивации равна: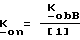
где [1] представляет собой концентрацию ингибирующего соединения.
Аналогичным образом определяли константу реактивации, 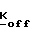 , после чего вычисляли константу ингибирования
, после чего вычисляли константу ингибирования 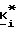 как:
как: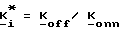
Величины 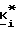 , определенные для соединений примеров 1-22, составляли в пределах от 0,035 до 100 нМ.
, определенные для соединений примеров 1-22, составляли в пределах от 0,035 до 100 нМ.
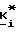 соединения примера 13 составляло 0,035.
соединения примера 13 составляло 0,035.
Новые 4-R1-R2-R3-2- сахаринилметил-, 4-R4-4-R5-6-R6- 4,5,6,7-тетрагидро-2-сахаринилметил- и 4,7-C-4,5,6,7-тетрагидро-2-сахаринилметилфосфаты, -фосфонаты, и -фосфинаты формул I, и II и IIA, соответственно, предназначенные для лечения дегенеративных значений; их содержащие композиции; и способы их использования для лечения дегенеративных заболеваний; а также способы получения указанных соединений посредством реакции соответствующих 2-галогенметилсахаринов с фосфатом, фосфонатом или фосфиновой кислотой формулы III в присутствии акцептора кислоты. 2 с.п. ф-лы, 5 ил., 8 табл.
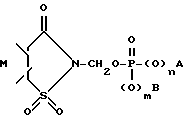
где М представляет собой группу формулы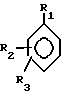
или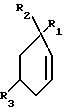
R1 водород, галоген, низший алкил, низший алкоксил, гидрокси, низший алкенил, низший алкинил или аминогруппа;
R2 водород, гидрокси, галоген, низший алкил, низший алкоксил;
R3 водород, галоген, низший алкил, циклоалкил, низший алкокси или карбоксигруппа или 1,3-диоксанзамещенный алкоксил;
m и n 0 или 1;
A и B гидрокси,
низший алкил, бензил или фенил, или их кислые аддитивные или основные аддитивные соли.
| Пожарный двухцилиндровый насос | 0 |
|
SU90A1 |
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| Способ приготовления консистентных мазей | 1919 |
|
SU1990A1 |
