Изобретение относится к производным хиноксалина, используемым в терапии.
L-Глутаминовая кислота является возбуждающим аминокислотным нейромедиатором, чья физиологическая роль в головном мозге связана с взаимодействием с четырьмя рецепторами, три из которых позднее были названы селективными агонистами: NMDA (N-метил-D-аспартат), AMPA (2-амино-3-гидрокси-5- метил-4-изоксазолпропионовая кислота) и каинат. Четвертый рецептор называют метаботропическим рецептором. В дополнение к участку для связывания глутаминовой кислоты NMDA-рецептор обладает характеризующимися высоким сродством участками для присоединения диссоциативных анестезирующих средств (например, кетамина), полиаминов (например, спермина), глицина и некоторых ионов металлов (например, Mg2+), Zn2+. Поскольку для NMDA-рецептора абсолютным требованием для возникновения возбуждения служит связывание глицина, то глициновые антагонисты могут действовать как функциональные NMDA-антагонисты.
Например, в области церебрального инфаркта, гипоксия вызывает высвобождение ненормально высоких концентраций глутаминовой кислоты, что приводит к повышенному раздражению NMDA-рецептора, приводящего к дегенерации и гибели нейронов. Таким образом, NMDA-рецепторные антагонисты, которые, как было показано, блокируют нейротоксичные воздействия глутаминовой кислоты in vitro и in vivo , могут быть полезны в лечении и/или профилактике патологических состояний, при которых представляется значительной активация NMDA-рецепторов. Примеры таких состояний включают нейродегенеративные расстройства, включающие старческое слабоумие и болезнь Альцгеймера, и заболевания головного и спинного мозга, являющиеся результатом таких случаев, как удар, преходящее нарушение мозгового кровообращения, связанная с оперативным вмешательством ишемия и травма головы. Они могут также быть полезны при состояниях, в которых ослаблена периферическая нервная функция, таких как ретинальная дегенерация и дегенерация желтого пятна.
Кроме того, показано, что NMDA-антагонисты обладают противосудорожной и анксиолитической активностью и поэтому могут быть использованы в терапии эпилепсии и тревоги. Они могут быть также полезны для снятия боли.
NMDA-антагонисты могут также снижать воздействие синдрома отмены алкоголя у животных с физической зависимостью (К.A. Grant et al. J. Pnarm. Exp. Ther. (1992), 260, 1017) и таким образом NMDA- антагонисты могут быть полезны в лечении хронического алкоголизма.
Описаны различные производные 1,2,3,4-тетрагидрохинолин-2,4-диона как NMDA (глициновый участок)-антагонисты (см. EP-A-0459561 и EP-A-0481676), тогда как WO-A-91/13878 и JP-A-3220124 описывают 1,4-дигидроксихиноксалин-2,3-дионы в качестве антагонистов глутаминовой кислоты.
WO-A-94/00124 описывает 1,4-дигидро-хиноксалин- 2,3-дионы (включая 6,7-дихлор-5-нитро-1,4-дигидрохиноксалин-2,3- дион), обладающие высоким сродством к глициновому участку связывания и используемые для лечения ударов и родственных нарушений.
Согласно данному изобретению предлагается соединение формулы 1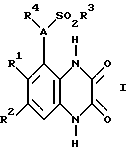
где A - N или CH;
R1 и R2 независимо C1-4-алкил, галоид или CF3;
R3 - C1-4алкил (необязательно замещенный C3-7циклоалкилом или арилом), C3-7циклоалкил, CF3 или арил;
R4 - H, C3-7циклоалкил или C1-6алкил [необязательно замещенный ОН, C1-4алкокси, арилом (необязательно замещенным до 3 заместителей, которые независимо выбирают из C1-4алкила, C 1-4алкокси, галоида и CF3), гетероциклом (необязательно замещенным до 3 заместителей, которые независимо выбирают из C1-4алкила, C1-4алкокси, ОН, галоида, CF3 и оксо, и необязательно бензоконденсированным), C2-6алкенилом, C2-6алкинилом, C2-6алканоилом, CO2H, C1-4алкоксикарбонилом, NH2, C1-4алкиламино, ди(C1-4алкил)амино, NHSO2CF3, CONR5R6, NHCONR5R6 или O(CH2)nNR5R6];
R5 и R6 независимо - H или C1-4алкил, или вместе в атомом азота, к которому они присоединены, могут представлять пирролидино-, пиперидино- или морфолиногруппу; и
n = 2, 3 или 4,
или его фармацевтически приемлемая соль (называемые здесь "соединения по изобретению").
Фармацевтически приемлемые соли включают соли основных или кислотных групп, которые могут быть представлены (например, натриевые соли карбоновых кислот и хлористоводородные соли амино-групп).
Предпочтительно, A обозначает H.
"Галоид" обозначает фтор, хлор, бром или йод. Предпочтительными группами являются фтор, хлор или бром.
Предпочтительными группами, которые независимо обозначают R1 и R2, являются галоид и C1-4алкил. Например, они оба могут обозначать хлор или один может обозначать хлор, а другой может представлять метил или этил.
"Арил" обозначает ароматический углеводород, такой как нафтил или, более желательно, фенил.
Предпочтительно, R3 обозначает C1-4алкил, более предпочтительно, метил.
"Гетероцикл" обозначает ароматический или неароматический гетероцикл, содержащий один или более гетероатомов, которые выбирают из O, S и N. Он может быть соединен с C1-6-алкильной группой посредством азота или, более предпочтительно, атомом углерода. В качестве гетероциклических групп могут быть упомянуты пиридинил, имидазолил, пиримидинил, пиразолил, триазолил, пиразинил, тетразолил, фурил, тиенил, изоксазолил и тиазолил. Гетероциклические группы, содержащие конденсированный бензольный цикл, включают бензимидазолил.
Предпочтительно, R4 обозначает C1-6алкил, замещенный OH или CO2H, более предпочтительно, он обозначает CH2CH2OH или CH2CO2H.
Соответствующие алкильная, алкокси-, алкинильная и алканоильная группы могут быть линейными или разветвленными.
Соединения формулы 1, в которых атом O или N в R4 связан с A посредством отдельного углеродного атома, могут быть недостаточно стабильны для того, чтобы использовать их в качестве лекарственных средств. Некоторые такие нестабильные соединения не являются составной частью изобретения.
В некоторых случаях соединения по данному изобретению могут существовать в виде таутомеров и все эти таутомеры входят в рамки объема изобретения и приложенных пунктов, независимо от того, раздельно или нет. В дополнение, соединения содержащие асимметрические центры, могут существовать как энантиомеры и дистереоизомеры и изобретение включает отдельные индивидуальные изомеры, равно как и смеси изомеров.
В частности, может быть ограничено вращение между A и 1,4-дигидро-2,3-диоксохиноксалиновым циклом и таким образом может возникать атропизомеризм. Предпочтительно, когда A обозначает N, R4 в формуле I располагается над плоскостью листа, a SO2R3 располагается за плоскостью листа, как показано ниже в формуле IA: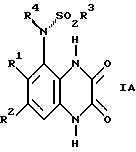
Например, когда R1 - C1, имеем (R) - стереохимическое отнесение этой связи, и (S)-, когда R1 - метил.
Оптические изомеры (включая атропизомеры) могут быть разделены обычными способами, такими как фракционированная кристаллизация диастереомерных производных [см., например, пример 80(b)].
Кроме того, изобретение касается способа получения соединения по изобретению, включающий удаление защитных групп соединения формулы II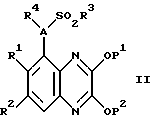
где A и R1-4 такие, как определено выше; P1 и P2 являются защитными группами для гидроксильных групп, связанных с ароматическими циклами, и где желательно или необходимо превращение полученного соединения в фармацевтически приемлемую соль или наоборот. Защитные группы, которые могут обозначать P1 и P2, включают бензил и C1-6алкил, в частности метил. Эти защитные группы могут быть удалены использованием обычных способов снятия защиты (см. "Protective Groups in Organic Syntnesis" by T.W. Greene and P.G.M.Wuts, Jonn Wiley and Sons Inc, 1991). Например, когда они обозначают метил, они могут быть удалены кислотным гидролизом с использованием разбавленной водной соляной кислоты (например, 2 молярн.). Реакцию обычно проводят нагреванием соединения формулы II, предпочтительно до температуры кипения с обратным холодильником, в смеси разбавленной водной соляной кислоты и подходящего органического растворителя, такого как диоксан или ацетон, скажем, в течение 2 - 48 часов до завершения реакции. Соединение по изобретению может быть выделено и очищено общепринятыми способами.
Соединения формулы II, как определено выше, составляют дальнейший аспект данного изобретения.
Соединения формулы II, в которых R4 отличается от водорода, могут быть получены взаимодействием соответствующего соединения формулы II, в котором R4 обозначает H, с соответствующим галогенидом формулы R4aX, где X - Cl, Br или I, и R4a имеет те же значения, что определены для R4 как указано выше, за тем исключением, что он не может обозначать H, в присутствии основания, такого как трет-бутилат калия. Обычно основание добавляют к раствору соединения формулы II, (где R4 обозначает H) в подходящем органическом растворителе, таком как диметилформамид. После перемешивания в течение нескольких минут добавляют галогенид R4X и смесь перемешивают в течение нескольких часов приблизительно при комнатной температуре [см., например, пример 7(a)]. Желаемое промежуточное соединение может быть затем выделено и очищено общепринятыми способами.
Кроме того, соединения формулы II могут быть получены из других соединений формулы II использованием общепринятых способов. Например, соединения, в которых A - CH, и R4 - аллил, могут быть превращены в соединения, в которых R4 - 2-гидроксиэтил, путем озонолиза с последующим восстановлением. Соединения формулы II, в которых A - CH, а R4 - аллил, могут также быть получены из соответствующих соединений формулы II, в которых R4 - H, взаимодействием с диаллилкарбонатом (например, см. пример 93).
В качестве альтернативы приведенному выше способу алкилирования, когда A обозначает N, может быть использована реакция Mitsunobu. Она включает взаимодействие спирта формулы R4aOH (где R4a - как определено выше) с диэтилазодикарбоксилатом, трифенилфосфином и соединением формулы II, в котором R4 - H. Реакцию обычно проводят в подходящем органическом растворителе, например тетрагидрофуране, приблизительно при комнатной температуре при перемешивании, скажем, в течение 6 - 12 часов [см., например, пример 49(a)].
Соединения формулы II, где R4 обозначает C1-C6 алкильную группу замещенную, гидроксильной, могут также быть получены по способам, или аналогично препаративным примерам 8 - 10, которые включают образование алканоилалкильного производного, которое либо восстанавливают, например, диизобутилалюминийгидридом, либо подвергают взаимодействию с алкилмагнийгалогенидом.
Соединения формулы II, где R4 - водород и A - N, могут быть получены сульфонилированием соответствующего хиноксалина формулы III,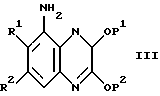
где R1, R2, P1 и P2 такие, как определено выше,
с использованием соответствующего сульфонилхлорида R3SO2Cl или ангидрида формулы (R3SO2)2O, где R3 такой, как определено выше, в подходящем органическом растворителе, например, дихлорметане или тетрагидрофуране, в присутствии кислотного акцептора, такого как пиридин (см., например, препаративный пример 5) три-триэтиламин. С некоторыми исходными веществами при использовании большого избытка сульфонилхлорида или ангидрида, может протекать дисульфонилирование или частичное ди-сульфонилирование. В этом случае, один из R3SO2 - заместителей может быть удален взаимодействием ди-сульфонилированного продукта с водной гидроокисью натрия (см., например, препаративный пример 3). Соединения формулы III могут быть получены общепринятыми способами, такими как способы, иллюстрируемые препаративными примерами 1 и 2.
Соединения формулы II, где R4 - водород и A - CH, могут быть получены взаимодействием соединения формулы IV,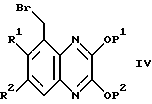
где R1, R2, P1 и P2 - как описано выше, с меркаптидом NaSR3, в котором R3 - как определен выше, с последующим окислением с использованием надкислоты, такой как 3-хлорнадбензойная кислота (см., например, препаративный пример 29). Соединения формулы IV могут быть получены общепринятыми способами (см., например, препаративный пример 28).
В синтезах соединений по изобретению может быть необходимо или желательно защитить чувствительные функциональные группы и затем снять с них защиту. Способы проведения таких операций известны специалистам в соответствующей области синтеза и описаны в "Protective Groups in Organic Synthesis" упомянутых выше.
Соединения по данному изобретению полезны, поскольку они обладают фармакологической активностью в отношении животных (включая человека). В частности, соединения полезны в лечении или профилактике нейродегенеративных расстройств (включая старческое слабоумие, болезнь Альцгеймера и заболевания головного и спинного мозга, являющиеся результатом таких случаев, как удар, преходящее нарушение мозгового кровообращения, связанная с оперативным вмешательством имешия и травма головы; и ретинальную дегенерацию и дегенерацию желтого пятна), судорог, боли и тревоги. В частности, интерес представляет лечение ударов.
Кроме того, согласно другому аспекту изобретения, дается анксиолитический, противосудорожный, аналгезирующий или нейрозащитный способ лечения, включающий введение соединения по изобретению пациенту, нуждающемуся в таком лечении. Также приводится применение соединений по изобретению в виде фармацевтических препаратов и применение соединений по изобретению в производстве анксиолитического, противосудорожного, аналгезирующего или нейрозащитного лекарственного средства.
Биологическую активность соединений по изобретению можно продемонстрировать следующими приведенными ниже примерами:
(a). Способность связывания по глициновому участку NMDA-рецептора.
Она может быть измерена испытанием способности соединения вытеснять радиолиганд с селективных глициновых участков из мембран головного мозга крыс, как описано в Brit. J. Pharm. (1991), 104, 74. В используемом варианте этого способа тщательно промытый мембранным белок инкубируют с [3H]-L-689,560 в течение 90 минут, используя трисацетатный буфер (pH 7,4). Вытеснение радиолиганда при использовании интервала концентраций испытуемого соединения, используют для получения IC50 значений (концентрация 50% ингибирования).
(b). Способность связывания AMPA-рецептором.
Может быть измерена испытанием способности соединений к вытеснению радиолиганда [3H] -AMPA из мембран головного мозга крыс. Мембранный гомогенат инкубируют с радиолигандом (10 нМ) в присутствии или в отсутствие соединений при различных концентрациях при 4oC в течение 45 мин. Свободную и связанную радиоактивную метку разделяют быстрой фильтрацией и радиоактивность измеряют с помощью жидкостного сцинтилляционного счетчика.
(c). Функциональный NMDA-антагонизм in vitro.
Демонстрируется способность соединений ингибировать деполяризацию в кортикальных крысиных срезах, индуцированную NMDA, аналогично способу, описанному в J. Med. Chem. (1990), 33, 789 и Brit J. Pharm. (1985), 84, 381. В данном варианте способа, оценивают реакцию на стандартную концентрацию NMDA в присутствии интервала концентраций испытуемого соединения, и полученные результаты используют для получения значений IC50 (концентрация 50% ингибирования).
(d). NMDA-Антагонизм in vivo.
Можно продемонстрировать по способности соединения ингибировать NMDA-индуцируемую беспорядочную беготню мышей, варьируя способ, описанный в Brit. J. Pharm " Proceedings Supplement (1992), 107 58P. В этой модели группы мышей обрабатывают испытуемыми соединениями при различных дозах перед введением NMDA (60 мг/кг, внутривенно). Регистрируют латентный период наступления беспорядочной беготни и наличие или отсутствие такого поведения используют для оценки ED50. Анализ единицы вероятности используют для определения дозы, при которой 50% мышей не обнаруживают способности к беспорядочной беготне спустя 10 минут после NMDA-введения.
(e). Блокирование кортикальной разрастающейся депрессии.
Активность соединения in vivo можно также продемонстрировать измерением его способности блокировать распространение электроинициируемой кортикальной разрастающейся депрессии у анестезированных крыс. Итак, самцов крыс анестезируют и вводят два стеклянных микроэлектрода в правую теменную зону коры головного мозга на глубину 0,5-1 мм для регистрации активности головного мозга. Кроме того, на твердой мозговой оболочке перед микроэлектродами помещают биполярный стимулирующий электрод. Затем твердую мозговую оболочку подвергают электростимуляции с 10-минутными интервалами, и с помощью микроэлектродов регистрируют волны разрастающейся депрессии, усиленные и воспроизведенные с помощью ленточного самописца. Испытуемые соединения растворяют в воде в виде их натриевых солей или хлористоводородных солей (где возможно) и вводят путем внутривенной инъекции при различных дозах, чтобы определить минимальную дозу, блокирующую распространение разрастающейся депрессии.
Соединения по изобретению можно вводить пациенту, нуждающемуся в лечении, разнообразными общепринятыми способами введения, включающими пероральное и внутривенное введение. Соединения обладают способностью адсорбироваться в желудочно-кишечном тракте и благодаря этому возможно также введение путем составов замедленного выделения.
В основном терапевтически эффективная пероральная доза находится приблизительно в интервале от 0,1 до 100 мг/кг веса тела, требующего обработки пациента, а внутривенная доза находится приблизительно в интервале 0,01 - 10 мг/кг веса тела обрабатываемого пациента, предпочтительно 0,1-5 мг/кг. При необходимости соединения могут также быть введены путем внутривенного вливания, при дозе, находящейся приблизительно в интервале от 0,01-1 мг/кг/час. На практике врач определяет фактическую дозу, наиболее подходящую для конкретного пациента, и она будет варьироваться с возрастом, весом и реакцией отдельного пациента. Конечно, вышеуказанные дозы являются типичными средними величинами, но возможны отдельные случаи, когда требуются более высокие или низкие интервалы доз, и они входят в рамки объема изобретения и приложенных пунктов.
Хотя соединения по изобретению можно вводить отдельно, обычно их вводят в смеси с фармацевтическим носителем, который выбирают в зависимости от предполагаемого способа введения и стандартной фармацевтической практики. Например, пероральное введение можно осуществлять в форме таблеток, содержащих такие эксципиенты, как крахмал или лактоза, в капсулах, либо в чистом виде, либо в смеси с эксципиентами, или в форме эликсиров или суспензий, содержащих отдушки или красители. Соединения можно вводить парентерально в виде инъекции, например внутривенно, внутримышечно или подкожно. Для парентерального введения их лучше использовать в форме стерильного водного раствора соответствующей соли соединения, и раствор может содержать другие вещества, такие как соли, для придания им изотоничности относительно крови.
Таким образом, приводится также фармацевтический состав, включающий соединение по изобретению в смеси с фармацевтически приемлемым адъювантом, разбавителем или носителем.
Соединения по данному изобретению обладают теми преимуществами, что они являются более сильными по действию, более растворимыми, более селективными [например, являются мощными антагонистами NMDA-(глициновый участок) - рецептора, но обладают слабым сродством, либо не обладают сродством к AMPA-рецептору] , менее токсичны или проявляют другие более желательные свойства, чем соединения, известные из уровня техники.
Изобретение иллюстрируется приведенными далее примерами. Промежуточные соединения могут быть получены, как описано в следующих ниже препаративных примерах.
Температуры плавления определяют, используя прибор Buchi'a, в стеклянных капиллярах и не корректируют. Спектроскопические данные получают на приборах. Perkin Elmer 983 (UK), Fisons Triо 1000 (масс-спектрометр, термонапыление, использующее в качестве носителя ацетат аммония в водном метаноле ), и Bruker AC 300 и Varian Uniti 300-ЯМР (оба 300 МГц), и они соответствуют приписываемым структурам. Колоночную хроматографию выполняют на Kieselgel 60, (230-400 меш) от E.Merck Darmstadt. Для тонкослойной хроматографии (ТСХ) используют Kieselgel 60 F254 - пластины от E.Merck, и соединения обнаруживают УФ-светом или раствором хлорплатиновой кислоты/йодида натрия. В случаях, когда соединения анализируют в виде гидратов, на протонном ЯМР-спектре очевидно присутствие воды в усиленном, благодаря воде, пике. Чистоту соединений тщательно оценивают, используя аналитическую ТСХ и протонный ЯМР (300 МГц), метод протонного ЯМР используют для расчета количества растворителя в сольватированных образцах. В многостадийных последовательных превращениях чистоту и структуру промежуточных соединений подтверждают спектроскопически путем протонного ЯМР. Сдвиги протонного ЯМР приведены в миллионных долях в низшей области спектра по отношению к тетраметилсилану.
В примерах и препаративных примерах использованы некоторые хорошо известные специалистам в соответствующей области обозначения, например Me (метил), Et (этил). Ac (ацетил), h (час), m (применительно к силикагелю - меш.)
В приведенных далее спектрах ЯМР приняты следующие обозначения:
м - мультиплет,
с - синглет,
кв - квартет,
т - триплет,
ушир. - уширенный сигнал,
д - дублет,
дд - двойной дублет,
дт - двойной триплет
Пример 1. N-(1,4-Дигидро-6,7-дихлор-2,3-диоксохиноксалин-5-ил)-этансульфонамид.
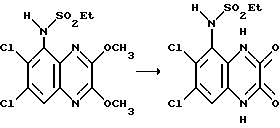
Смесь N-(6,7-дихлор-диметоксихиноксалин-5-ил)-этан- сульфонамида (препаративный пример 4) (100 мг, 0,273 ммоль), 2 М соляной кислоты (2 мл) и диоксана (4 мл) нагревают до температуры кипения с обратным холодильником в течение 2,5 часов, охлаждают и концентрируют при пониженном давлении. Твердый остаток суспендируют в воде, отфильтровывают и промывают водой и диэтиловым эфиром, получая указанное в заглавии соединение (90 мг, 98%) в виде твердого белого продукта, т. пл. 297oC (разл.).
Анализ%:
Найдено: C 33,97; H 2,97; N 11,68; для C10H9Cl2N3O4S•H2O рассчитано; C 33,72; H 3,11; N 11,79%.
Примеры 2-6. Следующие примеры, приведенные в таблице 1, выполнены по способу примера 1 с использованием соответствующего 2,3- диметоксихиноксалинового производного (препаративные примеры 3,5-7 и 12).
Пример 7. N-(1,4-Дигидро-6,7-дихлор-2,3- диоксохиноксалин-5-ил)-N-(метил)этансульфонамид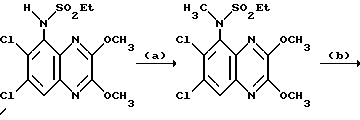
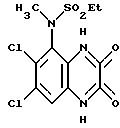
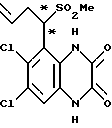
a). трет-Бутилат калия (67,5 мг, 1,1 ммоль) добавляют к перемешиваемому раствору N-(6,7-дихлор-2,3-диметоксихиноксалин-5- ил)этансульфонамида (препаративный пример 4) (200 мг, 0,55 ммоль) в сухом диметилформамиде (3 мл) в атмосфере азота при 20oC. Через 5 минут добавляют метилйодид (38 мкл, 1,1 ммоль) и смесь перемешивают при 20oC 2 часа. Смесь концентрируют при пониженном давлении, распределяют между этилацетатом и водой и объединенные органические экстракты промывают разбавленной водной гидроокисью натрия. Раствор сушат (MgSO4) и концентрируют при пониженном давлении. Остаток чистят флэш хроматографией (элюирование дихлорметаном), получая N- (6,7-дихлор-2,3-диметоксихиноксалин-5-ил)-N- (метил)этансульфонамид (150 мг, 79%).
1H-ЯМР (300 МГц, CDCl3): δ =1,51 (3H, т, J 7 Гц), 3,35 (3H, с), 3,37 (2H, кв., J 7 Гц); 4,14 (3H, с), 4,20 (3H, с), 7,92 (1H, с), m/z (термонапыление) 380 (MH+).
b). Смесь N-(6,7-дихлор-2,3-диметоксихиноксалин-5-ил)-N- (метил)-этансульфонамида (150 мг, 0,39 ммоль), 2 М соляной кислоты (4 мл) и диоксана (8 мл) нагревают до температуры кипения с обратным холодильником в течение 16 часов, охлаждают и концентрируют при пониженном давлении. Оставшийся твердый продукт суспендируют в воде, отфильтровывают и промывают водой и диэтиловым эфиром, получая указанное в заглавии соединение (140 мг, 99%) в виде белого твердого продукта, т. пл. > 300oC.
Анализ%:
Найдено: C 37,69; H 3,09; N 11,84. Для C11H11Cl2N3O4S рассчитано: C 35,51; H 3,15; N 11,93%.
Примеры 8 - 48. Соединения следующих примеров, приведенных в таблице 2, получают по способу примера 7, используют соответствующее 2,3-диметоксихиноксалиновое производное (препаративные примеры 3, 4, 6, 7, 11 - 14) и соответствующий алкил галогенид [например, метилйодид, этилйодид, н-бутилбромид, 3- (N,N-диметиламино)пропилхлорид, бензилхлорид, фенетилбромид, 2- пропилбромид, 2-метоксиэтилбромид, аллилбромид, циклопентилбромид, 2-(морфолин)этилхлорид, 4-пиколилхлорид, 2-гидроксиэтилбромид, н-пропилбромид, 2-пиколилхлорид, 3- гидроксипропилбромид, хлорацетон, пропаргилбромид и 2-(бромметил)-6- метоксипиридин (соединение препаративного примера 22)].
Примечания к таблице 2:
a) 1H-ЯМР (300 МГц, ДМСО-d6): 3,39 (3H, с), 4,74 (1H, д, J 14 Гц), 4,82 (1H, д, J 14 Гц), 7,20 (4H, м), 7,30 (2H, м), 10,24 (1H, ушир.с), 12,13 (1H, ушир. с).
b) Получено по способу примера 7 (b) использованием N-(6, 7-дихлор-2,3-диметоксихиноксалин-5-ил)-N-[(6-мeтoкcи- пиридин-2- ил)метил] метансульфонамида (препаративный пример 22). В ходе гидролиза диметоксихиноксалина, метоксипиридин превращают в 2- пиридон.
c) 1H-ЯМР (300 МГц, ДМСО-d6) δ = 0,95 (3H, т, J 8 Гц), 1,18 (3H, т, J 8 Гц), 2,70 (2H, кв, J 8 Гц), 3,20 (3H, с), 3,71 (2H, м), 7,05 (1H, с), 10,75 (1H, ушир. с), 12,09 (1H, ушир.с) m/z (термонапыление) 357 (MNH4 +), νmax (KBr) 3300, 2950, 1720, 1330 и 1150 см-1.
d) 1H-ЯМР (300 МГц, ДМСО-d6) δ = 0,80 (3H, т, J 8 Гц), 1,30 (2H, м), 2,19 (3H, с), 2,22 (3H, с), 3,19 (3H, невидим), 3,49 (2H, м), 6,98 (1H, с) 9.95 (1H, ушир.с), 11,83 (1H, ушир.с).
m/z (термонапыление) 326 (MH+), 343 (MNH4 +), νmax (KBr) 3380, 3220, 1720, 1680 и 1150 см-1.
e) 1H-ЯМР (300 МГц, ДМСО-d6) δ = 2,19 (3H, с), 2,21 (3H, с), 3,16 (3H, с), 6,95 (1H, с), 10,67 (1H, ушир.c). 11,82 (1H, ушир.с).
m/z (термонапыление 298 (MH+), 315 (MNH4 +, νmax (KBr) 3225, 1700, 1325, 1140 и 750 см-1.
f) 1H-ЯМР (300 МГц, ДМСО-d6) δ = 1,00 (3H, т, J 8 Гц), 2,35 (3H, с), 3,58 (3H, с), 3,72 (2H, м), 7,12 (1H, с.), 10,40 (1H, ушир.с), 12,01 (1H, ушир.с).
m/z (термонапыление) 349 (MNH4 +) νmax (KBr) 3450, 3260, 2950, 1700, 1380, 1330, 1150 и 520 см-1.
g) 1H-ЯМР (300 МГц, ДМСО-d6): δ = 2,31 (3H, с), 3,20 (3H, c), 3,34 (2H, м), 4,02 (2H, м), 7,10 (1H, с), 10,80 (1H, ушир.с), 12,10 (1H, ушир. с).
m/z (термонапыление ) 365 (MNH4 +)
h) 1H-ЯМР (300 МГц, ДМСО-d6) δ = 1,00 (3H, т, J 7 Гц), 2,30 (3H, с), 3,23 (3H, с), 3,65 (2H, кв. J 7 Гц), 7,24 (1H, с), 10,40 (1H, ушир.с), 11,93 (1H, ушир.с)
m/z (термонапыление) 349 (MNH4 +),
i) 1H-ЯМР (300 МГц, ДМСО-d6) δ = 2,30 (3H, с), 3,19 (3H, невидим), 3,34 (2H, м), 3,74 (1H, м), 4,05 (1H, м), 5,98 (1H, ушир.с), 7,23 (1H, с), 10.92 (1H, ушир.с), 11,91 (1H, ушир.с).
m/z (термонапыление) 348 (MH+), 365 (MNH4 +).
j) 1H-ЯМР (300 МГц, ДМСО-d6) δ = 1,05 (3H, т, J 8 Гц), 2,18 (3H, с), 2,22 (3H, с), 3,90 (2H, м), 7,10 (1H, c), 10,82 (1H, ушир.с), 11,94 (1H, ушир.с).
m/z (термонапыление 383 (MNH4 +).
k). 1H-ЯМР (300 МГц, ДМСО-d6) δ = 2,18 (3H, с), 2,22 (3H, с), 3,35 (1H, м), 3,50 (1H, м), 3,70 (1H, м), 4,16 (1H, м), 6,10 (1H, ушир.с). 7,05 (1H, с), 10,85 (1H, ушир.с), 11,95 (1H, ушир.с),
m/z (термонапыление) 382 (MH+), 399 (MNH4 +).
Или же иначе, соединение примера 17 может быть получено следующим образом:
(RS)-N-(1,4-Дигидро-6,7-дихлор-2,3-диоксо-хиноксалин-5-ил)-N- (2-гидроксиэтил) метансульфонамид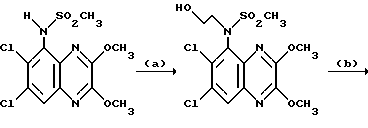
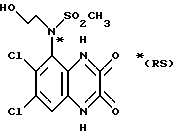
a) Смесь карбоната калия (25,81 г, 0,187 моль), 2-бромэтанола (13,26 мл, 0,187 моль) и N-(6,7-дихлор-2,3-диметоксихиноксалин-5-ил) метансульфонамида (препаративный пример 3) (55,0 г, 0,156 моль) в ацетоне (2,5 л), нагревают до температуры кипения с обратным холодильником в течение 20 час, охлаждают и удаляют ацетон при пониженном давлении. Остаток распределяют между дихлорметаном и 1 М гидроокисью натрия. Затем органический слой сушат (MgSO4), концентрируют при пониженном давлении и остаток чистят трехкратной перекристаллизацией из метанола, что дает (RS)-N-(6,7-дихлор-2, 3-диметоксихиноксалин-5-ил)-N-(2- гидроксиэтил)метансульфонамид (43,7 г, 70%) в виде белого твердого вещества, т. пл. 240-242oC.
Анализ %:
Найдено: C 39,35; H 3,78; N 10,55. Рассчитано для C13H15N3O5Cl2: C 39,41; H 3,82; N 10,61%.
b) Смесь (RS)-N-(6,7-дихлор-2,3-диметоксихиноксалин-5-ил)-N-(2- гидроксиэтил)метансульфонамида (11,41 г, 0,029 моль) и 2 М хлористоводородной (300 мл) кислоты нагревают до температуры кипения с обратным холодильником в течение 18 1/2 часа, затем охлаждают на ледяной бане. Твердый продукт отфильтровывают и промывают водой, что дает указанное в заглавии соединение (9,65 г, 91%) в виде белого твердого вещества. Т.пл. 272 - 274oC.
Анализ %:
Найдено: C 35,82; H 3,04; H 11,37;
Рассчитано для C11H11N3O5Cl2S: C 35,88; H 3,01; N 11,41%.
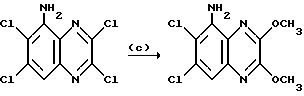
Пример 49. N-(1,4-Диулор-6,7-дихлор-2,3- диоксохиноксалин-5-ил)-N-(3-пиридилметил) метансульфонамида, гидрохлорид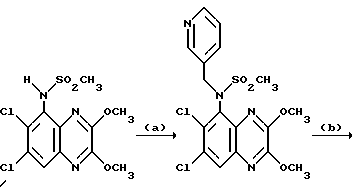
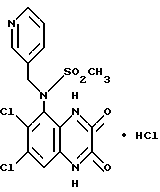
a) Диэтилазодикарбоксилат (90 мкл, 0,57 ммоль) добавляют к перемешиваемому раствору N-(6,7-дихлор-2,3-диметоксихиноксалин-5- ил)метансульфонамида (200 мг, 0,57 ммоль - см. препаративный пример 3), 3-(гидроксиметил) пиридина (55 мкл, 0,57 ммоль) и трифенилфосфина (149 мг, 0,57 ммоль) в сухом тетрагидрофуране (12 мл) в атмосфере азота при 23oC. Через 8 часов растворитель удаляют при пониженном давлении и остаток чистят флэш хроматографией (градиентное элюирование диэтиловым эфиром/метанолом), получая N-(6,7-дихлор-2, 3- диметоксихиноксалин-5-ил)-N-(3-пиридилметил)метансульфонамид (145 мг, 57%) в виде белого твердого продукта, т. пл. 217oC (разл.).
1H-ЯМР (300 МГц, CDCl3): δ = 3,18 (3H, с), 4,10 (3H, с), 4,14 (3H, с), 4,95 (2H, с), 7,17 (1H, дд. J 4 и 6 Гц), 7,68 (1H, дт, J 2 и 6 Гц), 7,90 (1H, с), 8,41 (1H, д, J 2 Гц), 8,48 (1H, дд, J 2 и 4 Гц).
m/z (термонапыление) 443 (MH+).
b) Смесь N-(6,7-дихлор-2,3-диметоксихиноксалин-5-ил)-N-(3- пиридилметил)-метансульфонамида (130 мг, 0,293 ммоль), 2 М соляной кислоты (2 мл) и диоксана (4 мл) нагревают до температуры кипения с обратным холодильником в течение 2,5 часов, охлаждают и концентрируют при пониженном давлении. Остаток суспендируют в воде (1 мл), отфильтровывают и промывают водой и диэтиловым эфиром, получая указанное в заглавии соединение (120 мг, 98%) в виде белого твердого вещества. Т. пл. 234 - 235oC (разл).
Анализ %:
Найдено: C 39,67; H 3,06; N 12,20; S, 7,05. Для C15H12Cl2N4O4S•HCl рассчитано: C 39,88; H 2,90; N 12,40; S 7,10%.
Примеры 50-65. Соединения следующих примеров, приведенных в таблице 3, получают по способу примера 49, используя соответствующее 2,3-диметоксихиноксалиновое производное (препаративные примеры 3 и 4) и соответствующий спирт (выпускаемый промышленно и/или полученный по способам препаративных примеров 15 - 19. Тритилзащитную группу в препаративных примерах 15 - 19 удаляют одновременно на конечной стадии кислотного гидролиза).
Примечания к таблице 3.
a) 1H-ЯМР (300 МГц, ДМСО-d6, уширенные сигналы благодаря таутомерному взаимному превращению) 2,10 (3H, с), 3,22 (3H, ушир. с), 4,80 (2H, ушир.с), 7,38 (1H, с), 8,65 (1H, ушир.с), 12,23 (1H, ушир.с), 14,0 (1H, ушир.с).
b) Как упомянуто выше для реакции Mitsunobu гетероциклы являются тритилзащищенными, как описано в препаративных примерах 15 - 19. Тритилзащитные группы удаляют одновременно с гидролизом диметоксихиноксалина, и побочный продукт, содержащий тритил, удаляют, растирая с ацетоном.
с) 1H-ЯМР (300 МГц, ДМСO-d6) δ = 3,22 (3H, с), 4,73 (1H, д, J 15 Гц), 4,89 (1H, д, J 15 Гц), 7,23 (1H, с), 7,47 (2H, с), 10,68 (1H, ушир.с), 12,10 (1H, ушир.с).
Пример 66. (RS), (RS)-N-(1,4-Дигидро-6,7-дихлор- 2,3-диоксохиноксалин-5-ил)-N-(2-гидроксипропил)метансульфонамид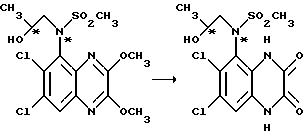
Указанное соединение получают из (RS)-N-(6,7-дихлор- 2,3-диметоксихиноксалин-5-ил)-N-(2-гидроксипропил)метансульфонамида (препаративный пример 9) по способу примера 1: выход 81%, белый твердый продукт (смесь диастереоизомеров), т. пл. 291-292oC (из воды).
Анализ %:
Найдено: C 37,77; H 3,15; N 10,63.
Рассчитано для C12H13Cl2N3O5S: C 37,71; H 3,43; N 10,99%.
Пример 67. (N-(1,4-Дигидро-6,7-дихлор-2,3- диоксохиноксалин-5-ил)-N-(2-гидрокси-2-метилпропил)метансульфонамид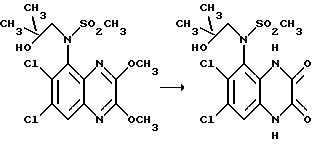
Указанное соединение получают из N-(6,7-дихлор-2,3- диметоксихиноксалин-5-ил)-N-(2-гидрокси-2-метилпропил)метансульфонамида (препаративный пример 10) по способу примера 1: выход 83%, белый продукт, т. пл. 252-253oC (разл.).
Анализ %:
Найдено: C 39,32; H 3,71; N 10,55,
Рассчитано для C13H15Cl2N3O5S 39,40; H 3,81; N 10,60%.
Пример 68. N-(1,4-Дигидро-6-хлор-7-трифторметил-2,3- диоксохиноксалин-5-ил)-N-этилметансульфонамид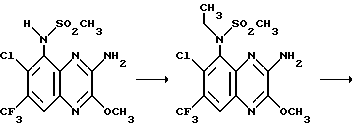
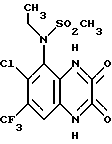
a) Смесь N-(3-амино-6-хлор-7-трифторметил-2-метокси-хиноксалин- 5-ил)метансульфонамида (препаративный пример 20, 73 мг, 0,2 ммоль) и безводного карбоната калия (33 мг, 0,24 ммоль) в ацетоне перемешивают при нагревании до температуры кипения с обратным холодильником в течение 20 мин. Добавляют йодэтан (32 мкл, 0,4 ммоль) и смесь нагревают дополнительно 2 часа. Добавляют дополнительное количество йодэтана (32 мкл, 0,4 ммоль) и нагревание продолжают еще 4 часа. Смесь концентрируют при пониженном давлении и остаток распределяют между водой и этилацетатом. Органический раствор сушат (MgSO4), концентрируют при пониженном давлении и остаток чистят флэш хроматогарфией (градиентное элюирование дихлорметан/метанолом), получая N-(3-амино-6-хлор-7- трифторметил-2-метоксихиноксалин-5-ил)-N-этилметансульфонамид (75 мг, 96%) в виде белого твердого продукта.
1H-ЯМР (300 МГц, CDCl3) δ/ = 1,16 (3H, т, J 7 Гц, (3,20 (3H, с), 3,86 (2H, м), 4,16 (3H, с), 5,50 (2H, ушир.с), 8,06 (1H, с).
m/z (термонапыление) 399 (MH+).
b) Смесь N-(3-амино-6-хлор-7-трифторметил-2-метоксихиноксалин-5-ил)-N-этил-метансульфонамида (стадия (a) приведенная выше, 70 мг, 0,18 ммоль), 2 М соляной кислоты (3 мл) и диоксана (6 мл) нагревают до температуры кипения с обратным холодильником в течение 2 час, охлаждают и концентрируют при пониженном давлении. Остаток суспендируют в воде, фильтруют и твердый продукт промывают водой. После сушки указанное в заглавии соединение (33 мг, 48%) получают в виде белого твердого продукта, т. пл. > 300oC.
Анализ %:
Найдено: C 37,61; H 2,73; N 10,80.
Рассчитано для C12H11ClF3N3O4S: C 37,36; H 2,87; N 10,89%.
Пример 69. N-(1,4-Дигидро-6-хлор-7-трифторметил-2,3- диоксохиноксалин-5-ил)-N-(2-гидроксиэтил)метансульфонамид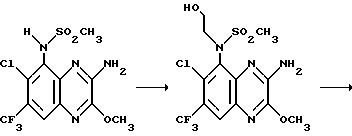
Указанное в заглавии соединение получают по способу примера 68, заменяя йодэтан на 2-бромэтанол. Указанное в заглавии соединение получают в виде белого твердого продукта (40 мг, 44% - выход для двух стадий), т. пл. 292-294oC.
Анализ %:
Найдено: C 36,17; H 2,73; H 10,26.
Рассчитано для C12H11ClF3N3O5S : C 35,88; H 2,76; N 10,46%.
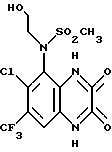
Пример 70. (RS)-N-(Карбоксиметил)-N-(1,4-дигидро-6,7- дихлор-2,3-диоксохиноксалин-5-ил)метансульфонамид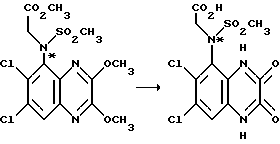
Смесь N-(6,7-дихлор-2,3-диметоксихиноксалин-5-ил)-N- (метоксикарбонилметил)метансульфонамида (препаративный пример 21, 3,17 г, 7,48 ммоль), 2 М соляной кислоты (80 мл) и диоксана (80 мл) нагревают до температуры кипения с обратным холодильником в течение 18 час, охлаждают и концентрируют при пониженном давлении, получая желтый твердый продукт (2,85 г, 100%), т. пл. 271oC (разл).
Анализ %:
Найдено: C 33,98; H 2,64; N 10,50.
Рассчитано для C11H9Cl2N3O6S • 1/2H2O: C 33,77; H 2,58; m 10,74%.
Пример 71. (RS)-N-(1,4-Дигидро-6,7-дихлор-2,3- диоксохиноксалин 5-ил)-N-(метоксикарбонилметил)метансульфонамид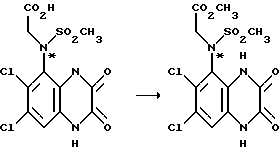
Раствор N-(карбоксиметил)-N-(1,4-дигидро-6,7-дихлор- -2,3-диоксохиноксалин-5-ил)метансульфонамида (пример 70, 2,85 г, 7,46 ммоль) в сухом метаноле (100 мл), насыщенном газообразным хлористым водородом нагревают до температуры кипения с обратным холодильником в течение 3 час, охлаждают и концентрируют при пониженном давлении, получая желтый твердый продукт (2,838 г, 96%), т. пл. 301oC (разл).
Анализ %:
Найдено: C 36,29; H 2,60; N 10,49.
Рассчитано для C12H11Cl2N3O6S: C 36,38, H 2,80; N 10,61%.
Пример 72. N-(1,4-Дигидро-6,7-дихлор-2,3-диоксохиноксалин-5-ил) - N-(N-метилкарбамоилметил)метансульфонамид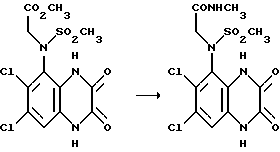
Смесь N-(1,4-дигидро-6,7-дихлор-2,3-диоксохиноксалин-5-ил) -N-(метоксикарбонилметил)метансульфонамида (из примера 71, 150 мг, 0,38 ммоль), этанола (3 мл) и метиламина (33% - раствор в этаноле, 3 мл) нагревают в закрытом сосуде при 75oC в течение 1 часа, затем при 90oC в течение 1,5 час. Смесь охлаждают и медленно выливают в избыток 2 М соляной кислоты. Белый осадок отфильтровывают и сушат, получая указанное в заглавии соединение (107 мг, 72%), т. пл. 289oC.
Анализ %:
Найдено: C 36,24; H 2,99; N 13,98,
Рассчитано для C12H12Cl2N4O5S: C 36,47; H 3,06; N 14,18%.
Соединения, приведенные ниже в таблице 4, получают из соединения примера 71 по способу примера 72, используя соответствующий амин вместо метиламина.
Пример 79. (RS), (RS)-N-(1-Карбоксиэтил)-N-(1,4-дигидро- 6,7-дихлор-2,3-диоксохиноксалин-5-ил)метансульфонамид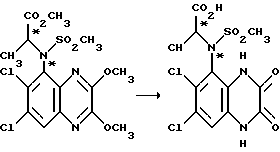
1: 1 смесь двух изомеров N-(6,7-дихлор-2,3-диметоксихиноксалин- 5-ил)-N-(1-метоксикарбонил-1-этил)-метансульфонамида (препаративный пример 23, 1,40 г, 32 ммоль), 2 М соляной кислоты (40 мл) и диоксана (40 мл) нагревают в автоклаве при 130oC в течение 48 часов и при 150oC в течение 24 часов. Смесь охлаждают, концентрируют до небольшого объема при пониженном давлении и твердый продукт отфильтровывают и промывают диэтиловым эфиром. Продукт растворяют в 1 М водной гидроокиси натрия (40 мл) и переосаждают, добавляя 2 М соляную кислоту (до pH 3), Белый твердый продукт отфильтровывают и сушат в вакууме, получая указанное в заглавии соединение (1,13 г, 89%), в виде смеси диастереоизомеров, т. пл. 282oC (разл.).
Анализ %:
Найдено: C 33,68; H 3,17; N 9,61.
Рассчитано для C12H11Cl2N3O6S•0,5H2O: C 34,06; H 3,33; N 9,93%.
Пример 80. (R) и (S)-N-(1,4-Дигидро-6,7-дихлор- 2,3-диоксохиноксалин-5-ил)-N-(2-гидроксиэтил)метансульфонамид
a) (RS)-N-(Карбоксиметил)-N-(1,4-дигидро-6,7-дихлор-2,3- диоксохиноксалин-5-ил)метансульфонамид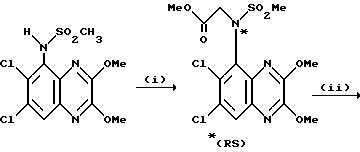
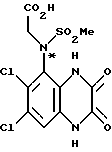
i) Смесь карбоната калия (42,37 г, 0,3 моль), метилбромацетата (48,4 мл, 0,51 моль) и N-(6,7-дихлор-2,3-диметоксихиноксалин-5-ил) метансульфонамида (препаративный пример 3) (90 г, 0,256 моль) в ацетоне (1,75 л), нагревают до температуры кипения с обратным холодильником в течение 8 1/2 часа, охлаждают и удаляют ацетон при пониженном давлении. Остаток перемешивают с водой (1,5 л) в течение 1/4 часа, отфильтровывают и твердый продукт промывают водой и затем диэтиловым эфиром, получают (RS)-N-(6,7-дихлор-2,3- диметоксихиноксалин-5-ил)-N-(метоксикарбонилметил)-метансульфонамид (108 г, 100%).
Анализ %:
Найдено: C 39,51; H 3,52; N 9,89. для C14H15Cl2N3O6S рассчитано: C 39,63; H 3,56; N 9,90%.
ii) Смесь (RS)-N-(6,7-дихлор-2,3-диметоксихиноксалин- 5-ил)-N-(метоксикарбонилметил)метансульфонамида из стадии, (i) приведенной выше, 2 М соляной кислоты (1 л) и диоксана (1 л) нагревают до температуры кипения с обратным холодильником в течение 18 часов, охлаждают и концентрируют при пониженном давлении. Твердый остаток суспендируют в воде (1,5 л), отфильтровывают и промывают водой и диэтиловым эфиром, получая указанное в подзаголовке соединение (95 г, 92%) в виде белого твердого продукта, т. пл. 271oC. (разл. ).
Анализ %:
Найдено: C 33,98; H 2,64; N 10,50.
Рассчитано для C11H9Cl2N3O6S•1/2H2O: C 33,77; H 2,58; N 10,74%.
b) (R)-N-(Карбоксиметил)-N-(1,4-дигидро-6,7-дихлор-2,3- диоксохиноксалин-5-ил)метансульфонамид и (S)-N-(карбоксиметил)-N- (1,4-дигидро-6,7-дихлор-2,3-диоксохиноксалин -5-ил)метансульфонамид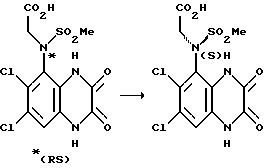
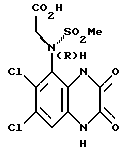
Хинин (25,48 г, 0,078 моль) в этаноле (300 мл) добавляют к нагреваемому до температуры кипения с обратным холодильником раствору (RS)-N-(карбоксиметил)-N-(1,4-дигидро-6,7-дихлор-2,3- диоксохиноксалин-5-ил)метансульфонамида (из стадии (a), 30 г, 0,078 моль) в этаноле (2,1 л). После 1/2 часа нагревания до температуры кипения с обратным холодильником суспензию отфильтровывают горячей и твердый продукт промывают этанолом, получая указанное в подзаголовке соединение (S) стереохимии в виде хининовой соли (23,2 г, 41,3%).
[α]
Фильтрату дают охладиться до комнатной температуры при перемешивании, оставляют дополнительно на 1 час, после чего отфильтровывают, получая указанное в подзаголовке соединение (R)-стереохимии в виде хининовой соли (19,1 г, 34%).
[α]
Хининовые соли отдельно суспендируют в воде (1,3 л) и обрабатывают концентрированной соляной кислотой (22 мл) при энергичном перемешивании, получая два указанных в подзаголовках соединения (после фильтрации).
Указанное в подзаголовке соединение (R)-стереохимии получают в виде белого твердого продукта (9,8 г, 95%) т. пл.>218oC (разл.).
[α]
Анализ %:
Найдено: C 33,51; H 2,32; N 10,46.
Рассчитано для C11H9Cl2N3O6S•1/2H2O: C 33,77; H 2,58; N 10,74%.
Указанное в подзаголовке соединение (S)-стереохимии получают в виде белого твердого продукта (11,9 г, 95%).
с) (R)-N-(1,4-Дигидро-6,7-дихлор-2,3-диоксохиноксалин- -5-ил)-N- (метоксикарбонилметил)метансульфонамид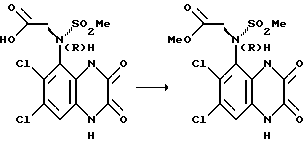
Смесь (R)-N- (карбоксиметил)-N-(1,4-дигидро-6,7-дихлор-2,3-диоксохиноксалин-5-ил) метансульфонамида (из стадии (b), 20,34 г, 0,053 моль) и метанола (266 мл) насыщают газообразным хлористым водородом, перемешивают 18 часов при комнатной температуре, упаривают досуха и остаток суспендируют в метаноле (300 мл). После перемешивания в течение 1/2 часа, твердый продукт отфильтровывают, получая указанное в подзаголовке соединение в виде атропизомера (17,5 г, 83%), т. пл. 290oC (разл.).
[α]
1H-ЯМР (300 МГц, ДМСО-d6): δ = 3,1 (3H, с), 3,75 (3H, с), 4,5 (1H, д), 4,85 (1H, д), 7,35 (1H, с), 10,8 (1H, ушир.с), 12,2 (1H, ушир. с). m/z (термонапыление) 396 (MH+).
d) (R)-N-(1,4-Дигидро-6,7-дихлор-2,3-диоксохиноксалин- 5-ил)-N- (2-гидроксиэтил)метансульфонамид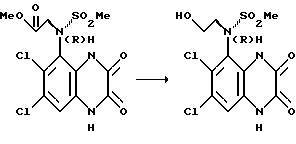
Алюмогидрид лития (39,4 мл, 1 молярн. в ТГФ, 39,4 ммоль) добавляют к (R)-N-(1,4-дигидро-6,7-дихлор-2,3-диоксохиноксалин-5-ил)-N- (метоксикарбонилметил)метансульфонамиду (из стадии (с), 9,75 г, 24,6 ммоль) в тетрагидрофуране (590 мл), охлаждают на ледяной бане до температуры между 0 - 5oC. Через 1/4 часа добавляют дополнительное количество алюмогидрида лития (2,4 мл, 2,46 ммоль), смесь перемешивают еще 1/2 часа и добавляют метанол в тетрагидрофуране (60 мл).
Смесь упаривают досуха при пониженном давлении и остаток распределяют между этилацетатом и 2 М соляной кислотой. Органические экстракты сушат над сульфатом натрия и концентрируют при пониженном давлении. Остаток чистят флэш хроматографией, используя градиентное элюирование (CH2Cl2:MeOH - смесь, содержащая 10% AcOH 100:0 ---> 95:5), получая первое указанное в заглавии соединение в виде отдельного атропизомера, (6,0 г, 66%), т. пл. 293 - 294oC.
[α]
Анализ %:
Найдено: C 35,89; H 2,83; N 11,42.
Рассчитано для C11H11Cl2N3O5S: C 35,88; H 3,01; N 11,41
e) (S)-N-(1,4-Дигидро-6, 7-дихлор-2,3-диоксохиноксалин-5-ил)-N- (метоксикарбонилметил)метансульфонамид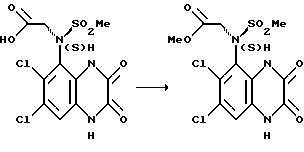
Указанное в подзаголовке соединение получают из (S)-N-карбоксиметил-N-(1,4-дигидро-6,7-дихлор-2,3-диоксохиноксалин-5-ил)метансульфонамида (стадия (b)) по способу стадии (c); выход 78% в виде белого твердого вещества.
1H-ЯМР (300 МГц, ДМСО-d6): δ = 3,30 (3H, с), 3,75 (3H, с), 4,32 (1H, д), 4,85 (1H, д), 7,35 (1H,с), 10,85 (1H, с), 12,60 (1H, с).
m/z (термонапыление) 396 (MH+).
f) (S)-N-(1,4-Дигидро-6,7-дихлор-2,3-диоксохиноксалин- 5-ил)-N-(2-гидроксиэтил)метансульфонамид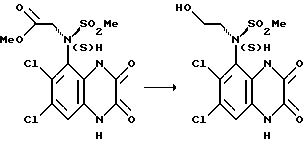
Второе указанное в заглавии соединение получают из (S)-N-(1,4-дигидро-6,7-дигидро-2,3-диоксохиноксалин-5-ил)-N- (метоксикарбонилметил)метансульфонамида (стадия (е)) по способу стадии (d): выход. 60% в виде белого твердого
продукта, т. пл. >300oC.
[α]
1H-ЯМР (300 МГц, ДМСО-d6): δ/ = 3,21 (5H, м), 3,65 (1H, м), 4,03 (1H, м), 6,02 (1H, ушир. с), 7,32 (1H, с), 11,00 (1H, ушир. с), 12,12 (1H, ушир. с).
m/z (термонапыление) 369 (MH+).
Пример 81. (RS), (RS)-N-(1,4-Дигидро-6,7-дихлор-2,3- диоксохиноксалин-5-ил)-N-(1-метоксикарбонилэтил)метансульфонамид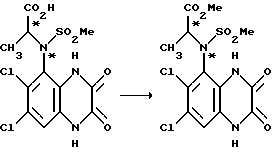
N-(1-Карбоксиэтил)-N-(1,4-дигидро-6,7-дихлор- 2,3- диоксохиноксалин-5-ил)метансульфонамид (из примера 79, 1 г, 2,53 ммоль) в метаноле (100 мл) насыщенный газообразным хлористым водородом, перемешивают при комнатной температуре 24 часа, затем 8 часов при 60oC. Твердый продукт отфильтровывают, получая указанное в заглавии соединение в виде смеси диастереомеров (431 мг, 42%).
1H-ЯМР (300 МГц, ДМСО-d6): δ = 1,70 (3H, д), 3,17 (3H, с), 3,77 (3H, с), 4,75 (1H, кв.), 7,39 (1H, с), 11,46 (1H, с), 12,20 (1H, с).
m/z (термонапыление) 413, 415 (MNH4 +).
Пример 82. (RS),(RS )-N-(1,4-Дигидро-6,7-дихлор-2,3-диоксохиноксалин-5-ил)-N- (1-(N'-метилкарбамоил)этил)метансульфонамид
N-(1,4-Дигидро-6,7-дихлор-2,3-диоксохиноксалин-5-ил)-N- (1-метоксикарбонилэтил)метансульфонамид (из примера 81, 110 мг, 0,27 ммоль) в 33% - метиламине в этаноле (6 мл) нагревают при 100oC 5 часов в запаянной ампуле, охлаждают и добавляют к 2 М соляной кислоте (400 мл). Полученный твердый продукт отфильтровывают, растворяют в 1 М гидроокиси натрия, осаждают 2 М соляной кислотой и отфильтровывают, получая указанное в заглавии соединение в виде смеси диастереомеров (63 мг, 57%), т. пл. 250oC (разл.).
Анализ %:
Найдено: C 37,98; H 3,37; N 13,19.
Рассчитано для C13H14Cl2N4O5S: C 37,66; H 3,55; N 13,51.
Пример 83. N-(1,4-Дигидро-7-хлор-6-фтор-2,3-диоксохиноксалин-5-ил)- N-(2-гидроксиэтил)метансульфонамид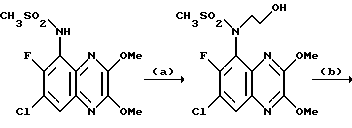
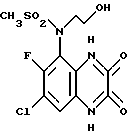
a) N- (7-Хлор-6-фтор-2,3-диметоксихиноксалин-5-ил)метансульфонамид (препаративный пример 25) превращают по способу примера 17 (a) в N-(7-Хлор-6-фтор-2,3-диметоксихиноксалин-5-ил)-N-(2-гидроксиэтил) метансульфонамид. Продукт получают в виде белого твердого вещества (91% выход), т. пл. 209 - 210oC.
1H-ЯМР (300 МГц, CDCl3): δ = 3,18 (3H, с), 3,32 (1H, м), 3,50 (1H, м), 3,74 (2H, м), 4,08 (1H, м), 4,14 (3H, с), 4,20 (3H, с), 7,90 (1H, д, J 8 Гц).
m/z (термонапыление) 380 (MH+).
b) N- (7-Хлор-6-фтор-2,3-диметоксихиноксалин-5-ил)-N-(2- гидроксиэтил)-метансульфонамида [из стадии (a)] превращают по способу примера 17 (b) в N-(1,4-дигидро-7-хлор-6-фтор-2,3- диоксохиноксалин-5-ил)-N-(2-гидроксиэтил) -метансульфонамид. Продукт получают в виде белого твердого вещества (86%), т. пл. 298 - 300oC.
Анализ %:
Найдено: C 37,44; H 3,00; N 11,79.
Рассчитано для C11H11ClFN3O5S: C 37,56; H 3,15; N 11,95%.
Пример 84. N-(1,4-Дигидро-6-хлор-7-фтор-2,3- диоксохиноксалин-5-ил)-N-(2-гидроксиэтил)метансульфонамид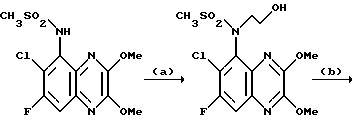
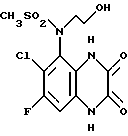
a) N- (6-Хлор-7-фтор-2,3-диметоксихиноксалин-5-ил)метансульфонамид (препаративный пример 26) превращают по способу примера 17(a) в N-(6-Хлор-7-фтор-2,3-диметоксихиноксалин-5-ил)-N-(2-гидроксиэтил) метансульфонамид. Продукт получают в виде белого твердого вещества (68% выход).
1H-ЯМР (300 МГц, CDCl3): δ/ = 3,26 (3H, с), 3,50-4,10 (4H, м), 4,16 (3H, с), 4,20 (3H, с), 7,60 (1H, д, J 10 Гц).
m/z (термонапыление) 380, 382 (MH+).
b) N-(6-Хлор-7-фтор-2,3-диметоксихиноксалин-5-ил)-N-(2- гидроксиэтил)метансульфонамид [из стадии (a)] превращают по способу примера 17(b) в N-(1,4-дигидро-6-хлор-7-фтор-2, 3-диоксохиноксалин-5- ил)-N-(2-гидроксиэтил)метансульфонамид. Продукт получают в виде белого твердого вещества (75% выход), т. пл. 290-291oC.
Анализ %:
Найдено: C 37,62; H 3,10; N 11,88.
Рассчитано для C11H11ClFN3O5S: C 37,56; H 3,15; N 11,95%.
Пример 85. N-(1,4-Дигидро-6,7-дихлор-2,3-диоксохиноксалин-5-ил)-N- (2-аминоэтил)метансульфонамид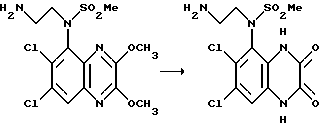
Указанное в заглавии соединение получают из N-(6,7-дихлор-2,3- диметоксихиноксалин-5-ил)-N-(2-аминоэтил)метансульфонамида (препаративный пример 27), 40 мг, 0,101 ммоль) по способу примера 7(b) в виде белого твердого соединения (18 мг, 48%), т. пл. >300oC.
Анализ %:
Найдено: C 31,85; H 3,74; N 13,15.
Рассчитано для C11H12Cl2N4O4S•HCl• 2/5H2O•1/10 CH2Cl2: C 31,79; H 3,36; N 13,36%.
Пример 86. N-(1,4-Дигидро-6,7-дихлор-2,3-диоксохиноксалин-5-ил)- N-(2-фталимидоэтил)метансульфонамид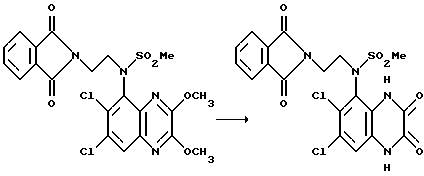
Указанное в заглавии соединение получают по способу примера 85 из N-(6,7-дихлор-2,3-диметоксихиноксалин-5- -5-ил)-N-(2-фталимидоэтил)-метансульфонамида (из препаративного примера 27 (a), 150 мг, 0,285 ммоль) в виде белого твердого вещества (131 мг, 92%), т. пл.>300oC.
1H-ЯMP (300 МГц, d6 - ДМСО); δ = 3,25 (3H, с), 3,70 - 3,82 (2H, м), 3,91 - 4,07 (2H, м), 7,25 (1H, с), 7,80 (4H, с), 11,09 (1H, с), 12,15 (1H, с).
m/z (термонапыление) 497 (MH+).
Пример 87. N-(1,4-Дигидро-6,7-дихлор-2,3-диоксохиноксалин-5-ил)-N-(2- (N'-трифторметансульфонил)аминоэтил)метансульфонамид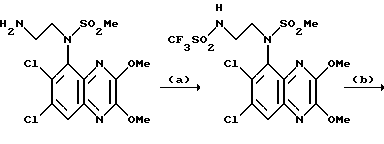
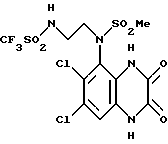
a) Триэтиламин (13 мкл, 0,139 ммоль) и затем ангидрид трифторметансульфоновой кислоты (23 мкл, 39 мг, 0,139 ммоль) добавляют по каплям к перемешиваемому раствору N-(6,7-дихлор-2,3-диметоксихиноксалин- 5-ил)-N-(2-аминоэтил)-метансульфонамида (из препаративного примера 27, 50 мг, 0,126 ммоль) в дихлорметане (1,5 мл) при -78oC в атмосфере азота. Смесь перемешивают в течение 30 минут и затем дают нагреться до комнатной температуры. Смесь промывают водой, насыщенным водным раствором бикарбоната натрия и раствором соли и затем сушат (MgSO4). Концентрация при пониженном давлении дает N-(6,7-дихлор-2,3-диметоксихиноксалин-5-ил)-N-(2-(N'-трифторметансульфонил) аминоэтил)метансульфонамид в виде бледно-желтого твердого продукта (50 мг, 75%).
1H-ЯМР (300 МГц, CDCl3): δ = 3,20 (3H, с), 3,20-3,30 (1H, м), 3,52-3,62 (1H, м), 3,86-3,96 (1H, м), 4,04-4,17 (1H, м), 4,18 (3H, с), 4,23 (3H, с), 8,00 (1H, с).
1 m/z (термонапыление) 527 (MH+).
b) Указанное в заглавии соединение получают по способу примера 7(b) из N-(6,7-дихлор-2,3-диметоксихиноксалин-5-ил)-N-(2-(N'- трифторметансульфонил)аминоэтил)метансульфонамида [из стадии (a)] в виде твердого вещества (60%) т. пл. 203,8 - 207,7oC.
Анализ %:
Найдено: C 29,13; H 2,77; N 9,96;
Рассчитано для C12H11N4S2O6Cl2F3 • H2O•3/10 Et2O: C 29,39; H 2,99; N 10,38.
Пример 88. N-(1,4-Дигидро-6,7-дихлор-2, 3-диоксохиноксалин-5-ил)-N-(2-[мeтилaминoкapбoнил]aминoэтил) метансульфонамид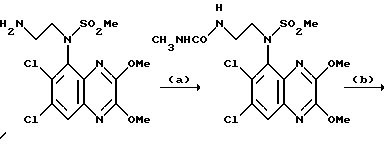
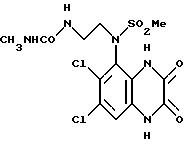
a) Метилизоцианат (8,2 мкл, 8,0 мг, 0,14 ммоль) добавляют к раствору N-(6,7-дихлор-2,3-диметоксихиноксалин-5-ил)-N-(2- аминоэтил)метансульфонамида (из препаративного примера 27, 50 мг, 0,127 ммоль) в дихлорметане (2 мл) при комнатной температуре и в атмосфере азота. Через 30 минут смесь концентрируют при пониженном давлении. Остаток растворяют в этилацетате и концентрируют при пониженном давлении, получая N-(6,7-дихлор-2,3-диметоксихиноксалин- 5-ил)-N-(2-[метиламинокарбонил]аминоэтил)метансульфонамид в виде бледно-желтой пены (52 мг, 91%).
1H-ЯМР (300 МГц, CDCl3): δ = 2,74 (3H, д, J 2 Гц), 3,18 (3H, с), 3,36 (2H, м), 3,92 (2H, м), 4,15 (3H, с), 4,17 (3H, с), 4,2 (1H, ушир.с), 5,14 (1H, ушир, с), 7,96 (1H, с).
m/z (термонапыление) 452 (MH+).
b) Указанное в заглавии соединение получают по способу примера 7(b) из N-(6,7-дихлор-2,3-диметоксихиноксалин-5-ил)-N-(2- [метиламинокарбонил] аминоэтил)метансульфонамида [из стадии (a)] в виде бледно-желтой пены (60%).
Анализ %:
Найдено: C 32,90; H 3,90; N 14,50.
Рассчитано для C13H15N5O5Cl2S•2 1/2 H2O: C, 33,27; H, 4,30; N, 14,92%.
1H-ЯМР (300 МГц, d16 - ДМСО): δ = 2,43 (3H, с), 3,21 (3H, с), 3,59-3,65 (2H, м), 3,75-3,86 (2H, м), 7,40 (1H, с), 10,60 (1H, с), 12,11 (1H,с).
Пример 89. N-(1,4-Дигидро-6,7-дихлор-2,3-диоксохиноксалин-5-ил)-N-(5-тетразолилметил) метансульфонамид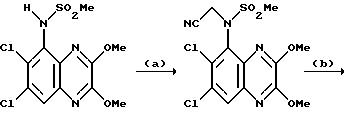
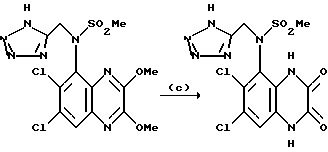
a) Хлорацетонитрил (233 мкл, 279 мг, 3,69 ммоль) добавляют к нагреваемой до температуры кипения с обратным холодильником смеси N-(6,7-дихлор-2,3-диметоксихиноксалин-5-ил)метансульфонамида (из препаративного примера 3, 1,00 г, 2,84 ммоль) и карбоната калия (0,47 г, 3,41 ммоль) в ацетоне (50 мл) в атмосфере азота. Смесь нагревают до температуры кипения с обратным холодильником 18 часов и затем дают охладиться и распределяют между этилацетатом (500 мл) и водой (500 мл). Органическую фазу сушат (MgSO4) и концентрируют при пониженном давлении. Остаток чистят флэш хроматографией (элюируя 0-100% этилацетата в гексане и затем 5%-метанолом в дихлорметане), получая N-(6,7-дихлор-2,3-диметоксихиноксалин-5-ил)-N-(цианометил) метансульфонамид в виде не совсем белого твердого продукта (600 мг, 54%).
1H-ЯМР (300 МГц, ДМСO-d6): δ = 3,31 (3H, с), 4,07 (3H, с), 4,08 (3H, с), 4,84 (1H, д, J = 19 Гц), 5,10 (1H, д, J = 19 Гц), 8,11 (1H, с).
b) Смесь N-(6,7-дихлор-2,3-диметоксихиноксалин-5-ил)-N- (цианометил)метансульфонамида (100 мг, 0,256 ммоль) и трибутилоловоазида (170 мг, 0,512 ммоль) (Synthesis, 1976, 329) в толуоле (10 мл) нагревают до температуры кипения с обратным холодильником в течение 18 часов. После охлаждения смесь концентрируют при пониженном давлении и остаток чистят флэш хроматографией (градиентное элюирование из смеси от дихлорметана до 90:10:1 дихлорметан: метанол: аммиак), что дает N-(6,7-дихлор-2,3- диметоксихиноксалин-5-ил)-N-(5-тетразолилметил) метансульфонамид в виде не совсем белого твердого вещества (78 мг, 70%).
1H-ЯМР (300 МГц, CDCl3): δ =3,30 (3H, с), 4,14 (3H, с), 4,18 (3H, с), 5,12 (1H, д, J 16 Гц), 5,31 (1H, д, J 16 Гц), 7,99 (1H, с),
m/z (термонапыление) 434 (MH+).
с) Указанное в заглавии соединение получают по способу примера 7(b) из N-(6,7-дихлор-2,3-диметоксихиноксалин-5-ил)-N-(5-тетразолилметил) метансульфонамида, но растирая с водой вместо диэтилового эфира, что дает белый твердый продукт (66%) т.пл. > 300oC.
Анализ %:
Найдено: C 32,79; H 2,15; N 23,25.
Рассчитано для C11H9N7O4Cl2S• 1/10 диоксан: C 32,99; H 2,38; N 23,62%.
Пример 90. (1,4-Дигидро-6,7-дихлор-2,3- диоксохиноксалин-5-ил)метил-метил сульфон
Смесь (6,7-дихлор-2,3-диметоксихиноксалин-5-ил)-метил метил сульфона (80 мг, 0,228 ммоль) (препаративный пример 29), 2 М соляной кислоты (1 мл) и диоксана (3 мл) нагревают до температуры кипения с обратным холодильником в течение 3 часов, охлаждают и концентрируют при пониженном давлении. Остаток разбавляют водой и полученный белый твердый продукт собирают фильтрацией, промывают водой и диэтиловым эфиром, и сушат при пониженном давлении при 60oC, получая указанное в заглавии соединение (58 мг, 79%) в виде белого твердого продукта, т. пл. > 300oC.
Анализ %:
Hайдено: C 37,35; H 2,35; N 8,44.
Рассчитано для C10H8N2O4Cl2S: C 37,17; H 2,50; N 8,67%.
Пример 91. (1,4-Дигидро-6,7-дихлор-2,3-диоксохиноксалин-5-ил) метил-этил сульфон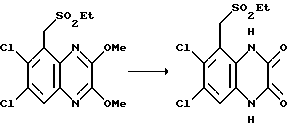
Указанное в заглавии соединение получают из (6,7-дихлор-2,3- диметоксихиноксалин-5-ил) метил-этил сульфона (препаративный пример 30) по способу примера 90 в виде белого твердого продукта (65%), т. пл. > 300oC.
Анализ %:
Найдено: C 39,21; H 2,99; N 8,25; S 9,70.
Рассчитано для C11H10N2O4Cl2S: C 39,18; H 2,99; N 8,31; S 9,51%.
Пример 92. (1,4-Дигидро-6,7-дихлор-2,3- диоксохиноксалин-5-ил)метилбензил сульфон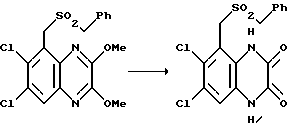
Указанное в заглавии соединение получают из (6,7-дихлор-2,3-диметоксихиноксалин-5-ил) метилбензил сульфона (препаративный пример 31) по способу примера 90 в виде белого твердого вещества (92%), т.пл. > 300oC.
Анализ %:
Hайдено: C 48,30; H 3,12; N 6,65.
Рассчитано для C16H12N2O4Cl2S•0,2 диоксан: C 48,40; H 3,29; N 6,72%.
Пример 93. 1-(1,4-Дигидро-6,7-дихлор-2,3-диоксохиноксалин-5-ил)-3-бутенил-метилсульфон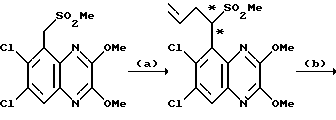
a) Раствор (6,7-дихлор-2,3-диметоксихиноксалин-5-ил)-метил метил сульфона (50 мг, 0,142 ммоль) (препаративный пример 29) и диаллил карбоната (41 мкл, 40 мг, 0,285 ммоль) в сухом тетрагидрофуране (0,8 мл) добавляют с помощью канюли к смеси трис(дибензилиденацетон) дипалладий(O)•хлороформ-аддукта (7,4 мг, 0,007 ммоль) и 1,2-бис(дифенилфосфино)этана (11,3 мг, 0,028 ммоль) в сухом тетрагидрофуране (0,6 мл) в атмосфере азота при комнатной температуре. Смесь перемешивают при комнатной температуре 5 минут и затем при нагревании до температуры кипения с обратным холодильником в течение 2 часов. Смеси дают охладиться, разбавляют дихлорметаном и концентрируют при пониженном давлении. Остаток чистят флэш хроматографией (элюирование 3:1 гексан:этилацетатом), получая 1-(6,7-дихлор-2,3- диметоксихиноксалин-5-ил)-3-бутенил метил сульфона в виде смеси диастереоизомеров (приблизительно 20:1 по 1H-ЯМР) в виде белой пены (29 мг, 52%).
1H-ЯМР (300 МГц, CDCl3): δ = (в основном только диастереоизомер) 2,93 (3H, с), 3,36 (1H, м), 3,84 (1H, м), 4,16 (3H, с), 4,25 (3H, с), 5,00 (2H, м), 5,45 (1H, м), 5,60 (1H, м), 7,96 (1H, с).
m/z (термонапыление) 391 (MH+).
b) Смесь 1-(6,7-дихлор-2,3-диметоксихиноксалин-5-ил)-3-бутенил метил сульфона (из стадии (a), 27 мг, 0,069 ммоль), 2 М соляной кислоты (0,5 мл) и диоксана (1 мл) нагревают при 90oC в течение 15 часов, охлаждают и концентрируют при пониженном давлении. Остаток обрабатывают ультразвуком с диэтиловым эфиром и несколькими каплями метанола и образовавшийся белый твердый продукт собирают фильтрацией, промывают диэтиловым эфиром и сушат, получая указанное в заглавии соединение (17 мг, 68%) в виде белого порошка, т. пл. 270,5 - 272oC.
Анализ %:
Найдено: C 42,98; H3,35; N 7,45.
Рассчитано для C13H12N2Cl2O4S: C 42,99; H 3,33; N 7,71%.
Пример 94. 1-(1,4-Дигидро-6,7-дихлор-2,3- диоксохиноксалин-5- ил)-3-гидроксипропил-метил сульфон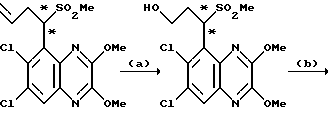
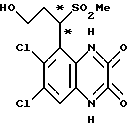
a) Озон барботируют через раствор 1-(6,7-дихлор-2,3-диметоксихиноксалин-5-ил)-3-бутенил метил сульфона (50 мг, 0,128 ммoль) [(пример 93(a)] в сухом дихлорметане (1,3 мл) при -78oC до появления синей окраски. Смесь перемешивают в течение 5 минут и затем продувают током кислорода и потом - азота. Добавляют метанол (1,3 мл) и смеси дают уравновеситься при -78oC перед добавлением гидроокиси натрия (12 мг, 0,319 ммоль) двумя порциями. Смесь перемешивают в течение 5 минут и затем дают нагреться до комнатной температуры. Смесь выливают в 2 М соляную кислоту (20 мл) и экстрагируют дихлорметаном (2 х 20 мл). Объединенные экстракты промывают солевым раствором (20 мл), сушат (MgSO4) и концентрируют при пониженном давлении. Остаток чистят флэш хроматографией (элюируя 1:1 гексаном:этилацетатом), что дает 1-(6,7-дихлор-2,3-диметоксихиноксалин-5-ил)-3-гидроксипропил метил сульфон (35,6 мг, 70%) в виде белой пены.
1H-ЯМР (300 МГц, CDCl3): δ = 2,96 (3H, с), 3,05 (2H, м), 3,51 (1H, м), 3,87 (1H, м), 4,15 (3H, с), 4,22 (3H, с), 5,18 (1H, дд, J 6,8 Гц), 7,98 (1H, с).
m/z (термонапыление) 395 (MH+).
b) Смесь 1-(6,7-дихлор-2,3-диметоксихиноксалин-5-ил)-3- гидроксипропилметилсульфона (стадии (a)) (32,4 мг, 0,082 ммоль), 2 М соляной кислоты (0,5 мл) и диоксана (1 мл) нагревают при температуре кипения с обратным холодильником в течение 16 часов, охлаждают и концентрируют при пониженном давлении. Остаток обрабатывают ультразвуком и образовавшийся твердый продукт собирают фильтрацией, промывают диэтиловым эфиром и сушат при пониженном давлении при 60oC, что дает указанное в заглавии соединение (24,7 мг, 82%) в виде бледно-желтого твердого продукта, т. пл. 267 - 269oC.
Анализ %:
Найдено: C 39,12; H 3,21; N 7,81.
Рассчитано для C12H12N2Cl2O5S: C 39,25; H 3,29; N 7,63%.
Пример 95. 1-(1,4-Дигидро-6,7-дихлор-2,3-диоксохиноксалин-5- ил)-4-гидроксибутилметилсульфон
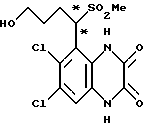
Раствор 9-борабицикло[3.3.1] нонана в тетрагидрофуране (0,5 М, 1,07 мл, 0,537 ммоль) добавляют к перемешиваемому раствору 1- (6,7-дихлор-2,3-диметоксихиноксалин-5-ил)-3-бутенилметилсульфона (пример 93 (a)) (200 мг, 0,511 ммоль) при комнатной температуре в атмосфере азота. Смесь перемешивают в течение 20 часов и затем добавляют порциями триметиламин N-оксид (119 мг, 1,58 ммоль). Смесь перемешивают при комнатной температуре 2 часа и затем нагревают до температуры кипения с обратным холодильником в течение 30 минут, охлаждают и концентрируют при пониженном давлении. Остаток чистят флэш хроматографией (элюируя 1:1 гексан:этилацетатом, затем чистым этилацетатом), получая 1-(6,7-дихлор-2,3-диметоксихиноксалин-5-ил) - 4-гидроксибутилметилсульфон (120 мг, 57%) в виде смеси диастереоизомеров (по 1H-ЯМР).
1H-ЯМР (300 МГц, CDCl3): δ = (в основном только диастереоизомер) 1,29 (1H, м), 1,40 (1H, м), 2,66 (1H, м), 2,89 (3H, с), 3,26 (1H, м), 3,65 (2H, м), 4,15 (3H, с), 4,22 (3H, с), 5,42 (1H, дд, J 6,8 Гц), 7,98 (1H, с).
m/z (термонапыление) 409 (MH+).
b) Указанное в заглавии соединение получают из 1-(6,7- дихлор-2,3-диметоксихиноксалин-5-ил)-4-гидроксибутилметилсульфона (стадия (a), 92 мг, 0,225 ммоль) по способу примера 94 (b) и обрабатывают ультразвуком со смесью диэтилового эфира, метанола и диизопропилового эфира, что дает бледно-серый твердый продукт (43 мг, 53%), т. пл. 306-307,5oC (по ЯМР-отдельный неотнесенный диастереоизомер).
Анализ %:
Найдено: C 41,06; H 3,76; N 7,26;
Рассчитано для C13H14Cl2N2O5S: C 40,96; H 3,70; N 7,35%
1H-ЯМР (300 МГц, d6-ДМСО) δ = 1,15 (1H, м), 1,32 (1H, м), 2,20 (1H, м), 2,37 (1H, м), 3,30 (5H, невидим., м), 5,32(1H, дд, J 6,8 Гц), 7,38 (1H, с), 10,31 (1H, с), 12,12 (1H, с).
Пример 96. Способность некоторых соединений из приведенных примеров к связыванию глициновых участков NMDA-рецептора определяют приведенным выше испытанием (a), и соединения, для которых найдено, что они имеют IC50 менее 50 нМ, включают соединения следующих примеров: 1, 8, 17, 29, 40, 56, 70, 80(b) (первое соединение) и 80(d).
Получение синтетических промежуточных соединений.
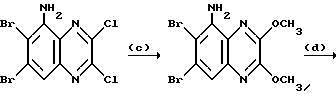
Препаративный пример 1. 5-Амино-6,7-дихлор-2,3- диметоксихиноксалин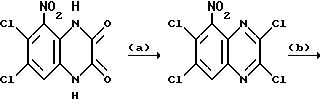
a) Смесь 6,7-дихлор-1,4-дигидро-5-нитрохиноксалин-2,3-диона (пример 1 из WO-A-94/00124, 84 г, 0,34 моль), тионилхлорида (840 мл) и диметилформамида (0,5 мл) нагревают при температуре кипения с обратным холодильником в течение 3 часов, охлаждают и концентрируют при пониженном давлении. Добавляют этилацетат (300 мл) и удаляют при пониженном давлении, после чего - петролейный эфир (т. кип. 100 - 120oC). Твердый остаток перекристаллизовывают из петролейного эфира (т. кип. 100-120oC), получая 2,3,6,7-тетрахлор-5-нитрохиноксалин (78 г, 73%) в виде бледно-желтого твердого вещества.
1H-ЯМР (300 МГц, CDCl3): δ = 8,6 (1H с).
b) Дигидрат хлорида олова (II) (346,3 г, 1,54 моль) добавляют к раствору продукта из стадии (a), приведенной выше (96,2 г, 0,31 моль) в этилацетате (1,8 л). Смесь нагревают до температуры кипения с обратным холодильником в течение 4 часов, охлаждают и осторожно выливают в избыток насыщенного водного бикарбоната натрия. Смесь фильтруют через "Целит", (Торговая марка), хорошо промывая этилацетатом. Плотный осадок на фильтре вымачивают в дополнительном количестве этилацетата и твердый продукт отфильтровывают. Объединенные этилацетатные растворы сушат (MgSO4) и концентрируют при пониженном давлении, получая 5-амино-2,3,6,7-тетрахлорхиноксалин (73,4 г, 84%) в виде желтого твердого продукта.
1H-ЯМР (300 МГц, CDCl3): δ = 5,45(2H, ушир, с), 7,47 (1H, с), m/z (термонапыление) 385 (MH+).
с) Раствор метилата натрия (25%-раствор в метаноле, 274 мл, 1,28 моль) добавляют к суспензии 5-амино-2,3,6,7-тетрахлорхиноксалина (72,4 г, 0,256 моль) в сухом метаноле (1 л) и образовавшуюся смесь нагревают до температуры кипения с обратным холодильником в течение 30 мин. Смесь охлаждают, концентрируют при пониженном давлении и остаток распределяют между водой и этилацетатом (всего 8 л). Органический раствор сушат (MgSO4) и концентрируют при пониженном давлении. Сырой продукт чистят, растирая в порошок с метанолом, с последующим растворением в дихлорметане (2 л) и фильтрацией. Фильтрат концентрируют при пониженном давлении, получая указанное в заглавии
соединение в виде желтого твердого вещества (55,0 г, 79%).
1H-ЯМР (300 МГц, CDCl3): δ = 4,13 (3H, с), 4,14 (3H, с), 5,07 (2H, ушир. с), 7,26 (1H, с).
m/z (термонапыление) 274 (MH+).
Препаративный пример 2. 5-Амино-2,3-диметокси-6,7-диметилхиноксалин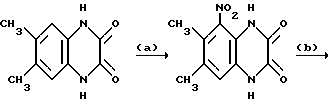
a) 1,4-Дигидро-6,7-диметилхиноксалин-2,3-дион (10,0 г, 52,6 ммоль - см. T. Liebigs Ann. Chem., 1982, 754-761) добавляют порциями в течение более 10 минут к концентрированной азотной кислоте (плотность 1,42 г•см-3, 100 мл) при 0oC. Через 5 минут охлаждающую баню удаляют и смесь перемешивают при 0oC в течение 7 часов, используя при необходимости охлаждение. Раствор выливают в ледяную воду и полученный твердый продукт отфильтровывают и сушат в вакууме при 75oC, получая 1,4-дигидро-6,7-диметил-5- нитрохиноксалин-2,3-дион (7,44 г, 60%) в виде бледно-желтого твердого вещества, т. пл. 280-290oC (разл.) (из диметилформамид/вода).
1H-ЯМР (300 МГц, ДМСО-d6): δ = 2,08 (3H, с), 2,25 (3H, с) 7,06 (1H, с), 11,70 (1H, ушир, с), 12,06 (1H, ушир, с), νmax (RBr) 3185, 1703, 1533, 1400, 1355 см-1.
m/z (термонапыление) 253 (MNH4 +).
b) Смесь 1,4-дигидро-6,7-диметил-5-нитрохиноксалин-2,3-диона (из стадии (a), 7,44 г, 31,6 ммоль), тионилхлорида (69,2 мл, 0,949 моль) и диметилформамида (0,25 мл, 3,16 ммоль) нагревают при температуре кипения с обратным холодильником в течение 3 часов и постепенно добавляют к энергично перемешиваемой смеси льда и воды (1,2 л) в течение более 15 минут. Полученный осадок отфильтровывают и сушат в вакууме при 80oC, что дает 2,3-дихлор-6,7-диметил-5-нитрохиноксалин (8,34 г, 97%), в виде бледно-оранжевого твердого соединения, т. пл. 133-134oC.
1H-ЯМР (300 МГц, ДМСО-d6): δ =2,38 (3H, с), 2,54 (3H, с), 8,12 (1H, с).
νmax (KBr) 1537, 1388, 1377, 1269, 1169 см-1
m/z (термонапыление) 289 (MNH4 +).
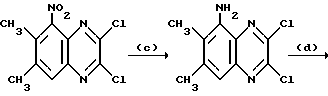
с) Смесь 2,3-дихлор-6,7-диметил-5-нитрохиноксалина (из стадии (b), 8,33 г, 30,6 ммоль) и дигидрата хлорида олова (34,54 г, 153 ммоль) в этилацетате (300 мл) нагревают до температуры кипения с обратным холодильником в течение 11 часов. Добавляют дополнительную порцию дигидрата хлорида олова (13,82 г, 61,2 ммоль) и смесь нагревают в течение 2 часов и разбавляют этилацетатом (500 мл). Смесь добавляют к насыщенному водному бикарбонату натрия (200 мл) и фильтруют, промывая хорошенько плотный осадок на фильтре этилацетатом. Органический слой отделяют, промывают насыщенным водным бикарбонатом натрия (3 х 100 мл), сушат (MgSO4) и концентрируют при пониженном давлении. Остаток чистят флэш хроматографией (градиентное элюирование метанол/дихлорметаном), получая 5-амино-2,3-дихлор-6,7- диметилхиноксалина (6,15 г, 83%), в виде оранжевого твердого продукта, т. пл. 178-180oC.
1H-ЯМР (300 МГц, ДМСО-d6): δ = 2,38 (3H, с), 2,54 (3H, с), 8,12 (1H, с) νmax (KBr) 3475, 1613, 1267, 1178 см-1.
m/z (термонапыление) 242 (MNH4 +).
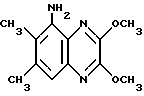
d) Метилат натрия (25% раствор в метаноле, 13,9 мл, 61 ммоль) добавляют за 12 минут к перемешанному раствору 5-амино-2,3-дихлор-6,7-диметилхиноксалин (из стадии (с), 6,15 г, 25,4 ммоль) в сухом тетрагидрофуране (250 мл) в атмосфере азота при 0oC. Смесь перемешивают при 0oC в течение 20 минут и при комнатной температуре в течение 72 часов. Смесь разбавляют этилацетатом (750 мл), промывают водой (2 х 250 мл) и раствор соли (250 мл), сушат (МgSO4) и концентрируют при пониженном давлении. Остаток чистят флэш хроматографией (градиентное элюирование гексан/дихлорметаном), что дает указанное в заглавии соединение в виде белого твердого продукта (4,55 г, 77%), т. пл. 166 - 167oC.
1H-ЯМР (300 МГц, CDCl3): δ =2,32 (3H, с), 2,35-(3H, с), 4,14 (3H, с), 4,15 (3H, с), 5,06 (2H, ушир, с), 7,06 (1H, с).
νmax (KBr) 3540, 2950, 1600, 1535, 1395, 1335, 1240 см-1
m/z (термонапыление) 234 (MH+).
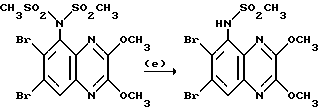
Препаративный пример 3. N-(6,7-Дихлор-2,3-диметоксихиноксалин- 5-ил)метансульфонамид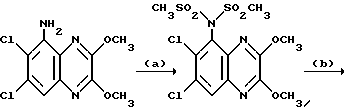
a) Смесь 5-амино-6,7-дихлор-2,3-диметоксихиноксалина (см. препаративный пример 1) (10,0 г, 36,5 ммоль), метансульфонового ангидрида (31,8 г, 183 ммоль) и пиридина (14,8 мл, 183 ммоль) в сухом дихлорметане (150 мл) перемешивают при 20oC 16 часов. Растворитель удаляют при пониженном давлении и остаток растворяют в смеси воды (5 мл) и тетрагидрофурана (50 мл). После перемешивания в течение 10 минут раствор распределяют между этилацетатом и 2 М соляной кислотой. Объединенные органические растворы промывают насыщенным водным бикарбонатом натрия, сушат (MgSO4) и концентрируют при пониженном давлении. Очистка остатка флэш хроматографией (градиентное элюирование гексан/дихлорметаном) дает 6,7-дихлор-5- ди(метансульфонил)амино-2,3-диметоксихиноксалина в виде не совсем белого твердого вещества (12,3 г, 78%), т. пл. 240-244oC.
1H-ЯМР (300 МГц, CDCl3): δ =3,62 (6H, с), 4,15 (3H, с), 4,18 (3H, с), 8,02 (1H, с).
m/z (термонапыление) 430, 432 (MH+).
b) Водную гидроокись натрия (1 М, 145 мл, 145 ммоль) добавляют к суспензии 6,7-дихлор-5-ди(метансульфонил)амино-2,3- диметоксихиноксалина (из стадии (a), 12,28 г, 28,6 ммоль) и смесь перемешивают при комнатной температуре 16 часов. Полученный органический раствор обрабатывают 2 М соляной кислотой (до pH 3) и осадившийся твердый продукт отфильтровывают, промывают водой и диэтиловым эфиром, сушат в вакууме при 80oC и получают N-(6,7-дихлор- 2,3-диметоксихиноксалин- 5-ил)метансульфонамид (8,46 г, 84%) в виде белого твердого вещества, т. пл. 225 - 227oC. 1H-ЯМР (300 МГц, CDCL3) δ = 3,42 (3H, с), 4,15 (3H, с), 4,20 (3H, с), 7,15 (1H, ушир, с), 8,02 (1H,с).
m/z (термонапыление) 352 (MH+).
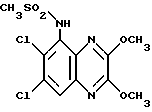
Препаративный пример 4. N-(6,7-Дихлор-2,3-диметоксихиноксалин-5-ил)этансульфонамид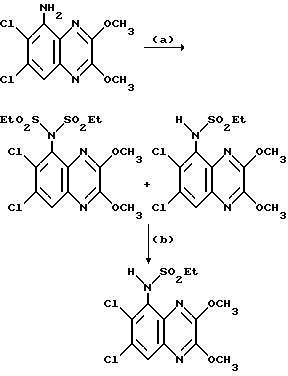
Указанное в заглавии соединение получают по способу препаративного примера 3 (a) и (b), используя этансульфоновый ангидрид. [J.Am.Chem. Soc., 76, 1222 (1954)] и 5-амино-6,7-дихлор-2,3- диметоксихиноксалин в виде бледно-желтого твердого продукта (47%-выход), т. пл. 174 - 176oC.
1H-ЯМР (300 МГц, CDCl3): δ =1,38 (3H, т, J 7 Гц), 3,56 (2H, кв., J 7 Гц), 4,13 (3H, с), 4,20 (3H, с), 6,97 (1H, ушир. с), 7,85 (1H, с).
m/z (термонапыление) 366 (MH+).
В этом случае стадия (a) приводит к смеси продуктов, которые обрабатывают как в стадии (b) препаративного примера 3.
Препаративный пример 5. N-(6,7-Дихлор-2,3-диметоксихиноксалин- 5-ил)-бензолсульфонамид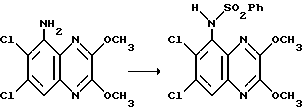
Смесь 5-амино-6,7-дихлор-2,3-диметоксихиноксалина (препаративный пример 1) (548 мг, 2,0 ммоль), бензолсульфонилхлорида (1,28 мл, 10 ммоль) и пиридина (0,8 мл, 10 ммоль) в сухом дихлорметане (30 мл) перемешивают при нагревании до температуры кипения с обратным холодильником в течение 100 часов. Смесь выливают в этилацетат/воду и белый твердый продукт отфильтровывают, промывают водой, этилацетатом, диэтиловым эфиром и сушат при 80oC в вакууме, получая указанное в заглавии соединение (250 мг, 30%), т.пл. 292-293oC.
1H-ЯМР (300 МГц, ДМСО-d6): δ = 3,53 (3H, с), 4,02 (3H, с), 7,48 (2H, J 8 Гц), 7,63 (3H, м), 8,00 (1H, с), 10,22 (1H, ушир, с), m/z (термонапыление) 414 (MH+).
Препаративный пример 6. N-(2,3-Диметокси-6,7-диметилхиноксалин-5- ил)-метансульфонамид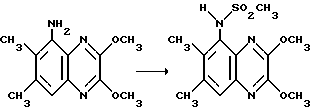
Смесь 5-амино-2,3-диметокси-6,7-диметилхиноксалина (из препаративного примера 2) (50 мг, 0,214 ммоль), метансульфонового ангидрида (187 мг, 1,07 ммоль) и пиридина (87 мл, 1,07 ммоль) в сухом тетрагидрофуране (1 мл) перемешивают при 20oC в течение 2,7 часа. Добавляют воду (0,3 мл) и смесь перемешивают 40 минут. Смесь распределяют между этилацетатом (15 мл) и 2 М соляной кислотой (5 мл). Органический раствор промывают насыщенным водным бикарбонатом натрия (5 мл), сушат (MgSO4) и концентрируют при пониженном давлении, получая указанное в заглавии соединение (63 мг, 94%) в виде белого твердого вещества, т.пл. 219oC.
1H-ЯМР (300 МГц, (CDCl3): δ = 2,46 (3H, с), 2,55 (3H, с), 2,87 (3H, с), 4,16 (6H, с), 7,00 (1H, ушир.с), 7,57 (1H, с).
νmax/ (KBr) 3545, 1480, 1160 см-1.
m/z (термонапыление) 312 (MH+).
Анализ %:
Найдено: C 50,02; H 5,48; N 13,35; S 10,51.
Рассчитано для C13H17N3SO4: C 50,15; H 5,50; N 13,50; S 10,30%.
Препаративный пример 7. N-(2,3-Диметокси-6,7- диметоксихиноксалин-5-ил)-этансульфонамид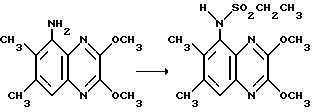
Смесь 5-амино-2,3-диметокси-6,7-диметилхиноксалина (из препаративного примера 2) (50 мг, 0,214 ммоль), этансульфонилхлорида (138 мг, 1,07 ммоль) и пиридина (87 мкл, 1,07 ммоль) в сухом тетрагидрофуране (1 мл) перемешивают при 20oC 3,5 часа. Добавляют дополнительные порции этансульфонилхлорида (138 мг, 1,07 ммоль) и пиридина (87 мл, 1,07 ммоль) и смесь перемешивают дополнительно 4 дня. Добавляют воду (0,6 мл) и смесь перемешивают 40 минут. Смесь распределяют между этилацетатом (15 мл) и 2 М соляной кислотой (5 мл). Органический раствор промывают водой (5 мл), насыщенным водным бикарбонатом натрия (5 мл), сушат (MgSO4) и концентрируют при пониженном давлении, получая указанное в заглавии соединение (67 мг, 96%) в виде твердого продукта соломенного цвета, т.пл. 201-203oC.
1H-ЯМР (300 МГц, CDCl3): δ = 1,35 (3H, т, J 7 Гц), 2,44 (3H, с), 2,54 (3H, с), 3,03 (2H, кв., J 7 Гц), 4,15 (3H, с), 4,16 (3H, с). 6,96 (1H, ушир, с), 7,56 (1H, с).
νmax (KBr) 3250, 2940, 1480, 1323, 1239, 1157 см-1
m/z (термонапыление) 326 (MH+).
Анализ %:
Найдено: C 51,88; H 6,02; N 12,43.
Рассчитано для C14H19N3SO4•0,15 этилацетат: C 51,79; H 6,01; N 12,41%.
Препаративный пример 8. N-(6,7-Дихлор-2,3-диметоксихиноксалин- 5-ил)-N-(2-оксопропил)метансульфонамид
Третбутилоксида калия (246 мг, 2,2 ммоль) добавляют к перемешиваемому раствору N-(6,7-дихлор-2,3- диметоксихиноксалин-5-ил)метансульфонамида (702 мг, 2,0 ммоль, см. препаративный пример 3) в сухом диметилформамиде (10 мл) в атмосфере азота при 20oC. Добавляют хлорацетон (175 мл, 2,2 ммоль) и смесь перемешивают в течение 4 часов. Смесь концентрируют при пониженном давлении и остаток распределяют между 1 М водной гидроокисью натрия и этилацетатом. Органические экстракты объединяют, промывают раствором соли, сушат (Na2SO4) и концентрируют при пониженном давлении, получая белый твердый продукт, который растирают диэтиловым эфиром, фильтруют и сушат, получая указанное в заглавии соединение (720 мг, 88%). т.пл. 247-248oC.
1H-ЯМР (300 МГц, CDCl3): δ = 2,23 (3H, с), 3,42 (3H, с), 4,17 (3H, с), 4,23 (3H, с), 4,45 (1H, д, J 18 Гц), 4,74 (1H, д, J 18 Гц), 7,95 (1H, с).
m/z (термонапыление) 408 (MH+).
Препаративный пример 9. (RS)-N-(6,7-Дихлор-2,3- диметоксихиноксалин-5-ил)-N-(2-гидроксипропил)метансульфонамид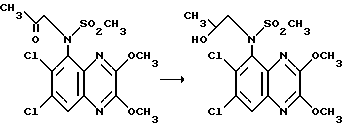
Гидрид диизобутилалюминия (1,0 М в дихлорметане, 1,0 мл, 1,0 ммоль) добавляют по каплям к перемешиваемому раствору N-(6,7- дихлор-2,3-диметоксихиноксалин-5-ил)-N-(2-оксопропил)- метансульфонамида (204 мг, 0,5 ммоль - препаративный пример 8) в сухом дихлорметане (10 мл) в атмосфере азота при 20oC. Через 2 часа добавляют насыщенный водный раствор хлорида аммония (2 мл) и смесь перемешивают в течение 15 минут перед фильтрацией через " Arbocel" (Торговая марка) ускоритель фильтрования. Плотный осадок на фильтре промывают дихлорметаном и фильтрат сушат (Na2SO4) и концентрируют при пониженном давлении. Остаток чистят флэш хроматографией (градиентное элюирование гексан/диэтиловым эфиром), получая указанное в заглавии соединение (182 мг, 89%) в виде белого твердого вещества, т. пл. 176- 177oC.
Анализ %:
Найдено: C 40,67; H 4,07; N 10,06.
Рассчитано для C14H17Cl2N3O5S: C 40,99; H 4,18; N 10,24 %.
Препаративный пример 10. N-(6,7-Дихлор-2,3-диметоксихиноксалин-5-ил)-N-(2- гидрокси-2-метилпропил)метансульфонамид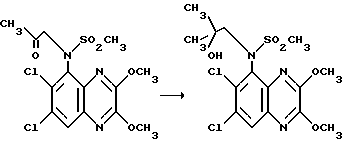
Метилмагнийбромид (1,4 мл, 1 М ди-н-бутиловом эфире, 1,4 ммоль) добавляют по каплям к раствору N-(6,7-дихлор-2,3- диметоксихиноксалин-5-ил)-N-(2-оксопропил)метансульфонамид (143 мг, 0,35 ммоль, см. препаративный пример 8) в сухом тетрагидрофуране (10 мл) в атмосфере азота при 5oC. Смеси дают медленно нагреться до комнатной температуры и перемешивают в течение 5 часов. Добавляют насыщенный водный хлорид аммония (1 мл) и тетрагидрофуран удаляют при пониженном давлении. Остаток распределяют между водой и этилацетатом (3 порции). Объединенные экстракты сушат (Na2SO4), концентрируют при пониженном давлении и остаток чистят флэш хроматографией (градиентное элюирование гексан/диэтиловый эфир), что дает указанное в заглавии соединение (105 мг, 75%) в виде белого твердого продукта, т.пл. 160oC.
1H-ЯМР (300 МГц, CDCl3): δ =0,97 (3H, с), 1,30 (3H, с), 3,22 (3H, с), 3,56 (1H, с), 3,79 (1H, д, J 15 Гц), 3,96 (1H, д, J 15 Гц), 4,14 (3H, с), 4,19 (3H, с), 7,96 (1H, с).
m/z (термонапыление) 424 (MH+).
Препаративный пример 11. N-(6,7-Дибром-2,3- диметоксихиноксалин-5-ил)-метансульфонамид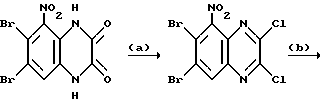
a) 1,4-Дигидро-6,7-дибром-5-нитрохиноксалин-2,3-дион (см. Пример 33, WO-A-94/00124) превращают в 6,7-дибром- 2,3-дихлор-5-нитрохиноксалин по способу препаративного примера 1(a). Продукт получают в виде не совсем белого твердого продукта (72%- выход), т.пл. 126-128oC (из гексана).
1H-ЯМР (300 МГц, CDCl3): δ = 8,50 (1H, с).
b) Промежуточный продукт из стадии (a) превращают в 5-амино-6,7-дибром-2,3-дихлорхиноксалин по способу препаративного примера 1(b). Продукт получают в виде желтого твердого вещества (61%-выход), т.пл. 108-110oC (после очистки флэш хроматографией).
1H-ЯМР (300 МГц, CDCl3): δ =5,55 (2H, ушир. с), 7,68 (1H, с).
с) Промежуточный продукт из приведенной выше стадии (b) превращают в 5-амино-6,7-дибром-2,3-диметоксихиноксалин по способу препаративного примера 1(с). Продукт получают в виде желтого твердого продукта, (59%-выход), т.пл. 148-150oC (после очистки флэш хроматографией).
1H-ЯМР (300 МГц, CDCl3): δ =4,13 (6H, с), 5,20 (2H, ушир.с), 7,51 (1H, с).
m/z (термонапыление) 364 (MH+).
d) Промежуточный продукт из приведенной выше стадии (с) превращают по способу препаративного примера 3(a) в 6,7-дибром-5-ди(метансульфонил) амино-2,3-диметоксихиноксалин (85%-выход), бледно-желтый твердый продукт, т.пл. 204-206 (после очистки флэш хроматографией).
1H-ЯМР (300 МГц, CDCl3): δ = 3,61 (6H, с), 4,15 (3H, с), 4,19 (3H, с), 8,20 (1H, с).
m/z (термонапыление) 520 (MH+).
е) Промежуточный продукт из приведенной выше стадии (d) превращают в N-(6,7-дибром-2,3-диметоксихиноксалин-5-ил)- метансульфонамид по способу препаративного примера 3(b). Продукт получают в виде бледно-желтого твердого вещества (86%-выход), т. пл. 186-187oC.
1H-ЯМР (300 МГц, CDCl3): δ = 3,45 (3H, с), 4,16 (8H, с), 4,21 (3H, с), 7,08 (1H, ушир.с), 8,09 (1H, с).
m/z (термонапыление) 442 (MH+).
Препаративный пример 12. N-(2,3-Диметокси-6,7-диметилхиноксалин- 5-ил)- трифторметансульфонамид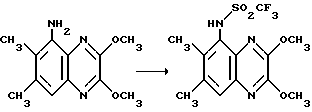
Трифторметансульфоновый ангидрид (126 мл, 0,75 ммоль) добавляют по каплям к раствору 5-амино-2,3-диметокси-6,7- диметилхиноксалина (препаративный пример 2) (170 мг, 0,73 ммоль) и триэтиламина (112 мл, 0,8 ммоль) в сухом дихлорметане (15 мл) в атмосфере азота при -50oC. Смесь перемешивают при -30oC в течение 2 часов, выливают в воду и экстрагируют тремя порциями дихлорметана. Затем продукт экстрагируют из дихлорметана, используя 1 М водную гидроокись натрия. Водную фазу подкисливают избытком 2 М соляной кислоты и продукт экстрагируют дихлорметаном (3 порции). Объединенный экстракт сушат (MgSO4) и концентрируют при пониженном давлении, получая белый твердый продукт (260 мг, 98%).
1H-ЯМР (300 МГц, CDCl3): δ = 2,44 (3H, с), 2,49 (3H, с), 4,14 (3H, с), 4,15 (3H, с), 7,13 (1H, ушир.с), 7,61 (1H, с).
m/z (термонапыление) 366 (MH+).
Препаративный пример 13. N-(6-Хлор-7-этил-2,3- диметоксихиноксалин-5-ил) метансульфонамид и N-(7-xлop-6-этил-2,3-димeтoкcиxинoкcaлин -5-ил)-метансульфонамид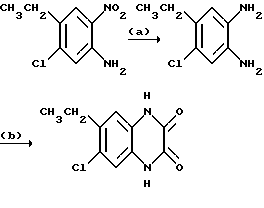
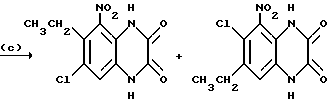
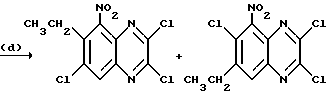
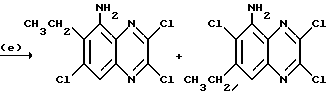
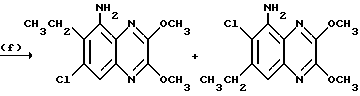
a) 1,2-Диамино-4-хлор-5-этилбензол.
Смесь 5-хлор-4-этил-2-нитроанилина (поставлен by the Sigma Aldrich Library of Rare Chemicals, 2.62 г, 13,1 ммоль), дигидрата хлорида двухвалентного олова (14,7 г, 65,3 ммоль) и этилацетата (130 мл) нагревают до температуры кипения с обратным холодильником в течение 22 часов. Смесь охлаждают и распределяют между 1 М водной гидроокисью натрия (500 мл) и этилацетатом (500 мл). Водный слой экстрагируют этилацетатом (250 мл) и объединенные органические растворы промывают насыщенным водным хлоридом натрия (100 мл), сушат (MgSO4) и концентрируют при пониженном давлении, получая белый твердый продукт (2,70 г, > 100%), который используют непосредственно, без дополнительной очистки.
1H-ЯМР (300 МГц, CDCl3) δ =1,19 (3H, т, J 7 Гц), 2,63 (2H, кв., J 7 Гц), 3,30 (4H, ушир.с), 6,57 (1H, с), 6,70 (1H, с).
b) 1,4-Дигидро-6-хлор-7-этилхиноксалин-2,3-дион.
Смесь 1,2- диамино-4-хлор-5-этилбензола (из стадии (a), 2,70 г, около 13 ммоль), щавелевой кислоты (1,65 г, 18,3 ммоль) и 4 М соляной кислоты (66 мл) нагревают до температуры кипения с обратным холодильником в течение 4,6 часа, охлаждают, и серый твердый продукт собирают фильтрацией и промывают водой. Твердый продукт сушат в вакууме при 50oC, получая указанное в заглавии соединение (2,34 г, 80%), т. пл. > 315oC.
Анализ %:
Найдено, C 53,60; H 3,87; N 12,40.
Рассчитано для C10H9ClN2O2: C 53,47; H 4,04; N 12,47%. 1H-ЯМР (300 МГц, ДМСО- d6) δ = 1,17 (3H, т, J 7 Гц), 2,66 (2H, кв., J 7 Гц), 7,05 (1H, с), 7,14 (1H, с), 11,78 (1H, ушир.с), 11,82 (1H, ушир.с).
с) 1,4-Дигидро-6-хлор-7-этил-5-нитрохиноксалин-2,3-дион и 1,4-дигидро-7-хлор-6-этил-5-нитрохиноксалин-2,3-дион.
1,4-Дигидро-6-хлор-7-этилхиноксалин-2, 3-дион (из стадии (b), 2,34 г, 10,4 ммоль) добавляют небольшими порциями за 10 минут к энергично перемешиваемой концентрированной азотной кислоте (20 мл) при комнатной температуре. Смесь нагревают затем при 40oC в течение 12 часов, охлаждают и выливают в ледяную воду. Образовавшийся твердый продукт отфильтровывают, промывают водой и сушат, получая указанные в заглавии соединения (2,55 г, 91%), в виде смеси (соотнош. 1,7:1).
1H-ЯМР (300 МГц, ДМСО-d6) δ = 1,09-1,19 (3H, м), 2,56 (1, 3H, кв., J 7 Гц), 2,71 (0,7, кв. J 7 Гц), 7,19 (0,4H, с), 7,29 (0,6 H, с); 11,95-12,15 (2H, ушир.м).
m/z (термонапыление) 287 (MNH4 +).
a) 2,3,7-Трихлор-6-этил-5-нитрохиноксалин и 2,3,6-трихлор-7- этил-5-нитрохиноксалин.
Смесь хиноксалинов из стадии (с), приведенной выше (2,75 г, 11 ммоль), тионилхлорида (28,6 мл, 0,305 моль) и N,N- диметилформамида (85 мкл, 1,0 ммоль) нагревают в атмосфере азота до температуры кипения с обратным холодильником в течение 24 часов. Раствор охлаждают и осторожно добавляют по каплям к перемешиваемой ледяной воде (600 мл). Через 1 час твердый продукт бежевого цвета отфильтровывают, промывают водой и сушат в вакууме, получая смесь указанных в заглавии соединений (2,26 г, 67%). Очистка смеси флэш хроматографией (элюирование гексан: дихлорметаном =3:1) позволяет получить в чистом виде небольшие количества изомеров для их охарактеризации. Первый выделенный изомер имеет температуру плавления 106-109oC.
Анализ %:
Найдено: C 39,21; H 1,99; N 13,71
Рассчитано для C10H6Cl3N3O2: C 39,18; H 1,97; N 13,71%.
1H-ЯМР (300 МГц, CDCl3) δ = 1,41 (3H, т, J 7 Гц), 3,06 (2H, кв. J 7 Гц), 8,02 (1H, с).
m/z (термонапыление) 323 (MH+).
Второй изомер имеет т.пл. 88-92oC.
Анализ %:
Найдено: C 39,06; H 1,87; N 13,85%.
1H-ЯМР (300 МГц, CDCl3) δ = 1,35 (3H, т, J 8 Гц), 2,98 (2H, кв, J 8 Гц), 8,19 (1H, с).
m/z (термонапыление) 323 (MH+).
е) 5-Амино-2,3,7-трихлор-6-этилхиноксалин и 5-амино-2,3,6- трихлор-7-этилхиноксалин.
Смесь хиноксалинов из стадии (d) приведенной выше (200 мг, 0,652 ммоль), дигидрата хлорида олова (II) (1,03 г, 4,57 ммоль) и этилацетата (6,5 мл) нагревают до температуры кипения с обратным холодильником в течение 4 часов, охлаждают и разбавляют этилацетатом. Раствор промывают 10% водным карбонатом натрия (25 мл). Водный слой экстрагируют этилацетатом (2 х 25 мл) и объединенные органические растворы промывают 10% водным карбонатом натрия (2 х 25 мл), насыщенным водным хлористым натрием (25 мл), сушат (MgSO4) и концентрируют при пониженном давлении, получая указанные в заглавии соединения в виде твердого вещества оранжевого цвета (174 мг, 91%), соотношение 1 : 1.
1H-ЯМР (300 МГц, CDCl3) δ =1,25 (1,5H, т, J 8 Гц), 1,37 (1,5H, т, J 8 Гц), 2,84-2,98 (2H, м), 5,05 (1H, ушир. с), 5,28 (1H, ушир. с), 7,22 (0,5H, с), 7,43 (0,5H, с).
f) 5-Амино-6-хлор-7-этил-2,3-диметоксихиноксалин и 5-амино-7-хлор-6-этил-2,3-диметоксихиноксалин.
Смесь трихлорхиноксалинов из стадии (е), приведенной выше (169 мг, 0,611 ммоль), в безводном тетрагидрофуране (6 мл) обрабатывают раствором метилата натрия (0,84 мл, 25% раствор в метаноле, 1,47 ммоль) при 0oC и при перемешивании. Через 3,5 ч раствор разбавляют этилацетатом, промывают водой (2х10 мл), насыщенным водным хлористым натрием (10 мл), сушат (MgSO4) и концентрируют при пониженном давлении. Очистка флэш хроматографией (элюирование гексан/этилацетатом 19:1) дает две фракции.
Первое вымываемое соединение, 5-амино-6-хлор-7- этил-2,3-диметоксихиноксалин (42 мг, 26%), получают в виде белого твердого продукта.
1H-ЯМР (300 МГц, CDCl3) δ = 1,32 (3H, т, J 8 Гц), 2,87 (2H, кв., J 7 Гц), 4,18 (6H, с), 4,90 (2H, ушир. с), 7,08 (1H, с). Второе вымываемое соединение, 5-амино-7-хлор-6-этил-2,3-диметоксихиноксалин (57 мг, 35%), получают в виде бледно-зеленого твердого продукта.
1H-ЯМР (300 МГц, CDCl3) δ = 1,14 (3H, т, J 7 Гц), 2,84 (2H, кв., J 7 Гц), 4,12 (3H, с), 4,14 (3H, с), 4,70 (2H, ушир.с), 7,22 (1H, с).
g) N-(6-хлор-7-этил-2,3-диметоксихиноксалин-5-ил)метансульфонамид.
Метансульфоновый ангидрид (671 мг, 3,85 ммоль) добавляют к перемешиваемому раствору 5-амино-6-хлор-7-этил-2,3- диметоксихиноксалина (207 мг, 0,77 ммоль) и безводного пиридина (305 мг, 3,85 ммоль) в безводном тетрагидрофуране (7,7 мл) при комнатной температуре. Через 72 часа добавляют воду (3 мл) и смесь перемешивают дополнительно 60 минут. Смесь разбавляют этилацетатом и промывают 2 М соляной кислотой (50 мл), водой (50 мл), насыщенным водным раствором бикарбоната натрия (50 мл) и насыщенным водным раствором хлористого натрия (50 мл). Органическую фазу сушат (MgSO4) и концентрируют при пониженном давлении, получая указанное в заглавии соединение (206 мг, 77%).
1H-ЯМР (300 МГц, CDCl3) δ = 1,37 (3H, т, J 8 Гц), 2,89-3,00 (2H, м), 3,39 (3H, с), 4,16 (3H, с), 4,19 (3H, с), 7,01 (1H, с), 7,60 (1H, с).
m/z (термонапыление) 346 (MH+).
h) N-(7-Хлор-6-этил-2,3-диметоксихиноксалин-5-ил)- метансульфонамид
5-Амино-7-хлор-6-этил-2,3-диметоксихиноксалин превращают в производное метансульфонамида по способу приведенной выше стадии (g). Получают продукт с 47%-выходом.
1H-ЯМР (300 МГц, CDCl3) δ = 1,25 (3H, т, J 8 Гц), 3,00 (3H, с), 3,28 (2H, кв, J 7 Гц), 4,17 (3H, с), 4,27 (3H, с), 6,87 (1H, с), 7,83 (1H, с).
m/z (термонапыление) 346 (MH+).
Препаративный пример 14. N-(6-хлор-2,3-диметокси-7- метилхиноксалин-5-ил) метансульфонамид и N-(7-хлор-2,3-диметокси-6-метилхиноксалин-5-ил) метансульфонамид.
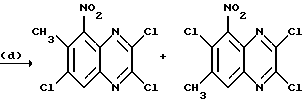
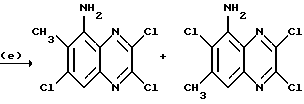
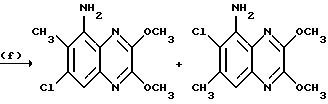
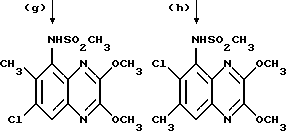
Следующие соединения, перечисленные ниже, получают способом, аналогичным используемому для 6-хлор-7-этил- и 7-хлор-6- этилпроизводных, полученных в препаративном примере 13.
1,4-Дигидро-6-хлор-7-метилхиноксалин-2,3-дион получают из 1,2-диамин-4-хлор-5-метилбензола (полученного by Maybridge Chemicals) в виде темно-серого твердого продукта, т. пл. > 330oC.
Анализ %:
Найдено: C 51,58; H 2,98; N 13,27;
Рассчитано для C9H7ClN2O2: C 51,32; H 3,35; N 13,30%.
1,4-Дигидро-6-хлор-7-метил-5-нитрохиноксалин-2,3-дион и 1,4- дигидро-7-хлор-6-метил-5-нитрохиноксалин-2,3-дион получают в виде желтого твердого продукта (1:2 соотношение).
1H-ЯМР (300 МГц, CDCl3) δ =2,23 (2H, с), 2,35 (1H, с), 7,19 (0,3H, с), 7,30 (0,7H, с), 11,9-12,25 (2H, ушир. м).
2,3,7-Трихлор-6-метил-5-нитрохиноксалин и 2,3,6-трихлор-7-метил-5-нитрохиноксалин получают в виде порошка соломенного цвета, соединения с трудом могут быть разделены для характеристики. Флэш хроматография (градиентное элюирование гексан/дихлорметаном) дает первым 6-метил-7-хлор-изомер в виде белого твердого вещества, т. пл. 164-165oC.
Анализ %:
Найдено: C 36,76; H l,37; H 14,43.
Рассчитано для C9H4Cl3N3O2: C 36,96; H 1,38; N 14,37%.
Элюируемый вторым (7-метил-6-хлор)-изомер представляет собой твердый продукт соломенного цвета, т. пл. 121 - 122oC.
Анализ %:
Найдено: C 39,78; H 2,02; N 13,23;
Рассчитано для C9H4Cl3N3O2•0,22 гексан: C 39,80; H 2,29; N 13,49%.
5-Амино-2,3,7-трихлор-6-метилхиноксалин и 5-амино-2,3,6-трихлор-7-метилхиноксалин получают в виде коричневого твердого продукта.
1H-ЯМР (300 МГц, CDCl3) δ =2,41 (2H, с), 2,55 (1H, с), 5,03 (1,3H, ушир. с), 5,08 (0,7H, ушир. с), 7,23 (0,3H, с), 7,44 (0,7H, с).
5-Амино-7-хлор-2,3-диметокси-6-метилхиноксалин и 5-амино-6-хлор- 2,3-диметокси-7-метилхиноксалин разделяют повторной колоночной хроматографией на силикагеле, элюируя гексан/этилацетатом = 1:1 и впоследствии дихлорметаном.
Первое вымываемое соединение, 6-хлор-7-метил-изомер, получают в виде не совсем белого твердого соединения, т.пл. 169-170oC.
Анализ %:
Найдено; C 53,80; H 5,16; N 16,18;
Рассчитано для C11H12ClN3O2•0,15 гексан: C 53,61; H 5,33; N 15,76%.
Вымываемое вторым соединение, 7-хлор-6-метил-изомер, получают в виде оранжевого твердого продукта, т.пл. 181-182oC.
Анализ %:
Найдено: C 52,55; H 4,72; N 16,61
Рассчитано для C11H12ClN3O2•0,5 гексан: C 52,61; H 4,96; N 16,29%.
N-(6-Хлор-2,3-диметокси-7-метилхиноксалин-6-ил)-метансульфонамид получают в виде белого твердого вещества, т.пл. 198oC.
1H-ЯМР (300 МГц, CDCl3) δ = 2,58 (3H, с), 3,38 (3H, с), 4,15 (3H, с), 4,18 (3H, с), 7,00 (1H, ушир.с), 7,60 (1H, с).
m/z (термонапыление) 322 (MH+) νmax/ (KBr) 3230, 2950, 1480 и 1150 см-1.
N-(7-хлор-2,3-диметокси-7-метилхиноксалин-5-ил) метансульфонамид получают в виде белого твердого продукта, т.пл. 228- 229oC.
Анализ %:
Найдено: C 43,51; H 3,98; N 12,60, C12H14ClN3O4S
Рассчитано: C 43,44; H 4,25; N 12,60%.
Препаративный пример 15. 4-Гидроксиметил-2-(трифенилметил) -1,2,3-триазол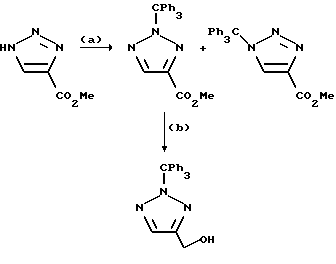
a) Метил 1H-1,2,3-триазол-4-карбоксилат [J. Org. Chem. 41, 1041 (1976)] (1,00 г, 7,09 ммоль) добавляют к перемешиваемой суспензии гидрида натрия (234 мг, 80% масляная дисперсия, 7,80 ммоль) в безводном тетрагидрофуране (100 мл) в атмосфере азота при комнатной температуре. Через 20 минут смесь охлаждают до 0oC и добавляют тритилхлорид (2,17 г, 7,80 ммоль). Смесь перемешивают 4 часа при 0oC. Добавляют воду (100 мл) и тетрагидрофуран удаляют при пониженном давлении. Оставшуюся смесь экстрагируют этилацетатом (100 мл). Органический слой сушат (MgSO4), фильтруют и концентрируют при пониженном давлении, что дает твердый продукт, который чистят флэш хроматографией на силикагеле, элюируя 5 - 30% этилацетатом в гексане. Вымываемый первым продукт экспериментально идентифицирован как метил 1-(трифенилметил)- 1,2,3-триазол-4-карбоксилат и получен в виде белого твердого продукта (600 мг, 21%).
1H-ЯМР (300 МГц, CDCl3) δ =3,92 (3H, с), 7,11 (6H, м), 7,32 (9H, м), 8,15 (1H, с).
Продукт, вымываемый вторым, экспериментально идентифицирован как метил 2-(трифенилметил)-1,2,3- триазол-4-карбоксилат и получен в виде белого твердого вещества. (500 мг, 18%).
1H-ЯМР (300 МГц, CDCL3) δ =3,94 (3H, с), 7,11 (6H, м), 7,36 (9H, м), 8,02 (1H, с).
b) Раствор алюмогидрида лития (1,04 мл, 1 М в тетрагидрофуране, 1,04 ммоль) добавляют по каплям при перемешивании к раствору метил 2- (трифенилметил)-1,2,3-триазол-4-карбоксилата (из стадии (a), 500 мг, 1,38 ммоль) в безводном тетрагидрофуране (30 мл) при 0oC в атмосфере азота. Смесь перемешивают в течение 1 часа при 0oC и затем добавляют воду (2 мл), 15% водную гидроокись натрия (4 мл) и далее, дополнительное количество воды (4 мл). Образовавшуюся суспензию фильтруют через Arbocel - ускоритель фильтрования и фильтрат сушат (MgSO4) и концентрируют при пониженном давлении. Остаток распределяют между дихлорметаном (150 мл) и водой (150 мл). Дихлорметановый слой промывают водой (600 мл) и затем сушат (MgSO4) и концентрируют при пониженном давлении, получая в остатке указанное в заглавии соединение (401 мг, 85%).
1H-ЯМР (300 МГц, CDCl3) δ =2,05 (1H, т, J 7 Гц), 4,81 (2H, д, J 7 Гц), 7,12 (6H, м), 7,35 (9H, м), 7,47 (1H, с).
Препаративный пример 16. 4-Гидроксиметил-5-метил-1- (трифенилметил)имидазол
a) Гидрид натрия (900 мг, 80% масляная дисперсия, 30 ммоль) добавляют порциями в течение более 5 минут к перемешиваемому раствору этил 4-метил-имидазол-5-карбоксилата (3,85 г, 25 ммоль) в сухом тетрагидрофуране (100 мл) при комнатной температуре в атмосфере азота. Через 10 минут добавляют за одну порцию тритилхлорид (8,36 г, 30 ммоль). Через 1 час смесь разбавляют этилацетатом (500 мл) и промывают водой (200 мл). Промывные воды экстрагируют этилацеатом (2 х 75 мл) и объединенные органические растворы сушат (MgSO4) и концентрируют при пониженном давлении. Остаток чистят флэш хроматографией (градиентное элюирование гексан/этилацетатом), получая этил 5-метил-1- (трифенилметил)имидазол-4-карбоксилат (1,01 г, 10%) в виде белого твердого продукта.
1H-ЯМР (300 МГц, CDCl3) δ = 1,38 (3H, т, J 6 Гц), 1,83 (3H, с), 4,36 (2H, кв, J 6 Гц), 7,14 (6H, м), 7,35 (10H, м).
b) Раствор алюмогидрида - лития (7,65 мл, 1 М в тетрагидрофуране, 7,65 ммоль) добавляют по каплям к перемешиваемой суспензии этил-5-метил-1-(трифенилметил)имидазол-4-карбоксилата (1,01 г, 2,55 ммоль) в безводном тетрагидрофуране (50 мл) в атмосфере азота при 0oC в течение более 1 минуты. Через 30 минут осторожно добавляют воду (300 мл), после чего - водную гидроокись натрия (1 М, 300 мкл) и воду (900 мкл). Суспензию фильтруют, используя Arbocel - ускоритель фильтрования, и плотный осадок на фильтре промывают тетрагидрофураном (2 х 30 мл). Фильтрат концентрируют при пониженном давлении и остаток растворяют в кипящем метаноле (50 мл). Добавляют дихлорметан (200 мл) и безводный сульфат магния и смесь фильтруют через Arbocel - ускоритель фильтрации. Фильтрат концентрируют при пониженном давлении, получая белый твердый продукт (325 мг, 36%).
1H-ЯМР (300 МГц, CDCl3) δ = 1,47 (3H, с), 4,05 (2H, с), 7,14 (6H, м), 7,35 (10H, м).
Препаративный пример 17. 4-Гидроксиметил-1-(трифенилметил)пиразол
a) Этил 1H-пиразол-4-карбоксилат (1,50 г, 10,7 ммоль) добавляют порциями к перемешиваемой суспензии гидрида натрия (353 мг, 80% масляная дисперсия, 11,8 ммоль) в безводном тетрагидрофуране (100 мл) в атмосфере азота при комнатной температуре. Через 20 минут смесь охлаждают до 0oC и добавляют тритилхлорид (3,28 г, 11,8 ммоль). Смесь перемешивают при 0oC в течение 4 часов и дают нагреться до комнатной температуры в течение ночи. 3атем смесь нагревают при 50oC 3 часа, охлаждают и добавляют дополнительные порции гидрида натрия (321 мг, 10,7 ммоль) и тритилхлорида (1,0 г, 3,59 ммоль). Смесь нагревают при 50oC в течение 2 часов, охлаждают, обрабатывают водой (50 мл) и концентрируют при пониженном давлении. Продукт экстрагируют этилацетатом (200 мл), раствор сушат (MgSO4) и концентрируют при пониженном давлении. Остаток чистят флэш хроматографией (градиентное элюирование гексан/этилацетатом), что дает этил 1-(трифенилметил)- пиразол-4-карбоксилат (2,273 г, 56%) в виде белого твердого вещества.
1H-ЯМР (300 МГц, CDCl3) δ = 1,31 (3H, т, J 7 Гц), 4,27 (2H, кв, 7 Гц), 7,13 (6H, м), 7,32 (9H, м), 7,94 (1H, с), 8,02 (1H, с).
b) Алюмогидрид лития (1 М раствор в тетрагидрофуране, 4,45 мл, 4,45 ммоль) добавляют по каплям к перемешиваемому раствору этил 1- (трифенилметил)пиразол-4-карбоксилата (2,27 г, 5,93 ммоль) в безводном тетрагидрофуране (300 мл) в атмосфере азота при 0oC. Через 1 час смесь обрабатывают последовательно водой (3 мл), 15% водной гидроокисью натрия (3 мл) и водой (6 мл). Смесь фильтруют через Arbocel - ускоритель фильтрования и фильтрат сушат (MgSO4) и концентрируют при пониженном давлении, получая 4-гидроксиметил-1-(трифенилметил)пиразол (1,746 г, 86%), в виде белого твердого продукта.
1H-ЯМР (300 МГц, CDCl3) δ = 1,43 (1H, ушир. с), 4,35 (2H, с), 7,16 (6H, м), 7,26 (9H, м), 7,37 (1H, с), 7,68 (1H, с).
Препаративный пример 18. 3-Гидроксиметил-1-(трифенилметил)- 1,2,4-триазол
Триэтиламин (1,13 мл, 8,08 ммоль) добавляют к суспензии 3- гидроксиметил-1H-l, 2,4-триазола [J. Am. Chem. Soc, 77, 1538, (1955)] (400 мг, 4,04 ммоль) в сухом дихлорметане (8 мл) при 0oC. Добавляют раствор тритилхлорида (1,24 г, 4,44 ммоль) в безводном тетрагидрофуране и смесь перемешивают 3 часа при комнатной температуре. Смесь распределяют между водой (75 мл) и дихлорметаном (75 мл). Органический слой сушат (MgSO4) и концентрируют при пониженном давлении, получая указанное в заглавии соединение (1,547 г, > 100%), которое используют без очистки.
1H-ЯМР (300 МГц, CDCl3) δ/ =4,77 (2H, с), 7,13 (6H, м), 7,32 (9H, м), 7,95 (1H, с).
Препаративный пример 19. 3-(Гидроксиметил)-1-(трифенилметил)пиразол
Данное соединение получают из 3-(гидроксиметил)-1H- пиразола [J. Am. Chem. Soc, 71, 3996, (1949)] , тритилхлорида и триметиламина, используя способ приведенного выше препаративного примера 18. Сырой продукт чистят флэш хроматографией (градиентное элюирование гексан/этилацетатом), получая белое твердое вещество (1,428 г, 60%).
1H-ЯМР (300 МГц, CDCl3) δ =1,14 (1H, т, J 5 Гц), 4,67 (2H, дд, 2 и 5 Гц), 6,22 (1H, д, J 2 Гц), 7,16 (6H, м), 7,30 (10H, м). m/z (термонапыление) 341 (MH+).
Препаративный пример 20. N-(3-Амино-6-хлор-7-трифторметил- 2-метоксихиноксалин-5-ил)метансульфонамид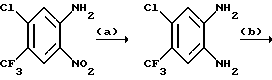
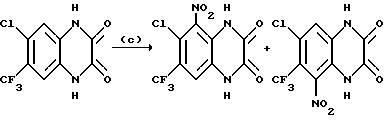
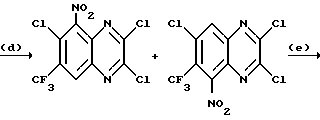
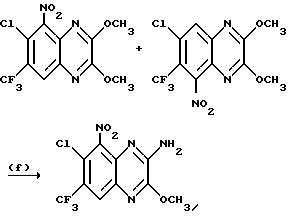
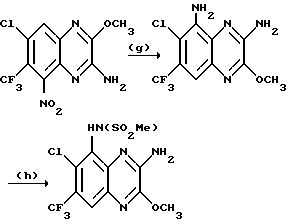
a) 1,2-Диамино-4-хлор-5-трифторметил бензол.
Раствор дитионита натрия (51,0 г, 293 ммоль) в воде (700 мл) добавляют к перемешиваемой смеси 5-хлор-4- трифторметил-2-нитроанилина [Khim. Geterotsikl. Soedin, 1136 (1976)] (23,5 г, 97,7 ммоль) бикарбоната калия (51,0 г) в метаноле (700 мл) при комнатной температуре. Через 30 минут смесь концентрируют при пониженном давлении и остаток распределяют между дихлорметаном и водой. Объединенные органические экстракты сушат (MgSO4) и концентрируют при пониженном давлении, получая оранжевый твердый продукт (13,3 г, 65%).
1H-ЯМР (300 МГц, CDCl3) δ =3,37 (2H, ушир.с), 3,70 (2H, ушир. с), 6,78 (1H, с), 6,98 (1H, с).
b) 1,4-Дигидро-6-хлор-7-трифторметилхиноксалин-2,3-дион.
Смесь 1,2-диамино-4-хлор-5-трифторметилбензола (13,4 г, 63,6 ммоль) и диэтилоксалата (100 мл) нагревают до температуры кипения с обратным холодильником в атмосфере азота при перемешивании в течение 5 час. После охлаждения образовавшийся твердый продукт отфильтровывают и хорошо промывают диэтиловым эфиром, получая бледно-оранжевый твердый продукт (14,5 г, 86%).
Анализ %:
Найдено: C 40,93; H 1,35; N 10,43;
Рассчитано для C9H4ClF3N2O2: C 40,85; H 1,52; N 10,59%.
с) 1,4-Дигидро-6-хлор-7-трифторметил-5-нитрохиноксалин-23-дион и 1,4-дигидро-7-хлор-6-трифторметил-5-нитрохиноксалин-2,3-дион.
1,4-Дигидро-6-хлор-7- трифторметилхиноксалин-2,3-дион (из стадии (b), 14,06 г, 53,1 ммоль) добавляют порциями к перемешиваемой дымящейся азотной кислоте (100 мл) при 0oC. Смеси дают нагреться до 20oC в течение более 30 минут (после завершения добавления) и образовавшийся раствор выливают в ледяную воду и осадившийся твердый продукт отфильтровывают, промывают водой и сушат, получая бледно-оранжевое твердое соединение (15,06 г, 92%), в виде смеси 2 : 1 изомеров.
1H-ЯМР (300 МГц, ДМСО-d6) δ =7,42 (1H, меньший, с), 7,62 (1H, основной, с), 12,33 (1H, основной, 1H меньший, ушир.с), 12,51 (1H основной, 1H меньший, ушир.с).
d) 2,3,6-Трихлор-7-трифторметил-5-нитрохиноксалин и 2,3,7-трихлор- 6-трифторметил-5-нитрохиноксалин.
Смесь хиноксалиндионов из стадии (с), приведенной выше, (14,7 г, 47,5 ммоль) тионилхлорида (140 мл) и сухого диметилформамида (1,4 мл) нагревают при температуре кипения с обратным холодильником в течение 30 минут, охлаждают и добавляют по каплям к ледяной воде. Образовавшийся твердый продукт экстрагируют этилацетатом и раствор промывают одной порцией насыщенного водного бикарбоната натрия и двумя порциями воды. Раствор сушат (MgSO4) и концентрируют при пониженном давлении, получая желтое масло (17,9 г, 100%).
1H-ЯМР (300 МГц, CDCl3) δ =8,33 (1H, меньший, с), 8,58 (1H основной, с).
е) 6-Хлор-7-трифторметил-2,3-диметокси-5- нитрохиноксалин и 7-хлор-6-трифторметил-2, 3-диметокси- 5-нитрохиноксалин
Гидрид натрия (0,95 г, 80% масляная дисперсия, 31,7 ммоль) добавляют порциями к перемешиваемому раствору смеси трихлорхиноксалинов из приведенной выше стадии (d) (5,0 г, 14,4 ммоль) в безводном метаноле (125 мл) при комнатной температуре. Через 18 часов смесь концентрируют при пониженном давлении, разбавляют водой и образовавшийся твердый продукт отфильтровывают, промывают водой и сушат, получая белое твердое соединение (4,4 г, 90%), в виде смеси 2:1 изомеров.
1H-ЯМР (300 МГц, CDCl3) δ =4,10 (3H, меньший, с), 4,15 (3H основной, с), 4,20 (3H основной, 3H меньший, с), 8,00 (1H, меньший, с), 8,25 (1H основной, с).
f) 3-Амино-6-хлор-7-трифторметил-2-метокси-5-нитрохиноксалин и 3-амино-7-хлор-6-трифторметил-2-метокси-5-нитрохиноксалин
Смесь диметоксихиноксалиновых изомеров из стадии (е), приведенной выше, (0,5 г, 1,5 ммоль) и насыщенного раствора аммиака в метаноле (7 мл) нагревают при 100oC в закрытом сосуде под давлением в течение 4 часов. Смесь охлаждают, выливают в воду и экстрагируют тремя порциями этилацетата. Объединенные экстракты сушат (MgSO4) и концентрируют при пониженном давлении. Реакцию повторяют в тех же масштабах и сырые продукты объединяют и чистят флэш хроматографией (элюируя гексан/этилацетатом = 3:1). Вымываемый первым продукт представляет собой 6-хлор-7-трифторметил-изомер (590 мг, 61%), получаемый в виде не совсем белого твердого соединения.
1H-ЯМР (300 МГц, CDCl3) δ =4,08 (3H, с), 7,88 (1H, ушир. с), 8,08 (1H, с), 8,43 (1H, ушир. с).
Вымываемый вторым продукт представляет собой 7-хлор-6-трифторметил-изомер (205 мг, 21%), получаемый в виде не совсем белого твердого соединения.
1H-ЯМР (300 МГц, CDCl3) δ =4,12 (3H, с), 5,65 (2H, ушир. с), 7,80 (1H, с).
q) 3,5-Диамино-6-хлор-7-трифторметил-2-метоксихиноксалин.
Гидразин гидрат (0,34 мл, 7,0 ммоль) добавляют при перемешивании к раствору 3-амино-6-хлор-7-трифторметил-2-метокси-5-нитрохиноксалина (0,55 г, 17 ммоль) в этаноле (25 мл), содержащему суспендированный 10% рутений-на-углероде (55 мг) в атмосфере азота при нагревании до температуры кипения с обратным холодильником. Через 20 минут добавляют дополнительное количество гидразин гидрата (0,17 мл, 3,4 ммоль) и смесь перемешивают при нагревании до температуры кипения в течение 30 минут, затем оставляют стоять при комнатной температуре в течение ночи. Смесь фильтруют через Arbocel - ускоритель фильтрования, и плотный осадок на фильтре промывают этанолом и дихлорметаном. Фильтрат концентрируют при пониженном давлении, получая указанное в заглавии соединение (0,50 г, 100%) в виде не совсем белого твердого соединения.
1H-ЯМР (300 МГц, CDCl3) δ = 4,13 (3H, с), 5,08 (2H, ушир. с), 5,30 (2H, ушир, с), 7,47 (1H, с).
h) N-(3-Амино-6-хлор-7-трифторметил-2-метоксихиноксалин-5-ил) метансульфонамид.
Метансульфоновый ангидрид (2,9 г, 16,6 ммоль) добавляют при перемешивании к раствору 3,5-диамино-6-хлор-2- метокси-7-трифторметилхиноксалина (0,48 г, 1,64 ммоль) и пиридина (1,34 мл, 16,6 ммоль) в безводном тетрагидрофуране в атмосфере азота при комнатной температуре. Через 18 часов смесь концентрируют при пониженном давлении, разбавляют водой и продукты экстрагируют этилацетатом (три порции). Объединенные органические растворы промывают 2 М соляной кислотой, насыщенным водным бикарбонатом натрия и насыщенным водным хлористым натрием, сушат (MgSO4) и концентрируют при пониженном давлении, получая желтый продукт (0,9 г). Это вещество суспендируют в 25% растворе этиламина в этаноле (20 мл) при 50oC в течение 2 часов. Смесь концентрируют при пониженном давлении и распределяют между этилацетатом и 2 М соляной кислотой. Экстракт сушат (MgSO4) и концентрируют при пониженном давлении. Остаток чистят флэш хроматографией (элюируя дихлорметаном, затем дихлорметан/метанолом = 99: 1 и окончательно дихлорметан/ метанолом = 98:2). Содержащие продукт фракции концентрируют при пониженном давлении, растворяют в этилацетате и экстрагируют 2 н. соляной кислотой. Кислотный раствор подщелачивают (pH 8), добавляя насыщенный водный бикарбонат натрия и продукт экстрагируют этилацетатом. Органический раствор сушат (MgSO4) и концентрируют, получая указанное в заглавии соединение (0,25 г, 41%), в виде белого твердого продукта.
1H-ЯМР (300 МГц, ДМСО-d6) δ = 3,29 (3H, с), 4,07 (3H, с), 7,70 (2H, ушир. с), 7,90 (1H, с), 9,35 (1H, с).
m/z (термонапыление) 371 (MH+).
Препаративный пример 21. N-(6,7-Дихлор-2,3-диметоксихиноксалин- 5-ил)-N-(метоксикарбонилметил)метансульфонамид.
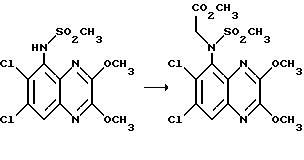
Безводный карбонат натрия (1,60 г, 11,2 ммоль) добавляют к суспензии N-(6,7-дихлор-2,3-диметоксихиноксалин-5-ил) метансульфонамида (см. препаративный пример 3, 3,0 г, 9,4 ммоль) в ацетоне (70 мл) при перемешивании. Смесь нагревают при температуре кипения с обратным холодильником в течение 15 минут, охлаждают и добавляют метилбромацетат (1,8 мл, 18,7 ммоль). 3атем смесь нагревают при температуре кипения с обратным холодильником 2 часа, охлаждают, разбавляют этилацетатом (300 мл) и промывают водой (2х100 мл). Органическую фазу сушат (MgSO4) и концентрируют при пониженном давлении. Остаток чистят флэш хроматографией (градиентное элюирование гексан/этилацетатом), получая указанное в заглавии соединение в виде белого твердого вещества (3,177 г, 80%).
1H-ЯМР (300 МГц, CDCl3) δ =3,44 (3H, с), 3,71 (3H, с), 4,14 (3H, с), 4,20 (3H, с), 4,37 (1H, д, J 8 Гц), 4,71 (1H, д, J 8 Гц), 7,95 (1H, с).
m/z (термонапыление) 424 (MH+).
Препаративный пример 22. N-(6,7-Дихлор-2,3-диметоксихиноксалин- 5-ил)-N-(6-метоксипиридин-2-ил)метил]метансульфонамид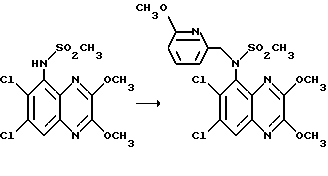
Данное соединение получают по способу препаративного примера 21, приведенного выше, используя 2-(бромметил)- 6-метоксипиридин [(см. Synth. Commun, 24, 1367 (1994)] вместо метилбромацетата. Соединение получают в виде белого твердого вещества ( 299 мг, 84%).
1H-ЯМР (300 МГц, CDCl3) δ = 3,38 (3H, с). 3,68 (3H, с), 4,07 (3H, с), 4,15 (3H, с), 4,85 (1H, д, J 14 Гц), 4,99 (1H, д, J 14 Гц), 6,58 (1H, д, J 8 Гц), 6,73 (1H, д. J 7 Гц), 7,40 (1H, дд, J 8 и 7 Гц), 7,92 (1H, с).
m/z (термонапыление) 473 (MH+).
Препаративный пример 23. N-(6,7-Дихлор-2,3-диметоксихиноксалин- 5-ил)-N-(1-метоксикарбонилэтил)-метансульфонамид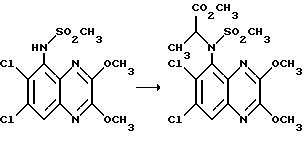
Соединение получают по способу препаративного примера 21, приведенного выше, используя метил 2-бромпропаноат вместо метилбромацетата. Продукт получают в виде смеси двух диастереоизомеров, которые разделяют флэш хроматографией (градиентное элюирование гексан/этилацетатом). Первый изомер получают в виде белого твердого продукта (1,456 г, 57%) (Rf 0,63, гексан/этилацетат = 1:1).
1H-ЯМР (300 МГц, CDCl3) δ/ =1,11 (3H, д, J 7 Гц), 3,36 (3H, с), 3,82 (3H, с), 4,15 (3H, с), 4,18 (3H, с), 5,10 (1H, кв., J 7 Гц), 7,96 (1H, с).
m/z (термонапыление) 438 (MH+).
Второй изомер получают также в виде белого твердого вещества (0,75 г, 29%) (Rf 0,45, гексан/этилацетат = 1:1).
1H-ЯМР (300 МГц, CDCl3) δ = 1,26 (3H, д, J 7 Гц), 3,36 (3H, с), 3,61 (3H, с), 4,15 (3H, с), 4,21 (3H, с), 4,98 (1H, кв, J 7 Гц), 7,97 (1H, с).
m/z (термонапыление) 438 (MH+).
Препаративный пример 24. 5-Амино-7-хлор-6-фтор-2,3- диметоксихиноксалин и 5-амино-6-хлор-7-фтор-2,3-диметоксихиноксалин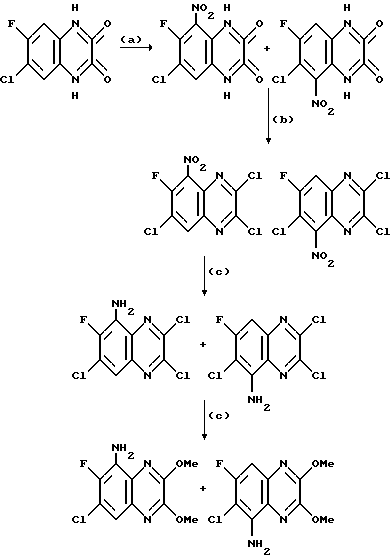
a) 1,4-Дигидро-6-хлор-7-фторхиноксалин-2,3-дион (см. пример 17, W094/00124) (0,200 г, 0,93 ммоль) добавляют порциями к дымящейся азотной кислоте (плотность 1,5 г•см-3, 5 мл) при комнатной температуре. Через 30 минут смесь добавляют к воде (50 мл). Образовавшийся твердый продукт отфильтровывают и сушат в вакууме, получая 6:1 смесь 1,4-дигидро-6-хлор-7-фтор-5-нитрохиноксалин-2,3- диона и 1,4-дигидро-7-хлор-6-фтор-5-нитрохиноксалин-2,3-диона (0,095 г, 39%) в виде бледно-оранжевого твердого соединения.
1H-ЯМР (300 МГц, ДМСО-d6) δ = 7,24 (1H основной, д, J 10 Гц), 7,39 (1H, основной, д, J 8 Гц), 12,17 (1H, основной, 1H меньший ушир.с), 12,29 (1H основной, 1H меньший, ушир.с).
m/z (термонапыление) 277, 279 (MH+).
b) Смесь промежуточных продуктов из приведенной выше стадии (a) превращают по способу препаративного примера 1(a) в 6:1 смесь 2,3,6-трихлор-7-фтор-5-нитрохиноксалина и 2,3,7-трихлор-6-фтор-5- нитрохиноксалина. Смесь продуктов получают в виде бледно-желтого твердого вещества (92% выход).
1H-ЯМР (300 МГц, CDCl3) δ =7,90 (1H основной, д, J 10 Гц), 8,28 (1H меньший, д, J 8 Гц).
с) Смесь промежуточных продуктов из стадии (b), приведенной выше, превращают в 6:1 смесь 5-амино-2,3,6-трихлор-7-фторхиноксалина и 5- амино-2,3,7-трихлор-6-фторхиноксалина по способу препаративного примера 1(b).
Смесь продуктов получают в виде желтого твердого вещества (100% выход).
1H-ЯМР (300 МГц, CDCl3) δ =4,92 (2H меньший, ушир.с), 5,45 (основной, ушир. с), 7,08 (1H основной, д, J 10 Гц), 7,38 (1H меньший, д, J 8 Гц).
m/z (термонапыление) 267 (MH+).
d) Смесь промежуточных продуктов из стадии (с), приведенной выше, превращают в смесь 5-амино-6-хлор-7-фтор-2,3-диметоксихиноксалина и 5-амино-7-хлор-6-фтор-2,3-диметоксихиноксалина по способу препаративного примера 1(с). Смесь продуктов разделяют флэш хроматографией (элюируя толуолом), что дает 5-амино-6-хлор-7-фтор-2,3- диметоксихиноксалин в виде белого твердого вещества (27% выход), т.пл. 199-200oC.
1H-ЯМР (300 МГц, CDCl3) δ =4,15 (6H, с), 5,04 (2H, ушир. с), 6,90 (1H, д, 10 Гц).
m/z (термонапыление) 258, 260 (MH+).
5-Амино-7-хлор-6-фтор-2,3-диметоксихиноксалин получают в виде белого твердого вещества (6% выход), т.пл. 198-199oC.
1H-ЯМР (300 МГц, CDCl3) δ =4,14 (3H, с), 4,18 (3H, с), 4,62 (2H, ушир. с), 7,40 (1H, д, J 8 Гц).
m/z (термонапыление): 258, 260 (MH+).
Препаративный пример 25. N-(7-Хлор-6-фтор-2,3-диметоксихиноксалин-5-ил) метансульфонамид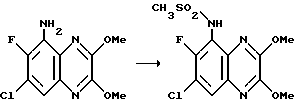
Метансульфоновый ангидрид (0,55 г, 3,16 ммоль) и пиридин (255 мкл, 3,15 ммоль) добавляют к перемешиваемой суспензии 5-амино-7-хлор- 6-фтор-2,3-диметоксихиноксалина (см. препаративный пример 24) (0,274 г, 1,06 ммоль) в дихлорметане (15 мл) при комнатной температуре в атмосфере азота. Смесь перемешивают при комнатной температуре в течение ночи. Концентрируют досуха и суспендируют в тетрагидрофуране (30 мл). Реакционную смесь обрабатывают 1 М гидроокисью натрия (5,3 мл, 5,3 ммоль) при охлаждении льдом с последующим перемешиванием при 5oC в течение 1,5 часа. Реакционную смесь подкисляют до pH 2 с помощью 2 М соляной кислоты, концентрируют и распределяют между водой и дихлорметаном. Слои разделяют и водный слой экстрагируют дважды дихлорметаном. Объединенные органические фазы сушат (MgSO4) и концентрируют, получая бледно-желтый твердый продукт. Суспендируют в ТГФ (30 мл) и обрабатывают 1 М гидроокисью натрия (5,3 мл, 5,3 ммоль) при 5oC и впоследствии перемешивают при комнатной температуре в течение 2 часов. Реакционную смесь подкисливают до pH 2 с помощью 2 М соляной кислоты, концентрируют до небольшого объема, разбавляют водой и фильтруют. Фильтрат промывают водой и охлажденным диэтиловым эфиром, получая белый твердый продукт (0,32 г, 90%), т.пл. 227-228oC.
1H-ЯМР (300 МГц, ДМСО-d6) δ = 3,12 (3H, с), 4,04 (3H, с), 4,12 (3H, с), 7,90 (1H, д, J 8 Гц), 9,50 (1H, с).
m/z (термонапыление) 336, 338 (MH+).
Препаративный пример 26. N-(6-Хлор-7-фтор-2,3- диметоксихиноксалин-5-ил)метансульфонамид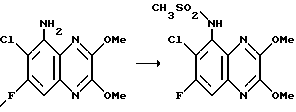
5-Амино-6-хлор-7-фтор-2,3-диметоксихиноксалин (см. препаративный пример 24) превращают по способу препаративного примера 25 в N-(6-Хлор-7-фтор-2,3-диметокси хиноксалин-5-ил) метансульфонамид. Продукт получают в виде белого твердого соединения (30% выход).
1H-ЯМР (300 МГц, ДМСО-d6) δ =3,16 (3H, с), 4,04 (3H, с), 4,12 (3H, с), 7,72 (1H, д, J 10 Гц), 9,60 (1H, с).
m/z (термонапыление) 336, 338 (MH+).
Препаративный пример 27.
N-(6,7-Дихлор-2, 3-диметоксихиноксалин-5-ил)-N- (2-аминоэтил)метансульфонамид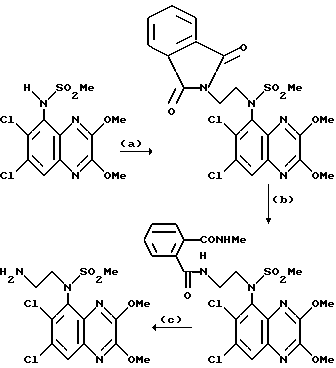
a) N-(2-бромэтил)фталимид (1,73 г, 6,81 ммоль) добавляют к нагреваемой до температуры кипения с обратным холодильником смеси N-(6,7-Дихлор-2,3- диметокси-хиноксалин-5-ил)метансульфонамида (препаративный 3, 2,00 г, 5,68 ммоль) и карбоната калия (1,88 г, 13,63 ммоль) в ацетоне (100 мл) в атмосфере азота. Через 48 часов добавляют дополнительное количество N-(2-бромэтил)фталимида (1,73 г, 6,81 ммоль) и нагревание до температуры кипения с обратным холодильником продолжают в течение 18 часов. После охлаждения смесь концентрируют при пониженном давлении и остаток растворяют в дихлорметане. Полученный раствор дважды промывают 1 н. раствором гидроокиси натрия, водой и раствором соли и затем сушат (MgSO4) и концентрируют при пониженном давлении. Остаток чистят флэш хроматографией (элюируя гексан:этилацетатом), что дает N-(6,7-дихлор-2,3- диметоксихиноксалин-5-ил)-N-(2-фталимидоэтил)метансульфонамид в виде бледно-желтого твердого вещества (2,55 г, 85%).
1H-ЯМР (300 МГц, CDCl3) δ =3,25 (3H, с), 3,64 (2H, т, J 8 Гц), 3,90-4,02 (2H, м), 4,12 (3H, с), 4,17 (3H, с), 7,65-7,80 (3H, м) 7,82-7,92 (2H, м).
m/z (термонапыление) 525 (MH+)
b) 33% раствор метиламина в IMS (промышленные денатурированные спирты) (1,77 мл, 19,03 ммоль) добавляют к перемешиваемому раствору N-(6,7-дихлор-2,3-диметоксихиноксалин-5-ил)-N-(2- фталимидэтил)метансульфонамида (2,00 г, 3,81 ммоль) в дихлорметане (38 мл) в сосуд, заполненный азотом, с резиновой прокладкой. Смесь перемешивают 72 часа и затем добавляют дополнительное количество 33% раствора метиламина в IMS (1,77 мл, 19,03 ммоль). Смесь перемешивают 18 часов и затем концентрируют при пониженном давлении. Остаток растворяют в дихлорметане и дважды экстрагируют 10% водной лимонной кислотой. Органический слой концентрируют при пониженном давлении, получая N-(6,7-дихлор-2, 3-диметоксихиноксалин-5-ил)-N-(N'-([2 - метиламинокарбонил] бензоил)аминоэтил)метансульфонамид в виде твердого вещества (996 мг, 49%).
1H-ЯМР (300 МГц, CDCl3) δ =2,92 (3H, д, J 5 Гц), 3,20 (3H, с), 3,45-3,62 (2H, м), 3,97 (3H, с), 3,98-4,10 (2H, м), 4,13 (3H, с), 6,60 (1H, ушир. д), 7,44 (1H, м), 7,60 (1H, м), 7,72 (1H, м), 7,82 (1H, м), 7,95 (1H, с).
m/z (термонапыление) 556 (MH+).
с) Гидразин гидрат (26 мкл, 26 мг, 0,531 ммоль) добавляют по каплям к раствору N-(6,7-дихлор-2,3-диметокси-хиноксалин-5-ил)-N-(N' -([2-метиламинокарбонил] бензоил)амино-этил)метансульфонамида (283 мг, 0,531 ммоль) в дихлорметане (5 мл) и смесь нагревают до температуры кипения 4 часа. Добавляют метанол (1 мл) и нагревание до температуры кипения с обратным холодильником продолжают в течение 18 часов. После охлаждения смесь концентрируют при пониженном давлении. Остаток растворяют в этилацетате и экстрагируют 10% лимонной кислотой. Водный слой доводят до pH 8 с помощью твердого карбоната калия и дважды экстрагируют дихлорметаном. Объединенные органические слои сушат (MgSO4) и концентрируют при пониженном давлении, получая N-(6,7-дихлор-2,3-диметоксихиноксалин-5-ил)-N- (2-аминоэтил)метансульфонамид в виде бледно-желтого твердого продукта (138 мг, 66%).
1H-ЯМР (300 МГц, CDCl3) δ =2,70-2,88 (2H, м), 3,11 (3H, с), 3,79 (2H, т, J 8 Гц), 4,17 (3H, с), 4,20 (3H, с), 7,95 (1H, с), m/z (термонапыление) 395 (MH+).
Препаративный пример 28.
5-Бромметил-6,7-дихлор-2, 3-диметоксихиноксалин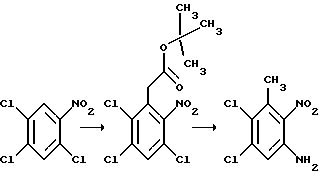
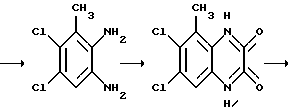
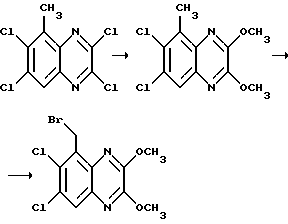
a) Раствор 2,4,5-трихлорнитробензола (Jpn. Kokai Tokyo Koho JP 81, 169, 651 (1980) Chem. Abstr. 1981, 96, 162307 g) (103 г, 0,46 моль) и трет-бутил хлорацетата (79 мл, 0,55 моль) в сухом тетрагидрофуране (400 мл) добавляют по каплям в течение более 30 минут к раствору трет-бутилата калия (128 г, 1,14 моль) в сухом тетрагидрофуране (800 мл) при перемешивании в атмосфере азота и при выдерживании температуры - 40oC. По завершении добавления образовавшийся темно-синий раствор перемешивают дополнительно 30 минут. Смесь выливают в 0,5 М соляную кислоту (2 л) и продукт экстрагируют этилацетатом (2,5 л и 1 л). Объединенные органические растворы сушат (MgSO4) и упаривают на силикагеле (70-200 меш, 200 г). Силикагель переносят в верхнюю часть силикагелевой хроматографической колонки (800 г) и продукт элюируют, используя градиент гексан/этилацетата. Содержащие продукты фракции объединяют и упаривают, получая желтый твердый продукт, который обрабатывают гексаном, что дает трет-бутил 2-нитро-3,5,6- трихлорфенилацетат (91,8 г, 59%) в виде белого твердого вещества. Найдено, 42,32; H, 3,50; N 4,03.
Рассчитано для C12H12Cl3NO4: С 42,32; H 3,55; N 4,11%. 1H-ЯМР (300 МГц, CDCl3) δ =1,42 (9H, с), 3,73 (2H, с), 7,60 (1H, с).
m/z (термонапыление) 357 (MNH4 +).
b) Смесь трет-бутил 2-нитро-3,5,6-трихлорфенилацетата (из стадии (a), 123 г, 0,361 моль) и насыщенного водного раствора аммиака (300 мл) в 2-метоксиэтаноле (360 мл) нагревают в автоклаве при 150oC в течение 72 час. Образовавшуюся вязкую черную смесь разбавляют водой (1 л) и этилацетатом (1 л) и фильтруют через Arbocel - ускоритель фильтрования. Темно-красный фильтрат отделяют и водный слой экстрагируют этилацетатом (2х1 л). Объединенные органические растворы промывают раствором соли (1 л), сушат (MgSO4) и упаривают на силикагеле (70-200 меш, 200 г). Силикагель переносят в верхнюю часть хроматографической колонки, содержащей силикагель (40-60 меш, 800 г). Элюирование гексан/этилацетатом (98:2-92:8) дает 3-амино-5,6-дихлор-2-нитротолуол (Европ. патент 385850) в виде ярко-оранжевого твердого вещества (39,7 г), загрязненного 5-амино-3,6-дихлор-2-нитротолуолом (14 %). Эту смесь используют в следующей стадии без дополнительной очистки.
1H-ЯМР (300 МГц, CDCl3) δ =2,48 (3H, с), 4,80 (2H, с), 6,82 (1H, с).
с) Раствор дитионита натрия (94 г, 0,54 моль) в воде (1 л) добавляют к перемешиваемой смеси 3-амино-5,6-дихлор-2-нитротолуола (из стадии (b), 39,7 г, 0,18 моль) и бикарбоната калия (94 г, 0,94 ммоль) в метаноле (1 л) при комнатной температуре. Через 30 минут смесь концентрируют при пониженном давлении и образовавшуюся суспензию экстрагируют этилацетатом (всего 700 мл). Экстракты сушат (MgSO4) и концентрируют при пониженном давлении, получая 2,3-диамино-5,6-дихлортолуол (26,1 г, 38% более 2 стадий) в виде коричневого твердого продукта.
1H-ЯМР (300 МГц, CDCl3) δ =2,28 (3H, ушир, с), 3,36 (2H, ушир.с), 3,42 (2H, ушир.с), 6,72 (1H, с).
d) Смесь 2,3-диамино-5,6-дихлортолуола (из стадии (с), 21,6 г, 0,137 моль) и щавелевой кислоты (18,45 г, 0,206 моль) в соляной кислоте (4 М, 900 мл) нагревают до температуры кипения с обратным холодильником в течение 6 часов, охлаждают и фильтруют. Темно-коричневый твердый продукт суспендируют в диэтиловом эфире, фильтруют и промывают добавочным количеством диэтилового эфира, получая 6,7-дихлор-5-метил-хиноксалин-2,3-дион (22,06 г, 66%).
1H-ЯМР (300 МГц, ДМСО) δ =2,40 (3H, с), 7,14 (1H, с), 11,37 (1H, с), 11,94 (1H, с).
е) Смесь 6,7-дихлор-5-метилхиноксалин-2,3-диона (из стадии (d) 22,06 г, 90 ммоль), тионилхлорида (300 мл) и диметилформамида (1 мл) нагревают до температуры кипения с обратным холодильником в течение 3 часов, охлаждают и медленно выливают в ледяную воду. Полученный темно-желтый осадок отфильтровывают, что дает 5-метил-2,3,6,7- тетрахлорхиноксалин (24,42 г, 96%)
1H-ЯМР (CDCl3) δ = 2,85 (3H, с), 8,02 (1H, с).
f) Раствор метилата натрия (38 мл, 25% раствор в метаноле, 175 ммоль) добавляют в течение более 10 минут к раствору 5-метил-2,3,6,7-тетрахлорхиноксалина (из стадии (е), 21 г, 74 ммоль) в сухом тетрагидрофуране (200 мг) при 20oC. Протекает слабоэкзотермическая реакция, сопровождающаяся образованием осадка. Через 1 час смесь разбавляют этилацетатом (3 л), промывают водой (1 л) сушат (MgSO4) и концентрируют при пониженном давлении, получая 6,7-дихлор-2,3-диметокси-5-метилхиноксалин (20,3 г, 100%).
1H-ЯМР (300 МГц, CDCl3) δ =2,75 (3H, с), 4,15 (3H, с), 4,18 (3H, с), 7,78 (1H, с).
m/z (термонапыление) 273 (MH+).
g) Смесь 6,7-дихлор-2,3-диметокси-5-метилхиноксалина (из стадии (f), 22,0 г, 80,5 ммоль), N-бромсукцинимида (17,2 г, 96,6 ммоль) и α,α- азоизобутиронитрила (1,3 г, 8,0 ммоль) нагревают до температуры кипения с обратным холодильником в 1,1,1-трихлорэтане (400 мл) в течение 4 час при облучении 500 W лампой дневного света. Смесь охлаждают, добавляют силикагель (50 г, 60-230 меш) и растворитель удаляют при пониженном давлении. Остаток загружают сверху в хроматографическую колонку с силикагелем и продукт элюируют, используя градиент гексан/этилацетата. Продукт растирают с гексаном, получая 5-бромметил-6,7-дихлор-2,3-диметоксихиноксалин (25,3 г, 87%) рыхлого белого твердого вещества.
Найдено: C 37,72; H 2,40; N 7,40; для C11H9BrCl2N2O2
рассчитано: C 37,53; H 2,58; N 7,96%.
1H-ЯМР (300 МГц, CDCl3) δ =4,15(3H, с), 4,22 (3H, с), 5,20 (2H, с), 7,89 (1H, с).
Препаративный пример 29. (6,7-Дихлор-2,3-диметоксихиноксалин-5- ил)метил метил сульфон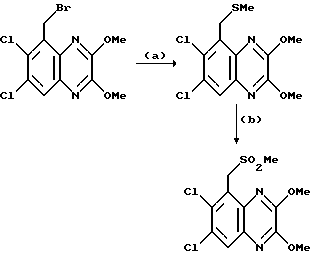
a) Метанмеркаптид натрия (22 мг, 0,312 ммоль) добавляют к перемешиваемому раствору 5-бромметил-6,7-дихлор-2,3- диметоксихиноксалина (см. препаративный пример 28) (100 мг, 0,284 ммоль) в сухом диметилформамиде (5 мл) в атмосфере азота при комнатной температуре. Смесь перемешивают 10 минут и затем реакцию обрывают добавлением охлажденного раствора соли и дважды экстрагируют дихлорметаном. Объединенные экстракты сушат (MgSO4) и концентрируют при пониженном давлении. Остаток чистят флэш хроматографией (элюируя 1:1 гексан: дихлорметаном), что дает 6,7- дихлор-2,3-диметокси-5-метил-тиометилхиноксалин (79 мг, 87%) в виде белого твердого вещества, т.пл. 143-5oC.
1H-ЯМР (300 МГц, CDCl3) δ = 2,10 (3H, с), 4,12 (3H, с), 4,15 (3H, с), 4,39 (2H, с), 7,81 (1H, с).
m/z (термонапыление) 319 (MH+).
b) 3-Хлорнадоксибензойную кислоту (50%, 1,904 г, 5,52 ммоль) добавляют порциями к перемешиваемому раствору 6,7-дихлор-2,3- диметокси-5-метилтиометилхиноксалина (стадия (a), 800 мг, 2,51 ммоль) в сухом дихлорметане при комнатной температуре в атмосфере азота. Смесь перемешивают 30 минут и затем реакцию обрывают добавлением 10% водного раствора сульфита натрия и органический слой отделяют. Раствор дихлорметана промывают 10% водным раствором карбоната калия, сушат (MgSO4) и концентрируют при пониженном давлении, получая в остатке (6,7-дихлор-2,3-диметокси-хиноксалин-5-ил)метил метил сульфон (980 мг, > 100%, содержит некоторое количество дихлорметана) в виде белого твердого продукта, т. пл. 161-3oC.
1H-ЯМР (300 МГц, CDCl3) δ =2,92 (3H, с), 4,13 (3H, с), 4,19 (3H, с), 5,16 (2H, с), 7,94 (1H, с).
m/z (термонапыление) 351 (MH+).
Препаративный пример 30. (6,7-Дихлор-2,3-диметоксихиноксалин- 5-ил) метил этил сульфон
Указанное в заглавии соединение получают из соединения препаративного примера 28 по способу препаративного примера 29 (a) и (b), используя этанмеркаптид натрия, в виде не совсем белого твердого вещества (31% из двух стадий), т.пл. 150-2oC.
1H-ЯМР (300 МГц, CDCl3) δ =1,43 (3H, т, J 8 Гц), 3,08 (2H, кв, J 8 Гц), 4,16 (3H, с), 4,21 (3H, с), 5,14 (2H, с), 7,96 (1H, с).
m/z (термонапыление) 365 (MH+).
Препаративный пример 31. (6,7-Дихлор-2,3- диметоксихиноксалин-5-ил)метил бензил сульфон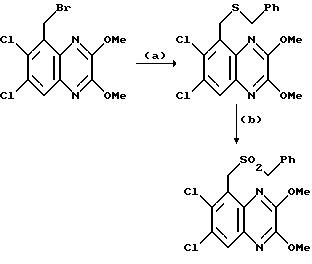
a) Карбонат калия (108 мг, 0,781 ммоль) и затем бензилмеркаптан (92 мкл, 97 мг, 0,781 ммоль) добавляют к перемешиваемому раствору 5-бромметил-6,7-дихлор-2,3-метоксихиноксалина (препаративный пример 28) (250 мг, 0,71 ммоль) в сухом диметилформамиде (10 мл) в атмосфере азота при комнатной температуре. Смесь перемешивают 30 минут и затем распределяют между раствором соли и этилацетатом. Органический слой отделяют и водную фазу экстрагируют дважды этилацетатом. Объединенные экстракты сушат (MgSO4) и концентрируют при пониженном давлении. Остаток чистят флэш хроматографией (элюируя 3:1, затем 2: 1 гексан: дихлорметаном), что дает 5-бензилтиометил-6,7-дихлор-2,3-диметоксихиноксалин (268 мг, 96%) в виде белого твердого вещества, т.пл. 121 - 122oC.
1H-ЯМР (300 МГц, CDCl3) δ =3,86 (2H, с), 4,00 (3H, с), 4,14 (3H, с), 4,43 (2H, с), 7,25 (5H, м), 7,80 (1H, с).
m/z (термонапыление) 395 (MH+).
b) Указанное в заглавии соединение получают из 5-бензилтиометил-6,7-дихлор-2,3-диметоксихиноксалина (стадия (a)) по способу препаративного примера 29 (b) в виде белого твердого вещества (93%), т.пл. 185-7oC.
1H-ЯМР (300 МГц, CDCl3) δ = 4,03 (3H, с), 4,12 (3H, с), 4,35 (2H, с), 5,12 (2H, с), 7,40 (5H, м), 7,93 (1H, с).
m/z (термонапыление) 427 (MH+.
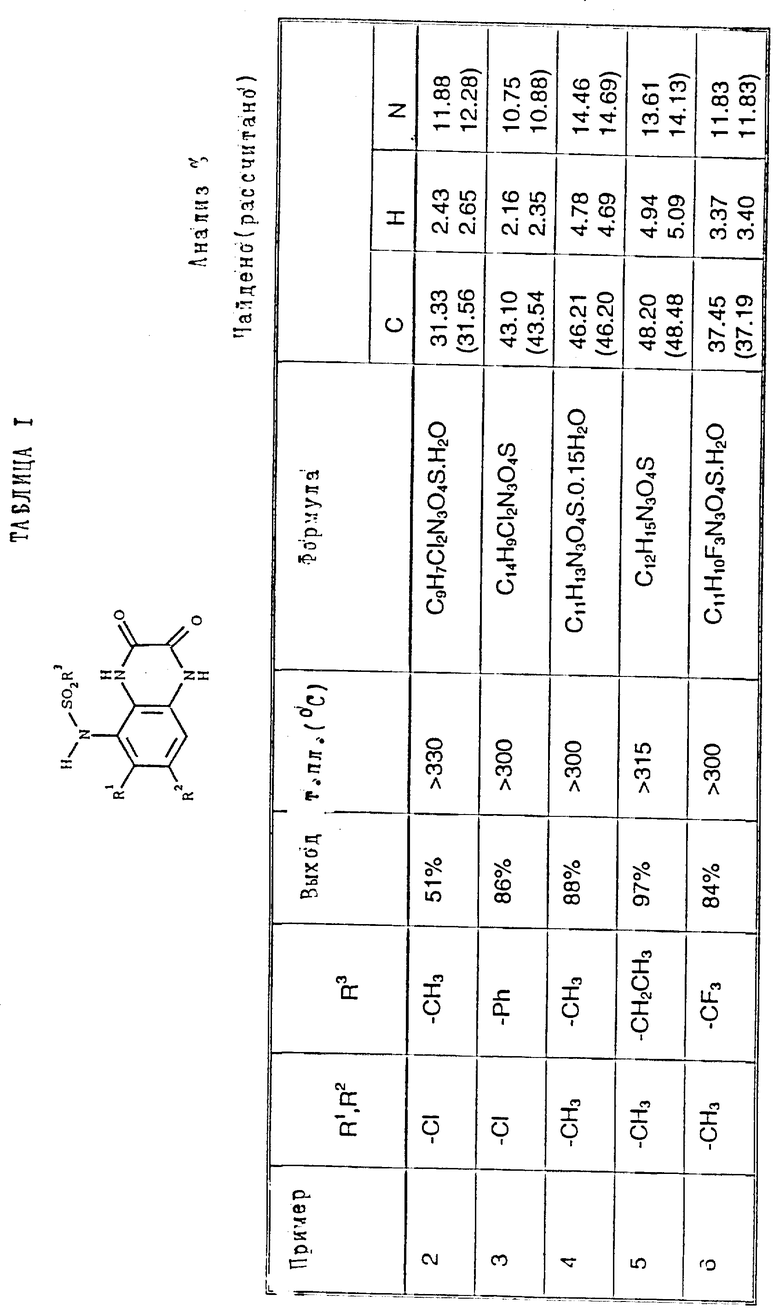
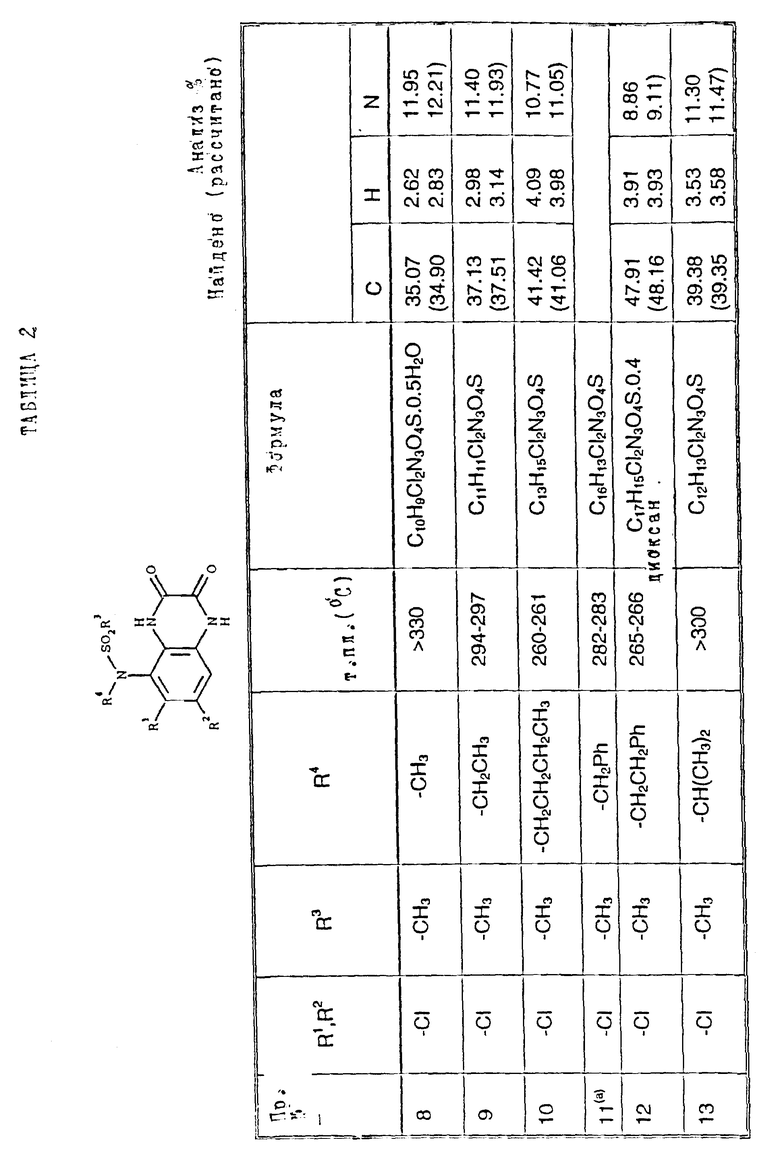
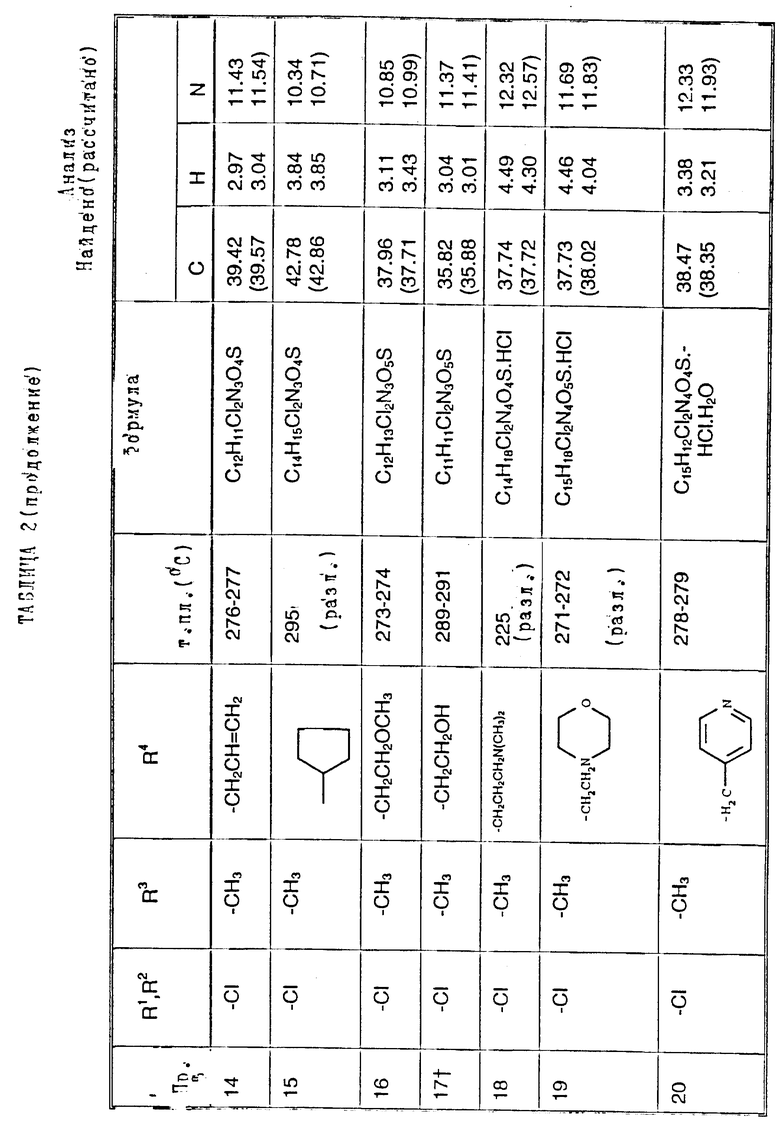
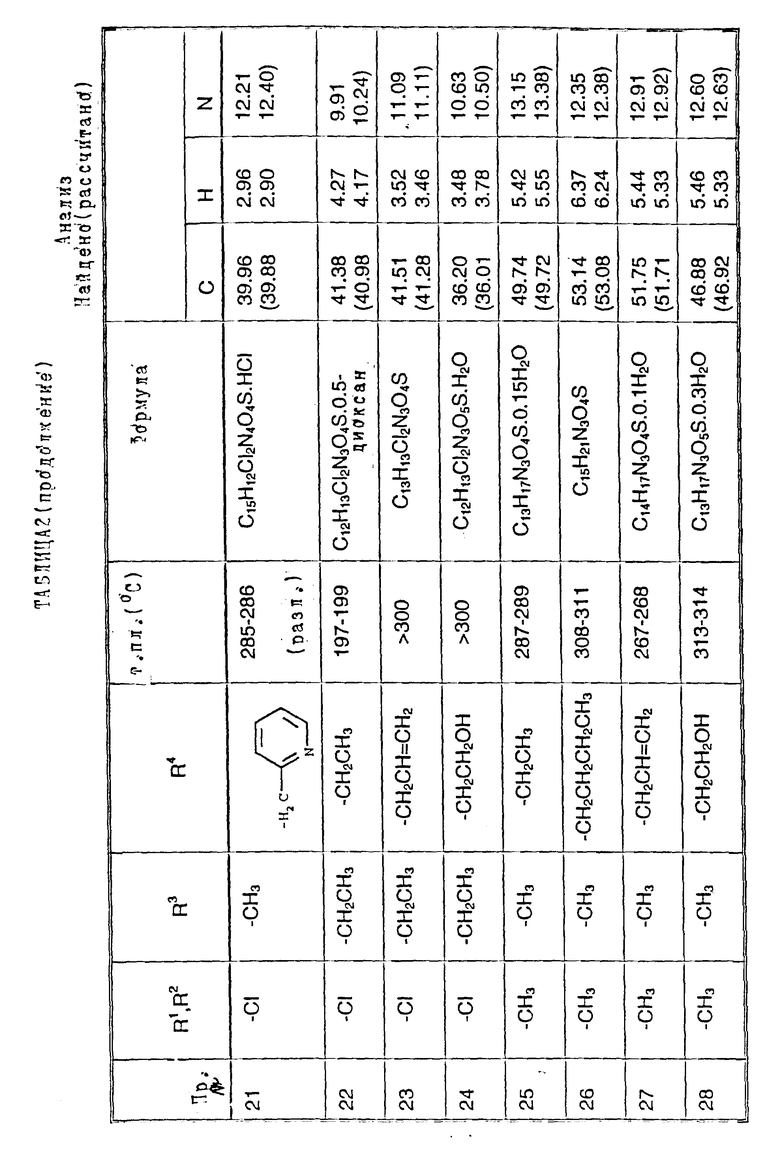
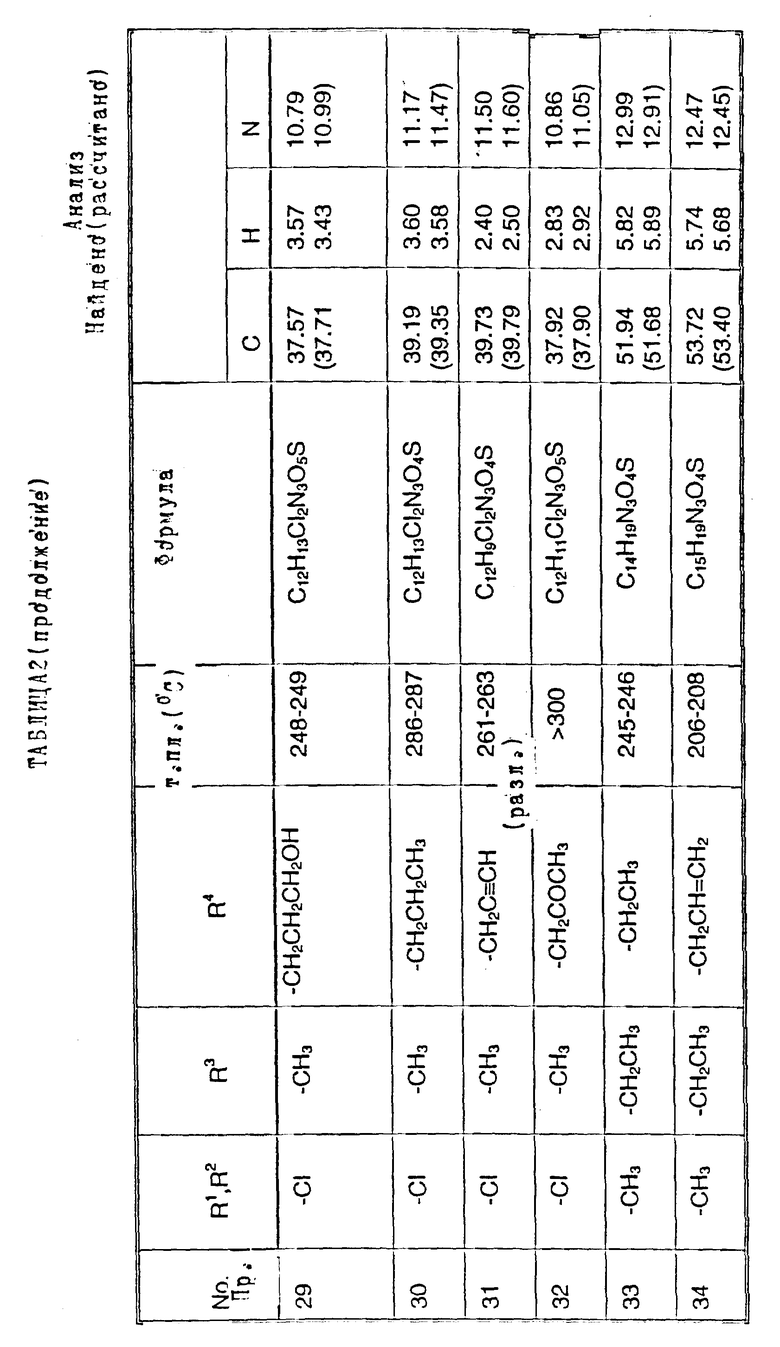
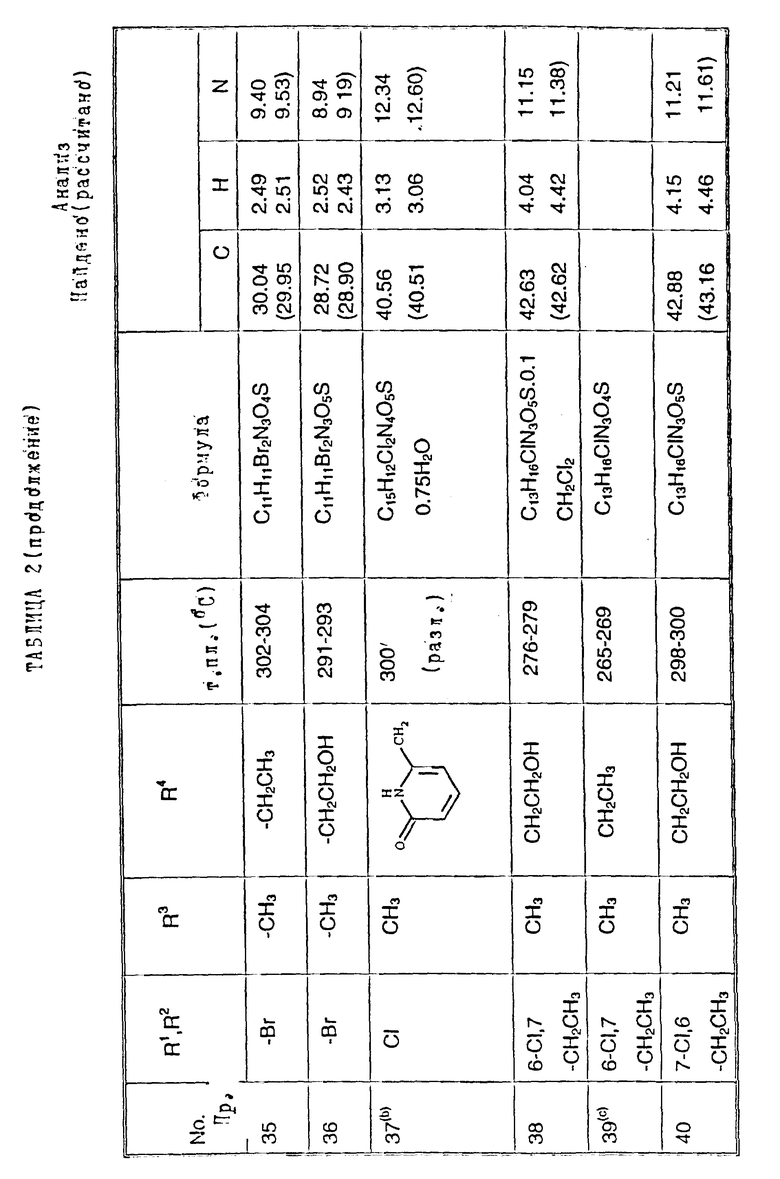
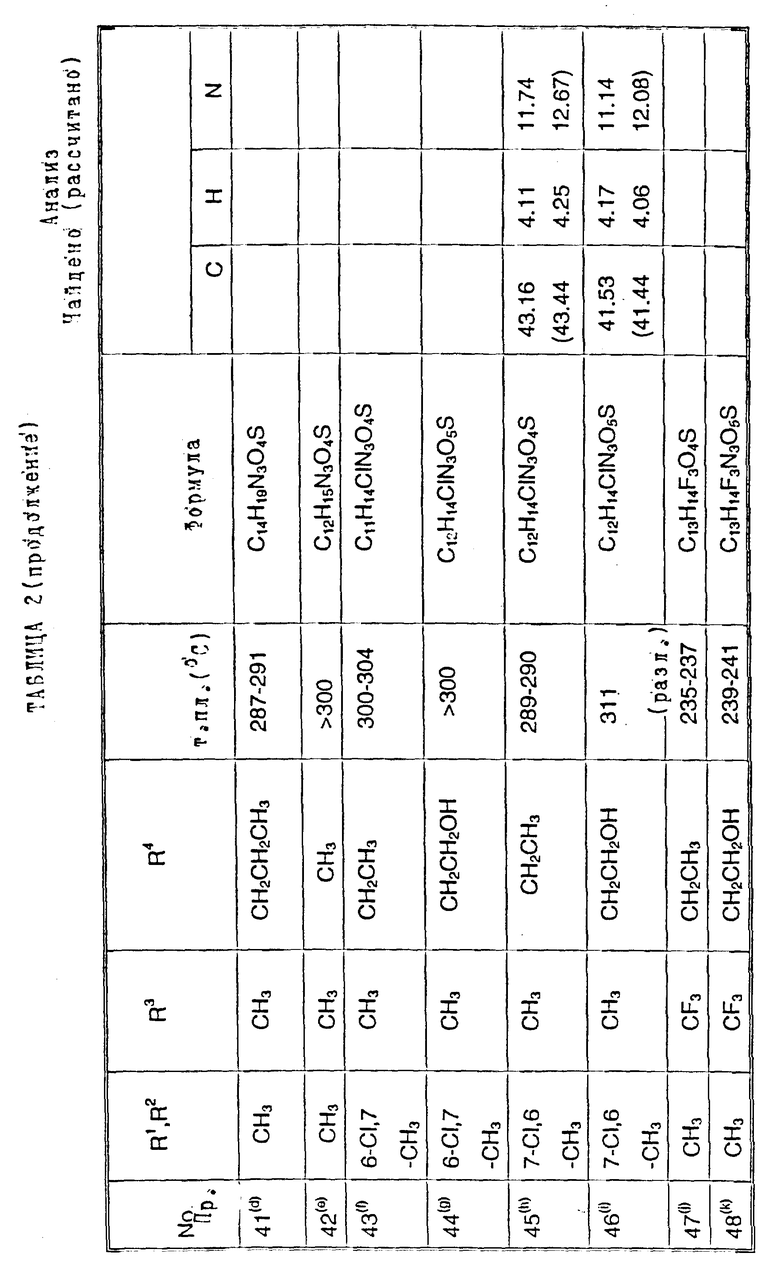

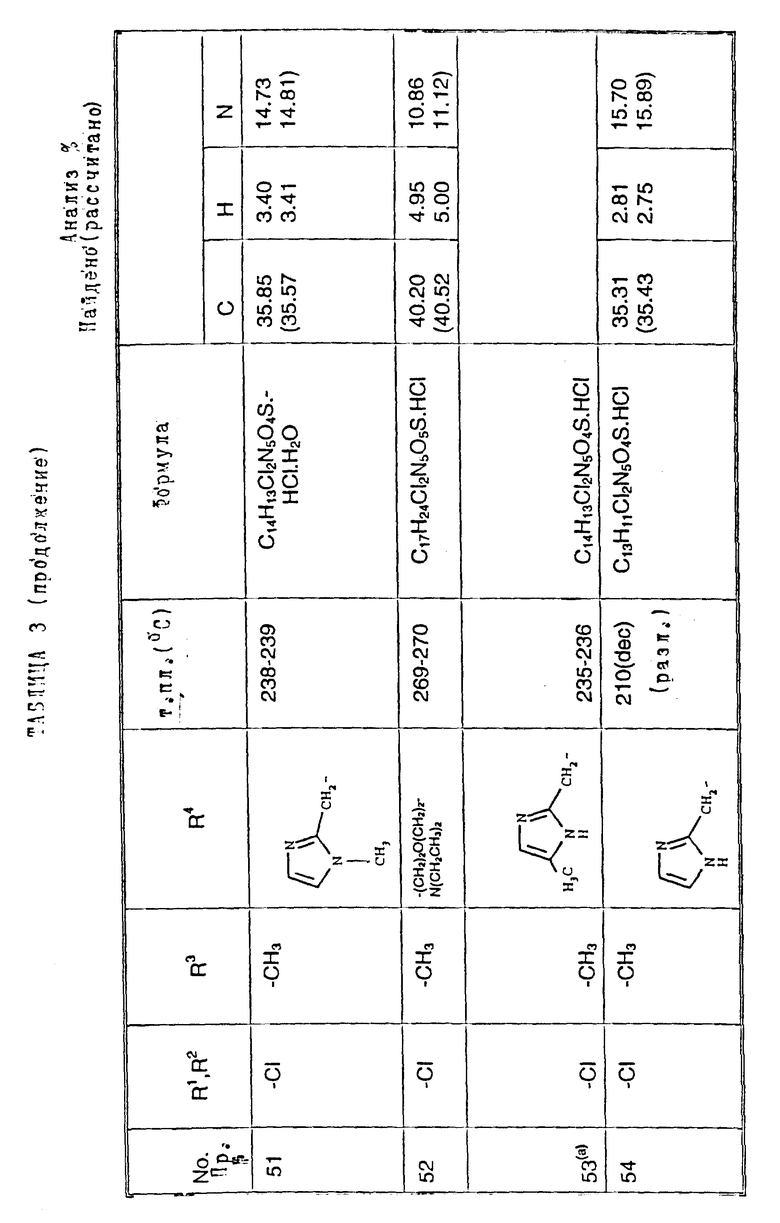
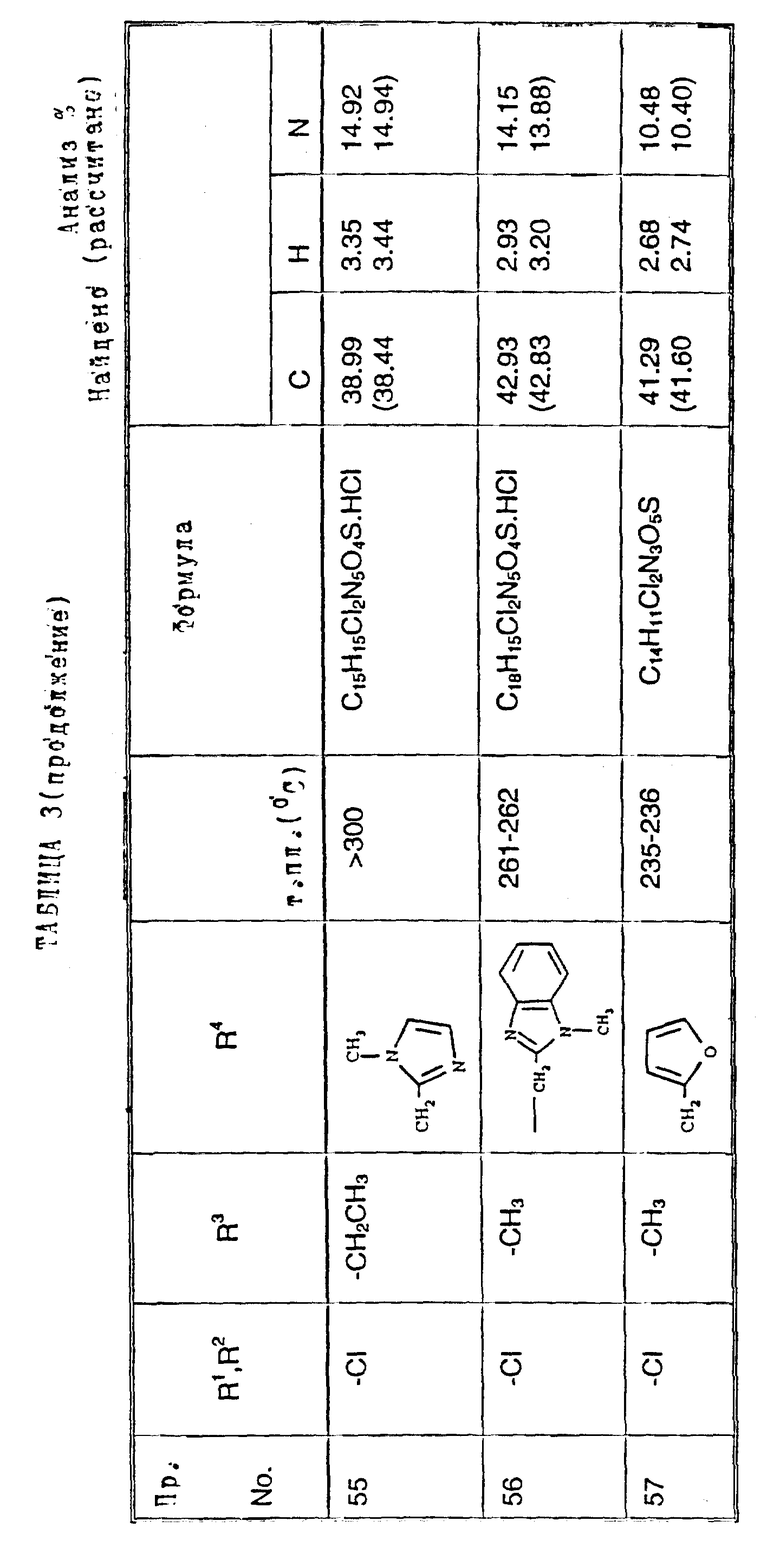
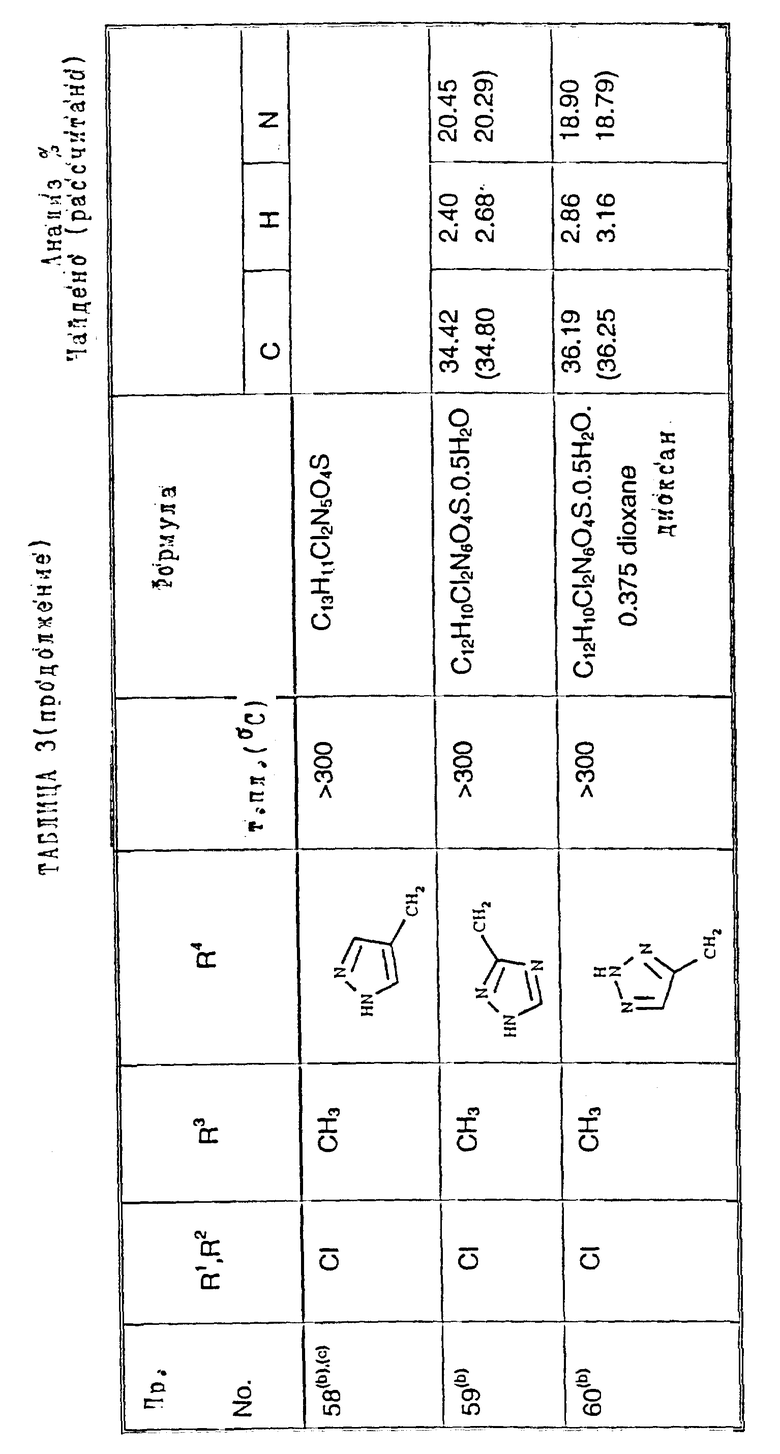
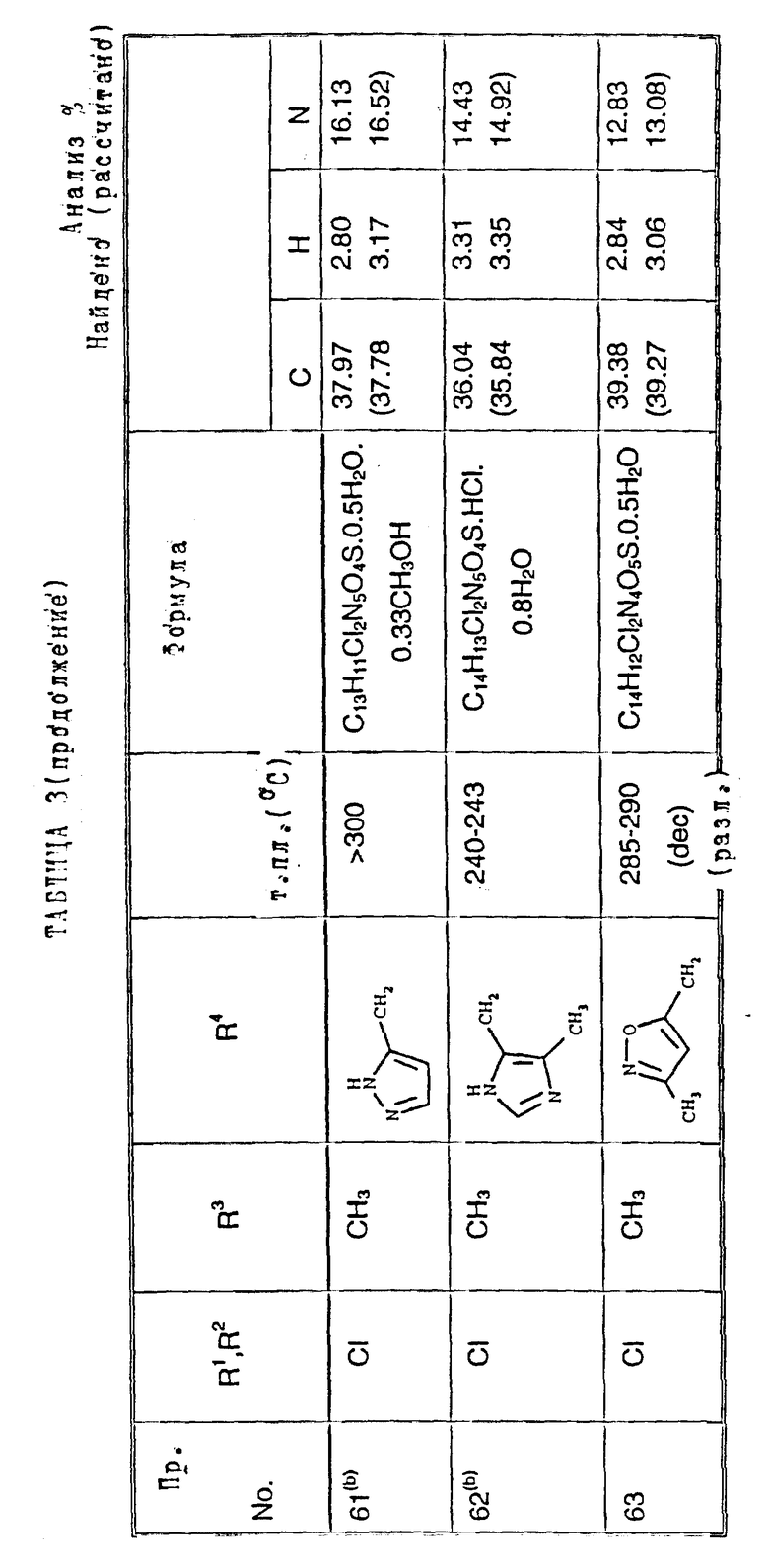
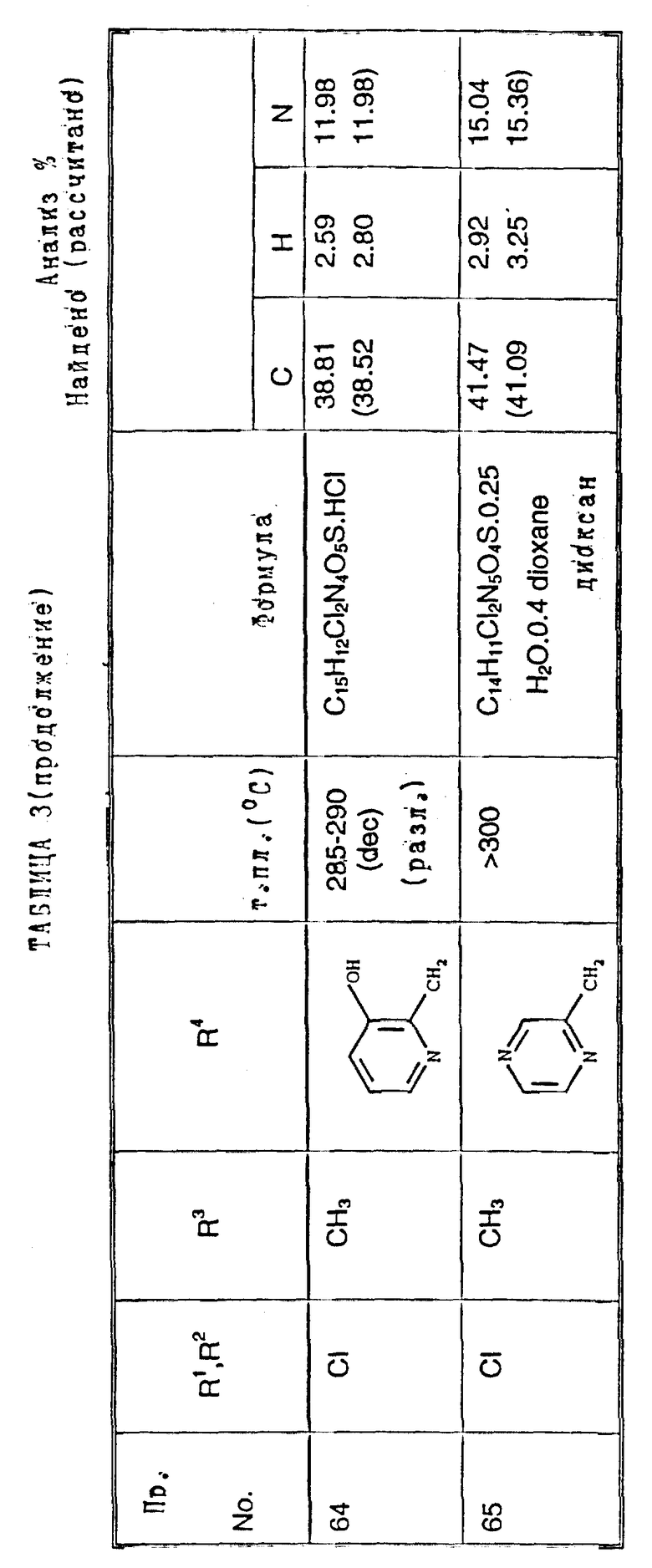
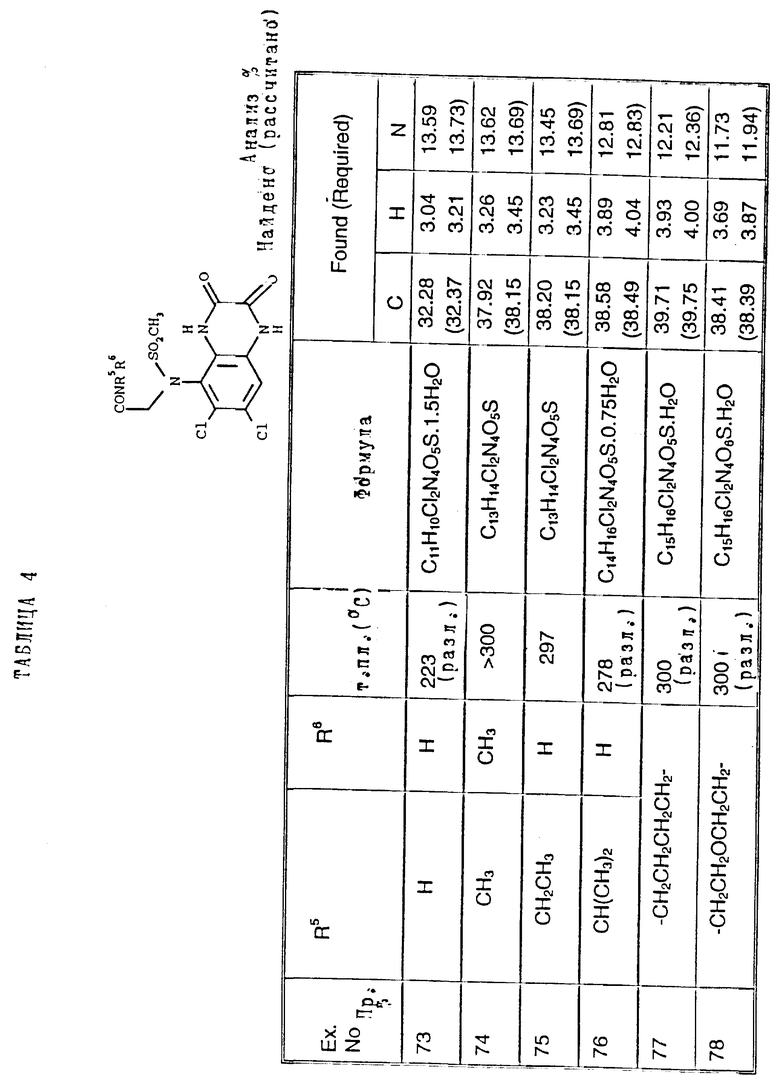
Производные хиноксалина общей формулы I, где A - азот или СН; R1, R2 - алкил, галоид или СF3; R4 - Н, циклоалкил или алкил, возможно замещенный гидрокси, алкокси, фенилом, пиридинилом, морфолинилом, бензимидазолилом, пиразолилом, озоксазолом или фталимидилом, С2-6алкенилом, C2-6алкинилом, С2-6алканоилом, карбокси, аминоалкоксикарбонилом, алкиламино, диалкиламино, NHSO2CF3, CONR5R6, O(CH2)nNR5R6, NHCO-NR5R6; R5, R6 - Н, алкил или вместе с атомом азота образуют пирролидино- или морфолиногруппу; n = 2, 3 или 4, и их фармацевтически приемлемые соли, обладают анксиолитической, противосудорожной аналгезирующей или нейрозащитной активностью. 4 с. и 11 з.п. ф-лы, 4 табл. 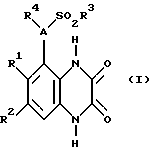
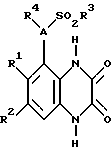
где A - N или n CH;
R1 и R2 независимо - C1-4алкил, галоид или CF3;
R3 - C1-4алкил, необязательно замещенный фенилом, CF3 или фенил;
R4 - водород, C3-7циклоалкил или C1-6алкил, [необязательно замещенный гидрокси, C1-4алкокси, фенилом, гетероциклической группой (выбранной из пиридинила, морфолинила, имидазолила, бензимидазолила, пиразолила, пиразинила, триазолила, тетразолила, фурила, изооксазолила и фталимидила, которая необязательно замещена до 3 заместителей, выбранных из C1-4алкила, гидрокси и оксо), C2-6алкенилом, C2-6алкинилом, C2-6алканоилом, карбокси, амино, C1-4алкоксикарбонил, C1-4алкиламино, ди(C1-4алкил)-амино, NHSO2CF3, CONR5R6, NHCONR5R6 или O(CH2)nNR5R6], где R5 и R6 независимо обозначают водород или C1-4алкил или вместе с атомом азота, к которому они присоединены, могут представлять пирролидино- или морфолиногруппу;
n = 2, 3 или 4,
или их фармацевтически приемлемые соли.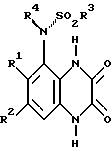
где R1-4 - такие, как определено в п.1.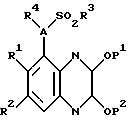
где A, R1 - R4 имеют значения, определенные в п.1;
P1 и P2 гидроксизащитная группа,
удаляют защитную группу и при необходимости полученное соединение превращают в его фармацевтически приемлемую соль.
в которой A, R1 - R4, P1 и P2 имеют значения, определенные в п.14.
| Способ получения замещенных хиноксалиндионов или их солей | 1974 |
|
SU584771A3 |
| ЗЕРНОУБОРОЧНЫЙ КОМБАЙН | 0 |
|
SU377112A1 |
| УСТРОЙСТВО для ФИКСИРОВАНИЯ ТЕРМОПАРНОЙПРОВОЛОКИ | 0 |
|
SU315959A1 |
| WO 9400124 А1, 06.01.94. | |||
