Изобретение связано с новыми производными 2-замещенного сахарина, которые ингибируют ферментативную активность протеолитических ферментов, с фармацевтическими композициями, содержащими эти производные, со способом использования их при лечении дегенеративных заболеваний и со способами получения этих производных.
При лечении заболеваний, связанных с перерождением тканей, таких как эмфизема, рематоидный артрит и панкреатит, при которых основной составной частью является протеолиз, используют ингибирование протеолитических ферментов нетоксичными реагентами. При биомедицинском исследовании широко применяются ингибиторы протеаз. Самым широко распространенным классом протеолитических ферментов являются серинпротеазы. Некоторые серинпротеазы исходя из специфичности их субстратов характеризуются как химотрипсинподобные или эластазаподобные. Химотрипсин или химотрипсинподобные ферменты обычно рвут пептидные связи в белках в том месте, где аминокислотный остаток на стороне карбонила обычно представляет собой Trp, Tyr, Phe, Met, Leu или другой аминокислотный остаток, который содержит ароматические или большие алкильные боковые цепи. Эластаза или эластазаподобные ферменты обычно рвут пептидные связи в том месте, где аминокислотный остаток на карбонильной стороне связи обычно представляет собой Ala, Val, Ser, Leu или другие аналогичные, меньшие но размерам аминокислоты. Как химотрипсинподобные, так и эластазаподобные ферменты были обнаружены в лейкоцитах, клетках молочной железы и в соке поджелудочной железы высших организмов, и они выделяются многими видами бактерий, дрожжей и паразитов.
Известны несколько классов соединений, которые являются ингибиторами серинпротеазы. Например, в патенте США 4 659 855 (заявитель Powers) предложено использовать в качестве ингибиторов эластазы производные арилсульфонилфторида. Doherty и др. в патентах США 4 547 371 и 4 623 645 предлагают использование соответственно цефалоспоринсульфонов и цефалоспоринсульфоксидов, которые, как утверждается, являются сильнодействующими ингибиторами эластазы, пригодными для лечения воспалительных состояний, особенно артрита и эмфиземы.
Teshima и др.[J.Biol. Chem. 257 (9), 5085-5091 (1982)] сообщают результаты исследований по воздействию на серинпротеазы (эластазу лейкоцитов человека, эластазу поджелудочной железы свиньи, катепсин G и бычий химотрипсин Aα ) 4-нитрофениловых сложных эфиров и тиоэфиров N-трифторацетилантранилатов, 2-замещенных-4Н-3,1-бензоксазин-4-онов, 2-замещенных-4-хиназолинонов и 2-замещенных-4-хлорхиназолинов.
Cha, Biochem. Pharmacol. 24, 2177-2185 (1975) обсуждает кинетические подходы по изучению связывания ингибиторов макромолекулами, такими как ферменты, и методы определения таких параметров, как константы ингибирования, скорости реакции и концентрации связанного и несвязанного фермента.
Известно, что некоторые производные 2-замещенного сахарина обладают ингибирующей активностью относительно протеазаподобных ферментов. Например, в патенте США 4 195 023 Mulvey предлагает R1-2-R2CO-1,2-бензизотиазол-3-оны, в бензоидном кольце которых R1 представляет собой галоген, алкоксил, алкоксикарбонил, водород, алкиламино-, диалкиламино-, амино- или нитрогруппу, а R2 является водородом, алкилом, алкенилом, алкинилом, циклралкилом, галогенсодержащим фенилом, гетероарилом или замещенным гетероарилом, и R1-2-A-CO-сахарины, где R1 имеет те же значения, что и заместители бензоидного кольца в 1,2-бензизотиазол-3-онах, а A представляет собой алкил, алкенил, алкинил, циклоалкил, фторфенил, гетероарил или замещенный гетероарил. Сказано, что эти соединения обладают ингибирующей активностью относительно эластазы и могут быть пригодны для лечения эмфиземы.
В патенте США 4 276 298 Jones с сотрудниками предлагает 2-R-1,2-бензизотиазолинон-1,1-диоксиды, где R представляет собой фенил, замещенный фтором, двумя нитрогруппами, трифторметилом, цианогруппой, алкоксикарбонилом, алкилкарбонилом, карбоксилом, карбамоилом, алкилациламиногруппой, алкилсульфонилом, N,N-диалкилсульфамоилом, трифторметоксилом, трифторметилтиогруппой, трифторметилсульфонилом и трифторметилтионилом или R представляет собой пиридил, замещенный группами, указанными выше для фенила, или нитрогруппой. Сказано, что эти соединения обладают ингибирующей активностью относительно фермента протеазы, особенно ингибирующей активностью относительно эластазы, и они могут быть использованы при лечении эмфиземы, ревматоидного артрита и других воспалительных заболеваний.
Powers [Biochem. 24, 2048-2058 (1985)] представил исследования по ингибированию четырех химотрипсинподобных ферментов, катепсина G, протеаз I и II клеток крысиных молочных желез, химазы кожи человека и химотрипсина Aα N-фуроилсахарином и N-(2,4-дицианофенил)сахарином.
Svotoda и др. [Coll. Czech. Chem. Commun. 51, 1133-1139 (1986)] описывают получение 4-окси-2Н-1,2-бензотиазин-3-карбоксилатов внутримолекулярной конденсацией Дикмана сложных эфиров 2Н-1,2-бензизотиазол-3-он-2-ацетат-1,1-диоксида.
Патент США 4 263 393 (заявитель Chen), патенты США 4 350 752 и 4 363 865 (заявитель Reczek и др.) и патент США 4 410 618 (заявители Vanmeter и др.) имеют отношение к фотографическим реагентам (Reczek 4 350 752 и патент Vanmeter и др.) фотографическим красителям (Reczek 4 363 865) и фотографическим элементам и пленочным установкам (Chen). Они предлагают различные 2-замещенные сахарины, пригодные для таких использований, например, 2-ароилметилсахарины (описанные Chen), фотографические реагенты, связанные с имидометильной блокирующей группой через гетероатом (описанные в патенте Reczek 4 350 752), диффундирующие в носителе фотографические красители, связанные с атомом азота имида через 1,1-алкиленовую группу (описанные Reczek в патенте 4363865), и N-ацилметилимиды, описанные в качестве защищенных фотографических реагентов, которые включают в себя остаток органического фотографического реагента, содержащий гетероатом, через который он связан с блокирующей группой (патент Vanmeter), Reczek в патенте 4 350 752, в частности, указал как соединение 28 разновидности 2-(1-фенил-1Н-тетразол-5-ил-тиометил)сахарина, а Vanmeter, в частности, предложил ряд 2-(R1-1Н-тетразол-5-ил-тиометил)сахаринов, замещенных по метиленовой функции ароилом или трет-бутилкарбонильной группой.
Freed в патенте США 3 314 960 предложил 2-(1,1,3-триоксо-1,2-бензизотиазол-2-ил)глутаримиды, которые, как утверждается, могут быть использованы в качестве седативных средств.
В японской патентной публикации 72/00419 описан ряд 2-RZ-сахаринов, которые, как заявлено, обладают сильной активностью против пирикуляриоза риса, бактерий, вызывающих завядание оболочки риса, гельминтоспоровой пятнистости листьев риса и бактериального заболевания, вызывающего завядание листьев риса, и в которых RZ представляет собой низший алкоксил, бутоксиэтоксил, этилтиоэтоксил, ди-(низший алкил)-аминоэтоксил, этилтиогруппу, 2-хлорэтоксил, 1-(2-пропенилокси)-группу, 1-(2-пропинилокси)-группу, 2-сахаринилметоксил, феноксигруппу (или феноксил, замещенный хлором, метилом, нитро- или метилтиогруппой), фенилтиогруппу, хлорфенилтиогруппу, бензилтиогруппу (или хлорбензилтиогруппу), ацетоксигруппу, дихлорацетоксигруппу, бензоилоксигруппу (или бензоилоксигруппу, замещенную хлором или нитрогруппой), ацетилтиогруппу, дихлорацетилоксигруппу, хлорбензоилтиогруппу, метил- или этилкарбамилоксигруппу, диметилкарбамилоксигруппу, фенилкарбамилоксигруппу, этилкарбамилтиогруппу, фенилкарбамилтиогруппу, диметилтиоилкарбамотиоилгруппу, этилтиотиоилтиогруппу, этоксикарбонилтиогруппу, этокситиоилтиогруппу и этилтиокарбонилтиогруппу.
В патенте Франции 1 451 417 предлагается 2-хлорметилсахарин в качестве промежуточного соединения при получении N-метилсахарин d,l-транс-хрисантемата, используемого в качестве инсектицида, а в патенте США 3 002 884 (заявитель Lo) описаны 2-хлор-, 2-бром- и 2-йодметилсахарины, используемые в качестве фунгицидных средств.
PCT заявка WO 90/13549 (заявители Dunlap и др.) описывает производные сахарина, используемые в качестве ингибиторов протеолитических ферментов и имеющие структурную формулу: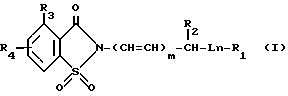
где:
L является -O, -S-, -SO- или -SO2-;
m и n каждый независимо представляет 0 или 1;
R1 представляет собой галоген, низший алканоил, 1-оксофеналенил, фенил [или фенил, замещенный галогеном, низшим алкилом, низшим алкоксилом, нитрогруппой, аминогруппой /низший алкил]-аминогруппой или ди-(низший алкил)-аминогруппой (или гетероцикл, выбранный из 1Н-(5-тетразолила), 5-оксо-1-тетразолила, 5-тиокос-1-тетразолила (если R2, определенный ниже, не является фенилтиогруппой), пиримидинила, 2-бензоксазолила, 2-бензотиазолила, 2-фталимидила, 2(1,3,4-тиадиазолила) 5-(1,2,4-тиадиазолила), 5-тиоксо-3-(1,2,4-тиадиазолила), 4-(5-оксо, 1,3,4-тиадиазолила), 4-(5-тиокос-1,3,4-тиадиазолила), 3-(1,2,4-триазолила), 4-(1,2,4-триазолила), 1-(1,2,3-триазолила), 2-имидазолила или 3-/1,2,4-триазоло-[4,3-а]-пиридинила, или такие гетероциклы, замещенные по любому доступному атому азота низшим алкилом, окси-(низшим алкилом), циклоалкилом, 2-, 3- или 4-пиридином, карбокси-(низшим алкилом), (низший алкокси)-карбонил-(низшим алкилом), аминокарбонил-(низшим алкилом), (низший алкил)-аминокарбонил(низшим алкилом), ди-(низший алкил)-аминокарбонил(низшим алкилом), амино-(низшим алкилом), (низший алкил)-амино-(низшим алкилом), ди-(низший алкил)-амино-(низшим алкилом), 4-морфолинил-(низшим алкилом), 1-пиперидинил-(низшим алкилом), 1-пирролидинил-(низшим алкилом) или фенилом [или фенилом, замещенным аминогруппой, (низший алкил)-аминогруппой, ди-(низший алкил)-аминогруппой, (низший алкан)-амидогруппой, N-(низший алкил)-(низший алкан)-амидогруппой, карбокси-(низший алкан)-амидогруппой, карбоксилом, карбо-(низшим алкоксилом), низшим алкоксилом или галогеном] или такие гетероциклы, замещенные по любому доступному атому углерода нитрогруппой, низшим алкилом, аминогруппой, (низший алкил)-аминогруппой, ди-(низший алкил)-аминогруппой, циклоалкил-аминогруппой, меркаптогруппой, (низший алкил)-тиогруппой, амино-(низший алкил)-тиогруппой, (низший алкил)-амино-(низший алкил)-тиогруппой, -ди-(низший алкил)-амино-(низший алкил)-тиогруппой, 4-морфолинил-(низший алкил)-тиогруппой, 1-пиперидинил-(низший алкил)-тиогруппой, 1-пирролидинил-(низший алкил)-тиогруппой, карбо-(низшим алкоксилом) или фенилом [или фенилом, замещенным амино-группой, (низший алкил)-аминогруппой, ди-(низший алкил)-аминогруппой, (низший алкан)-амидогруппой, N-(низший алкил)-(низший алкан)-амидогруппой, низшим алкилом, низшим алкоксилом, или галогеном]
R2 является водородом, карбо-(низшим алкоксилом), фенилом или фенилтиогруппой;
R3 является водородом, галогеном, первичным или вторичным низшим алкилом, низшим алкоксилом, карбо-(низшим алкоксилом), фенилом, фтор-(низшим алкилом), низшим алкенилом или цианогруппой;
R4 представляет собой водород или 1-2 заместители, выбранные из галогена, цианогруппы, нитрогруппы, аминогруппы, (низший алкан)-амидогруппы, фенил-(низший алкан)-амидогруппы, дифенил-(низший алкан)-амидогруппы, (низший алкил)-сульфониламиногруппы, полифтор-(низший алкил)-сульфониламиногруппы, аминосульфонила, низшего алкила, полигалоидированного низшего алкила, циклоалкила, полигалоидированного низшего алкоксила, гидроксила, низшего алкоксила, карбоксила, оксиметила, формила, аминометила, (низший алкил)-сульфонила, (полигалоидированный низший алкил)-сульфонила, (низший алкил)-сульфониламино-сульфонила и (низший алкокси)-поли-(низший алкилен)-оксигруппы; и где -CHR2-группа всегда присоединена или к гетероатому фрагмента L, определенного выше, или она присоединена к гетероатому фрагмента R1, при условии, что
(i) если m и n равны 0, а все R2, R3 и R4 представляют собой водород, то R1 не может быть галогеном;
(ii) если m=0, n=1, L является -S-, а каждый из R2, R3 и R4 представляет собой водород, то R1 не может быть 1-фенил-1Н-(5-тетразолилом);
(iii) если m=0, n=1, L является -O- или -S-, а все R2, R3 и R4 представляют собой водород, то R1 не может быть низшим алканоилом;
(iv) если m= 0, n= 1, L является -O-, -S- или -SO-, а все R2, R3 и R4 представляют собой водород, или m=0, n=1, L является -S-, R2, и R4 представляют собой водород, а R3 является галогеном, или если m=0, n=1, L является -SO- или -SO2-, R2 представляет собой карбо-(низший алкоксил), а R3 и R4 оба являются водородом, то R1 не может быть фенилом или замещенным фенилом.
В настоящее время было найдено, что следующие новые 2-замещенные соединения формулы I также обладают ингибирующей активностью относительно протеазных ферментов и они могут быть использованы при лечении дегенеративных заболеваний.
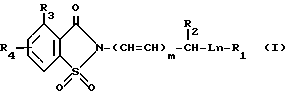
В соединениях формулы I L представляет собой -O-, -S-, -SO- или -SO2-; m и n независимо равны 0 или 1;
R1 представляет собой фенил, замещенный 1-[4-(низший алкил)-пиперазин-1-ил]-карбонилом, 4-морфолинилсульфонилом, формилом, (низший алкокси)-карбонилом, 4-тиаморфолинилсульфонилом, или его S-диоксидом, окси-(низшим алкилом), галогенсодержащим низшим алкилом, 4-морфолинил-(низший алкил)-аминокарбонилом, 4-морфолинил-(низший алкокси)-карбонилом, 1-[4-(низший алкил)-пиперазин-1-ил-(сульфонилом, 4-морфолин-(низшим алкоксилом), ди-(низший алкил)-амино-(низший алкил)-аминосульфонилом или его N-(низший алкил)-производным, низший алкил)-сульфонилом, 4,5-дигидрооксазол-2-илом, (низший алкил)-тетразол-5-илом, 4-морфолинилкарбонилом, нитрофенилазогруппой, карбоксилом, или ди-(низший алкил)-фосфонилом,
или представляет собой любую вышеуказанную группу в сочетании с замещением фенила галогеном, низшим алкилом, низшим алкоксилом, нитрогруппой, аминогруппой, (низший алкил)-аминогруппой или ди-(низший алкил)-аминогруппой, как определено в опубликованной PCT заявке WO/13549,
гетероцикл, выбранный из пирадазин-3-ила, 4-пирон-3-ила, хинолин-8-ила, 1,3,4-оксадиазол-2-ила, кумарин-7-ила, сахарин-6-ила, имидазол-1*-ила, 1,3,4-триазол-2-ила, тиазол-2-ила, 2-тиокос-2,3-дигидро-1,3,4-оксадиазол-3-ила, 1,2,5-триадиазол-3-ила, 2-тиоксо-2,3-дигидро-1,3,4-тиадиазол-3-ила, 2-тиоксо-2,3-дигидро-1,3,4-триадиазол-5-ила, 1,2,3-триазол-2-ила, 1,2,4-триазин-5-ила, 5-оксо-6-окси-4,5-дигидро-1,2,4-триазин-3-ила, изоксазол-5-ила, изоксазол-3-ила, 4,5-дигидро-5-оксо-1,2,4-оксадиазол-4-ила, пиридила, 1,1,3-триоксотетрагидро-1,2,5-триадиазол-2-ила, 6,7-дигидро-1Н-1,2,4-триазоло-[3,4-в] [1,3] триазин-3-ила, 4,5-дигидро-5-оксо-1,2,4-оксадиазол-4-ила, 2,5-диоксопирролидин-1-ила, 3-индолила, оксазол-2-ила, триазол-4-ила, 2,3-дигидро-2оксо-5-фенил-1,3,4-триадиазол-3-ила, 2,3-дигидро-2-оксо-5-фенил-1,3,4-оксадиазол-3-ила, 6-оксо-1,2-дигидро-1,2, -4-триазин-1-ила, 1,2,3-триазин-1-ила и 1-индолила,
или указанный гетероцикл, замещенный по любому доступному атому азота низшим алкилом, окси-(низшим алкилом), циклоалкилом, 2-, 3- или 4-пиридинилом, (низший алкокси)-карбонилом*/, (низший алкокси)-карбонил-(низшим алкилом), аминокарбонил-(низшим алкилом), (низший алкил)-аминокарбонил-(низшим алкилом), ди-(низший алкил)-амино-карбонил-(низшим алкилом), амино-(низшим алкилом), (низший алкил)-амино-(низшим алкилом), ди-(низший алкил-(амино-(низшим алкилом), 4-морфолинил-(низшим алкилом), 1-пиперидинил-(низшим алкилом), 1-пирролидинил-(низшим алкилом), или фенилом [или фенилом, замещенным аминогруппой, (низший алкил)-аминогруппой, ди-(низший алкил)-аминогруппой, (низший алкан)-амидогруппой, N-(низший алкил)-(низший алкан)-амидогруппой, карбокси-(низший алкан)-амидогруппой, карбоксилом, (низший алкокси)-карбонилом, низшим алкоксилом или галогеном]
или указанный гетероцикл, замещенный по любому доступному атому углерода нитрогруппой, низшим алкилом, аминогруппой, (низший алкил)-аминогруппой, ди-(низший алкил)-аминогруппой, циклоалкиламиногруппой, меркаптогруппой, (низший алкил)-тио-группой, амино-(низший алкил)-тиогруппой, (низший алкил)-амино-(низший алкил)-тиогруппой, ди-(низший алкил)-амино-(низший алкил)-тиогруппой, 4-морфолинил-(низший алкил)-тиогруппой, 1-пиперидинил-(низший алкил)-тиогруппой, 1-пирролидинил-(низший алкил)-тиогруппой) (низший алкокси)-карбонилом, ди-(низший алкил)-амино-(низшим алкилом), 4-морфолинил-(низший алкил)-аминогруппой, цианогруппой, 1-пиперидинил-(низшим алкилом), окси-(низшим алкилом), фенилсульфонилом, толуолсульфонилом, галогеном, три-(низший алкил)-силилом, карбоксилом или его солью с щелочным металлом, фурилом, трифторметилом, 2-бензотиазолилом, (низший алкил)-сульфонилом, аминокарбонилом, бензилом, 4-морфолинилом, пиридинилом, низшим алкоксилом, пиразинилом, (низший алкокси)-карбонил-(низшим алкилом, ди-(низший алкил)-амино-сульфонилом, 1-пиперидинилкарбонилом, бензилоксигруппой, гидроксилом, 4-морфолинил-(низшим алкилом, бензоилом или бензоилом, замещенным низшим, алкоксилом или галогеном, или фенилом [или фенилом, замещенным аминогруппой, (низший алкил)-аминогруппой, ди-(низший алкил)-аминогруппой, (низший алкан)-амидогруппой, N-(низший алкил)-(низший алкан)-амидогруппой, низшим алкилом, низшим алкоксилом, галогеном, трифторметилом, (низший алкокси)-поли-(низшим алкоксилом), метилендиоксигруппой или (низший алкокси)-карбонилом]
или гетероцикл, выбранный из 1Н-(5-тетразолила-),5-оксо-1-тетразолинила, 5-тиоксо-1-тетразолинила (если R2, определенный ниже, не является фенилтиогруппой), пиримидинила, 2-бензо-ксазолила, 2-бензотиазолила, 2-фталимидила, 2-(1,3,4-тиадиазолила), 5-(1,2,4-тиадиазолила), 5-тиоксо-3-(1,2)4-тиадиазолинила), 4-(5-оксо-1,3,4-тиадиазолинила), 4-(5-тиоксо-1,3,4-тиадиазолинила), 3-(1,2,4-триазолила), 4-(1,2,4-триазолила), 1,2,3-триазол-1-ила, 2-имидазолила и 3-(1,2,4-триазоло[4,3-а] пиридинила, будучи замещенный по любому доступному атому углерода ди-(низший алкил)-амино-(низшим алкилом), 4-морфолинил-(низший алкил)-аминогруппой, цианогруппой, 1-пиперидинил-(низшим алкилом), окси-(низшим алкилом), фенилсульфонилом, толуолсульфонилом, галогеном, три-(низший алкил)-силилом, карбоксилом или его солью с щелочным металлом, фурилом, трифторметилом, 2-бензотиазолилом, (низший алкил)-сульфонилом, аминокарбонилом, бензилом, 4-морфолинилом, пиридинилом, низшим алкоксилом, пиразинилом, (низший алкокси)-карбонил-(низшим алкилом), ди-(низший алкил)-аминосульфонилом, 1-пиперидинилкарбонилом, бензилоксигруппой, гидроксилом, 4-морфолинил-(низшим алкилом), бензоилом или бензоилом, замещенным низшим алкоксилом или галогеном или фенилом, замещенным трифторметилом, (низший алкокси)-поли-(низшим алкоксилом), метилендиоксигруппой или (низший алкокси)-карбонилом,
или, если R4 является карбокси-(низшим алкоксилом), (низший алкокси)-карбонил-(низшим алкоксилом) или ди-(низший алкил)-аминокарбонилоксигруппой, то гетероцикл представляет собой 1-фенил-тетразол-5-ил,
или, если L является -O-, а n равно 1, то R1 представляет собой циклогептатриенон-2-ил; если L является -S-, а n равно 1, то R1 представляет собой цианогруппу или (низший алкокси)-тиокарбонил; или, если L является -SO2-, а n равно 1, то R1 представляет собой низший алкил или трифторметил;
R2 является водородом, (низший алкокси)-карбонилом, фенилом или фенилтиогруппой;
R3 является водородом, галогеном, первичным или вторичным низшим алкилом, низшим алкоксилом, (низший алкокси)-карбонилом, фенилом, фторсодержащим низшим алкилом, низшим алкенилом, цианогруппой или ди-(низший алкил)-аминогруппой; а
R4 представляет собой водород или 1-3 заместителя, выбранные из галогена, цианогруппы, нитрогруппы, аминогруппы, (низший алкан)-амидогруппы, фенил-(низший алкан)-амидогруппы, дифенил-(низший алкан)-амидогруппы, (низший алкил)-сульфонил-аминогруппы, полифтор-(низший алкил)-сульфониламиногруппы, аминосульфонила, низшего алкила, полигалоидированного низшего алкила, циклоалкила, полигалоидированного низшего алкоксила, гидроксила, низшего алкоксила, карбоксила, оксиметила, формила, аминометила, (низший алкил)-сульфонила, (полигалоидированный низший алкил)-сульфонила, (низший алкил)-сульфониламиносульфонила, (низший алкокси)-(низшего алкоксила) (низший алкокси)-поли-(низший алкилен)-оксигруппы, карбокси-(низшего алкоксила), (низший алкокси)-карбонил-(низшего алкоксила) или ди-(низший алкил)-аминокарбонилоксигруппы; при условии, что
(1) когда n равно 0, то R1 может быть только гетероциклом, а CHR2 может быть только связан с кольцевым атомом азота R1 и
(2) когда m равно 0, n равно 1, L является -O-, -S- или -SO-, а R2, R3 и R4 все представляют собой водород, или когда m равно 0, n равно 1, L является -S-, R2 и R4 представляют собой водород, а R3 является галогеном, или когда m равно 0, n равно 1, L является -SO- или -SO2-, R2 представляет собой (низший алкокси)-карбонил, а R3 и R4 оба являются водородом, то тогда R1 не может быть замещенным фенилом.
Согласно изобретению предлагаются новые производные 2-замещенного сахарина формулы I, которые обладают ингибирующей активностью относительно протеазных ферментов и которые пригодны для лечения дегенеративных заболеваний.
С другой стороны, изобретение связано с композициями, используемыми для лечения дегенеративных заболеваний и содержащими фармацевтический носитель и новые производные 2-замещенного сахарина формулы I в количестве, эффективном для ингибирования протеолитических ферментов.
Дополнительным аспектом изобретения является предоставление способа применения указанных 2-замещенных сахаринов формулы I при получении лекарственного препарата, используемого для лечения дегенеративного заболевания у пациента, нуждающегося в таком лечении, и содержащего указанный 2-замещенный сахарин в количестве, эффективном для ингибирования протеолитических ферментов.
Предпочтительными соединениями формулы I, описанной выше, являются соединения формулы I, где m равно 0, R2 является водородом, R3 представляет собой водород, галоген, первичный или вторичный низший алкил или низший алкоксил, R4 представляет собой водород, гидроксил, низший алкоксил, (низший алкокси)-(низший алкоксил), (низший алкокси)-поли-(низший алкилен)-окси-группу, карбокси-(низший алкоксил) или (низший алкокси)-карбонил-(низший алкоксил), а
n равно 1, L является -O-, а R1 представляет собой фенил, замещенный 1-[4-(низший алкил)-пиперазин-1-ил] -карбонилом, 4-морфолинилсульфонилом, 4-тиаморфолинилсульфонилом или его S-диоксидом, 4-морфолинил-(низший алкил)-аминокарбонилом, 4-морфолинил(-низший алкокси)-карбонилом, 1-[4-(низший алкил)-пиперазин-1-ил] -сульфонилом, ди-(низший алкил)-амино-(низший алкил)-аминосульфонилом, и/или 4-морфолинилкарбонилом или 1,2,5-тиадиазол-З-ил или 1,2,5-тиодиазол-З-ил, замещенный 4-морфолинилом, или изоксазолил, замещенный (низший алкокси)-карбонилом; или
n равно 1, L является -S-, а R1 представляет собой 1,3,4-оксадиазол-2-ил, замещенный по любому доступному атому углерода фурилом, бензилом, пиридинилом, пиразинилом или фенилом; или
n равно 0, а R1 представляет собой 1,2,3-триазол-1-ил или 1,2,3-триазол-2-ил, замещенный по любому доступному атому углерода цианогруппой или (низший алкил)-сульфонилом.
В частности, предпочтительными соединениями являются соединения, в которых L представляет собой -O- или -S-, m равно 0, равно 0 или 1, а R2 является водородом, особенно в которых R3 представляет собой водород, хлор, бром, метил, этил, пропил, изопропил, втор-бутил, метоксил, этоксил, изопропоксил или фенил, и в которых R4 представляет собой водород, 7-хлор-5-нитрогруппу, 6-нитрогруппу, 5-аминогруппу, 5-ацетиоаминогруппу, 5-(3,3-дифенил-пропионамидо)-группу, 5-(1,1,3,3-тетраметилбутил), 6-гидроксил, 7-гидроксил, 5-метоксил, 6-метоксил, 7-метоксил, 5,6-диметоксил, 5,7-диметоксил, 6,7-диметоксил, 6[-(2-метоксиэтокси)этоксил] 7-[2-(2-метоксиэтокси)этоксил] 7-карбоксиметоксил, 7-(трет-бутоксикарбонил)метоксил, 7-диметиламинокарбонилоксигруппу, 6-пропил-7-метоксил, 5,7-диметокси-6-метил или 5-окси-6-метоксил.
Следует понимать, что соединения, имеющие общую структурную формулу I, в химической литературе обычно называют как 1,2-бензизотиазол-(2Н)-3-он,-1,1-диоксиды. Однако для краткости такие соединения называют производными сахарина, и в этом патенте при описании соединений изобретения и их биологических свойств используется эта терминология.
Используемые здесь термины "низший алкил", "низший алкоксил" и "низший алкал" обозначают одновалентные алифатические радикалы, включая радикалы с разветвленной цепью, содержащие от 1 до 10 атомов углерода. Таким образом, алкильный (или алкановый) фрагмент таких групп включает в себя, например, метил, этил, пропил, изопропил, н-бутил, втор-бутил, трет-бутил, н-пентил, 2-метил-З-бутил, 1-метилбутил, 2-метилбутил, неопентил, н-гексил, 1-ментилпентил, 3-метилпентил, 1-этилбутил, 2-этил-бутил, 2-гексил, 3-гексил, 1,1,3,3-тетраметилпентил, 1,1-диметилоктил и т.п. Низший алканоил содержит от 2 до 10 атомов углерода и является разветвленным или неразветвленным.
Используемый здесь термин "галоген" (или "гало") обозначает фтор, хлор, бром или йод.
Используемый здесь термин "циклоалкил" обозначает карбоциклические кольца, содержащие от 3 до 6 атомов углерода, включая циклопропил, циклобутил, циклопентил и циклогексил, которые могут быть замещены по любому углеродному атому кольца одной или несколькими низшими алкильными группами.
Используемый здесь термин "низший алкенил" означает одновалентные ненасыщенные радикалы, включая радикалы с разветвленной цепью, содержащие от 3 до 10 атомов углерода, и, таким образом, эти радикалы включают в себя 1-(2-пропенил), 1-(2-бутенил), 1-(1-метил-2-пропенил), 1-(4-метил-2-пентенил), 4,4,6-триметил-2-гептенил и т.п.
Соединения изобретения ингибируют активность серинпротеаз, в частности эластазы лейкоцитов человека и химотрипсинподобных ферментов, и поэтому они полезны при лечении дегенеративных заболеваний, таких как эмфизема, ревматоидный артрит и панкреатит. Предполагается, что в ходе связывания и ингибирования активности протеолитического фермента соединения изобретения расщепляются по связи между функциями метилен (CHP2) и ZnR1 и что группа ZnR1 отделяется в виде аниона, который, таким образом, может характеризоваться как уходящая группа. Предполагается, что это расщепление будет облегчаться присутствием электронно-акцепторной группы, такой как цианогруппа, галоген, нитрогруппа, карбоксил, (низший алкокси)-карбонил, ацил, или фенилтиогруппа в R1 функциональности, тем самым увеличивая ее электроотрицательность, которая может быть выражена в виде значений рКа кислотного вида уходящей группы, которые идеально должны быть меньше 7. Особенно предпочтительной группой таких соединений являются соединения формулы I, в которых R3 не является водородом.
Другой аспект изобретения связан со способом получения указанных производных 2-сахарина, который включает в себя взаимодействие 2-галометилсахарина или с щелочной солью фрагмента LnR1, или с фрагментом LnR1 в присутствии акцептора кислоты.
Следующий аспект изобретения связан со способом получения указанных производных 2-сахарина, который включает в себя взаимодействие щелочной соли или таллиевой соли 2-незамещенного сахарина или с фрагментом гало-CHR2-LnR1 с получением желаемого продукта, или с разновидностью 3-хлор-3-(фенилтио)-пропил-LnR1 с последующим окислением продукта надкислотой с получением 2[1-(фенилсульфинил)пропил-LnR1] сахарина, нагреванием последнего с получением 2-[1-(2-пропенил)-LnR1]сахарина.
Еще один аспект изобретения заключается в предоставлении способа получения 4-первичный- или вторичный-(низший алкил)-R4-2-незамещенных сахаринов, используемых в качестве промежуточных веществ при синтезе соответствующих производных 2-сахарина, который включает в себя взаимодействие 2-(первичный низший алкил)-N, N-ди-(низший алкил)-бензамида с (низший алкил) литием в инертном органическом растворителе; взаимодействие получающейся соли лития с (низший алкил/галогенидом; взаимодействие получающегося 2-первичный- или вторичный-(низший алкил)-R4-N,N-ди-(низший алкил)-бензамида с (низший алкил)-литием; взаимодействие получающейся соли лития с двуокисью серы с последующим взаимодействием в присутствии основания с гидроксиламиносульфокислотой; и нагревание продукта в кислотной среде.
Так, например, соединения формулы I, где m равно 0, R2 является водородом, а L представляет собой -O- или -S-, получают взаимодействием производного 2-галометилсахарина формулы I, где R1 является галогеном, R2 является водородом, m и n равны 0, R3 и R4 имеют значения, приведенные выше, с соответствующим фрагментом LnR1. Реакция может быть проведена или в присутствии акцептора кислоты, такого как карбонат щелочного металла, три-n(низший алкил)-амина, низшего алкоголята щелочного металла или таллия, или гидрида щелочного металла, или возможно использование щелочной соли фрагмента LnR1. Реакцию проводят в органическом растворителе, инертном в условиях реакции, например, в ацетоне, метилэтилкетоне (MEK), тетрагидрофуране (THF), диэтиловом эфире, диметилформамиде (DMF), хлористом метилене (MDC) или в низших спиртах, в диапазоне температур от комнатной до температуры кипения используемого растворителя. Соответствующие соединения, в которых L является -SO- или -SO2-, получают окислением соответствующих соединений формулы I, в которых L является -S-, одним или двумя мольными эквивалентами, что подходит, надкислоты, такой как 3-хлорнадбензойная кислота.
С другой стороны, соединения формулы I, в которых m равно 0, могут быть получены взаимодействием соли сахарина и щелочного металла или таллия (полученной реакцией соответствующего 4-R3-R4-2-незамещенного сахарина с алкоголятом щелочного металла или с низшим алкоголятом таллия) с гало-CHR2-LnR1 фрагментом, где R1, R2, R3, R4, L и n имеют значения, приведенные выше относительно формулы I. Соль таллия может быть использована, когда R2 имеет все вышеприведенные значения, однако соль щелочного металла может быть использована только тогда, когда R2 является водородом. Реакцию проводят в инертном органическом растворителе, например, в низшем спирте или диметилформамиде, при температурах в диапазоне от 20oC до температуры кипения используемого растворителя.
Соединения формулы I, где m равно 1, а R2 является водородом, получают взаимодействием соединения 3-(фенилтио)пропил-Ln-R1, полученного реакцией LnR1-пропилгалогенида с тиофеноксидом натрия в метилэтилкетоне (MEK), с N-хлорсукцинимидом с получением соединения вида 3-хлор-3-(фенилтио)пропил-LnR1. Реакция последнего с таллиевой солью соответствующего 4-R3-R4-сахарина и тех же самых условиях, что описаны выше при получении соединений формулы I из соли сахарина и гало-CHR2-LnR1 фрагмента, приводит к образованию 2-[1-(фенилтио/пропил)-LnR1]сахарина. Окисление этого соединения до соответствующего 2-[1-(фенилсульфинил)пропил-LnR1] сахарина с последующим нагреванием продукта в алкиленгликолевом эфире, например, этиленгликольдиметиловом эфире, приводит к образованию соединений формулы I, где m равно 1, а R1 является водородом.
Соединения формулы I, в которых R1 представляет собой низший алканоил, R2 является водородом, L является -O-, m равно 0, n равно 1, а R3 и R4 имеют значения, приведенные выше, получают взаимодействием соответствующего 2-оксиметилсахарина с подходящим ангидридом кислоты в присутствии каталитических количеств минеральной кислоты или сильной органической кислоты, например, в присутствии серной кислоты или пара-толуолсульфокислоты.
Соединение формулы I, где n равно 0, а R1 представляет собой 1,2,3-триазол-1-ил, получают конденсацией соответствующего соединения формулы I, где R1 представляет собой галоген, с азидом щелочного металла и затем циклоприсоединением получающегося азида с соответствующим замещенным или незамещенным ацетиленом. Предпочтительным азидом щелочного металла является азид натрия. Конденсацию проводят с или без нагревания или охлаждения, предпочтительно при комнатной температуре, в инертном растворителе, например, бензоле, толуоле или диметилформамиде, возможно использование краун-эфира, например, 18-краун-6-эфира. Циклоприсоединение предпочтительно проводят при нагревании в том же инертном растворителе 2-галометилсахарины формулы I, где R1 является галогеном R2 является водородом, m и n равны 0, R3 и R4 имеют значения, приведенные выше относительно формулы I, и соответствующие 4-R3-R4-2-незамещенные сахарины, необходимые для получения соединений формулы I, где R1, L, m и n имеют другие значения, приведенные выше, получают по способам, описанным D'Alelio и др. [J. Macromol. Sci-Chem. A3(5), 941 (1969)] и Saari и др. [Het. Chem. 23, 1253 (1986)] По способу, описанному Saari, метиловый эфир соответствующей антраниловой кислоты получают обычным способом из замещенной антраниловой кислоты и диазотированного сложного эфира. Затем соль диазония взаимодействует с двуокисью серы и хлористой медью с образованием хлористого сульфонила, который затем взаимодействует с концентрированной гидроокисью аммония с образованием производных замещенного сахарина формулы II. При реакции последних с формальдегидом в растворителе (низшем спирте) образуются 2-окси-метилсахарины формулы III, которые при реакции с тионилгалогенидом или тригалидом фосфора дают соответствующие производные 2-галометилсахарина. Этот метод можно проиллюстрировать следующим образом: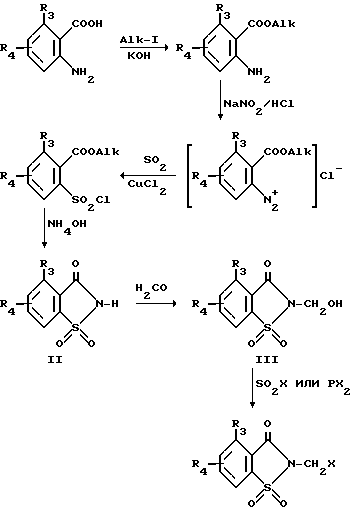
где R3 и R4 имеют значения, приведенные выше, а X представляет собой галоген.
Галометилсахарины формулы I, где R1 является галогеном, R2 является водородом, m и n равны 0, а R3 и R4 имеют значения, приведенные выше относительно формулы I, могут быть также получены взаимодействием соответствующего 2-фенилтиометилсахарина с сульфурилгалогенидом в инертном органическом растворителе, например, в хлористом метилене, дихлорэтане (EDC) или четыреххлористом углероде, при температуре от 0 до 30oC.
Соединения формулы II, где R3 представляет собой или первичный, или вторичный низший алкил, которые используют в качестве промежуточных веществ при синтезе соединений формулы I, описанной выше, получают одним из двух методов. Соединения формулы II, где R3 является первичным низшим алкилом, получают взаимодействием 4-метил-R4-2-незамещенного сахарина с двумя мольными эквивалентами (низший алкил)-лития в инертном органическом растворителе, например; THF и взаимодействием получающейся соли лития с одним мольным эквивалентом галогенида низшего алкила, причем обе реакции проводят при температуре от -50 до -80oC.
Соединения формулы II, где R3 является или первичным, или вторичным низшим алкилом, получают реакцией 2-(первичный низший алкил)-R4-N,N-ди-(низший алкил)-бензамида с одним мольным эквивалентом (низший алкил)-лития в присутствии тетра-(низший алкил)-этилендиамина в инертном органическом растворителе, например, THF, и взаимодействием получающейся соли лития с одним мольным эквивалентом галогенида низшего алкила при температуре в диапазоне от -50 до -80oC. Получающийся 2-первичный- или вторичный-(низший алкил)-R4-N, N-ди-(низший алкил)-бензамид затем взаимодействует с одним мольным эквивалентом (низший алкил)-лития в присутствии тетра-(низший алкил)-этилендиамина в инертном органическом растворителе, например, THE, а получающаяся соль лития взаимодействует с двуокисью серы при температуре от -50 до -80oC с последующей реакцией полученного продукта с гидроксиламиносульфокислотой в присутствии основания. После этого получающийся 2-(низший алкил)-R4-6-амино-сульфонил-N, N-ди-(низший алкил)-бензамид нагревают в кислотной среде для осуществления циклизации его до желаемого 4-первичный - или вторичный-(низший алкил)-R4-2-незамещенного-сахарина формулы II. Предпочтительно циклизацию проводить в кипящей ледяной уксусной кислоте. Если в исходном веществе 2-(низший алкил)-R4-N,N-ди-(низший алкил)-бензамиде низшая алкильная группа в положении 2 представляет собой метил, то алкилирование приводит к образованию соединений, в которых низшая алкильная группа в положении 2 является линейной или разветвленной, в зависимости от того, линейный или разветвленный (низший алкил)-галогенид был использован для алкилирования. С другой стороны, если в исходном веществе низшая алкильная группа в положении 2 содержит более одного атома углерода, то алкилирование проходит по атому углерода, примыкающему к бензольному кольцу, и приводит к образованию продуктов, содержащих в положении 2 вторичную низшую алкильную группу.
В некоторых случаях подход к созданию определенных необходимых промежуточных соединений формулы II требует построения двух колец, составляющих ядро сахарина. Так, для получения соединений, в которых R3 представляет собой низший алкоксил, а R4 является 7-гидроксилом, 3,3-дитио-биспропионовую кислоту превращают в хлорангидрид бис-кислоты реакцией этой кислоты с хлористым тионилом, а затем хлорангидрид взаимодействует с двумя мольными эквивалентами бензиламина с образованием бис-н-бензиламида. При взаимодействии этого амида с хлористым сульфурилом в растворителе, таком как MDC, EDC или четыреххлористый углерод, образуется 5-хлор-2-бензил-2Н-изотиазол-3-он, который окисляют одним мольным эквивалентном надкислоты, такой как надбензойная кислота или 3-хлор-надбензойная кислота, до 5-хлор-2-бензил-2Н-изотиазол-3-он-1-оксида. При взаимодействии этого соединения под давлением и при нагревании с 2-(низший алкокси)-фураном в органическом растворителе, таком как бензол, толуол или ксилол, образуется 4-(низший алкокси)-7-окси-2-бензил-1,2-бензизотиазол-2Н-3-он-1-оксид. Если необходимо, то можно осуществить взаимодействие между гидроксигруппой, находящейся в положении 7, и (низший алкил)-галогенидом или (низший алкокси)-поли-(низший алкокси)-(низший алкил)-галогенидом с получением соответствующего 4,7-ди-(низший алкокси)- или 4-(низший алкокси)-7-(низший алкокси)-поли-(низший алкилен)-окси-2-бензил-1,2-бензизотиазол-2Н-3-он-1-оксида. Дальнейшее окисление этого продукта одним мольным эквивалентом надкислоты, как описано выше, с последующим каталитическим дебензилированием приводит к образованию соответствующих 4-(низший алкокси)-7-R4-2-незамещенных сахаринов.
Для проведения изменений функциональных групп в соединениях изобретения могут быть использованы другие простые химические превращения, которые являются общепринятыми и хорошо известны специалистам, работающим в этой области химии. Например, когда необходимо, может быть проведено каталитическое восстановление нитрогрупп с получением соответствующим аминозамещенных соединений, ацилирование аминозамещенных соединений с получением соответствующих амидов или окисление сульфидов или сульфоксидов с получением соответствующих сульфоксидов или сульфонов.
Стандартные методики биологических испытаний показали, что соединения формулы I обладают ингибирующей активностью относительно эластазы лейкоцитов человека (HLE) и химотрипсина, и, следовательно, они могут быть использованы при лечении дегенеративных заболеваний, таких как эмфизема, ревматоидный артрит или панкреатит.
Соединения формулы I, содержащие основные функции, могут быть переведены в солевую форму (соль присоединения кислоты) взаимодействием этого основания с кислотой. Аналогичным образом свободное основание может быть обычным способом регенерировано из солевой формы (соли присоединения кислоты), т.е. путем обработки этой соли охлажденными) слабыми водными растворами оснований, например, растворами карбонатов и бикарбонатов щелочных металлов. Основания, регенерированные таким образом, могут быть приведены во взаимодействие с той же или другой кислотой, при этом снова получают ту же или другую соль присоединения кислоты. Таким образом, основания и все их соли присоединения кислоты легко взаимопревращаемы.
Таким образом, некоторые соединения формулы I, содержащие кислотные функции, т. е. функции карбоновой кислоты, могут быть переведены в их солевые формы взаимодействием этой кислоты с основанием, таким как гидроокись щелочного металла или гидроокись аммония, или с органическими основаниями, такими как алкил, диалкил-, или триалкиламины, и эти кислоты могут быть регенерированы из солей обработкой этих солей водными растворами кислот.
Поэтому следует принимать во внимание, что формула I не только изображает структурных конфигурацию оснований и кислот формулы I, но она также отражает структурные организации, которые являются общими для всех соединений формулы I, независимо от того, находятся ли эти соединения в виде свободного основания, свободных кислот или в виде солей этих оснований и кислот. Было найдено, что вследствие этих общих структурных организаций соединения формулы I и их соли обладают свойственной им фармакологической активностью типа, который более полно описан будет ниже. Эта неотъемлемая фармакологическая активность может быть использована в ценном виде для фармацевтических целей путем применения самих свободных оснований или свободных кислот, или солей, образованных из фармацевтически приемлемых кислот и оснований, т.е. кислот и оснований, чьи анионы или катионы в эффективных дозах солей безвредны для организма животного, таким образом, что целебные свойства, присущие общей структурной организации, представленной свободными основаниями и свободными кислотами, не искажаются побочными эффектами, приписываемыми этим анионам или катионам.
При использовании этой фармакологической активности солей конечно, предпочтительно применять фармацевтически приемлемые соли. Хотя нерастворимость в воде, высокая токсичность и отсутствие кристаллического характера могут сделать некоторые конкретные виды солей непригодными или менее желательными для использования как таковых в определенном фармакологическом применении, однако водонерастворимые или токсичные соли могут быть переведены в соответствующую фармацевтически приемлемую основу путем разложения этих солей водным раствором основания или водным раствором кислоты, как объяснено выше, или, с другой стороны, они могут быть переведена в любую подходящую фармацевтически приемлемую соль реакциями двойного разложения, включая анион или катион, например, ионообменными методами.
Кроме того, помимо полезности в фармацевтических применениях, эти соли пригодны в качестве характеризующих или идентифицирующих производных свободных оснований или свободных кислот, или в процессах очистки или выделения. Подобно всем солям такая характеризация или очистка может быть использована, если необходимо, для регенерирования фармацевтически приемлемых свободных оснований или свободных кислот взаимодействием солей с водным раствором основания или водным раствором кислоты, или, с другой стороны, они могут быть переведены в фармацевтически приемлемую соль, например, ионообменными методами.
В таком случае новая особенность этих соединений заключается в концепции основы и катионных и анионных форм 4-R3-R4-2-замещенных сахаринов формулы I, а не в каком-либо конкретном кислотном или основном фрагменте, т.е. не в анионе кислоты или катионе основания, связанных с солевыми формами этих соединений; вернее всего, кислотные или основные фрагменты, т.е. анионы, которые могут быть связаны с солевыми формами, сами по себе не являются ни новыми, ни определяющими, и, следовательно, могут быть любым анионом кислоты или катионом основания, способными к образованию соли при взаимодействии с основаниями или кислотами.
Соединения формулы I изобретения могут быть приготовлены для фармацевтического использования введением их в унифицированную лекарственную форму, такую как таблетки или капсулы для перорального применения, или в чистом виде, или в сочетании с подходящими адъювантами, такими как карбонат кальция, крахмал, лактоза, тальк, стеарат магния, аравийская камедь и т.п. Кроме того, эти соединения могут быть приготовлены для перорального, парентерального употребления или употребления в виде аэрозольной ингаляции, или в виде водных растворов водорастворимых солей этих соединений, или в виде спиртовых, гликолевых или масляных растворов, или в виде водомасляных эмульсий, тем же способом, каким получают обычные лекарственные вещества.
В таких композициях процентное содержание активного компонента можно варьировать так, чтобы получить приемлемую дозу. Дозу, вводимую конкретному пациенту, варьируют в зависимости от клинической оценки, используя в качестве критерия: способ применения, продолжительность лечения, вес и физическое состояние пациента, эффективность активного компонента и восприимчивость его пациентом. Следовательно, эффективное количество дозы активного компонента может быть определено только клиницистом после рассмотрения всех этих критерий и выбора наилучших показаний в интересах пациента.
Молекулярные структуры соединений этого изобретения были расшифрованы на основании ИК- и ЯМР-спектров. Эти структуры были подтверждены соответствием между вычисленными и найденными методом элементарного анализа величинами, определяющими содержание элементов.
Следующие примеры приведены для дальнейшей иллюстрации этого изобретения, а не для ограничения сферы его притязаний. Все температуры плавления являются неточными.
Пример 1. Порошкообразную гидроокись калия (7,4 г; 132 ммоль; 2 эквивалента) смешивали со 100 мл диметилсульфоксида (DMSO) и перемешивали эту смесь в течение 5 мин. Затем к смеси добавляли по каплям 6-метилантраниловую кислоту (10,0 г; 66 ммоль) и йодистый метил (4,52 мл; 73 ммоль; 1,1 эквивалента). Реакционную смесь перемешивали при комнатной температуре в течение 30 мин, затем разбавляли 250 мл диэтилового эфира, промывали водой (3 х 10 мл), сушили над MgSO4 и концентрировали. Неочищенный продукт фильтровали через слой отожженного силикагеля (32-63), элюировали смесью диэтиловый эфир: гексан 1:9 и получали 4,23 г метилового эфира 6-метилантраниловой кислоты в виде масла, выход 39% 1H ЯМР (300 МГц, CDCl3: 7,078 (1Н, триплет, J=7,67 Гц); 6,529 (2Н, дублет, J=7,79 Гц); 5,111 (2Н, широкий синглет); 3,867 (3Н, синглет), 2,424 (3Н, синглет), ИК (неразбавленный тонкий слой, см-1): 3480 (средняя), 3380 (средняя), 2950 (слабая), 1690 (сильная), 1605 (сильная).
Полученный на стадии (a) метиловый эфир 6-метилантраниловой кислоты (4,23 г; 25,6 ммоль) растворяли в 25 мл уксусной кислоты и охлаждали раствор до 0oC. К раствору добавляли 45 мл концентрированной соляной кислоты и получали шлам желтовато-коричневого цвета. К шламу добавляли по каплям при перемешивании раствор нитрата натрия (1,89 г; 27,4 ммоль; 1,07 эквивалента) в 8 мл воды, получающийся раствор оранжевого цвета перемешивали при 0oC в течение 1 ч, а затем добавляли его шестью порциями к смеси дигидрата хлорида меди (2) (2,18 г; 12,8 ммоль; 0,5 эквивалента) и двуокиси серы (6,3 г; избыток) в 33 мл уксусной кислоты 6 мл воды при 0oC. Темно-зеленый раствор перемешивали при комнатной температуре в течение ночи, выливали в 300 мл воды, охлажденной льдом, выделившееся твердое вещество собирали, сушили отсасыванием и получали 1,11 г хлористого сульфонила, которые сразу же добавляли к гидроокиси аммония (100 мл), охлажденной льдом, и перемешивали при комнатной температуре в течение ночи. Раствор подкисляли до pH 1 концентрированной HCl, получающийся осадок собирали, сушили на воздухе и получали 729 мг (выход 12% ) 4-метилсахарина, температура плавления 225-226oC. 1H ЯМР (300 МГц, CD3CN): 9,5 (1Н, широкий синглет); 7,782 (2Н, дублет, J=4,35 Гц); 7,644 (1Н, триплет, J= 4,20 Гц); 2,683 (3Н, синглет), ИК (KBr, см-1): 3400 (слабая), 3100 (сильная), 3000 (сильная); 1720 (сильная); 1580 (средняя). Масс-спектрометрия (FDMS): m/e 197 (M+).
Полученный на стадии (b) 4-метилсахарин (500 мг; 2,54 ммоль) растворяли в 2,53 мл подогретого этанола (паровая баня). После образования гомогенного раствора к нему по каплям добавляли формалин (37% в метаноле; 1,76 мл; избыток). Раствору давали возможность охладиться до комнатной температуры, а затем выдерживали его при 0oC в течение 4 дней. Образовавшееся твердое вещество собирали, сушили на воздухе и получали 476 мг (выход 82%) 2-оксиметил-4-метилсахарина, температура плавления 196-198oC. 1H ЯМР (300 МГц, CDCl3): 7,767 (1Н, триплет, J=6,75 Гц); 7,732 (1Н, дублет, J=7,72 Гц); 7,600 (1Н, дублет, J= 6,64 Гц); 5,361 (2Н, дублет, J=8,00 Гц); 3,296 (1Н, триплет, J= 8,16 Гц); 2,793 (3Н синглет). ИК (KBr, см-1): 3505 (сильная); 3070 (средняя); 1735 (сильная); 1580 (средняя).
Полученный на стадии (c) 2-оксиметил-4-метилсахарин (76 мг; 0,33 мл) смешивали с уксусным ангидридом (1 мл; избыток) и добавляли к нему 2 капли концентрированной серной кислоты. Реакционную смесь перемешивали при комнатной температуре в течение 2 ч, в это время методом тонкослойной хроматографии (TLC) наблюдали неполярное пятно. Реакционную смесь разбавляли хлористым метиленом (50 мл) и промывали насыщенным раствором бикарбоната натрия (2 x 15 мл). После сушки над Na2SO4 удаляли растворитель и получали 64 мг (выход 72%) 2-ацетоксиметил-4-метилсахарина, температура плавления 198-205oC (разл. ). 1H ЯМР (300 МГц, CDCl3): 7,8 (2Н, мультиплет); 7,64 (1Н, дублет, J 6,18 Гц); 5,84 (2Н, синглет); 2,82 (3Н, синглет); 2,15 (3Н, синглет), ИК (KBr, см-1): 2920 (слабая); 1745 (сильная); 1735 (сильная); 1630 (слабая). Масс-спектрометрия (FDMS): m/e 269 (M+).
Пример 2. Используя методику, описанную выше в примере 1, проводили реакцию между 6-хлорантраниловой кислотой (5 00 г; 29,2 ммоля) и йодистым метилом (2,75 мл; 44 ммоль; 1,5 эквивалента) в присутствии порошкообразной гидроокиси калия (4,08 г; 72,7 ммоль; 2,5 эквивалента) и получали 4,22 г (выход 78%) метилового эфира 6-хлорантраниловой кислоты в виде масла. 1H ЯМР (300 МГц, CDCl3): 7,077 (1Н, триплет, J=8,06 Гц); 6,744 (1Н, дублет, J=6,7 Гц); 6,575 (1Н, дублет, J=8,25 Гц); 4,871 (1Н, широкий синглет); 3,929 (3Н, синглет). ИК (неразбавленный тонкий слой, см-1): 3480 (средняя), 3380 (средняя); 2950 (слабая); 1705 (сильная); 1610 (сильная).
4-хлорсахарин получали по той же методике, что использовали при синтезе 4-метилсахарина, используя метиловый эфир 6-хлорантраниловой кислоты (4,22 г; 22,7 ммоль) в 22 мл уксусной кислоты, концентрированную HCl (40 мл) и нитрат натрия (1,68 г; 24,3 ммоль) в 7 мл воды, при этом образовывалась соль диазония, которую добавляли к смеси дигидрата хлорида меди (2) (1,93 г; 11,4 ммоль; 0,5 эквивалента) и диоксида серы (6,5 г; избыток) в 30 мл уксусной кислоты и 5 мл воды. Получающийся хлористый сульфонил обрабатывали, как описано выше, 150 мл гидроокиси аммония и получали 3,07 г бледно-желтого твердого вещества 4-хлорсахарина, температура плавления 245-246oC, выход 62% 1H ЯМР (300 МГц, CD3CN): 7,918 (1Н, двойной дублет, J=7,39, 1,91 Гц); 7,865 (1Н, триплет, J= 7,52 Гц); 7,829 (1Н, широкий дублет, J=7,30 Гц). ИК (KBr, см-1): 3570 (сильная); 3520 (сильная); 2950 (сильная, широкая); 1735 (сильная); 1630 (средняя). Масс-спектрометрия (FDMS): m/e 217 (M+).
2-оксиметил-4-хлорсахарин получали таким же способом, что и 2-оксиметил-4-метилсахарин в примере 1, из 4-хлорсахарина (1,00 г; 4,60 ммоль) и формалина (37% 3,22 мл; избыток). Все попытки выкристаллизовать вязкий маслянистый продукт приводили к разложению исходного вещества и поэтому на следующей стадии был использован этот продукт без кристаллизации.
2-ацетоксиметил-4-хлорсахарин получали таким же способом, что и 2-ацетоксиметил-4-метилсахарин в примере 1, из неочищенного 2-оксиметил-4-хлорсахарина (0,34 г; 1,4 ммоль) и уксусного ангидрида (2,5 мл) с 2 каплями серной кислоты. В этом случае после выделения продукт очищали фильтрацией через слой силикагеля и элюированием смесью диэтиловый эфир: гексан 1:1, после чего получали 2-ацетоксиметил-4-хлорсахарин (35 мг; выход 95%) в виде белого твердого вещества, температура плавления 138-142oC. 1H ЯМР (300 МГц, CDCl3): 7,921 (1Н, двойной дублет, J=6,54, 2,63 Гц); 7,874 (1Н, триплет, J=7,98Гц); 7,842 (1Н, двойной дублет, J=6,70, 2,20 Гц); 5,869 (2Н, синглет); 2,172 (3Н, синглет), ИК (KBr, см-1): 1745 (сильная); 1735 (средняя, плечо); 1575 (слабая). По данным анализа сжиганием:
Теоретич. C 41,46; H 2,78; N 4,83;
Найдено, C 41,17; H 2,81; N 4,75.
Пример 3. Неочищенный 2-оксиметил-4-хлорсахарин из примера 2 (609 мг; 2,46 ммоль макс.) смешивали с 5 мл диэтилового эфира и добавляли хлористый тионил (3 мл; избыток). Получающуюся смесь нагревали до полного растворения, перемешивали при комнатной температуре в течение ночи, разбавляли 20 мл диэтилового эфира, фильтровали через слой целита, сверху покрытый песком, и элюировали диэтиловым эфиром. После удаления растворителя получали 430 мг неочищенного хлорметилпроизводного. Часть вещества (225 мг) использовали в дальнейших реакциях. Остаток (205 мг) хроматографировали на испарительной колонке с силикагелем, элюировали смесью 40% диэтиловый эфир/пентан и получали 137 мг 2-хлорметил-4-хлорсахарина, температура плавления 135-136oC. 1H ЯМР (300 МГц, CDCl3): 7,925 (1Н, двойной дублет, J=6,62, 2,26 Гц); 7,882 (1Н, триплет, J=8,18 Гц); 7,846 (1Н, двойной дублет, J=7,42, 2,36 Гц); 5,561 (2Н, синглет). ИК (KBr, см-1): 3090 (слабая); 3050 (слабая); 1750 (сильная); 1575 (средняя). Масс-спектрометрия (FDMS): m/e 265 (M+).
Пример 4. Хлорметилпроизводное, полученное в примере 3 (225 мг; 0,85 ммоль) и натриевую соль 1-фенил-5-меркапто-1Н-тетразола (200 мг) 1,01 ммоль; 1,2 эквивалента) растворяли в 5 мл ацетона и получали раствор желтовато-коричневого цвета. Через 10 мин наблюдали осадок, а после перемешивания в течение ночи при комнатной температуре в смеси не был обнаружен (по данным анализа тонкослойной хроматографии) 2-хлорметил-4-хлорсахарин. Реакционную смесь выливали в воду и экстрагировали хлористым метиленом (3 x 25 мл). Объединенные экстракты сушили над Na2SO4 концентрировали, а остаток хроматографировали на испарительной колонке с силикагелем и элюировали смесью диэтиловый эфир:гексан 1:1. Основную фракцию собирали и получали 122 мг 4-хлор-2-/1-фенил-1Н-тетразол-5-илтиометил/сахарина в виде твердого вещества белого цвета, температура плавления 175-177oC, 1H ЯМР (300 МГц, CDCl3: 7,813 (3Н, мультиплет); 7,515 (5Н, синглет); 5,710 (2Н, синглет), ИК (KBr, см-1): 3080 (слабая); 1740 (сильная); 1590 (слабая). Масс-спектрометрия (FDMS): m/e 407 (M+; 230 (M+ PMT); 178 (1% PMT).
Пример 5. Хлорметилпроизводное (337 мг неочищенного вещества; максимум 1,27 ммоль), полученное по методике, описанной в примере 3, растворяли (насколько это возможно) в 10 мл ацетона. К раствору добавляли натриевую соль 1-фенил-5-меркапто-1Н-тетразола (304 мг, 1,52 ммоль; 1,2 эквивалента) и перемешивали реакционную смесь при комнатной температуре в течение 3 дней. Затем смесь разбавляли хлористым метиленом (50 мл), промывали водой (3 x 25 мл), сушили над Na2SO4 концентрировали и фильтровали через слой силикагеля (элюент смесь диэтилового эфира и гексана 1:1). Вещество, полученное таким образом, хроматографировали на испарительной колонке с силикагелем, элюировали смесью диэтиловый эфир:гексан 1:1 и получали 44 мг (выход 8,5%) 4-хлор-2-/4-фенил-5-тиоксотетразолин-1-илметил/сахарина, температура плавления 158-162oC. 1H ЯМР (300 МГц, CDCl3): 7,981 (1Н, дублет, J=7,12 Гц); 7,95 (2Н, мультиплет); 7,887 (1Н, триплет, J=6,74 Гц); 7,864 (1Н, дублет, J=7,32 Гц); 7,567 (3Н, мультиплет); 6,392 (2Н, синглет), ИК (KBr, см-1): 1745 (сильная); 1185 (сильная). Масс-спектрометрия (FDMS): m/e 407 (M+); 230 (M+ PMT).
Пример 6. Смесь 2-/хлорметил/сахарина (0,98 г, 4,2 ммоль), 1-(3-ацетамидофенил)-5-меркапто-1Н-тетразола (1 г, 4,2 ммоль) и бикарбоната калия (0,84 г, 8,4 ммоль) в 50 мл метилэтилкетона нагревали при 50oC в атмосфере азота в течение ночи. Реакционную смесь охлаждали, выливали в HCl, разбавленную охлажденной водой (300 мл), декантировали воду с полутвердого вещества, которое затвердевало при растирании с горячим этилацетатом. Полученное твердое вещество белого цвета рекристаллизовали из ацетонитрила (MeCN) с древесным углем и получали 0)82 г 2-[1-(3-ацетамидофенил]-1Н-тетразол-5-ил-тиометил)сахарина в виде небольших игл белого цвета, температура плавления 195-196oC (разл. ). 1H ЯМР (90 МГц, CDCl3): 2,05 (3Н, синглет); 5,65 (2Н, синглет). Масс-спектрометрия (FDMS): m/e 430 (M+).
Теоретич. C 47,43; H 3,28; N 19,52;
Найдено, C 47,02; H 3,27; N 19,53.
Пример 7. Смесь-2-(бромметил)сахарина (2,7 г, 9,8 ммоль), 1-(3-гептан-амидо-фенил)-5-меркапто-1Н-тетраза (3 г, 9,8 ммоль) и карбоната калия (3,4 г, 24,5 ммоль) нагревали с обратным холодильником в метилэтилкетоне (50 мл) в атмосфере азота в течение 1 ч. Затем смесь охлаждали и выливали в водный раствор бикарбоната натрия, охлажденный льдом. С получающегося полутвердого вещества белого цвета декантировали слой воды. Полутвердое вещество промывали водой, затем растворяли в горячем ацетонитриле, раствор обрабатывали активированным углем и фильтровали. Из фильтрата отгоняли в вакууме растворитель, а получающееся твердое вещество хроматографировали (силикагель-95: 5 CH2Cl2: ацетон) и получали светлое масло. Это масло кристаллизовали из горячего этанола и получали 1,6 г твердого вещества белого цвета 2[1-(3-гептанамидофенил)-1Н-тетразол-5-илтио-метил] сахарин, температура плавления 146-147,5oC. 1H ЯМР (90 МГц, CDCl3 (2Н, (синглет). Масс-спектрометрия (FDMS): m/e 500 (M+).
Теоретич. C 52,79; H 4,83; N 16,79;
Найдено, C 52,44; H 4,75; N 16,64.
Пример 8. Смесь 2-(бромометил)сахарина (3 г, 10,8 ммоль) и натриевой соли 5-меркапто-1-метил-1Н-тетразола (1,49 г, 10,8 ммоль) нагревали с обратным холодильником в метилэтилкетоне (75 мл) в течение 2 ч. Затем смесь охлаждали, выливали в разбавленный водный раствор бикарбоната натрия, осажденный льдом, и дважды экстрагировали хлористым метиленом. Объединенные органические экстракты сушили над Na2SO4 и в вакууме отгоняли из них растворитель. Неочищенный продукт хроматографировали (силикагель- 95:5 CH2Cl2: диэтиловый эфир), а получающееся масло кристаллизовали из горячего изопропанола и получали 2,7 г, (выход 80%) твердого вещества белого цвета 2-[1-метил-1Н-тетразол-5-илтио-метил]-сахарина, температура плавления 106-110oC. 1H ЯМР (90 МГц, CDCl3): 5,55 (2Н, синглет). Масс-спектрометрия (FDMS): m/e 311 (M+).
Теоретич. C 38,58; H 2,91; N 22,49;
Найдено, C 38,53; H 2,79; N 22,60.
Пример 9. Смесь 2-(хлорметил)сахарина (3 г, 12,9 ммоль), 1-циклогексил-5-меркапто-1Н-тетразола (2,37 г, 12,9 ммоль) и карбоната калия (4,45 г, 32,2 ммоль) нагревали с обратным холодильником в метилэтилкетоне (50 мл) в течение 1 ч. Затем реакционную смесь охлаждали, выливали в разбавленный водный раствор бикарбоната натрия, охлажденный льдом, и дважды экстрагировали этилацетатом. Объединенные органические экстракты промывали водой, сушили над Na2SO4 и в вакууме отгоняли из них растворитель. Остаток хроматографировали (силикагель; хлористый метилен) и получали 2 г 2-[1-циклогексил-1Н-тетразол-5-илтиометил]сахарина в виде белой пены, которую кристаллизовали из горячего циклогексана) температура плавления 103-105oC. 1H ЯМР (90 МГц, CDCl3): 5,65 (2Н, синглет). Масс-спектрометрия (FDMS): m/e 379 (M+).
Теоретич. C 47,48; H 4,52; N 18,46;
Найдено, C 47,84; H 4,61; N 18,36.
Пример 10. Смесь мета-хлорнадбензойной кислоты (0,43 г, 2,67 ммоль) и 2-(1-фенил-1Н-тетразол-5-илтиометил)сахарина (1 г, 2,67 ммоль) в хлористом метилене перемешивали при комнатной температуре в течение 24 ч. Анализ методом тонкослойной хроматографии (TLC) (95 5 CH2Cl2 диэтиловый эфир) показал присутствие исходного сульфида. Было добавлено дополнительное количество (0,2 г) надкислоты, после чего смесь перемешивали еще в течение двух дней. Затем реакционную смесь промывали раствором бикарбоната натрия, сушили над Na2SO4 и в вакууме отгоняли из нее растворитель. Остаток хроматографировали (силикагель 95 5 CH2Cl2 диэтиловый эфир) и получали пену, которую кристаллизовали из диэтилового эфира и выделяли 0,52 г 2-[1-фенил-1Н-тетразол-5-илсульфинил] сахарина в виде твердого вещества белого цвета, температура плавления 161-162oC. 1H ЯМР (90 МГц, CDCl3): 5,5-6,0 (2Н, квадруплет). Масс-спектроскопия (FDMS): m/e 196 (M+-PMT); 389 (M+).
Теоретич. C 46,27; H 2,85; N 17,98;
Найдено, C 46,00; H 2,83; N 17,76.
Пример 11. Смесь 2-бромометил-5-нитросахарина (2 г, 6,2 ммоля) и натриевой соли 1-фенил-5-меркапто-1Н-тетразола нагревали с обратным холодильником в течение 2 ч в смеси, содержащей 40 мл метилэтилкетона и 10 мл диметилформамида (DMF). Затем реакционную смесь охлаждали и выливали в разбавленный водный раствор бикарбоната натрия, охлажденный льдом. Получающееся твердое вещество белого цвета отфильтровывали, промывали водой и сушили на воздухе. Для удаления растворимых примесей это вещество гомогенизировали со смесью хлористого метилена и ацетона (50:50) и фильтровали. Оставшееся твердое вещество рекристаллизовали из смеси ацетонитрил:этанол 2:1 и получали 1,5 г 5-нитро-2-[1-фенил-1Н-тетразол-5-илтиометил]сахарина в виде твердого вещества не совсем белого цвета, температура плавления 189-190oC. 1H ЯМР (90 МГц, ДМСО-d6): 5,75 (2Н, синглет). Масс-спектрометрия (FDMS) m/e 418 (M+).
Теоретич. C 43,06; H 2,41; N 20,09;
Найдено, C 42,29; H 2,43; N 20,13.
Пример 12. Смесь мета-хлорнадбензойной кислоты (2,2 г, 12,8 ммоль) и 2-(фенилсульфинилметил)сахарина (3,75 г) 11)6 ммоль) перемешивали при комнатной температуре в 50 мл хлористого метилена в течение 2 ч. После добавления дополнительного количества надкислоты перемешивание продолжали в течение 1 ч. Метахлорбензойную кислоту удаляли фильтрацией, а твердое вещество промывали небольшим количеством хлористого метилена. Фильтрат промывали водным раствором бикарбоната натрия, сушили над Na2SO4 и в вакууме отгоняли из него растворитель. Получающееся твердое вещество рекристаллизовали из смеси этанол: ацетонитрил 50: 50 и получали 2-(фенилсульфонилметил)сахарин в виде твердого вещества белого цвета, температура плавления 169-171oC. 1H ЯМР (90 МГц, ДМСО-d6), CDCl3: 5,15 (2Н, синглет). Масс-спектрометрия (FDMS): m/e 196 (M+-PMT).
Теоретич. C 49,84; H 3,29; N 4,15;
Найдено, C 49,92; H 3,24; N 4,13.
Пример 13. Смесь мета-хлорнадбензойной кислоты (0,9 г, 5,37 ммоль) и 2-(2-пиримидилтиометил)сахарина (1,5 г, 4,8 ммоль), полученного по методикам, аналогичным тем, что описаны в примерах 9 и 11, в 75 мл хлористого метилена перемешивали при комнатной температуре в течение ночи. Реакционную смесь промывали водным раствором бикарбоната натрия, сушили над Na2SO4 и в вакууме отгоняли из нее растворитель. Часть этого неочищенного продукта (0,5 г) сохраняли для непосредственного превращения в сульфон, а оставшееся вещество хроматографировали (силикагель 95 5 CH2Cl2 ацетон). После рекристаллизации из смеси этанол: ацетонитрил получали 0,95 г 2-(2-пиримидил-сульфинилметил)сахарина в виде кристаллов) температура плавления 197-198oC (разл.). 1H ЯМР (90 МГц, CDCl3, ДМСО-d6): 5,1-5,5 (2Н, квадруплет).
Теоретич. C 44,57; H 2,81; N 13,00;
Найдено, C 44,67; H 2,84; N 12,97.
Пример 14. Смесь мета-хлорнадбензойной кислоты (0,4 г, 2,3 ммоль) и сульфоксида (0,75 г, 2,3 ммоль), полученного в примере 13, перемешивали при комнатной температуре в 50 мл хлористого метилена, наблюдая за реакцией с помощью метода тонкослойной хроматографии (TLC) (95 5 МДС:ацетон). После 2 ч перемешивания еще оставалось некоторое количество исходного сульфоксида; к реакционной смеси добавляли дополнительную порцию надкислоты и продолжали перемешивание в течение ночи. После чего добавляли 100 мл хлористого метилена и промывали смесь раствором бикарбоната натрия. Органический слой сушили над Na2SO4 а растворитель отгоняли в вакууме. После рекристаллизации остатка из смеси ацетонитрил: этанол получали 0,95 г 2-(2-пиримидилсульфонил-метил)-сахарина в виде твердого вещества белого цвета, температура плавления 225-227oC (разл.). 1H ЯМР (90 МГц, ДМСО-d6): 5,78 (2Н, синглет), Масс-спектрометрия (M+): m/e 196 (M+ PMT), 339 (M+).
Теоретич. C 42,47; H 2,67; N 12,38;
Найдено, C 42,20; H 2,62; N 12,46.
Пример 15. Смесь 2-(хлорметил)сахарина (3 г, 12,9 ммоль) и пара-нитрофенолята натрия (2,55 г, 12,9 ммоль) в 50 мл тетрагидрофурана (THF) нагревали при 50oC в течение ночи, а затем кипятили с обратным холодильником в течение 45 мин. Реакционную смесь охлаждали, выливали в разбавленный водный раствор бикарбоната натрия, охлажденный льдом, и дважды экстрагировали этилацетатом. Объединенные органические экстракты промывали раствором бикарбоната натрия и водой, сушили над Na2SO4, а затем досушивали в вакууме. Остаток хроматографировали (силикагель; хлористый метилен) и получали масло, которое кристаллизовали из горячей смеси циклогексана и диэтилового эфира. Образующееся твердое вещество рекристаллизовали из этанола и получали 0,92 г 2-(4-нитрофеноксиметил-)сахарина в виде блестящих пластинок белого цвета, температура плавления 162-164oC. 1H ЯМР (90 МГц CDCl3), ДМСО-d6): 5,95 (2Н, синглет). Масс спектроскопия (FDMS) m/e 334 (M+).
Теоретич. C 50,30; H 3,02; N 8,38;
Найдено, C 50,06; H 2,91; N 8,28.
Пример 16. 5-нитро-2-(1-фенил-1Н-тетразол-5-илтиометил)сахарин (4 г, 9,56 ммоля), полученный в примере 11, растворяли в 250 мл тетрагидрофурана и помещали раствор в качающуюся склянку Парра. В атмосфере азота к раствору добавляли две лопаточки 10%-ного палладия на угле, после чего смесь встряхивали в атмосфере водорода (давление водорода около 4 ат) в течение 2,5 дней. Реакционную смесь фильтровали через целит диатомовую землю, фильтрат смешивали с водой и экстрагировали хлористым метиленом. Органический слой сушили над Na2SO4, отгоняли из него в вакууме растворитель, а получающуюся пену желтого цвета гомогенизировали с теплым этанолом, охлаждали и фильтровали. Было выделено соответствующее производное 5-аминосахарина (0,5 г), которое представляло собой твердое вещество кремового цвета. Масс-спектроскопия (FDMS) m/e (М+).
Смесь вышеуказанного производного 5-аминосухарина (0,5 г, 1,29 ммоль) и 3,3-дифенилпропаноилхлорида (0,315 г, 1,29 ммоля) в 50 мл ацетонитрила нагревали с обратным холодильником в течение 2,5 ч. Анализ методом тонкослойной хроматографии показал присутствие некоторое количества исходного амина. После добавления небольшого количества хлорангидрида нагревание с обратным холодильником продолжали в течение 1,5 ч, а затем реакционную смесь охлаждали и выливали в воду, охлажденную льдом (400 мл). Через 30 мин смесь фильтровали, полученное твердое вещество желтовато-коричневого цвета промывали водой и сушили на воздухе. После хроматографирования на колонке с силикагелем (95: 5 хлористый метилен:диэтиловый эфир) получали пену, которую кристаллизовали из этанола и получали 0,68 г 5-(3,3-дифенилпропионамидо)-2-(1-фенил-1Н-тетразол-5-илтиометил)сахарина в виде твердого вещества белого цвета, температура плавления 92-93oC (разл.). Масс-спектрометрия (FDMS): m/e 696 (M+).
1H ЯМР (90 МГц, CDCl3): 3,25 (1Н, дублет); 4,8 (2Н, триплет); 5,6 (2Н, синглет); 6,9 8,2 (мультиплет, Ar). Анализ методом ЯМР также показал присутствие двух молекул кристаллизованного этанола 1,25 (триплет); 3,7 (квадруплет). По данным анализа сжигания: для C30H24N6O4 2 + 2C2H5OH
Теоретич. C 59,28; H 5,27; N 12,2;
Найдено, C 58,09; H 5,15; N 12,09.
Пример 17. Метиловый эфир 2-хлор-2-фенилтиоуксусной кислоты получали по методике, описанной в литературе [Fleming и J.Iqbal, Tetra Lett. 24, 327 (1983); M. Campbell и др. Tetra Lett, 21, 3305 (1980)]
Сахарин (10 г, 54,6 ммоль) растворяли при слабом нагревании в 500 мл этанола. К раствору добавляли по каплям этилат таллия (13,6 г, 54,6 ммоль), полученную гомогенную смесь перемешивали при комнатной температуре в течение 2 ч, затем охлаждали, фильтровали, а твердое вещество промывали холодным этанолом. Кристаллическое твердое вещество серовато-белого цвета сушили в эксикаторе под вакуумом и получали 19,4 г (выход 92%) таллиевой соли сахарина.
Смесь таллиевой соли сахарина (1,78 г, 4,6 ммоль) и метилового эфира 2-хлор-2-фенилтиоуксусной кислоты (1 г, 4,6 ммоль) в 25 мл диметилформамида перемешивали при 60oC в течение 7 ч. Затем смесь охлаждали и выливали в воду, охлажденную льдом (400 мл). Через 30 мин смесь фильтровали, твердое вещество промывали водой и сушили на воздухе. После хроматографирования на колонке с силикагелем (хлористый метилен) получали светлое масло, которое кристаллизовали из горячего этанола и получали 0,87 г (выход 51%) метилового эфира 2-фенилтио-2-(2-сахаринил)уксусной кислоты в виде игл белого цвета, температура плавления 144-146oC.
1H ЯМР (90 МГц, CDCl3): 3,8 (3Н, синглет); 5,95 (1Н, синглет); 7,2-8,15 (9Н, мультиплет). Масс-спектрометрия (FDMS): m/e 363 (M+).
Раствор метилового эфира 2-фенилтио-2-(2-сахаринил)уксусной кислоты (2 г, 5,5 ммоль) и хлористого сульфурила (0,74 г, 5,5 ммоль) в 50 мл хлористого метилена перемешивали при комнатной температуре в течение 2 ч. Растворитель удаляли в вакууме, а масло желтого цвета кристаллизовали из подогретого этанола и получали 0,94 г продукта. Анализ методом ЯМР показал присутствие более 50% исходного вещества. К смеси, содержащей сырой продукт, добавили еще исходного вещества (1 г, 2,75 ммоль) и растворили все в хлористом метилене. К раствору снова добавили хлористый сульфурил (0,5 мл) и смесь перемешивали при комнатной температуре в течение 12 ч. Обработав смесь, как описано выше, получали 0,66 неочищенного метилового эфира 2-хлор-2-(2-сахаринил)уксусной кислоты, который сразу же использовали в следующей стадии.
Смесь этого хлорида (0,66 г сырой смеси) и 1-фенил-5-меркапто-1Н-тетразола в виде натриевой соли (0,44 г, 2,2 ммоль) нагревали с обратным холодильником в 20 мл метилэтилкетона в течение 4 ч. После перемешивания при комнатной температуре в течение 2 дней реакционную смесь выливали в воду, охлажденную льдом. Отфильтрованное твердое вещество желтовато-коричневого цвета промывали водой и сушили на воздухе. После хроматографирования на колонке с силикагелем (хлористый метилен) получали пену не совсем белого цвета, которую кристаллизовали из этанола и получали 0,36 г метилового эфира 2-(1-фенил-1Н-тетразол-5-илтио)-2-(2-сахаринил)уксусной кислоты в виде кристаллического твердого вещества белого цвета, температура плавления 160-162oC.
1H ЯМР (90 МГц, CDCl3): 3,8 (синглет); 7,05 (1Н, синглет); 7,4-8,1 (9Н, мультиплет). Масс-спектрометрия (FDMS): m/e 431 (M+).
Для C17H13N5O52
Теоретич. C 47,33; H 3,04; N 16,23;
Найдено, C 47,15; H 3,09; N 16,30.
Пример 18. К суспензии 6,0 г (0,03 ммоль) йодида меди (1) в 100 мл тетрагидрофурана добавляли 25 мл диметилсульфида, полученный раствор желтого цвета охлаждали до -78oC и добавляли к нему по каплям 23 мл 3,0 М раствора магнийбромфенила (0,06 моль) в диэтиловом эфире. Получающийся раствор бледно-желто-оранжевого цвета перемешивали в атмосфере азота при -78oC в течение 1 ч, а затем обрабатывали 3,02 г (0,03 ммоль) 2-цикло-гексанона в 10 мл тетрагидрофурана. Полученной смеси давали возможность нагреться в течение 2 ч до 0oC, затем ее повторно охлаждали до -78oC, обрабатывали 15 мл гексаметилфосфорамида, перемешивали в течение 30 мин, обрабатывали 8,0 г (0,09 моль) метилового эфира цианомуравьиной кислоты, а затем оставляли на ночь при комнатной температуре. Реакционную смесь выливали в 100 мл 2N соляной кислоты, органический слой отделяли, а водную фазу снова экстрагировали хлористым метиленом. Объединенные органические экстракты сушили досуха в вакууме, а остаток растирали с хлористым аммонием, затем с водой, затем с рассолом, после чего снова сушили досуха и получали 3,2 г метилового эфира 2-фенилциклогексан-6-он-карбоновой кислоты в виде масла.
Последнее вещество (3,0 г, 0,013 моль), бензилмеркаптан 4,8 г (0,039 моля) и 1 г смолы Amberlyst® 15 (Pohm и Haas) нагревали в хлороформе с обратным холодильником в течение 20 ч, затем к смеси добавляли еще 1,5 г смолы и дополнительно нагревали в течение 4 ч. После этого смесь охлаждали до комнатной температуры, фильтровали, фильтрат сушили досуха в вакууме, остаток растирали с гексаном, твердое вещество отфильтровывали и получали 0,85 г (выход 19% ) смеси метилового эфира 2-бензилтио-6-фенилциклогекс-2-ен-карбоновой кислоты и метилового эфира-2-бензил-тио-6-фенилциклогекс-1-ен-карбоновой кислоты, 0,6 г (0,0018 моль) которой нагревали с 2,0 г 2,3-дихлор-5,6-дициано-бензохинона в 25 мл толуола при перемешивании в атмосфере азота в течение 24 ч.
Смесь фильтровали через слой силикагеля, элюировали 2:1 хлористый метилен:гексан, элюат сушили досуха и получали 0,3 г (выход 67%) метилового эфира 2-бензилтио-6-фенилбензойной кислоты.
Этот эфир (6,52 г, 0,0016 моля), растворенный в 10 мл хлористого метилена, разбавляли 20 мл уксусной кислоты и 5 мл воды, смесь охлаждали до -10oC и барботировали через нее хлор (газ) до тех пор, пока не прекратится экзотермическая реакция. Затем смесь перемешивали в течение 10 мин, сушили досуха в вакууме и получали 0,41 г (выход 85%) метилового эфира 2-хлорсульфонил-6-фенил-бензойной кислоты, которые растворяли в 10 мл тетрагидрофурана и добавляли при охлаждении на бане сухой лед/ацетон к 25 мл концентрированного раствора гидроокиси аммония. Реакционную смесь экстрагировали хлористым метиленом, органическую фазу убирали, а водный слой подкисляли до pH 1 концентрированной соляной кислотой и экстрагировали хлористым метиленом. Органические экстракты промывали рассолом, сушили, испаряли досуха и получали 0,33 г (выход 97%) 4-фенилсахарина.
Следуя методике, описанной в примере 21, проводили реакцию между 4-фенилсахарином (0,33 г, 0,0012 моль) и хлорметилфенилсульфидом (0,3 г, 0,0019 моль) в 15 мл толуола в присутствии 0,08 г (0,0025 моль) бромистого тетрабутиламмония и получали 0,48 г (выход 100%) 2-фенилтиометил-4-фенилсахарина, который обрабатывали в хлористом метилене хлористым сульфурилом и получали 0,36 г (выход 95%) 2-хлорметил-4-фенилсахарина.
Пример 19. Раствор 2-(фенилхлорметил)-сахарина (2б2 г, 0,0071 моль) и натриевой соли 1-фенил-5-меркапто-тетразола (1,4 г, 0,0071 моль) в 30 мл диметилформамида нагревали при 55oC в течение 3,5 ч, затем перемешивали при комнатной температуре в течение 16 ч и выливали в ледяную воду, содержащую разбавленный раствор бикарбоната натрия. Выделившееся твердое вещество собирали, промывали водой, сушили на воздухе, хроматографировали на колонке с силикагелем, элюируя смесью хлористый метилен:диэтиловый эфир 98:2, и получали 2 г (выход 63% ) 2-1(-фенил-1Н-тетразол-5-илтиофенилметил)сахарина, температура плавления 192-193oC.
Пример 20. Раствор натриевой соли сахарина (4,53 г, 0,022 моль) и 1-фенил-4-хлорметил-тетразолин-5-тиона (5 г, 0,022 моль) в 50 мл диметилформамида нагревали при 130oC в течение 4 ч, затем охлаждали и выливали в ледяную воду. Выделившееся твердое вещество собирали, промывали водой, сушили и хроматографировали на колонке с силикагелем, элюируя хлористым метиленом; получали 4,8 г (выход 58%) 2-(1-фенил-5-тиоксотетразолин-4-илметил)сахарина, температура плавления 140-142oC.
Пример 21. Смесь 3,27 г (0,012 моля) 4-бромсахарина [японская патентная публикация 58 (79,034, C.A. 7773W)1984] трет-бутилата калия (1,63 г, 0,015 моль), бромистого тетрабутиламмония (0,39 г, 0,0012 моль) и хлорметилфенилсульфида (3,0 мл, 0,022 мл) в 100 мл толуола нагревали с обратным холодильником в атмосфере азота в течение 8 ч, а затем при комнатной температуре в течение 16 ч. Затем реакционную смесь охлаждали, разбавляли этилацетатом и промывали органический слой бикарбонатом, водой и рассолом, затем сушили над сульфатом магния и испаряли досуха в вакууме. Оставшееся твердое вещество рекристаллизовали из смеси толуол-гексан и получали 3,86 г (выход 64%) 4-бром-2-фенил-тиометилсахарина, температура плавления 174,5-178oC.
К раствору этого соединения (3,27 г, 0,0085 моль) в 85 мл хлористого метилена добавляли по каплям при перемешивании 1,02 мл (0,0127 моль) хлористого сульфурила. Смесь перемешивали при комнатной температуре в течение 1,5 ч, концентрировали в вакууме, а остаток растирали с гексаном, фильтровали и получали 2,61 г сырого продукта, который рекристаллизовали из смеси толуол-гексан и получали 2,24 г (выход 85%) 2-хлорметил-4-бром-сахарина, температура плавления 157-159oC.
Пример 22A. К раствору 8,0 мл (0,053 моль) тетраметилэтилендиамина (TMEDA) в 350 мл тетрагидрофурана при -70oC добавляли 42 мл 1,3 М раствора втор-бутиллития (0,055 моль) в гексане и перемешивали смесь в течение 15 мин. К этому раствору добавляли по каплям при перемешивании раствор 2-метокси-N, N-диэтилбензамида (10,36 г, 0,050 моль) в 150 мл тетрагидрофурана, поддерживая температуру -60oC или ниже, а затем в реакционную смесь барботировали двуокись серы, поддерживая температуру реакции ниже -50oC до тех пор, пока реакционная смесь не стала кислой (анализ влажной лакмусовой бумагой). Затем эту смесь перемешивали при комнатной температуре в течение 2 ч, разбавляли 450 мл гексана, образовавшееся твердое вещество собирали, растворяли в 200 мл воды, обрабатывали смесь 65 г ацетата натрия и добавляли частями при перемешивании гидроксиламино-O-сульфокислоту (21,5 г, 0,19 моль). Выделившееся твердое вещество белого цвета собирали, сушили и получали 7,04 г (49%) 2-амино-сульфонил-6-метокси-N, N-диэтилбензамида, температура плавления 190-194,5oC. Смесь этого продукта (4,3 г, 0,015 моль) в 75 мл диоксана и 25 мл концентрированной соляной кислоты нагревали на паровой бане в течение 70 ч, затем охлаждали, концентрировали в вакууме, разбавляли водой и льдом и сильно подщелачивали концентрированным раствором гидроокиси натрия. Смесь экстрагировали хлористым метиленом, из органических экстрактов выделяли продукт и получали 1,29 г (выход 40%) 4-метоксисахарина. Другим, более предпочтительным способом циклизацию 2-аминосульфонил-6-метокси-N,N-диэтилбензамида в 4-метоксисахарин с выходом 65% проводили путем нагревания с обратным холодильником в ледяной уксусной кислоте в течение 6,5 ч.
Используя методику, приведенную в примере 21, проводили взаимодействие между 4-метоксисахарином (1,14 г, 0,0053 моль) и хлорметилфенилсульфидом (1,31 мл, 0,0097 моль) в толуоле в присутствии трет-бутилата калия (0,72 г, 0,0064 моль) и бромистого тетрабутиламмония (174 мг, 0,00054 моль) и получали 1,23 г (выход 69%) 4-метокси-2фенилтиометилсахарина, температура плавления 152,5-154,5oC (из смеси амилацетат-гексан). 1,02 г (0,003 моль) этого вещества обрабатывали хлористым сульфурилом (0,36 мл, 0,0045 моль) в хлористом метилене и получали 282 мг (выход 36%) 2-хлорметил-4-метоксисахарина, температура плавления 169-174oC.
Пример 22B. К раствору тетраметилэтилендиамина (4,74 мл, 0,031 моль) в 300 мл тетрагидрофурана (пропущенные перед использованием через окись алюминия) добавляли 2-этил-N,N-диэтилбензамид (5,8 г, 0,03 моль). Раствор охлаждали до -78oC и обрабатывали 34,9 мл 0,9М раствора втор-бутиллития (0,031 моль) в циклогексане. По окончании добавления смесь перемешивали в течение 20 мин, а затем обрабатывали ее раствором йодистого этила (3,2 мл, 0,04 моль), поддерживая температуру смеси -78oC. Затем давали возможность смеси нагреться до комнатной температуры, после чего перемешивали ее в течение 16 ч, а затем выливали в воду. Получающееся масло отделяли, хроматографировали на колонке с силикагелем, элюируя 10% этилацетат/гексан, и получали 2,86 г (выход 43%) 2-фтор-бутил-N,N-диэтилбензамида, который представлял собой масло желтого цвета.
Следуя методике, приведенной в примере 22A, 2-втор-бутил-N,N-диэтилбензамид (10,45 г, 0,045 моль), растворенный в 70 мл тетрагидрофурана, добавляли к 39,2 мл 1,2 М раствора втор-бутиллития (0,047 моль) в циклогексане и 7,1 мл (0,047 моль) тетраметилэтилендиамина в 250 мл тетрагидрофурана, поддерживая температуру -78oC.
По окончании добавления смесь перемешивали при -78oC еще в течение 0,5 ч, затем обрабатывали при -70oC двуокисью серы, после чего давали возможность смеси нагреться до комнатной температуры. Смесь высушивали в вакууме досуха, а остаток растворяли в воде, добавляли при перемешивании к охлажденному раствору гидроксиламиносульфокислоты (15,2 г, 0,134 моль) и 15,4 мл 35% -ной гидроокиси натрия (0,134 моль) и получали 10,1 г (выход 72%) 2- аминосульфонил-6-втор-бутил-N,N-диэтилбензамида.
Это вещество (6,83 г, 0,22 моль) растворяли в 100 мл ледяной уксусной кислоты, раствор нагревали с обратным холодильником в течение 13 ч, а затем испаряли досуха. Остаток растирали с диэтиловым эфиром и после фильтрования получали 5,7 г (выход 83%) диэтиламмониевой соли 4-втор-бутилсахарина.
При взаимодействии этой соли (3,0 г, 0,096 моль) с хлорметилфенилсульфидом (1,13 мл) 0,012 моль) в толуоле получали 3,47 г (выход 100%) 2-фенилтиометил-4-втор-бутилсахарина.
В результате реакции этого вещества (3,2 г, 0,0097 моль) с 2,3 мл (6,029 моль) хлористого сульфурила в 20 мл хлористого метилена получали 2,4 г (выход 87%) 2-хлорметил-4-втор-бутил-сахарина.
Пример 22С. К раствору тетраметилэтилендиамина (9,3 мл, 0,058 моль) в 340 мл тетрагидрофурана добавляли при -78oC 52 мл 1,1 М раствора втор-бутиллития (0,057 моль) в тетрагидрофуране. Затем к раствору добавляли при -78oC раствор 2-пропил-N,N-диэтилбензамида (11,37 г, 0,052 моль) в 75 мл тетрагидрофурана, после чего смесь перемешивали в течение 15 мин, а затем добавляли к ней раствор йодистого этила (8,3 мл, 0,104 моль) в тетрагидрофуране. Раствор перемешивали при -78oC в течение 1,5 ч, а затем охлаждали, добавляя по каплям насыщенный раствор хлористого аммония при -78oC. Затем давали возможность смеси нагреться до комнатной температуры, разбавляли диэтиловым эфиром, промывали сначала разбавленной соляной кислотой, затем водой, насыщенным раствором бикарбоната натрия, рассолом, после чего сушили досуха и получали 12,91 г неочищенного продукта, который хроматографировали на колонке с силикагелем, элюируя смесью 10% этилацетат/гексан, и получали 3,23 г (25%) 2-(3-пентил)-N,N-диэтилбензамида в виде масла желтого цвета.
Используя методику, описанную выше в примере 22A, проводили реакцию между 2-(3-пентил)-N,N-диэтилбензамидом (3,05 г, 0,0115 моль), растворенным в тетрагидрофуране, и 10,5 мл 1,2 М раствора втор-бутиллития (0,126 моль) в тетрагидрофуране в присутствии тетраметилэтилендиамина (2,1 мл, 0,014 моль). Затем образовавшуюся соль лития обрабатывали сначала двуокисью серы, а затем натриевой солью гидроксиламиносульфокислоты и получали 1,97 г (выход 52%) 2-амино-сульфонил-6-(3-пентил)-N, N-диэтилбензамида в виде кристаллов бледно-желтого вида, температура плавления 118-120oC (температура размягчения 102oC) 1,84 г (0,0056 моль), которого циклизовали, нагревая при кипении с обратным холодильником в 22 мл ледяной уксусной кислоты, и получали 1,28 г (выход 70% ) диэтиламмониевой соли 4-(3-пентил)-сахарина, температура плавления 107,5-109,5oC.
При взаимодействии этой соли (0,0037 моль) с хлорметилфенилсульфидом (0,74 мл, 0,0055 моль) в присутствии бромистого тетрабутиламмония (116 мг, 0,0004 моль) в 45 мл толуола получали 1,93 г 2-фенилтиометил-4-(3-пентил)-сахарина в виде масла бледно-желтого цвета, при взаимодействии которого (1,93 г, 0,0037 моль) с хлористым сульфурилом (0,59 мл, 0,0073 моль) в 37 мл хлористого метилена получали 1,2 г 2-хлорметил-4-(3-пентил)-сахарина в виде масла бледно-желтого цвета.
Примеры 22D-22N. По методике, описанной в примере 22A, заменяя используемый там 2-метокси-N, N-диэтилбензамид на соответствующий 2-R3-R4-замещенный-N,N-диэтилбензамид, были получены через соответствующие 2-фенилтиометилсахарины последующие 2-гомометил-4-R3R4-замещенные сахарины, которые перечислены в табл. 1. Для каждого 2-незамещенного сахарина, 2-фенилтиометилсахарина и 2-хлорметилсахарина в столбцах, озаглавленных "Тпл/раст." и "Выход", приведены температура плавления, растворитель, из которого проводили рекристаллизацию, и выход, если эти данные имелись. Во всех случаях промежуточные 2-фенил-тиометилсахарины сразу же использовали в следующей стадии без дополнительной характеристики или очистки.
Пример 23. Используя методику, описанную в примере 1, проводили реакцию сахарина (18,3 г, 0,1 моль) с 70 мл 37% формалина в этаноле и получали 3,58 г (выход 70%) 2-оксиметилсахарина, при взаимодействии которого (25 г, 0,117 моль) с трехбромистым фосфором (63,3 г, 0,234 моль) в диэтиловом эфире получали 29,8 г (92%) 2-бромметилсахарина, температура плавления 155-157oC.
Пример 24. К раствору 6-нитросахарина (4 г, 0,0175 моль) в 240 мл этанола добавляли этилат таллия (4,4 г, 0,0175 моль), смесь оставляли стоять при комнатной температуре в течение 1 ч, охлаждали в течение 16 ч, выпавшее в осадок твердое вещество собирали, сушили и получали 7,6 г (выход 100%) таллиевой соли 6-нитросахарина. Этот продукт суспендировали в 50 мл диметилформамида, смесь обрабатывали хлорметилфенилсульфидом (3,07 г, 0,0194 моль), затем нагревали при 6ЗoC в течение 5 ч, оставляли стоять при комнатной температуре в течение 16 ч, а затем выливали в ледяную воду. Отфильтрованный неочищенный продукт перемешивали в хлористом метилене и фильтровали для удаления солей таллия. Из фильтрата удаляли растворитель, а полученное твердое вещество бледно-желтого цвета гомогенизировали с подогретым этанолом, снова отфильтровывали, сушили и получали 4,6 г (выход 75%) 6-нитро-2-фенил-тиометилсахарина, температура плавления 161-163oC. Используя методику, описанную выше в примере 17, проводили взаимодействие этого вещества с хлористым сульфурилом в хлористом метилене и получали 3,7 г 2-хлорметил-6-нитросахарина.
Пример 25A. Раствор 2-окси-5-(1,1,3,3-тетраметилбутил)бензойной кислоты (49,8 г, 0,199 моль), в 200 мл метанола нагревали до 50oC, а затем добавляли к нему по каплям 80 г серной кислоты, скорость добавления была такова, чтобы поддерживать смесь при кипении. Затем смесь нагревали с обратным холодильником еще в течение 11 ч, после чего охлаждали и распределяли между водой и этилацетатом. Органический слой промывали насыщенным раствором бикарбоната натрия, затем рассолом, сушили над сульфатом натрия, испаряли досуха и получали 48,6 г (выход 92%) метилового эфира 2-окси-5-(1,1,3,3-тетраметилбутил)бензойной кислоты.
Это вещество растворяли в 250 мл диметилформамида и добавляли к нему сначала 1,4-диазабицикло [2,2,2] октан (40,4 г, 0,36 моль), а затем диметилтиокарбамоилхлорид (3,4 г, 0,27 моль, и 100 мл диметилформамида. Реакционную смесь нагревали при 45oC в течение 8 ч, охлаждали, выливали в смесь концентрированной соляной кислоты, воды и льда, а затем экстрагировали этилацетатом. Объединенные органические экстракты промывали разбавленной соляной кислотой, затем раствором бикарбоната натрия, рассолом, сушили, испаряли досуха и получали 48,2 г (выход 76%) метилового эфира 2-(N,N-диметилтиокарбамилокси)-5-(1,1,3,3-тетраметилбутил)-бензойной кислоты, которые нагревали при 200oC в течение 15 ч, затем охлаждали, растворяли в толуоле, хроматографировали на колонке с силикагелем, элюируя смесью этилацетат:толуол 1:9, и получали 3,6 г (выход 14%) метилового эфира 2-(N,N-диметил-карбамилтио)-5-(1,1,3,3-тетра-метилбутил)-бензойной кислоты.
К раствору этого соединения (0,025 моль) в 40 мл хлористого метилена добавляли при перемешивании 80 мл ледяной уксусной кислоты, а затем 16 мл воды. Реакционную смесь охлаждали до 0oC и барботировали через нее хлор в течение 5 мин, поддерживая температуру смеси от 5 до 24oC. Затем смесь перемешивали еще в течение 30 мин, концентрировали в вакууме, а оставшийся раствор выливали в ледяную воду. Смесь экстрагировали этилацетатом, из объединенных органических экстрактов выделяли продукт и получали 6,8 г (выход 78%) метилового эфира 2-хлорсульфонил-5-(1,1,3,3-тетраметил-бутил)бензойной кислоты.
Этот продукт (9,0 г, 0,026 моль) растворяли в тетрагидрофуране и добавляли при охлаждении на ледяной бане к 100 мл концентрированной гидроокиси аммония. Получающийся раствор перемешивали в течение 16 ч, а затем концентрировали в вакууме, после чего этот концентрированный раствор подкисляли до pH 3 концентрированной соляной кислотой. Смесь перемешивали в течение нескольких часов, выделившееся твердое вещество собирали, промывали водой, сушили и получали 9,0 г 5-(1,1,3,3-тетраметилбутил)сахарина), температура плавления 213-215oC.
По методике, описанной в примере 17, проводили реакцию этого сахарина (9,0 г, 0,30 моль) с этилатом таллия в этаноле, а затем осуществляли взаимодействие получающейся соли таллия с хлорметилфенилсульфидом (3,33 г, 0,021 моль) в диметилформамиде и получали 5,76 г (выход 66%) 2-фенилтиометил-5-(1,1,3,3-тетраметилбутил)сахарина, 3,3 г (0,007 моль) которого обрабатывали 0,944 г хлористого сульфурила в хлористом метилене и получали 1 г (выход 41% ) 2 хлорметил-5-(1,1,3,3-тетраметилбутил)сахарина.
Пример 25B. Используя методику, описанную 25A, проводили реакцию этилового эфира 2-окси-6-метилбензойной кислоты (15,5 г, 0,086 моль) с N,N-диметилхлортиокарбаматом (15,9 г, 0,129 моль) в присутствии 1,4-диазабицикло[2.2.2]октана (19,3 г, 0,172 моль) в диметилформамиде и получали 22,1 г (выход 96%) этилового эфира 2-(N,N-диметилтиокарбамилокси)-6-метилбензойной кислоты, которые нагревали при 220oC в течение 10 ч. Полученный продукт хроматографировали в хлористом метилене на колонке с силикагелем и получали этиловый эфир 2-(N, N-диметилкарбамилтио)-6-метилбензойной кислоты в виде масла красно-коричневого цвета.
Раствор этого эфира (22,6 г, 0,0844 моль) в хлористом метилене (170 мл) обрабатывали 340 мл ледяной уксусной кислоты и 68 мл воды, охлаждая на бане сухой лед/ацетон, а затем барботировали через реакционную смесь в течение 10-15 мин хлор (газ). Реакционный сосуд откачивали, чтобы удалить избыток хлора, а хлористый метилен и смесь выливали в воду и распределяли смесь между хлористым метиленом и водой. Органический слой сушили, испаряли досуха и получали 19 г этилового эфира 2-хлорсульфонил-6-метилбензойной кислоты, при взаимодействии которого (5 г, 0,019 моль) с концентрированной гидроокисью аммония в тетрагидрофуране получали 6,1 г (выход 67%) 4-метилсахарина.
Используя методику, описанную в примере 17, этот продукт (10,1 г, 0,0512 моль) переводили в соль таллия реакцией с этилатом таллия (12,8 г, 0,0512 моль) в этаноле, а затем при взаимодействии соли таллия с хлорметилфенилсульфидом (6,7 г, 0,0427 моль) в диметилформамиде получали 6,85 г (выход 50% ) 2 фенилтиометил-4-метилсахарина.
При реакции этого соединения (6,7 г, 0,021 моль) с хлористым сульфурилом в хлористом метилене получали 4,9 г (выход 95%) 2-хлорметил-4-метилсахарина.
Пример 26A. Смесь 3,3-дитиобиспропионовой кислоты (75 г, 0,36 моль) 102 мл хлористого тионила и каталитических количеств пиридина перемешивали в течение 24 ч, а затем испаряли досуха в вакууме. Остаток обрабатывали хлористым метиленом, для удаления остатков хлористого тионила и пиридина снова испаряли досуха и получали 87 г (выход 98%) соответствующего хлорангидрида бис-кислоты, 44,8 г (0,18 моль) которого растворяли в тетрагидрофуране и добавляли по каплям к раствору бензиламина (77,16 г, 0,72 моль) в тетрагидрофуране. Смесь перемешивали при 40-45oC в течение 2 ч, охлаждали, а выпавшее в осадок твердое вещество собирали, промывали водой. Сушили и получали 59 г (выход 84%) N,N'-дибензилкарбоксамида 3,3-дитиобиспропионовой кислоты.
При взаимодействии этого соединения (7,0 г, 0,018 моль) с хлористым сульфурилом (10,25 г, 0,076 моль) в хлористом метилене получали смесь 2-бензил-2Н-изотиазол-3-она и 5-хлор-2-бензил-2Н-изотиазол-3-она, которые преимущественно разделяли друг от друга гомогенизированием в хлористом метилене (который растворял исключительно первое соединение). Нерастворимое вещество отфильтровывали и хроматографировали на колонке с силикагелем хлористым метиленом. Таким образом, был получен 5-хлор-2-бензил-2Н-изотиазол-3-он, температура плавления 58-68oC.
Раствор этого соединения (10 г, 0,044 моль) в хлористом метилене охлаждали до 0oC и добавляли к нему 3-хлорнадбензойную кислоту 7,6 г (0,044 моль) смесь перемешивали в течение 10 мин, а затем добавляли к ней вторую порцию (7,6 г) надбензойной кислоты. Реакционную смесь фильтровали, фильтр промывали хлористым метиленом, а фильтрат промывами насыщенным раствором бикарбоната натрия, затем рассолом, сушили над сульфатом натрия, испаряли досуха, а остаток хроматографировали в хлористом метилене на силикагеле, элюируя продукт смесью гексан:хлористый метилен 50:50 и получали 7,15 г (выход 46%) 5-хлор-2-бензил-2Н-изотиазол-3-он-1оксида.
К раствору этого соединения (1,1 г, 0,0045 моль) в 8 мл бензола добавляли 2-метоксифуран (0,55 г, 0,0051 моль) и нагревали раствор в толстостенной склянке для реакций под давлением при 70oC в течение 1,5 ч, а затем охлаждали, твердое вещество собирали, промывали бензолом, сушили и получали 2-бензил-7-окси-4-метоксибензизотиазол-3-он-1-оксид, температура плавления 235-237oC.
Смесь этого продукта (1,85 г, 0,006 моль), карбоната калия (2,48 г, 0,018 моль) и йодистого метила (1,70 г, 0,012 моль) в ацетоне нагревали с обратным холодильником в течение 1,5 ч, а затем охлаждали и выливали в воду. Выпавшее в осадок твердое вещество выделяли фильтрацией, промывали водой, сушили и получали 1,70 г (выход 89%) 2-бензил-4,7-диметоксибензизотиазол-З-он-1-оксида, 1,13 г которого (0,0035 моль) окисляли в хлористом метилене 3-хлорнадбензойной кислотой (1,20 г, 0,007 моль) по методике, описанной выше, и получали 1,03 г (выход 88%) 2-бензил-4,7-диметоксисахарина.
Смесь 2-бензил-4,7-диметоксисахарина (2,07 г, 0,0062 моль) муравьинокислого аммония (1,37 г, 0,02 моль) и 1,5 г катализатора (10% палладия на угле) в 80 мл метанола нагревали с обратным холодильником в течение 1 ч, затем охлаждали, фильтровали, фильтрат испаряли досуха и получали 0,92 г (выход 57%) аммониевой соли 4,7-диметоксисахарина.
К раствору этой аммониевой соли (1,11 г, 0,0042 моль) в диметилформамиде добавляли хлорметилфенилсульфид (0,67 г, 0,0042 моль) и нагревали раствор с обратным холодильником в течение 8 ч, затем смесь охлаждали и выливали в ледяную воду. Выделившееся твердое вещество собирали, промывали водой, сушили и получали 0,50 г (выход 33%) 2-фенилтиометил-4,7-диметоксисахарина.
Проводя реакцию между 2-фенилтиометил-4,7-диметоксисахарином (0,5 г, 0,0013 моль) и хлористым сульфурилом в хлористом метилене по методике, описанной выше в примере 17, получали 0,22 г (выход 58%) 2-хлорметил-4,7-диметоксисахарина.
Примеры 26B и 26C. Используя методику аналогичную той, что описана в примере 26A, другие производные 2-хлорметилсахарина получали следующим образом.
Пример 26B. При взаимодействии 5-хлор-2-бензил-2Н-изотиазол-4-она (5,8 г, 0,024 моль) с 2-этоксифураном (3,76 г, 0,0335 моль) получали 3,05 г (выход 40%) 2-бензил-4 этокси-7-оксибензизотиазол-З-он-1-оксида, при реакции которого (5,7 г) с бромистым 2-[2-метоксиэтокси]этилом (3,6 г, 0,0197 моль) в присутствии карбоната калия (4,95 г, 0,0358 моль) в 125 мл метилэтилкетона и 25 мл диметилформамида получали 7,0 г (выход 93%) 2-бензил-4-этокси-7-[2-(2-метоксиэтокси)этокси]бензизотиазол-3-он-1-оксид, который окисляли, как описано выше, 3-хлорнадбензойной кислотой в хлористом метилене и получали 2-бензил-4-этокси-7-[2-(2-метоксиэтокси)этокси]сахарин. При дебензилировании этого соединения (6,6 г, 0,16 моль) с муравьинокислым аммонием (3,34 г, 0,053 моль) в присутствии 6,4 г катализатора (10% палладия на угле) в метаноле получали аммониевую соль 4-этокси-7-[2-(2-метоксиэтокси)этокси]сахарина, а затем проводили реакцию этой соли с хлорметилфенилсульфидом (2,38 г, 0,015 моль) в 100 мл диметилформамида и получали 1,46 г (выход 21%) 2-фенилтиометил-4-этокси-7-[2-(2-метоксиэтокси)этокси] сахарина, температура плавления 73-75oC (из изопропанола). При взаимодействии этого продукта (1,4 г, 0,0029 моль) с хлористым сульфурилом (0,4 г, 0,0029 моль) в хлористом метилене получали 1,16 г (выход 100% ) 2-хлорметил-4-этокси-7-[2-2(-метоксиэтокси)этокси]сахарина.
Пример 26C. При взаимодействии 2-бензил-7-окси-4-метокси-бензизотиазол-З-он-1-оксида (3,03 г, 0,01 моль) полученного в примере 26A, с бромистым 2-(2-метоксиэтокси)этилом (2,01 г, 0,011 моль) в метилэтилкетоне в присутствии карбоната калия (2 г, 0,015 моль) получали 2,58 г (выход 64%) 2-бензил-4-метокси-7-[2-(2-метокси)этокси] -бензизотиазол-3-он-1-оксида, который окисляли 3-хлорнадбензойной кислотой (1,1 г, 0,0063 моль) в хлористом метилене и получали 2-бензил-4-метокси-7-[2-(2-метоксиэтокси)этокси]сахарин. При дебензилировании этого продукта (0,25 г, 0,0006 моль) с муравьинокислым аммонием (0,13 г, 0,0021 моль) в присутствии 0,25 г катализатора (10% палладия на угле) в метаноле получали 0,21 г (выход 100%) аммониевой соли 4-метокси-7-[2-(2-метоксиэтокси)этокси]сахарина. При реакции этой аммониевой соли (1,4 г, 0,004 моль) с хлорметилфенилсульфидом (0,63 г, 0,004 моль) в диметилформамиде образовывался 2-фенилтио-метил-4-метокси-7-[2-(2-метоксиэтокси)этокси]-сахарин, при взаимодействии которого с хлористым сульфурилом в хлористом метилене получали 0,53 г (выход 35%) 2-хлор-метил-4-метокси-7-[2-(2-метоксиэтокси)этокси]сахарина.
Пример 27. Раствор 1,89 г (0,011 моль) диэтиламминотрехфтористой серы (DAST) в 20 мл хлористого метилена добавляли к суспензии 2-окси-метилсахарина (2,13 г, 0,01 моль) в 25 мл хлористого метилена, поддерживая реакционную смесь при температуре -78oC.
Реакционную смесь перемешивали при -78oC в течение 1 ч, затем давали возможность смеси постепенно нагреться до комнатной температуры, перемешивали при этой температуре в течение 16 ч, после чего выливали в ледяную воду. Органический слой отделяли, промывали водой, сушили над сульфатом магния, испаряли досуха и получали 2,2 г продукта, который рекристаллизовали из этилацетата и получали 1,6 г (выход 74%) 2-фторметилсахарина, температура плавления 96-98oC.
Пример 28A. К раствору 4-метилсахарина (0,5 г, 0,0025 моль) в тетрагидрофуране, охлажденному до -78oC на бане сухой лед/ацетон, добавляли по каплям при перемешивании 5,2 мл 1,3 М раствора вторбутиллития в тетрагидрофуране. Смесь перемешивали при -78oC еще в течение 1 ч, а затем обрабатывали в течение 1,5 ч йодистым метилом (0,16 мл, 0,025 моль). Смесь перемешивали в течение 1 ч и 45 мин, быстро охлаждали в 25 мл 1 N соляной кислоты, затем к реакционной смеси добавляли основание, водную смесь экстрагировали хлороформом, после чего подкисляли и экстрагировали этилацетатом. Объединенные органические экстракты промывали 10%-ным раствором тиосульфата натрия, затем рассолом, сушили над сульфатом натрия, испаряли досуха и получали продукт, который, по данным спектра электронного парамагнитного резонанса (ЭПР), представляет собой смесь, содержащую 74% 4-этилсахарина, и 21% 4,7-диметилсахарина.
Используя методику, описанную в примере 17, проводили взаимодействие этого неочищенного продукта (0,47 г, 0,022 моль) с хлорметилфенилсульфидом (0,24 мл, 0,0028 моль) в толуоле в присутствии бромистого тетрабутиламмония, полученный продукт хроматографировали на колонке с силикагелем, элюируя хлористым метиленом и собирая фракции по 5 мл. Первые 420 мл элюата отбрасывали. При испарении следующих 20 фракций получали 0,07 смеси, содержащей главным образом 4,7-диметилсахарин, который отбрасывали. После испарения последующих 25 фракций получали 0,37 г 2-фенилтиометил-4-этилсахарина, при взаимодействии которого с хлористым сульфурилом в хлористом метилене получали 0,29 г (выход 66%) 2-хлорметил-4-этилсахарина.
Пример 28B. Используя методику, описанную в примере 28A, проводили реакцию 4-метилсахарина (10 г, 0,051 моль) с 86 мл, 1,18 М раствора втор-бутиллития (0,10 моль) в тетрагидрофуране, получающийся раствор обрабатывали йодистым метилом (4,5 мл, 0,050 моль) и получали 10,15 г (выход 89%) 4-пропилсахарина, при взаимодействии которого с хлорметилфенилсульфидом (5,32 мл, 0,056 моль) в толуоле в присутствии бромистого тетрабутиламмония образовывался 2-фенилтиометил-4-пропилсахарин (выход 65%) в виде масла, после чего проводили реакцию этого вещества (1,8 г, 0,0052 моль) с хлористым сульфурилом (1,25 мл, 0,016 моль) в хлористом метилене и получали 0,94 г (выход 66%) 2-хлор-метил-4-пропилсахарина.
Пример 29. Проводили реакцию между веществом (проба 0,07 г), полученным из начальных фракций при хроматографическом разделении, описанном выше в примере 28A, и хлористым сульфурилом (0,05 мл) в хлористом метилене, продукт рекристаллизовали из смеси циклогексан этилацетат и получали 20 мг (выход 51%) 2-хлорметил-4,7-диметил-сахарина, температура плавления 107-108oC.
Примеры 30A-30BK. Используя методику аналогичную той, что описана в примере 4, и заменяя 2-хлорметил-4-хлорсахарин и натриевую соль 1-фенил-тетразола, используемые там, на эквимолярные количества соответственно подходящего 2-галометил-4-R3-R4-замещенного сахарина и подходящего фрагмента LnR1, получали соединения, приведенные в табл.2. В столбце, озаглавленном "X/основание", приведены галоген, содержащийся в 2-галометилсахарине, и основание, которые было использовано, чтобы катализировать реакцию, т.е. или натриевая или таллиевая соль LnR1 реагента, или добавленный основный катализатор, т.е. карбонат калия, триэтиламин (TEA), этилдиизопропиламин (EDIPA) или метилат натрия. В столбце, озаглавленном "Раст.", приведен используемый растворитель [DMF (диметилформамид), THE (тетрагидрофуран), MDC (хлористый метилен), MEK (метилэтилкетон) или ацетон] а в столбце, озаглавленном "Тпл/раст. ", приведены температура плавления получаемого вещества и растворитель, из которого проводили рекристаллизацию. В табл.2 и в табл.3-8 приведены следующие сокращения для гетероциклических или других групп, R1: тет.
тетразолил, триаз. триазолил, мор. морфолинил, тиадиаз. тиадиазолил, имидаз. имидазолил, бензтиаз. бензотиазолил, бензоксаз. бензоксазолил.
Примеры 31A-31C. Используя методику, описанную в примере 17, и заменяя сахарин и метиловый эфир 2-хлор-2фенилтиоуксусной кислоты, применяемые там, на эквимолярные количества соответственно подходящего 4-R3-R4-замещенного сахарина и подходящего Cl-CHR2-S-R1 фрагмента, аналогично получали соединения, приведенные в табл.3, причем в этих соединениях n 1, а L представляет собой -S-. В каждом случае использовали таллиевую соль производного сахарина, а реакции проводили в диметилформамиде.
Пример 32A. Раствор 2-(2,6-дихлорфенилтиометил)сахарина (0,28 г, 0,00067 моль) в 5 мл хлористого метилена обрабатывали при перемешивании 3-хлорнадбензойной кислотой (0,3 г, 0,0017 моль), затем смесь перемешивали в течение 16 ч, после чего быстро охлаждали 10%-ным водным раствором бисульфита натрия. Реакционную смесь разбавляли хлористым метиленом, соли разделяли, органический слой промывали последовательно водой, насыщенным раствором бикарбоната натрия и насыщенным раствором хлористого аммония, сушили над сульфатом натрия, испаряли досуха, а остаток хроматографировали на колонке с силикагелем, элюируя смесью хлористый метилен:диэтиловый эфир 10:1. Таким образом, было получено 0,1 г(выход 10%) 2-(-2,6-дихлорфенилсульфонилметил)сахарина, температура плавления 201,0-203,0oC.
Пример 32B. Используя методику, описанную в примере 32A, 2-(2-пиримидинил-сульфонилметил)сахарин (0,75 г, 0,0023 моль) окисляли 3-хлор-надбензойной кислотой (0,4 г, 0,0023 моль) в 50 мл хлористого метилена, а продукт рекристаллизовали из смеси акрилонитрил:этанол 75:25 и получали 2-(2-пиримидинилсульфонилметил)сахарин, температура плавления 225-227oC.
Пример 33A. К раствору 2-(5-меркапто-1,3,4-тиадиазол-2-илтиометил)сахарина (0,345 г, 0,001 моль) и триэтиламина (0,46 мл, 0,003 моль) в 2 мл диметилформамида добавляли хлоргидрат 4-(2-хлорэтил)морфолина (0,37 г, 0,002 моль). Реакционную смесь перемешивали при комнатной температуре в течение 24 ч, а затем быстро охлаждали, выливая в воду, и экстрагировали этилацетатом. Органический слой промывали водой, затем рассолом, испаряли досуха и получали масло желтого цвета, которое переносили в хлороформ и хроматографировали на колонке с силикагелем, элюируя этилацетатом. Таким образом было получено 0,225 г (выход 49%) 2-(5-[2-(4-морфолинил)-этилтио]-1,3,4-тиадиазол-2-илтиометил)сахарина, температура плавления 129-131oC.
Пример 33B. Используя методику, описанную выше в примере 33A, проводили реакцию 2-(5-меркапто-1,3,4-тиадиазол-2-илтиометил)-сахарина (1,72 г, 0,005 моля) с хлоргидратом 2-диметиламиноэтилхлорида (1,44 г, 0,01 моль) в 10 мл диметилформамида в присутствии триэтиламина (1,72 г, 0,017 моль) и получали 1,2 г (выход 58%) 2-/5-[N,N-диметиламино/этилтио]-1,3,4-тиадиазол-2-илтиометил/сахарина, температура плавления 90,5-91,5oC (из этилацетата).
Пример 33C. Используя методику, описанную в примере 33A, проводили реакцию 2-(5-меркапто-1,3,4-тиадиазо-2-илтиометил)сахарина (0,69 г, 0,002 моль) с хлоргидратом 1-(2-хлорэтил)пиперидина (0,74 г, 0,004 моль) в 4 мл диметилформамида в присутствии 0,4 г триэтиламина и получали 0,55 г (выход 60% ) 2-/5-[2-(1-пиперидинил)этилтио]-1,3,4-тиадиазол-2-илтиометил)сахарина, температура плавления 100,0-101,0oC (из этилацетата).
Пример 32Д. Используя методику, описанную в примере 33A, проводили реакцию 2-(5-меркапто-1,3,4-тиадиазол-2-илтиометил)сахарина с хлоргидратом 2-хлорэтилдиэтиламина в диметилформамиде в присутствии триэтиламина и получали 2-/5-[2-(диэтиламина)этилтио]-1,3,4-тиадиазол-2-илтиометил/сахарина, температура плавления 93,0-94,5oC (из смеси циклогексан:этилацетат).
Пример 33E. Используя методику, описанную в примере 33A, проводили реакцию 2-(5-меркапто-1,3,4-тиадиазол-2-илтиометил)сахарина (1,72 г, 0,005 моль) с хлоргидратом 1-(2 -хлорэтил)пиперидина (1,84 г, 0,01 моль) в 10 мл диметилформамида в присутствии 2,4 мл триэтиламина и получали 1,08 г (выход 47% ) 2-/5-[2-1-/-пиперидинилэтилтио] -2-тиоксо-1,3,4-тиадиазолин-3-илметил)сахарина, температура плавления 89,0-92,0oC, в котором 1,3,4-тиадиазол-2-илтиометильная группа во время реакции подвергается перегруппировке.
Пример 34. К раствору 2-[1-(2-оксиэтил)-1Н-тетразол-5-илтиометил]сахарина (0,44 г, 0,0013 моль) в 10 мл ацетона добавляли хромовую кислоту, полученную из разбавленной серной кислоты и дигидрата бихромата натрия (реактив Джонсона), поддерживая реакционную смесь при 0oC до тех пор, пока раствор не стал постоянно оранжево-коричневого цвета. Смесь перемешивали при 0oC еще в течение 1 ч, затем при комнатной температуре в течение 6 ч, разбавляли водой и экстрагировали этилацетатом. Объединенные органические экстракты промывали водой, затем рассолом, сушили и испаряли досуха, а остаток хроматографировали на колонке с силикагелем, элюируя смесью 5-15% метанола - хлористый метилен, и получали 0,29 г (выход 63%) 2-[1-/2-карбоксиметил/-1Н-тетразол 5-илтиометил]сахарина, температура плавления 173-175oC.
Пример 35. Раствор 2-[1-(3-аминофенил)-1Н-тетразол-5-илтиометил]сахарина (1 г, 0,0026 моль) и ангидрида янтарной кислоты (0,26 г, 0,0026 моль) в 50 мл диоксана перемешивали при комнатной температуре в течение 2 ч, нагревали с обратным холодильником в течение 6 ч, затем перемешивали при комнатной температуре в течение двух дней, после чего снова нагревали до кипения и добавляли в течение нескольких часов небольшие количества ангидрида янтарной кислоты до тех пор, пока анализ методом тонкослойной хроматографии (TLC) не показал отсутствие исходного вещества в реакционной смеси. Реакционную смесь охлаждали, выливали в разбавленную соляную кислоту и ледяную воду, а твердое вещество, выпавшее в осадок, собирали, сушили, рекристаллизовали из смеси этанол: ацетонитрил 50:50 и получали 0,5 г (выход 39%) 2-[1-/3-сукциноиламинофенил/-1Н-тетразол-5-илтилметил]сахарина, температура плавления 197-199oC.
Пример 36. 5-нитро-2-/1-фенил-1Н-тетразол-5-илтиометил/сахарин (1 г, 0,0024 моль) восстанавливали в 150 мл тетрагидрофурана при давлении водорода 3,6 ат в присутствии 3 ложечек никеля Ренея (промытого перед использованием тетрагидрофураном). По окончании восстановления (приблизительно через 5 ч) реакционную смесь фильтровали, слой фильтра промывали тетрагидрофураном, а фильтрат испаряли досуха и получали мутное масло желтого цвета, которое переносили в этанол и фильтровали. Продукт, выделившийся при охлаждении, собирали и получали 0,4 г (выход 43%) 5-амино-2-/1-фенил-1Н-тетразол-5-илтиометил/сахарина в виде кристаллов желтого цвета.
Пример 37. Суспензию 5-амино-2-/1-фенил-1Н-тетразол-5-илтиометил/сахарина (0,4 г) в 25 мл ацетонитрила обрабатывали хлорангидридом уксусной кислоты (6,08 г, 0,001 моль), смесь нагревали с обратным холодильником в течение 30 мин, затем добавляли еще каплю хлорангидрида уксусной кислоты и продолжали кипятить в течение 30 мин. Испарение смеси досуха приводило к образованию белой пены, которую хроматографировали на колонке с силикагелем, элюируя смесью хлористого этилена и этилацетата (95:5), и получали 200 мг (43%) 5-ацетиламино-2-(1-фенил-1Н-тетразол-5-илтиометил)сахарина.
Пример 38. Раствор натриевой соли 1-фенил-5-меркаптотетразола (5 г, 0,025 моль) в 50 мл метилэтилкетона добавляли по каплям при перемешивании к подогретому раствору 3-хлор-1-йодпропана в 300 мл метилэтилкетона. Смесь перемешивали при 40oC в течение 6 ч, затем оставляли стоять при комнатной температуре в течение двух дней, после чего реакционную смесь испаряли досуха в вакууме. Остаток растворяли в хлористом метилене, раствор промывали водой, а водные вытяжки снова экстрагировали хлористым метиленом. Объединенные органические экстракты сушили, испаряли досуха и получали масло желтого цвета, которое хроматографировали на колонке с силикагелем, элюируя хлористым метиленом. Таким образом было получено 3,6 г (выход 75%) 1-фенил-5-/3-хлорпропилтио/-1Н-тетразола в виде масла бледно-желтого цвета. (В другом опыте это же вещество было получено в виде твердого кристаллического вещества белого цвета, температура плавления 33-34oC.)
Этот продукт (6,5 г, 0,014 моль) растворяли в 100 мл метилэтилкетона, добавляли к нему натриевую соль тиофенола (1,7 г, 0,014 моль), смесь нагревали при 40oC в течение 3 ч, затем выливали в раствор бикарбоната калия и экстрагировали смесь хлористым метиленом. Объединенные органические экстракты сушили, испаряли досуха и получали масло бледно-желтого цвета, которое хроматографировали на колонке с силикагелем, элюируя хлористым метиленом, и получали 3,85 г (выход 86%) 1-фенил-5-[3-(фенилтио)пропилтио]-1Н-тетразола. (В другом опыте то же вещество было получено в виде белых кристаллов с температурой плавления 57-59oC).
Раствор этого продукта (3,8 г, 0,012 моль) в 100 мл четыреххлористого углерода обрабатывали N-хлорсукцинимидом (1,5 г, 0,012 моль), смесь оставляли стоять при комнатной температуре в течение 1 ч, затем фильтровали, фильтрат испаряли досуха и получали 4,3 г 1-фенил-5-[3-хлор-3-(фенилтио)пропилтио]-1Н-тетразола.
Этот продукт (0,012 моль) и таллиевую соль сахарина (4,58 г, 0,12 моль), растворенные в 75 мл диметилформамида, нагревали при 50oC в течение 3 ч, а затем оставляли стоять при комнатной температуре в течение 2 ч, после чего фильтровали и промывали фильтр диметилформамидом. Объединенный фильтрат выливали в воду, смесь экстрагировали хлористым метиленом, объединенные органические экстракты промывали рассолом, концентрировали досуха и получали масло бледно-желтого цвета, которое хроматографировали на колонке с силикагелем, элюируя хлористым метиленом, и получали 2,45 г (выход 41%) 2-[1-фенилтио-3-(1-фенил-1Н-тетразол-5-илтио)-пропил]сахарина.
Этот продукт (1 г, 0,002 моль) растворяли в хлористом метилене и окисляли 3-хлорнадбензойной кислотой (0,34 г, 0,002 моль) по методике, описанной в примере 32A. Таким образом, было получено 0,8 г (выход 76%) 2-[1-фенилсульфинил-3-/1-фенил-1Н-тетразол-5-илтил]пропил сахарина.
Этот продукт (1,6 г, 0,003 моль) нагревали при 120oC в течение 45 мин в 130 мл диметилового эфира диэтиленгликоля, затем смесь охлаждали и выливали в воду. Выделившееся твердое вещество собирали, сушили и растворяли в хлористом метилене, а раствор хроматографировали на колонке с силикагелем, элюируя хлористым метиленом. Таким образом, было получено 1,2 г транс-2-[3-/1-фенил-1Н-тетразол-5-илтио-/1-/пропенил] сахарина, температура плавления 191-193oC.
Пример 39. Другие 2-незамещенные сахарины формулы II, используемые в качестве промежуточных соединений при синтезе соединений формулы I, могут быть получены следующим образом.
Реакция 3-трифторметилбензойной кислоты с хлористым тионилом приводит к образованию хлорангидрида 3-трифторметилбензойной кислоты, при взаимодействии которого с диэтиламином получают 3-трифторметил-N,N-диэтилбензамид. Используя методику, описанную в примере 22A, проводят реакцию этого соединения с втор-бутиллитием с последующим взаимодействием получающейся соли лития с двуокисью серы, а затем с натриевой солью гидроксиаминосульфокислоты и получают 3-трифторметил-2-аминосульфонил-N,N-диэтилбензамид, при нагревании которого в ледяной уксусной кислоте образуется 7-трифторметил-сахарин.
Аналогично реакция 4-циклогексилбензойной кислоты с хлористым тионилом приводит к образованию хлорангидрида 4-циклогексилбензойной кислоты, при взаимодействии которого с диэтиламином получают 4-циклогексил-N,N-диэтилбензамид. Используя методику, описанную в примере 22A, проводят реакцию этого соединения с втор-бутиллитием с последующим взаимодействием получающейся соли лития с двуокисью серы, а затем с натриевой солью гидроксиламиносульфокислоты и получают 4-циклогексил-2-амино-сульфонил-N,N-диэтилбензамид, при нагревании которого в ледяной уксусной кислоте образуется 6-циклогексилсахарин.
При взаимодействии 6-аминосахарина с хлорангидридом метансульфокислоты или с хлористым трифторметилсульфонилом в хлористом метилене в присутствии пиридина образуются, соответственно 6-метилсульфониламиносахарин или 6-трифторметилсульфониламиносахарин.
Диазотирование 6-аминосахарина азотистой кислотой и разложение получающейся соли диазония в присутствии цианистой меди (2) или хлористой меди (2) и двуокиси серы, или хлористой меди (2) и щелочной соли метилмеркаптана или трифторметилмеркаптана приводит, соответственно, к образованию 6-цианосахарина, 6-хлорсульфонилсахарина, 6-метилтиосахарина или 6-трифторметилтиоса-арина. При взаимодействии 6-хлорсульфонилсахарина in situ с аммиаком или метансульфониламидом образуются соответственно 6-аминосульфонилсахарин и 6-метансульфониламиносульфонилсахарин. При окислении 6-метилтиосахарина и 6-трифторметилтиосахарина двумя мольными эквивалентами 3-хлорнадбензойной кислоты получают, соответственно, 6-метилсульфонилсахарин и 6-трифторметилсульфонилсахарин.
Гидролиз 6-цианосахарина путем нагревания с водным раствором гидроокиси натрия приводит к образованию сахарин-6-карбоновой кислоты. При взаимодействии 6-цианосахарина с каталитическим количеством серной кислоты, которое проводят в этанольном растворе при нагревании, получают этиловый эфир сахарин-6-карбоновой кислоты, при восстановлении которого боргидридом лития образуется 6-оксиметилсахарин. При окислении этого соединения комплексом пиридин: трехокись хрома (2: 1) (реагент Collin) в хлористом метилене получают 6-формилсахарин, восстановительное аминирование которого аммиаком и цианоборгидридом натрия приводит к образованию 6-аминометилсахарина.
Реакция каждого 2-незамещенного сахарина, полученного таким образом, с хлорметилфенилсульфидом в присутствии трет-бутилата калия и бромистого тетрабутиламмония и последующая реакция получающихся 2-фенилтиометилсахаринов с хлористым сульфурилом в хлористом метилене приводят к образованию R4-2-незамещенных сахаринов формулы I, приведенных ниже, где m и n в каждом случае равны 0, R1 является Cl, а R2 и R3 оба представляют собой водород:
Пример R4
39A 7-CF3
39B 6-циклогексил
39C 6-CH3SO2NH
39D 6-CF3SO2NH
39E 6-CN
39F 6-NH2SO2
39G 6-CH3SO2NHSO2
39H 6-CH3SO2
39I 6-CF3SO2
39J 6-HOOC
39K 6-HOCH2
39L 6-OHC
39M 6-NH2CH2
Пример 40. Используя методику, описанную в примере 4, и заменяя 2-хлорметил-4-хлорсахарин и натриевую соль 1-фенилтетразола, применяемые там, на эквимолярные количества соответственно подходящего 2-хлорметил-R4-сахарина, описанного выше, и подходящего LnR1фрагмента, получали соединения, приведенные в табл.4, в которых R3 в каждом случае, если не оговорено особо, является водородом.
В соединении 40N (a) R3 является изопропилом. Это соединение получают реакцией N, N-диэтилкарбамилхлорида с литиевой солью 2-бром-5-метокси-изопропилбензола; взаимодействием полученного (выход 79%) N,N-диэтил-2-изопропил-4-метоксибензамида с втор-бутиллитием и последующим взаимодействием с двуокисью серы и натриевой солью гидроксиламиносульфокислоты; нагреванием полученного (выход 56%) N,N-диэтил-2-аминосульфонил-4-метокси-6-изопропилбензамида в ледяной уксусной кислоте; реакцией полученной (выход 10%) диэтиламмониевой соли 4-изопропил-6-метоксисахарина с хлорметилфенилсульфидом; 6-метоксисахарина с хлористым сульфурилом и реакцией полученного (выход 88%) 2-хлорметил-4-изопропил-6-метоксисахарина с натриевой солью 1-фенил-5-меркаптотетразола.
Результаты биологических испытаний.
Измерение константы ингибирования, Ki, комплекса HLE ингибитор было описано Cha в Biochem. Pharmacol. 24, 2177-2185 (1975) для констант поистине обратимого ингибирования, имеющих отношение к конкурентным ингибиторам. Однако соединения данного изобретения не образуют истинно обратимые комплексы ингибитора, а потребляются в некоторой степени ферментом. Поэтому вместо измерения Ki вычисляются K
Скорость инактивации ферментативной активности (kon) определяют для испытуемых соединений, измеряя зависимость ферментативной активности аликвоты соответствующего фермента от времени после добавления испытуемого соединения. Построив зависимость log ферментативной активности от времени, получают наблюдаемую скорость инактивации kobs, которая может быть выражена как kobs ln2/t1/2, где t1/2 представляет собой время, необходимое для того, чтобы активность фермента упала на 50% Тогда скорость инактивации равна
kon koff/[J]
где [J] концентрация ингибирующего соединения.
Аналогично определяют константу реактивации, koff, а затем вычисляют константу ингибирования K
K
Значения, полученные для kon и K
Примеры 41A-41L. Дополнительные образцы соединений формулы I, где m равно 0, а R2, R3 и R4 каждый представляет собой водород, были получены из 2-(хлорметил)сахарина [или, если указано, из 2-(бромметил)сахарина] и в каждом примере соответствующего R1-Ln-H (за исключением отмеченных примеров), и эти соединения приведены в табл.6.
Замечания к табл.6:
A Алкилированием 1-(2-хлорэтил)пиперидином в диметилформамиде при комнатной температуре 2-[/5-меркапто-1,3,4-тиадиазол-2-ил/тиометил]сахарина, полученного в свою очередь конденсацией 2-(бромметил)сахарина и 1,3,4-тиадиазол-2,5-дитиола с метилатом натрия в этаноле при нагревании с обратным холодильником, выход 38% (температура плавления 204-206,5oC из 1,2-дихлорэтана, выход 38%).
C Из 2-(бромметил)сахарина, используя в качестве растворителя этанол, а в качестве основания метилат натрия.
D Из 2-(бромметил)сахарина, используя в качестве растворителя тетрагидрофуран.
E Окислением соответствующего сульфида.
G Восстановлением соединения примера 41F.
H Замещением гидроксила в соединении примера 41G на хлор.
I Используя в качестве растворителя ацетонитрил, а в качестве основания метилтриазабициклодецен.
J Используя в качестве растворителя диметилформамид, а в качестве основания этилат таллия.
L Используя в качестве растворителя метилэтилкетон, а в качестве основания карбонат калия.
* Хлористоводородная соль.
Примеры 42A-Bu. Дополнительные образцы соединений формулы I, где m равно 0 (за исключением примера 42B, где m равно 1), R2 и R4 представляют собой водород, а R3 является изопропилом, были получены из 2-хлорметил-4-изопропилсахарина (пример 22E, приведенный выше) (или, если указано, из 2-бромметил-4-изопропилсахарина) и в каждом примере соответствующего R1-Ln-H (за исключением отмеченных примеров), и эти соединения приведены в табл.7.
Замечания к таблице 7:
A: Используя в качестве растворителя диметилформамид, а в качестве основания триэтиламин.
B: Трехстадийный способ: на первой стадии конденсации таллиевой соли 4-изопропилсахарина, полученной из 4-изопропилсахарина и этилата таллия, с 1-фенил-5-(3-хлор-3-фенилтио-пропил)тетразолом в диметилформамиде с выходом 77% на второй стадии окисление полученного 2[3-(1-фенилтетразол-5-ил)-1-(фенилтио)пропил] -4-изопропилсахарина метахлорнадбензойной кислотой с выходом 87% и на третьей стадии термической выделение полученного 2[3-(1-фенилтетразол-5-ил)-1-(фенилтио)пропил]-4-изопропилсахарина, выход 59%
C: Двухстадийный способ: первая стадия конденсация 2-(хлор-метил)-4-изопропилсахарина с метилтиолатом натрия, вторая стадия окисление полученного 2-(метилтио)-4-изопропилсахарина.
D: Реакция с 1,1,3-триоксотетрагидро-1,2,5-тиадиазолом и бромистым тетрабутиламмонием, используя в качестве растворителя толуол.
E: Используя в качестве растворителя диметилформамид, а в качестве основания гидрид натрия.
F: Используя в качестве растворителя метилэтилкетон, а в качестве основания карбонат калия. *Хлористоводородная соль.
H: Двухстадийный способ: на первой стадии конденсация 2-(хлор-метил)-4-изопропилсахарина с монокалиевой солью 1,3,4-тиадиазол-2,5-дитиола в метаноле с выходом 83% на второй стадии конденсация полученного 2-[(5-меркапто-1,3,4-тиадиазол-2-ил)-тиометил] -4-изопропилсахарина с хлоргидратом 4-(2-хлорэтил)морфолина в диметилформамиде с использованием в качестве основания триэтиламина, выход 87% *Хлористоводородная соль.
I: Используя в качестве растворителя дихлорметан, а в качестве основания диазабициклоундецен.
J: Используя в качестве растворителя ацетонитрил, а в качестве основания метилтриазабициклодецен.
K: Алкилирование 2-[(5-меркапто-1,3,4-тиадиазол-2-ил)тиометил]-4-изопропилсахарина (пример 42H) хлоргидратом 1-(2-хлорэтил)-пиперидина с использованием в качестве растворителя диметилформамида, а в качестве основания триэтиламина.
L: Используя в качестве растворителя этанол, а в качестве основания метилат натрия.
M: Алкилирование 2-[(5-меркапто-1,3,4-тиадиазол-2-ил)тиометил]-4-изопропилсахарина (пример 42H) хлоргидратом (2-хлорэтил)-диметиламина в диметилформамиде с использованием в качестве основания триэтиламина. *Соль малеиновой кислоты.
N: Используя в качестве растворителя этанол, а в качестве основания метилат натрия. *Соль малеиновой кислоты.
O: Используя в качестве растворителя этанол, а в качестве основания триэтиламин. *Соль малеиновой кислоты.
Q: Двухстадийный способ: на первой стадии конденсация 2-(хлорметил)-4-изопропилсахарина с азидом натрия в присутствии каталитического количества 18-краун-6 эфира при комнатной температуре с использованием в качестве растворителя бензола или смеси бензол-диметилформамид или бензол-тетрагидрофуран на второй стадии циклоприсоединение получение 2-(азидометил)-4-изопропилсахарина с соответствующим ацетиленом, в этом примере с диметиловым эфиром ацетилендикарбоновой кислоты, реакцию проводят в том же растворителе с или без нагревания.
T: Двухстадийный способ: на первой стадии конденсация 2-(хлор-метил)-4-изопропилсахарина и 2-хлор-4-(4-тиоморфолинил)фенола в ацетонитриле с использованием в качестве основания метилтриазабициклодецена, выход 74,7% на второй стадии окисление полученного 2-[2-хлор-4-(4-тиоморфолинил)феноксиметил] -4-изопропил-сахарина мета-хлорнадбензойной кислотой с выходом 38,5%
X: Используя в качестве растворителя диметилформамид, а в качестве основания карбонат цезия.
AV: Двухстадийный способ: на первой стадии конденсация 2-(хлорметил)-4-изопропил-сахарина и 2-бензил-6-оксисахарина в метил-этилкетоне с использованием в качестве основания карбоната калия, выход 43% на второй стадии дебензилирование каталитическим гидрированием в присутствии палладия, нанесенного на угле, в метаноле, содержащем муравьинокислый аммоний, выход 26%
AX: Используя в качестве растворителя метилэтилкетон, а в качестве основания 0-этилдитиокарбонат калия.
BB: Окисление соединения примера 42AV одним мольным эквивалентом мета-хлорбензойной кислоты.
BC: Окисление соединения примера 42BB одним мольным эквивалентом мета-хлорнадбензойной кислоты.
BF: Получен неочищенным.
BJ: Используя в качестве растворителя ацетонитрил, а в качестве основания гидрид натрия.
BO: Используя в качестве растворителя диметилформамид, а в качестве основания карбонат калия.
BQ: Двухстадийный способ: на первой стадии конденсация 2-(хлорметил)-4-изопропил-сахарина и бензилового эфира 2,6-дихлор-З-оксибензойной кислоты в диметилформамиде с использованием в качестве основания гидрида натрия, на второй стадии дебензилирование каталитическим гидрированием в присутствии палладия, нанесенного на угле, в метаноле, содержащем уксусную кислоту.
BT: Используя в качестве растворителя тетрагидрофуран, а в качестве основания триэтиламин.
Примеры 43A-BV. Получение 2-хлорметил-4-изопропил-6-метоксисахарина.
К раствору 300 мл N,N,N',N'-тетраметилэтилендиамина (TMEDA, 1,99 моля) в 4 л безводного диэтилового эфира добавляли 1550 мл втор-BuLi (1,3 М) и охлаждали смесь в атмосфере азота -70oC. К этому раствору добавляли по каплям в течение 30 мин раствор 2-изопропил-4-метокси-N,N-диэтилбензамида (454 г, 1,82 моль) в 300 мл безводного диэтилового эфира. При добавлении температуру смеси поддерживали -60oC или ниже. После добавления смесь перемешивали при -70oC в течение 1 ч, затем давали ей возможность нагреться до -50oC, выдерживали при -50oC в течение 30 мин, после чего снова охлаждали до -70oC. Затем к смеси добавляли в течение 20 мин при повышенном давлении азота через канюлю раствор 200 г SO2 в 200 мл высушенного диэтилового эфира, предварительно охлажденный до -40oC.
При добавлении температуры реакционной смеси поддерживали ниже -40oC. Почти сразу же выделялся порошкообразный осадок белого цвета - ариллитийсульфинат. После добавления охлаждающую баню убирали и перемешивали смесь при комнатной температуре в течение 2 ч, после чего охлаждали до -5oC. Затем к смеси добавляли по каплям в течение 15 мин при непрерывном перемешивании 190 мл хлористого сульфурила (2,36 моль), поддерживая температуру ниже 10oC. Смесь перемешивали еще в течение 30 мин при 0-5oC, нерастворимый осадок белого цвета отфильтровывали и промывали 2 л безводного диэтилового эфира. После удаления растворителя при атмосферном давлении полученный хлористый сульфонил (темное неочищенное масло) растворяли в 1,4 л тетрагидрофурана. Раствор охлаждали до -10oC и добавляли к нему частями 540 мл концентрированного водного раствора аммиака (28%) в течение 15 мин. При добавлении температуру поддерживали 15oC или ниже. После перемешивания в течение 15 мин при комнатной температуре в вакууме удаляли тетрагидрофуран и избыток аммония и получали темное масло, которое растворяли в 6,0 л воды и подкисляли 3 NHCl до pH 1. Полученное твердое вещество бледно-желтого цвета отфильтровывали, промывали 800 мл воды, сушили в вакууме при 60oC в течение 18 ч, затем рекристаллизовали из смеси 800 мл этилацетата и 3 л гексана и получали 429 л (выход 72%) 2-амино-сульфонил-6-изопропил-4-метокси-N,N-диэтилбензамида, температура плавления 122-125oC.
Раствор этого диэтилбензамида (429,6 г, 1,31 моль) в 1,5 л уксусной кислоты нагревали с обратным холодильником в течение 20 ч, а затем охлаждали до комнатной температуры. Растворитель удаляли в вакууме. Маслянистый остаток растворяли в 6 л воды и доводили pH раствора 6NHCl до 1. Сырой продукт отфильтровывали, промывали 2 л воды, сушили в вакууме при 60oC в течение 18 ч, рекристаллизовали из смеси этилацетат/гексан и получали 303 г (выход 91%) 4-изопропил-6-метоксисахарина, температура плавления 188oC.
К суспензии параформальдегида (24 г, 0,08 моль) и хлор-триметилсилана (86,4 г, 1,6 моль) в 200 мл 1,2-дихлорэтана добавляли 0,8 мл безводного четыреххлористого олова и перемешивали полученный раствор на паровой бане в течение 1 ч. Затем к прозрачному раствору добавляли 4-изопропил-6-метоксисахарин (51,4 г, 0,2 моль) и нагревали смесь с обратным холодильником в течение 18 ч, затем охлаждали до комнатной температуры и выливали в воду. Органический слой отделяли, промывали 50 мл 2N раствора гидроокиси натрия, сушили над сульфатом магния и концентрировали в вакууме. Остаток очищали кристаллизацией из смеси этилацетат/гексан и получали 57 г (выход 87%) 2-хлорметил-4-изопропил-6-метоксисахарина, температура плавления 151oC.
Приведенные в примерах 43A-CA соединения, в которых m равно 0 (за исключением соединения примера 43F, где m равно 1), R1 является водородом, R3 представляет собой изопропил, а R4 является 6-метоксилом, были получены из 2-хлорметил-4-изопропил-6-метоксисахарина и в каждом примере соответствующего R1-Ln-H (за исключением отмеченных примеров), и эти соединения приведены в табл.8.
Замечания к таблице 8:
A: Используя в качестве растворителя диметилформамид и применяя натриевую соль 1-фенилтетразол-5-тиола.
B: Используя в качестве растворителя дихлорметан, а в качестве основания триэтиламин.
C: Используя в качестве растворителя диметилформамид, а в качестве основания метилат натрия.
D: Двухстадийный способ: на первой стадии конденсация 2-(хлор-метил)-4-изопропил-6-метоксисахарина и азида натрия в бензоле или толуоле в присутствии каталитического количества 18-краун-6-эфира при комнатной температуре, на второй стадии циклоприсоединение полученного 2-(азидометил)-4-изопрспилса-харина с соответствующим ацетиленом, в этом примере фенилсульфонилтриметилсилилацетиленом, реакцию проводят в том же растворителе с или без нагревания.
E: Используя в качестве растворителя метилэтилкетон, а в качестве основания карбонат калия. *Хлористоводородная соль.
F: Трехстадийный спос об: на первой стадии конденсация таллиевой соли 4-изопропил-6-метоксисахарина, полученной из 4-изопропил-6-метоксисахарина и этилата таллия, с 1-фенил-5-3-хлор-(3-фенилтиопропил)-тетразолом в диметилформамиде с выходом 57% на второй стадии окисление полученного 2[-3-(1-фенилтетразол-ил)-1-(фенилтио)пропил] -4-изопропил-6-метоксисахарина мета-хлорнадбензойной кислотой с выходом 60,5% на третьей стадии термическое выделение полученного 2-[3-(1-фенилтетразол-5-ил)-1-(фенилтио)-пропил] -4-изопропил-6-метоксисахарина, выход 75%
I: Используя в качестве растворителя диметилформамид, а в качестве основания гидрид натрия.
K: Используя в качестве растворителя ацетонитрил, а в качестве основания метилтриазабициклодецен. * Смесь геометрических изомеров (1:1).
AD: Используя в качестве растворителя метилэтилкетон и применяя калиевую соль З-фенил-2-тиоксо-2,3-дигидро-1,3,4-тиадиазол-5-тиола.
AH: Используя в качестве растворителя диметилформамид, а в качестве основания карбонат калия.
AV: Используя в качестве растворителя ацетон и применяя тиоцианат калия.
AW: Используя в качестве растворителя диметилформамид, а в качестве основания диизопропилэтиламин.
BB: Используя в качестве растворителя ацетонитрил, а в качестве основания диизопропилэтиламин.
BF: Используя в качестве растворителя тетрагидрофуран, а в качестве основания трет-бутилат калия.
BH: Используя в качестве растворителя смесь ацетонитрила и диметилформамида, а в качестве основания метилтриазабициклодецен.
Пример 44. Раствор 2-хлорметил-4-этил-5,7-диметоксисахарина (пример 22L, 0,4 г) и натриевой соли 1-фенилтетразол-5-тиола (0,28 г) в 3 мл диметилформамида нагревали при 110oC в течение 2 ч, а затем выливали в воду. После рекристаллизации образующегося твердого вещества из смеси этанол-вода получали 2-(1-фенилтетразол-5ил)тиометил-4-этил-5,7-диметоксисахарин (0,44 г, выход 74%), температура плавления 162-164oC.
Пример 45.
A. Раствор 5-хлор-2-бензил-2Н-изотиазол-3-он-1-оксида (пример 26A, 1,5 г) и 1-метокси-З-триметилсилилоксибутадиена (d 0,885, 1,30 мл) нагревали при 65oC в течение ночи в трубке для работы под давлением. Затем добавляли еще 0,74 мл 1-метокси-З-триметилсилилоксибутадиена, и снова нагревали раствор в этой трубке при 65oC в течение ночи, после чего десорбировали летучие вещества. К смеси добавляли дихлорметан, десорбировали из нее летучие вещества, и в заключение повторяли добавление и десорбцию при высоком вакууме. К маслу золотисто-янтарного цвета добавляли дихлорметан, через несколько часов это масло затвердевало. Получали 1,16 г (выход 68%) 2-бензил-6-окси-1,2-бензизотиазол-(1Н)-он-1-оксида.
B. Раствор 2-бензил-6-окси-1,2-бензизотиазол-(1Н)-3-он-1-оксида (3,75 г) в 75-100 мл метанола добавляли по каплям при комнатной температуре к раствору магниевой соли орто-мононадфталиевой кислоты (8,14 г) в 70-100 мл воды. Для растворения получающегося осадка добавляли 200-250 мл метанола и перемешивали раствор при комнатной температуре в течение ночи. Затем добавляли одинаковый объем воды и экстрагировали смесь дихлорметаном. Дихлорметановый экстракт промывали водой и насыщенным водным раствором хлористого натрия, сушили над сульфатом натрия и отгоняли из него дихлорметан. Раствор полученного твердого вещества (4,17 г) в дихлорметане промывали водой и насыщенным водным раствором хлористого натрия, сушили над сульфатом натрия, отгоняли из него дихлорметан и получали 3,87 г (выход 98%) 2-бензил-6-оксисахарина.
C. Смесь 2-бензил-6-оксисахарина (0,86 г) 2-бромэтил-2-метоксиэтилового эфира (d 1,347, 0,45 мл), карбоната калия (1, 4 г), метилэтилкетона (50 мл) и диметилформамида (2 мл) нагревали с обратным холодильником в течение 5 ч, а затем выливали в 500 мл воды, охлажденной льдом. Образующееся твердое вещество собирали, промывали водой, сушили и получали 0,92 г (выход 78%) 2 бензил-6-[2-(2-метоксиэтокси)этокси]сахарина, температура плавления 86-88oC.
D. Смесь 2-бензил-6-[2-(2-метоксиэтокси)этокси]сахарина (2,05 г) метанола (75-100 мл), муравьинокислого аммония (1,10 г) и 1,0 г катализатора (10% палладия на угле) нагревали с обратным холодильником в течение 40 мин, затем охлаждали и фильтровали. Из фильтрата отгоняли летучие вещества и получали аммониевую соль 6-[2-(2-метоксиэтокси)этокси]сахарина, смесь которой с хлорметилфенилсульфидом (0,79 г) и диметилформамидом нагревали при 10oC в течение 8 ч, затем перемешивали при комнатной температуре в течение ночи и выливали в 600 мл воды, охлажденной льдом. Полученную смесь водой и насыщенным водным раствором хлористого натрия, сушили над сульфатом натрия и отгоняли из него дихлорметан. Остаток (1,70 г) хроматографировали на колонке с силикагелем, элюируя сначала смесью дихлорметан ацетон (98:1), а затем смесью дихлорметан ацетон (99:1), и получали 0,96 г (выход 45%) 2-фенилтиометил-6-[2-(2-метоксиэтокси)этокси]сахарина.
E. К раствору 0,96 г 2-фенилтиометил-6-[2-(2-метоксиэтокси)-этокси]сахарина в дихлорметане добавляли по каплям при перемешивании 0,34 г хлористого сульфурила Перемешивание продолжали при комнатной температуре в течение 3 ч, а затем из раствора отгоняли летучие соединения. Затем добавляли дихлорметан и снова из раствора отгоняли летучие соединения. К остатку добавляли гексан и перемешивали смесь при комнатной температуре в течение ночи. Образующееся твердое вещество белого цвета собирали, сушили и получали 0,69 г (выход 89%) 2-хлорметил-6-[2-(2-метоксиэтокси)этокси] сахарина, температура плавления 113-115oC.
F. По методике, описанной в примере 44, проводили конденсацию 2-хлорметил-6-[2-(2-метоксиэтокси)этокси] сахарина (0,34 г) и натриевой соли 1-фенилтетразол-5-тиола (0,19 г) в метилэтилкетоне при 60oC. Продукт (470 мг) хроматографировали на колонке с силикагелем, используя сначала дихлорметан, а затем смесь дихлорметан ацетон (вплоть до 97:3), и получали 390 мг (выход 82%) 2-(1-фенилтетразол-5-ил)тиометил-6-[2-/2- метоксиэтокси/этокси]сахарина в виде масла.
Пример 46. Раствор 2-(5-фенил-1-тетразолил)тиометил-4-этилсахарина (пример 30AE, 0,020 г) и натриевой соли 1-фенилтетразол-5-тиола (0,0026 г) в 1 мл диметилформамида нагревали при 100oC в течение трех дней, а затем выливали в воду. После рекристаллизации твердого вещества из смеси этанол - вода получали 0,012 г (выход 60%) 2-(4-фенил-5-тиоксо-1-тетразолил)метил-4-этилсахарина, температура плавления 127-129oC.
Пример 47.
A. Используя методику, приведенную в примере 22A, 3,4-диметоксипропил, N-,N-диметилбензамид (9,2 г) аминосульфонилировали диоксидом серы и оксиамино-0-сульфокислотой 5,6 г и получали 7,4 г (выход 63%) 2-аминосульфонил-4,5-диметокси-6-пропил-N, N-диметилбензамида, который циклизовали с количественным выходом в 4-пропил-5,6-диметоксисахарин, при фенилтиометилировании которого хлорметил-фенилсульфидом (1,42 мл) получали 4,07 г 2-фенилтиометил-4-пропил-5,6-диметоксисахарин, а затем проводили реакцию 3,59 г этого вещества с 2,12 мл хлористого сульфурила и получали 2,84 г (выход 97%) 2-хлорметил-4-пропил-5,6-диметоксисахарина.
B. По методике, описанной в примере 44, проводили конденсацию 2-хлорметил-4-пропил-5,6-диметоксисахарина (0,6 г) и натриевой соли 1-фенилтетразол-5-тиола (0,36 г) в 5 мл диметилформамида, продукт очищали на колонке с силикагелем, элюируя смесью этилацетат гексан (6:7), затем растирали с гексаном и получали 0,65 г (выход 76%) 2-(фенилтетразол-5-ил)тиометил-4-пропил-5,6-диметоксисахарина, температура плавления 145-146oC.
Пример 48.
A. По методике, описанной в примере 22A, получали 2-аминосульфонил-4,5-диметокси-6-изопропил-N, N-диметилбензамид (10,75 г) и циклизовали его в 4-изопропил-5,6-диметоксисахарин (температура плавления 186-188oC из смеси диэтиловый эфир гексан), который (5 г) фенилтиометилировали хлорметилфенилсульфидом (2,48 мл) и получали 4,07 г 2-фенилтиометил-4-изопропил-5,6-диметоксисахарина, реакция которого с хлористым сульфурилом (три мольных эквивалента) приводила к образованию 2-хлорметил-4-изопропил-5,6-диметоксисахарина (выход 85%) температура плавления 117-119oC из смеси этилацетат гексан.
B. По методике, описанной в примере 44, проводили конденсацию 2-хлорметил-4-изопропил-5,6-диметоксисахарина (1,46 г) и натриевой соли 1-фенилтетразол-5-тиола (0,92 г) в диметилформамиде (5 мл), продукт рекристаллизовали из смеси этанол вода и получали 1,05 г (выход 51%) 2-(1-фенилтетразол-5-ил)тиометил-4-изопропил-5,6-диметоксисахарина, температура плавления 69-71oC.
Пример 49.
A. Этилмеркаптан (43,9 г) добавляли при перемешивании к суспензии хлористого алюминия (62,74 г) в 500 мл хлороформа при 0oC. К полученному раствору добавляли в течение 30 мин раствор 4-изопропил-6-метоксисахарина (20,0 г) в 550 мл хлороформа. Полученный раствор нагревали при 60oC в течение 3-4 ч, а затем выливали в ледяную воду, подкисленную соляной кислотой. Образующееся твердое вещество отфильтровывали, промывали водой, сушили и получали 18,4 г (выход 97%) 4-изопропил 6-оксисахарина.
В. По методике, описанной в примере 21, проводили фенилтиометилирование 4-изопропил-6-оксисахарина (0,004 моль) хлорметилфенилсульфидом (0,61 мл) и получали 0,32 г (выход 1%) 2-фенилтиометил-4-изопропил-6-оксисахарина, в результате которого с хлористым сульфурилом (0,73 г) получали 2-хлорметил-4-изопропил-6-оксисахарин (выход 84%), температура плавления 149-150oC.
C. По методике, приведенной в примере 44, проводили конденсацию 2-хлорметил-4-изопропил-6-оксисахарина (0,3 г) и натриевой соли 1-фенилтетразол-5-тиола (0,2 г) в 10 мл диметилформамида, образующийся продукт хроматографировали на испарительной колонке с силикагелем, элюируя смесью гексан этилацетат (7:3) и получали 0,3 (выход 67%) 2-/1-фенилтетразол-1-ил/тиометил/4-изопропил-6-оксисахарина, температура плавления 188,5-189,5oC.
Пример 50.
A. Раствор 5,8 г 5-хлор-2-бензил-2Н-изотиазол-3-он-1-оксида (пример 26A) и 3,76 г 2-этоксифурана в 40 мл бензола нагревали до 50oC. К отвердевшей реакционной смеси добавляли еще 30 мл бензола и нагревали ее с обратным холодильником в течение 15 мин, затем при 70oC в течение 45 мин, после чего охлаждали в холодильнике в течение ночи. Образующееся твердое вещество бледно-желтого цвета собирали, промывали холодным бензолом, сушили и получали 3,05 г (выход 40%) 2-бензил-4-этокси-7-окси-1,2-бензизотиазол-(1Н/-3-он-1-оксида.
B. К раствору 2-бензил-4-этокси-7-окси-1,2-бензизотиазол-(1Н)-3-он-1-оксида (7,9 г) в 500 мл дихлорметана и 31 мл метанола добавляли при перемешивании мета-хлорнадбензойную кислоту (8,6 г). Перемешивание продолжали в течение нескольких часов, затем добавляли еще мета-хлорнадбензойной кислоты (8,6 г) и продолжали перемешивание в течение нескольких дней. К смеси добавляли дихлорметан. Раствор промывали водой. Твердое вещество (2,33 г), выделившееся при повторном промывании раствора водой, собирали, температура плавления 196-199oC. Собирали также твердое вещество (4,10 г), выделившееся при промывании дихлорметанового слоя насыщенным водным раствором хлористого натрия, температура плавления 196-199oC. Было установлено, что оба твердых вещества представляют собой 2-бензил-4-этокси-7-оксисахарина.
C. Смесь 2-бензил-4-этокси-7-оксисахарина (1,0 г), бромистого аллила (0,36 г), карбоната калия (0,62 г) и метилэтилкетона (около 20 мл) нагревали при 80oC в течение 1 ч. Затем добавляли еще бромистый аллил (0,36 г) и продолжали нагревание при 80oC в течение 1,5 ч. Смесь охлаждали и выливали в воду, охлажденную льдом (500 мл). Твердое вещество собирали и получали 1,08 г (выход 96% ) 2-бензил-4-этокси-7-аллилоксисахарина, температура плавления 148-149oC.
D. Смесь 2-бензил-4-этокси-7-аллилоксисахарина (0,25 г) и триглима нагревали при 200oC в течение 20 мин, методом тонкослойной хроматографии анализировали глубину реакции, затем нагревали при 200oC еще в течение 20 мин, перемешивали в течение ночи и выливали в ледяную воду. Получающееся липкое твердое вещество коричневого цвета растворяли в дихлорметане. Дихлорметановый раствор промывали насыщенным водным раствором хлористого натрия, сушили над сульфатом натрия и отгоняли из него дихлорметан. Остаток (0,29 г) хроматографировали на колонке с силикагелем, элюируя дихлорметаном и смесью дихлорэтан: ацетон (вплоть до 99:1), и получали 90 мг (выход 36%) 2-бензил-4-этокси-6-аллил-7-оксисахарина, масс-спектр которого свидетельствовал о присутствии молекулярного иона с массой 373.
E. Смесь 2-бензил-4-этокси-6-аллил-7-оксисахарина (2,45 г), карбоната калия (2,76 г) и йодистого метила (1,84 г) в ацетоне нагревали при перемешивании и температуре 50oC в течение 2 ч, затем выливали при перемешивании в ледяную воду (500 мл). Образовавшийся липкий осадок кристаллизовался в течение ночи. Твердое вещество выделяли и получали 2,35 г (выход 92%) 2-бензил-4-этокси-6-аллил-7-метоксисахарина, температура плавления 92-94oC.
F. По методике, приведенной в части D примера 45, 2-бензил-4-этокси-6-аллил-7-метоксисахарин (2,35 г) одновременно дебензилировали и гидрировали в присутствии муравьинокислого аммония (1,51 г) и 1,25 г катализатора (10% палладия на угле) в 70 100 мл метанола и получали 1,94 г аммониевой соли 4-этокси-6-пропил-7-метоксисахарина, у фенилтиометилировали в диметилформамиде хлорметилфенилсульфидом (0,95 г), а продукт (масло желтого цвета, 2,67 г) очищали хроматографированием на колонке с силикагелем, элюируя смесью дихлорметан-гексан (80:20), и получали 0,8 г (выход 34%) 2-фенилтиометил-4-этокси-6-пропил-7-метоксисахарина.
G. По методике, приведенной в части E примера 45, проводили реакцию 2 фенилтиометил-4-этокси-6-пропил-7-метоксисахарина (0,85 г) с хлористым сульфурилом (0,30 г) в дихлорметане, продукт очищали гексаном и получали 0,62 г (выход 89% ) 2-хлорметил-4-этокси-6-пропил-7-метоксисахарина, температура плавления 131-133oC.
H. Используя методику, приведенную в части F примера 45, проводили конденсацию 2-хлорметил-4-этокси-6-пропил-7-метокси-сахарина (0,62 г) и натриевой соли 1-фенил-тетразол-5-тиола (сначала использовали 0,36 г, а через 3,5 ч добавляли еще небольшое количество соли, общее время реакции 8 ч маслянистый продукт выделяли экстракцией дихлорметаном, очищали сначала хроматографированием на колонке с силикагелем, элюируя дихлорметаном, а затем кристаллизацией полученного масла (0,62 г, выход 71%) из этанола и получали 2-(1-фенилтетразол-5-ил)тиометил-4-этокси-6-пропил-7-метоксисахарин, температура плавления 110-111oC.
Пример 51.
A. К раствору 2,2 г 4-этил-5,7-диметоксисахарина (пример 22L) в 100 мл тетрагидрофурана добавляли по каплям при перемешивании и температуре -78oC в течение 1 ч с втор-бутиллитий (0,87 М в циклогексане, 20,4 мл). Перемешивание продолжили при -78oC в течение 1 ч, после чего добавляли 1,5 мл йодистого метила. Перемешивание продолжали при -78oC в течение 15 мин, затем давали возможность смеси нагреться до комнатной температуры и охлаждали ее в воде. К смеси добавляли водный раствор гидроокиси натрия (0,5% 200 мл), после чего промывали ее 200 мл этилацетата, подкисляли концентрированной соляной кислотой и экстрагировали 200 мл этилацетата. Экстракт промывали водным раствором тиосульфата натрия (10% 50 мл) и насыщенным водным раствором хлористого натрия (50 мл) сушили над сульфатом натрия, отгоняли из него этилацетат и получали 0,73 г (выход 32%) 4-этил-5,7-диметокси-6-метилсахарина.
B. Используя методику, приведенную в примере 21, проводили фенилтиометилирование 4-этил-5,7-диметокси-6-метилсахарина (0,6 г) хлорметилфенилсульфидом (0,24 мл) и получали 2-фенилтиометил-4-этил-5,7-диметокси-6-метилсахарин, при взаимодействии которого с хлористым сульфурилом получали 0,16 г (выход 22%) 2-хлорметил-4-этил-5,7-диметокси-6-метилсахарина.
C. По методике, приведенной в примере 44, проводили конденсацию 2-хлорметил-4-этил-5,7-диметокси-6-метилсахарина (0,17 г) и натриевой соли 1-фенилтетразол-5-тиола (0,084 г) в 4 мл диметилформамида, продукт очищали хроматографированием на испарительной колонке с силикагелем, элюируя смесью гексан этилацетат (75: 25), и получали 0,15 г (выход 76%) 2-(1-фенилтетразол-5-ил)тиометил-4-этил-5,7-диметокси-6-метилсахарина, температура плавления 44-46oC.
Пример 52.
A. Смесь 2-бензил-4-этокси-7-оксисахарина (часть B примера 50, 0,5 г), трет-бутилового эфира бромуксусной кислоты (0,3 г, 0,25 мл), карбонат калия (0,30 г) и метилэтилкетон (около 10 мл) нагревали в течение 1 ч при 70-80oC, затем охлаждали и выливали в воду, охлажденную льдом (400 мл). Смесь перемешивали в течение ночи, твердое вещество собирали и получали 0,58 г (выход 86%), 2-бензил-4-этокси-7-(трет-бутокси-карбонилметокси)сахарина.
B. По методике, приведенной в части примера 45, проводили два последовательных синтезадебензилирование 2-бензил-4-этокси-7-(трет-бутоксикарбонилметокси)сахарина (0,5 г, 6,5 г) муравьинокислым аммонием (0,25 г, 3,66 г) в присутствии катализатора (10% палладия на угле, 0,25 г и 2 г) в метаноле с образованием аммониевой соли 4-этокси-7-(трет-бутоксикарбонилметокси)сахарина (0,44 г, 5,63 г); а затем фенилтиометилирование этой соли хлорметилфенилсульфидом (0,2 г, 2,38 г) в диметилформамиде с последующей очисткой объединенного продукта (0,46 г, 6,42 г) хроматографией на колонке с силикагелем (элюат дихлорметан смесь дихлорметан-ацетон 98:2) и получали 1,9 г (выход 40% ) 2-фенилтиометил-4-этокси-7-(трет-бутоксикарбонилметокси)сахарина.
C. По методике, приведенной в части E примера 45, проводили реакцию 2-фенилтиометил-4-этокси-7-(трет-бутоксикарбонилметокси)-сахарина (1,9 г) и хлористого сульфурила (0,54 г) в дихлорметане при 5oC, продукт очищали гексаном и получали 1,54 г (выход 95%) 2-хлорметил-4-этокси-7-(трет-бутоксикарбонилметокси)сахарина, температура плавления 117-119oC.
D. По методике, приведенной в части F, примера 45, проводили конденсацию 2-хлорметил-4-этокси-7-(третбутоксикарбонилметокси)-сахарина (1,42 г) и натриевой соли 1-фенилтетразол-5-тиола (сначала 0,70 г, а через 6 ч реакции добавляли еще небольшое количество соли, общее время реакции 8 ч), продукт (1,20 г) выделяли экстракцией дихлорметаном, часть его (0,42 г) очищали хроматографией на колонке с силикагелем, элюируя смесью дихлорметан ацетон (до 98: 2) и получали 300 мг (выход 71%) 2-/1-фенилтетразол-5-ил(тиометил-4-этокси-7-)трет-бутоксикарбонилметокси/сахарина, температура плавления 110-112oC.
Пример 53. Раствор 2-(1-фенилтетразол-5-ил)тиометил-4-этокси-7-(третбутоксикарбонилметокси)сахарина (пример 52, 0,47 г) в 5-10 мл трифторуксусной кислоты и 5-10 дихлорметана перемешивали при комнатной температуре в течение 2 ч, затем отгоняли из него летучие вещества, потом еще трижды десорбировали из него дихлорметан, после чего отгоняли из него ацетон. Раствор маслянистого остатка в ацетоне (1 мл) добавляли к 100 мл ледяной воды, содержащей концентрированную соляную кислоту (1 мл). Образовавшееся твердое вещество собирали, промывали водой и сушили (0,34 г). Раствор этого вещества в дихлорметане элюировали через силикагель на 15 мл воронке из спекшегося стекла смесью хлороформ метанол (95:5) и получали 70 мг (выход 17%) 2-(1-фенилтетразол-5-ил)тиометил-4-этокси-7-карбоксиметоксисахарина, температура плавления 187-189oC.
Пример 54.
A. 1,2-диметокси-4-изопропилбензол- (31 г) бромировали N-бромсукцинимидом на кизельгуре в четыреххлористом метане по методике Hisatoshi и др. (Bulletin of the Chemical Society of Japan, т.32, с.591-593, 1989) с получением 5-бром-1,2-диметокси-4-изопропилбензола, при литиировании которого н-бутиллитием в диэтиловом эфире образовывался 5-литий, 1,2-ди-метокси-4-изопропилбензол, затем это соединение диэтиламинокарбонилировали в том же растворителе и получали 15,2 г 2-изопропил-4,5-диметокси-N,N-диэтилбензамида в виде вязкого масла.
B. По методике, приведенной в примере 43, проводили аминосульфирование этого 2-изопропил-4,5-диметокси-N,N-сульфурилом и концентрированным аммиаком с образованием 4,5 г 2 изопропил-4,5-ди-метокси-6-аминосульфонил-N,N-диэтилбензамида, температура плавления 181-182oC, при циклизации которого в уксусной кислоте получали 2,86 г 4-изопропил-6,7-диметоксисахарина, температура плавления 210-212oC.
C. К раствору 4-изопропил-6,7-диметоксисахарина (0,5 г) в 3 мл диметилформамида добавляли 0,5 мл диизопропилэтиламина, нагревали при 80oC в течение 16 ч, разбавляли этилацетатом, промывали водным раствором карбоната натрия, 3N соляной кислотой и водным раствором хлористого натрия, сушили над сульфатом натрия и отгоняли из нее растворители. Остаток очищали хроматографированием на испарительной колонке с силикагелем с использованием в качестве элюента дихлорметана и получали 0,35 г 2-фенилтиометил-4-изопропил-6,7-диметоксисахарина, затем это вещество хлорировали хлористым сульфурилом (0,1 мл) в дихлорметане (3 мл), продут очищали растиранием с гексаном и получали 0,3 г 2-хлорметил-4-изопропил-6,7-диметоксисахарина. конденсацию 2 хлорметил-4-изопропил-6,7-диметоксисахарина.
D. Используя методику, приведенную в примере 44, проводили конденсацию 2-хлорметил-4-изопропил-6,7-диметоксисахарина (0,0095 г) и натриевой соли 1-фенилтетразол-5-тиола (0,120 г) в 2 мл ацетонитрила, продукт очищали кристаллизацией из этанола и получали 0,099 г (выход 70%) 2-(1-фенилтетразол-5-ил)тиометил-4-изопропил-6,7-диметокси-6-метилсахарина, температура плавления 169-171oC.
Пример 55.
A. По методикам, приведенным в частях D и E примера 45, проводили дебензилирование 2-бензил-4-этокси-7-оксисахарина (часть B примера 50) муравьинокислым аммонием в присутствии катализатора (палладий на угле) в метаноле и получали аммониевую соль 4-этокси-7-оксисахарина, которую фенилтиометилировали хлорметилфенилсульфидом в диметилформамиде до 2-фенилтиометил-4-этокси-7-оксисахарина, а затем проводили реакцию этого соединения с 4-этокси-7-оксисахарин.
B. По методике, описанной в части F примера 45, проводили конденсацию 2-хлорметил-4-этокси-7-оксисахарина (1,95 г) и натриевой соли 1-фенил-тетразол-5-тиола (1,5 г, время реакции около 8 ч), часть продукта (2,5 г, выход 88% ) очищали из этанола и получали 2-(1-фенилтетразол-5-ил)тиометил-4-этокси-7-оксисахарин, температура плавления 178-180oC.
Пример 56. Смесь 2-(1-фенилтетразол-5-ил)тиометил-4-этокси-7-окси-сахарина (пример 55, 0,5 г), хлорангидрида диметилкарбаминовой кислоты (0,14 мл, 0,16 г), диазабициклооктана (0,27 г) и N,N-диметилацетамида (20 мл) нагревали при 80oC в течение 4-5 ч, затем охлаждали и выливали при перемешивании в ледяную воду (400 мл). Образовавшееся твердое вещество хроматографировали на колонке с силикагелем, элюируя дихлорметаном и смесью дихлорметан ацетон до 98: 2, и получали сначала 2-(4-фенил-5-тиоксо-1-тетразолил)метил-4-этокси-7-диметиламинокарбонилсахарин (температура плавления 172-173oC после рекристаллизации из этанола), а затем 2-(1-фенилтетразол-5-ил)тиометил-4-этокси-7-диметиламинокарбонилоксисахарин (температура плавления 145-146oC после гомогенизирования в диэтиловом эфире).
Пример 57. 2-(1-фенилтетразол-5-ид)тиометил-4-изопропил-5,6-диметоксисахарин (0,48 г) добавляли при перемешивании и температуре 0oC к раствору хлористого алюминия (0,4 г) и этилмеркаптана (0,15 мл) в 3 мл хлороформа. По окончании добавления смесь выдерживали при комнатной температуре в течение 18 ч. Затем реакционную смесь пропускали через силикагель, элюируя смесью этилацетат гексан (2:3) и получали 0,3 г (выход 77%) 2-(1-фенилтетразол-5-ил)тиометил-4-изопропил-5-окси-6-метоксисахарина в виде бесцветного масла; структура этого соединения была подтверждена методами 13С ЯМР и протонного магнитного резонанса, данными масс-спектрометрическсго анализа и методом ядерного усиления. Последний метод показал 19% усиление сигнала протонов метоксигруппы и 10% усиление сигнала С-7 протона.
Пример 58. (Общая композиция для таблеток). Ингредиенты, мас.
Соединение примеров 41-57 32-62
Дезинтегрирующее вещество 28-36
Связывающее вещество 10-20
Смазывающее вещество 0,1-0,5
Другие наполнители 2-5
Примечания. Дезинтегрирующее вещество может быть представлено различными крахмалами. Связующее может быть представлено различными целлюлозами. Смазывающее вещество представляет собой тальк, силикагель и стеарат магния.
Пример 59. Ингредиенты, мг/таблетку:
Соединение из примера 57 150
Лактоза, моногидрат 100
Гидроксипропилцеллюлоза 50
Поливинилпирролидон 20
Стеарат магния 2
Пример 60. Ингредиенты, мг/таблетку:
Соединение из примера 50 140
Остальные ингредиенты те же самые, что и в примере 59.
Результаты биологических испытаний.
При исследовании ингибирования эластазы с использованием эластазы лейкоцитов человека описанные выше соединения формулы I примеров 41-57 показали величины K
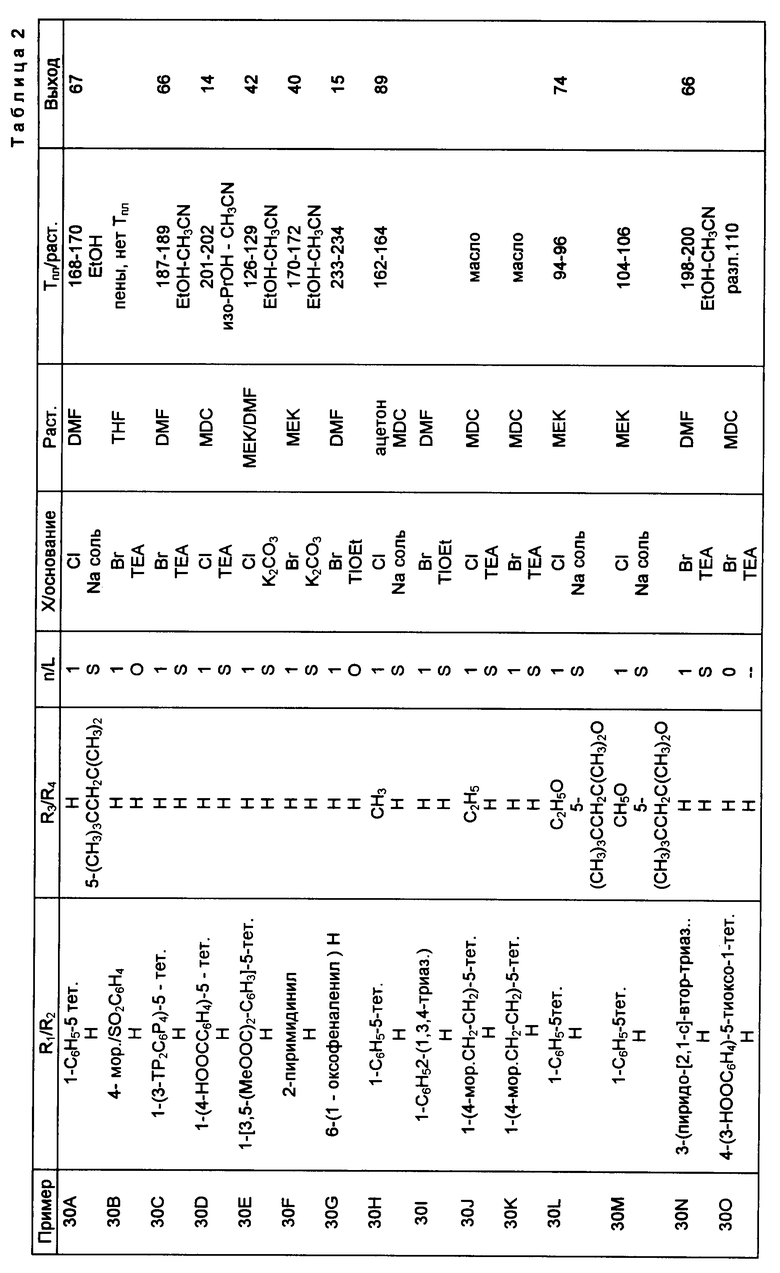


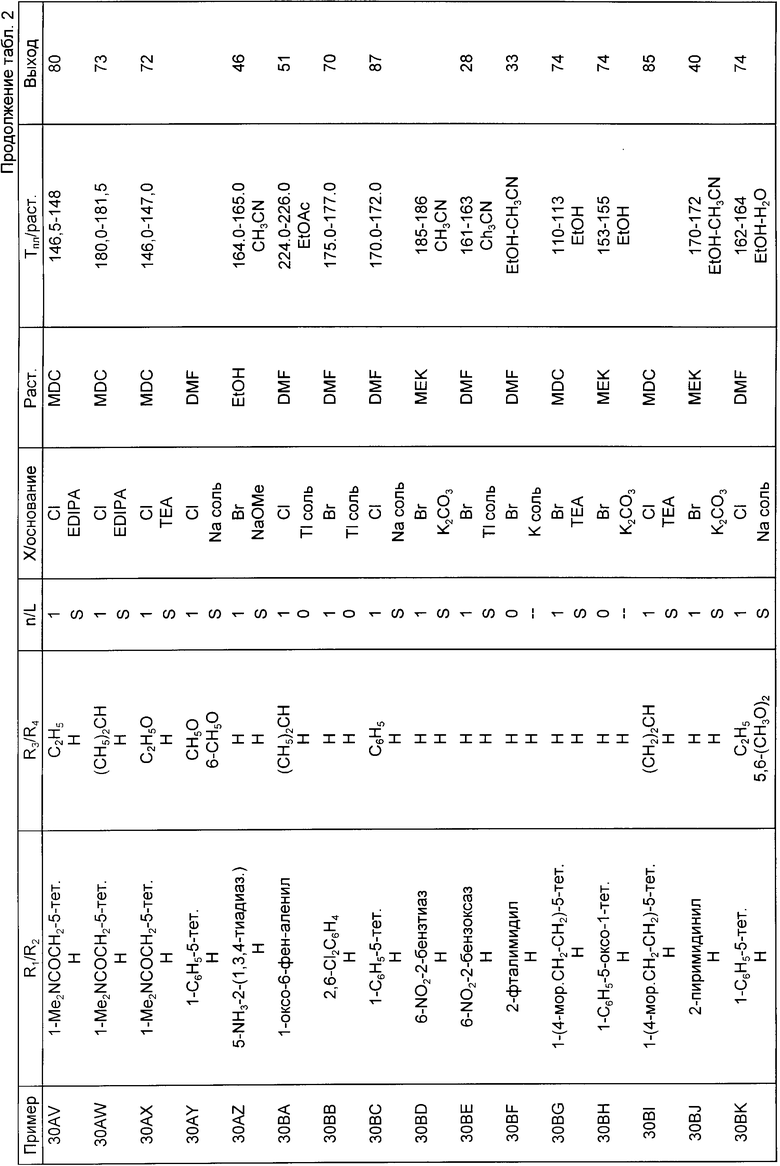
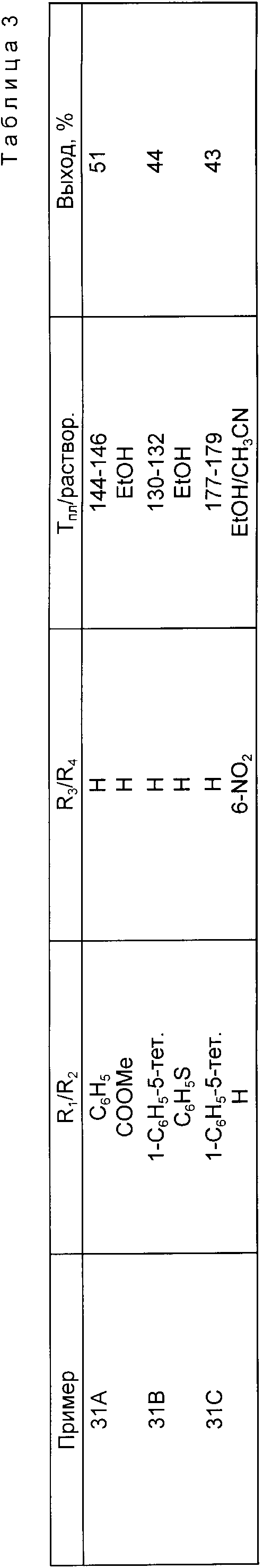
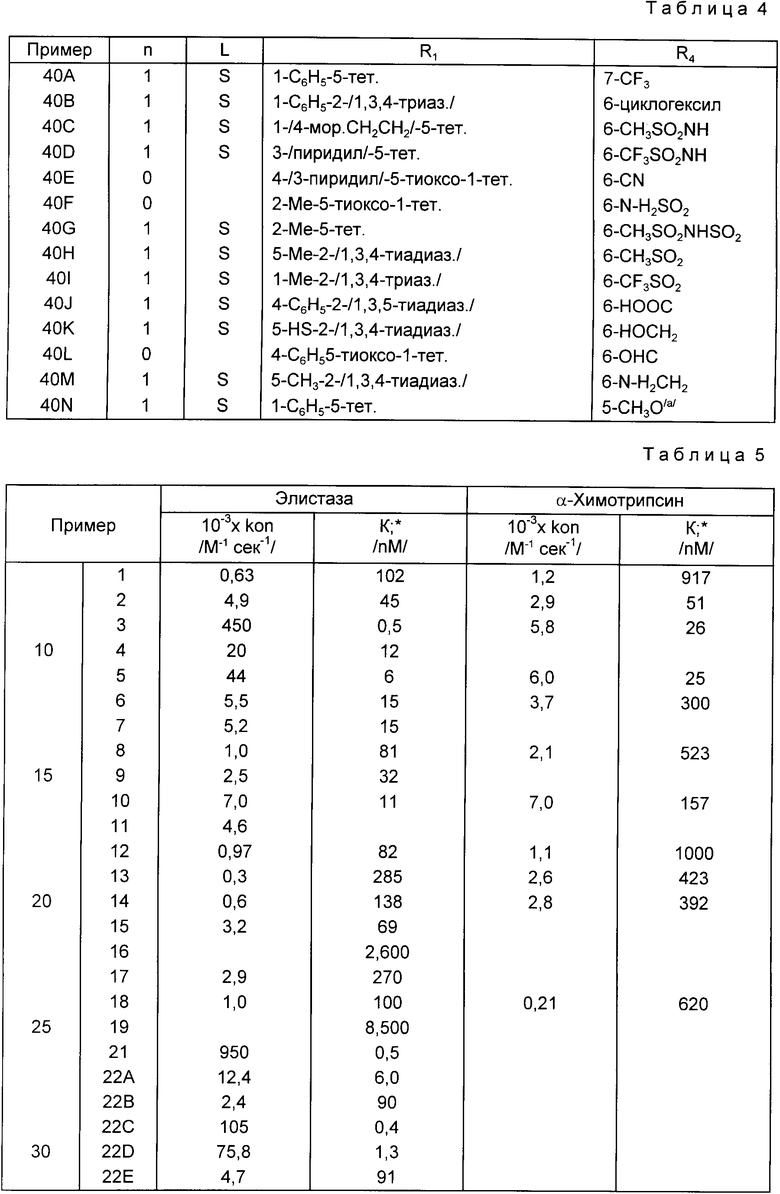
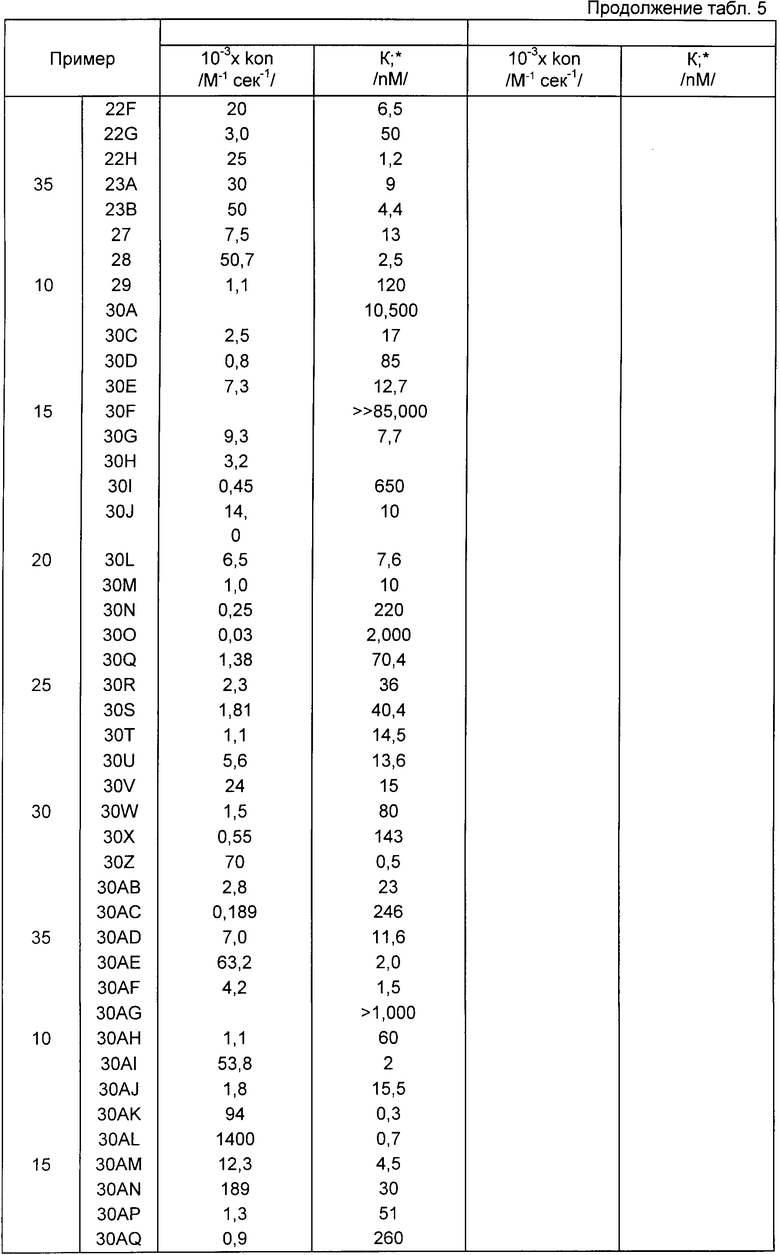
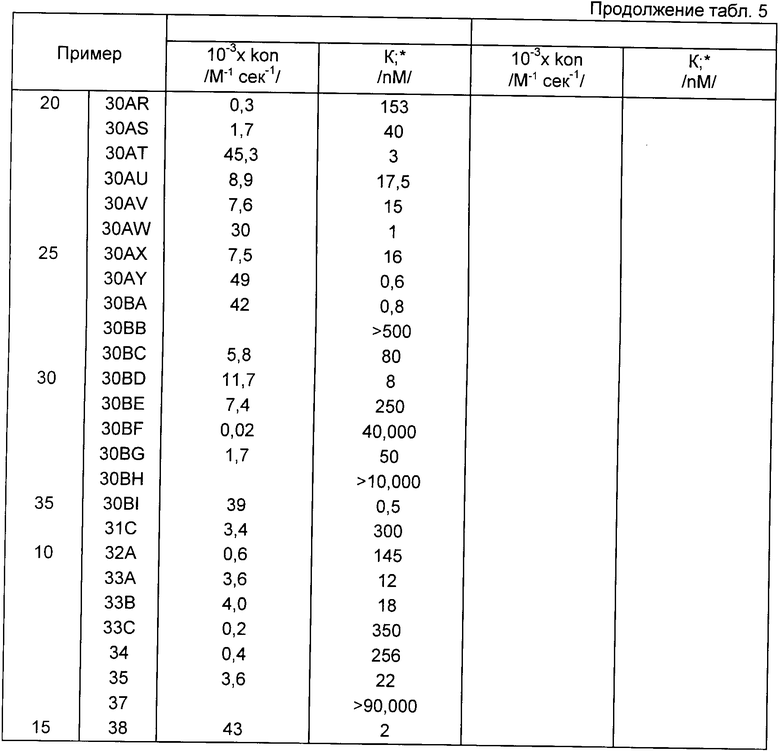
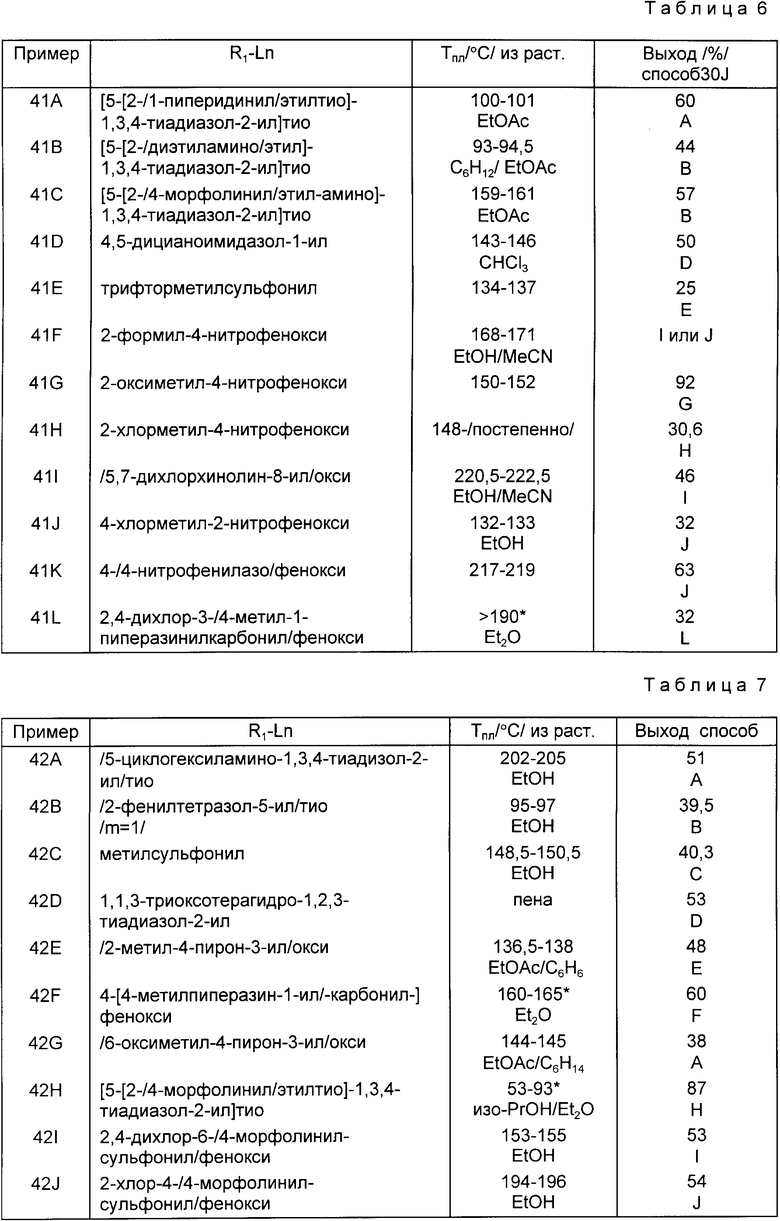
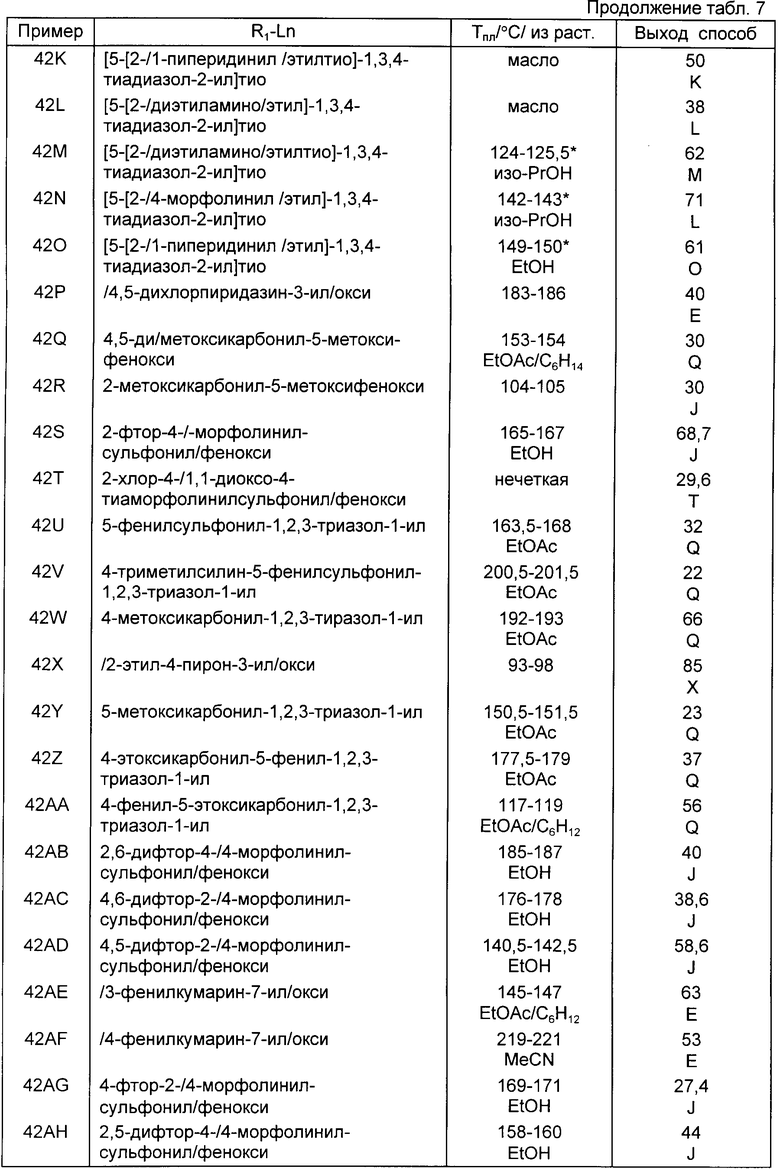
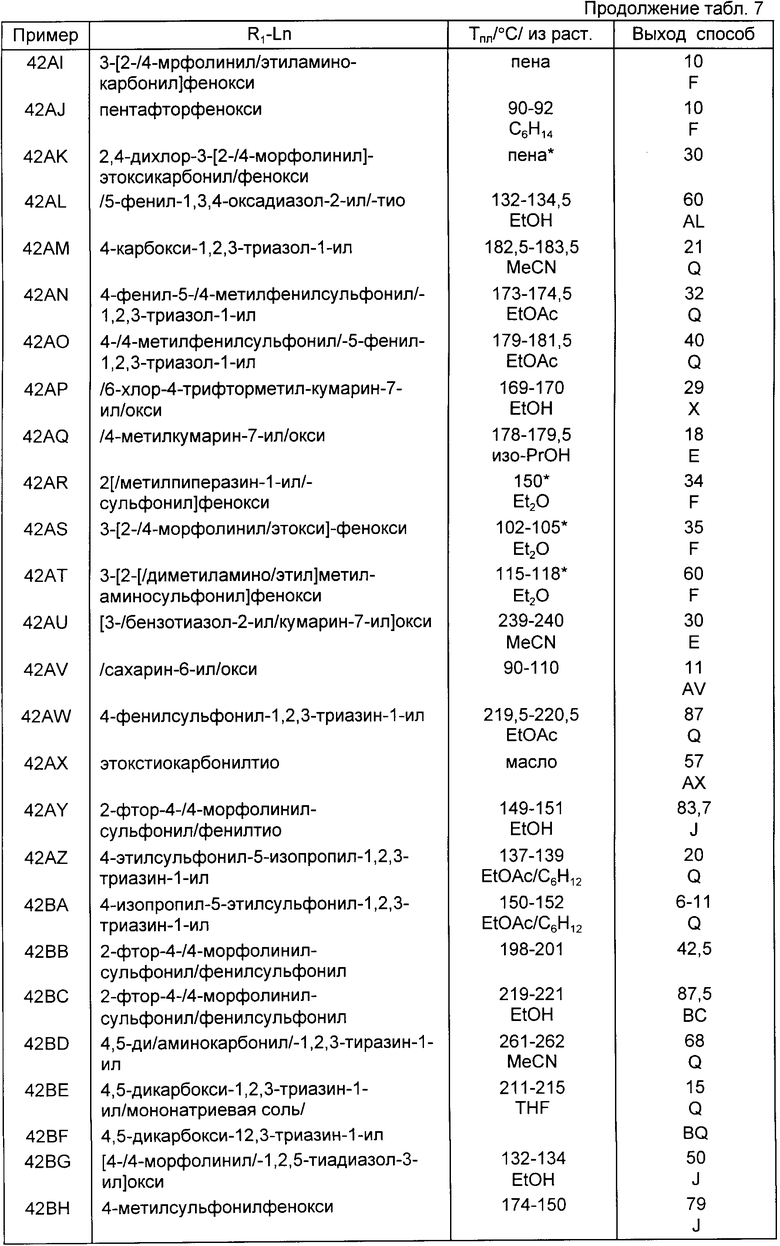
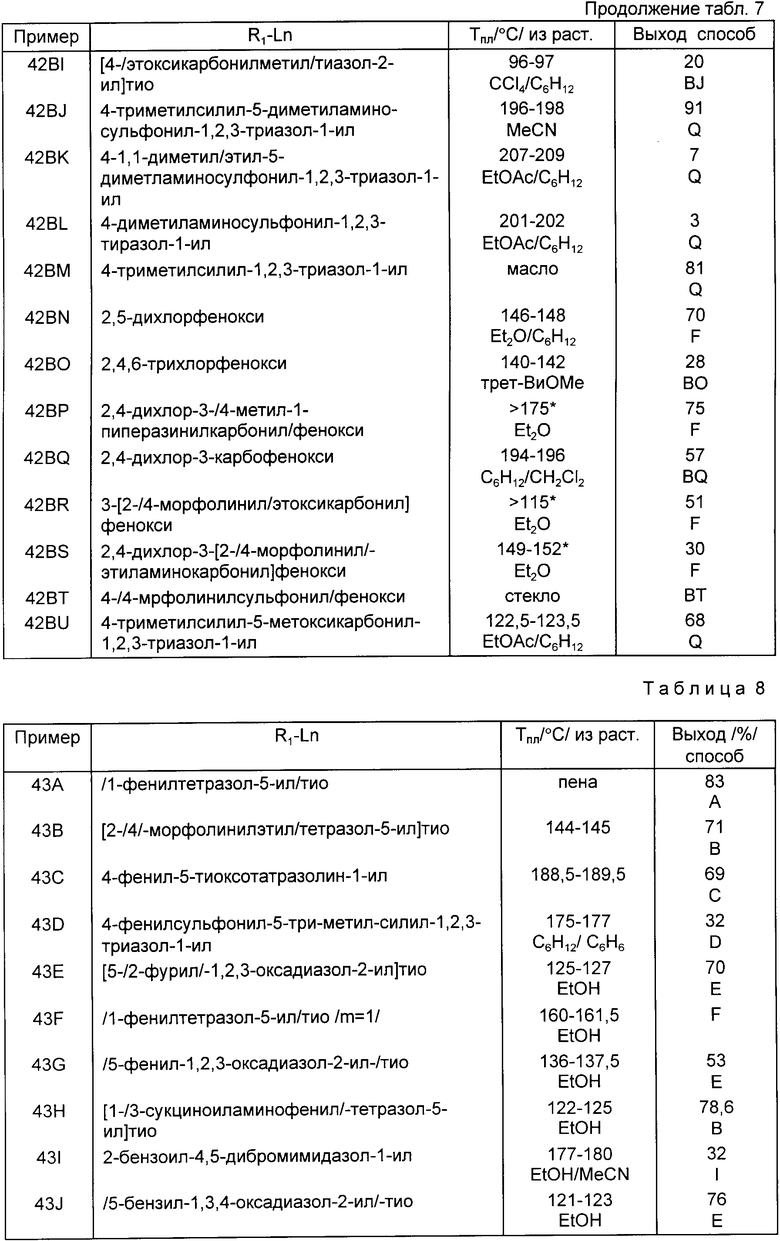
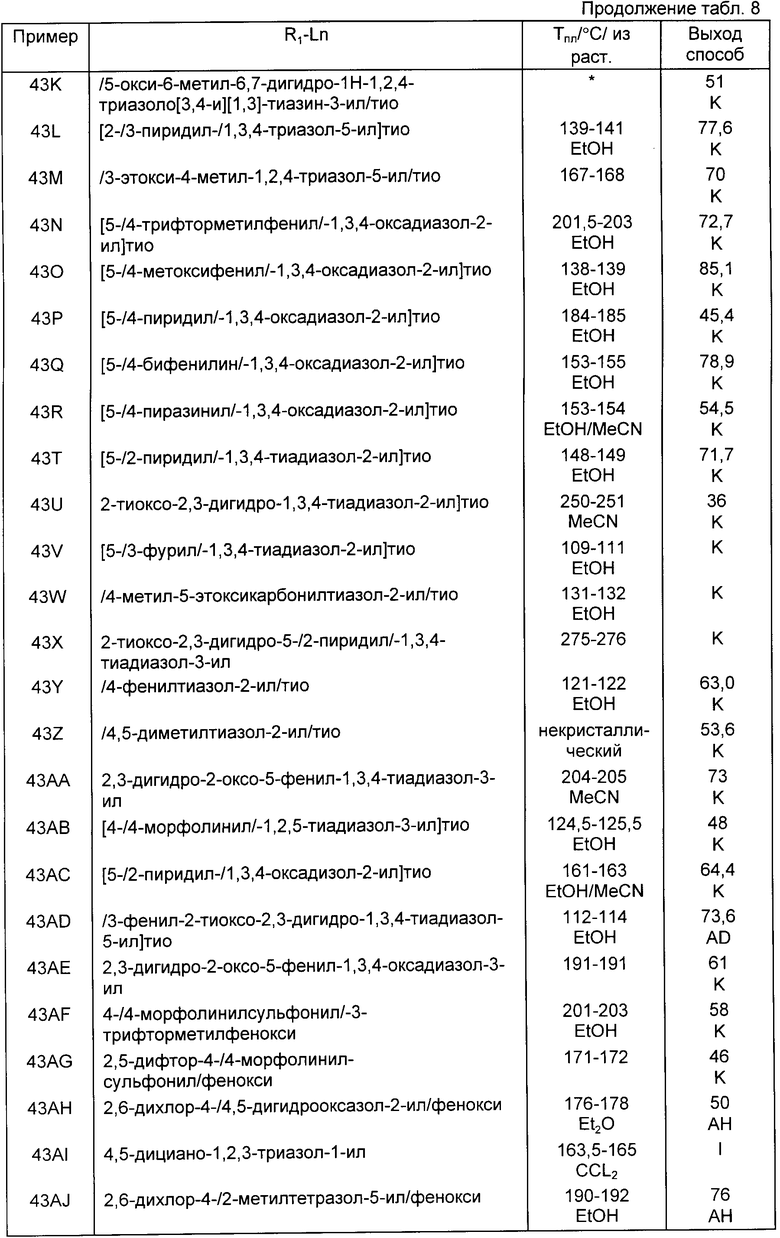
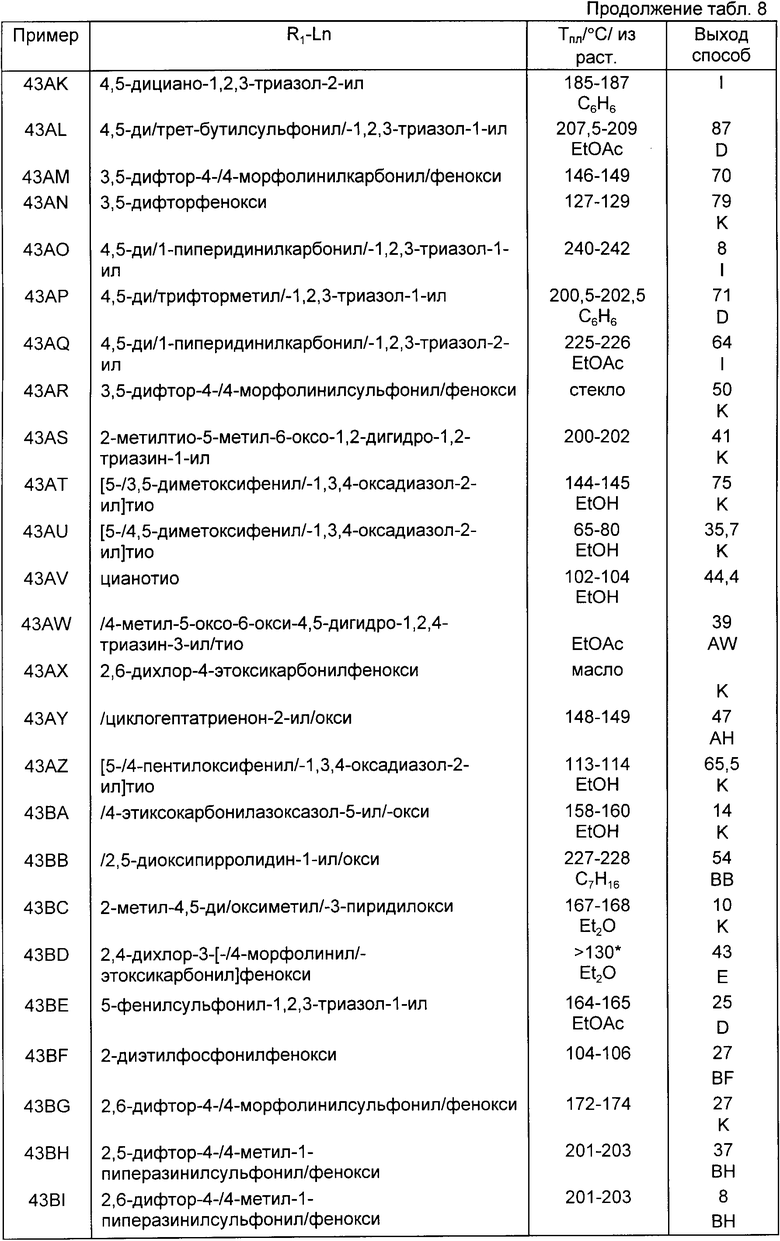
Изобретение относится к химии гетероциклических соединений, проявляющих ингибиторную активность против эластазы. Сущность изобретения: производное 2-замещенного сахарина формулы I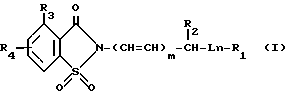
где L является -O-, -S-, -SO- или -SO2-; m и n независимо равны 0 или 1; R1 представляет собой замещенный фенил, гетероцикл или замещенный гетероцикл, или когда L является -O-, а n равно 1, то R1 представляет собой циклогептатриенон-2-ил, или когда L является -S-, а n равно 1, то R1 представляет собой цианогруппу или (низший алкокси)-тиокарбонил, или когда L является -SO2-, а n равно 1, то R1 представляет собой низший алкил или трифторметил; R2 представляет собой водород; R3 является водородом, первичным или вторичным низшим алкилом или низшим алкоксилом, R4 - водород или 1-3 различных заместителя, и фармацевтическая композиция на их основе. 2 с. и 9 з.п.ф-лы, 8 табл.
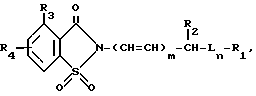
где L -O-, -S-, -SO- или -SO2-;
m и n 0 или 1;
R1 фенил, замещенный 1-[4-(низший алкил)-пиперазин-1-ил]-карбонилом, 4-морфолинилсульфонилом, формилом, (низший алкокси)-карбонилом, 4-тиаморфолинилсульфонилом или его S-диоксидом, окси-(низшим алкилом), галогенсодержащим низшим алкилом, 4-морфолинил-(низший алкил)-аминокарбонилом, 4-морфолинил-(низший алкокси)-карбонилом, 1-[4-(низший алкил)-пиперазин-1-ил)-сульфонилом, 4-морфолинил-(низший алкоксилом), ди-(низший алкил)-амино-(низший алкил)-аминосульфонилом или его N-(низший алкил)-производным, (низший алкил)-сульфонилом, 4,5-дигидрооксазол-2-илом, (низший алкил)-тетразол-5-илом, 4-морфолинилкарбонилом, нитрофенил-азогруппой, карбоксилом или ди-(низший алкил)-фосфонилом, или представляет собой любую указанную группу в сочетании с замещением фенила галогеном, низшим алкоксилом или нитрогруппой, или гетероцикл, выбранный из пиридазин-3-ила, 4-пирон-3-ила, хинолин-8-ила, 1,3,4-оксадиазол-2-ила, кумарин-7-ила, сахарин-6-ила, имидазол-1-ила, тиазол-2-ила, 2-тиоксо-2,3-дигидро-1,3,4-оксадиазол-3-ила, 1,2,5-тиадиазол-3-ила, 2-тиоксо-2,3-дигидро-1,3,4-тиадиазол-3-ила, 2-тиоксо-2,3-дигидро-1,3,4-тиадиазол-5-ила, 1,2,3-триазол-2-ила, 5-оксо-6-окси-4,5-дигидро-1,2,4-триазин-3-ила, изоксазол-5-ила, изоксазол-3-ила, 4,5-дигидро-5-оксо-1,2,4-оксадиазол-4-ила, пиридила, 1,1,3-триоксотетрагидро-1,2,5-тиадиазол-2-ила, 6,7-дигидро-1H-1,2,4- триазоло-[3,4-b] [1,3]тиазин-3-ила, 2,5-диоксопирролидин-1-ила, 3-индолила, оксазол-2-ила, тиазол-4-ила, 2,3-дигидро-2-оксо-5-фенил-1,3,4-тиадиазол-3-ила, 2,3-дигидро-2-оксо-5-фенил-1,3,4-оксадиазол-3-ила, 6-оксо-1,2-дигидро-1,2,4-триазин-1-ила, 1,2,3-триазин-1-ила и 1-индолила, или указанный гетероцикл, замещенный по любому доступному атому азота низшим алкилом, окси-(низший алкилом), 2-, 3- или 4-пиридинилом, (низший алкокси)-карбонилом или фенилом, или указанный гетероцикл, замещенный по любому доступному атому углерода низшим алкилом, циклоалкиламиногруппой, (низший алкил)-тиогруппой, 1-пиперидинил-(низший алкил)-тиогруппой, (низший алкокси)-карбонилом, цианогруппой, окси-(низший алкилом), фенилсульфонилом, галогеном, карбоксилом или его солью с щелочным металлом, фурилом, трифторметилом, 2-бензотиазолилом, (низший алкил)-сульфонилом, аминокарбонилом, бензилом, 4-морфолинилом, пиридинилом, пиразинилом, (низший алкокси)-карбонил-(низший алкилом), 1-пиперидинилкарбонилом, бензилоксигруппой, гидроксилом, бензоилом, или бензоилом, замещенным низшим алкоксилом или галогеном, или фенилом, или фенилом, замещенным низшим алкоксилом, трифторметилом, (низший алкокси)-поли(низший алкоксилом), метилендиоксигруппой или (низший алкокси)-карбонилом, или гетероцикл, выбранный из 1H-(5-тетразолила), 5-оксо-1-тетразолинила, 5-тиоксо-1-тетразолинила, 2-(1,3,4-тиадиазолила) или 1,2,3-триазол-1-ила, замещенного по любому доступному атому углерода ди-(низший алкил)-амино-(низшим алкилом), 4-морфолинил-(низший алкил)-аминогруппой, цианогруппой, 1-пиперидинил-(низший алкилом), фенилсульфонилом, толуолсульфонилом, три-(низший алкил)-силилом, карбоксилом или его солью с щелочным металлом, трифторметилом, (низший алкил)-сульфонилом, пирилинилом, ди-(низший алкил)-аминосульфонилом, 1-пиперидинилкарбонилом, 4-морфолинил-(низший алкилом) или фенилом, замещенным (низший алкокси)-карбонилом, или когда R4 является карбокси-(низший алкоксилом), (низший алкокси)-карбонил-(низший алкоксилом) или ди-(низший алкил)-аминокарбонилоксигруппой, то гетероцикл представляет собой 1-фенилтетразол-5-ил, или, когда L является -O-, а n 1, то R1 - циклогептатриенон-2-ил, или когда L является -S-, а n 1, то R1 цианогруппа или (низший алкокси)-тиокарбонил, или, когда L является -SO2-, а n 1, то R1 низший алкил или трифторметил;
R2 водород;
R3 водород, первичный или вторичный низший алкил или низший алкоксил;
R4 водород или 1 3 заместителя, выбранных из низшего алкила, гидоксила, низшего алкоксила, (низший алкокси)-поли-(низший алкилен)-оксигруппы, карбокси-(низшего алкоксила), (низший алкокси)-карбонил-(низший алкоксила) или ди-(низший алкил)-аминокарбонилоксигруппы, при условии, что (1) когда n 0, то R1 может быть только гетероциклом, а CHR2 может быть только связан с кольцевым атомом азота радикала R1, и (2) когда m 0, n 1, L является -O-, -S- или -SO2-, а R2 R3 и R4 - водород, то R1 не может быть замещенным фенилом.
| PCT, заявка, WO/13549, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| EP, патент, 236930, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
