Изобретение относится к новым химическим соединениям с ценными свойствами, в частности к производным хинолон- и нафтиридонкарбоновой кислоты общей формулы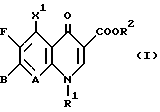
в которой
A CH, CF, CCl, C-OCH3, C-CH3, N;
X1 водород, галоген, NH2, CH3;
R1 алкил, содержащий 1 3 атома углерода, FCH2CH2-, циклопропил, фенил, который может быть от одного до трех раз замещен галогеном, или
A и R1 вместе могут означать мостик структуры C-O-CH2-CH(CH)3,
R2 водород, не замещенный или замещенный гидроксигруппой, галогеном или аминогруппой алкил, содержащий 1 3 атома углерода, или 5-метил-2-оксо-1,3-диоксол-4-ил-метил;
B остаток формул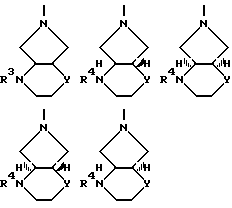
где
Y O или CH2;
R3 оксоалкил, содержащий 2 5 атомов углерода, CH2-CO-C6H5, CH2CH2CO2R', R'O2C-CH=  -CO2R', -CH=CH-CO2R' или CH2CH2-CN, где R' означает водород или алкил, содержащий 1 3 атома углерода;
-CO2R', -CH=CH-CO2R' или CH2CH2-CN, где R' означает водород или алкил, содержащий 1 3 атома углерода;
R4 водород, алкил, содержащий 1 3 атома углерода, оксоалкил, содержащий 2 5 атомов углерода, CH2-CO-C6H5, CH2CH2CO2R', R'O2C-CH=  CH=CH-CO2-R' или CH2CH2-CN или 5-метил-2-оксо-1,3-диоксол-4-ил-метил, где R' означает водород или алкил, содержащий 1 3 атома углерода,
CH=CH-CO2-R' или CH2CH2-CN или 5-метил-2-оксо-1,3-диоксол-4-ил-метил, где R' означает водород или алкил, содержащий 1 3 атома углерода,
в виде смеси изомеров или отдельных изомеров, их фармацевтически применимых гидратов и солей, например кислотно-аддитивных солей и щелочных, щелочноземельных, серебряных и гуанидиновых солей соответствующих карбоновых кислот.
Новые соединения обладают высокой антибактериальной активностью. Особенно они отличаются тем, что проявляют высокую активность против находящихся в состоянии покоя и устойчивых микроорганизмов.
Предпочтительными являются соединения формулы (I),
в которой A означает CH, CF, CCL, C-OCH3, N;
X1 водород, F, CL, Br, NH2, CH3;
R1 C2H5, циклопропил или 2,4-дифторфенил или A и R1 вместе могут означать мостик структуры -O-CH2-CH(CH3)-;
R2 водород, CH3, C2H5, 5-метил-2-оксо-1,3-диоксол-4-ил-метил;
B остаток формул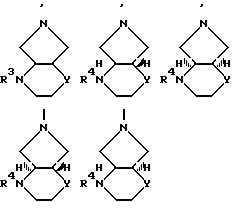
где
Y означает O или CH2;
R3 CH2-CO-CH3, CH2-CO-C6H5, CH2CH2-CO-CH3, CH2CH2CO2R', R'O2C-CH=  -CO2R', -CH=CH-CO2R' или CH2CH2-CN, где R' означает алкил, содержащий 1 2 атома углерода;
-CO2R', -CH=CH-CO2R' или CH2CH2-CN, где R' означает алкил, содержащий 1 2 атома углерода;
R4 водород, алкил, содержащий 1 3 атома углерода, 5-метил-2-оксо-1,3-диоксол-4-ил-метил, CH2-CO-CH3, CH2-CO-C6H5, CH2CH2CO-CH3, CH2CH2CO2R', R'O2C-CH=  -CO2R', -CH=CH-CO2R' или CH2CH2-CN,
-CO2R', -CH=CH-CO2R' или CH2CH2-CN,
где
R' означает алкил, содержащий 1 2 атома углерода.
Особенно предпочтительны соединения формулы (I),
где
A означает CH, CF, CCL, C-OCH3, N;
X1 водород, F, CL, Br, NH2, CH3;
R1 C2H5, циклопропил или 2,4-дифторфенил или A и R1 вместе могут означать мостик структуры -O-CH2-CH(CH3)-,
R2 водород, CH3, C2H5;
B остаток формул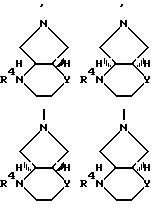
где
Y означает O или CH2;
R4 водород, алкил, содержащий 1 3 атома углерода, 5-метил-2-оксо-1,3-диоксол-4-ил-метил, CH2-CO-CH3, CH2-CO-C6H5, CH2CH2-CO-CH3, CH2CH2CO2R', R'O2C-CH=  -CO2R', -CH=CH-CO2R' или CH2CH2-CN, где R' означает алкил, содержащий 1 2 атома углерода.
-CO2R', -CH=CH-CO2R' или CH2CH2-CN, где R' означает алкил, содержащий 1 2 атома углерода.
Соединения формулы (I), в которой A, X1, R1, R2 имеют указанное выше значение и B означает остаток формулы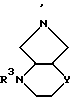
где
R3 и Y имеют указанное выше значение,
получают тем, что соединение формулы
где
A, Y, X1, R1 и R2 имеют указанное выше значение,
подвергают взаимодействию с соединением формулы,
R3-X3
где
R3 означает оксоалкил, содержащий 2 5 атомов углерода, CH2-CO-C6H5, CH2CH2-CO2R' или CH2CH2-CN, где R' водород или алкил, содержащий 1 3 атома углерода,
X3 галоген, в частности хлор, бром, или иод,
при необходимости, в присутствии связывающего кислоту средства. (Реакция А)
Соединения согласно формулы (I), где A, Y, X1, R1 и R2 имеют вышеуказанное значение и B означает остаток формулы
где
Y имеет вышеуказанное значение;
R3 означает CH2CH2-CO-CH3, CH2CH2-CO2R', R'O2C-CH=C-CO2R', -CH=CH-CO2R' или CH2CH2-CN, где R' означает водород или алкил, содержащий 1 3 атома углерода,
могут быть получены тем, что соединение формулы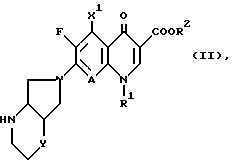
подвергают взаимодействию с акцептором Михаэля, например, диалкиловым эфиром ацетилендикарбоновой кислоты, алкиловым эфиром пропиоловой кислоты или соединением формулы
CH2 CH-R5,
в которой
R5 означает COCH3, CO2R' или CN (Реакция Б).
Для получения энантиомерночистых соединений формулы (I) соединение формулы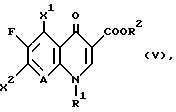
в которой
A, R1, R2 и X1 имеют вышеуказанное значение;
X2 означает галоген, в частности фтор или хлор,
подвергают взаимодействию с энантиомерночистыми соединениями формулы
где
Y O или CH2;
R4 водород или алкил, содержащий 1 3 атома углерода,
при необходимости в присутствии связывающего кислоту средства, и продукт реакции при желании, дальше подвергают превращению с соединением формулы
R4-X3,
где
X3 имеет указанное выше значение;
R4 означает оксоалкил, содержащий 2 5 атомов углерода, CH2-CO-C6H5, CH2CH2CO2R' или CH2CH2-CN, где R' означает водород или алкил, содержащий 1 3 атома углерода, или с акцептором Михаэля, например диалкиловым эфиром ацетилендикарбоновой кислоты, алкиловым эфиром пропиоловой кислоты или соединением формулы
CH2 CH R5
где
R5 означает COCH3, CO2R' или CN (Реакция В)
Если применяют, например, в качестве исходных соединений 8-хлор-1-циклопропил-6,7-дифтор-1,4-дигидро-4-оксо-3- хинолинкарбоновую кислоту и [S,S] -2,8-диазабицикло[4.3.0]нонан, то реакция протекает по следующей схеме: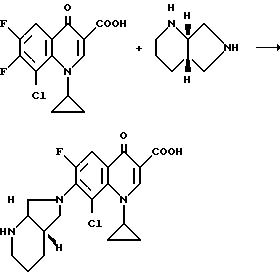
Если в качестве исходных соединений применяют, например, 6,8-дифтор-1-(2,4-дифторфенил)-1,4-дигидро-7-([1S, 6R] -2-окса-5,8-диазабицикло[4.3.0] нон-8-ил)-4-оксо-3-хинолинкарбоновую кислоту и диэтиловый эфир ацетилендикарбоновой кислоты, то реакция протекает по следующей схеме: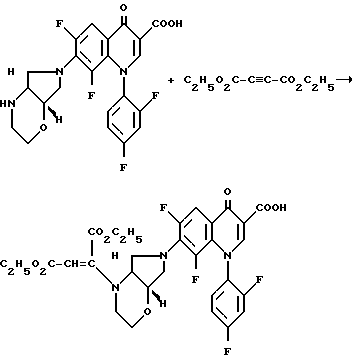
Применяемые в качестве исходных соединений рацемические соединения формулы (II) большей частью являются известными. Энантиомерночистые соединения формулы (II) являются новыми и могут быть получены различными путями.
1. Подвергают взаимодействию рацемическое промежуточное соединение формулы (II) с энантиомерночистым вспомогательным реагентом, разделяют полученные диастереомеры, например, хроматографически, и опять отщепляют у полученного диастереомера вспомогательную хиральную группу. В качестве примера следует привести следующую реакцию:
2. Бициклические амины (VI), как энантиомерночистые соединения, являются новыми. Они могут быть получены следующими способами:
2.1 Рацемические бициклические амины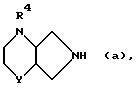
где
R4 означает водород или алкил, содержащий 1 3 атома углерода, могут подвергаться превращению с энантиомерночистыми кислотами, например карбоновыми кислотами или сульфокислотами, как N-ацетил-L-глутаминовая кислота, N-бензоил-L-аланин, 3-бромкамфар-9-сульфокислота, камфар-3-карбоновая кислота, цис-камфарная кислота, камфар-10-сульфокисота, O,O'-дибензоил-винная кислота, D- или L-винная кислота, миндальная кислота, α-метокси-фенилуксусная кислота. 1-фенил-этансульфокислота, α-фенил-янтарная кислота, в смесь диастереомерных солей, которые разделяю. т фракционной кристаллизацией на диастереомерночистые соли (см. П.Нейман. Optical Resolution Procedures for Chemical Compounds, т. 1).
Молярное соотношение амина и энантиомерночистой кислоты может изменяться в широком интервале. Обработкой этих солей гидроокисями щелочных и щелочноземельных металлов выделяют энантиомерночистые амины.
2.2 Аналогичным образом, как описано в п. 2.1, проводят расщепление рацематов основных промежуточных соединений, которые образуются при получении рацемических бициклических аминов, с указанными выше энантиомерночистыми кислотами. Примерами подобных основных промежуточных соединений являются: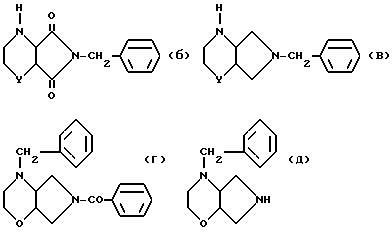
На схеме, приведенной ниже, в качестве примера расщепления рацематов приведено разделение 8-бензил-цис-2,8-диазабицикло[4.3.0] нонана через тартраты на энантиомеры и их перевод в энантиомерночистые цис-2,8-диазабицикло[4.3.0]нонаны:
2.3 Как рацемические амины (а), так и основные промежуточные соединения (б)-(д) могут быть при необходимости после ацилирования разделены хроматографически на хиральных носителях (см. например, г. Блашке. Angew. Chem. 92, 14 [1980]).
2.4 Как рацемические амины (а), так и основные промежуточные соединения (б), (с), (д), могут быть переведены химическим связыванием с хиральными ацильными остатками в смеси диастереомеров, которые разделяют дистилляцией, кристаллизацией или хроматографией на диастереомерночистые ацильные производные, из которых выделяют омылением энантиомерночистые амины. Примерами реагентов для связывания с хиральными ацильными остатками являются: α -метокси- a -трифторметил-фенилацетилхлорид, метилизоцианат, D- или L- a -фенил-этил-изоцианат, метиловый эфир хлормуравьиной кислоты, хлорангидрид камфар-10-сульфокислоты.
2.5. В ходе синтеза бициклических аминов (а) могут вводиться вместо ахиральных также хиральные группы. Таким образом получают диастереомеры, которые разделяют. Например, при синтезе цис-2,8-диазабицикло[4.3.0]нонана бензильный остаток может быть заменен a -фенилэтильным остатком с R- или S-конфигурацией: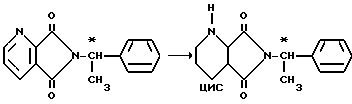
2.6 Энантиомерночистые амины (VI) могут быть также произведены из энантиомерночистых предшественников, например, [R,R] или [S,S]-дигидроксипирролидина, который у азота должен быть защищен защитной группой.
В качестве примера синтеза энантиомерночистого амина из энантиомерночистого 1-бензил-3,4-дигидрокси-пирролидина следует привести следующую схему: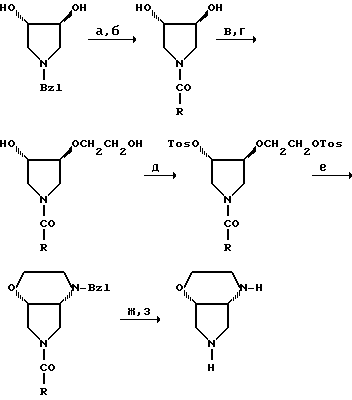
R означает, например, (CH3)3C-O,
а: водород, палладий/активированный уголь;
б: ацилирование;
в: NaH, BrCH2COOC2H5 или в: CH2=CH-CH2Br, NaH,
г: LiBH4 г: O3 NaBH4;
д: тозилхлорид, триэтиламин;
е: бензиламин, ксилол, нагревание до образования флегмы;
ж: гидролиз;
з: водород, палладий/активированный уголь.
В качестве примеров соединений формулы (VI) следует назвать:
цис-2,8-диазабицикло[4.3.0]нонан,
цис-2-окса-5,8-диазабицикло[4.3.0]нонан,
транс-2-окса-5,8-диазабицикло[4/3/0]нонан,
S,S-2,8-диазабицикло[4.3.0]нонан,
1R,6S-2-окса-5,8-диазабицикло[4.3.0]нонан,
1S,6R-2-окса-5,8-диазабицикло[4.3.0]нонан,
1R,6R-2-окса-5,8-диазабицикло[4.3.0]2нонан,
1S,6S-2-окса-5,8-диазабицикло[4.3.0]нонан.
Взаимодействие (V) с (VI), при котором соединения (VI) могут также использоваться в виде их солей, например гидрохлоридов, проводится предпочтительно в разбавителе, например диметилсульфоксиде, N,N-диметилформамиде, N-метилпирролидоне, триамиде гексаметилфосфорной кислоты, сульфолане, ацетонитриле, воде, спирте, например метаноле, этаноле, н-пропаноле, изопропаноле, монометиловом эфире гликоля или пиридине. Также могут применяться смеси этих растворителей.
В качестве связывающего кислоту средства можно применять все обычные неорганические или органические кислотосвязующие. К ним относятся, предпочтительно, гидроокиси щелочных металлов, карбонаты щелочных металлов, органические амины и амидины.
Особенно подходящими следует назвать: триэтиламин, 1,4-диазабицикло[2.2.2]октан, 1,8-диазабицикло[5.4.0]ундек-7-ен или избыток амина (VI).
Температура реакции может варьироваться в широком диапазоне. В основном работают при температуре 20 200oC, предпочтительно 80 180oC.
Взаимодействие можно проводить при нормальном давлении, но также и при повышенном давлении. В основном работают при давлении 1 100 бар, предпочтительно 1 10 бар.
При осуществлении этого способа вводят на 1 моль соединения (V) от 1 до 15 моль, предпочтительно от 1 до 6 моль, соединения (VI).
Исходные соединения формул (III) и (IV) известны.
В качестве примера можно назвать хлорацетон, 4-хлор-2-бутанон, 5-хлор-2-пентанон, 1-бром-2-бутанон, хлорацетофенон, метилакрилат, этилакрилат, акрилонитрил, метилвинилкетон, диметиловый эфир ацетилендикарбоновой кислоты, диэтиловый эфир ацетилендикарбоновой кислоты, метиловый эфир пропиоловой кислоты, этиловый эфир пропиоловой кислоты.
Взаимодействие (II) с (III) проводят предпочтительно в разбавителе, например диметилсульфоксиде, N,N-диметилформамиде, N-метилпирролидоне, триамиде гексаметилфосфорной кислоты, сульфолане, ацетонитриле, воде, спирте, напр. метаноле, этаноле, н-пропаноле, изопропаноле, монометиловом эфире гликоля или пиридине, в присутствии связывающего кислоту средства. Также могут применяться смеси этих растворителей.
В качестве связывающего кислоту средства можно применять все обычные неорганические и органические кислотосвязующие. К ним относятся предпочтительно гидроокиси щелочных металлов, карбонаты щелочных металлов, органические амины и амидины. Особенно подходящими следует назвать: триэтиламин, 1,4-диазабицикло[2.2.2] октан, 1,8-диазабицикло[5.4.0]ундек-7-ен или избыток амина (VI).
Температура реакции может варьироваться в широком диапазоне. В основном работают при температуре 20 200oC, предпочтительно 60 130oC.
Взаимодействие можно проводить при нормальном давлении, но также и при повышенном давлении. В основном работают при давлении 1 100 бар, предпочтительно 1 10 бар.
При осуществлении этого способа вводят на 1 моль соединения (II) от 1 до 15 моль, предпочтительно от 1 до 6 моль, соединения (III).
Взаимодействие соединений (II) с акцепторами Михаэля (IV) по способу Б проводят предпочтительно в разбавителе, как ацетонитрил, диметилсульфоксид, N, N-диметилформамид, спирт, например метанол, этанол, пропанол или изопропанол, или монометиловый эфир гликоля.
Температура реакции может колебаться в широком интервале. В основном работают при температурах около 20 150oC, предпочтительно 40 - 100oC.
Взаимодействие можно проводить при нормальном давлении, но также при повышенном давлении. В основном работают при давлениях между 1 и 100 бар, предпочтительно между 1 и 10 бар. При осуществлении способа согласно изобретению вводят на 1 моль соединения (II) от 1 до 5 моль, предпочтительно 1 2 моль, соединения (IV).
Получение кислотно-аддитивных солей соединений согласно изобретению происходит обычным образом, например, растворением бетаина в водной кислоте и осаждением соли смешивающимся с водой органическим растворителем, например метанолом, этанолом, ацетоном, ацетонитрилом. Можно также нагревать эквивалентные количества бетаина и кислоты в воде или спирте, например монометиловом эфире гликоля, и затем упарить досуха или отфильтровать выпавшую соль.
Под фармацевтически применимыми солями понимают, например, соли соляной кислоты, серной кислоты, уксусной кислоты, гликолевой кислоты, молочной кислоты, янтарной кислоты, лимонной кислоты, винной кислоты, метансульфокислоты, 4-толуолсульфокислоты, галактуроновой кислоты, эмбоновой кислоты, глутаминовой кислоты или аспарагиновой кислоты.
Щелочные и щелочноземельные соли кислот согласно изобретению получают, например, растворением бетаина в недостаточном количестве основания щелочного или щелочноземельного металла, фильтрацией нерастворившегося бетаина и упариванием фильтрата досуха.
Фармацевтически пригодными являются соли натрия, калия, или кальция. Обработкой щелочной или щелочноземельной соли подходящей серебряной солью, например нитратом серебра, получают соответствующие соли серебра.
Заявленные соединения формулы (I) проявляют сильную антибиотическую активность и при незначительной токсичности обладают широком спектром действия против грамположительных и грамотрицательных микробов, особенно против эндобактерий, прежде всего против таких, которые устойчивы к различным антибиотикам, например пенициллину, цефалоспорину, аминоглюкозидам, сульфамидам, тетрациклину.
Благодаря этим ценным свойствам соединения могут применяться в медицине, а также для консервирования органических и неорганических материалов, в особенности всех видов органических материалов, например полимеров, смазочных веществ, красок, волокон, кожи, бумаги и древесины, пищевых продуктов и воды. Соединения согласно изобретению активны против очень широкого спектра микроорганизмов.
С их помощью борются с грамположительными и грамотрицательными бактериями и бактериоподобными микроорганизмами, а также с вызываемыми ими болезнями.
Соединения согласно изобретению проявляют сильную активность в отношении покоящихся и резистентных микроорганизмов. В случае покоящихся бактерий, т. е. у которых не наблюдается роста, соединения активны при концентрациях значительно более низких, чем известные соединения. Это относится не только к вводимым количествам, но и к скорости умерщвления. Такие результаты наблюдались у грамположительных и грамотрицательных бактерий, в частности у Staphylococcus aureus, Pseudomonas aeruginosa, Enterococcus faecalis и Escherichia coli. Также по отношению к бактериям, значительно менее восприимчивым к веществам, взятым для сравнения, в особенности к резистентным Staphylococcus aureus, Escherichia coli, Pseudomonas aeruginosa и Enterjcoccus faecalis, заявленные соединения проявляют значительное повышение активности. Особенно активны соединения по отношению к бактериям и бактериоподобным микроорганизмам. Поэтому они особенно пригодны в медицине и ветеринарии для профилактики и химиотерапии локальных и системных инфекций, вызываемых этими возбудителями. Также соединения годятся для борьбы с протозоонозом и гельминтозом.
Заявленные соединения могут применяться в различных препаративных формах. Особенно предпочтительными являются таблетки, драже, капсулы, пилюли, гранулы, суппозитории, растворы, суспензии и эмульсии, пасты, мази, гели, кремы, лосьоны, пудры и аэрозоли.
Данные, приведенные в табл. 1, подтверждают неожиданное преимущество заявленных соединений по сравнению с ципрофлоксацином [1-циклопропил-6-фтор-1,4-дигидро-4-оксо-7-(1-пиперазинил)- 3-хинолинкарбоновая кислота (EP 0176846 B1, кл. C 07 C 101/34, 1986, кол. 4)] на инфицированных Staphylicoccus aureus мышах.
В табл. 2 даны минимальные концентрации торможения (МКТ) для заявленного соединения по примеру 2 и известного соединения ципрофлоксацина, полученные способом последовательного разбавления на Iso-sensitest агаре (оксоид). При этом готовили ряд агаровых пластин, которые при двойном разбавлении содержали уменьшающиеся концентрации активного вещества. Агаровые пластины прививали с помощью многоточечного инокулятора типа Денлей. Для прививки применяли выращенные в течение ночи культуры Еррегера, которые сначала разбавляли так, чтобы каждая точка прививки содержала около 104 образующих колонию частиц. Привитые агаровые пластины инкубировали при 37oC и спустя около 20 ч определяли рост микроорганизмов. Значение МКТ (μг/мл) дает самую низкую концентрацию активного вещества, при которой невооруженным глазом не наблюдалось никакого роста.
Нижеследующие примеры поясняют получение исходных и промежуточных продуктов.
Пример А. [S,S]-2,8-диазабицикло[4.3.0]нонан.
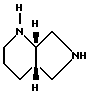
1) [S,S]-8-бензил-2,8-диазабицикло[4.3.0]нонан,
Способ I.
а) Разделение диастереомерных солей: 3,0 г (20 ммоль) D(-)-винной кислоты растворяют в 10 мл диметилформамида нагреванием до 80oC и смешивают с раствором из 2,16 г (10 ммоль) цис-8-бензил-2,8-диазабицикло[4.3.0]нонана в 3 мл диметилформамида. Перемешивают в течение часа при 0oC, затем отфильтровывают и промывают диметилформамидом и метоксиэтанолом.
Выход: 1,93 г; Т. пл. 146 151oC; [α]
Однократной перекристаллизацией из метоксиэтанола получают диастереомерночистый [S,S]-8-бензил-2,8-диазабицикло[4.3.0] нонан-D-тартрат.
[α]
б) Выделение основания: 40 г [S,S]-8-бензил-2,8-диазабицикло[4.3.0]нонан-D-тартрата растворяют в 250 мл воды и смешивают с 32 г 45-ной натриевой щелочи. Выделившееся масло поглощают в 150 мл трет.бутил-метилового эфира, водную фазу еще раз экстрагируют 150 мл трет.бутил-метилового эфира и объединенные органические фазы после сушки над сульфатом натрия сгущают. Затем отгоняют в вакууме.
Выход: 18,5 г [S,S]-8-бензил-2,8-диазабицикло[4.3.0]нонана.
Т. кип. 107 109oC/0,1 мбар; [α]
Способ II. 75,0 г (0,5 моль) L(+)-винной кислоты растворяют при 80oC в 250 мл диметилформамида и прикапывают раствор 54,1 г (0,25 моль) цис-8-бензил-2,8-диазабицикло[4.3.0]нонана в 75 мл диметилформамида. Медленно охлаждают до 20oC и перемешивают в течение часа суспензию кристаллов. Кристаллы ([R, R] -8-бензил-2,8-диазабицикло[4.3.0] нонан-L-тартрат) отфильтровывают и фильтрат сгущают в ротационном испарителе. Остаток растворяют в 500 мл воды и обрабатывают, как описано в способе I, с 63 г 45-ной натриевой щелочи.
Выход: 25,2 г [S,S]-8-бензил-2,8-диазабицикло[4.3.0]нонана; продукт содержит 3,6 R,R-энантиомера (определялся газовой хроматографией после получения производного с ментиловым эфиром хлормуравьиной кислоты).
Соединение может быть с D(-)-винной кислотой превращено в диастереомерночистый [S,S]-8-бензил-2,8-диазабицикло[4.3.0]нонан -D-тартарт. При этом перекристаллизация не требуется.
Способ III. К раствору 102,9 г (0,685 моль) L(+)-винной кислоты в 343 мл диметилформамида прикапывают при 80 90oC раствор 73,6 г (0,34 моль) цис-8-бензил-2,8-диазабицикло[4.3.0] нонана в 111 мл диметилформамида. Добавляют для затравки [R,R]-8-бензил-2,8-диазабицикло[4.3.0]нонан-L-тартрат и медленно охлаждают до внутренней температуры 18oC. Кристаллы отфильтровывают. Фильтрат затравляют [S,S]-8-бензил-2,8-диазабицикло[4.3.0]нонан-L-тартратом и перемешивают до полной кристаллизации (из маточника после сгущения и выделения основания, как описано в способе I, получают обработкой D(-)-винной кислотой [S,S]-8-бензил-2,8-диазабицикло[4.3.0]нонан- D-тартрат). Затем отфильтровывают, промывают диметилформамидом и изопропанолом и сушат на воздухе. Кристаллы перекристаллизовывают из 88%-ного этанола. Получают 52 г тригидрата [S,S]-8-бензил-2,8-диазабицикло[4.3.0]нонан-L-тартрата.
Т. пл. 201 204oC; [α]
Соль можно перерабатывать по способу I (выделение основания) в энантиомерночистый [S,S]-8-бензил-2,8-диазабицикло[4.3.0]нонан.
Способ IV.
а) Разделение энантиомеров цис-8-бензил-7,9-диоксо-2,8-диазабицикло[4.3.0]нонана на [1S,6R]-8-бензил-7,9-диоксо-2,8-диазабицикло[4.3.0]нонан. Поступают аналогично примеру Б (способ IIa), причем в качестве хирального вспомогательного реагента применяют D(-)-винную кислоту, или поступают следующим образом: сгущают маточник и промывочную жидкость, содержащую [1R, 6S]-8-бензил-7,9-диоксо-2,8-диазабицикло[4.3.0]нонан- L-тартрата (из примера Б, способ II/a), помещают в воду и трижды экстрагируют толуолом. Водную фазу смешивают с насыщенным раствором кислого углекислого натрия, доводят значение pH до 7 8, в заключение трижды экстрагируют метиленхлоридом. Объединенные метиленхлоридные фазы сушат над сульфатом магния и сгущают.
Выход: 14,4 г (60 от теории на первоначально введенный рацемический цис-8-бензил-7,9-диоксо-2,8-диазабицикло [4.3.0] нонан). [α]
Эти 14,4 г (50 ммоль) кристаллизуют с 8,6 г (57 ммоль) D(-)винной кислоты аналогично примеру Б (способ IIa).
Выход: 8,9 г (77 от теории) [1S,6R]-8-бензил-7,9-диоксо-2,8-диазабицикло[4.3.0] нонан-D-тартрата. [α]
5,0 г (12,7 ммоль) полученного таким образом диастереомерночистого тартрата, как описано в примере Б, способ IIa, переводят в свободный амин.
Выход: 3,0 г (96 от теории) [1S,6R]-8-бензил-7,9-диоксо-2,8- диазабицикло[4.3.0]нонана; Т. пл. 60 61oC; [α]
После получения производного с ментиловым эфиром хлормуравьиной кислоты был определен путем газовой хроматографии избыток энантиомера 96,6)
б) Восстановление [1S, 6R]-8-бензил-7,9-диоксо-2,8-диазабицикло[4.3.0] нонана до [S,S]-8-бензил-2,8-диазабицикло[4.3.0]нонана. Поступают аналогично примеру Б (способ II б), причем в качестве исходного продукта берут [1S,6R] -8-бензил-7,9-диоксо-2,8-диазабицикло[4.3.0]нонан.
Полученный после переработки сырой продукт оказался при получении производного с ментиловым эфиром хлормуравьиной кислоты [S,S]-8-бензил-2,8-диазабицикло[4.3.0]нонаном.
Рацемация при восстановлении не наблюдалась.
2) [S,S]-2,8-диазабицикло[4.3.0]нонан. 28,4 г (0,131 моль) [S,S]-8-бензил-2,8-диазабицикло[4.3.0] нонана гидрируют в 190 мл метанола на 5,8 г палладия на активированном угле (5) при 90oC и 90 бар в течение 5 ч. Затем отфильтровывают катализатор, промывают метанолом и фильтрат сгущают в ротационном испарителе. Остаток перегоняют без фракционирования.
Выход: 15,0 г (90,5% от теории) [S,S]-2,8-диазабицикло[4.3.0]нонана; Т. кип. 44 59oC/0,18 мбар; [α]
Способ V. Берут 3,75 г (25 ммоль) L(+)-винной кислоты, растворенной в 50 мл диметилформамида при 80oC, и прикапывают 10,82 г (50 ммоль) цис-8-бензил-2,8-диазабицикло[4.3.0] нонана в виде раствора в 15 мл диметилформамида. Дают затравку [R,R]-8-бензил-2,8-диазабицикло[4.3.0]нонан-L-тартрата и перемешивают один час при температуре около 72oC до завершения образования кристаллических зародышей. Затем медленно охлаждают до 15oC, отфильтровывают и дважды промывают по 13 мл диметилформамида. Объединенные фильтраты нагревают до 80oC и смешивают с дополнительными 3,75 г (25 ммоль) L(+)-винной кислоты. Нагревают еще до 119oC до образования прозрачного раствора и медленно опять охлаждают до комнатной температуры с затравкой [S,S]-8-бензил-2,8-диазабицикло[4.3.0] нонан-L-тартрата. Кристаллы отфильтровывают, промывают по очереди диметилформамидом, 2-этоксиэтанолом и этанолом и сушат на воздухе.
Выход: 9,59 г; Т. пл. 188 192oC.
Кристаллы перекристаллизовывают из 95 мл 80%-ного этанола. Получают 8,00 г тригидрата [S,S]-8-бензил-2,8-диазобицикло[4.3.0]нонан-L-тартрата (76% от теории), который плавится при 112 118oC со вспениванием, затем опять затвердевает и снова плавится при 199 201oC.
[α]
Пример Б. [R,R]-2,8-диазабицикло[4.3.0]нонан.
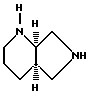
1) [R,R]-8-бензил-2,8-диазабицикло[4.3.0]нонан.
Способ I. полученные по примеру А, способ II кристаллы (49,2 г) [R,R]-8-бензил-2,8-диазабицикло[4.3.0] нонана промывают диметилформамидом и метоксиэтанолом и перекристаллизовывают из 300 мл метоксиэтанола. Получают 45,6 г энантиомерночистого [R,R]-8-бензил-2,8-диазабицикло[4.3.0]нонан-L-тартрата. Чистота энантиомера определялась газовой хроматографией после получения производного с ментиловым эфиром хлормуравьиной кислоты.
Т. пл. 121 124oC; [α]
Соль (44,5 г) переводят в свободное основание, как описано в примере А, способ 1б. Получают 20,2 г [R,R]-8-бензил-2,8-диазабицикло[4.3.0]нонана.
Т. кип. 107 111oC/0,04 мбар; [α]
Способ II.
а) Разделение энантиомеров цис-8-бензил-7,9-диоксо-2,8-диазабицикло[4.3.0]нонана на [1R,6S]-8-бензил-7,9-диоксо-2,8-диазабицикло[4.3.0]нонан. 24,1 г (98,8 ммоль) цис-8-бензил-7,9-диоксо-2,8-диазабицикло[4.3.0]нонана нагревают с смесью из 410 мл этанола и 25 мл ацетонитрила в трехгорлой колбе при перемешивании до кипения с обратным холодильником. В заключение одноразово прибавляют 14,8 г (98,8 ммоль) L(+)-винной кислоты. После того, как все количество винной кислоты полностью растворится, прекращают сначала обогрев, но оставляют колбу в масляной бане. Когда система охладится настолько, что раствор больше не кипит, прекращают перемешивание. При температуре 50oC происходит кристаллизация при внесении затравочных кристаллов. После выдерживания в течение ночи и охлаждения до комнатной температуры отфильтровывают выпавшие кристаллы, промывают небольшим количеством смеси этанол/петролейный эфир (1 1) и сушат два часа при 80oC.
Выход: 9,8 г 50% от теории) [1R,6S]-8-бензил-7,9-диоксо-2,8-диазабицикло[4.3.0]нонан-L-тартрата; [α]
Двукратной перекристаллизацией из смеси этанола и монометилового эфира гликоля соединение подвергают дополнительной очистке: [α]
1H-ЯМР (диметилсульфоксид): 7,22 7,35 (2м, 2H. арил-H); 4,55 (с, 2H, бензил-CH2); 4,28 (с, 2H, винная кислота-CH); 3,91 (д, 1H, 1-CH); 2,97 (дд, 1H, 6-CH); 2,53 2,66 (м, 2H, 3-CH2); 1,78 и 1,68 (2м, 2H, 5-CH2); 1,42 и 1,28 ч/милл (2м, 2H, 4-CH2).
C18H22N2O8 (394).
Рассчитано: C 54,4; H 5,6; N 7,1; O 32,5.
Найдено: C 54,7; H 5,8; N 7,1; O 32,4.
Определение полной конфигурации происходило при помощи рентгеноструктурного анализа: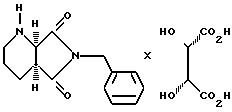
3,6 г (9,1 ммоль) полученного таким путем диастереомерночистого тартрата растворяют в воде для выделения основания и смешивают с насыщенным раствором кислого углекислого натрия до установления значения pH от 7 до 8. Водный раствор экстрагируют четыре раза по 20 мл метиленхлоридом. Объединенные метиленхлоридные фазы сушат над сульфатом магния и сгущают.
Выход: 2,2 г (99% от теории) [1R,6S]-8-бензил-7,9-диоксо-2,8-диазабицикло[4.3.0]нонана.
Т. пл. 60 61oC; [α]
Газовая хроматография дает после получения производного с ментиловым эфиром хлормуравьиной кислоты избыток энантиомера 93,8
б) Восстановление [1R, 6S]-8-бензил-7,9-диоксо-2,8-диазабицикло[4.3.0] нонана до [R,R]-8-бензил-2,8-диазабицикло[4.3.0]нонана. В подогретую колбу помещают в атмосфере азота 0,34 г (9 ммоль) литийалюминий гидрида в 18 мл безводного тетрагидрофурана и прикапывают 0,73 (3 ммоль) [1R,6S]-8-бензил-7,9-диоксо-2,8-диазабицикло[4.3.0] нонана в виде раствора в 3 мл безводного тетрагидрофурана. Затем кипятят 16 ч с обратным холодильником. Обработку проводят прикапыванием 0,34 мл воды в 10 мл тетрагидрофурана, 0,34 мл 10%-ной натриевой щелочи, а также 1,02 мл воды. Осадок отфильтровывают, промывают тетрагидрофураном и фильтрат сгущают. Выделяют 0,7 г сырого [R,R]-8-бензил-2,8-диазабицикло[4.3.0]нонана (состав газовой хроматографией: 99%).
При газохроматографическом определении чистоты энантиомера не наблюдалась рацемация.
2) [R, R]-2,8-диазабицикло[4.3.0]нонан. По прописи примера А2 гидрируют 19,4 г (0,09 моль) [R,R]-8-бензил-2,8-диазабицикло[4.3.0]нонана.
Выход: 9,61 г (85) [R,R]-2,8-диазабицикло[4.3.0]нонана. Т. кип. 45 - 58oC/0,08 мбар; [α]
Пример В. [S,S]-2-метил-2,8-диазабицикло[4.3.0]нонан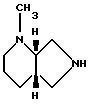
1) [S, S] -8-бензил-2-метил-2,8-диазабицикло[4.3.0] нонан. 43,2 г (0,2 ммоль) [S,S]-8-бензил-2,8-диазабицикло[4.3.0]нонана смешивают с 20 мл 37%-ного раствора формальдегида, 40 мл воды и 24 г уксусной кислоты и гидрируют 10 ч при 20oC и 20 бар над 2 г палладия на активированном угле (5). Затем фильтруют, фильтрат подщелачивают карбонатом калия и продукт экстрагируют трет. -бутилметиловым эфиром. После сушки над сульфатом натрия сгущают и остаток перегоняют в вакууме. Выход: 14,8 г, Т. кип. 114 124oC/0,14 мбар.
2) [S, S]-2-метил-2,8-диазабицикло[4.3.0]нонан. 12,9 г (56 ммоль) [S,S] -8-бензил-2-метил-2,8-диазабицикло[4.3.0] нонана гидрируют в 90 мл метанола при 90oC и 90 бар над 1,1 г палладия на активированном угле (5). Затем фильтруют, фильтрат упаривают в ротационном испарителе и остаток перегоняют в вакууме.
Выход: 5,5 г энантиомерночистого [S,S]-2-метил-2,8-диазабицикло[4.3.0] нонана (определено получением производного с реактивом Мошера). Т. кип. 78 81oC/14 мбар.
Пример Г. [R,R]-2-метил-2,8-диазабицикло[4.3.0]нонан.
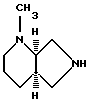
Соединение получают по описанному в примере В способу, исходя из 43,2 г (0,2 моль) [R,R]-8-бензил-2,8-диазабицикло[4.3.0]нонана.
Выход: 4,9 г [R, R]-метил-2,8-диазабицикло[4.3.0]нонана; Т. кип. 30 - 33oC/0,12 мбар
Пример Д. Цис-7,9-диоксо-8-([S] 1-фенил-этил)-2,8-диазабицикло[4.3.0] нонан.
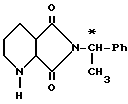
1) ([S]1-фенил-этил)имид пиридин-2,3-дикарбоновой кислоты. Берут 74,5 г (0,5 моль) ангидрида пиридин-2,3-дикарбоновой кислоты, растворенного при 20oC в 500 мл диоксана, и прикапывают 60,5 г (0,5 моль) S(-)1-фенил-этиламина, после чего повышают температуру до 33oC. Перемешивают еще один час, затем сгущают в ротационном испарителе и удаляют оставшийся растворитель при 40oC/0,1 мбар. Остаток поглощают в 245 г (2,4 моль) уксусного ангидрида, смешивают с 4,9 г (0,006 моль) безводного ацетата натрия и перемешивают 1 ч при 100oC. После охлаждения при хорошем перемешивании выливают на 1 л ледяной воды, отсасывают, промывают холодной водой и сушат на воздухе. Сырой продукт (114 г, т. пл. 112 114oC) перекристаллизовывают из 285 мл метанола.
Выход: 96,3 г (76%); Т. пл. 115 117oC; [α]
2) Цис-7,9-диоксо-8-([S] )1-фенил-этил)-2,8-диазабицикло[4.3.0] нонан. 79,7 г (0,316 моль) ([S]1-фенил-этил)-имида пиридин-2,3-дикарбоновой кислоты гидрируют в 600 мл тетрагидрофурана при 90oC/100 бар над 10 г палладия на активированном угле. После окончания поглощения водорода отфильтровывают катализатор и фильтрат полностью сгущают. Получают 83,7 г вязкого остатка. Содержание 95%
1H-ЯМР (дейт. хлороформ, 200 МГц): 1,4 1,7 (м, 3H); 1,82 и 1,83 (2д, 3H); 1,9 2,05 (м, 1H); 2,28 (шир, с, 1H); 2,54 2,86 (м, 3H); 3,77 (д, 1H); 5,39 (кв, 1H); 7,24 7,48 ч/милл (м, 5H).
Пример Е. Цис-2-окса-5,8-диазабицикло[4.3.0]нонан.

1) Транс-1-бензоил-3-бром-4-(2-гидроксиэтокси)-пирролидин. Растворяют 95 г (0,55 моль) 1-бензоил-3-пирролина в 380 г этиленгликоля и добавляют при комнатной температуре 101 г (0,57 моль) N-бромсукцинимида пятиграммовыми порциями в течение 2 ч. После этого перемешивают в течение ночи при комнатной температуре, выливают на воду, экстрагируют метиленхлоридом, сушат над сульфатом магния и раствор сгущают. Остаток (188 г) хроматографируют на силикагеле с этилацетатом.
Выход: 136,5 г (78% от теории): Содержание по газовой хроматографии: 99%
2) Транс-1-бензоил-3-бром-4-(2-тозилоксиэтокси)-пирролидин. Растворяют 92 г (0,239 моль) транс-1-бензоил-3-бром-4-(2-гидроксиэтокси)пирролидина, 32 г (0,316 моль) триэтиламина и 1 г 4-диметиламинопиридина в 750 мл толуола и прикапывают 60 г (0,31 моль) тозилхлорида в 450 мл толуола. Перемешивают два дня при комнатной температуре, добавляют воду, отделяют водную фазу и экстрагируют ее толуолом. Толуольные растворы промывают 10%-ной соляной кислотой, сушат над сульфатом магния, сгущают, растворяют в этилацетате и фильтруют через силикагель. Фильтрат сгущают.
Выход: 125 г (91% от теории). Тонкослойная хроматография показывает однородное соединение.
3) Цис-8-бензоил-2-окса-5,8-диазабицикло[4.3.0] нонан. Нагревают 124 г (0,265 моль) транс-1-бензоил-3-бром-4-(2-тозилэтокси)-пирролидина с 86 г (0,8 моль) бензиламина в 1,5 л ксилола в течение ночи с обратным холодильником. Отфильтровывают соль бензиламина и фильтрат сгущают.
Выход сырого продукта: 91,2 г.
4) Цис-5-бензил-2-окса-5,8-диазабицикло[4.3.0] нонан. Нагревают 91 г (0,265 моль) цис-8-бензоил-5-бензил-2-окса-5,8-диазабицикло[4.3.0]нонана с 200 мл концентрированной соляной кислоты и 140 мл воды в течение ночи с обратным холодильником. После охлаждения отфильтровывают бензойную кислоту, сгущают до половины объема, подщелачивают раствор карбонатом калия, экстрагируют хлороформом, сушат над карбонатом калия, сгущают и перегоняют.
Выход: 30,7 г (48,8% от теории); Т. кип. 134 142oC/0,6 мбар. Содержание по газовой хроматографии 92%
5) Дигидрохлорид цис-2-окса-5,8-диазабицикло[4.3.0]нонана. Гидрируют 26 г (0,11 моль, 92%-ного) цис-5-бензил-2-окса-5,8-диазабицикло[4.3.0]нонана в 180 мл этанола и 19 мл концентрированной соляной кислоты с помощью 3 г палладия на активированном угле (10% Pd) при 100oC и давлении водорода 100 бар. Отфильтровывают катализатор, сгущают фильтрат и сушат выделившиеся кристаллы в эксикаторе над пятиокисью фосфора.
Выход: 17,1 г (77% от теории). Т. пл. 244 250oC.
Пример Ж. Разделение энантиомеров цис-5-бензил-2-окса-5,8-диазабицикло[4.3.0]нонана.
Помещают 150,1 г (1 моль) D(-)-винной кислоты при 60 65oC в 700 мл метанола и прикапывают раствор 218,3 г (1 моль) цис-5-бензил-2-окса-5,8-диазабицикло[4.3.0] нонана в 300 мл метанола. Затем дают медленно охладиться до 49oC до тех пор, пока раствор не помутнеет, затравляют полученными в предыдущем опыте кристаллами 1R,6S-5-бензил-2-окса-5,8-диазабицикло[4.3.0]нона-D-тартрата, перемешивают 30 мин при этой температуре до образования зародышей кристаллов и медленно охлаждают до 0 3oC. После фильтрации промывают охлажденной до 0oC смесью из 200 мл этанола и 100 мл метанола и затем три раза по 300 мл этанола, в заключение продукт сушат на воздухе.
Выход: 160 г 1R,6S-5-бензил-2-окса-5,8-диазабицикло[4.3.0]нонан-тратрата (87% от теории). Т. пл. 174,5 176,5oC.
Чистота изомера > 97% (после превращения с 1-фенил-этилизоцианатом и определения методом жидкостной хроматографии под давлением). [α]
156,9 г первого кристаллизата перекристаллизовывают из 1500 мл метанола.
Выход: 140,0 г (90% от теории) Т. пл. 176 177oC. [α]
Метанольные маточники первой кристаллизации упаривают в ротационном испарителе. Сиропообразный остаток (236 г) растворяют в 500 мл воды, с помощью 250 мл 6 н. натриевой щелочи устанавливают pH 12 13, трижды экстрагируют толуолом по 350 мл, экстракт сушат над карбонатом натрия и упаривают в вакууме. Остаток, 113,1 г коричневого масла, которое содержит, по данным газохроматографического анализа, 97% цис-5-бензил-2-окса-5,8-диазабицикло[4.3.0] нонана, без очистки используется для получения 1S,6R-энантиомера.
113,1 г (0,518 моль) сырого обогащенного 1S,6R-5-бензил-2-окса-5,8-диазабицикло[4.3.0] нонана растворяют в 155 мл метанола и прикапывают к кипящему раствору 77,8 г (0,518 моль) L(+)-винной кислоты в 363 мл метанола. Уже во время прикапывания образуется постепенно каша кристаллов. Перемешивают 1 ч при 60oC и затем медленно в течение двух часов охлаждают до 0oC. Кристаллы отфильтровывают и промывают охлажденной до 0oC смесью (2 1) из этанола и метанола и в заключение промывают трижды этанолом. Затем сушат на воздухе.
Выход: 145,5 г 1S,6R-5-бензил-2-окса-5,8-диазабицикло[4.3.0]нонан-L-тартрата (79% от теории). Т. пл. 174,5 176,5oC.
Чистота изомера > 97% (после превращения с 1-фенил-этилизоцианатом по данным жидкостной хроматографии под давлением) [α]
Выделение энантиомерночистого основания:
144 г (0,39 моль) 1S,6R-5-бензил-2-окса-5,8-диазабицикло[4.3.0]нонан-тартрата растворяют в 250 мл воды и обрабатывают 175 мл (1,05 моль) 6 н. натриевой щелочи. Выделившееся масло помещают в 500 мл толуола, отделяют органическую фазу и водную фазу трижды экстрагируют по 250 мл толуола. Объединенные органические фазы сушат над углекислым натрием. Фильтруют и упаривают в ротационном испарителе. Остаток перегоняют в глубоком вакууме на колонке (20 см) типа Vigreux.
Выход: 81,6 г (96 от теории) 1S,6R-5-бензил-2-окса-5,8-диазабицикло[4.3.0] нонана. Т. кип. 120 - 139oC/0,04 0,07 мбар. Содержание: 100% (определено газохроматографически). Плотность: δ = 1,113 г/мл. [α]
Подобным образом было получено из 139,2 г (0,376 моль) 1R,6S-5-бензил-2-окса-5,8-диазабицикло[4.3.0] нонан-тартрата 76,0 г (93 от теории) 1R,6S-5-бензил-2-окса-5,8-диазабицикло[4.3.0]нонана. [α]
Описанное для цис-5-бензил-2-окса-5,8-диазабицикло[4.3.0]нонана разделение энантиомеров проводят аналогично с транс-5-бензил-2-окса-5,8-диазабицикло[4.3.0]нонаном до R,R- и S,S-5-бензил-2-окса-5,8-диазабицикло[4.3.0]нонана.
Пример 3.
1) Трет.бутиловый эфир 3S,4S-4-аллилокси-3-гидроксипирролидин- 1-карбоновой кислоты. Помещают 16,5 г (0,55 моль) 80-ного гидрида натрия в 500 мл абсолютного диоксана и прикапывают при 60oC раствор из 107,5 г (0,53 моль) трет. бутилового эфира S,S-3,4-дигидрокси-пирролидин-1-карбоновой кислоты, растворенного в горячем виде в абсолютном диоксане. Перемешивают еще час при 60oC и затем прикапывают 64 г (0,53 моль) аллилбромида. В заключение перемешивают три часа при 60oC. Упаривают и остаток растворяют в 200 мл воды и 600 мл метанола. Трижды экстрагируют по 200 мл пентана, отгоняют метанол в ротационном испарителе, разбавляют 200 мл воды и экстрагируют метиленхлоридом. Метиленхлоридный раствор сушат над сульфатом магния, упаривают и растворяют в трет.бутилметиловом эфире (200 мл). Спустя ночь выкристаллизовывается 9 г продукта (44 ммоль). Эфирный раствор сгущают и перегоняют.
Выход: 83 г (80 от теории, считая на возвращенный исходный продукт и диаллиловый эфир). Т. кип. 149oC/0,7 мбар до 159oC/0,9 мбар.
Дистиллят содержит 5 исходного продукта реакции и 4 диаллилового эфира.
[α]
2) Трет. бутиловый эфир 3S,3S-3-гидрокси-4-(2-гидроксиэтокси)-пирролидин-1-карбоновой кислоты. Растворяют 64 г (0,34 моль, 91-ного) трет.бутилового эфира 3S,4S-4-аллилокси-3-гидроксипирролидин-1-карбоновой кислоты в 250 мл метанола, охлаждают до 0oC и пропускают через раствор озон до тех пор, пока подключенная промывочная емкость с раствором иодистого калия не покажет появления озона и тем самым завершение реакции. Остатки озона вытесняют азотом.
Затем восстанавливают полученный озонид при 0oC с 18 г натрийборгидрида, который прибавляют порциями по 1 г. В заключение перемешивают в течение ночи при комнатной температуре, смесь сгущают, разбавляют водой, смешивают с 20 г карбоната калия и экстрагируют пять раз по 100 мл метиленхлоридом. Органические растворы сушат над сульфатом магния и сгущают.
Выход: 65,8 г (100 от теории). Продукт 91-ный (газохроматогр.). [α]
3) 3S,4S-1-трет.бутоксикарбонил-3-тозилокси-4-(2-тозилоксиэтокси)- пирролидин. 2,7 г (10 ммоль, 91-ного) трет.бутилового эфира 3S,4S-3-гидрокси-4-(2-гидроксиэтиокси)-пирролидин-1-карбоновой кислоты вносят в 30 мл метиленхлорида, прибавляют 6 мл 45-ной натриевой щелочи и 0,1 г бензилтриэтиламмонийхлорида и прикапывают затем при охлаждении раствор 2,86 г (20 ммоль) тозилхлорида в 10 мл метиленхлорида. В заключение перемешивают еще один час при комнатной температуре, выливают на 20 мл воды, отделяют органическую фазу и экстрагируют водную фазу метиленхлоридом. Органические фазы сушат над сульфатом натрия и сгущают.
Выход: 5 г (90 от теории). Продукт однородный (тонкослойная хроматография)
4) трет.бутиловый эфир 1S,6R-5-бензил-2-окса-5,8-диазабицикло[4.3.0]нонан-8-карбоновой кислоты. Нагревают 87 г (156 ммоль) 3S,4S-1-трет.бутоксикарбонил-3-тозилокси-4-(2-тозилоксиэтокси)-пирролидина с 58 г (0,54 моль) бензиламина в 1 л ксилола в течение ночи с обратным холодильником. Охлаждают, отфильтровывают выпавшую соль бензиламина и остаток сгущают.
Выход: 43 г (58 от теории). Продукт 67-ный (газохроматографически).
5) 1S, 6R-5-бензил-2-окса-5,8-диазабицикло[4.3.0] нонан. Нагревают 43 г (90 ммоль) трет. бутилового эфира 1S, 6R-5-бензил-2-окса-5,8-диазабицикло[4.3.0] нонана в 35 мл концентрированной соляной кислоты и 35 мл воды с обратным холодильником до окончания выделения двуокиси углерода. Подщелачивают карбонатом калия, экстрагируют хлороформом, сушат органические растворы над сульфатом магния, сгущают и дважды перегоняют на колонке (20 мл) типа Vigreux.
Выход: 11,1 г (55 от теории). Т. кип. 108 115oC/0,07 мбар, [α]
Пример И.
1) Трет. бутиловый эфир 3R,4R-4-аллилокси-3-гидроксипирролидин -1-карбоновой кислоты. Взаимодействие происходит аналогично примеру 3 1) с трет. бутиловым эфиром R,R-3,4-дигидроксипирролидин-1-карбоновой кислоты.
Т. кип. 145oC/0,1 мбар. [α]
2) Трет. бутиловый эфир 3R,4R-3-гидрокси-4-(2-гидроксиэтокси)- пирролидин-1-карбоновой кислоты. Взаимодействие происходит аналогично примеру 3 2) с трет.бутиловым эфиром 3R,4R-4-аллилокси-3-гидроксипирролидин-1-карбоновой кислоты.
Выход: 99 от теории (0,175 молярная загрузка). [α]
3) 3R,4R-1-трет.бутоксикарбонил-3-тозилокси-4-(2-тозилоксиэтокси)- пирролидин. Взаимодействие происходит аналогично примеру 3 3) с трет.бутиловым эфиром 3R,4R-3-гидрокси-4-(2-гидроксиэтокси)-пирролидин-1- карбоновой кислоты.
Выход: количественный (0,11 молярная загрузка).
4) Трет. бутиловый эфир 1R,6S-5-бензил-2-окса-5,8-диазабицикло [4.3.нонан-8-карбоновой кислоты. Взаимодействие происходит аналогично примеру 3 4) с 3R,4R-1-бутоксикарбонил-3-тозилокси-4 -(2-тозилоксиэтокси)-пирролидином.
Выход: 40% от теории (0,1 молярная загрузка).
5) 1R, 6S-5-бензил-2-окса-5,8-диазабицикло[4.3.0] нонан. Взаимодействие происходит аналогично примеру 3 5) с трет.бутиловым эфиром 1R,6S-5-бензил-2-окса-5,8-диазабицикло[4.3.0]нонан-8- карбоновой кислоты.
Выход: 63 от теории (40 μ молярная загрузка). Т. кип. 120oC/0,06 мбар. Продукт 95-ный (газохроматографически). [α]
Пример К.
1) Дигидрохлорид 1S,6R-2-окса-5,8-диазабицикло[4.3.0]нонана. Гидрируют 7,5 г (34,4 ммоль) 1S,6R-5-бензил-2-окса-5,8-диазабицикло [4.3.0]нонана в 200 мл этанола при добавлении 7 мл концентрированной соляной кислоты с 1 г палладия на активированном угле (10% Pd) при 100oC и 100 бар. Отфильтровывают катализатор и промывают его многократно водой. Водный фильтрат сгущают, после чего остаток кристаллизуется. Кристаллы тщательно перемешивают с этанолом, отфильтровывают и сушат на воздухе.
Выход: 4,6 г (66,5% от теории). Т. пл. 233 235oC.
2) 1S,6R-2-окса-5,8-диазабицикло[4.3.0]нонан. Гидрируют 59 г (0,27 моль) 1S, 6R-5-бензил-2-окса-5,8-диазабицикло[4.3.0] нонана в 500 мл этанола с 5 г палладия на активированном угле (10% Pd) при 120oC и 120 бар. Катализатор отфильтровывают, фильтрат сгущают и остаток перегоняют.
Выход: 32,9 г (95 от теории). Т. кип. 65oC/0,03 мбар. [α]
Чистота ≥99,5% (после дериватизации с реактивом Мошера).
Пример Л.
1) Дигидрохлорид 1R,6S-2-окса-5,8-диазабицикло[4.3.0]нонана. Взаимодействие происходит аналогично примеру К1 с 1R,6S-5-бензил-2-окса-5,8-диазабицикло[4.3.0]нонаном.
Выход: 77 от теории (23,8 ммолярная загрузка).
Т. пл. 230 232oC.
2) 1R,6S-2-окса-5,8-диазабицикло[4.3.0]нонан. Взаимодействие происходит аналогично примеру К2 с 1R,6S-бензил-2-окса-5,8-диазабицикло[4.3.0]нонаном.
Выход: 93,3% от теории (1,58 молярная загрузка). Т. кип. 63 - 65oC/0,03 мбар. [α]
Чистота изомера: ≥99,5 (дериватизация с реактивом Мошера).
1R, 6R- соответ. 1S, 6S-2-окса-5,8-диазабицикло[4.3.0]нонан могут быть получены аналогично.
Пример М. Дигидробромид 1R,6S-2-окса-5,8-диазабицикло[4.3.0]нонана.
1) 1R. 6S-(1R-фенилэтил)-8-тозил-2-окса-5,8-диазабицикло [4.3.)нонан. Нагревают 101,8 г (0,196 моль) транс-3-бром-1-тозил-4-(2-тозилоксиэтокси)-пирролидина и 72 г (0,584 моль) R-(+)-1-фенилэтиламина в 900 мл ксилола в течение ночи с обратным холодильником. Промывают охлажденный раствор 2 н. натриевой щелочью, сушат над карбонатом калия, удаляют осушитель и раствор сгущают. При охлаждении из остатка выделяются кристаллы, которые отфильтровывают и перекристаллизовывают из смеси 750 мл промывочного бензина и 200 мл н. бутанола.
Выход: 16 г (39 от теории, оптически чистый материал).
Т. пл. 188oC. [α]
2) 1R, 6S-8-тозил-2-окса-5,8-диазабицикло[4.3.0] нонан. Гидрируют 13 г (33,6 ммоль) 1R,6S-5-(1R-фенилэтил)-8-тозил-2-окса-5,8-диазабицикло[4.3.0] нонана в 200 мл этанола над 2,5 г палладия на активированном угле (10 Pd) при 100oC и 100 бар. Отфильтровывают катализатор, фильтрат сгущают и перекристаллизовывают из 30 мл толуола.
Выход: 7,5 г (79 от теории). Т. пл. 160 161oC. [α]
3) Дигидробромид 1R,6S-2-окса-5,8-диазабицикло[4.3.0]нонана. Растворяют 7 г (24,8 ммоль) 1R,6S-8-тозил-2-окса-5,8-диазабицикло[4.3.0]нонана в 25 мл 33-ного раствора бромистого водорода в ледяной уксусной кислоте, прибавляют 5 г фенола и перемешивают в течение ночи при комнатной температуре. Разбавляют диизопропиловым эфиром, отфильтровывают выкристаллизовавшуюся соль и сушат ее на воздухе.
Выход: 5,5 г.
Дериватизация с реактивом Мошера и газохроматографический анализ показывают лишь определяемым энантиомер (чистота изомера ≥99,5)
Пример Н. 5-бром-1-циклопропил-6,7,8-трифтор-1,4-дигидро- 4-оксо-3-хинолинкарбоновая кислота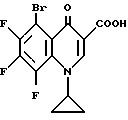
1) 2-бром-3,4,5,6-тетрафтор-бензоилхлорид. 365 г (1,33 моль) 2-бром-3,4,5,6-тетрафторбензойной кислоты вносят в 2 л тионилхлорида и смесь нагревают 11 ч с обратным холодильником до прекращения выделения газа. Избыточный тионилхлорид отгоняют в вакууме и остаток перегоняют.
Выход: 330 г (85% от теории). Т. кип. 81 85oC/3 5 мбар,
2) Диэтиловый эфир (2-бром-3,4,5,6-тетрафтор-бензоил)-малоновой кислоты. Вносят 15,9 г (0,167 моль) хлорида магния в 150 мл безводного ацетонитрила (высушенного над цеолитом) и при охлаждении прикапывают 26,9 г (0,167 моль) диэтилового эфира малоновой кислоты. Охлаждают до 0oC, прикапывают 46 мл (33,7 г 0,33 моль) триэтиламина и перемешивают 30 мин. В заключение прикапывают 48,9 г (0,168 моль) 2-бром-3,4,5,6-тетрафторбензоилхлорида, еще перемешивают один час при 0oC и смесь выдерживают в течение ночи при комнатной температуре. Смешивают с 100 мл 5 н. соляной кислоты, трижды экстрагируют метиленхлоридом, сушат над сульфатом натрия и сгущают в вакууме. Сырой продукт: 62,7 г
3) Этиловый эфир (2-бром-3,4,5,6-тетрафтор-бензоил)-уксусной кислоты. 60 г сырого диэтилового эфира (2-бром-3,4,5,6-тетрафтор-бензоил)-малоновой кислоты вносят в 150 мл воды, смешивают с 0,6 г толуолсульфокислоты и нагревают 6 ч с обратным холодильником. Экстрагируют метиленхлоридом, промывают водой, сушат над сульфатом натрия и сгущают.
Выход сырого продукта: 46 г.
Т. кип. (пробная дистилляция в шаровой трубке): 150 160oC (печь)/3 мбар;
Масс-спектр: м/е 342 (M+), 279 (M+-OC2H5), 263 (M+-Br), 257, 255 (M+-CH2CO2C2H5), 235 (263-28).
4) Этиловый эфир 2-(2-бром-3,4,5,6-тетрафтор-бензоил)-3- этоксиакриловой кислоты. Вносят 45 г сырого этилового эфира (2-бром-3,4,5,6-тетрафтор-бензоил)-уксусной кислоты в 32,2 г (0,31 моль) уксусного ангидрида и 28,4 г (0,19 моль) триэтилового эфира ортомуравьиной кислоты и нагревают 2 ч с обратным холодильником. Избыточный реагент удаляют сначала в вакууме, затем в глубоком вакууме (температура бани 120 130oC) и сырой продукт подают на следующую стадию.
Выход сырого продукта: 50,7 г
5) Этиловый эфир 2-(2-бром-3,4,5,6-тетрафтор-бензоил)-3-циклопропиламино-акриловой кислоты. 50,7 г сырого продукта со стадии 4) в 90 мл этанола по каплям смешивают при охлаждении льдом с 8,6 г (0,15 моль) циклопропиламина, смесь перемешивают при комнатной температуре, оставляют на ночь, еще раз хорошо охлаждают, отфильтровывают кристаллизат, промывают холодным метанолом и сушат.
Выход: 29 г (42 после четырех стадий).
Т. пл. 103 105oC (из этанола)
6) Этиловый эфир 5-бром-1-циклопропил-6,7,8-трифтор-1,4-дигидро-4-оксо-3- хинолинкарбоновой кислоты. 28 г (68 ммоль) этилового эфира 2-(2-бром-3,4,5,6-тетрафтор-бензоил)-3-циклопропиламино-акриловой кислоты нагревают в 88 мл диметилформамида с 6,9 г (164 ммоль) фторида натрия 6 ч с обратным холодильником. Смесь после охлаждения выливают в воду, отфильтровывают выпавший осадок (красный), промывают большим количеством воды и сушат при 80oC в воздушном шкафу.
Выход сырого продукта: 27,3 г.
Т. пл. 150 170oC: после перекристаллизации из монометилового эфира гликоля т. пл. 187 191oC
7) 5-Бром-1-циклопропил-6,7,8-трифтор-1,4-дигидро-4- оксо-3-хинолинкарбоновая кислота. Вносят 26,7 г (68 ммоль) сырого этилового эфира 5-бром-1-циклопропил-6,7,8-трифтор-1,4-дигидро-4-оксо-3- хинолинкарбоновой кислоты в смесь из 165 мл уксусной кислоты, 110 мл воды и 18 мл концентрированной серной кислоты и нагревают 2 ч с обратным холодильником. Охлажденную реакционную смесь выливают в ледяную воду, отфильтровывают выпавший осадок, промывают большим количеством воды и сушат в воздушном шкафу при 80oC.
Выход: 19,7 (80% от теории).
Т. пл. 208 210oC (с разложением); после перекристаллизации из монометилового эфира гликоля т. пл. 212 214oC (с разложением).
1H-ЯМР (диметилсульфоксид: 8,73 с (1H y C-2), 4,16 м (1H, циклопропил), 1,2 м (4H, циклопропил) [ч/милл]
Масс-спектр: м/е 361 (M+-H2O), 317 (M-CO2), 41 (100 C3H5).
Нижеследующие примеры поясняют получение новых соединений формулы (I).
Пример 1.
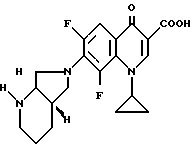
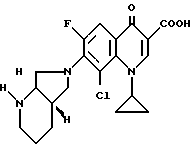
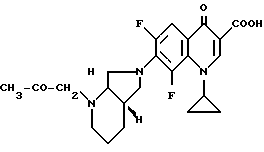
А. 1-Циклопропил-7-([S, S] -2,8-диазабицикло[4.3.0] нон-8-ил)- 6,8-дифтор-1,4-дигидро-4-оксо-3-хинолинкарбоновая кислота. 141,5 г (0,5 моль) 1-циклопропил-6,7,8-трифтор-1,4-дигидро-4-оксо-3- хинолинкарбоновой кислоты нагревают 1 ч с обратным холодильником в смеси из 1500 мл ацетонитрила и 750 мл диметилформамида в присутствии 55 г (0,5 моль) 1,4-диазабицикло[2.2.2]октана с 69,25 г (0,55 моль) (+)-[S,S]-2,8-диазабицикло[4.3.0]нонана (чистота изомера 99,5% газовая хроматография: 99,8%-ный). Суспензию охлаждают, осадок отфильтровывают, промывают водой и затем перемешивают еще с 1 л воды (pH 7). Отфильтровывают и сушат при 60oC в воздушном шкафу.
Выход: 163,4 г (84 от теории).
Т. пл. 249 251oC (с разложением).
Б. Гидрохлорид (-)-1-циклопропил-7-([S,S]-2,8-диазабицикло [4.3.0]нон-8-ил)-6,8-дифтор-1,4-дигидро-4-оксо-3- хинолинкарбоновой кислоты. 6,0 г (15,4 ммоль) 1-циклопропил-7-([S,S]-2,8-диазабицикло[4.3.0]нон-8-ил)- 6,8-1,4-дигидро-4-оксо-3-хинолинкарбоновой кислоты растворяют в 40 мл полуконцентрированной соляной кислоты при 60oC и фильтруют раствор гидрохлорида. Фильтрат сгущают наполовину, охлаждают льдом и смешивают с 40 мл этанола. Отфильтровывают желтый кристаллизат, промывают этанолом и сушат при 60oC в глубоком вакууме, при этом окраска осветляется. Получают 5,51 г (84 от теории) гидрохлорида, который уже очень чистый. Для дальнейшей очистки растворяют его в 50 мл воды при нагревании. Желтый раствор смешивают с 5 мл полуконцентрированной соляной кислоты, охлаждают льдом, отфильтровывают выпавший кристаллизат, хорошо промывают этанолом и сушат сначала при комнатной температуре и затем в глубоком вакууме при 100oC.
Выход: 4,64 г (70,8% от теории). Т. пл. 324 325oC (с разложением).
Тонкослойная хроматография (силикагель, дихлорметан/метанол/17%-ный водный аммиак 30 8 1); однородный, Rf 0,3. [α]
Состав: 99,4% (жидкостная хроматография под давлением)
C20H21F2N3O3 • HCL (425,5)
Рассчитано: C 56,4; H 5,2; N 9,9; CL 8,3.
Найдено: C 56,3; H 5,4; N 9,8; CL 8,3.
Пример 2.
А. 8-хлор-1-циклопропил-7-([S, S]-2,8-диазабицикло[4.3.0]нон-8- ил)-6-фтор-1,4-дигидро-4-оксо-3-хинолинкарбоновая кислота. Параллельно готовят и одновременно перерабатывают 2 реакционные смеси следующего состава: 180 г (0,6 моль) 8-хлор-1-циклопропил-6,7-дифтор-1,4-дигидро-4-оксо-3- хинолинкарбоновой кислоты нагревают в смеси из 1,8 л ацетонитрила и 900 мл диметилформамида в присутствии 99 г (0,88 моль) 1,4-диазабицикло[2.2.2]октана с 84 г (0,67 моль) (+)-[S,S]-2,8-диазабицикло[4.3.0]нонана 1 ч с обратным холодильником (внутренняя температура: 90,5oC). Желтый раствор охлаждают и смешивают с затравочными кристаллами (получены из 5 мл пробы, которую сгустили; остаток растерли с ацетонитрилом). Это перемешивают 2 ч при температуре около 3oC, выпавший осадок из двух смесей быстро отфильтровывают, промывают ацетонитрилом и вносят в 1,5 л ледяной воды. Сначала жидкая, хорошо перемешиваемая суспензия спустя приблизительно 10 мин превращается в трудно перемешиваемую массу, которая разбавляется дополнительными 150 мл воды. Отфильтровывают, промывают водой и сушат в воздушном сушильном шкафу при 80oC.
Выход: 402 г (82,7% от теории), светло-желтый продукт.
Т. пл. 193 196oC (с разложением).
Rf (силикагель; метилхлорид/метанол/17%-ный водный аммиак 30 8 1) 0,4.
Б. Гидрохлорид 8-хлор-1-циклопропил-7-([S,S]-2,8-диазабицикло[4.3.0]нон-8-ил)- 6-фтор-1,4-дигидро-4-оксо-3-хинолинкарбоновой кислоты. 13,1 г (32 ммоль) 8-хлор-1-циклопропил-7-([S, S]-2,8-диазабицикло[4.3.0] нон-8-ил)-6-фтор-1,4-дигидро-4-оксо-3-хинолинкарбоновой кислоты суспендируют в 50 мл воды и переводят в раствор добавкой 50 мл полуконцентрированной соляной кислоты. Фильтруют через стеклянный фильтр, сгущают в вакууме и перемешивают остаток с приблизительно 300 мл абсолютного этанола. Суспензию охлаждают льдом, отфильтровывают осадок, промывают этанолом и сушат сначала при комнатной температуре и затем при 100oC в вакууме.
Выход: 13,4 г (93,8 от теории).
Т. пл. 328 330oC (с разложением).
Rf (силикагель; метиленхлорид/метанол/17%-ный водный аммиак 30 8 1) 0,4.
Состав: 99,9%-ный (жидкостная хроматография под давлением).
[α]
C20H21CLFN3O3 • HCL 9442,3).
Рассчитано: C 54,3; H 5,0; N 9,5; CL 16,0.
Найдено: C 54,3; H 5,0; N 9,5; CL 16,0.
В. Аналогично могут быть получены, например, следующие соли:
Метансульфонат 8-хлор-1-циклопропил-7-([S, S] -2,8-диазабицикло [4.3.0] нон-8-ил)-6-фтор-1,4-дигидро-4-оксо-3-хинолинкарбоновой кислоты.
Толуолсульфонат 8-хлор-1-циклопропил-7-([S, S]-2,8-диазабицикло [4.3.0] нон-8-ил)-6-фтор-1,4-дигидро-4-оксо-3-хинолинкарбоновой кислоты.
Сульфат 8-хлор-1-циклопропил-7-([S, S] -2,8-диазабицикло [4.3.0]нон-8-ил)-6-фтор-1,4-дигидро-4-оксо-3-хинолинкарбоновой кислоты.
Ацетат 8-хлор-1-циклопропил-7-([S, S] -2,8-диазабицикло [4.3.0]нон-8-ил)-6-фтор-1,4-дигидро-4-оксо-3-хинолинкарбоновой кислоты.
Лактат 8-хлор-1-циклопропил-7-([S, S] -2,8-диазабицикло [4.3.0]нон-8-ил)-6-фтор-1,4-дигидро-4-оксо-3-хинолинкарбоновой кислоты.
Цитрат 8-хлор-1-циклопропил-7-([S, S] -2,8-диазабицикло [4.3.0]нон-8-ил)-6-фтор-1,4-дигидро-4-оксо-3-хинолинкарбоновой кислоты.
Эмбонат 8-хлор-1-циклопропил-7-([S, S] -2,8-диазабицикло [4.3.0]нон-8-ил)-6-фтор-1,4-дигидро-4-оксо-3-хинолинкарбоновой кислоты.
Пример 3.
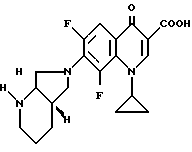
Аналогично примеру 1 получают с 1-циклопропил-6,7-дифтор-1,4-дигидро-4-оксо-3-хинолинкарбоновой кислотой:
А. 1-Циклопропил-7-([S,S]-2,8-диазабицикло[4.3.0]нон-8-ил)- 1,4-дигидро-4-оксо-3-хинолинкарбоновую кислоту.
Т. пл. 256 258oC (с разложением).
Б. Гидрохлорид 1-циклопропил-7-([S,S]-2,8-диазабицикло- [4.3.0]нон-8-ил)-6-фтор-1,4-дигидро-4-оксо-3-хинолинкарбоновой кислоты.
Т. пл. >320oC (с разложением).
[α]
Пример 4.
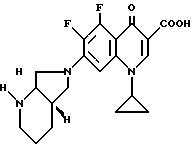
А. 6 г (20 ммоль) 1-циклопропил-5,6,7,8-тетрафтор-1,4-дигидро- 4-оксо-3-хинолинкарбоновой кислоты нагревают в смеси из 40 мл ацетонитрила и 20 мл N-метилпирролидона в присутствии 2,2 г (20 ммоль) 1,4-диазабицикло[2.2.2] октана с 2,7 г (21,4 ммоль) (+)-[S,S]-2,8-диазабицикло[4.3.0]нонана один час с обратным холодильником. Полученную суспензию охлаждают, осадок отфильтровывают, промывают ацетонитрилом и сушат при 100oC/12 мбар.
Выход: 6,7 г (82,3 от теории) 1-циклопропил-7-([S,S]-диазабицикло[4.3.0] нон-8-ил)-5,6,7- трифтор-1,4-дигидро-4-оксо-3-хинолинкарбоновой кислоты.
Т. пл. 257 259oC (с разложением); после перекристаллизации из монометилового эфира гликоля т. пл. 260 265oC (с разложением).
Б. 1,5 г (3,7 ммоль) продукта со стадии А вносят в 6 мл 1н. соляной кислоты. Вскоре выпадает гидрохлорид, который отфильтровывают, дважды промывают этанолом по 2,5 мл и сушат при 100oC/12 мбар.
Выход: 1,4 г (85,7% от теории) гидрохлорида 1-циклопропил-7-([S,S]-2,8-диазабицикло[4.3.0] нон-8-ил)- 5,6,8, -трифтор-1,4-дигидро-4-оксо-3-хинолинкарбоновой кислоты.
Т. пл. > 310oC (с разложением).
[α]
Пример 5.
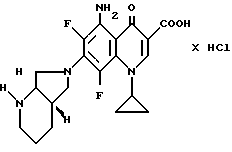
5,2 г (13 ммоль) продукта из примера 4А смешивают в 80 мл пиридина в автоклаве с 15 мл жидкого аммиака и нагревают 12 ч до 130oC. Затем охлаждают, спускают давление в автоклаве, смесь концентрируют, остаток обрабатывают ацетонитрилом в ультразвуковой бане. Нерастворившийся осадок отфильтровывают, остаток растворяют в приблизительно 150 мл воды при нагревании, раствор фильтруют и осаждают гидрохлорид 10 мл полуконцентрированной соляной кислоты, отфильтровывают и сушат при 100oC в воздушном сушильном шкафу. Полученный продукт суспендируют в 100 мл монометилового эфира гликоля при 110 115oC и переводят в раствор добавкой 38 мл полуконцентрированной кислоты. Раствор фильтруют в горячем виде через стеклянный фильтр, охлаждают и отфильтровывают выпавшие кристаллы, промывают этанолом и сушат при 120oC/12 мбар.
Выход: 2,5 г (44 от теории) гидрохлорида 5-амина-1-циклопропил-7-([S,S] -2,8-диазабицикло[4.3.0] нон-8-ил)- 6,8-дифтор-1,4-дигидро-4-оксо-3-хинолинкарбоновой кислоты.
Т. пл. > 335oC (с разложением; темнеет уже ниже 335oC).
[α]
Пример 6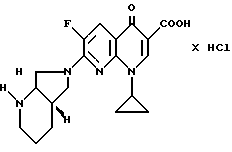
1,4 г (5 ммоль) 7-хлор-1-циклопропил-6-фтор-1,4-дигидро-4- оксо-1,8-нафтиридин-3-карбоновой кислоты перемешивают в 15 мл ацетонитрила с 1,3 г (10,3 ммоль) (+)-[S,S]-2,8-диазабицикло [4.3.0]нонана в отсутствии воды 1 ч при комнатной температуре. Спустя ночь отфильтровывают, промывают ацетонитрилом и очищают путем хроматографии на силикагеле (растворитель: метиленхлорид/метанол/17% -ный водный аммиак 30 8 1; Rf 0,4). Полученную 1-циклопропил-7-([S, S] -диазабицикло[4.3.0] нон-8-ил) -6-фтор-1,4-дигидро-4-оксо-1,8-нафтиридин-3-карбоновую кислоту растворяют в 15 мл полуконцентрированной соляной кислоты, раствор упаривают и остаток размешивают с этанолом. Осадок отфильтровывают, промывают этанолом и сушат при 120oC/12 мбар.
Выход: 960 мг (47 от теории) гидрохлорида 1-циклопропил-7-([S,S]-2,8-диазабицикло[4.3.0] нон-8-ил)-6-фтор- 1,4-дигидро-4-оксо-1,8-нафтиридин-3-карбоновой кислоты.
Т. пл. 345 346oC (с разложением).
[α]
Пример 7.
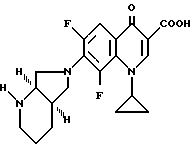
Аналогично примеру 1 получают с (-)-[R,R]-2,8-диазабицикло[4.3.0]нонаном:
А. 1-Циклопропил-7-([R, R] -2,8-диазабицикло[4.3.0] нон-8-ил)- 6,8-дифтор-1,4-дигидро-4-оксо-3-хинолинкарбоновую кислоту.
Т. пл. 247 249oC (с разложением).
Б. Гидрохлорид 1-циклопропил-7-([R, R]-2,8-диазабицикло[4.3.0] нон-8-ил)-6,8-дифтор-1,4-дигидро-4-оксо-3-хинолинкарбоновой кислоты.
Т. пл. 322 326oC (с разложением).
Состав (жидкостная хроматография под давлением): 99,4
Чистота изомера 98,6
[α]
Пример 8.
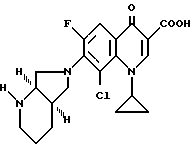
Аналогично примеру 2 получают с (-)-[R,R]-2,8-диазабицикло[4.3.0]нонаном:
А. 8-Хлор-1-циклопропил-7-[R, R] -2,8-диазабицикло[4.3.0] нон-8-ил)-6-фтор-1,4-дигидро-4-оксо-хинолинкарбоновую кислоту.
Т. пл. 192 195oC (с разложением).
Б. Гидрохлорид 8-хлор-1-циклопропил-7-[R, R] -2,8- диазабицикло[4.3.0] нон-8-ил)-6-фтор-1,4-дигидро-4-оксо-3- хинолинкарбоновой кислоты.
Т. пл. 323 324oC (с разложением)
Состав (жидкостная хроматография под давлением): 99,9%
[α]
C20H21CLFN3O3 • HCL (442,3)
Рассчитано: C 54,3; H 5,0; N 9,5; CL 16,0.
найдено: C 54,2; H 5,0; N 9,5; CL 16,1.
Пример 9.
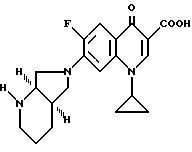
Аналогично примеру 1 получают из 1-циклопропил-6,7-дифтор-1,4- дигидро-4-оксо-3-хинолинкарбоновой кислоты и (-)-[R,R]-2,8-диазабицикло[4.3.0]нонана:
А. 1-Циклопропил-7-([R, R] -2,8-диазабицикло[4.3.0] нон-8-ил)-6-фтор-1,4-дигидро-4-оксо-3-хинолинкарбоновую кислоту.
Т. пл. 254 258oC (с разложением).
Б. 1-Циклопропил-7-([R, R] -2,8-диазабицикло[4.3.0] нон-8-ил)- 6-фтор-1,4-дигидро-4-оксо-3-хинолинкарбоновую кислоту.
Т. пл. выше 320oC разложение.
[α]
Пример 10.
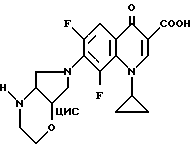
А. 1-Циклопропил-6,8-дифтор-1,4-дигидро-7-(цис-2-окса-5,8- диазабицикло[4.3.0] нон-8-ил)-4-оксо-3-хинолинкарбоновая кислота. 1,43 г (5 ммоль) 1-циклопропил-6,7,8-трифтор-1,4-дигидро-4-оксо-3- хинолинкарбоновой кислоты нагревают с обратным холодильником в течение одного часа в смеси из 15 мл ацетонитрила и 75 мл диметилформамида в присутствии 0,67 г (6 ммоль) 1,4-диазабицикло[2.2.2] октана с 0,74 г (5,4 ммоль) 93%-ного цис-2-окса-5,8-диазабицикло[4.3.0] нонана. Суспензию сгущают, остаток перемешивают с водой, осадок отфильтровывают и сушат при 80oC в вакууме.
Выход: 1,67 г (85,4% от теории).
Т. пл. 210 212oC (с разложением).
Б. Гидрохлорид 1-циклопропил-6,8-дифтор-1,4-дигидро-7- (цис-2-окса-5,8-диазабицикло[4.3.0] нон-8-ил)-4-оксо-3- хинолинкарбоновой кислоты. 1,6 г (4 ммоль) продукта со стадии А растворяют в 120 мл полуконцентрированной соляной кислоты при 60oC, раствор сгущают, остаток промывают этанолом и осадок отфильтровывают и сушат при 90oC в вакууме.
Выход: 1,57 г.
Т. пл. 300 303oC (с разложением).
Состав по жидкостной хроматографии под давлением: 97%
В. Аналогично примеру 10А получают с 1R, 6S-2-окса-5,8-диазабицикло[4.3.0] нонаном 1-циклопропил-6,8-дифтор-1,4-дигидро-7-(1R,6S-2-окса-5,8-диазабицикло [4.3.0] нон-8-ил)-4-оксо-3-хинолинкарбоновую кислоту с т. пл. 204 206oC (с разложением).
Г. Аналогично примеру 10Б получают с бетаином из примера 10В гидрохлорид 1-циклопропил-6,8-дифтор-1,4-дигидро-7- (1R, 6S-2-окса-5,8-диазабицикло[4.3.0]нон-8-ил)-4-оксо-3- хинолинкарбоновой кислоты с т. пл. 324 325oC (с разложением). [α]
Д. Аналогично примеру 10А получают с 1S, 6R-2-окса-5,8-диазабицикло[4.3.0] нонаном 1-циклопропил-6,8-дифтор- 1,4-дигидро-7-(1S, 6R-2-окса-5,8-диазабицикло[4.3.0] нон-8-ил)-4- оксо-3-хинолинкарбоновую кислоту с т. пл. 204 206oC (с разложением).
[α]
Е. Аналогично примеру 10Б получают с бетаином из примера 10Д гидрохлорид 1-циклопропил-6,8-дифтор-1,4-дигидро-7-(1S, 6R-2- окса-5,8-диазабицикло[4.3.0] нон-8-ил)-4-оксо-3-хинолинкарбоновой кислоты с т. пл. 323oC (с разложением).
[α]
Пример 11.
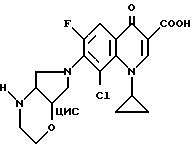
Аналогично примеру 10 получают с 8-хлор-1-циклопропил-6,7-дифтор-1,4-дигидро-4-оксо-3- хинолинкарбоновой кислотой:
А. 8-Хлор-1-циклопропил-6-фтор-1,4-дигидро-7-(цис-2-окса- 5,8-диазабицикло[4.3.0] нон-8-ил)-4-оксо-3-хинолинкарбоновую кислоту. Т. пл. 180 185oC (с разложением).
Б. Гидрохлорид 8-хлор-1-циклопропил-6-фтор-1,4-дигидро-7-(цис- 2-оксо-5,8-диазабицикло[4.3.0]нон-8-ил)-4-оксо-3- хинолинкарбоновой кислоты. Т. пл. 227 232oC (с разложением).
В. 8-Хлор-1-циклопропил-6-фтор-1,4-дигидро-7-(1R, 6S-2- окса-5,8-диазабицикло[4.3.0]нон-8-ил)-4-оксо-3-хинолинкарбоновую кислоту. Т. пл. 186 188oC (с разложением). [α]
Г. Гидрохлорид 8-хлор-1-циклопропил-6-фтор-1,4-дигидро-7- (1R,6S-2-окса-5,8-диазабицико[4.3.0]нон-8-ил)-4-оксо-3- хинолинкарбоновой кислоты. Т. пл. 278 280oC (с разложением). [α]
Д. 8-хлор-1-циклопропил-6-фтор-1,4-дигидро-7-(1S, 6R-2- окса-5,8-диазабицикло[4.3.0]нон-8-ил)-4-оксо-3-хинолинкарбоновую кислоту. Т. пл. 188 190oC (с разложением). [α]
Е. Гидрохлорид 8-хлор-1-циклопропил-6-фтор-1,4-дигидро-7-(1S, 6R-2-окса-5,8- диазабицикло[4.3.0]нон-8-ил)-4-оксо-3-хинолинкарбоновой кислоты. Т. пл. 292 294oC (с разложением). [α]
Пример 12.

Аналогично примеру 10А получают с 1-циклопропил-6,7-дифтор-1,4-дигидро-4-оксо-3-хинолинкарбоновой кислотой:
А. 1-Циклопропил-6-фтор-1,4-дигидро-7-(цис-2-окса-5,8- диазабицикло[4.3.0]нон-8-ил)-4-оксо-3-хинолинкарбоновую кислоту. Т. пл. 246 - 249oC (с разложением, из монометилового эфира гликоля).
Б. 1-Циклопропил-6-фтор-1,4-дигидро-7-(1R, 6S-2-окса-5,8- диазабицикло[4.3.0]нон-8-ил)-4-оксо-3-хинолинкарбоновую кислоту. Т. пл. 243 - 245oC (с разложением).
В. Гидрохлорид 1-циклопропил-6-фтор-1,4-дигидро-7-(1R,6S-2- окса-5,8-диазабицикло[4.3.0] нон-8-ил)-4-оксо-3-хинолинкарбоновой кислоты. Т. пл. 300oC (с разложением). [α]
Пример 13.
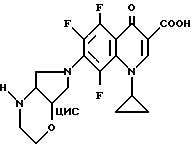
Аналогично примеру 10А получают с 1-циклопропил-5,6,7,8- тетрафтор-1,4-дигидро-4-оксо-3-хинолинкарбоновой кислотой:
А. 1-Циклопропил-5,6,8-трифтор-1,4, -дигидро-7-(цис-2- окса-5,8-диазабицикло[4.3.0]нон-8-ил)-4-оксо-3-хинолинкарбоновую кислоту. Т. пл. 210 216oC (с разложением).
Б. 1-циклопропил-5,6,8-трифтор-1,4-дигидро-7-(1R, 6S-2-окса-5,8- диазабицикло[4.3.0] нон-8-ил)-4-оксо-3-хинолинкарбоновую кислоту. Т. пл. 234 - 237oC (с разложением). [α]
В. 1-Циклопропил-5,6,8-трифтор-1,4-дигидро-7-(1S, 6R-2-окса-5,8- диазабицико[4.3.0] нон-8-ил)-4-оксо-3-хинолинкарбоновую кислоту. Т. пл. 236 - 237oC (с разложением). [α]
Пример 14.
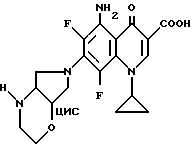
А. 4,1 г (10 ммоль) продукта из примера 13А смешивают в 40 мл пиридина с 5 мл жидкого аммиака и нагревают в автоклаве 10 ч до 130oC. После охлаждения осадок отфильтровывают, промывают водой и сушат при 100oC в воздушном сушильном шкафу. Сырой продукт (2 г) очищают перекристаллизацией из монометилового эфира гликоля (желтые кристаллы).
Выход: 1,3 г (31 от теории) 5-амино-1-циклопропил-6,8-дифтор- 1,4-дигидро-7-(цис-2-окса-5,8-диазабицикло[4.3.0] нон-8-ил)-4- оксо-3-хинолинкарбоновой кислоты.
Т. пл. 233 240oC (с разложением).
Б. Аналогично получают с продуктом из примера 13В 5-амино-1-циклопропил-6,8-дифтор-1,4-дигидро-7-(1R, 6S-2-окса-5,8- диазабицикло[4.3.0]нон-8-ил)-4-оксо-3-хинолинкарбоновую кислоту. Т. пл. 212 - 214oC (с разложением). [α]
В. Аналогично получают с продуктом из примера 13В 5-амино-1-циклопропил-6,8-дифтор-1,4-дигидро-7-(1S, 6R-2-окса-5,8- диазабицикло[4.3.0]нон-8-ил)-4-оксо-3-хинолинкарбоновую кислоту. Т. пл. 213 - 215oC (с разложением). [α]
Масс-спектр: м/е 406 (M+, 95%), 346, 249, 98, 41, 28 (100).
Пример 15.
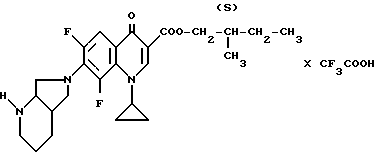
А. 7-(2-трет. -бутоксикарбонил-2,8-диазабицикло[4.3.0]нон-8- ил)-1-циклопропил-6,8-дифтор-1,4-дигидро-4-оксо-3- хинолинкарбоновая кислота. 7,8 г (20 ммоль) 1-циклопропил-7-(2,8-диазабицикло[4.3.0]нон-8-ил)-6,8-дифтор-1,4- дигидро-4-оксо-3-хинолинкарбоновой кислоты растворяют в смеси из 60 мл диоксана и воды (2 1) и 20 мл 1 н. натриевой щелочи и смешивают при охлаждении льдом и перемешивании с 5,24 г (24 ммоль) ди-трет.-бутилового эфира пироугольной кислоты. Перемешивают 1 ч при комнатной температуре и оставляют на ночь. Выпавший осадок отфильтровывают, промывают 250 мл воды и сушат ночь при 50oC в воздушном сушильном шкафу.
Выход: 9,34 г (95,5 от теории).
Т. пл. 216 219oC (с разложением).
Б. 2S-метил-1-бутиловый эфир 7-(2-трет.-бутоксикарбонил-2,8- диазабицикло[4.3.0] нон-8-ил)-1-циклопропил-6,8-дифтор-1,4- дигидро-4-оксо-3-хинолинкарбоновой кислоты. 2,15 г (4,4 ммоль) продукта со стадии А суспендируют в 60 мл смеси тетрагидрофурана и воды (1 1) при комнатной температуре и прибавляют 1,65 г (5 ммоль) карбоната цезия. Оставляют реагировать на 20 мин в ультразвуковой бане при температуре около 40oC, отгоняют около 40 мл растворителя при 40oC/12 мбар и лиофилизируют оставшийся раствор, при этом получают легко растворимую сырую соль цезия: 3,3 г этой сырой соли растворяют в 40 мл диметилформамида и смешивают с 1,4 г S(+)-1-бром-2-метил-бутана и подвергают взаимодействию в течение ночи в ультразвуковой бане при 40 50oC. Полученную суспензию сгущают, остаток смешивают с водой и экстрагируют метиленхлоридом. После сушки над сульфатом натрия раствор сгущают и остаток подвергают хроматографической очистке (силикагель, растворитель: метиленхлорид/метанол 95 5).
Выход: 950 мг (38% от теории).
Т. пл. 72 83oC (с разложением).
В. Трифторацетат (2S-метил-1-бутиловый эфир) 1-циклопропил-7-(2,8-диазабицикло[4.3.0]нон-8-ил)-6,8-дифтор-1,4- дигидро-4-оксо-3-хинолинкарбоновой кислоты. 570 мг (1 ммоль) продукта со стадии Б растворяют в 3 мл трифторуксусной кислоты при комнатной температуре и раствор сгущают при 60oC/12 мбар. Полученное вязкое масло перемешивают с 5 мл диэтилового эфира, при этом выпадает твердый продукт. Его отфильтровывают, промывают эфиром и сушат при 80oC в глубоком вакууме.
Выход: 450 мг (78 от теории).
Т. пл. 214 216oC (с разложением); [α]
Пример 16.
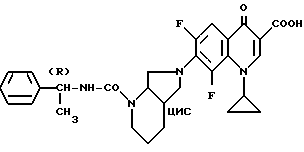
390 мг (1 ммоль) 1-циклопропил-7-(2,8-диазабицикло[4.3.0] нон-8-ил)-6,8-дифтор-1,4-дигидро-4-оксо-3-хинолинкарбоновой кислоты растворяют в растворе 40 мг гидроокиси натрия в 3 мл воды при комнатной температуре в ультразвуковой бане и раствор смешивают при охлаждении льдом с раствором 160 мг (1,1 ммоль) R(+)- -метил-бензил-изоцианата. Выпавший осадок отфильтровывают, промывают диоксаном и сушат при 100oC в глубоком вакууме.
Выход: 530 мг (99 от теории) 1-циклопропил-6,8-дифтор- 1,4-дигидро-4-оксо-7-(2-[1R-фенил-этил-амино-карбонил] -2,8- диазабицикло[4.3.0] нон-8-ил)-3-хинолинкарбоновой кислоты.
Т. пл. 208 210oC (с разложением); [α]
Реакционный продукт разделяют хроматографически на диастереомеры и опять удаляют карбамоильный остаток кислым гидролизом, при этом получают соединения примеров 1 и 7.
Пример 17.
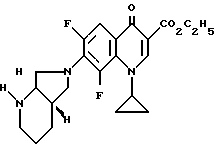
1,52 г (5 ммоль) этилового эфира 1-циклопропил-6,7,8-трифтор-1,4-дигидро-4-оксо-3-хинолинкарбоновой кислоты подвергают взаимодействию в 30 мл ацетонитрила с 550 мг (5 ммоль) 1,4-диазабицикло[2.2.2]октана и 760 мг (6 ммоль) (+)-[S,S]-2,8-диазабицикло[4.3.0]нонана 2 ч при 50oC и 2 ч при 60oC. После охлаждения отфильтровывают полученную суспензию, осадок промывают водой и сушат при 90oC в вакууме.
Выход: 0,99 г (47,5 от теории) этилового эфира 1-циклопропил-7-([S,S]-2,8-диазабицикло[4.3.0] нон-8-ил)-6,8- дифтор-1,4-дигидро-4-оксо-3-хинолинкарбоновой кислоты.
Т. пл. 194 195oC (из ацетонитрила). [α]
Пример 18.
1,4 г (5 ммоль) 9,10-дифтор-2,3-дигидро-3-метил-7-оксо-7Н- пиридо[1,2,3-де] [1,4]бензоксазин-6-карбоновой кислоты подвергают взаимодействию аналогично примеру 1 в смеси из 15 мл ацетонитрила и 7,5 мл диметилформамида с 0,85 г (7,7 ммоль) 1,4-диазабицикло[2.2.2] октана и 0,7 г (5,6 ммоль) (+)-[S,S]-2,8-диазабицикло[4.3.0]нонана.
Выход: 1,24 г (64% от теории) 10-([S,S]-2,8-диазабицикло[4.3.0] нон-8-ил)-9-фтор-2,3-дигидро-3-метил-7-оксо-7Н-пиридо [1,2,3-де] [1,4] бензоксазин-6-карбоновой кислоты.
Т. пл. 265 268oC (с разложением). [α]D = -232,2o (с 0,58, метиленхлорид).
Аналогично получают также 3S-10-([S,S]-2,8-диазабицикло- [4.3.0]нон-8-ил)-9-фтор-2,3-дигидро-3-метил-7-оксо-7Н-пиридо [1,2,3-де] [1,4] бензоксазин-6-карбоновую кислоту.
Пример 19.

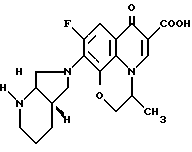
1-Циклопропил-6,7-дифтор-1,4-дигидро-8-метокси-4-оксо-3- хинолинкарбоновую кислоту подвергают взаимодействию аналогично примеру 1 и продукт превращения подвергают хроматографической очистке (силикагель, растворитель: метиленхлорид/метанол/17%-ный водный аммиак 30 8 1).
Получают 1-циклопропил-7-)[S, S] -2,8-диазабицикло[4.3.0]нон- 8-ил)-6-фтор-1,4-дигидро-8-метокси-4-оксо-3-хинолинкарбоновую кислоту с т. пл. 203 208oC (с разложением).
[α]
Пример 20.
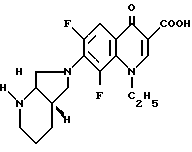
Аналогично примеру 1А подвергают превращению 1-этил-6,7,8-трифтор-1,4-дигидро-4-оксо-3-хинолинкарбоновую кислоту и получают 1-этил-7-([S,S]-2,8-диазабицикло[4.3.0] нон-8-ил)-6,8- дифтор-1,4-дигидро-4-оксо-3-хинолинкарбоновую кислоту с т. пл. 236 239oC (с разложением, из монометилового эфира гликоля).
[α]
Пример 21,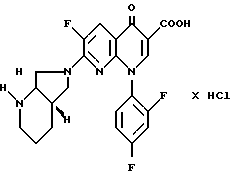
A. Этиловый эфир 7-([S, S]-2,8-диазабицикло[4.3.0]нон-8-ил)- 1-(2,4-дифторфенил)-6-фтор-1,4-дигидро-4-оксо-1,8-нафтиридин-3- карбоновой кислоты. 1,9 г (5 ммоль) этилового эфира 7-хлор-1-(2,4-дифторфенил)-6-фтор-1,4-дигидро-4-оксо-1,6-нафтиридин- 3-карбоновой кислоты перемешивают в 20 мл ацетонитрила в присутствии 560 мг (5 ммоль) 1,4-диазабицикло[2.2.2]октана с 680 мг (5,4 ммоль) [S,S]-2,8-диазабицикло[4.3.0]нонана 3 ч при 10oC. Суспензию отфильтровывают, промывают водой и сушат. Получают 0,35 г продукта. После сгущения маточников, размешивания остатка с водой, выделения нерастворившегося продукта и хроматографической очистки (силикагель, растворитель: дихлорметан/метанол/17%-ный водный аммиак) получают дополнительно 0,7 г продукта.
Общий выход: 1,05 г (44 от теории).
Т. пл. 184 185oC (с разложением). [α]
Б. Гидрохлорид 7-([S,S]-2,8-диазабицикло[4.3.0]нон-8-ил)-1- (2,4-дифторфенил)-6-фтор-1,4-дигидро-4-оксо-1,8-нафтиридин-3-карбоновой кислоты. 0,8 г (1,7 ммоль) продукта со стадии А нагревают в смеси из 10 мл уксусной кислоты и 8 мл полуразбавленной соляной кислоты 4 ч с обратным холодильником. Смесь сгущают, остаток размешивают с небольшим количеством воды, осадок отфильтровывают, промывают охлажденным льдом этанолом и сушат.
Выход: 0,67 г (83 от теории).
Т. пл. 324 326oC (с разложением). [α]
Пример 22.
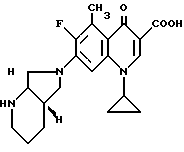
0,56 г (2 ммоль) 1-циклопропил-6,7-дифтор-1,4-дигидро-5-метил- 4-оксо-3-хинолинкарбоновой кислоты нагревают с 0,38 г (3 ммоль) [S,S]-2,8-диазабицикло[4.3.0] нонана и 0,45 г (4 ммоль) 1,4-диазабицикло[2.2.2]октана в 3,5 млдиметилсульфоксида 2 ч до 120oC. После охлаждения растворитель удаляют в глубоком вакууме. Остаток поглощают ацетонитрилом. Отделяют твердое вещество, промывают его ацетонитрилом и сушат при 60 80oC.
Выход: 0,5 г (65 от теории) 1-циклопропил-7-([S,S]02,8-диазабицикло[4.3.0] нон-8-ил)-6-фтор-1,4- дигидро-5-метил-4-оксо-3-хинолинкарбоновой кислоты.
Т. пл. 217 219oC (с разложением), [α]D = -199o (с 0,5, диметилформамид).
Пример 23.
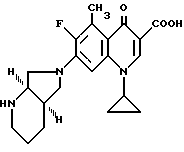
А. 873 мг (3 ммоль) 1-циклопропил-6,7-дифтор-1,4-дигидро-5- метил-4-оксо-3-хинолинкарбоновой кислоты нагревают в смеси из 10 мл ацетонитрила и 5 мл диметилформамида с 1,1 г (10 ммоль) 1,4-диазабицикло[2.2.2]октана и 665 мг (3,3 ммоль) дигидрохлорида 1R,6S-окса-5,8-диазабицикло[4.3.0]нонана 2 ч с обратным холодильником, смесь упаривают, остаток перемешивают с 30 мл воды, осадок отфильтровывают и сушат при 80oC в вакууме.
Выход: 400 мг (34 от теории) 1-циклопропил-6-фтор-1,4- дигидро-5-метил-7-(1R, 6S-2-окса-5,8-диазабицикло[4.3.0] нон-8-ил)- 4-оксо-3-хинолинкарбоновой кислоты.
Т. пл. 213 214oC (с разложением).
Б. 0,4 г бетаина со стадии А растворяют в 5 мл полуконцентрированной соляной кислоты при комнатной температуре, раствор сгущают и остаток размешивают с 3 мл этанола. Осадок отфильтровывают и сушат при 80oC/12 мбар.
Выход: 290 мг (66% от теории) гидрохлорида 1-циклопропил-6-фтор-1,4-дигидро-5-метил-7-(1R, 6S-2-окса-5,8- диазабицикло[4.3.0]нон-8-ил)-4-оксо-3-хинолинкарбоновой кислоты.
Т. пл. 305 308oC (с разложением), [α]
Пример 24.
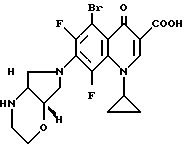
362 г (1 ммоль) 5-бром-1-циклопропил-6,7,8-трифтор-1,4-дигидро-4-оксо-3- хинолинкарбоновой кислоты нагревают в смеси из 3 мл ацетонитрила и 1,5 мл диметилформамида с 220 мг (2 ммоль) 1,4-диазабицикло[2.2.2]октана и 220 мг (1,1 ммоль) дигидрохлорида 1S,6R-2-окса-5,8-диазабицикло[4.3.0]нонана 1,5 ч с обратным холодильником.
Суспензию охлаждают, осадок отфильтровывают, перемешивают с 30 мл воды и сушат при 90oC в глубоком вакууме.
Выход: 320 мг (68% от теории) 5-бром-1-циклопропил-6,8-дифтор- 1,4-дигидро-7-(1S, 6R-2-окса-5,8-диазабицикло[4.3.0] нон-8-ил)-4-оксо- 3-хинолинкарбоновой кислоты.
Т. пл. 263 264oC (с разложением), [α]
Пример 25.
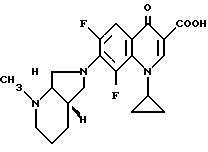
Аналогично примеру 1 получают с [S,S]-2-метил-2,8-диазабицикло[4.3.0] нонаном:
А. 1-Циклопропил-6,8-дифтор-1,4-дигидро-7-([S, S]-2-метил-2,8- диазабицикло[4.3.0]нон-8-ил)-4-оксо-3-хинолинкарбоновую кислоту.
Т. пл. 230 233oC (с разложением, перекристаллизация из монометилового эфира гликоля).
Б. Гидрохлорид 1-циклопропил-6,8-дифтор-1,4-дигидро-7- ([S,S]-2-метил-2,8-диазабицикло[4.3.0]нон-8-ил)-4-оксо-3- хинолинкарбоновой кислоты.
Т. пл. 258 260oC (с разложением), [α]
Пример 26.
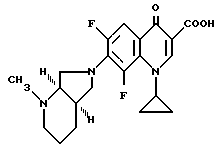
Аналогично примеру 1 получают с [R,R]-2-метил-2,8-диазабицикло[4.3.0] нонаном:
А. 1-Циклопропил-6,8-дифтор-1,4-дигидро-7-([R, R]-2-метил- 2,8-диазабицикло[4.3.0]нон-8-ил)-4-оксо-3-хинолинкарбоновую кислоту.
Т. пл. 228 230oC (с разложением, перекристаллизация из моноэтилового эфира гликоля).
Б. Гидрохлорид 1-циклопропил-6,8-дифтор-1,4-дигидро-7- 9[R,R]-2-метил-2,8-диазабицикло[4.3.0]нон-8-ил)-4-оксо-3- хинолинкарбоновой кислоты.
Т. пл. 258 260oC (с разложением), [α]
Пример 27.
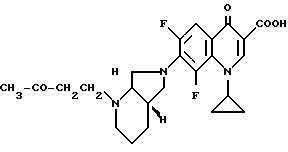
1,95 г (5 ммоль) продукта из примера 1А нагревают в 50 мл этанола с 2,1 г (30 ммоль) метилвинилкетона 4 ч с обратным холодильником. Смесь сгущают, остаток перемешивают с водой, осадок отфильтровывают, промывают этанолом и сушат при 100oC/12 мбар.
Выход: 2,1 г (91,5% от теории) 1-циклопропил-6,8-дифтор-1,4- дигидро-4-оксо-7-([S, S] -2-[3-оксо-1-бутил] -2,8-диазабицикло- [4.3.0]нон-8-ил)-3-хинолинкарбоновой кислоты.
Т. пл. 181 183oC (с разложением, перекристаллизация из монометилового гликоля), [α]
Пример 28.
1,95 г (5 ммоль) продукта из примера 1А нагревают в 30 мл диметилформамида с 1,0 г (10,8 ммоль) хлорацетона и 1,3 г (13 ммоль) триэтиламина 3 ч при 50 80oC. Раствор сгущают, остаток перемешивают с водой (pH 6), нерастворившийся осадок отфильтровывают, промывают водой и сушат в воздушном сушильному шкафу при 100oC.
Выход сырого продукта: 1,3 г; после перекристаллизации из монометилового эфира гликоля:
выход: 1,12 г (50% от теории) 1-циклопропил-6,8-дифтор-1,4- дигидро-4-оксо-7-([S, S] -2-[2-оксопропил] -2,8-диазабицикло- [4.3.0]нон-8-ил)-3-хинолинкарбоновой кислоты.
Т. пл. 181 184oC (с разложением),
[α]
Пример 29.
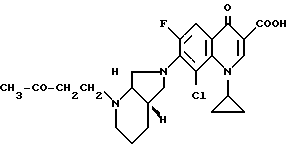
А. Аналогично примеру 27 подвергают превращению продукт из примера 2А и получают 8-хлор-1-циклопропил-6-фтор-1,4-дигидро-4- оксо-7-([S,S]-2-[3-оксо-1-бутил]-2,8-диазабицикло[4.3.0]нон-8-ил)- хинолинкарбоновую кислоту с т. пл. 107 109oC.
[α]
Состав: 99,2%-ный (жидкостная хроматография высокого давления).
Б. Аналогично получают с 8-хлор-1-циклопропил-7-(цис-2,8- диазабицикло[4.3.0] нон-8-ил)-6-фтор-1,4-дигидро-4-оксо-3- хинолинкарбоновой кислотой рацемическую 8-хлор-1-циклопропил-6-фтор-1,4-дигидро-4-оксо-7-(цис-2-[3-оксо-1- бутил]-2,8-диазабицикло[4.3.0]нон-8-ил)-3-хинолинкарбоновую кислоту с т. пл. 124 125oC.
Пример 30.
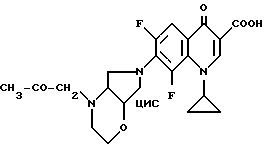
1,56 г (4 ммоль) продукта из примера 10А смешивают в 30 мл диметилформамида с 0,82 г (8,8 моль) хлорацетона и 1,05 г (10,4 ммоль) триэтиламина и нагревают 3 ч при 50 80oC. Полученный желтый раствор сгущают при 80oC/15 мбар, маслянистый остаток перемешивают с водой до тех пор, пока он не затвердеет. Твердый продукт отфильтровывают, промывают водой и перекристаллизовывают из монометилового эфира гликоля.
Выход: 830 мг (47% от теории) 1-циклопропил-6,8-дифтор- 1,4-дигидро-4-оксо-7-(цис-5-[2-оксопропил] -2-окса-5,8- диазабицикло[4.3.0]нон-8-ил)-3-хинолинкарбоновой кислоты.
Т. пл. 192 193oC (с разложением).
Пример 31.
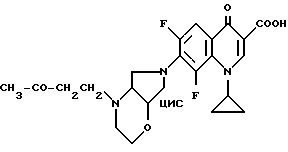
1,56 г (4 ммоль) продукта из примера 10А нагревают в 50 мл этанола с 1,8 г (25,6 ммоль) метилвинилкетона 3 ч с обратным холодильником. Суспензию сгущают при 90oC/12 мбар, остаток перемешивают с водой и перекристаллизовывают из монометилового эфира гликоля.
Выход: 1,33 г (72% от теории) 1-циклопропил-6,8-дифтор-1,4- дигидро-4-оксо-7-(цис-5-[3-оксо-1-бутил] -2-окса-5,8-диазабицикло [4.3.0]нон-8-ил)-3-хинолинкарбоновой кислоты.
Т. пл. 188 189oC (с разложением).
Пример 32.
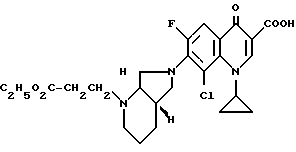
1,95 г (4,8 ммоль) продукта по примеру 2А нагревают в 30 мл монометилового эфира гликоля с 3 г (30 ммоль) этилового эфира акриловой кислоты 2 ч с обратным холодильником. Смесь упаривают, остаток перемешивают с водой, осадок отфильтровывают, сушат (выход сырого продукта: 1,9 г) и перекристаллизовывают из монометилового эфира гликоля.
Выход: 1,45 г (60% от теории) 8-хлор-1-циклопропил-7- ([S,S]-2-[2-эткосикарбонил-этил] -2,8-диазабицикло[4.3.0] нон-8-ил)- 6-фтор-1,4-дигидро-4-оксо-3-хинолинкарбоновой кислоты.
Т. пл. 117 118oC (с разложением), [α]
Состав: 99,6%-ный (жидкостная хроматография высокого давления).
Пример 33.
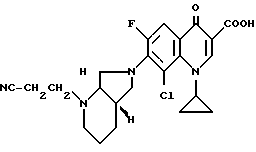
1,95 г (4,8 ммоль) продукта по примеру 2А нагревают в 30 мл этанола с 0,8 г (15 ммоль) акрилнитрила 5 ч с обратным холодильником. Смесь упаривают, остаток перемешивают с водой, сушат (выход сырого продукта: 1,9 г) и перекристаллизовывают из монометилового эфира гликоля.
Выход: 1,6 г (73% от теории) 8-хлор-7-([S,S]-2-[2-цианоэтил]-2,8-диазабицикло[4.3.0] нон-8-ил)- 1-циклопропил-6-фтор-1,4-дигидро-3-хинолинкарбоновой кислоты.
Т. пл. 153 155oC (с разложением),
[α]
Состав: 96%-ный (жидкостная хроматография высокого давления).
Масс-спектр: м/е 458 (M+), 250, 149 (100% C9H13N2), 110,49
Пример 34.
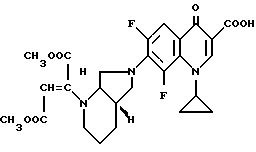
1,95 г (5 ммоль) продукта по примеру 1А нагревают в 60 мл этанола с 1,2 г (8 ммоль) диметилового эфира ацетилендикарбоновой кислоты 2 ч с обратным холодильником. Суспензию сгущают, остаток перемешивают с водой, осадок отфильтровывают и сушат. Сырой продукт (2,3 г) перекристаллизовывают из смеси монометилового эфира гликоля и диметилформамида.
Выход: 2 г (74% от теории) 1-циклопропил-7-[2-(1,2-бис-метоксикарбонил-винил)-1S, 6R-2,8- диазабицикло[4.3.0]нон-8-ил]-6,8-дифтор-1,4-дигидро-4-оксо-3- хинолинкарбоновой кислоты.
Т. пл. 262 264oC (с разложением), [α]
Пример 35.
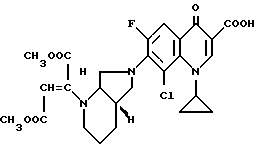
Аналогично примеру 34 продукт по примеру 2А подвергают превращению с диметиловым эфиром ацетилендикарбоновой кислоты. Получают с выходом 87% 8-хлор-циклопропил-7-[2-(1,2-бис-метоксикарбонил-винил)- 1S, 6S-2,8-диазабицикло[4.3.0]нон-8-ил]-6-фтор-1,4-дигидро-4-оксо-3- хинолинкарбоновую кислоту с т. пл. 210 212oC (с разложением), [α]
Пример 36.
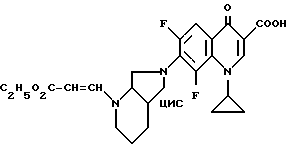
780 мг (2 ммоль) 1-циклопропил-7-(цис-2,8-диазабицикло [4.3.0]нон-8-ил)-6,8-дифтор-1,4-дигидро-4-оксо-3- хинолинкарбоновой кислоты нагревают в 15 мл этанол с 500 мг (5 ммоль) этилового эфира пропиоловой кислоты в течение 1 ч с обратным холодильником. Суспензию охлаждают, осадок отфильтровывают, промывают 25 мл этанола и сушат при 0oC в глубоком вакууме.
Выход: 880 мг (90% от теории) 1-циклопропил-7-[2-(транс-2-этоксикарбонил-винил)-цис-2,8- диазабицикло[4.3.0] нон-8-ил]-6,8-дифтор-4-оксо-3-хинолинкарбоновой кислоты.
Т. пл. 244 246oC
Аналогично примеру 36 получают приведенные в табл. 3 и 4 соединения формулы (I).
Пример 48.
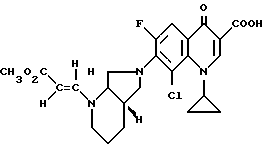
Аналогично примеру 36 подвергают превращению 8-хлор-1-цикопропил-7-9[S, S] -2,8-диазабицикло[4.3.0] нон-8-ил)- фтор-1,4-дигидро-4-оксо-3-хинолинкарбоновую кислоту с метиловым эфиром пропиоловой кислоты в этаноле или метаноле и получают 8-хлор-1-циклопропил-6-фтор-1,4-дигидро-7-[2-(транс-2- метоксикарбонил-винил)-[S, S] -2,8-диазабицикло-[4.3.0]нон-8-ил)-4- оксо-3-хинолинкарбоновую кислоту с т. пл. 220 222oC (с разложением), [α]
Пример 49.

407,5 г (1 ммоль) 8-хлор-1-циклопропил-6-фтор-1,4-дигидро- 7-([1S,6R-2-окса-5,8-диазабицикло[4.3.0] нон-8-ил)-4-оксо-3- хиноликарбоновой кислоты (из примера 11Д) нагревают в 10 мл метанола с 210 мг (2,5 ммоль) метилового эфира пропиоловой кислоты 1 ч с обратным холодильником. Сгущают и перекристаллизовывают выделенный сырой продукт (450 мг) из 4 мл ацетонитрила.
Выход: 8-хлор-1-циклопропил-6-фтор-1,4-дигидро-7-[5-(транс-2- метоксикарбонил-винил)-1S, 6R-2-окса-5,8-диазабицикло[4.3.0] нон- 8-ил)-4-оксо-3-хинолинкарбоновая кислота.
Т. пл. 153 156oC (с разложением), [α]
Пример 50.
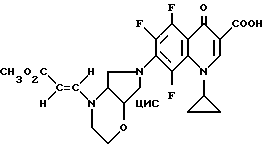
Аналогично примеру 49 подвергают превращению соединение по примеру 13А и получают 1-циклопропил-5,6,8-трифтор-1,4- дигидро-7-[5-(транс-2-метоксикарбонил-винил)-цис-2-окса-5,8- диазабицикло[4.3.0]нон-8-ил]-4-оксо-3-хиноликарбоновую кислоту с т. пл. 169 - 170oC (с разложением, из монометилового эфира гликоля).
Пример 51.
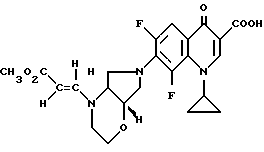
Аналогично примеру 49 подвергают превращению соединение по примеру 10Д и получают 1-циклопропил-6,8-дифтор-1,4-дигидро-7- [5-(транс-2-метоксикарбонил-винил)-1S, 6R-2-окса-5,8-диазабицикло [4.3.0]нон-8-ил]-4-оксо-хинолинкарбоновую кислоту с т. пл. 230 234oC (с разложением, из монометилового эфира гликоля).
[α]
Пример 52.
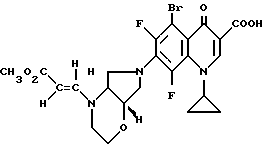
Аналогично примеру 49 подвергают взаимодействию соединение по примеру 24 и получают 5-бром-1-циклопропил-6,8-дифтор-1,4-дигидро-7-[5-(транс-2- метоксикарбонил-винил)-1S, 6R-2-окса-5,8-диазабицикло[4.3.0] нон-8-ил] -4-оксо-3-хинолинкарбоновую кислоту с т. пл. 158 160oC (с разложением, из изопропанола).
[α]
Пример 53.
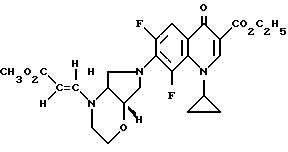
Аналогично примеру 36 подвергают превращению соединение по примеру 17 и получают этиловый эфир 1-циклопропил-7-[2-(транс-2-этоксикарбонил-винил)-1S, 6S-2,8- диазабицикло[4.3.0] нон-8-ил] -6,8-дифтор-1,4-дигидро-4-оксо-3- хинолинкарбоновой кислоты с т.пл. 168 169oC.
Пример 54.
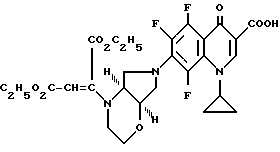
818 мг (2 ммоль) 1-циклопропил-5,6,8-трифтор-1,4-дигидро-7- (1R,6S-2-окса-5,8-диазабицикло[4.3.0] нон-8-ил)-4-оксо-3- хинолинкарбоновой кислоты (по примеру 13Б) смешивают в 15 мл этанола с 680 г (4 ммоль) диэтилового эфира ацетилендикарбоновой кислоты и обрабатывают 1 ч при 30oC в ультразвуковой бане. Суспензию отфильтровывают, промывают этанолом и сушат при 70oC в глубоком вакууме.
Выход: 890 мг (77% от теории) 1-циклопропил-7-[5-(1,2-бис-этоксикарбонил-винил)-1R, 6S-2-окса- 5,8-диазабицикло[4.3.0] нон-8-ил)-5,6,8-трифтор-1,4-дигидро-4- оксо-3-хинолинкарбоновой кислоты.
Т. пл. 220 222oC (с разложением, из монометилового эфира гликоля), [α]
Пример 55.
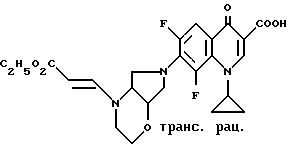
Аналогично примеру 36 подвергают превращению 1-циклопропил- 6,8-дифтор-1,4-дигидро-7-(транс-2-окса-5,8-диазабицикло[4.3.0] нон-8-ил)-4-оксо-3-хинолинкарбоновую кислоту и получают 1-циклопропил-7-[5-(транс-2-этоксикарбонил-винил] -транс-2- окса-5,8-диазабицикло[4.3.0]нон-8-ил]-6,8-дифтор-1,4 дигидро-4-оксо-3-хинолинкарбоновую кислоту.
Т. пл. 266 268oC (с разложением, из монометилового эфира гликоля).
Пример 56.
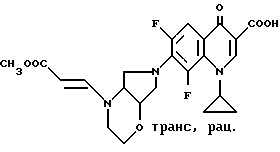
Аналогично примеру 36 подвергают превращению 1-циклопропил- 6,8-дифтор-1,4-дигидро-7-(транс-2-окса-5,8-диазабицикло[4.3.0] нон-8-ил)-4-оксо-3-хинолинкарбоновую кислоту с метиловым эфиром пропиоловой кислоты и получают 1-циклопропил-7-[5-транс-2-метоксикарбонил-винил)-транс-2-окса- 5,8-диазабицико[4.3.0]нон-8-ил]-6,8-дифтор-1,4-дигидро-4-оксо-3- хинолинкарбоновую кислоту.
Т. пл. 275 277oC (с разложением).
Пример 57. Известными приемами готовят таблетки состава, мг: 500 соединения примера 19, 55 микрокристаллической целлюлозы, 72 влажного кукурузного крахмала, 30 поперечно сшитого поливинилпирролидона, 5 двуокиси кремния и 5 стеарата магния.
Пример 58. Известными приемами готовят инфузионный раствор, состоящий из 1,27 г соединения примера 19, 1,45 г молочной кислоты, 1,80 г 2-М. натрового щелока (для установления pH 3,6), 1,37 маннита и воды до 100 мл.
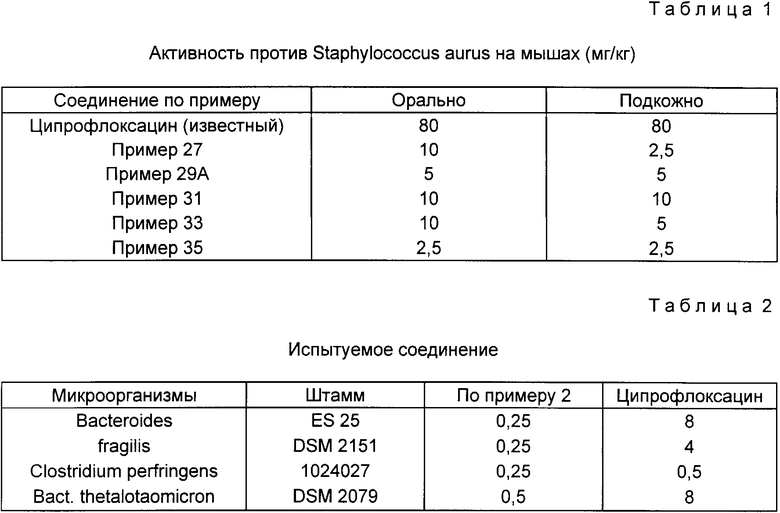

Объектом изобретения являются производные хинолон- и нафтиридонкарбоновой кислоты формулы (I)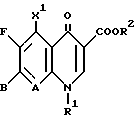
в которой A означает CH, CF, CCL, C - OCH3, C - CH3, N;
X1 - водород, галоген, NH2, CH3;
R1 - алкил, содержащий 1 - 3 атома углерода, FCH2CH2-, циклопропил, фенил, который может быть от одного до трех раз замещен галогеном, или
A и R1 вместе могут означать мостик структуры C - O - CH2 - CH(CH)3
R2 - водород, алкил, содержащий 1 - 3 атома углерода, который может быть замещен гидроксигруппой, галогеном или аминогруппой, или 5-метил-2-оксо 1,3-диоксол-4-ил-метил;
B - остаток формул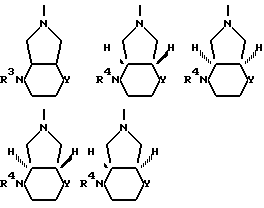
где Y означает O или CH2;
R3 - оксоалкил, содержащий 2 - 5 атома углерода, CH2 - CO - C6H5, CH2CH2CO2R', R'O2C - CH = C - CO2R', -CH = CH - CO2R' или CH2CH2 - CN,
где R' - водород или алкил, содержащий 1 - 3 атома углерода;
R4 - водород, алкил, содержащий 1 - 3 атома углерода, оксоалкил, содержащий 2 - 5 атомов углерода, CH2 - CO - C6H5, CH2CH2CO2R', R'O2C - CH = CO2R', -CH = CH - CO2-R или CH2CH2 - CN или 5-метил-2-оксо-1,3 диоксол-4-ил-метил, где R' - водород или алкил, содержащий 1 - 3 атома углерода, в виде смеси изомеров или отдельных изомеров, их гидраты и соли. Другими объектами изобретения являются способ получения соединений формулы (I) и содержащее их лекарственное средство. 2 с. и 2 з.п. ф-лы, 1 табл., 1 ил.
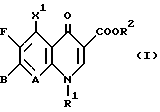
где А СН, CF, CCl, C-OCH3, N;
Х1 водород, галоген, NH2, CH3;
R1 алкил, содержащий 1 3 атома углерода, циклопропил, фенил, который может быть от одного до трех раз замещен галогеном, или А и R1 вместе могут означать мостик структуры С-О-СН2-СН(СН3)-;
R2 водород, алкил, содержащий 1 3 атома углерода,
В остаток формулы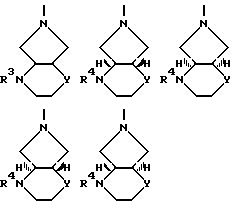
где Y означает O или СН2;
R3 оксоалкил, содержащий 2 5 атомов углерода, СН2СН2СО2R', 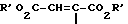 , -CH=CH-CO2R' или СН2СН2-СN,
, -CH=CH-CO2R' или СН2СН2-СN,
где R' водород или алкил, содержащий 1 3 атома углерода,
R4 водород, алкил, содержащий 1 3 атома углерода, оксоалкил, содержащий 2 5 атомов углерода СН2СН2СО2R', 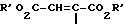 , -СН=СН-СО2-R' или СН2СН2-СN,
, -СН=СН-СО2-R' или СН2СН2-СN,
где R' имеет вышеуказанное значение,
в виде смеси изомеров или отдельных изомеров, их гидраты и соли.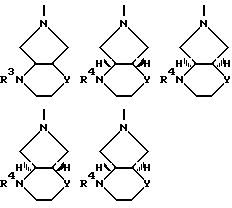
где Y означает О или СН2,
R3 СН2-СО-СН3, СН2СН2-СО -СН3, СН2СН2СО2R', 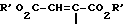 , -СН= СН-СО2 R' или СН2СН2-СN, где R' означает алкил, содержащий 1 2 атома углерода,
, -СН= СН-СО2 R' или СН2СН2-СN, где R' означает алкил, содержащий 1 2 атома углерода,
R4 водород, алкил, содержащий 1 3 атома углерода, СН2-СО-СН3, СН2СН2СО-СН3, СН2СН2СО2R', 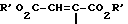 , -СН СН-СО2R' или СН2СН2-СN, где R' имеет вышеуказанное значение,
, -СН СН-СО2R' или СН2СН2-СN, где R' имеет вышеуказанное значение,
в виде смеси изомеров или отдельных изомеров, их гидраты и соли.
где Y означает О или СН2;
R4 водород, алкил, содержащий 1 3 атома углерода, СН2-СО-СН3, СН2СН2СО -СН3, СН2СН2 СО2R', 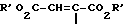 -СН СН СО2R' или СН2СН2-СN, где R' означает алкил, содержащий 1 2 атома углерода,
-СН СН СО2R' или СН2СН2-СN, где R' означает алкил, содержащий 1 2 атома углерода,
в виде смеси изомеров или отдельных изомеров и их гидраты и соли.
Приоритет по признакам:
10.01.92. Все значения А, В, Х1, R1, R2, R3, R' R4 водород, СН2-СО-СН3, СН2СН2СО-СН3, СН2СН2СО2R', R'О2С-СН= С-СО2R', -СН=ОН-СО2R' или СН2СН2-СN; 11.01.93. R4 алкил, содержащий 1 3 атома углерода.
| ЕР, заявка N 023088, кл | |||
| Устройство для сортировки каменного угля | 1921 |
|
SU61A1 |

