Изобретение относится к 8-оксо-5-тиа-1-изабицикло(4.2.0)-окт-2-ен-5,5-диоксид-2-карботионовым кислотам, способу их получения и содержащим их фармацевтическим и ветеринарным препаратам. Соединения изобретения применимы в качестве ингибиторов протеаз, в особенности лейкоцитэластазы человека (ЛЭЧ), а также для профилактики, контроля и лечения вызываемых протеолитическими ферментами воспалительных и перорожденных заболеваний, в частности эмфиземы, респираторной дистресс-синдром у взрослых, ревматоидный артрит, остероартит, инфекционный артрит, ревматическая атака, спондилит, подагра, волчанка и псориаз.
В данном описании использованы следующие сокращения:
Ac ацетил,
Boc трет-бутоксикарбонил,
Cnз бензоксикарбонил,
Cuc 3-карбоксипропионил,
Glu 4-карбоксибутирил,
NPG аминозащитная группа, выбранная из вышеприведенных групп.
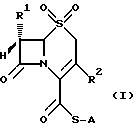
Изобретением обеспечиваются соединения общей формулы (I)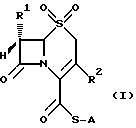
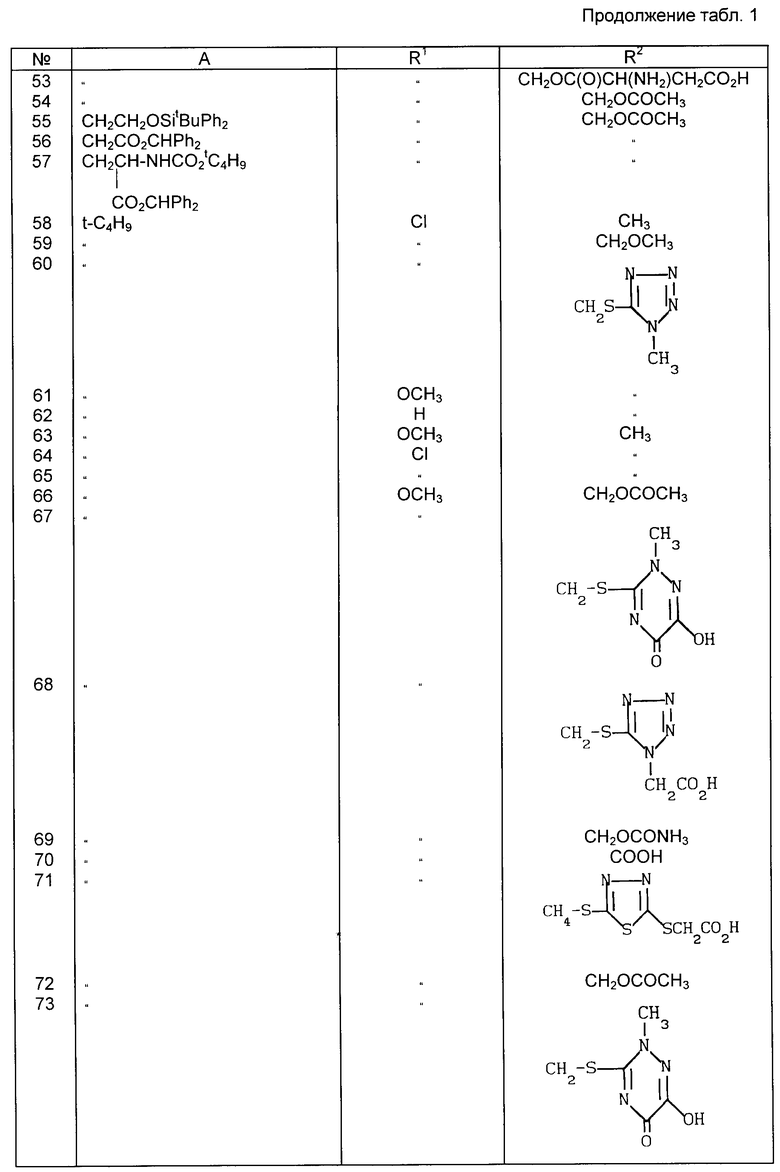
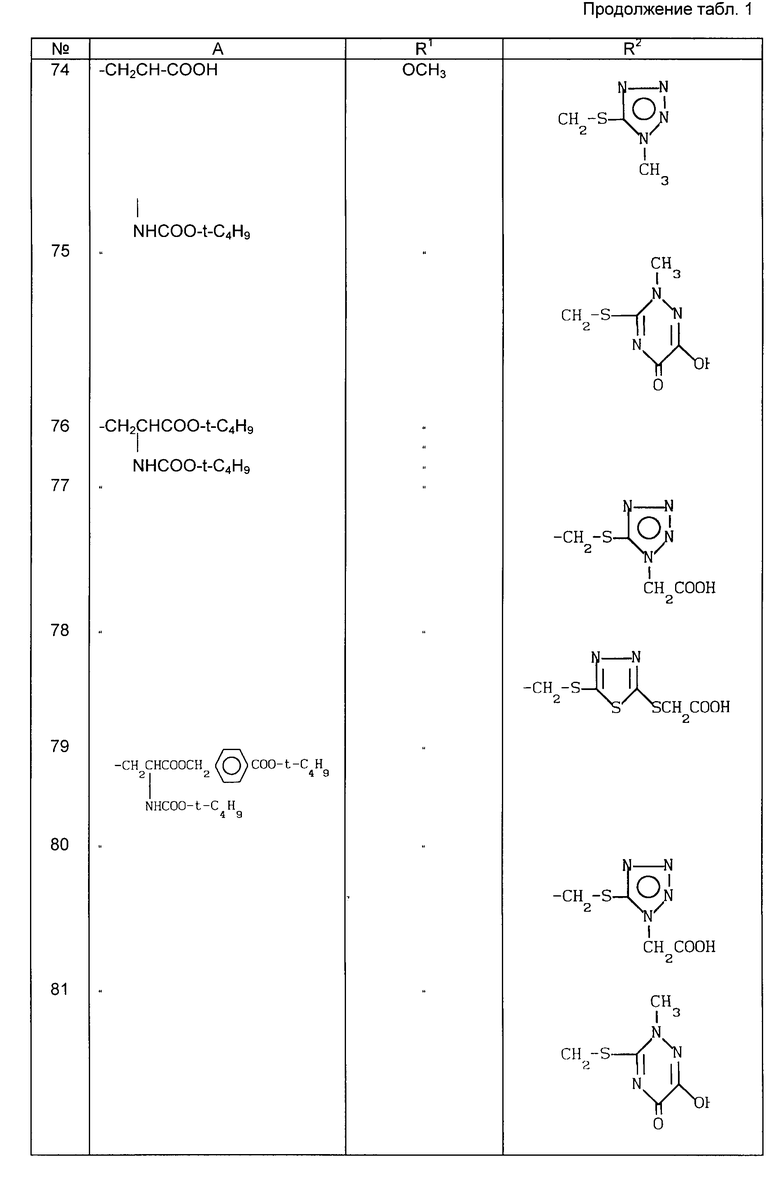
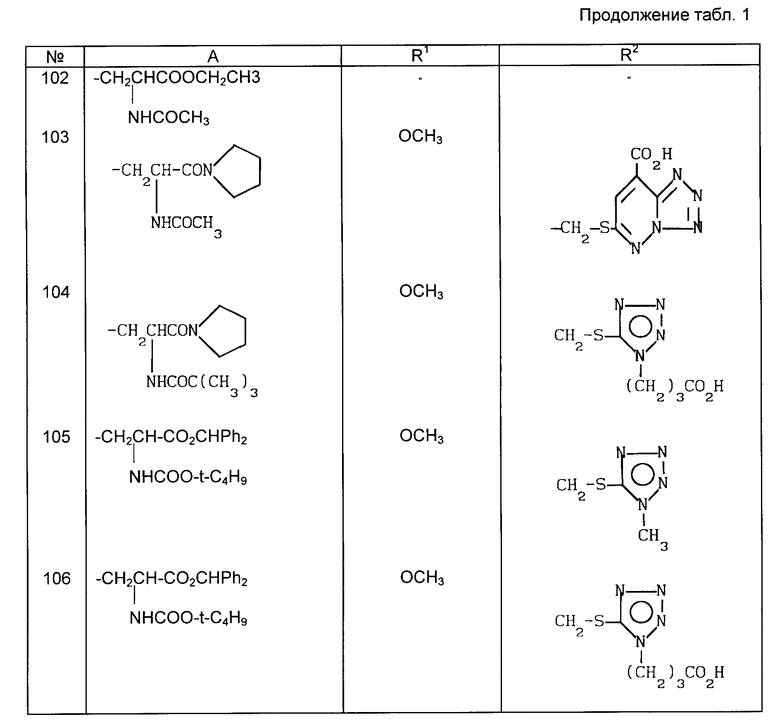
в которой A неразветвленная или разветвленная (C1-C4) алкильная группа, необязательно замещенная одним или двумя заместителями, выбранными из гидрокси-, карбокси-, амино-, ацетиламино-, трет-бутоксикарбониламино-, дифенилметоксикарбонильной, трет-бутилфенилсилилокси- и фенильной групп, или представляет собой метоксифенольную группу;
R1 хлор или водород или метокси-, аллильная или пропильная группа и
R2 метильная, метоксиметильная, ацетоксиметильная или (1-метил-1,2,3,4-тетразол-5-ил)тиометильная группа, или его фармацевтически приемлемой соли.
Изобретение также касается способа получения соединения формулы (I) или его соли, в котором соединение общей формулы (II)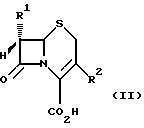
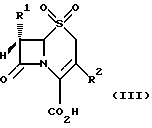
в которой группа R1 является такой, какой она была определена выше, и группа R2 представляет собой метильную, метоксиметильную или ацетоксиметильную группу, подвергается окислению обработкой надкислотой для получения соединения формулы (III)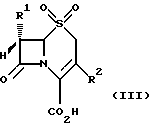
в которой группы R1 и R2 являются такими, какими они были определены выше, с превращением, при желании, полученного соединения формулы (III), в которой R2 ацетоксиметильная группа, в другое соединение формулы (III), в котором R2 (1-метил-1,2,3,4-тетразол-5-ил)тиометильная группа, обработкой N-бромсукцинимидом и AIBN (азо-бис-изобутиронитрилом) или эфиратом трехфтористого бора и 5-меркапто-1-метил-1,2,3,4-тетразолом;
обработкой карбоксигруппы в положении 4 соединения формулы (III), определенного выше, в присутствии пиридина полифосфатным сложным эфиром или дициклогексилкабодиимидом и меркаптаном формулы HS A', в котором группа A' является такой, какой она была определена выше в случае группы A, исключая неразветвленную или разветвленную (C1-C4)алкильную группу, замещенную одним или двумя заместителями, выбранными из гидрокси-, карбокси- и аминогруппы;
при желании, для получения соединения формулы (I), в которой A - неразветвленная или разветвленная (C1-C4)-алкильная группа, замещенная одним или несколькими заместителями, выбранными из гидрокси-, карбокси- и аминогрупп, проводят гидролиз соединения (III), и/или, при желании, превращают соединение формулы (I) в его фармацевтически приемлемую соль.
На стадии реакции с меркаптаном или его производным общей формулы III;
H S A'
где A' принимает вышеуказанные значения, может оказаться необходимой или желательной предварительная или in situ активация.
Примеры предварительной активации включают превращение карбоновых кислот в их хлорангидриды, ангидриды или смешанные ангидриды с карбоновыми кислотами (например, в реакции с пивалоилхлоридом), производными карбоновых кислот (например, в реакции с галоидформатами, такими как этилхлоридформат), сульфокислотами (например, в реакции с мезилхлоридом или тозилхлоридом) и производными фосфиновой кислоты (например, в реакции с дифенилхлорформатом, диэтилхлорфосфатом, фенилдихлорфосфатом, N, N-диметилфосфорамиддихлоридом, диэтилфосфорилцианидом или дифенилфосфонилазидом). Примеры активации in situ включают проведение реакции в присутствии примерно одного молярного эквивалента карбодиимида (например, дициклогексил карбодиимида) или 1-гидроксибензтриазола и дициклогексилкарбодиимида в присутствии большого избытка полифосфатного эфира (ПФЭ, получение см. W. Pollman, G. Schramm, Biochem. Biophys. Acta, 80, 1, 1964). Общие условия проведения подобной активации превращения известны (E. Haslam. Последние достижения в способах этерификации и защиты карбоксильных групп. Tetrahedron, 36, 2409, 1980). Приемлемые растворители включают органические апротонные полярные и неполярные растворители, такие как дихлорметан, хлороформ, тетрагидрофуран, ацетонитрил, диэтиловый эфир, бензол, диметилформамид, диметилсульфоксид, пиридин и этилацетат, приемлем температурный интервал от -78 до +80oC, предпочтительно от -30oC до комнатной температуры.
На стадии окисления соединения окисляют в соответствующие сульфоны. В качестве окислителей рекомендуются перекиси в инертном органическом растворителе или в смеси воды с органическим растворителем. Приемлемые перекиси включают, например, надуксусную кислоту, м-хлорнадбензойную кислоту (МХНБК) и мононадфталевую кислоту. В качестве растворителя приемлемы хлороформ, дихлорметан, тетрагидрофуран и этанол.
Необходимо подчеркнуть, что в вышеописанных способах любая функциональная группа при необходимости или при желании может быть защищена обычными методами с удалением защитной группы в конце процесса или при удобном случае. Также необходимо указать, что группа R2 может быть превращена обычными методами в группу R2 с другим значением, но в пределах вышеуказанных значений, и такое превращение может быть осуществлено при желании в конце или на любой стадии процесса. Указанные превращения или введение/удаление защитных групп хорошо известны для цефемов формулы II.
Соединения формулы II и III являются известными соединениями или могут быть синтезированы из известных соединений по известным методикам.
Примеры рекомендуемых соединений (табл.1)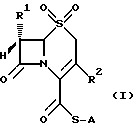
Возможность лечения ингибиторами протеазы состояний, возникающих при разрезе соединительных тканей, в последнее время привлекает особое внимание. Многочисленные попытки были направлены на поиски ингибиторов лейкоцитэластазы человека (ЛЭЧ), являющихся основными разрушительными агентами при эмфиземе легких и, вероятно, участвующих в ревматоидном артрите (J.C. Pewer, Am. Res. Resp. Discases, 127, 54-58, 1983; C.H. Hassal и др. FEBS Letters, 183, N 2, 201, 1985, G. Weinbaum, V.V. Damiano, TIP S 8, 6, 1987, M. Velvart, Rheymatol, Int. 1, 121, 1981). Низкомолекулярные ингибиторы, вероятно, имеют ряд преимуществ в сравнении с природными высокомолекулярными ингибиторами протеазы растительного или животного происхождения, заключающимися в следующем:
они могут быть получены в больших количествах;
они могут быть рационально спроектированы и оптимизированы;
они не антигенны;
они могут быть использованы перорально или в аэрозолях.
Многие открытые к настоящему времени ингибиторы эластазы содержат реакционноспособные функциональные группы (хлорметилкетоны, изоцианаты и т.д.), они могут реагировать с функциональными группами белков, вследствие чего могут оказаться токсичными. В этой связи особый интерес представляют производные β--лактама, которые хотя и реакционноспособны по отношению к серинпротеазе, как известно, нетоксичны в очень больших концентрациях.
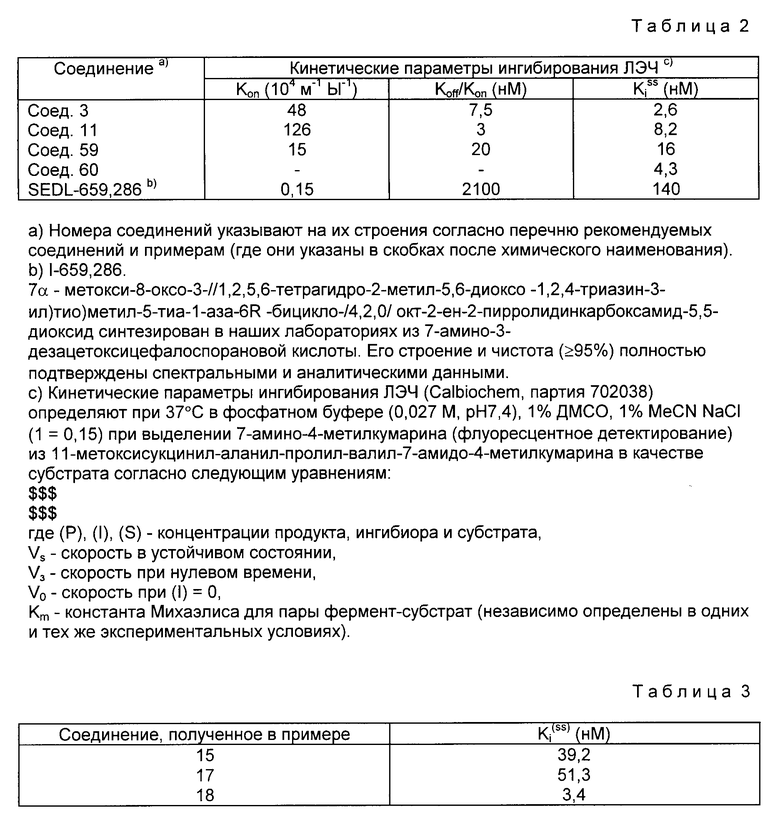
Соединения настоящего изобретения характеризуются высоким уровнем ингибирующей активности по отношению к ЛЭЧ. В частности, они превосходят наиболее активные цефалоспориновые эфиры и амиды (Nature, 1986, 332, 192) с точки зрения скорости образования (Kon) константы равновесия (Koff) Kon и кажущиеся константы диссоциации комплекса ЛЭЧ-ингибитор в устойчивом состоянии (Kiss). С целью иллюстрации данного утверждения приводится табл. 2, в которой даются вышеуказанные кинетические параметры, определенные для некоторых соединений настоящего изобретения в сравнении с Meret. SEDL-659, 286-цефалоспорановым производным, проходящим по сообщениям предварительные клинические испытания (Am. Rev. Pepiy. Dis. 1988, 137, 204; Agents and Actions 1988, 25, 60).
Вследствие их высокой активности по ингибированию эластазы и совершенно ничтожной токсичности соединения изобретения могут быть использованы для приготовления лекарств, применимых для предотвращения или прекращения развития заболеваний, вызванных протеолитическим распадом легких и соединительных тканей, уменьшения воспалений и жара, а также облегчения боли. Такие заболевания включают эмфизему, респираторный дистресс-синдром, воспаление бронхов, ревматоидный артрит, остеоартрит, инфекционный артрит, ревматическую атаку, спондилит, подагру, волчанку и псориаз.
Соответственно, изобретением предлагается фармацевтическая или ветеринарная композиция, содержащая соединение общей формулы I в смеси с фармацевтически или ветеринарно приемлемым разбавителем или носителем. Фармацевтические и ветеринарные композиции изобретения могут быть получены обычным путем в различных дозировочных формах, предназначенных для различных способов введения. В частности, композиции изобретения могут быть введены следующим образом.
а) Перорально, например, в виде таблеток, пастилок, ложенджис, водных или масляных суспензий, диспергируемых порошков или гранул, эмульсий, твердых или мягких капсул, сиропов или эликсиров. Композиции, предназначенные для перорального введения, могут быть приготовлены любым известным способом приготовления фармацевтических композиций, и такие композиции могут содержать одно или более средств, выбранных из группы, включающей: подслащивающие средства, ароматизирующие средства, красители и консерванты, с получением привлекательных и аппетитных препаратов.
Таблетки содержат активный компонент в смеси с неядовитыми фармацевтически приемлемыми наполнителями, применимыми для изготовления таблеток. Такие наполнители могут представлять собой, например, инертные разбавители, такие как карбонат кальция, карбонат натрия, лактоза, фосфат кальция или фосфат натрия, гранулирующие и размельчающие средства, например кукурузный крахмал или альгиновая кислота, связующие средства, например крахмал, желатин или камедь акации, и смазки, например стеарат магния, стеариновая кислота или тальк. Таблетки могут иметь или не иметь покрытия, которое может быть нанесено обычными методами с целью замедлить разрушение и адсорбцию в желудочно-кишечном тракте и тем самым обеспечить отложенное действие в течение более длительного периода. Например, может быть использовано вещество, обеспечивающее отложенное действие, такое как моностеарат глицерина и дистеарат глицерина.
Составы для перорального введения могут быть также приготовлены в виде твердых желатиновых капсул, в которых активный компонент смешивают с инертным твердым разбавителем, например карбонатом кальция, фосфатом кальция или каолином, или в виде мягких желатиновых капсул, в которых активный компонент смешивают с водой или масляной средой, например арахисовым маслом, жидким парафином или оливковым маслом.
Водные суспензии содержат активные вещества в смеси с наполнителями, пригодными для получения водных суспензий. К таким наполнителям относятся суспендирующие средства, например натрийкарбоксиметилцеллюлоза, метилцеллюлоза, гидроксипропилметилцеллюлоза, альгинат натрия, поливинилпирролидон, трагакантовая камедь, камедь акации, диспергирующие и смачивающие средства могут быть природными фосфатидами, к примеру, лецитином или продуктами конденсации алкиленоксида с жирными кислотами, к примеру полиоксиэтиленстеаратом, или продуктами конденсации окиси этилена с длиноцепными алифатическими спиртами, к примеру гептадекаэтиленоксицетанолом, или продуктами конденсации окиси этилена с частичными эфирами, полученными из жирных кислот и гексита, таким как полиоксиэтиленсорбитмоноолеат, или продуктами конденсации окиси этилена с ангидридами гексита, к примеру полиоксиэтиленсорбитмоноолеатом. Такие водные суспензии могут также включать один или несколько консервантов, например этил- или н-пропил-гидроксибензоат, один или несколько красителей, одно или несколько ароматизирующих веществ и одно или несколько подслащивающих средств, таких как сахароза или сахарин.
Суспензии в масле могут быть приготовлены суспендированием активного компонента в растительном масле, например арахисовом масле, оливковом масле, кунжутном масле или кокосовом масле или в минеральном масле, таком как жидкий парафин. Суспензии в масле могут включать загуститель, например пчелиный воск, твердый парафин или цетиловый спирт. Подслащивающие средства, типа вышеупомянутых, а также ароматизирующие вещества могут быть добавлены с получением аппетитных пероральных препаратов. В этом состоянии препараты могут быть сохранены добавлением антиокислителя, такого как аскорбиновая кислота.
Диспергируемые порошки и гранулы, пригодные для приготовления водных суспензий при добавлении воды, представляют собой смесь активного компонента с диспергирующим или смачивающим средством, суспендирующим средством и одним или несколькими консервантами. Приемлемые суспендирующие, диспергирующие и смачивающие средства приведены в виде примеров выше. Могут также присутствовать дополнительные наполнители, например подслащивающие и ароматизирующие добавки и красители.
Фармацевтические композиции изобретения могут также иметь вид эмульсий типа масла в воде. Масляная фаза может состоять из растительного масла, например оливкового масла или арахисового масла, или минерального масла, например жидкого парафина, или представлять собой смесь указанных масел. В качестве приемлемых эмульгаторов могут быть использованы природные камеди, например камедь акации или трагакантовая камедь, природные фосфатиды, например соевые бобы, лецитин, а также эфиры или частичные эфиры, полученные на основе жирных кислот и ангидридов гексита, например сорбитмоноолеат, и продукты конденсации вышеуказанных частичных эфиров с окисью этилена, например полиоксиэтиленсорбитмоноолеат. Эмульсия может также включать подслащивающие и ароматизирующие добавки.
Сиропы и элексиры могут быть приготовлены с использованием подслащивающих средств, например глицерина, сорбита и сахарозы. Такие составы могут также включать антиэмульгатор, консервант, ароматизирующую добавку и краситель.
b) Парентерально, т.е. подкожно, внутривенно, внутримышечно, внутригрудинно или вливанием в виде стерильных инъектируемых водных или масляных суспензий. Фармацевтические композиции могут иметь вид стерильных инъектируемых водных или масляных суспензий. Такие суспензии могут быть получены известными способами с использованием приемлемых диспергирующих или смачивающих средств, или суспендирующих средств, указанных выше. Стерильный инъектируемый препарат может также представлять собой стерильный инъектируемый раствор или суспензию в неядовитом парентерально приемлемом разбавителе или растворителе, например в 1,3-бутандиоле. К числу применимых носителей или растворителей относятся вода, раствор Ринджера и изотонический раствор хлористого натрия. Кроме того, в качестве растворяющей или суспендирующей среды часто применяют стерильное нелетучее масло. Для этой цели может быть использовано любое нелетучее масло, в том числе синтетические моно- или диглицериды. Кроме того, жирные кислоты, такие как олеиновая кислота находят применение для приготовления инъектируемых препаратов.
c) Ингаляцией, т.е. в виде аэрозолей или растворов для получения паров.
d) Ректально, т.е. в виде свеч, полученных смешиванием лекарства с приемлемым не вызывающим раздражения наполнителем, который при обычной температуре является твердым веществом, но превращается в жидкость при температуре прямой кишки, т.е. будет плавиться в прямой кишке с выделением лекарства. К таким материалам относятся масло-какао и полиэтиленгликоли.
e) Локально в виде кремов, мазей желе, растворов или суспензий.
Ежедневные дозировки составляют 0,5-100 мг на кг массы тела в зависимости от активности конкретного соединения, возраста, массы и состояния подвергаемого лечению субъекта, типа и тяжести заболевания, частоты и пути введения. Рекомендуемые ежедневные дозировки для человека составляют от 50 мг до 2 г. Количество активного компонента, которое может быть смешано с материалом носителя с получением единичной дозировочной формы, меняется в зависимости от объекта лечения и конкретного пути введения. К примеру, состав, предназначенный для перорального введения человеку, может содержать от 5 до 2 г активного компонента в смеси с приемлемым и обычным количеством носителя, количество которого может составлять 5-95% от всей композиции. Дозировочная единичная форма обычно содержит 25-500 мг активного компонента.
Пример 1. Трет-бутиловый эфир 3-ацетоксиметил-7S-хлор-8-оксо-5-тиа-1-азабицикло /4,2.0/окт-2-ен-5,5-диоксид-2-карботиоловой кислоты
Соединение 3
Раствор 5 г (7α)-хлорцефалоспорановой кислоты в 45 мл дихлорметана и 15 мл этанола перемешивают сутки при 5-10oC с 8 г м-хлорнадбензойной кислоты. Реакционную смесь промывают водным бисульфитом натрия и растворитель органического слоя удаляют в вакууме. Остаток растворяют в 40 мл диэтилового эфира и оставляют на сутки для кристаллизации. Получают 2,8 г 2-карбокси-3-ацетоксиметил-7S-хлор-8-оксо-5-тиа-1-азабицикло (4.2.0)окт-2-ен-5,5-диоксида в виде белого порошка.
Полученный продукт (323 мг, 1 ммоль) последовательно обрабатывают при -20oC 0,6 мл пиридина, 0,135 мл (1,2 ммоль) трет-бутилмеркаптана и 4 мл полифосфатного эфира (ПФЭ).
Реакционную смесь оставляют на 5 ч при 0oC, после чего переносят в смесь этилацетата и водного раствора бикарбоната натрия и интенсивно перемешивают. Органическую фазу сушат над безводным сульфатом натрия и концентрируют в вакууме. Хроматографией остатка на силикагеле (смеси н-гексана с этилацетатом в качестве элюентов) получают заглавное вещество в виде белого порошка (290 мг).
ИК (Kbr) nmax: 1790, 1735, 1665, 1650 см-1.
ЯМР (90 МГц, CDCl3) δ (ч/млн): 1,6 (9H, с), 2,11 (3H, с), 3,92 (2H, ABk, J= 18 Гц), 4,81 (2H, ABk, J=14 Гц), 4,85 (1H, д, J=2 Гц), 5,37 (1H, д, J=2 Гц).
Пример 2. н-Бутиловый эфир 3-ацетоксиметил-7S-хлор-8-оксо-5-тиа-1-азабицикло /4.2.0/окт-2-ен-5,5-диоксид-2-карботиоловой кислоты
Соединение 2
К перемешиваемому раствору 323 мг 2-карбокси-3-ацетоксиметил-7S-хлор-8-оксо-5-тиа-1- азабицикло/4.2.0/окт-2-ен-5,5-диоксида (см. пример 1) в 12 мл дихлорметана при комнатной температуре прибавляют 0,107 мл н-бутантиола и 206 мг дициклогексилкарбодиимида. Через 10 мин растворитель удаляют в вакууме, а остаток очищают колоночной хроматографией на силикагеле с использованием в качестве элюента смесей н-гексана с этилацетатом. Заглавное соединение получено в виде воскоподобного вещества (90 мг).
ИК (KBr) nmax 1812, 1735, 1650 см-1.
ЯМР (90 МГц, CD Cl3) δ (ч/млн): 0,93 (3H, т, J=6,2 Гц), 1,1-2 (4H, м), 2,11 (3H, с), 3,08 (2H, д, J=6,2 Гц), 3,92 (2H, ABk, J=17,5 Гц), 4,83 (1H, д, J=1,8 Гц), 4,84 (2H, ABk, J=14,5 Гц), 5,34 (1H, д, J=1,8 Гц).
МС(FD): 395 м/з (M+).
Пример 3. п/Метоксифениловый эфир 3-ацетоксиметил-7S-хлор-оксо-5-тиа-1-азабицикло/4.2.0/окт-2-ен-5,5-диоксид-2-карботиоловой кислоты
Соединение 1
По методике, аналогичной методике примера 2, но с заменой н-бутантиола п-метокситиофенолом получают заглавное соединение в виде белого порошка (выход 25%).
ИК (KBr) nmax: 1815, 1740, 1670 см-1.
ЯМР (90 МГц, CD Cl3) δ (ч/млн): 2,12 (3H, с), 3,87 (3H, с), 4,02 (2H, ABk, J=18 Гц), 4,82 (2H, ABk, J=14,5 Гц), 4,93 (1H, д, J<2 Гц), 5,41 (1H, д, 1<2 Гц), 7,02 (2H, д, J=8 Гц), 7,45 (2H, д, J=8 Гц).
МС (FD): 445 м/з (M+).
Пример 4. По методике, аналогичной методике примера 1, но использованием соответствующего меркаптана вместо трет-бутилмеркаптана получены:
Изопропиловый эфир 3-ацетоксиметил-7-S хлор-8-оксо-5-тиа-1-азабицикло /4.2.0/окт-2-ен-5,5-диоксид-2-карботиоловой кислоты
Соединение 4
ИК (KBr) nmax: 1805, 1735, 1650, 1620 см-1
ЯМК (90 Мгц, CD Cl3) δ (ч/млн): 1,41 (6H, д, д, J 6,5 Гц), 2,1 (3H, с), 3,7-5 (1H, м), 3,93 (2H, ABk, J 17,8 Гц), 4,83 (1H, д, J 1,6 Гц), 4,85 (2H, ABk, J 14,3 Гц).
Трет-бутилдифенилсилилоксиметиловый эфир 3-ацетокси-метил-7S-хлор-8-оксо-5-тиа-1-азабицикло /4.2.0/окт-2-ен-5,5-диоксид-2-карботиоловой кислоты
Соединение 55
ИК (KBr) nmax: 1810, 1740, 1660 см-1
ЯМК (90 Мгц, CDCl3) δ (ч/млн): 1,07 (9H, с), 2,05 (3H, с), 3,28 (2H, т, J 6 Гц), 3,88 (2H, т, J 6 Гц), 3,95 (2H, ABk, J 18 Гц), 4,83 (2H, ABk, J 14,2 Гц), 4,86 (1H, д, J 1,7 Гц), 5,38 (1H д, J 1,7 Гц), 7,3 7,6 (10H, м).
Бензиловый эфир 3-ацетоксиметил-7S-хлор-8-оксо-5-тиа-1-азабицикло-/4.2.0/окт-2-ен-5,5-диоксид-2-карботиоловой кислоты
Соединение 11
ИК (KBr) nmax: 1812, 1738, 1655 см-1.
ЯМР (90 МГц, CD Cl3) δ (ч/млн): 2,07 (3H, с), 3,96 (2H, ABk, J 18,5 Гц), 4,32 (2H, с), 4,86 (2H, ABk, J 13,5 Гц), 4,87 (1H, д, J 1,8 Гц), 5,35 (1H, д, J 1,8 Гц), 7,37 (5H, с).
Дифенилметоксикарбонилметиловый эфир 3-ацетоксиметил-7S хлор-8-оксо-5-тиа-1-азабицикло /4.2.0/окт-2-ен-5,5-диоксид-2-карботиоловой кислоты
Соединение 56
ИК (KBr) nmax: 1808, 1740, 1665 см-1.
ЯМР (90 МГц, CD Cl3): 2,07 (3H, с), 3,93 (2H, ABk, J 18 Гц), 3,97 (2H, с), 4,8 (2H, ABk, J 14,5 Гц), 4,8 (1H, д, J 1,7 Гц), 5,34 (1H, д, J 1,7 Гц), 6,93 (1H, с), 7,39 (10H, с).
2-Дифенилметоксикарбонил-2-(трет-бутоксикарбониламино)этиловый эфир 3-ацетоксиметил-7S-хлор-8-оксо-5-тиа-1-азабицикло /4.2.0/окт-2-ен-5,5-диоксид-2-карботиоловой кислоты
Соединение 57
ИК (KBr) νmax: 1807, 1738, 1710 (уш,), 1660 (сдв.) см-1
ЯМР (200 МГц, CD Cl3): 1,41 (9H, с), 2,08 (3H, с), 3,38 (1H, дв.д, J 5,5 и 14,1 Гц), 3,73 (1H, д, J 18,2 Гц), 3,86 (1H, дв. д, J 4,3 и 14,1 Гц), 4 (1H, д, J 18,2 Гц), 4,54 (1H, д, J 14 Гц), 5,33 (1H, д, J 1,7 Гц), 6,9 (1H, с), 7,33 (10H, с).
Пример 5. 2-Гидроксиэтиловый эфир 3-ацетоксиметил-7S-хлор-8-оксо-5-тиа-1-азабицикло /4.2.0/окт-2-ен-5,5-диоксид-2-карботиоловой кислоты
Соединение 19
К перемешиваемому раствору 60 мг 2-трет-бутилдифенилсилилоксиметилового эфира 3-ацетоксиметил-7S-хлор-8-оксо-5-тиа-1-азабицикло /4.2.0/окт-2-ен-5,5-диоксид-2-тиоловой кислоты в 6 мл сухого тетрагидрофурана при комнатной температуре последовательно прибавляют 300 мл уксусной кислоты и 250 мг тригидрата тетрабутиламмонийфторида. Полученный раствор оставляют на 20 ч и после удаления растворителя и колоночной хроматографии остатка получают 25 мг заглавного соединения.
ЯМР (90 МГц, CD Cl3): 2,1 (3H, с), 3,27 (2H, м), 3,85 (2H, м), 3,91 (2H, ABk, J 18 Гц), 4,81 (2H, ABk, J 14 Гц), 4,87 (1H, д, J 1,6 Гц), 5,36 (1H, д, J 1,6 Гц).
Пример 6. 2-Амино-2-карбоксиэтиловый эфир 3-ацетоксиметил-7S-хлор-8-оксо-5-тиа-1-азабицикло /4.2.0/окт-2-ен-5,5-диоксид-2-карботиоловой кислоты
Соединение 34
Раствор 120 мг 2-дифенилметоксикарбонил-2-(трет-бутоксикарбониламино) этилового эфира 7S-хлор-8-оксо-5-тиа-1-азабицикло/4.2.0/окт-2-ен-5,5-диоксид-2-карботиоловой кислоты в 8 мл дихлорметана и 1 мл анизола обрабатывают 1 мл трифторуксусной кислоты и оставляют на 5 ч. После удаления растворителя остаток обрабатывают 15 мл дихлорметана и 5 мл диизопропилового эфира и перемешивают десять минут при комнатной температуре. Центрифугированием суспензии получают 42 мг белого порошка.
ИК (KBr) νmax/ : 1808, 1742, 1670, 1620 см-1
ЯМР (200 МГц, ДМCO- D2O): 5,92 (1H, д, J 1,6 Гц), 5,66 (1H, д, J 1,6 Гц), 4,7 (2H, ABk J 13,6 Гц), 3,74 (1H, м), 3,66 (2H, с, невидима), 3,52 (1H, дв. д, J 5,2 и 13,8 Гц), 3,35 (1H, дв. д, J 6,6 и 13,8 Гц), 2,01 (3H, с).
Пример 7. Карбоксиметиловый эфир 3-ацетокси-7S-хлор-8-оксо-5-тиа-1-азабицикло /4.2.0/окт-2-ен-5,5-диоксид-2-карботиоловой кислоты
Соединение 22
В 25 мл смеси дихлорметан-анизол-трифторуксусная кислота (8:1:1 по объему) растворяют 180 мг дифенилметоксикарбонилметилового эфира 3-ацетоксиметил-7S-хлор-8-оксо-5-тиа-1-азабицикло /4.2.0/окт-2-ен-5,5-диоксид-2-карботиоловой кислоты и оставляют на 2,5 ч при комнатной температуре. Удалением растворителя получают остаток, который хроматографируют на силикагеле с применением в качестве элюента смесей н-гексана с этилацетатом и получают 105 мг заглавного соединения в виде белого порошка.
ИК (KBr) νmax 1810-115, 1720-1740, 1655 см-1
ЯМР (200 МГц, ДМСО): 2,03 (3H, с), 3,88 (2H, с), 4,42 (2H, ABk, J 18 Гц), 4,7 (2H, ABk, J 13,5 Гц), 5,72 (1H, д, J 1,5 Гц), 5,96 (1H, д, J 1,5 Гц).
Пример 8. 2-Фенилэтиловый эфир 3-ацетоксиметил-7S-хлор-8-оксо-5-тиа-1-азабицикло /4.2.0/окт-2-ен-5,5-диоксид-2-карботиоловой кислоты
Соединение 15
К раствору 3-ацетоксиметил-2-карбокси-7S-хлор-8-оксо-5-тиа-1-азабицикло /4.2.0/окт-2-ен-5,5-диоксида (0,323 г) в дихлорметане (4 мл) прибавляют пиридин (0,6 мл) и 2-фенил-этилмеркаптан (0,14 мл). Затем добавляют 4 мл полифосфатного эфира (ПФЭ) при -10oC и реакционную смесь перемешивают при -10oC 6 ч. После распределения между этилацетатом и водным раствором бикарбоната натрия органическую фазу отделяют, сушат над безводным сульфатом натрия и концентрируют в вакууме. Остаток хроматографируют на силикагеле (смесь н-гексана с этилацетатом) с отбором более быстро вымываемого продукта, который (0,13 г) растворяют в дихлорметане (20 мл) и обрабатывают при -40oC 70%-ной м-хлорнадбензойной кислотой (0,11 г). Смесь перемешивают 1 ч при -10oC и затем последовательно промывают водными растворами бисульфита и бикарбоната. Органическую фазу отделяют и концентрируют в вакууме. Хроматография остатка на силикагеле (н-гексан-этилацетат) получают 80 мг заглавного соединения в виде белого вещества, т.пл. 133-135oC.
ИК (KBr) νmax 1815, 1730, 1640 см-1
ЯМР (90 МГц, CD Cl3) δ (ч/млн): 2,1 (3H, с), 3 и 3,3 (4H, каждая д, J 7 Гц), 3,75 и 4,05 (2H, каждая д, J 18 Гц), 4,65 и 4,97 (2H, каждая д, J 14 Гц), 4,81 (1H, д, J 1,8 Гц), 5,35 (1H, д, J 1,8 Гц).
МС (FD): 443 (M+), 384 (-CH3CO2)
Пример 9. Трет-бутиловый эфир 7S-хлор-3-метил-8-оксо-5-тиа-1-азабицикло-/4.2.0/окт-2-ен-5,5-диоксид-2-карботиоловой кислоты
Соединение 58
Раствор 7S-хлордезацетоксицефалоспорановой кислоты (20 г) в смеси метанола с водой (1:1, 800 мл) обрабатывают пероксимоносульфатом калия (Оксон). После нагревания 105 мин при 55oC реакционную смесь охлаждают и фильтруют от нерастворимых в EtOAc продуктов. Фильтрат распределяют между EtOAc-H2O, органическую фазу отделяют, промывают водой (200 мл), сушат и после концентрирования получают сырой 2-карбокси-7S хлор-3-метил-8-оксо-5-тиа-1-азабицикло /4.2.0/окт-2-ен-5,5-диоксида в виде желтоватого масла (15 г).
Порцию полученного продукта (3,2 г) в дихлорметане (40 мл) обрабатывают трет-бутилмеркаптаном (1,35 мл) и ПФЭ (40 мл) по методике, приведенной в примере 1, с получением в результате 1,7 г заглавного соединения в виде белого порошка.
ИК (KBr) nmax 1800, 1660 см-1
ЯМР (90 МГц, CD Cl3) δ (ч/млн): 1,55 (9H, с), 2,07 (3H, с), 3,77 (2H, ABk, J 18 Гц), 4,73 (1H, д, J 1,5 Гц), 5,32 (1H, д, J 1,5 Гц).
МС (FD) 337 (M+), 280 (-C4H9), 248 (-SC4H9)м/з.
Пример 10. Трет-бутиловый эфир 7S-хлор-3-метоксиметил-8-оксо-5-тиа-1-азабицикло /4.2.0/ окт-2-ен-5,5-диоксид-2-карбоновой кислоты
Раствор сырой 7S-хлорцефалоспорановой кислоты (10 г) в метаноле (110 мл) и воде (50 мл) обрабатывают водным раствором бикарбоната натрия (2,7 г в 120 мл). Добавляют хлорид кальция (187 г) и реакционную смесь нагревают 1,5 ч при 70oC. После охлаждения до 10oC добавляют конц. HCl (7 мл) и раствор экстрагируют этилацетатом (2 х 350 мл). Экстракты сушат рассолом, сушат (Na2SO4) и концентрируют в вакууме. Остаток переносят в этиловый эфир (150 мл) и перемешивают 30 мин. Нерастворимые вещества, в основном 7S-хлорцефалоспорановый лактон отбрасывают. Концентрированием раствора получают 4 г 2-карбокси-3-метоксиметил-7S-хлор-8-оксо-5-тиа-1-азабицикло /4.2.0/окт-2-ена в виде желтоватого масла.
Порцию полученного продукта (3,5 г) в 50%-ном водном метаноле (120 мл) обрабатывают пероксимоносульфатом калия (14 г) 90 мин при 60oC. После концентирования в вакууме реакционную смесь распределяют между этилацетатом и водой. Испарением растворителя из экстракта получают 3 г сырого 2-карбокси-3-метоксиметил-7S-хлор-8-оксо-5-тиа-1-азабицикло /4.2.0/- окт-2-ен-5,5-диоксида в виде пены. Порцию полученного продукта (295 мл) обрабатывают трет-бутилмеркаптаном и ПФЭ по методике примера 1 с получением в результате 120 мг заглавного соединения в виде белого порошка.
ИК (KBr) nmax 1790, 1665 см-1.
Пример 10. Трет-бутиловый эфир 7S-хлор-3-метоксиметил-8-оксо-5-тиа-1-азабицикло[4.2.0]окт-2-ен-5,5-диоксид-2-карбоновой кислоты
Соединение 59
Раствор сырой 7S-хлорцефалоспорановой кислоты (10 г) в метаноле (110 мл) и воде (50 мл) обрабатывают водным раствором бикарбоната натрия (2,7 г в 120 мл). Добавляют хлорид кальция (187 г) и реакционную смесь нагревают 1,5 ч при 70oC. После охлаждения до 10oC добавляют конц. HCl (7 мл) и раствор экстрагируют этилацетатом (2 х 350 мл). Экстракты сушат рассолом, сушат (Na2SO4) и концентрируют в вакууме. Остаток переносят в этиловый эфир (150 мл) и перемешивают 30 мин. Нерастворимые вещества, в основном 7S-хлорцефалоспорановый лактон отбрасывают. Концентрированием раствора получают 4 г 2-карбокси-3-метоксиметил-7S-хлор-8-оксо-5-тиа-1-азабицикло[4.2.0] окт-2-ена в виде желтоватого масла.
Порцию полученного продукта (3,5 г) в 50%-ном водном метаноле (120 мл) обрабатывают пероксимоносульфатом калия (14 г) 90 мин при 60oC. После концентрирования в вакууме реакционную смесь распределяют между этилацетатом и водой. Испарением растворителя из экстракта получают 3 г сырого 2-карбокси-3-метоксиметил-7S-хлор-8-оксо-5-тиа-1-азабицикло-[4.2.0] окт-2-ен-5,5-диоксида в виде пены. Порцию полученного продукта (295 мг) обрабатывают трет-бутилмеркаптаном и ПФЭ по методике примера 1 с получением в результате 120 мг заглавного соединения в виде белого порошка.
ИК (KBr) νmax 1790, 1665 см-1.
ЯМР (90 МГц, CD Cl3) δ (ч/млн): 1,55 (9H, с), 3,95 (3H, с), 3,96 2H, AB, J 16,5 Гц), 4,17 (2H, с), 4,8 (1H, д, J 1,8 Гц), 5,34 (1H, д, J 1,8 Гц).
Пример 11. Трет-бутиловый эфир 7S-хлор-3-(1-метил-1,2,3,4-тетразол-5-ил)-тиометил-8-оксо-5-тиа-1-азабицикло /4.2.0/окт-2-ен-5,5 диоксид-2-карботиоловой кислоты
Соединение 60
Раствор сульфона 7S-хлорцефалоспорановой кислоты (400 мг) в ацетонитриле (15 мл) обрабатывают 6-меркапто-1-метил-1,2,3,4-тетразолом (200 мг) и эфиратом трехфтористого бора (0,5 мл). После нагревания 6 ч при 50oC реакционную смесь испаряют в вакууме с получением в результате сырого 2-карбокси-7S-хлор-3-(1-метил-1,2,3,4-тетразол-5-ил) тиометил-8-оксо-5-тиа-1-азабицикло /4.2.0/окт-2-ен-5,5-диоксида в виде аморфного вещества.
Обработкой полученного продукта трет-бутилмеркаптаном и ПФЭ по методике примера 1 получают заглавное соединение в виде аморфного вещества (110 мг).
ИК (KBr) nmax 1810, 1655 см-1
ЯМР (90 МГц, CD Cl3) δ (ч/млн): 1,55 (9H, с), 3,98 (3H, с), 4,1-4,5 (4H, два ABk), 4,87 (1H, J 1,7 Гц), 5,35 (1H, J 1,7 Гц).
МС (FD): 451 (M+), 394 (-C4H9) м/з
Пример 12. Трет-бутиловый эфир 3-(1-метил-1,2,3,4-тетразол-5-ил) тиометил-8-оксо-5-тиа-1-азабицикло /4.2.0/окт-2-ен-5,5-диоксид-2- карботиоловой кислоты
Соединение 62
Раствор 7-аминодезацетоксицефалоспорановой кислоты (7-АДЦК) (42,9 г) в метаноле (1,2 л) последовательно обрабатывают при -15oC (внутри реакционного сосуда) 48% -ным водным HBr (70 мл), 70%-ной водной HClO4 (115 мл) и водным нитритом натрия (35 г в 100 мл). Смесь интенсивно перемешивают 35 мин с повышением температуры внутри сосуда от -15 до +20oC. Добавляют воду (1,5 л) и этиловый эфир (0,8 л), органическую фазу отделяют, промывают рассолом (0,4 л), сушат над Na2SO4 и после концентрирования в вакууме получают сырой 7S-бром-2-карбокси-3-метил-8-оксо-5-тиа-1-азабицикло /4.2.0/окт-2-ен в виде желтого сиропа. Полученный продукт окисляют пероксимоносульфатом калия (1,7 мол. экв. ) по методике, приведенной в примере 9. Полученный сырой продукт растворяют в дихлорметане (200 мл) и уксусной кислоте (20 мл) и при перемешивании в течение 1 одного часа обрабатывают Zn-пылью (30 г), добавляемой порциями. Реакционную смесь фильтруют и распределяют между дихлорметаном и водой. Водную фазу подкисляют конц. HCl, насыщают NaCl и экстрагируют этилацетатом. Испарением растворителя получают желтоватое твердое вещество, которое переносят в этиловый эфир и фильтруют. Собранный продукт растворяют в безэтанольном дихлорметане и обрабатывают 2 ч при 0oC оксалилхлоридом (4,6 мл) в присутствии ДМФА в качестве катализатора (0,05 мл). Реакционную смесь испаряют в высоком вакууме и переносят в дихлорметан. Добавляют трет-бутанол (30 мл), затем триэтиламин (4 мл). Через 15 мин раствор промывают водным раствором бикарбоната натрия, органический слой сушат (Na2SO4) и растворитель удаляют в вакууме. Хроматографией на силикагеле (смеси гексанэтилацетат) вымывают в следующей последовательности:
трет-бутиловый эфир 7S-метокси-3-метил-8-оксо-5-тиа-1-азабицикло /4.2.0/-окт-2-ен-5,5-диоксид-2-карбоновой кислоты (0,75 г) и трет-бутиловый эфир 3-метил-8-оксо-5-тиа-1-азабицикло /4.2.0/окт-2-ен-5,5-диоксид-2-карбоновой кислоты (9,2 г).
Порцию последнего соединения (0,7 г) растворяют в четыреххлористом углероде (50 мл), кипятят 4 ч с N-бромсукцинимидом (0,86 г) и азо-бисизобутиронитрилом (АИБН, 15 мг). Реакционную смесь разбавляют дихлорметаном и интенсивно перемешивают 15 мин с насышенным раствором бисульфита натрия. Органический слой отделяют, сушат (CaCl2) и концентрируют. Хроматографией на силикагеле получают 0,55 г трет-бутилового эфира 3-бромметил-8-оксо-5-тиа-1-азабицикло /4.2.0/окт-2-ен-5,5-диоксид-2-карбоновой кислоты. Полученное соединение растворяют в ацетонитриле и обрабатывают 5-меркапто-1-метил-1,2,3,4-тетразолом (0,19 г) и спустя 10 мин растворитель удаляют в вакууме. Хроматографией на силикагеле получают 400 мг трет-бутилового эфира 3-(1-метил-1,2,3,4 -тетразол-5-ил)тиометил-8-оксо-5-тиа-1-азабицикло /4.2.0/ окт-2-ен-5,5-диоксид-2-карбоновой кислоты в виде белого твердого вещества. Полученное соединение растворяют в смеси трифторуксусной кислоты, дихлорметана и анизола (1:1:0,5). Через 30 мин раствор концентрируют и остаток переносят в дихлорметан (5 мл). После охлаждения до -20oC прибавляют пиридин (0,7 мл), трет-бутилмеркаптан (0,16 мл) и ПФЭ (5 мл) и смесь оставляют на 5 ч при 0oC. Обработкой и хроматографией по методике примера 1 получают 120 мг заглавного соединения в виде желтоватого порошка.
ИК (KBr) nmax: 1807, 1638 см-1
ЯМР (90 МГц, CD Cl3) δ (ч/млн): 1,53 (9H, с), 3,48 и 3,55 (1H, каждая, д, J 4,9 Гц), 3,59 и 3,67 (1H, каждая д, J 2,5 Гц), 3,93 (3H, с), 3,98 (1H, д, J= 17 Гц), 4,24 (1H, дв.д, J 17 и 0,9 Гц), 4,02 и 4,33 (2H, каждая, д, J 14 Гц), 4,76 (1H, м).
Пример 13. Трет-бутиловый эфир 7S-метокси-3-(1-метил-1,2,3,4-тетразол-5-ил)тиометил-8-оксо-5-тиа-1-азабицикло /4.2.0/окт-2-ен-5,5-диоксид-2-карботиоловой кислоты.
Соединение 61
Раствор трет-бутилового эфира 7S-метокси-3-метил-8-оксо-5-тиа-1-азабицикло /4.2.0/окт-2-ен-5,5-диоксид-2-карбоновой кислоты (800 мг) в четыреххлористом углероде (100 мл) обрабатывают НБС (550 мг) и АИБН (50 мг), после чего кипятят 3 ч. Реакционную смесь концентрируют в вакууме и фракционированием остатка хроматографией на силикагеле получают 700 мг трет-бутилового эфира 3-бромметил-7S-метокси-8-оксо-5-тиа-1-азабицикло/4.2.0/окт-2-ен-5,5-диоксид-2-карбоновой кислоты в виде сиропа.
Полученный продукт растворяют в ацетонитриле (20 мг) и ДМФА (5 мл) и после добавления 5-меркапто-1-метил-1,2,3,4-тетрахлора (500 мг) смесь перемешивают 10 мин, большую часть растворителя удаляют в вакууме, а остаток распределяют между этилацетатом и водой. Органическую фазу сушат (Na2SO4) концентрируют и очисткой остатка хроматографией на силикагеле получают 605 мг трет-бутилового эфира 7S-метокси-3-(1-метил-1,2,3,4, -тетразол-5-ил)тиометил-8-оксо-5-тиа-1-азабицикло(4.2.0)окт-2-ен-5,5-диоксид-2-карбоновой кислоты в виде твердого вещества: МС(FD):431(М+)m/з.
Полученное соединение растворяют в смеси трифторуксусной кислоты, дихлорметана и анизола (1,1:0,5) и через 30 мин раствор концентрируют в вакууме, остаток промывают этиловым эфиром. Фильтрованием получают 400 мг 2-карбокси-7S- метокси-3-(1-метил-1,2,3,4-тетразол-5-ил)тиометил- 8-оксо-5-тиа-1-азабицикло/4.2.0/окт-2-ен-5,5-диоксида в виде светло-коричневого порошка.
Полученное соединение растворяют в дихлорметане, обрабатывают при -20oC пиридином (0,5 мл), трет-бутилмеркаптаном (0,135 мл) и ПФЭ (3,5 мл) и оставляют реагировать на 5 ч при 0oС. Обработкой и хроматографией по методике примера 1 получают 250 мг заглавного соединения.
ИК (CHCl3) nmax 1800, 1650 см-1
ЯМР (90 МГц, CD Cl3) δ (ч/млн): 1,55 (9H, с), 3,58(3H, с) 3,88 (1H, д, J 17,5 Гц), 3,93 (3H, с.), 4,04 и 4,38 (2H, каждая д, J 14 Гц), 4,27(1H, дв.д, J 17,5 и 1,1 Гц), 4,63(1H, дв. д, J=1,7 и 1,1 Гц), 5,13(1H, д, J 1,7 Гц).
Пример 14. 2-Дифенилметоксикарбонил-2-(трет-бутоксикарбониламино)этиловый эфир 7S-метокси-3-(1-метил-1,2,3,4-тетразол-5-ил)тиометил-8-оксо-5-тиа- 1-азабицикло/4.2.0/окт-2-ен-5,5-диоксид-2-карботиоловой кислоты.
Соединение 105
Дихлорметановый раствор 2-карбокси-7S-метокси-3-(1-метил-1,2,3,4-тетразол-5-ил)тиометил-8-оксо-5-тиа-1-азабицикло/4.2.0/окт-2-ен-5,5-диоксида (получение см. пример 13) вводят в реакцию с пиридином, бензгидриловым эфиром N-третбутоксикарбонил-1-цистеина и ПФЭ в условиях, приведенных в примере 1. В результате получают заглавное соединение (75% после хроматографии на силикагеле) в виде желтоватого масла.
ИК (CHCl3) nmax: 1800, 1735, 1705, 1665 см-1
ИК (CHCl3) νmax: 1800, 1735, 1705, 1665 см-1
ЯМР (200 МГц, CDCl3 δ (ч/млн): 1,39 (9H, с), 3,36 (1H, дв. д, J 6,8 и 14,1 Гц), 3,9 (1H, д, J 17,2 Гц), 3,94 (1H, д, J=14,5 Гц), 4,23 (1H, д, J 17,2 Гц), 4,53 (1H, уш. H6), 4,59 (1H, д, J=14,5 Гц) 4,7 (1H, м, C a цистеина), 5,13 (1H, д, J=1,6 Гц), 5,75 (1H, д, J 8,1 Гц, обмен D2O), 6,9 (1H, с), 7,27-7,35 (1OH, м).
Пример 15. Бензил-7S-аллил-3-(1-метил-2,3,4-тетразол-5-ил)тиометил-8-оксо-5-тиа-1-азабицикло [4,2,0]окт-2-ен-5,5-диоксид-2-карботиолат
Раствор трет-бутил-7S-аллил-3-ацетоксиметил-8-оксо-5-тиа-1 азабицикло[4,2,0] окт-2-ен-2-карбоксилата (20 ммоль) в дихлорметане (150 мл) охлаждался до 0oC. Добавляли 55% МСРВА (20 ммоль) и смесь перемешивали в течение 6 ч при комнатной температуре.
Нерастворимое вещество отфильтровывалось промыванием холодным CH2Cl2. Фильтрат промывался водным NaHSO3, затем водой, водным NaHCO3 и рассолом. После высушивания над Na2SO4 органическая фаза концентрировалась в вакууме. Остаток хроматографировался на силикагеле (смеси этилацетат-гексан в качестве элюента), обеспечивая выделение трет-бутил-7S-аллил-3-ацетоксиметил-8-оксо-5-тиа-1-азабицикло[4,2,0] окт-2-ен-5,5-диоксид-2-карбоксилат в виде белого твердого вещества.
ЯМР (CDCl3, 200 МГц) d 1,54 (9H, с.); 2,08 (3H, с.); 2,65 (2H, м.); 3,68 (1H, д, J= 18,4 Гц); 3,93 (1H, широкий д, J=18,4 Гц); 3,97 (1H, м.); 4,50 (1H, м.); 4,67 и 5,05 (2H, два д. J= 13,5 Гц); 5,25 (2H, м.); 5,80 (1H, м.).
ИК (KBr) 1810, 1745, 1725 см -1.
Трет-бутил-7S-аллил-3-ацетоксиметил-8-оксо-5-тиа-1-азабицикло[4,2,0] окт-2-ен-2-карбоксилат-5,5-диоксид (2 г) выдерживался в смеси CH2Cl2-TFA-анизол 8: 2:1 (5 мл) в течение 2 ч при 5oC. Смесь концентрировалась досуха. Остаток поглощался ацетонитрилом (40 мл) и обрабатывался 5-меркапто-1-метил-1,2,3,4-тетразолом (0,8 г) и 50% BF3•Et2O (2 мл). Затем смесь нагревалась при 55oC в течение 6 ч, затем концентрировалась в вакууме. Мгновенная хроматография остатка обеспечивала выделение 1,1- диоксида-7-a-аллил-3-(1-метил-1,2,3,4-тетразол-5-ил)тиометил-3-цефем-4-карбоновой кислоты (1,7 г).
386 г этого вещества последовательно обрабатывалось при -20oC 0,6 мл пиридина, 0,14 мл бензилмеркаптана и 4 мл полифосфатного сложного эфира. Реакционная смесь оставлялась стоять при 0oC в течение 5 ч, затем выливалась в смесь этилацетата и водного раствора NaHCO3 и энергично перемешивалась. Органическая фаза сушилась над Na2SO4 и концентрировалась в вакууме. Хроматография остатка над силикагелем (н-гексан: этилацетатные смеси в качестве элюента) давала целевой продукт в виде белого порошка (120 мг).
ИК (KBr): 1795, 1650 см-1
ЯМР (CDCl3, 200 МГц) d 2,64 (2H, м.); 3,86 (3H, с.); 3,9-4,1 (1H, м.); 3,96 (1H, д, J= 17,7 Гц); 3,98 (1H, д, J=14,2 Гц); 4,21 и 4,31 (2H, два д. J= 13,8 Гц); 4,25 (1H, широкий д, J= 17,7 Гц); 4,39 (2H, д, J=14,1 Гц); 4,44 (1H, широкий с.); 5,2-5,3 (2H, м.); 5,7-5,9 (1H, м.); 7,1-7,3 (5H, м.).
Пример 16. Трет-бутил-3-ацетоксиметил-7S-пропил-8-оксо-5-тиа-азабицикло-[4,2,0]окт-2-ен-5,5-диоксид-2-карботиолат.
Раствор трет-бутил-3-ацетоксиметил-7S-пропил-8-оксо-5-тиа-азабицикло[4,2,0] окт-2-карбоксилата (100 мг) в дихлорметане и этаноле окислялся в соответствии с процедурой, описанной в примере 1. Очистка остатка с помощью мгновенной хроматографии (SiO2, EtOAc, н-гексан в качестве элюента) давала трет-бутил-3-ацетоксиметил-7S-пропил-8-оксо-5-тиа-1-азабицикло-[4,2,0] окт-2-ен-5,5-диоксид-2-карбоксидат в виде воскообразного твердого вещества (85 мг).
FD-MS: 387 (М+), 328 (М CH3COO), 286 (М-BOC)+
ИК (KBr): 1805, 1740, 1720 см-1
ЯМР (CDCl3, 200 МГц) d: 0,99 (3H, т. J=7,3 Гц); 1,4-2,0 (4H, м.); 1,54 (9H, с.); 3,66 (1H, д, J=18,3 Гц); 3,8-3,9 (1H, м.); 3,93 (1H, широкий д, J= 18,3 Гц); 4,49 (1H, м.); 4,64 и 5,04 (2H, два д. J=13,5 Гц).
Обработка этого вещества CH2Cl2-TFA-анизолом (8:2:1), как в предыдущем примере, и затем трет-бутилмеркаптаном и PPE в соответствии с процедурой, описанной в примере 1, давала указанное в заголовке соединение в виде аморфного твердого вещества (20 мг).
ИК (KBr) Vmax: 1805, 1660 см-1.
Пример 17. Трет-бутил-7S-аллил-3-(1 метил-1,2,3,4-тетразол-5-ил)-тиометил-8-оксо-5-тиа-1-азабицикло[4,2,0]окт-2-ен-2-5,5-диоксид-2-карботиолат.
Согласно процедуре, описанной в примере 15, но с применением t-бутилмеркаптана вместо бензилмеркаптана, получалось названное соединение в виде твердого вещества.
1H ЯМР (CDCl3, 200 МГц): 1,53 (9H, с.); 2,64 (2H, м.); 3,91 (3H, с.); 3,9-4,4 (4H, м. ); 3,90 (1H, м.); 4,45 (1H, шир. с.); 5,23 (1H, д, J=10,2 Гц); 5,25 (1H, д, J= 17,1 Гц); 5,7-5,9 (1H, м.).
ИК (KBr): 1792, 1640, 1615 см-1.
Пример 18. 2-Дифенилметоксикарбонил-2-ацетиламино-этил-7S-аллил-3-(1-метил-1,2,3,4-тетразол-5-ил)-тиометил-8-оксо-5-тиа-1-азабицикло[4,2,0] окт-2-ен-5,5-диоксид-2-карботиолат.
Следуя процедуре, описанной в предыдущем примере, но применяя N, ацетил-L-цистеин бензагидриловый сложный эфир вместо бензилмеркаптана, получали названное в заголовке соединение в виде белого твердого вещества.
1H ЯМР (CDCl3, 200 МГц): 1,96 (3H, с.); 2,60 (2H, м.); 3,47 (1H, д. J= 7,2 и 14,1 Гц); 3,73 и 4,76 (2H, два д. J=14,5 Гц); 3,79 (1H, дд, J=4,2 и 14,1 Гц); 3,83 (1H, д. J=17,1 Гц); 3,91 (3H, с.); 3,94 (1H, м); 4,13 (1H, шир. д, J=17,1 Гц); 3,91 (3H, с.); 3,94 (1H, м.); 4,13 (1H, шир. д. J=17,1 Гц); 4,29 (1H, шир. с.); 4,97 (1H, м.), 4,19 (1H, д. J=10,0 Гц); 5,21 (1H, д. J= 17,0 Гц); 5,6-5,9 (1H, м.); 6,90 (1H, с.); 7,04 (1H, д. J=8,1 Гц); 7,2-7,3 (10H, м.).
ИК (KBr): 1800, 1745, 1650 см-1.
Пример 19. Получение натриевой соли карбоксиметил 3-ацетоксиметил-7S-хлор-8-оксо-5-тиа-1-азабицикло[4,2,0]окт-2-ен-5,5-диоксид-2-карботиолата.
Раствор карбоксиметил 3-ацетоксиметил-7S-хлор-8-оксо-5-тиа-1-азабицикло[4,2,0]окт-2-ен-5,5-диоксид-2-карботиолата в CH3CN обрабатывали эквимолярным количеством водного 4 мас. раствора NaHCO3. После удаления растворителя остаток очищали хроматографией с обращенной фазой (колонка Мерк Лихропреп® C18), элюируя H2O/CH3CN с получением указанного соединения.
ИК (KBr) νmax: 1810, 1740, 1670-1580 (широкий) см-1.
| название | год | авторы | номер документа |
|---|---|---|---|
| СОЕДИНЕНИЕ ЦЕФЕМА И ЕГО ФАРМАЦЕВТИЧЕСКИ ИЛИ ВЕТЕРИНАРНО ПРИЕМЛЕМЫЕ СОЛИ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 1990 |
|
RU2074186C1 |
| Способ получения производных бета-лактама | 1989 |
|
SU1750430A3 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ЗАМЕЩЕННОГО БЕНЗОФУРАНА ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫХ СОЛЕЙ | 1993 |
|
RU2098415C1 |
| N-ИМИДАЗОЛИЛЬНЫЕ ПРОИЗВОДНЫЕ ЗАМЕЩЕННЫХ АЛКОКСИИМИНОТЕТРАГИДРОНАФТАЛИНОВ ИЛИ ХРОМАНОВ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ПРОЯВЛЯЮЩАЯ АКТИВНОСТЬ АНТАГОНИСТА ТРОМБОКСАНА И ИНГИБИТОРА ТРОМБОКСАНСИНТАЗЫ | 1992 |
|
RU2083566C1 |
| СПОСОБ ПОЛУЧЕНИЯ 4-ЗАМЕЩЕННЫХ АНТРАЦИКЛИНОНОВ | 1989 |
|
RU2071463C1 |
| СОЕДИНЕНИЯ ПЕНЕМА И ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1992 |
|
RU2079498C1 |
| 17БЕТА-ЗАМЕЩЕННЫЕ 3-КАРБОКСИСТЕРОИДЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМКОМПОЗИЦИЯ | 1992 |
|
RU2104283C1 |
| ДИГИДРОБЕНЗОФУРАНОВЫЕ КАРБОКСАМИДЫ ИЛИ ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1991 |
|
RU2043991C1 |
| ПРОИЗВОДНОЕ 3-ДЕЗОКСИМАННОЗАМИНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ ИНГИБИРУЮЩЕЙ АКТИВНОСТЬЮ К РАЗВИТИЮ СОСУДОВ И МЕТАСТАЗОВ | 1992 |
|
RU2099333C1 |
| СПОСОБ ПОЛУЧЕНИЯ СОЕДИНЕНИЯ ПЕНЕМА И СПОСОБ ПОЛУЧЕНИЯ СОЕДИНЕНИЯ АЗЕТИДИН-2-ОНА | 1991 |
|
RU2086553C1 |



Сущность изобретения: производные 1,1-диоксоцефем-4-карботиоловой кислоты общей формулы /I/ и их фармацевтически приемлемые соли, где A означает /C1-C4/алкил, необязательно замещенный одним или двумя заместителями, или метоксифенильную группу; R1 означает атом H, Cl, метокси, алкильную или пропильную группу; R2 означает метил, метоксиметил, ацетоксиметил или /1-метил-1,2,3,4-тетразол-5-ил/тиометил; получают путем обработки соединения общей формулы /II/ надкислотой с получением соединения общей формулы /III/. Полученное соединение общей формулы /III/, где R2 означает метил или ацетоксиметил в случае необходимости переводят в другое соединение общей формулы /III/, где R2 означает /1-метил-1,2,3,4-тетразол-5-ил/, тиометильную группу. Затем соединение общей формулы /III/, где R1 и R2 имеют перечисленные выше значения, подвергают взаимодействию с соединением A1SH, где A1 - то же, что и A, кроме замещенного гидрокси-, карбокси- или аминогруппой /C1-C4/алкила. В случае необходимости получения целевого продукта общей формулы /I/, где A - замещенный гидрокси-, карбокси или аминогруппой /C1-C4/алкил, удаляют в соответствующем замещенном продукте защитные группы кислотным гидролизом. 2 с.п. ф-лы, 3 табл.

 и
и

где A неразветвленная или разветвленная C1 C4-алкильная группа, необязательно замещенная одним или двумя заместителями, выбранными из гидрокси-, карбокси-, амино-, ацетиламино-, трет-бутоксикарбониламино-, дифенилметоксикарбонильной, трет-бутилфенилсилилокси- и фенильной групп, или представляет собой метоксифенильную группу;
R1 хлор или водород или метокси-, аллильная или пропильная группа;
R2 метильная, метоксиметильная, ацетоксиметильная или (1-метил-1,2,3,4-тетразол-5-ил)тиометильная группа,
и их фармацевтически приемлемые соли.
где R1 имеет указанные значения;
R2 метильная, метоксиметильная или ацетоксиметильная группа,
подвергают окислению обработкой надкислотой с образованием соединения общей формулы III
где R1 и R2 имеют указанные для соединения общей формулы II значения,
и при необходимости полученное соединение общей формулы III или его защищенное по карбоксильной группе производное, в котором R2 - метильная или ацетоксиметильная группа, переводят в другое соединение общей формулы III, в котором R2 (1-метил-1,2,3,4-тетразол-5-ил)тиометильная группа, обработкой эфиратом треххлористого бора и 5-меркапто-1-метил-1,2,3,4-тетразолом или обработкой N-бромсукцинимидом и азо-бис-изобутиронитрилом с последующей обработкой 5-меркапто-1-метил-1,2,3,4-тетразолом и удалением в случае необходимости карбоксизащитной группы и полученное соединение общей формулы III, в котором R2 метильная, метоксиметильная, ацетоксиметильная или (1-метил-1,2,3,4-тетразол-5-ил) тиометильная группа, подвергают взаимодействию с соединением общей формулы IV A1-SH, где A1 имеет указанные для A значения, кроме прямого или разветвленного C1 C4-алкила, замещенного одним или двумя разными заместителями, выбранными из ряда, содержащего гидрокси-, карбокси- или аминогруппы, в присутствии пиридина и эфира полифосфорной кислоты или в присутствии дициклогексилкарбодиимида, и в случае необходимости получения целевого продукта общей формулы I, где A - прямой или разветвленный C1 C4-алкил, замещенный одним или двумя разными заместителями, выбранными из ряда, содержащего гидрокси-, карбокси- и аминогруппы, удаляют в соответствующем замещенном продукте защитные группы кислотным гидролизом и при необходимости выделяют соединения общей формулы I в виде их фармацевтически приемлемых солей.
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Патент Великобpитании N 1187323, кл | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| УСТРОЙСТВО для УПРАВЛЕНИЯ ВЗРЫВОМ ТОНКОЙ ПРОВОЛОЧКИ Б МАГНИТНОМ ПОЛЕ | 0 |
|
SU207447A1 |
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
