Данное изобретение относится к производным триазола, которые могут использоваться в терапии (в частности, для лечения грибковых инфекций людей и других млекопитающих), способу их применения, содержащим их препаратам и способам их получения.
Известно большое количество противогрибковых соединений триазола. Например, в Европейской патентной заявке 0440372, пример 7, описан (2R,3S)-2-(2,4-дифторфенил)-3-(5-фтор-4- пиримидинил)-1-(1H-1,2,4-триазол-1- ил)бутан-2-ол (так же известный, как вориконазол), который обладает особо хорошей активностью против клинически важных грибков Aspergillus spp. Однако это соединение имеет низкую растворимость в водной среде, что делает необходимым применение комплексообразующих агентов для получения удовлетворительных водных препаратов, таких как препараты для внутривенного введения. В Европейской патентной заявке 0440372 предлагаются совместные препараты с производными циклодекстрина для повышения растворимости; однако всегда желательно свести число компонентов в препарате до минимума для минимизации возможных вредных реакций у пациентов.
В патентной заявке Великобритании 2128193 описаны эфиры фосфорной кислоты для применения в качестве фунгицидов и инсектицидов растений.
Maurine и др. [Int. J. Pharm, 1993, 94 (1- 3), 11-14] описывают бисмезилат α -(2,4-дифторфенил)- α - [(l-(2-(3- пиридил)фенилэтенил)]-1H-1,2,4-триазол-1-этанола, который, как установлено, является противогрибковым агентом с высокой растворимостью.
Другие триазольные противогрибковые средства известны из Европейской патентной заявки 0576201 и Международной патентной заявки WO 97/01552.
В Европейской патентной заявке 0413674 описано получение пролекарств терапевтических ингибиторов гликозидазы путем фосфорилирования свободной гидроксильной группы этой молекулы. Однако фосфорилирование третичных гидроксильных групп не описано.
В настоящее время обнаружено, что противогрибковые соединения триазола типа соединений, содержащих третичную гидроксильную группу, включая (2R, 3S)-2-(2, 4-дифторфенил)-3-(5- фтор-4-пиримидинил)-1-(1H-1,2,4-триазол-1-ил)-бутан-2- ол, могут быть преобразованы в пролекарства со значительно большей растворимостью, которые легко преобразуются in vivo, давая желаемый активный радикал.
Согласно данному изобретению предложено соединение формулы I
R1-OP(O)(OH)2,
где R1 представляет негидроксильную часть противогрибкового соединения триазола типа соединения, содержащего третичную гидроксильную группу;
или его фармацевтически приемлемая соль (здесь обозначены как "соединения по изобретению").
Соединения по изобретению отличаются от известных соединений, так как третичная гидроксильная группа в противогрибковых соединениях триазола данного типа раньше не подвергалась функционализации.
Фармацевтически пригодные соли, которые можно указать, включают соли щелочных металлов фосфатной группы, например, динатриевые или дикалиевые соли; и соли с противоионом амина, например соли этилендиамина, глицина или холина.
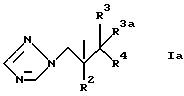
R1, предпочтительно, представляет группу формулы Ia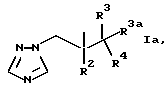
где R2 представляет фенил, замещенный одним или несколькими атомами галогена;
R3 представляет H или CH3;
R3a представляет H или вместе с R3 может представлять =CH2;
и R4 представляет 5- или 6-членное азотсодержащее гетероциклическое кольцо, которое необязательно замещено одной или несколькими группами, выбранными из галогена, =O, фенила [замещенного группой, выбранной из CN и (C6H4)-OCH2CF2CHF2] или CH= CH-(С6H4)-OCH2CF2CHF2; или фенил, замещенный одной или несколькими группами, выбранными из галогена и метилпиразолила.
Когда R1 представляет группу формулы Ia, как определено выше, R2, предпочтительно, является 2,4-дифторфенилом и R3, предпочтительно, является H или метилом.
Азотсодержащие гетероциклические кольца, которые может представлять или содержать R4, включают триазолил, пиримидинил и тиазолил.
Предпочтительные конкретные группы, которые может представлять R1, включают;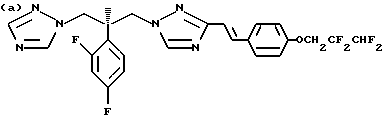
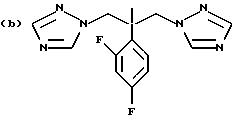
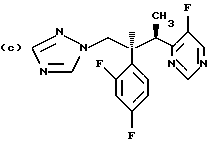
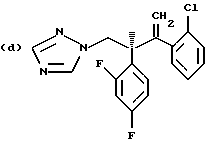
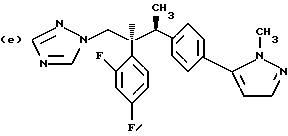
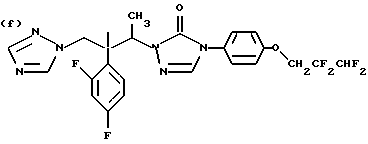
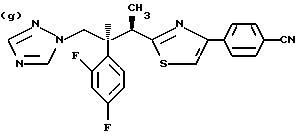
Противогрибковыми соединениями триазола, соответствующими указанным выше группам (а)-(g), являются:
(a) D-0870 (в разработке у Zeneca, смотри также пример 19 Европейской патентной заявки 0472392); (b) флуконазол (fluconazole) (продает Pfizer, смотри также патентную заявку Великобритании 2099818); (с) пример 7 Европейской патентной заявки 0440372, также известен как вориконазол (voriconazole); (d) пример 35 патентной заявки Великобритании 4952232; (е) соединение примера 8 настоящей заявки; (f) соединение А из WO 95/22973 (смотри стр. 29), первоначально описанное как соединение 30 в примере 27 ЕР 567982; и (g) ER- 30346 (смотри Drugs of the Future, 1996, 21(1): 20-24, Tetrahedron Letters, Vol. 37, 45, pp. 8117-8120, 1996 и Европейскую патентную заявку 0667346, пример 88).
Как указано выше, настоящее изобретение относится также к способу получения соединения формулы I или его фармацевтически приемлемой соли, который включает фосфорилирование соединения формулы II
R1OH,
где R1 определено выше;
и, когда желательно или необходимо, преобразование полученного соединения в фармацевтически приемлемую соль или обратно.
Фосфорилирование может быть осуществлено, используя следующие стадии (1)-(3):
(1) Взаимодействие соединения формулы II, указанной выше, с соединением формулы III
RaRbN-P(ORc)(ORd),
где Ra и Rb, независимо, представляют C1-6-алкил, фенил или замещенный фенил, или вместе с атомом азота, к которому они присоединены, они могут представлять кольцо, такое как морфолиновое кольцо; и Rc и Rd, независимо, представляют гидроксизащитные группы, выбранные из бензила, необязательно замещенного одним или несколькими атомами галогена; с получением фосфитного соединения формулы IV
R1-O-P(ORc) (ORd),
где R1, Rc и Rd определены выше.
Реакцию можно проводить в растворителе, который не оказывает вредного влияния на реакцию (например, метиленхлорид), в присутствии слабой кислоты (например, тетразол, 5- метилтетразол или гидробромид пиридиния) и, необязательно, 4- диметиламинопиридина при комнатной температуре или более высокой.
(2) Взаимодействие полученного фосфита формулы IV с окислителем (например, перкислота, такая как 3-хлорпероксибензойная кислота или H2O2) с получением фосфата формулы V
R1-OP(O)(ORc)(ORd),
где R1, Rc и Rd определены выше. Реакцию можно проводить в растворителе, который не оказывает вредного влияния на реакцию (например, метиленхлорид или этилацетат) при температуре ниже комнатной (например, 0 - -20oC).
(3) Удаление гидроксизащитных групп соединения формулы V с получением соединения формулы I, указанного выше.
В качестве альтернативы стадии (1) фосфиты формулы IV можно получить согласно стадии (1A) и (1B):
(1A) Взаимодействие указанного выше соединения формулы II с PCl3 в присутствии основания с получением постулированного промежуточного соединения формулы VI
R1-O-PCl2,
где R1 определено выше. Реакцию можно проводить в растворителе, который не оказывает вредного влияния на реакцию (например, метиленхлорид или этилацетат) при температуре в диапазоне от -20oC до +20oC (например, 0oC). Подходящие основания включают пиридин и N-метилимидазол.
(1B) Взаимодействие соединения формулы VI с соединением формулы RcOH и/или RdOH (где Rc и Rd определены выше), с получением указанного выше соединения формулы IV. Эту реакцию проводят без выделения соединения формулы VI при температуре около комнатной.
Гидроксизащитные группы, которые могут представлять Rc и Rd, включают 2,6-дихлорбензил и 2-хлор-6-фторбензил. Бензильные группы можно удалить путем каталитического гидрирования (например, над катализатором Перлмана "Pearlman's catalyst" или палладием-на- угле) или с помощью бромтриметилсилана.
Если стадию (3) проводят в присутствии ацетата натрия или гидроксида натрия, можно сразу получить динатриевую соль.
Указанная выше стадия (3) способа и промежуточные соединения формулы V являются дополнительными аспектами данного изобретения. Соединения формулы II и III либо являются известными, либо их можно получить, применяя известные методы.
Специалистам понятно, что в процессе синтеза соединения данного изобретения может потребоваться защита чувствительных функциональных групп и удаление защиты. Этого можно достичь общеизвестными способами, например, как описано Т. W. Green и P.G.M.Wuts "Protective Groups in Organic Synthesis", John Wiley and Sons Inc, 1991.
Соединения по изобретению могут использоваться для животных, включая человека, вследствие своей фармакологической активности. В частности, соединения могут использоваться при лечении или профилактике локальных грибковых инфекций у людей, вызванных, среди прочих организмов, штаммами Candida, Trichophyton, Microsporum или Epidermophyton или инфекцией слизистой оболочки, вызванных Candida albicans (например, кандидозного стоматита и вагинального кандидоза). Их также можно применять при лечении грибковых инфекций организма, вызванных, например, штаммами Candida (например, Candida albicans), Cryptococcus neoformans, Aspergillus flavus, Aspergillus fumigatus, Coccidioides, Paracoccidiodes, Histoplasma или Blastomyces.
Таким образом, согласно другому аспекту данного изобретения предложен способ лечения или профилактики грибковой инфекции, который включает введение пациенту терапевтически эффективного количества соединения по изобретению. Предложено также применение соединений по изобретению в качестве фармацевтических препаратов и применение соединений по изобретению при получении лекарственных препаратов для лечения или профилактики грибковых инфекций.
Оценку противогрибковой активности соединений данного изобретения in vitro проводили путем определения минимальной ингибирующей концентрации (м.и. к. ), которая является концентрацией исследуемого соединения в подходящей среде, при которой не происходит роста конкретного микроорганизма. На практике серии чашек с агаровой средой, каждая из которых включает конкретную концентрацию исследуемого соединения, засевают стандартной культурой, например, Candida albicans, и затем каждую чашку инкубируют в течение 48 часов при 37oC. Затем чашки исследуют на присутствие или отсутствие роста грибков и отмечают соответствующее значение минимальной ингибирующей концентрации, м. и.к. Другие используемые в этом тесте микроорганизмы могут включать Aspergillus fumigatus, Trichophyton spp., Microsporum spp., Epidermophyton floccosum, Coccidioides immitis и Torulopsis glabrata.
Некоторые соединения по изобретению, хотя и являются активными in vivo, могут не проявлять активность в этих тестах in vitro.
Оценить соединения данного изобретения in vivo можно по сериям доз при внутрибрюшинной или внутривенной инъекции или при оральном введении мышам, зараженным, например, штаммом Candida albicans или fumigatus. Активность основана на продолжительности жизни обработанной группы мышей после смерти мышей из необработанной группы. Отмечают величину дозы, при которой данное соединение обеспечивает 50% защиту против летального действия инфекции (PD50). Для моделей инфекции Aspergillus spp. количество мышей, прошедших курс лечения после установленной дозы, позволяет дополнительно оценить активность.
Людям можно вводить соединения по изобретению сами по себе, но обычно в смеси с фармацевтически пригодным носителем, выбранным в соответствии с предполагаемым способом введения и обычной фармацевтической практикой. Например, их можно принимать орально в виде таблеток, содержащих такой наполнитель, как крахмал или лактоза, или в виде капсул или драже, самих по себе или в смеси с наполнителем, или в виде эликсиров, растворов или суспензий, содержащих вкусовые или подкрашивающие агенты. Их можно вводить парентерально, например, внутривенно, внутримышечно или подкожно. Для парентерального введения лучше всего применять их в виде стерильных водных растворов, которые могут содержать другие вещества, например, соли или глюкозу в количестве, достаточном для достижения изотоничности раствора с кровью.
Ежедневная доза соединений данного изобретения для орального и парентерального введения людям составляет от 0,01 до 20 мг/кг (в виде разовой дозы или разделенной на несколько раз), когда введение производят орально или парентерально. Следовательно, таблетки или капсулы этих соединений содержат от 5 мг до 0,5 г активного соединения для введения одного соединения или двух или более одновременно, как предусмотрено. В любом случае врач определяет фактическую дозировку, которая наиболее подходит конкретному пациенту, и она меняется в зависимости от возраста, веса и реакции конкретного пациента. Указанная выше дозировка является примером среднего случая, конечно, могут быть отдельные случаи, когда предпочтительны диапазоны больших или меньших доз, и такие случаи входят в объем данного изобретения.
Альтернативно, соединения по изобретению можно вводить в виде суппозиториев или пессария, или их можно наносить наружно в виде лосьона, раствора, крема (например, включающего водную эмульсию полиэтиленгликолей или вазелинового масла), или их можно ввести в состав при концентрации от 1 до 10% мазей, содержащих основу из белого воска или бесцветного мягкого парафина вместе со стабилизаторами и консервантами, которые могут потребоваться.
Таким образом, согласно еще одному аспекту, данное изобретение относится к фармацевтической композиции, содержащей предпочтительно, менее 50 мас.% соединения по изобретению в смеси с фармацевтически пригодным адъювантом, разбавителем или носителем. Особый интерес представляют водные препараты для внутривенного введения.
Приведенные далее примеры иллюстрируют данное изобретение.
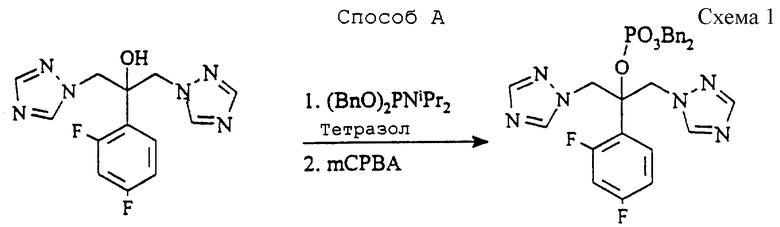
Пример 1. 2-(2,4-Дифторфенил)-1,3-бис(1H-1,2,4-триазол-1-ил)- 2-пропилдигидрофосфат
(а) Дибензил 2-(2,4-дифторфенил)-1,3-бис (1H-1,2,4-триазол- 1-ил) -2-пропилфосфат
Способ А (см. схему 1 в конце описания).
Раствор 2-(2,4-дифторфенил)1,3-бис(1H-1,2,4-триазол- 1-ил)-2-ола (известного так же, как флуконазол, 10,0 г, 32,6 ммоль), 1H-тетразола (6,85 г, 97,8 ммоль), дибензилдиизопропилфосфорамидита) (phosphoramidite) (22,55 г, 65,2 ммоль) в метиленхлориде (100 мл) перемешивают при комнатной температуре в атмосфере азота в течение 2 часов. Затем смесь охлаждают до 0oC и добавляют раствор 3-хлорпероксибензойной кислоты (13,5 г, 50-55% мас/мас, 39,1 ммоль) в метиленхлориде (50 мл), поддерживая температуру при 0oC. Полученной смеси дают нагреться до комнатной температуры в течение 1 часа, затем промывают водным метабисульфитом натрия и бикарбонатом натрия. После сушки (MgSO4) растворитель удаляют и заменяют метилизобутилкетоном (37 мл) и трет-бутилметиловым эфиром (74 мл). После гранулирования при -10oC в течение 1 часа продукт фильтруют, промывают охлажденными на льду метилизобутилкетоном и трет-бутилметиловым эфиром (1:3, 15 мл) и сушат при 50oC в вакууме в течение 18 часов, получая указанное в подзаголовке соединение (16,05 г, 87%), т.пл.93oC.
Найдено: С 57,12; H 4,46; N 14,85.
Рассчитано для C27H25F2N6O4P: С 57,24; H 4,46; N 14,84% m/z 567(MH+)
1H ЯМР (300 МГц, CDCl3) δ = 4,90 (д, 2H), 4,95 (д, 2H), 5,05 (д, 2H), 5,19 (д, 2H), 6,58-6,73 (м, 2H), 6,88-6,95 (м, 1H), 7,20-7,30 (м, 4H), 7,32-7,38 (м, 6H), 7,80 (с, 2H), 8,36 (с, 2H).
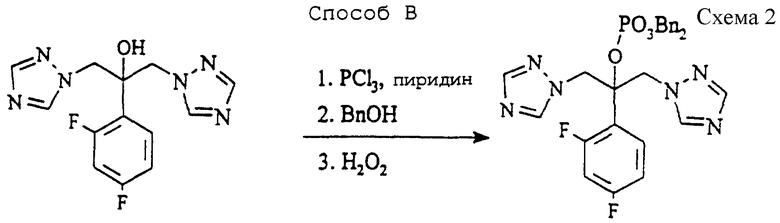
Способ В (см. схему 2 в конце описания).
К перемешиваемому этилацетату (1530 мл) добавляют 2-(2,4-дифторфенил)-1,3-бис(1H-1,2,4- триазол-1-ил)-2-ол (известный так же, как флуконазол, 306,0 г, 1,00 ммоль) и пиридин (237,3 г, 3,00 моль) перед охлаждением до 0oC. К реакционной смеси добавляют по каплям треххлористый фосфор (137,4 г, 1,00 моль), поддерживая температуру между 0 и 5oC, затем дают реакционной смеси нагреться до 15oC за 30 минут. Затем добавляют бензиловый спирт (216 г, 2,00 моль) за 30 минут при 15- 20oC. Еще через 30 минут добавляют перекись водорода (27,5% мас/мас в воде, 373 г), поддерживая температуру при 15-20oC. Через 30 минут отделяют водную фазу, и органическую фазу промывают водным метабисульфитом натрия, разбавленной соляной кислотой и водой. Растворитель удаляют при пониженном давлении и заменяют метилизобутилкетоном (850 мл) и трет- бутилметиловым эфиром (1132 мл). После гранулирования при 20oC в течение 1 часа и при 0oC в течение 1 часа продукт фильтруют и промывают охлажденными на льду трет-бутилметиловым эфиром (2х220 мл) и сушат при 50oC в вакууме в течение 18 часов, получая указанное в подзаголовке соединение (358 г, 63%). Точка плавления и спектроскопические данные идентичны данным, установленным в способе А.
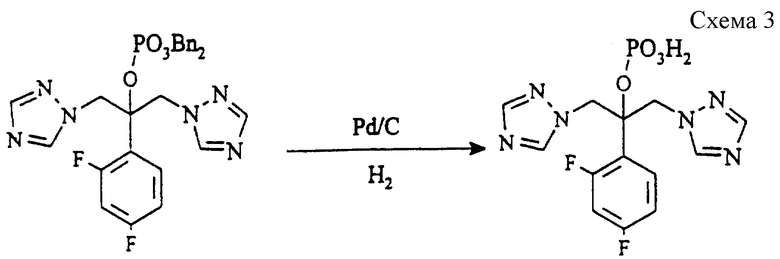
(б) 2-(2,4-Дифторфенил)-1,3-бис(1H-1,2,4- триазол-1-ил) -2-пропилдигидрофосфат (см. схему 3 в конце описания).
Пасту соединения стадии (а) (9,80 г, 17,3 ммоль), катализатора 5% палладия-на-угле (50% влажность, 1,0 г) и гидроксида натрия (1,38 г, 34,6 ммоль) в воде (26 мл) гидрируют при комнатной температуре и давлении 414 килопаскалей (60 psi = 4,219 кг/см2) в течение 20 часов. Раствор фильтруют через набивку из целита (торговая марка) и промывают водой (5 мл). Отделяют толуол и охлаждают водную фазу до 0oC, после чего добавляют серную кислоту (1,70 г, 17,3 ммоль). Полученную пасту гранулируют при 0oC в течение 1 часа и затем отфильтровывают, промывают водой (2х5 мл) и сушат в вакууме при 50oC, получая указанное в заголовке соединение (5,80 г, 87%), т.пл.223-224oC.
Найдено: С 40,28; H 3,39; N 21,63.
Рассчитано для C13H13F2N6O4P: С 40,43; H 3,39; N 21,76%.
1H ЯМР (300 МГц, ДМСО) δ = 5,07 (д, 2H), 6,77-6,83 (м, 1H), 7,00-7,18 (м, 2H), 7,75 (с, 2H), 8,53 (с, 2H).
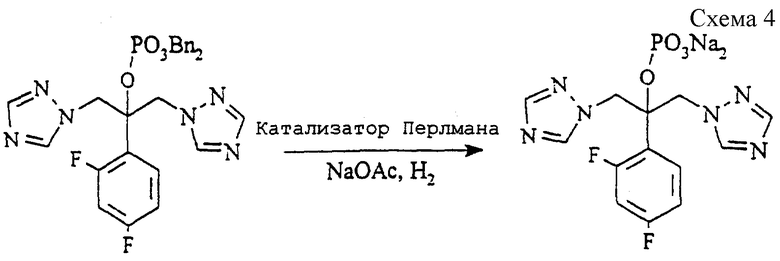
Пример 2. 2-(2,4-Дифторфенил)-1,3-бис (1H-1,2,4-триазол-1-ил) - 2-пропилдинатрийфосфат (см. схему 4 в конце описания).
Раствор соединения примера 1(a) (10,0 г, 17,7 ммоль) и ацетата натрия (2,90 г, 35,3 ммоль) в этаноле (160 мл) и воде (20 мл) гидрируют на катализаторе Перлмана (1,00 г) при комнатной температуре и давлении 345 килопаскалей (50 psi = 3,515 кг/см2) в течение 16 часов. Раствор фильтруют через набивку из целита (торговая марка), и растворители удаляют при пониженном давлении, оставляя густой сироп. Его растворяют в этаноле (100 мл) при помощи ультразвука и нагревают до кипения с обратным холодильником. Полученному раствору дают медленно остыть и гранулируют в течение 1 часа при комнатной температуре. Продукт отфильтровывают, промывают этанолом (10 мл) и сушат в вакууме при 50oC, получая указанное в заголовке соединение (4,48 г, 59%), т. пл.160-162oC.
1H ЯМР (300 МГц, D2O) δ = 5,01 (д, 2H), 5,40 (д, 2H), 6,60 (м, 1H), 6,79 (м, 1H), 7,11 (м, 1H), 7,63 (с, 2H), 8,68 (с, 2H).
Пример 3. (2R, 3S) 2-(2,4-дифторфенил)-3- (5-фтор-4-пиримидинил-1-(1H-1,2,4-триазол-1-ил)-2-бутилдигидрофосфат
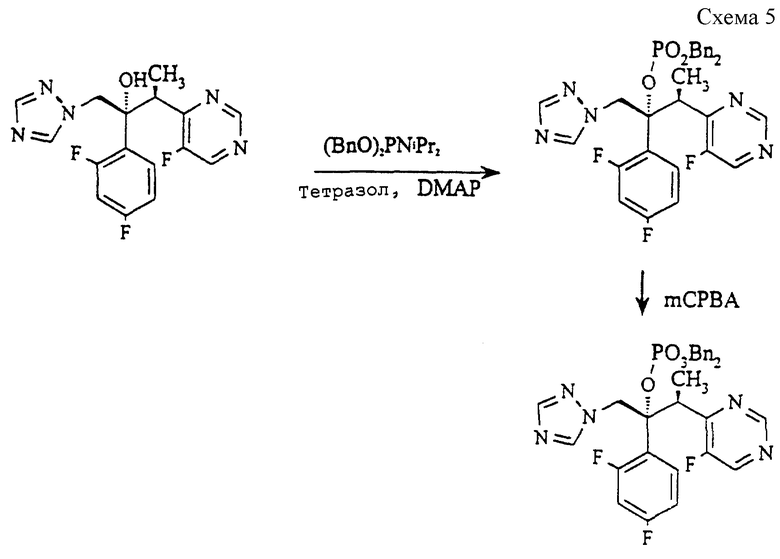
(а) Дибензил (2R, 3S) 2-(2,4-дифторфенил)-3-(5-фтор-4-пиримидинил)-1-(1H-1,2, 4-триазол-1-ил)-2-бутилфосфат (см. схему 5 в конце описания).
Раствор (2R, 3S) 2-(2,4-дифторфенил)-3-(5-фтор-4-пиримидинил)-1-(1H-1,2,4-триазол-1-ил)-2-ола (соединение примера 7. EP 0440372, известное так же, как вориконазол, 17,0 г, 48,7 ммоль), 4-диметиламинопиридина (10,2 г, 83,5 ммоль), 1H тетразола (10,2 г, 146 ммоль) и дибензилдиизопропилфосфорамидита) (33,6 г, 97,4 ммоль) в метиленхлориде (100 мл) перемешивают при кипячении с обратным холодильником в течение 2 часов и еще 16 часов при комнатной температуре в атмосфере азота. После этого реакционную смесь промывают соляной кислотой и затем бикарбонатом натрия, сушат (MgSO4) и концентрируют. Сырой продукт (фосфит) очищают методом колоночной хроматографии (силикагель - 300 г, элюирование гексаном:этилацетатом от 3:1 до 1:1), получая светло-желтое масло. Его растворяют в метиленхлориде (100 мл) и охлаждают до -10oC, после чего добавляют раствор 3-хлорпероксибензойной кислоты (14,8 г, 57% мас/мас, 48,9 ммоль) в метиленхлориде (100 мл), поддерживая температуру ниже 0oC. Полученной смеси дают нагреться до комнатной температуры в течение 10 минут и затем промывают водным метабисульфитом натрия и бикарбонатом натрия. После сушки (MgSO4) и концентрирования сырой продукт очищают методом колоночной хроматографии (силикагель - 300 г, элюирование этилацетатом), получая указанное в подзаголовке соединение в виде вязкого сиропа (17,86 г, 60%).
m/z 610 (MH+)
1H ЯМР (300 МГц, CDCl3) δ = 1,39 (д, 3H), 4,41 (квартет, 1H), 4,79 (д, 2H), 4,96 (д, 2H), 5,34 (д, 1H), 5,40 (д, 1H), 6,59-6,66 (м, 1H), 6,72-6,82 (м, 1H), 7,02-7,18 (м, 3H), 7,23-7,37 (м, 8H), 7,79 (с, 1H), 8,46 (д, 1H), 8,52 (с, 1H), 8,90 (д, 1H).
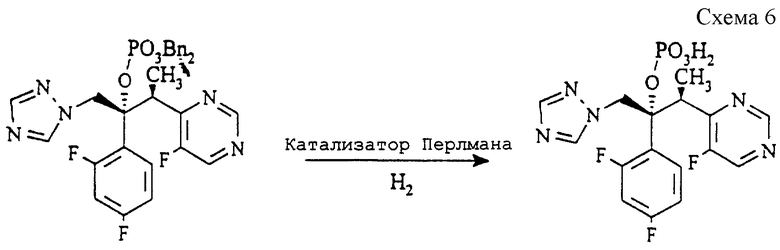
(б) 2R, 3S) 2-(2,4-Дифторфенил)-3-(5-фтор-4-пиримидинил-1-(1H-1,2,4-триазол-1-ил)-2-бутилдигидрофосфат (см. схему 6 в конце описания).
Раствор соединения со стадии (а) (5,0 г, 8,83 ммоль) в метаноле (100 мл) гидрируют на катализаторе Перлмана (Pearlman's catalyst) (1,0 г) при комнатной температуре и давлении 414 килопаскалей (60 psi = 4, 219 кг/см2) в течение 16 часов. Раствор фильтруют через набивку из целита (торговая марка) и концентрируют. Сырой продукт снова растворяют в горячем метаноле (20 мл) и гранулируют при 0oC в течение 1 часа. После фильтрования и промывания метанолом (5 мл) продукт сушат в вакууме при 50oC, получая указанное в заголовке соединение (1,72 г, 49%), т.пл.145-146oC.
1H ЯМР (300 МГц, ДМСО) δ = 1,31 (д, 3H), 4,01 (квартет, 1H), 5,31 (д, 1H), 5,42 (д, 1H), 6,90-6,97 (м, 1H), 7,04-7,14 (м, 1H), 7,20-7,30 (м, 1H), 7,95 (с, 1H), 8,70 (д, 1H), 8,73 (с, 1H), 8,89 (д, 1H).
Пример 4. (2R, 3S) 2-(2,4-дифторфенил)-3-(5-фтор-4-пиримидинил-1-(1H-1,2,4 -триазол-1-ил)-2-бутилдигидрофосфат
(Альтернативный способ получения)
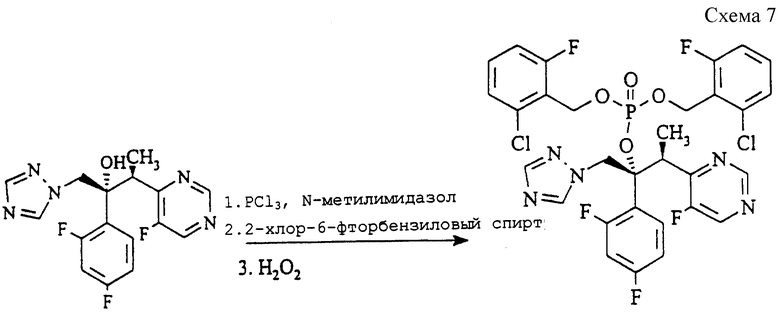
(а) Бис(2-хлор-6-фторбензил) (2R, 3S) 2-(2,4-дифторфенил)-3-(5-фтор-4-пиримидинил)-1-(1H-1,2,4-триазол-1-ил)-2- бутилфосфат (см. схему 7 в конце описания).
Раствор (2R, 3S) 2-(2,4-дифтopфeнил)-3-(5-фтop-4-пиpимидинил)-1-(1H-1,2,4-триазол-1-ил)-2-ола (соединение примера 7, ЕР 0440372, известное так же, как вориконазол, 10,0 г, 28,6 ммоль) и 1- метилимидазола (9,40 г, 114 ммоль) в метиленхлориде (30 мл) охлаждают до 0oC, после чего добавляют раствор треххлористого фосфора (4,73 г, 34,4 ммоль) в метиленхлориде (20 мл), поддерживая температуру ниже 10oC. Через 15 минут добавляют раствор 2-хлор-б-фторбензилового спирта (12,0 г, 74,4 ммоль) в метиленхлориде (40 мл) при температуре между 0 и 10oC. Через 30 минут добавляют по каплям перекись водорода (25 мл, 30% раствор в воде), поддерживая температуру ниже 20oC при охлаждении. Еще через 1 час реакционную смесь разделяют, и органическую фазу промывают водой (20х100 мл), сушат (MgSO4) и концентрируют. Полученное вязкое масло гранулируют с трет-бутилметиловым эфиром (60 мл) в течение 2 часов при 0oC. Продукт отфильтровывают, промывают трет-бутилметиловым эфиром (20 мл) и сушат при 50oC в течение 18 часов, получая указанное в заголовке соединение в виде белого твердого кристаллического вещества (18,1 г, выход 88%), т.пл. 140-141oC.
1H ЯМР (300 МГц, CDCl3) δ = 1,39 (д, 3H), 4,33 (квартет, 1H), 5,08 (д, 1H), 5,13 (д, 1H), 5,27 (д, 1H), 5,31 (д, 1H), 5,32 (д, 1H), 5,42 (д, 1H), 6,60-6,75 (м, 2H), 6,92-7,07 (м, 2H), 7,11-7,37 (м, 5H), 7,81 (с, 1H), 8,44 (д, 1H), 8,61 (с, 1H), 8,91 (с, 1H).
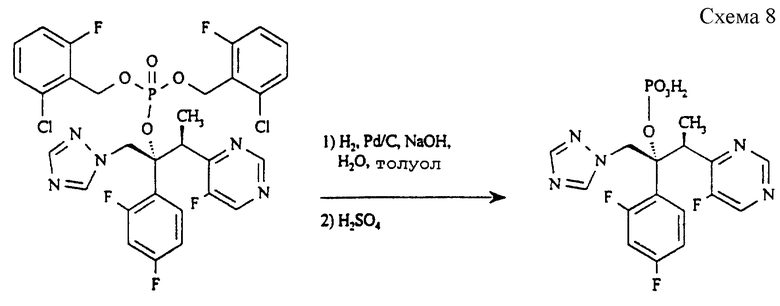
(б) (2R, 3S) 2-(2,4-Дифторфенил)-3-(5-фтор-4- пиримидинил-1-(1H-1,2,4-триазол-1-ил)-2-бутилдигидрофосфат (см. схему 8 в конце описания).
Смесь соединения со стадии (а) (50 г, 70 ммоль), гидроксида натрия (8,40 г, 210 ммоль) и катализатора 5% палладия-на-угле (10 г) в толуоле (450 мл) и воде (150 мл) гидрируют при комнатной температуре и давлении 414 килопаскалей (60 psi = 4,219 кг/см2) в течение 24 часов. Реакционную смесь фильтруют через целит (торговая марка), и толуольный слой отделяют и выбрасывают. Водный слой промывают метиленхлоридом (2х75 мл) и толуолом (2х75 мл) и затем охлаждают до 0oC, после чего добавляют серную кислоту (10,3 г, 105 ммоль). После гранулирования при 0oC в течение 1 часа продукт отфильтровывают, промывают водой (60 мл) и сушат в вакууме при 50oC в течение 16 часов, получая указанное в заголовке соединение (20,5 г, 68%). Данные протонного ЯМР идентичны данным, полученным в примере 3 (б).
Найдено: С 44,48; H 3,45; N 16,19.
Рассчитано для C16H15F3N5O4P: С 44,77; H 3,52; N 16,31%.
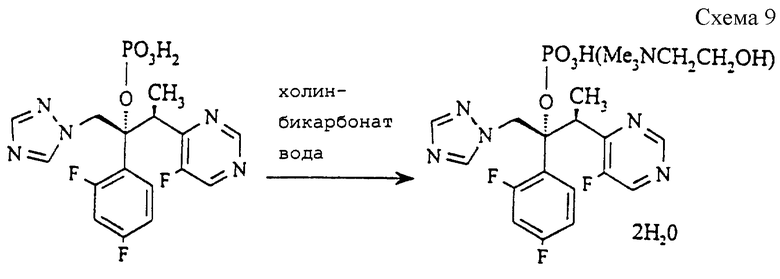
Пример 5. Дигидрат (2R, 3S) 2-(2,4-дифторфенил)-3-(5- фтор-4-пиримидинил-1-(1H-1,2,4-триазол-1-ил)-2-бутил (2- гидроксиэтил)триметиламмонийгидрофосфата (см. схему 9 в конце описания).
К перемешиваемой пасте из соединения примера 3(б) (214,7 г, 500 ммоль) в ацетоне (2070 мл) добавляют в течение 10 минут раствор холинбикарбоната (75% мас/мас в воде, 110 г, 500 ммоль), фильтруют через набивку из целита (торговая марка) для удаления нерастворимых продуктов, затем охлаждают до 20oC и гранулируют в течение 1 часа. Полученный продукт собирают фильтрацией, промывают ацетоном (2х250 мл) и сушат при 20oC в вакууме в течение 18 часов, получая указанное в заголовке соединение (233,3 г, 74%), т.пл. 114- 115oC.
1H ЯМР (300 МГц, ДМСО) δ = 1,23 (д, 3H), 3,07 (с, 9H), 3,38 (т, 2H), 3,60 (квартет, 1H), 3,78 (квартет, 2H), 5,50 (с, 2H), 6,72-6,80 (м, 1H), 6,94-7,02 (м, 1H), 7,36-7,42 (м, 1H), 7,82 (с, 1H), 8,59 (д, 1H), 8,78 (д, 1H), 9,35 (с, 1H).
Пример 6. 2-(2,4-Дифторфенил)-1-{ 3-[(Е)-4- (2,2,3,3-тетрафторпропокси)стирил] -1H-1,2,4-триазол-1-ил} -3-(1H-1,2,4-триазол-1-ил)-2-пропилдигидрофосфат
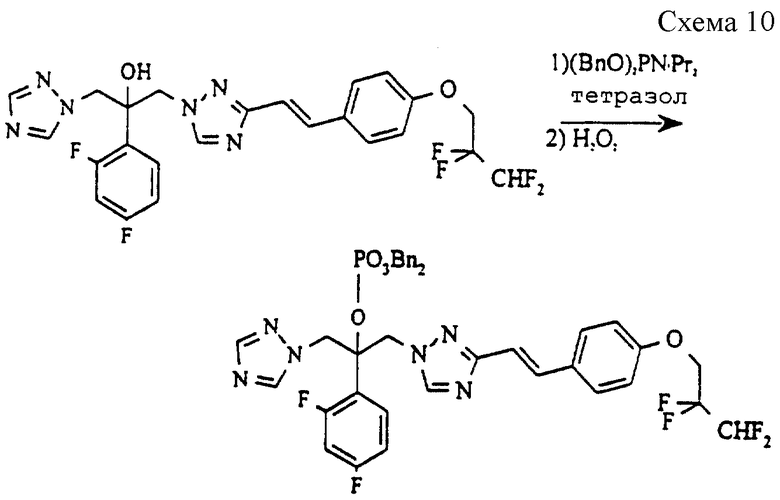
(а) (Дибензил) 2-(2,4-дифторфенил)-1-{3- [(Е)-4-(2,2,3,3-тетрафторпропокси) стирил] -1H-1,2,4-триазол-1-ил} -3- (1H-1,2,4-триазол-1-ил) -2-пропилфосфат (см. схему 10 в конце описания)
2-(2,4-Дифторфенил)-1-{ 3-[(E)-4-(2,2,3,3- тетрафторпропокси)стирил] -1H-1,2,4-триазол-1-ил} -3-(1H-1,2, 4-триазол-1-ил)пропан-2-ол (рацемат по примеру 19, ЕР 0472392, 475 мг, 0,88 ммоль), 1H-тетразол (185 мг, 2,64 ммоль) и дибензилдиизопропилфосфорамидит (607 мг, 1,76 ммоль) в метиленхлориде (5 мл) перемешивают при комнатной температуре в атмосфере азота в течение 20 часов. Затем смесь охлаждают до 0oC и добавляют по каплям перекись водорода (1,0 мл, 30% раствор в воде), поддерживая температуру ниже 20oC. Полученную смесь перемешивают при 20oC в течение 30 минут, затем отделяют органический слой, который промывают водой, сушат (MgSO4), и выпаривают растворитель. Полученное светло-желтое масло очищают методом колоночной хроматографии (силикагель, элюирование этилацетатом/гексаном), получая указанное в подзаголовке соединение в виде вязкого сиропа (595 мг, 84%).
1H ЯМР (300 МГц, CDCl3) δ = 4,37 (т, 2H), 4,91 (д, 2H), 4,97 (д, 2H), 5,02 (д, 1H), 5,07 (д, 1H), 5,16 (д, 1H), 5,18 (д, 1H), 6,05 (тт, 1H), 6,59-6,78 (м, 2H), 6,82 (д, 1H), 6,90 (д, 2H), 6,91-7,00 (м, 1H), 7,21-7,38 (м, 10H), 7,42 (д, 2H), 7,42 (д, 1H), 7,79 (с, 1H), 8,28 (с, 1H), 8,39 (с, 1H).
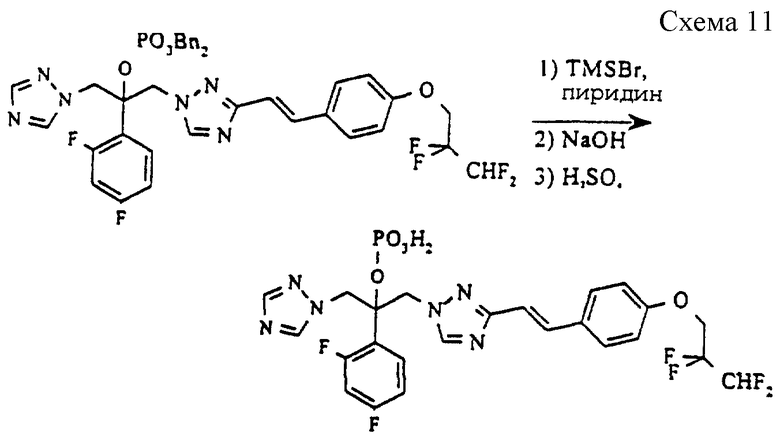
(б) 2-(2,4-Дифторфенил)-1-{3-[(Е)-4-(2,2, 3,3-тетрафтор-пропокси)стирил] -1H-1,2,4-триазол-1-ил} -3- (1H-1,2,4-триазол-1-ил)-2-пропилдигидрофосфат (см. схему 11 в конце описания).
Раствор соединения стадии (а) (298 мг, 0,37 ммоль) в метиленхлориде (5 мл) охлаждают до 0oC и затем обрабатывают бромтриметилсиланом (254 мг, 1,66 ммоль) и пиридином (180 мг, 3,10 ммоль). Полученную смесь перемешивают при 0oC в течение 3 часов и затем резко охлаждают водой (1 мл), содержащей гидроксид натрия (96 мг, 2,41 ммоль). Затем смесь подкисляют разбавленной серной кислотой, и продукт экстрагируют в этилацетат. После промывания солевым раствором фазу этилацетата сушат (MgSO4) и выпаривают растворитель, получая указанное в заголовке соединение в виде светло-желтой пены (202 мг, 88%).
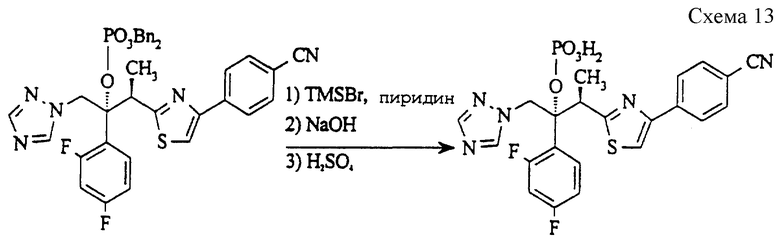
Пример 7. (2RS, 3RS)-3-(4-[4-цианфенил]тиазол-2-ил)-2-(2,4- дифторфенил)-1-(1H-1,2,4-триазол-1- ил)-2-бутилдигидрофосфат
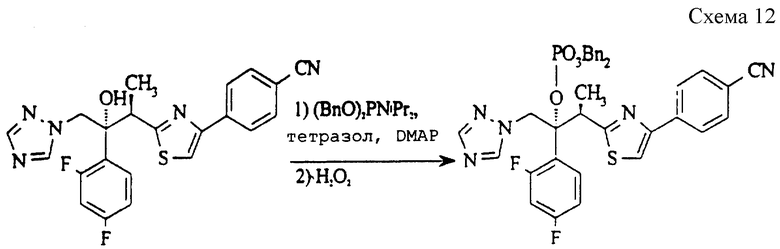
(а) Дибензил (2RS, 3RS)-3-(4-[4-цианфенил]тиазол-2-ил)-2- (2,4-дифторфенил)-1-(1H-1,2,4-триазол-1-ил)-2-бутилфосфат (см. схему 12 в конце описания).
3-[4-(4-Цианфенил)тиазол-2-ил] -2- (2,4-дифторфенил)-1-(1H-1,2,4-триазол-1-ил)бутан-2-ол (пример 88, ЕР 0667346, 900 мг, 2,06 ммоль), 1H-тетразол (432 мг, 6,18 ммоль), 4-диметиламинопиридин (100 мг, 0,82 ммоль) и дибензилдиизопропилфосфорамидит (1,42 г, 4,12 ммоль) в метиленхлориде (10 мл) кипятят с обратным холодильником в атмосфере азота в течение 20 часов. Затем смесь охлаждают до 0oC и добавляют по каплям перекись водорода (2,5 мл, 30% раствор в воде), поддерживая температуру ниже 20oC. Полученную смесь перемешивают при 20oC в течение 30 минут и затем отделяют органический слой, который промывают водой, сушат (MgSO4) и выпаривают растворитель. Полученное светло-желтое масло очищают методом колоночной хроматографии (силикагель, элюирование этилацетатом/гексаном), получая указанное в подзаголовке соединение в виде вязкого сиропа (732 мг, 51%).
1H ЯМР (300 МГц, CDCl3) δ = 1,40 (д, 3H), 4,38 (квартет, 1H), 4,81-4,96 (м, 4H), 5,40 (д, 1H), 5,43 (д, 1H), 6,62-6,71 (м, 1H), 6,74-6,82 (м, 1H), 7,15-7,37 (м, 10H), 7,58 (с, 1H), 7,62 (д, 2H), 7,73 (с, 1H), 7,97 (д, 2H), 8,48 (с, 1H).
(б) (2RS, 3RS)-3-(4-[4-Цианфенил]тиазол-2-ил)-2-(2, 4-дифторфенил)-1-(1H-1,2,4-триазол-1-ил)-2-бутилдигидрофосфат (см. схему 13 в конце описания).
Раствор соединения стадии (a) (310 мг, 0,44 ммоль) в метиленхлориде (5 мл) охлаждают до 0oC и затем обрабатывают бромтриметилсиланом (303 мг, 1,98 ммоль) и пиридином (215 мг, 3,7 ммоль). Полученную смесь перемешивают при 0oC в течение 3 часов и потом резко охлаждают водой (1 мл), содержащей гидроксид натрия (115 мг, 2,87 ммоль). Получают желтый осадок, который выделяют путем фильтрации и затем делят между разбавленной серной кислотой и метиленхлоридом. Органическую фазу промывают солевым раствором, сушат (MgSO4), и выпаривают растворитель, получая указанное в заголовке соединение в виде светло- желтого твердого вещества (80 мг, 35%).
1H ЯМР (300 МГц, ДМСО) δ = 1,38 (д, 3H), 4,22 (квартет, 1H), 5,37 (д, 1H), 5,41 (д, 1H), 6,88-6,97 (м, 1H), 7,09-7,19 (м, 1H), 7,31-7,40 (м, 1H), 7,80 (с, 1H), 7,87 (д, 2H), 8,05 (д, 2H), 8,32 (с, IH), 8,65 (с, 1H).
Пример 8. (2R, 3S)-2-(2,4-дифторфенил)-3-[4-(1-диметилпиразол-5-ил) фенил]-1-(1,2,4-триазол-1-ил)-2-бутилдинатрийфосфат
(а) О,N-диметил-4-йодбензолгидроксамовая кислота
Раствор пиридина (104 г, 1,32 моль) в дихлорметане (150 мл) добавляют по каплям к суспензии 4-йодбензоилхлорида (251 г, 0,94 моль) и N,O-диметилгидроксиамингидрохлорида (97 г, 0,94 моль) в дихлорметане (850 мл) при 0oC. Смеси дают нагреться до комнатной температуры и перемешивают в течение 18 часов. Раствор выпаривают при пониженном давлении, остаток растворяют в этилацетате (1 л) и затем промывают разбавленной соляной кислотой (2H, 3х400 мл) и насыщенным раствором бикарбоната натрия (300 мл) и сушат (Na2SO4). Органический экстракт выпаривают при пониженном давлении. Остаток чистят путем перегонки, получая указанное в подзаголовке соединение (241 г, 93%) в виде желтого масла, т. кип. 130oC (0,1 мм рт. ст.), которое характеризуют методом 1H ЯМР.
(б) 2-(2,4-Дифторфенил)-1-(4-йодфенил)этанон
2,4-Дифторбензилбромид (23,7 мл, 0,114 моль) добавляют по каплям к перемешиваемой смеси магниевых стружек (8,1 г, 0,183 моль) в сухом эфире (300 мл) под азотом. Сначала смесь нагревают до тех пор, пока не начнется реакция, и после этого добавляют бромид с такой скоростью, чтобы поддерживать спокойное кипение с обратным холодильником. Через 1 час полученный раствор реактива Гриньяра добавляют по каплям при -78oC к раствору O,N-диметил-4- йодбензолгидроксамовой кислоты [см. Стадию (а)] (45,7 г, 0,157 моль) в сухом эфире (300 мл), и смеси дают медленно нагреться до комнатной температуры в течение ночи. Смесь делят между насыщенным водным хлоридом аммония и этилацетатом, органический раствор отделяют, сушат (MgSO4) и концентрируют при пониженном давлении, получая указанное в заголовке соединение в виде белого твердого вещества (38,71 г, 69%), которое характеризуют методом 1H ЯМР-спектроскопии.
(в) 2-(2,4-Дифторфенил)-1-(4-йодфенил)проп-2-енон
Бис(диметиламино)метан (8,78 мл, 0,075 моль) добавляют по каплям к перемешиваемой суспензии 2-(2,4- дифторфенил)этанона [17,73 г, 0,04595 моль из стадии (б)] в уксусном ангидриде (23,1 мл, 0,248 моль) при комнатной температуре. Эта реакция является экзотермической и температура смеси возрастает до 60oC. По окончании добавления смесь перемешивают при комнатной температуре в течение 35 минут и затем добавляют ледяную воду для гидролиза избытка уксусного ангидрида. Еще через 30 минут продукт экстрагируют в этилацетат, и экстракты промывают разбавленной соляной кислотой, насыщенным водным бикарбонатом натрия, сушат (MgSO4) и концентрируют при пониженном давлении, получая указанное в заголовке соединение в виде белого твердого вещества (17,03 г, 93%), которое характеризуют методом 1H ЯМР-спектроскопии.
(г) 2-(2,4-Дифторфенил)-2-(4-йодбензоил) оксиран
Гидроксид бензилтриметиламмония (3,44 мл, 40% водный раствор, 8,2 ммоль) добавляют одной порцией к раствору 2- (2,4-дифторфенил)-1-(4-йодфенил)проп-2-енона [37,3 г, 100,8 ммоль из стадии (в)] и трет-бутилгидропероксида (36,6 мл, 3 M в триметилпентане, 109 ммоль) в толуоле (500 мл) при комнатной температуре. Через два часа смесь промывают водой (2 х 500 мл), сушат (MgSO4) и концентрируют при пониженном давлении, получая указанное в заголовке соединение в виде белого твердого вещества (37,46 г, 96%), которое характеризуют методом 1H ЯМР-спектроскопии.
(д) (2,4-Дифторфенил)-2-[1-(4-йодфенил) этенил] оксиран
н-Бутиллитий (50 мл, 2,5 М в гексане, 125 ммоль) добавляют по каплям за 10 минут к перемешиваемой суспензии метилтрифенилфосфонийбромида (45,0 г, 126 ммоль) в сухом ТГФ (600 мл) под азотом при -70oC. Смеси дают нагреться до -20oC за 20 минут, затем добавляют за 5 минут раствор 2-(2,4- дифтофенил)-2-(4-йодбензоил)оксирана [37,46 г, 97 ммоль из стадии (г)] в сухом ТГФ (200 мл). Смеси дают нагреться до комнатной температуры и перемешивают в течение 84 часов. Добавляют 10% водный хлорид аммония (500 мл) и смесь концентрируют при пониженном давлении. Продукт экстрагируют в этилацетат и объединенный экстракт сушат (MgSO4) и концентрируют при пониженном давлении. Твердый остаток обрабатывают кипящим гексаном (3 х 500 мл), и выбрасывают оставшееся твердое вещество. Гексановые растворы объединяют, фильтруют через небольшую набивку из силикагеля и концентрируют при пониженном давлении, получая указанное в заголовке соединение в виде желтого масла (34,3 г, 92%), которое характеризуют методом 1H ЯМР-спектроскопии.
(е) 2-(2,4-Дифторфенил)-3-(4-йодфенил)-1-(1H-1,2,4-триазол-1-ил)-3-бутен-2-ол
Натрий (1,2,4-триазол) (12,15 г, 133 ммоль) добавляют к раствору (2,4-дифторфенил) -2-[1-(4-йодфенил)этенил] оксирана [34,3 г, 89 ммоль из стадии (д)] в сухом ДМФ (350 мл) в атмосфере с азотом при 70oC. Смесь перемешивают в течение 5 часов, охлаждают и удаляют растворитель при пониженном давлении. Остаток делят между эфиром (800 мл) и водой (2 х 500 мл). Органический раствор сушат (MgSO4), фильтруют и добавляют силикагель (60-200 m, 75 г). Эфир удаляют при пониженном давлении, и оставшееся твердое вещество наносят на верхний конец колонки с силикагелем (40-60 m, 75 г) и элюируют продукт, используя гексан и возрастающее количество этилацетата (0-75%). Получают продукт в виде белой пены (23,8 г, 61%), который характеризуют методом 1H ЯМР-спектроскопии.
(ж) [(R)-2-(2,4-Дифторфенил)-3-(4-йодфенил)-1-(1H-1,2,4-триазол-1-ил)-3-бутен-2-ол](+)-3-бромкамфор-10-сульфонат
Раствор (+)-3-бромкамфор-10- сульфоновой кислоты (36,3 г, 0,110 моль) в IMS (40 мл) добавляют к раствору продукта стадии (е) (50 г, 0,110 моль) в IMS (300 мл). После внесения затравки полученную пасту гранулируют в течение 20 часов при комнатной температуре. Белое твердое вещество (22 г, 0,03 моль) собирают посредством фильтрации после дополнительного гранулирования в течение 1 часа при низкой температуре. Хиральная чистота, оцененная способом хиральной высокоэффективной жидкостной хроматографии с применением колонки Chiralcel OD и элюирования этанолом/гексаном [40:60], составляет 95% (95% ее).
(з) (R)-2-(2,4-Дифторфенил)-3-(4-йодфенил)-1-(1H-1,2,4-триазол-1-ил)-3-бутен-2-ол
Продукт стадии (ж) (206,5 г, 0,27 моль) добавляют к метиленхлориду (620 мл) и подщелачивают 40% NaOH. Смесь перемешивают в течение 15 минут при комнатной температуре и делят. Водную фазу повторно экстрагируют метиленхлоридом (310 мл). Органический раствор продукта промывают водой (620 мл) и концентрируют до объема 245 мл. К перемешиваемому концентрату с затравкой добавляют при комнатной температуре гексан (2450 мл) с постоянной скоростью. Полученную пасту гранулируют при 5oC в течение 1 часа. Фильтрация дает белое твердое вещество (117,4 г, 0,26 моль), которое характеризуют методом 1H ЯМР-спектроскопии.
(и) (2R)-2-(2,4-Дифторфенил)-3-[4-(1- метилпиразол-5-ил)фенил] -1-(1,2,4-триазол-1-ил)-3-бутен-2-ол
н-BuLi (1,6H, 24,1 мл, 0,04 моль) добавляют к раствору 1-метилпиразола (3,28 г, 0,04 моль) в ТГФ (370 мл) при -70oC, сохраняя температуру ниже -60oC, и перемешивают в течение 30 минут. Поддерживая температуру ниже -40oC, добавляют раствор хлорида цинка (0,5H, 77,1 мл, 0,04 моль), а затем палладий-тетракис (трифенилфосфин) (15% мас/мас, 0,9 г). Поддерживая температуру ниже -40oC, добавляют с постоянной скоростью раствор стадии (з) (6 г, 0,013 моль) в ТГФ (36 мл). Реакционной смеси дают нагреться до комнатной температуры и затем кипятят с обратным холодильником в течение 2 часов. После охлаждения до комнатной температуры реакцию резко охлаждают уксусной кислотой (12 мл) и водой (120 мл), поддерживая температуру ниже 25oC. Реакционную смесь выпаривают при пониженном давлении для удаления ТГФ. Продукт экстрагируют метиленхлоридом (120 мл), и затем водную фазу экстрагируют метиленхлоридом (50 мл). Объединенные органические экстракты промывают водой (2 х 120 мл) и концентрируют, получая масло. К перемешиваемому раствору масла в этилацетате (100 мл) добавляют 5-сульфосалициловую
кислоту (3,3 г, 0,13 моль) в IPA (10 мл). Полученную смесь перемешивают в течение 1/2 часа при комнатной температуре. Полученное отфильтрованное твердое соединение повторно суспендируют в этилацетате (50 мл) и перекристаллизовывают из IPA (60 мл), получая белое твердое вещество (7,2 г, 0,01 моль). Это твердое вещество добавляют к метиленхлориду (35 мл) и воде (50 мл) и подщелачивают 40% NaOH. Смесь перемешивают при комнатной температуре в течение 15 минут и делят. Водную фазу повторно экстрагируют метиленхлоридом (25 мл), и объединенные органические экстракты промывают водой (35 мл). Органический раствор продукта концентрируют до пены и характеризуют.
[α]D = -25,6
Рассчитано для C27H25F2N6O4P: С 63,45; H 4,84; N 16,82%.
Найдено: С 63,92; H 4,86; N 16,64.
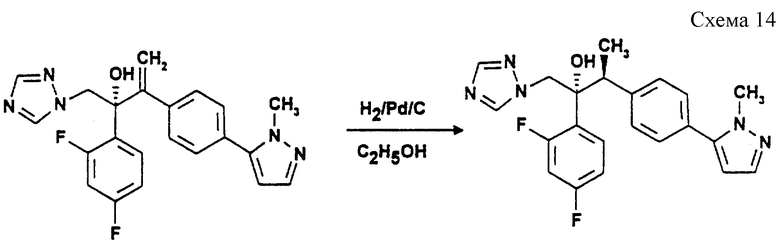
(к) (2R, 3S)-2-(2,4-Дифторфенил)-3-[4-(1-метилпиразол- 5-ил)фенил]-1-(1,2,4-триазол-1-ил)-3-бутан-2-ол (см. схему 14 в конце описания)
Раствор продукта стадии (и) (2,0 г, 5 ммоль) в этаноле (50 мл) гидрируют при 50oC и давлении 50 psi (333 килопаскаля = 3,515 кг/см2) над 5% палладием-на-угле (0,2 г) в течение 18 часов. Добавляют еще порцию катализатора (0,2 г) и продолжают гидрирование еще в течение 18 часов. Смесь фильтруют через ArbacelTM, и выпаривают фильтрат при пониженном давлении. Остаток хроматографируют на диоксиде кремния при градиентном элюировании этилацетатом/гексаном/диэтиламином (0:95:5 - 65:33:2). Фракции, содержащие требуемый продукт, объединяют и выпаривают при пониженном давлении. Остаток растворяют в диэтилацетате и повторно выпаривают его (х3), затем растворяют в эфире и выпаривают (х3), получая бесцветное твердое вещество. Это твердое вещество перекристаллизовывают из водного этанола, получая указанное в подзаголовке соединение (1,25 г, 62%) в виде бесцветного твердого вещества, т.пл. 144-145oC
[α]D = -107 (с=0,1%, CH2Cl2, 25oC)
Элементный анализ (%)
Найдено: С 64,26; H 5,13; N 17,07.
Рассчитано для C27H25F2N6O4P: С 64,54; H 5,17; N 17,10.
[Это соединение раскрыто так же, как пример 67 в совместно поданной международной патентной заявке РСТ/ЕР96/02470].
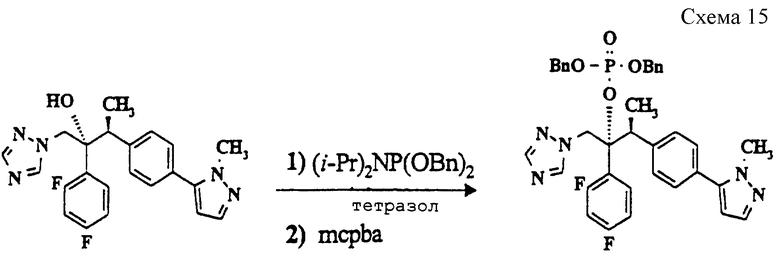
(л) (Дибензил) (2R, 3S)-2-(2,4-дифторфенил) -3-[4-(1-метилпиразол-5-ил)фенил] -1-(1,2,4-триазол-1-ил)-2-бутилфосфат (см. схему 15 в конце описания).
Раствор продукта со стадии (к) (2,0 г, 4,4 ммоль), дибензилдиизопропилфосфорамидита (2,28 г, 6,6 ммоль), тетразола (0,92 г, 13,2 ммоль) и 4- диметиламинопиридина (50 мг) в дихлорметане (30 мл) нагревают при кипячении с обратным холодильником в течение 13 часов. Реакционную смесь охлаждают (0oC) и добавляют метахлорпербензойную кислоту (1,52 г, 8,8 ммоль). Раствор перемешивают при 0oC в течение часа, затем дают нагреться до комнатной температуры. Реакционную смесь промывают водным раствором сульфита натрия (10%, 30 мл), насыщенным раствором бикарбоната натрия (30 мл) и солевым раствором (10%, 30 мл). Органический слой сушат (Na2SO4), и выпаривают растворитель в вакууме, получая масло. Очистка методом колоночной хроматографии (силикагель - 45 г, градиентное элюирование от толуола до 3,5% диэтиламина в толуоле) дает требуемый продукт в виде бесцветного масла (0,8 г, 27%), m/z 671 (M++1).
1H ЯМР (CDCl3) δ = 1,3 (д, 3H), 3,8 (с, 3H), 3,85 (квартет, 1H), 4,8 (м, 2H), 4,9 (м, 2H), 5,2 (с, 2H), 6,25 (с, 1H), 6,6 (м, 1H), 6,8 (м, 1H), 7,05 (м, 1H), 7,15 (м, 2H), 7,2-7,35 (м, 12H), 7,5 (с, 1H), 7,8 (с, 1H), 8,23 (с, 1H).
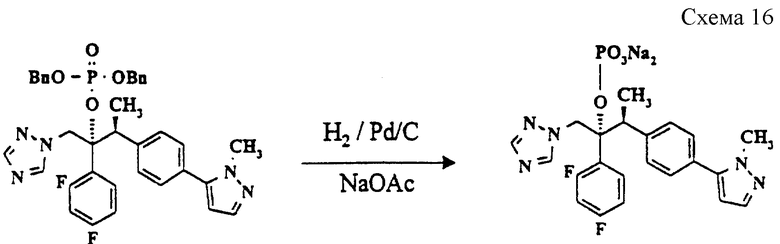
(м) (2R, 3S)-2-(2,4-Дифторфенил)-3-[4-(1-метилпиразол-5-ил) фенил] -1-(1,2,4-триазол-1-ил)-2-бутилдинатрийфосфат (см. схему 16 в конце описания).
Суспензию продукта стадии (л) (0,5 г, 0,75 ммоль) и ацетата натрия (0,14 г, 1,65 ммоль) в этаноле (20 мл) гидрируют над 5% палладием-на-угле (75 мг) при комнатной температуре и давлении 333 килопаскалей (50 psi = 3,515 кг/см2) в течение 24 часов. Тонкослойная хроматография показывает неполное прохождение реакции, и катализатор отфильтровывают через набивку фильтра Arbacel (торговая марка). Затем добавляют катализатор Перлмана (Perlman's catalyst) и продолжают гидрирование еще в течение 72 часов. Катализатор отфильтровывают через набивку Arbacel, и удаляют растворитель в вакууме. Остаток растворяют в дихлорметане (20 мл), и фильтруют через набивку фильтра (Hiflow, торговая марка) для удаления избытка ацетата натрия. Растворитель выпаривают в вакууме, и после обработки диэтиловым эфиром получают указанное в заголовке соединение в виде белого твердого вещества (0,250 г, 68%).
Найдено: С 46,66; H 4,87; N 11,82.
Рассчитано для C22H22F2N5Na2O4P• 0,09Et2O: С 46,62; H 4,36; N 12,16%.
1H ЯМР (ДМСО) δ = 1,2 (д, 3H), 3,75 (квартет, 1H), 3,8 (с, 3H), 5,1, 5,5 (AB система, 2H), 6,3 (с, 1H), 6,9 (м, 1H), 7,2, 7,4 (AB система, 4H), 6,6 (м, 1H), 6,8 (м, 1H), 7,05 (м, 1H), 7,15 (м, 2H), 7,4 (м, 3H), 7,45 (м, 1H), 7,6 (с, 1H), 9,1 (с, 1H).
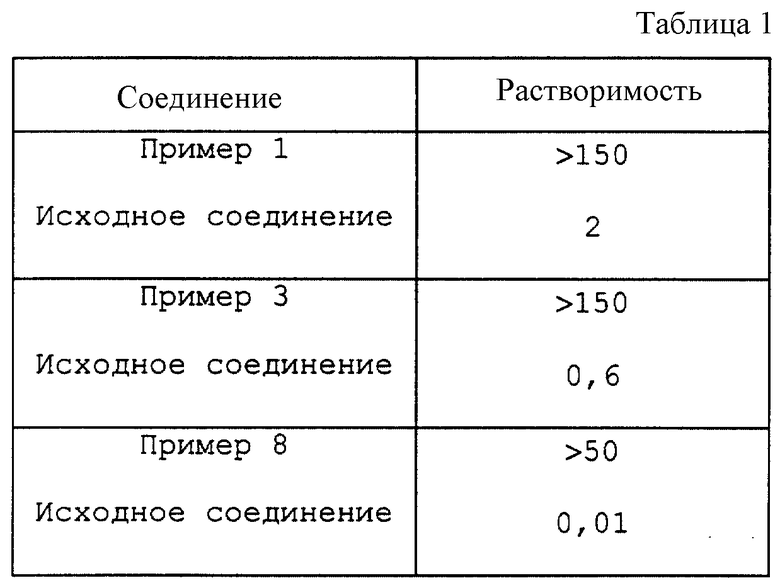
Пример 9
Сравнивают растворимость в воде соединений примеров 1, 3 и 8 (в виде их динатриевых солей) с растворимостью соответствующих им исходных (нефосфорилированных) соединений (в виде свободных оснований). Результаты представлены в табл. 1 (см. в конце описания).
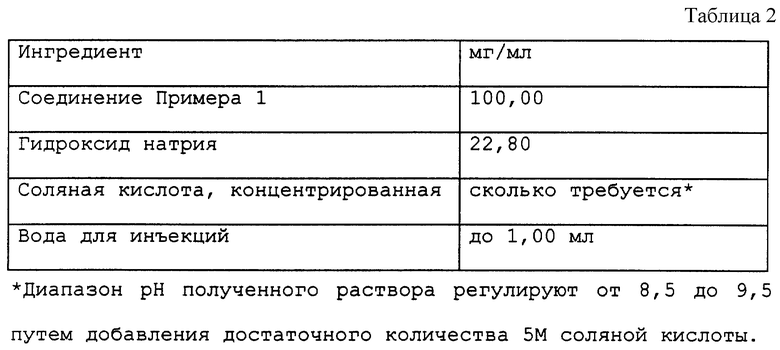
Пример 10
Водные препараты для внутривенных инъекций представлены в табл. 2 в конце описания.
Исследования, демонстрирующие биологическую равноценность инъецируемого в небольшом объеме пролекарственного препарата фосфата флуконазола и вводимого путем инфузии большого объема флуконазола.
Объекты исследования и планирование эксперимента
Основным объектом исследования было изучение фармакокинетики флуконазола, содержащегося в лекарственной форме в виде болюсов для внутривенных инъекций 1000 мг (эквивалентной 800 мг флуконазола) пролекарственного препарата фосфата флуконазола в сравнении с внутривенным вливанием 800 мг флуконазола. Вторичным объектом исследований было сравнение безопасности и толерантности к внутривенному введению болюсов пролекарственного препарата фосфата флуконазола и внутривенному вливанию флуконазола.
Это был рандомизированный дважды повторенный слепой метод исследования с дважды повторенной имитацией способа, с введением единичной дозы, включающий два периода перекрестного исследования с периодом в 14 дней между введениями лекарства для вымывания лекарства из организма.
Субъекты
В исследовании принимали участие 24 здоровых мужчин (в возрасте от 18 до 42 лет; весом от 58,7 до 96,9 кг). Все принимающие участие в исследовании были представителями белой расы. Пятеро были курильщиками.
Введение лекарства
Пролекарственный препарат фосфат флуконазола был предоставлен в виде растворов 100 мг/мл (эвивалент 80 мг/мл флуконазола) в 10 мл ампулах (см. пример 10 настоящей заявки). Плацебо был предоставлен в 10 мл ампулах 0,9%-ного раствора хлорида натрия.
Флуконазол был предоставлен в виде раствора 2 мг/мл в 100 мл пробирках. Плацебо для флуконазола предоставляли в виде 0,9% раствора хлорида натрия в 500 мл пакетах. Флуконазол или плацебо асептическим способом переносили в 500 мл инфузионные пакеты, используемые для введения.
Дважды повторенная имитация способа включала введение через одну и ту же постоянную канюлю (1) болюсной инъекции пролекарственного препарата фосфата флуконазола (1000 мл) с немедленным введением после этого плацебо для флуконазола путем инфузии; или (2) болюсной инъекции плацебо для пролекарственного средства фосфата флуконазола с немедленным после этого введением 800 мг флуконазола путем непрерывной инфузии в течение 4 часов.
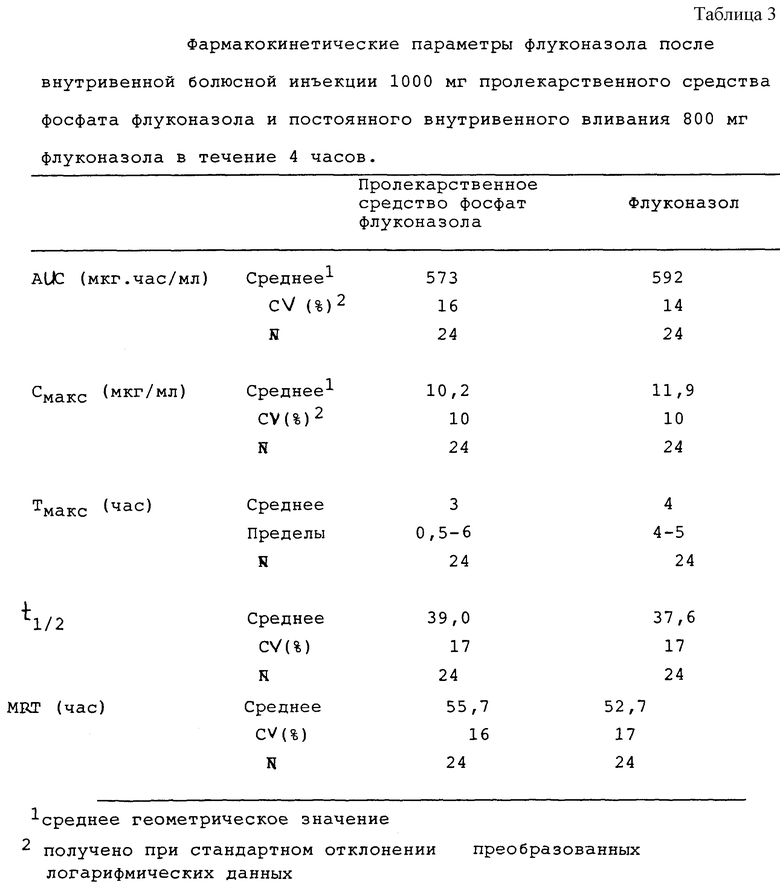
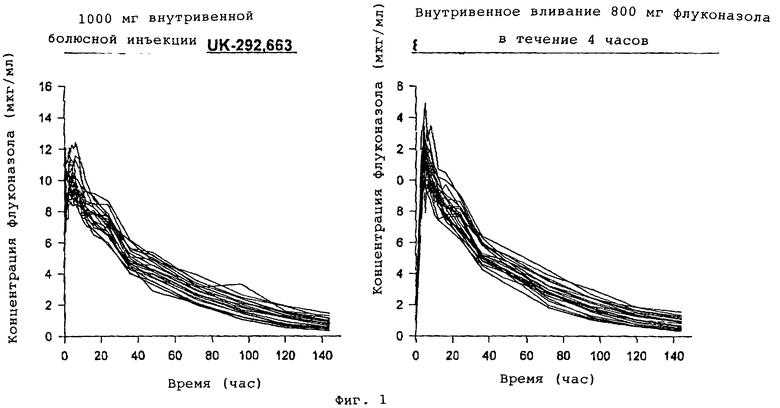
фармакокинетические результаты
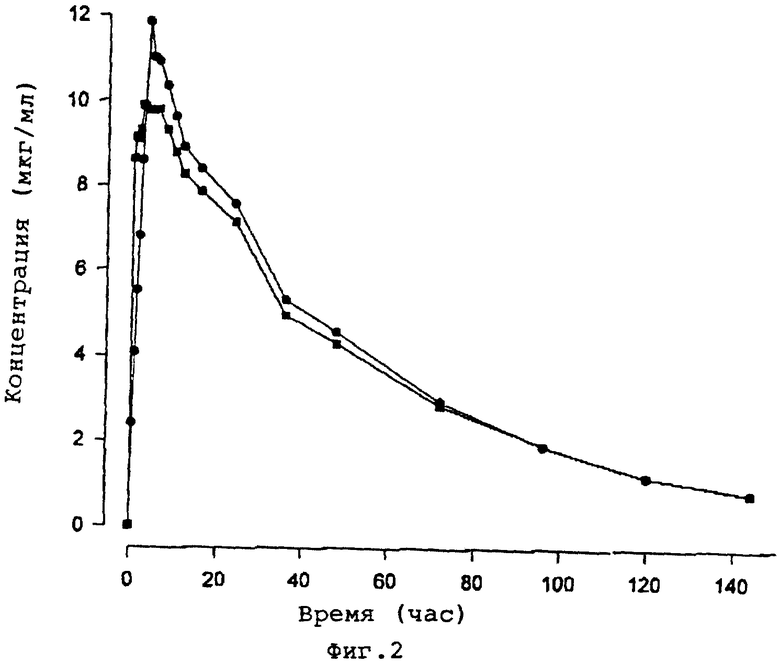
Индивидуальные и усредненные фармакокинетические профили флуконазола с последующим введением пролекарственного препарата фосфата флуконазола и флуконазола представлены на фиг. 1 и 2 соответственно. Краткая характеристика фармакокинетических параметров флуконазола представлена в табл. 1.
Среднее геометрическое значение биодоступности флуконазола из пролекарственного средства фосфата флуконазола составило 97% (90% CI 95%-99%). Концентрация флуконазола в плазме крови первоначально была выше после внутривенной болюсной инъекции пролекарственного средства фосфата флуконазола в сравнении с внутривенной инфузией флуконазола, но максимальная концентрация (CМАКС) флуконазола была выше после инфузионного введения флуконазола. Среднее геометрическое значение флуконазола СМАКС после введения пролекарственного средства фосфата флуконазола в сравнении с флуконазолом составило 86% (90% CI 84%-88%). Среднее время, необходимое для максимальной концентрации (ТМАКС) флуконазола после введения пролекарственного средства фосфата флуконазола, составило 3 часа (от 0,5 до 6 часов). Предельный период полувыведения из организма (t1/2) и среднее время пребывания в организме (MRT) флуконазола были одинаковыми после 2 обработок.
Выводы
Биодоступность флуконазола из пролекарственного средства - фосфата флуконазола составила 97% (90% CI 95%-99%). Поэтому эквивалентное системное воздействие флуконазола можно получить при болюсной внутривенной инъекции пролекарственного средства фосфата флуконазола. Болюсная внутривенная инъекция 1000 мг пролекарственного препарата фосфата флуконазола является хорошо переносимой в сравнении с внутривенным вливанием 800 мг флуконазола, при одинаковом профиле побочных явлений. Далее см. табл. 3 в конце описания.
Описываются соединения формулы (I) R1-ОР(О)(ОН)2, где R1 представляет собой группу формулы Iа, в которой R2 представляет фенил, замещенный одним или несколькими атомами галогена; R3 представляет Н или СН3; R3a представляет Н или вместе с R3 может представлять =СН2; и R4 представляет 5- или 6-членное азотсодержащее гетероциклическое кольцо, которое произвольно замещено одной группой или более, выбранное из галогена, =О, фенила [замещенного группой, выбранной из CN и (C6H4)-OCH2CF2CHF2] или СН= СН(С6Н4)ОСН2СF2СНF2; или фенила, замещенного одной или несколькими группами, выбранными из галогена и метилпиразолила, или его фармацевтически приемлемая соль. Вышеуказанные производные триазола могут использоваться в терапии, в частности для лечения грибковых инфекций людей и других млекопитающих. Описываются также способы их получения, фармацевтическая композиция, способ лечения или профилактики грибковых инфекций и промежуточный продукт. 6 с. и 4 з.п.ф-лы, 2 ил., 3 табл.
R1-OP(O)(ОН)2 (I),
где R1 представляет собой группу формулы Iа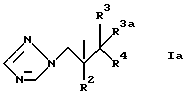
где R2 представляет фенил, замещенный двумя атомами галогена;
R3 представляет Н или СН3;
R3a представляет Н;
R4 представляет триазолил, необязательно замещенный группой CH= CH(C6H4)OCH2CF2CHF2; пиримидинил, замещенный одним атомом галогена; фенил, замещенный метилпиразолилом; тиазолил, замещенный фенилом, который в свою очередь замещен цианогруппой,
или его фармацевтически приемлемая соль.
R'-OH, II
где R' имеет значения, определенные в п.1,
соединением формулы III
RaRbN-P(ORc)(ORd), III
где Ra и Rb независимо представляют C1-С6 алкил, фенил, замещенный фенил или вместе с атомом N, к которому они присоединены, могут представлять кольцо;
Rc и Rd независимо представляет гидроксизащитную группу,
с получением соединения формулы IV
R1-OP(ORc)(ORd), IV
где R1, Rc и Rd имеют указанные значения;
b) окисление полученного соединения формулы IV с получением фосфатного соединения формулы V
R1-OP(O)(ORc)(ORd), V
где R1, Rc и Rd имеют указанные значения;
с) удаление гидроксизащитных групп Rc и Rd в соединении формулы V с получением соединения формулы I и d) необязательное превращение полученного соединения формулы I в его фармацевтически приемлемую соль.
R'-OH, II
где R' имеет значения, определенные в п.1,
обрабатывают РСl3 в присутствии основания, с получением соединения формулы VI
R1-OPCl2, VI
где R1 имеет значения, указанные выше,
с последующей реакцией с соединением формулы
RcОН и/или RdOH,
где Rc и Rd определены выше,
с получением соединения формулы IV
R1-OP(ORc)(ORd), IV
где R1, Rc и Rd имеют указанные значения;
b) окисление полученного соединения формулы IV с получением фосфатного соединения формулы V
R1-OP(O)(ORc)(ORd), V
где R1, Rc и Rd имеют указанные значения;
c) удаление гидроксизащитных групп Rc и Rd в соединении формулы V с получением соединения формулы I и d) необязательное превращение полученного соединения формулы I в его фармацевтически приемлемую соль.
R1-OP(O)(ORc)(ORd), V
где R1 определено в п.1,
Rc и Rd независимо представляет гидроксизащитные группы, выбранные из бензила, необязательно замещенного одним или несколькими атомами галогена.
| Контактная система электромагнитного реле | 1973 |
|
SU472392A1 |
| Устройство для балансировки гироскопического прибора | 1975 |
|
SU567982A1 |
| Бесколесный шариковый ход для железнодорожных вагонов | 1917 |
|
SU97A1 |
| ПРЕОБРАЗОВАТЕЛЬ СВЕТОВОЙ ЭНЕРГИИ В ЭЛЕКТРИЧЕСКУЮ НА ОСНОВЕ P-N-ПЕРЕХОДА С ПОВЕРХНОСТНЫМ ИЗОТИПНЫМ ГЕТЕРОПЕРЕХОДОМ | 1996 |
|
RU2099818C1 |
| US 4952232 А, 1990 | |||
| СПОСОБ ПОЛУЧЕНИЯ ГЕТЕРОЦИКЛИЧЕСКИХ СЛОЖНЫХ ЭФИРОВ ТИОФОСФОРНОЙ или ТИОФОСФОНОВОЙкислоты | 0 |
|
SU268302A1 |
| Лекарственные препараты зарубежных фирм в России | |||
| - М.: Астрафармсервис, 1993, с | |||
| ПАРОВАЯ ИЛИ ГАЗОВАЯ ТУРБИНА | 1914 |
|
SU278A1 |

