Изобретение относится к новым биологически активным производным 5'-Н-фосфоната 3'-азидо-3'-дезокситимидина формулы I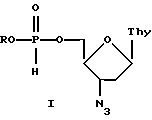
где R представляет собой изопропил, неопентил или циклогексил, и содержащим их фармацевтическим композициям.
Новые соединения обладают противовирусным действием, в первую очередь против вируса иммунодефицита человека (ВИЧ).
Известно, что ингибировать репродукцию вируса иммунодефицита можно на разных стадиях его жизненного цикла, но очевидно, что целесообразно использовать ингибиторы наиболее ранних процессов. Именно поэтому обратная транскриптаза ВИЧ, первый по времени функционирования фермент в цепи репликации вируса, является наиболее привлекательной мишенью для подавления размножения вируса. В настоящее время основную группу лекарств, используемых для лечения СПИД, представляют нуклеозидные ингибиторы обратной транскриптазы. К ним относятся 3'-азидо-3'-дезокситимидин (АЗТ, AZT, зидовудин, "Ретровир", "Тимазид"), 2',3'-дидезоксицитидин (ddC, зальцитабин, "Гивид"), 2', 3'-дидезоксиинозин (ddl, диданозин, "Видекс"), 3'-дезокси-2',3'-дидегидротимидин (d4T, ставудин "Зерит") и 3'-тиоцитидин (ЗТС, ламивудин, "Эпивир"). Их главным недостатком является необходимость клеточного фосфорилирования до соответствующих трифосфатов, эффективность которого крайне невелика. Так, процесс превращения АЗТ в организме человека в соответствующий 5'-трифосфат занимает около 1,5-2 часов. За это время проникший в клетки вирус успевает в форме провирусной ДНК интегрировать в геном человека. Поскольку выход трифосфата достигает лишь нескольких сотых долей процента, для лечения необходимо использовать большие дозы препарата. Это, в свою очередь, приводит к многочисленным побочным эффектам и быстрому вырабатыванию вирусной и клеточной резистентности.
Использование в качестве лекарственных препаратов нуклеозид-5'-трифосфатов с немодифицированной трифосфатной частью невозможно из-за их низкой стабильности к действию ферментов гидролиза и вследствие этого низкой способности проникать внутрь клетки. Были предприняты попытки синтеза модифицированных нуклеозид-5'-трифосфатов (WO 98/20017). Однако соединения оказались недостаточно активны для создания лекарственного препарата.
Более перспективными оказались такие формы активных нуклеозидов, которые позволили сократить процесс фосфорилирования в клетке до двух стадий, были проницаемы для клеточных мембран и достаточно долго были стабильны в кровотоке. Одним из таких соединений, описанных в Европейском патенте 0 354 246, является 5'-Н-фосфонат 3'-азидо-3'-дезокситимидина (Фосфазид) формулы II
В настоящее время Фосфазид используется в качестве лекарственного средства под названием "Никавир" для лечения СПИД (ВИЧ-инфекций).
Известны производные 5'-Н-фосфоната 3'-азидо-3'-дезокситимидина формулы I, в которых R выбирается из группы: метил, этил, гексил, гептил или октадецил, которые показали повышенную активность и меньшую токсичность по сравнению с азидотимидином в опытах с клетками С8166, инфицированными ВИЧ-1 (McGuigan С. , Bellevergue P. et al, Alkyl hydrogen phosphonate derivatives of anti-HIV agent AZT may be less toxic than parent nucleosides. Antiviral Chem., Chemother., 1994, 5, 271-277).
В работе Cardona V.M.F, Ayi A.I. и др. (Antiviral Research, 1999, 42, 189-196) было показано, что соединение формулы I, в котором R - бензил, активно как анти-ВИЧ препарат в испытаниях в CEM-SS и МТ-4 клетках, инфицированных ВИЧ, причем в последнем случае его активность в 5 раз превышала таковую для AZT. В СЕМ клетках, дефицитных по тимидинкиназе, соединение не проявило активность.
В соответствии с настоящим изобретением предлагаются производные 5'-Н-фосфоната 3'-азидо 3'-дезокситимидина общей формулы I, где R представляет собой изопропил, неопентил или циклогексил.
Соединения формулы I имеют пониженную гидрофобность и вследствие этого повышенную проницаемость через клеточные мембраны. Механизм превращения соединений формулы I в активную форму внутри клетки на сегодняшний день неизвестен. Один из предполагаемых механизмов заключается в том, что благодаря способности соединений I к легкому окислению до фосфата АЗТ или его производных возможно протекание сокращенного процесса внутриклеточного фосфорилирования. Другим вариантом механизма превращения соединений формулы I в активную форму является их гидролиз либо до фосфазида, либо до АЗТ.
Для получения соединений формулы I могут быть использованы два синтетических подхода. Первый основан на взаимодействии спирта ROH с треххлористым фосфором и последующем прибавлении в реакционную смесь 5'-Н-фосфоната 3'-азидо-3'-дезокситимидина (метод А). Второй путь включает реакцию 5'-Н-фосфоната 3'-азидо-3'-дезокситимидина и спирта ROH в присутствии пивалоил хлорида как активирующего агента (метод В).
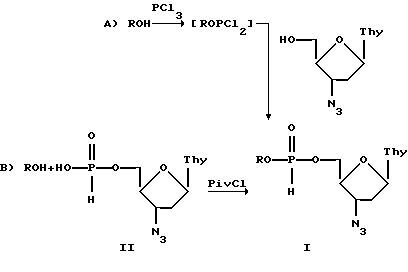
Соединения формулы I могут быть выделены и очищены обычными способами, например, экстракцией, осаждением, хроматографией и т.д.
Согласно настоящему изобретению соединения формулы I могут быть использованы для получения противовирусных фармацевтических композиций с подходящим носителем. Фармацевтические композиции могут быть получены известными в данной области техники способами.
В качестве носителей могут быть использованы различные виды носителей, обычно используемых в лекарственных средствах, например наполнители, связывающие вещества, разрыхляющие вещества, смазывающие вещества, красители, вкусовые вещества, ароматизирующие вещества, поверхностно-активные вещества и т.п.
Фармацевтические композиции на основе соединений формулы I могут быть использованы в качестве лекарственного средства в любой лекарственной форме. Лекарственная форма может быть правильно выбрана в соответствии с объектом лечения. Конкретные примеры лекарственных форм включают пероральные препараты, такие как таблетки, гранулы, капсулы, и парентеральные препараты, такие как, например, растворы для инъекций.
Количество соединения формулы I по настоящему изобретению, которое должно содержать вышеописанные фармацевтические композиции, изменяется в соответствии с типом композиции, способом введения, схемой дозирования и поэтому не может быть точно определено, а выбирается в соответствии с потребностью из широкого диапазона. Однако композиция может предпочтительно содержать примерно 1-80 мас.% соединения формулы I от веса композиции.
Настоящее изобретение проиллюстрировано нижеследующими примерами получения конкретных соединений, фармацевтических композиций и фармакологических испытаний.
Пример 1. 3'-Азидо-3'-дезокситимидин, 5'-(2,2-диметилпропил)фосфит (Iа).
Раствор РСl3 (135 мг, 88 мкл, 1 ммоль) в дихлорметане (2 мл) охлаждают до 1-2oС и при перемешивании добавляют неопентиловый спирт (88 мг, 1 ммоль) и пиридин (80 мкл, 1 ммоль). Раствор AZT (130 мг, 0,5 ммоль) в пиридине (80 мкл) и ацетонитриле (3 мл) добавляют при 4-5oС. Смесь перемешивают 3 ч при комнатной температуре и разбавляют холодным раствором насыщенного NаНСО3 (3 мл) и хлороформом (5 мл). Органический слой промывают водой, сушат Nа2SO4 и упаривают. Остаток хроматографируют на колонке с силикагелем (2•25 см), элюируя системой хлороформ-метанол 96: 4. Получают соединение Iа (94 мг). Выход 47%. УФ (H2O, λмакс): 266 нм. 1Н ЯМР (СD3СN), δ, м.д.: 7.28с, 7.31с (1Н, Н6), 6.87д, 6.85д (1Н, J706 Гц, Р-Н), 6.15т (1Н, J 6.0 Гц, Н1'), 5.90д (1Н, J 697 Гц, Н-Р), 4.35м (1H, Н3'), 4.19м (2Н, Н5'), 4.01м (1H, Н4'), 3.73дб, 3.70д (2Н, J6 Гц), 2.38т (2Н, J 6.0 Гц, Н2'), 1.85с, 1.86с (3Н, СН3), 0.92с (9Н, СН3С). 31P ЯМР (CD3CN), δ, ppm: 8.99с, 8.40с.
Пример 2. 3'-Азидо-3'-дезокситимидин, 5'-изопропилфосфит (Iб).
Водный раствор Na-соли 5'-Н-фосфоната 3'-азидо-3'-дезокситимидина (176 мг, 0,5 ммоль) пропускают через колонку с Dowex-50 (Ру+) (1х8 см), элюируя водой. Элюат упаривают досуха и переупаривают с пиридином (3•2 мл). Остаток растворяют в MeCN (3,5 мл) и пиридине (2 мл) и добавляют при перемешивании изопропиловый спирт (23 мкл, 0,5 ммоль). Смесь охлаждают до -10oС и добавляют пивалоилхлорид (180 мкл, 0,75 ммоль). Охлаждение снимают и смесь перемешивают 60 мин. Смесь разбавляют хлороформом (5 мл) и промывают насыщенным раствором NaHCO3 (3 мл) и водой (3•3 мл). Органический слой сушат Na2SO4, упаривают и переупаривают с толуолом. Продукт хроматографируют на колонке с силикагелем (15•2,5 см), элюируя системой хлороформ-метанол 96:4. Получают соединение Iб (156 мг). Выход 84%. УФ (Н2О, λмакс): 266 нм. 1Н ЯМР (СD3СМ), δ, м.д.: 7.38с, 7.39с (1Н, Н6), 6.86д (1Н, J 700 Гц, Р-Н), 6.15т (1Н, J 6.0 Гц, Н1'), 4.75т (1Н, СН), 4.35м (1Н, Н3'), 4.19м (2Н, Н5'), 4.01м (1Н, Н4'), 2.38т (2Н, J 6.0 Гц, Н2'), 1.87с (3Н, СН3), 1.35 д (6Н, J 6.6 Гц, СН3). 31Р ЯМР (СD3CN), δ, ppm: 10.09 с, 10.01 с.
Пример 3. 3'-Азидо-3'-дезокситимидин, 5'-циклогексилфосфит (Iв).
Водный раствор Na-соли 5'-Н-фосфоната 3'-азидо-3'-дезокситимидина (176 мг, 0,5 ммоль) пропускают через колонку с Dowex-50 (Ру+) (1•8 см), элюируя водой. Элюат упаривают досуха и переупаривают с пиридином (3•2 мл). Остаток растворяют в MeCN (3.5 мл) и пиридине (2 мл) и добавляют при перемешивании циклогексиловый спирт (60 мкл, 0,55 ммоль). Смесь охлаждают до -10oС и добавляют пивалоилхлорид (180 мкл, 0,75 ммоль). Охлаждение снимают и смесь перемешивают 60 мин. Смесь разбавляют хлороформом (5 мл) и промывают насыщенным раствором NаНСО3 (3 мл) и водой (3•3 мл). Органический слой сушат Na2SO4, упаривают и переупаривают с толуолом. Продукт хроматографируют на колонке с силикагелем (15•2,5 см), элюируя системой хлороформ-метанол 96:4. Получают соединение Iв (128 мг). Выход 62%. УФ (Н2О, λмакс): 266 нм. 1Н ЯМР (CD3CN). δ, м.д.: 7.38с, 7.37с (1Н, Н6,), 6.86д (1Н, J 700 Гц, Р-Н), 6.15т (1Н, J 6.0 Гц, Н1'), 4.75т (1Н, СH), 4.35м (1H, Н3'), 4.19м (2Н, Н5'), 4.01м (1Н, Н4'), 2.38т (2Н, J 6.0 Гц, Н2'), 1.92с (3Н, СН3), 1.90т, 1.71т, 1.50т, 1.24т (10Н, циклогексил). 31Р ЯМР (CD3CN), δ, м.д.: 7.60 с, 8.04 с.
Пример 4. Таблетка.
Для приготовления таблетки используют следующий состав: соединение Iв - 200 мг, кальций карбонат осажденный - 110 мг, целлюлоза микрокристаллическая - 40 мг, аэросил - 13 мг, кальция стеарат - 12 мг.
Соединение Iв смешивают с аэросилом и микрокристаллической целлюлозой, просеивают и добавляют карбонат кальция. Полученную смесь опудривают стеаратом кальция и перемешивают до получения однородной массы. Полученную таблеточную смесь прессуют в брикеты на таблеточном прессе. Полученные брикеты измельчают на грануляторе. Гранулы прессуют на таблеточном прессе до получения таблеток массой 375 мг.
Была изучена стабильность синтезированных соединений формулы I в водных растворах при рН 7,5 и в сыворотке крови человека. Синтезированные эфиры гидролизуются до азидотимидина, причем качественный состав продуктов в обеих исследованных жидкостях совпадает.
Пример 5. Химический гидролиз соединений формулы I.
Эфиры I (15-20 мг) растворяют в ацетонитриле (0.25 мл) и раствор добавляют к 0.1 М фосфатному буферу (рН 7.5, 0.25 мл) при комнатной температуре. Через определенные промежутки времени отбирают аликвоты и исследуют их методом ТСХ и ВЭЖХ. Условия ВЭЖХ: колонка (150•4 мм) Durasil C-8 (7 мкм); градиент буфера Б в буфере А. Буфер А: 5 мМ калий-фосфатный буфер (рН 6.2); буфер Б: 80% МеОН. 0,5 мин - буфер А; 5 мин - 40 мин - 0-->100% буфер Б; 40-45 мин - буфер Б. Данные приведены в таблице 1.
Пример 6. Гидролиз соединений формулы I в сыворотке крови человека.
К 100 mM раствору соединений I в формамиде (0.5 мкл) прибавляют сыворотку (100%, 99 мкл) и смесь инкубируют при 37oС. Через определенные промежутки времени к аликвотам (10 мкл) прибавляют ацетон (40 мкл), смесь выдерживают 20 мин при -20oС и центрифугируют. Супернатант упаривают, остаток растворяют в воде (50 мкл) и анализируют методом ВЭЖХ или ТСХ. Условия ВЭЖХ, как в примере 5. Результаты приведены в таблице 1.
Была исследована биологическая активность соединений формулы I, a именно их способность ингибировать репродукцию вируса иммунодефицита человека и цитотоксичность.
Пример 7. Исследование противовирусной активности соединений формулы I в отношении ВИЧ.
Исследование проводили с использованием штамма ВИЧ-1 ГКВ 4046 на перевиваемой линии чувствительных клеток МТ-4. Для заражения использовали супернатант инфицированных клеток, хранившийся в жидком азоте, множественность заражения составляла 0,2-0,5 инфекционных единиц на клетку. Суспензию клеток МТ-4 с концентрацией 2,0•106 в миллилитре и жизнеспособностью не менее 90% помещали в лунки 96-луночного планшета ("Сеl-Cult", Великобритания) непосредственно после внесения вируссодержащего материала и сразу же вносили исследуемые препараты, разведенные в среде RPMI-1640 без добавок до конечных концентраций 0,001-100 мкг/мл (по три лунки на каждую дозу). Контролями служили инфицированные ВИЧ-1 клетки МТ-4 без добавления препаратов (вместо препарата вносили такое же количество среды RPMI-1640 без добавок) и неинфицированные клетки.
Планшет инкубировали в течение часа при 37oС для адсорбции вируса, затем клетки разводили до посевной концентрации (0,5•106 в миллилитре) питательной средой RPMI-1640 с добавлением 300 мг/мл L-глютамина и 100 мкг/мл гентамицина, а также 10% фетальной сыворотки КРС, предварительно инактивированной прогреванием при 56oС в течение 30 минут. Планшет помещали в термостат на 37oС в атмосфере 5% СО2. На 3-4 сутки культивирования подсчитывали концентрацию и жизнеспособность клеток методом исключения трипанового синего. По полученным данным определяли прирост жизнеспособности клеток относительно контроля под действием возрастающих доз препаратов.
В лунки планшета добавляли Твин-80 до конечной концентрации 0,1% и помещали его на 24 часа при 4oС для инактивации ВИЧ. Затем проводили оценку анти-ВИЧ активности препаратов с использованием количественного определения вирусспецифического белка р24 методом прямого иммуноферментного анализа. Дозы препаратов, на 50 и 90% подавляющие прирост вирусного антигена (ID50 и ID90), определяли по графику. Полученные данные представлены в таблице 2. На основании этих количественных показателей ингибирования можно судить об эффективности противовирусного действия препаратов, заключающейся в высокой степени подавления размножения ВИЧ-1 в культуре клеток МТ-4.
Пример 8. Исследование цитотоксичности соединений формулы I.
Оценку цитотоксичности препаратов проводили путем добавления их разведений в среде RPMI-1640 к клеточной суспензии МТ-4, помещенной в лунки 96-луночного планшета, до конечных концентраций 0,001-100 мкг/мл (в трех повторах) с последующим культивированием при 37oС в течение 3-4 суток. Контролем служили клетки без добавления препаратов. Посевная концентрация составляла 0,5•106 в миллилитре. Жизнеспособность клеток подсчитывали в камере Горяева с предварительным окрашиванием трипановым синим. По полученным результатам были рассчитаны дозы препаратов, на 50% снижающие жизнеспособность клеток по сравнению с контролем (CD50), данные приведены в таблице 2. Следует отметить, что токсическое действие на клетки МТ-4, выражающееся в изменении их морфологии, снижении репродуктивности и жизнеспособности, оказывали только максимальные дозы препаратов (100 мкг/мл), на 4-5 порядков превышающие эффективные в отношении ВИЧ-1 концентрации.
Таким образом, показано, что испытанные соединения формулы I обладают способностью ингибировать репродукцию вируса иммунодефицита 1 типа в культуре клеток МТ-4, причем эффективность этих препаратов сравнима с эффективностью используемого в клинической практике препарата. Это говорит о перспективности соединения формулы I для создания новых лекарственных форм, которые могут пополнить арсенал средств, используемых в терапии СПИД.
| название | год | авторы | номер документа |
|---|---|---|---|
| МОДИФИЦИРОВАННЫЕ 5'-ФОСФОНАТЫ АЗТ В КАЧЕСТВЕ АКТИВНЫХ КОМПОНЕНТОВ ДЛЯ ПОТЕНЦИАЛЬНЫХ ПРОТИВОВИРУСНЫХ ПРЕПАРАТОВ | 2004 |
|
RU2322450C2 |
| ЗАМЕЩЕННЫЕ АММОНИЕВЫЕ СОЛИ 5'-Н-ФОСФОНАТА 3'-АЗИДО-3'-ДЕЗОКСИТИМИДИНА, ЯВЛЯЮЩИЕСЯ СЕЛЕКТИВНЫМИ ИНГИБИТОРАМИ ПРОДУКЦИИ ВИРУСА ИММУНОДЕФИЦИТА ЧЕЛОВЕКА ВИЧ-1 И ВИЧ-2 | 1998 |
|
RU2207343C2 |
| 5`-ХОЛИНФОСФАТ 3`-АЗИДО-3`-ДЕЗОКСИТИМИДИНА КАК АНТИВИРУСНЫЙ АГЕНТ | 2002 |
|
RU2293739C2 |
| СОЛИ 5'-АМИНОКАРБОНИЛФОСФОНАТА 3'-АЗИДО-3'-ДЕЗОКСИТИМИДИНА, ЯВЛЯЮЩИЕСЯ СЕЛЕКТИВНЫМИ ИНГИБИТОРАМИ ПРОДУКЦИИ ВИРУСА ИММУНОДЕФИЦИТА ЧЕЛОВЕКА ВИЧ-1 | 2010 |
|
RU2441016C1 |
| МОДИФИЦИРОВАННЫЕ НУКЛЕОЗИД-5'-ТРИФОСФАТЫ КАК АНТИВИРУСНЫЕ АГЕНТЫ | 1996 |
|
RU2183213C2 |
| СПОСОБ ПОЛУЧЕНИЯ 5'-АМИНОКАРБОНИЛФОСФОНАТОВ НУКЛЕОЗИДОВ И СПОСОБ ПОЛУЧЕНИЯ ХЛОРАНГИДРИДА ТРИМЕТИЛСИЛИЛЬНОГО ЭФИРА ЭТОКСИКАРБОНИЛФОСФОНОВОЙ КИСЛОТЫ | 2010 |
|
RU2446169C2 |
| 5'-ФОСФОРСОДЕРЖАЩИЕ ПРОИЗВОДНЫЕ 2',3'-ДИДЕЗОКСИ-3'-ТИАЦИТИДИНА НОВЫЕ ПРОТИВОВИРУСНЫЕ АГЕНТЫ | 2007 |
|
RU2373218C2 |
| ФОСФОРАМИДАТЫ НУКЛЕОЗИДНЫХ АНАЛОГОВ - ИНГИБИТОРЫ РЕПРОДУКЦИИ ВИРУСА ИММУНОДЕФИЦИТА ЧЕЛОВЕКА | 2003 |
|
RU2243972C1 |
| 2',3'-ДИДЕГИДРО-2',3'-ДИДЕЗОКСИТИМИДИН-5'[(ЭТОКСИКАРБОНИЛ)(ЭТИЛ)ФОСФОНАТ]- ИНГИБИТОР РЕПРОДУКЦИИ ВИРУСА ИММУНОДЕФИЦИТА ЧЕЛОВЕКА | 2000 |
|
RU2188203C2 |
| УРЕТАНОВЫЕ ПРОИЗВОДНЫЕ АЗТ - ПОТЕНЦИАЛЬНЫЕ ПРОТИВОВИРУСНЫЕ ПРЕПАРАТЫ | 2009 |
|
RU2430103C1 |


Изобретение относится к новым противовирусным производным 5'-Н-фосфоната 3'-азидо 3'-дезокситимидина общей формулы I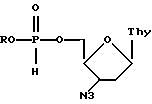
где R представляет собой изопропил, неопентил или циклогексил,
и содержащим их фармацевтическим композициям. Соединения формулы I получают взаимодействием спирта ROH с треххлористым фосфором и последующем прибавлении в реакционную смесь 5'-Н-фосфоната 3'-азидо-3'-дезокситимидина или взаимодействием 5'-Н-фосфоната 3'-азидо-3'-дезокситимидина и спирта ROH в присутствии пивалоил хлорида как активирующего агента. Соединения формулы I имеют пониженную гидрофобность и повышенную проницаемость через клеточные мембраны. Фармацевтические композиции, содержащие соединения I в эффективной терапевтической дозе и фармацевтически приемлемый носитель, обладают противовирусной активностью, в том числе против ВИЧ. 2 с.п. ф-лы, 2 табл.
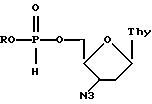
где R представляет собой изопропил, неопентил или циклогексил.
| СОЛИ 5'-Н-ФОСФОНАТА 3'- АЗИДО-3'-ДЕЗОКСИТИМИДИНА, ЯВЛЯЮЩИЕСЯ СПЕЦИФИЧЕСКИМИ ИНГИБИТОРАМИ ПРОДУКЦИИ ВИРУСА ИММУНОДЕФИЦИТА ЧЕЛОВЕКА ВИЧ-1 И ВИЧ-2 | 1996 |
|
RU2106353C1 |
| СПОСОБ ПОЛУЧЕНИЯ 3'-АЗИДО- 2', 3'-ДИДЕЗОКСИТИМИДИНА | 1997 |
|
RU2135512C1 |
| Домовый номерной фонарь, служащий одновременно для указания названия улицы и номера дома и для освещения прилежащего участка улицы | 1917 |
|
SU93A1 |
| УСТРОЙСТВО для РЕМОНТА МЕТАЛЛУРГИЧЕСКИХ ПЕЧЕЙ | 0 |
|
SU354246A1 |

