Изобретение относится к способу выделения энантиомеров из рацемической смеси противоточной экстракцией с применением по меньшей мере двух жидкостей, по меньшей мере одна из которых хиральная или содержит хиральное вспомогательное средство. Такой способ известен из статьи Jakeuchi et al, b Sep. Sci. and Jechnol., 25(7 &, 8), 941 - 951 (1990). В этой статье описан следующий способ: смесь энантиомеров, а именно DL-валин, разделяют на D-валин и L-валин соответственно, противоточной экстракцией при помощи по меньшей мере двух несмешиваемых жидкостей различной хиральности.
С этой целью в случае DL-валина применяют систему для энантиоселективной экстракции растворителями, состоящую из н-бутанола и воды, содержащей N-н-алкил-L-пролин или N-н-алкил-L-гидроксипролин и ионы меди (II).
Недостаток способа, описанного в этой статье, состоит в том, что может быть такое отклонение от 1 в распределении разделяемых энантиомеров между двумя несмешиваемыми жидкостями, которое может вызвать увеличение числа основных технических проблем при его проведении. Хотя можно получить подходящие результаты при применении известного способа в препаративных целях, однако, его применение в технических масштабах все же требует дальнейшей значительной разработки. Кроме того, для разделения других энантиомеров нужно предложить новые системы для энантиоселективной экстракции растворителями.
Поэтому существует большая практическая потребность в разработке способа, который позволит эффективно разделить также другие группы энантиомеров. Это относится в равной мере к пестицидам, душистым веществам и большому числу фармацевтических препаратов, где отсутствие соответствующего способа разделения обозначает, что в некоторых случаях неизвестно, различаются или вообще известные энантиомерные соединения в эффективности действия. В случае некоторых фармацевтических прерпаратов, состоящих из рацемической смеси, известно, что один оптический изомер будет обладать конкретной желаемой физиологической активностью, тогда как другой оптический изомер во многих случаях не только будет неактивным, но может даже вызывать нежелательные вредные побочные действия. Поскольку все более необходимы знания о таких побочных действиях, самоочевидна необходимость разработки способа стереоселективного получения коммерческих фармацевтических продуктов.
Настоящее изобретение теперь предлагает способ, при помощи которого эту необходимость можно в наибольшей части удовлетворить.
Изобретение характеризуется тем, что указанные в первом абзаце известные типы жидкостей способа разделения полностью смешиваются и разделены друг от друга фазой, с которой они не смешиваются.
Новый предложенный способ имеет двойное преимущество: во-первых, стадию разделения двух частично смешиваемых фаз можно опустить, и, во-вторых, способ настоящего изобретения позволяет создать большое число равновесных ступеней в экстракционной колонне.
В соответствии с изобретением подходящие результаты можно получить при помощи способа, в котором разделяющую, несмешиваемую фазу вводят в твердый носитель.
Этот твердый носитель может состоять из пористого материала или твердой сплошной мембраны.
В качестве разделяющей несмешиваемой фазы рекомендуется применять жидкость. Полярность этой жидкости обычно противоположна полярности двух экстрагирующих жидкостей.
Разделение энантиомерных смесей с помощью жидкости, с которой они не смешиваются, было уже предложено в WO-91/17816. В отличие от способа настоящего изобретения, разделение в этом изобретении осуществляют не экстракцией с применением двух жидкостей, одна из которых хиральная или содержит хиральное вспомогательное средство, а введением хирального носителя в несмешиваемую жидкость.
Способ, предложенный в этом изобретении, имеет недостаток, выражающийся в том, что требуется большое число стадий для хорошо выполненного разделения, поэтому, как можно ожидать, применение его для разделения в коммерческих масштабах вызовет проблемы.
Подходящие результаты можно подобным образом получить при применении газа в качестве разделяющей фазы. В частности, когда разделяемые энантиомерные материалы летучие, заманчиво применять заполненный газом твердый пористый носитель. В качестве летучих энантиомерных материалов можно указать несколько душистых веществ и вкусовых веществ.
Твердые носители можно изготовлять из неорганического или органического материала.
Примеры неорганических носителей включают α - оксид алюминия, γ - оксид алюминия, уголь, керамический материал или их комбинации, например, оксид алюминия на носителе из пористого угля.
Возможные примеры твердых носителей из органических материалов, которые можно применять в соответствии с изобретением, включают микропористые, а также сплошные мембраны. Минимальный размер пор в первых мембранах определяется гидродинамическим объемом молекул переносимого вещества. Следует отметить, что, как правило, минимальный размер пор должен превышать гидродинамический объем переносимого вещества. На практике размер пор всегда будет меньше 0,1 мм, предпочтительно менее 10 мкм. Если применяют твердые, сплошные мембраны, разделяемые вещества должны растворяться в них.
Форма мембраны не имеет существенного значения. В одном возможном примере осуществления изобретения мембрана имеет форму ровной пластины, разделяющей жидкие фазы. В другом примере две мембраны в форме пленок свернуты спирально вокруг друг друга таким образом, чтобы между двумя мембранами оставалось достаточное пространство. Ввиду основных технических преимуществ предпочтительно применение мембран в виде полых волокон.
В частности, если полярность экстрагирующих жидкостей противоположна полярности стороны мембраны, обращенной к этим жидкостям, то приемлемые результаты можно получить с применением способа, в котором твердому носителю, в зависимости от того, обладает ли жидкость, текущая вокруг (около) него, полярными или неполярными свойствами, придают гидрофильность или гидрофобность химическим путем, например прививанием акрилатного соединения.
Предпочтение здесь отдается способу, в котором твердому носителю, в зависимости от того, имеет ли текущая вокруг него жидкость полярные или неполярные свойства, придают гидрофильность или гидрофобность физической модификацией, например, обработкой поверхностно-активным соединением.
Для получения мембран, применяемых в настоящем изобретении, применяют как нетермопластичные полимеры, например купрофан, ацетат целлюлозы, триацетат целлюлозы, нитрат целлюлозы, политетрафторэтилен, полиакрилонитрил или (регенерированная) целлюлоза, так и термопластичные материалы, например полиолефины, конденсационные полимеры, окислительные полимеры и их смеси. Примеры полимеров, пригодных для изготовления мембран, включают полиэтилен низкого давления и высокого давления, полипропилен, полистирол, поливинилхлорид, поливинилиденхлорид, терполимер акрилонитрила, бутадиена и стирола, сополимер стирола и акрилонитрила, сополимер стирола и бутадиена, поли-(4-метилпентен-1), полибутен, поливинилбутираль, хлорированный полиэтилен, поливинилацетат, поливиниловый спирт, полиметилметакрилат, полиимид, поливинилдисульфид, полифениленоксид, полиэтилентерефталат, полибутилентерефталат, полиарамид, сополимеры простых и сложных эфиров на основе бутилентерафталата и полиэтиленоксидгликоля, имеющего молекулярную массу в приделах 800 - 6000, полиамид 6, полиамид 6,6, полиамид 11, полиамид 12, поликарбонат, полиэфирокарбамид, полипиперазин, полипиперазинамид, поливинилпирролидон, полиэфиросульфон, полисульфон и полидиметилсилоксаны (PDMS).
Получение микропористых мембран из термопластичных полимеров описано в патенте MSA-4098 901. Получение мембран в виде полых волокон из термопластичных полимеров описано в патенте USA-4 564 488.
Очень благоприятные, вплоть до настоящего времени, результаты были получены также с применением полых волокон целлюлозы.
Мембраны из полых волокон обычно приготовляют в виде модуля из полых волокон. Здесь можно выделить два различных варианта. В первом варианте одна хиральная фаза течет через полые волокна, а другая течет вокруг (около) них. Другой вариант, описанный Sengupta et al. , b Sep. Sci. and Teсhnol/23 (128<13) pp. 1735-1751, 1988, характеризуется тем, что одна хиральная фаза течет через одну часть волокон, тогда как другая хиральная фаза течет через другую часть. Пространство между волокнами в этом случае является частью разделяющей фазы. В зависимости от гидрофильного или гидрофобного характера волокон и от полярности хиральной фазы стенки волокон заполняются хиральными жидкостями или разделяющей, несмешиваемой фазой.
В частности, когда разделяемые энантиомерные вещества летучие, может быть заманчиво применение мембраны, заполненной газом. Примеры летучих энантиомерных веществ включают различные душистые вещества и вкусовые вещества. Когда последние, например, растворимы в воде, можно применять микропористую гидрофобную мембрану, поры которой заполнены газом.
В соответствии с настоящим изобретением предпочтение отдано мембранной фазе с минимальной толщиной. Толщина мембраны должна быть в пределах от 0,1 мкм до 1 мм, предпочтительно в пределах от 1 мкм до 100 мкм. Когда применяют модуль с полыми волокнами, где одна хиральная фаза течет через одну часть волокон, а другая хиральная фаза течет через другую часть, слой несмешиваемой фазы занимает место между двумя типами волокон, иногда увеличивая толщину за счет двойной толщины стенок пористого носителя.
По меньшей мере одна из экстрагирующих жидкостей должна быть хиральной или должна содержать хиральное вспомогательное средство. Предпочтителен способ, в котором каждая из экстрагирующих жидкостей содержит хиральное вспомогательное средство, причем каждое из этих вспомогательных средств имеет отличающееся от другого зеркальное изображение.
Что касается экстрагирующих жидкостей, то предпочтение отдано способу, в котором каждая из экстрагирующих жидкостей имеет в основе один и тот же растворитель.
Благоприятные результаты можно получить, когда экстрагирующие жидкости являются полярными жидкостями, более конкретно, водными жидкостями. В этом случае в качестве разделяющей фазы предпочтительно применение неполярной жидкости или газа.
Очень благоприятные результаты получали при применении неполярных органических, несмешиваемых с водой или частично смешиваемых с водой жидкостей в качестве экстрагирующих жидкостей. В этом случае рекомендуется применять полярную жидкость, более конкретно водную жидкость в качестве разделяющей фазы.
Примеры ахиральных экстрагирующих жидкостей, пригодных для применения по способу настоящего изобретения, включают: воду, трихлорфторметан, хлороформ, четыреххлористый углерод, трихлорэтилен, дихлорметан или различные углеводородные соединения группы (цикло)алканов, имеющих по меньшей мере 5 атомов углерода, спиртов, имеющих по меньшей мере 6 атомов углерода, (цикло)алканонов, имеющих по меньшей мере 5 атомов углерода, и алкенов, имеющих по меньшей мере 6 атомов углерода. Примеры подходящих органических соединений включают н-бутилбензол, о-н-бутилтолуол, п-цимол, 1,4-диэтилбензол, дифенил, дифенилметан, этилбензол, o-этилтолуол, изобутилбензол, изопропилбензол, 1-метилнафталин, н-нонилбензол, н-пропилбензол, м-пропилтолуол, o-пропилтолуол, дифениловый эфир, o-ксилол, ди-( α -метилбензиловый)эфир, 2-этил-2-метил-1,3-диоксолан, 4-метилтетрагидропиран, α -метилбензилдиметиламин, п-винилтолуол, бромбензол, п-хлорстирол, o-дибромбензол, п-дихлорбензол, перхлорэтилен, 1,1,1-трихлорэтан, тиациклопентан, N-этилморфолин, N-изопропилморфолин, нитробензол, нитроэтан, триглицерид линолевой кислоты, метилакрилат, хлористый винил, бензилацетон, метилизопропенилкетон, двухлористый пропилен, N-этиланилин, этилбензоат, метакриловый альдегид, гескаметоксиметилмеламин, тиациклопентан, диэтилфталат, бромхлорметан, этилацетоацетат, циклопентанон, 1,2-дихлорэтан, тетрагидрофуран, ацетофенон, бензиламин, тиофен, аминодифенилметан, уайт-спирит, (цикло)гексан, бензол или толуол. Во все эти жидкости следует ввести по меньшей мере одно хиральное вспомогательное средство. В качестве хирального вспомогательного средства обычно применяют низкомолекулярные соединения, но для такого применения пригодны также растворенные хиральные полимеры или хиральные биомолекулы.
В качестве хиральных вспомогательных средств можно применять очень широкий диапазон оптически активных соединений. Примеры этих соединений, среди которых можно указать также соответствующие соли и производные, включают: 1-аминоэтилфосфоновую кислоту, 2-бромпропионовую кислоту, эпихлоргидрин, серин, 2,3-диаминопропионовую кислоту, пропиленгликоль, аланинол, 1-амино-2-пропанол, аспарагиновую кислоту, яблочную кислоту, винную кислоту, 5-(гидроксиметил)-2-пирролидинон, пролин, цис-3-гидроксипролин, транс-1,2-циклопентандиол, 2-метилмасляную кислоту, α -гидроксиизовалериановую кислоту, метил-3-гидроксибутират, метил- β -гидроксиизобутират, арабинозу, ликсозу, рибозу, ксилозу, пролинол, этиловый эфир аланина, норвалин, валин, метионин, пеницилламин, метионинсульфоксид, 2-пентанол, 2,4-пентандиол, арабит, 2-метил-1-бутиламин, валинол, 1,2-диаминоциклогексан, 1-амино-2-(метоксиметил)пирролидин, лизин, аргинин, 2-гексанол, 2-метил-2,4-пентандиол, лейцинол, 2-фторфенилаланин, 3-фторфенилаланин, 5-фторфенилтриптофан, 5-гидрокситриптофан, 2-бензилокси-1,3,4-бутантриол, изопропилнорадреналин, 1-(1-нафтил)этанол, 1-(1-нафтил)этиламин, транс-2-фенил-1-циклогексанол, тироксин, метилацетат, N-(3,5-динитробензоил)- α - фенилглицин, N-метилэфедрин, 3,5-динитро-N-(1-фенилэтил)бензамид, α,α -дифенилпролинол.
Способ настоящего изобретения позволяет разделять рацематы соединений природы самого широкого диапазона. Рацематы могут быть соединениями группы фармации, а также синтонами, душистыми веществами и вкусовыми веществами или пестицидами.
Хотя, согласно изобретению, можно применять очень широкий диапазон оптически активных соединений в качестве хиральных вспомогательных средств для разделения даже более широкого ряда рацемических смесей, наиболее предпочтительно применение некоторых комбинаций хиральных вспомогательных средств и разделяемых рацемических смесей. Обычно предпочтительны те хиральные вспомогательные средства, которые проявляют способность к сильному взаимодействию с разделяемыми рацемическими веществами. Такое сильное взаимодействие может быть обусловлено, например, образованием водородного мостика между хиральным вспомогательным средством и рацемическим соединением. Альтернативно сильное взаимодействие может быть результатом кулоновских или ван-дервальсовых сил.
Для разделения рацемических смесей, имеющих первичную или вторичную аминогруппу, гидроксигруппу и/или меркаптогруппу, лучше использовать хиральные вспомогательные средства, имеющие (тио)карбонил, аминогруппу, простую (тио)эфирную группу, гидрокси- или меркаптогруппу. Рацематы, имеющие третичную аминогруппу, легко разделимы при помощи хирального вспомогательного средства, содержащего первичную или вторичную аминогруппу, гидроксигруппу или меркаптогруппу.
Примеры рацемических смесей, которые можно разделить этим способом, включают:
а) β -миметики и β -блокаторы, например изопреналин, орципреналин, тербуталин, фенотерол, салбутамол, гексопреналин, ритодрин, добутамин, изоетарин, пирбутерол, римитерол, формотерол, изоксуприн, карбутерол, ксамотерол, салметерол, пропранолол, атенолол, метопролол, тимолол, пиндолол, ацебутолол, алпренолол, буфенолол, бетаксолол, бевантолол, биспролол, бориндолол, картеолол, карведилол, целипролол, эпанолол, эсмолол, лабетолол, левобунолол, метипранолол, оксипренолол, пенбутолол, соталол и тертатолол;
б) α -гидроксиамины, например норэфедрин, ψ -норэфедрин, эфедрин, ψ -эфедрин, эритро-2-амино-1,2-дифенилэтанол, трес-2-амино-1,2-дифенилэтанол, эритро-1-амино-1-фенил-2-пропанол, трео-1-амино-1-фенилпропанол, фенилглицин, трео-2-амино-1-фенил-1,3-пропандиол и аналогичные соединения;
с) антагонисты кальция, например амлодипин, верапамил, дилтиазем, бепридил, галлопамил, фелодипин, израдипин, никардипин, нимодипин, низолдипин и нитрендипин;
d) антидепрессанты, например бутриптилин, циталопрам, флуоксетин, миансерин, миртазапин, окситриптан, пароксетин, сертралин, транилципромин и тримипрамин.
Для разделения указанных выше рацемических смесей благоприятно применение следующих хиральных вспомогательных средств или полученных из них соединений: камфора-10-сульфокислоты, малеиновой кислоты, α -метоксифенилуксусной кислоты, α -метокси- α -трифторметилфенилуксусной кислоты, винной кислоты, ментола, изопинокамфеола, α -терпинеола, камфоры, фенхона, α -амино- ε -капролактама, α -метилбензиламина, эфедрина, норэфедрина и аналогичных соединений.
Для разделения рацемических смесей карбоновых кислот, тиокарбоновых кислот и сульфокислот благоприятно применение хиральных вспомогательных средств, содержащих карбонил, аминогруппу или гидроксигруппу. Предпочтение отдано в этом случае соединениям, содержащим как карбонил, так и гидроксигруппу, или аминогруппу, а также карбонил. В качестве примеров можно указать производные винной кислоты, миндальной кислоты, аминокислоты, амиды и органические кислоты.
Примеры рацемических смесей, разделяемых этим способом, включают:
a) противовоспалительные средства, например ибупрофен, кетопрофен, пирпрофен, фенопрофен, флурбипрофен, тиапрофеновую кислоту и напроксен;
b) ингибиторы ферментов, превращающих ангиотензин (ACE), например беназеприл, каптоприл, целазаприл, еналаприл, еналаприлат, лизоноприл, цилазаприл, фосиноприл, периндоприл, хинаприл, рамприл и спираприл;
c) душистые вещества, например цитронелловую кислоту, 2-метилгептановую кислоту, 2-метилгексановую кислоту, 3-метилпентановую кислоту, малеиновую кислоту и тиомолочную кислоту;
d) пестициды, например 2-бромметилпропионовую кислоту, 2,4-дихлорфеноксимасляную кислоту, 2,4-дихлорфеноксипропионовую кислоту, камфановую кислоту, 2-гидроксиметилпропановую кислоту и o-метилминдальную кислоту;
e) аминокислоты.
Для разделения указанных выше смесей благоприятно применение следующих хиральных вспомогательных средств или полученных из них соединений: α -метилбензиламина, 2-аминобутана, 2-аминобутанола, 1-(1-нафтил)этанола, 1-(2-нафтил)этанола, 1-(1-нафтил)этиламина, эфедрина и норэфедрина и аналогичных соединений. Очень благоприятные результаты были получены с применением винной кислоты, миндальной кислоты, из эфиров и аминокислот и их производных.
Рацемические смеси аминокислот можно разделить с применением аминокислот в качестве хиральных вспомогательных средств. Другие подходящие хиральные вспомогательные средства включают хиральные амины, кислоты, спирты, кетоны и альдегиды. Особенно благоприятные результаты получали при применении хиральных аминокислот и их производных, причем в этом случае комплекс образуется с помощью ионов меди (II), а также с применением винной кислоты, миндальной кислоты и их производных.
Для разделения рацемических смесей (тио)эфиров, эфиров фосфорной кислоты, (тио)кетонов и (тио)альдегидов благоприятно применение хиральных вспомогательных средств, содержащих гидрокси- или аминогруппу.
Примеры рацемических смесей, которые можно разделить этим способом, включают:
а) борниловые сложные эфиры, карвиловые сложные эфиры, цитронеллиловые сложные эфиры, изоборниловые сложные эфиры, линалиловые сложные эфиры, терпиниловые сложные эфиры и родиниловые сложные эфиры;
b) 3-бензил-4-гептанон, хиральные лактоны, 2-метил-2-бутеналь, 3-меркапто-2-пентанон и 3-меркапто-2-бутанон;
c) антагонисты кальция, например амлодипин, верапамил, дилтиазем, бепридил, галлопамил, фелодипин, израдипин, никардипин, нимодипин, низолдипин и нитрендипин;
d) пестициды, имеющие группу эфира фосфорной кислоты, например цианофенфос, диэтилмалатион, фонофос и лептофос.
Для этой цели в качестве хиральных вспомогательных средств можно применять удачно следующие соединения или их производные:
α -фенилэтиламин, α -метилбензиламин, винную кислоту, миндальную кислоту, эфедрин, норэфедрин, аминосоединения.
Разделение по способу настоящего изобретения можно проводить при помощи только однократной экстракции, но предпочтительно применение ряда последовательных экстракционных ступеней. В случае противоточного фракционирования (жидкости жидкостью) эквимолярной рацемической смеси при помощи двух жидких фаз, по меньшей мере одна из которых хиральная или содержит хиральное вспомогательное средство, соотношение энантиомеров разделяемого вещества в выходящем потоке (R/S) можно рассчитать при помощи формулы (I):
в которой
NTU обозначает число ступеней переноса в устройстве и LR и LS представляют собой коэффициент извлечения энантиомеров R и соответственно. LR и LS можно рассчитать при помощи формул (2) и (3),
LR= mRFe/Ff (2),
LS= mSFe/Ff (3),
где
Fe и F обозначают объемы потоков экстрагирующей фазы и сырьевой фазы, в которой растворен разделяемый рацемат, соответственно и mR является коэффициентом распределения энантиомера R между сырьевой и экстрагирующей фазами и mS является коэффициентом распределения энантиомера S между сырьевой и экстрагирующей фазами.
Изобретение дополнительно иллюстрируется следующими примерами, в которых показано, что при разделении смеси энантиомеров способом настоящего изобретения можно достичь чистоту выше 99%. Кроме того, для дальнейшего разъяснения изобретения включен ряд фигур.
Нет необходимости заявлять, что не ограничивающие изобретение примеры представлены только для лучшего понимания изобретения.
На фиг.1 приведена структурная формула смеси энантиомеров соединения A.
На фиг. 2 показано устройство для определения коэффициентов распределения mR и mS при помощи модуля с полыми волокнами.
На фиг. 3 показано устройство для противоточной обработки однократным прохождением в модуле с полыми волокнами.
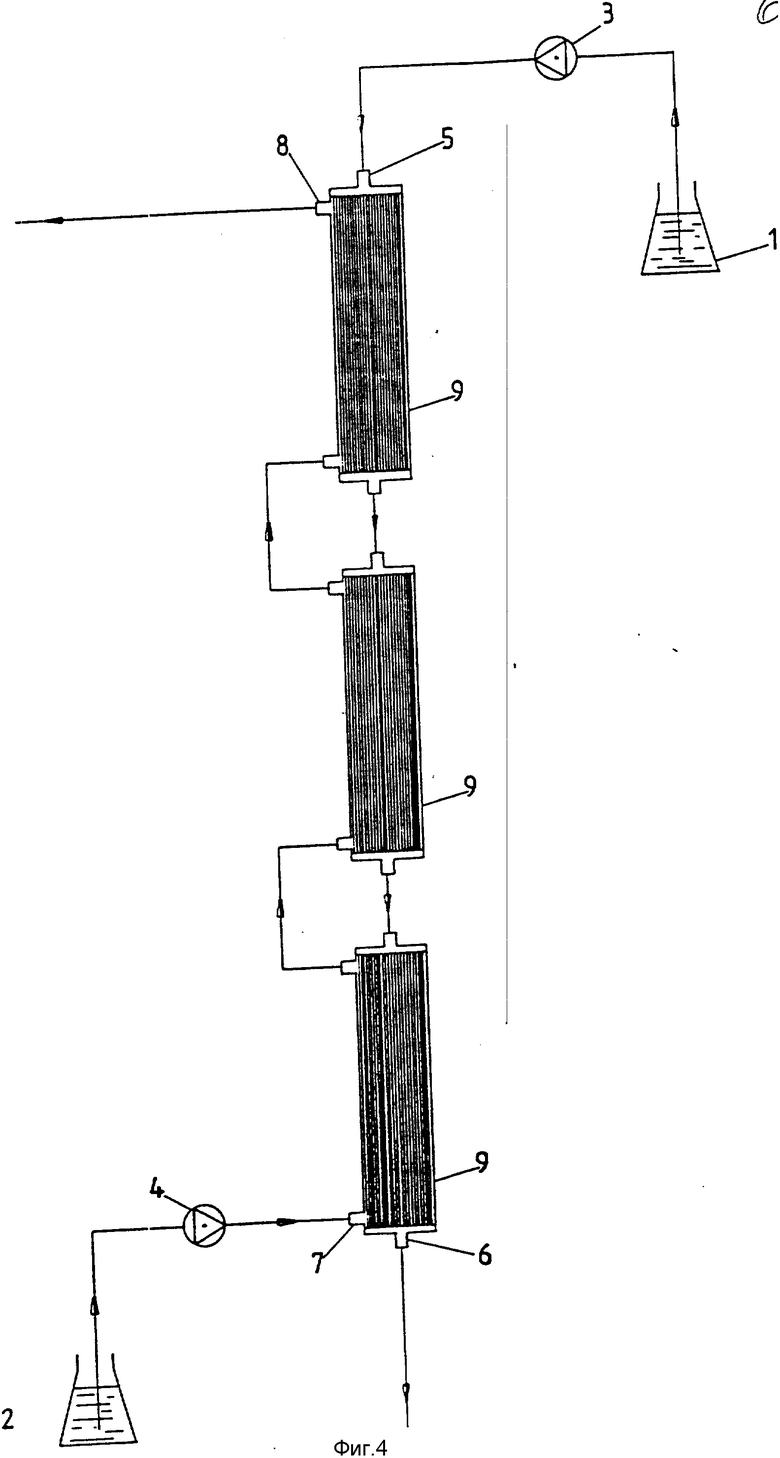
На фиг. 4 показано устройство, аналогичное устройству фиг. 3, за исключением того, что здесь последовательно соединен ряд модулей с полыми волокнами.
На фиг. 5 показано устройство для разделения смеси энантиомеров на два отдельных энантиомера.
На фиг. 6 приведены аналитические результаты разделения, проведенного с применением устройства, указанного на фиг. 5.
На фиг. 2 указанный выше модуль волокон обозначается позицией 9. В процессе измерения модуль расположен вертикально и заполнен в продольном направлении полыми волокнами, у которых концы открыты в сторону штуцеров 5 и 6 Штуцер 5 соединен через насос 3 проходом, открывающимся в сосуд 1, в котором находится жидкость, содержащая хиральное вспомогательное средство (1). После прохождения через внутренний канал волокон эта жидкость течет назад в сосуд 1 через штуцер 6. Жидкость, которая содержит хиральное вспомогательное средство (2), и разделяемый рецемат присутствуют в сосуде 2, из которого смесь подают в модуль полых волокон через насос 4 и штуцер 7. Эта смесь возвращается в сосуд 2 через штуцер 8.
На фиг. 3 показано устройство, сравнимое с устройством фиг. 2. при условии, что жидкости, содержащие хиральные вспомогательные средства, имеют противоположные направления течения в модуле полых волокон, благодаря чему не возникают сомнения в том, что из содержащих хиральные вспомогательные средства жидкостей возвращаются в один из сосудов 1 и 2. Через насос 3 штуцер 5 соединен с проходом, открывающимся в сосуд 1, в котором находится жидкость, содержащая хиральное вспомогательное средство (1) и разделяемый рецемат. Сосуд 2 содержит такую же жидкость, которая на этот раз содержит хиральное вспомогательное средство (2). Эту жидкость подают в модуль полых волокон через насос 4 и штуцер 7 и затем разгружают снова через штуцер 8. Частично разделенная смесь покидает модуль через штуцер 6.
На фиг. 4 показано устройство, сравнимое с устройством фиг. 3, при условии, что в этом случае имеется три последовательно соединенных модуля полых волокон.
На фиг. 5 показано устройство, сравнимое с устройством фиг. 4, при условии, что на этот раз системе для разделения смеси энантиомеров на индивидуальные компоненты имеет симметричную конструкцию. Другое отличие от устройства, изображенного на фиг. 4, состоит в том, что жидкости, содержащие хиральное вспомогательное средство и рецемическую смесь, не проходят через внутренние каналы волокон, а вокруг (около) волокон, способом, аналогичным способу, указанному на фиг. 2. Таким образом, имеются жидкости, содержащие хиральные вспомогательные средства, которые проходят через внутренние каналы волокон. Разделенные вещества ( 1 и 2) в конце покидают модули через штуцеры 8а и 8b соответственно.
Пример 1. Смесь энантиомеров соединения А структурной формулы, приведенной на фиг. 1, сначала имела коэффициент распределения m, определенный на устройстве, например, указанным схематично на фиг. 2. С этой целью сначала буферную жидкость с pH 1,7, содержащую водную 0,15 М лимонную кислоту /Na2HPO4, пропускали через модуль полых волокон купрофана. Затем буферную жидкость заменяли на раствор 10 мас.% S,S-дибензоилвинной кислоты в деканоле. В пространстве вокруг волокон также находился в деканоле, но содержащий 10 мас.% R,R-дибензоилвинной кислоты. После добавления 10 мг/мл соединения А в последний раствор в сосуде 2 два раствора рециркулировали. Периодически определяли распределение двух энантиомеров соединения А. Коэффициенты распределения двух энантиомеров, а именно, 1,06 и 0,94, можно получить из величин, измеренных при равновесии. По причине симметрии (mR=1/mS) все приведенные ниже примеры включают результат определения только одного коэффициента распределения.
Пример 2. Было найдено, что величина коэффициента распределения, определенная в соответствии с примером 1, полностью сходна с величиной, полученной совершенно другим методом, а именно, определением равновесного распределения рецемической смеси между двумя несмешиваемыми фазами, одна из которых содержит растворенное в нем хиральное вспомогательное средство. Затем коэффициент распределения рассчитывали делением соотношения энантиомеров в одной фазе на соотношение энантиомеров в другой фазе.
Определение коэффициента распределения соединения А в системе вода/деканол в присутствии 10 мас. % R,R-дибензоилвинной кислоты проводили следующим образом. Сначала 5 мл буферного раствора, содержащего водную 0,15 М лимонную кислоту /Na2HPO4, тщательно смешали в плотно закрываемой склянке на 15 мл с 5 мл раствора, содержащего 10 мг/мл соединения А и 10 мас.% R, R-дибензоилвинной кислоты в деканоле. После разделения фаз определяли соотношение энантиомеров соединения А в двух фазах. Было найдено, что полученные таким образом величины соотношения полностью соответствуют величинам 1,06 и 0,94 соответственно, указанным в примере 1.
Пример 3. Способом, аналогичным способу, описанному в примере 2, определяли коэффициент распределения норэфедрина в системе, состоящей из водной фазы, содержащей 0,1 М фосфатного буфера (pH 7), и органический фазы, представляющей собой раствор 10 мас.% D-дигексилтартрата в гептане.
Определение проводили способом, аналогичным способу, описанному в примере 2. С этой целью 4 мг норэфедрина растворяли в 5 г фосфатного буфера в плотно закрываемой склянке на 15 мл. В раствор добавляли 5 мг органической фазы, после чего склянку встряхивали в течение ночи в вибраторе (Mini-Sharer, Aclolf Kuhner AG, Surtgerland) при 300 об/мин. Было найдено, что содержание активного компонента в водной фазе и органической фазе достигло равномерного содержания. После полного расслоения две фазы разделяли и норэфедрин экстрагировали из органической фазы при помощи 1,3 г цитратного буфера (0,1 М; pH 3). При помощи капиллярного зонального электрофореза определяли соотношение энантиомеров норэфедрина как в водной, так и в экстрагированной органической фазе. В этих условиях содержание норэфедрина в органической фазе было 5% и рассчитанная величина коэффициента распределения была 1,19.
Аналогичным образом определяли коэффициент распределения норэфедрина в системе, состоящей из водной фазы, содержащей 0,04 М натриевой соли фосфор( V)-фтористоводородной кислоты (NaPF6) в 0,1 М цитратном буфере (pH3), и органической фазы, представляющей собой раствор 10 мас% D-дигексилтартрата в гептане. После достижения равновесия и разделения слоев фазы разделяли и норэфедрии экстрагировали из органической фазы при помощи 1,3 г фосфатного буфера (0,1 М, pH 2). Снова с применением капиллярного зонного электрофореза определяли соотношение энантиомеров как в водной, так и в органической фазе. В этих условиях содержание норэфедрина в органической фазе было 4%, а величина коэффициента распределения была 1,1.
Аналогичным образом рассчитывали коэффициент распределения норэфедрина в системе, состоящей из водной фазы, содержащей 0,1 М фосфатный буфер (pH7), и органической фазы, представляющей собой раствор 10 мас.% поли-L-молочной кислоты в хлороформе.
Измерения показали, что коэффициенты распределения составляли 1,07 при комнатной температуре и 1,10 при 4oC.
Пример 4. Способом, аналогичным описанному в примере 3, коэффициент распределения рацемической смеси ибупрофена определяли в системе, состоящей из водной фазы, содержащей 0,1 М фосфатный буфер (pH 7), и органической фазы, представляющей собой раствор 4 мас.% D-дигексилтартрата в гептане. Снова соотношение энантиомеров определяли как в водной, так и органической фазах при помощи капиллярного зонного электрофореза.
В этих условиях измеренное содержание ибупрофена в водной фазе было 76% и рассчитанная величина коэффициента распределения была 1,10.
Пример 5. Способом, аналогичным описанному в примере 3, определяли коэффициент распределения рацемической смеси тербуталина в системе, состоящей из водной фазы, содержащей 5 мас.% метилсульфита натрия в 1N буфере хлористого аммония (pH 9,5) и органической фазы, представляющей собой раствор 50 мас.% диметил-2,3-0-изопропилиден-L-тартрата в циклопентаноне.
Определение коэффициента распределения тербуталина в системе вода/раствор 50 мас.%диметил-2,3-0-изопропилиден-l-тартрата в циклопентаноне проводили следующим образом. Сначала 5 мг тербуталина растворяли в 5 г водной фазы в плотно закрываемом сосуде на 15 мл. После добавления в раствор 5 г органической фазы всю систему встряхивали в течение ночи в вибраторе при 300 об/мин. Было найдено, что содержание активного компонента в водной фазе и в органической фазе достигло равновесия. После полного расслаивания фазы разделяли и из органической фазы тербуталин экстрагировали при помощи буфера 0,05 М три(гидроксиметил)аминометан и установили pH 2,5 добавлением фосфорной кислоты. Как в водной, так и в экстрагированной органической фазе, при помощи капиллярного зонного электрофореза определяли соотношение энантиомеров тербуталина. В этих условиях содержание тербуталлина в органической фазе было 25%, а селективность была 1,02.
Пример 6. Способом, аналогичным описанному в примере 5, определяли коэффициент распределения рацемической смеси тербуталина в системе, состоящей из водной фазы, содержащей 5 мас.% метабисульфита натрия в буфере IN хлористого аммония (pH 9,5), и органической фазы, представляющей собой раствор 10 мас.% фенхона в пентаноне.
Определение проводили следующим образом. Сначала 5 мг тербуталина растворяли в 5 г водной фазы в плотно закрываемом сосуде на 15 мл. После добавления в раствор 5 г органической фазы всю систему встряхивали в течение ночи в вибраторе при 300 об/мин. Нашли, что содержание активного компонента в водной фазе и органической фазе соответствует равновесному. После полного разделения слоев фазы разделяли и тербуталин экстрагировали из органической фазы при помощи буфера 0,05М три (гидроксиметил)аминометана и устанавливали pH 2,5 добавлением фосфорной кислоты. Капиллярным зонным электрофорезом определяли соотношение энантиомеров тербуталина как в водной фазе, так и экстрагированной органической фазе. В этих условиях содержание тербуталина в органической фазе было 30% и селективность была 1,02.
Пример 7. Способом, аналогичным описанному в примере 5, определяли коэффициент распределения рецемической смеси фенилглицина в системе, состоящей из водной фазы, содержащей 0,1 M карбонатный буфер (pH 9,5), и органической фазы, представляющей собой раствор 50 мас.% дигексилтартрата в гептане. Определение проводили следующим образом. Сначала 5 мг фенилглицина растворяли в 5 г водной фазы в плотно закрываемом сосуде на 15 мл. После добавления в раствор 5 г органической фазы всю систему встряхивали в течение ночи в вибраторе при 300 об/мин. Установили, что содержание активного компонента в водной фазе и органической фазе достигло равновесия. После полного расслоения фазы разделяли и из органической фазы экстрагировали фенилглицин при помощи 0,01 М хлорной кислоты (pH 2). Соотношение энантиомеров фенилглицина в водной и органической фазах определяли жидкостной хроматографией при высоком давлении, применяя в качестве стационарной фазы Crownpak CR +(Daicel Chem. Iud., Japan), в качестве подвижной фазы 0,01 М хлорную кислоту (pH 2), подаваемую со скоростью 1 мл/мин, и УФ-детектирование 254 нм. В этих условиях содержание фенилглицина в органической фазе было 3% и селективность была 1,06.
Пример 8. Способом, аналогичным описанному в примере 3, определяли коэффициент распределения рацемической смеси пропранолола в системе, состоящей из водной фазы, содержащей раствор буфера 0,1 M лимонная кислота/цитрат (pH 3,5), и органической фазы, представляющей собой раствор 10 мас.% D-дигексилтартрата в гептане. И в этом случае определяли соотношение энантиомеров в водной и органической фазах при помощи капиллярного зонного электрофореза.
В этих условиях измеренное содержание пропранолола в органической фазе составляло 40%, а рассчитанная величина коэффициента распределения была 1,03.
Пример 9. Способом, аналогичным описанному в примере 3, определяли коэффициент распределения рацемической смеси салбутамола. Для того, чтобы было возможно применять такую сильно неполярную фазу, как гептан, применяли липофильный анион, образующий комплекс с разделяемым компонентом. В этом случае в водную фазу вводили 0,018 M тетрафенилбората натрия (NaTPB), тогда как органическая фаза содержала 10 мас.% D-дигексилтартрата в гептане. Снова соотношение энантомеров в водной и органической фазах определяли при помощи капиллярного зонного электрофореза.
В этих условиях измеренное содержание салбутамола в органической фазе было 24%, а рассчитанная величина коэффициента распределения была 1,06.
Пример 10. Способом, аналогичным описанному в примере 3, определяли коэффициент распределения рацемической смеси салбутамола в водной фазе, содержащей 0,0035 M тетрафенилбората Na(NaTFB), и органической фазе, содержащей 10 мас. % D-дигексилтартрата в циклогексане. После полного расслоения фазы разделяли и из органической фазы экстрагировали салбутамол при помощи водного раствора 1,3 г NaOH (0,1 M). И в этом случае содержание энантиомеров в водной и органической фазах определяли капиллярным зонным электрофорезом.
В этих условиях измеренное содержание салбутамола в органической фазе было 6%, а коэффициент распределения был 1,04.
Пример 11. Повторяли эксперимент примера 1, за исключением того, что на этот раз коэффициент распределения соединения A определяли в растворе гексана, в который были введены 10 мас.% R, R -дигексилтартрата и 10 мас.% S, S-дигексилтартрата соответственно. Из величин, измеренных в состоянии равновесия, можно было рассчитать коэффициент распределения, который был 1,05.
Пример 12. Способом, аналогичным описанному в примере 1, сначала через модуль с полыми волокнами купрофана пропускали водный раствор, содержащий 0,5 моль/л NaPF6. Затем эту водную фазу заменили раствором 10 мас.% S, S -дигексилтартрата в гексане. В местах вокруг волокон был также раствор в гексане, содержащий 10 мас.% R,R -дигексилтартрата. После добавления в последний 10 мг/мл норэфедрина два раствора рециркулировали. Из величин, измеренных в равновесных условиях, можно было рассчитать коэффициент распределения, который был 1,2.
Пример 13. Приведенный ниже пример показывает, что возможно разделение 50: 50 смесь энантиомеров соединения A при помощи модуля с полыми волокнами супрофана, если две органические фазы не рециркулировали для определения коэффициента распределения как в примерах 1, 7 и 9, но поток пропускали противоточно через модуль с полыми волокнами путем одноразового прохождения, как указано на фиг.3. Применяемый модуль имел длину 18 см и содержал 4200 волокон. После пропускания через полые волокна буферной жидкости с pH 3,5, которая содержала водную 0,15 M лимонную кислоту Na2HPO4 и в которую добавляли также 0,1 мМ бромистого диметиламмония, водную фазу между волокнами заменяли на насыщенный водой раствор 10 мас.% R,R-дигексилтартрата в гептане. Водную фазу во внутренних каналах волокон заменяли на насыщенный водой раствор S, S -дигексилтартрата в гептане, в который ввели также 2 мг/мл смеси энантиомеров соединения А.Затем через модуль (через волокна и вокруг волокон) противоточно и без рециркуляции пропустили два этих гептановых раствора со скоростью течения через волокна и вокруг волокон 11,7 и 12,8 мл/ч соответственно. В выходящем потоке раствора, содержащего S, S-дигексилтартрат, соотношение двух энантиомеров соединения A было 82:18.
Пример 14. Способом, аналогичным описанному в примере 13, противоточную экстракцию проводили с применением смеси энантиомеров норэфедрина. Сначала через полые волокна купрофана пропускали водный раствор, содержащий фосфатный буфер (pH 7,8) и 0,1 MDTAB. Раствор S,S-дигексилтартрата в этом случае содержал 4 мг/г (50:50) рацемического норэфедрина. Через волокна и вокруг волокон пропускали жидкости со скоростью 10,3 и 18,1 мл/ч соответственно. В выходящем, содержащем S, S-дигексилтартрат, растворе соотношение энантиомеров было 82:18.
Пример 15. Повторяли экстракцию по примеру 14, но вместо модуля с полыми волокнами применяли три последовательно соединенных модуля с полыми волокнами, как схематично показано на фиг. 4. В выходящем, содержащем S, S-дигексилтартрат, растворе соотношение энантиомеров было 98:2.
Пример 16. Приведенный ниже пример показывает, что можно разделить смесь энантиомеров на R- и S-компоненты при помощи устройства, например приведенного схематично на фиг.5. Каждая из колонок, обозначенных числами 9a и 9b, в этом случае содержала 7 последовательно соединенных модулей с полыми волокнами. Модули с полыми волокнами были такие же, как в примере 10. Сосуды 1a и 1b были заполнены растворами в гептане 10 мас.% R, R-дигексилтартрата и S, S-дигексилтартрата соответственно. Сосуды 2a и 2b были заполнены растворами 10 мас.% S,S-дигексилтартрата и R, R-дигексилтартрата соответственно в гептане, в каждом из которых было растворено 4 мг/мл рацемического норэфедрина. Подачу потоков регулировали насосами следующим образом: 3a: 97 мл/ч; 3b: 100 мл/ч; 4b: 4 мл/ч; 4a: 4 мл/ч.
Определяли соотношение энантиомеров в потоках 1 и 2, выходящих через штуцеры 8b и 8a. Результаты определения приведены на фиг.6, из которой становится ясно, что после достижения равновесия каждый из потоков имеет оптическую чистоту выше 99%. Один поток содержит 99% D-норэфедрина и другой содержит 99% L-норэфедрина. Эта установка позволяет разделять в день 768 мг рацемического норэфедрина на его энантиомеры.




Изобретение относится к способу выделения энантиомеров из рацемической смеси противоточной экстракцией при помощи по меньшей мере двух жидкостей, имеющих взаимно различную хиральность, причем эти жидкости полностью смешиваются и разделены друг от друга фазой, с которой они не смешиваются. Разделяющую, несмешиваемую фазу можно вывести в твердый носитель, который может быть пористым или непористым и предпочтительно имеет форму полого волокна. Жидкости со взаимно различающейся хиральностью могут буть хирально активными по своей природе или альтернативно их можно получить введением хирального вспомогательного средства по меньшей мере в одну из экстрагирующих жидкостей. 12 з.п.ф-лы, 6 ил.
| WO, заявка, 91/17816, кл | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| US, патент, 4960762, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
