Изобретение относится к производным азетидина и пирролидина, содержащим их фармацевтическим композициям, способу их получения, а также использованию производных азетидина и пирролидина для получения лекарства, которое действует на центральную нервную систему.
В последние годы хорошо описан вклад серотонинэргической активности в механизм действия антидепрессантов и разработаны соединения, усиливающие активность системы серотонина и успешно использующиеся в качестве антидепрессантов. Ингибиторы обратного захвата серотонина (SRI) работают, повышая количество серотонина, доступного при синапсе. Хотя (SRI) имеют менее выраженные побочные эффекты по сравнению с предшествующими поколениями, но и они не лишены недостатков и отличаются замедленным развитием эффекта [Andrews and Nemeroff, "Contemporary management of depression" - American Journal of Medicine 97(6A): 24S-32S (1994); Leonard, "The comparative pharmacology of new antidepressants" - Journal of Clinical Psychiatry 54(Suppl): 3-15 (1993)] . Более того, механизм действия SRI, хотя и специфический для серотонина, не является избирательным, поскольку они влияют на активность при множестве различных подтипов рецепторов серотонина. Такой широкий спектр активности может привести ко многим побочным эффектам, связанным с (SRI), например, тошнота от активирования 5-НТ3, головная боль вследствие активирования 5-НТ2В. Таким образом, SRI могут менять действие нескольких подтипов рецепторов 5-НТ2, однако эффективность этих препаратов может наиболее сильно коррелировать с их действием на систему 5-НТ2С [(Broekkamp and Berendsen "The importance of 5-HT1C receptors for antidepressant effects" - Polish Journal of Pharmacology and Pharmacy 44 (Suppl): 20(1992); Cesana et al. "Mesulergine antagonism towards the fluoxetine antiimmobility effect in the forced swimming test in mice" -Journal of Pharmacy and Pharmacology 45: 473-475 (1993); Berendsen and Broekkamp "Comparison of stimulus properties of fluoxetine and 5-HT receptor agonists in a conditioned taste aversion procedure" - European Journal of Pharmacology 253: 83-89(1994)].
Эти данные позволяют предположить, что соединения, которые избирательно активируют рецептор 5-НТ2С, будут эффективными при лечении эмоциональных расстройств и связанных с этим состояний.
Настоящее изобретение относится к соединениям формулы I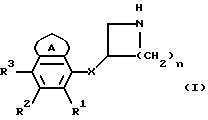
в которой А означает необязательно насыщенное 5- или 6-членное кольцо, которое может содержать гетероатом, выбранный из N, О и S, и которое может быть замещено оксо или (1-6С)алкилом; R1, R2 и R3 независимо представляют собой Н, (1-6C)алкил, (1-6С)алкокси, (1-6С)алкокси-(1-6С)алкил, или атом галогена; Х означает О или S и n = 1 или 2; или их фармацевтически приемлемым солям, за исключением 3-(нафт-1-ил-окси)-пирролидина и 3-(5,6,7,8-тетрагидронафт-1-ил-окси)-пирролидина. Эти соединения обладают селективным действием на рецепторы 5-НТ2С в центральной нервной системе.
В патенте США 4452809 [поданном 22 апреля 1983] раскрыты 3-арилокси-4-гидроксипирролидины и было установлено, что 3-нафтил или 3-инденилокси-4-оксипирролидины обладают антиаритмическим действием, тогда как 3-фенокси-4-гидроксипирролидины обладают антидепрессантным действием. Несколькими годами раньше в патенте Германии 2738477 [дата приоритета 1 сентября 1977] также были раскрыты 3-арилокси-4-гидроксипирролидины, и предпочтительными соединениями с антидепрессантным действием также были 3-фенокси производные. Другие 3-арилоксипирролидины, где пирролидиновая группа во всех соединениях является N-замещенной, влияющие на рецептор серотонина, были раскрыты в Европейском патенте 0338331 [с приоритетом от 19 апреля 1988].
Неожиданно, после многих лет исследований было установлено, что соединения формулы I (бициклические арил)окси-замещенные пирролидины и (бициклические арил)окси-замещенные азетидины, в которых 5- или 4-членный гетероцикл не замещен ни в одном из положений кольца, обладают селективным действием на рецепторы 5-НТ2С в центральной нервной системе. Также было установлено, что соединения 3-(нафт-1-ил-окси)-пирролидин и 3-(5,6,7,8-тетрагидро-нафт-1-ил-окси)пирролидин, известные как промежуточные соединения, но не заявленные в Европейском патенте 0338331, обладают этим действием. Поэтому, следует добиться защиты на использование этих соединений и на содержащие их фармацевтические композиции. Таким образом, настоящее изобретение также относится к первому медицинскому использованию соединений формулы I, т. е. включая соединения 3-(нафт-1-ил-окси)-пирролидин, 3-(5,6,7,8-тетрагидро-нафт-1-ил-окси)-пирролидин для использования в качестве лекарства (или, другими словами, для использования в терапии).
Использование селективного агониста 5-НТ2С обеспечивает немедленное проявление фармакологической активности и преимущественно при рецепторах 5-НТ2С, позволяя достичь значительно более быстрого нарастания селективной фармакологической активации, чем наблюдаемой при использовании SRI. Более того, селективность такого соединения снижает вероятность проявления отрицательных эффектов, вызываемых другими рецепторами серотонина, например, тошноты, головной боли, эффектов, которые могут мешать выявлению осложнений и таким образом отрицательно сказываться на эффективности действия препарата.
Соединения настоящего изобретения действуют на центральную нервную систему, в частности, как антидепрессанты и против навязчивых эмоциональных расстройств, тревоги, включая общее беспокойство, панические атаки, агорафобия, расстройства, связанные с приемом пищи, такое как ожирение, недержание мочи, импотенция, агрессия и злоупотребления лекарствами, такие как пристрастие к алкоголю или наркотикам.
Предпочтительные соединения настоящего изобретения имеют формулу I, в которой гетероатом в радикале А, если таковой присутствует, представляет собой атом N или S; R1 означает атом водорода, (1-6С)алкил, (1-6C)алкокси, (1-6С)алкокси-(1-6С)алкил; R2 означает атом водорода, (1-6С)алкокси или галоген, и R3 представляет собой атом водорода, (1-6С)алкил, (1-6С)алкокси или галоген. Более предпочтительными являются соединения формулы (Iа)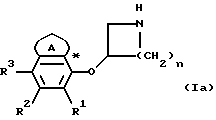
в которой А представляет собой незамещенное насыщенное 5-членное или необязательно ароматическое 6-членное кольцо, которое может содержать атом азота, соседний к положению, обозначенному звездочкой; R1 означает атом водорода или (1-6С)алкокси; R2 означает атом водорода, (1-6С)алкокси или атом галогена; R3 представляет собой атом водорода или атом галогена; и n = 1 или 2. Более предпочтительными являются соединения формулы (Iа), в которой А означает незамещенное насыщенное 5-членное или необязательно ароматическое 6-членное кольцо; радикал R1 означает (1-6С)алкокси и радикалы R2 и R3 представляют собой атом водорода. Наиболее предпочтительно, когда А в формуле (Iа) представляет собой 5-членное кольцо и радикал R1 представляет собой метоксигруппу, особенно, когда n = 2.
Термин (1-6С)алкил означает разветвленную или неразветвленную алкильную группу, содержащую от 1 до 6 атомов углерода, такую как метил, этил, t-бутил, изопентил и тому подобное. Наиболее предпочтительной алкильной группой является метил.
Термин (1-6С)алкокси означает алкоксигруппу, содержащую 1-6 атомов углерода, алкильный остаток, который имеет вышеуказанные значения. Наиболее предпочтительной алкоксигруппой является метокси.
Термин атом галогена означает атом фтора, хлора или брома.
Соединения формулы I могут быть получены способом, известным для таких соединений. На этой стадии соединения общей формулы II, в которых А, R1, R2, R3, Х и n имеют значения, определенные выше, и Р представляет собой любую N-защитную группу, стабильную в щелочных условиях [пригодные для использования N-защитные группы можно найти в публикации Т.W. Green and P.G.M. Wuts: Protective Groups in Organic Synthesis, Second Edition (Wiley, NY, 1991)], подвергают реакции вывода защитной группы, используя для этого соответствующие условия, такие как каталитическая гидрогенизация или образование промежуточного карбамата, с последующим взаимодействием со спиртами. Необязательно в то же время может образоваться соль.

Соединения общей формулы (II) могут быть получены путем образования простого арилэфира соответствующих N-защищенных 3-гидрокси-азетидинов или -пирролидинов, в которых защитная группа представляет собой группу, указанную выше, с соответствующим образом замещенными ароматическими или гетероароматическими соединениями, содержащими приемлемую отщепляемую группу. В другом варианте, N-защищенные-азетидины или пирролидины, содержащие соответствующую отщепляемую группу в положении 3, такую как атом галогена, трифлат, тозилат или мезилат, могут провзаимодействовать с соответствующим образом замещенными ароматическими или гетероароматическими соединениями, содержащими гидрокси или меркаптогруппу. Соединения настоящего изобретения, которые могут быть в форме свободного основания, можно выделить из реакционной смеси в виде фармацевтически приемлемой соли. Фармацевтически приемлемые соли также могут быть получены путем обработки свободного основания формулы I органической или минеральной кислотой, такой как хлористоводородной, бромистоводородной, иодистоводородной, серной кислотой, фосфорной кислотой, уксусной кислотой, пропионовой кислотой, гликолевой кислотой, малеиновой кислотой, малоновой кислотой, метансульфокислотой, сукциновой кислотой, щавелевой кислотой, лимонной кислотой, бензойной кислотой, аскорбиновой кислотой и тому подобное. Соединения настоящего изобретения могут содержать один или несколько хиральных атомов углерода и поэтому могут быть получены в виде чистого энантиомера или в виде смеси энантиомеров, или в виде смеси, содержащей диастереомеры. Методы получения чистых энантиомеров хорошо известны, например, кристаллизация солей, которые получают из оптически активных кислот и рацемической смеси, или хроматография с использованием хиральных колонок.
Соединения настоящего изобретения можно вводить энтерально или парэнтерально, и для людей предпочтительно при дневной дозе 0,001-100,0 мг на 1 кг массы тела, предпочтительно 0,01-10,0 мг на 1 кг массы тела. Будучи смешанными с фармацевтически приемлемыми ингредиентами, например, как описано в стандартной ссылке, Gennaro et al. , Remington's Pharmaceutical Sciences (18th ed. , Mack Publishing Company, 1990, Part 8: Pharmaceutical Preparations and their Manufacture), эти соединения могут быть спрессованы в твердые разовые дозы, такие как пилюли, таблетки, или же могут быть использованы для изготовления капсул или суппозиториев. При помощи фармацевтически приемлемых жидкостей эти соединения могут быть введены в виде раствора, суспензии, эмульсии, например, для использования в качестве препаратов для инъекций, или в качестве спреев, например, для использования в качестве назального спрея. Для изготовления разовых доз, например, таблеток, рассматривается использование традиционных добавок, таких как наполнители, красители, полимерные связующие и тому подобное. В общем, может быть использована любая фармацевтически приемлемая добавка, которая не влияет на действие активнодействующих соединений.
Приемлемые носители, с которыми композиции могут быть введены, включают лактозу, крахмал, производные целлюлозы и тому подобное, или их смеси, в соответствующих количествах.
Далее настоящее изобретение поясняется с помощью следующих примеров.
Примеры
Экспериментальная часть
Общие методы
Первая стадия общего процесса состоит в получении приемлемого N-защищенного-3-гидрокси-азетидина или - пирролидина с последующим образованием простого арилэфира.
Подойдет любая защитная группа, стабильная в щелочных условиях реакции сочетания. Это применимо также к рядам пирролидинов, где бензильная группа оказывается наиболее приемлемой и наиболее удобной защитной группой. Что касается азетидинов, для более легкого образования азетидинового кольца в процессе синтеза могут быть использованы объемные группы, такие как трифенилметил, 4,4'-двузамещенный дифенилметил, α-метилбензил и, на выбор, дифенилметил. Эти соединения могут быть получены по реакции соответствующих первичных аминов с эпихлоргидрином в среде полярного растворителя, такого как метанол или диметилформамид, при повышенных температурах, лежащих в диапазоне значений от комнатной температуры до температуры кипения с обратным холодильником, обычно в течение нескольких дней.
N-Защищенные-3-гидрокси-азетидины и - пирролидины могут быть использованы как таковые в реакции конденсации с широким рядом соединений, содержащих приемлемую отщепляемую группу на ароматическом остатке, с целью образования простого эфира. Когда отщепляемой группой является, например, атом галогена, то реакцию можно провести в среде такого полярного растворителя, как диметилформамид, и приемлемого основания, например, карбоната калия, в присутствии каталитически подобной активированной меди, при повышенных температурах, лежащих в диапазоне от комнатной температуры до температуры кипения с обратным холодильником.
На предпочтительной второй стадии общего процесса гидроксигруппу N-защищенного-3-гидрокси-азетидинов и - пирролидинов преобразуют с помощью хорошо известных методов в реакционноспособную отщепляемую группу, такую, как например, атом галогена, трифлат, тозилат и, на выбор, мезилат, с проведением последующей реакции конденсации с широким рядом соединений, содержащих в ароматической части гидрокси (или меркапто)группу, для получения широкого ряда простых ариловых эфиров, а также простых ариловых тиоэфиров.
Мезилаты удобно получать путем добавления метансульфонилхлорида к 3-гидроксисоединениям в среде неполярного растворителя, такого как толуол, в присутствии органического основания, такого, как триэтиламин, при температуре от -30oС до температуры кипения с обратным холодильником, обычно при пониженной температуре.
Хотя могут быть использованы все общие реакции образования простых арилалкиловых эфиров, известные из литературы, но большинство соединений настоящего изобретения было получено с использованием трех основных методов.
(i) Первый общий метод, пригодный для получения, состоит из гетерогенной двухфазной реакции между мезилатом и соответствующим нуклеофилом, предпочтительно бициклическим соединением, содержащим ароматическую гидрокси или меркаптогруппу. Двухфазная система состоит из водного раствора неорганического основания, такого как гидроксид натрия, и органического слоя, предпочтительно 4-метил-2-пентанона. Реакцию осуществляют при температуре от 25oС до температуры кипения с обратным холодильником, предпочтительно при повышенной температуре.
(ii) Второй метод получения состоит в реакции конденсации обоих субстратов, мезилата и нуклеофила, в среде полярного органического растворителя, такого, как трет-бутанол или диметилсульфоксид или их смесей, и соответствующего основания, подобного трет-бутоксиду калия, обычно при повышенных температурах, лежащих в диапазоне 25 - 100oС.
(iii) Третий метод получения состоит в образовании аниона подобным основанию гидридом натрия и последующей реакции с мезилатом в среде полярного органического растворителя, такого как диметилформамид, обычно при повышенных температурах, лежащих в пределах от 25oС до кипения с обратным холодильником.
Третья стадия общего процесса касается методов отщепления защитных групп всех типов, приводящих к образованию (циклических) вторичных аминов. К рассмотрению должны быть привлечены все традиционные методы, касающиеся выбранной защитной группы. Что касается аралкильных групп, широко использованных в настоящем изобретении, предпочтительными являются методы отщепления защитных групп двух типов. Первый метод заключается в удалении защитной группы с помощью каталитической гидрогенизации при давлении, меняющемся от атмосферного до 420 кПа в среде полярного растворителя, такого как этанол или метанол, в присутствии широко используемого катализатора, такого, как палладий на активированном углероде или гидроксид палладия на углероде, при 25-60oС. Второй метод состоит в замене исходной защитной группы на промежуточную карбаматную функциональную группу, которая затем удаляется. Пригодными для использования реагентами являются, например, 1-хлорэтил хлорформиат или винилхлорформиат в апротонном растворителе, таком как 1,2-дихлорэтан, при температуре от -15oС до температуры кипения с обратным холодильником и последующая реакция со спиртом, таким как метанол или этанол при температуре от -15oС до температуры кипения с обратным холодильником.
Четвертая стадия общего процесса касается получения или разделения стереоизомеров, включая диастереомеры и энантиомеры, как следствие присутствия одного или нескольких центров хиральности.
Энантиоселективные методы получения могут быть осуществлены, начиная с энантиочистых (R) и (S)субстратов, таких как например, (R)- или (S)-1-бензил-3-пирроидинол.
Из смеси стереоизомеров также могут быть получены индивидуальные энантиомеры с помощью хорошо известных методов разделения таких изомеров на составляющие эту смесь энантиомеры. Например, используя методы, описанные в Stereochemistry of Organic compounds, E.L. Eliel and S.H. Wilen, chapter 7, 1994. В частности, такие методы, как образование соли с оптически активными кислотами и последующей фракционной кристаллизацией или дифференциальной абсорбцией с использованием колонок, заполненных хиральным материалом, например, хиральной жидкостной или газовой хроматографией.
Пятая стадия общего процесса включает преобразование вторичных или третичных аминов, полученных в процессе синтеза, в любую соль или сольватную форму, предпочтительно фармацевтически приемлемые соли или сольваты, такие как гидрохлориды, а именно, полученные добавлением выбранной кислоты к свободному основанию в таком растворителе, как этанол, и выделение в виде твердого вещества.
Получение исходных материалов (азетидиновые субстраты)
Надежный метод, пригодный для широкомасштабного производства как 1-(дифенилметил)-3-азетидинола, так и его метансульфоната (модификация патента США 4183923, E.H. Gold с соавт., январь 1980):
1-(Дифенилметил)-3-азетидинол.
К раствору эпихлоргидрина (34,7 мл) в 1 л безводного диметилформамида под атмосферой азота добавляют дифенилметиламин (34,5 мл). Реакционную смесь нагревают при 95oС в течение 64 ч. После этого ее охлаждают до 5oС и по каплям добавляют смесь 20 мл водного раствора концентрированной хлористоводородной кислоты и 20 мл воды. После выпаривания под вакуумом остаток перемешивают с диэтиловым эфиром и профильтровывают. Твердый остаток промывают диэтиловым эфиром и затем разделяют между диэтиловым эфиром и 2н. водным раствором гидроксида натрия. Органический слой высушивают сульфатом натрия, профильтровывают и концентрируют под вакуумом. Остаток кристаллизуют из смеси толуола и петролейного эфира, в результате чего получают 36,3 г 1-(дифенилметил)-3-азетидинола, т.пл. 107oС.
1-(Дифенилметил)-3-метансульфонилокси-азетидин
К суспензии 1-(дифенилметил)-3-азетидинола (29,3 г) и триэтиламина (14 мл) в 220 мл сухого толуола при 15oС и под атмосферой азота по каплям медленно добавляют метансульфонилхлорид (7,8 мл). Температуру медленно повышают до комнатной температуры и реакционную смесь перемешивают в течение 17 ч. Затем добавляют 220 мл сухого диэтилового эфира и выпавший в осадок гидрохлорид триэтиламина отфильтровывают и промывают смесью диэтиловый эфир/дихлорметан (4: 1). Органический слой промывают 100 мл 1,1 М раствора бикарбоната натрия, а затем крепким раствором соли. Его высушивают сульфатом натрия, фильтруют и выпаривают под вакуумом, в результате чего получают 29,4 г 1-(дифенилметил)-3-метансульфонилоксиазетидина, M. S. (C.I.) (M/Z): 318 [М+Н]+.
Получение других исходных материалов.
5-Хлор-2,3-дигидро-1Н-инден-4-ол (Стадии а, б, в)
а) Сложный 2-хлорфениловый эфир 3-хлор-пропановой кислоты
К 2-хлорфенолу (18,18 г) добавляют 3-хлорпропионилхлорид (14 мл) и смесь перемешивают и нагревают при 60oС в течение 1 ч, при 75oС в течение 1 ч и оставляют при комнатной температуре на конец недели. Соединение очищают перегонкой под вакуумом и получают 19,7 г (т.к. 91-94oС, 0,08 мм рт.ст) сложного 2-хлорфенилового эфира 3-хлор-пропановой кислоты.
б) 6-Хлор-2,3-дигидро-7-гидрокси-1Н-инден-1-он.
К сложному 2-хлорфениловому эфиру 3-хлор-пропановой кислоты (19,6 г) добавляют 1 эквивалент хлорида алюминия (11,93 г) и смесь перемешивают под атмосферой азота в течение 2,5 ч при 100oС, охлаждают, добавляют вторую порцию хлорида алюминия (14 г) и нагревают при 170oС в течение 2 ч. Реакционную смесь охлаждают до 70-80oС и осторожно добавляют воду. Затем при перемешивании добавляют этилацетат и разделяют слои. Этилацетатный раствор промывают водой, сушат сульфатом магния, фильтруют и концентрируют под вакуумом. Выпавший осадок отфильтровывают, а оставшийся фильтрат выпаривают досуха. Остаток пропускают через хроматографическую колонку с диоксидом кремния, используя в качестве элюента толуол, и получают 3,2 г 6-хлор-2,3-дигидро-7-гидрокси-1Н-инден-1-она. M.S. (C.I.) (M/Z): 183 [M+H]+.
в) 5-Хлор-2,3-дигидро-1Н-инден-4-ол.
3,2 г 6-Хлор-7-гидрокси-1Н-инден-1-она в 16,8 мл воды и 67,2 мл концентрированной водной хлористоводородной кислоты перемешивают и нагревают со свежеприготовленной амальгамой цинка (из 26,88 г цинковой ваты) на масляной бане при 120oС в течение 16 ч. Реакционную смесь охлаждают, декантируют и обрабатывают этилацетатом и дихлорметаном. Органический слой выпаривают под вакуумом. Остаток очищают методом кислотно-щелочного разделения и получают 2,08 г 5-хлор-2,3-дигидро-1Н-инден-4-ола, M.S. (С.I.) (M/Z): 169 [М+Н]+.
Аналогичным образом получили 2,3-дигидро-5-метил-1Н-инден-4-ол, M.S. (C. I. ) (M/Z): 149 [М+Н]+, используя в качестве исходного соединения сложный 2-метилфениловый эфир 3-хлор-пропановой кислоты.
2,3-Дигидро-5-метокси-1Н-инден-4-ол (стадии а, б, в, г).
а) 3-(2,3-Диметоксифенил)-пропановая кислота.
В течение 15 мин через перемешиваемую суспензию (Z)-3-(2,3-диметоксифенил)-2-пропеновой кислоты (14,67 г) в 400 мл метанола пропускают азот. Затем добавляют 1,4 г 10% палладия на активированном углероде и через реакционную смесь в течение 16 ч пропускают поток водорода. После удаления палладиевого катализатора методом фильтрования фильтрат выпаривают и получают 14,2 г 3-(2,3-диметоксифенил)-пропановой кислоты, M.S. (C.I.) (M/Z): 211 [М+Н]+.
б) 2,3-Дигидро-4,5-диметокси-1Н-инден-1-он.
Раствор 3-(2,3-диметоксифенил)-пропановой кислоты (2 г) в 50 мл метансульфоновой кислоты, находящийся под атмосферой азота, нагревают до 60oС и поддерживают его при этой температуре в течение 2 ч. Реакционную смесь охлаждают до комнатной температуры и выливают в воду со льдом. После экстракции этилацетатом органический слой промывают 1н. водным раствором гидроксида натрия, сушат сульфатом магния, профильтровывают и выпаривают под вакуумом, в результате чего получают 1,2 г 2,3-дигидро-4,5-диметокси-1Н-инден-1-она, M.S. (C.I.) (M/Z): 193 [M+H]+.
в) 2,3-Дигидро-4-гидрокси-5-метокси-1Н-инден-1-он.
Под атмосферой азота 2,3-дигидро-4,5-диметокси-1H-инден-1-он (31,7 г) растворяют в 600 мл безводного 1,2-дихлорэтана и охлаждают до 0oС. Частями добавляют хлорид алюминия (44 г) и реакционную смесь нагревают до 60oС в течение 17 ч, охлаждают до комнатной температуры и выливают в воду со льдом. После экстракции дихлорметаном органический слой сушат и выпаривают. Остаток перекристаллизовывают из этилацетата и получают 20,5 г 2,3-дигидро-4-гидрокси-5-метокси-1H-инден-1-она, M.S. (C.I.) (M/Z): 179 [M+H]+.
г) 2,3-Дигидро-5-метокси-1Н-инден-4-ол.
20,5 г 2,3-Дигидро-4-гидрокси-5-метокси-1Н-инден-1-она суспендируют в смеси 310 мл концентрированной водной хлористоводородной кислоты и 53 мл воды. К смеси добавляют свежеприготовленную цинковую амальгаму (из 87 г цинковой ваты) и смесь перемешивают в течение 3 ч при температуре окружающей среды. После декантации оставшуюся цинковую амальгаму трижды промывают диэтиловым эфиром и кислый водный раствор экстрагируют диэтиловым эфиром. Объединенные эфирные растворы промывают 1н. водным раствором хлористоводородной кислоты, сушат сульфатом магния, профильтровывают и выпаривают под вакуумом, в результате чего получают 15,0 г 2,3-дигидро-5-метокси-1Н-инден-4-ола, M.S. (С.I.) (M/Z): 165 [М+H]+.
6-Фтор-1-метил-1H-инден-4-ол (стадии а, б, в).
а) Сложный 3-фторфениловый эфир 4-хлор-бутановой кислоты
4-Хлорбутирилхлорид (35,3 г) добавляют к 3-фторфенолу (25 г). Эту смесь перемешивают в течение 48 ч при комнатной температуре. После завершения реакции целевой продукт очищают вакуумной перегонкой. Выход: 35,3 г (т.к. 106oС, 3 мм рт.ст.) сложного 3-фторфенилового эфира 4-хлор-бутановой кислоты в виде белого масла.
б) 5-Фтор-2,3-дигидро-7-гидрокси-3-метил-1Н-инден-1-он.
После нагревания полученного сложного 3-фторфенилового эфира 4-хлор-бутановой кислоты (35,33 г) до 80oС добавляют хлорид алюминия (24,0 г). Реакционная смесь начинает вспениваться. По окончании вспенивания смесь перемешивают в течение 2 ч при 100oС. После добавления охлаждающей воды и этилацетата смесь нагревают на паровой бане. После растворения всего масла органический слой отделяют и промывают водой и крепким рассолом. Растворитель удаляют, а остаток перекристаллизовывают из 2-пропанола, в результате чего получают 21,2 г 5-фтор-2,3-дигидро-7-гидрокси-3-метил-1Н-инден-1-она. M.S. (C.I.) (M/Z): 181 [M+H]+.
в) 6-Фтор-2,3-дигидро-1-метил-1Н-инден-4-ол.
5 г 5-Фтор-2,3-дигидро-7-гидрокси-3-метил-1Н-инден-1-она нагревают при 80oС до полного плавления твердого вещества. После добавления к этому расплаву хлорида алюминия (9,3 г) реакционную смесь нагревают до 170oС в течение 17 ч. После добавления охлаждающей воды и этилацетата смесь нагревают на паровой бане до полного растворения содержимого. Органический слой отделяют и промывают водой и рассолом. После удаления растворителя соединение очищают на хроматографической колонке с помощью гептана/этилацетата (9: 1), в результате чего получают 2,2 г 6-фтор-2,3-дигидро-1-метил-1Н-инден-4-ола в виде полутвердого вещества, M.S. (С.I.) (M/Z): 167 [M+H]+.
СПОСОБЫ ПОЛУЧЕНИЯ
Пример 1.
3-[(5-Хлор-2, 3-дигидро-1Н-инден-4-ил) окси]-1-(дифенилметил)-азетидин.
а) 2 г 5-Хлор-2,3-дигидро-1Н-инден-4-ола перемешивают в 75 мл 2н. раствора гидроксида натрия в течение 1 ч. К прозрачному раствору добавляют 75 мл 4-метил-2-пентанона и 3,76 г 1-(дифенилметил)-3-метансульфонилоксиазетидина и смесь нагревают на масляной бане при 120oС в течение 3,5 ч. Затем добавляют еще 2 г мезилата и нагревание продолжают в течение 64 ч. Верхний слой отделяют и промывают водой. После выпаривания под вакуумом и хроматографии толуолом/этилацетатом (95: 5) получают 4,22 г 3-[(5-хлор-2,3-дигидро-1Н-инден-4-ил)окси] -1-(дифенилметил)-азетидина в виде прозрачного масла, которое самопроизвольно отверждается. M.S. (C.I.) (M/Z): 391 [M+Н]+.
Аналогичным образом были получены следующие соединения:
б) 3-[(2,4-дихлор-1-нафталенил)окси]-1-(дифенилметил)-азетидин, М.S. (С. I.) (M/Z): 435 [М+Н]+, исходя из 2, 4-дихлор-1-нафтола,
в) 1-(дифенилметил)-3-[(4-метил-1-нафталенил)окси]-азетидин, M.S. (C.I.) (M/Z): 380 [М+Н]+, исходя из 4-метил-1-нафтола,
г) 1-(дифенилметил)-3-[(2-метокси-1-нафталенил)окси]-азетидин, M.S. (С. I.) (M/Z): 393 [M+H]+, исходя из 2-метокси-1-нафтола,
д) 1-(дифенилметил)-3-[(5,6,7,8-тетрагидро-1-нафталенил) окси]-азетидин, М.S. (С.I.) (M/Z): 370 [M+H]+, исходя из 5,6,7,8-тетрагидро-1-нафтола,
е) 1-(дифенилметил)-3-[(2,3-дигидро-5-метокси-1Н-инден-4-ил)окси] -азетидин, M. S. (С.I.) (M/Z): 386 [M+Н]+, исходя из 2,3-дигидро-5-метокси-1H-инден-4-ола,
ж) 3-[(7-бром-2,3-дигидро-1Н-инден-4-ил)окси]-1-(дифенилметил)-азетидин, M.S. (C.I.) (M/Z): 435 [М+Н]+, исходя из 7-бром-2,3-дигидро-1Н-инден-4-ола,
з) 1-(дифенилметил)-3-[(6-фтор-2,3-дигидро-1-метил-1Н-инден-4-ил)окси] -азетидин, M.S. (С.I.) (M/Z): 388 [M+H]+, исходя из 6-фтор-2,3-дигидро-1-метил-1Н-инден-4-ола,
и) 3-[(2,3-дигидро-5-метил-1Н-инден-4-ил)окси] -1-(дифенилметил)-азетидин, M. S. (C. I.) (M/Z): 370 [M+H]+, исходя из 2,3-дигидро-5-метил-1Н-инден-4-ола,
к) 1-(дифенилметил)-3-[(2,3-дигидро-1Н-инден-4-ил)окси] азетидин, M.S. (C.I.) (M/Z): 356 [М+Н]+, исходя из 2,3-дигидро-1H-инден-4-ола,
л) 3-[(бензо(b)тиен-4-ил)-1-(дифенилметил)-азетидин, M.S. (C.I.) (M/Z): 372 [М+Н]+, исходя из бензо (b) тиофен-4-ола,
м) 5-(3-азетидинилокси)-1-(дифенилметил)-изохинолин, M.S. (C.I.) (M/Z): 367 [M+Н]+, исходя из 5-гидроксиизохинолина,
н) 8-(3-азетидинилокси)-1-(дифенилметил)-хинолин, M.S. (C.I.) (M/Z): 371 [М+Н]+, исходя из 8-оксихинолина.
Пример 2.
1-(Дифенилметил)-3-(1-нафталенилокси)-азетидина гидрохлорид.
а) К раствору 3,37 г трет-бутоксида калия в 71 мл трет-бутанола под атмосферой азота добавляют 1,44 г 1-нафтола. После перемешивания в течение получаса добавляют 4,33 г малеата 1-(дифенилметил)-3-метансульфонилокси-азетидина.
Чтобы увеличить растворимость, добавляют 71 мл диметилсульфоксида. Смесь нагревают на масляной бане при 80oС в течение 40 ч. t-Бутанол отгоняют под вакуумом и остаток распределяют между водой и этилацетатом. Этилацетатные экстракты промывают водой, сушат сульфатом магния, фильтруют и выпаривают под вакуумом. Остаток пропускают через хроматографическую колонку с помощью толуола и получают 2,7 г целевого продукта. Его обрабатывают раствором хлористоводородной кислоты в метаноле, выпаривают под вакуумом и перекристаллизовывают из абсолютного этанола, в результате чего получают 2,09 г гидрохлорида 1-(дифенилметил)-3-(1-нафталенилокси)-азетидина, т.пл. 182oС.
Аналогичным образом были получены следующие соединения:
b) 1-(дифенилметил)-3-[(2-метил-1-нафталенил)окси]-азетидин, M.S. (C.I.) (M/Z): 380 [М+Н]+, исходя из 2-метил-1-нафтола,
в) 1-(дифенилметил)-3-[(4-метокси-1-нафталенил)окси]-азетидин, M.S. (С. I.) (M/Z): 396 [M+Н]+, исходя из 4-метокси-1-нафтола,
г) 1-(дифенилметил)-3-(1-нафталенилтио)-азетидин, M.S. (C.I.) (M/Z): 382 [M+H]+, исходя из 1-нафталентиола,
д) 1-(дифенилметил)-3-(2-нафталенилокси)-азетидин, M. S. (C.I.) (M/Z): 366 [M+H]+, исходя из 2-нафтола.
Пример 3.
1-(Дифенилметил)-3-[(2-метокси-1-нафталенил)окси]-азетидина гидрохлорид
а) Смесь 6,89 г 1-(дифенилметил)-3-азетидинола, 25 мл безводного диметилформамида, 10,37 г карбоната калия, 5,93 г 1-бром-2-метоксинафталина и 200 мг активированной меди при перемешивании нагревают в течение 40 ч на масляной бане при 170oС. Реакционную смесь разделяют между водой и толуолом. Смесь сырого продукта из органических экстрактов пропускают через хроматографическую колонку, используя толуол и толуол/этилацетат (95:5). Целевой продукт растворяют в простом диэтиловом эфире и очищают путем добавления раствора хлористоводородной кислоты в метаноле. Выход: 1,96 г гидрохлорида 1- дифенилметил)-3-[(2-метокси-1-нафталенил)окси] -азетидина, M.S. (C.I.) (M/Z): 400 [M+H]+.
Аналогичным способом было получено следующее соединение;
б) 1-(дифенилметил)-3-[(2-метоксиметил)-1-нафталенил)окси]-азетидина гидрохлорид, M.S. (C.I.) (M/Z): 410 [М+Н]+, исходя из 1-бром-2-(метоксиметил)-нафталина.
Пример 4.
3-[(2,3-Дигидро-1Н-инден-4-ил)окси]азетидина гидрохлорид.
а) К суспензии 3 г 3-[(2,3-дигидро-1Н-инден-4-ил)окси]-1-(дифенилметил)-азетидина гидрохлорида в 250 мл этанола добавляют 600 мг гидроксида палладия на порошкообразном углероде и смесь гидрируют на установке Парра при давлении 420 кПа в течение 16 ч. После удаления катализатора и выпаривания растворителя под вакуумом остаток промывают несколько раз диэтиловым эфиром и декантируют с целью удаления образовавшегося дифенилметана. Оставшееся твердое вещество перекристаллизовывают из смеси этанол/диэтиловый эфир, в результате чего получают 1,27 г гидрохлорида 3-[(2,3-дигидро-1Н-инден-4-ил)окси]азетидина, т.пл. 65oС.
Аналогичным способом получают следующие соединения:
б) гидрохлорид 3-[(2-метил-1-нафталенил)окси] -азетидина, т.пл. 171oС, исходя из 1-(дифенилметил)-3-[(2-метил-1-нафталенил)окси]-азетидина,
в) гидрохлорид 3-(1-нафталенилокси)-азетидина, т.пл. 292oС, исходя из 1-(дифенилметил)-3-[(1-нафталенил)окси]-азетидина,
г) гидрохлорид 3-[(4-метокси-1-нафталенил)окси]-азетидина, т.пл. 198oС, исходя из 1-(дифенилметил)-3-[(4-метокси-1-нафталенил)окси]-азетидина,
д) гидрохлорид 3-[(5,6,7,8-тетрагидро-1-нафталенил)окси]-азетидина, т. пл. 187oС, исходя из 1-(дифенилметил)-3-[(5,6,7,8-тетрагидро-1-нафталенил)окси]-азетидина,
е) гидрохлорид 3-[(5,6,7,8-тетрагидро-2-метокси-1-нафталенил) окси]-азетидина, т. пл. 164oС, исходя из 1-(дифенилметил)-3-[(2-метокси-1-нафталенил)окси]-азетидина,
ж) гидрохлорид 3-(2-нафталенилокси)-азетидина, т.пл. 168oС, исходя из 1-(дифенилметил)-3-(2-нафталенилокси)-азетидина,
з) гидрохлорид 8-(3-азетидинилокси)-1,2,3,4-тетрагидрохинолина, т.пл. >250oС, исходя из 8-(3-азетидинилокси)-1-(дифенилметил)-хинолина.
Пример 5.
Гидрохлорид 3-[(5-хлор-2,3-дигидро-1Н-инден-4-ил)окси]-азетидина.
а) 4,22 г 3-[(5-Хлор-2,3-дигидро-1H-инден-4-ил)окси]-1-((дифенилметил))-азетидина растворяют в 71 мл 1,2-дихлорэтана. Добавляют 1,58 г 1-хлорэтилхлорформиата. Смесь кипятят с обратным холодильником на масляной бане при 120oС в течение 2,5 ч. После выпаривания под вакуумом остаток кипятят с обратным холодильником в 71 мл безводного метанола в течение 2 ч. После выпаривания под вакуумом полутвердый остаток смешивают с диэтиловым эфиром и фильтруют. Остаток перекристаллизовывают из этанола/диэтилового эфира и получают 1,57 г гидрохлорида 3-[(5-хлор-2,3-дигидро-1Н-инден-4-ил)окси]-азетидина, т.пл. 188oС.
Аналогичным способом получают следующие соединения:
б) гидрохлорид 3-[(2,4-дихлор-1-нафталенил)окси]-азетидина, т.пл. 187oС, исходя из 3-[(2,4-дихлор-1-нафталенил)окси]-1-(дифенилметил)-азетидина,
в) гидрохлорид 3-[(4-метил-1-нафталенил)окси] -азетидин, т.пл. 180oС, исходя из 1-(дифенилметил)-3-[(4-метил-1-нафталенил)окси]-азетидина,
г) гидрохлорид 3-[(2,3-дигидро-5-метокси-1Н-инден-4-ил)окси]-азетидина, т. пл. 166oС, исходя из 1-(дифенилметил)-3-[(2,3-дигидро-5-метокси-1Н-инден-4-ил)окси]-азетидина,
д) гидрохлорид 3-[(7-бром-2,3-дигидро-1Н-инден-4-ил)окси]-азетидина, т. пл. 203oС, исходя из 3-[(7-бром-2,3-дигидро-1Н-инден-4-ил)окси] -1-(дифенилметил)-азетидина,
ж) гидрохлорид 3-[(6-фтор-2,3-дигидро-1-метил-1Н-инден-4-ил)окси]-азетидина, т.пл. 170oС, исходя из 1-(дифенилметил)-3-[(6-фтор-2,3-дигидро-1-метил-1Н-инден-4-ил)окси]-азетидина,
з) гидрохлорид 3-[(2,3-дигидро-5-метил-1Н-инден-4-ил)окси]азетидина, т. пл. 184oС, исходя из 1-(дифенилметил)-3-[(2,3-дигидро-5-метил-1Н-инден-4-ил)окси]-азетидина,
и) гидрохлорид 3-[(бензо[b]тиен-4-ил)окси]-азетидина, т.пл. 203oС, исходя из 3-[(бензо[b]тиен-4-ил)окси]-1-(дифенилметил)-азетидина,
к) дигидрохлорид 5-(3-азетидинилокси)-изохинолина, т.пл. 198oС, исходя из 5-(3-азетидинилокси)-1-(дифенилметил)-изохинолина.
Пример 6.
Гидрохлорид 3-[(2-метокси-1-нафталенил)окси]-азетидина.
а) К раствору 2,07 г 1-(дифенилметил)-3-[(2-метокси-1-нафталенил)окси] -азетидина в виде свободного основания в 20 мл 1,2-дихлорэтана при -15oС по каплям добавляют раствор 0,58 мл винилоксикарбонилхлорида в 20 мл 1,2-дихлорэтана в течение 15 мин и реакционную смесь поддерживают при этой температуре еще в течение получаса. Через 16 ч при температуре окружающей среды из капельной воронки добавляют этанол. Реакционную смесь выпаривают под вакуумом и остаток очищают на хроматографической колонке, используя толуол/этилацетат (95: 5), в результате чего получают 1,55 г твердого вещества, которое растворяют в 25 мл 2М раствора хлористоводородной кислоты в метаноле. После выстаивания при температуре окружающей среды в течение 16 ч раствор выпаривают под вакуумом и продукт перекристаллизовывают из этанола/диэтилового эфира. Выделяют 1,02 г гидрохлорида 3-[ (2-метокси-1-нафталенил)окси]-азетидина, т.пл. 187oС.
Аналогичным способом получают следующие соединения:
б) гидрохлорид 3-(1-нафталенилтио)-азетидина, т. пл. 159oС, исходя из 1-(дифенилметил)-3-(1-нафталенилтио)-азетидина,
в) гидрохлорид 3-[(2-метоксиметил)-1-нафталенил)окси] -азетидин, т.пл. 127oС, исходя из 1-(дифенилметил)-3-[(2-(метоксиметил)-1-нафталенил) окси] -азетидина.
Пример 7.
(R)-3-Метансульфонилокси-1-(фенилметил)-пирролидин.
а) 10 г (R)-1-(Фенилметил)-3-пирролидинола растворяют в 160 мл безводного толуола. Раствор перемешивают под током азота, охлаждают в этанольно-ледяной бане и добавляют 8,7 мл триэтиламина. При температуре -5oС в течение 1,5 ч по каплям добавляют 4,9 мл раствора метансульфонилхлорида в 110 мл безводного толуола, и реакционную смесь перемешивают в течение 1 ч при 0oС. Осадок отфильтровывают и промывают этилацетатом. Фильтрат промывают водой, сушат и выпаривают под вакуумом, в результате чего получают 13,9 г (R)-3-метансульфонилокси-1-(фенилметил)-пирролидина в виде почти бесцветного масла. M.S. (С.I.) (M/Z): 256 [М+Н]+.
Аналогичным способом получают следующие соединения:
б) (S)-3-метансульфонилокси-1-(фенилметил)-пирролидин, M. S. (C. I. ) (M/Z): 256 [М+Н]+, исходя из (S)-1-(фенилметил)-3-пирролидинола,
в) (рац)-3-метансульфонилокси-1-(фенилметил)-пирролидин, M. S. (C.I.) (M/Z): 256 [M+H]+, исходя из (рац)-1-(фенилметил)-3-пирролидинола.
Пример 8.
(S) -3-[(2,3-Дигидро-5-метокси-1Н-инден-4-ил)окси] -1-(фенилметил)-пирролидин.
а) В 540 мл безводного диметилформамида растворяют 5 г 2,3-дигидро-5-метокси-1Н-инден-4-ола. Раствор перемешивают, помещают в поток азота и добавляют 1,5 г 60% дисперсии гидрида натрия в масле. Реакционную смесь перемешивают при комнатной температуре в течение получаса. Температуру повышают до 100oС и по каплям в течение 1 ч добавляют раствор 7,78 г (R)-3-метансульфонилокси-1-(фенилметил)-пирролидина в 78 мл безводного диметилформамида. По каплям в течение 0,5 ч добавляют еще 3,0 г мезилата в 30 мл безводного диметилформамида и реакцию продолжают еще в течение 1,5 ч при 100oС. После выпаривания под вакуумом получают полутвердое вещество, которое распределяют между водой и этилацетатом. Этилацетатный экстракт сушат и выпаривают под вакуумом. Полученный продукт выделяют хроматографически, над диоксидом кремния, используя в качестве элюента толуол/этанол, в результате чего получают 9,45 г (S)-3-[(2,3-дигидро-5-метокси-1Н-инден-4-ил)окси] -1-(фенилметил)-пирролидина в виде масла. M.S. (C.I.) (M/Z): 324 [M+H]+.
Аналогичным способом получают следующие соединения:
б) (R)-3-[(2,3-дигидро-5-метокси-1Н-инден-4-ил)окси] -1-(фенилметил)-пирролидин, M. S. (С.I.) (M/Z): 324 [М+Н]+, исходя из 2,3-дигидро-5-метокси-1Н-инден-4-ола и (S)-3-метансульфонилокси-1-(фенилметил)-пирролидина,
в) (рац)-3-[(2, 3-дигидро-5-метокси-1Н-инден-4-ил)окси]-1-(фенилметил-пирролидин M.S. (С.I.) (M/Z): 324 [M+H]+, исходя из 2,3-дигидро-5-метокси-1Н-инден-4-ола и (рац)-3-метансульфонилокси-1-(фенилметил)-пирролидина,
г) 3-(1-нафталенилокси)-1-(фенилметил)-пирролидин M.S. (C.I.) (M/Z): 304 [М+Н]+, исходя из 1-нафтола,
д) 3-[(5,6,7,8-тетрагидро-1-нафталенил)окси]-1-(фенилметил)-пирролидин, M.S. (C.I.) (M/Z): 308 [M+H]+, исходя из 5,6, 7,8-тетрагидро-1-нафтола.
Пример 9.
(S)-(+)-3-[(2,3-Дигидро-5-метокси-1Н-инден-4-ил)окси]пирролидина гидрохлорид.
а) 9,4 г (S)-3-[(2,3-Дигидро-5-метокси-1Н-инден-4-ил)окси]-1-(фенилметил)-пирролидина растворяют в 300 мл безводного метанола и добавляют 2,0 г гидроксида палладия на углероде. Смесь гидрируют в установке Парра в течение 16 ч при давлении 350 кПа. Катализатор отфильтровывают через дикалит и промывают метанолом. Фильтрат концентрируют до его первоначального объема и добавляют 1 г свежего гидроксида палладия на углероде. Гидрирование продолжают в течение 3 ч.
Катализатор снова отфильтровывают, и фильтрат обрабатывают избытком 1М раствора хлористоводородной кислоты в диэтиловом эфире. После выпаривания и перекристаллизации из метанола/этилацетата/диэтилового эфира получают цветные кристаллы, которые промывают ацетоном и диэтиловым эфиром, в результате чего получают 3,95 г гидрохлорида (S)-(+)-3-[(2,3-дигидро-5-метокси-1Н-инден-4-ил)окси]-пирролидина, т.пл. 176oС.
Аналогичным способом получают следующее соединение:
б) гидрохлорид 3-[(5,6,7,8-тетрагидро-1-нафталенил)окси]-пирролидина, т. пл. 207oС, исходя из 3-[(5,6,7,8-тетрагидро-1-нафталенил)окси]-1-(фенилметил)-пирролидина
Пример 10
(R)-(-)-3-[(2,3-Дигидро-5-метокси-1Н-инден-4-ил)окси] -пирролидина гидрохлорид
а) 800 мг (R)-3-[(2,3-Дигидро-5-метокси-1Н-инден-4-ил)окси] -1-(фенилметил)-пирролидина растворяют в 150 мл безводного метанола и добавляют 1,5 эквивалента хлористоводородной кислоты, растворенной в этилацетате. Добавляют приблизительно 80 мг палладия на углероде 10% и через смесь при перемешивании пропускают ток водорода. Через 24 ч добавляют новый катализатор и гидрирование продолжают в течение 17 ч. Катализатор отфильтровывают, фильтрат выпаривают под вакуумом и продукт перекристаллизовывают из этанола/этилацетата/диэтилового эфира, в результате чего получают 360 мг гидрохлорида (R)-(-)-3-[(2, 3-дигидро-5-метокси-1Н-инден-4-ил)окси]-пирролидина, т. пл. 174oС.
Аналогичным способом получают следующие соединения:
б) (рац)-3-[(2,3-дигидро-5-метокси-1Н-инден-4-ил)окси] пирролидина гидрохлорид, т. пл. 154oС, исходя из (рац)-3-[ (2,3-дигидро-5-метокси-1Н-инден-4-ил)окси]-1-(фенилметил)-пирролидина,
в) (рац)-3-[(1-нафталенил)окси] пирролидина гидрохлорид, т.пл. 222oС, исходя из (рац)-3-[(1-нафталенил)окси]-1-(фенилметил)-пирролидина.
Пример 11
(+)-3-[(1-Нафталенил)окси]-пирролидина гидрохлорид.
(-)-3-[(1-Нафталенил)окси]пирролидина гидрохлорид.
3-[(1-Нафталенил)окси] -пирролидин (80 мг) разделяют методом препаративной хиральной ВЖХЛ на отдельные энантиомеры. Разделение осуществляют при комнатной температуре на колонке с Chiracel OD размером 240•4,6 мм с помощью гексана/этанола (80:20) и 0,15% диэтиламина, скорость течения 1 мл/мин.
(+)-3-[(1-Нафталенил)окси]-пирролидин собрали при tR 7,4 мин,
(-)-3-[(1-нафталенил)окси]-пирролидин собрали при tR 9,8 мин.
Растворы немедленно выпаривают под вакуумом и превращают в их гидрохлориды, в результате чего получают по 10 мг каждого. Определенная энантиочистота для обоих этих энантиомеров составила >99,5%.
ИСПЫТАНИЯ
Активность соединений настоящего изобретения по отношению к центральной нервной системе была подтверждена с помощью нижеописанных фармакологических испытаний; эти испытания продемонстрировали серотонинэргическую активность и эффекты, аналогичные антидепрессантам, для соединений настоящего изобретения.
ИСПЫТАНИЯ НА СВЯЗЫВАНИЕ
Эти испытания проводят, используя клонированные рецепторы человека, выраженные в ЗТЗ клетках, в соответствии с протоколами, описанными в публикации Stam с соавт. "Genomic organization, coding sequence and functional expression of human 5-HT2 and 5-HT1A receptor genes" - European Journal of Pharmacology - Molecular Pharmacology Section 227: 153-162 (1993) и Stam с соавт. "Genomic organization and functional expression of the gene encoding the human serotonin 5-HT2C receptors" - European Journal of Pharmacology - Molecular Pharmacology Section 269: 339-348 (1994). Сходство к 5-НТ2А и 5-HT2C рецепторам детерминируется способностью соединений настоящего изобретения вытеснять [3Н]-кетансерин и [3Н]-мезулергин из соответствующего рецептора, табл. 1, 2.
ЭРЕКТАЛЬНЫЙ ТЕСТ
Этот тест, проводимый в соответствии с протоколом Beredsen с соавт. ["In volment of 5-HT1C receptors in drug-induced penile erection in rats" - Psychopharmacology 101: 57-61 (1990)] дает указание на потенциальную активность, аналогичную антидепрессантам.
Полученные результаты показывают, что соединения настоящего изобретения имеют большее сродство к 5-НТ2С рецепторам человека, чем к 5-НТ2А рецепторам человека: и это сродство коррелирует с активностью агониста in vivo, а также с активностью, аналогичной антидепрессантам, в живых моделях для эффективности антидепрессанта.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЗАМЕЩЕННЫЕ БЕНЗИЛАМИНЫ И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ЛЕЧЕНИЯ ДЕПРЕССИИ | 1997 |
|
RU2179553C2 |
| ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ ПИПЕРИДИНА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1998 |
|
RU2198172C2 |
| ИНГИБИТОРЫ СЕРИНОВЫХ ПРОТЕАЗ | 1998 |
|
RU2183642C2 |
| ИНГИБИТОРЫ СЕРИНОВЫХ ПРОТЕАЗ | 1997 |
|
RU2172321C2 |
| ИНГИБИТОР СЕРИНОВЫХ ПРОТЕАЗ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ | 1999 |
|
RU2232760C2 |
| ИНГИБИТОРЫ ПРОТЕАЗЫ СЕРИНА | 1997 |
|
RU2178419C2 |
| СТЕРОИДНОЕ СОЕДИНЕНИЕ, СПОСОБЫ ЕГО ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1998 |
|
RU2182153C2 |
| ПРОИЗВОДНЫЕ ИМИДАЗО[1,5-А]ПИРИДИНА КАК ИНГИБИТОРЫ СЕРИНПРОТЕАЗ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1996 |
|
RU2175327C2 |
| СТЕРОИД С 17-СПИРОМЕТИЛЕНЛАКТОНОВОЙ ГРУППОЙ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 1995 |
|
RU2168516C2 |
| ПРОИЗВОДНЫЕ 11,21-БИСФЕНИЛ-19-НОРПРЕГНАНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 1995 |
|
RU2152952C2 |



Изобретение относится к новым производным азетидина и пирролидина общей формулы
где А представляет необязательно ненасыщенное 5- или 6-членное кольцо, которое может содержать гетероатом, выбранный из N и S, и которое может быть замещено оксо- или (1-6С) алкилом; R1, R2 и R3 независимо друг от друга представляют собой Н, (1-6С)алкил, (1-6С)алкокси, (1-6С)алкокси-(1-6С)алкил, атом галогена; Х означает атом О или S и n = 1 или 2, или его фармацевтически приемлемая соль, за исключением 3-(нафт-1-ил-окси)-пирролидина и 3-(5,6,7,8-тетрагидро-нафт-1-ил-окси)-пирролидина. Соединения I обладают сродством к 5-НТ2С-рецептору, что позволяет использовать их в фармацевтической композиции для лечения или предотвращения серотонинзависимых расстройств. 2 с. и 5 з.п. ф-лы, 3 табл.

где А - необязательно ненасыщенное 5- или 6-членное кольцо, которое может содержать гетероатом, выбранный из N и S, и которое может быть замещено оксо- или (1-6С)алкилом;
R1, R2 и R3 независимо друг от друга - атом Н, (1-6С)алкил, (1-6C)алкокси, (1-6С)алкокси-(1-6C)алкил, атом галогена;
Х - атом О или S;
n = 1 или 2,
или его фармацевтически приемлемая соль, за исключением 3-(нафт-1-ил-окси)-пирролидина и 3-(5,6,7,8-тетрагидро-нафт-1-ил-окси)-пирролидина.
где А - незамещенное насыщенное 5-членное или необязательно ароматическое 6-членное кольцо, которое может содержать атом азота, соседний с положением, обозначенным звездочкой;
R1 - атом Н или (1-6С)алкокси;
R2 - атом Н, (1-6С)алкокси или галогена;
R3 - атом Н или галогена;
n = 1 или 2;
4. Соединение по п.3, отличающееся тем, что А - незамещенное насыщенное 5-членное или необязательно ароматическое 6-членное кольцо, R1 - (1-6С) алкокси и R2 и R3 - атом Н.
| ЕР 0338331 A1, 25.10.1989 | |||
| DE 1964510, 23.12.1969 | |||
| US 4452809, 05.06.1984 | |||
| Способ получения производных пирролидин (или пиперидин)-карбоксальдегида | 1979 |
|
SU791227A3 |

