Изобретение относится к некоторым дизамещенным бензолалкановым кислотам.
Такие соединения способны селективно противодействовать действию тромбоксана A2(TXA)2 и его предшественника простагландина H2(PGH2) у рецептора тромбоксана. Эти соединения, таким образом, пригодны в качестве терапевтических средств, их можно применять или отдельно, или (что предпочтительно) в комбинации с ингибитором тромбоксан-синтетазы, например, для лечения атеросклероза и нестабильной стенокардии и для предотвращения реокклюзии (как острой, так и хронической) после чрескожной пластической операции на коронарных и бедренных сосудах.
Комбинация может найти также клиническое применение в других видах патологических состояний, в которых участвует тромбоксан A2, например при лечении инфаркта миокарда, удара, сердечной аритмии, переходящего нарушения мозгового кровообращения, опухолевых метастаз, периферического сосудистого заболевания, бронхиальной астмы, заболевания почек, индуцированной циклоспорином непротоксичности, отторжения почечного трансплантата, сосудистых осложнений диабета и эндотоксинового шока, травмы преэклампсии и в обходном шунтировании коронарных артерий и гемодиализе.
В EP-A-0031954 описаны некоторые сульфамидозамещенные бензолалкановые кислоты, применяемые в качестве ингредиентов агрегирования тромбоцитов.
Изобретение относится к соединениям формулы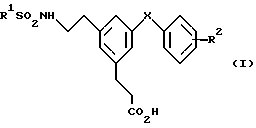
и их фармацевтически приемлемым солям и боилабильным эфирам,
где R1 представляет собой фенил, необязательно замещенный атомам галогена, CF3 или нафтил;
R2 - H, C1-C4-алкоксигруппу, галоген, CN или S(O)n (C1-C4-алкил);
X - CH2-, -CHCH3-, CH(OH)-, -CO- или -O-;
n = 2.
В указанном выше определении галоген обозначает фтор, хлор, бром или иод.
Соединения, содержащие асимметрические центры, могут существовать как энантиомеры и диастереомеры, и настоящее изобретение включает выделенные индивидуальные изомеры, а также смеси изомеров.
В изобретении включены также меченные радиоактивными изотопами производные соединений формулы (I), которые пригодны для биологических исследований.
Термин биолабильный эфир в указанном выше определении обозначает фармацевтически пригодный, биологически разрушаемый эфир соединения формулы (I), являющейся пролекарством, которое при введении животному или человеку превращается в организме в соединение формулы (I). В случае соединений формулы (I) такие биолабильные эфирные пролекарственные производные, в частности, удобные для образования соединений формулы (I), пригодны для перорального введения. Пригодность любой конкретной эфирообразующей группы можно оценить путем изучения in vivo (животные) или in vitro ферментативного гидролиза
Таким образом, для получения оптимального эффекта желательно, чтобы эфир гидролизовался только после завершения поглощения. В соответствии с этим эфир должен быть устойчив к преждевременному гидролизу пищеварительными ферментами до поглощения, но должен продуктивно гидролизоваться, например, ферментами стенок кишечника, плазмы или печени. Таким путем активная кислота высвобождается в ток крови после пероральной абсорбции пролекарственного производного.
Пригодные биолабильные эфиры могут включать алкиловые, алканоилоксиалкиловые, циклоалканоилоксиалкиловые, ароилоксиалкиловые и алкоксикарбонилоксиалкиловые эфиры, включая их циклоалкил- и арилзамещенные производные, ариловые эфиры и циклоалкиловые эфиры, у которых алкилы, алканоилы и алкоксигруппы могут содержать 1 - 8 атомов углерода и имеют нормальное или разветвленное строение, причем циклоалкилы могут содержать 3 - 7 атомов углерода и циклоалканоилы содержат 4 - 8 атомов углерода, и те и другие могут быть бензоконденсированы. Арилы и ароилы включают замещенные фениловые, нафтиловые или инданиловые циклические системы. Предпочтительно биолабильные эфиры изобретения являются C1-C4-алкиловыми эфирами. Более предпочтительно они являются метиловыми, этиловыми и трет-бутиловыми эфирами.
Фармацевтически пригодные соли соединений формулы (I) образованы основаниями, которые образуют нетоксичные соли. Примеры их включают соли щелочных и щелочноземельных металлов, например натрия, калия или кальция, и соли с аминами, например диэтиламином.
Предпочтительна группа соединений формулы (I), у которых каждый X представляет собой CH2, CH(CH3), CO или O. R1 предпочтительно является фенилом, замещенным галогеном или CF3, наиболее предпочтительно 4-хлорфенилом. R2 предпочтительно является H, F, OCH3, SO2CH3 или CN.
В частности, предпочтительные соединения включают
3-[4-фторфенил)метил] -5-[2-[(4-трифторметилфенил)- сульфониламино]этил] бензолпропановую кислоту;
3-[2-[(4-хлорфенил)сульфониламино]этил]-5-[1-(4- фторфенил)этил]бензолпропановую кислоту;
3-[2-[(4-хлорфенил)сульфониламино] этил]-5-[(4-фторфенил)метил] бензолпропановую кислоту;
3-[2-[(4-хлорфенил)сульфониламино] этил] -5-(4-фторфенокси) бензолпропановую кислоту;
3-[2-[(4-хлорфенил)сульфониламино] этил]-5-(4-метоксибензоил) бензолпропановую кислоту и
3-[2-[(4-хлорфенил)сульфониламино] этил]-5-(4-цианобензоил) бензолпропановую кислоту.
Соединение формулы (I) изобретения получают гидролизом являющихся их предшественниками низших алкиловых эфиров формулы (II)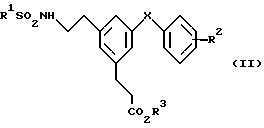
в которой R1, R2 и X имеют значения, указанные выше для формулы (I) и R3 представляет собой C1-C4-алкил, предпочтительно метил, этил или трет-бутил.
Реакцию можно проводить в основных или кислотных условиях, например, с применением избытка водной щелочи, предпочтительно раствора едкого натра, или избытка соляной кислоты соответственно, возможно с подходящим сорастворителем, например C1-C4-алканалом, предпочтительно метанолом, при температуре от около 20o до температуры кипения реакционной среды.
Некоторые соединения формулы (I) можно также превратить в другие соединения формулы (I) стандартными взаимными превращениями функциональных групп. Например, соединения формулы (I), у которых R3 является 2F, 2-Cl, 4-F или 4-Cl и X является CO, можно превратить в соответствующие соединения, у которых R2 является 2-(C1-C4)алкокси- или 4-(C1-C4)алкоксигруппой, обработкой алкоксидом щелочного металла в подходящем растворителе, например N,N-диметилформамиде или избытке (C1-C4)алканола, при температурах, вплоть до точки кипения растворителя. Такие же взаимопревращения можно также проводить нагреванием фтор- или хлорсоединения с избытком C1-C4-алканола в присутствии основания, например карбоната натрия или калия.
Соединения формулы (I), у которых X представляет собой CO и R2 представляет собой 2- или 4-(C1-C4)алкилтиогруппу, можно также получить из соответствующих соединений, у которых R2 представляет собой 2-F, 2-Cl, 4-F или 4-Cl, обработкой (C1-C4)алкилтиолатом (солью) щелочного металла в подходящем растворителе, например N,N-диметилформамиде или диметилсульфоксиде, при 50 - 150oC. Аналогично, с примерами (C1-C4)алкилсульфината щелочного металла в растворителе, например N,N-диметилформамиде или диметилсульфоксиде, при 50 - 150oC получают соответствующее соединение формулы (I), у которого R2 является (C1-C4)алкилсульфонилом.
Соединения формулы (I), у которых R2 является группой SO2 (C1-C4)алкил, можно также получить окислением соответствующего соединения, у которого R2 является S(C1-C4)алкилом, при помощи, например, пероксида водорода в подходящем растворителе, например уксусной кислоте. Регулируемое окисление при помощи, например, стехиометрического количества пероксида водорода в уксусной кислоте или метаиодата натрия в водном метаноле позволяет получить соответствующие соединения формулы (I), у которых R2 является группой SO2 (C1-C4)алкил.
Соединения формулы (I), у которых R2 является CONH2, предпочтительно получают из соответствующего соединения, у которого R2 является CN, обработкой пероксидом водорода и водным основанием (например, едким натром), обычно при температуре около 50oC. В качестве сорастворителя можно добавить этанол. Амидный продукт можно превратить в соответствующее соединение, у которого R2 является NH2, при помощи реакции Гофмана, т.е. обработкой гипохлоритом натрия в водных щелочных условиях. Аминосоединения пригодны в качестве промежуточных продуктов для некоторых других соединений формулы (I). Например, диазотированием с последующей обработкой хлористой медью (1) или бромистой медью (1) получают соответствующие продукты, у которых R2 представляет собой Cl или Br соответственно. Альтернативно, обработкой диазониевого соединения иодидом (солью), например, иодидом калия, получают соответствующий продукт формулы (I), у которого R2 является I. Предпочтительны соединения формулы (I), у которых R2 является Br или I, поскольку последние заместители будут реагировать в нескольких промежуточных стадиях, описанных ниже, образуя другие соединения формулы (I).
Соединения формулы (I), в которых X является CO и R2 является 2-CN или 4-CN, можно получить обработкой соответствующего соединения, у которого R2 является 2F, 2-Cl, 4-F или 4-Cl, цианидом щелочного металла в подходящем растворителе, например N,N-диметилформамиде или диметилсульфоксиде, при 50 - 150o.
Соединения формулы (I), у которых X является CH2 и R7 имеет указанные выше значения, можно также получить восстановлением соответствующего соединения, у которого X представляет собой CO или CH(OH), с применением триэтилсилана в трифторуксусной кислоте. Кроме того, соединения формулы (I), у которых X является CHOH и R7 имеет указанные выше значения, можно также получить восстановлением соответствующего соединения, у которого X является CO, при помощи боргидрида натрия в подходящем растворителе, например метаноле или этаноле.
Соединения формулы (I), у которых X является CO, можно также получить окислением соответствующего соединения, у которого X является CHOH. Окисление можно проводить при помощи, например, хлористого оксалила и диметилсульфоксида в дихлорметане (окисление Сверна).
Соединения формулы (II) также можно получить сульфонилированием или ацилированием соответствующего амина формулы (III)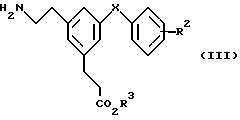
в которой R2, R3 и X имеют значения, указанные выше для формулы (II). Сульфонилирование можно проводить реакцией амина формулы (III) с ангидридом сульфокислоты формулы (R1SO2)2O или сульфонилгалогенидом (предпочтительно хлоридом) формулы R1SO2 галоген, где галоген и R1 имеют указанные выше значения. Для ацилирования применяют соответствующий ангидрид кислоты формулы (R1CO)2 или ацилгалогенид (предпочтительно хлорид) формулы R1CO галоген, где галоген и R1 имеют указанные выше значения. Эти реакции обычно проводят в присутствии избытка третичного амина, например триэтиламина, 4-диметиламинопиридина (DMAP) или пиридина, который действует в качестве акцептора кислоты, возможно в присутствии DMAP в качестве катализатора, если его не применяют в качестве акцептора кислоты, в подходящем растворителе, например дихлорметане, при температуре от около -75 до около 40oC. Альтернативно пиридин можно применять как в качестве акцептора кислоты, так и в качестве растворителя.
Ацилирование можно также проводить с применением ацилимидазолида в растворителе, например тетрагидрофуране или диоксане. Альтернативно можно применять стандартную методику пептидного взаимодействия. В типичной методике смесь амина формулы (III) и карбоновой кислоты формулы R1CO2H обрабатывают гидрохлоридом 1-(3-диметиламинопропил)-3-этилкарбодиимида, 1-гидроксибензотриазолом и основанием, например N,N-диэтилизопропиламином, в подходящем растворителе, например дихлорметане.
Соединения общей формулы (II), у которых R1, R2, R3 и X имеют указанные выше значения, можно также получить этерификацией соединения общей формулы (I) спиртом формулы R3OH в присутствии кислотного катализатора, например хлористого водорода или серной кислоты.
Соединения общей формулы (II) можно также получить с применением реакций взаимопревращения функциональных групп, как описано выше для получения соединений формулы (I). Например соединения, у которых X является CO, можно восстановить в соединения, у которых X является CHOH или CH2, и соединения, у которых X является CHOH, можно окислить в соединения, у которых X является CO. В случае, когда X является CO, можно также проводить нуклеофильное замещение группы 2-F, 2-Cl, 4-F или 4-Cl для получения, например, соответствующего соединения, у которого R2 является 2-CN или 4-CN, как описано ранее.
Соединения формулы (II), у которых X является CHCH3, можно получить восстановлением соответствующего соединения формулы (II), у которого X является C = CH2. Такое восстановление может быть осуществлено каталитическим гидрированием с применением в качестве катализатора палладия на угле в подходящем растворителе, например этаноле. Соединения формулы (II), у которых X представляет собой группу C = CH2, можно, в свою очередь, синтезировать дегидратацией соответствующего соединения формулы (II), у которого X представляет собой C(OH)CH3, обработкой этого третичного спирта кислотой, например трифторуксусной кислотой, при температуре около 50oC.
Указанные выше соединения формул (II) и (III) также являются частью настоящего изобретения. Первые могут быть активными in vivo благодаря медиированному эстеразой гидролизу с выделением соответствующей кислоты формулы (I), тогда как последние являются ключевыми промежуточными продуктами.
В альтернативном способе получения соединения формулы (I), у которых R1, R2 и X имеют указанные выше значения и можно также получить из соединений формулы (IV) способом "в единой реакционной смеси", в которой амид расщепляют по Гофману в присутствии гипохлорита натрия и водного основания (например, едкого натра) в растворителе, например диоксане или тетрагидрофуране. Полученный амин затем обрабатывают (без выделения) подходящим сульфонирующим или ацилирующим агентом, как указано выше. Избыток основания обеспечивает как акцептирование кислоты в процессе сульфонилирования или ацилирования, так и вызывает in situ гидролиз эфира в соответствующую кислоту формулы (I)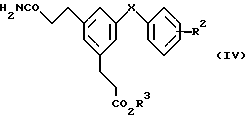
Соединения формулы (III), у которых R2, R3 и X имеют значения, указанные выше для формулы (III), можно получить из соответствующих карбаматов формулы (V)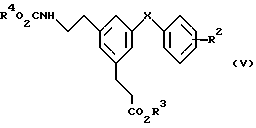
в которой R4 является группой, которую можно селективно удалить в присутствии R3, например бензилом или трет-бутилом, R1, R2, R3 и X имеют значения, указанные выше для формулы (III). Когда R4 является бензилом, удаление защитного радикала амина предпочтительно проводят каталитическим переносным гидрированием субстрата при помощи формиата аммония и палладия на угле в качестве катализатора в подходящем растворителе, например смеси метанолтетрагидрофуране, при температуре кипения реакционной среды. Альтернативно, когда R4 является трет-бутилом, можно применять хлористый водород или трифторуксусную кислоту в подходящем растворителе, например дихлорметане, при температуре от около 0 до около 20oC для требуемого удаления защитной группы.
Соединения формулы (V), у которых R2, R3, R4 и X имеют значения, указанные выше для формулы (V), можно также синтезировать непосредственно способом "в одной реакционной смеси" из карбоновых кислот формулы (VIa)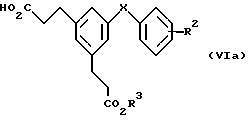
в которой R2, R3 и X имеют значения, указанные выше для формулы (V). Реакцию проводят нагреванием при кипячении раствора соединения формулы (VIa), "реагента-переносчика азида", например дифенилфосфорилазида, третичного амина, например триэтиламина, и избытка требуемого спирта формулы R4OH, например бензилового спирта или трет-бутанола, в инертном растворителе, например 1,4-диоксане; альтернативно, в качестве подходящего растворителя можно применять избыток спирта. В первой фазе реакции образуется производное ацилазида (VIa), которое в условиях реакции подвергается перегруппировке Курциуса, генерируя промежуточный вариант. Последний затем улавливается in situ присутствующим бензиловым спиртом или трет-бутанолом, в результате чего образуется бензил- или трет-бутилкарбамат соответственно формулы (V).
Соединения формулы (IV), у которых R2, R3 имеют значения, указанные выше для формулы (II), можно синтезировать из карбоновых кислот формулы (IVa) превращением их в активированную форму, например хлорангидрид, обработкой, например, хлористым оксалилом, хлористым тионилом или пятихлористым фосфором, с последующей реакцией с аммиаком в подходящем растворителе, например диэтиловом эфире или ацетоне.
Альтернативно соединения формулы (IV) можно синтезировать гидрированием соединения формулы (VII)
Гидрирование проводят в присутствии катализатора, например палладия на угле, в подходящем растворителе, например этаноле, этилацетате или уксусной кислоте, при давлении 1 - 10 атмосфер и температуре 20 - 100oC. Альтернативно можно применять каталитическое переносное гидрирование и проводить его в условиях, описанных для превращения (V) в (III).
В варианте указанного выше способа гидрирование амида формулы (VII), у которого R2 и R3 имеют указанные выше значения и X является группой CH(OCOR5), где R5 представляет собой C1-C4-алкил или фенил, приводит к одновременному гидрированию двойных связей и гидрогенолизу ацилокси-заместителя и образованию, следовательно, соответствующего соединения формулы (VI), у которого X является CH2. Аналогично, гидрирование амида формулы (VII), у которого X является группой C = CH2, приводит к одновременному восстановлению всех связей и образованию продукта формулы (IV), у которого X представляет собой CH(CH3).
В случаях, когда, например, R3 является метилом или этилом, многокислотные промежуточные продукты формулы (VIa) можно получить из диэфиров формулы (VIIIa)
в которой R6 является группой, например трет-бутилом, которую можно селективно удалить в присутствии R3 (R3 является метилом или этилом) и R2, R3 и X имеют значения, указанные выше для формулы (VIa).
До такого селективного удаления защитной эфирной группы одновременно восстанавливают две алкенильные группы, предпочтительно каталитическим переносным гидрированием, которое можно проводить в условиях, описанных выше для превращения (V) в (III), когда R4 является бензилом, но предпочтительно при температуре около 60oC. Альтернативно можно применять обычное гидрирование в подходящем растворителе, например этаноле, этилацетате или уксусной кислоте, в присутствии катализатора, например палладия на угле. После этой стадии проводят удаление трет-бутила (R6) при помощи, например, хлористого водорода или трифторуксусной кислоты при температуре от около 0 до около 20oC в растворителе, например дихлорметане. Очевидно, что в случаях, когда R6 является бензилом, восстановление двух алкенильных групп и удаление R6 можно проводить в одну стадию, например в условиях каталитического переносного гидрирования.
В случаях, когда, например, R6 является трет-бутилом, (VIa) можно получить из (VIIIa) при гарантии, что R6 можно селективно удалить в присутствии R3, например, если R6 является метилом или этилом. Таким образом, после восстановления двух алкенильных групп проводят основной гидролиз в мягких условиях с применением, например, около одного эквивалента неорганического основания, например, едкого натра или едкого кали в водном 1,4-диоксане, применяемом в качестве растворителя при температуре от около 20 до около 100oC.
В альтернативном способе соединения формулы (IV), у которых R2, R3 и X имеют указанные выше значения, можно синтезировать также из монокислот формулы (VIb)
в которой, R2, R3 и X имеют значения выше значения, способами, аналогичными способам, описанным выше для превращения (VIa) в (IV).
Соединения формулы (V), у которых R2, R3, R4 и X имеют значения, указанные выше для формулы (V), можно синтезировать из монокислот формулы ( VIb). Монокислоты формулы (VIb) получают также двухстадийным способом из симметричных ненасыщенных диэфиров формулы (VIIIb).
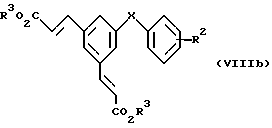
в которой R2, R3 и X имеют значения, указанные выше для формулы (VIb), каталитическим переносным гидрированием или обычным гидрированием, как ранее описано, для образования соответствующего диэфира (IX)
с последующим селективным удалением эфирной защитной группы, предпочтительно основным гидролизом с применением, например, около одного эквивалентного неорганического основания, например едкого натра или едкого кали, в водном растворе вместе с соответствующим сорастворителем при температуре от около 20oC до температуры кипения реакционной среды.
В варианте указанного выше способа в результате гидрирования диэфира формулы VIIIb, у которого R2 и R3 имеют значения, указанные выше, и X является группой CH(OCOR5), в которой R5 является C1-C4-алкилом или фенилом, происходит одновременно восстановление двойных связей и гидрогенолиз ацилоксизаместителя.
Соединения формулы (III), у которых R2, R3 и X имеют значения, указанные выше для формулы (III), можно получить непосредственно восстановлением соединений формулы (VIIIc)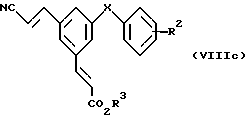
в которой
R2, R3 и X имеют значения, указанные выше для формулы (III). Одностадийное восстановление нитрильной группы и обеих алкенильных групп соединений формулы (VIIIc) можно достичь медиированным кобальтом (II) (Cobalt (II)-mediated) способом, в котором смесь хлорида кобальта (II), борогидрида натрия и соединения формулы (VIIIc) в подходящем растворителе, например этаноле, выдерживают для реакции при температуре около 0oC.
Соединения формулы (VIIIa) можно получить различными синтетическими способами, зависящими от природы X. Например, если X представляет собой CH2, CH(OH), C(OH)CH3, CO или O и R2, R3 и R6 имеют значения, указанные выше для формулы (VIIIa), их можно получить из алкеновых эфиров формулы (X)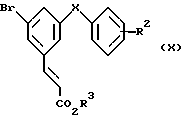 в которой X является CH2, CH(OH), C(OH)CH3, CO или C и R2 и R3 имеют значения, указанные выше для формулы (VIIIa), с использованием стандартной методики реакции Хека. Эта методика включает обработку соединения (X) избытком алкенового эфира формулы (XI)
в которой X является CH2, CH(OH), C(OH)CH3, CO или C и R2 и R3 имеют значения, указанные выше для формулы (VIIIa), с использованием стандартной методики реакции Хека. Эта методика включает обработку соединения (X) избытком алкенового эфира формулы (XI)
в которой R6 имеет значения, указанные выше для формулы (VIIIa), в присутствии ацетата палладия (II), три-о-толилфосфина и триэтиламина в подходящем растворителе, например ацетонитриле или диметилформамиде при температуре от около 80 до около 160oC.
Соединения формулы (VII) можно получить из соединения формулы (X), у которого X представляет собой CH2, CH(OH), C(OH)CH3, CO или O и R2 и R3 имеют значения, указанные выше для формулы (VII), обработкой ненасыщенным амидом формулы (XII)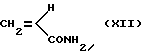
в присутствии ацетата палладия (II), три-о-толилфосфина и триэтиламина в подходящем растворителе, например аценонитриле или диметилформамиде, при температуре от около 80 до около 160oC. В условиях продолжительного нагревания (до 18 ч) продукт формулы (VII), у которого X представляет собой C(OH)CH3, может дегидратироваться с образованием соответствующего продукта, у которого X является C = CH2.
Алкеновый эфир формулы (X) можно синтезировать реакцией при температуре от около 20 до около 100oC соответствующего альдегида или кетона формулы (XIII)
в которой R2 имеет значения, указанные выше для формулы (X), с фосфонатом формулы (XIV)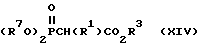
в которой R7 является C1-C4-алкилом, предпочтительно метилом или этилом, R3 имеет значения, указанные выше для формулы (X). Промежуточный фосфористый илид образует in situ из (XIV), применяя основание, например гидрид натрия, в подходящем сухом растворителе, например тетрагидрофуране, 1,2-диметоксиэтане или диметилформамиде.
Соединения формулы (XIII), у которых X представляет собой CH2, CH(OH), C(OH)CH3 или O, можно получить
из соответствующих дибромареновых предшественников формулы (XV)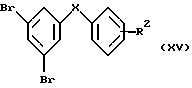
в которой R2 имеет значения, указанные выше для формулы (XIII), следующим образом:
(i) обменом одного атома брома на литий при помощи н-бутиллития в сухой смеси простой эфир-гексан, применяемый в качестве растворителя, при температуре около -70oC;
(ii) реакцией полученного ариллития с соответствующим третичным амидом, например N,N-диметиламидом формулы RCON(CH3)2, при температуре от около -70 до около 0oC.
Соединения формулы (XV) можно получить из 1,3,5-трибромбензола одной из нескольких различных методик. Например, если X является CH2, их получают следующим образом:
(i) обменом одного атома брома на литий при помощи н-бутиллития в сухой смеси простой эфир-гексан при температуре около -70oC;
(ii) реакцией образованного 3,5-дибромфениллития с подходящим ароматическим нитрилом при температуре от -78 до 0oC; и
(iii) быстрым прекращением реакции и гидролизом промежуточной соли литий-имин соляной кислотой при температуре от около 0 до около 100oC. Эти три стадии дают возможность получить кетопредшественники соединений (XV), т.е. соединения формулы (XV), у которых X является CO и которые восстанавливают в обычных условиях реакции Вольфа-Кижнера (модификация Ханга-Минлона) обработкой гидразингидратом и затем едким кали при кипячении в этиленгликоле.
Альтернативно кетопредшественник можно получить обработкой 3,5-дибромбензонитрила ариллитием в тех же условиях с последующим гидролизом соли литий-имин.
Когда X представляет собой CH(OH) или C(OH)CH3, соединения формулы (XV) можно синтезировать реакцией 3,5-дибромфениллития (получен как указано выше) с альдегидом или кетоном при температуре от около -78 до около 0oC.
Альтернативно промежуточные продукты формулы (XV), у которых X представляет собой CH(OH) или C(OH)CH3, можно синтезировать обработкой 3,5-дибромбензальдегида или 3,5-дибромацетофенона ариллитием в тех же условиях. Вместо ариллития можно применять арилмагнийгалогенид, в этом случае реакцию можно проводить в диэтиловом эфире, тетрагидрофуране или их смеси при температуре от 25oC до температуры кипения растворителя.
Альтернативно соединения формулы (XIII), у которых X является CH(OH) или C(OH)CH3, можно получить способом "в одной реакционной массе" из 3,5-дибромфениллития (получен как указано выше) реакцией с альдегидом или кетоном при температуре около -70oC. Через соответствующий отрезок времени (от 15 мин до 2 ч) добавляют другой эквивалент н-бутиллития и затем N,N-диметиламид, получая целевой альдегидный или кетонный промежуточный продукт. В варианте этого способа порядок добавления изменяют таким образом, что N,N-диметиламид добавляют после первой стадии введения лития и альдегид и кетон добавляют после второй стадии введения лития. Соединения формулы (XIII), у которых X является CO, можно также получить способом "в одной реакционной массе" из 3,5-дибромфениллития реакцией с N,N-диметиламидом при температуре от -78 до -50oC. Через соответствующий отрезок времени (1-4 ч) добавляют другой эквивалент н-бутиллития, после чего ароматический нитрил, смесь перемешивают при температуре от -78 до 0oC в течение до 4 ч и затем реакцию резко прекращают и промежуточный продукт гидролизуют для получения декетона формулы (XIII), у которого X является CO.
Когда X представляет собой 0, соединения формулы (XV) можно получить реакцией 1,3,5-трибромбензола с анионом фенола, генерированного при помощи основания, например гидрида натрия, в присутствии окиси меди (I) в подходящем растворителе, например коллидине, при температуре около 200oC. Альтернативно соединения, у которых X является 0, можно получить из аниона 3,5-дибромфенола и производного галогенбензола, у которого галоген предпочтительно является бромом.
Альтернативно соединение (XIII) можно превратить в соединение (VIIIa) реакцией (реакция Хека) с соединением (XI) с последующим превращением полученного ациларилалкеноата реакцией Виттига-Хорнера с соединением (XIV).
Соединения формулы (VIIIb) можно также получить различными синтетическими способами, зависящими от природы X. Например, когда X представляет собой CH2, CH(OH)CH3 или O и R2 имеет значения, ранее указанные для формулы (VIIIb), их можно получить из (XV) путем "двойной реакции Хека" с применением требуемого избытка алкеноата (XVI) в условиях, ранее описанных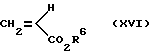
Соединения формулы (VIIIb), у которых R2 и R3 имеют указанные выше значения, и X является группой CH(OCOR5), где R5 является C1-C4-алкилом или фенилом, можно получить обработкой соответствующего соединения, у которого X является CH(OH), ацилирующим агентом, например ангидридом кислоты формулы (R5CO)2O или ацилгалогенидом (предпочтительно хлоридом) формулы R5COHal, где галоген и R5 имеет ранее указанные значения. Эти реакции обычно проводят в присутствии избытка третичного амина, например триэтиламина, 4-диметиламинопиридина (DMAP) или пиридина, который действует в качестве акцептора кислоты, в подходящем растворителе, например дихлорметане, при температуре от около -75 до 40oC. Альтернативно пиридин можно применять как в качестве акцептора кислоты, так и в качестве растворителя.
Соединения формулы (VII), у которых R2 и R3 имеют значения, указанные выше, и X является CH(OCOR5), где R5 является C1-C4-алкилом или фенилом, можно получить ацилированием соответствующего соединения, у которого X является группой CH(OH), методиками, аналогичными методиками, описанным выше для получения соединений формулы (VIIIb).
Соединение формулы (VIIIc) можно получить способами, полностью аналогичными способам, описанным для генерирования (VIIIa) и (VIIIb) применения соответствующий α , β -ненасыщенный нитрил для реакции Хека или соответствующий цианоалкилфосфат для реакции Виттига-Хорнера.
Несколько возможных превышений функциональных групп, включая заместитель R2 и соединяющую группу X, описанные выше для соединений формулы (I), можно также проводить на промежуточных стадиях, при условии совместимости реакционных условий с другими функциональными группами, присутствующими в промежуточном продукте. Например, окислением формулы (IX), у которого X является CH(OH) и R2 и R3 имеют указанные ранее значения, в условиях реакции Сверна получают соответствующее соединение, у которого X является CO.
Алкеновые эфиры формул (XI) и (XVI), фосфонат формулы (XIV), α,β- -ненасыщенные нитрилы или цианоалкилфосфаты, необходимые для получения соединений формулы (XIII), и сульфонилгалогениды, ацилгалогениды и ангидриды кислот, необходимые в ранее описанных способах получения, когда они коммерчески недоступны или потом не описаны, можно получить обычным синтетическими методиками в соответствии с предшествующей литературой или легко доступных исходных материалов, применяя соответствующие реагенты и условия реакции.
Специалисты, работающие в данной области, должны знать, что алкены, описанные ранее, можно получить в виде различных геометрических изомеров или смесей геометрических изомеров, они представлены в одной такой форме только в интересах ясности и удобства.
Биолабильные эфиры, отличающиеся от эфиров, представленных ранее формулой (II), можно получить из кислот формулы (I) стандартными реакциями. Например, ариловые и алкиловые эфиры можно синтезировать путем активирования карбоновой кислоты формулы (I) различными методиками, например образованием ацилхлорида, с последующей реакцией его с требуемым фенолом или спиртом. Альтернативно алкиловые эфиры можно получить алкилированием подходящего карбоксилата щелочного или щелочноземельного металла, образованного из карбоновой кислоты формулы (I).
Фармацевтически пригодные соли соединений формулы (I) можно также получить обычным способом. Например, раствор свободной кислоты обрабатывают соответствующим основанием (либо при нагревании, либо в соответствующем растворителе) и образованную соль выделяют фильтрованием или выпариванием растворителя при пониженном давлении.
Все указанные выше реакции вполне удобны в проведении, и необходимые реагенты и условия для их проведения можно легко установить по ссылкам на книги стандартных методик и по приведенным ниже примерам. Изменения и варианты должны быть также понятны специалистам, что дает возможность получить все соединения, определенные формулой (I).
Как указано выше, соединения изобретения являются антагонистами действия тромбоксана A2 и простагландина H2 у рецептора тромбоксана A2.
Тромбоксан A2 (TXA2) является природным простаноидом, который, как известно, является сильным сосудосуживающим фактором и агентом, агрегирующим тромбоциты. Считается, что TXA2 связан с рядом болезненных состояний, включающих атеросклероз, ишемическую болезнь сердца, периферическое сосудистое заболевание и инфаркт миокарда. TXA2 действует у рецептора тромбоксана A2, у которого другие простаноиды, особенно простагландин H2, могут также быть антагонистами.
Ингибиторы TXA2-синтетазы предотвращают образование TXA2 из предшественника PGH2, который может быть отвлечен на продуцирование дополнительно сосудорасширяющего и антиагрегирующего PGI2. Однако возможный недостаток этого типа агента состоит в том, что субстрат аккумулированного PGH2 может активировать рецептор TXA2, таким образом частично исключая или нейтрализуя пользу от подавления образования TXA2. Кроме того, если ингибирование TXA2-синтетазы неполное, то может быть доступно достаточное для некоторого индуцирования активирования тромбоцитов количество ТХА2. Оба эти недостатка можно преодолеть, если присутствует антагонист рецептора ТХА2, блокирующий действие ТХА2 или субстрата аккумулированного PGI2. Было показано, что комбинация антагониста ТХА2 и ингибитора ТХА2-синтетазы проявляют синергизм действия на агрегирование тромбоцитов in vitro (Watts et al., Brit. J. Pharmacol. , 102, 497, 1991). Кроме того, введение сулотроба (антагониста ТХА2) и диазоксибена (ингибитора ТХА2-синтетазы) добровольцам показало более сильное ингибирования тромбоцитов, чем при введении только одного такого агента (Gresele et al., J. Clin. Invest., 80, 1435, 1987).
Таким образом, соединения изобретения особенно ценны, когда их применяют в комбинации с селективным ингибитором фермента тромбоксан-синтетаза, и полученная комбинация найдет применение при лечении уже указанных болезненных состояний, а также болезненных состояний, в которые PGD2 и PGF2α могут быть вовлечены в качестве медиаторов, например диабета, бронхиальной астмы и других воспалительных состояний.
Поэтому в настоящее изобретение включен также фармацевтический препарат, содержащий в качестве активного компонента новый антагонист рецептора ТХА2 указанной выше формулы (I) и ингибитор ТХА2-синтетазы вместе с фармацевтически приемлемым разбавителем или носителем.
Подходящие ингибиторы ТХА2-синтетазы для включения в качестве активных компонентов в препараты в соответствии с настоящим изобретением включают, например, известные соединения:
1) 4-[2-(1H-имидазол-1-ил)этокси] бензойную кислоту (диазоксибен, R.P. Dickinson, et al., J. Med.Chem., 1985, 28, 1427-1432);
2) 3-(1H-имидазол-1-илметил)-2-метил-1H-индол-1-пропановую кислоту (dazmegel, R.P. Dickinson, et al., J. Med. Chem., 1986, 29, 342-346);
3) 2-метил-3-(3-пиридилметил)-1H-индол-2-пропановую кислоту (European Patent, 0054417);
4) 3-метил-2-(3-пиридилметил)бензо [b] тиофен-5-карбоновую кислоту (UK-49883, P. E. Cross, R.P. Dickinson, Spec. Pebl. Royal Soc.Chem., N 50, 1984, p.268-286.
5) 1,3-диметил-2-(1H-имидазол-1-илметил)-1H-индол-5-карбоновую кислоту (R.P. Dickinson et al., J. Med. Chеm., 1986, 29, 1643 - 1650);
6) карбокси-, низший алкоксикарбонил- или карбамоилзамещенный бензотиофен, бензофуран или индол, как заявленные в Европейском патенте 0073663;
7) 2-метил-3-(3-пиридил)-1H-индол-1-пентановую кислоту или другой ингибитор тромбоксан-синтетазы, который действует синергическим образом и химически совместим с новыми соединениями формулы (I).
Биологическую активность соединений изобретения можно показать при помощи следующих методик анализа in vitro и in vivo.
1. Антагонизм рецептору тромбоксана А2
Спирально нарезанные полоски аорты крыс, установленные для изометрического измерения напряжения в 20 мл органические ванны, выдерживают в бикарбонатном растворе Кребса при 37oC. После периода инкубирования 2 ч при остаточном напряжении 1 г ткани сначала обрабатывают U-46619 (агонист рецептора тромбоксана А2) в течение 10 мин, затем промывают и оставляют ткань для достижения равновесия на 1 ч. В жидкость ванны последовательно вводят кумулятивные дозы U-46619 в пределах 1 - 100 нМ и отмечают повышение значений тканевого тонуса.
Испытываемые соединения инкубируют в ткань в течение 15 мин до повторения кумулятивного дозирования U-46619 и определяют способность соединения противодействовать рецептору тромбоксана А2 из дозозависимых кривых для U-46619 в присутствии изменяемых концентраций испытываемого соединения.
2. Анастезированные кролики
Антагонизм рецептору тромбоксана А2 оценивают (ex vivo) в анастезированных кроликах следующим образом.
Белых кроликов Новой Зеландии (2 - 2,5 кг) анастезируют фентанилцитратом (0,189 мг) и флуанизоном (6 мг), вводимыми внутримышечного, и мидазоламом (3 мг), вводимым внутривенно, и анастезию поддерживают внутривенной инфузией фентанилцитрата (0,315 мг), флуанизона (1 мг) и мидазолама (1 мг) в течение часа. После канюлилирования трахеи сонную артерию канюлилируют для отбора проб крови. Катетер поддерживают проходимым путем присутствия в нем солевого раствора, содержащего гепарин (50 мкг/мл). Контрольные пробы крови сонной артерии берут за 25 и 5 мин до введения испытываемого соединения через краевую ушную вену. Применяли две группы кроликов. Первая группа получила сразу 0,01 мг/кг испытываемого соединения и затем с интервалами в 10 мин дозы 0,03, 0,1, 0,3, 1,0, 3,0 и 10 мг/кг. Вторая группа кроликов контрольная. Пробы крови сонной артерии берут через 5 мин после введения всех доз. В каждой точке времени 900 мкг пробы крови сразу смешивают с 100 мкл тринатриевой соли лимонной кислоты (3,15%). После инкубирования в течение 90 мин при комнатной температуре эту пробу смешивают в равным количеством буфера для агрегометрии (J. Pharmacol. Methods, 1981, 6, 315) и доводят до 37oC. В кровь помещают электроды для измерения электрического сопротивления и затем добавляют в нее U-46619 (конечная концентрация 3 мкМ). Противодействие испытуемого соединения тромбоцитной активности рецепторов тромбоксана А2 оценивают сравнением изменения электрического сопротивления, продуцированного U-46619, у кроликов, которым ввели соединение изобретения, с контрольной группой кроликов, которым не вводили это соединение.
3. Находящиеся в создании собаки
Антагонизм рецепторам тромбоксана А2 можно также оценивать ex vivo в ограниченных движениях, находящимся в сознании собаках после перорального (p. o. ) или внутривенного (i.v.) введения соединения изобретения. Способы отбора проб и анализа аналогичны тем, которые описаны для экспериментов ex vivo на анастезированных кроликах.
Для введения человеку с целью лечения или предупреждения заболеваний или неблагоприятных медицинских состояний, в которые ТХА2 вовлечен в качестве этиологического фактора, пероральные дозы соединений, как можно ожидать, находятся в пределах 2 - 200 мг ежедневного для среднего взрослого пациента (70 кг). Таким образом, отдельные таблетки и капсулы для типичного взрослого пациента содержат 1 - 200 мг активного соединения в подходящем фармацевтически пригодном наполнителе или носителе и пригодны для введения в виде разовой дозы или нескольких доз один или несколько раз в день. Дозы для внутривенного введения должны быть обычно в пределах 1 - 200 мг на требуемую одинарную дозу. На практике врач определит действительную дозу, которая будет наиболее пригодна для конкретного пациента, и она будет изменяться с возрастом, весом и восприимчивостью отдельного пациента и состоянием пациента. Указанные выше дозы являются усредненными, но могут быть, конечно, отдельные примеры, когда необходимы более высокие или более низкие пределы доз, и они являются объектом настоящего изобретения.
Соединения формулы (I) можно вводить человеку отдельно, но обычно их будут вводить в смеси с фармацевтическим носителем, выбранным с учетом предполагаемого способа введения и обычной фармацевтической практики. Например, их можно вводить перорально в форме таблеток, содержащих наполнители, например крахмал или лактозу, или капсул или шариков, которые можно применять отдельно или в смеси с наполнителями, или в форме элексиров или суспензий, содержащих придающее вкус или окрашивающее средство. Их можно ввести парентерально инъекцией, например внутривенно, внутримышечно или подкожно. Для парентерального введения их лучше всего применять в виде стерильного водного раствора, который может содержать другие вещества, например достаточное количество солей или глюкозы для того, чтобы раствор был изотоническим с кровью.
Таким образом, изобретение предлагает фармацевтический препарат, содержащий соединение формулы (I) или его фармацевтически пригодную соль или биолабильный эфир вместе с фармацевтически пригодным разбавителем или носителем.
Изобретение предлагает также соединение формулы (I) или его фармацевтически пригодную соль или биолабильный эфир или фармацевтический препарат, содержащий любое из этих соединений, для применения в медицине.
Изобретение включает также применение соединения формулы (I) или его фармацевтически пригодной соли или биолабильного эфира для получения лекарственного препарата, предназначенного для лечения болезненных состояний, в которых тромбоксан А2 является этиологическим фактором.
Еще одним объектом изобретения способ лучения или предупреждения болезненных состояний, в которых тромбоксан А2 является этиологическим фактором, у млекопитающего (включая человека), который предусматривает введение этому млекопитающему терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли или биолабильного эфира.
Изобретение включает также любые новые промежуточные продукты, описанные в нем, например промежуточные продукты формул (II), (III), и (IV).
Синтезы соединений изобретения и промежуточных продуктов для их получения иллюстрируют следующие Примеры и Получения. Чистоту соединений обычно контролировали тонкослойной хроматографией (ТСХ) с применением пластин Merck Kieselgel 60 F254 и следующих систем растворителей (SS):
SS1. Дихлорметан/гексан (1:1)
SS2. Гексан
SS3. Дихлорметан
SS4. Дихлорметан/метанол (95:5)
SS5. Дихлорметан/метанол/0,880 аммиак (90:10:1)
SS6. Этилацетат/гексан (1:5)
SS7. Этилацетат/гексан (1:1)
SS8. Дихлорметан/метанол (9:1)
SS9. Дихлорметан/метанол/уксусная кислота (100:5:0,5)
SS10. Этилацетат/гексан/уксусная кислота (10:10:1)
SS11. Дихлорметан/метанол/уксусная кислота (90:10:1)
SS12. Этилацетат/гексан (5:1)
SS13. Дихлорметан/этанол (20:1)
SS14. Этилацетат/гексан/уксусная кислоты (50:50:1)
SS15. Этилацетат/гексан/уксусная кислота (70:30:1)
Спектры ядерного магнитного резонанса на ядрах 1H регистрировали с применением спектрометра Nicolet QE-300 или Bruker AC-300, во всех случаях спектры соответствовали предполагаемым структурам. Химические сдвиги даны в частях на миллион, считая от тетраметилсилана. При этом применяли обычную аббревиатуру для обозначения основных пиков: с, синглет, д, дублет, т, триплет, м, мультиплет и ш. широкий.
Получение 1
3,5-Дибром- α- -(4-фторфенил)бензолметанол
2,5 М раствор н-бутиллития в гексане (44,0 мл) добавляли по каплям в перемешиваемую суспензию 1,3,5-трибромбензола (31,5 г) в сухом простом эфире (1000 мл) при -78oC в атмосфере сухого азота. Полученную смесь перемешивали при этой температуре 30 мин и затем добавляли по каплям 4-фторбензальдегид (13,65 г). Перемешивание при -78oC продолжали еще 30 мин и затем реакцию обрывали добавлением воды. Реакционную смесь оставляли для нагревания до комнатной температуры и отделяли органический слой и сушили над (MgSO4). Выпариванием растворителя получили масло, которое хроматографировали на силикагеле. Колонку элюировали смесью дихлорметан/гексан (1:5), постепенно повышая соотношение растворителей до 2:3. Фракции продукта объединяли, выпаривали растворитель и остаток растирали с гексаном, получая целевое соединение (31,42 г) с т.пл. 92 - 93oC.
Найдено: C 43,75; H 2,43. Для формулы C13H9Br2FO требуется:C 43,37; H 2,52%.
Следующие соединения были получены аналогично из соответствующего альдегида или кетона:
3,5-Дибром- α -(2-фторфенил)бензолметанол (получен в виде масла) Rf0,2(SS 1). δ (CDCl3): 2,40 (1H, ш.с.), 6,08 (1H, с), 7,03 - 7,09 (1H, м), 7,16 - 7,22 (1H, м), 7,27 - 7,33 (1H, м), 7,42 - 7,48 (1H, м), 7,50 (2H, д), 7,58 (1H, д).
3,5-Дибром- α -(4-фторфенил)- α -метилбензолметанол, т.пл. 72 - 73oC. Найдено: C 44,95, H 2,90. Для формулы C14H11Br2FO требуется C 44,95, H 2,96%.
Получение 2
3,5-Дибром- α -(2-метоксифенил)бензолметанол
1-Бром-2-метоксибензол (7,48 г) растворяли в сухом диэтиловом эфире (50 мл) и 5 мл аликвоту раствора отобрали и добавили в смесь магниевой стружки (1,0 г) и кристалла йода. Смесь нагревали до кипячения для инициирования реакции и источник нагревания удаляли. Затем добавляли остальное количество раствора бромметоксибензола со скоростью, достаточной для поддерживания кипения, и смесь нагревали при кипячении в течение еще 1 ч. Смесь затем несколько охлаждали и добавляли по каплям раствор 3,5-дибромбензальдегида (10,0 г) в сухом тетрагидрофуране (40 мл). Смесь нагревали при кипячении в течение 30 мин, затем охлаждали до комнатной температуры и добавляли при быстром перемешивании раствор хлористого аммония (8,0 г) в воде (40 мл). Смесь разбавляли простым эфиром и органический слой отделяли, промывали водой и сушили над (MgSO4). Раствор выпаривали и остаток хроматографировали на силикагеле. Элюирование начинали смесью гексан/дихлорметан (5:1) и содержание дихлорметана постепенно повышали до соотношения 1:4. Последние фракции, содержащие продукт, объединяли и выпаривали, получая целевое соединение (11,85 г), т.пл. 112-116oC (из циклогексана). Найдено: C 45,52; H 3,09. Для C14H12Br2O2 требуется: C 45,19, H 3,25%.
Получение 3
1,3-Дибром-5-(4-фторфенокси)бензол
Гидрид натрия (3,24 г 60 %-ной суспензии в минеральном масле) добавляли по частям в перемешиваемую смесь 1,3,5-трибромбензола (76,4 г) 4-фторфенола (18,16 г) и оксида меди (1) (11,6) в коллидине (400 мл) при комнатной температуре. После прекращения выделения водорода смесь нагревали при кипячении и перемешивании 8 ч, затем охлаждали и фильтровали. Остаток последовательно промывали этилацетатом и концентрированным водным аммиаком и объединенный фильтрат и провывание жидкости распределяли между этилацетатом и водой. Органический слой промывали два раза соляным раствором и выпаривали. Остаток растворяли в этилацетате и раствор фильтровали, промывали несколько раз раствором лимонной кислоты и сушили над (MgSO4). Растворитель выпаривали и остаток растворяли в горячем гексане. Раствор фильтровали и фильтрат выпаривали. Остаток хроматографировали на силикагеле с применением в качестве элюента гексана, получая целевое соединение в виде масла (18,2 г) с Rf0,31 (SS 2). Найдено: C 41,71, H 1,97. Для C12H7Br2FO требуется:C 41,66, H 2,04%.
Получение 4
3-Бром-5-[(4-фторфенил)гидроксиметил]бензальдегид
2,5 М раствор н-бутиллития в гексане (22,0 мл) добавляли по каплям в перемешиваемую суспензию 1,3,5-трибромбензола (15,74 г) в сухом диметиловом эфире (500 мл) при -78oC в атмосфере сухого азота. Полученную смесь перемешивали при этой температуре 30 мин и затем добавляли по каплям 4-фторбензальдегид (6,83 г). Перемешивание при -78oC продолжали в течение еще 30 мин и затем добавляли по каплям еще один эквивалент н-бутиллития (22,0 мл 2,5 М раствора в гексане). Смесь перемешивали при -78oC еще 15 мин и затем добавляли N,N-диметилформамид (15,45 г). Перемешивание продолжали в течение 1 ч и затем добавляли воду (200 мл). Смесь оставляли для нагревания до комнатной температуры и органический слой отделяли и сушили над (MgSO4). После выпаривания растворителя получали масло, которое хроматографировали на силикагеле. Элюированием дихлорметаном получили примеси, дальнейшим элюированием смесью дихлорметан/метанол (99: 1) выделяли целевое соединение в виде смолы (10,94 г) с Rf0,2 (SS 3). Найдено: C 53,99, H 3,41. Для C14H10BrFO2 требуется C 54,39, H 3,26%.
Аналогично с применением соответствующего альдегида получали следующие соединения:
3-Бром-5-[гидрокси(фенил)метил] бензальдегид, Rf 0,2 (SS 3). δ (CDCl3): 2,43 (1H, с), 5,89 (1H, с), 7,32 - 7,38 (5H, м), 7,83 (2H, м), 7,90 (1H, м), 9,91 (1H, с).
3-Бром-5-[(3-фторфенил)гидроксиметил] бензальдегид, Rf 0,15 (SS 3). δ (CDCl3):2,62 (1H, ш.с.), 5,86 (1H, с), 6,98 - 7,15 (3H, м), 7,31 - 7,38 (1H, м), 7,80 (2H, м), 7,91 (1H, м), 9,91 (1H, с).
Получение 5
3-Бром-5-[1-(4-фторфенил)-1-гидроксиэтил] бензальдегид
2,5 М Раствор н-бутиллития в гексане (10,7 мл) добавляли по каплям в перемешиваемую суспензию 3,5-дибром- α -(4-фторфенил)- α -метилбензолметанола (5,90 г) в сухом диметиловом эфире (160 мл) при -78oC в атмосфере сухого азота. Полученную смесь перемешивали при этой температуре в течение 30 мин и затем добавляли по каплям N, N-диметилформамид (2,93 г). Перемешивание при -78oC продолжали еще 1 ч и затем реакцию прерывали давлением воды. Смесь оставляли для нагревания до комнатной температуры и органический слой отделяли, промывали водой и сушили над (MgSO4). После выпаривания растворителя получали масло, которое хроматографировали на силикагеле. Элюирование начинали смесью гексан-дихлорметан (1:1) и соотношение их постепенно изменяли до достижения 1: 5. Последние фракции продукта объединяли и выпаривали, получая целевое соединение в виде масла (2,10 г), Rf 0,15 (SS 1). Найдено: C 55,39, H 3,78. Требуется для C15H12BrO2F: C 55,75, H 3,74%.
Получение 6
Этил-3-[3-бром-5-[(4-фторфенил)гидроксиметил]фенил]-2-пропеноат
Триэтилфосфоноацетат (8,30 г) добавляли по каплям в перемешиваемую, охлаждаемую суспензию гидрида натрия (1,48 г 60 %-ной суспензии в минеральном масле) в сухом тетрагидрофуране (50 мл) и смесь перемешивали 30 мин, получая прозрачный раствор. По каплям добавляли раствор 3-бром-5-[(4-фторфенил)гидроксиметил)] бензальдегида (10,38 г) в сухом тетрагидрофуране (50 мл). Раствор перемешивали при 0oC 1 ч и затем выливали в смесь простого эфира и воды. Водный слой отделяли, экстрагировали простым эфиром, органические слои объединяли, промывали водой и сушили над (MgSO4). Растворитель выпаривали и остаток хроматографировали на силикагеле с применением дихлорметана в качестве элюента. Фракции продукта объединили и выпаривали. Остаток кристаллизовали из смеси эфир/гексан, получая целевое соединение (10,22 г) с т. пл. 82 - 84oC. Найдено: C 57,35, H 4,04. Для C18H16BrO3 требуется C 57,01, H 4,25%.
Аналогично из соответствующего альдегида получали следующие соединения:
Этил-3-[3-бром-5-[гидрокси(фенил)метил] фенил] -2-пропеноат, т.пл. 104 - 105oC. Найдено: C 60,04, H 4,62. Для C18H17BrO3 требуется: C 59,85, H 4,7%.
Этил-3-[3-бром-5-[3-фторфенил)гидроксиметил]фенил]-2-пропеноат, т.пл. 86 - 87oC. Найдено: C 57,04, H 4,26. Для C18H16BrFO3 требуется: C 57,01, H 4,25%.
Этил-3-[3-бром-5-[1-(4-фторфенил)-1-гидроксиэтил] фенил]-2-пропеноат, т. пл. 97 - 98oC. Найдено: C 57,58, H 4,56. Для C19H18BrFO3 требуется: C 58,03, H 4,61%.
Получение 7
Этил-3-[3-(2-карбамоилэтенил)-5-[(4-фторфенил)гидроксиметил] -фенил]-2-пропеноат
Смесь этил-3-[3-бром-5-[(4-фторфенил)гидроксиметил] фенил]-2-пропеноата (9,50 г), акриламида (2,67 г), ацетата палладия (II) (0,306 г), три-о-толилфосфина (0,763 г) и триэтиламина (3,80 г) в ацетонитриле (10 мл) нагревали до 100oC в течение 4 ч в атмосфере азота и затем охлаждали. Добавляли воду (150 мл) и дихлорметан (150 мл) и смесь нагревали при перемешивании для диспергирования твердого остатка. Смесь затем охлаждали и фильтровали, твердую часть растворяли в горячем изопропаноле (500 мл). Раствор фильтровали в горячем состоянии с применением вспомогательного порошка и фильтрат концентрировали до начала кристаллизации. Затем добавляли равный объем эфира и смесь оставляли для охлаждения. Твердую часть отделяли фильтрованием и сушили, получая целевое соединение (5,50 г), т.пл. 197 - 199oC. Найдено: C 68,55, H 5,17, N 4,01. Требуется для C21H20FNO4: C 68,28, H 5,46, N 3,79%.
Высушиванием над (MgSO4) и выпариванием раствора в дихлорметане с последующей кристаллизацией остатка из смеси изопропанол/эфир получали дополнительно 2,21 г продукта с т.пл. 196 - 198oC.
Аналогично получали следующие соединения:
Этил-3-[3-(2-карбамоилэтенил)-5-[гидрокси(фенил)метил] фенил] -2-пропеноат, т.пл. 179 - 180 oC. Найдено: C 71,26, H 6,36, N 3,92.
Требуется для C21H22NO4: C 71,57, H 6,29, N 3,97%.
Этил-3-[3-(2-карбамоилэтенил)-5-[(3-фторфенил)гидроксиметил] -фенил] -2-пропеноат, т. пл. 166 - 168oC. Найдено: C 67,49, H 5,43, N 3,77. Для C21H21FNO4 требуется: C 68,10, H 5,71, N 3,78%.
Этил-3-[3-(2-карбомоилэтенил)-5-[1-(4-фторфенил)этенил] -фенил]-2-пропеноат, полученный с применением этил-3-[3-бром-5-[1-(4-фторфенил)гидроксиэтил] фенил]-2-пропеноата в качестве исходного соединения, время реакции 18 ч, смола, Rf 0,3 (SS4). δ (CDCL3):1,35 (3H, т), 4,27 (2H, к), 5,47 (1H, с), 5,52 (1H, с), 5,63 (2H, ш.), 6,43 (1H, д), 7,02 - 7,08 (2H, м), 7,27 - 7,33 (2H, м), 7,46 (2H, с), 7,61 - 7,65 (3H, м), 7,69 (1H, д).
Получение 8
Диэтил-3,3'-[5-(4-фторфенил)гидроксиметил-1,3-фенилен]бис-2-пропеноат
Смесь 3,5-дибром- α - 4-фторфенил)бензолметанола (13,8 г), этилакрилата (11,5 г), ацетата палладия (II) (469 мг), три-о-толилфосфина (1,17 г), триэтиламина (16 мл) и ацетонитрила (25 мл) нагревали при кипячении в атмосфере сухого азота в течение 4 ч. Смесь охлаждали и распределяли между простым эфиром и водой. Водный слой экстрагировали несколько раз эфиром. Объединенный органический слой промывали водой и сушили над (MgSO4). После выпаривания растворителя получали масло, которое хроматографировали на силикагеле. Элюированием дихлорметаном выделяли сначала примеси, а затем чистый продукт. Фракции продукта объединяли и выпаривали, получив целевое соединение (9,32 г), т.пл. 98 - 100oC. Найдено: C 69,40, H 5,85. Для C23H23FO5 требуется: C 69,33, H 5,82%.
Аналогично были получены следующие соединения:
Диэтил-3,3'-[5-(2-фторфенил)гидроксиметил-1,3-фенилен] бис-2-пропеноат, т. пл. 102 - 103oC. Найдено: C 69,45, H 5,83. Для C23H23FO5 требуется: C 69,33, H 5,82%.
Диэтил-3,3'-[5-(2-метоксифенил)гидроксиметил-1,3-фенилен] бис-2-пропеноат, т.пл. 104 - 107oC. Найдено: C 70,36, H 6,46%.
Для C24H26O6: C 70,23, H 6,39%.
Диэтил-3,3'-[5-(4-фторфенокси)-1,3-фенилен] бис-2-пропеноат, масло, Rf 0,25 (SS3). Найдено: C 68,47, H 5,40%. Требуется для C22H21FO5: C 68,74, H 5,06%.
Получение 9
Диэтил-3,3'-[5-[ α -ацетокси-(4-фторфенил)метил]-1,3-фенилен]бис-2-пропеноат
Диэтил-3,3'-[5-(4-фторфенил)гидроксиметил-1,3-фенилен] бис-2-пропеноата (9,30 г), уксусного ангидрида (4,40 мл), пиридина (50 мл) и 4-диметиламинопиридина (50 мг) в дихлорметане (50 мл) перемешивали при комнатной температуре 2 ч. Раствор затем промывали последовательно водой, 1 N соляной кислотой и раствором бикарбоната натрия и затем сушили над (MgSO4). После выпаривания растворителя получали масло, которое растирали с простым эфиром для индуцирования кристаллизации. Смесь разбавляли гексаном и фильтровали, получая целевое соединение (9,50 г), т.пл. 123 - 125oC. Найдено: C 68,47, H 5,79. Для C25H25FO6 требуется: C 68,17, H 5,72%.
Аналогично получали следующие соединения:
Диэтил-3,3'-[5-[ α -ацетокси-(2-фторфенил)метил]-1,3-фенилен]бис-2-пропеноат, т.пл. 83 - 85oC. Найдено: C 68,15, H 5,42.
Для C25H25FO6 требуется: C 68,17, H 5,72%.
Диэтил-3,3'-[5-[ α -ацетокси-(2-метоксифенил)метил]-1,3-фенилен]бис-2-пропеноат, т.пл. 128 - 132oC. Найдено: C 69,22, H 6,38. Для C26H28O7 требуется: C 69,01, H 6,24%.
Получение 10
Этил-3-[3-[ α -ацетокси(фенилметил)]-5-[(2-карбамоил)этенил]фенил]-2-пропеноат
Смесь этил-3-[3-(2-карбамоил)этенил-5-[гидрокси(фенил)-метил]фенил]-2-пропеноата (4,46 г) уксусного ангидрида (3,12 г), 4-диметиламинопиридина (30 мг) и пиридина перемешивали при комнатной температуре 18 ч. Образованный раствор разбавляли этилацетатом и промывали два раза водой, два раза 1 N соляной кислотой и два раза раствором бикарбоната натрия. Его сушили над (MgSO4) и выпаривали. Смолистый остаток растворяли в небольшом объеме простого эфира, затем добавляли равный объем гексана и смесь выдерживали при 0oC 18 ч. Твердую часть отделили фильтрованием и сушили, получая целевое соединение (3,98 г), т.пл. 101 - 102oC. Найдено: C 70,29 H 6,00, N 3,48. Для C23H23NO5F требуется: C 70,21, H 5,89, N 3,56%.
Аналогично получили следующие соединения:
Этил-3-[3-[ α -ацетокси-(4-фторфенил)метил]-5-(2-карбамоилэтенил)фенил-2-пропеноат, т. пл. 89 - 91oC. Найдено: C 67,09, H 5,21, N 3,15. Для C23 H22FNO5 требуется: C 67,14, H 5,39, N 3,41%.
Этил-3-[3-[ α -ацетокси-(3-фторфенил)метил]-5-(2-карбамоилэтенил)фенил] -2-пропеноат, т.пл. 161 - 162oC. Найдено: C 66,55, H 5,45, N 3,18. Требуется для C23H22FNO5: C 67,14, H 5,39, N 3,41%.
Получение 11
Диэтил-5-[(4-фторфенил)метил]-1,3-бензолдипропаноат
Смесь диэтил-3,3'-[5-[ α -ацетокси-(4-фторфенил)метил] -1,3-фенилен] бис-2-пропеноата (9,20 г) и 10% палладия на угле (1,0 г) в этилацетате (100 мл) гидрировали при давлении 50 фунтов на квадратный дюйм (3,45 бар) при комнатной температуре в течение 6 ч. Затем смесь фильтровали и фильтрат промывали раствором бикарбоната натрия и сушили над (MgSO4). После выпаривания растворителя получали целевое соединение в виде смолы (8,07 г), Rf 0,5 (SS3). Найдено: C 72,22, H 6,95. Для C23H27FO4 требуется: C 71,48, H 7,04%.
Аналогично получали следующие соединения:
Диэтил-5-[(2-фторметил)метил]-1,3-бензолдипропаноат, масло Rf 0,8 (SS5), δ (CDCl3) : 1,23 (6H, т), 2,57 (4H, т), 2,89 (4H, т), 3,93 (2H, с), 4,11 (4H, к), 6,89 (3H, с), 4,01 - 4,20 (4H, м).
Диэтил-5-[(2-метоксифенил)метил] -1,3-бензолдипропаноат, масло, Rf 0,3 (SS6). Найдено: C 72,15, H 7,67. Для C24H30O5 требуется: C 72,33, H 7,59%.
Получение 12
Диэтил-5-[(4-фторфенил)гидроксиметил]-1,3-бензолдипропаноат
Смесь диэтил-3,3'-[5-(4-фторфенил)гидроксиметил-1,3-фенилен]бис-2-пропеноата (30,6 г), формиата аммония (48,0 г) и 10% палладия на угле (3,0 г) в этаноле (250 мл) и тетрагидрофуране (250 мл) нагревали при 60oC в течение 1,5 ч и затем охлаждали. Смесь фильтровали и остаток промывали этанолом. Затем объединяли фильтрат и промывные жидкости и выпаривали. Остаток распределяли между диэтиловыми эфиром и водой. Органический слой отделяли, сушили над (MgSO4) и выпарили, получая целевое соединение в виде масла (30,9 г). Найдено: C 68,60, H 6,89. Для C23H27FO5 требуется: C 68,64, H 6,76%.
Получение 13
Диэтил-5-(4-фторфеноски)-1,3-бензолдипропаноат
Обработкой диэтил-3,3'-[5-(4-фторфенокси)-1,3-фенилен] бис-2-пропеноата (17,0 г) формиатом аммония (27,85 г) и 10% палладия на угле (1,70 г) по методике Получения 12 получали целевое соединение в виде масла (12,12 г), R1 0,30 (SS1). Найдено: C 68,01, H 6,46. Для C22H25FO5 требуется C 68,03, H6,49%.
Получение 14
Диэтил-5-(4-фторбензоил)-1,3-бензолдипропаноат
Диметилсульфоксид (16,2 мл) добавляли по каплям в перемешиваемый раствор хлористого оксалила (11,55 г) в сухом дихлорметане (500 мл) при -78oC в атмосфере сухого азота. Смесь перемешивали 10 мин и затем в него по каплям добавляли раствор диэтил-5-[(4-фторфенил)гидроксиметил]-1,3-бензолдипропаноата (30,65 г) в сухом дихлорметане (150 мл). Смесь перемешивали при -78oC 30 мин добавляли триэтиламин (23,03 г) и перемешивание продолжали еще 10 мин. Смесь оставляли для нагревания до комнатной температуры, затем промывали водой. Органический слой отделяли, сушили над (MgSO4) и выпаривали. Остаток хроматографировали на силикагеле. Элюированием дихлорметаном и затем смесью дихлорметан/метанол (50:1) получали целевое соединение в виде масла (28,92 г), Rf 0,75 (SS 4). Найдено: C 69,08, H 6,46. Для C23H25Fo5 требуется: C 68,98, H 6,29%.
Получение 15
Моноэтил-5-[(4-фторфенил)метил]-1,3-бензолдипропаноат
2N раствор едкого натра (10 мл) добавляли в перемешиваемый раствор диэтил-5-[(4-фторфенил)метил]-1,3-бензолдипропаноата (7,70 г) в этаноле (50 мл) и смесь оставляли на 2 ч и затем выпаривали. Остаток распределяли между простым эфиром и водой. Эфирный слой отделяли, сушили над (MgSO4) и выпаривали, получая исходный материал (3,35 г). Водный слой подкисливали 2N соляной кислотой и смесь экстрагировали несколько раз этилацетатом. Объединенные экстракты сушили над (MgSO4) и выпаривали. Остаток хроматографировали на силикагеле. Колонку элюировали смесью этилацетата/гексан (1:1), постепенно повышая полярность элюента до чистого этилацетата. Фракции продукта объединяли и выпаривали, получая целевое соединение в виде смолы (3,12 г), Rf 0,5 (SS7). Найдено C 70,17, H 6,60. Для C21H23FO4 требуется: C 70,37, H 6,47%.
Аналогично получали следующие соединения:
Моноэтил-5-[(2-фторфенил)метил] -1,3-бензолдипропаноат, масло, Rf 0,65 (SS8), δ (CDCL3): 1,22 (3H, т), 2,60 (2H, т), 2,68 (2H, т), 2,90 (4H, м), 3,95 (2H, c), 4,14 (2H, к), 6,30 (3H, ш), 7,0 - 7,25 (4H, м), 11,0 (1H, ш.с. ).
Моноэтил-5-[(2-метоксифенил)метил] -1,3-бензолдипропаноат, масло, Rf, 0,30 (SS9). Найдено: C 70,84, H 6,85. Для C22H26O5 требуется: C 71,33, H 7,00%.
Моноэтил-5-(4-фторбензоил)-1,3-дипропаноат, масло, Rf 0,25 (SS8). Найдено: C 67,47, H 5,76. Для C21H21FO5 требуется C 67,73, H 5,68%.
Моноэтил-5-(4-фторфенокси)-1,3-дипропаноат, масло, Rf 0,4 (SS10). Найдено: C 66,69, H 6,22. Требуется C 66,66, H 5,87%.
Получение 16
Этил-3-(2-карбамоилэтил)-5-[(4-фторфенил)метил]бензолпропаноат
В перемешиваемый раствор моноэтил-5-[(4-фторфенил)метил]-1,3- бензолдипропаноата в сухом дихлорметана (200 мл) при комнатной температуре добавляли хлористый оксалил (17,8 г) и затем N,N-диметилформамид (5 капель). Раствор перемешивали при комнатной температуре в течение 3 ч и затем выпаривали. Остаток растворяли в диэтиловом эфире (150 мл) и раствор добавляли постепенно в интенсивно перемешиваемую смесь концентрированного водного аммиака (500 мл) и диэтилового эфира (200 мл). Смесь перемешивали 3 ч и водный слой отделяли и экстрагировали два раза диэтиловым эфиром (300 мл) и один раз этилацетатом (200 мл). Органические слои объединяли, сушили над (MgSO4), выпаривали и остаток кристаллизовали из смеси эфир/гексан, получая целевое соединение (42,5 г), т. пл. 69 - 70oC. Найдено: C 70,05, H 7,13, N 4,02. Для C21H24FNO3 требуется: C 70,57, H 6,77, N 3,92%.
Аналогично из соответствующих кислот получали следующие соединения:
Этил-3-(2-карбамоилэтил)-5-[(2-фторфенил)метил]бензолпропаноат, масло Rf 0,55 (SS 11), δ (CDCl3): 1,11 (3H, т), 2,48 (2H,т), 2,57 (2H, т), 2,85 - 2,93 (4H, м), 3,95 (2H, c), 4, 11 (2H, к), 5,40 (2H, ш.д.), 6,91 (3H, с), 7,00 - 7,25 (4H, м).
Этил-3-(2-карбамоилэтил)-5-[(2-метоксифенил)метил]бензолпропаноат, т.пл. 70 - 75oC. Найдено: C 71,23, H 7,40, N 3,63. Требуется для C22H27NO4: C 71,52, H 7,37, N 3,79%.
Этил-3-(2-карбамоилэтил)-5-(4-фторбензоил)бензолпропаноат, т. пл. 96 - 98oC. Найдено: C 68,06, H 5,89, N 3,68. Для C21H22FNO4 требуется: C 67,91, H 5,97, N 3,77%.
Этил-3-(2-карбамоилэтил)-5-(4-фторфенокси)бензолпропаноат, т. пл. 74 - 75oC. Найдено: C 67,06, H 6,32, N 3,95. Для C20H22FNO4 требуется: C 66,84, H 6,17, N 3,90%.
Получение 17
Этил-3-(2-карбамоилэтил)-5-[(4-фторфенил)метил]бензолпропаноат
Раствор этил-3-[3-[α - ацетокси-(4-фторфенил)метил] -5-[(2- карбамоил)этенил] фенил] -2-пропеноата (7,50 г) в этилацетате (75 мл) гидрировали в течение 5 ч при комнатной температуре и давлении 1,5 атмосфер в присутствии 100 палладия на угле (0,70 г). Смесь фильтровали с применением вспомогательного порошка и остаток промывами этилацетатом. Фильтрат и промывную жидкость объединяли, промывали раствором карбоната натрия и сушили над (MgSO4). Растворитель выпаривали и остаток растирали с гексаном, получая целевое соединение (6,05 г), т.пл. 68 - 70oC, идентичное с продуктом Получения 16.
Аналогично получили следующие соединения:
Этил-3-(2-карбамоилэтил)-5-(фенилметил)бензолпропаноат, масло, Rf 0,35 (SS12), δ (CDCl3): 1,11 (3H, т), 2,48 (2H, т), 2,57 (2H, т), 2,87 - 2,93 (4H, м), 3,92 (2H, с), 4,09 (2H, к), 5,35 (2H, ш.с.), 6,89 (3H, c), 7,15 - 7,30 (5H, м).
Этил-3-(2-карбамоилэтил)-5-(3-фторфенилметил)бензолпропаноат, т.пл. 66 - 67oC. Найдено: C 70,84, H 6,97, N 3,62. Для C21H24FNO3 требуется: C 70,57, H 6,77, N 3,92%.
Этил-3-(2-карбамоилэтил)5-[1-(4-фторфенил)этил]бензолпропаноат (с применением этил-3-[3-[(2-карбамоил)этенил] -5-[1-(4-фторфенил)этенил] фенил]-2- пропеноата в качестве исходного соединения), масло, δ (CDCL3): 1,11 (3H, т), 1,59 (3H, д), 2,48 (2H, т), 2,57 (2H, т), 2,84 - 2,92 (4H, м), 4,08 (2H, к), 4,05 (1H, к), 5,46 (1H, ш.с.), 6,70 (1H, ш.с.), 6,90 - 7,0 (5H, м), 7,12 - 7,17 (2H, м).
Получение 18
Этил-3-[2-(трет-бутоксикарбониламино)этил]-5-[(4-фторфенил) метил]бензолпропаноат
Раствор моноэтил-5-[(4-фторфенил)метил]-1,3-бензолдипропаноата (3,0 г), дифенилфосфорилазида (2,51 г) и триэтиламина (1,3 мл) в сухом трет-бутаноле (25 мл) перемешивали при комнатной температуре 15 мин и затем нагревали при кипении в течение 20 ч. Раствор выпаривали и остаток распределяли между эфиром и водой. Эфирный слой отделяли, сушили над (MgSO4) и выпаривали, получая масло, которое хроматографировали на силикагеле. Элюированием дихлорметаном выделяли целевое соединение в виде смолы (2,41 г), Rf 0,2 (SS3). ЯМР-спектр, δ (CDCl3): 1,23 (3H, т), 1,44 (9H, с), 2,58 (2H, т), 2,72 (2H, т), 2,28 (2H, т), 3,32 (2H, м), 3,89 (2H, с), 4,10 (2H, к), 4,50 (1H, ш), 6,82-6,90 (3H), 6,94-7,0 (2H, м), 7,09 - 7,15 (2H, м).
Получение 19
Этил-3-(2-аминоэтил)-5-[(4-фторфенил)метил]бензолпропаноат
Раствор этил-3-[2-(трет-бутоксикарбониламино)этил] -5-[(4- фторфенил)метил]бензолпропаноата (2,36 г) и трифторуксусной кислоты (5 мг) в дихлорметане (25 мл) выдерживали при комнатной температуре в течение ночи и затем выпаривали. Остаток распределяли между простым эфиром и разбавленным раствором едкого натра. Органический слой отделяли, промывали водой и высушивали над (MgSO4). Раствор выпаривали и остаток хроматографировали на силикагеле, элюируя смесями дихлорметана, метанола и концентрированного раствора гидроксида аммония. Фракции, содержащие продукт, объединяли и выпаривали растворитель, получая целевое соединение в виде смолы (1,13 г), Rf 0,1 (SS4). ЯМР - спектр, δ (CDCL3): 1,24 (3H, т), 1,48 (2H, с), 2,61 (2H, т), 2,73 (2H, т), 2,90-2,98 (4H, м), 3,93 (2H, с), 4,14 (2H, к), 6,88 (2H, с), 6,91 (1H, с), 6,96-7,03 (2H, м), 7,12 - 7,18 (2H, м).
Пример 1. Этил-3-[2-[(4-хлорфенил)сульфониламино]этил]-5-(4-фторфенилметил) бензолпропаноат.
4-Хлорбензолсульфонилхлорид (352 мг) добавляли по каплям в перемешиваемый раствор этил-3-(2-аминоэтил)-5-[(4-фторфенил)метил] бензолпропаноата (500 мг) и триэтиламина (0,25 мл) в сухом дихлорметане (5 мл) при комнатной температуре. Раствор перемешивали при комнатной температуре 5 ч и затем промывали водой и сушили над (MgSO4). Растворитель выпаривали и остаток хроматографировали на силикагеле. Элюированием дихлорметаном сначала выделяли примеси и затем чистый продукт. Фракции продукта объединяли и выпаривали, получая целевое соединение в виде масла (560 мг), Rf 0,2 (SS3). Найдено: C 62,08, H 5,47, N 2,73. Для C26H27CLFNO 4S требуется: C 61,96, H 5,40, N 2,78%.
Пример 2. 3-[2-[(4-Хлорфенил)сульфониламино] этил] -5- [(4-фторфенил)метил]бензопропановая кислота.
Смесь этил-3-[2-[(4-хлорфенил)сульфониламино] этил] -5- [(4-фторфенил)метил] бензопропаноата (500 мг), 2N раствора едкого натра (1,5 мл) и метанола (5 мл) нагревали при кипячении 1 ч и затем выпаривали. Остаток растворяли в небольшом объеме воды и раствор подкисляли 2N соляной кислотой. Полученную смесь экстрагировали несколько раз эфиром и объединенный экстракт сушили над (MgSO4) и выпаривали, получая целевое соединение (390 мг), т. пл. 85 - 87oC. Найдено: C 60,78, H 5,06, N 2,87. Для C24H33ClFNO4S требуется: C 60,56, H 4,87, N 2,94%.
Пример 3. 3-[2-[(4-Хлорфенил)сульфониламино] этил] -5- [(4-фторфенил)метил]-бензолпропановая кислота.
2N раствор едкого натра (60 мл) добавили в перемешиваемый раствор этил-3-(2-карбамоилэтил)-5-[(4-фторфенил)метил]бензол- пропаноата (8,50 г) в диоксане (100 мл) и перемешивание продолжали 30 мин при комнатной температуре. Раствор охлаждали до 0oC и добавляли раствор гипохлорита натрия (15,2 мл 14%-ного раствора). Раствор перемешивали 1 ч при 0oC, затем 3 ч при комнатной температуре и еще 30 мин при 100oC. Его затем охлаждали до 0oC и добавляли 4-хлорбензосульфонилхлорид (7,50 г). Смесь перемешивали 2 ч, постепенно повышая ее температуру до комнатной. После этого добавили еще 4-хлорбензолсульфонилхлорид (5,0 г) и смесь перемешивали в течение дополнительных 30 мин, затем подкисливали концентрированной соляной кислотой. Смесь разбавляли водой и экстрагировали несколько раз диэтиловым эфиром. Объединенный эфирный экстракт промывали водой, сушили над (MgSO4) и выпаривали. Остаток хроматографировали на силикагеле. Элюирование начинали смесью дихлорметан/метанол (99: 1), постепенно изменяя их соотношение до 95:5. Фракции продукта объединяли и выпаривали. Остаток кристаллизовали из смеси эфир/гексан, получая целевое соединение (6,24 г) в виде полиморфа с т. пл. 107 - 110oC. Найдено: C 60,94, H 5,05, N 2,91. Для C23H23CIFNO4S требуется: C 60,56, H 4,87, N 2,94%.
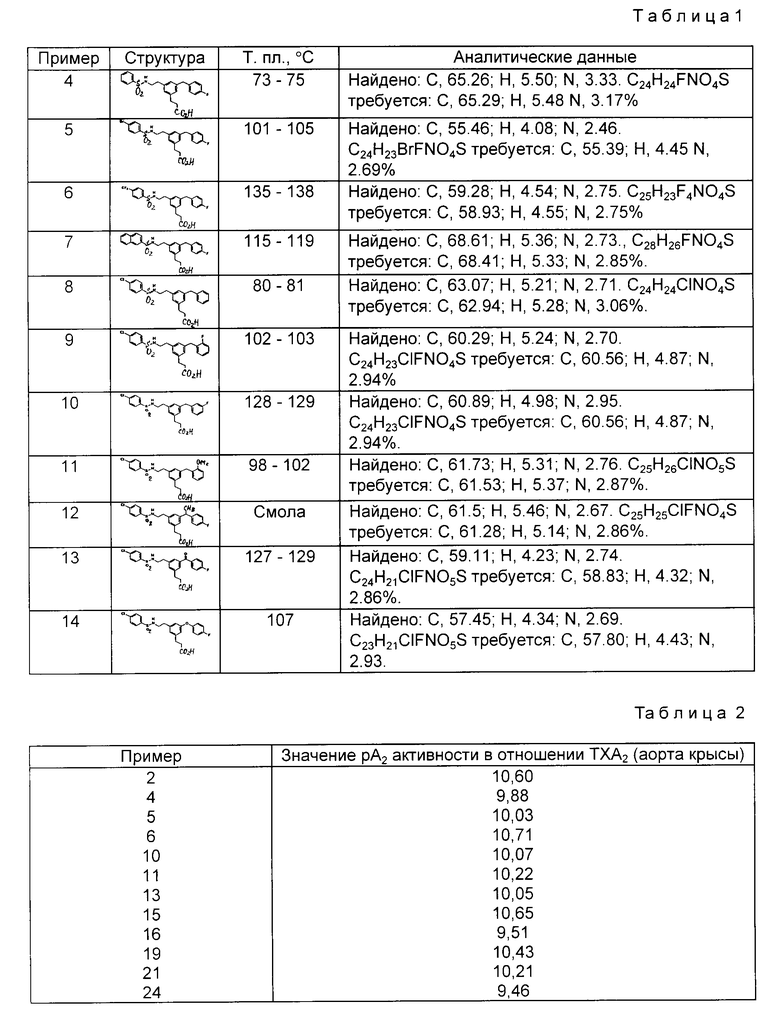
Аналогично из соответствующего карбоксамида получали следующие соединения (табл. 1):
Пример 15. 3-[2-[(4-Хлорфенил)сульфониламино]этил]-5- (4-метоксибензоил)-бензолпропановая кислота.
Смесь 3-[2-[(4-хлорфенил)сульфониламино]этил]-5-(4-фторбензоил)бензолпропановой кислоты (0,25 г) и карбоната калия (0,212 г) в безводном метаноле (5,0 мл) нагревали при кипении в течение 18 ч, затем добавляли дополнительное количество карбоната калия (0,212 г) и смесь нагревали при кипении еще 20 ч и затем охладили. Добавили воду (15 мл) и раствор выпаривали до около 10 мл и затем подкислили 2N соляной кислотой. Смесь экстрагировали несколько раз этилацетатом и объединенный экстракт промыли водой и сушили над (MgSO4). После выпаривания растворителя получили целевое соединение в стекловидной форме (0,22 г). Найдено: C 60,25, H 4,92, N 2,51. Для C25H24CINO6S требуется: C 59,81, H 4,82, N 2,79%.
Пример 16. 3-[2-[(4-Хлорфенил)сульфониламино] этил]-5- (4-метилсульфонилбензоил)бензолпропановая кислота.
Раствор 3-[2-[(4-хлорфенил)сульфониламино] этил] -5-(4-фторбензоил)бензолпропановой кислоты (0,25 г) и метансульфината натрия (0,61 г) в диметилсульфоксиде (1,0 мл) нагревали при 130oC в атмосфере сухого азота 30 ч, после добавляли дополнительное количество (1,2 г) метансульфината натрия и нагревание продолжали еще 20 ч. Раствор разбавляли водой, подкисливали 2N соляной кислотой и экстрагировали несколько раз этилацетатом. Объединенный экстракт промывали водой, сушили над (MgSO4) и выпаривали. Остаток хроматографировали на силикагеле, применяя в качестве элюента смесь дихлорметан/метанол (97:3). Фракции продукта объединили и выпарили, получая целевое соединение в виде пены (0,115 г), Rf 0,25 (SS8). Найдено: C 54,96, H 4,53, N 2,18. Для C25H24CINO7S2 требуется: C 54,59, H 4,40, N 2,55%.
Пример 17. Этил-3-[2-[(4-хлорфенил)сульфониламино]этил]-5- (4-фторбензол)-бензолпропаноат.
Смесь 3-[2-[(4-хлорфенил)сульфониламино]этил]-5-(4- фторбензоил)бензолпропановой кислоты (1,0 г), этанола (25 мл) и концентрированной серной кислоты (5 капель) перемешивали при комнатной температуре 72 ч. Образованный раствор выпаривали до одной трети первоначального объема и распределяли между этилацетатом (100 мл) и насыщенным раствором бикарбоната натрия (100 мл). Водный слой отделяли, экстрагировали этилацетатом и органические слои объединяли, промывали водой и сушили над (MgSO4). После выпаривания растворителя получали масло, которое хроматографировали на силикагеле. Элюированием дихлорметаном и затем смесью дихлорметан/метанол (50:1) выделяли целевое соединение в виде масла (0,83 г). Rf 0,6 (SS13), δ, (CDCl3): 1,13 (3H, т), 2,63 (2H, т), 2,82 (2H, т), 2,98 (2H, т), 3,26 (2H, м), 4,13 (2H, к), 4,67 (1H, т), 7,17 - 7,22 (3H, м), 7,33 (1H, с), 7,43 - 7,48 (3H, м), 7,72 - 7,83 (4H, м).
Пример 18. Этил-3-[2-[(4-хлорфенил)сульфониламино]этил]-5- (4-цианобензоил)бензолпропаноат.
Раствор этил-3-[2-[(4-(хлорфенил)сульфониламино] этил]-5-(4- фторбензоил)бензолпропаноата (0,78 г) и цианида калия (0,39 г) в сухом диметилсульфоксиде (5 мл) нагревали при 150oC в течение 17 ч. Раствор распределяли между этилацетатом (100 мл) и водой (100 мл) и добавляли твердый хлористый натрий в количестве, достаточном для разделения фаз. Органический слой отделяли, промывали водой и высушивали над (MgSO4). Растворитель выпаривали и остаток хроматографировали на силикагеле. Элюированием смесью гексан/этилацетат (10: 1) с постепенным изменением соотношения их до 2:1 получали целевое соединение в виде смолы (0,09 г) с Rf 0,40 (SS7). Найдено: C 61,99, H 4,71, N 5,07. Для C27H25CLN2O5S требуется:C 61,77, H 4,80, N 5,36%.
Пример 19. 3-[2-[(4-Хлорфенил)сульфониламино] этил] -5- (4-цианобензоил)бензолпропановая кислота.
1N раствор гидроксида натрия (0,6 мл) добавили в раствор этил-3-[2-[(4-хлорфенил)сульфониламино] этил] -5-(цианобензоил) бензолпропаноата (0,15 г) в этаноле (5 мл) и раствор перемешивали при комнатной температуре 4 ч. Добавили еще 1N раствор едкого натра (0,4 мл) и перемешивание продолжали еще 17 ч. Раствор разбавляли водой (35 мл), подкисливали 2N соляной кислотой и экстрагировали три раза дихлорметаном. Объединенный экстракт сушили над (MgSO4) и выпаривали. Остаток хромаграфировали на силикагеле. Колонку элюировали смесью дихлорметан/метанол/уксусная кислота (99:1:0,1) и получали смолу, которую повторно хроматографировали на силикагеле с применением смеси гексан/этилацетат/уксусная кислота (100:50:2), получая целевое соединение в виде смолы (0,055 г), Rf 0,20 (SS14). Найдено: C 60,53, H 4,21, N 5,07. Для C25H21CLN2O5S требуется: C 60,42, H 4,26, N 5,64%.
Пример 20. 3-[2-[(4-Хлорфенил)сульфониламино] этил] -5- [(4-метоксифенил)метил]бензолпропановая кислота.
Раствор 3-[2-[(4-хлорфенил)сульфониламино] этил] -5- [(4-метоксифенил)метил]бензолпропановой кислоты (0,95 г) и триэтиламина (1,20 мл) в трифторуксусной кислоте (20 мл) перемешивали при комнатной температуре в течение 18 ч и затем выпаривали. Остаток растирали с диэтиловым эфиром и смесь фильтровали, получая целевое соединение (0,81 г), т.пл. 106 - 108oC. Найдено: C 61,68, H 5,02, N 2,91. Для C25H26ClNOS требуется: C 61,53, H 5,37, N 2,87.
Пример 21. 3-[2-[(4-Хлорфенил)сульфониламино]этил]-5- [(4-метилсульфонилфенил)метил]бензолпропановая кислота.
Обработкой 3-[2-[(4-хлорфенил)сульфониламино]этил]-5- [(4-метилсульфонилфенил)метил] бензолпропановой кислоты (0,825 г) триэтилсиланом (0,86 мл) в трифторуксусной кислоте (15 мл) по методике примера 20 получали целевое соединение (0,51 г), т.пл. 105 - 107oC. Найдено: C 55,84, H 4,87, N 2,45. Для C25H26ClNO6S2 требуется: C 56,01, H 4,89, N 2,61%.
Пример 22. 3-[2-[(4-Хлорфенил)сульфониламино]этил]-5- [(4-фторфенил)-гидроксиметил]бензолпропановая кислота.
Борогидрид натрия (0,077 г) добавили по порциям в суспензию 3-[2-[(4-хлорфенил)сульфониламино] этил] -5- (4-фторбензоил)бензолпропановой кислоты (0,50 г) в этаноле (12 мл) и образованный раствор перемешивали при комнатной температуре 18 ч. В раствор добавляли по каплям 2N соляную кислоту (5 мл), затем этилацетат (50 мл) и смесь перемешивали 1 ч. Органический слой отделяли, промывали водой и сушили над (MgSO4). Растворитель выпаривали и остаток хроматографировали на силикагеле. Колонку элюировали смесью гексан/этилацетат (5 : 1), затем смесью гексан/этилацетат/уксусная кислота (50 : 50 : 1), постепенно повышая полярность до соотношения их 10 : 90 : 1. Фракции продукта объединяли и выпаривали, получая целевое соединение в виде смолы (0,36 г), Rf 0,45 (SS15). Найдено: C 58,58, H 4,71, N 2,65. Для C24H23ClNO5S требуется: C 58,59, H 4,71, N 2,85%.
Пример 23. Этил-3-[2-[z-2-(4-хлорфенил)циклопроп-1-илкарбоксамидо]этил] -5- [(4-фторфенил)метил]бензолпропаноат.
Хлоргидрат 1-(3-диметиламинопропил)-3-этилкарбодиимида (0,575 г) добавляли в раствор эти-3-(2-аминоэтил)-5-[(4-фторфенил)метил]бензолпропаноата (0,50 г), z-2-(4-хлорфенил)циклопропан-1-карбоновой кислоты (EP 487 095) (0,295 г), гидрата 1-гидроксибензотриазола (0,203 г) и N,N-диизопропилэтиламина (0,194 г) в сухом дихлорметане (5 мл) и раствор оставляли при комнатной температуре на ночь. Остаток хроматографировали на силикагеле с применением дихлорметана в качестве элюента, постепенно повышая полярность до смеси дихлорметан/метанол (99 : 1). Фракции продукта объединяли и выпаривали, получая целевое соединение в виде смолы (0,69 г), Rf 0,15 (SS2). Найдено: C 70,85, H 6,08 N 2,71. Для C30H31ClFNO3 требуется: C 70,82, H 6,15, N 2,76%.
Пример 24. 3-[2-[z-2-(4-Хлорфенил)циклопроп- 1-илкарбоксамидо]этил]-5-[(4-фторфенил)метил]бензолпропановая кислота.
Гидролизом этил-3-[2-[z-2-(4-Хлорфенил)циклопроп- 1-илкарбоксамидо]этил] -5-[(4-фторфенил)метил] бензолпропаноата (0,60 г) по методике Примера 2 получили целевое соединение в виде смолы (507 г), Rf 0,7 (SS5). Найдено: C 69,99, H 5,88, N 2,84. Для C28H27ClFNO3 требуется: C 70,07, H 5,67, N 2,92%.
Пример 25.
Фармацевтические капсулы - кг/капсулу
Антагонист тромбоксана A2 - 25,0
Ингибитор тромбоксан-синтетазы - 150,0
Крахмал - 74,0
Стеарат магния ВР - 1,0 - 250 мг
Антагонист тромбоксана A2 и ингибитор тромбоксан-синтетазы просеивают и смешивают с крахмалом и наполнителями. Смесью заполняют твердые желатиновые капсулы размер N 2 при помощи подходящего устройства. Аналогичным образом можно получить капсулы другой прочности и с другим соотношением активных компонентов.
Пример 26.
Определяют способность соединения противодействовать рецептору тромбоксана A2 по методике, описанной выше.
Данные по антагонистической активности соединений формулы (1) в отношении рецептора тромбоксансинтетазы (ТХА2) представлены в табл.2.
Пример 27. Исследование эффективности фармацевтической композиции, содержащей ингибитор тромбоксансинтетазы.
Готовят смесь, содержащую антагонист тробмоксансинтетазы по примеру 2 и ингибитор тромбоксансинтетазы.
Смесь исследуют на увеличение образования PGl2 и ингибирование агрегации тромбоцитов в крови человека in vitro следующим образом.
Кровь человека разбавляют в соотношении 1 : 1 гепаринизированным буфером, содержащим кальций, непосредственно перед использованием.
Затем измеряют способность к агрегации раствора кровь/буфер методом измерения полного электрического сопротивления, вызванного агрегированием в ответ на введение 5 мг/мл коллагена, дозы, которая вызывает агрегацию тромбоцитов посредством механизма в основном, не зависящем от тромбоксана.
Вышеуказанная смесь ингибирует агрегацию тромбоцитов, вызванную коллагеном, в зависимости от дозы, причем максимальное ингибирование проявляется при концентрации от 1 мкмоль ТХА2 ингибитора до 0,15 мкмоль антагониста тромбоксан синтетазы.
При использовании вышеупомянутых компонентов раздельно при концентрации 10 мкмоль наблюдается незначительное ингибирование агрегации тромбоцитов, вызванной коллагеном.
Данный пример показывает преимущество, достигаемое при введении фармацевтической композиции по п.9, обусловленное синергическим эффектом.
Сущность изобретения: предлагаются соединения формулы (I)
в которой R1 представляет собой фенил, не обязательно замещенный атомом галогена, CF3 или нафтил; R2 - H, C1 - C4-алкоксигруппу, галоген или S(O)n(C1 - C4 - алкил); Х представляет собой -CH2-, -CHCH3-, -CH(OH)-, -CO- или -O-; n равно 2,
и их фармацевтически приемлемые соли. Кроме того, предлагаются фармацевтические композиции, содержащие в качестве активного ингредиента соединения формулы (I). 2 с. и 6 з.п. ф-лы, 2 табл.
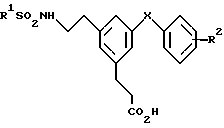
где R1 - фенил, необязательно замещенный атомом галогена, CF3 или нафтил;
R2 - Н, С1 - С4-алкоксигруппа, галоген, CN или S(O)n - С1 - С4-алкил;
X - СН2-, -СНСН3-, -СН(ОН)-, СО- или -О-;
n = 2,
и их фармацевтически приемлемые соли.
3-[(4-фторфенил)метил] -5-[2-[(4-трифторметилфенил) сульфониламино]этил] бензолпропановой кислоты,
3-[2-[(4-хлорфенил)сульфониламино] этил] -5-[1- (4-фторфенил)этил] бензолпропановой кислоты,
3-[2-[(4-хлорфенил) сульфониламино] этил] -5-[(4-фторфенил)метил] бензолпропановой кислоты,
3-[2-[(4-хлорфенил) сульфониламино] этил]-5-(4-фторфенокси) бензолпропановой кислоты;
3-[2-[(4-хлорфенил) сульфониламино]этил]-5-(4-метоксибензоил) бензолпропановой кислоты и
3-[2-[(4-хлорфенил)сульфониламино] этил-5-(4-цианобензоил)- бензолпропановой кислоты.
4-[2-(1Н-имидазол-1-ил)этокси] бензойной кислоты,
3-(1Н-имидазол-1-илметил)-2-метил-1Н-индол-1-пропановой кислоты,
2-метил-3-(3-пиридилметил)-1Н-индол-1-пропановой кислоты,
3-метил-2-(3-пиридилметил)бензо[b]тиофен-5-карбоновой кислоты,
1,3-диметил-2-(1Н-имидазол-1-илметил)-1Н-индол-5-карбоновой кислоты,
2-метил-3-(3-пиридил)-1Н-индол-1-пентановой кислоты.
| ЕР, патент, 0253321, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| US, патент, 59130812, кл | |||
| Устройство для сортировки каменного угля | 1921 |
|
SU61A1 |
