Изобретение относится к ряду циклоалкилзамещенных производных глутарамида, которые являются гепотензивными агентами, используемыми для лечения различных заболеваний сердечно-сосудистой системы, включая гипертонию и разрыв сердца.
Согласно описанию Европейской патентной заявки 274234 мы раскрыли некоторые циклоалкил-замещенные производные глутарамида, которые являются ингибиторами цинк-зависимой нейтральной эндопептидазы E.C.3.4.24.11 которые, таким образом, могут снижать биологическое действие атриального натриуретического фактора и, в частности, являются нитриуретиками, гипотензивными и диуретическими агентами, ценными для лечения различных заболеваний сердечно-сосудистой системы.
Соединения данного изобретения являются также ингибиторами энзима E.C. 3.4.24.11 и, кроме того, они также могут ингибировать ангеотензивный конвертирующий энзим, другой энзим, который участвует в процессе регулирования давления крови. Таким образом, данные соединения обладают двойным фармакологическим действием, связанным с ингибированием двух ключевых энзимов, принимающих участие в процессе регулирования давления крови, что делает эти соединения особенно ценными для лечения различных форм гипертонии и связанных заболеваний сердечно-сосудистой системы и глаукомы.
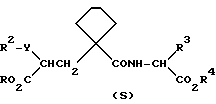
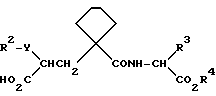
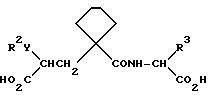
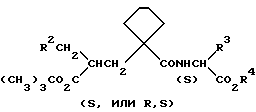
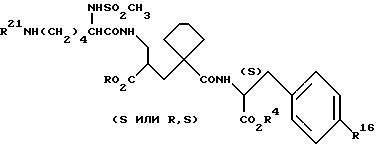
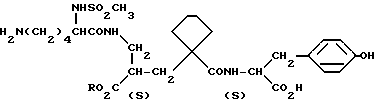
Данные соединения описываются формулой (I)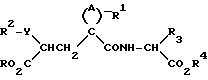
где
A означает (CH2)4 или (CH2)5 или образует с атомом углерода насыщенный или мононенасыщенный цикл; R1 означает водород или (C1-C4)алкил; R и R4 каждый независимо обозначает водород, (C1-C6)алкил, (C3-C7) циклоалкил, бензил или биолабильную эфирообразующую группу;
Y обозначает простую связь или (C1-C6) алкилен с прямой или разветвленной цепью;
R2 обозначает водород, фенил, R6CONR5- или R7NR5CO-;
R5 представляет водород, (C1-C6) алкил или фенил (C1C6)алкил;
при условии, что Y не обозначает простую связь, когда R2 обозначает водород или фенил, R6 и R7 каждый обозначает группу формулы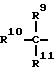
где
R9 обозначает H, OH, (C1-C6) алкил, фенил (C1-C6) алкил, гидрокси (C1-C6) алкил, R12SO2NH, (R13)2N;
R10 и R11 обозначают каждый, независимо, H или (C1-C6) алкил, или R10 обозначает H, R11 обозначает амино (C1-C6) алкил, имдазолилметил, фенил, 4-гидроксифенил, бензилокси (C1-C6) алкил, гидрокси (C1-C6) алкил, метилтио (C1-C6) алкил, или две группы R10 и R11 вместе с атомом углерода, к которому они присоединены, образуют 3-6-членное карбоциклическое кольцо или пирролидиновое, которое может быть необязательно замещено аминогруппой или бензилом, R12 обозначает (C1-C6) алкил, (C3-C7) циклоалкил, фенил, 4-хлорфенил, фенил (C1-C6) алкил, фурил или пиридил;
R13 обозначает H, (C1-C6) алкил, фенил (C1-C6) алкил;
R3 обозначает группу формулы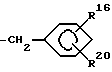
где R16 обозначает H, гало-ид, 4-OH, 4-(C1-C6) алкокси, 4-[(C1-C6) алкоксикарбонилокси] , 3-[(C1-C4) алкил SO2NH], 4-[(C3-C7) циклоалкоксикарбонилокси], R20 обозначает H, (C1-C4) алкил, (C1-C4) алкоски, (C2-C6) алканоил или галоид, или R3 обозначает группу формулы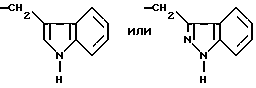
или их фармацевтически приемлемые соли, или их биолабильные эфиры.
В вышеприведенных значениях, за исключением особо оговоренных случаев, алкильные группы, имеющие от трех или больше атомов углерода, могут быть прямыми или разветвленными.
Соединения формулы (I) могут содержать несколько центров асимметрии и таким образом они могут существовать в виде энантиомеров и диастереомеров. Данное изобретение включает оба отдельных изомеров, а также смеси изомеров.
Фармацевтически приемлемыми солями соединений формулы (I), содержащими кислотные центры, являются те соли, которые образованы основаниями и являются нетоксичными. Примеры включают соли щелочных и щелочно-земельных металлов, такие как натриевые, калиевые или кальциевые соли и соли с аминами, такими как диэтиламин. Соединения, содержащие основной центр, также могут образовывать соли при добавлении фармацевтически приемлемых кислот. Примеры включают гидрохлорид, гидробромид, сульфат или бисульфат, фосфат или кислый фосфат, ацетат, цитрат, фумарат, глюконат, лактат, малеат, сукцинат, тартрат, тозилат и лаурилсульфат.
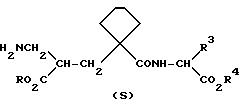
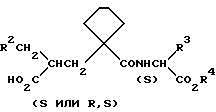
Предпочтительными группами соединений формулы (I) являются те, в которых символ A означает (CH2)4 и радикал R1 представляет собой H, например соединения формулы (II), в которых радикалы R, R2, R3 и R4 имеют значения, указанные выше для формулы (I)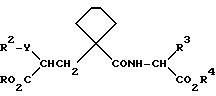
Предпочтительными являются также те соединения формулы (I) и (II), в которых оба радикала R и R4 представляют собой H (двухосновные кислоты), а также их биолабильные моно и ди-сложноэфирные производные, в которых один или оба радикала R и R4 представляют собой биолабильную, образующую сложный эфир группу.
Термин "биолабильная, образующая сложный эфир группа" хорошо ясен специалистам и обозначает группу, которая обеспечивает образование сложного эфира, который может легко расщепляться в организме и выделять соответствующую двухосновную кислоту формулы (I), где оба радикала R и R4 представляют собой H. Количество таких сложноэфирных групп хорошо известно, например, в области пенициллина или в случае ACE-ингибирующих гипотензивных агентов.
В случае соединений формул (I) и (II) такие биолабильные, предшествующие препарату сложные эфиры являются особенно предпочтительными для получения соединений формулы (I), пригодных для орального введения. Применимость любой конкретной сложноэфирообразующей группы можно оценить с помощью традиционных методов гидролиза энзима в организме животного или in vitro. Таким образом, для достижения оптимального эффекта желательно, чтобы сложный эфир гидролизовался только после поглощения, соответственно, сложный эфир должен обладать гидролитической стабильностью к пищеварительным энзимам до поглощения, но должен легко гидролизоваться, например, под воздействием энзимов желудка, плазмы или печени. Таким образом происходит выделение активной двуосновной кислоты в кровь после орального приема.
Помимо низших алкиловых сложных эфиров (особенно этиловых) и бензиловых сложных эфиров приемлемые биолабильные сложные эфиры включают алканоилоксиалкиловые сложные эфиры, включая их алкил, циклоалкил и арил замещенные производные, ароилоксиалкиловые сложные эфиры, арилэфиры, аралкилэфиры, галоилкиловые сложные эфиры и оксиалкиловые сложные эфиры, включая их кетальные производные, в которых указанные алканоильные или алкильные группы содержат от 1 до 8 атомов углерода и которые являются прямыми или разветвленными и указанными арильными группами являются фенил, нафтил или инданил, необязательно замещенный одним или несколькими C1-C4 алкилом, C1-C4 алкокси или C1-C4 алкоксикарбонильной группами или атомами галогенов.
Примеры радикалов R и R 4, когда они являются биолабильными сложноэфирными группами, включают этил, инданил, изопропил, н-бутил, втор-бутил, трет-бутил, циклогексил, бензил, фенетил, фенипропил, ацетонил, глицерил, пивалоилоксиметил, 5-/4-метил-1,3-диоксолен-2-онил/метил, циклогексилметил, циклогексилкарбоксиэтил, циклогексилацетоксиэтил, пропионилоксиизобутил, гексаноилоксиэтил, пентаноилоксиэтил, ацетоксиэтил, ацетоксибензил, пентаноилоксибензил, циклогексилоксикарбонилоксиэтил, бутилоксикарбонилоксиэтил, изобутилоксикарбонилэтил и этоксикарбонилоксиэтил.
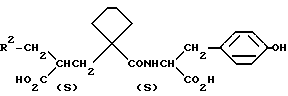
В одном из предпочтительных аспектов данного изобретения группа R4 представляет собой 4-оксибензил и атом углерода, к которому она присоединена, является (S) стереохимическим; группа NHCH (R3) CO2R4 является производной от L-тирозина. Предпочтительными являются также соединения, в которых R3 представляет 4-метоксибензил или 3-метансульфонамидобензил.
В других аспектах данного изобретения R2 представляет собой H и Y - (CH2)3) или R2 означает фенил и Y -(CH2)2.
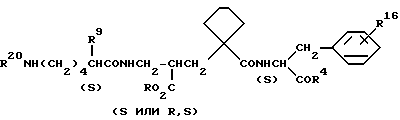
В другом аспекте изобретения радикал R2 обозначает R6CONR5 и Y означает CH2, радикал R3 представляет собой 4-оксибензил, 4-метоксибензил или 3-метансульфонамидобензил и R6 представлен формулой R9 R10 R11 C-, где R9 обозначает (R13)2N-, (R12)CO2NH- или R12CONH-, где R12 и R13 определены выше, R10 обозначают H и R11 обозначает амино (C1-C6) алкил.
Особенно предпочтительными являются соединения формулы 1, где R6CO обозначает (S)-лизил или N2 - замещенный -(S)- лизил формулы R9 R10 R11 CCO -, где R9 обозначает NH2 R12 CONH или R12SO2NH и R12 определен выше, R10 обозначает H и R11 обозначает 4-аминобутил.
Предпочтительными соединениями формулы (I) являются соединения, выбранные из
N-[1-(2-(S)-карбокси-3-(S)-дизиламинопропил)-1- циклопентанкарбонил] -(S)-тирозина,
N-{ 1-[2(S)-карбокси-3-(N2-метансульфонил-(S)-лизиламино) пропил]-1-циклопентанкарбонил}-(S)-тирозина,
N-{ 1-[(2S)-карбокси-3-(N2-2-фуроил-(S)-лизиламино)-пропил]-1- циклопентанкарбонил}-(S)-тирозина,
N-{ 1-[2(S)-карбокси-3-(N2-ацетил-(S)-лизиламино)пропил] - 1-циклопентанкарбонил}-(S)-4-метоксифенилаланина,
N-[1-(2-карбокси-3-(S)-лизиламинопропил-1-циклопентанкарбонил] -(S)-3-метансульфонамидофенилаланина,
N-{ 1-(2-карбокси-3-(N2-метансульфонил-(S)-лизиламино)- пропил] -1-циклопентанкарбонил}-(S)-метанльсуфонамидофениланина.
N-{ 1-[2(S)-карбокси-3-(N2-ацетил-(S)-лизиламино)- пропил] 1-циклопентаткарбонил}-(S)-3-метансульфонамидофенилаланина и
N-{1-[2(S)-карбокси-3-(N2-фенилсульфонил-(S)-лизиламино) пропил}-1-циклопентанкарбонил}-(S)-тирозина.
Соединения формулы (I) получают различными способами.
а) Один из способов включает синтез частично защищенного циклоалкил-замещенного производного глутаровой кислоты, которое при взаимодействии со сложноэфирным производным аминокислоты образует целевой глутарамид. Любые реакционноспособные группы в R2 и R3' могут потребовать защиты на стадии реакции сочетания, и такие защитные группы удаляют на конечной стадии процесса.
Схема синтеза проиллюстрирована приведенной ниже схемой реакций, где радикалы A и R1 имеют значения, указанные выше, радикалы R2' и R3' имеют те же значения, что и радикалы R2 и R3 с любыми содержащимися в них реакционноспособными группами, при необходимости защищенными, и радикалы R17 и R18 имеют значения, указанные выше для радикалов R и R4, исключен H, или они являются традиционными защитными группами карбоновых кислот.
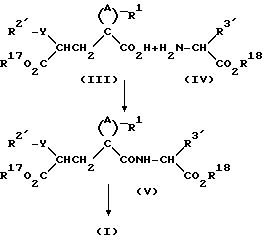
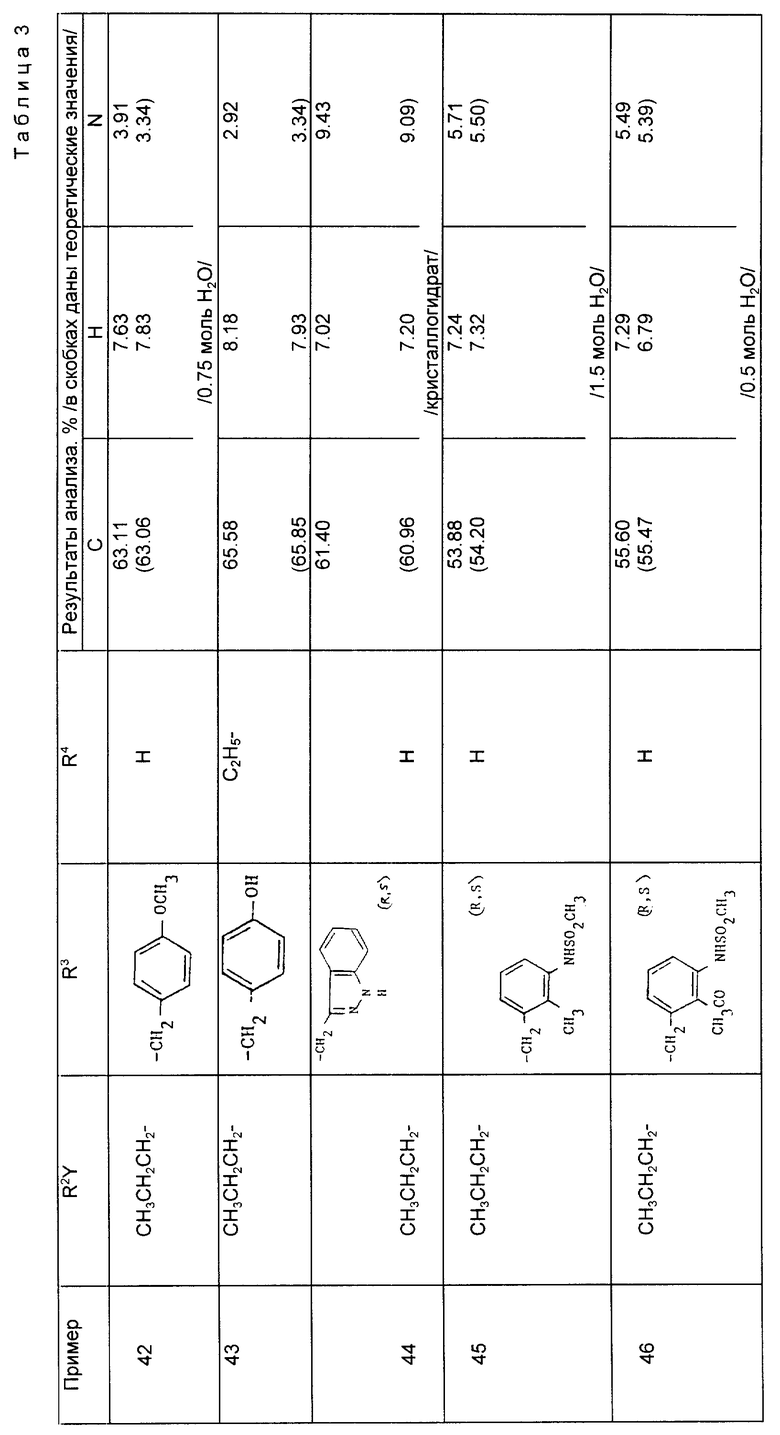
Реакцию соединений формул (III) и (IV) осуществляют с помощью традиционных методов сочетания амида. Таким образом, в одном способе реакции осуществляют с помощью реактантов, растворенных в органическом растворителе, например дихлорметане, используя димидный агент конденсации, например 1-этил-3-/диметиламинопропил/- карбодиимид, или N, N'-дициклогексилкарбодиимид, предпочтительно в присутствии 1-оксибензотриазола и органического основания, такого как N-метилморфолин. Реакция обычно завершается в течение 12-24 ч при комнатной температуре, в продукт затем выделяют обычными методами, например промыванием водой или фильтрованием с целью удаления побочного продукта - мочевины - и выпариванием растворителя. После этого продукт можно очистить кристаллизацией или хроматографически, если в этом есть необходимость.
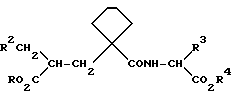
Соединения формулы (V) включают соединения формулы (I), где R и R4 представляют собой C1-C6 алкил или бензил.
Далее осуществляют реакцию сложных диэфиров формулы (V) с получением сложного моноэфира или производных двуосновной кислоты формулы (I), где один или оба радикала R и R4 представляют собой H. Условия проведения реакции будут зависеть от природы групп R17 и R18, присутствующих в соединении формулы (V), и здесь возможны варианты. Так, например, когда обе радикала R17 и R18 представляют собой бензил, в результате гидрогенизации продукта будет образовываться двуосновная кислота формулы (I), где оба радикала R и R4 означают H. В другом варианте, если один из радикалов R17 и R18 означает бензил, а другой - алкил, в результате гидрогенизации будет образовываться сложный моноэфир. Его затем можно подвергнуть гидролизу, если необходимо, и вновь получить двуосновную кислоту. Когда один из радикалов R17 и R18 представляет собой трет-бутил, в результате обработки соединения формулы (V) трифторуксусной кислотой или хлористым водородом образуется соответствующая кислота. Если для радикалов R17 и R18 используются другие защитные группы карбоновой кислоты, то для их удаления на конечной стадии должны быть использованы совершенно определенные условия, чтобы получить сложный эфир или двуосновную кислоту формулы (I). Например, когда радикал R17 или R18 представляет собой триметилсилилэтил, его можно удалить путем обработки фторидом тетрабутиламмония. Любые защитные группы, содержащиеся в R2' и R3', также должны быть удалены, и это можно осуществить одновременно с удалением защитных групп, содержащихся в радикалах R17 и R18, или на отдельной стадии с помощью методов, соответствующих конкретному типу используемых защитных групп. Так например, когда R2' содержит замещенную или защищенную аминогруппу (например, бензиламино, дибензиламино, бензилоксикарбониламино или трет-бутилоксикарбониламиногруппу), то соединения можно превратить в свободные амины путем гидрогенизации или гидролиза.
б) В другом варианте процесса соединения формулы (I), где R1 означает R6CO NR5-, получают способом, который предусматривает реакцию амина формулы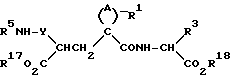 (IV)
(IV)
где радикалы A, Y, R1, R3, R5, R17 и R18 имеют значения, указанные выше, с карбоновой кислотой R6CO2H
или реакционноспособным производным карбоновой кислоты, где радикал R6 имеет значения, указанные выше, и где любые реакционноспособные группы необязательно защищены, в результате чего образуется, например, соединение формулы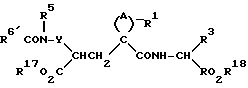
где радикалы R6' имеют те же значения, что указаны для радикалов R6, с любыми реакционноспособными группами в нем, необязательно защищенными, их последующим удалением любых защитных групп, если они присутствуют, и, если необходимо, гидролизом сложного эфира с получением соединений формулы (I), где радикалы R и R4 обозначают H.
Аналогичным образом, в результате реакции с сульфонилхлоридом образуются соответствующие сульфонамиды.
Реакцию амина формулы (VI) в соединения формулы R6CO2H осуществляют с помощью традиционного метода сочетания амида, как описано выше. Последующее удаление защитных групп достигается с помощью соответствующих методов, как указано выше.
Амины формулы (VI) получают по той же методике, что описана в пункте (а) выше, не используют кислоту формулы (Е), где радикал R2' означает защищенный амин формулы R19NR5-, где радикал R5 имеет значения, указанные выше, а радикал R19 представляет собой амино-защитную группу.
Таким образом, в одном варианте этого процесса реакцию сочетания с производным аминокислоты осуществляют, используя соединение формулы (III), где радикал R2' представляет собой R19R5N- и радикалы R19 и R5 означают оба бензил. В другом варианте оба радикала R19 и R5 представляют собой 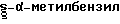 , что обеспечивает возможность выделения S - изомера соединения формулы (V). Гидрогенизация продукта сочетания формулы (V) дает амин формулы (VI), где радикал R5 представляет собой H. Затем осуществляют его взаимодействие, например, с защищенным производным лизина формулы R6'CO2H (где R6' представляет собой R9R10R11C-, радикал R9 означает защищенную аминогруппу или R12CO NH-, R12SO2 NH-, радикал R10 представляет собой N-зазищенный -4-аминобутил и R11 означает H, в результате удаления защитных групп у полученного продукта образуется соответствующий продукт формулы (I), где R6CO представляет собой (S)-лизин или N2-защищенный -(S)-лизил.
, что обеспечивает возможность выделения S - изомера соединения формулы (V). Гидрогенизация продукта сочетания формулы (V) дает амин формулы (VI), где радикал R5 представляет собой H. Затем осуществляют его взаимодействие, например, с защищенным производным лизина формулы R6'CO2H (где R6' представляет собой R9R10R11C-, радикал R9 означает защищенную аминогруппу или R12CO NH-, R12SO2 NH-, радикал R10 представляет собой N-зазищенный -4-аминобутил и R11 означает H, в результате удаления защитных групп у полученного продукта образуется соответствующий продукт формулы (I), где R6CO представляет собой (S)-лизин или N2-защищенный -(S)-лизил.
в) Соединения формулы (I), где радикал R2 представляет собой R7NR6CO- получают совершенно аналогичным методом, что и метод, описанный выше, но используя в качестве исходных соединений карбоновую кислоту формулы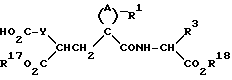
где радикалы A, Y, R1, R17 и R18 имеют значения, указанные выше, и осуществляя взаимодействие с амином формулы R17R5NH, с последующим удалением защитных групп, если таковые присутствуют, и, при необходимости, гидролизом или гидрогенизацией сложного эфира с получением соединений формулы (I), где радикалы R и R4 представляют собой H.
г) В другом варианте этих способов сочетание осуществляют, используя соединение формулы (IV), где радикал R3' представляет собой остаток формулы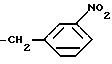
В результате последующего восстановления нитрогруппы и сульфонирования продукта сульфонилгалогенидом формулы (C1-C4) алкил SO2Cl образуется соответствующее соединение формулы (V), где радикал R3' представляет собой остаток формулы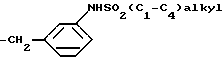
Соединения формулы (I), где один или оба радикала R и R4 представляют собой биолабильную, образующую сложный эфир группу, получают по методикам, аналогичным тем, что описаны выше, используя соответствующие сложноэфирные группы для R или R4.
Кроме удаления любой защитной группы, которую может содержать радикал R2', возможно протекание реакций различных химических превращений конечного сложного моноэфира или двуосновной кислоты, как это описано выше. В любом случае целевой продукт может быть получен в виде свободной карбоновой кислоты или она может быть нейтрализована соответствующим основанием и выделена в виде соли.
Соответствующие методики осуществления реакций сочетания и защиты для всех описанных выше стадий и возможные варианты и методики хорошо известны специалистам из соответствующих справочников и будут ясны из приведенных ниже примеров.
Исходные спирозамещенные сложные моноэфиры глутаровой кислоты формулы (III) можно получить, как описано в нашей Европейской патентной заявке 274234. Сложные эфиры аминокислот формулы (IV) обычно являются хорошо известными соединениями, которые либо производятся в промышленности, либо могут быть получены с помощью стандартных методов, описанных в литературе.
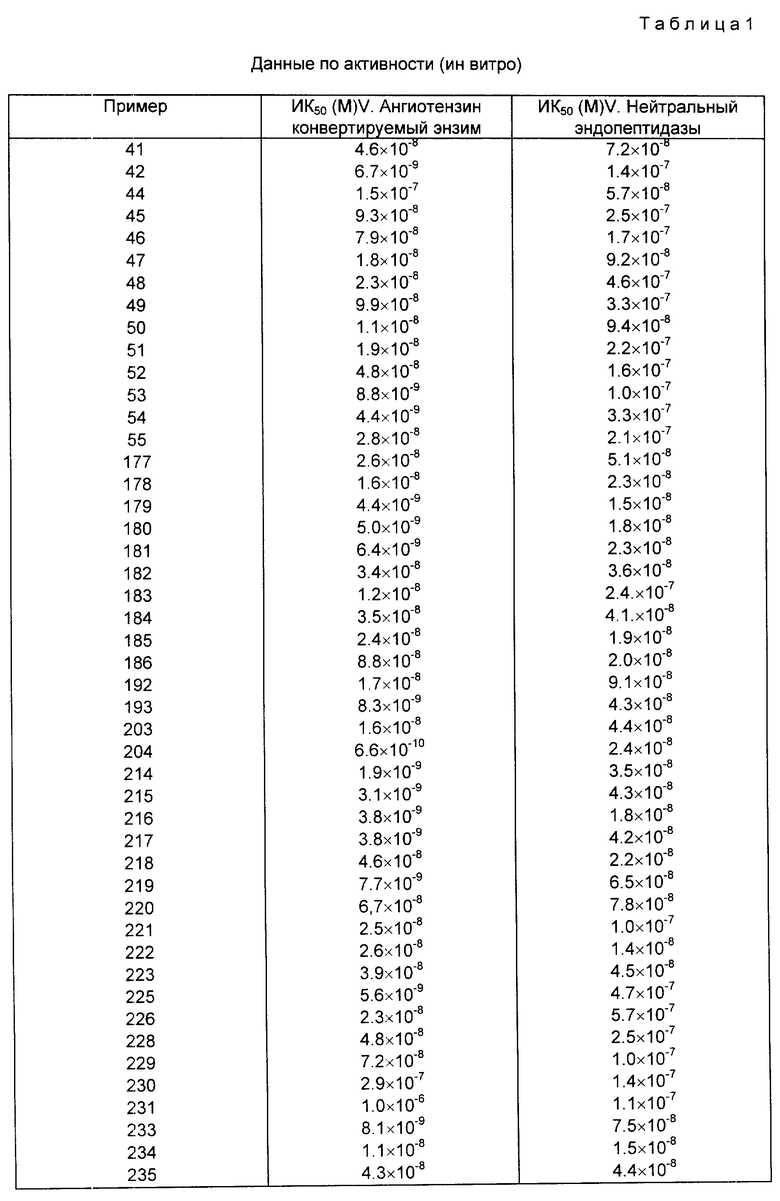
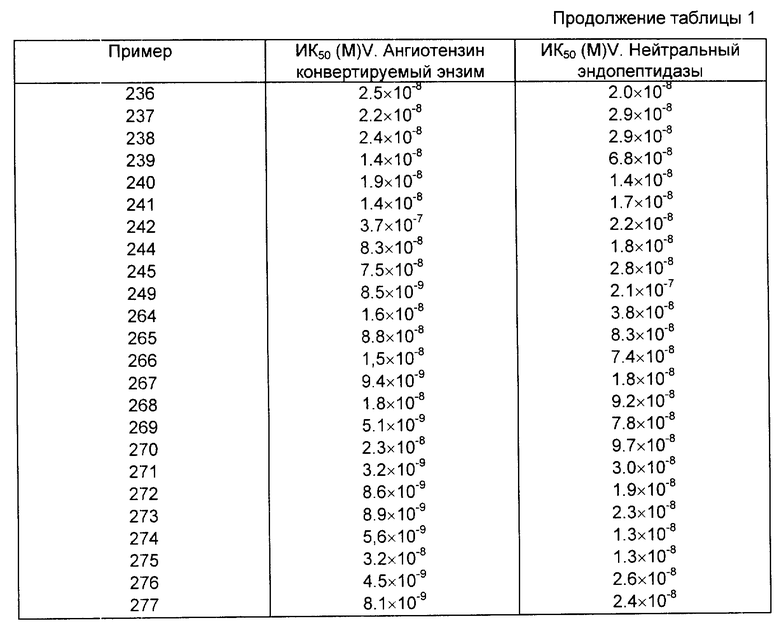
Как отмечалось выше, соединения данного изобретения являются эффективными ингибиторами нейтральной эндопептидазы (Е.С.3.4.24.11). Этот энзим участвует в разрушении ряда пептидных гормонов, включая, в частности, разрушение атриального натриуретического фактора (АНФ). Таким образом, соединения данного изобретения, предотвращая разрушение АНФ под действием эндопептидазы Е. С. 3.4.24.11, могут усиливать его биологическое действие и являются таким образом диуретиками, натриуретиками и гипотензивными агентами, которые могут использоваться при ряде заболеваний, в том числе гипертонии, сердечной недостаточности, ангине, почечной недостаточности, при менструальном синдроме, циклических отеках, болезни Меньереса, гиперальдостеронеизме (первичном и вторичном) и гиперкальциурии. Кроме того, вследствие своей способности усиливать действие АНФ соединения данного изобретения могут быть использованы для лечения глаукомы. Другим следствием их способности ингибировать нейтральную эндопептидазу Е.С.3.4.24.11 является эффективность соединений данного изобретения в других областях терапии, включая например, лечение астмы, воспалительных заболеваний, болей, эпилепсии, болезненных расстройств, слабоумия и герметрического смешения, ожирения и болезней желудочно-кишечного тракта (особенно диарееи и воспалительного синдрома кишечника), снижение секреции желудочного сока и лечение гиперренинамии.
Активность против нейтральной эндопептидазы Е.С.3.4.24.11 оценивается по методике, основанной на пробе, описанной J.T.Gafford, R.A. Skidgel, E.G. Erdos and L.B. Hersch, Biochemistry, 1983, 32, 3265 - 3271.
Этот метод предусматривает определение концентрации соединения, необходимой для снижения на 50% скорости выделения радиомеченной гиппуровой кислоты из гиппурил-L-фенилаланил-L-аргинина под действием препарата нейтральной эндопептидазы из почки крысы.
Как указано выше, соединения данного изобретения являются также ингибиторами ангиотензивного ковертирующего энзима. Как таковые они могут быть использованы при лечении других различных состояний, для которых используются ингибиторы АСЕ, включая ограничение ишемического поражения миокардом, защита почек от повреждения гиперфильтрации, предотвращение обратимой левой вентрикулярной гипертрофии, улучшение памяти, контроль познавательной функции, слабоумие и предотвращение реокклюзин с последующей коронарной ангиопластомией или операцией обходной коронарной артерии. ИК действие против этого энзима оценивают с помощью модифицированной методики, основанной на пробе, описанной Rohrbach, M.S., Anal. Biochem., 1978, 84, 272. Этот метод предусматривает определение концентрации соединения, необходимой для снижения на 50% степени выделения радиомеченной гиппуровой кислоты из гиппурил-L-гистидил-L-лейцина под действием ангиотензивного конвертирующего энзима, выделенного из крысиных почек (табл. 1).
Ингибирующую активность измеряют также in vivo после внутривенного вливания в организм анестезированных крыс, используя методы, описанные I.L.Notoff et al., Journal of Pharmacological Metods, 1981, 5, 305; D.M. Gross et al. J. Pharmacol. Exp. Ther., 1981, 216, 552, и определяют дозировку ингибитора, необходимого для снижения отклика, вызванного внутривенным вливанием ангиотензина 1 (50 нг пищевой массы) на 50%.
Активность соединений данного изобретения как диуретиков определяется путем измерения их способности увеличивать выход мочи и ионов натрия из организма мышей, насыщенного солевым раствором. В этом опыте самцы мышей (Charles River [D], 22 - 28 г) выдерживают без пищи в течение ночи в клетках.
Мышам вводят внутривенно через хвостовую вену испытуемое соединение, растворенное в объеме солевого раствора, эквивалентном 2,5% массы тела. Пробы мочи собирают каждый час в течение двух часов в предварительно взвешенные пробирки и анализируют их на концентрацию электролита. Объем мочи и концентрацию ионов натрия у подопытных животных сравнивают с контрольной группой, которая получала только солевой раствор.
Гипотензивное действие данных соединений оценивается путем измерения падения кровяного давления после орального или внутривенного введения в организм обессоленных, подвергнутых воздействию двуретика крыс с самопроизвольным повышением давления, обессоленных собак с почечно-индуцированной гипертонией или крыс с индуцированной ДОСА/соль гипертонией.
Для введения в организм человека для лечения или профилактики гипертонии, острой сердечной недостаточности или почечной недостаточности дозировки для орального введения обычно будут составлять от 3 до 1500 мг ежедневно для пациента среднего возраста (70 кг). Таким образом, для взрослого пациента отдельные таблетки или капсулы содержат от 1 до 500 мг активнодействующего соединения, в соответствующем фармацевтически приемлемом носителе или таковые для введения в виде единичной или множественной дозы, один или несколько раз в день. Дозировки для внутривенного введения обычно будут лежать в пределах от 1 до 500 мг на дозу. На практике врач определит действующую дозировку, которая будет наилучшим образом соответствовать конкретному пациенту и будет зависеть от возраста, веса и отклика конкретного пациента. Вышеназванные дозировки являются примерами среднего случая и, безусловно, в конкретных случаях могут быть выше или ниже, а такие дозировки также входят в объем изобретения.
Для использования в медицинской практике соединения формулы (I) могут вводиться отдельно, но обычно их вводят в смеси с фармацевтическим носителем, выбираемым в зависимости от предполагаемого способа введения и стандартной фармацевтической практики. Например, они могут вводиться орально в виде таблеток, содержащих такие ингредиенты, как крахмал или лактоза, или в капсулах либо самостоятельно, либо в смеси с ингредиентами, или же в виде элексиров или суспензий, содержащих отдушки или окрашивающие добавки. Их можно вводить парэнтерально, например внутривенно, внутримышечно или подкожно. Для парэнтерального введения их лучше всего использовать в виде стерильного водного раствора, который может содержать другие вещества, например достаточное количество солей или глюкозу, обеспечивающее изотоничность раствора с кровью.
Соединения данного изобретения можно вводить совместно с другими агентами, что может быть необходимо для регулирования кровяного давления или лечения сердечного состояния или легочной недостаточности. Так, например, их можно вводить совместно с дигиталисом или другим стимулирующим сердечную деятельность препаратом либо с альфа-блокатором, бета-блокатором, эксогенным АНФ или с калиевым канальным активатором или другим дауретиком, что определяет врач для конкретного пациента или степени заболевания.
Таким образом, в другом аспекте данное изобретение касается фармацевтической композиции, содержащей соединение формулы (I) или (II), или его фармацевтически приемлемую соль, или его биопредшественник, вместе с фармацевтически приемлемым разбавителем или носителем.
Данное изобретение также включает соединения формулы (I) или (II), или их фармацевтически приемлемые соли, или их биопредшественники, предназначенные для использования в медицине, в частности для лечения гипертонии, острой сердечной недостаточности или легочной недостаточности.
Способы получения соединений данного изобретения и промежуточных соединений, используемых при их получении, проиллюстрированы следующими примерами.
Пример 1. N-[1-/2-трет-Бутилоксикарбонил-3-дибензиламинопропил/-1-циклопентанкарбонил]- O-трет-бутил-/S/-тирозин-третбутиловый сложный эфир.
К охлажденному льдом раствору 1-/2-трет-бутилоксикарбонил-3-дибензиламинопропил/-1-циклопентан карбоновой кислоты (12,7 г, 27 ммоль) в сухом дихлорметане (100 мл) добавили 1-окибензтриазол (4,2 г, 31 ммоль) и 1-метил-5-/диметиламинопропил/-карбодиимид (7 г, 36 ммоль) и полученный раствор перемешивали при 0oC в течение 30 мин. К этому раствору добавляли 0-трет-бутилтирозин трет-бутиловый сложный эфир (8,4 г, 28,6 ммоль) и N-метилморфолин (5,25 г, 52 ммоль) и раствор оставили стоять в течение ночи при комнатной температуре. Растворитель выпаривали при пониженном давлении и оставшееся жидкое масло растворили в хлористом метилене и промывали водой (2x), 2M соляной кислотой и насыщенным водным раствором бикарбоната натрия (1x), осушали (MgSO4), раствор отфильтровывали и выпаривали, в результате чего получили сырой целевой продукт в виде смолы. После перекристаллизации из н-гексана получили указанное соединение в виде кристаллического вещества (13 г, 69), т. пл. 82 - 87oC. Другую партию материала получили после выпаривания верхних фракций и дальнейшей перекристаллизации.
Анализ для C45H62N2O6
Найдено, %: C 74,12; H 8,69; N 3,87;
Рассчитано, %: C 74,34; H 8,59; N 3,85.
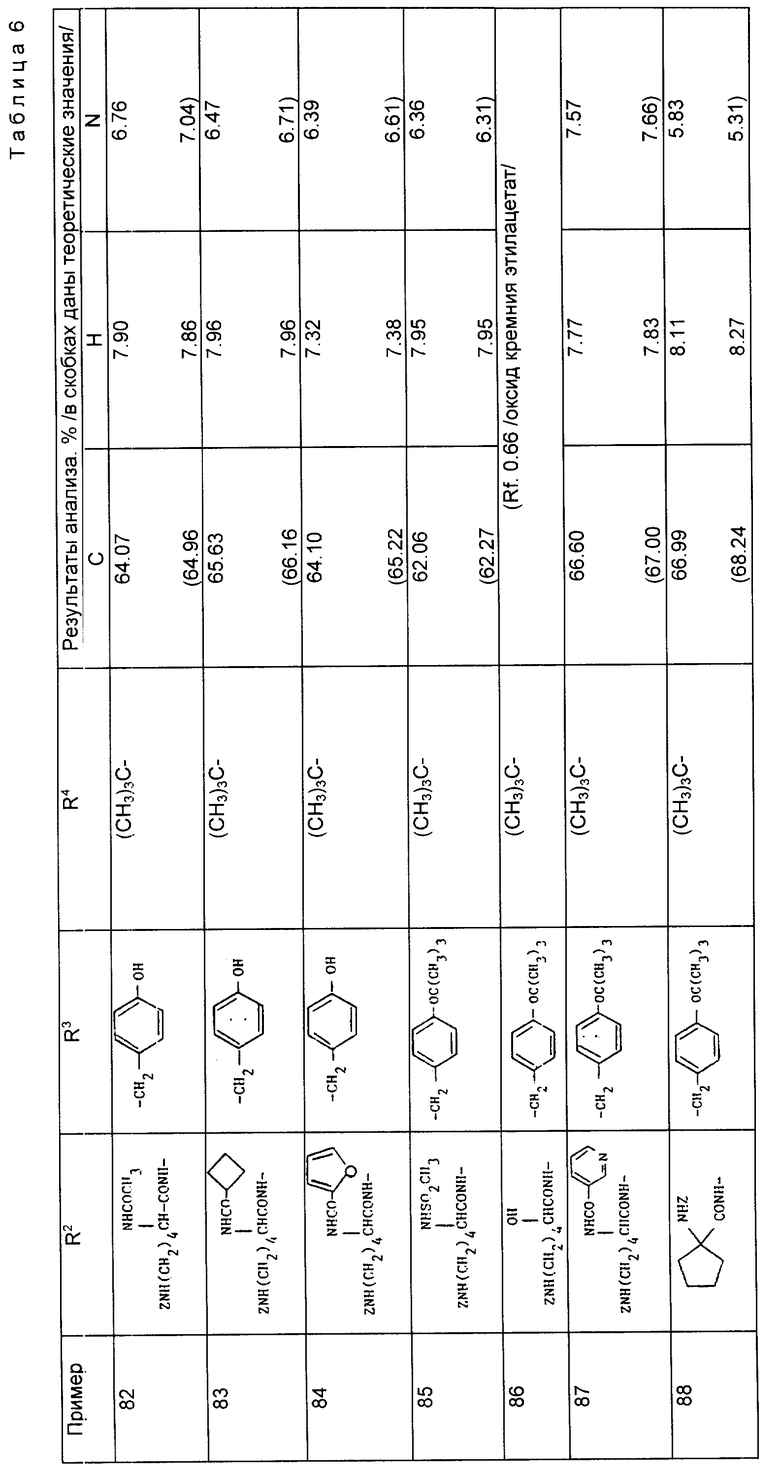
Примеры 2 - 38 (таблица 2). По общей методике, описанной в примере 1, получили следующие соединения, используя в качестве исходных материалов соответствующую карбоновую кислоту и осуществляя реакцию сочетания с соответствующим сложным эфиром аминокислоты. За исключением специально оговоренных случаев группа -NHCH(R3)CO2R4 является производной для встречающихся в природе аминокислот, имеющих S-стереохимию.
Соединения примеров 33 - 35 являются отдельными изомерами, имеющими S, S-стереохимию.
Пример 39. 1-/2-Бензилоксикарбонилпентил/-1-циклопентанкарбонил-3-метансульфонамидо- [R,S] фенилаланин бензиловый сложный эфир.
(а) Смесь 1-/2-безилоксикарбонилпентил/-1-циклопентанкарбонил-3-нитро/R, S/-фенилаланин бензилового сложного эфира (3 г, 499 ммоль), цинкового порошка (7 г, 107 ммоль) в хлориде аммония (7 г, 131 ммоль) в метаноле (200 мл) кипятили в течение 24 ч. Растворитель удаляли при пониженном давлении, остаток подщелачивали до pH 12 путем добавления 2H раствора гидроксида натрия и полученную смесь проэкстрагировали этилацетатом (3 x 75 мл). Объединенные экстракты промывали крепким раствором соли, осушали (MgSO4) и растворитель выпаривали, в результате чего получили 1-/2- бензилоксикарбонилпентил/-1-циклопентанкарбонил-3-амино-(R,S)-фенилаланин бензиловый сложный эфир в виде масла (2,36 г).
(б) Метансульфонилхлорид (0,56 г, 0,49 ммоль) в пиридин (0,039 г, 0,49 ммоль) добавляли к раствору амина из части (A), описанной выше (0,236 г, 0,41 ммоль), в дихлорметане (5 мл) и раствор перемешивали при комнатной температуре в течение 1 ч. Раствор разбавляли дихлорметаном (50 мл), промывали лимонной кислотой (1H, 3 x 5 мл), насыщенным водным раствором бикарбоната натрия (3 x 35 мл) и водой, осушали в растворитель выпаривали при пониженном давлении. Полученное масло пропустили через хроматографическую колонку с силикагелем, элюируя дихлорметаном, а затем смесью дихлорметана и метанола (98: 3), в результате чего получили указанный продукт в виде вязкого масла (0,17 г).
Пример 40. 1-/2-трет-Бутилоксикарбонил-3-дибензиламинопропил/-1-циклопентанкарбонил- 3-метансульфонамидо-/R,S/-фенилаланин этиловый сложный эфир.
Использовали методику, описанную в примере 39, взяв в качестве исходного соединения 1-/2-трет-бутилоксикарбонил-3-дибензиламинопропил/-1-циклопентанкарбонил- 3-нитро-/R,S/-фенилаланин этиловый сложный эфир (из примера 5), и получили указанное соединение в виде масла (3,17 г, 72%).
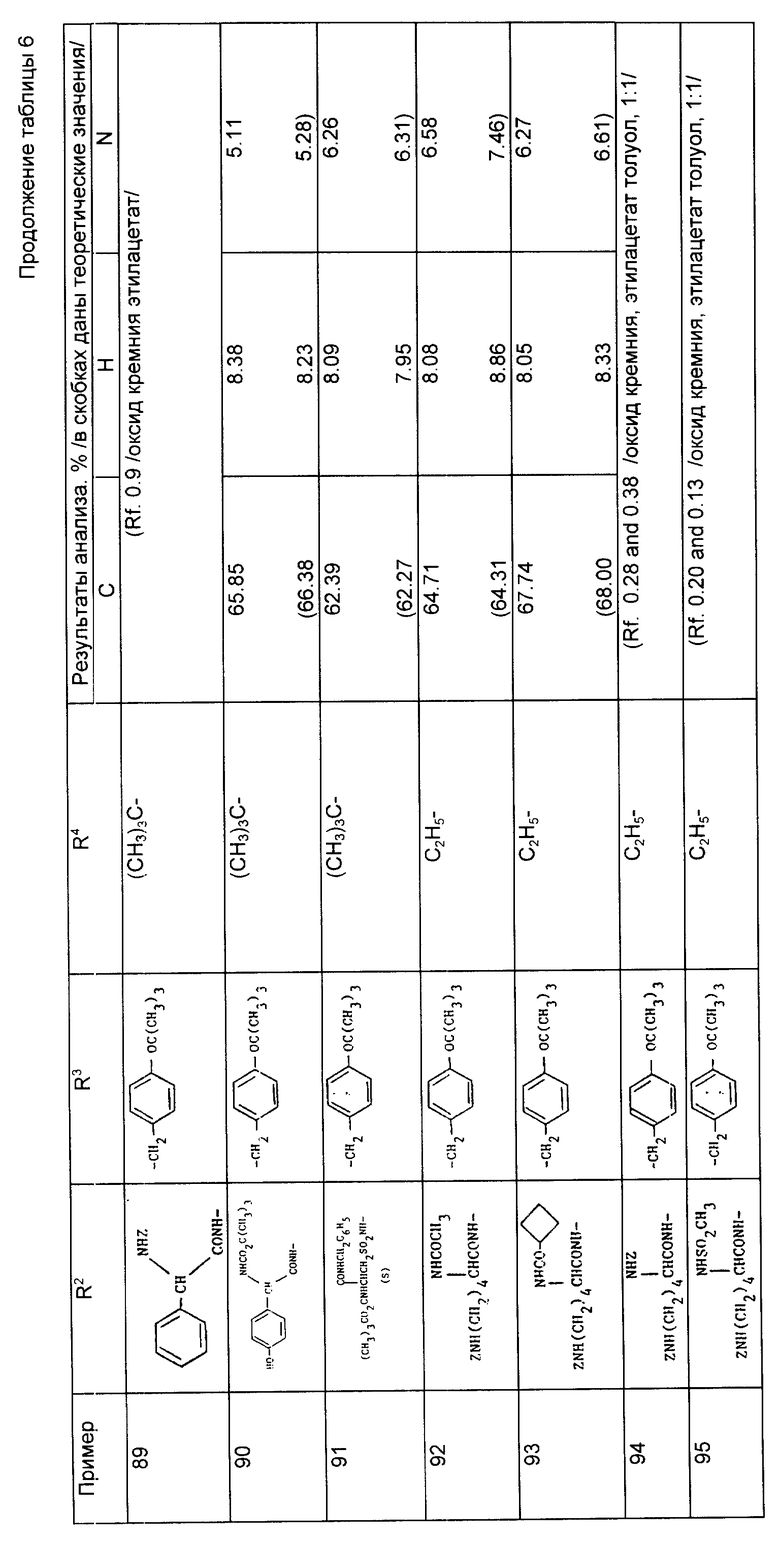
Пример 41. 1-/2-Карбоксипентил/-1-циклопентанкарбонил-3-метансульфонамидо-/R, S/-фенилаланин.
Раствор 1-/2-бензилоксикарбонилпентил/-1-циклопентанкарбонил-3-метансульфонамидо-/R, S/-фенилаланин бензилового сложного эфира (0,16 г) в этаноле (5 мл) в воде (1 мл) гидрировали над палладиевым катализатором на активированном угле (10%, 0,016 мг) при давлении 2,1 кг/см2 и комнатной температуре в течение 3 ч. Катализатор отфильтровали и растворитель выпарили, в результате чего получили пенистый продукт. Этот продукт растерли с диэтиловым эфиром, а затем высушили под вакуумом и получили указанный продукт в виде аморфного вещества (0,45 г).
Анализ для С22H32N2O7•0,5 H2O
Найдено, %: C 55,37; H 6,97; N 5,69;
Рассчитано, %: C 55,33; H 6,96; N 5,87.
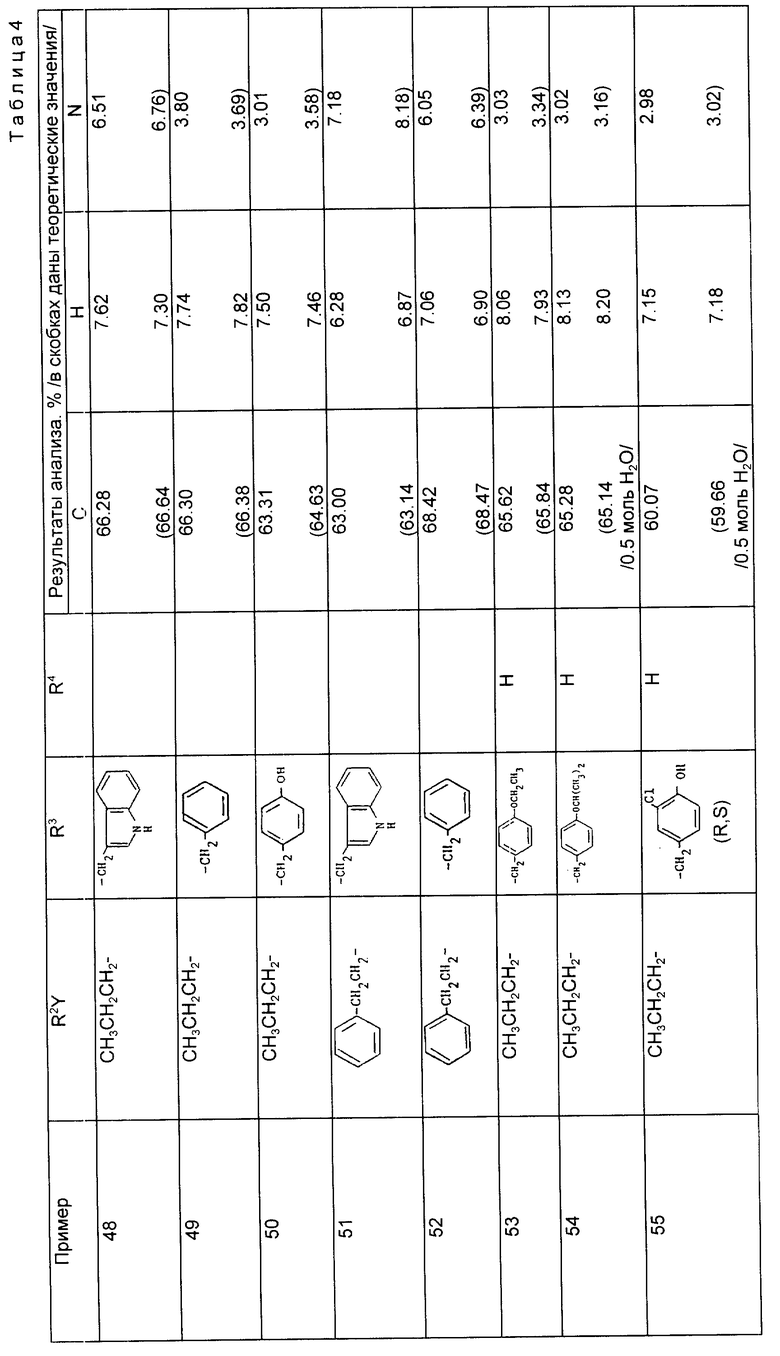
Примеры 42 - 47 (табл. 3). Следующие соединения получили путем каталитического гидрирования соответствующего сложного бензилового эфира по методике, описанной в примере 41.
Пример 47. N-[1-/2-карбокси-4-фенилбутил/-1-циклопентанкарбонил]-/S/-тирозин.
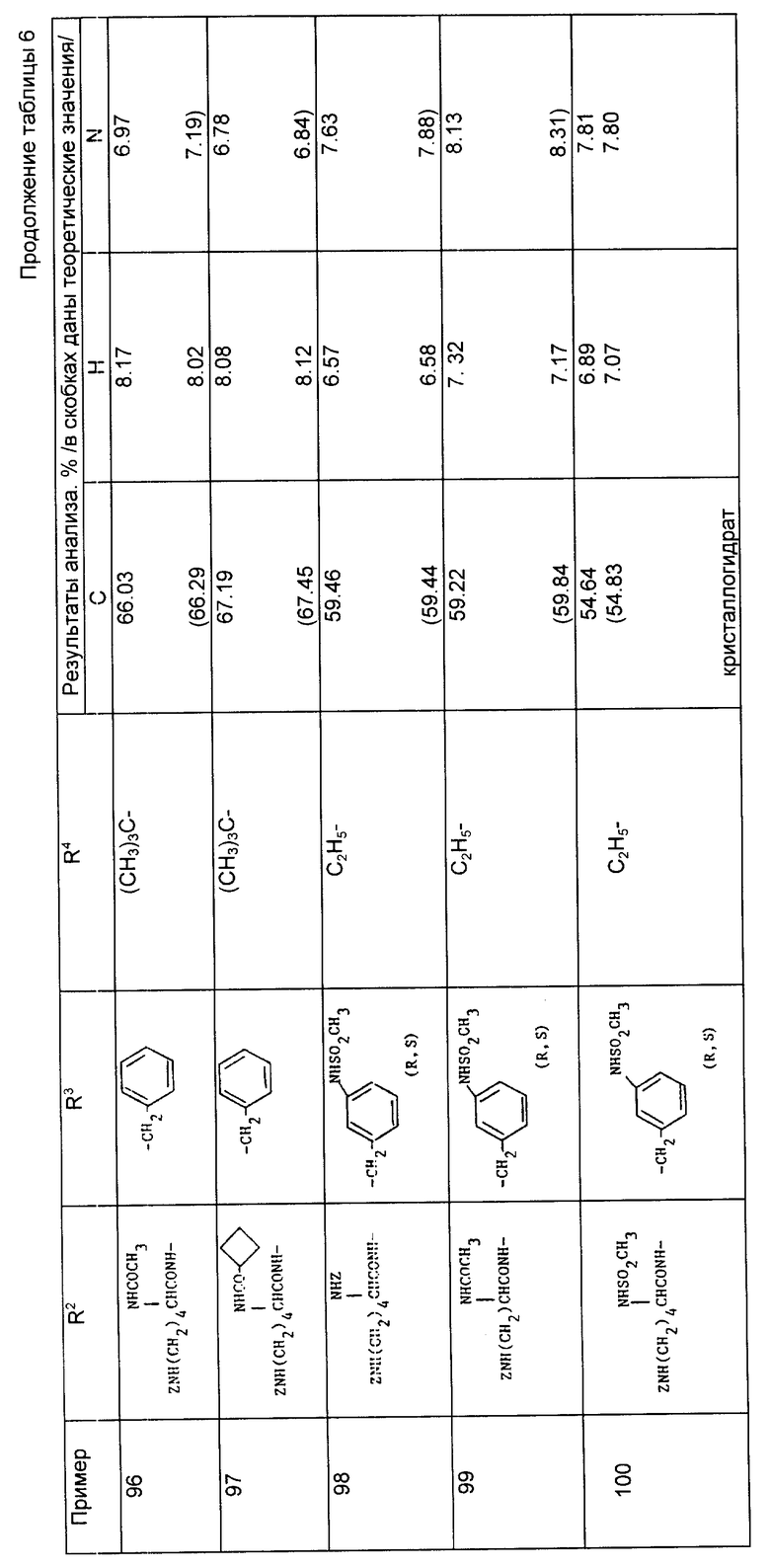
Раствор N-[1-/2-бензилоксикарбонил-4-фенилбутил/-1-циклопентанкарбонил] -/S/-тирозин метилового сложного эфира (0,8 г, 1,47 ммоль) в метаноле (8 мл) гидрировали над 10% палладием на активированном угле (100 мг) под атмосферой водорода 1,8 кг/см2 при комнатной температуре в течение 2 ч. Реакционную смесь профильтровали через воронку c "ar bacel" и выпаривали досуха. Остаток повторно растворили в водном растворе гидроксида натрия (0,5 М, 10 мл) и перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь промыли диэтиловым эфиром и подкисляли до pH 1 с помощью соляной кислоты (10%). Водную фазу проэкстрагировали диэтиловым эфиром (x2) и объединенные органические фазы осушили (MgSO4) и выпарили, в результате чего получали указанный продукт в виде пенистого вещества (0,27 г, 40%).
Анализ для C26H31NO6•0,25H2O
Найдено, %: C 67,24; H 6,85; N 3,26;
Рассчитано, %: C 67,54; H 6,97; N 3,03.
Примеры 48 - 55 (табл. 4). Следующие соединения получали методом каталитического гидрирования с последующим гидролизом образующегося сложного моноэфира по методике, описанной в примере 47.
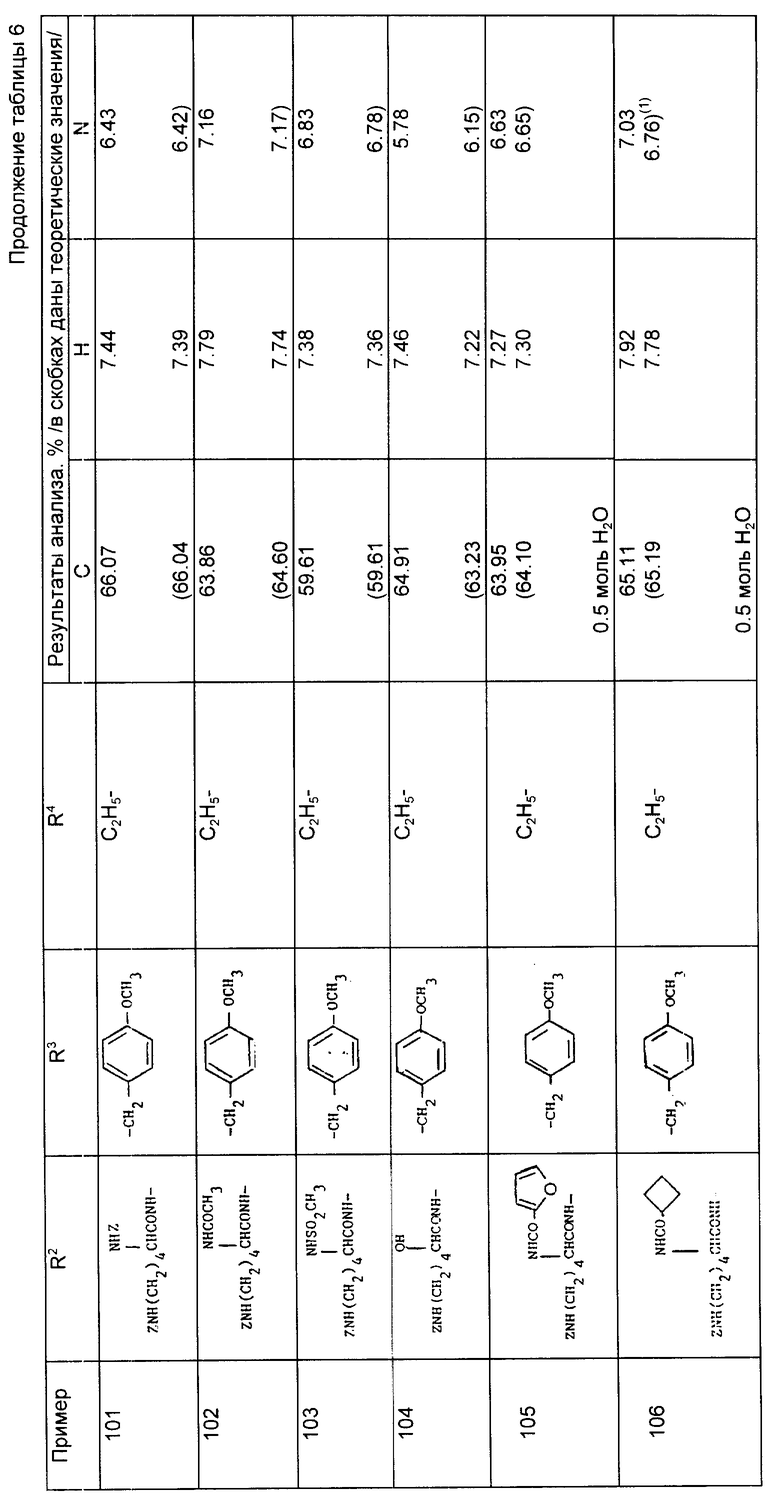
Пример 56. N-[1-/3-аминопропил-2-/S/-трет-бутилоксикарбонил/1- циклопентан-карбонил] -0-третбутил-/S/-тирозин трет бутиловый сложный эфир.
N-[1-/2-трет-бутилоксикарбонил-3-дибензиламинопропил/-1- циклопентакарбонил] -0-трет-бутил-/S/-тиразин трет-бутиловый сложный эфир (на примере 1, 19 г) растворили в смеси этанол:вода (8:1, 300 мл) и гидрировали под атмосферой водорода (4,2 кг/см2) при комнатной температуре над 20% гидроксидом палладия на активированном угле (2 г). Через 24 ч раствор профильтровали через воронку с "solkafloc" и фильтрат выпарили, в результате чего получили масло, которое кристаллизовалось. Его растерли с гексаном, охладили и профильтровали, в результате чего получили чистый энантиомер указанного соединения в виде кристаллического вещества (6 г, 42%), т. пл. 122 - 127oC.
Анализ для C31H50N2O6
Найдено,%: C 67,90; H 9,33; N 5,08;
Рассчитано,%: C 68,09; H 9,22; N 5,12.
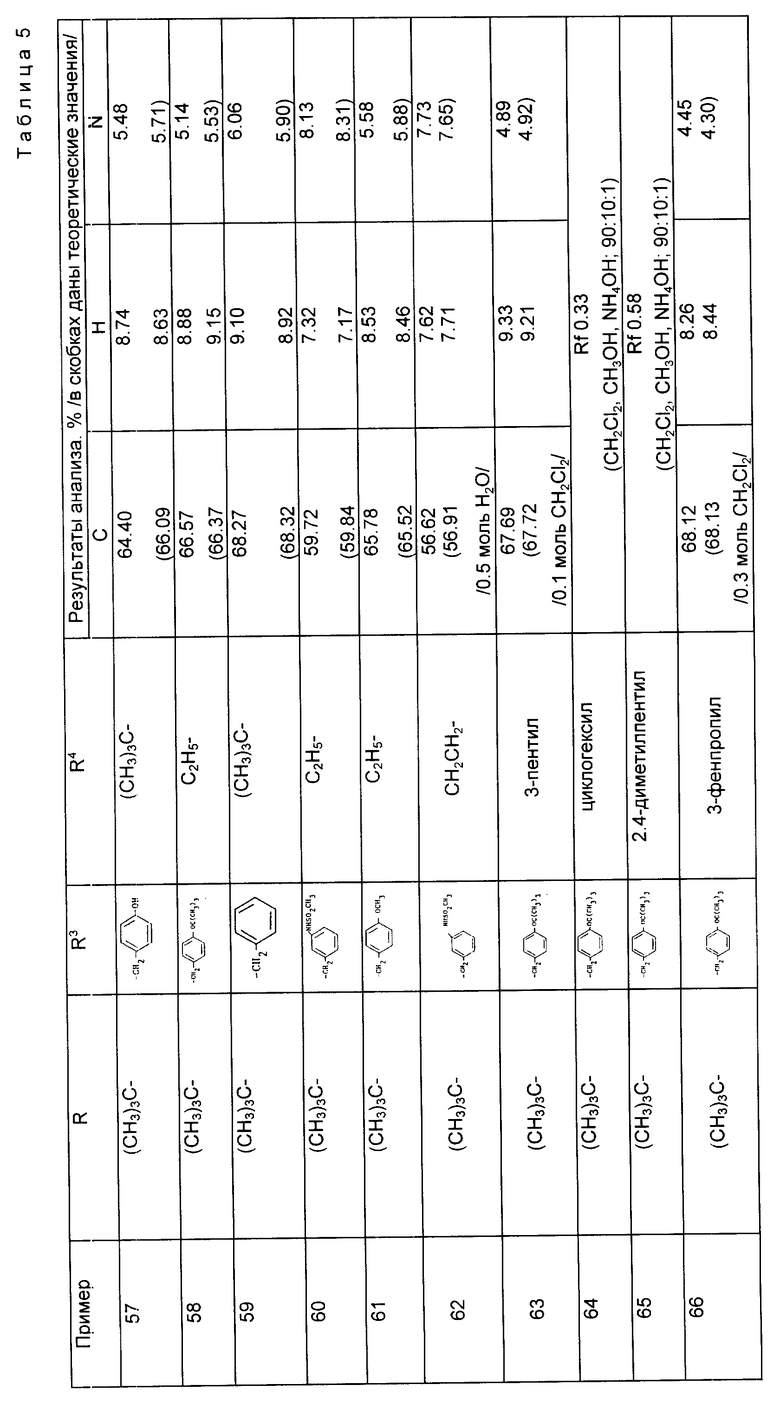
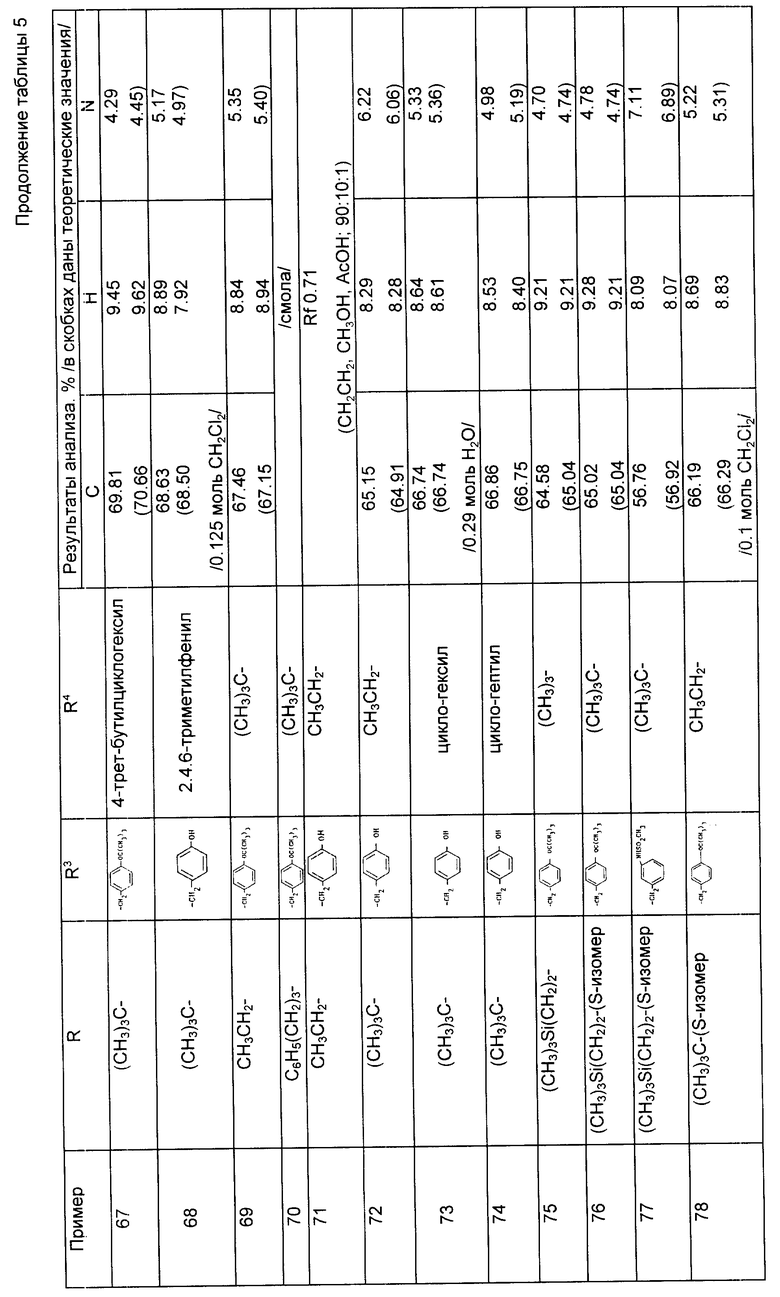
Примеры 57 - 78 (табл. 5). Следующие соединения получили из соответствующего дибензиламинопропилового исходного материала по методике, описанной в примере 56.
Пример 79. N-[1-/2-/S/-трет-бутилоксикарбонил-3-N- метиламинопропил/-1-циклопентан-карбонил] -0-трет-бутил-/S/-тирозин трет-бутиловый сложный эфир.
а) Раствор N-[1-/3-аминопропил-2-/S/-трет-бутилоксикарбонил/- 1-циклопентанкарбонил] -0-трет-бутил-/S/-тирозин трет-бутилового сложного эфира (2,0 г, 1 экв) и N-метилморфолина (0,55 г, 1,5 экв) в сухом дихлорметане (17 мл) при перемешивании охладили на льду и по каплям, в течение 20 мин добавили трифторуксусный ангидрид (1,0 г, 1,3 экв) в дихлорметане (3 мл). Раствор перемешивали в течение 30 мин и в этот период добавили еще одну аликвоту трифторуксуного ангидрида (0,5 г) и раствор перемешивали еще в течение 30 мин. Реакционную смесь разбавили диэтиловым эфиром (10 мл), промыли водой (2 х 10 мл), разбавили соляной кислотой (2 х 10 мл), осушили (MgSO4), отфильтровали и растворитель выпарили, в результате чего получили N-[1-/2-/S/-трет-бутилоксикарбонил-3-трифторацетамидопропил/-1- циклопентан-карбонил] -0-трет-бутил-/S/-тирозин трет-бутиловый сложный эфир в виде желтой смолы (2,2 г, 94%).
б) Сухой карбонат калия (1 г, 2,0 экв) добавили к охлажденному, перемешиваемому раствору вышеназванного продукта (2,2 г, 1,0 экв) и метилиодида (2,0 г, 0,9 мл, 4,0 экв) в сухом диметилформамиде (10 мл) и смеси дали нагреться до комнатной температуры и перемешивали ее в течение ночи. Реакционную смесь разбавили этилацетатом (20 мл) и промыли водой (10 мл), разбавленной соляной кислотой (5 х 5 мл), осушили (MgSO4), отфильтровывали и растворитель выпарили, в результате чего получили 3-метилтрифторацемидное производное в виде желтой смолы (1,95 г, 87%).
в) Гидроксил натрия (0,14 г, 1,2 экв) добавили к охлажденному льдом, перемешиваемому раствору вышеназванного трифторацетамида (1,94 г, 1,0 экв) в этаноле (10 мл), реакционной смеси дали нагреться до комнатной температуры путем выпаривания растворителя при пониженном давлении и разбавили смесью этилацетата (20 мл) и воды (5 мл). Органическую фазу отделили, а водную фазу повторно экстрагировали этилацетатом (10 мл). Объединенные органические экстракты осушили (MgSO4), отфильтровали и растворитель выпарили, в результате чего получили масло, которое при стоянии закристаллизовалось. После перекристаллизации из гексана получили указанный продукт (1,24 г, 75%), т. пл. 105 - 109oC.
Анализ для C32H52N2O6
Найдено,%: C 68,85; H 9,41; N 4,90;
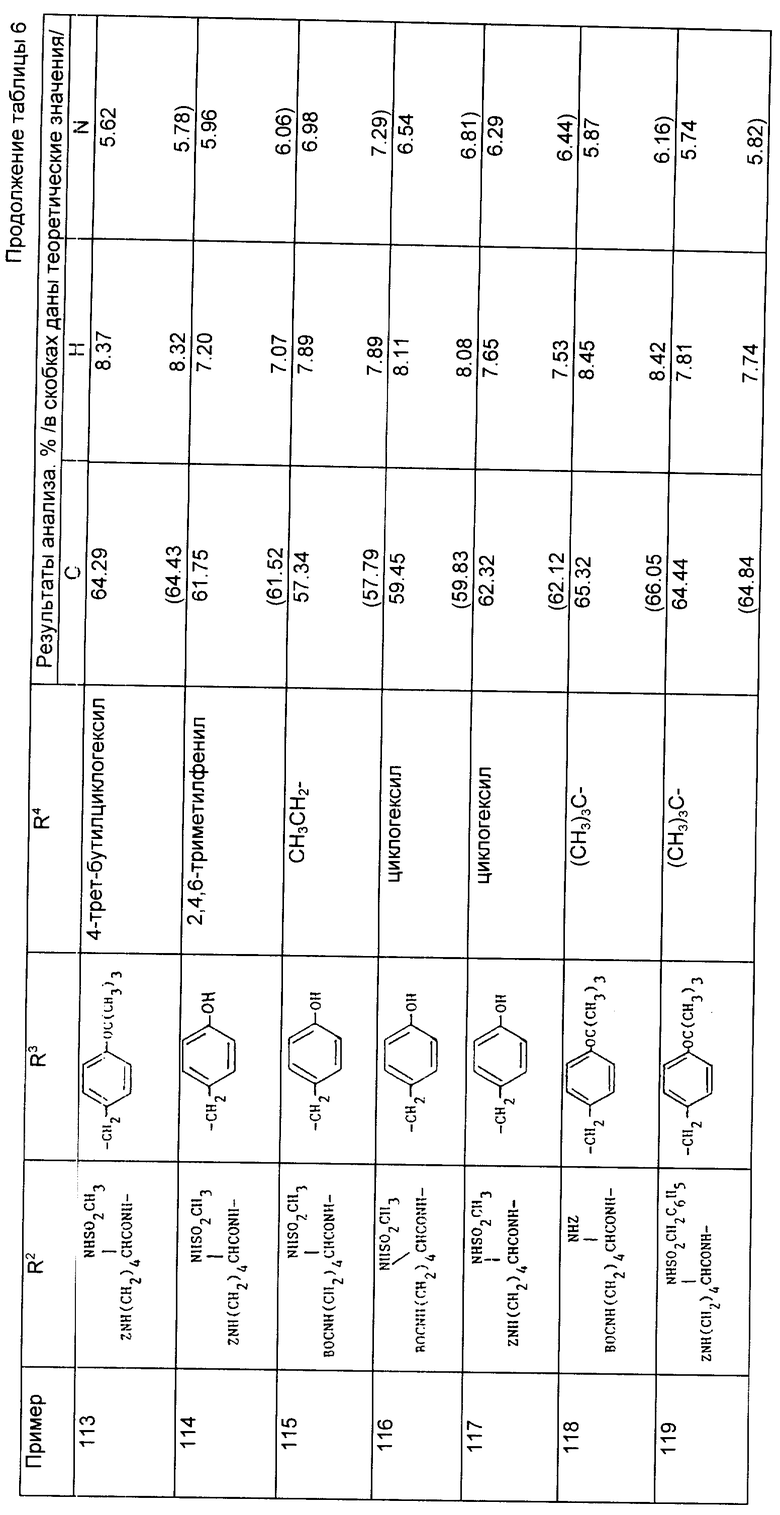
Рассчитано,%: C 68,54; H 9,35; N 4,99.
Пример 80. N-{1-[3-карбокси-2-/R,S/-трет-бутилоксикарбонилпропил]- 1-циклопентан-карбонил}-0-трет-бутил-/S/-тирозин этиловый сложный эфир.
а) Раствор 1-[3-бензилоксикарбонил-2-трет-бутилоксикарбонилпропил] -1- циклопентакарбоновой кислоты (2, 55 г, 6,53 ммоль) в сухом дихлорметане (40 мл) охладили до 0oC, обработали 1-гидроксибензтриазолом (0,97 г, 7,18 ммоль), N-метилморфолином (0,86 г, 8,32 ммоль) и 1-этил-3- /диметиламинопропил/карбодиимидом (1,64 г, 8,32 ммоль) и смесь перемешивали при 0oC в течение 10 мин. Добавили 0-трет-бутил- /S/-тирозин-этиловый сложный эфир (1,73 г, 6,53 ммоль), реакционной смеси дали нагреться до комнатной температуры и перемешивали ее в течение ночи. Затем из реакционной смеси удалили растворитель при пониженном давлении и полученную смолу оставили стоять еще на 48 ч при комнатной температуре. Затем реакционную смесь обработали этилацетатом (100 мл) и водой (50 мл). Органический слой отделили и промыли водой (2 х 30 мл), насыщенным раствором соли (30 мл), осушили (MgSO4), отфильтровали и растворитель выпарили, в результате чего получали сырой продукт в виде масла. После хроматографирования на силикагеле с использованием в качестве элюента смеси гексана и диэтилового эфира получили N-{ 1-[3-бензилоксикарбонил-2/R,S/-трет- бутилоксикарбонилпропил]-1-циклопентан} карбонил-0-трет-бутил-/S/- тирозин этиловый сложный эфир в виде желтого масла (2,56 г, 60%).
Анализ для C37H51NO8
Найдено,%: C 69,31; H 8,49; N 2,49;
Рассчитано,%: C 69,67; H 8,06; N 2,20.
б) Полученный выше продукт (2,48 г, 3,89 ммоль) растворили в этанол:водной смеси (9:1,66 мл) и гидрировали при комнатной температуре под атмосферой водорода (4,2 кг/см2) над 10% палладием на активированном угле (250 мг) в течение 5 ч. Реакционную смесь профильтровали через воронку с "solkoflok" фильтра, выпарили досуха. Из остатка приготовили азеотропную смесь с дихлорметаном (3x) и получили сырой продукт в виде белого вспененного вещества. После хроматографирования на силикагеле и элюирования смесью гексана с этилацетатом получили указанное соединение в виде белого вспененного вещества (1,83 г, 96%).
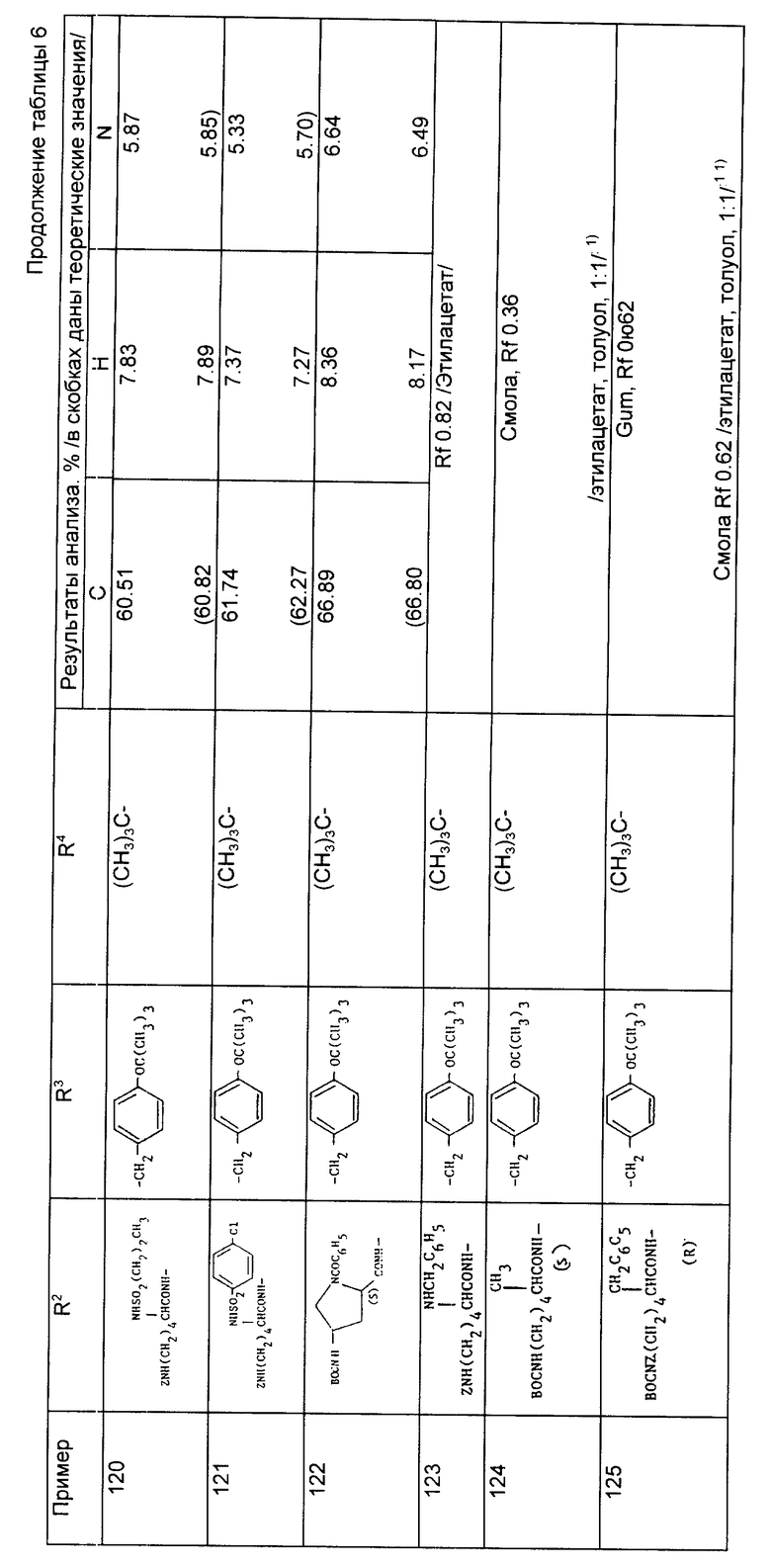
Анализ для C30H45NO8
Найдено,%: C 65,48; H 8,33; N 1,92;
Рассчитано,%: C 65,79; H 8,28; N 2,56.
Пример 81. N-{1-[3-/N2,N6-дибензилоксикарбонил- /S/-лизиламино/-2/S/-трет-бутилоксикарбонилпропил] -1- цилопентанкарбонил} -0-трет-бутил-/S/-тиразин-трет-бутиловый сложный эфир.
Раствор N-[1-/3-аминопропил-2/S/-трет-бутилоксикарбонил/-1- циклопентанкарбонил] -0-трет-бутил-/S/-тирозин-трет-бутилового сложного эфира (из примера 56, 0,4 г, 0,73 ммоль) в сухом дихлорметане (10 мл) охладили до 0oC, обработали 1-оксибензтриазолом (0,13 г, 0,88 ммоль) и 1-этил-3-/диметиламинопропил карбодиимидом (0,21 г, 0,88 ммоль) и смесь перемешивали при 0oC в течение 30 мин. Добавили N2,N6-дибензилоксикарбонил-/S/лизин (0,33 г, 0,80 ммоль) и реакционной смеси дали нагреться до комнатной температуры и перемешивали ее в течение ночи. Реакционную смесь разбавили хлористым метиленом (5 мл) и промыли водой (2 х 10 мл), разбавленной соляной кислотой (1М, 2 х 10 мл), водным раствором бикарбоната натрия (10 мл) и насыщенным раствором соли (10 мл), сушили (MgSO4), профильтровали и растворитель выпарили, в результате чего получили сырой продукт в виде масла. После хроматографирования на силикагеле и элюирования смесью гексана с этилацетатом получили указанное соединение в виде вспененного материала (0,55 г, 85%).
Анализ для C53H14N4O11
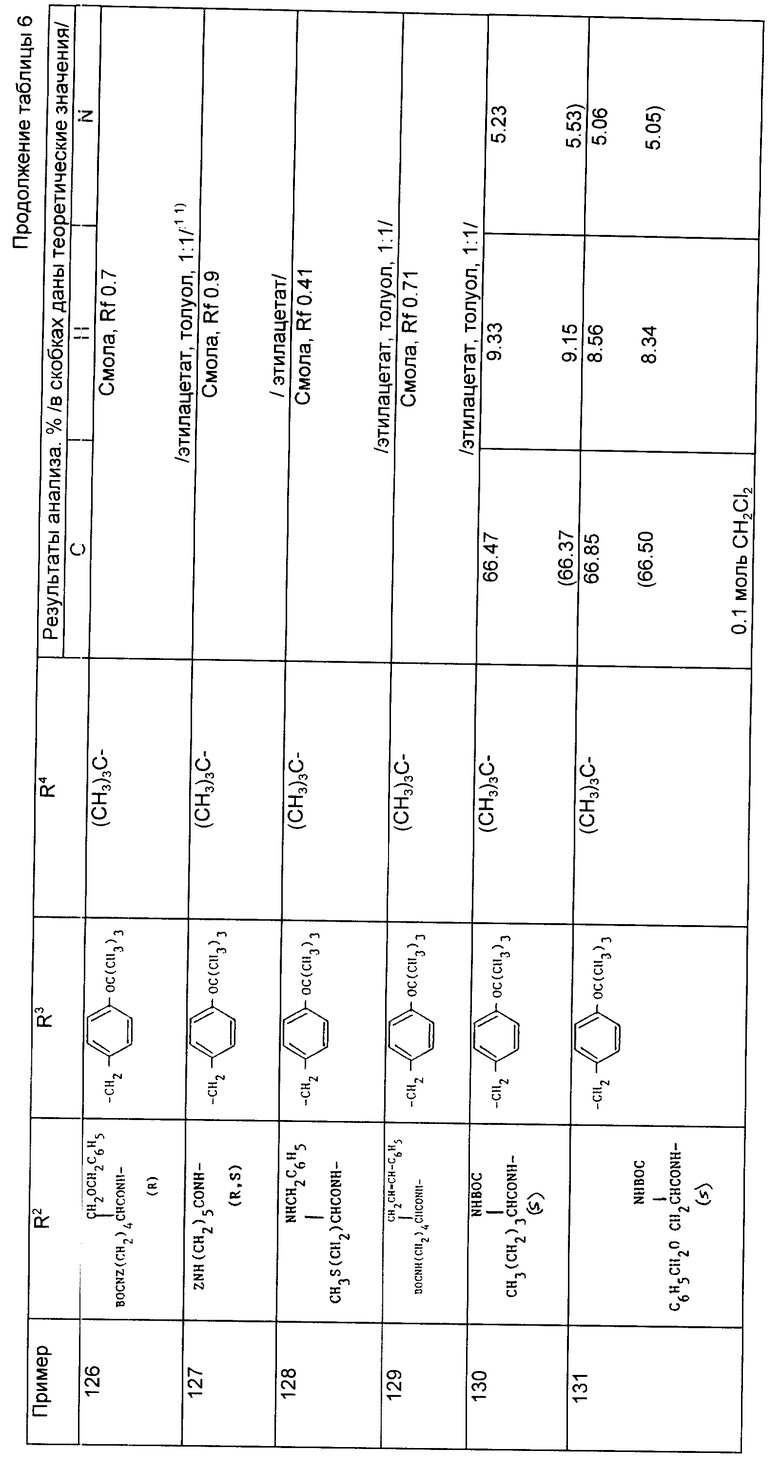
Найдено,%: C 67,47; H 7,99; N 5,74;
Рассчитано,%: C 67,49; H 7,91; N 5,94.
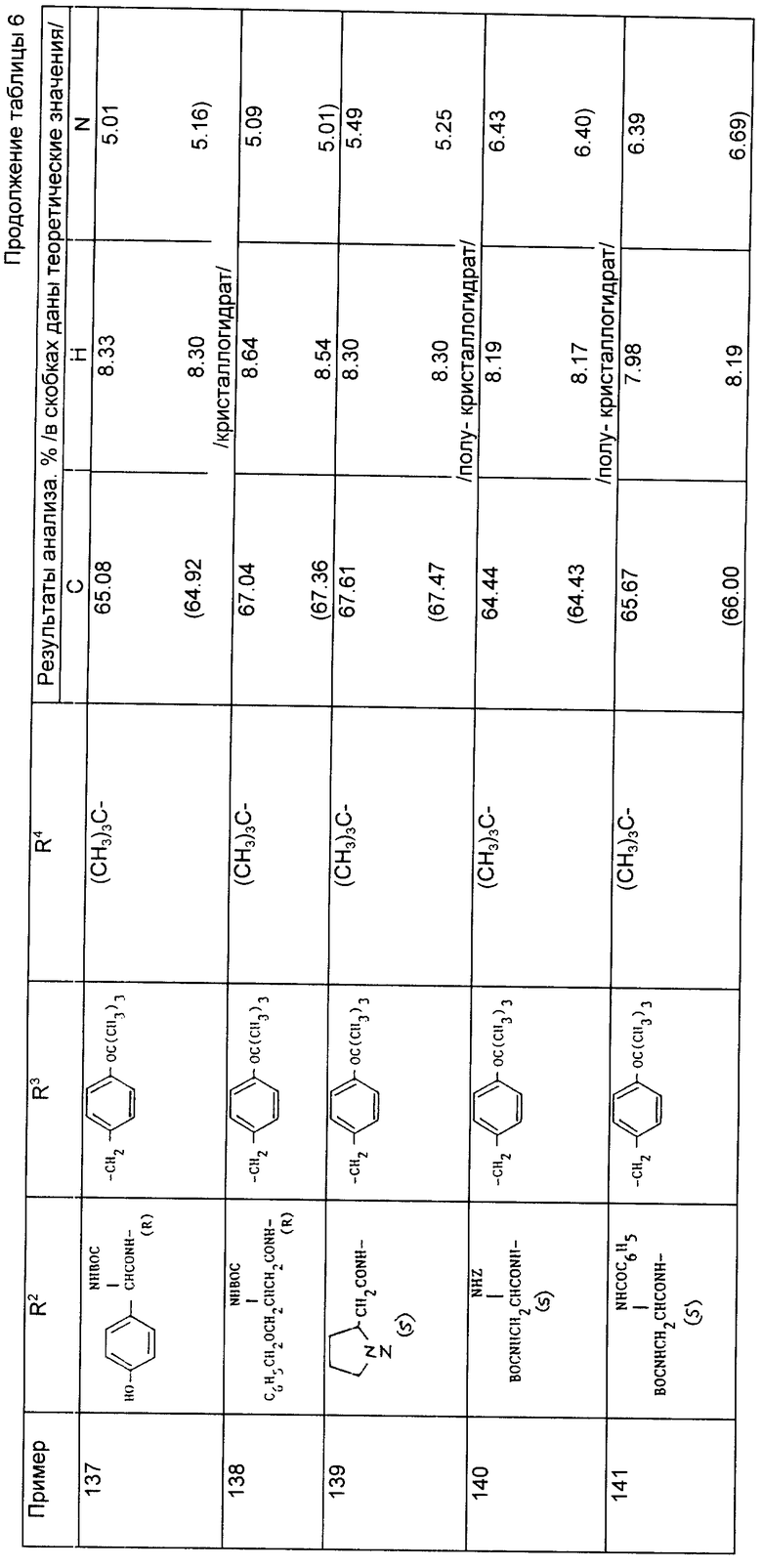
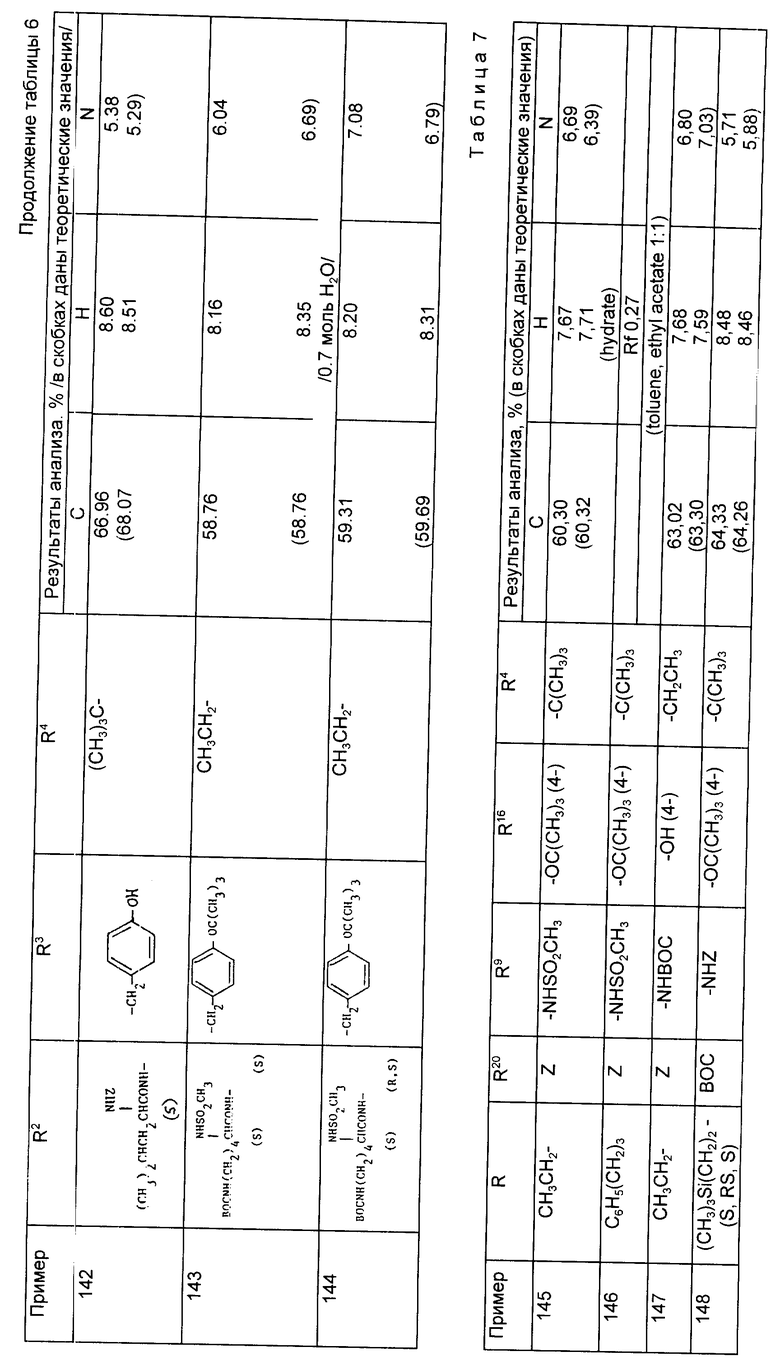
Примеры 82 - 144 (табл. 6). Следующие соединения получили по методике, описанной в примере 81, используя соответствующие амины из примеров 56 - 79 и осуществляя реакцию сочетания с соответствующей аминокислотой. Символ Z означает бензилоксикарбонил N - защитную группу и символ ВОС означает трет-бутилоксикарбонильную группу. Если специально не указано, то радикалы R2 и R3 являются производными встречающихся в природе аминокислот, имеющих S-стереохимию.
При осуществлении примеров 85 - 91, 107, 108, 118 - 141 и 143 использовали соответствующие амины формулы (VI) из примеров 76 - 78, имеющие S стереохимию.
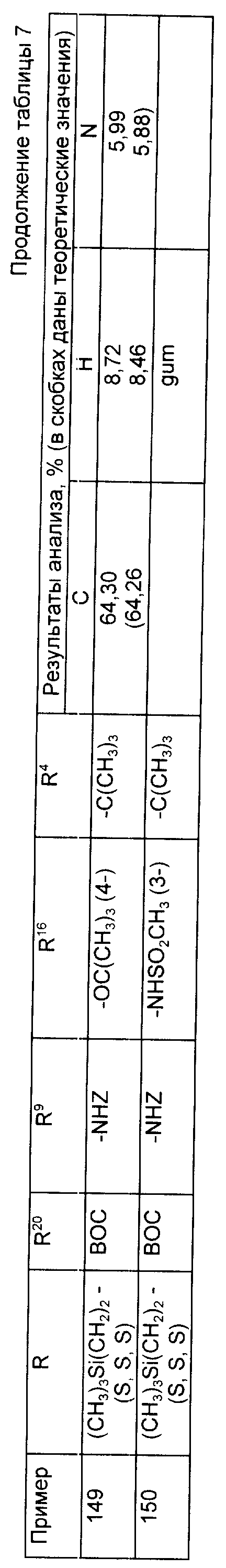
Примеры 145 - 150 (табл. 7). Следующие соединения были получены по методике, описанной в примере 81, при использовании соответствующих аминов.
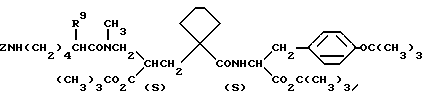
Примеры 151-152. (табл. 8). Следующие соединения были получены по методике, описанной в примере 81, при использовании в качестве исходного соединения N-метил амина из примера 79.
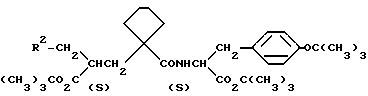
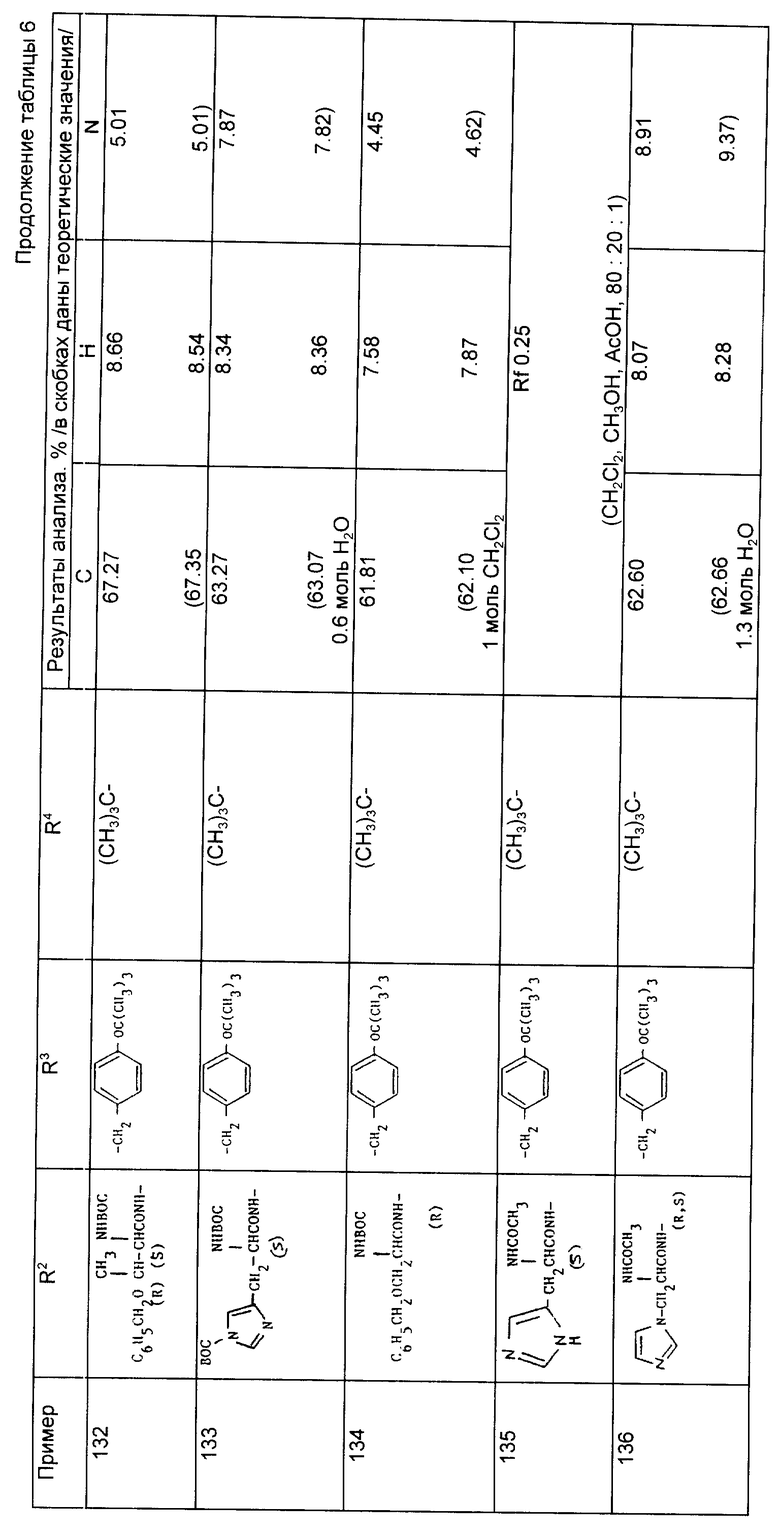
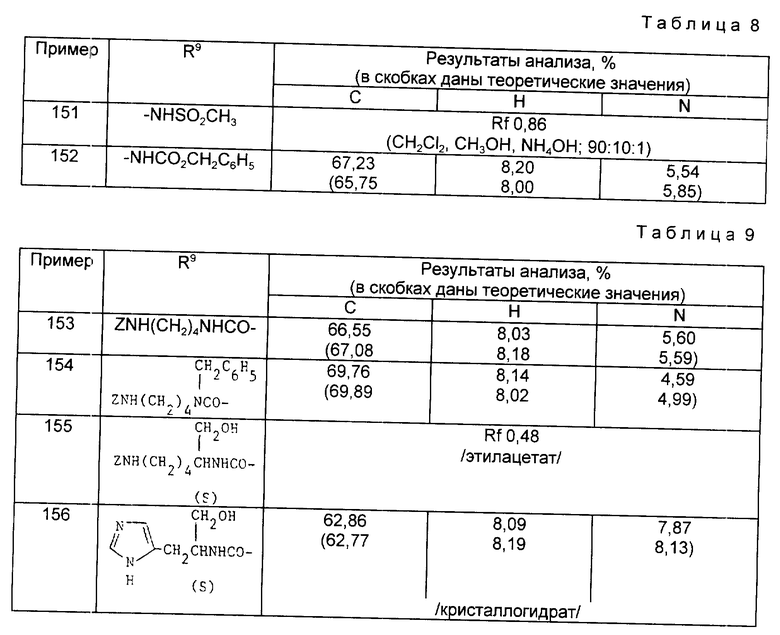
Примеры 153-156 (табл. 9). Следующие соединения были получены по методике, описанной в примере 81, при использовании в качестве исходного соединения кислоты из примера 80 и осуществлении реакции сочетания с соответствующим амином.
Пример 157. N-{1-[3-/N2-метансульфонил-N6- третбутилоксикарбонил-/S/-линил-амино/-2-(R,S)- триметилсилилэтоксикарбонилпронил]-1-циклопентанкарбонил}-O-трет- бутил-/S/-тирозин-трет-бутиловый сложный эфир.
Раствор N-{1-[3-/N6-трет-бутилоксикарбонил-/S/-лизил- амино-2/R,S/-триметилсилилэтоксикарбонил пропил]-1- циклопентанкарбонил}-O-трет-бутил-/S/-тирозин-трет-бутилового сложного эфира (2,5 г, 3,1 ммоль) в охлажденном на льду дихлорметане (50 мл) обработали пиридином (1,25 г, 15,8 ммоль) и метансульфонилхлоридом (860 мг, 7,5 ммоль) и перемешивали в течение ночи при комнатной температуре. Растворители удалили при пониженном давлении и остаток обработали этилацетатом и разбавленной лимонной кислотой. Объединенные экстракты промыли разбавленным водным раствором бикарбоната натрия и насыщенным раствором соли, осушили и выпарили, в результате чего получили желтое вспененное вещество, которое после хроматографирования на силикагеле и элюировании смесью гексан, этилацетат и метанол (80:20:5) позволило получить указанное соединение в виде бесцветного вспененного вещества (1,92 г, 69%).
Анализ для C44H76N4O11S Si:
Найдено,%: C 58,64; H 8,50; N 6,01;
Рассчитано,%: C 58,89; H 8,54; N 6,24.
Пример 158. Полностью разделенный продукт получили аналогично тому, как описано выше в примере, используя в качестве исходного S,S,S-изомер, и получили N-[1-[3-/N2-метансульфонил- N6-трет-бутоксикарбонил-/S/-лизиламино/-2/S/- триметилсилилэтоксикарбонилпропил] -1-циклопентанкарбонил} -O-трет- бутил/S/-тирозин-трет-бутиловый сложный эфир.
Найдено,%: C 59,20; H 8,60; N H 6,23.
Пример 159. N-{ 1-[3-/N6-бензилоксикарбонил-N2- метансульфонил-/S/-лизиламино/-2-/R, S/-этоксикарбонилпропил]-1- циклопентанкарбонил}-/S/-тирозин этиловый сложный эфир.
Использовали описанную выше методику, использовав в качестве соединения соответствующее N6-бензилоксикарбонильное производное, в результате чего получили указанный продукт.
Анализ для C38H54N4O11S(0,75•CH2Cl2):
Найдено,%: C 56,61; H 6,80; N 6,67;
Рассчитано,%: C 55,50; H 6,67; N 6,88.
Пример 160. N-{1-[3-/N6-бутоксикарбонил-N2- ацетил-/S/-лизиламино/-2-/S/-триметилсилилэтоксикарбонилпропил] -1- циклопентанкарбонил}-3-метансульфонамидо-/S/-фенилаланин трет-бутиловый сложный эфир.
Использовали методику, описанную в примере 157, использовав соответствующий сложный диэфир и осуществив реакцию с ацетилхлоридом вместо метансульфонилхлорида, в результате чего получали указанное N2-ацетильное производное в виде бесцветного вспененного вещества.
Пример 161. N-{1-[3-/N2-метансульфонил-N6- третбутилоксикарбонил-/S/-лизиламино/-2-/R, S/карбоксипропил] -1- циклопентанкарбонил}-O-трет-бутил-/S/-тирозин-трет-бутиловый сложный эфир.
Раствор N-{ 1-[3-/N2-метенсульфонил-N6- третбутоксикарбонил-/S/-лизиламино-2/R, S/- триметилсилилэтоксикарбонилпропил]-1-циклопентанкарбонил}-O-трет- бутил-/S/-тирозин-трет-бутилового сложного эфира (1,80 г, 2,0 ммоль) в тетрагидрофуране (20 мл) обработали раствором фторида тетрабутиламмония в тетрагидрофуране (1М, 8 мл, 3,0 ммоль) и нагрели до 60oC под атмосферой азота. Растворитель удалили при пониженном давлении, остаток обработали этилацетатом и разбавленной лимонной кислотой, объединенные экстракты промыли насыщенным раствором соли, осушили и растворитель выпарили, в результате чего получили вспененное вещество, которое после хроматографирования на силикагеле и элюирования этилацетатом, метанолом, гексаном (4:1:5) позволило получить указанный продукт в виде вспененного вещества (1,17 г, 74%).
Анализ для C39H64N4O11S•H2O:
Найдено,%: C 57,49; H 7,89; N 6,93;
Рассчитано,%: C 57,46; H 8,16; N 6,87.
Пример 162. Полностью разделенный материал получили аналогично тому, как описано выше, из S,S,S-изомера, полученного в примере 158, в результате чего образовался N-{ 1-[3-/N2-метансульфонил- N6-трет-бутоксикарбонил-/S/-лизиламино- 2-/S/-карбоксипропил]-1-циклопентанкарбонил}-O-трет-бутил-/S/-тирозин- трет-бутиловый сложный эфир.
Анализ для C39H64N4O11S:
Найдено,%: C 59,01; H 8,21; N 6,87;
Рассчитано,%: C 58,77; H 8,09; N 7,03.
Пример 163. N-{1-[3-/N2-метансульфонил-N6- третбутилоксикарбонил-/S/-лизил-амино/-2-/R, S/трет- бутилоксикарбонилпропил]-1-циклопентанкарбонил}-O-трет- бутил-/S/-тирозин.
N-{ 1-[3-/N2-метансульфонил-N6-трет- -бутилоксикарбонил-/S/-лизил-амино-2/R, S'/-трет- -бутилоксикарбонилпропил]-1- циклопентанкарбонил}-O-трет-бутил-/S/-тирозин этиловый сложный эфир (2,21 г, 2,68 ммоль) растворили в ацетоне (5,5 мл), а затем добавили 1H водный раствор гидроксида натрия (5,36 г, 5,38 ммоль). После перемешивания в течение 10 мин при комнатной температуре раствор подкисляли до pH с помощью водного раствора лимонной кислоты (10%). Затем ацетон удаляли на ротационном испарителе и остаток проэкстрагировали этилацетатом (50 мл). Органический слой отделили, промыли насыщенным раствором соли, осушили сульфатом магния и растворитель удалили при пониженном давлении, в результате чего получили указанное соединение в виде белого вспененного вещества (1,89 г, 88%).
Анализ для C39H64N4O11S:
Найдено,%: C 58,49; H 8,01; N 6,64;
Рассчитано,%: C 58,77; H 8,09; N 7,08.
Пример 164. N-{1-[3-N2-метансульфонил-N6-трет- бутилоксикарбонил-/S/-лизил-амино-2-/S/-трет-бутилоксикарбонилпропил] - 1-циклопентанкарбонил}-O-трет-бутил-/S/-тирозин.
Использовали методику, описанную в примере 163, использовав разделенные исходные материалы на примере 143, в результате чего получили указанное соединение.
Анализ для C39H64N4O11S•(0,66 H2O):
Найдено,%: C 58,17; H 8,09; N 6,42;
Рассчитано,%: C 57,89; H 8,14; N 6,93.
Пример 165. N-{1-[3-/N6-бензилоксикарбонил-N2- метансульфонил-/S/-лизиламино/-2-/S/-карбоксипропил] -1- циклопентанкарбонил}-O-бензил-/S/-тирозин бензиловый сложный эфир.
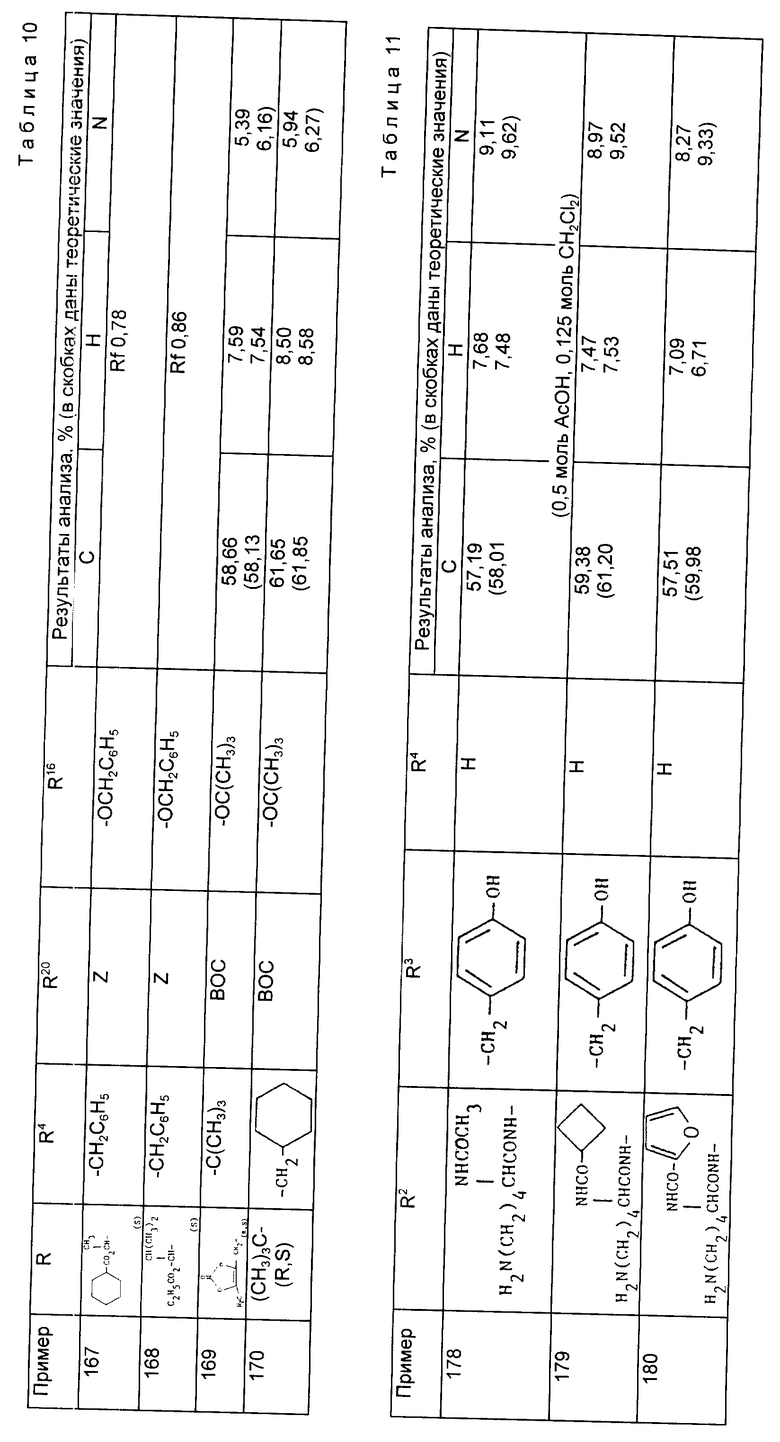
а) Водный раствор гидроксила натрия (1H, 9,2 мл, 1 экв) добавили к раствору 1-/3-бис/S/-альфа-метилбензил/-амино-2-/S/- бутоксикарбонилпропил/-циклопентан карбоновой кислоты (4,5 г, 1 экв) в водном этаноле (9:1, 80 мл) и полученную смесь гидрировали над 20% гидроксидом палладия (0,5 г) при давлении 4,2 кг/см2 и комнатной температуре в течение ночи. Добавили еще 0,5 г катализатора и гидрирование осуществляли дополнительно в течение пяти часов, когда т.с.х. показала завершение реакции. Катализатор отфильтровывали и реакционную смесь выпарили при пониженном давлении. Остаток дважды промыли дихлорметаном, и окончательно аминный продукт собрали в дихлорметане и использовали непосредственно для осуществления последующей реакции.
б) К охлажденному на льду раствору N2-трихлорэтоксикарбонил-N6-бензилоксикарбонил-/S/ -лизина (4,17 г) в сухом дихлорметане (20 мл) добавляли 1-окси-бенэтриазол (1,49 г), 1-этил-3-/диметиламинопропил/-карбодиимид (4,46 г) и полученный раствор перемешивали при 0oC в течение 30 мин. К нему добавляли раствор натриевой соли 1-(2-(S)-трет-бутоксикарбонил-3-аминопропил/-циклопентан карбоновой кислоты в дихлорметане (10 мл) из части (a) и реакционной смеси дали нагреться до комнатной температуры и перемешивали ее в течение ночи. Реакционную смесь выпарили досуха и остаток обработали этилацетатом (20 мл) и водой (20 мл). Слои разделяли и органический слой промывали водой (2 x 10 мл), 1H соляной кислотой (2 x 10 мл), водным раствором бикарбоната натрия, насыщенным раствором соли, а затем осушили (MgSO4), отфильтровали и выпарили, в результате чего получили сырой продукт в виде масла. Его подвергли хроматографической очистке на силикагеле (160 г), элюируя смесями гексана и этилацетата. Необходимые фракции соединили, сконцентрировали, а затем промыли толуолом, в результате чего получили чистый продукт в виде вспененного вещества (4,28 г, 66%).
в) Активированный сложный эфир этого соединения (4,63 г) в дихлорметане (20 мл) получили, как описано в части (б) и обработали при 0oC раствором соли тозилата О-бензил-/S/-тирозин бензилового сложного эфира (3,48 г) и N-метилморфолином (1,33 г) в дихлорметане (20 мл). Реакционной смеси дали нагреться до комнатной температуры и перемешивали ее в течение ночи. Затем раствор выпарили досуха, остаток растворили в этилацетате и промыли водой (2 x 10 мл). 1H соляной кислотой (2 x 10 мл), водным раствором бикарбоната натрия, насыщенным раствором соли, осушили (MgSO4), профильтровали и выпарили, в результате чего получили сырой продукт в виде масла (8,02 г). Его подвергли хроматографической очистке на силикагеле (130 г), элюируя смесями гексана и этилацетата. Соответствующие фракции соединили и выпарили, в результате чего получили чистый продукт в виде вспененного материала (4,32 г, 68%).
г) К охлажденному раствору продукта на части (B) (4,32 г) в уксусной кислоте (25 мл) добавили активированный цинковый порошок (4 г) единоразово, реакционной смеси дали нагреться до комнатной температуры и перемешивали ее. Через 90 мин твердый остаток отфильтровали и промыли его водой. Объединенный фильтр и промывочные воды выпарили при пониженном давлении и остаток затем промыли толуолом (x3), после чего растворили в этилацетате и промыли водным раствором бикарбоната натрия. Органический слой осушили, профильтровали и выпарили, в результате чего получили аминный продукт в виде смолы.
д) К раствору аминного продукта из части г) (3,38 г) и N-метилморфолина (0,48 г) в сухом дихлорметане (20 мл) при перемешивании и после охлаждения до 0oC добавили метансульфонилхлорид (0,49 г), реакционной смеси дали нагреться до комнатной температуры и перемешивали ее в течение ночи. Реакционную смесь разбавили дихлорметаном (20 мл) и промыли водой (2 x 10 мл), 0,1 M соляной кислотой (10 мл), насыщенным раствором соли и осушили (MgSO4), профильтровали и выпарили, в результате получили сырой сульфонамид в виде вспененного материала (4 г). Его подвергли хроматографической очистке на силикагеле (65 г), элюируя смесями гексана и этилацетата, в результате чего получили целевой N2-метансульфонильный продукт в виде вспененного соединения (2,9 г, 79%).
е) Трифторуксуснуя кислоту (15 мл) по каплям при перемешивании добавляли к раствору продукта на части (д) (2,87 г) и анизола (0,4 г) в сухом дихлорметане (15 мл), охлажденному до 0oC. Через 3 ч реакционную смесь выпарили досуха при пониженном давлении. Остаток растворяли в этилацетате (30 мл) и промыли водным раствором бикарбоната натрия (2 x 10 мл), 0,1M соляной кислотой и насыщенным раствором соли, осушили (MgSO4), отфильтровывали и выпаривали, в результате чего получили сырой продукт в виде желтого масла (3,5 г). Его подвергли хроматографической очистке на силикагеле (60 г), элюируя смесями гексана и этилацетата с 1% уксусной кислотой, в результате чего получили указанную кислоту в виде вспененного материала (2,6 г, 97%). Часть ее превратили в цезиевую соль, используя для этого водный этанольный раствор карбоната цезия.
Анализ для C48H57N4O11SCs:
Найдено, %: C 54,81; H 5,70; N 5,21;
Рассчитано, %: C 55,92; H 5,57; N 5,43.
Пример 166. N-{1-[3-/N6-бензилоксикарбонил-N2 -метан-сульфонил-/S/-лизиламино-2-/S/-пиралоксиметоксикарбонилпропил] -1-циклопентан-карбонил}-O-бензил-/S/-тирозин бензиловый сложный эфир.
Пивалоилоксиметилхлорид (0,12 г) при перемешивании добавляли к раствору цезиевой соли из примера 165 (0,55 г) в сухом диметилформамиде (6 мл), и реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавили этилацетатом (20 мл) и промыли водой (5 x 10 мл), 1H соляной кислотой (2 x 10 мл), водным раствором бикарбоната (10 мл), насыщенным раствором соли и осушили (MgSO4), профильтровали и вырапили, в результате чего получили сырой продукт в виде бледного желтоватого масла (0,7 г). После хроматографирования на силикагеле (12 г) и элюирования смесями гексана и этилацетата получали указанный сложный эфир в виде вспененного материала (0,465 г, 88%).
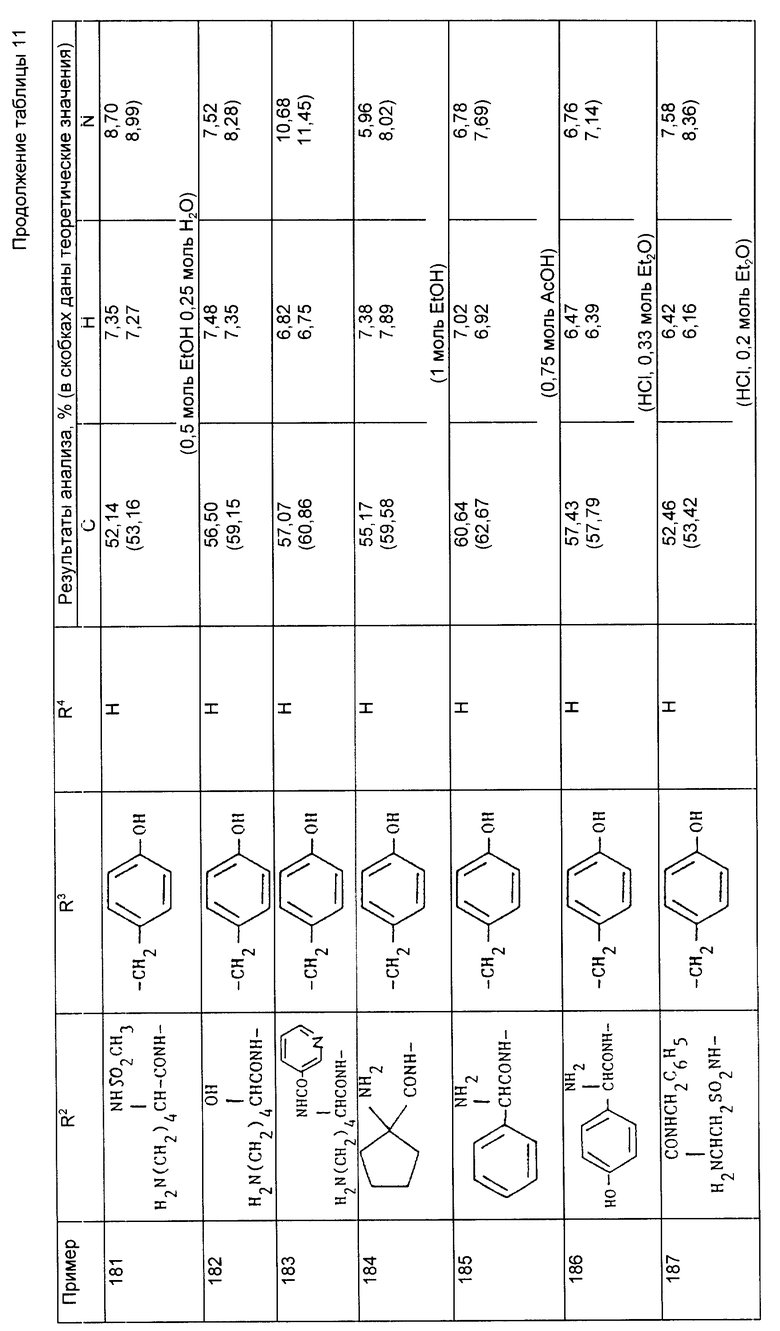
Примеры 167-170 (табл. 10). Следующие соединения были получены по методике, описанной в примере 166, при использовании цезиевой соли из примера 165 и осуществляли реакции с соответствующим хлоридом.
Пример 171. N-{ 1-[3-N6-бензилоксикарбонил-N2 -метан-сульфонил-/S/-лизиламино-2-/S/-инданил-оксикарбонилпропил] -1-циклопентанкарбонил] -O-бензил-/S/-тирозин бензиловый сложный эфир.
1-Этил-3-/диметиламинопропил/-карбодиимид (0,28 г) добавляли к раствору кислоты из примера 165 (e) (1,0 г) и оксибензтриазола (0,17 г) в дихлорметане (25 мл), охлажденному до 0oC.
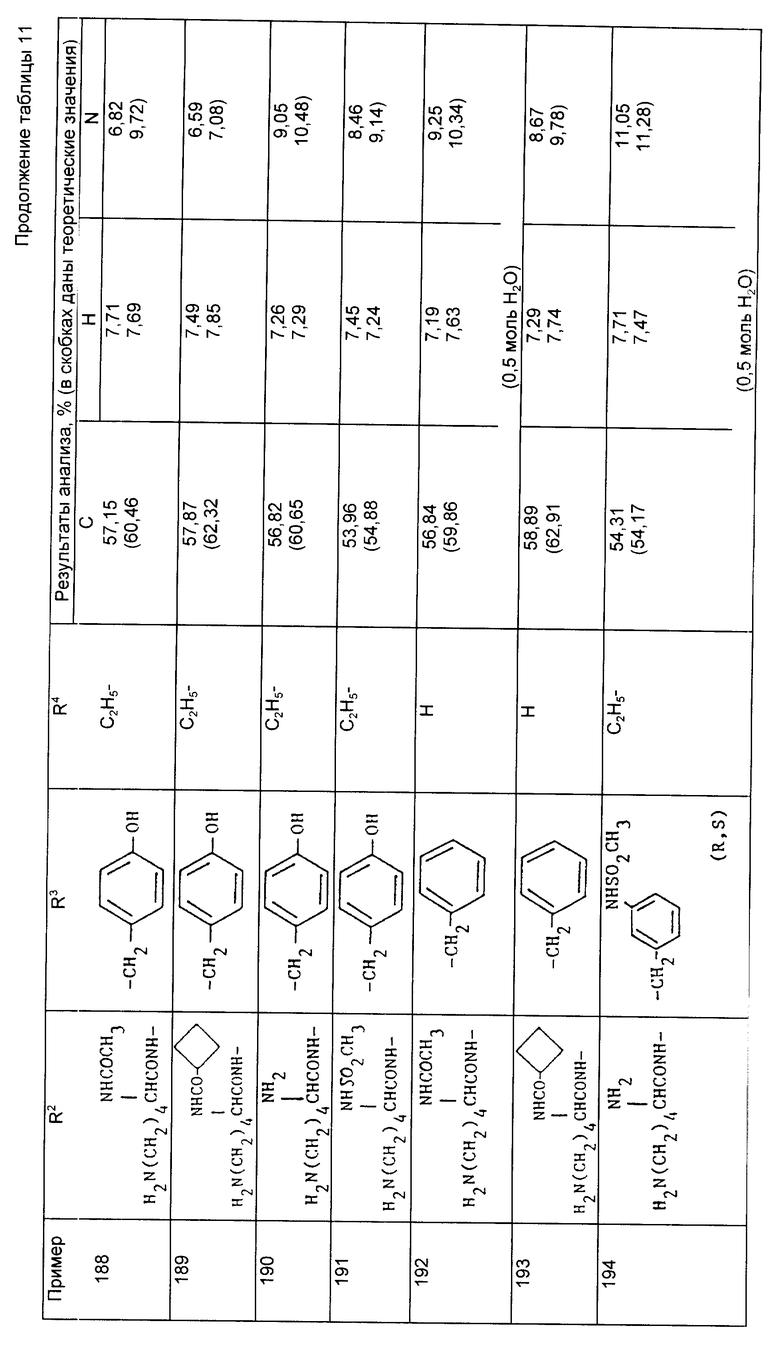
Через 10 мин добавили N-метилморфолин (0,42 г), инданол (0,42 г) и диметиламинопиридин (10 мг) и смесь перемешивали в течение 72 ч. Реакционную смесь разбавили дихлорметаном, промыли водой (2 x 10 мл), 2M соляной кислотой (2 x 10 мл), насыщенным раствором соли (10 мл), осушили (MgSO4), профильтровали и выпарили, в результате чего получили сырой продукт в виде масла. Его подвергли хроматографической очистке на силикагеле, элюируя смесями этилацетата и гексана, и получили указанный инданиловый сложный эфир в виде вспененного материала (0,98 г, 69%).
Пример 172. N-{1-[3-/N6-трет-бутилоксикарбонил-N2 -метансульфонил-/S/-лизиламино/-2-/S/-трет-бутилоксикарбонилпропил] -1-циклопентанкарбонил}-O-трет-бутил-/S/-тирозин 5-индениловый сложный эфир.
По описанной выше методике, используя в качестве исходного соединения кислоту из примера 164, приготовили тирозин 5-инданиловый сложный эфир в виде вспененного материала.
Анализ для C48H72N4O11S:
Найдено,%: C 62,37; H 8,04; N 5,95;
Рассчитано,%: H 7,95; N 6,14.
Пример 173. N-{1-[3-/N2-метансульфонил-N6-трет -бутилоксикарбонил-/S/-трет-бутилоксикарбонилпропил] -1-циклопентакарбонил}-O-этоксикарбонил-/S/-тирозин-этиловый сложный эфир.
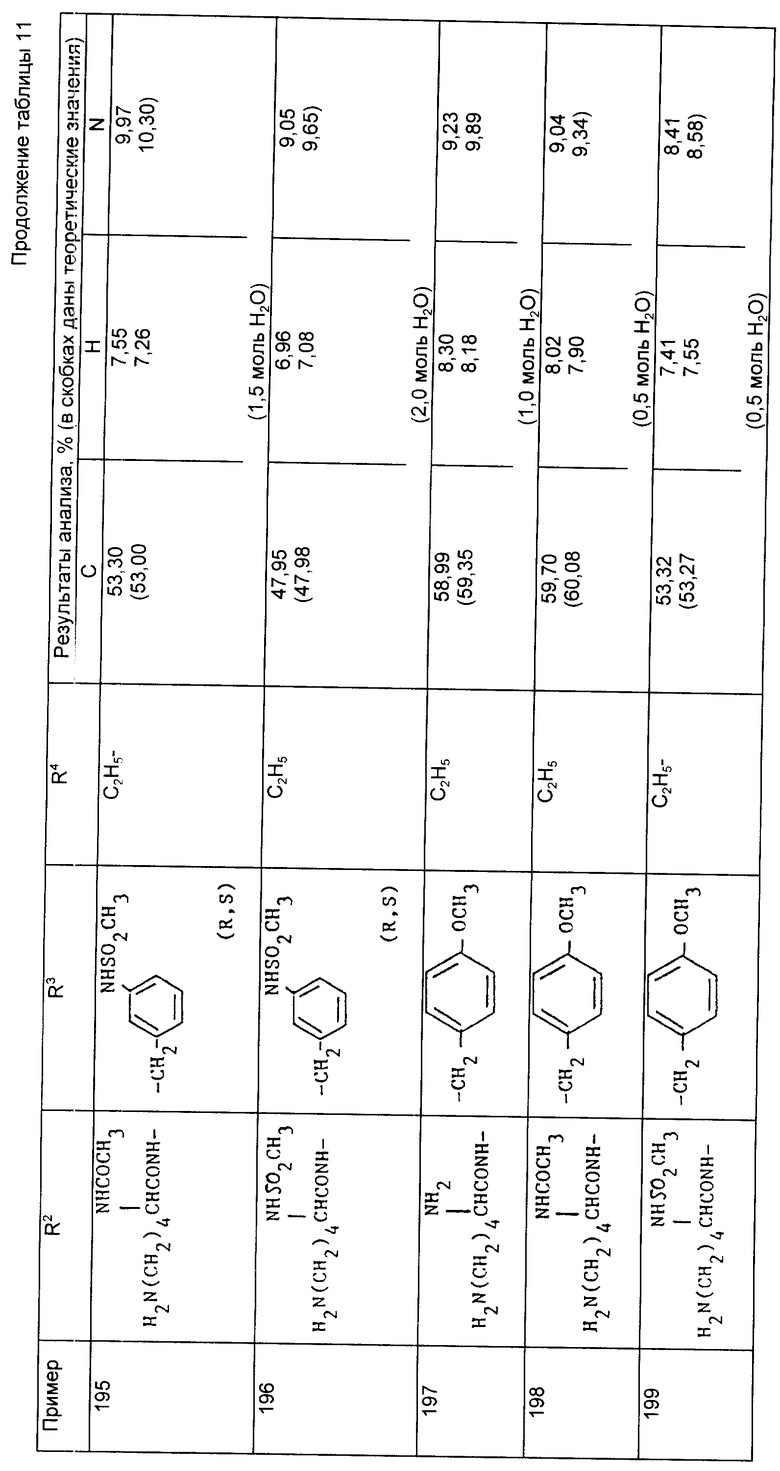
Этилхлорформиат (0,103 г, 1,007 ммоль) добавили к охлажденному на льду раствору N-{ 1-[3-/N2метансульфонил- N6-трет-бутилоксикарбонил-/S/-дизиламино-2-/S/-трет- бутилоксикарбонилпропил]-1-циклопентакарбонил}-/S/-тирозинэтилового сложного эфира (0,7041 г, 0,916 ммоль), триэтиламина (0,2781 г, 2,75 ммоль) и 4-диметиламинопиридина (0,0112 г) в сухом дихлорметане (20 мл). Через 30 мин ледяную баню удалили и реакционную смесь перемешивали в течение ночи при комнатной температуре. Растворитель выпарили при пониженном давлении и оставшееся масло обработали этилацетатом (50 мл) и 2H соляной кислотой (50 мл). Фазы разделили и органический слой промыли насыщенным раствором бикарбоната натрия (50 мл), затем насыщенным раствором соли (50 мл) и окончательно осушили сульфатом магния, прежде чем удалить растворитель при пониженном давлении. В результате получили сырой продукт в виде масла. После хроматографирования на силикагеле и элюирования смесями дихлорметана и диэтилового эфира получили указанный продукт в виде белого вспененного вещества (0,367 г, 48%).
Анализ для C39H64N4O13S:
Найдено,%: C 56,68; H 7,36; N 6,65;
Рассчитано,%: C 56,50; H 7,78; N 6,76.
Пример 174. N-{1-[3-/N2-метансульфонил-N6-трет- бутилоксикарбонил-/S/-лизиламино/2-/R, S/-трет-бутоксикарбонилпропил]-1- циклопентанкарбонил}-о-циклогексилоксикарбонил-/S/-тирозин-циклогенсиловый сложный эфир.
Указанное соединение получили аналогично тому, как описано в примере 173, но используя в качестве исходного соединения из примера 116 и осуществляя взаимодействие с циклогексилхлорформиатом, в результате чего получили указанный продукт в виде белого вспененного вещества (1,672 г,81%).
Анализ для C48H76N4O13S:
Найдено,%: C 60,69; H8,16; N 6,14;
Рассчитано,%: C 60,73; H 8,07; N 5,90.
Пример 175. N-{ 1-[2-/S/-трет-бутилоксикарбонил-3-/N6- трет-бутилоксикарбонил-N2-этил-/S/-лизиламино/пропил] -1- циклопетанкарбонил}-о-трет-бутил-/S/-тирозин-трет-бутиловый сложный эфир.
Цианоборгидрид натрия (45 мг) единоразово добавили при перемешивании к охлажденному на льду раствору N-{ 1-[2-/S/-трет-бутилоксикарбонил-3-/N6-трет-бутоксикарбонил- /S/-лизиламино/пропил] -1-циклопентанкарбонил} -о-трет-бутил-/S/- тирозин-трет-бутилового сложного эфира (507 мг) и ацетильдегида (31 мг) в водном этаноле (80%, 10 мл) и pH раствора довели до 5 с помощью 1H соляной кислоты. Полученному раствору дали нагреться до комнатной температуры и перемешивали в течение 1,5 ч. Реакционную смесь выпарили досуха и остаток обработали водой и этилацетатом. Фазы разделили и органический слой промыли небольшим количеством водного раствора бикарбоната натрия, осушили (Mg SO4), профильтровали и выпарили. Остаток подвергли хроматографической очистке на силикагеле, элюируя смесями гексана и этилацетата, содержащими 1 % диэтиламина, в результате чего получили указанное соединение в виде масла (370 мг, 64 %), Rf 0,55 (оксид кремния, CH2Cl2, CH3OH, NH4OH, 90:10:1).
Пример 176. N-{ 1-[3-/N2, N6-дибензилоксикарбонил-/S/- лизиламино/-2-/S/-карбоксипропил]-1-циклопентанкарбонил}-/S/- тирозин.
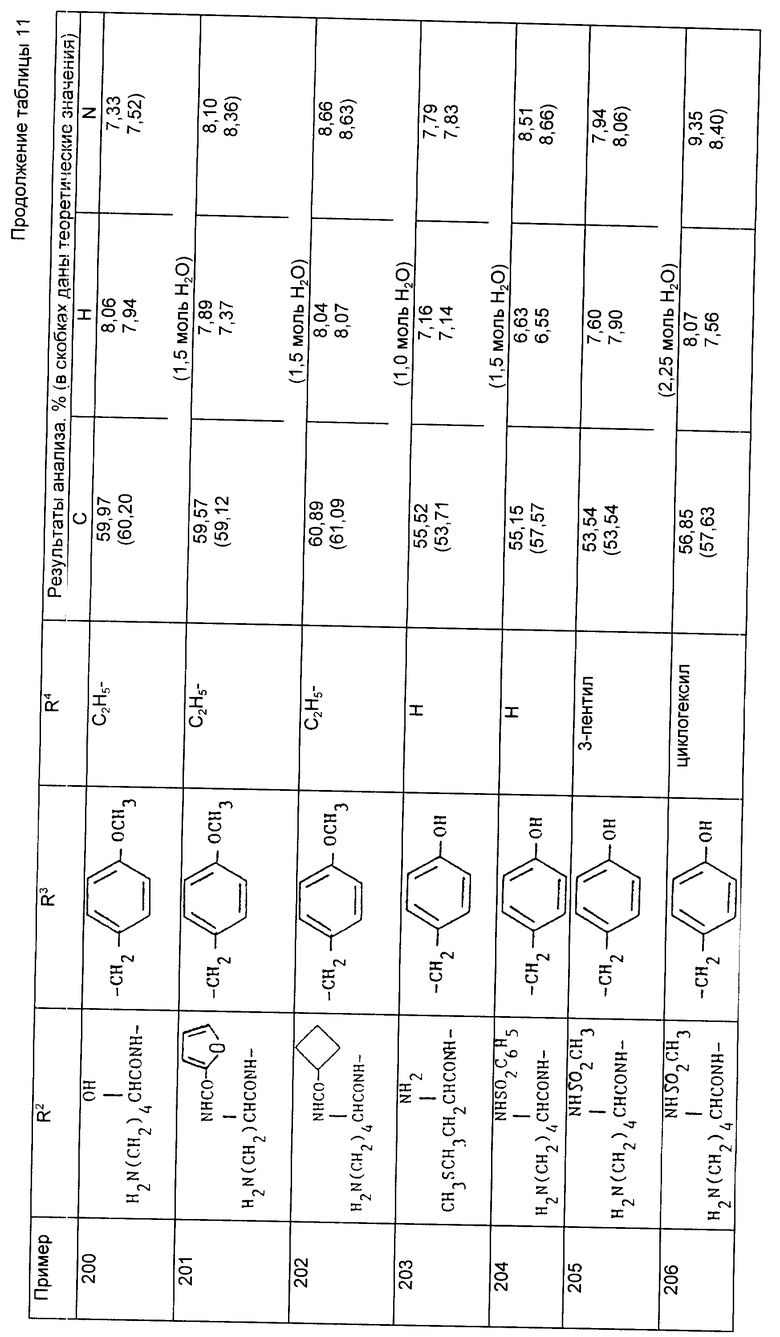
Хлористый водород пропустили при перемешивании через охлажденный на льду раствор N-{1-[3-/N2,N6-дибензилоксикарбонил-/S/- лизиламино/-2-трет-бутилоксикарбонилпропил] -1-циклопентанкарбонил} - о-трет-бутил-/S/-тирозин-трет-бутилового сложного эфира (из примера 81, 0,445 г, 0,47 ммоль) и анизола (0,765 г, 7,1 ммоль) в сухом дихлорметане (10 мл) до полного насыщения. Образовался осадок. После перемешивания в течение 1,5 ч растворитель выпарили при пониженном давлении и остаток обработали сухим дихлорметаном. Остаток распределили между этилацетатом и водным раствором бикарбоната натрия. Фазы разделили и органический слой промыли двумя порциями водного раствора бикарбоната натрия. Объединенные водные фазы повторно проэкстрагировали диэтиловым эфиром и затем подкислили 1M соляной кислотой до pH 2. Водную фазу проэкстрагировали этилацетатом (2х) и объединенные органические фазы осушили (MgSO4), профильтровали и растворитель выпаривали, в результате чего получили вспененное вещество, которое обработали хлористым метиленом и получили указанное соединение в виде твердого вспененного вещества (0,325 г, 89%).
Анализ для C41H50N4O11 • 0,4CH2C2
Найдено,%: C 62,81; N 6,92; H 6,68;
Рассчитано,%: C 61,17; H 6,33; N 6,93.
Пример 177. N-{ 1-/2/S/-карбокси-3-/S/-лизиламино-пропил-1- циклопентанкарбонил]-/S/-тирозин.
Продукт, полученный в примере 176 (0,247 г, 0,32 ммоль), растворили в смеси этанол: вода (9:1, 20 мл) и гидрировали при комнатной температуре под давлением водорода, 4,2 кг/см2 над 10% палладием на активированном угле (100 мг) в течение ночи. Реакционную смесь профильтровали через воронку с "solkaflok" и фильтрат выпарили досуха. Остаток обработали дихлорметаном (3х) и получили указанное соединение в виде вспененного соединения (0,12 г, 74%).
Анализ для C25H38N4O7 • 0,65H2O;
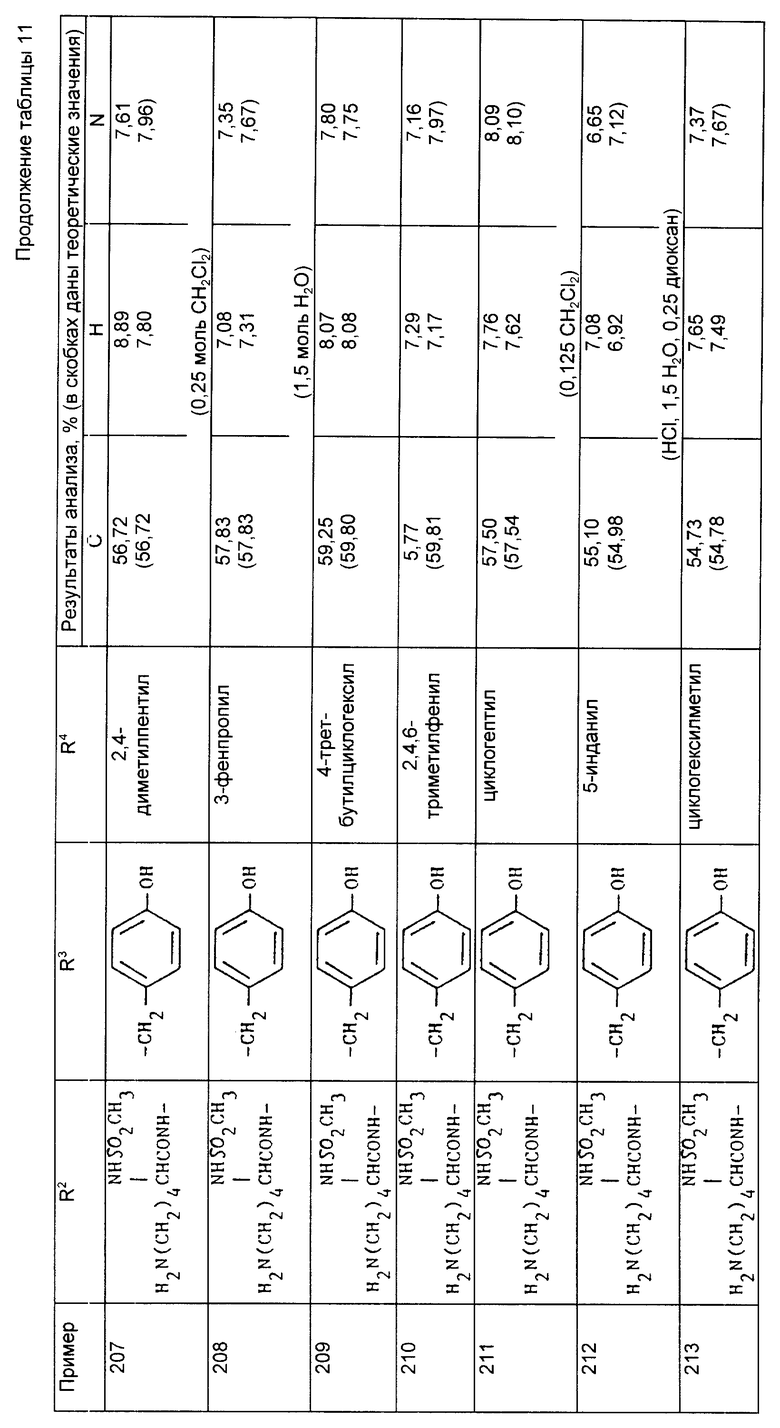
Найдено,%: C 56,87; H 7,76; N 10,36;
Рассчитано,%: C 57,93; H 7,64; N 10,81.
Примеры 178-213 (табл. 11). Следующие соединения получили по методикам удаления защитных групп, описанных в примерах 176 и 177, используя в качестве исходного соединения соответствующий трет-бутиловый или бензиловый сложный эфир (трет-бутилоксикарбонил или бензилоксикарбонил защищенное соединение). Если специально не указано, то соединения на основе лизина и тирозина имеют (S)-стереохимию.
При осуществлении примеров 178-187, 203 и 204 использовали разрешенные соединения, имеющие S, S - стереохимию.
Примеры 214-245 (табл. 12). Следующие соединения получили из соответствующих трет-бутил или бензиловых сложных эфиров (трет-бутоксикарбонил или бензилоксикарбонил замещенных соединений) путем обработки HCl или гидрированием по методике, описанной в примерах 176 и 177.
Производные лизина имеют (S) стереохимию, если это специально не оговорено.
(1) Пример 217 осуществили путем удаления защиты в положении с помощью H в уксусной кислоте.
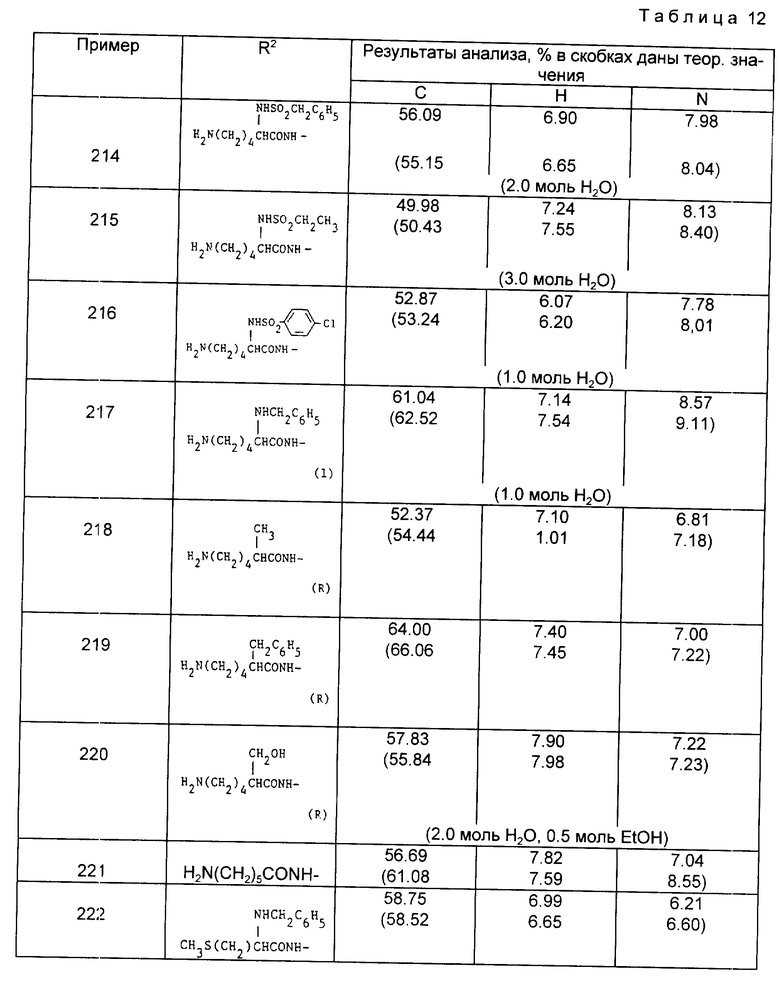
(2) За исключением примеров 228-229 и 245 соединения являются разрешенными S,S-изомерами.
Примеры 246-259 (табл. 13). Следующие соединения получили из соответствующих трет-бутиловых или бензиловых сложных эфиров (трет-бутоксикарбонил или бензилоксикарбонил защищенного соединения путем обработки HCl и/или гидрированием по методикам, описанным в примерах 176 и 177. Остатки от лизина и тирозина имеют (S)-стереохимию, если нет специальных указаний, примеры 249, 251, 252, 258 и 259 являются полностью разрешенными S,S,S-изомерами.
Примеры 260-263 (табл. 14). Следующие соединения получили путем удаления защитных групп из соответствующего N-бутилоксикарбонильного или N-бензилоксикарбонильного производного по методике, описанной в примере 176 или 177, исходя из соответствующего S,S,S-изомера.
Пример 264. N-{ 1-[3-/N2-ацетил-/S/-лизиламино/-2- карбоксипропил]-1-циклопентанкарбонил}-3-метансульфонамидо-/R,S/-фенилаланин.
Раствор N-{ 1-[3-N2-ацетил-/S/-лизиламино-2-карбоксипропил] -1- циклопентанкарбонил} -3-метансульфонамидо-/R, S/-фенилаланин этилового сложного эфира (из примера 195, 0,21 г) в эталоне (10 мл) обработали раствором гидроксида натрия (5 мл 2H) и раствор перемешивали при комнатной температуре в течение 3,5 ч. Реакционную смесь вылили в колонку с сильно кислотной ионно-обменной смолой, которую промыли до нейтральной реакции и продукт затем элюировали водным пиридином (3%). После выпаривания фракций, содержащих продукт, получили указанную двуосновную карбоновую кислоту в виде аморфного вещества (0,092 г, 46%). Т.пл. 160-164oC.
Анализ для C28H43N5O9S•1.5H2O;
Найдено, %: C 51,32; H 6,86; N 10,75;
Рассчитано, %: C 51,52; H 7,10; N 10,73.
Примеры 265-273 (табл. 15). Следующие соединения были получены по методике, описанной в примере 264, где в качестве исходного соединения был использован соответствующий сложный этиловый эфир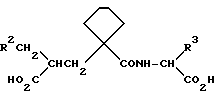
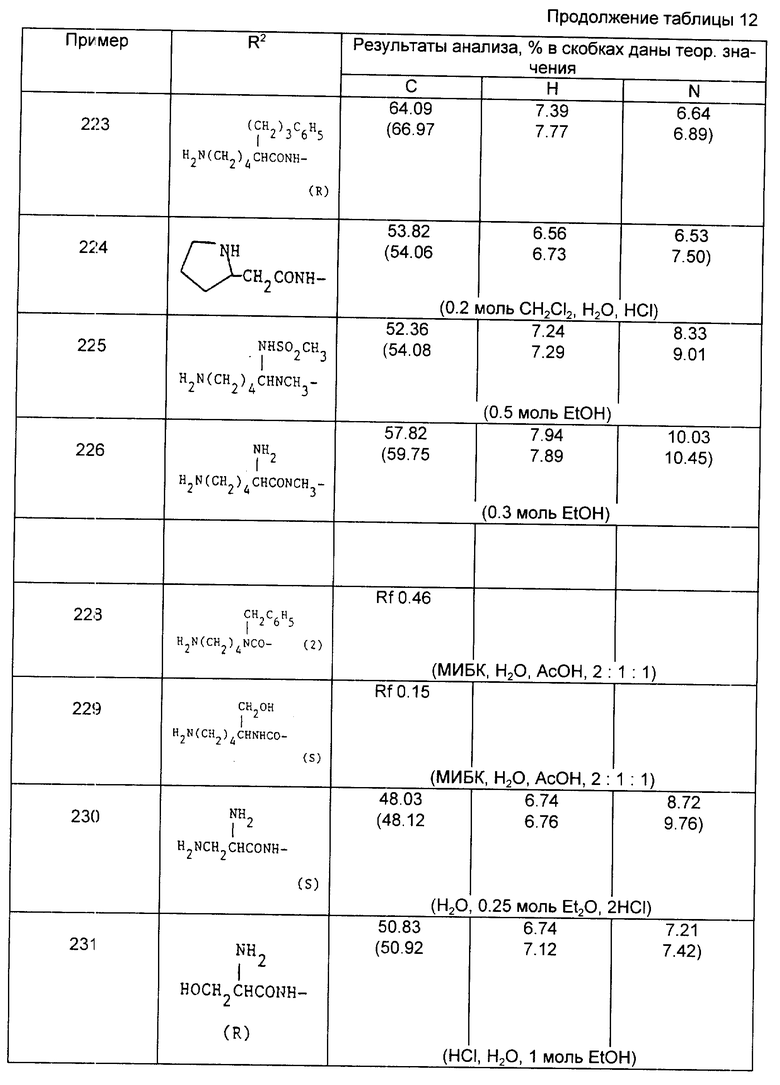
Соединения примеров 267 и 273 являются разрешенными соединениями, имеющими S,S,-стереохимию.
Следующие три примера фармацевтических композиций приводятся в отношении соединения из примера 181.
10 мг/мл раствор для внутривенной инъекции
Ингредиент - мг/единица
Соединение изобретения - 10,0
Na2HPO4 - 8,10
KH2PO4 - 1,90
NaCl - 3,54
Вода для инъекций - до 1,00 мл - Всего 1,00 мл
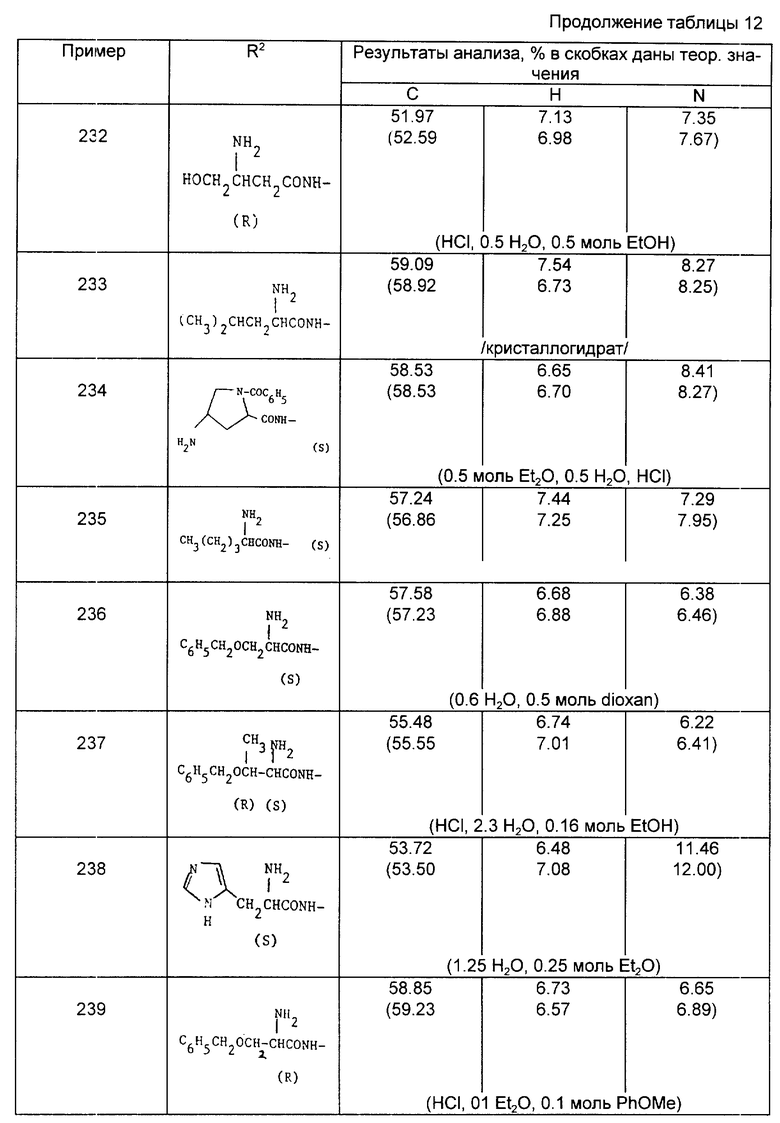
25 мг капсула
Ингредиент - мг/единица
Соединение изобретения - 25,00
Двухосновный фосфат кальция - 222,00
Предварительно желатинизированный крахмал - 35,00
Кросповидон - 15,00
Стеарат магния - 3,00 - Всего 300,00
Плюс белая непрозрачная жесткая желатиновая оболочка капсулы 2 размера.
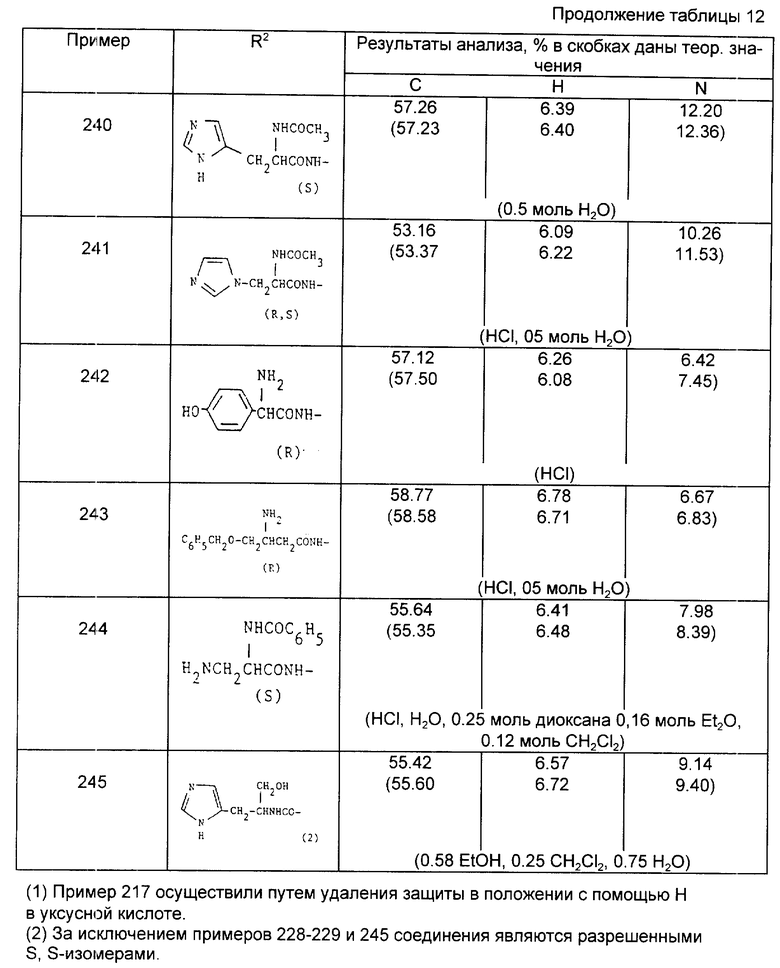
Сердцевина 100 мг таблетки
Ингредиент - мг/единица
Соединение изобретения - 100,00
Микрокристаллическая целлюлоза - 106,00
Двухосновный фосфат кальция - 53,20
Повидон - 15,00
Кросповидон - 22,50
Стеарат магния - 3,00
Пропан-2-ол - 236,00 (Это количество может меняться в зависимости от размера загрузки и теряться в процессе переработки). - Всего 300,00
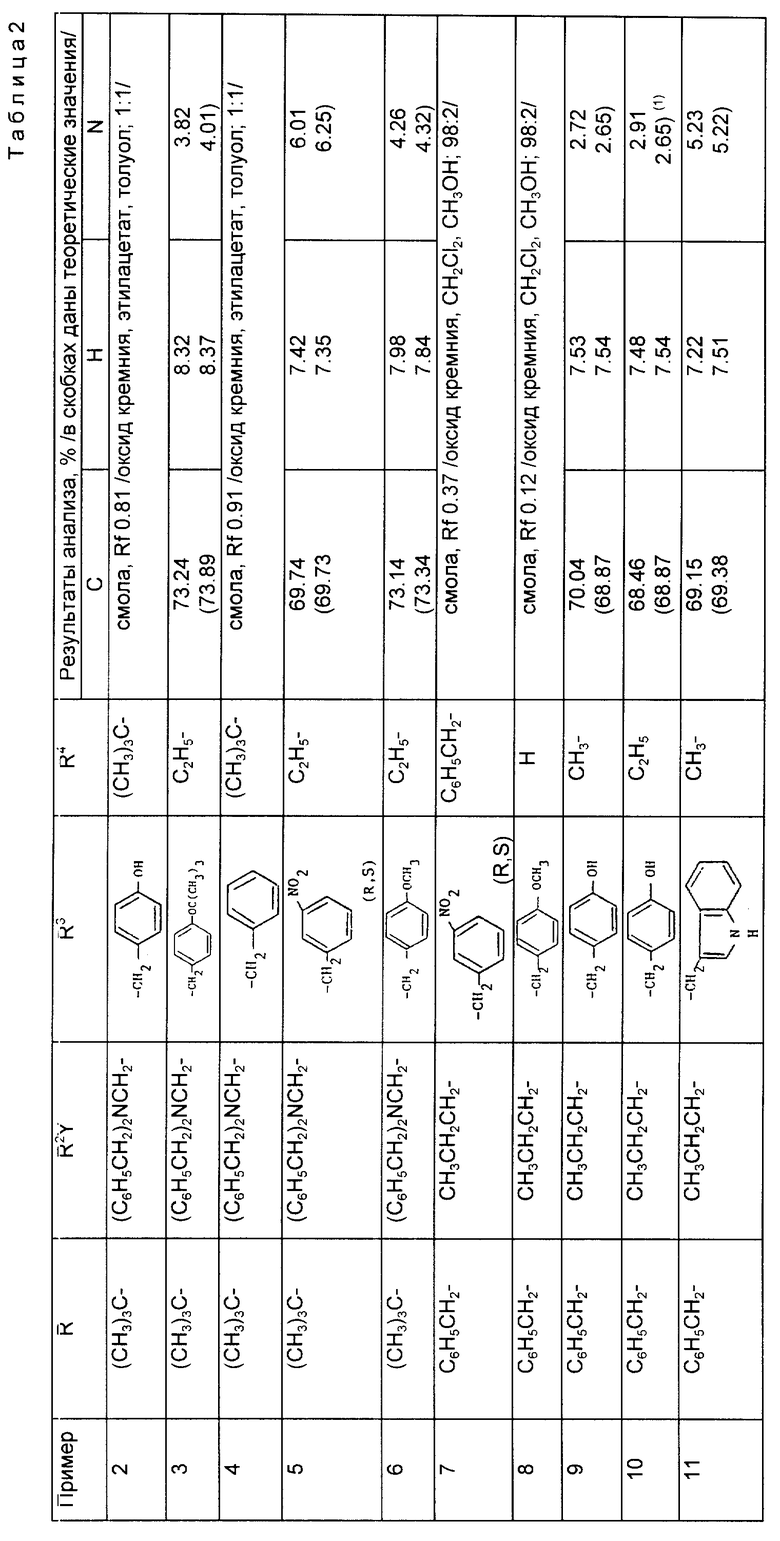
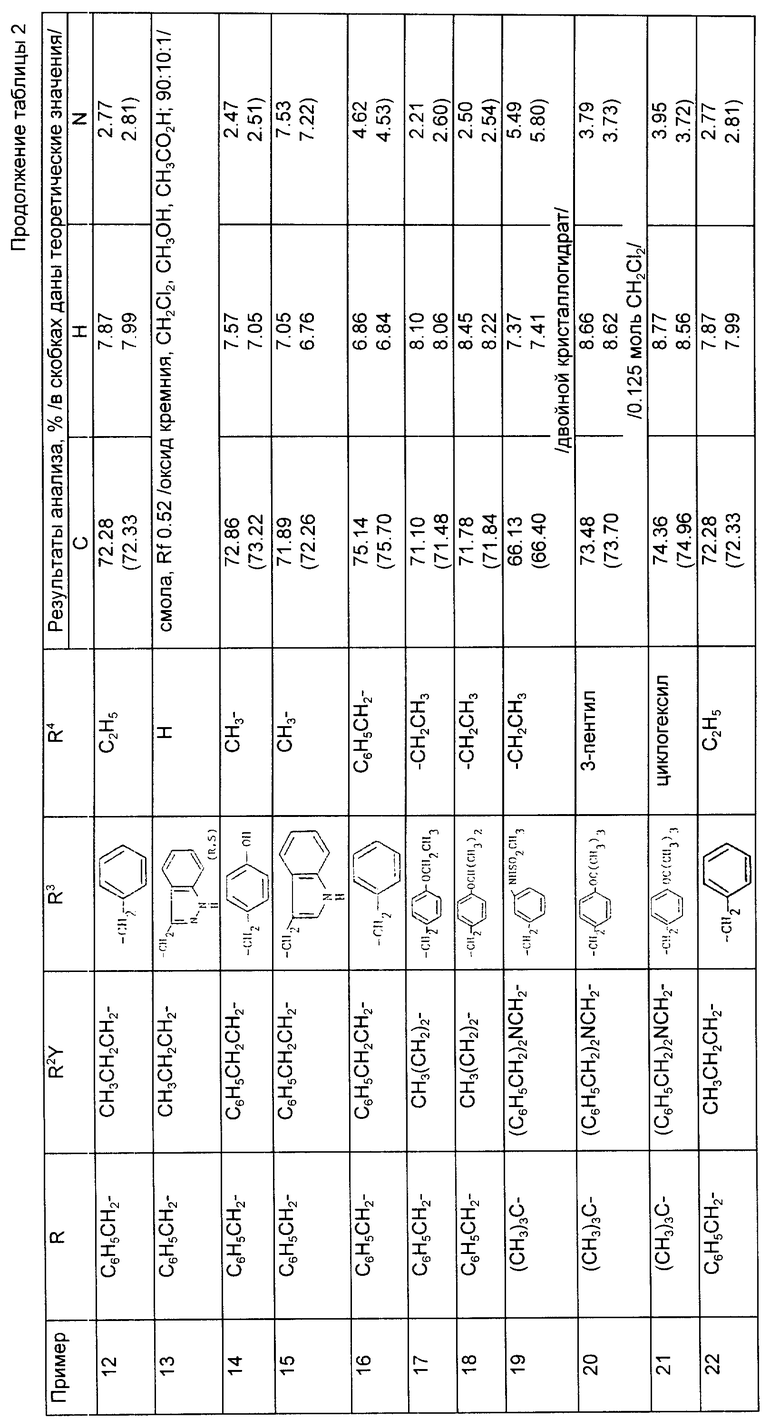
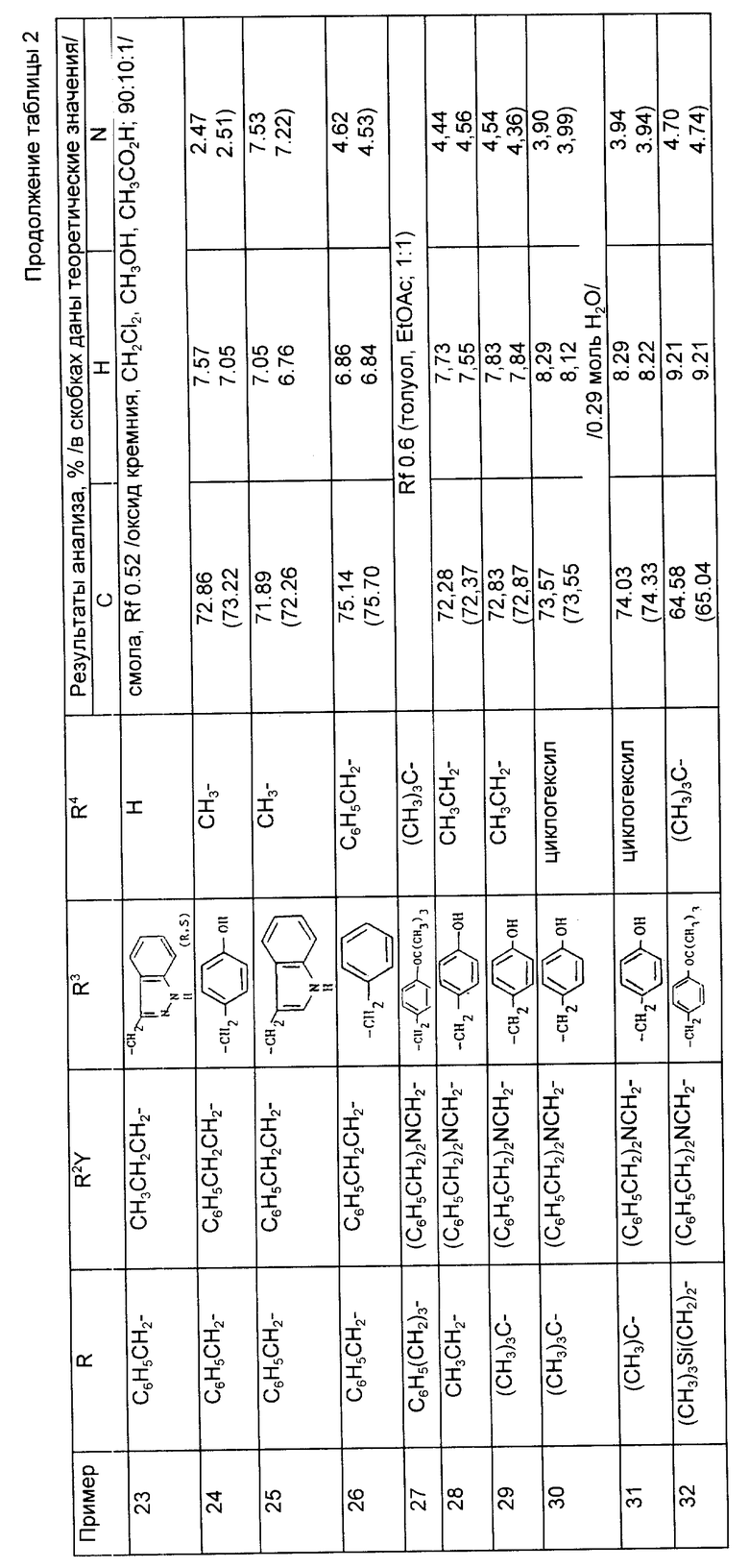
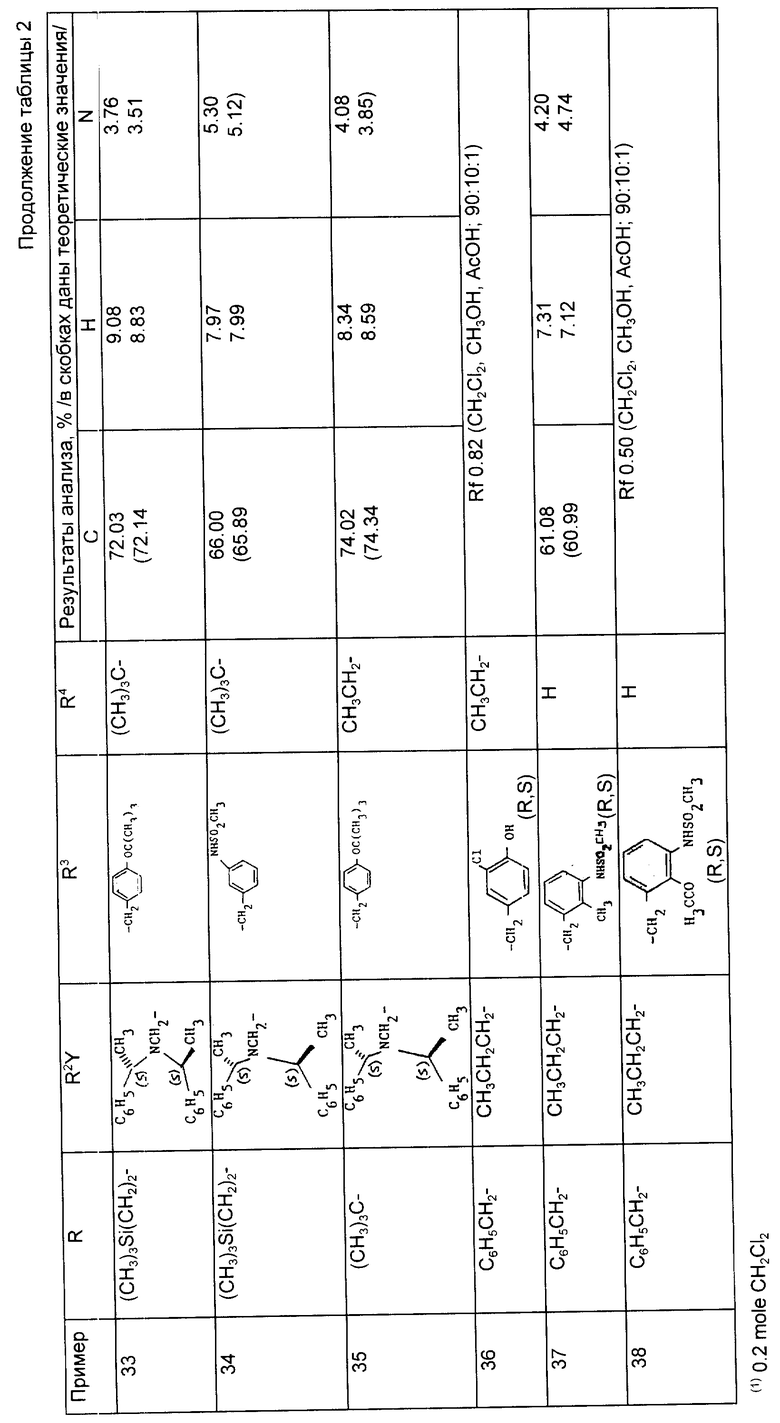

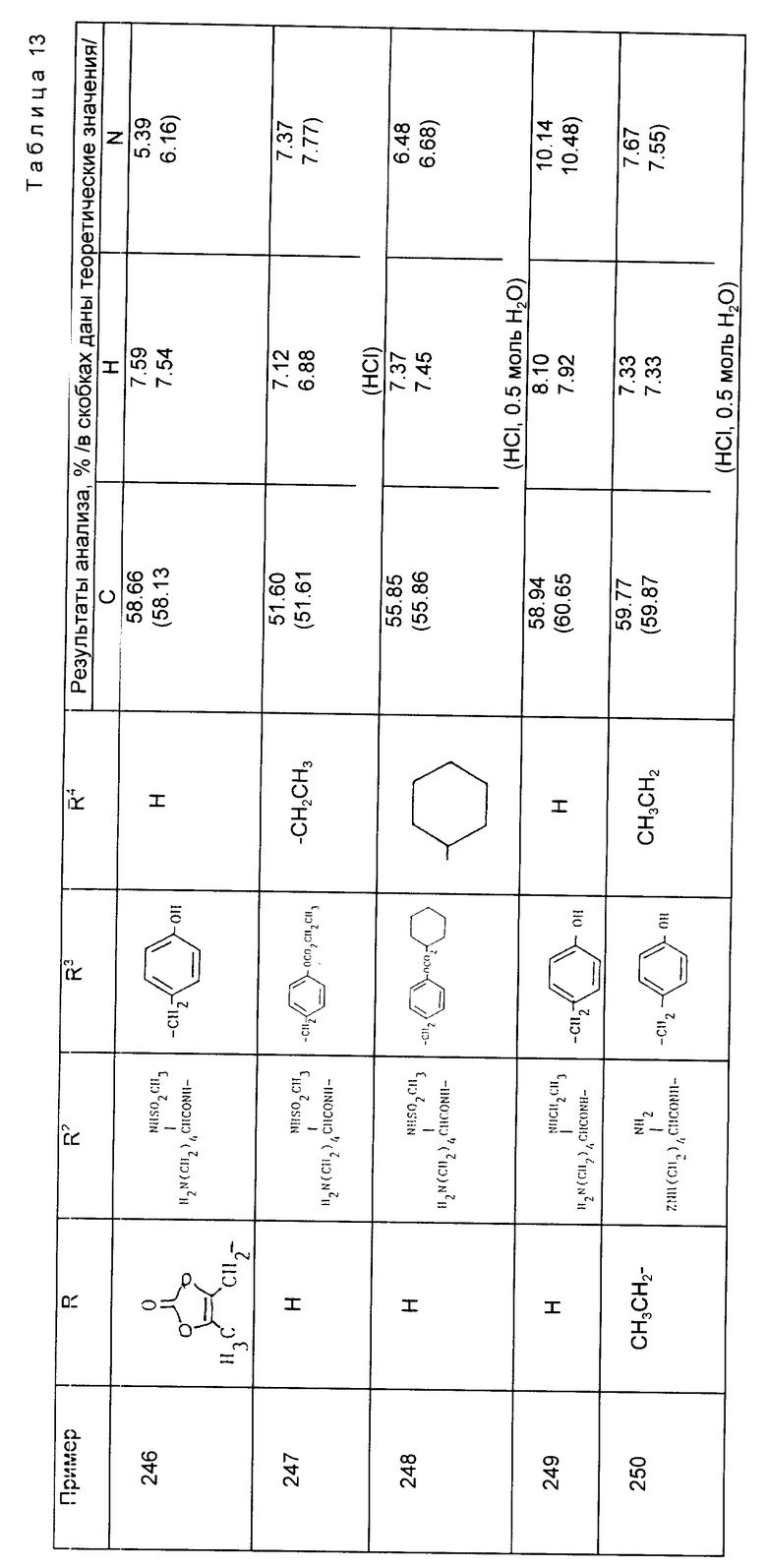
Предложены производные циклоалкилзамещенного глутарамида общей формулы (I) 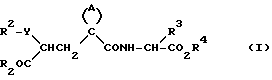
где A-(СН2)4, (СН2)5; R1-Н, (С1 - С4)алкил; R и R4-Н, (С1 - С4)алкил, (С3 - С7)циклоалкил, бензил или биолабильная эфирообразующая группа; Y - простая связь, (С1 - С6)алкилен; R2-Н, фенил, R6CONR5- или R7NR5СО-; R3 обозначает группу формулы (II), или их фармацевтически приемлемые соли, или биолабильный эфир. Предложена также фармацевтическая композиция, обладающая ингибирующей ангиотензинпревращающей фермент и нейтральную эндопептидазу активностью, на основе соединений формулы (I). 3 с. и 11 з.п. ф-лы, 15 табл.

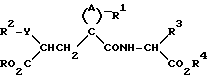
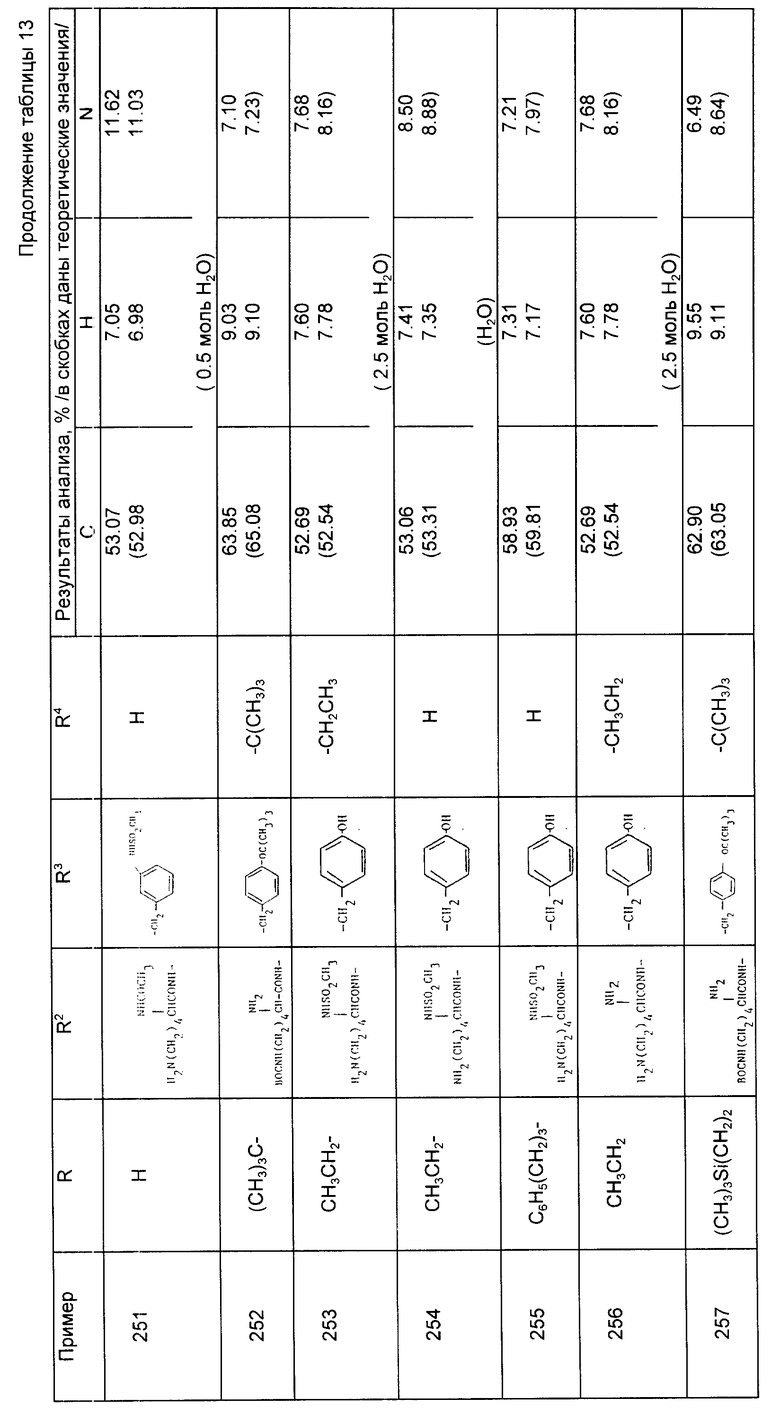
где А - (СН2)4 или (СН2)5 и образует с атомом углерода насыщенный или мононенасыщенный цикл;
R1 - водород или С1 - С4-алкил;
R и R4 каждый независимо - водород, С1 - С6-алкил, С3 - С7-циклоалкил, бензил или биолабильная эфирообразующая группа;
Y - простая связь или С1 - С6-алкилен с прямой или разветвленной цепью;
R2 - водород, фенил, R6CONR5- или R7NR5CO-, где R5 - водород, С1 - С6-алкил или фенил-С1 - С6-алкил, при условии, что Y не обозначает простую связь, когда R2 - водород или фенил, R6 и R7 каждый - группа формулы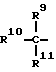
где R9 - водород, ОН, С1 - С6-алкил, фенил-С1 - С6-алкил, гидрокси-С1 - С6-алкил, R1 2SO2NH, R1 2CONH, (R1 3)2N;
R1 0 и R1 1 каждый независимо - водород или С1 - С6-алкил или R1 0 - водород, R1 1 - амино-С1 -С6-алкил, имидазолилметил, фенил, 4-гидроксифенил, бензилокси-С1 - С6-алкил, гидрокси-С1 - С6-алкил, метилтио-С1 - С6-алкил или две группы R1 0 и R1 1 вместе с атомом углерода, к которому они присоединены, образуют 3 - 6-членное карбоциклическое кольцо или пирролидиновое, которое может быть необязательно замещено аминогруппой или бензилом, R1 2 - С1 - С6-алкил, С3 - С7-циклоалкил, фенил, 4-хлорфенил, фенил-С1 - С6-алкил, фурил или пиридил, R1 3 - водород, С1 - С6-алкил, фенил-С1 - С6-алкил;
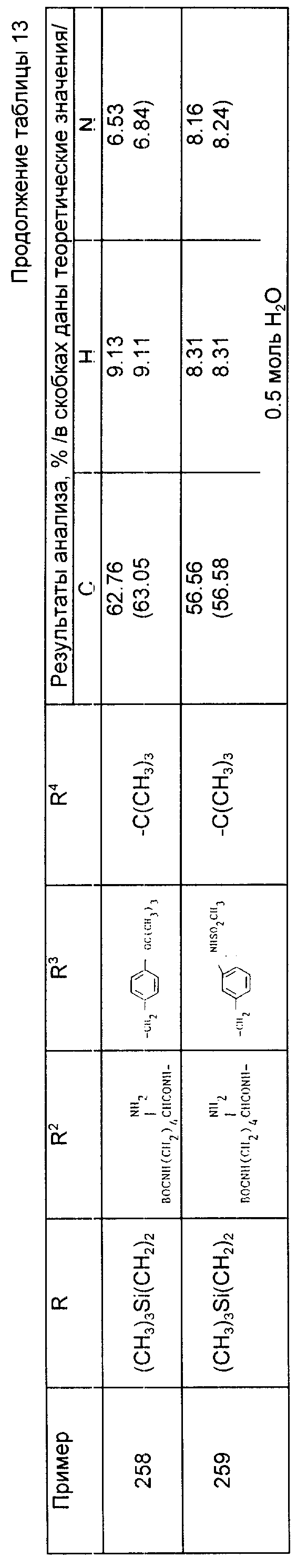
R3 - группа формулы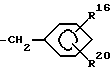
где R1 6 - водород, галоид, 4-ОН, 4-С1 - С6-алкокси, 4-[(С1 - С6-алкокси)карбонилокси],3 - [(С1 - С4-алкил)SO2NH-], 4-[(С3 - С7-циклоалкокси) карбонилокси], R2 0 - водород, С1 - С4-алкил, С1 - С4-алкокси, С2 - С6-алканоил или галоид,
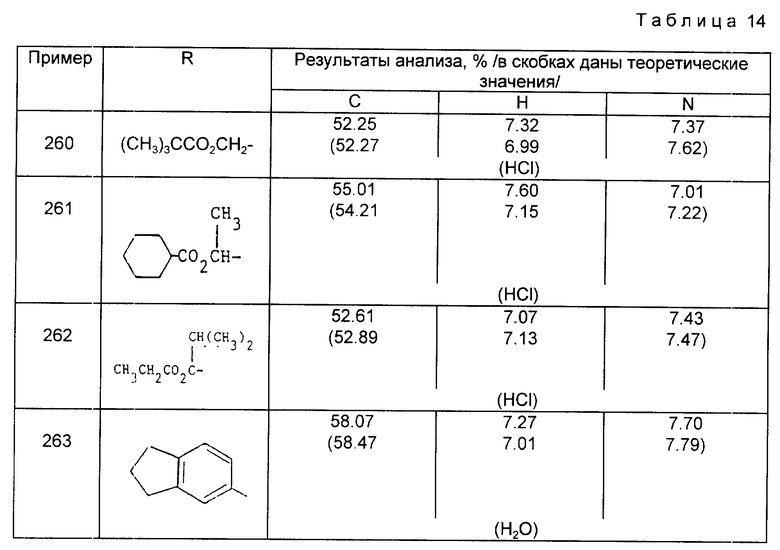
или R3 - группа формулы
или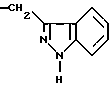
или его фармацевтически приемлемая соль, или его биолабильный эфир.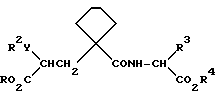
где R, R2, R3, R4 и Y указаны в п.1.
| EP, заявка, 274234, кл | |||
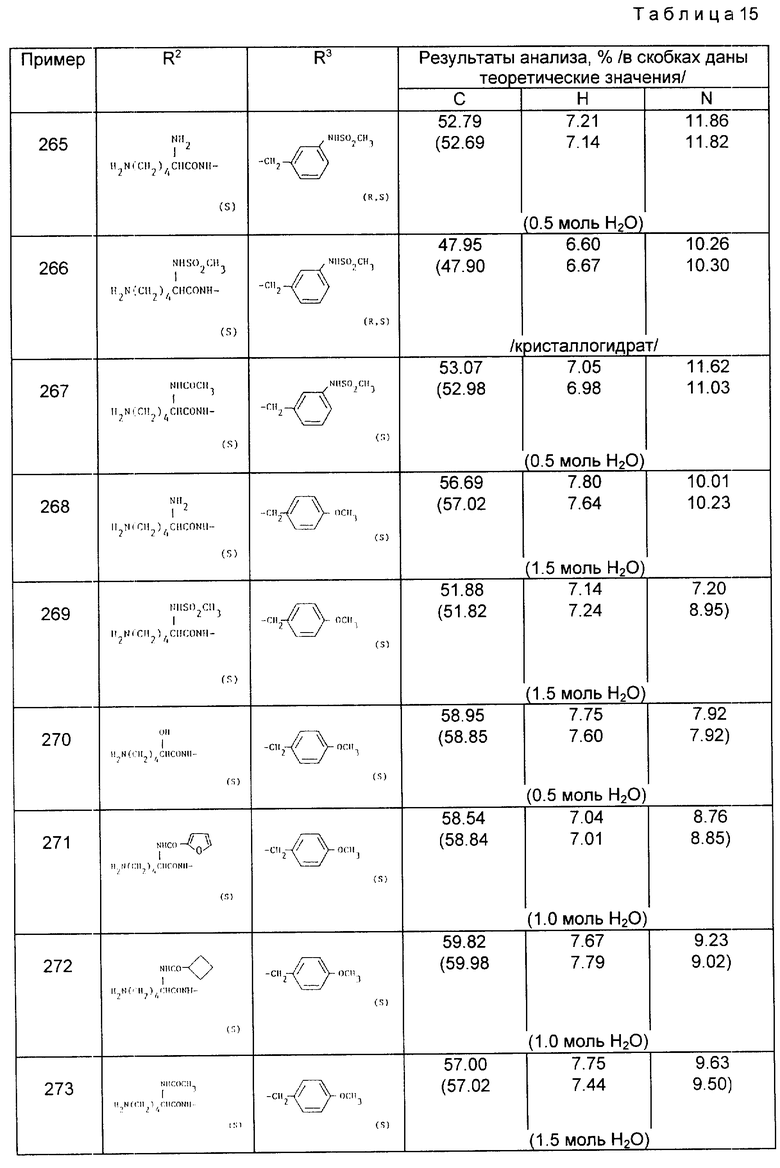
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
