Настоящее изобретение относится к кристаллической, полиморфной форме (S, S, S)-(N-(1-[2-карбокси-3-(N-мезиллизиламино)пропил] -1- циклопентилкарбонил)тирозина, который имеет формулу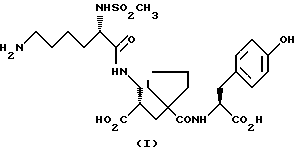
далее обозначаемую как α-форма соединения формулы (I).
Более конкретно, изобретение относится к α-форме соединения (I) и способу ее получения, к промежуточным продуктам, используемым для ее получения, к содержащим ее композициям и к применению α-формы
Аморфная форма (далее обозначаемая как β-форма) соединения формулы (I) была описана в Европейской патентной публикации N EP-A-0358398 в примере 181. Соединение является эффективным ингредиентом цинкзависимой нейтральной эндопептидазы E.C.3.4.24.11. и поэтому способно потенцировать биологические эффекты натрийуретического фактора предсердия. Таким образом, оно является натрийуретическим, противогипертоническим и диуретическим агентом, который применим для лечения различных сердечно-сосудистых заболеваний. Соединение также является эффективным ингредиентом ангиотензин превращающего фермента, другого фермента, который участвует в регулировании кровяного давления. Соединение, следовательно, имеет двойное фармакологическое действие, посредством чего способно ингибировать два ключевых фрагмента, участвующих в регулировании кровяного давления. Следовательно, оно, по-видимому, будет полезно при лечении различных форм гипертонии и связанных с ней сердечно-сосудистых заболеваний, таких как сердечная недостаточность и глаукома.
β-Форма может быть получена способом, таким как высушивание замораживанием раствора соединения формулы (I), быстрым выпариванием растворителя из раствора или осаждением из такого раствора путем добавления слабого растворителя. β-Форма не расплавляется в точке, но обыкновенно "размягчается" при приблизительно 160oC.
Однако у β-формы были обнаружены некоторые свойства, которые делают ее, в частности, не приемлемой для фармацевтического состава. А именно, она является гигроскопической по природе, она имеет низкую объемную плотность и плохие прессовочные свойства. Проведенные технологические эксперименты по использованию β-формы выявили проблемы в производстве таблеток из композиций, содержащих эту форму.
Целью настоящего изобретения является создание формы соединения формулы (I), которая может быть эффективно подвергнута технологическому процессу для получения стабильного и эффективного состава лекарственного препарата.
Эта цель решена путем неожиданного обнаружения того, что может быть получена α-форма соединения формулы (I), являющаяся негигроскопической, кристаллической, и по сравнению с β-формой имеющая более высокую объемную плотность и лучшие прессовочные свойства. α-Форма, в частности, является подходящей для использования в фармацевтическом составе лекарственного препарата.
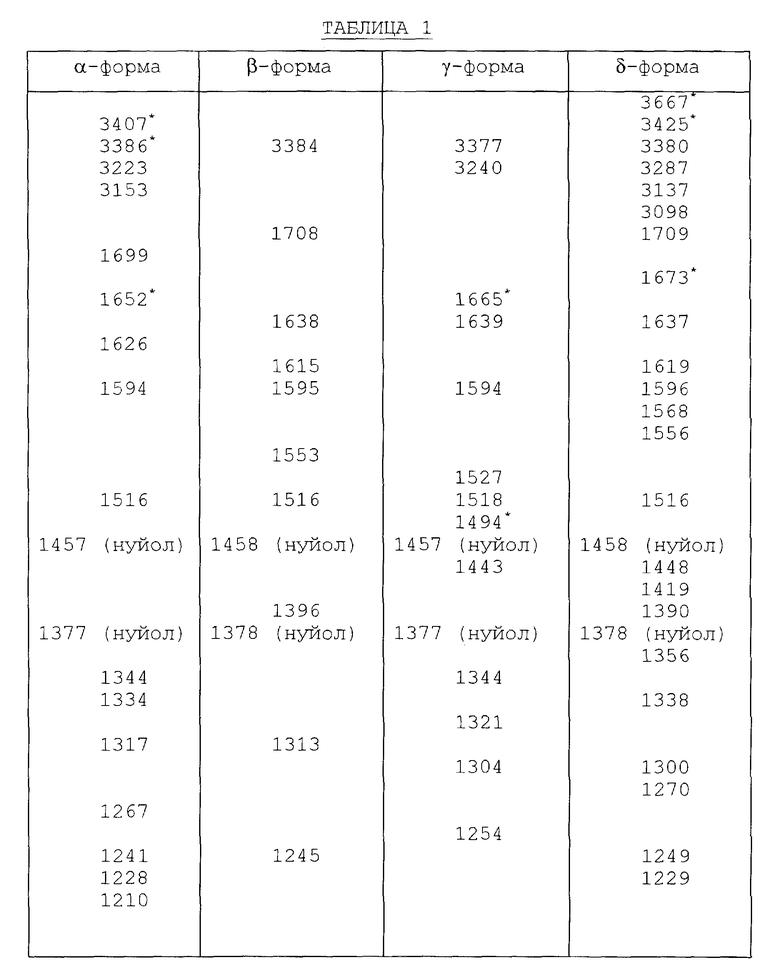
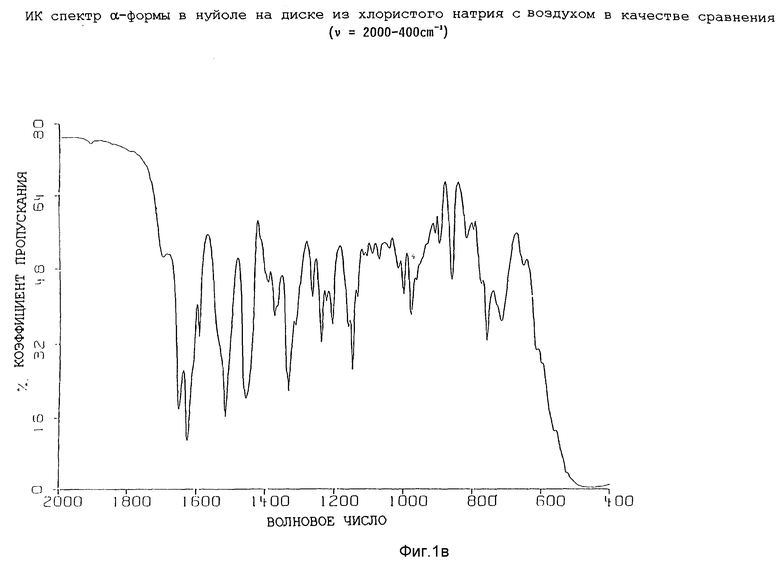
Таким образом, настоящее изобретение относится к кристаллической, полиморфной α-форме соединения формулы (I), обладающей инфракрасным спектром в минеральном масле (нуйол), проявляющим полосы поглощения при ν = 3407, 3386, 3153, 1699, 1652, 1626, 1594, 1516, 1457 (нуйол), 1377 (нуйол), 1344, 1334, 1317, 1267, 1241, 1228, 1210, 1164, 1151, 1137, 1118, 1109, 1093, 1074, 1045, 1019, 1003, 981, 965, 911, 897, 862, 818, 800, 778, 762, 720 и 655 см-1.
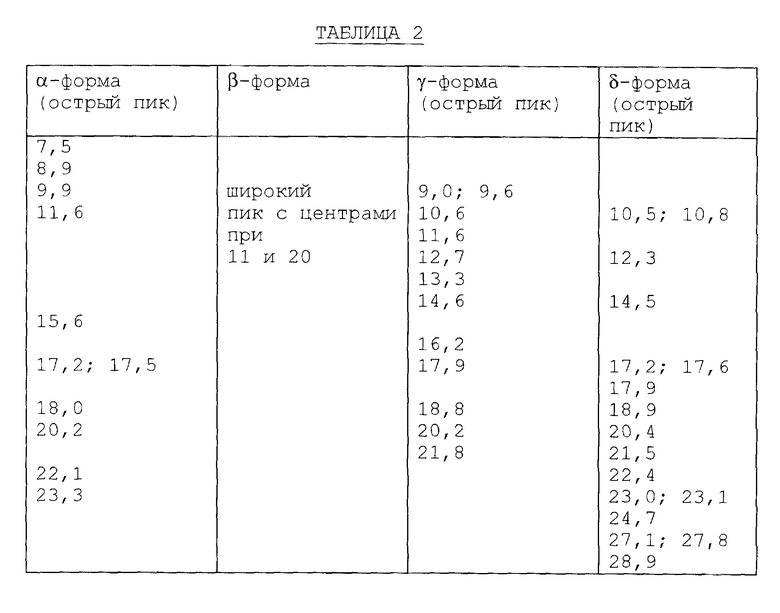
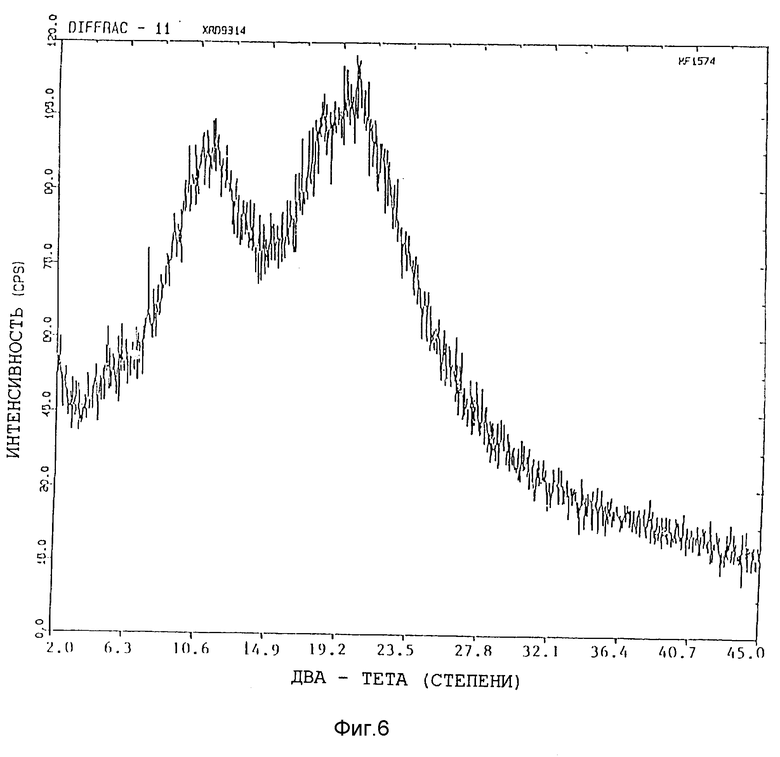
Далее α-форма охарактеризована картиной дифракции рентгеновский лучей на порошке, полученной при использовании графитового монохроматора с медным радиационным фильтром (λ = 0,15405 нм) которая проявляет следующие основные пики при 7,5; 8,9; 9,9; 11,6; 15,6; 17,2; 18,0; 20,2; 22,1 и 23,3 степенях 2θ.
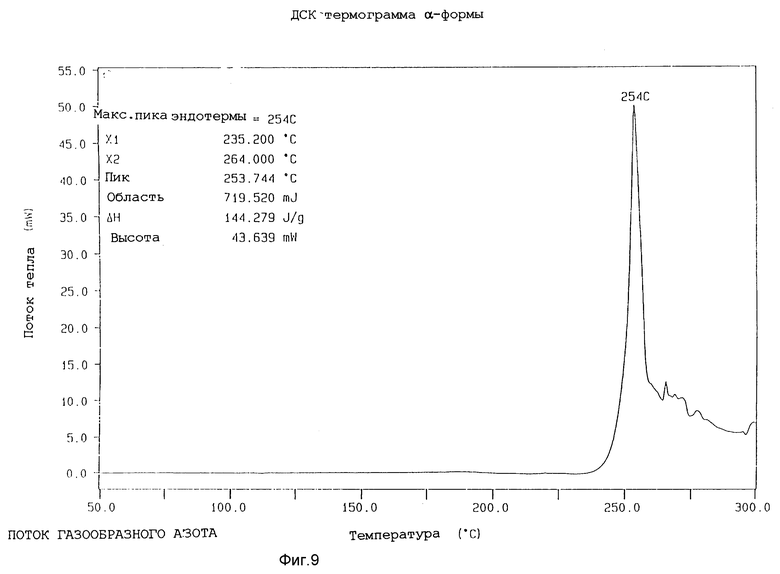
Далее α-форма охарактеризована методом дифференциальной сканирующей калориметрии, в которой она проявляет острую эндотерму в районе 248 - 259oC и разлагается при около 260oC, когда ее подвергают скорости сканирования 20oC в минуту.
α-Форма обычно резко плавится в районе 242 - 252oC, хотя была зарегистрирована и более низкая точка плавления.
Были также получены другие формы (далее обозначаемые как γ- и δ-формы) соединения формулы (I), которые также составляют часть настоящего изобретения, поскольку они могут быть использованы в качестве промежуточных продуктов при получении α-формы.
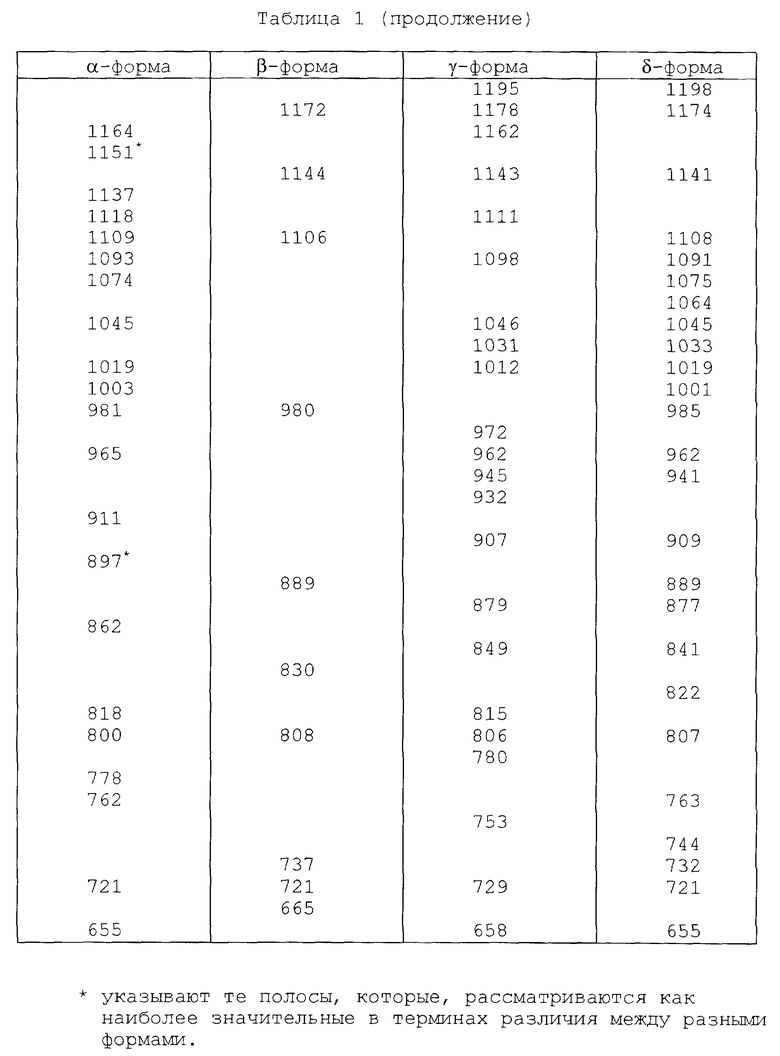
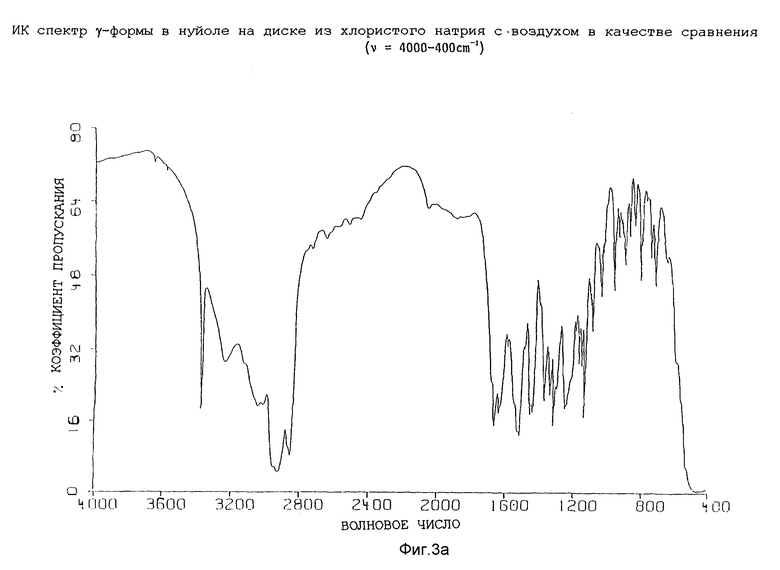
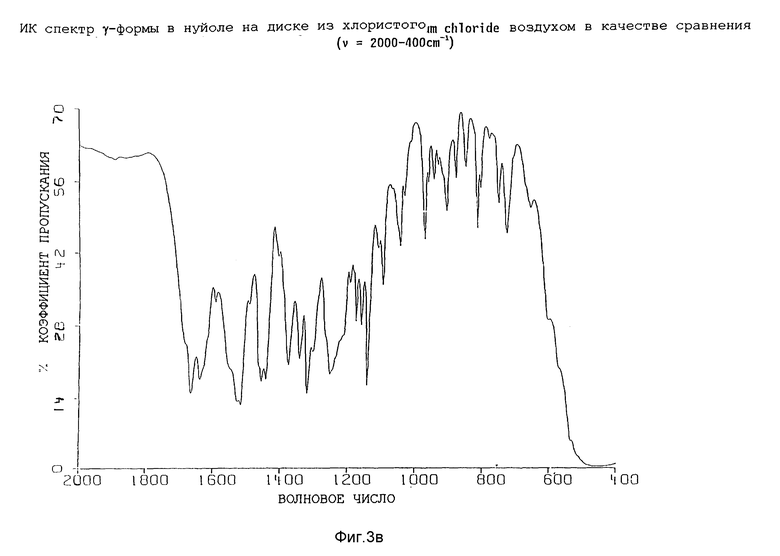
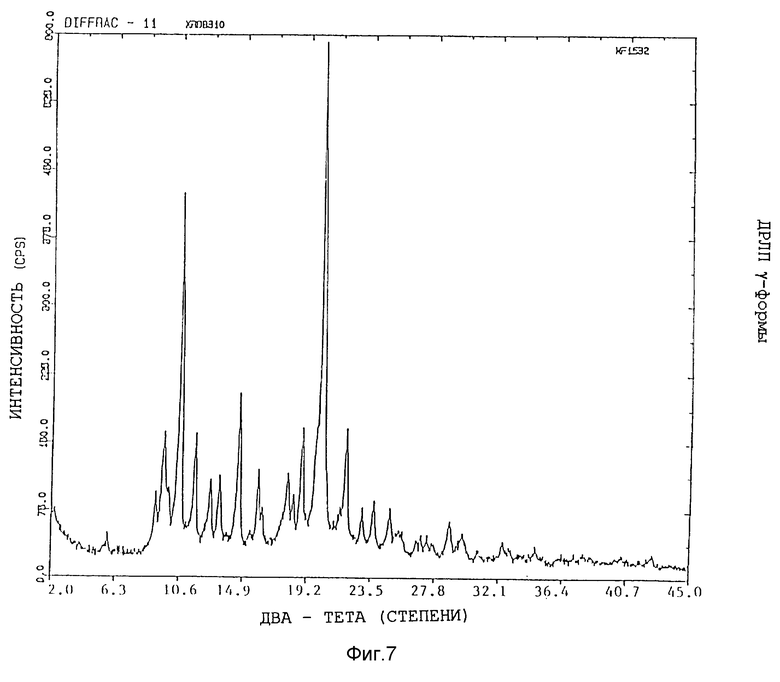
Таким образом, изобретение далее относится к полиморфной γ-форме соединения формулы (I), обладающей инфракрасным спектром в минеральном масле (нуйол), проявляющим полосы поглощения при ν = 3377, 3240, 1665, 1639, 1594, 1527, 1518, 1494, 1457 (нуйол), 1443, 1377 (нуйол), 1344, 1321, 1301, 1254, 1195, 1178, 1162, 1143, 1111, 1098, 1046, 1031, 1012, 972, 962, 945, 932, 907, 879, 849, 815, 806, 780, 753 и 658 см-1.
Далее γ-форма охарактеризована картиной дифракции рентгеновский лучей на порошке, полученной при использовании графитового монохроматора с медным радиационным фильтром (λ = 0,15405 нм), которая проявляет следующие основные пики при 9,6; 10,6; 11,6; 12,7; 13,3; 14,6; 16,2; 17,9; 18,8; 20,2 и 21,8 степенях 2θ.
Далее γ-форма охарактеризована методом дифференциальной сканирующей калориметрии, в которой она проявляет острую эндотерму в районе 176 - 186oC, экзотерму при около 207oC и слабую эндотерму при около 213oC и разлагается при около 250oC, когда ее подвергают скорости сканирования 20oC в минуту.
γ-Форма обычно резко плавится в области 170 - 185oC.
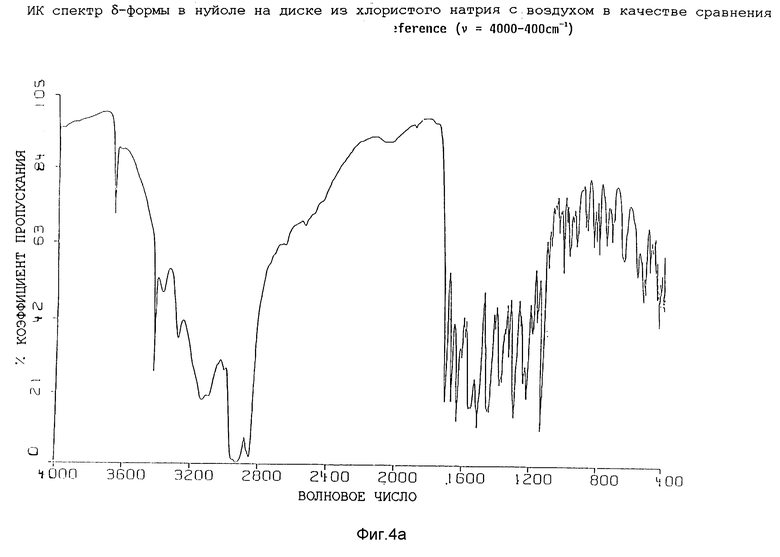
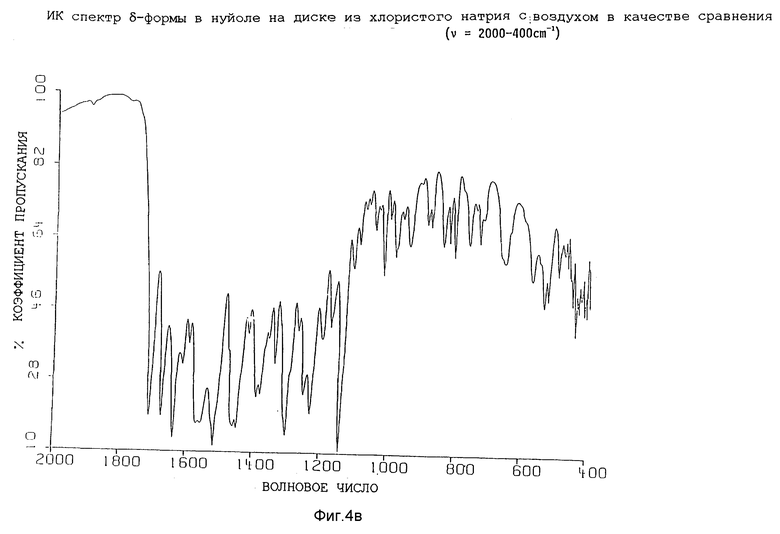
Изобретение, таким образом, также относится к гидратной δ-форме соединения формулы (I), имеющей содержание воды от 1 до 7%, предпочтительно от 2 до 4% по весу, как определено согласно Karl Fisher анализу, и обладающей инфракрасным спектром в минеральном масле (нуйол), проявляющим полосы поглощение при ν = 3667, 3425, 3380, 3287, 3137, 3098, 1709, 1673, 1937, 1619, 1596, 1568, 1556, 1516, 1458 (нуйол), 1448, 1419, 1390, 1378 (нуйол), 1356, 1338, 1300, 1270, 1249, 1229, 1198, 1174, 1141, 1108, 1091, 1075, 1064, 1045, 1033, 1019, 1001, 985, 632, 941, 909, 889, 841, 822, 807, 763, 744, 732,721 и 655 см-1.
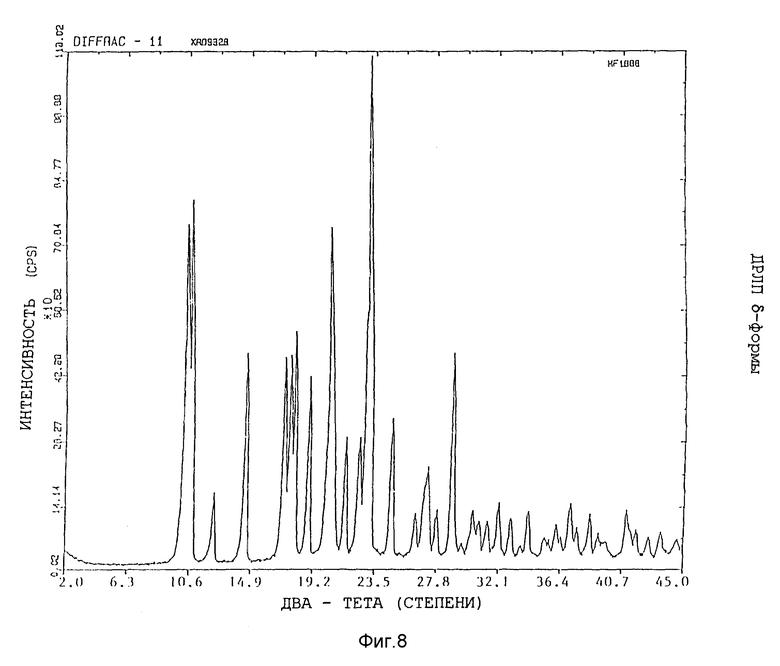
Далее δ-форма охарактеризована картиной дифракции рентгеновских лучей на порошке, полученной при использовании графитового монохроматора с медным радиационным фильтром (λ = 0,15405 нм), которая проявляет следующие основные пики при 10,5; 10,8; 12,3; 14,5; 17,2; 17,6; 17,9; 18,9; 20,4; 21,5; 22,4; 23,0; 23,1; 24,7; 27,1; 27,8 и 28,9 степенях 2θ.
Далее δ-форма охарактеризована методом дифференциальной сканирующей калориметрии, в которой она проявляет острые эндотермы при около 162oC и в районе около 166 - 168oC и разлагается при около 200oC, когда ее подвергают скорости сканирования 20oC.
δ-Форма обычно резко плавится в районе 165 - 175oC.
Хотя γ- и δ-формы соединений формулы (I) обладают теми же видами фармакологической активности, что и α- и β-формы, они являются не столь подходящими, как α-форма, для цели фармацевтического состава.
α-Форма соединения формулы (I) может быть получена следующими способами:
1) α-Форма может быть получена путем каталитического гидрирования в водном растворе соли натрия, магния, аммония или (C1-C4 алкил) аммония соединения формулы: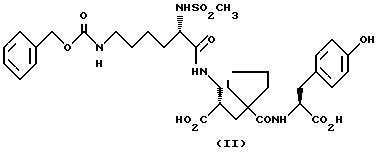
применяя подходящий катализатор для удаления бензилоксикарбонильной защитной группы, например палладий-на-угле, с последующим подкислением полученной основной соли соединения формулы (I), при pH от 3 до 5, предпочтительно при pH около 4 и предпочтительно при от 35 до 45oC, с получением α-формы. Предпочтительно используют динатриевую соль соединения формулы (II). Далее, подходящими катализаторами для удаления бензилоксикарбонильной защитной группы являются хорошо известные специалистам, например см. T.W. Green and P.G.Wuts, "Protective Groops in Organic Syntesis", второе издание, 1991, указание на которое включено здесь в качестве ссылки. В обычном способе раствор соединения формулы (II) в подходящем органическом растворителе, например этилацетате, встряхивают с водным раствором гидроксида натрия для получения его динатриевой соли. Водный раствор, содержащий натриевую соль, затем отделяют и гидрируют в присутствии 5% катализатора палладия-на-угле при давлении около 414 kPa (60 psi) при комнатной температуре для удаления бензилоксикарбонильной защитной группы. Катализатор затем удаляют путем фильтрации и pH фильтрата доводят до 4, используя подходящую кислоту, например водный раствор соляной кислоты. α-Форма высаживается из раствора и может быть собрана путем фильтрации.
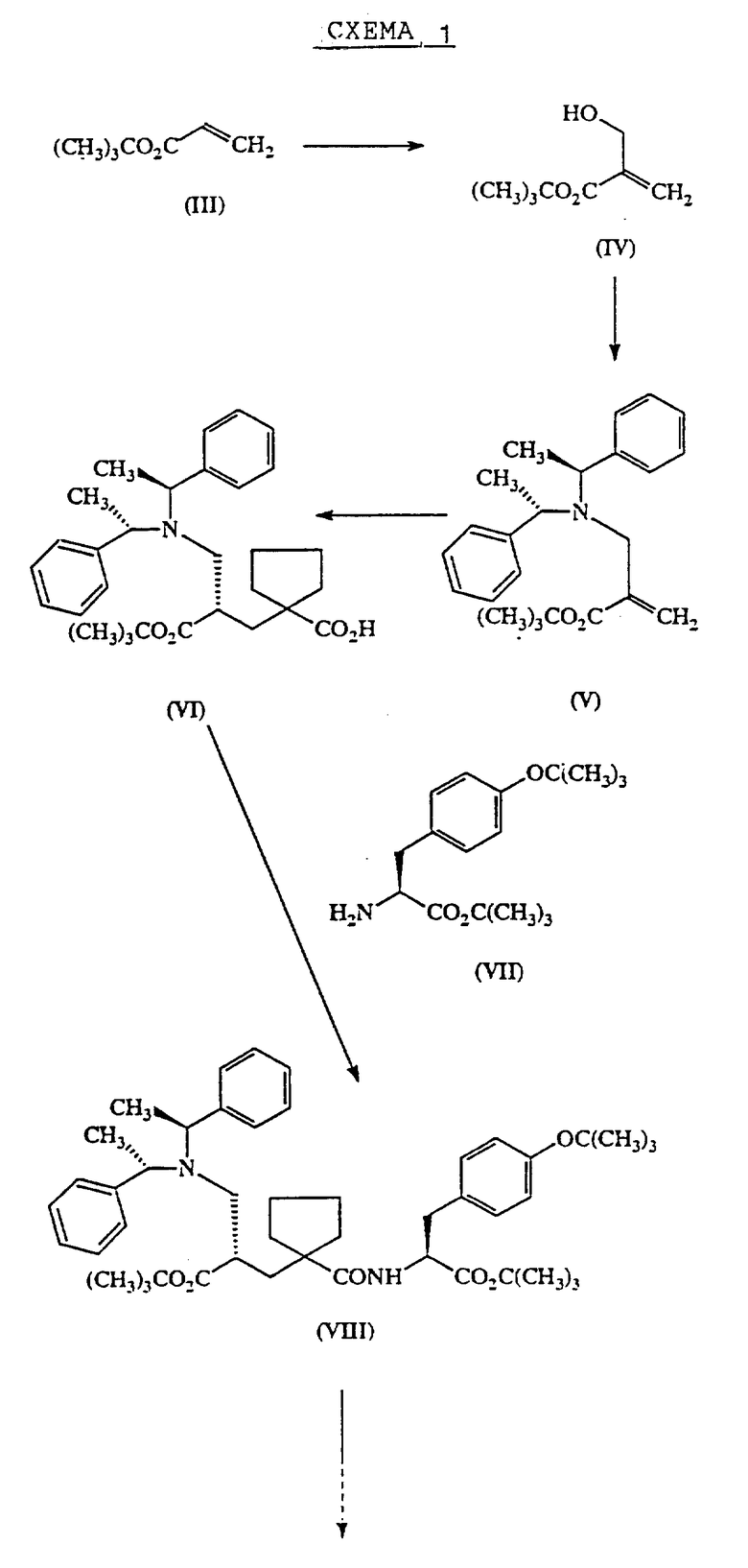
Соединение формулы (II) может быть получено путем, представленным на схеме 1 (см. в конце описания). Обычным способом, трет-бутилакрилат (III) подвергают взаимодействию с параформальдегидом в присутствии 3-хинуклидинола с получением трет-бутилдигидроксиметилакрилата (IV). Его сначала обрабатывают тионилхлоридом в присутствии триэтиламина и пиридина с получением соответствующего хлорметилакрилата, который затем подвергают взаимодействию с (S, S) -α-α′- диметилдибензиламином с получением акрилата формулы (V). Это соединение преобразуют в соединение формулы (IX) способом, представленным в Tet. Lett. , 1993, 34 (8), 1323-6. Соединение формулы (IX) затем конденсировали с производным лизина формулы (X) способом, аналогичным описанному в EP-A-0358398 для получения соединения формулы (XI). Соединение формулы (XI) затем преобразуют в соединение формулы (II), используя раствор трихлоруксусной кислоты и анизола в дихлорметане.
2) α-Форма может быть получена из δ-формы путем перемешивания раствора δ-формы в воде или в водном растворе подходящего органического растворителя, например C1 -C4 алканола, такого как метанол или изопропанол, или C3-C6 алканона, такого как ацетон.
По обычному способу δ-форму растворяют в смеси 1:5 вода/метанол или в смеси 1:10 вода/ацетон и раствор перемешивают в течение нескольких дней при комнатной температуре. α-Форма высаживается из раствора и может быть собрана фильтрацией.
3) α-Форма может быть получена из γ-формы путем перемешивания раствора γ-формы в воде или водном растворе подходящего органического растворителя, например C1-C4 алканола, такого как пропанол или изопропанол, или C3-C6 алканона, такого как ацетон.
По обычному способу γ-форму растворяют в смеси 1:1 вода/метанол и раствор перемешивают в течение около 17 часов при комнатной температуре. α-Форма высаживается из раствора и может быть собрана фильтрацией.
4) α-Форма может быть получена из β-формы по способу, аналогичному представленному выше в способе (3).
5) α-Форма может быть получена путем удаления защиты, предпочтительно в кислых условиях, у соединения формулы: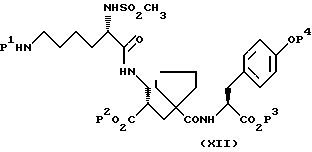
где P1, P2, P3 и P4 все являются подходящими защитными группами, которые способны к удалению, предпочтительно в кислых условиях, с получением после установления pH от 3 до 5, предпочтительно около 4, α-формы.
Подходящие защитные группы для этой цели и условия для их удаления хорошо известны специалисту, а именно см. T.W.Green, and P.G.Wuts, "Protective Groups in Organic Syntesis", Second Edition, Wiley-Interscience. P1 предпочтительно является формилом или бензилоксикарбонилом. P2, P3 и P4 каждый предпочтительно является трет-бутилом.
По обычному способу, где P1 представляет формил или бензилоксикарбонил и P2, P3 и P4 каждый представляет трет-бутил, раствор соединения формулы (XII) в подходящем растворителе, например 1,4-диоксане или этилацетате, обрабатывали подходящей кислотой, а именно соляной, для удаления защитных групп и установления pH до около 4 с получением α-формы.
Промежуточные продукты формулы (XII) могут быть получены общепринятыми способами. Соединение формулы (XII), где P1 является бензилоксикарбонилом и P2, P3 и P4 каждый является трет-бутилом, соответствует соединению формулы (XI) на схеме 1, синтез которого далее описан в способе (1). Соединение формулы (XII), где P1 представляет формил, P2, P3 и P4 каждый является трет-бутилом, может быть получено путем удаления сначала бензилоксикарбонильной группы из соединения формулы (XI) путем гидрирования с использованием подходящего катализатора, а именно палладия-на-угле, и последующего формилирования полученного амина, например, используя ангидрид муравьиной кислоты.
6) α-Форма может быть получена путем удаления защиты, предпочтительно в кислых условиях, у соединения формулы:
где P5 представляет подходящую защитную группу, которая способна к удалению в кислых условиях с получением после установления pH от 3 до 5, предпочтительно около 4, α-формы. Подходящие защитные группы для этих целей и условия для их удаления хорошо известны специалисту, например см. T.W. Green, and P. G.Wuts, "Protective Groups in Organic Syntesis", Second Edition, Wiley-Interscience. P5 предпочтительно является формилом и другими примерами P5 являются бензилоксикарбонил и третбутилоксикарбонил. По обычному способу, где P5 представляет формил, раствор соединения формулы (XIII) в подходящем растворителе, например 1,4-диоксане, обрабатывают подходящей кислотой, например соляной, для удаления защитной группы и установления pH около 4 с образованием α-формы.
Промежуточные продукты формулы (XIII) могут быть получены общепринятым способом, таким как селективное удаление защиты у соединения формулы (XII) с удалением только P2, P3 и P4 защитных групп. Например, когда P1 представляет формил, а P2, P3 и P4 каждый является трет-бутилом, защитная группа трет-бутил может быть селективно удалена путем обработки соединения формулы (XII) трифторуксусной кислотой в подходящем растворителе, например дихлорметане.
β-, γ- и δ-Формы, которые используют в качестве промежуточных продуктов для получения α-формы, могут быть получены следующим образом:
i) β-Форма может быть получена методом каталитического гидрирования раствора соединения формулы (II) в подходящем растворителе и в присутствии подходящего катализатора для удаления защитной группы, например палладия-на-угле. По обычному способу раствор соединения формулы (II) в водном этаноле гидрируют при около 414 kPa (60 psi) и комнатной температуре в присутствии катализатора палладия-на-угле. Катализатор затем удаляют фильтрацией либо концентрируют при пониженном давлении с получением пены, которую перемешивают с C3-C6 алканоном, например ацетоном, либо высушивают при заморозке с получением β-формы, которую можно собрать фильтрацией.
Если применяют обработку C3-C6 алканоном, это приготовление может также случайно дать α-форму.
(ii) δ-Форма может быть получена методом каталитического гидрирования раствора соединения формулы (II) в смеси подходящего не смешивающегося с водой органического растворителя, например этилацетата, и воды в присутствии подходящего катализатора для удаления защитной группы, например палладия-на-угле, с последующим удалением катализатора, отделением водного слоя и осаждением продукта из водного слоя, используя C1-C4 алканол, например метанол. По обычному способу раствор соединения формулы (II) в водном этаноле гидрируют при около 414 kPa (60 psi) и комнатной температуре в присутствии катализатора палладия-на-угле. Затем катализатор удаляют путем фильтрации, водную фазу отделяют от фильтрата, концентрируют при пониженном давлении до небольшого объема и выливают в метанол. δ-Форма медленно высаживается из раствора и может быть собрана путем фильтрации.
Это приготовление может также случайно дать α-форму.
(iii) β-Форма может быть получена путем сначала вымораживания водного раствора δ-формы и затем высушиванием при замораживании полученной твердой массы.
(iv) γ-Форма может быть получена путем перемешивания δ-формы с н-пропанолом или ацетонитрилом.
По обычному способу смесь перемешивают в течение около 24 часов при комнатной температуре и собирают γ-форму путем фильтрации.
(v) γ-Форма может быть получена путем перемешивания взвеси β-формы в ацетонитриле или н-пропаноле, обычно в течение 5 дней при комнатной температуре. γ-Форму собирают путем фильтрации.
(vi) γ-Форма может быть получена обработкой водного раствора δ-формы C3-C6 алканоном, например ацетоном.
По обычному способу водный раствор δ-формы выливают в энергично перемешиваемый избыток ацетона при комнатной температуре. γ-Форма высаживается из раствора и может быть собрана фильтрацией.
Это приготовление может также случайно дать α-форму.
(vii) β-Форма может быть получена путем высушивания при заморозке концентрированного водного раствора α-формы. По обычному способу получали концентрированный раствор α-формы в горячей воде, раствор фильтровали для удаления любого нерастворимого материала, затем охлаждали, замораживали и, наконец, высушивали с получением β-формы.
Как указывалось ранее, α-форма является эффективным ингибитором нейтральной эндопептидазы (E. C. 3.4.24.11). Этот фермент участвует в распаде многочисленных пептидных гормонов и пептидных аутокоидных субстанций, включая, в частности, предсердный натрийуретический фактор (ANF). Таким образом, α-форма, предотвращая деградацию ANF под действием нейтральной эндопептидазы E. C.3.4.24.11, может потенцировать биологические факторы ANF и поэтому использоваться в качестве диуретического, натрийуретического и противогипертонического агента при лечении многих заболеваний, включая гипертонию, сердечную недостаточность, грудную жабу, почечную недостаточность, хроническую почечную недостаточность, предменструальный синдром, периодические отеки, болезнь Меньера, гиперальдостеронизм (первичный и вторичный) и гиперкальциурию. Кроме этого, благодаря своей способности потенцировать эффекты ANF, α-форма используется при лечении глаукомы. Далее, в результате своей способности ингибировать нейтральную эндопептидазу E.C.3.4.24.11, α-форма может быть полезной при лечении астмы, воспаления, боли, эпилепсии, аффективных нарушений, слабоумия, старческого слабоумия, ожирения, желудочно-кишечных заболеваний (особенно диареи и синдрома раздражения кишечника) и гиперренинемии (повышенной секреции ренина) и регуляции секреции желудочной кислоты.
Активность по отношению к нейтральной эндопептидазе может быть определена, используя метод, основанный на определении, описанном Barclay, P.L., et al, Biochem. Biophys. Res. Comm., 1989, 164, 58 - 65. Метод включает определение концентрации соединения, требующегося для уменьшения на 50% степени высвобождения меченной радиоактивной меткой гиппуровой кислоты из гиппуринил-L-фенилаланил-L-аргинина под действием препарата нейтральной эндопептидазы из почек крысы.
Как указано выше, α-форма является также ингибитором ангиотензин превращающего фермента (АПФ). Как и ингибиторы АПФ, она будет полезной при лечении различных состояний, для которых известно, что ингибиторы АПФ являются полезными, включая гипотонию, сердечную недостаточность, ограниченные ишемические повреждения миокарда, защита почек от гиперфильтрационных нарушений, защита или обращение гипертрофии левого желудочка, улучшение памяти, контроль распознавательной функции, слабоумие и защита от реокклюзии после коронарной ангиопластики или операции по коронарному шунтированию. Ее активность относительно этого фермента может быть определена, используя метод, который основан на модификации определения, описанного Rohrbach, M.S., Anal. Biochem. , 1978, 84, 272. Метод включает определение концентрации соединения, требующегося для подавления на 50% степени высвобождения меченной радиоактивной меткой гиппуровой кислоты из гиппурил-L-гистидил-L-лейцина под действием ангиотензин превращающего фермента, выделенного из почек крысы.
Ингибирующая активность может также быть изменена in vivo после внутривенной инъекции анестезированным крысам, используя методы, описанные I.L. Natoff et al, Journal of Pharmacological Metods, 1981, 5 305 и D.M.Gross et al, J. Pharmacol. Exp. Ther., 1981, 216, 552. Определяются дозы ингибитора, которые требуются для подавления на 50% прессорного ответа, вызываемого внутривенной инъекцией ангиотензина I (50 нг болюс).
Активность α-формы, как диуретического агента, может быть определена путем измерения ее способности увеличивать выделение мочи и экскрецию ионов натрия у бодрствующих с AV-блоком собак, используя методы, описанные Alabaster, C.T., et al, Brit. J. Pharmacol., 1989, 98, 823P.
Противогипертоническая активность α-формы может быть оценена путем измерения понижения давления крови после приема или внутривенного введения у обедненных солью, подготовленных диуретиком, спонтанно гипертензивных крыс, у обедненных солью почечно гипертензивных собак или у дезоксикортикостеронацетат (ДОКА)/соль обработанных гипертензивных крыс.
При назначении животному при лечении гипертонии, сердечной недостаточности или почечной недостаточности пероральные дозы обычно будут в области от 1 - 500 мг ежедневно и предпочтительно 5 - 200 мг ежедневно для лечения людей, в расчете на взрослого пациента. Таким образом, для типичного взрослого пациента отдельные таблетки или капсулы содержат от 1 до 200 мг соединения в фармацевтически подходящем приемлемом наполнителе или носителе для одноразового введения или для многоразовых доз, один или несколько раз в день. Дозы для внутривенного введения могли бы быть от 0,01 до 50 мг, предпочтительно от 0,1 до 10 мг соединения на единичную дозу, как требуется. На практике врач будет определять действительную дозу, которая будет наиболее подходящей для отдельного пациента и будет изменяться в зависимости от возраста, веса, чувствительности конкретного пациента. Вышеуказанные дозы даны как примеры для среднего случая, но могут конечно быть отдельные случаи, где необходимыми являются более высокие или более низкие области доз, и это входит в объем данного изобретения.
При использовании для человека α-форма может быть введена сама по себе, но обычно вводится в смеси с фармацевтически приемлемым разбавителем или носителем, выбранным в соответствии с предлагаемым путем введения и стандартной фармацевтической практикой. Например, она может быть введена перорально в виде таблеток, содержащих такие добавки, как крахмал или двухосновный фосфат кальция, или в капсулах, или в овулах, либо сам по себе, либо в смеси с добавками, или в виде эликсира или суспензии, содержащей ароматизирующие или подкрашивающие агенты. Она может быть инъецирована парентерально, например внутривенно, внутримышечно или подкожно. Для парентерального введения наилучшее использование в виде стерильного водного раствора, который может содержать другие вещества, например достаточное количество солей или глюкозы для создания изотонического с кровью раствора.
α-Форма может быть введена совместно с другими агентами, которые используются для понижения кровяного давления, лечения сердечных состояний или почечной недостаточности. Так, например, она может быть совместно введена с сердечным стимуляторами, например дигиталисом, альфа-блокатором, например доксазозином, бета-блокатором, блокатором кальциевых каналов, например амлодипином, экзогенным ANF, активатором калиевых каналов или с другим диуретическим агентом, как будет определено врачом в отношении конкретного пациента или состояния заболевания.
Терапевтическое лечение путем использования α-формы, как это описано здесь, может подразумевать текущее или профилактическое лечение конкретного заболевания.
Таким образом изобретение далее включает:
(a) фармацевтическую композицию, содержащую α-форму, γ-форму или гидратированную δ-форму соединения формулы (I) вместе с фармацевтически приемлемым разбавителем или носителем;
(b) α-форму, γ-форму или гидратированную δ-форму соединения формулы (I) или его фармацевтическую композицию для использования в качестве лекарственного средства;
(c) применение α-формы, γ-формы или гидратированной δ-формы соединения формулы (I) или его фармацевтической композиции для получения лекарственного средства для лечения заболевания, которое зависит от ингибирования ангиотензин превращающего фермента и/или цинкзависимой нейтральной эндопептидазы E. C.3.4.24.11;
(d) использование по п. (c), где заболеванием является сердечно-сосудистое заболевание, такое как гипертония, сердечная недостаточность, почечная недостаточность или глаукома;
(e) способ лечения животных, включая человека, лечение заболевания, которое зависит от ингибирования ангиотензин превращающего фермента и/или цинкзависимой нейтральной эндопептидазы E.C.3.4.24.11, который включает введение указанным животным ингибирующего количества α-формы, γ-формы или гидратированой δ-формы соединения формулы (I), ингибиторов указанного фермента и/или указанной эндопептидазы, или фармацевтической композиции, содержащей их;
(f) способ по п. (e), где заболеванием является заболевание по п. (d);
(g) натриевая, калиевая, аммониевая или (C1-C4 алкил) аммониевая соль соединения формулы (II);
(h) γ-форма соединения формулы (I);
(i) гидратированная δ-форма соединения формулы (I);
(j) соединение формулы (XII) при условии, что P1 не является бензилоксикарбонилом, когда P2, P3 и P4 каждый является трет-бутилом;
(k) соединение формулы (XIII) при условии, что P5 не является бензилоксикарбонилом.
Получение α-формы проиллюстрировано следующими примерами.
Пример 1
(S, S, S)-N-(1-[2-карбокси-3-(N2-метиллизиламино)пропил]-1- циклопентилкарбонил)тирозин, α-форма
Раствор (S,S,S)-N-(1-[3-(N6-бензилоксикарбонил-N2- метиллизиламино)-2-карбоксипропил] -1-циклопентилкарбонил)тирозина в этилацетате (1190 мл) (часть раствора получена в соответствии со способом приготовления 9 и содержит 219 г исходного материала) встряхивали с раствором гидроксида натрия (23,1 мл) в воде (503 мл). Водную фазу отделили и гидрировали при 414 kPa (60 psi) и комнатной температуре над 5% катализатором палладий-на-угле (20 г) в течение 5 часов. Катализатор затем отфильтровывали и фильтрат доводили до pH 4 водным раствором 5 Н соляной кислоты и высаживали более твердое вещество. После гранулирования в течение 18 часов при комнатной температуре твердый продукт отфильтровывали, промывали водой и высушивали с получением указанного в заголовке соединения в виде твердого белого продукта (124,4 г), т. пл. 248 - 250oC.
Найдено, %: C 53,47; H 7,25; N 9,50.
C26H40N4O9S
Вычислено, %: C 53,41; H 6,90; N 9,58.
Пример 2
(S,S,S)-N-(1-[2-карбокси-3-(N2-метиллизиламино)пропил]-1- циклопентилкарбонил)тирозин, α-форма
Раствор (S,S,S)-N-(1-[2-карбокси-3-(N2-метиллизиламино)пропил]-1- циклопентилкарбонил)тирозингидрата (δ-форма, см. приготовление 2) (3,0 г) на смеси 1:5 вода/метанол (18 мл) или 1:10 вода/ацетон (33 мл) перемешивали в течение 3 дней при комнатной температуре. Полученное твердое вещество собирали путем фильтрации и высушивали с получением указанного в заголовке соединения в виде твердого белого продукта, т. пл. 246 - 8oC (по водно-метанольному способу), т. пл. 242 - 3oC (по водно-ацетоновому способу).
Пример 3
(S,S,S)-N-(1-[2-карбокси-3-(N2-метиллизиламино)пропил]-1- циклопентилкарбонил)тирозин, α-форма
Раствор (S,S,S)-N-(1-[2-карбокси-3-(N2-метиллизиламино)пропил]-1- циклопентилкарбонил)тирозин, γ-форма (см. приготовления 4, 5, 7 и 8) (0,5 г) растворяли в воде (4 мл) и добавляли к метанолу (4 мл). Конечный раствор перемешивали в течение 17 часов при комнатной температуре. Образовывалось белое твердое вещество, которое собирали путем фильтрации и высушивали с получением указанного в заголовке соединения в виде твердого белого продукта (0,43 г), т. пл. 250 - 252oC.
Пример 4
(S,S,S)-N-(1-[2-карбокси-3-(N2-метиллизиламино)пропил]-1- циклопентилкарбонил)тирозин, α-форма
(S,S,S)-N-(1-[2-карбокси-3-(N2-метиллизиламино)пропил]-1- циклопентилкарбонил)тирозин, β-форма (см. приготовления 1, 3 и 6) (0,5 г) растворяли в воде (4 мл) и добавляли к 4 мл метанола. Конечный раствор перемешивали в течение 17 часов при комнатной температуре. Образовывалось белое твердое вещество, которое собирали путем фильтрации и высушивали с получением указанного в заголовке соединения (0,43 г), т. пл. 249 - 251oC.
Пример 5
(S, S, S)-N-(1-[2-карбокси-3-(N2-метиллизиламино)пропил]-1- циклопентилкарбонил)тирозин, α-форма
К раствору соединения по приготовлению 12 (2,50 г, 3,20 ммоль) в 1,4-диоксане (20 мл) добавили раствор 1,4-диоксана (20 мл), насыщенного газообразным HCl. Через 30 минут прозрачный сначала раствор образует масло, которое перемешивали в течение 24 часов при комнатной температуре. Добавляли воду (20 мл) для получения прозрачного раствора, который перемешивали при комнатной температуре в течение 60 часов. Выпаривание конечного раствора при пониженном давлении давало масло, которое растворяли в воде и подщелачивали водным раствором гидроксида натрия до получения pH 4. Растворитель удалили выпариванием при пониженном давлении, а гранулирование конечного материала в метаноле давало более твердое вещество, которое собирали путем фильтрации и ресуспендировали в воде (4 мл) в течение ночи. Твердый остаток отфильтровывали и высушивали с получением указанного в заголовке соединения (0,97 г), т. пл. 225 - 230oC. ИК и ДРЛП анализы подтвердили, что продукт является требуемой α-формой.
Пример 6
(S, S, S)-N-(1-[2-карбокси-3-(N2-метиллизиламино)пропил]-1- циклопентилкарбонил)тирозин, α-форма
К раствору соединения препарата 13 (1,78 г) в диоксане (18 мл) добавляли водный раствор 4 М соляной кислоты (18 мл). Прозрачный водный раствор перемешивали при комнатной температуре в течение 60 часов и затем 18 часов при 35oC. Удаление растворителя при пониженном давлении дало 5,42 г материала, 4,22 г которого растворяли в воде (10 мл), раствор подщелачивали водным раствором гидроксида натрия до pH 4,0, вносили затравку соединения по примеру 1 и перемешивали при комнатной температуре в течение 18 часов. Полученный прозрачный раствор концентрировали до объема около 10 мл при пониженном давлении, разбавили метанолом (15 мл) и гранулировали в течение 48 часов. Твердое вещество собирали путем фильтрации и высушивали с получением указанного в заголовке соединения (1,25 г), т. пл. 232 - 235oC.
ИК и ДРЛП анализы подтвердили, что продукт является требуемой α-формой.
Пример 7
(S, S, S)-N-(1-[2-карбокси-3-(N2-метиллизиламино)пропил]-1- циклопентилкарбонил)тирозин, α-форма
К охлажденному (10oC) раствору трет-бутил (S,S,S)-N-(1-[3-(N6-бензилоксикарбокси-N2-метиллизиламино) -2-(третбутоксикарбонил)пропил] -1-циклопентилкарбонил)-O4- третбутилтирозината (13,3 г, 15 ммоль) в этилацетате (27 мл) добавляли 5,1 М раствор соляной кислоты в этилацетате (70 мл) (357 ммоль HCl). Через 30 минут исходный прозрачный раствор выделил смолистое вещество. Смесь перемешивали при комнатной температуре в течение 18 часов. Прозрачный раствор декантировали от смолистого вещества, а смолистое вещество растирали с этилацетатом (75 мл) до получения клейкого вещества. Декантирование и растворение повторяли 5 раз для получения гигроскопического твердого вещества, которое растворяли в воде (12 мл). Конечный водный раствор дважды промывали этилацетатом, подщелачивали водным раствором гидроксида натрия до pH 4,0, внесли затравку соединения по примеру 1 и перемешивали при 45 - 50oC в течение 42 часов. Белое твердое вещество собирали путем фильтрации, промывали водой и ацетоном и высушивали с получением соединения, указанного в заголовке (1,95 г), т. пл. 237 - 238oC. ИК и ДРЛП анализы подтвердили, что продукт является требуемой α-формой.
Следующие приготовления иллюстрируют получение некоторых промежуточных соединений, используемых при синтезе α-формы.
Приготовление 1
(S, S, S)-N-(1-[2-карбокси-3-(N2-метиллизиламино)пропил]-1- циклопентилкарбонил)тирозин, β-форма
Раствор (S,S,S)-N-(1-[3-(N6-бензилоксикарбонил-N2- метиллизиламино)-2-карбоксипропил] -1- циклопентилкарбонил)тирозина (см. приготовление 9) (371 г) в смеси 9:1 этанол/вода (2,225 л) гидрировали при 414 kПа (60 psi) и комнатной температуре на 10% катализаторе палладий-на-угле в течение 4 часов. Катализатор отфильтровывали, а фильтрат выпаривали с получением сырого продукта в виде пены. Это вещество перемешивали с ацетоном (3,13 л) в течение 24 часов с получением соединения, указанного в заголовке в виде белого аморфного продукта (283 г).
Найдено, %: C 52,97; H 7,02; N 8,97.
C26H40N4O9S
Вычислено, %: C 53,41; H 6,90; N 9,58.
Приготовление 2
(S,S,S)-N-(1-[2-карбокси-3-(N2-метиллизиламино)пропил]-1- циклопентилкарбонил)тирозин, (δ-форма)
Раствор (S,S,S)-N-(1-[3-(N6-бензилоксикарбонил-N2- метиллизиламино)-2-карбоксипропил] -1- циклопентилкарбонил)тирозина (см. приготовление 9) (351 г) на этилацетате (1300 мл) добавляли к воде (385 мл) и гидрировали двухфазную смесь при 414 kPa (60 psi) при комнатной температуре на 5% катализаторе палладий-на-угле (35 г) в течение 20 часов. Катализатор отфильтровывали, водную фазу отделяли и концентрировали до небольшого объема при пониженном давлении. Вязкий раствор вносили в метанол (2,85 л) и перемешивали при комнатной температуре в течение 18 часов, в течение которых происходило медленное осаждение твердого вещества. Твердое вещество гранулировали при 5 - 10oC в течение 2 часов, фильтровали, промывали метанолом и высушивали с получением указанного в заголовке в виде твердого белого продукта (178,1 г), т. пл. 168 - 171oC.
Найдено, %: C 51,37; H 7,47; N 9,06.
C26H40O9S•χH2O (где χ = 1)
Вычислено, %: C 51,81; H 7,02; N 9,30.
Содержание воды = 3,6% по весу, что определено согласно Karl Fischer анализу (χ = 1 требует 3,0% по весу).
Приготовление 3
(S,S,S)-N-(1-[2-карбокси-3-(N2-метиллизиламино)пропил]-1- циклопентилкарбонил)тирозин, β-форма
(S,S,S)-N-(1-[2-карбокси-3-(N2-метиллизиламино)пропил]-1- циклопентилкарбонил)тирозингидрат, (δ-форма см. приготовление 2) (20,0 г) растворили в воде (250 мл) при комнатной температуре и полученный прозрачный раствор замораживали, используя баню из сухой двуокиси углерода/ацетона. Твердая масса была высушена при заморозке с получением указанного в заголовке соединения в виде твердого белого продукта (19,0 г). Этот продукт медленно разлагался при температуре выше 155 - 170oC.
Приготовление 4
(S,S,S)-N-(1-[2-карбокси-3-(N2-метиллизиламино)пропил]-1- циклопентилкарбонил)тирозин, γ-форма
(S,S,S)-N-(1-[2-карбокси-3-(N2-метиллизиламино)пропил]-1- циклопентилкарбонил)тирозингидрат, (δ-форма, см. приготовление 2) (1,0 г) перемешивали или с н-пропанолом или с ацетонитрилом (10 мл) в течение 24 часов при комнатной температуре. В каждом случае полученное белое твердое вещество собирали путем фильтрации и высушивали с получением соединения, указанного в заголовке, т. пл. 172 - 176oC.
Приготовление 5
(S, S, S)-N-(1-[2-карбокси-3-(N2-метиллизиламино)пропил]-1- циклопентилкарбонил)тирозин, γ-форма
(S, S, S)-N-(1-[2-карбокси-3-(N2-метиллизиламино)пропил]-1- циклопентилкарбонил)тирозингидрат, (δ-форма, см. приготовление 2) (847,0 г) растворяли в воде (762 мл) и раствор разбавляли ацетоном (1,0 л). Этот раствор медленно добавляли к интенсивно перемешиваемому ацетону (18,05 л) при комнатной температуре и высаживали белое твердое вещество. Смесь перемешивали при комнатной температуре в течение 18 часов, а твердое вещество собирали путем фильтрации, промывали ацетоном и высушивали с получением указанного в заголовке соединения в виде твердого белого продукта (775 г), т. пл. 179 - 181oC.
Найдено, %: C 53,45; H 6,88; N 9,37; S 5,49.
C26H40N4O9S
Вычислено, %: C 53,41; H 6,90; N 9,58; S 5,48.
Приготовление 6
(S, S, S)-N-(1-[2-карбокси-3-(N2-метиллизиламино)пропил]-1- циклопентилкарбонил)тирозин, β-форма
(S, S, S)-N-(1-[2-карбокси-3-(N2-метиллизиламино)пропил]-1- циклопентилкарбонил)тирозин, α-форма (см. примеры 1 - 4) (4 г) добавляли к воде (200 мл) и смесь перемешивали при 90 - 95oC в течение 30 минут. Нерастворимый материал отфильтровывали, фильтрат далее разбавляли водой (50 мл) и охлаждали до комнатной температуры. После фильтрования для удаления легкого помутнения прозрачный фильтрат замораживали, используя баню из сухой двуокиси углерода/ацетона. Полученную твердую массу высушивали при замораживании с получением указанного в заголовке соединения в виде твердого белого продукта (3,0 г). Этот продукт медленно разлагается при температуре выше 155 - 165oC.
Приготовление 7
(S, S, S)-N-(1-[2-карбокси-3-(N2-метиллизиламино)пропил]-1- циклопентилкарбонил)тирозин, γ-форма
(S, S, S)-N-(1-[2-карбокси-3-(N2-метиллизиламино)пропил]-1- циклопентилкарбонил)тирозин, β-форма (см. приготовления 1, 3 и 6) (0,3 г) диспергировали в ацетонитриле (15 мл) и перемешивали в течение 5 дней. Конечное твердое белое вещество собрали путем фильтрации и высушивали при пониженном давлении с получением соединения, указанного в заголовке (0,26 г).
Приготовление 8
(S, S, S)-N-(1-[2-карбокси-3-(N2-метиллизиламино)пропил]-1- циклопентилкарбонил)тирозин, γ-форма
(S, S, S)-N-(1-[2-карбокси-3-(N2-метиллизиламино)пропил]-1- циклопентилкарбонил)тирозин, β-форма (см. приготовления 1, 3 и 6) (0,3 г) суспендировали в н-пропаноле (10 мл) и перемешивали в течение 5 дней. Конечное твердое белое вещество собирали путем фильтрации и высушивали при пониженном давлении с получением соединения, указанного в заголовке (0,26 г), т. пл. 175 - 180oC.
Приготовление 9
(S, S, S)-N-(1-[3-(N6-бензилоксикарбокси-N2- метиллизиламино)-2-(карбоксипропил]-1-циклопентилкарбонил)тирозин
Трет-бутил (S, S, S)-N-(1-[3-(N6-бензилоксикарбокси-N2- метиллизиламино)-2-(трет-бутоксикарбонил)пропил] -1-циклопентилкарбонил)- O4-трет-бутилтирозинат (404 г) растворяли в дихлорметане (810 мл). В одну часть по каплям в течение 10 минут добавляли анизол (769 г), а затем трифторуксусную кислоту (1,158 кг). По завершении добавления реакционную смесь оставляли перемешиваться при 35oC в течение 6 часов и затем при комнатной температуре перемешивали в течение ночи. После добавления воды (1000 мл) образовалось три слоя. Слои со дна и поверхности объединяли и растворяли в этилацетате (2 л) и конечный раствор промывали солевым раствором. Органическую фазу смешивали с солевым раствором, устанавливали pH до 3, оставляли разделяться на слои. Образовалось три слоя. Органическую фазу отделяли, обрабатывали этилацетатом и экстрагировали насыщенным водным раствором бикарбоната натрия (1,6 л) и солевым раствором (0,5 л). Объединенные водные слои промывали этилацетатом, затем подкисляли и экстрагировали этилацетатом с получением этилацетатного раствора (1,54 л) указанного в заголовке соединения. Этот раствор либо использовали непосредственно (например, см. пример 1), либо растворитель удаляли получением соединения, указанного в заголовке.
Приготовление 10
(S)-N6-Бензилоксикарбонил-N2-мезиллизин
(S)-N6-Бензилоксикарбониллизин (1,5 кг) суспендировали в метиленхлориде (7,5 л) и добавляли хлортриметилсилан (1,36 л) в течение 10 минут. Смесь нагревали с обратным холодильником в течение 30 минут с получением раствора, который охлаждали до 3oC перед одновременным прибавлением диизопропилэтиламина (1,87 л) и метансульфонилхлорида (435 мл) с такой скоростью, чтобы поддерживать температуру ниже 25oC. Реакционную смесь далее перемешивали 2,5 часа, затем вливали в 2 М водный раствор соляной кислоты. Слои разделяли и метиленхлоридную фазу промывали 2 М водным раствором соляной кислоты с последующим промыванием водой. Растворитель удаляли при пониженном давлении и заменяли н-бутилацетатом. Раствор охлаждали и конечный кристаллический материал собирали путем фильтрации, промывали н-бутилацетатом и высушивали при пониженном давлении с получением соединения, указанного в заголовке (1,63 кг), т. пл. 83,5 - 84oC. [α]
Найдено, %: C 50,23; H 6,40; N 7,76.
C15H22N2O6S
Вычислено, %: C 50,27; H 6,19; N 7,82.
1H-ЯМР (300 МГц, d6-ДМСО): δ = 1,23 - 1,78 (6H, м), 2,85 (3H, с), 2,98 (2H, кв), 3,80 (1H, дт), 5,00 (2H, с), 7,25 (1H, т), 7,30 - 7,43 (5H, м), 7,51 (1H, д) м.д.
Приготовление 11
Трет-бутил(S, S, S)-N-(1-[2-трет-бутоксикарбонил-2-(N2- метиллизиламино)пропил]-1-циклопентилкарбонил)-O4-трет- бутилтирозинат
К раствору трет-бутил(S,S,S)-N-(1-[3-(N6-бензилоксикарбокси-N2- метиллизиламино)-2-(трет-бутоксикарбонил)пропил] -1-циклопентилкарбонил)- O4-трет-бутилтирозината (48,64 г. 54,8 ммоль) в техническом метиловом спирте (1,0 л) прибавляют 5% палладия-на-угле (5 г) (увлажненный водой) и смесь гидрируют при 345 - 414 кПа (50 - 60 psi) при комнатной температуре в течение 19 часов. После удаления катализатора фильтрацией полученный раствор концентрируют при пониженном давлении с получением указанного в заголовке соединения в виде бесцветного масла (46,56 г), которое содержит этанол.
1H-ЯМР (300 МГц, CDCl3): δ 1,27 (9H, с), 1,41 (9H, с), 1,44 (9H, с), 1,45 - 1,62 (14H, широкий м), 1,8 - 2,05 (4H, широкий м), 2,21 (2H, м), 2,72 (2H, т), 2,79 (3H, широкий), 2,96 (3H, с), 3,0 (2H, м), 3,59 (1H, м), 3,96 (1H, т), 4,73 (1H, м), 6,43 (1H, дт), 3,89 (2H, дт), 7,09 (2H, дт), 7,51 (1H, дт) м.д.
Приготовление 12
Трет-бутил(S, S,S)-N-(1-[3-(N6-бензилоксикарбокси-N2- метиллизиламино)-2-(трет-бутоксикарбонил)пропил] -1-циклопентилкарбонил)- O4-трет-бутилтирозинат
Охлажденный (0oC) раствор ангидрида муравьиной кислоты в уксусной кислоте (полученной путем объединения 45,3 мл уксусного ангидрида и 22,8 мл муравьиной кислоты, нагревание полученного раствора до 50 - 60oC в течение 15 минут, затем охлаждение до 0oC) добавляли к раствору соединение по приготовлению 11 (27,3 г, 36,3 ммоль) в муравьиной кислоте (33,7 мл) при 0oC в течение более 10 минут. Раствор оставляли нагреваться при комнатной температуре в течение 45 минут и затем резко охлаждали на льду. Конечный раствор нейтрализовали водным раствором гидроксида натрия и экстрагировали дихлорметаном (х 2). Объединенные органические слои дважды промывали солевым раствором и выпаривали при пониженном давлении с получением соединения, указанного в заголовке в виде желтой пены (28,0 г).
1H-ЯМР (300 МГц, CDCl3): δ = 1,26 (9H, с), 1,41 (18H, с), 1,45 - 2,03 (16H, широкий м), 2,23 (H,широкий м), 2,97 (3H, с), 3,08 (2H, м), 3,28 (2H, м), 3,51 (1H, м), 3,98 (1H, широкий м), 4,73 (1H, кв), 5,57 (1H, широкий дт), 5,91 (1H, широкий), 6,32 (1H, дт), 6,90 (2H, дт), 7,08 (2H, дт), 7,29 (1H, широкий), 8,17 (1H, с) м.д.
Приготовление 13
(S, S, S)-N-(1-[2-карбокси-3-(N6-формил-N2- метиллизиламино)-1-циклопентилкарбонил)тирозин
К охлажденному (0oC) раствору соединения по приготовлению 12 (2,71 г, 3,46 ммоль) в дихлорметане (4,8 мл) добавляли трихлоруксусную кислоту (4,8 мл). Реакционную смесь оставляли прогреваться до комнатной температуры и перемешиваться в течение 48 часов. Затем смесь концентрировали при пониженном давлении с получением соединения, указанного в заголовке в виде твердого продукта (2,4 г), т. пл. 56 - 60oC.
1ЯМР (300 МГц, d6-МДСО): δ = 1,2 - 1,6 (14H, широкий м), 1,71 - 1,86 (3H, м), 1,86 - 1,99 (1H, м), 2,28 - 2,41 (1H, м), 2,78 (3H, с), 2,8 - 3,09 (4H, м), 3,12 - 3,25 (2H, м), 3,7 (1H, м), 4,35 (1H, м), 6,6 (2H, дт), 6,98 (2H, дт), 7,25 (1H, дт), 7,50 (1H, дт), 7,91 (2H, м), 7,97 (1H, с) м.д.
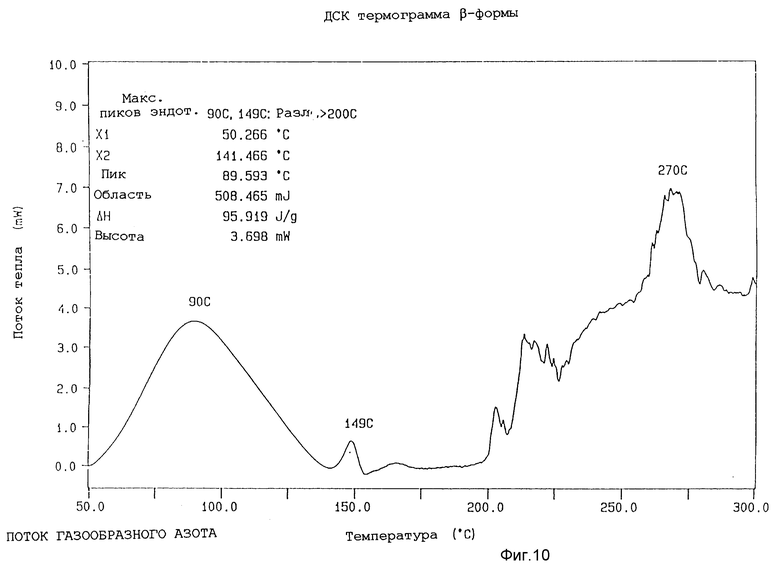
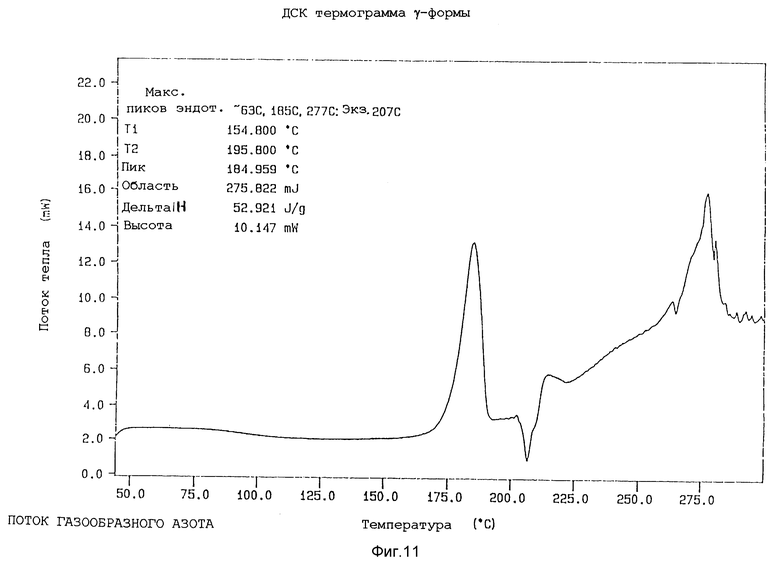
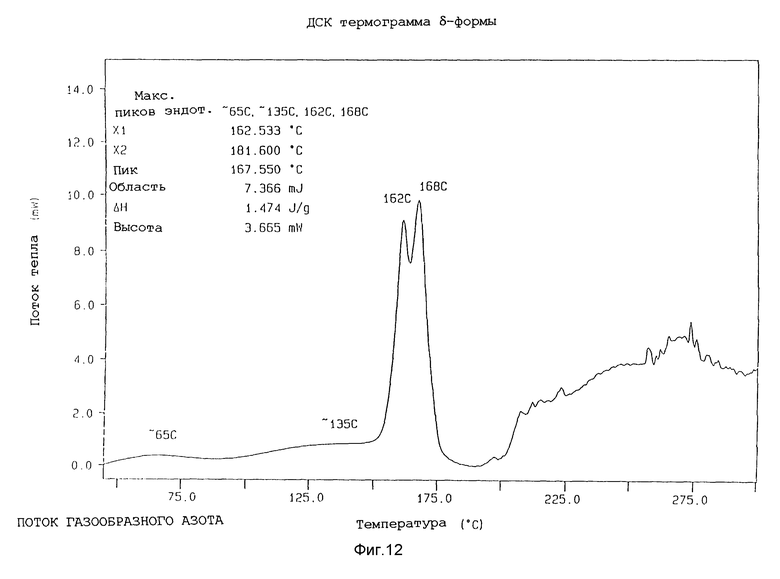
Характеристики α- β- γ- и δ-форм методами ИК, ДРЛП и ДСК анализа и по определению точки плавления
a) Инфракрасная спектроскопия (ИК)
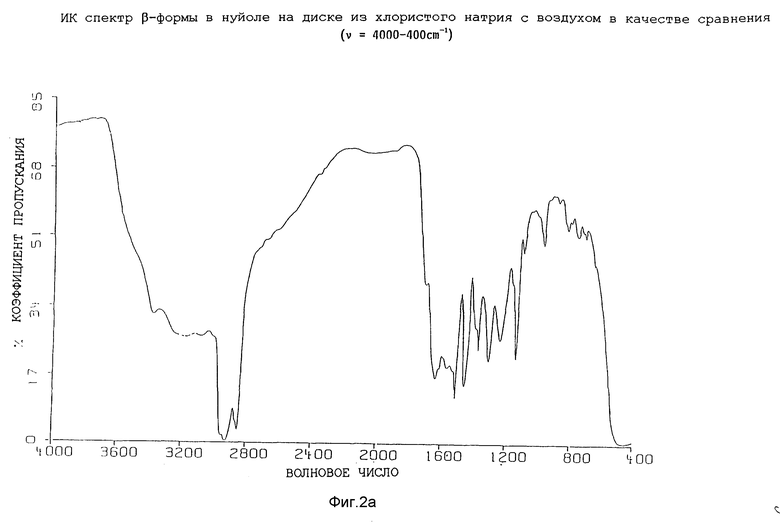
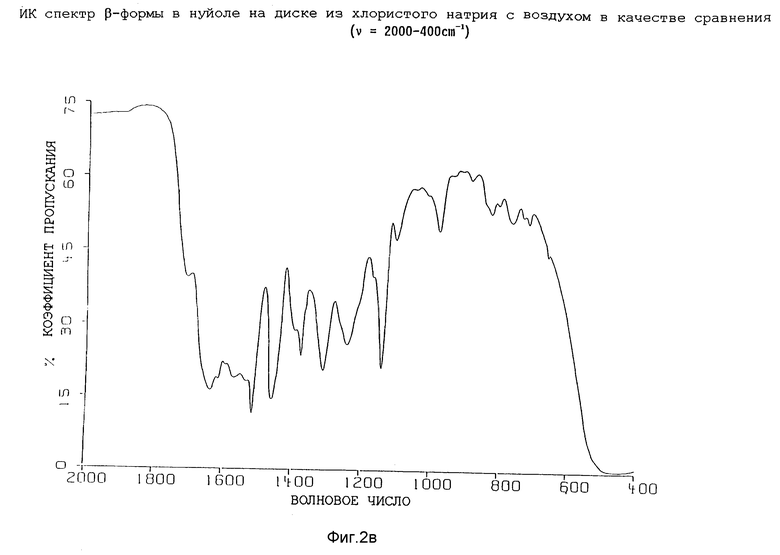
Инфракрасные спектры различных форм были определены в видах в минеральном масле (нуйол), используя Nicolet 800 FT-IR спектрометр. Для каждой формы волновые числа (ν [см-1]) полосы поглощения перечислены в таблице 1.
Представленные инфракрасные спектры для различных форм указаны на фиг. 1а, 1в, 2а, 2в, 3а, 3в, 4а и 4в.
b) Дифракция рентгеновских лучей на порошке (ДРЛП)
Картина дифракции рентгеновских лучей на порошке различных форм была получена при использовании Siemens D500 дифрактометра, который работает при 40 kV/30 mA, с использованием графитового монохроматора с медным радиационным фильтром (λ = 0,15405 нм) и сцинтилляционным датчиком-детектором. Для каждой формы интенсивность луча, выраженную в виде функции угла 2θ, записывали в интервале 2o - 45o 2θ, используя для подсчета шаг сканирования для шести секунд, как шаг интервала 0,03o2θ. Для каждой формы основные пики (степени 2θ), видные на картине, перечислены в таблице 2.
Представленные картины дифракции рентгеновских лучей на порошках различных форм показаны на фиг. 5 - 8.
c) Дифференциальная сканирующая колориметрия (ДКС)
Образцы (около 5 г) различных форм анализировали, используя температурный анализатор Perkin-Elmer 7-й серии, при скорости сканирования 20oC в минуту. Данные, полученные для различных форм, собраны в таблице 3.
Представленные термограммы ДСК для различных форм показаны на фиг. 9 - 12.
d) Точка плавления
Точку плавления различных форм определяли с помощью микроскопии горячей стадии, используя аппарат Mettler FP5/FP52 при скорости нагревания 2oC в минуту. Типичные интервалы, при которых расплавляются различные формы, представлены в таблице 4.
Сравнительные исследования
α- и β-Формы сравнили, используя технологическое и гигроскопическое исследования.
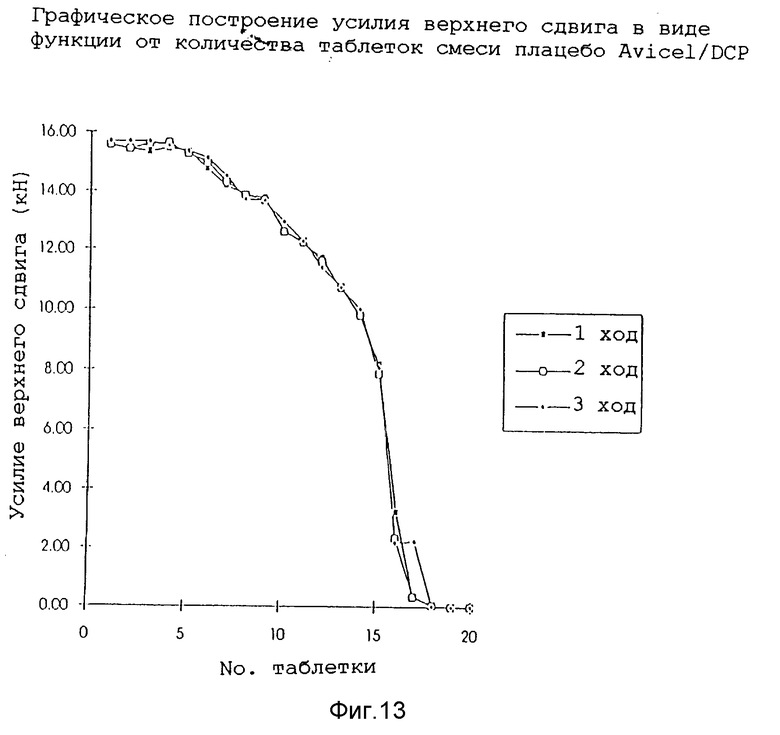
a) Технологическое исследование
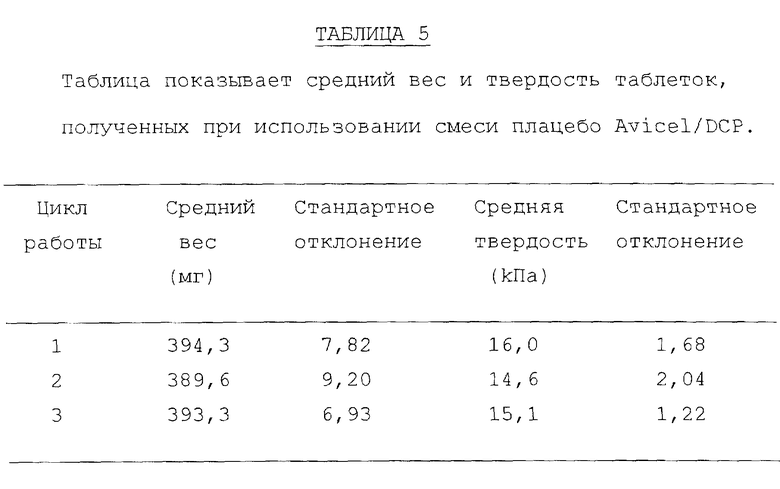
Инструментальную таблеточную машину (Manesty Machines Limited, Model F3) соответствующим образом откалибровали на усилие верхнего прессовочного сдвига.
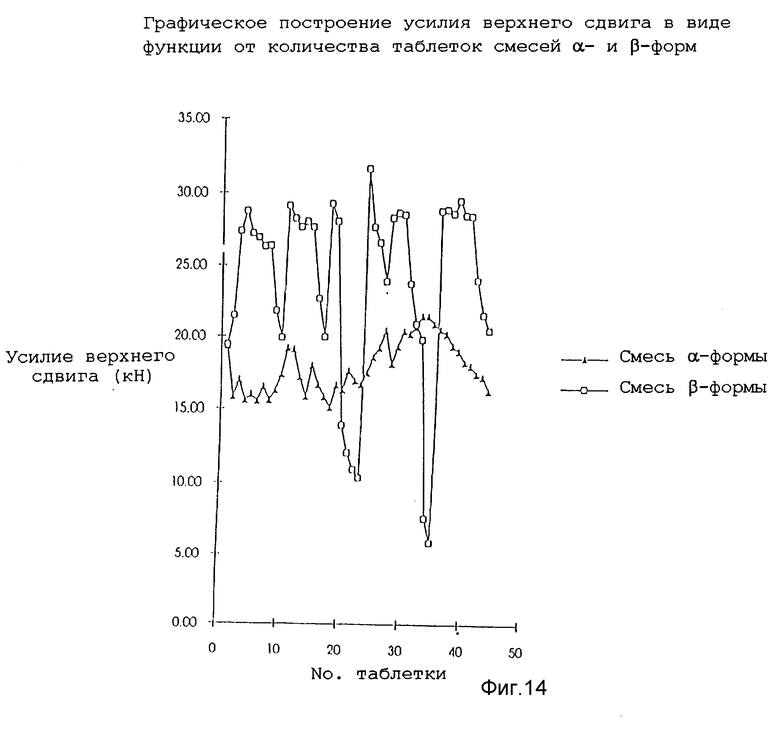
После калибровки машины подвергли технологической обработке смесь плацебо Avicel (торговая марка)/DCP (двухосновный фосфат кальция), используя 13 мм плоские лицевые штампы для измерения воспроизводимости технологии. Используя разные порции смеси, машину настроили подходящим образом для достижения усилия сжатия в мишени (400 мг) и достаточной твердости. Затем раздельно взвесили двадцать равных порций и загрузили в штамподержатель машины. Машиной управляли до тех пор, пока в штамподержателе не истощилась смесь и таблетки больше не производились. На фиг. 13 показан график зависимости усилия верхнего прессовочного сдвига в виде функции от количества таблеток для трех смесей плацебо Avicel/DCP для каждых двадцати комплектов и в таблице 5 показан средний вес и твердость десяти наиболее тяжелых таблеток (предположительно первых десяти произведенных). Как можно видеть из данных, представленных на фиг. 13, общие процессы для этих смесей были хорошо воспроизводимы. Снижение усилия верхнего прессовочного сдвига, которое наблюдалось в конце работы, может быть сопоставлено с уменьшением количества смеси в штамподержателе и соответствующим неполным заполнением пресс-формы.
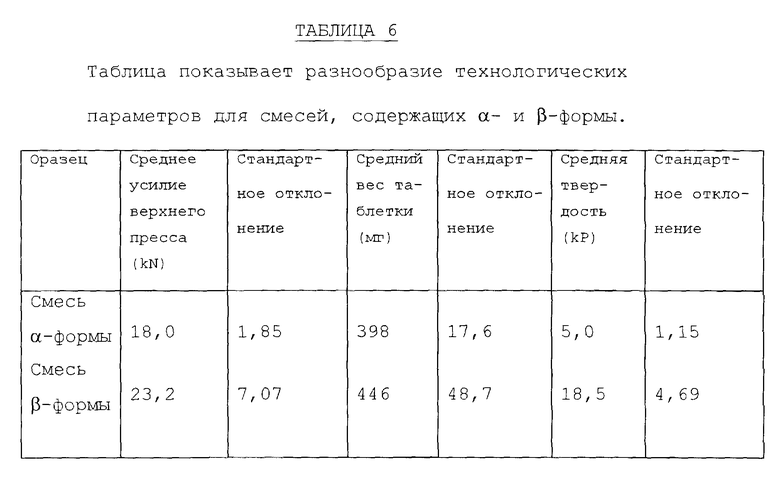
Для определения воспроизводимости технологии в последующих экспериментах раздельно приготовили смеси, содержащие α-форму и β-форму, согласно следующему составу: α или β-форма (100 мг), предварительно желатинизированный крахмал (40 мг), двухосновный фосфат кальция (безводная форма) (256 мг) и стеарат магния (2 мг). Для производства 20 г смеси была использована технология смешивания/просеивания/смешивания перед загрузкой машины. На основании предыдущего опыта загрузка составляла 100 мг, поскольку, чем более высокая загрузка, тем с большими технологическими трудностями приходилось сталкиваться. Машина была настроена таким образом для смеси, чтобы было произведено 50 таблеток из конкретной смеси в одной нераздельной партии.
Оптимизация машины была более сложной для смеси β-формы из-за ее плохих текучих свойств. Несмотря на внимательное управление отклонениями в процессе не представлялось возможным поддерживать усилие верхнего прессовочного сдвига на постоянном уровне между обоими смесями и соответственно смесь β-формы спрессовалась с более высокой твердостью.
Данные по верхнему сжатию обоих смесей представлены на фиг. 14. Значительная вариабельность усилия верхнего пресса (и веса таблеток) для смеси β-формы была связана с неравномерным заполнением пресс-формы данным составом. Данные, представленные в таблице 6, подтверждают, что производственная технология состава β-формы представляла большие сложности и была предметом большего разнообразия, чем при использовании состава α-формы.
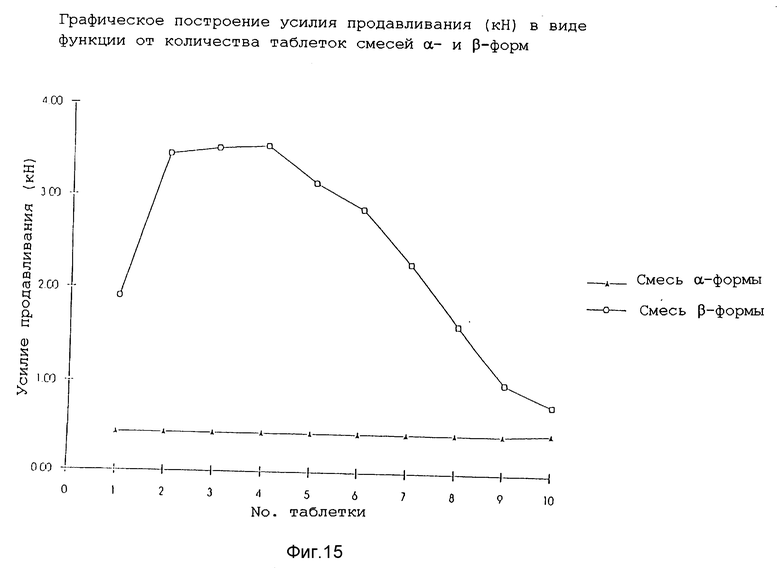
Измеренное усилие продавливания для последних десяти таблеток каждой смеси представлено на фиг. 15. Таблетки, полученные из β-формы, требовали значительно большего усилия для удаления из пресс-формы. Это показывает эффект выщелкивания таблеток самих по себе из пресс-формы штамподержателя.
Полученные данные демонстрируют плохие технологические свойства β-формы по сравнению с α-формой. β-Форма имеет низкую объемную плотность (пористая плотность = 0,09 г/мл, по сравнению с 0,36 г/мл у α-формы) и худшие текучие свойства, и если таблетируют смесь, содержащую ее, проявляется значительное разнообразие веса таблеток и требуется большее усилие продавливания. По отношению ко всему этому, α-форма проявляет превосходные свойства, делающие ее, в частности, применимой в фармацевтических свойствах.
(b) Исследование гигроскопичности
(i) Гигроскопичность α- и β-форм определяли методом гравиметрического анализа следующим образом. Образцы α- и β-форм раздельно помещали в сосуды Kilner (торговая марка) в следующие условия: 40oC; 40oC и 75% ОВ (относительной влажности); 40oC и 95% ОВ. Поглощение воды каждым образцом определяли гравиметрически в трипликатах через определенные интервалы времени.
Образцы β-формы, выдержанные при 40oC/75% ОВ или 40oC/95% ОВ в течение одного дня, претерпевали морфологическое изменение. Образцы β-формы, выдержанные при 40oC/95% ОВ в течение одного дня претерпевали небольшую потерю веса (предположительно, после увеличения веса в результате поглощения воды с последующим морфологическим изменением и затем потерей влажности), тогда как образцы, хранившиеся при 40oC/75% ОВ, приобретали в среднем 6% от своего нормального веса.
На фиг. 16 показаны результаты, полученные по проведению гравиметрических анализов. Было обнаружено, что α-форма не является гигроскопичной. Тогда, как было обнаружено, β-форма является очень гигроскопичной при 40oC/75% ОВ.
(ii) Эксперименты по микробалансу влажности α- и β-форм подтверждают, что α-форма не является гигроскопичной, тогда как β-форма была очень гигроскопичной. Образцы α- и β-форм раздельно помещали в аппараты при 40oC, чтобы установить равновесие с окружающей средой, перед тем как конкретный образец выдерживали в условиях повышенной относительной влажности, с периодами установления равновесия между каждыми увеличениями влажности. Результаты показаны на фиг. 17. Они отмечают, что не менее 8% по весу воды (относительно оригинального веса) было поглощено β-формой в течение эксперимента.
Было обнаружено и затем исследовано морфологическое изменение трансформации β-формы, подвергнутой испытанию при высокой влажности, из состояния с очень низкой объемной плотностью в плотное стекловидное твердое вещество.






Изобретение относится к кристаллической α-полиморфной форме соединения формулы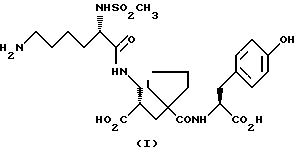
характеризующейся инфракрасным спектром в минеральном масле (нуйол), проявляющим полосы поглощения при ν = 3407, 3386, 3223, 3153, 1699, 1652, 1626, 1594, 1516, 1457 (нуйол), 1377 (нуйол), 1344, 1334, 1317, 1267, 1241, 1228, 1210, 1164, 1151, 1137, 1118, 1109, 1093, 1074, 1045, 1019, 1003, 981, 965, 911, 897, 862, 818, 800, 778, 762, 721 и 655 см-1, а также способу ее получения, промежуточным продуктам, используемым при ее получении, композициям, содержащим ее и к применению α- полиморфной формы, например композиции, ингибирующей ангиотензин. 23 c. и 20 з.п. ф-лы, 17 ил., 6 табл.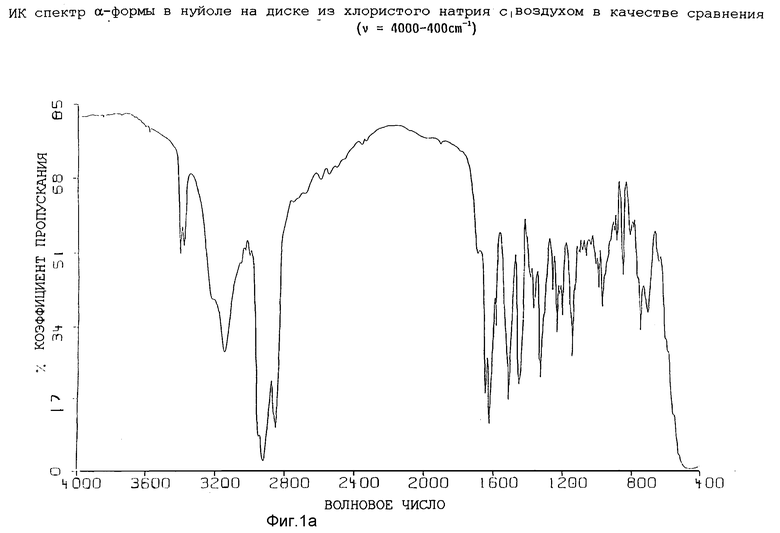
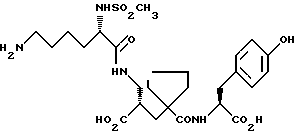
характеризующаяся инфракрасным спектром в минеральном масле (нуйол), проявляющим полосы поглощения при ν =3407, 3386, 3223, 3153, 1699, 1652, 1626, 1594, 1516, 1457 (нуйол), 1377 (нуйол), 1344, 1334, 1317, 1267, 1241, 1228, 1210, 1164, 1151, 1137, 1118, 1109, 1093, 1074, 1045, 1019, 1003, 981, 965, 911, 897, 862, 818, 800, 778, 762, 721 и 655 см-1.
3. γ-Полиморфная форма (S,S,S)-(N-(1-[2-карбокси-3-(N2-мезиллизиламино)пропил] -1-циклопентилкарбонил)тирозина формулы I, как определено в п. 1, характеризующаяся инфракрасным спектром в минеральном масле (нуйол), проявляющим полосы поглощения при ν =3377, 3240, 1665, 1639, 1594, 1527, 1518, 1494, 1457 (нуйол), 1443, 1377 (нуйол), 1344, 1321, 1304, 1254, 1195, 1178, 1162, 1143, 1111, 1098, 1046, 1031, 1012, 972, 962, 945, 932, 907, 879, 849, 815, 806, 780, 753, 729 и 658 см-1.
5. Гидратная δ -форма (S,S,S)-(N-(1-[2-карбокси-3-(N2-мезиллизиламино)пропил] -1-циклопентилкарбонил)тирозина формулы I, как определено в п. 1, характеризующаяся содержанием воды 1 - 7% по весу, как определено согласно Karl Fischer анализу и инфракрасным спектром в нуйоле, проявляющим полосы поглощения при ν =3667, 3425, 3380, 3287, 3137, 3098, 1709, 1673, 1637, 1619, 1596, 1568, 1556, 1516, 1458 (нуйол), 1448, 1419, 1390, 1378 (нуйол), 1356, 1338, 1300, 1270, 1249, 1229, 1198, 1174, 1141, 1108, 1091, 1075, 1064, 1045, 1033, 1019, 1001, 985, 962, 941, 909, 889, 877, 841, 822, 807, 763, 744, 732, 721 и 655 см-1.
7. Соединение по пп.5 и 6, которое имеет содержание воды 2 - 4% по весу, как определено согласно Karl Fischer анализу.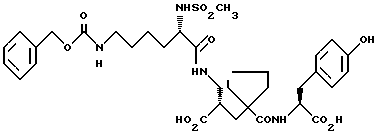
18. Натриевая соль соединения формулы II по п.17.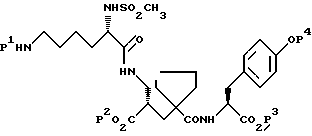
где P1, P2, P3, P4, которые могут быть одинаковыми или различными, все являются защитными группами, которые способны к удалению предпочтительно в кислых условиях, с получением соединения формулы I, как определено в п.1, при условии, что P1 не обязательно является бензилоксикарбонилом, когда P2, P3 и P4 каждый представляет трет-бутил.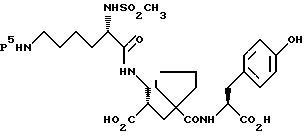
где P5 является защитной группой, которая способна к удалению предпочтительно в кислых условиях, с получением соединения формулы I, как определено в п.1, при условии, что P5 не является бензилоксикарбонилом.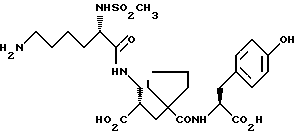
которое характеризуется инфракрасным спектром в минеральном масле (нуйол), проявляющим полосы поглощения при ν = 3407, 3386, 3223, 3153, 1699, 1652, 1626, 1594, 1516, 1457 (нуол), 1377 (нуйол), 1344, 1334, 1317, 1267, 1241, 1228, 1210, 1164, 1151, 1137, 1118, 1109, 1093, 1074, 1045, 1019, 1003, 981,965, 911, 897, 862, 818, 800, 778, 762, 721 и 655 см-1, который заключается в каталитическом гидрировании водного раствора натриевой, калиевой, аммониевой или (C1-C4 алкил) аммониевой соли соединения формулы (II)
используя катализатор, способный к удалению бензилоксикарбонильной защитной группы, с последующим подкислением полученной соли основания соединения формулы I до pH 3 - 5, предпочтительно около pH 4, с получением требуемой α-полиморфной формы.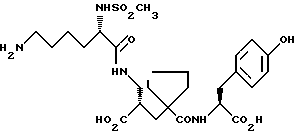
которое характеризуется инфракрасным спектром в минеральном масле (нуйол), проявляющим полосы поглощения при ν = 3407, 3386, 3223, 3153, 1699, 1652, 1626, 1594, 1516, 1457 (нуйол), 1377 (нуйол), 1344, 1334, 1317, 1267, 1241, 1228, 1210, 1164, 1151, 1137, 1118, 1109, 1093, 1074, 1045, 1019, 1003, 981, 965, 911, 897, 862, 818, 800, 778, 762, 721 и 655 см-1, который заключается в обработке гидратной δ-формы соединения формулы I, приведенной выше, характеризующейся содержанием воды 1 - 7% по весу, согласно определению Karl Fischer анализа, и инфракрасным спектром в нуйоле, проявляющим полосы поглощения при ν =3667, 3425, 3380, 3287, 3137, 3098, 1709, 1673, 1637, 1619, 1596, 1568, 1556, 1516, 1458 (нуйол), 1448, 1419, 1390, 1378 (нуйол), 1356, 1338, 1300, 1270, 1249, 1229, 1198, 1174, 1141, 1108, 1091, 1075, 1064, 1045, 1033, 1019, 1001, 985, 962, 941, 909, 889, 877, 841, 822, 807, 763, 744, 732, 721 и 655 см-1, водой водным раствором C1-C4-алканола, такого, как метанол или 2-пропанол, или водным раствором C3-C6 алканона, такого, как ацетон.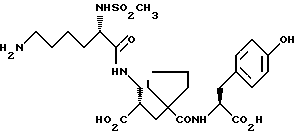
которое характеризуется инфракрасным спектром в минеральном масле (нуйол), проявляющим полосы поглощения при ν = 3407, 3386, 3223, 3153, 1699, 1652, 1626, 1594, 1516, 1457 (нуйол), 1377 (нуйол), 1344, 1334, 1317, 1267, 1241, 1228, 1210, 1164, 1151, 1137, 1118, 1109, 1093, 1074, 1045, 1019, 1003, 981, 965, 911, 897, 862, 818, 800, 778, 762, 721 и 655 см-1, который заключается в обработке γ-полиморфной формы соединения формулы (I), приведенной выше, характеризующейся инфракрасным спектром в минеральном масле (нуйол), проявляющим полосы поглощения при ν =3377, 3240, 1665, 1639, 1594, 1527, 1518, 1494, 1457 (нуйол), 1443, 1377 (нуйол), 1344, 1321, 1304, 1254, 1195, 1178, 1162, 1143, 1111, 1098, 1046, 1031, 1012, 972, 962, 945, 932, 907, 879, 849, 815, 806, 780, 753, 729 и 658 см-1, водой, водным раствором C1-C4-алканола, такого как метанол или 2-пропанол, или водным раствором C3-C6 алканона, такого, как ацетон. полиморфной формы соединения формулы I
полиморфной формы соединения формулы I
которое характеризуется инфракрасным спектром в минеральном масле (нуйол), проявляющим полосы поглощения при ν =3407, 3386, 3223, 3153, 1699, 1652, 1626, 1594, 1516, 1457 (нуйол), 1377 (нуйол), 1344, 1334, 1317, 1267, 1241, 1228, 1210, 1164, 1151, 1137, 1118, 1109, 1093, 1074, 1045, 1019, 1003, 981, 965, 911, 897, 862, 818, 800, 778, 762, 721 и 655 см-1, который заключается в обработке аморфной β-формы соединения формулы I, указанной в данном пункте, характеризующейся инфракрасным спектром в минеральном масле (нуйол), проявляющим полосы поглощения при ν =3384, 1708 1638, 1615, 1595, 1533, 1516, 1458 (нуйол), 1396, 1378 (нуйол), 1313, 1245, 1172, 1144, 1106, 980, 889, 830, 808, 737, 721 и 665 см-1, водой, водным раствором C1-C4-алканола, такого как метанол или 2-пропанол, или водным раствором C3-C6 алканона, такого, как ацетон.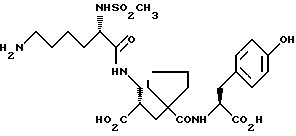
которое характеризуется инфракрасным спектром в минеральном масле (нуйол), проявляющим полосы поглощения при ν = 3407, 3386, 3223, 3153, 1699, 1652, 1626, 1594, 1516, 1457 (нуйол), 1377 (нуйол), 1344, 1334, 1317, 1267, 1241, 1228, 1210, 1164, 1151, 1137, 1118, 1109, 1093, 1074, 1045, 1019, 1003, 981, 965, 911, 897, 862, 818, 800, 778, 762, 721 и 655 см-1, который заключается в каталитическом гидрировании раствора соединения формулы II, указанного в данном пункте, в водном органическом растворителе в присутствии катализатора, способного к удалению бензоксикарбонильной защитной группы, удалении катализатора и растворителя из смеси, по существу полном удалении бензилоксикарбонильной группы и перемешивании остатка с C3-C6 алканоном, таким, как ацетон.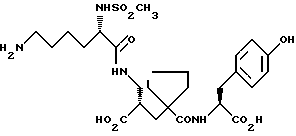
которое характеризуется инфракрасным спектром в минеральном масле (нуйол), проявляющим полосы поглощения при ν =3407, 3386, 3223, 3153, 1699, 1652, 1626, 1594, 1516, 1457 (нуйол), 1377 (нуйол), 1344, 1334, 1317, 1267, 1241, 1228, 1210, 1164, 1151, 1137, 1118, 1109, 1093, 1074, 1045, 1019, 1003, 981, 965, 911, 897, 862, 818, 800, 778, 762, 721 и 655 см-1, который заключается в удалении защиты, предпочтительно в кислых условиях, у соединения формулы XII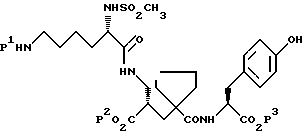
где P1, P2, P3 и P4, которые могут быть одинаковыми или различными, все являются защитными группами, которые способны к удалению, предпочтительно в кислых условиях, с последующим установлением pH 3 - 5, предпочтительно около 4, с получением требуемой α-полиаморфной формы.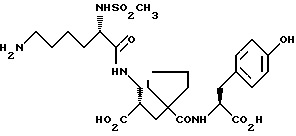
которое характеризуется инфракрасным спектром в минеральном масле (нуйол), проявляющим полосы поглощения при 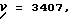 3386, 3223, 3153, 1699, 1652, 1626, 1594, 1516, 1457 (нуйол), 1377 (нуйол), 1344, 1334, 1317, 1267, 1241, 1228, 1210, 1164, 1151, 1137, 1118, 1109, 1093, 1074, 1045, 1019, 1003, 981, 965, 911, 897, 862, 818, 800, 778, 762, 721 и 655 см-1, который заключается в удалении защиты, предпочтительно в кислых условиях, у соединения формулы XIII
3386, 3223, 3153, 1699, 1652, 1626, 1594, 1516, 1457 (нуйол), 1377 (нуйол), 1344, 1334, 1317, 1267, 1241, 1228, 1210, 1164, 1151, 1137, 1118, 1109, 1093, 1074, 1045, 1019, 1003, 981, 965, 911, 897, 862, 818, 800, 778, 762, 721 и 655 см-1, который заключается в удалении защиты, предпочтительно в кислых условиях, у соединения формулы XIII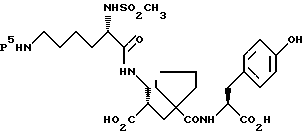
где P5 является защитной группой, которая способна к удалению, предпочтительно в кислых условиях, с последующим установлением pH 3 - 5, предпочтительно около 4, с получением требуемой α-полиаморфной формы.
38. Способ получения γ-полиморфной формы соединения формулы I, отличающийся тем, что включает обработку гидратной δ-формы соединения формулы I 1-пропанолом или ацетонитрилом.
| ЛИТЕЙНЫЙ СПЛАВ НА ОСНОВЕ АЛЮМИНИЯ | 0 |
|
SU358398A1 |
| Сдвигающий регистр | 1973 |
|
SU478363A1 |
| Экономайзер | 0 |
|
SU94A1 |
| Способ получения производных феноксиалкилкарбоновой кислоты,их солей,сложных эфиров и амидов | 1979 |
|
SU1052157A3 |
| Машковский М.Д | |||
| Лекарственные средства | |||
| - М.: Медицина, 1985, ч | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Способ получения твердых неплавких и нерастворимых продуктов уплотнения формальдегида с фонолами | 1925 |
|
SU435A1 |

