Изобретение касается ранее неизвестных 2-сахаринилметиларилкарбоксилатов, которые ингибируют ферментативную активность протеолитических ферментов, составов, содержащих таковые, способа использования таковых в терапии дегенеративных заболеваний и способов получения таковых.
Ингибирование действия протеолитических ферментов нетоксическими препаратами является полезным в терапии дегенеративных расстройств, таких как эмфизема, ревматоидный артрит и панкреатит, в случае которых существенным элементом является протеолиз.
Ингибиторы протеазы широко используют при проведении биомедицинских исследований. Сериновые протеазы являются наиболее широко распространенным классом протеолитических ферментов. Некоторые сериновые протеазы характеризуют, основываясь на их субстатной специфичности, как химотрипсиноподобные или эластазаподобные вещества.
Химотрипсин и химотрипсиноподобные ферменты обычно разрывают пептидные связи у белков там, где аминокислотным остатком на карбоксильной стороне связи в типичном случае являются Трип (триптофан), Тир (тирозин), Фен (фенилаланин), Мет (метионин), Лей (лейцин) или какой-то другой аминокислотный остаток, который содержит ароматические или большие алкильные боковые цепи.
Эластаза и эластазаподобные ферменты обычно разрывают пептидные связи там, где аминокислотным остатком на карбоксильной стороне связи в типичном случае являются Ала (аланин), Вал (валин), Сер (серин), Лей (лейцин) или другие аналогичные меньшие аминокислоты.
И химотрипсиноподобные, и эластазаподобные ферменты обнаруживают в лейкоцитах, в тучных клетках и в панкреатическом соке у высших организмов, и они вырабатываются многими видами бактерий, дрожжами и паразитами.
В патенте Японии N 7200419 раскрывается ряд 2-сахаринилметилбензоатов, включая 2-сахаринилметилбензоат как таковой и 2-сахаринилметил-2,4-дихлорбензоат и 4-нитробензоат. Утверждается, что эти соединения "обладают высокой активностью в отношении парикуляриоза риса, гнили рисовой оболочки, рисовой гельминтоспоровой листовой пятнистости и рисового бактериального листового гнилостного заболевания".
Санкел (Sunkel) и др. в журнале J. Med. Chem. 31, 1886-1890 (1988) установили ряд 2-сахарининил(низший)алкил-1,4-дигидропиридин-3-карбоксилатов, обладающих тромбоцитной агрегационной задерживающей активностью и антитромботической активностью.
Чен (Chen) в патенте США N 4263393 раскрыл различные 2-ароилметилсахарины, пригодные для использования в качестве "фотографических элементов и пленочных устройств".
Малвей (Mulvey) и др. в патенте США N 4195023 раскрыли R1-2-R2CO-1,2-бензизотиазол-3-оновые соединения, где R1 представляет собой галоген, алкоксигруппу, алкиламиногруппу, диалкиламиногруппу, алкоксикарбонильную группу, аминогруппу, нитрогруппу или водород в бензольном кольце и R2 представляет собой водород, алкильную группу, алкенильную группу, алкинильную группу, циклоалкильную группу, галофенильную группу, гетероарильную группу или замещенную гетероарильную группу, и R1-2-A-CO-сахарины, где группа R1 имеет тот же смысл, что и в случае заместителей бензольного кольца у 1,2-бензизотиазол-3-оновых соединений, и A представляет собой алкильную, алкенильную, алкинильную, циклоалкильную, фторфенильную, гетероарильную или замещенную гетероарильную группу. Утверждается, что эти соединения обладают эластазной задерживающей активностью и могут оказаться полезными в терапии эмфиземы.
Циммерман (Zimmerman) и др. в журнале J. Biol. Chem., 225 (20), 9848-9851 (1980) раскрыли N-ацилсахарины, где ацильная группа представляет собой фуроильную, теноильную, бензоильную, циклопропаноильную, этилбутирильную и акрилоильную группу, которые обладают сериновой протеазной задерживающей активностью.
В патентной публикации Японии N 73/35457 раскрывается 4-метилфенил-2-сахаринилкарбоксилат, который, как утверждают, обладает бактерицидной и фунгицидной активностями.
Известно, что несколько классов соединений являются сериновыми протеазными ингибиторами. Например, Пауэра (Powers) в патенте США N 4659855 раскрывает арилсульфонильные фтористые производные, пригодные для использования в качестве ингибиторов эластазы. Доуэрти (Doherty) и др. в патентах США N 4547371 и 4623645 раскрывают цефалоспориновые сульфоны и сульфоксиды соответственно, которые, как утверждается, являются сильнодействующими эластазными ингибиторами, пригодными для использования в терапии воспалительных состояний, особенно артрита и эмфиземы.
Тешима (Teshima) и др. в журнале J. Biol. Chem., 257 (9), 5085-5091 (1982) сообщают результаты исследований, проведенных на сериновых протеазах (человеческая относящаяся к лейкоцитам эластаза, свиная панкреатическая эластаза, катепсин-C и бычий химотрипсин Aα с использованием сложных 4-нитрофенилэфиров и тиоэфиров N-трифторацетилантранилатов, 2-замещенных-4H-3,1-бензоксазин-4-онов, 2-замещенных-4-хиназолинов и 2-замещенных-4-хлорхиназолинов.
Ча (Cha) в журнале Biochem. Pharmacol. 24, 2177-2185 (1975) обсуждает кинетические подходы к исследованию процесса связывания ингибиторов с макромолекулами, такими как ферменты, и методы определения таких параметров, как константы ингибирования, скорости реакций и концентрации у связанного и несвязанного фермента.
Джоунз (Jones) и др. в патенте США N 4276298 раскрывают 2-R-1,2-бензизотиазолинон-1,1-диоксиды, где группа R-фенильная группа, замещенная фтором, динитрогруппой, трифторметильной группой, цианогруппой, алкилкарбонильной группой, карбоксильной группой, карбамоильной группой, алкилациламиногруппой, алкилсульфонильной группой, N,N-диалкилсульфамоильной группой, трифторметоксигруппой, трифторметилтиогруппой, трифторметилсульфонильной группой и трифторметилсульфинильной группой, или пиридильная группа, замещенная такими же группами, что и в случае группы R, когда она представляет собой фенильную группу, исключая лишь то, что пиридильная группа может быть также мононитрозамещенной. Эти соединения, как утверждают, обладают протеазной ферментной ингибирующей активностью, особенно эластазной ингибирующей активностью, и могут оказаться полезными в терапии энфиземы, ревматоидного артрита и других воспалительных заболеваний.
Пауэрз (Powers) в журнале Biochem., 24, 2048-2058, (1985) описывает исследования по изучению ингибирующей способности у четырех химотрипсиноподобных ферментов - катепсина-G, крысиных тучных клеточных протеаз 1 и 22, человеческой кожной химазы и химотрипсина Aα - при воздействии N-фуроилсахарина и N-(2,4-дицианофенил)сахарина.
Свобода (Svoboda) и др. в журнале Coll. Czech. Chem., Commun, 51, 1133-1139 (1986) раскрывают способ получения 4-гидрокси-2H-1,2-бензотиазин-3-карбоксилатов посредством межмолекулярной конденсации Дикмана у сложных 2H-1,2-бензизотиазол-3-он-2-ацетат-1,1-диоксидэфиров.
Рецек (Reczek) и др. в патентах США N 43507562 и 4363865 и Ванметер (Vanmeter) и др. в патенте США N 4410618 касаются фотографических реактивов (Рецек, патент N 4350752 и Ванметер и др.) и фотографических красителей (Рецек, патент N 4363865) и раскрывают различные 2-замещенные сахарины, пригодные для таких применений, например, в качестве "фотографических реактивов", присоединяемых через гетероатом к "имидометильной блокирующей" группе (Рецек, патент N 4350752), в качестве "способных диффундировать на носителе фотографических красителей", присоединяемых к атому азота имида через 1,1-алкиленовую группу (Рецек, патент N 4363865), и в виде N-ацилметилимидов, которые описываются как "блокированные фотографические реактивы", у которых имеется "остаток от органического фотографического реактива, содержащий гетероатом, через который он присоединяется к блокирующей группе" (Ванметер).
Фрид (Freed) в патенте США N 3314960 раскрывает 2-(1,1,3-триоксо-1,2-бензизотиазол-2-ил)глутаримиды, которые, как утверждается, могут оказаться полезными в качестве седативных средств.
Соединение 2-хлорметилсахарин описывается во Французском патенте N 1451417 в качестве промежуточного продукта при получении N-метилсахарин-d, 1-трансхризантемата, полезного в качестве инсектицида; и Ло (Lo) в патенте США N 3002884 раскрывает 2-хлор, 2-бром- и 2-иодметилсахарины, пригодные для использования в качестве фунгицидных средств.
Настоящее изобретение касается 4-R4-R5-2-сахаринилметиларилкарбоксилатов и 4,5,6,7-тетрагидро-2-сахаринилметиларилкарбоксилатов, которые обладают ингибирующей активностью в отношении протеазного фермента и которые являются полезными в терапии дегенеративных заболеваний.
Изобретение касается составов, ингибирующих активность протеолитического фермента, которые включают в себя фармацевтический носитель и эффективно действующее количество (ингибирующее протеолитический фермент) 2-сахаринилметиларилкарбоксилата, в частности 4-R4-R5-2-сахаринилметиларилкарбоксилата или 4,5,6,7-тетрагидро-2-сахаринилметиларилкарбоксилата.
Изобретениекасаетсяспособовполученияупомянутых4-R4-R5-2-сахаринилметиларилкарбоксилатов и 4,5,6,7-тетрагидро-2-сахаринилметиларилкарбоксилатов, которые включают в себя 1) взаимодействие 2-галометилсахарина с арилкарбоновой кислотой в присутствии кислоты-акцептора, или 2) взаимодействие сахарина со сложным хлорметильным эфиром арилкарбоновой кислоты в присутствии акцептора кислоты или 3) взаимодействие соли щелочного металла или таллия для надлежащей кислоты с надлежащими галометильными частицами.
Более конкретно изобретение касается соединений общей формулы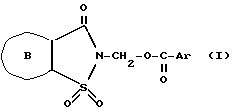 ,
,
где
кольцо B выбирают из следующих групп, сконденсированных с изотиазольным циклом стороной а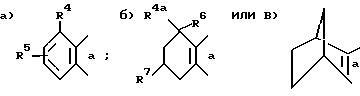 ,
,
где
Ar - фенильная, нафтильная или антрильная группа, замещенная одной - тремя, одинаковыми или разными заместителями, выбранными из группы, включающей: низший алкил, перфтор (низший) алкил, перхлор (низший) алкил, (низший)алкокси, галоген, PO-(низший)алкокси)2, амино, (низший)алкиламино, ди(низший)алкиламино, (низший)алканоиламино, (низший)алкоксикарбонил, гидрокси, бензилокси, карбокси(низший)алкокси, -SO2-N=B, -алкилен)-N=B или -O-(алкилен)-N=B, где в каждом случае N=B представляет собой амино, (низший)алкиламино, ди(низший)алкиламино, 1-азетидинол,1-пирролидинил, 1-пиперидинил, 4-морфолинил, 1-пиперазинил, 4-(низший)-алкил-1-пиперазинил, 4-бензил-1-пиперазинил, 1-имидазолил,
R4 выбран из группы, включающей водород, галоген, низший алкил, перфтор(низший)алкил, перхлор(низший)алкил, (низший)алкенил, (низший)алкинил, амино, (низший)алкиламино, ди(низший)алкиламино, (низший)алкокси, (низший)алкоксикарбонил, фенил или карбоксамидо и
R5 - водород или один-два одинаковых или разных заместителя в любом из положений 5, 6 или 7, выбранных из группы, включающей: галоген, циано, нитро, N=B, (низший)алкилсульфониламино, полифтор(низший)алкилсульфониламино, полихлор(низший)алкилсульфониламино, аминосульфонил, (низший)алкил, полифтор(низший)алкил, полихлор(низший)алкил, циклоалкил, (низший)алкокси, гидрокси, карбокси, гидрокси(низший)алкил, метилендиокси, формил, аминометил, (низший)алкилсульфонил, полифтор(низший)алкилсульфонил, полихлор(низший)алкилсульфонил, (низший)алкилсульфониламиносульфонил, -SR, -SOR, -SO2R, -OCOR, -O-(алкилен)-N= B, где R - представляет собой низший алкил, фенил, бензил или нафтил либо фенил или нафтил, замещенный одним-двумя заместителями, выбранными из группы, состоящей из: низшей алкильной группы, низшей алкоксигруппы или галогена, а N=B имеет вышеуказанное значение;
или R5 представляет собой 5- или 6-членное насыщенное кольцо, сконденсированное с сахарином в положениях 5, 6 или 6, 7, причем упомянутое кольцо содержит два гетероатома, выбранных из группы, состоящей из азота, кислорода или серы, либо метилированное производное упомянутого кольца;
R4a - водород, низшая алкильная группа или фенильная группа;
R6 - водород или первичная низшая алкильная группа либо R4a и R6 вместе образуют спироциклопропильное кольцо;
R7 - водород или низший алкокси;
A - метилен, этилен или диметилметилен, или их кислотно-аддитивные соли основного фрагмента либо соли присоединения основания кислотного фрагмента
при условии, что, когда обе группы R4 и R5 представляют собой водород, группа Ar не может быть фенильной, 2,4-дихлорфенильной или 4-нитрофенильной группой.
Предпочтительно соединение формулы I, в котором, если R5 представляет собой водород, группа R4 отлична от водорода или от метильной группы.
Еще одной группой предпочтительных соединений являются соединения, где R5-H, а R4 отлична от H и CH3, а
Ar - фенильная, нафтильная или антрильная группа, замещенная одной- тремя одинаковыми или разными группами, выбранными из группы, включающей : низший алкил, перфтор(низший)алкил, низший алкокси, галоген, PO(низший)алкокси)2, (низший)алканоиламино, гидрокси, карбокси(низший)алкокси, бензилокси, -SO2-N= B или -O-(алкилен)-N= B, где N=B представляет собой - ди(низший)-алкиламино, 1-пирролидинил, 1-пиперидинил, 4-морфолинил, 1-пиперазинил, 4-(низший)алкил-1-пиперазинил, 4-бензил-1-пиперазинил.
Еще более предпочтительными соединениями из вышеуказанной группы являются соединения, где
Ar - фенильная группа или фенильная группа, замещенная одной- тремя одинаковыми или разными группами, выбранными из группы, включающей: низший алкил, низший алкокси, галоген, гидрокси, карбокси(низший)алкокси, бензилокси, -SO2-N=B, или -O-(алкилен)-N=B, где N=B -ди(низший)алкиламино, 4-морфолинил, 1-пиперазинил, 4-(низший)алкил-1-пиперазинил, 1-пирролидинил, 1-пиперидинил, 4-бензил-1-пиперазинил,
R4 - первичная или вторичная низшая алкильная группа или низшая алкоксигруппа и
R5 - водород, низшая алкоксигруппа, метилендиоксигруппа.
Из вышеуказанных наиболее предпочтительны следующие:
4-изопропил-5-метокси-2-сахаринилметил-2,6-диметилбензоат,
4-изопропил-6-метокси-2-сахаринилметил-2,6-диметоксибензоат,
4-изопропил-6-метокси-2-сахаринилметил-2,6-дихлор-3-[2-(4- морфолинил)этокси]бензоат,
4-изопропил-6-метокси-2-сахаринилметил-2,6-дихлорбензоат,
4-изопропил-6-метокси-2-сахаринилметил-2,6-дихлор-3-(1- пиперидинилэтокси)бензоат,
4-изопропил-6-метокси-2-сахаринилметил-2,6-дихлор-3-(1- пирролинилэтокси)бензоат,
4-изопропил-6-метокси-2-сахаринилметил-2,6-дихлор-3-[2- (N,N-диэтиламино)этокси)бензоат,
4-изопропил-6-метокси-2-сахаринилметил-2,6-дихлор-3-[2- (1-пирролидинил)этокси)бензоат,
4-изопропил-6-метокси-2-сахаринилметил-2,6-дихлор-3- (4-метил-1-пиперазинилсульфонил)бензоат,
4-изопропил-6-метокси-2-сахаринилметил-2,6-дихлор-3-[N-[2- (диметиламино)этил-]-метиламиносульфонил)бензоат,
4-изопропил-6-метокси-2-сахаринилметил-2,6-дихлор-3- (гидроксибензоат,
4-изопропил-6-изопропокси-2-сахаринилметил-2,6-дихлорбензоат,
4-изопропил-6-[2-(2-метоксиэтокси)этокси]-2-сахаринилметил-2,6- дихлорбензоат,
4-изопропил-6-[2-(4-морфолинил)этокси] -2-сахаринилметил-2,6- дихлорбензоат,
4-изопропил-6,7-метилендиокси-2-сахаринилметил-2,6-дихлор-3-2- (4-морфолинил)этокси бензоат,
4-пропил-5,6-диметокси-2-сахаринилметил-2,6-дихлор-3-[2- (4-морфолинил)этокси]бензоат,
6-этокси-4-изопропил-2-сахаринилметил-2,6-дихлорбензоат,
5,6-диметокси-4-изопропил-2-сахаринилметил-1-нафтилкарбоксилат,
4-изопропил-6,7-метилендиокси-2-сахаринилметил-2,6-дихлорбензоат,
6-(2,3-дигидроксипропокси)-4-изопропил-2-сахаринилметил-2,6- дихлорбензоат и
6-(2,3-диметоксипропокси)-4-изопропил-2-сахаринилметил-2,6- дихлорбензоат,
особенно предпочтительны следующие:
6-(бензилоксикарбонил)метокси-4-изопропил-2-сахаринилметил-2,6- дихлорбензоат,
6-(трет-бутоксикарбонил)метокси-4-изопропил-2-сахаринилметил-2,6- дихлорбензоат,
4-изопропил-6-(метоксикарбонил)метокси-2-сахаринилметил-2,6- дихлорбензоат и
6-гидрокси-4-изопропилсахаринилметил-2,6-дихлорбензоат.
Также предпочтительная группа соединений I включает 2-сахаринилметиларилкарбоксилаты, в которых
R4a - водород, низший алкил,
R6 - водород, низший алкил,
Ar - фенил, замещенный один - тремя атомами галогена, из которых более предпочтительны
4,5,6,7-Тетрагидро-2-сахаринилметил-2,6-дихлорбензоат, 4-метил-4,5,6,7-тетрагидро-2-сахаринилметил-2,6-дихлорбензоат, 4-этил-4,5,6,7-тетрагидро-2-сахаринилметил-2,6-дихлорбензоат, 4-изопропил-4,5,6,7-тетрагидро-2-сахаринилметил-2,6-дихлорбензоат или 4,4-диметил-4,5,6,7-тетрагидро-2-сахаринилметил-2,6-дихлорбензоат, наиболее предпочтительным же является соединение, представляющее собой 2-(2,6-Дихлорбензоилоксиметил)-4,5,6,7-тетрагидро-4,7-метано- 1,2-бензизотиазол-3(2H-)-он-1,1-диоксид.
Следующим аспектом данного изобретения является способ получения 2-сахаринилметиларилкарбоксилатов формулы I, где B - кольцо а или б, по п. 1, отличающийся тем, что соединение общей формулы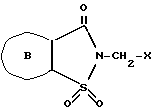 ,
,
где
B - кольцо а или б, X - галоген, подвергают взаимодействию с арилкарбоновой кислотой формулы ArCOOH,
где
Ar имеет указанные в п. 1 значения, в присутствии акцептора кислоты, или с солью щелочного металла или таллия указанной кислоты.
Еще один аспект изобретения включает способ получения 2-сахаринилметиларилкарбоксилатов формулы I, где B - кольцо а, заключающийся в том, что соединение общей формулы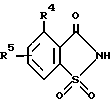 ,
,
где
R4 и R5 определены в п. 1, подвергают взаимодействию с хлорметиловым эфиром арилкарбоновой кислоты формулы
ArCOOCH2Cl, где Ar имеет указанные в п. 1 значения, в присутствии акцептора кислоты.
Из перечисленных выше соединений I предпочтительны 2-сахаринилметиларилкарбоксилаты формулы I, где B - кольцо а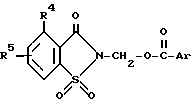 ,
,
в которой
Ar - фенильная, нафтильная или антрильная группа, замещенные одной-тремя, одинаковыми или разными заместителями, выбранными из группы, включающей: низший алкил, перфтор(низший)алкил, перхлор(низший)алкил, низший алкокси, галоген, амино, (низший)алкиламино, ди(низший)алкиламино, (низший)алканоиламино, карбо(низший)алкокси, гидрокси, бензилокси, карбокси(низший)алкокси, -SO2-N=B, (алкилен)-N=B или -O-(алкилен)-N=B, где группа N= B в каждом случае представляет собой: амино, (низший)алкиламино, ди(низший)алкиламино, 1-азетидинил, 1-пирролидинил, 1-пиперидинил, 4-морфолинил, 1-пиперазинил, 4-(низший)алкил-1-пиперазинил, 4-бензил-1-пиперазинил, 1-имидазолил;
R4 выбран из группы, включающей: водород, галоген, первичная или вторичная низшая алкильная группа, перфтор(низший)алкил, перхлор(низший)алкил, низший алкенил, низший алкинил, амино, (низший)алкиламино, ди(низший)алкиламино, низший алкокси, карб(низший)алкокси или фенил;
R5 - водород или один - два заместителя в любом из положений 5, 6 или 7, выбранных из группы, включающей: галоген, циано, нитро, N=B, (низший)алкилсульфонил, полифтор(низший)алкилсульфониламино, полихлор(низший)алкилсульфониламино, аминосульфонил, низший алкил, полифтор(низший)алкил, полихлор(низший)алкил, циклоалкил, низший алкокси, гидрокси, карбокси, гидрокси(низший)алкил, формил, аминометил, (низший)алкилсульфонил, полифтор(низший)алкилсульфонил, полихлор(низший)алкилсульфонил, (низший)алкоксиполи(низший)алкиленокси, -SR, -SOR, -SO2R или -OCOR, где R - представляет собой низший алкил, фенил или нафтил либо фенил или нафтил, замещенный одним-двумя заместителями, выбранными из группы, состоящей из низшей алкильной группы, низшей алкоксигруппы или галогена, группа N=B имеет вышеуказанное значение или их кислотно-аддитивные соли основных фрагментов, либо соли либо присоединения оснований к кислотным фрагментам при условии, что, когда обе группы R4 и R5 представляет собой водород, группа Ar не может быть фенильной, 2,4-дихлорфенильной, 4-нитрофенильной группой.
Частным случаем способа получения 2-сахаринилметиларилкарбоксилатов таким образом является способ получения вышеуказанных соединений, где соединение формулы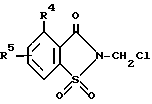 ,
,
в которой
R4 и R5 имеют указанные выше значения, подвергают взаимодействию с арилкарбоновой кислотой формулы ArCOOH, в которой Ar имеет указанные выше значения, в присутствии акцептора кислоты.
Еще одним аспектом изобретения является уже упомянутая выше композиция, ингибирующая активность протеолитического фермента, включающая активное вещество и фармацевтически приемлемые носители, которая в качестве активного вещества содержит соединение формулы I в эффективном количестве.
Понятно, что предпочтительными являются композиции, включающие в качестве активного вещества предпочтительные соединения из вышеперечисленных.
Здесь далее термины (низшая)алкильная группа, (низшая)алкоксигруппа и (низшая)алкановая группа относятся к моновалентным алифатическим радикалам, включая радикалы с разветвленной цепью, с числом атомов углерода от одного до десяти. Таким образом, к (низшей)алкильной (или(низшей)алкановой) части таких групп относятся, например, метильная, этильная, пропильная, изопропильная, н-бутильная, втор-бутильная, трет-бутильная, н-пентильная, 2-метил-3-бутильная, 1-метилбутильная, 2-метилбутильная, неопентильная, н-гексильная, 1-метилпентильная, 3-метилпентильная, 1-этилбутильная, 2-этилбутильная, 2-гексильная, 1,1,3,3-тетраметилпентильная, 1,1-диметилоктильная и им подобные группы.
Здесь далее термин галоген (или гало-) обозначает фтор, хлор, бром или иод.
Здесь далее термины (низшая)алкенильная и (низшая)алкинильная группы относятся к моновалентным ненасыщенным радикалам, включая радикалы с разветвленной цепью, с числом атомов углерода от одного до десяти; и таким образом, сюда подпадают 1-этенильная, 1-(2-пропенильная), 1-(2-бутенильная), 1-(1-метил-2-пропенильная), 1-(4-метил-2-пентенильная), 4,4,6-триметил-2-гептенильная, 1-этинильная, 1-(2-пропинильная), 1-(2-бутинильная), 1-(1-метил-2-пропинильная), 1-(4-метил-2-пентинильная) и им подобные группы.
Здесь далее термин алкилен относится к двухвалентным насыщенным радикалам, включая радикалы с разветвленной цепью, с числом атомов углерода от одного до десяти, у которых их свободные валентности приходятся на разные углеродные атомы; и, таким образом, термин распространяется на 1,2-этиленовую, 1,3-пропиленовую, 1,4-бутиленовую, 1-метил-1,2-этиленовую, 1,8-октиленовую и им подобные группы.
Здесь далее циклоалкильная группа относится к насыщенным моноциклическим углеводородным остаткам с числом атомов углерода от трех до семи; и, таким образом, это понятие распространяется на циклопропильную, циклобутильную, циклопентильную, циклогексильную и циклогептильную группы.
Соединения, отвечающие настоящему изобретению, ингибируют активность сериновых протеаз, особенно человеческой, относящейся к лейкоцитам эластазы и химотрипсиноподобных ферментов; и, тем самым, они оказываются полезными в терапии дегенеративных болезненных состояний, таких как эмфизема, ревматоидный артрит, панкреатит, муковисцидоз, хронический бронхит, респираторный дистресс-синдром у взрослых, воспалительное кишечное заболевание, псориаз, бычий пемфигоид и альфа-1-антитрипсиновая недостаточность.
Соединения с формулой I и формулой VI получают взаимодействием 2-галометилсахарина или 2-галометил-4,5,6,7-тетрагидросахарина с надлежащей арилкарбоновой кислотой Ar-COOH или взаимодействием сахарина или тетрагидросахарина со сложным хлорметиловым эфиром арилкарбоновой кислоты (см. схему 1). Реакцию можно вести либо в присутствии акцептора кислоты, таких как карбонат щелочного металла, три(низший)алкиламин, либо в присутствии 1,8-диазабицикло-[5.4.0]ундек-7-ена, именуемого далее ДБУ. Или же может быть использована соль щелочного металла, особенно цезия, или талиевая (I) соль арилкарбоновой кислоты, полученная взаимодействием кислоты со щелочным металлом. Реакцию проводят в органическом растворителе, инертном в условиях протекания реакции, например в ацетоне, метилэтилкетоне, ацетонитриле, тетрагидрофуране, простом диэтиловом эфире, диметилформамиде, N-метилпирролидиноне, двуххлористом метилене, ксилоле, толуоле или в (низших)алканолах, что делают при температуре, находящейся в области от температуры окружающей среды до температуры кипения используемого растворителя.
Соединения 4-R4-R5-2-галометилсахарины, необходимые для получения соединений с формулой I, готовят способами, описанными в журналах (D' Alelio et al., J. Macromol, Sci-Chem., A3(5), 941 (1969) и Saari et al., J. Het, Chem. 23, 1253 (1986)) (см. схему 2).
По методу, описанному Сарри (Saari), сложный метиловый эфир надлежащей антраниловой кислоты готовят обычными способами из замещенной антраниловой кислоты и диазотированного сложного эфира. Диазониевую соль затем подвергают взаимодействию с диоксидом серы и хлоридом меди (2), чтобы образовывался хлористый сульфонил, который затем взаимодействует с концентрированным гидроксидом аммония с образованием замещенных производных сахарина с формулой II. Последние при взаимодействии с формальдегидом в (низшем)алканольном растворителе дают 4-R4-R5-2-гидроксиметилсахарины с формулой III, которые при взаимодействии с тионилгалогенидом или с фосфорным тригалогенидом дают соответствующие 4-R4-R5-2-галометилсахариновые производные с формулой IV.
Соединения 4-R4-R5-2-галометилсахарины с формулой IV, где X - хлор или бром, могут быть также получены взаимодействием соответствующего 3-R4-R5-2-фенилтиометилсахарина с сульфурилгалогенидом в инертном органическом растворителе, например в двуххлористом метилене, двуххлористом этилене или четыреххлористом углероде при температурах в области от примерно 0oC и примерно до 30oC. Соединения 4-R4-R5-2-фенилтиометилсахарины получают, в свою очередь, взаимодействием 4-R4-R5-сахарина с формулой II с галометилфенилсульфидом в инертном органическом растворителе, таком как толуол, ксилол, диметилформамид или двухлористый метилен, что делают при температуре, находящейся в области от температуры окружающей среды до температуры кипения используемого растворителя. Реакцию можно проводить путем взаимодействия галометилфенилсульфида с талиевой (3) солью сахаринового производного с формулой II (полученного взаимодействием производного сахарина с таллиевым (2) (низшим)алкоксидом в (низшем)алканоле), или с ди(низший)алкиламмониевой солью сахариновых производных (полученных как это описано ниже) в присутствии тетра(низший)алкиламмониевого галогенида, такого как тетрабутиламмониевый бромид (именуемый далее ТБАБ), или с производным сахарина с формулой II, как таковым, в присутствии тетра(низший)алкиламмониевым галогенидом или с производным сахарина с формулой II, как таковым, в присутствии тетра(низший)алкиламмониевого галогенида и (низшего)алкоксида щелочного металла, такого как трет-бутоксид калия.
Сахарины с формулой II могут быть также превращены в хлорметильные сахарины с формулой IV, где X - хлор, в одну стадию путем взаимодействия с избыточным количеством формальдегида или равноценного ему вещества, такого как п-формальдегид или 1,3,5-триоксан, и хлорсилана, желательно хлортриметилсилана, в присутствии кислоты Люиса, желательно каталитически достаточного количества хлорида олова (4) в инертном растворителе, желательно 1,2-дихлорэтане (двуххлористом этилене).
Все способы превращения сахаринов с формулой II в 2-хлорметилсахарины с формулой IV одинаково пригодны для осуществления превращения тетрагидросахаринов с формулой VII в 2-хлорметилтетрагидросахарины с формулой VIII.
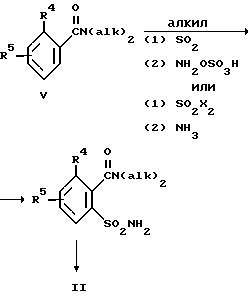
Соединения с формулой II могут быть также получены взаимодействием 2-R4-R5-N,N-ди(низший)алкилбензамида с формулой V с одним молярным эквивалентом (низшего)алкильного соединения щелочного металла, такого как литий, желательно в присутствии тетра(низший)алкилэтилендиамина, в инертном органическом растворителе, например в тетрагидрофуране, и взаимодействием образующейся соли щелочного металла либо с диоксидом серы при температуре в области от -50 до -80oC с последующим взаимодействием образующегося сульфината щелочного металла с гидроксиламин-0-сульфоновой кислотой в присутствии основания, либо с сульфурилгалогенидом с последующим воздействием аммиака. Если идут по пути использования диоксида серы и гидроксиламин-0-сульфоновой кислоты, то тогда особенно желательно провести нейтрализацию гидроксиламин-0-сульфоновой кислоты одним эквивалентом гидроксида натрия перед добавлением сульфината щелочного металла. Образующийся 2-R4-R5-6-аминосульфонил-N,N-ди(низший)алкилбензамид нагревают затем в кислой среде с целью инициирования циклизации последнего, чтобы образовалась ди(низший)алкиламмониевая соль требуемого 4-R4-R5-сахарина с формулой II, который может быть использован как таковой в последующей реакции или же, при желании, может быть гидролизован разбавленной кислотой и может быть выделен свободный сахарин. Циклизацию желательно проводить в ледяной уксусной кислоте с использованием сосуда с обратным холодильником. Способ проиллюстрирован ниже, где группа R4, R5 и Alk имеют тот же смысл, что и выше, а щелочным металлом является литий.
 .
.
Соединения с формулой II, где R4 - либо первичная либо вторичная (низшая)алкильная группа и которые могут быть использованы в качестве промежуточных продуктов при получении соединений с формулой I, как это описано выше, готовят одним из двух способов. Соединения с формулой II, где R4 - первичная (низшая)алкильная группа, получают взаимодействием 4-метил-R5-сахарина (формула II, R4 - группа CH3) с двумя молярными эквивалентами (низшего)алкильного соединения щелочного металла, такого как литий, в инертном органическом растворителе, например в тетрагидрофуране, и взаимодействием образующейся соли щелочного металла с одним молярным эквивалентом (низшего)алкилгалогенида, причем обе реакции ведут при температуре, находящейся в области примерно от -50 до -80oC.
Еще один способ получения соединений с формулой II, где R4 - либо первичная, либо вторичная (низшая)алкильная группа, сводится к взаимодействию 2-перв-(низший)алкил-R5-N, N-ди(низший)алкилбензамида (формула V, R4 - перв-(низшая)алкильная группа) с одним молярным эквивалентом (низшего)алкильного соединения щелочного металла или ди(низшего)алкиламида щелочного металла, где особенно желательным щелочным металлом является литий, желательно в присутствии тетра(низший)алкилэтилендиамина, в инертном органическом растворителе, например в тетрагидрофуране, и взаимодействию образующейся соли щелочного металла с одним молярным эквивалентом (низшего)алкильного галогенида при температуре, находящейся в области примерно от -50 до -80oC. Образующийся 2-первичный или вторичный (низший)алкил-R5-N,N-ди(низший)алкилбензамид превращают затем в соединения с формулой II, где R4 - первичная или вторичная (низшая)алкильная группа, проводя такую же последовательность реакций, что и описанная выше, т.е. путем взаимодействия 2-первичного или вторичного (низший)алкил-R5-N,N-(низший)алкилбензамида с одним молярным эквивалентом (низшего)алкильного соединения щелочного металла, такого как литий, взаимодействия образующейся соли щелочного металла либо с диоксидом серы с последующим воздействием гидроксиламин-0-сульфоновой кислоты в присутствии основания или с сульфурилгалогенидом с последующим воздействием аммиака и циклизации продукта с образованием требуемого 4-первичного или вторичного (низший)алкил-R5-сахарина с формулой II. Если 2-(низшая)алкильная группа у исходного 2-(низший)алкил-R5-N,N-ди(низший)алкилбензамида представляет собой метильную группу, то тогда при алкилировании получаются частицы, у которых 2-(низшая)алкильная группа характеризуется либо прямой, либо разветвленной цепью, что зависит от наличия прямой или разветвленной цепи у (низшего)алкилгалогенида, использованного при алкилировании. С другой стороны, когда 2-(низшая)алкильная группа у исходного вещества содержит не один, а несколько атомов углерода, алкилирование происходит на атоме углерода, примыкающем к бензольному кольцу, и получаются продукты, содержащие втор-(низшую)алкильную группу в положении 2.
Особенно полезным способом получения соединений с формулой II, где R4-н-(низшая)алкильная группа и R5 - водород, является способ с защитой бензильных протонов у исходного вещества с формулой V триалкилсилильной группой, чем обеспечивается возможность введения металла, например лития, в положение 6 и образования сульфонамида, как это описано выше.
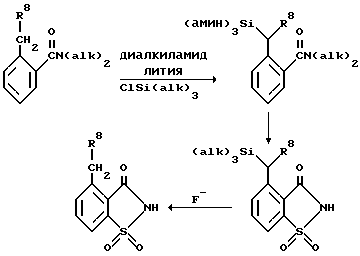
Соединение 2-н-(низший)алкилбензамид, где R8 - (низшая)алкильная группа, подвергают силилированию, образуя бензильный анион при использовании алкильного соединения щелочного металла или, что более желательно, диалкиламида щелочного металла, где особенно желательным щелочным металлом является литий, в инертном растворителе, желательно в тетрагидрофуране, и обрабатывая подходящим хлортриалкилсиланом, желательно хлортриметилсиланом. Сахарин синтезируют как и ранее, и силильную группу удаляют, воздействуя источником фтористого аниона, желательно фторидом цезия в диметилформамиде или фтористым тетра-н-бутиламмонием в инертном растворителе.
При наличии некоторых требуемых промежуточных продуктов в ряде случаев возникает необходимость построения двух колец с образованием ядер сахарина или тетрагидросахарина. Так, для получения сахаринов, где R4 - (низшая)алкоксигруппа и R5 - 7-гидроксигруппа, или тетрагидросахаринов, где R7 - (низшая)алкоксигруппа, может быть проведен следующий синтез: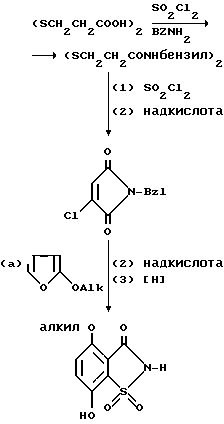
3,3-Дитиобиспропионовую кислоту превращают в хлорид бискислоты, проводя взаимодействие кислоты с хлористым тионилом, и хлорид кислоты затем подвергают взаимодействию с двумя молярными эквивалентами бензиламина, получая бис-N-бензиламид. Последний, взаимодействуя с хлористым сульфурилом в органическом растворителе, таком как двухлористый метилен, двухлористый этилен или четыреххлористый углерод, дает 5-хлор-2-бензил-2H-изотиазол-3-он, который окисляют одним молярным эквивалентом надкислоты, такой как надбензойная кислота или 3-хлорнадбензойная кислота, получая 5-хлор-2-бензил-2H-изотиазол-3-он-1-оксид. Последний, нагревая под давлением с 2-(низший)алкоксифураном в органическом растворителе, таком как бензол, толуол или ксилол, превращают в 4-(низший)алкокси-7-гидрокси-2-бензил-1,2- бензизотиазол-2H-3-он-1-оксид. 7-Гидроксигруппа может быть, при желании, подвергнута затем взаимодействию с (низшим)алкилгалогенидом или с (низшим)алкоксиполи(низший)алкокси(низший)алкилгалогенидом, в результате чего получают соответствующий 4,7-ди(низший)алкокси- или 4-(низший)алкокси-7-(низший)алкоксиполи(низший)алкокси-2-бензил- 1,2-бензизотиазол-2H-3-он-1-оксид. Дальнейшее окисление продукта одним молярным эквивалентом надкислоты, как это было описано выше, сопровождаемое каталитическим дебензилированием посредством гидрогенизации с переносом, ведет к получению соответствующих 4-(низший)алкокси-7-гидроксисахаринов.
При необходимости получения тетрагидросахарина используют следующий вариант синтеза: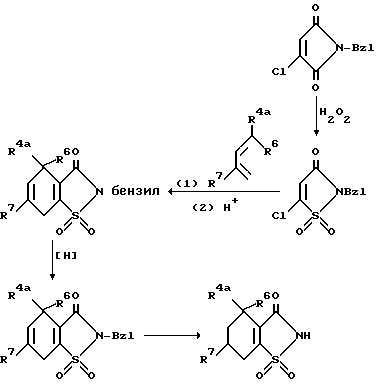 .
.
5-Хлор-2-бензил-2H-изотиазол-3-он-1-оксид может быть окислен надлежащим окисляющим веществом, желательно пероксидом водорода в уксусной кислоте, до 1,1-диоксида, который затем взаимодействует в типичных условиях Дильса-Альдера с надлежащим диеном и восстанавливается с образованием 2-бензил-тетрагидросахарина, который подвергают гидролизу, как и ранее, получая тетрагидросахарин.
Соединения с формулой II, где R4 - (низшая)алкильная или фенильная группа и R5 - водород, могут быть синтезированы иным способом из 2-циклогексенона: .
.
2-Циклогексенон взаимодействует с надлежащим купратом, затем с метилцианоформатом по способу, описанному Уинклером (Winkler) и др. [Tet. Lett. 1987, 1051 и J. Org. Chem. , 54, 4491 (1989)]. Образующийся β -кетоэфир подвергают взаимодействию с бензилмеркаптаном в присутствии кислого глинистого монтмориллонита KSF, в результате чего получают смесь пространственных изомеров простого бензилтиоенольного эфира. Смесь ароматизируют, обрабатывая дихлордицианобензохиноном, и окисляют газообразным хлором в водном растворе кислоты, получая сложный сульфонильный хлористый эфир, который может быть затем превращен в промежуточный продукт с формулой II, как это было показано ранее.
Соединения 4,5,6,7-тетрагидросахарины, которые являются исходными веществами для соединений с формулой VI, где R7 - водород, синтезируют способом, аналогичным описанному выше (см. схему 3).
3-Алкил-2-циклогексенон подвергают взаимодействию с надлежащим алкиллитиевым купратом в эфирном растворителе, желательно в диэтиловом эфире, что делают в области температур от -50 до +20oC, желательно при температуре порядка 0oC, и образующийся аддукт обрабатывают тут же метилцианоформатом и гексаметилфосфорамидом. Образовавшийся при этом 6,6-диалкил-2-оксоциклогексанкарбоксилат подвергают взаимодействию с бензилмеркаптаном, как это было описано выше, и смесь 2-(бензилтио)-циклогексанкарбоксилатов окислительно хлорируют, как это было описано выше, получая смесь сложных хлорсульфонильных эфиров, которые, как и ранее, обрабатывают аммиаком, получая требуемые 4,4-диалкил-4,5,6,7-тетрагидросахарины.
Арилкарбоновые кислоты Ar-COOH, используемые для получения конечных продуктов с формулами I и VI, являются членами хорошо известного класса соединений и могут быть приготовлены хорошо известными общепринятыми синтетическими способами.
Сложные хлорметиловые эфиры арилкарбоновой кислоты могут быть получены обработкой карбоновой кислоты формальдегидом или равноценным ему веществом, желательно п-формальдегидом, в присутствии 1) хлоркислоты, желательно хлорида цинка или хлористоводородной кислоты, или 2) триметилсилилхлорида и одновременно хлорида олова (4).
Простые химические превращения, которые являются обычными и хорошо известными специалистам, работающим в этой области химии, могут быть использованы для проведения изменений у функциональных групп соединений, отвечающих настоящему изобретению. Например, при необходимости могут быть проведены каталитическое восстановление нитрогрупп с образованием соответствующих аминозамещенных соединений, ацилирование аминозамещенных образований с получением соответствующих амидов, окисление сульфидов или сульфоксидов с образованием соответствующих надлежащих сульфоксидов или сульфонов, омыление сложных эфиров с образованием соответствующих карбоновых кислот, каталитическое дебензилирование простых фенольных эфиров или бензиламинов с образованием соответствующих фенолов или дебензилированных аминов или взаимодействие фенолов с алкилирующим веществом в присутствии основания с получением простых эфиров.
Применением стандартных биологических тест-методик установлено, что соединения с формулами I и VI обладают ингибирующей активностью в отношении человеческой лейкоцитной эластазы и химотрипсина и, тем самым, оказываются полезными в терапии дегенеративных заболеваний, таких как эмфизема, ревматоидный артрит, панкреатит, муковисцидоз, хронический бронхит, респираторный дистресс-синдром у взрослых, воспалительное кишечное заболевание, псориаз, бычий пемфигоид и альфа-1-антритрипсиновая недостаточность.
Соединения с формулами I и VI, обладая основными свойствами, могут быть присоединением кислотных остатков превращены в соли, образующиеся при взаимодействии основания с кислотой. Аналогичным образом, свободное основание может быть регенерировано из соли, полученной присоединением кислотного остатка, что достигается обычным способом, т.е. обработкой солей холодным слабым водным раствором оснований, например карбонатами щелочного металла и бикарбонатами щелочного металла. Основания, полученные при такой регенерации, могут быть подвергнуты взаимодействию с той же или иной кислотой с обратным образованием той же или иной соли, получаемой присоединением кислотного остатка. Таким образом, основания и все их соли, образующиеся присоединением кислотного остатка, являются взаимообратимыми.
Аналогичным образом, некоторые соединения с формулами I и VI, обладающие свойствами кислоты, т.е. карбоновой кислоты, могут быть превращены в солевые формы таковых взаимодействием кислоты с основанием, таким как гидрооксиды щелочного металла или аммония, с органическими основаниями, такими как алкил-, диалкил- или триалкиламины; и кислоты могут быть регенерированы из солей обработкой солей водными растворами кислот.
Формулы I и VI не только характеризуют структурную конфигурацию оснований и кислот, но они также отображают структурные единицы, которые являются общими у всех соединений с формулами I и VI независимо от того, находятся ли они в форме свободного основания, свободных кислот или в форме солей оснований и кислот. Установлено, что в силу наличия этих общий структурных единиц соединения с формулами I и VI и их соли обладают присущей им фармакологической активностью того типа, который более подробно будет описан ниже. Эта неотъемлемая фармакологическая активность может быть облечена в полезную для фармацевтических целей форму с применением свободных оснований и свободных кислот самих по себе или же солей, образованных из фармацевтически приемлемых кислот и оснований, т.е. кислот и оснований, анионы или катионы которых являются безвредными для организма животного при введении эффективных доз солей, так что полезные свойства, присущие общей структурной единице, представленной свободными основаниями и свободными кислотами, оказываются не перекрытыми побочными эффектами, свойственными анионам или катионам.
При использовании этой фармакологической активности соли желательно использовать фармакологически приемлемые соли. Хотя нерастворимость в воде, высокая токсичность или отсутствие кристалличности и могут делать некоторые отдельные солевые частицы неприемлемыми или менее желательными для использования как таковых в данном фармацевтическом применении, не растворимые в воде или токсичные соли могут быть превращены в соответствующие фармацевтически приемлемые основания, что достигается разложением солей водным раствором основания или водным раствором кислоты, о чем говорили выше, или же они могут быть превращены в какую-либо желаемую фармацевтически приемлемую соль посредством двойных реакций разложения с участием аниона или катиона, например применением методик ионного обмена.
Более того, помимо их полезности в фармацевтических применениях, соли являются полезными в качестве характеризующих или идентифицирующих производных свободных оснований или свободных кислот или при оценке методик выделения или очистки. Как и в случае всех солей, такие производные солей, применяемые для установления характеристик или для оценки процесса очистки, могут быть, при желании, использованы для регенерации фармацевтически приемлемых свободных оснований или свободных кислот, что достигается путем взаимодействия солей с водным раствором основания или водным раствором кислоты, или же они могут быть превращены в фармацевтически приемлемую соль с применением, к примеру, методик ионного обмена.
В таком случае ранее неизвестная особенность соединений состоит в самом существовании 2-сахаринилметиларилкарбоксилатов с формулами I и VI, а не в наличии какой-либо кислотной или основной части молекулы или аниона кислоты или катиона основания у солевых форм соединений.
Соединения с формулами I и VI, отвечающие настоящему изобретению, могут быть приготовлены для фармацевтического применения посредством введения их в единичную дозированную форму в виде таблеток или капсул, предназначенных для орального введения, либо как таковых, либо в сочетании с подходящими адъювантами, такими как карбонат кальция, крахмал, лактоза, тальк, стеарат магния, аравийская камедь и им подобные вещества. Более того, соединения могут быть введены в рецептуры, предназначенные для орального, парентерального или аэрозольного ингаляционного введения, либо в виде водных растворов растворимых в воде солей таких соединений, либо в виде водно-спиртовых, водно-гликолевых или водно-масляных растворов или масляно-водных эмульсий, которые готовят тем же способом, что и обычно медицинские препараты.
Процентное содержание активного компонента в таких составах может претерпевать изменение, чем обеспечивается получение требуемой дозировки. Дозировка, используемая при введении некоторому данному пациенту, изменяется с учетом клинического заключения, основанного на следующих критериях: способ введения, продолжительность лечения, размер и физическое состояние пациента, эффективность действия активного компонента и реакция пациента на него. Эффективное дозированное количество активного компонента может, тем самым, определить только клиницист после рассмотрения всех критериев с принятием решения исключительно в интересах пациента.
Молекулярные структуры у соединений, отвечающих настоящему изобретению, были установлены на основе изучения их инфракрасных спектров и спектров ядерного магнитного резонанса. Структуры были подтверждены наличием соответствия между расчетными и найденными величинами, полученными при проведении элементарного анализа на элементы, или данными анализа масс-спектров высокого разрешения.
Следующие примеры дополнительно иллюстрируют настоящее изобретение, не ограничивая, однако, его рамки. Во всех случаях в температуры плавления поправку не вносили.
Приготовление исходных веществ.
Синтез 1.
Измельченный в порошок гидроксид калия (7,4 г, 0,132 моль) смешивали с диметилсульфоксидом (100 мл), смесь перемешивали в течение 5 мин. Затем к смеси добавляли 6-метилантраниловую кислоту (10,0 г, 0,066 моль) и по каплям добавляли иодистый метан (4,52 мл, 0,073 моль). Реакционную смесь перемешивали в течение 30 мин при комнатной температуре, затем разбавляли 250 мл простого эфира, промывали водой (3 раза по 100 мл), сушили над сульфатом магния и концентрировали. Сырой продукт фильтровали через набивку из силикагеля квалификации "для испарительной хроматографии" (32-63) и элюировали смесью простого эфира с гексаном, взятыми в соотношении 1:9, в результате чего получали 4,23 г (выход 39%) метил-6-метилантранилата, имеющего вид маслянистой жидкости.
Приготовленный таким способом метил-6-метилантранилат (4,23 г, 0,026 моль) растворяли в 25 мл уксусной кислоты, и раствор охлаждали до 0oC. Добавляли концентрированную хлористоводородную кислоту (45 мл), получая дубильный шлам. Раствор, состоящий из 1,89 г (0,027 моль) нитрита натрия в 8 мл воды, добавляли по каплям при перемешивании, образующийся органический раствор перемешивали при 0oC в течение 1 ч и затем добавляли шестью порциями к смеси, состоящей из 2,18 г (0,013 моль) дигидрата хлорида меди (2) и диоксида серы (6,3 г) в 33 мл уксусной кислоты и 6 мл воды, находящихся при 0oC. Темно-зеленый раствор перемешивали при комнатной температуре на протяжении ночи, выливали в 300 мл воды со льдом, и твердое вещество, которое выпадало, собирали и сушили отсосом, получая 1,11 г метил-2-хлорсульфонил-6-метилбензоата, который сразу же добавляли к 100 мл охлажденному льдом гидроксиду аммония и перемешивали при комнатной температуре в течение ночи. Раствор подкисляли до величины pH 1 концентрированной хлористоводородной кислотой, и образующийся осадок собирали и сушили на воздухе, получая 729 мг (выход 12%) 4-метилсахарина; температура плавления составляла 224-226oC.
Смесь, состоящую из 1,0 г (0,005 моль) 4-метилсахарина, 0,33 г (0,001 моль) бромистого тетрабутиламмония и 1,2 г (0,0075 моль) хлорметилфенилсульфида в 25 мл толуола, грели в сосуде с обратным холодильником на протяжении примерно 16 ч и затем охлаждали, разбавляли этилацетатом, и раствор промывали водным раствором бикарбоната и водой. Органический слой сушили, и, осушив досуха, получали 0,74 г 2-фенилтиометил-4-метилсахарина. Последний (0,74 г, 0,002 моль) растворяли в 25 мл двухлористого метилена, и раствор обрабатывали, вводя по каплям на протяжении примерно двух часов при перемешивании раствор, состоящий из 0,47 г (0,003 моль) хлористого сульфурила в двухлористом метилене, и реакционную смесь сушили досуха. Оставшееся желтое твердое вещество растирали с гексаном, фильтровали и сушили, получая 0,46 г 2-хлорметил-4-метилсахарина в виде бледно-желтого твердого вещества.
Синтез 2.
По методике, описанной в синтезе 1, 5,0 г (0,029 моль) 6-хлорантраниловой кислоты и 2,75 мл (0,044 моль) иодистого метана подвергали взаимодействию в присутствии 4,08 г (0,073 моль) превращенного в порошок гидроксида калия, в результате чего получали 4,22 г (выход 78%) метил-6-хлорантранилата, имевшего вид маслянистой жидкости.
4-Хлорсахарин готовили тем же способом, что и использованный при приготовлении 4-метилсахарина, беря 4,22 г (0,023 моль) метил-6-хлорантранилата в 22 мл уксусной кислоты и 40 мл концентрированной хлористоводородной кислоты, и 1,68 г (0,024 моль) нитрита натрия в 7 мл воды с целью получения диазониевой соли, которую добавляли к 1,93 г (0,011 моль) дигидрата хлорида меди (2) и 6,5 г диоксида серы в 30 мл уксусной кислоты и 5 мл воды. Образующийся метил-2-хлорсульфонил-6-хлорбензоат обрабатывали 150 мл гидроксида аммония, как это описано выше, в результате чего получали 3,07 г (выход 62%) 4-хлорсахарина в виде бледно-желтого твердого вещества; температура плавления составляла 245-246oC.
2-Гидроксиметил-4-хлорсахарин готовили нагреванием раствора, состоящего из 1,00 г (0,0046 моль) 4-хлорсахарина и 3,22 мл водного 37%-ного формалина в этаноле. Все попытки закристаллизовать вязкий маслянистый продукт приводили к разложению исходного вещества и на следующей стадии продукт использовали, не устанавливая его характеристики.
Сырой 2-гидроксиметил-4-хлорсахарин, полученный указанным способом (609 мг, 0,0025 моль), смешивали с 5 мл диэтилового эфира и добавляли 3 мл хлористого тионила. Образующуюся смесь грели для обеспечения полного растворения, перемешивали при комнатной температуре на протяжении ночи, разбавляли 20 мл простого эфира и фильтровали через набивку из цеолита с верхним слоем песка и элюировали простым эфиром. Удаляя растворитель, получали 430 мг сырого хлорметильного производного. Часть (225 мг) убирали, предназначая для проведения дальнейших реакций. Оставшуюся часть (205 мг) подвергали испарительной хроматографии на силикагеле и элюировали 40%-ной смесью простого эфира с пентаном, в результате чего получали 137 мг 2-хлорметил-4-хлорсахарина; температура плавления составляла 135-136oC.
Синтез 3A.
К суспензии, состоящей из 6,0 г (0,03 моль) иодида меди (I) в 100 мл тетрагидрофурана, добавляли 25 мл диметилсульфида, и образующийся желтый раствор охлаждали до -78oC и обрабатывали по каплям раствором, состоящим из 23 мл (0,06 моль) 3,0 М раствора бромистого фенилмагния в диэтиловом эфире. Образующийся бледно-желто-оранжевый раствор перемешивали при -78oC в среде азота в течение одного часа и затем обрабатывали 3,02 г (0,03 моль) 2-циклогексенона в 10 мл тетрагидрофурана. Образовавшейся смеси давали нагреться до 0oC на протяжении промежутка времени в два часа, ее вновь охлаждали до -78oC, обрабатывали 15 мл гексаметилфосфорамида, перемешивали в течение 30 мин, обрабатывали 8,0 г (0,09 моль) метилцианоформата и давали за ночь нагреться до температуры окружающей среды. Реакционную смесь выливали в 100 мл 2 н. раствора хлористоводородной кислоты, органическую фазу отделяли, водную фазу обратно экстрагировали двухлористым метиленом. Соединенные органические экстракты сушили досуха в вакууме, остаток растирали с насыщенным раствором хлорида аммония, затем с водой, затем с рассолом и затем еще раз сушили досуха, получая 3,2 г метил-2-фенилциклогексан-6-он-карбоксилата в виде маслянистой жидкости. Последнее вещество (3,0 г, 0,013 моль), 4,8 г (0,039 моль) бензилмеркаптана и 1,0 г смолы амерлист®-15 (продукт с зарегистрированным фирменным названием; фирма "Ром энд Хаас") в хлороформе грели в сосуде с обратным холодильником на протяжении 20 ч, смесь дополнительно обрабатывали 1,5 г смолы и дополнительно грели в течение четырех часов. Смесь затем охлаждали до температуры окружающей среды, фильтровали, фильтрат сушили досуха в вакууме, остаток растирали с гексаном, и твердое вещество собирали фильтрованием, получая 0,85 г (выход 19%) смеси метил-2-бензилтио-6-фенилциклогекс-2-енкарбоксилата с метил-2-бензилтио-6-фенилциклогекс-1-енкарбоксилатом, 0,6 г (0,0018 моль) которой грели с 2,0 г 2,3-дихлор-5,6-дицианобензохинона в 25 мл толуола при перемешивании в среде азота в течение 24 ч. Смесь фильтровали через набивку из силикагеля, элюируя смесью двухлористого метилена с гексаном, взятыми в соотношении 2:1, и элюат сушили досуха, получая 0,3 г (выход 67%) метил-2-бензилтио-6-фенилбензоата. Последнее вещество (0,52 г, 0,0016 моль), растворенное в 10 мл двухлористого метилена, разбавляли 20 мл уксусной кислоты и 5 мл воды, смесь охлаждали до -10oC, газообразный хлор пробулькивали через смесь до тех пор, пока не начинала ослабевать экзотермическая реакция. Смесь затем перемешивали в течение 10 мин и сушили досуха в вакууме, получая 0,41 г (выход 85%) метил-2-хлорсульфонил-6-фенилбензоата, который растворяли в 10 мл тетрагидрофурана и добавляли к 25 мл концентрированного раствора гидроксида аммония с охлаждением на бане из ацетона со льдом. Реакционную смесь экстрагировали двухлористым метиленом, органическую фазу отбрасывали, водный слой подкисляли до величины pH 1 концентрированной хлористоводородной кислотой и экстрагировали двухлористым метиленом. Органические экстракты после промывки рассолом сушили и упаривали досуха, получая 0,33 г (выход 97%) 4-фенилсахарина.
Последующая методика является аналогичной методике, описанной в синтезе 1; последнее вещество (0,33 г, 0,0012 моль) подвергали взаимодействию с 0,3 г (0,0019 моль) хлорметилфенилсульфида тетрабутиламмония, и продукт, представляющий собой 2-фенилтиометил-4-фенилсахарин (0,48 г, выход 100%), обрабатывали хлористым сульфурилом в двухлористом метилене, получая 0,36 г (выход 95%) 2-хлорметил-4-фенилсахарина.
Синтез 3В.
К суспензии, состоящей из безводного цианида меди (I) Cu CN (2,16 г, 0,025 моль) в безводном простом эфире (100 мл), при -78oC добавляли трет-бутиллитий (29,0 мл 1,7 М раствора в пентане, 0,05 моль). После перемешивания при -78oC в течение 1 ч и при -45oC в течение 30 мин реакционную смесь охлаждали до -78oC. Добавляли раствор циклогексенона (2,4 г, 0,025 моль) в простом эфире (25 мл), перемешивание продолжали в течение 15 мин при -78oC и при -45oC в течение 30 мин. Образовавшуюся смесь вновь охлаждали до -78oC, добавляли гексаметилфосфорамид (10 мл) в виде раствора в простом эфире (25 мл). Через 5 мин добавляли метилцианоформат (2,55 г, 0,03 моль) в простом эфире (25 мл), реакционную смесь нагревали до 0oC на протяжении промежутка времени в 2 ч. Состав у реакционной смеси резко фиксировали 2 н. раствором HCl (100 мл), слои разделяли, и органический слой промывали насыщенным раствором хлорида аммония (3 раза по 50 мл), водой (2 раза по 50 мл), рассолом (1 раз по 50 мл) и сушили (сульфатом натрия). Удалением растворителя в вакууме и очисткой посредством дистилляции по Кугельрору (Kugelrohr) (температура куба 100-115oC при давлении 0,6 мм рт.ст.), в результате чего получали 4,7 г (выход 88%) метил-2-(1,1-диметилэтил)циклогексан-6-он-карбоксилата.
Циклогексанон (4,6 г, 0,022 моль) смешивали с бензилмеркаптаном (2,95 г, 0,024 моль) и кислым глинистым монтмориллонитом KSF (7,5 г) в безводном толуоле (7,5 мл). Смесь грели в сосуде с обратным холодильником в среде азота в условиях азеотропного удаления воды на протяжении 6 ч, охлаждали до комнатной температуры и оставляли стоять на всю ночь. Твердые вещества отфильтровывали и промывали простым эфиром. Объединенный фильтрат промывали 10%-ным раствором карбоната натрия, водой, рассолом и сушили. Удалением растворителя в вакууме и очисткой остатка испарительной хроматографией на силикагеле (10%-ный раствор простого эфира в гексанах) получали 4,4 г (выход 66%) смеси метил-2-бензилтио-6-(1,1-диметилэтил)циклогекс-2-ен-карбоксилата с 2-бензилтио-6-(1,1-диметилэтил)циклогекс-1-ен-карбоксилатом, которую перемешивали с дихлордицианобензохиноном (17,5 г, 0,077 моль) в толуоле (50 мл) в течение 16 ч. Красную реакционную смесь фильтровали через набивку из силикагеля длиной 15 см, элюируя смесью гексанов с двухлористым метиленом и простым эфиром (1000 мл), взятыми в соотношении 6:3:1. Элюенты промывали 10%-ным раствором гидроксида натрия, водой, рассолом и сушили. Удалением растворителя в вакууме и очисткой хроматографией на силикагеле (5%-ный раствор простого эфира в гексанах) получали 1,6 г (выход 40%) метил-2-бензилтио-6-(1,1-диметил)бензоата.
Бензилтиобензоат (1,3 г, 0,004 моль), растворенный в двухлористом метилене (5 мл), разбавляли уксусной кислотой (25 мл) и водой (2 мл), смесь охлаждали до -10oC, и газообразный хлор пробулькивали до тех пор, пока не прекращалась экзотермическая реакция. Смесь затем перемешивали в течение 10 мин и сушили досуха в вакууме. Очисткой остатка испарительной хроматографией на силикагеле (элюировали смесью гексанов с двухлористым метиленом, взятыми в соотношении 1: 1) получали 0,8 г (выход 67%) метил-2-хлорсульфонил-6-(1,1-диметилэтил)бензоата, который растворяли в тетрагидрофуране (5 мл) и добавляли к раствору концентрированного гидроксида аммония (25 мл) при охлаждении на бане из ацетона и льда. После перемешивания при комнатной температуре в течение 16 ч реакционную смесь концентрировали в вакууме и подкисляли до величины рН I 2 н. HCl. Выпавшие твердые вещества собирали фильтрованием и кристаллизовали из простого эфира, получая 0,64 г (выход 95%) 4-(1,1-диметилэтил)-сахарина; температура плавления составляет 185-187oC.
4-(1,1-диметилэтил)сахарин (0,025 г, 1,0 моль) смешивали с хлорметилфенилсульфидом (0,25 г, 1,5 моль) и бромистым тетрабутиламмонием (0,2 г, 0,6 ммоль) в толуоле (25 мл) и грели в сосуде с обратным холодильником в среде азота на протяжении 16 ч. Образовавшуюся смесь охлаждали до комнатной температуры, упаривали досуха и очищали хроматографией на силикагеле (алюировали 80%-ным раствором двухлористого метилена в гексанах), получая 0,35 г (выход 98%) 2-фенилтиометил-4-(1,1-диметилэтил)-сахарина, который обрабатывали хлористым сульфурилом (0,25 г, 1,8 ммоль) в двухлористом метилене, в результате чего получали 0,21 г (выход 75%) 2-хлорметил-4-(1,1-диметилэтил)сахарина.
Синтез 4.
Смесь, состоящую из 3,22 г (0,012 ммоль) 4-бромсахарина [патентная публикация Японии, 58/79, 034, С.А. 100, 7773 W (1984)], 1,63 г (0,015 моль) трет-бутоксида калия, 0,39 г (0,0012 моль) бромистого тетрабутиламмония и 3,0 мл (0,022 моль) хлорметилфенилсульфида в 100 мл толуола, грели в сосуде с обратным холодильником в среде азота в течение 8 ч и затем перемешивали при температуре окружающей среды в течение примерно 16 ч. Реакционную смесь затем разбавляли этилацетатом, и органический слой промывали разбавленным раствором карбоната калия, водой и рассолом, сушили над сульфатом магния и доводили до сухого состояния в вакууме. Оставшееся твердое вещество выкристаллизовывали из смеси толуола с гексаном, в результате чего получали 3,86 г (выход 84%) 4-бром-2-фенилтиометилсахарина; температура плавления составляла 174,5-178oC.
К раствору последнего вещества (3,27 г, 0,0085 моль) в 85 мл двухлористого метилена добавляли по каплям при перемешивании 1,02 мл (0,0127 моль) хлористого сульфурила. Смесь перемешивали при температуре окружающей среды в течение 1 ч. и 30 мин, концентрировали в вакууме, остаток растирали с гексаном и фильтровали, получая 2,61 г сырого продукта, который выкристаллизовали из смеси толуола с гексаном, получая 2,24 г (выход 85%) 2-хлорметил-4-бромсахарина, температура плавления составляла 157-159oC.
Синтез 5.
К раствору, состоящему из 8,0 мл (0,053 моль) тетраметилэтилендиамина в 350 мл тетрагидрофурана, при -70oC добавляли 42 мл (0,055 моль) 1,3 M раствора втор-бутиллития в циклогексане, и смесь перемешивали в течение 15 мин. К раствору по каплям при перемешивании добавляли раствор, состоящий из 10,36 г (0,050 моль) 2-метокси-N,N-диметилбензамида в 150 мл тетрагидрофурана, поддерживая при этом температуру на уровне -60oC или менее. После перемешивания в течение 20 мин через реакционную смесь пробулькивали диоксид серы, поддерживая температуру реакции ниже -50oC, что делали до тех пор, пока реакционная смесь не становилась кислой, о чем судили по мокрой лакмусовой бумаге. Смесь перемешивали при температуре окружающей среды в течение 2 ч, разбавляли 450 мл гексана, и твердое вещество, которое выделялось, собирали, растворяли в 200 мл воды, смесь обрабатывали 65 г ацетата натрия и 21,5 г (0,19 моль) гидроксиламин-0-сульфоновой кислотой, что делали порциями при перемешивании. Выпавшее белое твердое вещество собирали и сушили, получая 7,04 г (выход 49%) 2-аминосульфонил-6-метокси-N,N-диэтилбензамида; температура плавления составляла 190-194,5oC.
Смесь, состоящую из этого продукта (4,3 г, 0,015 моль) в 75 мл диоксана и 25 мл концентрированной хлористоводородной кислоты, грели на паровой бане в течение 70 ч, затем охлаждали, концентрировали в вакууме, разбавляли водой и льдом, и среду делали сильно основной, добавляя концентрированный раствор гидроксида натрия. Смесь промывали двухлористым метиленом, и водный слой подкисляли разбавленной хлористоводородной кислотой и экстрагировали двухлористым метиленом. Экстракты сушили над сульфатом магния и доводили до сухого состояния, в результате чего получали 1,29 г (выход 40%) 4-метоксисахарина. При использовании альтернативной и более предпочтительной методики циклизацию 2-аминосульфонил-6-метокси-N,N-диэтилбензамида до образования 4-метоксисахарина с выходом 65% проводят нагреванием в сосуде с обратным холодильником при использовании ледяной уксусной кислоты, на что затрачивали 6,5 ч.
Следуя методике, аналогичной описанной в синтезе 4, 1,14 г (0,0053 моль) последнего вещества приводили во взаимодействие с 1,31 мл (0,0097 моль) хлорметилфенилсульфида в толуоле в присутствии 0,72 г (0,0064 моль) трет-бутоксида калия и 174 мг (0,00054 моль) бромистого тетрабутиламмония, в результате чего получали 1,23 г (выход 69%) 4-метокси-2-фенилтиометилсахарина; температура плавления составляла 152,5-154,5oC (из смеси этилацетата с гексаном); 1,02 г (0,003 моль) этого продукта обрабатывали 0,36 мл (0,0045 моль) хлористого сульфурила в двухлористом метилене, в результате чего получали 282 мг 2-хлорметил-4-метоксисахарина; температура плавления составляла 169-174oC.
Синтез 6А.
К раствору, состоящему из 4,74 мл (0,031 моль) тетраметилэтилендиамина в 300 мл тетрагидрофурана (пропущенного перед использованием через оксид алюминия), добавляли 5,8 г (0,03 моль) 2-этил-N,N-диэтилбензамида. Раствор охлаждали до -78oC и обрабатывали 34,9 мл (0,031 моль) 0,9 М раствора втор-бутиллития в циклогексане. После завершения добавления смесь перемешивали в течение 20 мин и затем обрабатывали раствором, состоящим из 3,2 мл (0,04 моль) йодистого этила, что делали в условиях поддержания температуры на уровне -78oC. Затем температуре давали подняться до температуры окружающей среды, смесь перемешивали в течение примерно 16 ч и затем выливали в воду. Образовавшуюся маслянистую жидкость отделяли и подвергали хроматографии на силикагеле, элюируя 10%-ным раствором этилацетата в гексане, в результате чего получали 2,86 г (выход 43%) 2-втор-бутил-N,N-диэтилбензамида, имеющего вид желтой маслянистой жидкости.
Следуя методике, аналогичной описанной в синтезе 5, последнее вещество (10,45 г, 0,045 моль), растворенное в 70 мл тетрагидрофурана, добавляли к раствору, состоящему из 39,2 мл (0,047 моль) 1,2 M раствора втор-бутиллития в циклогексане и 7,1 мл (0,047 моль) тетраметилэтилендиамина в 250 мл тетрагидрофурана, что делали в условиях поддержания температуры на уровне -78oC. После завершения добавления смесь перемешивали еще в течение 1,5 ч при -78oC, затем обрабатывали диоксидом серы при -70oC и затем давали нагреться до комнатной температуры. Смесь доводили до сухого состояния в вакууме, остаток растворяли в воде и добавляли при перемешивании к холодному раствору, состоящему из 15,2 г (0,134 моль) гидроксиламин-0-сульфоновой кислоты и 15,4 мл (0,134 моль) 35%-ного раствора гидроксида натрия, в результате чего получали 10,1 г (выход 72%) 2-аминосульфонил-6-втор-бутил-N,N-диэтилбензамида.
Последнее вещество (6,83 г, 0,22 моль) растворяли в 100 мл ледяной уксусной кислоты, раствор грели в сосуде с обратным холодильником в течение 13 ч и затем доводили до сухого состояния. Остаток растирали с диэтиловым эфиром и собирали фильтрованием, получая 5,7 г (выход 83%) диэтиламмониевой соли 4-вторбутилсахарина. Последнее вещество (3,0 г, 0,0096 моль) после взаимодействия с 1,13 мл (0,012 моль) хлорметилфенилсульфида в толуоле давало 3,47 г (выход 100%) 2-фенилтиометил-4-втор-бутилсахарина. После взаимодействия последнего вещества (3,2 г, 0,0097 моль) с 2,3 мл (0,029 моль) хлористого сульфурила в 20 мл двухлористого метилена получали 2,4 г (выход 87%) 2-хлорметил-4-втор-бутилсахарина.
Синтез 6B. По методике, аналогичной описанной в синтезе 6А, 9,2 г (32,9 моль) 3,4-диметокси-2-пропил-N,N-диэтилбензамида приводили во взаимодействие с диоксидом серы и 5,6 г (49,4 моль) гидроксиламин-0-сульфоновой кислоты, в результате чего получали 7,4 (выход 63%) 2-аминосульфонил-4,5-диметокси-6-пропил-N, N-диметилбензамида, который подвергали циклизации с количественным выходом в уксусной кислоте и фенилтиометилированию с использованием 1,42 мл (15 ммоль) хлорметилфенилсульфида, в результате чего получали 4,07 г 5,6-диметокси-2-фенилтиометил-4-пропилсахарина. Подвергая взаимодействию 3,59 г (8,8 ммоль) простого фенилтиоэфира с 2,12 мл (26,4 ммоль) хлористого сульфурила, получали 2,84 г (выход 97%) 2-хлорметил-5,6-диметокси-4-пропилсахарина.
3,4-Диметокси-2-пропил-N,N-диэтилбензамид получали следующим образом.
К раствору, содержащему 0,216 моль н-бутиллития в 250 мл простого эфира, при температуре окружающего воздуха добавляли по каплям 138,2 г (0,216 г) вератрола, растворенного в 100 мл простого эфира и 32,6 мл (0,216 моль) тетраметилендиамина. Реакционную смесь перемешивали при температуре окружающей среды в течение 14 ч и при охлаждении добавляли 21,9 мл (0,225 моль) иодистого н-пропила. Реакционную смесь перемешивали в течение 1 ч при комнатной температуре и обрабатывали 1 н. раствором HCl, в результате чего получали 14 г (выход 36%) 2,3-диметоксибензолпропана, который бромировали 14,52 (81,6 ммоль)N-бромсукцинимидом на 36 г кизельгеля в 400 мл CCl4 по методу Хисатоши (Hisatoshi) и др. (Bull Chem. Soc. Jap., 32, 591-593, 1989), в результате чего получали 19,6 г (выход 98%) 6-бром-2,3-диметоксибензолпропропана.
Бромбензол (14,2 г, 54,8 ммоль) растворяли в 200 мл простого эфира, охлаждали до -78oC и добавляли 25,2 мл (63 ммоль) 2,5 н. раствора н-бутиллития в гексане. Реакционную смесь нагревали до 0oC, выдерживали в течение 1 ч и охлаждали до -70oC, к ней добавляли 9 мл (71,2 ммоль) хлористого диэтилкарбамида. Реакционную смесь оставляли стоять при комнатной температуре, реакцию тушили насыщенным раствором хлорида аммония. После проведения экстракции и сушки продукт кристаллизовали из гексана, получая 9,5 г (выход 62%) 3,4-диметокси-2-пропил-N, N-диэтилбензамида, температура плавления составляла 65-67oC.
Синтез 6C.
По способу, аналогичному описанному в синтезе 6B, 10,75 г (30 ммоль) 6-аминосульфонил-3,4-диметокси-2-изопропил-N, N-диэтилбензамида подвергали циклизации, получая 6,43 г 5,6-диметокси-4-изопропилсахарина (температура плавления составляла 186-188oC; из смеси простого эфира с гексаном), 5 г (17,5 ммоль) которого фенилтиометилировали, воздействуя 2,48 мл (26,3 ммоль) хлористым фенилтиометилом, что делали по методике, описанной в синтезе 5, и хлорировали тремя эквивалентами хлористого сульфурила, в результате чего с выходом 85% получали 2-хлорметил-5,6-диметокси-4-изопропилсахарин; температура плавления составляла 117-119oC; из смеси этилацетата с гексаном.
Требуемый бензамид получали из 2,3-диметокси- α -метилбензолэтана, проводя бромирование с последующим карбамилированием, как это описано в синтезе 6B, в результате чего получали промежуточное соединение 3,4-диметокси-2-изопропил-N,N-диэтилбензамид. Раствор, состоящий из 66 мл 0,96 раствора втор-бутиллития, добавляли к 16,1 г (57,6 ммоль) бензамида в 400 мл тетрагидрофурана при -78oC в среде азота. После перемешивания в течение 2 ч оранжевый раствор по трубке подавали в избыточное количество диоксида серы, находящегося при -60oC. Реакционную смесь оставляли стоять при комнатной температуре и перемешивали в течение 18 ч с целью удаления диоксида серы. При 0oC добавляли несколько миллилитров хлористого сульфурила, и реакционную смесь подвергали разгонке. Хлористый сульфурил экстрагировали смесью этилацетата с простым эфиром, промывали водой, сушили и удаляли легкие компоненты. Остаток растворяли в 80 мл тетрагидрофурана и при 0oC добавляли 17 мл концентрированного гидроксида аммония. Реакционную смесь недолго перемешивали при комнатной температуре, удаляли легкие компоненты и растирали со смесью простого эфира с гексаном, взятых в соотношении 2:1, в результате чего получали 12,89 г (выход 62%) 6-аминосульфонил-3,4-диметокси-2-изопропил-N,N-диэтилбензамида; температура плавления составляла 138 - 140oC.
Синтез 7.
К раствору, состоящему из 9,3 мл (0,058 моль) тетраметилэтилендиамина в 340 мл тетрагидрофурана, при -78oC добавляли 52 мл 1,1 М раствора (0,057 моль) втор-бутиллития в циклогексане. Этот раствор затем обрабатывали раствором, состоящим из 11,37 г (0,052 моль) 2-пропил-N,N-диэтилбензамида в 75 мл тетрагидрофурана, при -78oC, раствор перемешивали в течение 15 мин и затем обрабатывали раствором, состоящим из 8,3 мл (0,104 моль) иодистого этила в тетрагидрофуране. Раствор перемешивали в течение 1 ч и 30 мин при -78oC и затем реакцию тушили добавлением насыщенного раствора хлорида аммония, который вводили по каплям при -78oC. Смеси затем давали нагреться до температуры окружающей среды, ее разбавляли диэтиловым эфиром, промывали сначала разбавленным раствором хлористоводородной кислоты, затем водой, затем насыщенным раствором бикарбоната натрия, затем рассолом, сушили и доводили до сухого состояния, в результате чего получали 12,91 г сырого продукта, который подвергали хроматографии на силикагеле, элюируя 10%-ным раствором этилацетата в гексане, что давало 3,23 г (выход 25%) 2-(3-пентил)-N, N-диэтилбензамида в виде желтой маслянистой жидкости.
Следуя методике, аналогичной описанной в синтезе 5, последнее вещество (3,05 г, 0,0115 моль) в тетрагидрофуране приводили во взаимодействие с 10,5 мл (0,126 моль) 1,2 М раствора втор-бутиллития в циклогексане в присутствии 2,1 мл (0,014 моль) тетраметилэтилендиамина. Образовавшуюся литиевую соль затем подвергали взаимодействию сначала с диоксидом серы, а затем с гидроксиламин-O-сульфонатом натрия, в результате чего получали 1,97 г (выход 52%) 2-аминосульфонил-6-(3-пентил)-N,N-диэтилбензамида в виде бледно-желтых кристаллов; температура плавления составляла 118 - 120oC (температура размягчения 102oC); 1,84 г этого продукта (0,0056 моль) подвергали циклизации в 22 мл ледяной уксусной кислоты при нагревании в сосуде с обратным холодильником, в результате чего получали 1,28 г (выход 70%) диэтиламмониевой соли 4-(3-пентил)сахарина; температура плавления составляла 107,5 - 109,5oC.
Из последнего вещества (0,0037 моль) после взаимодействия с 0,74 мл (0,0055 моль) хлорметилфенилсульфида в присутствии 116 мг (0,0004 моль) бромистого тетрабутиламмония, находящегося в 45 мл толуола, получали 1,93 г 2-фенилтиометил-4-(3-пентил)сахарина в виде бледно-желтой маслянистой жидкости, из 1,93 г (0,0037 моль) которой после взаимодействия с 0,59 мл (0,0073 моль) хлористого сульфурила в 37 мл двухлористого метилена получали 1,2 г 2-хлорметил-4-(3-пентил)сахарина в виде бледно-желтой маслянистой жидкости.
Синтез 8.
Раствор, состоящий из 50,0 г (0,27 моль) 2,4-диметоксибензойной кислоты в 60 мл (98,0 г, 0,82 моль) хлористого тионила, грели в сосуде с обратным холодильником на протяжении 3 ч, затем охлаждали и избыточные количества хлористого тионила отгоняли. Образовавшийся хлористый 2,4-диметоксибензоил растворяли в 150 мл двухлористого метилена, и раствор обрабатывали раствором, состоящим из 68 мл (48 г, 0,66 моль) диэтиламина в 500 мл двухлористого метилена, и охлаждали до 0oC. После завершения добавления смесь перемешивали в течение 15 ч при температуре окружающей среды, затем промывали насыщенным раствором бикарбоната натрия, водой и рассолом, доводили до сухого состояния, и остаток подвергали отгонке в вакууме, в результате чего получали 44,78 г (выход 69%) 2,4-диметокси-N,N-диэтилбензамида; температура кипения составляла 155 - 163oC при давлении 0,4 мм рт. ст.
Следуя методике, аналогичной описанной в синтезе 5, 10,0 г (0,042 моль) этого продукта, находящегося в 250 мл тетрагидрофурана, подвергали взаимодействию с 40,57 мл 1,1 М раствора (0,044 моль) втор-бутиллития в циклогексане и 6,35 мл (0,042 моль) тетраметилэтилендиамина в тетрагидрофуране. Образующуюся литиевую соль затем подвергали сначала взаимодействию примерно с 40 мл диоксида серы, а затем с водным раствором (0,13 моль) гидроксиламин-O-сульфоната натрия, в результате чего получали 8,26 г 2-аминосульфонил-4,6-диметокси-N,N-диэтилбензамида, 7,0 г (0,022 моль) которого подвергали циклизации в 80 мл ледяной уксусной кислоты, делая это при нагревании в сосуде с обратным холодильником, в результате чего получали 6,6 г (выход 94%) диэтиламмониевой соли 4,6-диметоксисахарина, который использовали на следующей стадии, не подвергая дополнительной очистке.
Из последнего вещества (6,0 г, 0,019 моль) после взаимодействия с 3,82 мл (0,028 моль) хлорметилфенилсульфида в присутствии 0,611 г (0,019 моль) бромистого тетрабутиламмония в 200 мл толуола получали 6,2 г (выход 89%) 2-фенилтиометил-4,6-диметоксисахарина, из 5,82 г (0,016 моль) которого после взаимодействия с 3,23 г (0,0019 моль) хлористого сульфурила в 100 мл двухлористого метилена получали 4,63 г (выход 100%) 2-хлорметил-4,6-диметоксисахарина; температура плавления составляла 185-187oC.
Синтезы 9А-9G.
Следуя методике, аналогичной описанной в синтезе 5, но используя надлежащий 2-R4-R5-замещенный-N, N-диэтилбензамид вместо 2-метокси-N,N-диэтилбензамида, получали через соответствующие 2-фенилтиометилсахарины следующие 2-галометил-4-R4-R5-сахарины, перечисленные в табл. 1. При наличии в колонках с названием "mp/Solv." ("температура плавления/растворитель") и "Yield" ("Выход") для каждого из 2-незамещенных сахаринов, 2-фенилтиометилсахаринов и 2-хлорметилсахаринов указывали температуру плавления, перекристаллизационный растворитель и выход. Во всех случаях промежуточные соединения 2-фенилтиометилсахарины использовали непосредственно на следующей стадии без определения характеристик и дополнительной очистки.
Синтез 10.
Следуя методике, аналогичной описанной в синтезе 2, взаимодействием 18,3 г (0,1 моль) сахарина с 70 мл 37%-ного раствора формалина в этаноле получали 3,58 г (выход 70%) 2-гидроксиметилсахарина. Последнее вещество (25 г, 0,117 моль) подвергали взаимодействию с 63,3 г (0,234 моль) трехбромистого фосфора в диэтиловом эфире, в результате чего получали 29,8 г (выход 92%) 2-бромметилсахарина; температура плавления составляла 155-157oC.
Синтез 11.
К раствору, состоящему из 4 г (0,0175 моль) 6-нитросахарина в 240 мл этанола, добавляли 4,4 г (0,0175 моль) этоксида таллия (3), и смеси позволяли стоять при комнатной температуре в течение 1 ч, ее держали в холодном состоянии в течение 16 ч, и осевшее твердое вещество собирали с сушили, в результате чего получали 6,6 г (выход 100%) таллиевой (3) соли 6-нитросахарина. Этот продукт суспендировали в 50 мл диметилформамида, и смесь обрабатывали 3,07 г (0,0194 моль) хлорметилфенилсульфида, смесь грели примерно при 63oC в течение 5 ч, оставляли стоять при температуре окружающей среды в течение примерно 16 ч, а затем выливали в воду со льдом. Сырой продукт, полученный фильтрованием, перемешивали в двухлористом метилене и фильтровали для удаления солей таллия (3). Фильтрат освобождали от растворителя, и полученное бледно-желтое твердое вещество подвергали ультразвуковому воздействию в теплом этаноле, его еще раз собирали и сушили, получая 4,6 г (выход 75%) 6-нитро-2-фенилтиометилсахарина; температура плавления составляла 161-163oC. Из последнего вещества после взаимодействия с хлористым сульфурилом в двухлористом метилене по методике, описанной в синтезе 4, получали 3,7 г 2-хлорметил-6-нитросахарина.
Синтез 12.
Раствор, состоящий из 49,8 г (0,199 моль) 2-гидрокси-5-(1,1,3,3-тетраметилбутил)бензойной кислоты в 200 мл метанола, нагревали до 50oC и затем по каплям обрабатывали примерно 80 г серной кислоты, делая это с такой скоростью, чтобы происходила конденсация компонентов реакционной смеси в обратном холодильнике. Реакционную смесь грели в сосуде с обратным холодильником еще в течение 11 ч, затем охлаждали и подвергали разделению между водой и этилацетатом. Органический слой промывали насыщенным раствором бикарбоната натрия, затем рассолом, сушили над сульфатом натрия и доводили до сухого состояния, получая 48,6 г (выход 92%) метил-2-гидрокси-5-(1,1,3,3-тетраметилбутил)бензоата.
Последнее вещество, растворенное в 250 мл диметилформамида, обрабатывали сначала 40,4 г (0,36 моль) 1,4-диазобицикло[2.2.2]октана, а затем 33,4 г (0,27 моль) N,N-диметилхлортиокарбамата и 100 мл диметилформамида. Реакционную смесь грели при 45oC в течение примерно 8 ч, выливали в воду со льдом и концентрированной хлористоводородной кислотой и затем экстрагировали этилацетатом. Объединенные органические экстракты промывали разбавленной хлористоводородной кислотой, затем бикарбонатом натрия и затем рассолом, сушили и доводили до полностью сухого состояния, получая 48,2 г (выход 76%) метил-2-(N, N-диметилтиокарбамилокси)-5-(1,1,3,3-тетраметилбутил)бензоата, который грели при 220oC в течение 15 ч, затем охлаждали, растворяли в толуоле и подвергали хроматографии на кремнеземе, элюируя смесью этилацетата с толуолом, взятым в соотношении 1:9, в результате чего получали 3,6 г (выход 14%) метил-2-(N,N-диметилкарбамилтио)-5-(1,1,3,3-тетраметилбутил)бензоата.
Раствор последнего вещества (0,025 моль) в 40 мл двухлористого метилена обрабатывали при перемешивании 80 мл ледяной уксусной кислоты, а затем 16 мл воды. Реакционную смесь охлаждали до 0oC, и через реакционную смесь в течение примерно 5 мин пробулькивали газообразный хлор, поддерживая при этом температуру в области от 5 до 24oC. Реакционную смесь перемешивали еще в течение 30 мин, концентрировали в вакууме, и образовавшийся раствор выливали в воду со льдом. Экстракцией смеси этилацетатом и выделением продукта из объединенных органических экстрактов получали 6,8 г (выход 78%) метил-2-хлорсульфонил-5-(1,1,3,3-тетраметилбутил)бензоата.
Продукт (9,0 г, 0,026 моль) растворяли в тетрагидрофуране и добавляли к 100 мл концентрированного гидроксида аммония при охлаждении на ледяной бане. Образовавшийся раствор перемешивали примерно в течение 16 ч, затем концентрировали в вакууме и концентрированный раствор подкисляли до величины pH 3, добавляя концентрированную хлористоводородную кислоту. Смесь перемешивали в течение нескольких часов, и выпавшее твердое вещество собирали, промывали водой и сушили, получая 9,0 г 5-(1,1,3,3-тетраметилбутил)сахарина; температура плавления составляла 213-215oC.
Следуя методике, аналогичной описанной в синтезе 11, 9,0 г (0,30 моль) этого продукта подвергали взаимодействию с этоксидом таллия в этаноле, и образовавшуюся соль таллия подвергали взаимодействию с 3,33 г (0,021 моль) хлорметилфенилсульфида в диметилформамиде, получая 5,76 г (выход 66%) 2-фенилтиометил-5-(1,1,3,3-тетраметилбутил)сахарина, 3,3 г (0,007 моль) которого обрабатывали 0,944 г хлористого сульфурила в двухлористом метилене, в результате чего получали 1 г (выход 41%) 2-хлорметил-5-(1,1,3,3-тетраметилбутил)сахарина.
Синтез 13.
Следуя методике, аналогичной описанной в синтезе 12, 15,5 г (0,086 моль) этил-2-гидрокси-6-метилбензоата подвергали взаимодействию с 15,9 г (0,129 моль) N, N-диметилхлортиокарбамата в присутствии 19,3 г (0,172 моль) 1,4-диазабицикло[2.2.2] октана в диметилформамиде, получая 22,1 г (выход 96%) этил-2-(N,N-диметилтиокарбамилокси)-6-метилбензоата, который грели при 220oC в течение примерно 10 ч. Этот продукт очищали хроматографией на силикагеле в двухлористом метилене, получая этил-2-(N,N-диметилкарбамилтио)-6-метилбензоат в виде красно-коричневой жидкости.
Раствор последнего вещества (22,6 г, 0,0844 моль) в 170 мл двухлористого метилена обрабатывали 340 мл ледяной уксусной кислоты и 68 мл воды в условиях охлаждения на бане из льда и ацетона, и через реакционную смесь в течение 10-15 мин пробулькивали газообразный хлор. Реакционный сосуд подвергали откачке для удаления избыточных количеств хлора и двухлористого метилена, смесь выливали в воду и производили разделение между двухлористым метиленом и водой. Из органического слоя после сушки и упаривания досуха получали 19 г этил-2-хлорсульфонил-6-метилбензоата, 5 г (0,019 моль) которого подвергали взаимодействию с концентрированным гидроксидом аммония в тетрагидрофуране, в результате чего получали 6,1 г (выход 67%) 4-метилсахарина.
Следуя методике, аналогичной описанной в синтезе 11, этот продукт (10,1 г 0,0512 моль) превращали в соль таллия взаимодействием с 12,8 г (0,0512 моль) этоксида таллия в этаноле, соль таллия подвергали взаимодействию с 6,7 г (0,0427 моль) хлорметилфенилсульфида в диметилформамиде, в результате чего получали 6,85 г (выход 50%) 2-фенилтиометил-4-метилсахарина. Подвергая последнее вещество (6,7 г, 0,021 моль) взаимодействию с хлористым сульфурилом в двухлористом метилене, получали 4,9 г (выход 95%) 2-хлорметил-4-метилсахарина.
Синтез 14А.
Смесь, состоящую из 75 г (0,36 моль) 3,3-дитиобиспропионовой кислоты, 102 мл хлористого тионила и каталитически достаточного количества пиридина, перемешивали в течение примерно 24 ч и затем упаривали досуха в вакууме. Остаток обрабатывали двухлористым метиленом и снова упаривали досуха, удаляя остаточные хлористый тионил и пиридин, в результате чего получали 87 г (выход 98%) соответствующего хлорида бискислоты, 44,8 г (0,18 моль) которого растворяли в тетрагидрофуране и по каплям добавляли к раствору, состоящему из 77,16 г (0,72 моль) бензиламина в тетрагидрофуране. Смесь перемешивали в течение 2 ч при 40-45oC, охлаждали и осевшее твердое вещество собирали, промывали водой и сушили, получая 59 г (выход 84%) N,N'-дибензилкарбоксамида 3,3-дитиобиспропионовой кислоты; температура плавления составляла 162-165oC.
Проводя взаимодействие 7,0 г (0,018 моль) последнего вещества с 10,25 г (0,076 моль) хлористого сульфурила в двухлористом метилене, получали смесь 2-бензил-2H-изотиазол-3-она и 5-хлор-2-бензил-2H-изотиазол-3-она, которые в основном отделяли друг от друга, проводя ультразвуковую обработку в двухлористом метилене (который стабилизирует большую часть первого вещества). Нерастворимое вещество собирали фильтрованием и подвергали хроматографии на силикагеле с двухлористым метиленом. Тем самым получали 5-хлор-2-бензил-2H-изотиазол-3-он; температура плавления составляла 58-68oC.
Раствор, состоящий из 10 г (0,044 моль) последнего вещества в двухлористом метилене, охлаждали до 0oC, и раствор обрабатывали 7,6 г (0,044 моль) 3-хлорнадбензойной кислотой, смесь перемешивали в течение 10 мин и затем обрабатывали второй порцией надбензойной кислоты, взятой в количестве 7,6 г. Реакционную смесь фильтровали, фильтр промывали двухлористым метиленом и фильтрат промывали насыщенным раствором бикарбоната натрия, затем рассолом, сушили над сульфатом натрия и доводили до сухого состояния, остаток подвергали хроматографии на силикагеле с использованием двухлористого метилена, причем продукт элюировали смесью гексана с двухлористым метиленом, взятыми в соотношении 50:50, в результате чего получали 7,15 г (выход 46%) 5-хлор-2-бензил-2H-изотиазол-3-он-1-оксида.
Раствор, состоящий из 1,1 г (0,0045 моль) последнего вещества в 8 мл бензола, обрабатывали 0,55 г (0,0051 моль) 2-метоксифурана, раствор грели в толстостенной склянке при 70oC в течение 1,5 ч и затем охлаждали, твердое вещество собирали, промывали бензолом и сушили, получая 2-бензил-7-гидрокси-4-метоксибензизотиазол-3-он-1-оксид; температура плавления составляла 235-237oC.
Смесь, состоящую из этого продукта (1,85 г, 0,006 моль), 2,48 г (0,018 моль) карбоната калия и 1,70 г (0,012 моль) иодистого метила в ацетоне, грели в сосуде с обратным холодильником в течение 1,5 ч и затем охлаждали и выливали в воду. Твердое вещество, которое выпадало, собирали фильтрованием, промывали водой и сушили, получая 1,70 г (выход 89%) 2-бензил-4,7-диметоксибензизотиазол-3-он-1-оксида, 1,13 г (0,0035 моль) которого окисляли 1,20 г (0,007 моль) 3-хлорнадбензойной кислоты в двухлористом метилене, делая это по методике, описанной выше, в результате чего получали 1,03 г (выход 88%) 2-бензил-4,7-диметоксисахарина.
Смесь, состоящую из 2,07 г (0,0062 моль) этого продукта, 1,37 г (0,02 моль) формата аммония и 1,5 г 10%-ного палладиевого катализатора, нанесенного на древесный активированный уголь, в 80 мл метанола, грели в сосуде с обратным холодильником в течение 1 ч, затем охлаждали и фильтровали, из фильтрата, доведенного до сухого состояния, получали 0,92 г (выход 57%) аммониевой соли 4,7-диметоксисахарина.
Раствор, состоящий из 1,11 г (0,0042 моль) аммониевой соли, растворяли в диметилформамиде, добавляли 0,67 г (0,0042 моль) хлорметилфенилсульфида, раствор грели в сосуде с обратным холодильником в течение 8 ч, затем охлаждали и выливали в воду со льдом. Твердое вещество, которое отделялось, собирали, промывали водой и сушили, получая 0,50 г (выход 33%) 2-фенилтиометил-4,7-диметоксисахарина. Проводя взаимодействие последнего вещества (0,5 г, 0,0013 моль) с хлористым сульфурилом в двухлористом метилене согласно методике, описанной в синтезе 4, получали 0,22 г (выход 58%) 2-хлорметил-4,7-диметоксисахарина.
Синтезы 14B и 14C.
Следуя методике, аналогичной описанной в синтезе 14А, приведенным ниже способом готовили другие производные 2-хлорметилсахарина.
Синтез 14B. Проводя взаимодействие 5,8 г (0,024 моль) 5-хлор-2-бензил-2H-изотиазол-3-он-1-оксида с 3,76 г (0,0335 моль) 2-этоксифурена, получали 3,05 г (выход 40%) 2-бензил-4-этокси-7-гидроксибензизотиазол-3-он-1-оксида, 5,7 г которого подвергли взаимодействию с 3,6 г (0,0197 моль) 2-[2-метоксиэтокси] этилового бромида в присутствии 4,95 г (0,0358 моль) карбоната калия в 125 мл метилэтилкетона и 25 мл диметилформамида, в результате чего получали 7,0 г (выход 95%) 2-бензил-4-этокси-7-[2-(2-метоксиэтокси)этокси] бензизотиазол-3-он-1-оксида, который, как и ранее, окисляли 3-хлорнадбензойной кислотой в двухлористом метилене, в результате чего получали 2-бензил-4-этокси-7-[2-(2-метоксиэтокси)этокси] сахарин. Проводя дебензилирование 6,6 г (0,015 моль) последнего вещества, 3,34 г (0,053 моль) формата аммония в присутствии 6,4 г 10%-ного палладиевого катализатора, нанесенного на активированный древесный уголь, в метаноле, получали аммониевую соль 4-этокси-7-[2-(2-метоксиэтокси)этокси] сахарина, который подвергали взаимодействию с 2,38 г (0,015 моль) хлорметилфенилсульфида в 100 мл диметилформамида, в результате чего получали 1,46 г (выход 21%) 2-фенилтиометил-4-этокси-7-[2-(2-метоксиэтокси)этокси] сахарина; температура плавления которого составляла 73-75oC (при кристаллизации из изопропанола). Обрабатывая 1,4 г (0,0029 моль) этого продукта 0,4 г (0,0029 моль) хлористого сульфурила в двухлористом метилене, получали 1,16 г (выход 100%) 2-хлорметил-4-этокси-7-[2-(2-метоксиэтокси)этокси]сахарина.
Синтез 14C. Проводя взаимодействие 3,03 г (0,01 моль) 2-бензил-7-гидрокси-4-метоксибензизотиазол-3-он-1-оксида (синтез 14A) с 2,01 г (0,011 моль) 2-(2-метоксиэтокси)этилового бромида в метилэтилкетоне в присутствии 2 г (0,015 моль) карбоната калия, получали 2,58 г (выход 64%) 2-бензил-4-метокси-7-[2-(2-метоксиэтокси)этокси]бензизотиазол-3-он-1- оксида, из которого при окислении 1,1 г (0,0063 моль) 3-хлорнадбензойной кислотой в двухлористом метилене получали 2-бензил-4-метокси-7-[2-(2-метоксиэтокси)этокси]сахарин. Дебензилированием 0,25 г (0,0006 моль) этого продукта 0,13 г (0,0021 моль) формата аммония в присутствии 0,25 г 10%-ного палладиевого катализатора, нанесенного на активированный древесный уголь, получали 0,21 г (выход 100%) аммониевой соли 4-метокси-7-[2-(2-метоксиэтокси)этокси] сахарина. Взаимодействием 1,4 г (0,004 моль) аммониевой соли с 0,63 г (0,004 моль) хлорметилфенилсульфида в диметилформамиде получали 2-фенилтиометил-4-метокси-7-[2-(2-метоксиэтокси)этокси] сахарин, из которого при взаимодействии с хлористым сульфурилом в двухлористом метилене получали 0,53 г (выход 35%) 2-хлорметил-4-метокси-7-[2-(2-метоксиэтокси)этокси]сахарина.
Синтез 15.
Раствор, состоящий из 1,89 г (0,011 моль) диэтиламинового трифторида серы в 20 мл двухлористого метилена, добавляли к суспензии, состоящей из 2,13 г (0,01 моль) 2-гидроксиметилсахарина в 25 мл двухлористого метилена, поддерживая температуру реакционной смеси на уровне -78oC.
Реакционную смесь перемешивали при -78oC в течение 1 ч, позволяя температуре медленно приближаться к температуре окружающей среды; смесь перемешивали в течение 16 ч и затем выливали в воду со льдом. Органический слой отделяли и промывали водой, сушили над сульфатом магния и доводили до сухого состояния, получая 2,2 г продукта, который перекристаллизовывали из этилацетата, в результате чего получали 1,6 г (выход 74%) 2-фторметилсахарина; температура плавления составляла 96-98oC.
Синтез 16A.
К раствору, состоящему из 0,5 г (0,0025 моль) 4-метилсахарина в тетрагидрофуране, который на бане из сухого льда с ацетоном был охлажден до -78oC, по каплям при перемешивании добавляли раствор, состоящий из 5,2 мл 1,3 М раствора втор-бутиллития в циклогексане. Смесь при -78oC дополнительно перемешивали еще в течение 1 ч и затем обрабатывали 0,16 мл (0,025 моль) иодистого метила, что делали на протяжении промежутка времени в 1,5 ч. Смесь перемешивали в течение 1 ч 45 мин, реакцию тушили, добавляя 25 мл 1 н. хлористоводородной кислоты, реакционной смеси придавали основной характер, водную смесь экстрагировали хлороформом и затем подкисляли и экстрагировали этилацетатом. Объединенные органические экстракты промывали 10%-ным раствором тиосульфата натрия, затем рассолом, сушили над сульфатом натрия и доводили до сухого состояния, в результате чего получали продукт, спектр протонного магнитного резонанса которого указывал на наличие смеси, состоящей из 74% 4-этилсахарина и 21% 4,7-диметилсахарина.
Следуя методике, аналогичной описанной в синтезе 4, сырое вещество (0,47 г, 0,0022 моль) подвергали взаимодействию с 0,24 мл (0,0028 моль) хлорметилфенилсульфида в толуоле в присутствии бромистого тетрабутиламмония, и продукт подвергали хроматографии на силикагеле, элюируя двухлористым метиленом, причем собирали фракции по 5 мл. Первые 420 мл элюата отбрасывали. Из следующих 20 фракций после упаривания получали 0,07 г вещества, преимущественно представляющего собой 2-фенилтиометил-4,7-диметилсахарин, которое откладывали. Из следующих 25 фракций получали 0,37 г 2-фенилтиометил-4-этилсахарина, который подвергали взаимодействию с хлористым сульфурилом в двуххлористом метилене, в результате чего получали 0,19 г (выход 66%) 2-хлорметил-4-этилсахарина.
Синтез 16B.
Следуя методике, аналогичной описанной в синтезе 16A, 10 г (0,051 моль) 4-метилсахарина в тетрагидрофуране подвергали взаимодействию с 86 мл (0,10 моль) 1,18 М раствора втор-бутиллития в циклогексане, образовавшийся раствор обрабатывали 4,6 мл (0,050 моль) иодистого этила, в результате чего получали 10, 15 г (выход 89%) 4-пропилсахарина, из которого при взаимодействии с 5,32 мл (0,056 моль) хлорметилфенилсульфида в толуоле в присутствии бромистого тетрабутиламмония получали сырую смесь, из которой испарительной хроматографией на силикагеле выделяли 2-фенилтиометил-4-пропилсахарин, имеющий вид маслянистой жидкости, из 1,8 г (0,0052 моль) которой при взаимодействии с 1,25 мл (0,016 моль) хлористого сульфурила в двухлористом метилене получали 0,94 г (выход 66%) 2-хлорметил-4-пропилсахарина.
Синтез 16C.
Предпочтительный иной способ проведения синтеза 16A состоит в следующем.
К раствору, состоящему из 5,13 г (25 ммоль) N,N, 2-триэтилбензамида в тетрагидрофуране (50 мл), при -78oC добавляли раствор ЛДА (Олдрич (Aldrich), 2,0 M раствор, 15,63 мл, 31,25 ммоль). Раствор нагревали до -10oC на водяной бане со льдом в течение часа, затем охлаждали до -78oC на бане из ацетона с сухим льдом. Точно при -78oC добавляли TMCCl (6,34 мл, 50 ммоль), затем через час температуру реакционной смеси поднимали до комнатной температуры. Реакцию тушили насыщенным раствором хлорида аммония и экстрагировали простым эфиром (два раза по 100 мл), сушили над сульфатом магния, удаляли легкие компоненты, и остаток подвергали разгонке в аппарате Кугельрора (130-140oC, давление 0,65 мм рт.ст.), в результате чего получали 6,51 г (выход 94%) N, N-диэтил-2-[1-(триметилсилил)этил]бензамида.
К раствору, состоящему из втор-бутиллития (0,97 M, 5,10 мл, 4,96 ммоль) и тетраметилендиамина (0,75 мл, 4,96 ммоль) в тетрагидрофуране, при -78oC добавляли амид (1,25 г, 4,50 ммоль), находящийся в тетрагидрофуране. Быстро при -78oC добавляли избыточные количества диоксида серы в тетрагидрофуране, затем раствор нагревали до комнатной температуры. Тетрагидрофуран удаляли в вакууме, и остаток обрабатывали при 0oC двумя эквивалентами раствора (при соотношении 1: 1) гидроксида натрия (0,36 г, 9,0 ммоль) и гидроксиламин-O-сульфоновой кислоты (1,0 г, 9,0 ммоль) в воде. Реакционную смесь перемешивали при комнатной температуре в течение 4 ч, экстрагировали этилацетатом, сушили над сульфатом магния, концентрировали и подвергали испарительной хроматографии на силикагеле при использовании 20%-ной смеси этилацетата в гексане, в результате чего получали 0,62 г (выход 41%) 2-аминосульфонил-N,N-диэтил-6-[1-(триметилсилил)этил] бензамида. Бензамид (0,95 г, 2,66 моль) грели в сосуде с обратным холодильником с ледяной уксусной кислотой (20 мл) в течение 18 ч, удаляли легкие фракции до достижения сухого состояния, растирали с горячим циклогексаном (30 мл) и следовыми количествами этилацетата (3 мл), охлаждали с растиранием и фильтровали. В результате получали 0,81 г (выход 85%) 4-[1-триметилсилил)этил]сахарина; температура плавления составляла 123-125oC.
К триметилсилилэтилсахарину (0,25 г, 0,70 ммоль), находящемуся в диметилформамиде (9 мл), при комнатной температуре добавляли воду (1 мл) и фторид цезия (0,75 г, 4,94 ммоль, 7 эквивалентов). Через 7 ч реакционную смесь выливали в 4%-ный раствор гидроксида натрия и экстрагировали этилацетатом. Водный слой подкисляли 12 н. хлористоводородной кислотой и экстрагировали смесью диэтилового эфира с этилацетатом (взятыми в соотношении 1:1), сушили над сульфатом натрия, фильтровали и удаляли легкие компоненты, в результате чего с количественным выходом получали бесцветное твердое вещество. Перекристаллизацией из 5%-ной смеси диэтилового эфира с гексанами получали 0,091 г (выход 64%) 4-этилсахарина; температура плавления составляла 183-185oC.
Синтез 17.
Образец вещества в количестве 0,07 г, полученного из ранних фракций при проведении хроматографического разделения, описанного в синтезе 16A, которое преимущественно представляло собой 2-фенилтиометил-4,7-диметилсахарин, подвергали взаимодействию с 0,05 мл хлористого сульфурила в двухлористом метилене, продукт перекристаллизовывали из смеси циклогексана с этилацетатом, в результате чего получали 20 мг (выход 51%) 2-хлорметил-4,7-диметилсахарина; температура плавления составляла 107 - 108oC.
Синтез 18A.
К раствору, состоящему из 40,0 г (0,174 моль) 2-изопропил-4-метоксибромбензола в 600 мл диэтилового эфира, при 0oC добавляли 103,68 мл (0,175 моль) 1,69 М раствора бутиллития в диэтиловом эфире. После завершения добавления раствор охлаждали до 0oC в течение 1 ч, перемешивание продолжали еще 5 ч, делая это при температуре окружающей среды, затем раствор охлаждали до -78oC и обрабатывали раствором, состоящим из 23,68 г (0,175 моль) N,N-диэтилкарбамилхлорида, находящегося в 80 мл диэтилового эфира. Образовавшийся раствор перемешивали в течение примерно 12 ч, позволяя температуре реакционной смеси повышаться, затем реакцию тушили, добавляли насыщенный раствор хлорида аммония. Водный и органический слои разделяли, водный слой обратно экстрагировали этилацетатом, и соединенные органические экстракты промывали один раз рассолом, затем сушили, из раствора, доведенного до сухого состояния, получали сырой продукт, который подвергали хроматографии на силикагеле, элюируя 30%-ным раствором этилацетата в гексане, в результате чего получали 34,4 г (выход 79%) 2-изопропил-4-метокси-N,N-диэтилбензамида, имевшего вид маслянистой жидкости, которую использовали как таковую на следующем этапе, не подвергая дополнительной очистке. Маслянистая жидкость может быть, при желании, разогнана; при давлении 0,2 - 0,3 мм рт.ст. она кипит при температуре 123 - 129oC.
Следуя методике, аналогичной описанной в синтезе 5, последнее вещество (15,0 г, 0,060 моль), растворенное в 100 мл диэтилового эфира, подвергали взаимодействию с 77,8 мл (0,784 моль) 1,2 М раствора втор-бутиллития в циклогексане в присутствии 6,98 г (0,06 моль) тетраметилэтилендиамина. Образовавшуюся литиевую соль затем подвергали взаимодействию сначала с 50 мл диоксида серы, а затем с 0,181 моль гидроксиламин-O-сульфоната натрия, в результате чего получали 11,6 г (выход 59%) 2-аминосульфонил-6-изопропил-4-метокси- N, N-диэтилбензамида; температура плавления составляла 103 - 105oC (при кристаллизации из смеси этилацетата с гексаном). Бензамид в количестве 11 г (0,034 моль) подвергали циклизации в 200 мл ледяной уксусной кислоты с нагреванием в сосуде с обратным холодильником, в результате чего получали 10,3 г диэтиламмониевой соли 4-изопропил-6-метоксисахарина; температура плавления составляла 132 - 135oC.
Из последнего вещества (0,030 моль), подвергнув его взаимодействию с 6,14 мл (7,25 г, 0,046 моль) хлорметилфенилсульфида в присутствии 0,98 г (0,003 моль) бромистого тетрабутиламмония, находящимися в 250 мл толуола, получали 10,1 г (выход 88%) 2-фенилтиометил-4-изопропил-6-метоксисахарина, имевшего вид маслянистой жидкости, из 9,7 г (0,026 моль) которого при взаимодействии с 3,1 мл (5,21 г, 0,039 моль) хлористого сульфурила в двуххлористом метилене получали 6,9 г (выход 88%) 2-хлорметил-4- изопропил-6-метоксисахарина; температура плавления составляла 151 - 152oC.
Синтез 18B.
Использовали также иную методику проведения синтеза. К раствору, состоящему из 300 мл N,N,N',N'-тетраметилэтилендиамина (1,99 моль) в 4 л безводного простого эфира, добавляли 1550 мл втор-бутиллития (при концентрации 1,3 М), систему охлаждали до -70oC в среде азота. Раствор, состоящий из 454,2 г 2-изопропил-4-метокси-N, N-диэтилбензамида (1,82 моль) в 300 мл безводного простого эфира, добавляли по каплям, делая это на протяжении 30 мин (при добавлении температуру поддерживали на уровне -60oC или менее). После завершения добавления реакционную смесь перемешивали при -70oC в течение 1 ч, смеси давали нагреться до -50oC. После выдержки при -50oC в течение 30 мин смесь снова охлаждали до -70oC. К этому перемешиваемому раствору по трубке добавляли раствор, состоящий из 200 г диоксида серы в 200 мл сухого простого эфира, предварительно охлажденного до -40oC, причем происходило это под избыточным давлением азота на протяжении промежутка времени в 20 мин. Температуру реакционной смеси при добавлении поддерживали ниже -40oC. (Почти сразу начинал выпадать белый порошкообразный ариллитиевый сульфинат). После добавления баню со льдом удаляли, реакционную смесь перемешивали при температуре окружающей среды в течение 2 ч. Смесь охлаждали до -5oC, к этому перемешиваемому раствору добавляли 190 мл хлористого сульфурила (2,36 моль), делая это по каплям на протяжении промежутка времени в 15 мин с поддержанием температуры при добавлении ниже 10oC. После дополнительного перемешивания в течение 30 мин при 0 - 5oC белый нерастворимый осадок отфильтровывали и промывали 2 л безводного простого эфира. Удалением растворителя при атмосферном давлении получали хлористый сульфонил в виде сырой темной маслянистой жидкости. Этот сырой хлористый сульфонил растворяли в 1,4 л тетрагидрофурана, охлаждали до -10oC, 540 мл концентрированного гидроксида аммония (с концентрацией 28%) добавляли порциями на протяжении промежутка времени в 15 мин (на протяжении всего периода добавления температуру поддерживали на уровне 15oC или менее). После перемешивания в течение 15 мин при температуре окружающей среды тетрагидрофуран и избыточный аммиак удаляли под вакуумом, в результате чего получали темную маслянистую жидкость, которую разбавляли 6,0 л воды и подкисляли 3 н. раствором хлористоводородной кислоты до величины pH 1. Светло-желтое твердое вещество собирали фильтрованием и промывали 800 мл воды. Твердое вещество сушили при 60oC под вакуумом в течение 18 ч и подвергали перекристаллизации из смеси 800 мл этилацетата и 3 л гексана, в результате чего получали 429 г (выход 72%) 2-аминосульфонил-6-изопропил-4-метокси-N,N-диэтилбензамида; температура плавления составляла 122-125oC.
Раствор, состоящий из 429,6 г диэтилбензамида (1,31 моль) в 1,5 г уксусной кислоты, грели в сосуде с обратным холодильником в течение 20 ч. Его охлаждали до комнатной температуры, растворитель удаляли под вакуумом. Маслянистый остаток растворяли в 6 л воды, величину pH доводили 6 н. хлористоводородной кислотой до единицы. Сырой продукт собирали фильтрованием и промывали 2 л воды. Твердое вещество сушили при 60oC под вакуумом в течение 18 ч и перекристаллизовывали из смеси этилацетата с гексаном, в результате чего получали 303 г (выход 91%) 4-изопропил-6-метоксисахарина; температура плавления составляла 188oC.
К суспензии, состоящей из 24 г п-формальдегида (0,8 моль) и 86,4 г хлортриметилсилана (1,6 моль) в 200 мл 1,2-дихлорэтана, добавляли 0,8 мл безводного хлорида олова (IV), и образовавшийся раствор перемешивали на паровой бане в течение 1 ч. В конце этого промежутка времени 51 г 4-изопропил-6-метоксисахарина (0,2 моль) добавляли к прозрачному раствору, реакционную смесь дополнительно грели в сосуде с обратным холодильником еще в течение 18 ч. Ее охлаждали до комнатной температуры, выливали в воду, органический слой отделяли и промывали 50 мл 2 н. раствора гидроксида натрия. Органический слой сушили над сульфатом магния и концентрировали под вакуумом, получая сырой продукт. Его очищали кристаллизацией из смеси этилацетата с гексаном, в результате чего получали 57 г (выход 87%) 2-хлорметил-4-изопропил-6-метоксисахарина; температура плавления составляла 151oC.
Синтез 19.
К раствору, состоящему из 1,0 г (0,0039 моль) 4-изопропил-6-метоксисахарина в 15 мл двухлористого метилена, добавляли при температуре окружающей среды 1,28 г (5,12 мл) 1 М раствора трибромида бора в двухлористом метилене. После завершения добавления реакционную смесь грели в сосуде с обратным холодильником в течение примерно 5 ч, охлаждали, доводили до сухого состояния в вакууме, остаток обрабатывали льдом и насыщенным раствором бикарбоната натрия. Водный раствор экстрагировали один раз этилацетатом и затем подкисляли до величины pH 1 концентрированной хлористоводородной кислотой. Проводя экстракцию смеси смесью этилацетата с диэтиловым эфиром (взятыми в соотношении 8:2), сушку органических экстрактов и удаление растворителя в вакууме, получали 0,9 г (выход 96%) 6-гидрокси-4-изопропилсахарина в виде белого кристаллического вещества, которое на следующем этапе использовали как таковое.
Использовали также иную методику синтеза. К перемешиваемой суспензии, состоящей из 62,74 г (0,47 моль) хлорида алюминия в 500 мл хлороформа, при 0oC добавляли 43,9 г (0,7 моль) этантиола. За несколько минут образовывался прозрачный раствор. К этому раствору добавляли раствор, состоящий из 20,0 г (0,078 моль) 4-изопропил-6-метоксисахарина в 550 мл хлороформа, что делали на протяжении промежутка времени в 30 мин. Этому раствору давали нагреться до комнатной температуры, и его перемешивали в течение 3-4 ч при 60oC. После охлаждения смесь выливали в воду со льдом и подкисляли разбавленной хлористоводородной кислотой. Твердое вещество, которое выпадало, собирали фильтрованием, промывали водой и сушили, получая 18,4 г (выход 97%) 6-гидрокси-4-изопропилсахарина.
Следуя методике, аналогичной описанной в синтезе 4, последнее вещество (0,004 моль) подвергали взаимодействию с 0,61 мл (0,0046 моль) хлорметилфенилсульфида в толуоле в присутствии 0,133 г (0,004 моль) бромистого тетрабутиламмония, в результате чего получали 0,32 г (выход 21%) 4-изопропил-6-гидрокси-2-фенилтиометилсахарина; температура плавления составляла 127-129,5oC. Это вещество в количестве 1,78 г обрабатывали 0,43 мл (0,73 г) хлористого сульфурила в двухлористом метилене, в результате чего получали 1,2 г (выход 84%) 2-хлорметил-4-изопропил-6-гидроксисахарина; температура плавления составляла 149-150oC.
Синтез 22.
Вещество 6-гидрокси-4-изопропилсахарин в количестве 5 г (0,0207 моль) растворяли в 150 мл метанола и добавляли 3,4 г (0,0104 моль) карбоната цезия. Смесь перемешивали в течение 3-4 ч при комнатной температуре. Избыточный метанол удаляли при пониженном давлении, остаток сушили в течение 2 ч при глубоком вакууме. Остаток затем растворяли в 110 мл диметилформамида и добавляли 0,32 г (0,0209 моль) хлорметилфенилсульфида. Перемешиваемую смесь грели при 70-75oC в течение 12 ч, охлаждали, обрабатывали водой со льдом и экстрагировали 600 мл смеси этилацетата с простым эфиром, взятыми в соотношении 4:1. Органический слой промывали водой, насыщенным раствором хлорида натрия и сушили. Растворитель удаляли при пониженном давлении. Остаток очищали испарительной хроматографией, элюируя 20%-ным раствором простого эфира в двухлористом метилене. Получали 4,5 г (выход 60%) 4-изопропил-6-гидрокси-2-фенилтиометилсахарина; температура плавления составляла 150-151oC; это вещество подвергали взаимодействию с хлористым сульфурилом, как это описано в синтезе 19, в результате чего получали, как и ранее, 2-хлорметил-4-изопропил-6-гидроксисахарин.
Синтез 23.
К раствору 5-хлор-2-бензил-4-изотиазолин-3-он (J Het. Chem., 8, 571, 1971) (9,4 г, 0,04 моль) в двухлористом метилене (100 мл) добавляли одну порцию 80-85%-ной 3-хлорпероксибензойной кислоты (10,8 г, 0,06 моль), образовавшуюся смесь перемешивали при комнатной температуре на протяжении ночи в среде азота. Осевшие твердые вещества отфильтровывали и промывали двухлористым метиленом (50 мл). Объединенный фильтрат упаривали почти до сухого состояния, остаток парционировали между этилацетатом (300 мл) и насыщенным раствором бикарбоната натрия (100 мл). Слои разделяли, и органическую фазу промывали насыщенным раствором бикарбоната натрия (2 раза по 100 мл), рассолом (1 раз по 100 мл) и сушили. Удаляя растворитель в вакууме, получали 10,0 г (выход 99%) 5-хлор-2-бензил-4-изотиазолин-3(2H)-он-1- оксида в виде бледно-желтой маслянистой жидкости.
Соединение 1-оксид (10,0 г, 0,04 моль), находящееся в ледяной уксусной кислоте (200 мл), обрабатывали 30%-ным пероксидом водорода (100 мл, 0,88 моль) и грели на паровой бане в течение 2 ч, причем в этот промежуток времени дополнительно вводили 30 мл (0,26 моль) 30%-ного раствора пероксида водорода. После прогрева на паровой бане еще в течение 1 ч реакционную смесь охлаждали до комнатной температуры и выливали в охлажденную льдом воду (1 л); снова перемешивали. Выпавшие твердые вещества собирали фильтрованием, промывали водой (2 раза по 100 мл), гексанами и сушили на воздухе, в результате чего получали 4,8 г (выход 45%) 5-хлор-2-бензил-4-изотиазолин-3(2H)-он-1,1-диоксида, имевшего вид бесцветного твердого вещества.
Диоксид (1,2 г, 4,7 ммоль) смешивали с 2,02 (11 ммоль) 2-триметилсилилокси-5-метилгекса-1,3-диена (приготовленного из 5-метил-гекс-3-ена по способу Кори (E.J. Corey) и др. Tet. Lett. 495, 1984) в толуоле (50 мл) и грели в сосуде с обратным холодильником в течение 30 ч в среде азота. Образовавшуюся смесь охлаждали до комнатной температуры и концентрировали в вакууме. Остаток растворяли в тетрагидрофуране (25 мл) и обрабатывали 2 н. раствором хлористоводородной кислоты (10 мл). После перемешивания в среде азота при комнатной температуре в течение 10 мин добавляли простой эфир (100 мл), и слои разделяли. Органическую фазу промывали водой, рассолом, сушили и упаривали до сухого состояния, получая бледно-желтое пенистое вещество. Это вещество растворяли в толуоле (30 мл), добавляли DBN (1,5 мл) и перемешивали при комнатной температуре в течение 2 ч. Добавляли двухлористый метилен (100 мл) и 2 н. раствор хлористоводородной кислоты (50 мл), перемешивание продолжали в течение 5 мин. Слои разделяли, и органическую фазу промывали водой, рассолом и сушили. Удалив растворитель в вакууме и подвергнув остаток очистке испарительной хроматографией на силикагеле (элюировали смесью, состоящей из гексанов, двухлористого метилена и простого эфира, взятых в соотношении 5: 4: 1), получали 0,6 г (выход 39%) 2-бензил-4-изопропил-6-оксотетрагидросахарина в виде бледно-желтого пенистого вещества.
Тетрагидросахарин (0,59 г, 1,7 ммоль) растворяли в толуоле (50 мл), добавляли диметиламингидрохлорид (1,5 г, 18,0 ммоль) и сита 4 А (2,0 г). Образовавшуюся смесь грели в сосуде с обратным холодильником в условиях азеотропного удаления воды в течение 96 ч. На протяжении этого промежутка времени в 96 ч через каждые 12 ч приходилось дополнительно вводить диметиламингидрохлорид (0,8 г, 10,0 ммоль) и молекулярные сита 4 А; в конце этого промежутка времени реакционную смесь охлаждали до комнатной температуры и фильтровали. Фильтровальную лепешку промывали диэтиловым эфиром (100 мл), и объединенные фильтраты концентрировали в вакууме, получая 0,63 г (выход 99%) 2-бензил-4-изопропил-6-диметиламино(4,5)-дигидросахарина в виде бледно-желтого твердого вещества.
К раствору дигидросахарина (0,63 г, 1,7 ммоль) в хлороформе в условиях нагревания с конденсацией последнего в обратном холодильнике (50 мл) добавляли активированный диоксид марганца (4,3 г, 49,5 ммоль), делая это порциями на протяжении промежутка времени в 4 ч. После добавления последней порции диоксида марганца реакционную смесь грели в сосуде с обратным холодильником еще в течение часа, охлаждали до комнатной температуры и фильтровали через набивку из суперцеля, элюируя этилацетатом. Объединенные элюенты концентрировали в вакууме, и остаток очищали испарительной хроматографией на силикагеле (элюировали смесью гексанов с двухлористым метиленом и простым эфиром, взятыми в соотношении 5:4:1), в результате чего получали 0,32 г (выход 50%) 2-бензил-4-изопропил-6-диметиламиносахарина, имевшего вид бесцветного твердого вещества.
2-Бензилсахарин (0,32 г, 0,9 ммоль), находящийся в метаноле (20 мл), обрабатывали форматом аммония (0,24 г, 3,8 ммоль) и 10%-ным палладиевым катализатором, нанесенным на углерод (0,25 г), грели в сосуде с обратным холодильником в течение 1 ч, охлаждали до комнатной температуры и фильтровали через набивку из суперцеля, элюируя метанолом (100 мл). Объединенные элюенты концентрировали в вакууме. Остаток растворяли в двухлористом метилене (10 мл), добавляли ледяную уксусную кислоту (0,25 мл), перемешивали в течение 5 мин и упаривали до сухого состояния в вакууме, в результате чего получали 0,25 г (выход 100%) 4-изопропил-6-диметиламиносахарина в виде бесцветного пенистого вещества.
Следуя методике, аналогичной описанной в синтезе 1, смесь, состоящую из 4-изопропил-6-диметиламиносахарина (0,27 г,1 ммоль), хлорметилфенилсульфида (0,32 г, 2,0 ммоль) и бромистого тетрабутиламмония (0,1 г, 0,2 ммоль) в толуоле, превращали в 0,22 г (выход 56%) 2-фенилтиометил-4-изопропил-6-диметиламиносахарина, который обрабатывали хлористым сульфурилом (1,86 мл 0,31 М раствора, 0,6 ммоль), в результате чего получали желтую смолу, на 25% (по данным ядерного магнитного резонанса) состоящую из 2-хлорметил-4-изопропил-6-диметиламино-7-хлорсахарина.
Синтез 28А.
Соединение 4-изопропил-1,2-диметоксибензол, взятое в количестве 31 г, обрабатывали N-бромсукцинимидом, после чего воздействовали бутиллитием и хлористым диэтилкарбамилом, как это делали в синтезе 6B, в результате чего получали 15,2 г 2-изопропил-4,5-диметокси-N,N-диэтилбензамида, имевшего вид вязкой маслянистой жидкости. Бензамид обрабатывали так, как это делали в синтезе 18B, воздействуя бутиллитием и диоксидом серы, после чего хлористым сульфурилом, а затем аммиаком, в результате чего получали 4,5 г сульфонамида; температура плавления составляла 181 - 182oC при кристаллизации из простого эфира. Это вещество подвергали циклизации в уксусной кислоте, как это делали в синтезе 18B, в результате чего получали 2,86 г 6,7-диметокси-4-изопропилсахарина; температура плавления составляла 210 - 212oC при кристаллизации из смеси этилацетата с гексаном.
К раствору, состоящему из 0,5 г 4-изопропил-6,7-диметоксисахарина, находящегося в 3 мл диметилформамида, добавляли 0,5 мл диизопропилэтиламина, что делали при комнатной температуре. По истечении 15 мин добавляли 0,35 г хлорметилфенилсульфида, и смесь грели при 80oC в течение 16 ч. Реакционную смесь выливали в этилацетат и промывали водным раствором карбоната натрия, водным раствором 2 н. хлористоводородной кислоты, насыщенным водным раствором хлорида натрия. Органический слой сушили над сульфатом натрия, растворители удаляли. Хроматографическим способом с использованием двухлористого метилена получали 0,35 г требуемого продукта, который сразу же использовали. Проводя обработку 0,35 г образца фенилтиометилсахарина в 3 мл двухлористого метилена 0,1 мл хлористого сульфурила в течение 30 мин при 20oC с последующим удалением растворителей и растиранием с гексаном, получали 0,3 г 2-хлорметил-6,7-диметокси-4-изопропилсахарина.
Синтез 28B.
К раствору, состоящему из 5,7 г метилпиперонилата, находящегося в 20 мл сухого простого эфира, добавляли 30 мл бромистого метилмагния с концентрацией в простом эфире 3,0, что делали при 0oC в течение промежутка времени в 20 мин. Смесь перемешивали в течение 20 ч, затем разбавляли 200 мл простого эфира и промывали водой. Органический слой сушили над сульфатом натрия, растворители удаляли, получая 5,6 г сырого 3,4-диметокси-(1'-гидрокси-1'-метилэтил)бензола. Это вещество сразу же обрабатывали в 50 мл уксусной кислоты 1 г 10%-ного палладиевого катализатора на углероде, что делали при давлении 344,5 кН/м2 (50 фунт/кв.дюйм) у водорода в течение 20 ч. Катализатор удаляли фильтрованием, после удаления растворителя получали 4,5 г 5-изопропил-1,3-бензодиоксола. Изопропилдиоксол бромировали, амидировали, сульфонировали и подвергали циклизации, как это делали в синтезе 28A, в результате чего получали 700 мг 4-изопропил-6,7-метилендиоксисахарина; температура плавления составляла 226 - 228oC при кристаллизации из смеси этилацетата с гексаном. Сахарин в количестве 500 мг подвергали хлорметилированию, как это делали в синтезе 28A, в результате чего получали 300 мг 2-хлорметил-4-изопропил-6,7-метилендиоксисахарина, температура плавления составляла 174 - 176oC.
Другие 4-R4-R5-сахарины с формулой II, используемые в качестве промежуточных продуктов при получении соединений с формулой I, могут быть получены следующим образом.
Проводя взаимодействие 2-трифторметилбензойной кислоты с хлористым тионилом, получают 2-трифторметилбензоилхлорид, который при взаимодействии с диэтиламином дает 2-трифторметил-N,N-диэтилбензамид. Следуя методике, аналогичной описанной в синтезе 5, при взаимодействии последнего вещества с втор-бутиллитием и взаимодействии образовавшейся литиевой соли с диоксидом серы с последующим воздействием гидроксиламин-O-сульфоната натрия получают 2-трифторметил-6-аминосульфонил-N,N-диэтилбензамид, который при нагревании в ледяной уксусной кислоте дает 4-трифторметилсахарин.
Аналогичным образом, проводя взаимодействие 2-трихлорметилбензойной кислоты с хлористым тиолом, получали 2-трихлорметилбензоилхлорид, из которого при взаимодействии с диэтиламином получали 2-трихлорметил-N,N-диэтилбензамид. Следуя методике, аналогичной описанной в синтезе 5, взаимодействием последнего вещества с втор-бутиллитием и взаимодействием образовавшейся литиевой соли с диоксидом серы с последующим воздействием гидроксиламина-O-сульфоната натрия получали 2-трихлорметил-6-аминосульфонил-N,N-диэтилбензамид, из которого при нагревании в ледяной уксусной кислоте получали 4-трихлорметилсахарин.
Взаимодействием 4-циклогексилбензойной кислоты с хлористым тионилом получали 4-циклогексилбензоилхлорид, из которого при взаимодействии с диэтиламином получали 4-циклогексил-N,N-диэтилбензамид. Следуя методике, аналогичной описанной в синтезе 5, взаимодействием последнего вещества с втор-бутиллитием и взаимодействием образовавшейся литиевой соли с диоксидом серы с последующим воздействием гидроксиламин-O-сульфоната натрия получали 4-циклогексил-2-аминосульфонил-N,N-диэтилбензамид, из которого при нагревании в ледяной уксусной кислоте получали 6-циклогексилсахарин.
Взаимодействием 2-бензил-6-аминосахарина с хлористым метансульфонилом, хлористым трифторметилсульфонилом или хлористым трихлорметилсульфонилом в двухлористом метилене в присутствии пиридина с последующим передаточным гидрогенолизом 2-бензильной блокирующей группы соответственно получали 6-метилсульфониламиносахарин, 6-трифторметилсульфониламиносахарин или 6-трихлорметилсульфониламиносахарин.
Диазотированием 6-аминосахарина с воздействием азотистой кислоты в кислой среде и разложением образовавшейся диазониевой соли в присутствии цианида меди (2) или хлорида меди (2) и диоксида серы, или хлорида меди (2) и щелочной соли метилмеркаптана или трифторметилмеркаптана соответственно получали 6-цианосахарин, 6-хлорсульфонилсахарин, 6-метилтиосахарин или 6-трифторметилтиосахарин. Взаимодействием 6-хлорсульфонилсахарина в момент его образования с аммиаком или метансульфониламидом соответственно получали 6-аминосульфонилсахарин и 6-метансульфониламиносульфонилсахарин. Окислением 6-метилтиосахарина и 6-трифторметилтиосахарина двумя молярными эквивалентами 3-хлорнадбензойной кислоты соответственно получали 6-метилсульфонилсахарин и 6-трифторметилсульфонилсахарин.
Гидролизом 6-цианосахарина посредством нагревания с водным раствором гидроксида натрия получали сахарин-6-карбоновую кислоту. Взаимодействием 6-цианосахарина в условиях нагревания с каталически достаточным количеством серной кислоты в этанольном растворе получали этилсахарин-6-карбоксилат, из которого при восстановлении боргидридом лития получали 6-гидроксиметилсахарин. Окислением последнего вещества пиридин-триоксидхромовым (2 : 1) комплексом (реактив Коллинза (Collins)) в двухлористом метилене получали 6-формилсахарин, из которого при восстановительном аминировании аммиаком и цианоборгидридом натрия получали 6-аминометилсахарин.
Взаимодействием 4-трифторметилбензойной кислоты с хлористым тионилом получали хлористый 4-трифторметилбензоил, из которого при взаимодействии с диэтиламином получали 4-трифторметил-N, N-диэтилбензамид. Следуя методике, аналогичной описанной в синтезе 5, взаимодействием последнего вещества с втор-бутиллитием и взаимодействием образовавшейся литиевой соли с диоксидом серы с последующим воздействием гидроксиламин-O-сульфоната натрия получали 4-трифторметил-2-аминосульфонил-N, N-диэтилбензамид, из которого при нагревании в ледяной уксусной кислоте получали 6-трифторметилсахарин.
Взаимодействием 4-трифторметилбензойной кислоты с хлористым тионилом получали хлористый 4-трихлорметилбензоил, из которого при взаимодействии с диэтиламином получали 4-трихлорметил-N, N-диэтилбензамид. Следуя методике, аналогичной описанной в синтезе 5, взаимодействием последнего вещества с втор-бутиллитием и взаимодействием образовавшейся литиевой соли с диоксидом серы с последующим воздействием гидроксиламин-O-сульфоната натрия получали 4-трихлорметил-2-аминосульфонил-N, N-диэтилбензамид, из которого при нагревании в ледяной уксусной кислоте получали 6-трихлорметилсахарин.
Взаимодействием 2-этенилбензойной кислоты с хлористым тионолом получали 2-этенилбензоилхлорид, из которого при взаимодействии с диэтиламином получали 2-этенил-N, N-диэтилбензамид. Взаимодействием последнего вещества с втор-бутиллитием и взаимодействием образовавшейся литиевой соли с диоксидом серы с последующим воздействием гидроксиламин-O-сульфоната натрия получали 2-этенил-6-аминосульфонил-N, N-диэтилбензамид, из которого при нагревании в ледяной уксусной кислоте получали 4-этиленсахарин.
Взаимодействием 2-этенил-6-аминосульфонил-N,N-диэтил-бензамида с бромом получали 2-(1,2-дибромэтил)-6-аминосульфонил-N,N-диэтилбензамид, из которого при взаимодействии с амидом натрия в аммиаке получали 2-этинил-6-аминосульфонил-N, N-диэтилбензамид, из которого при нагревании в ледяной уксусной кислоте получали 4-этинилсахарин.
Взаимодействием этил-2-аминобензоата с двумя молярными эквивалентами хлористого бензила в ацетоне в присутствии карбоната калия получали 2-(N, N-дибензиламино)бензоат, из которого при омылении в водном этанольном растворе гидроксида калия и выделении продукта из нейтральной среды получали 2-(N,N-дибензиламино)бензойную кислоту.
Взаимодействием последнего вещества с хлористым тионилом получали 2-(N, N-дибензиламино)бензоилхлорид, из которого при взаимодействии с диэтиламином получали 2-(N, N-дибензиламино)-N,N-диэтилбензамид. Взаимодействием последнего вещества с втор-бутиллитием и взаимодействием образовавшейся литиевой соли с диоксидом серы с последующим воздействием гидроксиламин-O-сульфоната натрия получали 2-(N, N-дибензил)-6-аминосульфонил-N, N-диэтилбензамид, из которого при нагревании в ледяной уксусной кислоте получали 4-(N,N-дибензиламино)сахарин, из которого при каталитическом дебензилировании с воздействием водорода на палладиевом катализаторе, нанесенном на древесный уголь, получали 4-аминосахарин. Восстановительным алкилированием последнего вещества одним молярным эквивалентом формальдегида в муравьиной кислоте получали 4-метиламиносахарин.
Взаимодействием 4-изопропил-6-гидроксисахарина (синтез 19) с N,N-диэтилтиокарбамилхлоридом в диметилформамиде, что делали по методике, описанной в синтезе 12, получали 4-изопропил-6-(N,N-диэтилтиокарбамилокси)сахарин, из которого при нагревании с протеканием перегруппировки получали 4-изопропил-6-(N,N-диэтилкарбамилтио)сахарин. Последнее вещество, подвергая гидролизу щелочью, превращали в 4-изопропил-6-меркаптосахарин, из которого после бензилирования, взаимодействия с иодистым метилом и гидрогенолиза с переносом получали 4-изопропил-6-метилтиосахарин. Окислением последнего вещества одним или двумя молярными эквивалентами 3-хлорнадбензойной кислоты получали 4-изопропил-6-метилсульфинилсахарин и 4-изопропил-6-метилсульфонилсахарин.
Взаимодействием 2-изопропил-4-фторбензойной кислоты с хлористым тионилом получали 2-изопропил-4-фторбензоилхлорид, из которого при взаимодействии с диэтиламином получали 2-изопропил-4-фтор-N,N-диэтилбензамид. Взаимодействием последнего вещества с втор-бутиллитием и взаимодействием образовавшейся литиевой соли с диоксидом серы с последующим воздействием гидроксиламина-O-сульфоната натрия получали 2-изопропил-4-фтор-6-аминосульфонил-N,N-диэтилбензамид, из которого при нагревании в ледяной уксусной кислоте получали 4-изопропил-6-фторсахарин.
Взаимодействием последнего вещества с тиофенолом, 4-метилфенилтиофенолом, 4-метоксифенилтиофенолом, 4-хлорфенилтиофенолом, 1-меркапто-4-метилнафталином или 1-меркаптонафталином с нагреванием реагирующих веществ в диметилформамиде соответственно получали 4-изопропил-6-фенилтиосахарин, 4-изопропил-6-(4-метилфенилтиоэсахарин, 4-изопропил-6-(4-метоксифенилтио)-сахарин, 4-изопропил-6-(4-хлорфенилтио)сахарин, 4-изопропил-6-(4-метил-1-нафтилтио)сахарин и 4-изопропил-6-(1-нафтилтио)сахарин. Окислением последних веществ одним или двумя молярными эквивалентами 3-хлорнадбензойной кислоты получали 4-изопропил-6-фенилсульфинилсахарин, 4-изопропил-6-фенилсульфонилсахарин, 4-изопропил-6-(4-метилфенилсульфинил)сахарин, 4-изопропил-6-(4-метилфенилсульфонил)сахарин, 4-изопропил-6-(4-метоксифенилсульфинил) сахарин, 4-изопропил-6-(4-метоксифенилсульфонил)сахарин, 4-изопропил-6-(4-хлорфенилсульфинил)сахарин, 4-изопропил-6-(4-хлорфенилсульфонил)сахарин, 4-изопропил-6-(4-метил-1-нафтилсульфинил)сахарин, 4-изопропил-6-(4-метил-1-нафтилсульфинил)сахарин, 4-изопропил-6-(1-нафтилсульфинил)сахарин и 4-изопропил-6-(1-нафтилсульфонил)сахарин.
Взаимодействием 4-изопропил-6-гидроксисахарина (синтез 19) с одним молярным эквивалентом уксусного ангидрида, хлористым бензоилом или 1-нафтилкарбоновокислым хлоридом соответственно получали 4-изопропил-6-ацетоксисахарин, 4-изопропил-6-бензоилоксисахарин и 4-изопропил-6-(1-нафтилкарбонилокси)сахарин.
Нагреванием 4-изопропил-6-фторсахарина в диметилформамиде с азетидином, пирролидином, пиперидином, морфолином, 1-бензилпиперидином, 1-метилпиперазином, имидазолом, трет-бутил-альфа-аминоацетатом или аммиаком соответственно получали 4-изопропил-6-(1-азетидинил)-сахарин, 4-изопропил-6-(1-пирролидинил)сахарин, 4-изопропил-6-(1-пиперидинил)сахарин, 4-изопропил-6-(4-морфолин)сахарин, 4-изопропил-6-(4-бензил-1-пиперазинил)сахарин, 4-изопропил-6-(4-метил-1-пиперазинил)сахарин, 4-изопропил-6-(1-1H-имидазолил)сахарин, 4 изопропил-6-(карбо-трет-бутоксиметиламино)сахарин и 4-изопропил-6-амино-сахарин.
Каталитическим дебензилированием 4-изопропил-6-(4-бензил-1-пиперазинил)сахарина водородом над палладиевым катализатором, нанесенным на древесный активированный уголь, получают 4-изопропил-6-(1-пиперазинил)сахарин.
Гидролизом 4-изопропил-6-(карбо-трет-бутоксикарбонилметиламино)сахарина разбавленной хлористоводородной кислотой и выделением продукта из нейтральной среды получали 4-изопропил-6-карбоксиметиламиносахарин.
Взаимодействием 4-изопропил-6-аминосахарина с одним молярным эквивалентом хлористого ацетила получали 4-изопропил-6-ацетиламиносахарин.
Омылением 4-карбометоксисахарина (синтез 9D) до соответствующей сахарин-4-карбоновой кислоты посредством щелочного гидролиза, превращением кислоты в соответствующий хлорид кислоты взаимодействием кислоты с хлористым тионилом и взаимодействием хлорида кислоты с аммиаком получали сахарин-4-карбоксамид.
Взаимодействием каждого из приготовленных таким способом 4-R4-R5-сахаринов с п-формальдегидом и хлортриметилсиланом в присутствии хлорида олова (4) в двухлористом этилене получали 4-R4-R5-2-хлорметилсахарины с формулой IV, перечисленные в табл. 2, где в каждом случае X представляет собой хлор.
Синтез 19BI
Взаимодействием изотиазол-5-карбоксальдегида с 3-(трифенилфосфораналиден)пропаноатом лития в стандартных условиях перегруппировки Виттинга получали 4-(5-изотиазолил)-3-бутеновую кислоту, которую подвергали восстановлению и циклизации хлоридом алюминия с получением 4-оксо-4,5,6,7-тетрагидробензизотиазола. 4-Оксосоединение подвергали взаимодействию с метилентрифенилфосфораном в стандартных условиях перегруппировки Виттинга, и метилен встраивали в образующееся 4-метиленовое соединение, прибегая к реакции Симмонса-Смита, в результате чего получали 6,7-дигидроспиро[бензотиазол-4(5H), 1'- циклопропан] , который окисляли пероксидом водорода в уксусной кислоте с получением 6,7-дигидроспиро[бензизотиазол-4(5H),1'-циклопропан-1,1- диоксид(4-спироциклопропилтетрагидросахарина). Это соединение хлорметилировали по методике синтеза 1А, в результате чего получали 2-хлорметил-4-спироциклопропил-4,5,6,7-тетрагидросахарин.
Синтез 19BJ.
Полученный по методике синтеза 23 2-бензил-4-изопропил-6- оксотетрагидросахарин подвергали восстановлению боргидридом натрия и метилировали иодистым метилом в присутствии гидрида натрия, в результате чего получали 2-бензил-4-изопропил-6-метокситетрагидросахарин. Это соединение дебензилировали и хлорметилировали, как это делали в синтезе 23, в результате чего получали 2-хлорметил-4-изопропил-6-метокси-4,5,6,7-тетрагидросахарин.
Синтез 20А.
Смесь, состоящую из 10,0 г (0,063 моль) 2,6-дифторбензойной кислоты и 66,0 г (0,57 моль) хлорсульфоновой кислоты, грели при 155-160oC и затем осторожно выливали в 100 мл воды со льдом. Твердые вещества, которые выпадали, собирали фильтрованием, сушили на воздухе и перекристаллизовывали из хлороформа, в результате чего получали 7,0 г 3-хлорсульфонил-2,6-дифторбензойной кислоты, 0,64 г (0,0025 моль) которой растворяли в двухлористом метилене и обрабатывали при -10oC раствором, состоящим из 0,25 г (0,0025 моль) 1-метилпиперазина и 0,33 г (0,0026 моль) диизопропилэтиламина. Продукт, который выпадал, собирали фильтрованием, промывали двухлористым метиленом и сушили, получая 0,4 г (выход 50%) 2,6-дифтор-3-(4-метил-1-пиперазинил)сульфонилбензойной кислоты.
Синтез 20B-20G.
Следуя методике, аналогичной описанной в синтезе 20А, 3-хлорсульфонил-2,6-дихлорбензойную кислоту с температурой плавления 172-175oC (из хлороформа) получали с выходом 56% нагреванием смеси 2,6-дихлорбензойной кислоты с хлорсульфоновой кислотой при 150-160oC. Реакцией последнего вещества с надлежащим амином (N=B) получали 3-аминосульфонил-2,6-дихлорбензойные кислоты, перечисленные в табл. 3. В каждом случае продукты не подвергали дальнейшей очистке, а использовали на следующем этапе как таковые.
Синтез 21А.
К смеси, состоящей из 1,0 г (0,003 моль) бензил-2,6-дихлор-3-гидробензоата и 0,18 г 60%-ной дисперсии гидрида натрия в минеральном масле, введенных в 30 мл диметилформамида, добавляли раствор, состоящий из 0,008 моль 4-(2-хлорэтил)морфолина в 20 мл простого трет-бутилметилового эфира, и смесь грели при 70oC в течение 3 ч. Реакционную смесь затем доводили до сухого состояния, остаток переводили в этилацетат и органический раствор промывали водой и рассолом, затем сушили и концентрировали в вакууме до сухого состояния, в результате чего получали 1,25 г (выход 83%) бензил-2,6-дихлор-3-[2-(4-морфолинил)этокси] бензоата, который растворяли в смеси этилацетата с метанолом (50 мл), взятых в соотношении 1:1, и восстанавливали водородом на 10%-ном палладиевом катализаторе, нанесенном на древесный активированный уголь, который брали в количестве 0,25 г. После завершения восстановления катализатор удаляли фильтрованием, промывали диметилформамидом, и объединенные фильтраты доводили до сухого состояния, получая 0,75 г (выход 75%) 2,6-дихлор-3-[2-(4-морфолинил)этокси]бензойной кислоты.
Синтез 21B.
Следуя методике, аналогичной описанной в синтезе 21А, 1,0 г (0,003 моль) бензил-2,6-дихлор-3-гидроксибензоата подвергали взаимодействию с 0,092 моль N-(2-хлорэтил)-N, N-диметиламина в 30 мл диметилформамида и 20 мл простого трет-бутилметилового эфира в присутствии 0,18 г 60%-ной дисперсии гидрида натрия в минеральном масле, в результате чего с количественным выходом получали бензил-2,6-дихлор-3-[2-(диметиламино)этокси] бензоат, который каталитически восстанавливали в растворе этилацетата с метанолом, взятых в соотношении 5: 2, на 10%-ном палладиевом катализаторе, нанесенном на древесный активированный уголь, количество которого составляло 0,2 г. Тем самым получали 0,2 г (выход 22%) 2,6-дихлор-3-[2-(диметиламино)этокси]бензойной кислоты.
Синтез 21C.
К раствору метил-2,6-дихлор-4-метоксибензоата (J. Org. Chem., 50, 408, 1985) (5,5 г, 0,023 моль) в метаноле (50 мл) добавляли 5 н. раствор гидроксида натрия (20 мл). Образовавшуюся смесь грели в сосуде с обратным холодильником в течение 20 ч, охлаждали до комнатной температуры, концентрировали в вакууме и подкисляли до величины pH 12 н. хлористоводородной кислотой. Выпавшие твердые вещества собирали фильтрованием, получая 5,2 г (выход 100%) 2,6-дихлор-4-метоксибезойной кислоты, которую обрабатывали 60 мл 1 M раствора трибромида бора (0,06 моль) в дихлорэтане (100 мл), делая это при нагревании в сосуде с обратным холодильником в течение 2 ч. Образовавшуюся смесь охлаждали до комнатной температуры и выливали в смесь воды с этанолом (50 мл смеси с соотношением 9:1). После перемешивания в течение 10 мин смесь экстрагировали простым эфиром (400 мл), органическую фазу промывали водой, рассолом и сушили. Удаляя растворитель в вакууме, получали 4,0 г (выход 80%) 2,6-дихлор-4-гидроксибензойной кислоты.
Последнее вещество (1,05 г, 0,005 моль) растворяли в 95%-ном этаноле (25 мл) и обрабатывали хлористым бензилом (0,71 г, 0,006 моль) и 1 н. раствором гидроксида натрия (5 мл). После нагревания в сосуде с обратным холодильником в течение 2 ч реакционную смесь охлаждали до комнатной температуры и концентрировали в вакууме. Остаток подкисляли 2 н. хлористоводородной кислотой и экстрагировали простым эфиром. Органическую фазу промывали насыщенным раствором бикарбоната натрия, водой и 10%-ным раствором гидроксида натрия. После удаления органической фазы растворы от промывки гидроксидом натрия подкисляли 2 н. хлористоводородной кислотой и вновь экстрагировали простым эфиром (2 раза по 50 мл). Эфирные экстракты затем сушили и концентрировали в вакууме, получая 0,63 г (выход 42%) бензил-2,6-дихлор-4-гидроксибензоата.
Следуя методике, аналогичной описанной в синтезе 21А, 630 мг бензил-2,6-дихлор-4-гидроксибензоата превращали в 350 мг 2,6-дихлор-4-[2-(4-морфолинил)этокси]бензойной кислоты.
Синтез 24А.
Раствор, состоящий из 1,9 г (0,01 моль) 2,6-дихлор-3- гидроксибензальдегида в 10 мл сухого диметилформамида, продували азотом, добавляли 0,3 г 97%-ного гидрида натрия при перемешивании магнитной мешалкой. С выделением водорода образовывался прозрачный красно-коричневый раствор. К этому раствору добавляли раствор 2-диметиламиноэтилхлорида (полученного из 2 г гидрохлорида) в 6 мл простого трет-бутилметилового эфира. Раствор грели в сосуде с обратным холодильником в течение 30 мин. Выпадал хлорид натрия. Холодильник удаляли, и нагревание продолжали в течение 30 мин. Реакционную смесь концентрировали до достижения сухого состояния, переводили в разбавленный раствор хлористоводородной кислоты и экстрагировали хлористым метиленом. Водный слой делали основным, добавляя 10%-ный раствор карбоната натрия, экстрагировали тремя порциями двухлористого метилена, и экстракты упаривали до образования коричневой маслянистой жидкости, которую разгоняли в аппарате Кугельрора; температура кипения составляла 155-160oC при давлении 0,12 мм рт. ст. Желтый дистиллят претерпевал кристаллизацию, и его превращали в гидрохлорид, воздействуя эфирной хлористоводородной кислотой. Перекристаллизацией из CH3CN получали 661 мг 2,6-дихлор-3-[2-диметиламино)этокси]бензальдегидрохлорида; температура плавления составляла 177-178oC.
Свежеприготовленный оксид серебра (из 1,7 г нитрата серебра) суспендировали в 1,0 мл 10%-ного раствора гидроксида натрия, который затем нагревали до 55oC. В условиях перемешивания магнитной мешалкой добавляли альдегид (2,62 г, 0,01 моль). За счет протекания экзотермической реакции температура возрастала до 65oC, и серебро выпадало. Нагревание продолжали при 60oC в течение 1/4 ч. Реакционную смесь фильтровали, и фильтрат экстрагировали двумя порциями двухлористого метилена. Испарением двухлористого метилена получали 0,804 г исходного альдегида. Водную фазу подкисляли 3 н. хлористоводородной кислотой и упаривали в вакууме до получения белого твердого вещества, которое перекристаллизовывали из 10 мл воды. Тем самым получали 1,065 г (выход 34%) 2,6-дихлор-3-[2- (диметиламино)этокси]бензойной кислоты; температура плавления составляла 234-236oC.
Синтезы 24B-24D.
Следуя методике, аналогичной описанной в синтезе 24A, готовили альдегиды и кислоты, указанные в табл. 4.
Синтез 25.
К раствору хлора (15,7 г, 0,22 моль) в ледяной уксусной кислоте (250 мл) при 0oC добавляли метил-3-гидроксибензоат (15,2 г, 0,1 моль). Образовавшийся раствор нагревали до комнатной температуры, перемешивали в течение 1 часа, упаривали до сухого состояния в вакууме, в результате чего получали 21,4 г желтой маслянистой жидкости, которая, как было установлено, содержала по данным ядерного магнитного резонанса 75% метил-2,6-дихлор-3-гидроксибензоата. Маслянистую жидкость (21,4 г) растворяли в ацетоне (600 мл), добавляли бромистый бензил (19,9 г, 0,12 моль) и карбонат калия (22,7 г, 0,16 моль) и грели в сосуде с обратным холодильником в среде азота в течение 16 ч. Реакционную смесь охлаждали до комнатной температуры, твердые вещества отфильтровывали. Фильтрат концентрировали в вакууме, остаток переводили в 10%-ный раствор этилацетата в гексанах (100 мл) и охлаждали на бане со льдом. Твердые вещества, которые выпадали, собирали фильтрованием и сушили на воздухе, в результате чего получали 14,3 г (выход 47%) метил-2,6-дихлор-3-бензилоксибензоата.
Раствор, состоящий из метил-2,6-дихлор-3-бензилоксибензоата (2,1 г, 6,7 ммоль) и 10%-ного водного раствора гидроксида натрия (25 мл) в метаноле (25 мл), грели в сосуде с обратным холодильником в среде азота в течение 24 ч и охлаждали до комнатной температуры. Образовавшуюся смесь концентрировали наполовину в вакууме и подкисляли до величины pH 12 н. хлористоводородной кислотой. Твердые вещества, которые выпадали, собирали фильтрованием, промывали водой, гексанами и сушили на воздухе, в результате чего получали 2,0 г (выход 100%) 2,6-дихлор-3-бензилоксибензойной кислоты, имевшей вид белого твердого вещества.
Синтез 26.
Раствор, состоящий из 5 г 2,6-диметокси-3-нитробензойной кислоты в тетрагидрофуране, подвергали гидрогенизации в присутствии 10%-ного палладиевого катализатора, нанесенного на углерод, и образовавшийся амин тут же ацетилировали уксусным ангидридом и пиридином, в результате чего получали 0,9 г 3-ацетил-амино-2,6-диметоксибензойной кислоты.
Синтез 27.
К суспензии, состоящей из 3,6 г (0,12 моль) п-формальдегида в 50 мл 1,2-дихлорэтана и 30 мл (26 г, 0,24 моль) хлористого триметилсилила, в среде азота добавляли 0,2 мл хлорида олова (4), и образовавшийся раствор перемешивали на паровой бане. По истечении 30 мин добавляли 9,55 г (0,05 моль) 2,6-дихлорбензойной кислоты, и реакционную смесь грели еще в течение 20 ч. Летучие вещества удаляли, остаток растворяли в двухлористом метилене и промывали бикарбонатом натрия, сушили и удаляли легкие компоненты до получения маслянистой жидкости, которую растирали с гексаном и фильтровали, в результате чего получали 8,5 г хлорметил-2,6-дихлорбензоата.
Приготовление конечных продуктов.
Пример 1А. Смесь, состоящую из 0,5 г (0,0017 моль) 2-хлорметил-4,6-диметоксисахарина, 0,33 г (0,0017 моль) 2,6 -дихлорбензойной кислоты и 17 г (0,25 мл, 0,0017 моль) триэтиламина в 15 мл толуола, грели в сосуде с обратным холодильником в течение примерно 6 ч, затем охлаждали и концентрировали в вакууме до сухого состояния. Остаток подвергали хроматографии на силикагеле, элюируя 40%-ным раствором этилацетата в гексане, в результате чего получали 0,44 г (выход 53%) 4,6-диметокси-2-сахаринметил-2,6-дихлорбензоата; температура плавления составляла 200-201oC.
Следуя методике, описанной в примере 1А, аналогичным образом готовили соединения с формулой I, перечисленные в табл. 5. Реакции проводили либо в присутствии карбоната цезия, карбоната калия, триэтиламина (TEA), диизопропилэтиламина (DIPEA) или 1,8-диазабицикло-[5.4.0]ундек-7-ена (DBU) в качестве основного катализатора, либо при использовании цезиевой или таллиевой соли бензойной кислоты и, при желании, в присутствии бромистого тетрабутиламмония (TBAB) в надлежащем органическом растворителе, как это указано в колонке с названием "Solv./Cat" ("Растворитель/Катализатор"). Сокращение NMP означает N-метилпирролидинон. В каждом из примеров 1D, 1I, 1N, 1AI и 1AJ-1AN продукты получали из 4-R4R5-2-бромметилсахарина. Во всех других примерах в качестве исходного материала использовали надлежащий 4-R4-R5-2-хлорметилсахарин.
Здесь и в иных местах этого описания различные гетероциклические или другие группы сокращены: A - ацетил, Mor - морфолинил, pip - пиперазинил, Bzl - бензил, azet - азетидинил, imidazol - имидазолил, pyr - пирролидинил, pid - пиперидинил.
Примечания к табл. 5.
a/ 2-Хлорметилсахарин подвергали взаимодействию с 2,6-дихлор-3-карбо-трет-бутоксикарбонилметиламиносульфонилбензойной кислотой, и продукт гидролизовали трифторуксусной кислотой в двухлористом метилене, в результате чего получали соответствующий 2-сахаринилметилкарбоксиметиламиносульфонилбензоат с выходом 76%.
b/ Соль хлористоводородной кислоты.
c/ 2-Хлорметил-4-изопропилсахарин подвергали взаимодействию с 2,6-дихлор-3-бензилоксикарбонилметиламиносульфонилбензойной кислотой, и продукт каталитически дебензилировали при давлении водорода 1 ат на палладиевом катализаторе, нанесенном на древесный активированный уголь, в этилацетате с уксусной кислотой при концентрации 17%, в результате чего получали соответствующую кислоту с выходом 80%.
d/ 2-Хлорметил-4-изопропилсахарин подвергали взаимодействию с 2,6-дихлор-3-бензилоксибензойной кислотой и получали два продукта, у одного из которых часть молекулы, отвечающая бензойной кислоте, подвергалась дехлорированию.
e/ Смесь хлористоводородной кислоты с водой при соотношении 5:2.
f/ Смесь хлористоводородной кислоты с водой при соотношении 3:2.
g/ Соль соединения CH3SO3H.
Пример 1AW. Цезиевую соль 2,6-дихлорбензойной кислоты готовили из 4,48 г (0,0235 моль) 2,6-дихлорбензойной кислоты и 3,82 г (0,0117 моль) карбоната цезия в метаноле. Соль выделяли, удаляя растворитель при пониженном давлении и производя сушку в глубоком вакууме в течение 1/2 ч. Высушенную соль суспендировали, перемешивая в 10 - 15 мл диметилформамида, и добавляли 3,4 г (0,0117 моль) 2-хлорметил-6-гидрокси-4-изопропилсахарина. Смесь грели при 80oC в течение 2 - 3 ч, охлаждали, разбавляли водой и экстрагировали 200 мл смеси простого эфира с этилацетатом, находящимися в соотношении 7:3. Органический слой промывали водой, насыщенным раствором хлорида натрия и сушили. Растворитель удаляли, и остаток очищали испарительной хроматографией на силикагеле, элюируя смесью этилацетата с гексаном, в результате чего получали 4,53 г (выход 87%) 6-гидрокси-4-изопропил-2-сахаринилметил-2,6-дихлорбензоата (без данных по температуре плавления).
Пример 2A. Раствор, состоящий из 1,4 г (0,0026 моль) 4-изопропил-2-сахаринилметил-2,6-дихлор-3-бензилоксибензоата в 50 мл этилацетата, обрабатывали 0,3 г 10%-ного палладиевого катализатора, нанесенного на древесный активированный уголь, и 0,5 мл уксусной кислоты, смесь перемешивали при давлении водорода 1 ат в течение 16 ч. Катализатор удаляли фильтрованием, и фильтрат доводили до сухого состояния в вакууме, в результате чего получали 1,16 г (выход 100%) 4-изопропил-2-сахаринилметил-2,6-дихлор-3-гидроксибензоата; температура плавления составляла 78 - 80oC.
Пример 2B. Следуя методике, аналогичный описанной в примере 2A, 1,2 г (0,0018 моль) 4-изопропил-2-сахаринилметил-2,6-дихлор-3-(4-бензил-1-пиперазинилсульфонил) бензоата (пример 1Z) восстанавливали водородом в 50 мл этилацетата и 2 мл уксусной кислоты на 0,3 г 10%-ного палладиевого катализатора, нанесенного на древесный активированный уголь, и продукт превращали в гидрохлоридную соль, в результате чего получали 0,5 г (выход 68%) 4-изопропил-2-сахаринилметил-2,6-дихлор-3-(1-пиперазинилсульфонил) бензоатгидрохлорида; температура плавления превышала 171oC.
Пример 2C. Смесь, состоящую из 4-изопропил-6-метокси-2-сахаринилметил-2,6-дихлор-3-бензилоксибензоата (2,5 г, 4,4 моль), взятую из примера 1BU, 10%-ного палладиевого катализатора, нанесенного на углерод (0,7 г), и ледяной уксусной кислоты (1 мл) в этилацетате (100 мл), перемешивали при давлении водорода 344,5 кН/м2 (50 фунт/кв.дюйм) в гидрогенизаторе Парра (Parr) в течение 1,5 ч. Образовавшуюся смесь фильтровали через набивку из суперцеля, элюируя этилацетатом (100 мл). Объединенный фильтрат промывали насыщенным раствором бикарбоната натрия, водой, рассолом и сушили. Удалением растворителя в вакууме и кристаллизацией из смеси простого эфира с гексанами, взятыми в соотношении 1:1, получали 2,1 г (выход 100%) 4-изопропил-6-метокси-2-сахаринилметил-2,6-дихлор-3-гидроксибензоата; температура плавления составляла 152-154oC.
Пример 2D. По способу, аналогичному описанному в примере 2A, 0,41 г 4-изопропил-6-метокси-2-сахаринилметил-2,6-дихлор-3- бензилокси-карбонилметиламиносульфонилбензоата, взятого из примера 1ВV, каталитически дебензилировали при давлении водорода 1 ат на палладиевом катализаторе, нанесенном на древесный активированный уголь, в этилацетате, содержанием 20% уксусной кислоты, в результате чего получали 0,16 г (выход 45%) 4-изопропил-6-метокси-2- сахаринилметил-2,6-дихлор-3-карбоксиметиламиносульфонил-бензоата; температура плавления составляла 204-206oC.
Пример 3А. Раствор, состоящий из 1,05 г (0,0024 моль) 4-изопропил-2-сахаринилметил-2,6-дихлор-3-гидроксибензоата (пример 2А), 0,50 г (0,0026 моль) трет-бутил-альфа-бромацетата и 0,48 г (0,0035 моль) карбоната калия в 25 мл ацетона, грели в сосуде с обратным холодильником в течение 7 ч, затем охлаждали до температуры окружающей среды, фильтровали, и из фильтрата, доведенного до сухого состояния, получали 0,32 г (выход 24%) 4-изопропил-2-сахаринилметил- 2,6-дихлор-3-трет-бутоксикарбонилметоксибензоата, который растворяли в 10 мл двухлористого метилена, содержащего 2 мл трифторуксусной кислоты. Раствор перемешивали при температуре окружающей среды в среде азота в течение 2 ч, доводили до сухого состояния, и остаток растирали со смесью гексана с простым эфиром. Образовавшееся твердое вещество собирали фильтрованием, в результате чего получали 0,18 г (выход 64%) 4-изопропил-2-сахаринилметил-2,6-дихлор-3-карбоксиметоксибензоата; температура плавления составляла 210-212oC.
Пример 3B. Раствор, состоящий из 0,78 г (1,6 ммоль) 4-изопропил-6-метокси-2-сахаринилметил-2,6-дихлор-3-гидроксибензоата, 0,38 г (2,0 моль) трет-бутил- α -бромацетата и 0,3 г (2,1 ммоль) карбоната калия в 50 мл ацетона, грели в сосуде с обратным холодильником в течение 16 ч, затем охлаждали до комнатной температуры, фильтровали, и фильтрат доводили до сухого состояния. Очисткой остатка испарительной хроматографией на силикагеле (элюировали смесью гексанов с этилацетатом, взятым в соотношении 4:2) получали 0,65 г (выход 67%) 4-изопропил-6-метокси-2-сахаринилметил- 2,6-дихлор-3-трет-бутоксикарбонилметоксибензоата. Сложный трет-бутиловый эфир (0,55 г, 0,9 ммоль) растворяли в 15 мл двуххлористого метилена, содержащего 5 мл трифторуксусной кислоты. Раствор перемешивали при комнатной температуре в среде азота в течение 2 ч, упаривали до сухого состояния, и остаток растирали со смесью гексана с простым эфиром. Образовавшееся твердое вещество собирали фильтрованием, в результате чего получали 0,4 г (выход 82%) 4-изопропил-6-метокси-2-сахаринилметил-2,6-дихлор-3-карбоксиметоксибензоата; температура плавления составляла 206-208oC.
Пример 4. Взаимодействием надлежащего 4-R4-R5-2- галометилсахарина с формулой IV с надлежащей арилкарбоновой кислотой при использовании методики, описанной в примере 1А, или взаимодействием надлежащего сахарина с формулой II с надлежащим хлорметилбензоатом при использовании методики, описанной в примере 11, могут быть получены соединения с формулой I, перечисленные в табл. 6.
Пример 4СВ. Следуя методике примера 4,2-хлорметил-4-спироциклопил- 4,5,6,7-тетрагидросахарин, полученный в синтезе 19BI, сочетали с 2,6-диметилбензойной кислотой, в результате чего получали 4-спироциклопропил-4,5,6,7-тетрагидро-2-сахаринилметил-2,6-диметилбензоат.
Пример 4СС. Следуя методике примера 4,2-хлорметил-4-изопропил-6-метокси-4,5,6,7-тетрагидросахарин, полученный в синтез 19BJ, сочетали с 2,6-диметилбензойной кислотой, в результате чего получали 4-изопропил-6-метокси-4,5,6,7-тетрагидро-2- сахаринилметил-2,6-диметилбензоат.
Пример 5A. К раствору, содержащему 500 мг (1,1 моль) 6-гидрокси-4-изопропил-2-сахаринилметил-2,6-дихлорбензоат в 10-15 мл тетрагидрофурана, добавляли 298 мг (1,14 ммоль) трифенилфосфина, 52 мг (1,13 ммоль) этанола и 198 мг (1,14 ммоль) диэтилазодикарбоксилата, что делали при комнатной температуре. Смесь перемешивали в течение 1 ч 30 мин и затем подвергали хроматографии на силикагеле с использованием 10%-ного раствора этилацетата в гексане, в результате чего получали 370 мг (выход 70%) 6-этокси-4-изопропил-2-сахаринилметил-2,6-дихлорбензоата, имевшего вид белого порошка; температура плавления составляла 140-141oC.
Следуя методике примера 5A, соединения, приведенные в табл. 7, готовили из 6-гидроксисоединения, взятого из примера 1AW.
Защищенный глицерол, использованный при проведении синтеза в примере 5F, получали следующим образом.
Раствор, состоящий из 10,0 г (0,055 моль) DL- α -O-бензилгицерола в небольшом количестве тетрагидрофурана, добавляли к суспензии, состоящей из 15,38 г (0,137 моль) трет-бутоксида калия в 300 мл тетрагидрофурана. Смесь перемешивали в течение 1 ч при комнатной температуре и добавляли 18,72 г (0,132 моль) иодметана. Белое твердое вещество сразу же отделяли. Реакционную смесь перемешивали в течение 10 ч при комнатной температуре, охлаждали, осторожно разбавляли раствором хлорида натрия и экстрагировали простым эфиром. Органический слой промывали водой, 5%-ным раствором хлористоводородной кислоты, водой, насыщенным раствором хлорида натрия и сушили. Растворитель удаляли, и остаток очищали испарительной хроматографией, в результате чего получали 9,16 г (выход 79%) 1-бензилокси-2,3-диметоксипропана, имевшего вид маслянистой жидкости.
Раствор, состоящий из 8,8 г (0,042 моль) этого вещества в 200 мл метонола, подвергали гидрогенизации, используя 1,1 г 10%-ного палладиевого катализатора, нанесенного на углерод, при давлении 344,5 кН/м2 (50 фунт/кв.дюйм). Катализатор удаляли фильтрованием, и растворитель удаляли при пониженном давлении, в результате чего получали 4,4 г (выход 87%) 2,3-диметокси-1-пропанола.
Пример 5I. Соединение 6-этокси-4-изопропил-2-фенилтиометилсахарин готовили из 6-гидроксианалога (синтез 19) по методике примера 5A, в результате чего с выходом 85% его получали в виде твердого вещества; температура плавления составляла 111,5-112,5oC; это вещество, следуя методике синтеза 18А, с выходом 91% превращали в 2-хлорметил-6-этокси-4-изопропилсахарин; температура плавления составляла 127-128oC.
Пример 5J. К раствору 4-изопропил-6-гидроксисахаринилметил-2,6- дихлорбензоата, взятого из примера 1C (0,44 г, 1,0 ммоль), в двухлористом метилене (20 мл) добавляли при 0oC триэтиламин (0,3 г, 3,0 ммоль) и трифторметансульфоновый ангидрид (0,37 г, 1,3 ммоль). После перемешивания при 0oC в течение 10 мин реакционную смесь разбавляли двухлористым метиленом (50 мл), промывали насыщенным раствором бикарбоната натрия, рассолом и сушили. Удалением растворителя в вакууме и очисткой остатка хроматографией на силикагеле (элюировали 5%-ным раствором этилацетата в двухлористом метилене) получали 0,53 г (выход 88%) 4-изопропил-6- трифторметансульфонилоксисахаринилметил-2,6-дихлорбензоата в виде бесцветного пенистого вещества.
Трифторметансульфонат (0,28 г, 0,49 ммоль) смешивали с 1-метил-2-триметилстаннилпирролом (0,19 г, 0,78 ммоль), тетракисом (трифенилфосфином) палладия (O) (0,012 г, 0,01 ммоль), хлоридом лития (0,062 г, 1,5 ммоль) и 2,6-ди-трет-бутил-4-метилфенолом (0,01 г, 0,05 ммоль) в п-диоксане, и смесь грели в сосуде с обратным холодильником в среде азота в течение 30 мин. Образовавшуюся темную реакционную смесь охлаждали до комнатной температуры, разбавляли простым эфиром (50 мл) и фильтровали через набивку из суперцеля. Фильтрат промывали водой, рассолом и сушили. Удалением растворителя в вакууме и очисткой остатка испарительной хроматографией на силикагеле (при элюировании смесью из гексанов, двухлористого метилена и простого эфира, взятых в соотношении 7:2:1:) получали 0,22 г (выход 92%) 4-изопропил-6-[2-(1-метил)пирролидинил] сахаринилметил- 2,6-дихлорбензоата в виде бледно-желтого вещества; температура плавления составляла 125-127oC.
Пример 5K. Соединение 4-изопропил-6- трифторметансульфонилоксисахаринилметил-2,6-дихлорбензоат, приготовленное как в примере 5J, (0,7 г, 1,2 ммоль) в тетрагидрофуране (10 мл) охлаждали до -5oC, обрабатывали 40%-ным водным раствором диметиламина (0,6 мл, 5,3 ммоль) и перемешивали при комнатной температуре на протяжении ночи. Образовавшуюся смесь разбавляли насыщенным раствором бикарбоната натрия (20 мл) и двухлористым метиленом (250 мл). Слои разделяли, и органическую фазу промывали водой, рассолом и сушили. Удалением растворителя в вакууме и очисткой остатка хроматографией на силикагеле (при элюировании смесью гексанов, двухлористого метилена и простого эфира, взятых в соотношении 6:3:1) получали 0,2 г (выход 35%) 4-изопропил-6-диметиламиносахаринилметил-2,6-дихлорбензоата; температура плавления составляла 177-179oC.
Пример 5L. Раствор, состоящий из 42 мг 4-изопропил-6-гидроксисахаринилметил-2,6-дихлорбензоата, взятого из примера 1C, ди-(втор-бутоксиметил)метиламина и толуола, грели при 80oC в течение 1 ч, охлаждали и удаляли летучие вещества. Готовя шлам в гексане, получали 30 мг 2-(2,6-дихлорбензоилоксиметил)-8-метил- 2,3,7,8-тетрагидро-9H-[1,3] оксазино[6,5-g]бензизотиазол-3-он-1,1-диоксида.
Пример 6. Раствор, состоящий из 600 мг (1,1 ммоль) изопропилидена, взятого из примера 5C (табл. 7), и 176 мг (0,9 ммоль) п-толуолсульфоновой кислоты в виде моногидрата в метаноле с хлороформом перемешивали на протяжении ночи. Смесь подвергали хроматографии на силикагеле, в результате чего получали 290 мг (выход 5,%) 6-(2,3-дигидроксипропокси)-4-изопропилсахаринилметил-2,6- дихлорбензоата, имевшего вид пенистого вещества.
Пример 7A. К раствору, состоящему из 1,0 г (2,3 ммоль) 6-гидрокси-4-изопропил-2-сахаринилметил-2,6-дихлорбензоата в 40 мл ацетона, при комнатной температуре добавляли 0,62 г (4,5 ммоль) безводного карбоната калия и 0,66 г (93,4 ммоль) трет-бутилбромацетата. Смесь перемешивали в течение 4-5 ч и фильтровали. Фильтрат концентрировали при пониженном давлении, и остаток очищали испарительной хроматографией, получая 1,13 г (выход 90%) 6-(2-трет-бутокси-2-оксоэтокси)-4-изопропил-2-сахаринилметил- 2,6-дихлорбензоата, имеющего вид стеклообразного вещества.
Пример 7B. Аналогичным образом из 6-гидроксисоединения и бензилбромацетата с выходом 61% получали в виде стеклообразного вещества 6-(2-бензилокси-2-оксоэтокси)-4-изопропил-2-сахаринилметил- 2,6-дихлорбензоат.
Пример 8. К свежеперегнанному циклопентадиену (25 мл) при 0oC добавляли 4-бром-2-(трет-бутил)изотиазиол-3(2H)-он-1,1- диоксид (Helv. Chim. Aeta, 72, 1416, 1989) (7,9 г, 0,03 моль). После перемешивания при 0oC в среде азота в течение 16 ч реакционную смесь концентрировали в вакууме. Остаток очищали фильтрованием через силикагель, элюируя гексанами (500 мл), а затем 20%-ным раствором этилацетата в гексанах (500 мл). Последние элюенты концентрировали в вакууме, получая 9,8 г (выход аддукта норборнена составляет 100%) 3а-бром-2-трет-бутил-3а, 4,7,7а-тетрагидро-4,7-метано-1,2- бензизотиазол-3(2H)-он-1,1-диоксида, имеющего вид белого твердого вещества.
Аддукт (0,4 г, 1,2 ммоль), находящийся в 25 мл этилацетата, содержащего 5%-ный палладиевый катализатор, нанесенный на карбонат кальция (0,2 г), перемешивали при давлении водорода 1 ат в течение 4 ч, и реакционную смесь фильтровали через набивку из силикагеля, элюируя этилацетатом (100 мл). Элюенты концентрировали в вакууме, и остаток кристаллизовали из гексанов, получая 0,4 г (выход 100%) бромнорборнана в виде белого кристаллического твердого вещества.
К раствору бромнорборнана (3,7 г, 0,011 моль) в толуоле (25 мл) при 0oC добавляли диазабициклононен (1,37 г, 0,011 моль) в толуоле (10 мл). После перемешивания при 0oC в течение 20 мин к реакционной смеси добавляли силикагель (25 г). Образовавшийся шлам помещали на набивку из силикагеля длиной 15 см и элюировали 20%-ным раствором этилацетата в гексанах (800 мл). Элюенты концентрировали в вакууме, в результате чего получали 2,8 г (выход 100%) дегидробромированного соединения в виде белого твердого вещества.
Соединение 2-трет-бутил-4,5,6,7-тетрагидро-4,7-метано-1,2- бензизотиазол-3(2H)-он-1,1-диоксид (2,8 г, 0,011 моль) в трифторуксусной кислоте (30 мл) грели в сосуде с обратным холодильником в течение 48 ч и оставляли стоять при комнатной температуре в течение четырех суток. Образовавшуюся смесь концентрировали в вакууме, обрабатывали метанолом (20 мл) и упаривали досуха. Остаток переносили в простой эфир (100 мл) и промывали насыщенным раствором бикарбоната натрия (1 раз, 50 мл). Слои разделяли, водный слой подкисляли до величины pH 1 хлористоводородной кислотой с концентрацией 2 н. и экстрагировали двухлористым метиленом (2 раза по 100 мл). Объединенные органические экстракты сушили и концентрировали в вакууме, в результате чего получали 0,9 г (выход 42%) бицикло-(2,2,1)-сахаринпроизводного в виде белого твердого вещества.
Смесь производного бицикло-(2,2,1)-сахарина (0,9 г, 5 ммоль), хлорметилфенилсульфида (0,07 г, 7 ммоль) и бромистого тетрабутиламмония (0,36 г, 0,16 ммоль) в толуоле (50 мл) грели в сосуде с обратным холодильником в среде азота в течение 16 ч, охлаждали до комнатной температуры и упаривали досуха под вакуумом. Остаток очищали испарительной хроматографией на силикагеле (100 г), используя чистый двухлористый метилен в качестве элюента, в результате чего получали 1,05 г (выход 72%) сульфида, имеющего вид вязкой маслянистой жидкости.
Сульфид (1,05 г, 3 ммоль), находящийся в дихлорметане (100 мл), обрабатывали хлористым сульфурилом (0,66 г, 5 ммоль) и перемешивали в течение 2 ч. Образовавшийся желтый раствор разбавляли двухлористым метиленом (100 мл), промывали насыщенным раствором бикарбоната натрия, сушили и концентрировали в вакууме. Остаток очищали испарительной хроматографией на силикагеле (элюируя 33%-ным раствором двухлористого метилена в гексанах), в результате чего получали 0,66 г (выход 81%) 2-хлорметил-4,5,6,7- тетрагидро-4,7-метано-1,2-бензизотиазол-3(2H)-он-1,1-диоксида.
2-Хлорметильное соединение (0,66 г, 2,7 ммоль) обрабатывали 2,6-дихлорбензойной кислотой (0,56 г, 2,9 ммоль), безводным карбонатом калия (0,55 г, 4,0 ммоль) и бромистым тетрабутиламмонием (0,2 г, 0,6 ммоль) в диметилформамиде (2,5 мл) при 70oC в течение 1 ч. Образовавшуюся смесь концентрировали в вакууме, разбавляли этилацетатом (100 мл) и фильтровали. Фильтрат промывали водой, насыщенным раствором бикарбоната натрия, водой и рассолом. Органическую фазу концентрировали в вакууме, и остаток очищали испарительной хроматографией на силикагеле (элюируя смесью двухлористого метилена с гексанами и простым эфиром, взятыми в соотношении 3:6:1), в результате чего получали 0,5 г (выход 47%) 2-(2,6-дихлорбензоилоксиметил)-4,5,6,7-тетрагидро-4,7-метано-1,2 -бензизотиазол-3(2H)-он-1,1-диоксида, имеющего вид бесцветного пенистого вещества.
Примеры 8B и 8C. Считается, что способом, аналогичным описанному в примере 8A, циклогексадиен и 1,1-диметилциклопентадиен могут быть превращены соответственно в 4,5,6,7-тетрагидро-4,7-этано-1,2- бензизотиазол-3(2H)-он-1,1-диоксид и 8,8-диметил-4,5,6,7-тетрагидро- 4,7-метано-1,2-бензизотиазол-3(2H)-он-1,1-диоксид.
Примеры 9A - 9D. Общая методика приготовления метил-2-алкилциклогексан-6-он-карбоксилата. К суспензии, состоящей из безводного иодида меди (I) (10 ммоль) в безводном тетрагидрофуране (100 мл), добавляли диметилсульфид Me2S (100 ммоль), и образовавшийся раствор охлаждали до -78oC. На протяжении промежутка времени в 15 мин добавляли реактив Гриньяра (20 ммоль). Подвергнув раствор перемешиванию при -78oC в течение 1 ч, к нему добавляли раствор циклокесенона (10 ммоль) в тетрагидрофуране и перемешивание продолжали еще 15 мин. К образовавшейся смеси добавляли гексаметилфосфорамид (5 мл) и по истечении 15 мин добавляли метилцианоформат (30 ммоль) в тетрагидрофуране (20 мл), и реакционную смесь нагревали до комнатной температуры и подвергали перемешиванию на протяжении ночи. Реакцию тушили, добавляя 2 н. хлористоводородную кислоту (50 мл). Слои разделяли, и водную фазу экстрагировали диэтиловым эфиром (1 раз, 100 мл). Объединенные органические экстракты промывали насыщенным раствором хлорида аммония (3 раза по 50 мл), водой (2 раза по 50 мл), рассолом (1 раз, 50 мл) и сушили (над сульфатом натрия). Удаляя растворитель в вакууме и производя очистку либо посредством перегонки по Кугельрору, либо проведением испарительной хроматографии, получали требуемый метил-2-алкилциклогексан-6-он-карбоксилат (табл.8).
Общая методика приготовления метил-2-бензилтио-6-алкил-циклогекс-2-ен-карбоксилата и 2-бензилтио-6-алкилциклогекс-1-ен-карбоксилата. Смесь, состоящую из метил-2-алкилциклогексан-6-он-карбоксилата (1 эквивалент), бензилмеркаптана (1,1-эквивалента) и кислого глинистого монтмориллонита KSF (вес которого в 1,5 раза превышает вес метил-2-алкилциклогексан-6-он-карбоксилата) в безводном толуоле (50 - 100 мл), грели в сосуде с обратным холодильником в среде азота в условиях азеотропного удаления воды в течение 12-14 ч и охлаждали до комнатной температуры. Твердые вещества отфильтровывали и промывали простым эфиром. Объединенный фильтрат промывали 10%-ным раствором карбоната натрия, водой, рассолом и сушили. Удалением растворителя в вакууме и очисткой остатка испарительной хроматографией на силикагеле (при элюировании 10%-ным раствором простого эфира в гексанах) получали смесь метил-2-бензилтио-6-алкилциклогекс-2-ен-карбоксилата с 2-бензилтио-6-алкилциклогекс-1-ен-карбоксилатом (табл.9), которые использовали на следующем этапе в виде смеси.
Общая методика приготовления 4-алкилтетрагидросахаринов. Раствор метил-2-бензилтио-6-алкилциклогекс-2-ен-карбоксилата и 2-бензилтио-6-алкилциклогекс-1-ен-карбоксилата (1 - 10 ммоль в виде смеси) в 10 мл двухлористого метилена разбавляли 20 - 50 мл ледяной уксусной кислоты и 1 - 5 мл воды, смесь охлаждали до -10oC, и газообразный хлор пробулькивали через смесь до прекращения экзотермической реакции. Смесь перемешивали в течение еще 10 мин и доводили до сухого состояния, получали смесь метил-2-хлорсульфонил-6-алкилциклогекс-1-ен-карбоксилатом, которую растворяли в 10 мл тетрагидрофурана и добавляли к 25 мл концентрированного раствора гидроксида аммония с охлаждением на бане из ацетона со льдом. После перемешивания в течение 2 ч реакционную смесь концентрировали в вакууме, остаток переносили в воду, подкисляли до величины pH 1 хлористоводородной кислотой с концентрацией 2 н. и экстрагировали двухлористым метиленом. Органическую фазу сушили и концентрировали в вакууме, в результате чего получали смесь метил-2-аминосульфонил-6-алкилциклогекс-2-ен-карбоксилата с 2-аминосульфонил-6-алкилциклогекс-1-ен-карбоксилатом. Смесь растворяли в метаноле и добавляли к свежеприготовленному раствору метоксида натрия (10 - 50 ммоль), и его перемешивали при температуре окружающей среды в течение 12 ч. Реакционную смесь концентрировали в вакууме, разбавляли водой и экстрагировали простым эфиром. Органическую фазу отбрасывали, а водную фазу подкисляли до величины pH 1 концентрированной хлористоводородной кислотой и экстрагировали двухлористым метиленом. Из органических экстрактов после промывки рассолом, сушки и упаривания досуха получали 4-алкил-4,5,6,7-тетрагидробензизотиазол-3-он-1,1-диоксид или 4-алкилтетрагидросахарин (табл. 10).
Смесь, состоящую из 4-алкил-4,5,6,7-тетрагидробензизотиазол-3-он-1,1-диоксида (4-алкилтетрагидросахарина) (1,0 эквивалент), хлорметилфенилсульфида (1,5 эквивалента) и бромистого тетрабутиламмония (0,2 эквивалента) в толуоле (25 мл на 1 г сахарина), грели в сосуде с обратным холодильником в среде азота в течение 16 - 24 ч и затем охлаждали до комнатной температуры. Образовавшуюся смесь упаривали досуха, и остаток подвергали хроматографии на силикагеле, элюируя смесь гексанов с двухлористым метиленом (взятыми в соотношении от 1: 1 до 1:3), в результате чего получали соответствующий 2-фенилтиометил-4- алкил-4,5,6,7-тетрагидробензизотиазол-3-он-1,1-диоксид или 2-фенилтиометил-4-алкилтетрагидросахарин (табл. 11).
Раствор 2-фенилтиометил-4-алкилтетрагидросахарина (1,0 эквивалент) обрабатывали хлористым сульфурилом (1,5 эквивалента) и перемешивали в течение 2 ч. Образовавшийся желтый раствор переводили в сухое состояние, получая 2-хлорметил-4-алкилтетрагидросахарин, который обрабатывали 2,6-дихлорбензойной кислотой (1,1 эквивалента), безводным карбонатом калия (1,5 эквивалента) и бромистым тетрабутиламмонием (0,2 эквивалента) в диметилформамиде (25 мл) при 70oC в течение 1 ч. Образовавшуюся смесь концентрировали в вакууме, разбавляли этилацетатом (100 мл) и фильтровали. Фильтрат промывали водой, насыщенным раствором бикарбоната натрия, водой и рассолом. Органическую фазу концентрировали в вакууме, и остаток очищали испарительной хроматографией на силикагеле (элюируя смесью двухлористого метилена с гексанами, взятыми в соотношении 2:1), в результате чего получали 4-алкил-4,5,6,7-тетрагидро-2-сахаринилметил-2,6-дихлорбензоат (табл. 12).
Следуя методике, аналогичной описанной в примере 1AA, 2-хлорметил-4-изопропил-4,5,6,7-тетригдробензизотиазол-3-он-1,1- диоксид обрабатывали 2,6-дихлор-3[[2-(N, N-диметиламино)этил] -N-метиламиносульфонил] бензойной кислотой (синтез 20F), в результате чего получали 4-изопропил-4,5,6,7-тетрагидро-2-сахаринилметил-2,6-дихлор-3- [[2-(N,N-диметиламино)-этил]-N-метиламиносульфонил] бензоатгидрохлорид; температура плавления составляла 121oC (идет разложение).
Пример 10. Получение метил-2,2-диметилциклогексан-6-он-карбоксилата. К суспензии, состоящей из безводного иодида меди (I) (70,0 г, 0,37 моль) в безводном этиловом эфире (500 мл), при 0oC добавляли свободный от галогенида метиллитий (520 мл в виде 1,4 М раствора в простом эфире, 0,73 моль). После перемешивания при 0oC в течение 15 мин добавляли раствор 3-метил-2-циклогексенона (20,0 г, 0,18 моль) в простом эфире (50 мл), и перемешивание продолжали еще в течение часа. К образовавшейся смеси добавляли тетрагидрофуран (50 мл) и гексаметилфосфорамид, а спустя 15 мин, метилцианоформат (45,0 г, 0,53 моль) в тетрагидрофуране (20 мл), и реакционную смесь нагревали до комнатной температуры и перемешивали в течение 3 ч. Реакцию тушили хлористоводородной кислотой с концентрацией 2 н. (50 мл). Слои разделяли, и водный слой экстрагировали диэтиловым эфиром (1 раз, 500 мл). Объединенные органические экстракты промывали насыщенным раствором хлорида аммония (3 раза по 50 мл), водой (2 раза по 50 мл), рассолом (1 раз, 50 мл) и сушили (над сульфатом натрия). Удалением растворителя в вакууме и очисткой посредством разгонки по Кугелрору получали 34,0 г (выход 99%) метил-2,2-диметилциклогексан-6-он-карбоксилата; температура кипения при давлении 0,6 мм рт.ст. составляла 80-84oC.
Следуя методике, описанной выше в примере 9D, циклогексанон превращали в 4,4-диметил-4,5,6,7-тетрагидро-2-сахаринил-метил-2,6- дихлорбензоат; температура плавления составляла 121-123oC.
Пример 11. Следуя методике, описанной в синтезе 18A, 5 г 2-бром-N,N-диметиланилин превращали в 3,5 г N,N-диэтил-2-диметиламинобензамида. Амид подвергали взаимодействию по методике, описанной в синтезе 18B, в результате чего получали 65 мг 4-диметиламиносахарина с температурой плавления 228-229oC при кристаллизации из смеси простого эфира с гексаном. Смесь, состоящую из 11,1 г 2,6-дихлорбензоилхлорида, 1,9 г п-формальдегида и 0,1 г плавленого хлорида цинка, грели при 100oC в течение 2 ч и затем подвергали вакуумной перегонке, в результате чего получали 3,5 г хлорметил-2,6-дихлорбензоата, собранного при температуре выше 145oC в условиях разрежения, который отверждался при охлаждении; температура плавления составляла 70-72oC. К раствору, состоящему из 4-диметиламиносахарина и 0,1 мл диизопропилэтиламина в 1 мл сухого ацетонитрила, добавляли 100 мг хлорметил-2,6-дихлорбензоата. Смесь перемешивали при комнатной температуре в течение 48 ч и затем при 50oC в течение 24 ч, делая это до момента, когда тонкослойная хроматография (при использовании двухлористого метилена) начинала указывать на завершение реакции. Смесь выливали в этилацетат и экстрагировали насыщенным раствором бикарбоната натрия. Органический слой сушили, и растворитель удаляли при пониженном давлении. Хроматографическим методом с использованием двухлористого метилена получали 15 мг 4-диметиламино-2-сахаринилметил-2,6-дихлорбензоата.
Результаты биологического тестирования.
При проведении измерений константы ингибирования Ki у комплекса, ингибирующего человеческую лейкоцитную эластазу, рассматривают "истинно обратимые константы ингибирования", обычно относящиеся к конкурирующим ингибиторам. [Cha, Biochem. Pharmacol, 24, 2177-2185, 1975]. Соединения, отвечающие настоящему изобретению, не образуют, однако, истинно обратимые ингибирующие комплексы, но, в какой-то мере, потребляются ферментом. Таким образом, вместо измерения константы Ki, рассчитывали константу K
Скорость инактивации kon у ферментативной активности определяли для испытуемых соединений, измеряя активность фермента у аликвоты соответствующего фермента в функции от времени после добавления испытуемого соединения. Строя логарифмическую зависимость активности фермента от времени, получали наблюдаемую скорость инактивации kobs, которая может быть выражена соотношением kobs = ln2/t1/2, где t1/2 - время, необходимое для падения активности фермента на 50%. Тогда скорость инактивации оказывается равной kon = kobs/[I], где [I] - концентрация ингибирующего соединения.
Константу реактивации koff определяли аналогичным образом, и тогда константа ингибирования K
K
Значения, полученные для kon и K
Примеры рецептур для инъекций приведены в табл.14.
Пример E. Рецептура, содержащая раствор активного соединения (100 мг) и сахарозу в ампуле объемом 20 мл.
Эксципиент - Количество, мг на ампулу
Активное соединение - 100,0
Сахароза - 250,0
Хлористый натрий - 5,0
0,5 М раствор молочной кислоты - 1,0 мл
1,0 М раствор гидроксида натрия - 57,0 мкл
Вода для инъекций до - 5,0
pH (интервал 3,70 - 4,30) - 4,00
Указанные выше рецептуры получали сушкой вымораживанием, т.е. без воды. Указанный выше объем воды используют для восстановления высушенной вымораживанием рецептуры непосредственно перед инъекцией пациенту.
В указанных выше рецептурах сахароза и хлористый натрий выполняют функции стабилизаторов, а молочная кислота и гидроксид натрия - функции буфера.
Способ сушки вымораживанием в получении вышеуказанной рецептуры и аналогичных составов следующий.
Ампулы требуемого размера выбирали таким образом, чтобы заполнить их рецептурой в соответствии с требуемой дозировкой. При выборе ампул, соответствующих дозировке, объем заполнения не должен превышать определенную часть объема ампулы. Так, например, заполнение смесью объемом в 5 мл должно производиться в ампулу объемом не менее 10 мл. После заполнения ампулы загружали в сушильную камеру и помещали непосредственно на полки холодильника, предварительно охлажденные до 4oC. В ряд ампул помещали термопары для слежения за температурой составов в ходе процесса лиофилизации. Затем температуру ампул уравновешивали с температурой полок (4oC) перед тем как охладить их до температуры -40oC в случае рецептуры на основе сахарозы и -30oC для рецептуры на основе маннита. При достижении температур -40oC и -30oC соответственно для рецептур на основе сахарозы и маннита ампулы выдерживали при таких температурах в течение 2 ч для обеспечения полного замораживания составов. (На этой стадии для рецептуры на основе маннита осуществляли также процедуру отжига). По истечении указанного периода времени змеевик конденсатора охлаждали до -60oC и включали вакуумный насос с целью откачки камеры конденсатора с последующим проведением процесса первичной и вторичной сушки. В ходе процесса первичной сушки главный клапан между конденсатором и сушильной камерой открывали и сушильную камеру откачивали до остаточного давления 100 микрон при продувке газообразным азотом. Такая часть лиофилизационного цикла (первичная сушка) требует от 40 до 50 ч. Процесс первичной сушки завершался при полном исчезновении льда с замороженной матрицы. В ходе процесса вторичной сушки температуру повышали с -20oC или -30oC до +25oC с целью удаления всей остаточной влаги, которая не удалялась в ходе первичной сушки. Время проведения такой вторичной сушки составляет примерно 15 ч.
После завершения процесса вторичной сушки главный клапан закрывали и сушильную камеру заполняли азотом с целью поддержания небольшого вакуума в камере. Вводили в действие запорный плунжер и ампулы закрывали пробками. Затем сушильную камеру уравновешивали до атмосферного давления, дверь камеры открывали, ампулы доставали и упаковывали в гофрированные упаковки. Затем ампулы хранили при предписанной температуре до заполнения водой для инъекций.
Твердые дозированные рецептуры.
Твердые дозированные формы для орального применения включают капсулы, таблетки, пилюли, порошки и гранулы. В такой твердой дозированной форме активное соединение смешано, по крайней мере, с одним общепринятым инертным эксципиентом (или носителем) таким, как цитрат натрия или кальций ди фосфатом, или (a) такими наполнителями, или объемными агентами, как, например, крахмалы, лактоза, сахароза, глюкоза, маннит и кремниевая кислота, (b) такими связующими агентами, как, например, карбоксиметилцеллюлоза, альгинаты, желатин, поливинилпирролидон, сахароза и аравийская камедь, (c) такими увлажнителями, как глицерин, (d) такими дезинтегрирующими агентами, как, например, агар-агар, карбонат кальция, крахмал картофельный или Тапиоки, альгиновая кислота, некоторые сложные силикаты и карбонат натрия, (e) такими замедлителями растворения, как, например, парафин, (f) такими ускорителями абсорбции, как, например, соединения четвертичного аммония, (g) такими смачивающими агентами, как, например, цетиловый спирт и моностеарат глицерина, (h) такими адсорбентами, как, например, каолин и бентонит, и (i) такими смазывающими агентами, как, например, тальк, стеарат кальция, стеарат магния, твердые полиэтиленгликоли, лаурил сульфат натрия или их смесь. В случае капсул, таблеток и пилюль дозированные формы могут также содержать буферные агенты.
Пример E. Мягкие или твердые капсулы.
Компоненты - Количества
Активное соединение или его соль - 1,00 мг
Крахмал - 51,00 мг
Моногидрат лактозы - 203,33 мг
Поливинилпирролидон - 4,30 мг
Лаурилсульфат натрия - 0,17 мг
Карбоксиметилцеллюлоза - 8,50 мг
Очищенная вода для влажного гранулирования - q.s.
Стеарат магния - 1,70 г
Указанной выше рецептурой заполняли капсулу таким образом, чтобы она содержала 170 мг рецептуры.
Примеры G и H приведены в табл. 15.
Указанными выше рецептурами заполняли капсулу так, чтобы она содержала 170 мг рецептуры.
Жидкие дозированные рецептуры.
Жидкие дозированные формы для орального применения включают фармацевтически приемлемые эмульсии, растворы, суспензии, сиропы и эликсиры. В качестве основы для жидких рецептур могут также готовиться натриевые рецептуры. Помимо активных соединений, жидкие дозированные формы могут содержать инертные разбавители, традиционно используемые в данной области техники, такие как вода или другие растворители, такие солюбилизирующие агенты и эмульгаторы, как, например, этиловый спирт, изопропиловый спирт, этил карбонат, этил ацетат, бензиловый спирт, бензил бензоат, пропиленгликоль, 1,3-бутиленгликоль, диметилформамид, масла, особенно масло хлопкового семени, масло арахиса, масло пшеничного семени, оливковое масло, касторовое масло и кунжутное масло, глицерин, тетрагидрофурфуриловый спирт, полиэтиленгликоли и сложные эфиры жирных кислот и сорбита либо смеси таких веществ и т.п. Помимо таких инертных разбавителей, композиция может также включать такие адъюванты, как смачивающие агенты, эмульгирующие и суспендирующие агенты, подслащивающие, вкусовые и ароматизирующие агенты.
Суспензии, помимо активного соединения, могут содержать такие суспендирующие агенты, как, например, этоксилированные изостеариловые спирты, полиоксиэтилен сорбит, полиэтиленгликоли различной молекулярной массы, а также сложные эфиры сорбита, микрокристаллическую целлюлозу, метагидроксид алюминия, бентонит, агар-агар и трагант, или смеси указанных веществ и т.п.
Аэрозольные рецептуры.
Композиции, предназначенные для применения в виде аэрозолей, готовят растворением активного соединения в воде или таком подходящем растворителе, как, например, простой эфир спирта или другие инертные растворители, и смешиванием с летучим пропеллентом с последующим помещением в резервуар, находящийся под давлением, снабженный дозирующим клапаном, обеспечивающим выделение содержимого в виде капель подходящего размера.
В качестве сжиженного пропеллента обычно используют вещество, имеющее точку кипения ниже температуры окружающей среды при атмосферном давлении. При применении в композициях, предназначенных для получения аэрозолей медицинского назначения, сжиженный пропеллент должен быть нетоксичным веществом. Среди подходящих сжиженных пропеллентов, которые могут использоваться, можно отметить низшие алканы, содержащие до пяти углеродных атомов, такие как бутан и пентан, или такой алкилхлорид, как метил, этил или пропилхлориды. Другими подходящими сжиженными пропеллентами являются фторированные и фторхлорзамещенные алканы, например, такие, которые выпускаются под торговой маркой "Freon" и "Genetron". Могут также использоваться смеси упомянутых выше пропеллентов.
Предпочтительными сжиженными пропеллентами являются не содержащие хлора пропелленты, например, 134a (тетрафторэтан) и 227c (гептафторпропан), которые могут использоваться в соответствии с описанным выше. Обычно в таких аэрозольных рецептурах используются такие сорастворители, как эфир, спирт или гликоль.


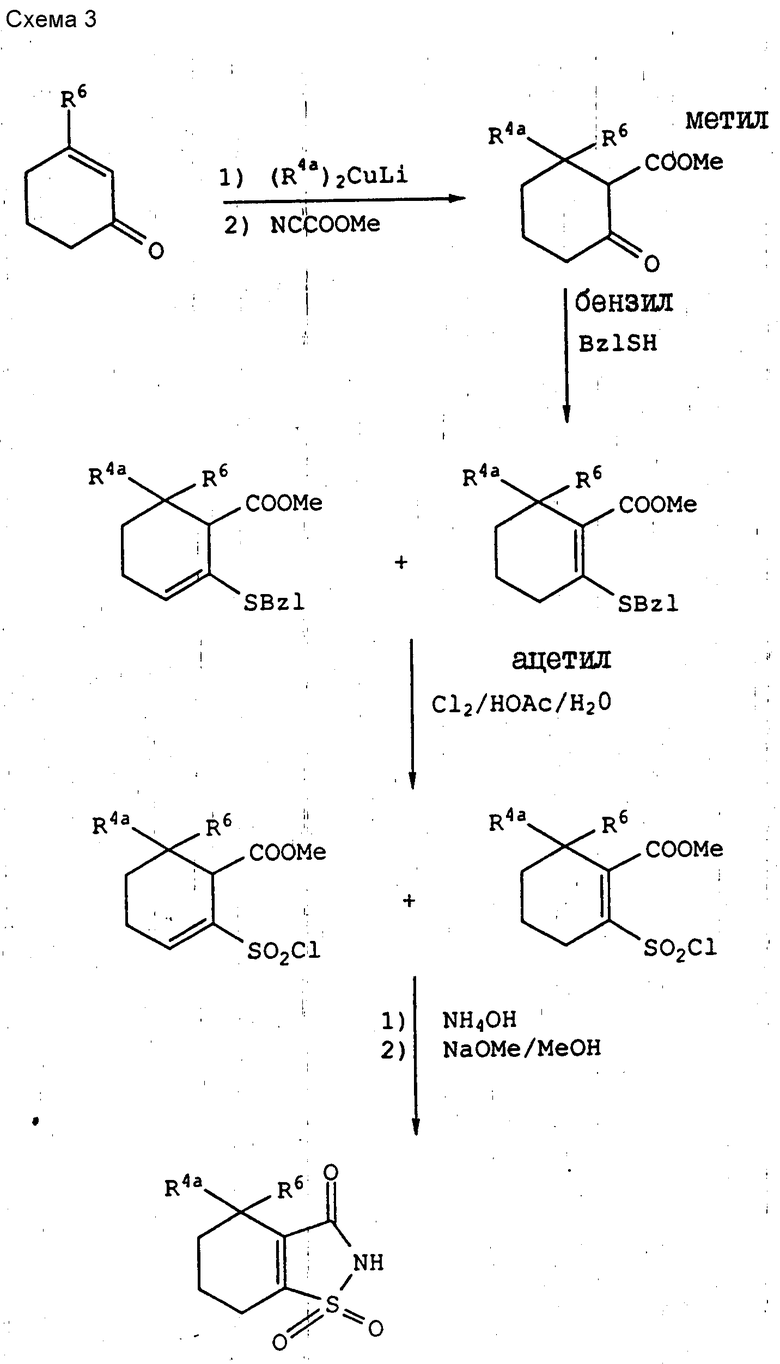
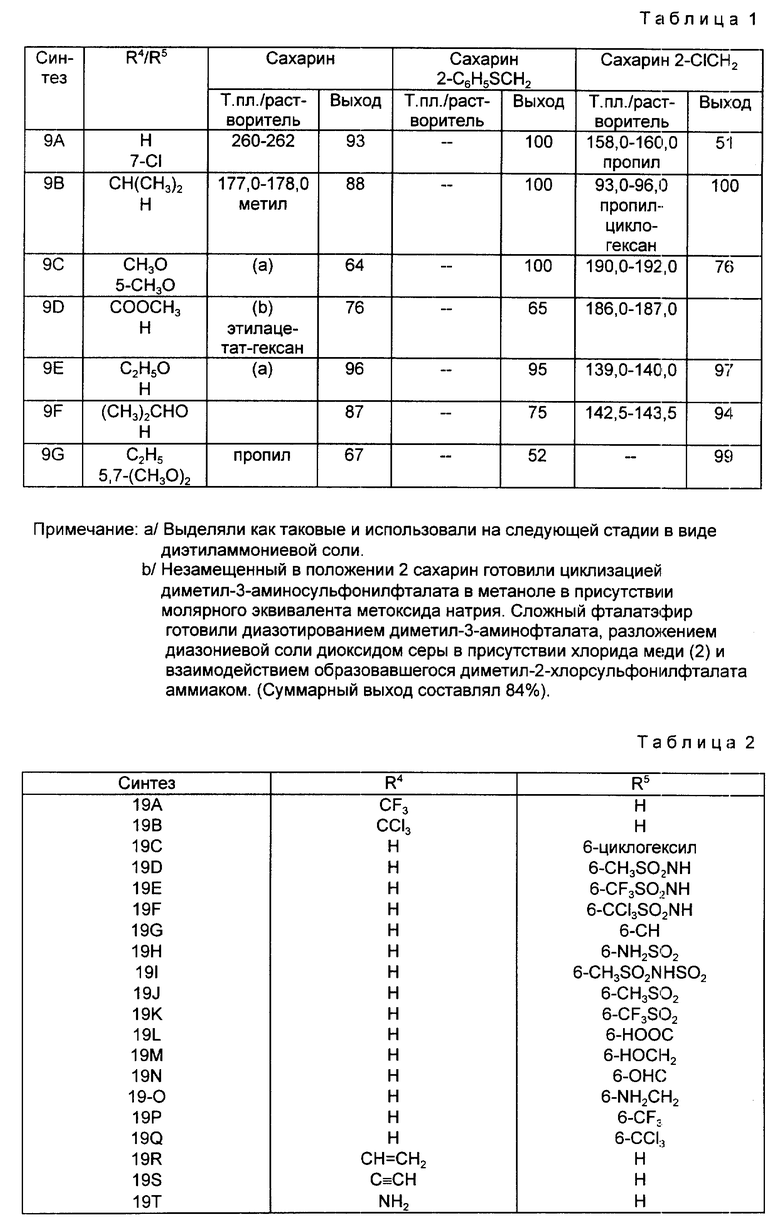
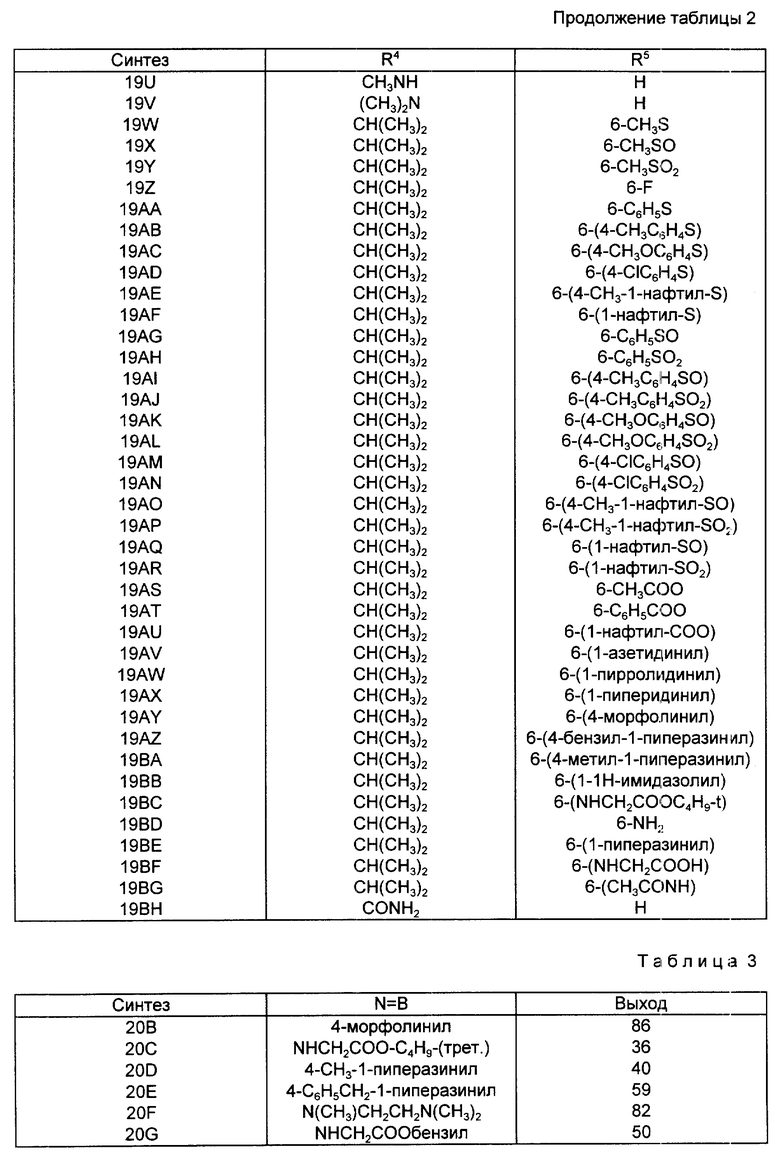
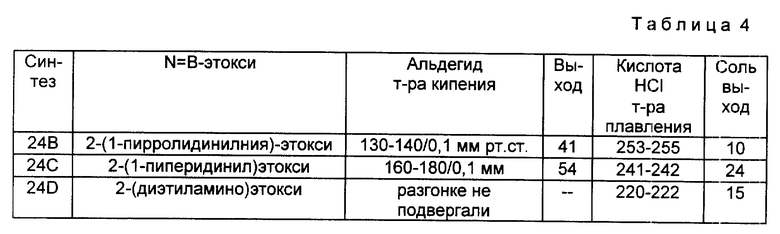
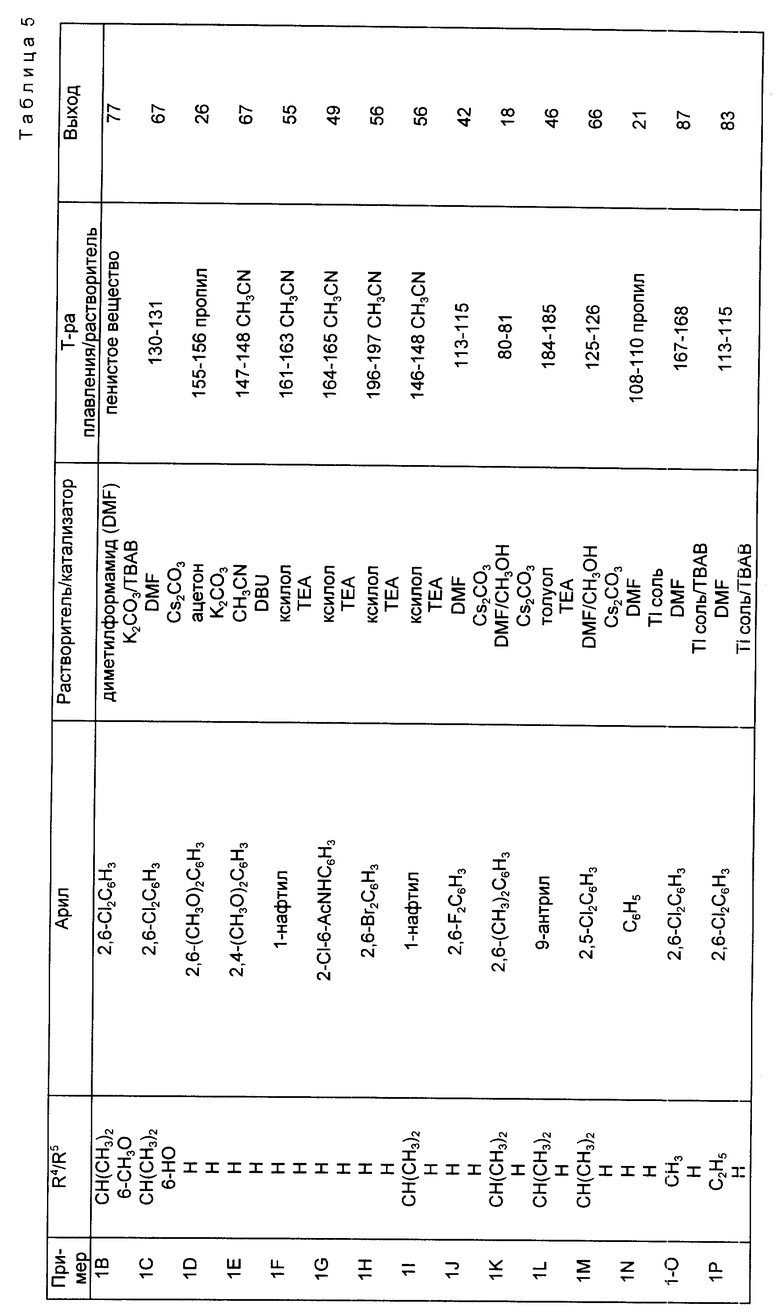

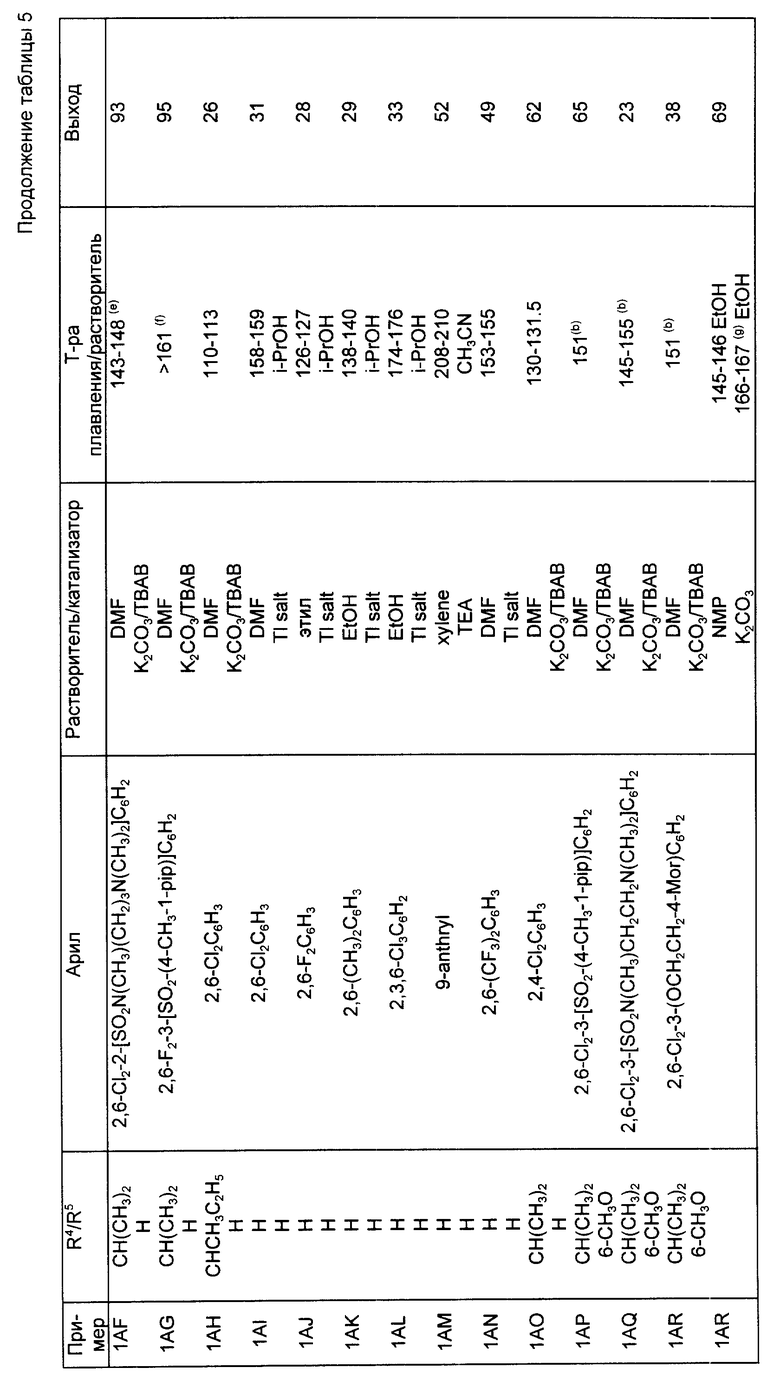
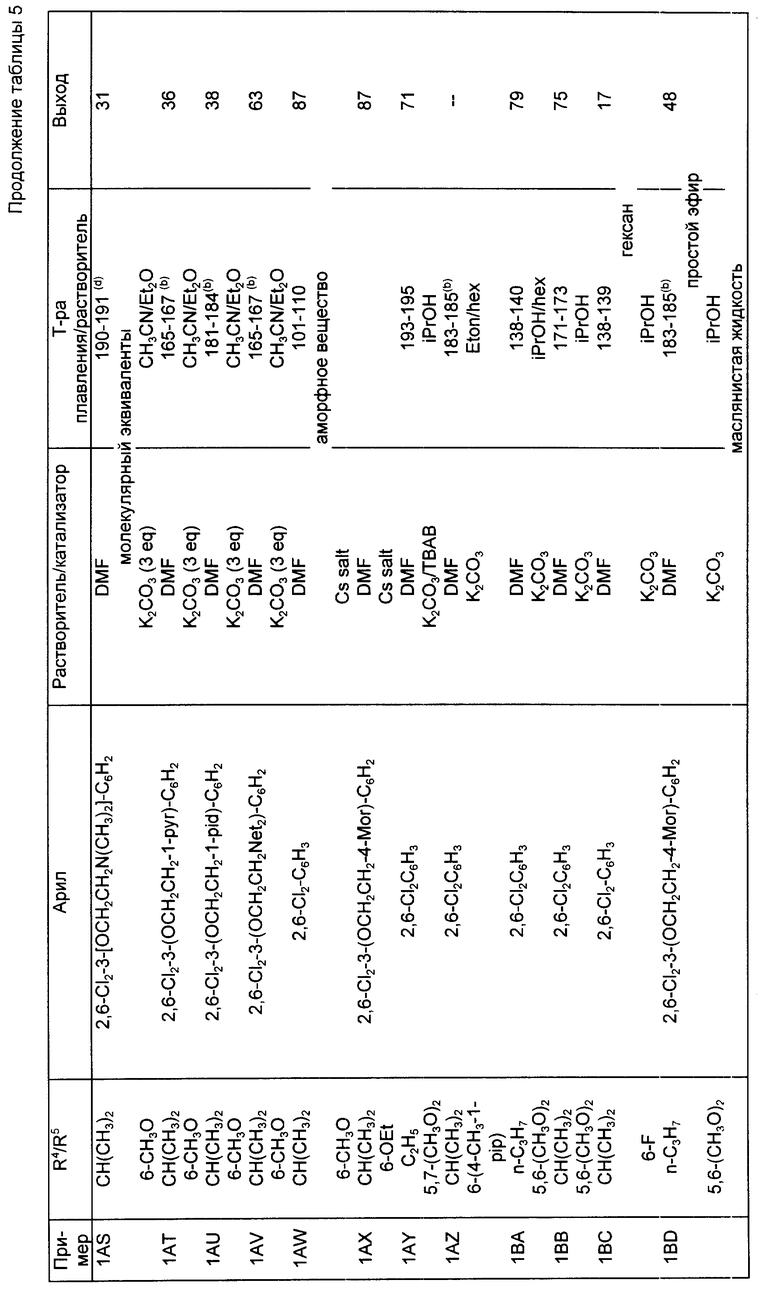
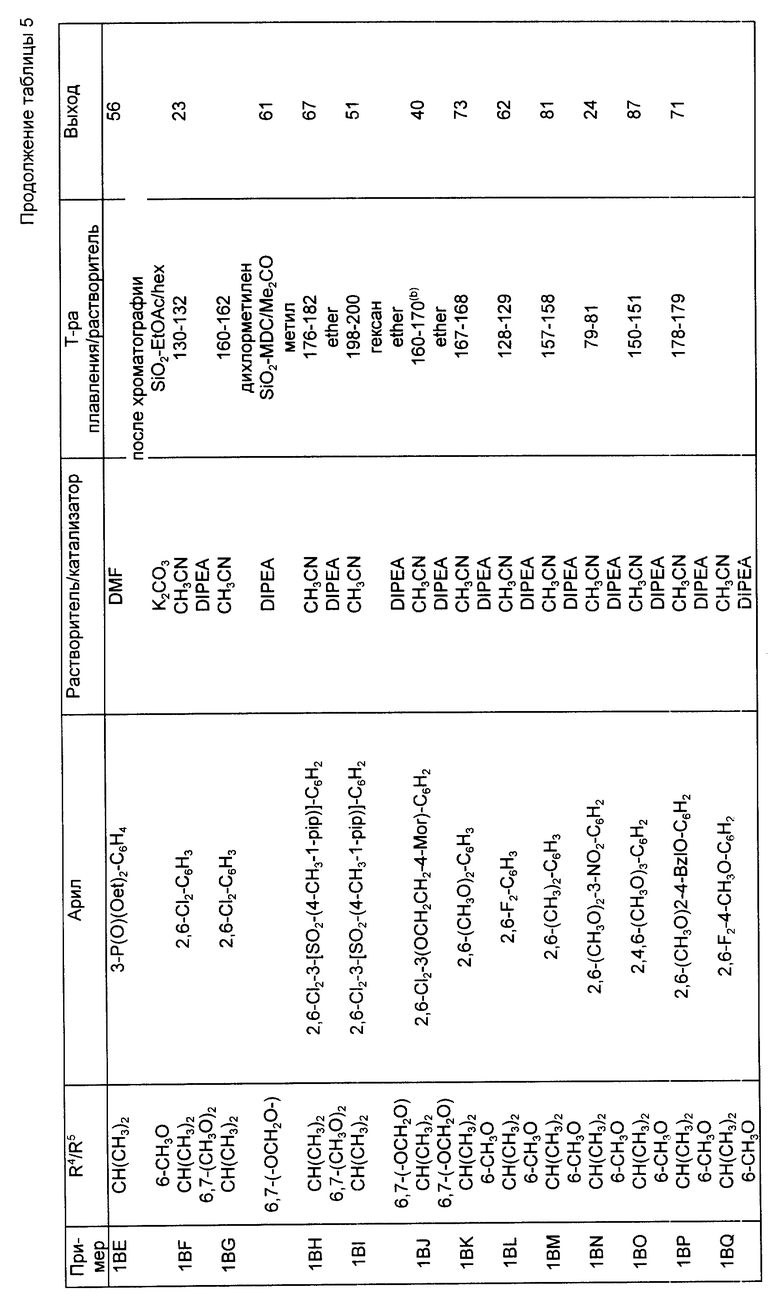

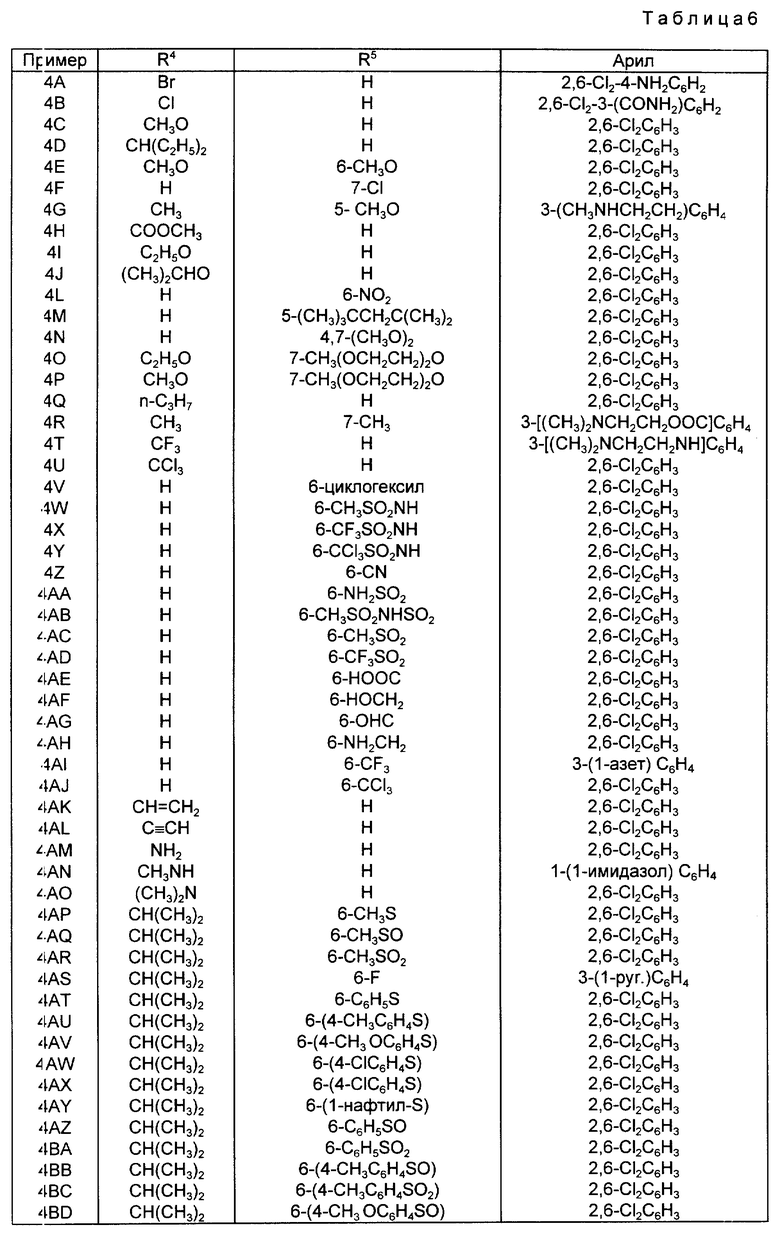
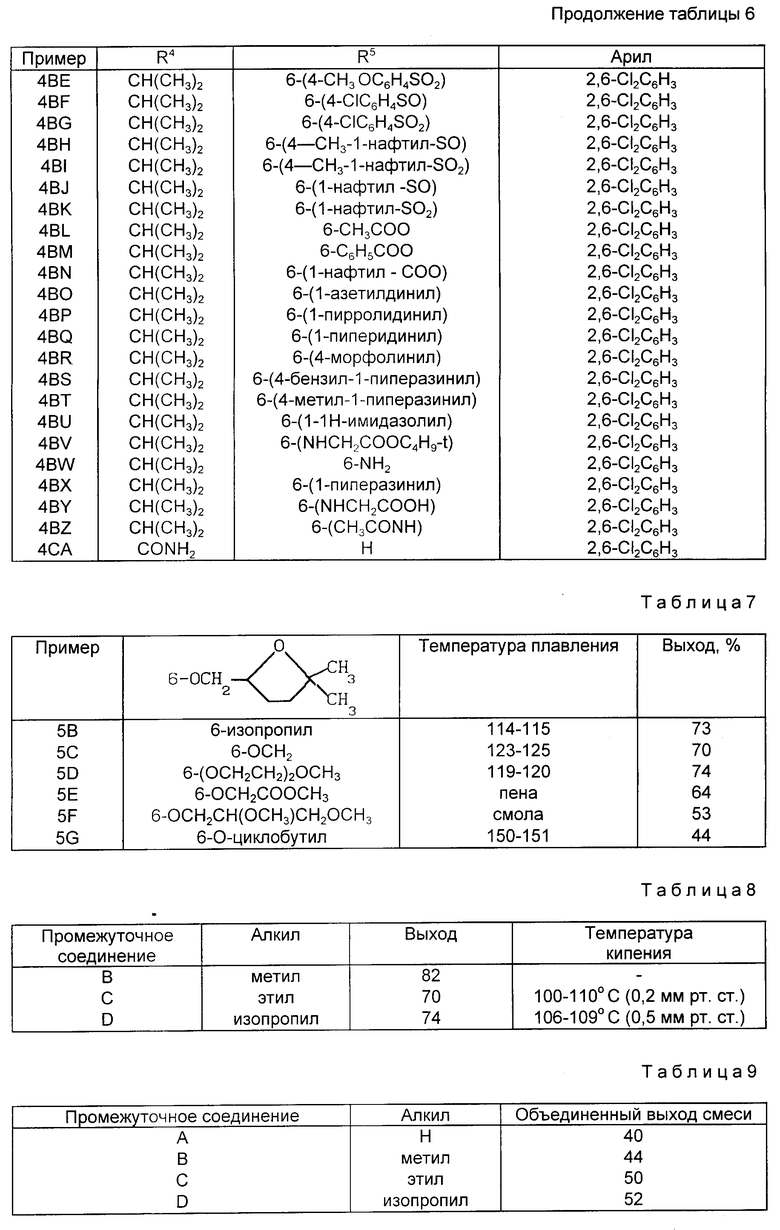
2-Сахаринилметиларилкарбоксилаты получают взаимодействием галометилсахарина с соответствующей кислотой или ее солью. Согласно второму способу сахарин подвергают взаимодействию со сложным хлорметиловым эфиром алкилкарбоновой кислоты. 4-Изопропил-6-метокси-2-сахаринилметил-2,6-дихлор-3-карбоксиметиламиносульфонилбензоат, выход 45%, т.пл. 204 - 206oC. Соединения ингибируют ферментативную активность протеолитических ферментов. 5 с. и 10 з.п. ф-лы, 15 табл.
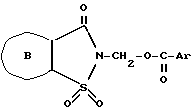
где кольцо B выбирают из следующих групп, сконденсированных с изотиазольным циклом стороной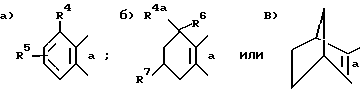
где Ar - фенильная, нафтильная или антрильная группа, замещенная одним - тремя одинаковыми или разными заместителями, выбранными из группы, включающей низший алкил, перфтор(низший)алкил, перхлор(низший)алкил, (низший)алкокси, галоген, PO-((низший)алкокси)2, амино, (низший)алкиламино, ди(низший)алкиламино, (низший)алканоиламино, (низший)алкоксикарбонил, гидрокси, бензилокси, карбокси(низший)алкокси, -SO2-N = B, -(алкилен)-N = B или -O-(алкилен)-N = B, где в каждом случае N = B представляет собой амино, (низший)алкиламино, ди(низший)алкиламино, 1-азетидинил, 1-пирролидинил, 1-пиперидинил, 4-морфолинил, 1-пиперазинил, 4-(низший)-алкил-1-пиперазинил, 4-бензил-1-пиперазинил, 1-имидазолил;
R4 выбран из группы, включающей водород, галоген, низший алкил, перфтор(низший)алкил, перхлор(низший)алкил, (низший)алкенил, (низший)алкинил, амино, (низший)алкиламино, ди(низший)алкиламино, (низший)алкокси, (низший)алкоксикарбонил, фенил или карбоксамидо;
R5 - водород или один - два одинаковых или разных заместителя в любом из положений 5, 6 или 7, выбранных из группы, включающей галоген, циано, нитро, N = B, (низший)алкилсульфониламино, полифтор(низший)алкилсульфониламино, полихлор(низший)алкилсульфониламино, аминосульфонил, (низший)алкил, полифтор(низший)алкил, полихлор(низший)алкил, циклоалкил, (низший)алкокси, гидрокси, карбокси, гидрокси(низший)алкил, метилендиокси, формил, аминометил, (низший)алкилсульфонил, полифтор(низший)алкилсульфонил, полихлор(низший)алкилсульфонил, (низший)алкилсульфониламиносульфонил, -SR, -SOR, -SO2R, -OCOR, -O-(алкилен)-N = B, где R - низший алкил, фенил, бензил или нафтил, либо фенил или нафтил, замещенный одним - двумя заместителями, выбранными из группы, состоящей из низшей алкильной группы, низшей алкоксигруппы или галогена, а N = B имеет указанное значение, или R5 представляет собой 5- или 6-членное насыщенное кольцо, сконденсированное с сахарином в положениях 5, 6 или 6, 7, причем упомянутое кольцо содержит два гетероатома, выбранных из группы, состоящей из азота, кислорода или серы, либо метилированное производное упомянутого кольца;
R4a - водород, низшая алкильная группа или фенильная группа;
R6 - водород или первичная низшая алкильная группа либо R4a и R6 вместе образуют спироциклопропильное кольцо;
R7 - водород или низший алкокси;
A - метилен, этилен или диметилметилен,
или их кислотно-аддитивные соли основного фрагмента, либо соли присоединения основания кислотного фрагмента при условии, что когда обе группы R4 и R5 представляют собой водород, группа Ar не может быть фенильной, 2,4-дихлорфенильной или 4-нитрофенильной группой.
4-изопропил-6-метокси-2-сахаринилметил-2,6-диметилбензоат,
4-изопропил-6-метокси-2-сахаринилметил-2,6-диметоксибензоат,
4-изопропил-6-метокси-2-сахаринилметил-2,6-дихлор-3[2-(4-морфолинил)этокси]бензоат,
4-изопропил-6-метокси-2-сахаринилметил-2,6-дихлорбензоат,
4-изопропил-6-метокси-2-сахаринилметил-2,6-дихлор-3-(1-пиперидинилэтокси)бензоат,
4-изопропил-6-метокси-2-сахаринилметил-2,6-дихлор-3-(1-пирролидинилэтокси)бензоат,
4-изопропил-6-метокси-2-сахаринилметил-2,6-дихлор-3[2-(N, N-диэтиламино)этокси]бензоат,
4-изопропил-6-метокси-2-сахаринилметил-2,6-дихлор-3[2-(1-пирролидинил)этокси]бензоат,
4-изопропил-6-метокси-2-сахаринилметил-2,6-дихлор-3-(4-метил-1-пиперазинилсульфонил)бензоат,
4-изопропил-6-метокси-2-сахаринилметил-2,6-дихлор-3-[N-2-(диметиламино)этил-N-метиламиносульфонил]бензоат,
4-изопропил-6-метокси-2-сахаринилметил-2,6-дихлор-3-гидроксибензоат,
4-изопропил-6-изопропокси-2-сахаринилметил-2,6-дихлорбензоат,
4-изопропил-6-[2-(2-метоксиэтокси)этокси] -2-сахаринилметил-2,6-дихлорбензоат,
4-изопропил-6-[2-(4-морфолинил)этокси] -2-сахаринилметил-2,6-дихлорбензоат,
4-изопропил-6,7-метилендиокси-2-сахаринилметил-2,6-дихлор-3-[2-(4-морфолинил)этокси]бензоат,
4-пропил-5,6-диметокси-2-сахаринилметил-2,6-дихлор-3-[2-(4-морфолинил)этокси]бензоат,
6-этокси-4-изопропил-2-сахаринилметил-2,6-дихлорбензоат,
5,6-диметокси-4-изопропил-2-сахаринилметил-1-нафтилкарбоксилат,
4-изопропил-6,7-метилендиокси-2-сахаринилметил-2,6-дихлорбензоат,
6-(2,3-дигидроксипропокси)-4-изопропил-2-сахаринилметил-2,6-дихлорбензоат и
6-(2,3-диметоксипропокси)-4-изопропил-2-сахаринилметил-2,6-дихлорбензоат.
6-(бензилоксикарбонил)метокси-4-изопропил-2-сахаринилметил-2,6-дихлорбензоат,
6-(трет-бутоксикарбонил)метокси-4-изопропил-2-сахаринилметил-2,6-дихлорбензоат,
4-изопропил-6-(метоксикарбонил)метокси-2-сахаринилметил-2,6-дихлорбензоат и
6-гидрокси-4-изопропилсахаринилметил-2,6-дихлорбензоат.
4,5,6,7-тетрагидро-2-сахаринилметил-2,6-дихлорбензоат,
4-метил-4,5,6,7-тетрагидро-2-сахаринилметил-2,6-дихлорбензоат,
4-этил-4,5,6,7-тетрагидро-2-сахаринилметил-2,6-дихлорбензоат,
4-изопропил-4,5,6,7-тетрагидро-2-сахаринилметил-2,
6-дихлорбензоат или
4,4-диметил-4,5,6,7-тетрагидро-2-сахаринилметил-2,6-дихлорбензоат.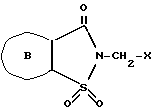
где B - кольцо а или б;
X - галоген,
подвергают взаимодействию с арилкарбоновой кислотой формулы
ArCOOH,
где Ar имеет указанные в п.1 значения,
в присутствии акцептора кислоты или с солью щелочного металла или таллия указанной кислоты.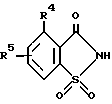
где R4 и R5 имеют указанные в п.1 значения,
подвергают взаимодействию с хлорметиловым эфиром арилкарбоновой кислоты формулы
ArCOOCH2Cl,
где Ar имеет указанные в п.1 значения,
в присутствии акцептора кислоты.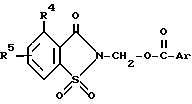
где Ar - фенильная, нафтильная или антрильная группа, замещенные одним - тремя одинаковыми или разными заместителями, выбранными из группы, включающей низший алкил, перфтор(низший)алкил, перхлор(низший)алкил, низший алкокси, галоген, амино, (низший)алкиламино, ди(низший)алкиламино, (низший)алканоиламино, карбо(низший)алкокси, гидрокси, бензилокси, карбокси(низший)алкокси, -SO2-N = B, (алкилен)-N = B или -O-(алкилен)-N = B, где группа N = B в каждом случае представляет собой амино, (низший)алкиламино, ди(низший)алкиламино, 1-азетидинил, 1-пирролидинил, 1-пиперидинил, 4-морфолинил, 1-пиперазинил, 4-(низший)алкил-1-пиперазинил, 4-бензил-1-пиперазинил, 1-имидазолил;
R4 выбран из группы, включающей водород, галоген, первичная или вторичная низшая алкильная группа, перфтор(низший)алкил, перхлор(низший)алкил, низший алкенил, низший алкинил, амино, (низший)алкиламино, ди(низший)алкиламино, низший алкокси, карб(низший) алкокси или фенил;
R5 - водород или один - два заместителя в любом из положений 5, 6 или 7, выбранных из группы, включающей галоген, циано, нитро, N = B, (низший)алкилсульфонил, полифтор(низший)алкилсульфониламино, полихлор(низший)алкилсульфониламино, аминосульфонил, низший алкил, полифтор(низший)алкил, полихлор(низший)алкил, циклоалкил, низший алкокси, гидрокси, карбокси, гидрокси(низший)алкил, формил, аминометил, (низший)алкилсульфонил, полифтор(низший) алкилсульфонил, полихлор(низший)алкилсульфонил, (низший) алкоксиполи(низший)алкиленокси, -SR, -SOR, -SO2R или OCOR, где R - низший алкил, фенил или нафтил, либо фенил или нафтил, замещенный одним - двумя заместителями, выбранными из группы, состоящей из низшей алкильной группы, низшей алкоксигруппы или галогена, группа N = B имеет указанное значение, или их кислотно-аддитивные соли основных фрагментов, либо соли, либо присоединения оснований к кислотным фрагментам при условии, что, когда обе группы R4 и R5 представляют собой водород, группа Ar не может быть фенильной, 2,4-дихлорфенильной, 4-нитрофенильной группой.
где R4 и R5 имеют указанные в п.12 значения,
подвергают взаимодействию с арилкарбоновой кислотой формулы
ArCOOH,
где Ar имеет указанные в п.12 значения,
в присутствии акцептора кислоты.
| WO, 9013549, 01.05.90, C 07 D 417/06 | |||
| US, 4783473, 08.11.88, A 61 K 31/34 . |
