Изобретение относится к новым противоопухолевым соединениям, в частности к хиназолиновым производным или их фармацевтически приемлемым солям или сложным эфирам, обладающим противоопухолевой активностью. Изобретение включает новые производные хиназолина и способы их получения, новые фармацевтические композиции, содержащие указанные хиназолиновые производные, использование этих производных для получения новых лекарственных препаратов, обладающих способностью продуцировать противоопухолевый ответ у теплокровных животных, например у человека.
Одна из групп противоопухолевых соединений представляет собой антиметаболиты, такие как аминоптерин и метотрексан, которые являются ингибиторами ферментов, таких как дигидрофолат-редуктаза, воздействующая на производные фолиевой кислоты. Соединение, известное как CB3717, которое было описано и заявлено в патенте Великобритании N 2065653B, показало многообещающие результаты при его клиническом испытании. Несмотря на значительную эффективность CB3717 в использовании его против рака молочной железы, рака яичника и рака печени это средство оказалось токсичным для человека, особенно в отношении печени и почек [Calvert, Alison, Harland, Robinson, Jackman, Jones, Newell, Siddik, Whiltshaw, Me Elwain, Swith and Harrap, J. Clin. Oncol., 1986, 4, 1245; Cantwell, Earnshaw and Harris, Cancer Treatment Reports, 1986, 70, 1335; Bassendine, Curtin, Loose, Harris and James, J. Hepatol., 1987, 4, 39; Vest, Bork and Hasen, Eur. J. Cancer. Clin. Oncol., 1988, 24, 201; Cantwell, Macaulay, Harris, Kaye, Smith, Milsted and Calvert, Eur. J. Cancer Clin. Oncol. , 1988, 24, 733; Sessa, Zucchetti, Ginier, Willems, D'Incalci and Cavalli, Eur. J. Cancer Clin. Oncol., 1988, 24, 769).
Указанное побочное действие является меньшим у соединений, в которых 2-амино-заместитель CB3717 либо отсутствует, либо замещен одним из различных альтернативных заместителей, как описано в патентах Великобритании N 2175903 и 2188319 соответственно.
Очевидно, противоопухолевое действие соединений типа CB3717 осуществляется не путем ингибирования дегидрофолатредуктазы, а путем ингибирования тимидилат-синтазы. Тимидилат-синтаза катализирует метилирование дезоксиуридинмонофосфата для получения тимидинмонофосфата, который необходим для синтеза ДНК. Противораковая активность CB3717 может быть оценена in vitro путем определения его ингибирующего действия на указанный фермент и в клеточных культурах путем определения его ингибирующего действия на линии раковых клеток, таких как клеточная линия L1210 лейкоза мышей, клеточные линии лимфомы мыши L 5178У TK-/- и L 5178У TK+/ и клеточная линия MCF-7 рака молочной железы человека.
Противораковую активность других соединений CB3717-типа можно поэтому определить и сравнить с активностью CB3717, например, против того же самого фермента и тех же самых клеточных линий.
Антиметаболиты, такие как аминоптерин и метотрексат, являющиеся ингибиторами ферментов, таких как дигидрофолатредуктаза, которая воздействует на производные фолиевой кислоты, могут быть также использованы для лечения различных аллергических заболеваний, таких как ринит, атопический дерматит и псориаз. Поэтому хиназолиновые производные настоящего изобретения, которые также являются антиметаболитами, представляют собой ценные терапевтические средства, которые могут быть использованы для лечения, например, аллергических состояний, таких как псориаз.
Антиметаболиты, такие как метотрексат, также показали активность при лечении различных воспалительных заболеваний, таких как воспаление суставов (в частности, ревматоидный артрит, остеоартрит, и подагра) и воспаление желудочно-кишечного тракта (в частности, кишечные воспалительные заболевания, язвенные колиты и гастриты) (Weinblatt и др., New England J. Med., 1985, 312, 818; Andersen и др. Ann. Internat. Med. 1985, 103, 489; Healey, Bull. Rheum. Dis., 1986, 36, 1). Таким образом, хиназолиновые производные настоящего изобретения являются ценными терапевтическими средствами, которые могут быть использованы, например, для лечения воспалительных заболеваний, таких как ревматоидный артрит.
В заявках на Европатент N 0239362 и 0284338 раскрываются две серии хиназолиновых производных, которые обладают противораковыми свойствами благодаря своей способности ингибировать тимидилат-синтазу. Примерами таких производных являются производные N-{p-[N-(4-оксо-3,4-дигидрохиназолин-6-илметил)-N-алкиамино] бензол} - L-глутаминовой кислоты. Очевидно, эти соединения, также как описанное ранее соединение CB3717, действуют посредством метаболита, продуцируемого при гамма-полиглутамилировании (Sikora и др., Biochem. Pharmacol. , 1988, 34, 4047; Jackman и др., Cancer Research, 1991, 51, 5579).
Мы неожиданно обнаружили, что структуры известных производных L-глутаминовой кислоты могут быть изменены путем удаления гамма-карбоксильной группы или путем ее замены на тетразол-5-ильную группу с получением соединений, обладающих повышенной ингибирующей активностью против тимидилат-синтазы. Кроме того, соединения настоящего изобретения, очевидно, действуют непосредственно, в случае, если процесс гамма-глутамилирования не может иметь место. Такой альтернативный способ действия соединений настоящего изобретения позволяет осуществлять более точный контроль противоракового действия, так как начало этого действия проявляется более быстро вследствие его независимости от метаболического процесса, который может значительно варьироваться у различных пациентов. Кроме того, ожидается, что соединения настоящего изобретения будут более полезными при лечении тех случаев раковых заболеваний, где не происходит процесса гамма-глутаминирования, чем ранее описанные соединения, которые для усиления своего противоракового действия, очевидно, требуют гамма-полиглутамилирования. Более того, полиглутамилирование обычно проводят для получения полиглутамилированных производных антиметаболитов, которые не могут легко диффундировать через клеточные мембраны. В случае, когда полезное противораковое действие соединения перекрывается его неблагоприятным токсическим действием, крайне нежелательно, если антиметаболит остается в нормальных клетках вследствие процесса полиглутамилирования. Поэтому альтернативный механизм действия соединений настоящего изобретения позволяет осуществлять точный контроль противораковой терапии пациента.
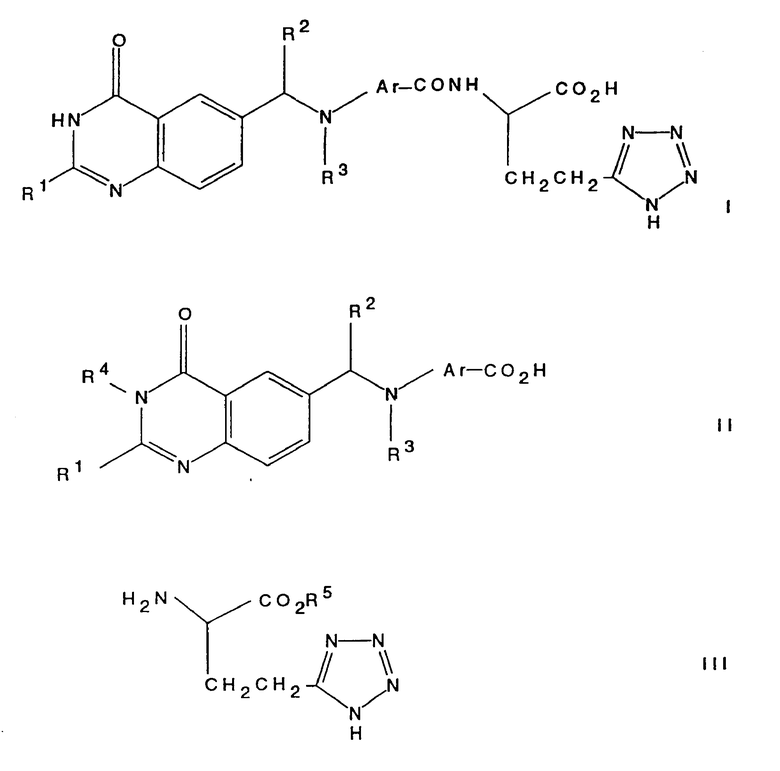
Настоящее изобретение относится к хиназолиновому производному формулы 1 (см. в конце текста), где R1 является (1-4C)алкилом; хиназолиновое кольцо может, но необязательно, содержать (в одном или двух из 5-, 7- и 8-положений) один или два дополнительных заместителя, выбранных из галогена (1-4C)алкила и (1-4C)алкокси; R2 является водородом или (1-4C) алкилом; R3 представляет собой (1-4C)алкил, (3-4C)алкенил, (3-4C)алкинил, гидрокси-(2-4C)алкил, галогено-(2-4C)алкил или циано-(1-4C)алкил; а Ar представляет собой фенилен или гетероциклен, который может, но необязательно, иметь один или два заместителя, выбранных из галогено, (1-4C)алкила и (1-4C)алкокси; либо настоящее изобретение относится к фармацевтически приемлемой соли или сложному эфиру указанного производного.
Используемый в настоящем описании термин "алкил" означает алкильные группы с прямой или разветвленной цепью, однако обозначения отдельных алкильных групп, таких как "пропил", являются специфическими лишь для групп с прямой цепью. Те же самые замечания относятся и к другим основным терминам.
Следует отметить, что хиназолиновое производное настоящего изобретения обладает одним или несколькими асимметрическими атомами углерода и поэтому оно существует в оптически активных формах. Отсюда очевидно, что настоящее изобретение включает любую оптически активную форму, обладающую противоопухолевой активностью; причем указанные оптически активные формы могут быть разделены общеизвестными методами. Предпочтительной формой хиназолинового производного настоящего изобретения является производное, обогащенное формой, имеющей (S)-конфигурацию у атома углерода, который несет карбоксильную группу, т. е. имеющей отношение (S):(R), превышающее 1:1. Более предпочтительно, если хиназолиновое производное настоящего изобретения имеет преимущественно (S)-конфигурацию в данном атоме углерода, т.е. отношение (S):(R) превышает 3: 2. И еще более предпочтительно, если указанное хиназолиновое производное в основном не содержит форму, имеющую (R)-конфигурацию в данном атоме углерода. Используемый выше термин "в основном не содержит..." указывает на присутствие не более чем 10%, предпочтительно не более чем 5%, и еще предпочтительней не более чем 2 мас.% любой (R)-формы.
Следует также отметить, что хиназолиновые производные настоящего изобретения формулы (I) могут обнаруживать явление таутомерии. Формулы, приведенные в настоящем описании, могут представлять лишь одну из возможных таутомерных форм. В частности, тетразол-5-ильная группа может быть, например, в виде 1H-тетразол-5-ильной или 3H-тетразол-5-ильной группы. Поэтому настоящее изобретение включает в себя любую таутомерную форму, которая обладает противоопухолевой активностью, и не ограничивается какой-либо одной таутомерной формой, изображенной в представленных формулах.
Следует также указать, что некоторые хиназолиновые производные формулы (I) могут существовать в сольватированных и несольватированных формах, например, таких как гидраты. При этом следует отметить, что настоящее изобретение включает все сольватированные формы, обладающие противоопухолевой активностью.
Ниже приводятся подходящие значения для основных радикалов, указанных выше.
Подходящим значением для R1, R2 и R3, если он является (1-4C)алкилом, или для (1-4C)алкильного заместителя, который может присутствовать на хинахолиновом кольце или на Ar, является, например, метил, этил, пропил, изопропил или бутил.
Подходящим значением для (1-4C)алкокси-заместителя, который может присутствовать на хиназолиновом кольце или на Ar, является, например, фторо-, хлоро-, или бромогруппа.
Подходящим значением для радикала R3, если он представляет собой (3-4C)алкенил, является, например, проп-2-еннил, бут-2-енил, бут-3-енил или 2-метилпропил-2-енил; если он представляет собой (3-4C)алкинил, то подходящим значением является, например, проп-2-инил или бут-3-инил; если он представляет собой гидрокси-(2-4C)алкил, то подходящим значением является, например, 2-гидроксиэтил или 3-гидроксиэтил; если он представляет собой галогено-(2-4C)алкил, то подходящим значением является, например, 2-фтороэтил, 2-хлороэтил, 2-бромоэтил, 3-фторопропил, 3-хлоропропил или 3-бромопропил; и, наконец, если он представляет собой циано-(1-4C)алкил, то подходящим значением, например, является цианометил, 2-цианоэтил или 3-цианопропил.
Если Ar представляет собой фенилен, то его подходящим значением является 1,3- или 1,4-фенилен.
Если Ar представляет собой гетероциклен, то его подходящим значением является, например, 5- или 6-членное (то есть полностью ненасыщенное) гетероциклическое кольцо, которое содержит до 3 гетероатомов, выбранных из атомов азота и серы, например, такое как тиофенедиил, пиридинедиил, пиримидинедиил или тиазоледиил. Если Ar представляет собой гетероциклен, то подходящим значением также является, например, тиофен-2,2-диил, тиофен-2,5-диил, пиридин-2,5-диил или тиазол-2,5-диил.
Подходящие фармацевтически приемлемые соли хиназолинового производного настоящего изобретения, которое является достаточно основным, представляют собой кислые аддитивные соли, образованные с неорганическими или органическими кислотами, например, такими как соляная, бромистоводородная, серная, фосфорная, трифторуксусная, лимонная или малеиновая кислота. Кроме того, подходящие фармацевтически приемлемые соли хиназолинового производного настоящего изобретения, которое является достаточно кислотным, представляют собой соли щелочного металла, например соли калия или натрия; соли щелочно-земельного металла, например соли кальция или магния; соли аммония или тетра-(2-гидроксиэтил)аммония; либо соли, образованные с органическим основанием, несущим физиологически приемлемый катион, например метиламиновая соль, триметиламиновая соль или трис-(2-гидроксиэтил)иминовая соль.
Подходящими фармацевтически приемлемыми сложными эфирами хиназолинового производного настоящего изобретения являются, например, сложные эфиры, образованные с (1-6C)спиртом, например, такие как метиловый, этиловый или трет-бутиловый эфир.
Конкретными новыми соединениям настоящего изобретения являются, например, хиназолиновые производные формулы (I), где
a) R1 представляет собой метил или этил; а заместители хиназолинового кольца R2, R3 и Ar имеют любое из значений, определенных выше или в данном параграфе, где определяются конкретные новые соединения настоящего изобретения;
b) хиназолиновое кольцо имеет в 7-положении еще один заместитель, выбранный из фторо, хлоро, бромо и метила; а R1, R2, R3 и Ar имеют любое из значений, определенных выше или в данном параграфе, где определяются конкретные новые соединения настоящего изобретения;
c) R2 является водородом; а R1, заместители хиназолинового кольца, R3 и Ar имеют любое из значений, определенных выше или в данном параграфе, где определяются конкретные новые соединения настоящего изобретения;
d) R3 является метилом, этилом, пропилом, проп-2-енилом, проп-2-инилом, 2-гидроксиэтилом, 2-фтороэтилом, 2-бромоэтилом или цианометилом; а R1, заместители хиназолинового кольца, R2 и Ar имеют любое из значений, определенных выше или в данном параграфе, где определяются конкретные новые соединения настоящего изобретения;
e) Ar является 1,4-фениленом, который может иметь, но необязательно, один или два заместителя, выбранные из фторо, хлоро, метила и метокси, либо Ar представляет собой тиофен-2,5-диил, тиазол-2,5-диил, или пиридин-2,5-диил; а R1, заместители хиназолинового кольца, R2 и R3 имеют любое из значений, определенных выше, или в данном параграфе, где определяются конкретные новые соединения настоящего изобретения; или их фармацевтически приемлемые соли или сложные эфиры.
Другим конкретным соединением настоящего изобретения является хиназолиновое производное формулы (I), где R1 является метилом; хиназолиновое кольцо может, но необязательно, иметь 7-фторо-, 7-хлоро-, 7-бромо- или 7-метиловый заместитель; R2 представляет собой водород; R3 представляет собой метил, этил, пропил, проп-2-энил или проп-2-инил; и Ar представляет собой 1,4-фенилен, который может, но необязательно, иметь фторо-заместитель, или Ar представляет собой тиофен-2,5-диил, тиазол-2,5-диил (с -CONH-группой в 2-положении) или пиридин-2,5-диил (с -CONH-группой во 2-положении);
или его фармацевтически приемлемая соль.
Еще одним предпочтительным соединением настоящего изобретения является хиназолиновое производное формулы (I), где R1 является метилом; хиназолиновое кольцо может иметь, но необязательно, 7-фторо-, 7-хлоро-, 7-бромо- или 7-метиловый заместитель; R2 представляет собой водород; R3 представляет собой метил, этил или проп-2-инил; и Ar представляет собой 1,4-фенилен, 2-фторо-1,4-фенилен (с -CONH-группой в 1-положении), или пиридин-2,5-диил (с -CONH-группой во 2-положении);
или его фармацевтически приемлемая соль.
Еще одним предпочтительным соединением настоящего изобретения является хиназолиновое производное формулы (I), где R1 представляет собой метил; хиназолиновое кольцо может, но необязательно, иметь 7-фторо-, 7-хлоро-, 7-бромо- или 7-метиловый заместитель; R2 представляет собой водород; R3 представляет собой метил, этил, пропил, проп-2-енил или проп-2-инил; и Ar представляет собой 1,4-фенилен, который может, но необязательно иметь фторо-заместитель, либо Ar представляет собой тиофен-2,5-диил или тиазол-2,5-диил (с -CONH-группой во 2-положении);
или его фармацевтически приемлемая соль.
Конкретным предпочтительным хиназолиновым производным настоящего изобретения является, например, одно из следующих хиназолиновых производных формулы (I), или его фармацевтически приемлемая соль
2-{ п-[N-(2-метил-4-окси-3,4-дигидрохиназолин-6-илметил)-N-(пропил-2-инил)амино]бензамидо}-4-(тетразол-5-ил)масляная кислота;
2-{ о-фторо-п-[N-(2-метил-4-оксо-3,4-дигидрохиназолин-6-илметил)-N-(проп-2-инил)амино]бензамидо}-4-тетразол-5-ил)масляная кислота;
2-{ п-[N-(2,7-диметил-4-оксо-3,4-дигидрохиназолин-6-илметил)-N- метиламино]бензамидо}-4-тетразол-5-ил)масляная кислота;
2{ о-фторо-п-[N-(2,7-диметил-4-оксо-3,4-дигидрохиназолин-6-ил- метил)-N-метиламино]бензаимидо}-4-тетразол-5-ил)масляная кислота;
2-{ п-[N-(2,7-диметил-4-оксо-3,4-дигидрохиназолин-6-илметил)-N-(пропил-2-инил)амино]бензамидо}-4-(тетразол-5-)масляная кислота;
2-{ о-фторо-п-[N-2,7-диметил-4-оксо-3,4-дигидрохиназолин-6-илметил)-N-(проп-2-инил)амино]бензамидо}-4-(тетразол-5-ил)-масляная кислота;
2-{ п-[N(7-хлоро-2-метил-4-оксо-3,4-дигидрохзиназолин-6-илметил)-N-(проп-2-инил)амино]бензамидо}-4-(тетразол-5-ил)- масляная кислота;
2-{ о-фторо-п-[N 7-хлоро-2-метил-4-оксо-3,4-дигидрохиназолин-6-илметил)-N-(проп-2-инил)амино]бензамидо}-4-(тетразол-5-ил)масляная кислота или
2{ п-[N-7-бромо-2-метил-4-оксо-3,4-дигидрохиназолин-6-илметил)-N-(проп-2-инил)амино]бензамидо}-4-тетразол-5-ил)-масляная кислота.
Другими характерным предпочтительным хиназолиновым производным настоящего изобретения является, например, одно из следующих хиназолиновых производных формулы (I), его фармацевтически приемлемая соль
(2S)-2-{ п-[N-(2-метил-4-оксо-3,4-дигидрохиназолин-6-илметил-N-(проп-2-инил)амино]бензамидо}-4-(тетразол-5-ил)масляная кислота;
(2S)-2-{ о-фторо-п-[N-(2-метил-4-оксо-3,4-дигидрохиназолин-6-илметил)-N-(проп-2-инил)амино]бензамидо}-4-(тетразол-5-ил)масляная кислота;
(2S)-2-{ п-[N-(2,7-диметил-4-оксо-3,4-дигидрохиназолин-6-илметил)-N-метиламино]бензамидо}-4-тетразол-5-ил)масляная кислота;
(2S)-2-{ о-фторо-п-[N-(2,7-диметил-4-оксо-3,4-дигидрохиназолин-6-илметил)N-метиламино]бензамидо}-4-тетразол-5-ил)масляная кислота;
(2S)-2-{ п-[N-(2,7-диметил-4-оксо-3,4-дигидрохиназолин-6-илметил)-N-(проп-2-инил)амино]бензамидо}-4-(тетразол-5-ил)масляная кислота;
(2S)-2-{ о-фторо-п-[N-(2,7-диметил-4-оксо-3,4-дигидрохиназолин-6-илметил)-N-(проп-2-инил)амино]бензамидо}-4-(тетразол-5-ил)масляная кислота;
(2S)-2-{ п-[N-(7-хлоро-2-метил-4-оксо-3,4-дигидрохиназолин-6-илметил)-N-(проп-2-инил)амино]бензамидо}-40(тетразол-5-ил)-масляная кислота;
(2S)-2-{ о-фторо-п-[N-(7-хлоро-2-метил-4-оксо-3,4-дигидрохиназолин-6- илметил)-N-(проп-2-инил)амино] бензамидо} -4-(тетразол-5-ил)масляная кислота или
(2S)-2-{ п-[N-(7-бромо-2-метил-4-оксо-3,4-дигидрохиназолин-6-илметил-N-(проп-2-инил)амино]бензамидо}-4-тетразол-5-ил)масляная кислота.
Хотя указанное хиназолиновое производное (S)-конфигурации может существовать в виде смеси с соответствующим производным (R)-конфигурации, однако, как указывалось выше, предпочтительно, чтобы производное (S)-конфигурации присутствовало в большем количестве.
Следующим характерным предпочтительным хиназолиновым производным настоящего изобретения является, например, одно из следующих хиназолиновых производных формулы (I) или его фармацевтически приемлемая соль
(2S)-2-{ о-фторо-п-[N-(2,7-диметил-4-оксо-3,4-дигидрохиназолин-6-илметил)-N-(проп-2-инил)амино]бензамидо}-4-(тетразол-5-ил)масляная кислота или
(2S)-2-{ 5[N-(2,7-диметил-4-оксо-3,4-дигидрохиназолин-6-илметил)-N-(проп-2-инил)амино] пиридин-2-карбоксамидо} -4-(тетразол-5-ил)масляная кислота.
В своем другом варианте настоящее изобретение относится к группе хиназолиновых производных, которые обладают улучшенной противоопухолевой активностью и которые, кроме того, могут обладать лучшими терапевтическими индексами по сравнению с активностью и терапевтическими индексами известных и структурно родственных производных N-{п-[N-(4-оксо-3,4-дигидрохиназолин-6-илметил)-N- алкиламино]бензоил}-L-глутаминовой кислоты.
Эта группа хиназолиновых производных предпочтительно включает хиназолиновое производное формулы (I), где R1 представляет собой метил; хиназолиновое кольцо имеет 7-метиловый заместитель; R2 представляет собой водород; R3 представляет собой метил, этил или проп-2-инил; и Ar представляет собой 1,4-фенилен или 2-фторо-1,4-фенилен (с -CONH-группой в 1-положении); или его фармацевтически приемлемую соль.
Альтернативно эта группа хиназолиновых производных включает хиназолиновое производное формулы (I), где R1 является метилом; хиназолиновое кольцо имеет 7-метиловый заместитель; R2 представляет собой водород; R3 представляет собой метил или проп-2-инил; и Ar представляет собой 1,4-фенилен, 2-фторо-1,4-фенилен (с -CONH-группой в 1-положении) или пиридин-2,5-диил (с -CОNH-группой в 2-положении); или его фармацевтически приемлемую соль.
Предпочтительным соединением этой группы хиназолиновых производных является 2-{ о-фторо-п-[N-(2,7-диметил-4-оксо-3,4- дигидрохиназолин-6-илметил)-N-метиламино]бензамидо}-4-тетразол-5- ил)масляная кислота или ее фармацевтически приемлемая соль.
Альтернативным соединением данной группы хиназолиновых производных является (2S)-2-{ о-фторо-п-[N-(2,7-диметил-4-оксо-3,4- дигидрохиназолин-6-илметил)-N-(проп-2-инил)амино] бензамидо}-4- (тетразол-5-ил)масляная кислота или ее фармацевтически приемлемая соль.
Альтернативным соединением данной группы хиназолиновых производных является (2S)-2-{ 5-N-(2,7-диметил-4-оксо-3,4-дигидрохиназозил-6-илметил)-N-(проп-2-инил)амин] пиридин-2-карбоксамидо}-4-(тетразол-5-ил)масляная кислота или ее фармацевтически приемлемая соль.
Терапевтический индекс хиназолинового производного настоящего изобретения может быть определен, например, путем сравнения дозы, обеспечивающей эффективную противоопухолевую активность в соответствующий in vivo - модели, такой как опухоль L 5178У TK +/- (Fisсher и др. Methods in Medical Research, 1964, 10, 247), в подходящих видах животных, например мышах, с дозой, вызывающей значительную потерю в весе испытуемого животного.
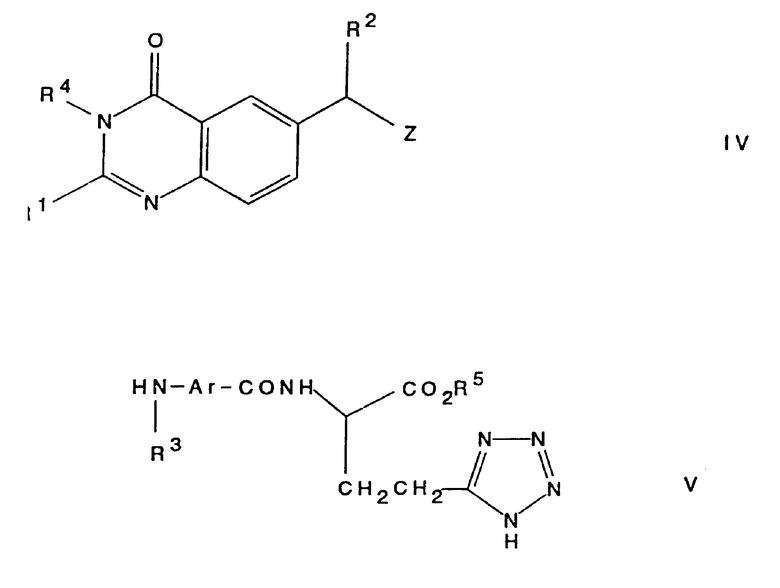
Соединение настоящего изобретения, представляющее собой хиназолиновое производное формулы (I), или его фармацевтически приемлемую соль, или его сложный эфир, может быть получено любым известным способом, который обычно используют для получения структурно родственных соединений. Указанные процедуры рассматриваются как дополнительный отличительный признак настоящего изобретения и проиллюстрированы ниже характерными примерами, где R1, R2, R3 и Ar (если это не оговорено особо) имеют значения, определенные выше. Альтернативно там, где это необходимо, на функциональных группах используются стандартные защитные группы, если только они не препятствуют нужному процессу. Примеры таких стандартных защитных групп приведены ниже. Любые указанные защитные группы могут быть удалены, если это необходимо, любым традиционным способом.
a) Реакция кислоты формулы (II) (представленной ниже) или ее реактивного производного, где R4 является водородом или защитной группой, с соединением формулы (III), где R5 является защитной группой, такой как (1-4C) алкильная группа, с последующим удалением защитных групп традиционным способом. Подходящим реактивным производным кислоты формулы (II) может быть, например, ацилгалид, такой как ацилхлорид, образованный с помощью реакции кислоты и неорганического хлорангидрида, например тионилхлорида; смешанного ангидрида, например ангидрида, образованного с помощью реакции кислоты и хлороформата, такого как изобутилхлороформат; активного сложного эфира, образованного с помощью реакции кислоты и фенола, такого как 1-гидроксибензотриазол; ацилазида, например азида, образованного с помощью реакции кислоты и азида, такого как дифенилфосфорилазид; ацилцианида, например цианида, образованного с помощью реакции кислоты и цианида, такого как диэтилфосфорилцианид; или продукта реакции кислоты с карбодиимидом, таким, как дициклогексилкарбодиимидом.
Эту реакцию осуществляют предпочтительно в присутствии соответствующего основания, например, такого как карбонат, алкоксид, гидроксид или гидрид щелочного или щелочно-земельного металла, например карбоната натрия, карбоната калия, этоксида натрия, бутоксида калия, гидроксид калия, гидрид натрия или гидрид калия; или органического аминового основания, такого как, например, пиридин, 2,6-лутидин, коллидин, 4-диметиламинопиридин, триэтиламин, морфолин или диазабицикло 5.40 унлек-7-ен. Реакцию также предпочтительно осуществляют в подходящем инертном растворителе, таком как тетрагидрофуран, 1,2-диметоксиэтан, N,N-диметилформамид, N,N-диметилцетамид, N-метилпирролидин-2-он, диметилсульфоксид или ацетон, при температуре порядка -78oC - 150oC, а предпочтительно при температуре, близкой к температуре окружающей среды.
Подходящим значением для радикала R4, если он представляет собой защитную группу, является, например, пивалоилметильная группа, которая может быть затем удалена путем гидролиза в присутствии основания, такого как гидроксид натрия или аммиак, в соответствующем инертном растворителе или разбавителе, например в метаноле или этаноле.
Подходящим значением для радикала R5, если он представляет собой (1-4C)алкильную группу, является, например, метил, этил, пропил, изопропил, бутил, изобутил или трет-бутил. R5 может быть удалена, например, путем гидролиза предпочтительно в присутствии основания, такого как гидроксид щелочного или щелочно-земельного металла, например гидроксид лития, гидроксид натрия или гидроксид калия. Альтернативно, если R5 представляет собой трет-бутильную группу, то она может быть удалена, например, путем обработки соответствующей неорганической кислотой, такой как соляная, серная или фосфорная кислота, либо соответствующей органической кислотой, такой как трифторуксусная кислота.
Подходящей защитной группой для гидрокси-(2-4C)алкильной группы является, например, алканоильная группа, такая как ацетильная группа; ароильная группа, такая как бензоильная группа; или арилметильная группа, такая как бензильная группа. Условия разблокирования для вышеуказанных защитных групп будут варьироваться в зависимости от выбора защитной группы. Так, например, ацильная группа, такая как алканоильная или ароильная группа, могут быть удалены, например, путем гидролиза в присутствии подходящего основания, такого как гидроксид щелочного металла, например гидроксид лития или натрия. Альтернативно, арилметильная группа, такая как бензильная группа, может быть удалена, например, путем гидрогенизации в присутствии катализатора, такого как палладированный уголь.
Исходные соединения формулы (II) и формулы (III) могут быть получены традиционными методами органической химии. Примеры получения указанных исходных материалов описаны в примерах, приведенных ниже, которые служат лишь иллюстративным целям и не должны рассматриваться как некое ограничение настоящего изобретения. Другие необходимые исходные материалы получают с помощью процедур, аналогичных описанным, или с помощью модификации стандартных процедур, известных специалистам в органической химии. Так, например, исходное соединение формулы (II) может быть получено с помощью реакции соединения формулы (IV), где Z является замещаемой группой, с амином формулы
HNR3-Ar1-CO2R5,
где R5 является защитной группой, определенной выше, которая затем может быть удалена с получением карбоновой кислоты.
Кроме того, исходное соединение формулы (III), где R5 является водородом, имеющее преимущественно (S)-конфигурацию у атома углерода, который несет амино- и карбоксигруппы, является известным соединением (Tetrahedron, 1977, 33, 2299). Это соединение может быть подвергнуто этерификации традиционным способом с образованием соединения формулы (III), где R5 является (1-4C)алкильной группой. Альтернативно, раскрытое здесь соединение, которое имеет структуру соединения формулы (III), где R5 является метилом, за исключением того, что аминогруппа является защищенной бензилоксикарбонильной группой, может быть разблокировано, например, путем гидрогенолиза.
Подходящим значением для замещаемой группы Z является, например, галогено- или сульфонилоксигруппа, такая как хлоро-, бромо-, метилсульфонилокси- или 4-толуолсульфонилоксигруппа.
b) Реакция соединения формулы (IV), где R4 является водородом или защитной группой, определенной выше, а Z является замещаемой группой, определенной выше, с амином формулы (V), где R5 является защитной группой, определенной выше, с последующим удалением защитных групп традиционными методами. Эту реакцию осуществляют предпочтительно в присутствии соответствующего основания, определенного выше, в соответствующем инертном растворителе или разбавителе, определенных выше, и при температуре порядка, например, 25 - 150oC, а предпочтительно примерно при 90oC.
Исходные соединения формул (IV) и (V) могут быть получены стандартными способами, обычно используемыми в органической химии. Примеры получения соединений формулы (IV) описаны путем ссылок на сопровождающие описание примера, которые являются лишь иллюстрированными и никоим образом не должны рассматриваться как ограничение изобретения. Другие необходимые исходные материалы могут быть получены с помощью процедур, аналогичных описанным, или с помощью модификации стандартных процедур, хорошо известных специалистам.
Если необходимо получить фармацевтически приемлемую соль нового соединения формулы (I), то такая соль может быть получена, например, с помощью реакции указанного соединения с соответствующей кислотой или основанием в соответствии с традиционной техникой. Если необходимо получить фармацевтически приемлемый сложный эфир нового соединения формулы (I), то такой эфир может быть получен, например, с помощью реакции указанного соединения с соответствующим (1-6C) спиртом в соответствии с традиционной техникой. Если необходимо получить оптически активную форму соединения формулы (I), то она может быть получена с помощью одного из вышеописанных способов с использованием оптически активного исходного материала либо путем разделения рацемической формы указанного соединения с использованием стандартной процедуры.
Как указывалось выше, хиназолиновое производное настоящего изобретения обладает противоопухолевой активностью. Эта активность может быть оценена, например, с использованием описанных ниже процедур.
a) In vitro - анализ для определения способности испытуемого соединения ингибировать тимидилат-синтазу (фермент). Тимидилат-синтазу получали из клеток L 1210 мышиного лейкоза в частности очищенном виде, используя процедуры, описанные Jackman и др. (Cancer Res., 1986, 46, 2810) и Sikora и др. , (Biochem. Pharmacol., 1988, 37, 4047).
в) Анализ для определения способности испытуемого соединения ингибировать рост клеточной линии лейкоза L1210 в клеточной культуре. Испытания проводили в соответствии с описаниями в патенте Великобритании N 206565B и Jones и др. в J. Med. Chem. 1985, 28, 1468.
с) Анализ для определения способности испытуемого соединения ингибировать рост клеточной линии MCF-7 рака молочной железы человека в культуре клеток. Испытания проводили в соответствии с описанием Lippman и др. (Cancer Res., 1976, 36, 4595).
Хотя фармакологические свойства хиназолинов настоящего изобретения варьируются в зависимости от структурных измерений, однако в испытаниях (a) - (c) (см. выше) хиназолины настоящего изобретения в основном имеют следующую активность:
тест (a) IC50, например, в пределах 1 - 100 нМ;
тест (b) IC50, например, в пределах 0,01 - 10 мкМ;
тест (c) IC50, например, в пределах 0,01 - 10 мкМ.
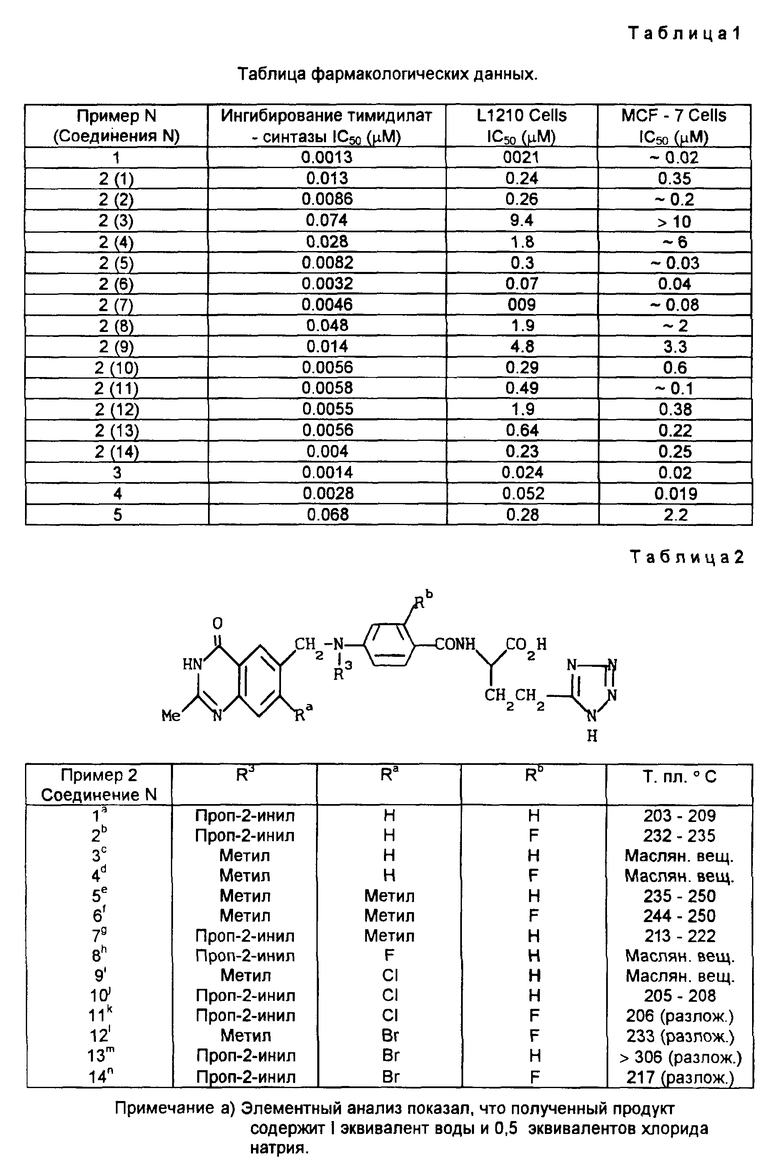
Результаты фармакологических свойств хиназолинов приведены в табл. 1.
В описанных выше испытаниях (a) - (c) те хиназолины настоящего изобретения, которые являются предпочтительными, в основном обладают следующей активностью:
тест (a) IC50, например, в пределах 1 - 20 нМ;
тест (b) IC50, например, в пределах 0,01 - 1 мкМ;
тест (c) IC50, например, в пределах 0,01 - 1 мкМ;
Так, например, соединение (2S)-2-{п-[N-(2-метил-4-оксо-3, 4-дигидрохиназолин-6-илметил)-N-(проп-2-инил)амино]бензамидо}-4- (третразол-5-ил) масляная кислота имеет IC50 ≈ 13 нМ в тесте (a), IC50 ≈ 0,12 мкМ в тесте (b) и IC50 ≈ 0,04 мкМ в тесте (c); соединение (2S)-2-{о-фторо-п-[N-(2,7-диметил-4-оксо-3,4-дигидрохиназолин-6-илметил)-N-метиламино]бензамидо}-4-(тетразол-5-ил) масляная кислота имеет IC50 ≈ 2 нМ в тесте (a), IC50 ≈ 0,07 мкМ в тесте (b) и IC50 ≈ 0,04 мкМ в тесте (c); соединение (2S)-2-{о-фторо-п-[N-(2,7-диметил-4-оксо-3,4-дигидрохиназолин -6-илметил)-N-(проп-2-инил)амино] бензамидо}-4-(тетразол-5-ил) масляная кислота имеет IС50 ≈ 1 нМ в тесте (a), IC50 ≈ 0,02 мкМ в тесте (b) и IC50 ≈ 0,01 мкМ в тесте (c).
Хиназолиновое производное настоящего изобретения, или его фармацевтически приемлемая соль, или сложный эфир могут быть введены теплокровному животному, включая человека, в виде фармацевтической композиции, которая содержит хиназолиновое производное, или его фармацевтически приемлемую соль, или сложный эфир в сочетании с фармацевтически приемлемым разбавителем или носителем.
Эта композиция может быть изготовлена в форме, предназначенной для перорального введения, например, в виде таблетки, капсулы, водных или масляных растворов, суспензий или эмульсий; для наружного применения, например, в виде кремов, мазей, гелей либо водных, либо масляных растворов или суспензий; для интраназального введения, например, в виде порошка для вдыхания через нос, аэрозоля и капель через нос; для вагинального или ректального введения, например, в виде суппозитория; для введения путем ингаляции, например, в виде аэрозолей для распыления тонкоизмельченных порошков или жидких аэрозолей; для подъязычного или трансбукального введения, например, в виде таблеток или капсул; или, что особенно предпочтительно, для парентерального введения (включая, внутривенное, подкожное, внутримышечное и интраваскулярное введение или вливание), например, в виде стерильных водных или масляных растворов, эмульсий или суспензий. Вышеуказанные композиции могут быть получены в основном, традиционными способами с использованием стандартных наполнителей.
Помимо хиназолинового производного настоящего изобретения композиция может содержать и другие противоопухолевые агенты, выбранные из таких средств, как митотические ингибиторы, например, винбластин; алкилирующие агенты, такие как цисплатин, карбоплатин и циклофосфамид; другие антиметаболиты, например 5-фтороурацил, цитозинарабинозид и гидроксимочевина; интеркалярные антибиотики, например адриамицин и блеомицин; ферменты, например аспарагиназа; ингибиторы топоизомеразы, например этопосид; и модификаторы биологического ответа, например интерферон.
Теплокровному животному хиназолин вводят в основном в виде разовой дозы, составляющей 50 - 500 мг/м2 поверхности тела животного, т.е. приблизительно 1 - 100 мг/кг, и представляющей собой нормальную терапевтически эффективную дозу. Разовая лекарственная форма, такая как таблетка или капсула, может в основном содержать, например, 1 - 250 мг активного ингредиента. Предпочтительная дневная доза составляет 1 - 50 мг/кг, а более предпочтительно 1 - 15 мг/кг. Однако эта дневная доза может варьироваться в зависимости от обрабатываемого индивидуума, конкретного способа введения и тяжести заболевания. Поэтому при лечении каждого конкретного пациента оптимальная доза назначается лечащим врачом.
Другим отличительным признаком настоящего изобретения является получение хиназолинового производного формулы (I), или его фармацевтически приемлемой соли, или сложного эфира для использования в способе лечения человека либо животного путем терапии.
Еще одним отличительным признаком настоящего изобретения является способ продуцирования противоопухолевого ответа у теплокровного животного, например человека, нуждающегося в таком лечении, путем введения указанному животному эффективного количества хиназолинового производного настоящего изобретения, или его фармацевтически приемлемой соли, или его сложного эфира.
Настоящее изобретение также относится к использованию хиназолинового производного настоящего изобретения, или его фармацевтически приемлемой соли, или сложного эфира в изготовлении нового лекарственного средства, предназначенного для продуцирования противоопухолевого ответа у теплокровных животных, например у человека.
Можно ожидать, что хиназолин настоящего изобретения обладает широким спектром противоопухолевой активности. СВ3717 показало многообещающую активность в отношении рака молочной железы, яичника и печени человека, и поэтому предполагается, что хиназолин настоящего изобретения также будет обладать противоопухолевой активностью для этих видов рака. Кроме того, предполагается, что хиназолин настоящего изобретения будет обладать активностью против лейкозов, злокачественных опухолей лимфоидной ткани и твердых опухолей, таких как карциномы и саркомы. Указанные опухоли требуют присутствия тимидинмонофосфата как одного из главных нуклеотидов, необходимых для синтеза клеточной ДНК. Предполагается, что в присутствии эффективного количества ингибитора тимидилат-синтазы, такого как хиназолинового производного настоящего изобретения, рост опухоли будет ингибироваться.
Как указывалось выше, хиназолиновое производное настоящего изобретения, или его фармацевтически приемлемая соль, или его сложный эфир может быть также использовано в лечении, например, аллергических состояний, таких как псориаз, или воспалительных заболеваний, таких как ревматоидный артрит. При использовании хиназолина настоящего изобретения для этих целей данное соединение в основном вводят в дозе, составляющей 500 - 50000 мг/м2 поверхности тела животного. При лечении аллергических состояний, таких как псориаз, хиназолин настоящего изобретения предпочтительно использовать путем наружного применения. Так, например, для местного применения дневная доза может в основном составлять 1 - 150 мг/кг, а предпочтительно 1 - 80 мг/кг.
В целях иллюстрации настоящего изобретения ниже приводятся примеры, которые, однако, не должны рассматриваться как некое ограничение настоящего изобретения и в которых (если это не оговорено особо)
I) выпаривание осуществляли в вакууме с помощью роторного испарителя, а процедуры обработки осуществляли после удаления оставшегося твердого вещества путем фильтрации;
II) операции проводили при температуре лаборатории (18 - 20oC) и в атмосфере инертного газа, такого как аргон;
III) колоночную хроматографию (посредством флеш-процедуры) и жидкостную хроматографию среднего давления (ЖХСД) проводили на двуокиси кремния Merck Kieselgel (Art, 9385) или на обращенно-фазовой двуокиси кремния Merck Lichroprep RP-18 (Art. 9303), полученной от E.Merck, Дармштадт, Зап. Германия;
IV) выводы даны лишь для иллюстрации и необязательно представляют максимально достижимый уровень;
V) конечные продукты формулы (I) показали удовлетворительные результаты при микроанализе, и их структуры были подтверждены ЯМР-анализом и масс-спектроскопией [спектры протонного магнитного резонанса определяли с использованием спектрометра JeoI FX 900 или Bruker AM200, работающего при напряженности поля 200 МГц; химические сдвиги в ЯМР-спектрах приведены в частях на миллион относительно тетраметилсилана, используемого в качестве внутреннего стандарта (δ-шкала), а мультиплетности пиков обозначены следующим образом: с (синглет); д (дублет); дд (дублет дублетов); т (триплет); м (мультиплет); данные масс-спектроскопии путем бомбардировки быстрыми атомами (FAB) были получены с использованием аналитического спектрометра (VG Analytical MS9) и газа ксенона; и там, где это было возможно, получали данные о положительных или отрицательных ионах];
VI) промежуточные соединения обычно полностью не характеризовали, а чистоту оценивали с помощью тонкослойной хроматографии, инфракрасного анализа (ИК) или ЯМР-анализа;
VII) точки плавления, которые приводятся без поправок, были определены с помощью автоматического устройства для определения температур плавления Mettler SP62; устройства с нагретыми пластинами Koffler или устройства с масляной баней;
VIII) хиральную чистоту конечных продуктов формулы (I) и промежуточных соединений, таких как соединения формулы (III), оценивали с помощью ЯМР- и хроматографического анализа;
IX) были использованы следующие сокращения:
ТГФ - тетрагидрофуран;
ДМФ - N, N-диметилформамид;
ДМА - N, N-диметилацетамид;
NМП - N -метилпирролидин-2-он;
ДМСО - диметилсульфоксид.
Пример 1. Смесь пентафторофенил о-фторо-п-[N-(2,7-диметил-4-оксо -3-(пивалоилоксиметил)-3,4-дигидрохиназолин-6-илметил)-N- (проп-2-инил)амино] бензоата (2,5 г), метил (2S)-2-амино-4-(тетразол-5-ил)бутирата (0,32 г), N-гидроксибензотриазола (0,05 г) и диметилформамида (ДМФ) (100 мл) перемешивали при комнатной температуре в течение 24 ч. Полученную смесь выпаривали и остаток очищали с помощью колоночной хроматографии, элюируя смесью метиленхлорида и метанола (19:1 об/об). Эту смесь, полученную в виде камеди, перетирали с диэтиловым эфиром, в результате чего получают твердое вещество (1,92 г), метил (2S)-2-{о-фторо-п-[N-(2,7-диметил-4-оксо-3-(пивалоилоксиметил)-3,4- дигидрохиназолин-6-илметил)-N-(проп-2-инил)амино] бензамидо} -4-(тетразол-5-ил)бутирата с выходом 79%.
Смесь полученного продукта, 2 н. раствора гидроксида натрия (20 мл) и метанола (5 мл) перемешивали при комнатной температуре в течение 1 ч. Полученную смесь концентрировали путем выпаривания из метанола, а водный остаток подкисляли до pH 4 добавлением концентрированной соляной кислоты. Полученный осадок выделяли, промывали водой и осушали, в результате чего получали (2S)-2-{ о-фторо-п-[N-(2,7-диметил-4-оксо-3,4-дигидрохиназолин-6-илметил) -N-(проп-2-инил)амино] бензамидо} -4-(тетразол-5-ил)масляную кислоту (1,21 г, 79%), т.пл. 232-240oC.
ЯМР-спектр (CD3SOCD3); 2,12-2,21 (мн, 2H); 2,24 (c, 3H); 2,30 (c, 3H); 2,94 (м, 2H), 3,20 (c, 1H); 4,28 (c, 2H); 4,41 (м, 1H); 4,68 (c, 2H); 6,62 (м, 2H); 7,42 (c, 1H); 7,56 (т, 1H); 7,67 (c, 1H); 8,05 (т, 1H); 12,03 (c, 1H).
Масс-спектр: масс-спектроскопия путем бомбардировки быстрыми атомами положительными ионами (MC-FAB) m/e(P+I) 533.
Элементный анализ для C26H25FN8O4 0,9 NaCl:
Найдено, %: C 53,1; H 4,4; N 19,5;
Вычислено, %: C 53,4; H 4,3; N 19,2.
В этом примере метил (2S)-2-амино-4-(тетразол-5-ил)-бутират был обогащен формой, имеющей (S)-конфигурацию у атома углерода, который несет метоксикарбонильную группу, в отношении (S):(R)=7:3, как было определено с помощью хроматографического анализа. Это изомерное соотношение оставалось в продукте примера 1.
Пентафторфенил о-фторо-п-[N-(2,7-диметил-4-оксо-3-(пивалоил -оксиметил)-3,4-дигидрохиназолин-6-илметил)-N-(проп-2-инил) амино]бензоат, используемый в качестве исходного материала, получали следующим образом.
Смесь трет-бутил о-фторо-п-(проп-2-инил)аминобензоата [0,882 г; полученного с 56%-ным выходом с помощью реакции трет-бутил- р-амино-о-фторобензоата (пример 3 заявки на Европатент N 0373891) с пропаргилбромидом], 6-бромометил-2,7-диметил-3-(пивалоилоксиметил) -3,4-дигидрохиназолин-4-она [0,9 г; пример 13 заявки на Европатент N 0459730], карбоната калия (0,691 г), 18-краун-6-(0,005 г) и NМП (20 мл) перемешивали и нагревали до 90oC в течение 6 ч. Эту смесь выпаривали и остаток распределяли между этилацетатом и водой. Органическую фазу промывали водой, солевым раствором, высушивали (MgSO4) и выпаривали. Полученный остаток очищали с помощью колоночной хроматографии, элюируя постоянно возрастающими полярными смесями метиленхлорида и этилацетата, в результате чего получали трет-бутил-о-фторо-п-[N-(2,7-диметил-4-оксо-3-(пиволоилоксиметил) -3,4-динидрохиназолин-6-илметил)-N-(проп-2-инил)амино]бензоат (0,9 г).
Смесь полученного таким образом продукта и трифторуксусной кислоты (20 мл) перемешивали при комнатной температуре в течение 1 ч. Полученную смесь выпаривали, а остаток перетирали с диэтиловым эфиром, в результате чего получали о-фторо-п-[N-(2,7-диметил-4-оксо -3-(пивалоилоксиметил)-3,4-дигидрохиназолин-6-илметил-N-(проп-2-инил) амино] бензойную кислоту в виде твердого вещества (0,64 г).
Элементный анализ для C27H28FN3O5 0,1 CF3CO2H:
Найдено, %: C 64,7; H 5,5; N 8,2;
Вычислено, %: C 64,7; H 5,6; N 8,3.
Дициклогексилкарбодиимид 14,9 г добавляли к суспензии о-фторо-п- [N-(2,7-диметил-4-оксо-3-(пивалоилоксиметил)3,4-дигидрохиназолин-6 -илметил)-N-(проп-2-инил)амино] бензойной кислоты (23,8 г) пентафторофенола (26,6 г) в ДМФ (200 мл) и полученную смесь перемешивали при комнатной температуре в течение 18 ч. Эту смесь фильтровали, а фильтрат выпаривали. Полученный остаток очищали с помощью колоночной хроматографии, элюируя смесью гексана и этилацетата (1:1 об/об). Таким образом получали требуемый исходный материал (11 г), т.пл. 163-165oC.
Этот метил(2S)2-амино-4-(тетразол-5-ил)бутират (в отношении (S):(R) = 7: 3), используемый в качестве исходного материала, получали следующим образом.
Сульфурилхлорид (1 г) добавляли к перемешанной смеси N-бензилоксикарбонил-L-глутамина (100 г) и метанола (1300 мл) и полученную смесь перемешивали при комнатной температуре в течение 24 ч. Эту смесь выпаривали и получали N-бензилоксикарбонил-L-глютаминметиловый сложный эфир (105 г).
п-Тозилхлорид (85,8 г) добавляли порциями к перемешанной смеси полученного продукта и пиридина (200 мл) так, чтобы температура реакционной смеси не превышала 35oC. Эту смесь нагревали до 65oC в течение 90 мин. После этого полученную смесь концентрировали путем выпаривания из пиридина и остаток распределяли между этилацетатом и водой. Органическую фазу промывали 2 н. водным раствором соляной кислоты и водой, затем высушивали (MgSO4) и выпаривали. Полученный остаток очищали с помощью колоночной хроматографии, элюируя смесью гексана и этилацетата (1:1 об/об). Таким образом получали метил (2S)-2-бензилоксикарбониламино-4-цианобутират (81 г).
Смесь полученного продукта, азида натрия (22,2 г), хлорида аммония (18,3 г) и ДМФ (400 мл) нагревали на паровой бане в течение 24 ч. Эту смесь концентрировали и к полученному остатку добавляли воду (55 мл). Полученную смесь подкисляли до pH 1 путем добавления концентрированной соляной кислоты и экстрагировали этилацетатом. Этот органический экстракт высушивали (MgSO4) и выпаривали. Полученный остаток перетирали диэтиловым эфиром, в результате получали метил(2S)-2-бензилоксикарбониламино-4-(тетразол-5-ил)бутират (36 г; в отношении (S):(R)=7:3).
ЯМР-спектр (CD3SOCD3); 1,95-2,35 (м, 2H); 2,95 (т, 2H); 3,64 (c, 3H); 4,15 (м, 1H); 5,04 (c, 2H); 7,36 (c, 5H); 7,88 (д, 1H); 13,0 (c, 1H).
Смесь части (17,1 г) полученного продукта, 10% палладированного угля (2,2 г) и этанола (300 мл) перемешивали при комнатной температуре в атмосфере водорода в течение 24 ч. Полученную смесь фильтровали и фильтрат выпаривали. Затем полученный остаток перетирали с диэтиловым эфиром и получали метил(2S)-2-амино-4-(тетразол-5-ил)бутират (11,75 г) в отношении (S):(R)=7: 3), т.пл. 177-182oC.
Пример 2. Повторяли процедуру, описанную в примере 1, за исключением того, что соответствующий пентафторобензоат подвергали реакции с (2S)-2-амино-4-(тетразол-5-ил)бутиратом. Таким образом получали соединения, описанные в табл. 2, структуры которых были подтверждены протонным магнитным резонансом, масс-спектроскопией и элементным анализом. Если это не указано особо, использовали партию метил(2S)-2-амино-4-(тетразол-5-ил)бутирата, обогащенную формой, имеющей (S)-конфигурацию с отношением (S):(R)=7:3.
Пентафторофенил п-[N-(2-метил-4-оксо-3-(пивалоилоксиметил)- 3,4-дигидрохиназолин-6-илметил)-N-(проп-2-инил)-амино]бензоат, используемый в качестве исходного материала, получали с помощью реакции п-[N-(2-метил-4-оксо-3-(пивалоилоксиметил)-3,4- дигидрохиназолин-6-илметил)-N-(проп-2-инил)-амино] бензойной кислоты (пример 1 заявки на Европатент N 459730) и пентафторофенола, используя процедуру, аналогичную описанной в той части примера 1, которая относится к получению исходных материалов.
Этот метил(2S)-2-амино-4-(тетразол-5-ил)бутират, использованный при получении этого продукта, получали в форме, в основном несодержащей (R)-конфигурацию, в соответствии с процедурой, описанной Tran и др. Tetrahedron, 1977, 33, 2299.
b) Элементный анализ показал, что полученный продукт содержит 0,5 эквивалентов воды и 0,5 эквивалентов хлорида натрия.
Этот пентафторофенил о-фторо-п-[N-(2-метил-4-оксо-3- пивалоилоксиметил)-3,4-дигидрохиназолин-6-илметил)-N-(проп-2-инил) амино] бензоат, использованный в качестве исходного материала, получали с помощью реакции о-фторо-п-[N-(2-метил-4-оксо-3- (пивалоилоксиметил)-3,4-дигидрохиназолин-6-илметил)-N-проп-2-инил) амино]бензойной кислоты (пример 13 заявки на Европатент N 0459730) и пентафторофенола, используя процедуру, аналогичную описанной в той части примера 1, которая относится к получению исходных материалов.
c) Полученный продукт имел следующие характеристики ЯМР-спектров (CD3SOCD3): 2,21 (м, 2H); 2,33 (с, 3H); 2,96 (т, 2H); 3,13 (с, 3H); 4,4 (м, 1H); 4,78 (с, 2H); 6,78 (д, 2H); 7,5-7,65 (м, 2H); 7,74 (д, 2H); 7,85 (д, 1H); 8,3 (д, 1H).
Этот пентафторофенил п-[N-(2-метил-4-оксо-3-(пивалоилоксиметил)- 3,4-дигидрохиназолин-6-илметил)-N-метиламино] бензоат, использованный в качестве исходного материала, получали из 6-брометил-2-метил-3- (пивалоилоксиметил)-3,4-дигидрохиназолин-4-она [пример 1 заявки на Европатент N 0239362] и третбутил п-метиламинобензоата [полученного путем реакции трет-бутил п-метиламинобензоата с метилиодидом], используя аналогичную процедуру, которая описана в той части примера 1, которая относится к получению исходных материалов.
d) Полученный продукт имел следующие характеристики ЯМР-спектров (CD3SOCD3): 2,0-2,25 (м, 2H); 2,3 (с,3H); 2,9-3,0 (т, 2H); 3,1 (с, 3H); 4,35-4,5 (м, 1H); 4,8 (с, 2H); 6,7 (м, 2H); 7,57 (м, 3H); 7,9 (м, 2H); 12,08 (с, 1H).
Этот пентафторофенил о-фторо-п-[N-(2-метил-4-оксо-3-(пивалоилоксиметил)-3,4-дигидрохиназолин-6-илметил)-N-метил-амино] бензоат, использованный в качестве исходного материала, получали из 6-бромометил-2-метил-3-(пивалоилоксиметил)-3,4-дигидрохиназолин-4-она и трет-бутил о-фторо-п-метиламинобензоата [полученного с помощью реакции трет-бутил п-амино-о-фторобензоата с метилиодидом], используя процедуру, аналогичную описанной в той части примера 1, которая относится к получению исходных материалов.
Этот метил (2S)-2-амино-4-(тетразол-5-ил)бутират, использованный при получении этого продукта, получали в форме, в основном несодержащей (R)-конфигурацию, в соответствии с процедурой, описанной Tran и др., Tetrahedron, 1977, 33, 2299.
e) Элементный анализ показал, что этот продукт содержит 1,2 эквивалента воды.
Полученный пентафторофенил п-[N-(2,7-диметил-4-оксо-3- (пивалоилоксиметил)-3,4-дигидрохиназолин-6-илметил)-N- метиламино] бензоат, используемый в качестве исходного материала, получали из 6-бромометил-2,7-диметил-3-(пивалоилоксиметил)-3,4- дигидрохиназолин-4-она и трет-бутил п-метиламинобензоата, используя процедуру, аналогичную описанной в той части примера 1, которая относится к получению исходных материалов.
f) Элементный анализ показал, что продукт содержит 0,5 эквивалентов воды и 0,45 эквивалентов хлорида натрия.
Этот пентафторофенил о-фторо-п-[N-(2,7-диметил-4-оксо-3-(пивалоилоксиметил)-3,4-дигидрохиназолин-6-илметил)- N-метиламино] бензоат, используемый в качестве исходного материала, получали из 6-бромометил-2,7-диметил-3-(пивалоилоксиметил)- 3,4-дигидрохиназолин-4-она и трет-бутил-о-фторо-п-метиламинобензоата, используя процедуру, аналогичную описанной в той части примера 1, которая относится к получению исходных материалов.
g) Элементный анализ показал, что полученный продукт содержит 1 эквивалент воды.
Этот пентафторофенил п-[N-2,7-диметил-4-оксо-3-(пивалоилоксиметил)- 3,4-дигидрохиназолин-6-илметил)-N-(проп-2-инил-амино] бензоат, используемый в качестве исходного материала, получали с помощью реакции п-[N-(2,7-диметил-4-оксо-3-(пивалоилоксиметил)-3,4- дигидрохиназолин-6-илметил)-N-(проп-2-инил)-амино] бензойной кислоты (пример 13 заявки на Европатент N 0459730) и пентафторофенола, используя процедуру, аналогичную описанной в той части примера 1, которая относится к получению исходных материалов.
h) Полученный продукт имел следующие характеристики ЯМР-спектров (CD3SOCD3): 2,2 (м, 2H); 2,33 (с, 3H); 2,95 (т, 2H); 3,15 (с, 1H); 4,28-4,45 (м, 3H); 4,78 (с, 2H); 6,86 (д, 2H); 7,35 (д, 1H); 7,77 (д, 2H); 7,92 (д, 1H); 8,47 (м, 1H).
Этот пентафторофенил п-[N-(7-фторо-2-метил-4-оксо-3- (пивалоилоксиметил)3,4-дигидрохиназолин-6-илметил)-N-(проп-2- инил)амино] бензоат, используемый в качестве исходного материала, получали с помощью реакции п-[N-(7-фторо-2-метил-4-оксо-3- (пивалоилоксиметил)-3,4-дигидрохиназолин-6-илметил)-N-(проп-2- инил)амино]бензойной кислоты (пример 26 заявки на Европатент N 0373891) и пентафторофенола, используя процедуру, аналогичную описанной в той части примера 1, которая относится к получению исходных материалов.
i) Полученный продукт имел следующие характеристики ЯМР-спектров (CD3SOCD3): 2,2(м, 2H); 2,32 (с, 3H); 2,95 (т, 2H); 3,18 (с, 3H); 4,4 (м, 1H); 4,76 (с, 2H): 6,72 (д, 2H); 7,72 (кв, 4H); 8,36 (д, 1H).
Полученный пентафторофенил п-[N-(7-хлоро-2-метил-4-оксо- 3-(пивалоилоксиметил)-3,4-дигидрохиназолин-6-илметил)-N-метиламино] бензоат, используемый в качестве исходного материала, получали из 6-бромометил-7-хлоро-2-метил-3-(пивалоилоксиметил)-3,4- дигидрохиназолин-4-она и трет-бутил-п-метиламинобензоата, используя процедуру, аналогичную описанной в той части примера 1, которая относится к получению исходных материалов.
Этот 6-бромометил-7-хлоро-2-метил-3-(пивалоилоксиметил)- 3,4-дигидрохиназолин-4-он, использованный выше, получали из 7- хлоро-2,6-диметил-3,4-дигидрохиназолин-4-она (пример 2 заявки на Европатент N 0284388), в соответствии со следующими процедурами:
7-Хлоро-2,6-диметил-3,4-дигидрохиназолин-4-он (17 г) добавляли частями к перемешанной суспензии гидрида натрия (60% м/м дисперсия в минеральном масле (2,9 г), промытая гексаном для удаления минерального масла) в ДMCO (200 мл), которую охлаждали до 20oC. Полученную смесь перемешивали при комнатной температуре в течение 1 ч. Затем добавляли порциями хлорометилпивалат (23,7 мл) и эту смесь перемешивали при комнатной температуре в течение 15 ч. Полученную смесь выливали в воду (150 мл) и экстрагировали этилацетатом (3 • 70 мл). Эти объединенные экстракты промывали водой, осушали (MgSO4) и выпаривали. Полученный остаток перетирали с диэтиловым эфиром, в результате чего получали 7-хлоро-2,6-диметил- 3-(пивалоилоксиметил)-3,4-дигидрохиназолин-4-он в виде твердого вещества (17 г).
Смесь полученного продукта, N-бромосуцинимид (9,8 г), бензоилпероксид (0,1 г) и тетрахлорметан (400 мл) нагревали с обратным холодильником в течение 4 ч. Полученную смесь охлаждали до комнатной температуры. Фильтровали и фильтрат концентрировали до объема 200 мл. Эту смесь оставляли при комнатной температуре в течение 16 ч. Полученный осадок выделяли и получали требуемый исходный материал в виде твердого вещества (15 г), т.пл. 160-164oC.
ЯМР-спектр (CDCl3): 1,22 (с, 9H); 2,66 (с, 3H); 4,68 (с, 2H); 6,09 (с, 2H); 7,68 (с, 1H); 8,31 (с, 1H).
j) Элементный анализ показал, что полученный продукт содержит 1 эквивалент воды и 0,5 эквивалентов хлорида натрия.
Этот пентафторофенил п-[N-(7-хлоро-2-метил-4-оксо-3-(пивалоилоксиметил) -3,4-дигидрохиназолин-6-илметил)-N-(проп-2-инил)амино] бензоат, используемый в качестве исходного материала, получали из 6-бромометил -7-хлоро-2-метил-3-(пивалоилоксиметил)-3,4-дигидрохиназолин-4-она и трет-бутил п-(проп-2-инил)аминобензоата [полученного с помощью реакции трет-бутил п-аминобензоата с пропаргилбромидом] , используя процедуру, аналогичную описанной в той части примера 1, которая относится к получению исходных материалов.
k) Элементный анализ показал, что этот продукт содержит 1 эквивалент воды, 0,25 эквивалентов диэтилового эфира и 1,5 эквивалентов хлорида натрия.
Этот пентафторофенил-п-[N-(7-хлоро-2-метил-4-оксо-3-(пивалоилоксиметил)- 3,4-дигидрохиназолин-6-илметил)-N-(проп-2-инил)амино] -o-фторобензоат, используемый в качестве исходного материала, получали из 6-бромометил-7 -хлоро-2-метил-3-(пивалоилоксиметил)-3,4-дигидрохиназолин-4-она и трет-бутил о-фторо-п(проп-2-инил)аминобензоата, используя процедуру, аналогичную описанной в той части примера 1, которая относится к получению исходных материалов.
l) Элементный анализ показал, что полученный продукт содержит 1,5 эквивалентов воды и 1 эквивалент хлорида натрия.
Этот пентафторофенил п-[N(7-бромо-2-метил-4-оксо-3-(пивалоилоксиметил)-3,4-дигидрохиназолин-6-илметил)-N-метиламино]-o-фторобензоат, используемый в качестве исходного материала, получали из 7-бромо-6-бромометил-2-метил-3-(пивалоилоксиметил)-3,4-дигидрохиназолин-4-она (пример 23 заявки Европатента N 0459730) и трет-бутил o-фторо-п-метиламинобензоата, используя процедуру, аналогичную описанной в той части примера 1, которая относится к получению исходных материалов.
m) Элементный анализ показал, что полученный продукт содержит 1 эквивалент воды и 0,6 эквивалентов хлорида натрия.
Полученный продукт имел следующие характеристики ЯМР-спектров (CD3SOCD3): 2,2 (м, 2H); 2,34 (с, 3H); 2,95 (т, 2H); 3,2 (с, 1H); 4,4 (шир. с, 3H); 4,7 (с, 2H); 6,79 (д, 2H); 7,72-7,8 (кв, 4H); 8,46 (д, 1H); 12,28 (с, 1H).
Этот пентафторофенил п-[N-(7-бромо-2-метил-4-оксо-3- (пивалоилоксиметил)-3,4-дигидрохиназолин-6-илметил)-N-проп- 2-инил)амино] бензоат, используемый в качестве исходного материала, описывается в примере 23 заявки на Европатент N 0459730.
n) Элементный анализ показал, что полученный продукт содержит 2 эквивалента воды и 3 эквивалента хлорида натрия.
Этот пентафторофенил п-[N-(7-бромо-2-метил-4-оксо-3-(пивалоилоксиметил)-3,4-дигидрохиназолин-6-илметил)-N-(проп-2-инил)амино] -o-фторобензоат, используемый в качестве исходного материала, получали из 7-бромо-6-бромометил-2-метил-3- (пивалоилоксиметил)-3,4-дигидрохиназолин-4-она и трет-бутил-o-(проп-2-инил)аминобензоата, используя процедуру, аналогичную описанной в той части примера 1, которая относится к получению исходных материалов.
Пример 3. Используя аналогичные процедуры, описанные в примере 1, пентафторофенил o-фторо-п-[N(2,7-диметил-4-оксо-3-(пивалоилоксиметил)- 3,4-дигидрохиназолин-6-илметил)-N-(проп-2-инил)-амино] бензоат подвергали реакции с метил (2S)-2-амино-4-(тетразол-5-ил)бутиратом, полученный метилбутират гидролизовали и получали (2S)-2-{o-фторо-п- [N-(2,7-диметил-4-оксо-3,4-дигидрохиназолин-6-илметил)-N-(проп- 2-инил)амино] бензамидо} -4-(тетразол-5-ил)масляную кислоту (осушенную с помощью азеотропной дистилляции воды из смеси толуола) с выходом 76%.
ЯМР-спектр (CD3SOCD3): 2,06-2,28 (м, 2H); 2,31 (с, 3H); 2,44 (с, 3H); 2,97 (м, 2H); 3,21 (с, 1H); 4,30 (с, 2H); 4,42 (м, 1H); 4,70 (с, 2H); 6,65 (м, 2H); 7,43 (с, 1H); 7,59 (т, 1H); 7,71 (с, 1H); 8,05 (т, 1H); 12,06 (с, 1H).
MC: (положительный ион FAB) m/e (P + I) 533.
Элементный анализ для C26H25FN8O4 0,38H2O 0,14Na+ 0,045Cl- x 0,04CH3C6H5:
Найдено,%: C 57,8; H 4,8; N 20,3;
Вычислено,%: C 57,6; H 4,8; N 20,4.
В этом примере метил (2S)-2-амино-4-(тетразол-5-ил)-бутират был обогащен формой, имеющей (S)-конфигурацию в отношении (S):(R) = 99:1 или более, как было определено с помощью хроматографического анализа. Это изомерное соотношение сохранялось и в продукте примера 3.
Этот метил (2S)-2-амино-4-(тетразол-5-ил)бутират (в отношении (S):(R) = 99:1 или более), используемый в качестве исходного материала, получали следующим образом.
Раствор N-бензилоксикарбонил-L-глютаминметилового сложного эфира (25 г) в ТГФ (500 мл) добавляли по капле к перемешанному раствору трифенилфосфина (44,5 г) в тетрахлорметане (1 л). Полученную смесь нагревали до 50oC в течение 2 ч. Затем эту смесь выпаривали. Полученное маслянистое вещество перетирали в этилацетате. После этого смесь фильтровали, а фильтрат выпаривали. Полученный остаток очищали с помощью колоночной хроматографии, элюируя смесью гексана и этилацетата (1:1), и получали метил (2S)-2-бензилоксикарбониламино-4-цианобутират (19,38 г, 83%).
Смесь метил (2S)-2-бензилоксикарбониламино-4-цианобутирата (10 г), азида три-п-бутилолова [полученного в соответствии с методом в Рес. Trav. Chim. Pay - Вas., 1963, 81, 286; 12 г] и ТГФ (60 мл) перемешивали и нагревали до температуры перегонки в течение 40 ч. Затем эту смесь выпаривали. Полученное маслянистое коричневое вещество перетирали в диэтиловом эфире, который был насыщен газообразным хлороводородом. Этот осадок выделяли и промывали диэтиловым эфиром, в результате чего получали метил (2S)-2-бензилоксикарбониламино-4-(тетразол-5-ил)-бутират (2,23 г, 32%).
ЯМР-спектр (CD3SOCD3): 1,95-2,35 (м, 2H); 2,95 (т, 2H); 3,64 (с, 3H); 4,15 (м, 1H); 5,04 (с, 2H); 7,36 (с, 5H); 7,88 (д, 1H); 13,0 (с, 1H).
Смесь полученного продукта гидрогенизировали, используя аналогичную процедуру, описанную в последнем параграфе примера 1, и получали метил (2S)-2-амино-4-(тетразол-5-ил)бутират с выходом 88%.
ЯМР-спектр (CD3SOCD3): 1,82-2,26 (м, 2H); 2,86-2,94 (т, 2H); 3,67 (с, 3H); 3,7-3,85 (м, 1H); 5,28 (шир. с, 2H).
Пример 4. Диэтилфосфорилцианид (0,18 г) добавляли к смеси 5-[N-(2,7-диметил-4-оксо-3,4-дигидрохиназолин-6-илметил)-N-(проп- 2-инил)амино] пиридин-2-карбоновой кислоты (0,135 г), N-метилморфолина (0,113 г) и ДМФ (10 мл). Эту смесь перемешивали при комнатной температуре в течение 1 ч. Затем добавляли раствор смеси метил (2S)-2-амино-4-(тетразол-5-ил)бутирата (0,14 г) и N-метилморфолина (0,113 г) в ДМФ (5 мл). Полученную смесь перемешивали при комнатной температуре в течение 64 ч. Затем эту смесь выпаривали и остаток распределяли между этилацетатом и водой. Органическую фазу промывали водой, высушивали (MgSO4) и выпаривали, в результате чего получали метил (2S)-2-{ 5-N(2,7-диметил-4-оксо-3,4-дигидрохиназолин-6-илметил)-N- (проп-2-инил)амино]пиридин-2-карбоксамидо}-4-(тетразол-5-ил)бутират (0,16 г).
ЯМР-спектр (CD3SOCD3): 2,31 (м, 5H); 2,44 (с, 3H); 2,92 (т, 2H); 3,63 (с, 3H); 4,30 (с, 2H); 4,50 (м, 1H); 4,75 (с, 2H); 7,20-7,81 (м, 1H); 7,44 (с, 1H); 7,72 (с, 1H); 7,80-7,90 (д, 1H); 8,13-8,20 (д, 1H); 8,65-8,77 (д, 1H); 12,1 (шир. с, 1H).
Смесь полученного материала и 2 н. раствора гидроксида натрия (3 мл) перемешивали при комнатной температуре в течение 1 ч. Эту смесь подкисляли до значения pH 4 путем добавления концентрированной соляной кислоты. Полученный осадок выделяли, затем промывали последовательно водой, ацетоном и диэтиловым эфиром, после чего высушивали и получали (2S)-2-{5-[N-(2,7-диметил-4-оксо-3,4-дигидрохиназолин-6-илметил)-N-(проп- 2-инил)амино] пиридин-2-карбоксамидо}-4-(тетразол-5-ил)-масляную кислоту (0,124 г).
ЯМР-спектр (CD3SOCD3): 2,30 (м, 5H); 2,45 (с, 3H); 2,82-2,89 (т, 2H); 4,39 (с, 2H); 4,41-4,60 (м, 1H); 4,76 (с, 2H); 7,2-7,31 (м, 1H); 7,45 (с, 1H); 7,73 (с, 1H); 7,8-7,9 (д, 1H); 8,15 (д, 1H); 8,32-8,61 (д, 1H).
МС: (положительный ион, FAB) m/e (P + 1) 515;
Элементный анализ для C25H25N9O4 1,5 NaCl 1,25 H2O:
Найдено, %: C 48,0; H 4,3; N 19,8;
Вычислено, %: C 48,0; H 4,4; N 20,15.
В этом примере метил (2S)-2-амино-4-(тетразол-5-ил)бутират был обогащен формой, имеющей (S)-конфигурацию в отношении (S):(R)=99:1 или более, как было определено с помощью хроматографического анализа. Это изомерное соотношение сохранялось и в продукте примера 4.
Эту 5-[N-(2,7-диметил-4-оксо-3,4-дигидрохинозолин-6-ил-метил)-N-(проп-2-инил) амино] пиридин-2-карбоновую кислоту, используемую в качестве исходного материала, получали следующим образом.
Смесь 6-бромометил-2,7-диметил-3-(пивалоилоксиметил)-3,4-дигидрохиназолин-4-она (1,11 г), метил 5-[N-(проп-2-инил)-амино]пиридин-2-карбоксилата (0,61 г), полученного с количественным выходом путем обработки метил 5-[N-(трет-бутоксикарбонил)-N-(проп-2-инил)амино] пиридин-2-карбоксилата (J. Med. Chem, 1991 1594) трифторуксусной кислотой при 0oC в течение 1 ч, 2,6 лутидина (0,62 г), йодина натрия (0,005 г) и ДМА (20 мл) перемешивали и нагревали до 95oC в течение 7 ч. Полученную смесь выпаривали, а остаток распределяли между этилацетатом и 2 н. раствором соляной кислоты. Кислотность водного слоя понижали до pH 4 путем добавления 2 н. раствора гидроксида натрия и этот раствор экстрагировали этилацетатом. Органический слой осушали сульфатом магния и выпаривали. Остаток очищали с помощью колоночной хроматографии, элюируя этилацетатом. В результате этого получали метил 5-[N-(2,7-диметил-4-оксо-3-(пивалоилоксиметил) -3,4-дигидрохиназолин-5-илметил-N-(проп-2-инил)амино]пиридин-2-карбоксилат в виде камеди (0,262 г).
Смесь полученного таким образом сложного эфира, 2 н. раствора гидроксида натрия (20 мл) и метанола (10 мл) размешивали 16 ч при комнатной температуре. Метаноловую фазу выпаривали, а оставшийся водный раствор подкисляли до pH 6 путем добавления концентрированной соляной кислоты. Полученный осадок выделяли, промывали поочереди водой и диэтиловым эфиром, а затем осушали. В результате описанной процедуры получали 5-[N(2,7-диметил-4-оксо-3,4-дигидрохиназолин-6-илметил)-N-(проп-2-инил) амино] пиридин-2-карбоновую кислоту (0,143 г).
ЯМР-спектр (CD3SOCD3): 2,30 (с, 3H); 2,45 (с, 3H); 3,28 (с, 1H); 4,35 (с, 2H); 7,12-7,25 (м, 1H); 7,45 (с, 1H); 7,71 (с, 1H); 7,82-7,91 (д, 1H); 8,17-8,22 (д, 1H).
Пример 5. В соответствии с процедурой, описанной в примере 1, пентафторофенил п-{N-[1-(2,7-диметил-4-оксо-3-(пивалоилметил)-3,4-дигидрохиназолин-6-ил) этил]-N-(проп-2-инил)амино] бензоат подвергали реакции с метил (2S)-2-амино-4-(тетразол-5-ил) бутиратом и полученный метилбутират гидролизовали с получением 2-п-N-[1-(2,7-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)-этил] -N-(проп-2-инил) амино бензамидо-4-(тетразол-5-ил) масляной кислоты с выходом 25%, т. пл. 207oC.
ЯМР-спектр (CD3SOCD3): 1,5 (д, 3H); 2,25 (м, 2H); 2,27 (с, 1H); 2,34 (с, 3H); 2,9 (с, 1H); 3,0 (т, 2H); 3,7 (д, 1H); 3,95 (д, 1H); 4,4 (м, 1H); 5,5 (м, 1H); 7,0 (д, 2H); 7,4 (с, 1H); 7,8 (д, 2H); 8,1 (с, 1H); 8,4 (д, 1H);
Элементный анализ C27H28N8O4 1H2O 0,25NaCl:
Найдено, %: C 57,9; H 5,2; N 19,5;
Вычислено, %: C 57,7; H 5,3; N 19,9.
В этом примере метил (2S)-2-амино-4-(тетразол-5-ил)бутират был обогащен формой, имеющей (S)-конфигурацию в отношении (S) : (R) = 7:3. Это изомерное отношение сохранялось в продукте примера 5.
Пентафторофенил п-{ N-[1-(2,7-диметил-4-оксо-3-(пивалоилоксиметил)- -3,4-дигидрохиназолин-6-ил)этил]-N-(проп-2-инил)-амино}бензоат, использованный в качестве исходного материала, получали следующим образом.
Бром (9,4 г) добавляли по капле к размешанному раствору 4'-этил-3'-метилацетанилида в уксусной кислоте (100 мл), который был нагрет до 45oC. Смесь размешивали 30 мин при 45oC. После этого смесь выпаривали, а остаток распределяли между этилацетатом и насыщенным водным раствором бикарбоната натрия. Органическую фазу промывали водой, осушали сульфатом магния и выпаривали. Остаток очищали с помощью колоночной хроматографии, элюируя смесью 5:1 (по объему) гексана и этилацетата. В результате этого получали 2'-бромо-4'-этил-5-метилацетанилид (13,2 г), т.пл. 92oC.
Смесь полученного соединения, цианида меди (6,8 г) и МП (100 мл) размешивали и нагревали до 120oC в течение 1 ч. Затем эту смесь охлаждали до комнатной температуры, выливали в смесь водного гидроксида аммония (0,88 г/мл, 300 мл) и льда (600 мл) и размешивали 10 мин. Осадок выделяли и поочереди промывали водой и этилацетатом. Органические промывки промывали водой и солевым раствором, осушали сульфатом магния и выпаривали. В результате этого получали 2'-циано-4'-этил-5-метилацетанилид (8 г), т.пл. 121oC.
Смесь полученного продукта, перекиси водорода (30%, 150 мл), гидроксида натрия (2,33 г) и воды (23 мл) размешивали и нагревали до 55oC в течение 2 ч. Затем смесь охлаждали до комнатной температуры и выпаривали. К остатку добавляли воду (200 мл) и полученный раствор подкисляли до pH путем добавления разбавленной водной соляной кислоты. Остаток выделяли, промывали и осушали. В результате этого получали 6-этил-2,7-диметил-3,4-дигидрохиназолин-4-он (6,7 г), т.пл. 288o (разложение).
ЯМР-спектр (CD3SOCD3): 1,2 (т, 3H), 2,3 (c, 3H); 2,35 (c, 3H); 2,7 (кв, 2H); 7,3 (c, 1H); 7,8 (c, 1H); 11,95 (шир.с, 1H).
Раствор полученного продукта в DMCO (50 мл) добавляли к размешанной смеси гидрида натрия (80%-ная дисперсия в минеральном масле, 1,5 г, где масло было промыто гексаном) и DMCO (50 мл). Полученную смесь размешивали при комнатной температуре в течение 30 мин. Затем добавляли хлорометилпивалат (9,7 г) и смесь размешивали при комнатной температуре в течение 20 ч. Смесь распределяли между этилацетатом и смесью льда и воды. Органическую фазу промывали водой, осушали сульфатом магния и выпаривали. Продукт очищали с помощью колоночной хроматографии, элюируя возрастающими количествами полярных смесей гексана и этилацетата. Таким образом получали 6-этил-2,7-диметил-3-(пивалоилоксиметил)-3,4-дигидрохиназолин-4-он (4,2 г), т.пл. 104oC.
Смесь части полученного продукта (3,2 г), N-бромосукцинимида (1,9 г), бензоилпероксида (0,01 г) и тетрахлорметана (300 мл) размешивали и нагревали с обратным холодильником в течение 3 ч. Затем смесь охлаждали до комнатной температуры и фильтровали. Фильтрат выпаривали и остаток очищали с помощью колоночной хроматографии, элюируя возрастающими количествами полярных смесей гексана и этилацетата. Таким образом получали 6-(1-бромоэтил)-2,7-диметил-3-(пивалоилоксиметил)-3,4-дигидрохиназолин-4-он (3,35 г), т.пл. 132oC.
ЯМР-спектр (CDCl3): 1,25 (c, 9H); 2,2 (д, 3H); 2,55 (c, 3H); 2,65 (c, 3H); 3,4 (кв, 1H); 6,1 (кв, 2H); 7,43 (c, 1H); 8,4 (c, 1H).
Смесь полученного продукта, трет-бутил п-аминобензоата (4,8 г), карбоната кальция (3,3 г) и ДМА (80 мл) размешивали и нагревали до 110oC в течение 3 ч. Смесь выпаривали и остаток распределяли между этилацетатом и водой. Органическую фазу осушали сульфатом магния и выпаривали. Остаток очищали с помощью колоночной хроматографии, элюируя возрастающими количествами полярных смесей гексана и этилацетата. Таким образом получали трет-бутил п-{N-[1-(2,7-диметил-4-оксо-3-(пивалоилоксиметил)-3,4-дигидрохиназолин- -6-ил)этил]амино}бензоат (2,75 г), т.пл. 220oC.
Смесь части (1,5 г) полученного продукта, пропаргилбромида (80% раствор в толуоле, 3,3 мл), карбоната кальция (1,5 г) и ДМА (50 мл) размешивали и нагревали до 110oC в течение 8 ч. Смесь выпаривали, а остаток распределяли между этилацетатом и водой. Органическую фазу осушали сульфатом магния и выпаривали. Остаток очищали с помощью колоночной хроматографии, элюируя смесью (1: 1, по объему) гексана и этилацетата. Таким образом, получали трет-бутил-п-{ N-[1-(2,7-диметил- -4-оксо-3-(пивалоилоксиметил)-3,4-дигидрохиназолин-6-ил)этил]-N-(проп-2-инил)амино}бензоат в виде камеди (0,78 г).
ЯМР-спектр (CDCl3): 1,23 (c, 9H); 1,57 (c, 9H); 1,72 (д, 3H); 2,0 (т, 1H); 2,3 (c, 3H); 2,63 (c, 3H); 3,7 (м, 1H); 3,9 (м, 1H); 5,3 (кв, 1H); 6,1 (c, 2H); 6,95 (д, 2H); 7,42 (c, 1H); 7,95 (д, 2H); 8,3 (c, 1H).
Смесь полученного продукта и трифторуксусной кислоты (20 мл) размешивали при комнатной температуре в течение 1 ч. Затем смесь выпаривали. К полученной смеси добавляли диэтиловый эфир (100 мл) и осадок выделяли. Таким образом получали п-{ N-[1-(2,7-диметил-4-оксо-3-(пивалоилоксиметил)-3,4-дигидрохиназолин-6-ил)этил] -N-(проп-2-инил)амино} бензойной кислоты трифторуксуснокислую соль (0,73 г), т.пл. 217oC.
Смесь полученного продукта, пентафторофенола (0,64 г), дициклогексилкарбодиимида (0,717 г) и этилацетата (120 мл) размешивали при комнатной температуре в течение 20 ч. Смесь фильтровали и фильтрат выпаривали. Остаток очищали с помощью колоночной хроматографии, элюируя смесью (3:1, по объему) гексана и этилацетата. Таким образом получали п-{N-[1-(2,7-диметил-4-оксо-3-(пивалоилоксиметил)-3,4-дигидрохиназолин-6-ил)этил] -N-(проп-2- -инил)амино}бензоат (0,68 г), т.пл. 112oC.
Пример 6. Ниже, в целях иллюстрации, представлены характерные фармацевтические формы, содержащие соединение формулы (I) или его фармацевтически приемлемую соль (соединение X) и предназначенные для профилактики или терапевтического лечения человека:
a) Таблетка I - мг/таблетка
Соединение X - 100
Лактоза Ph. Eur. - 182,75
Кроскармелоза-Na - 12,0
Таблетка I - мг/таблетка
Кукурузный крахмал, паста (5 мас.%/об.) - 2,25
Стеарат магния - 3,0
b) Таблетка II - мг/таблетка
Соединение X - 50
Лактоза Ph.Eur. - 223,73
Кроскармелоза-Na - 6,0
Кукурузный крахмал - 15,0
Поливинилпирролидон паста (5 мас.%/об.) - 2,25
Стеарат магния - 3,0
c) Таблетка III - мг/таблетка
Соединение X - 1,0
Лактоза Ph.Eur. - 93,25
Кроскармелоза-Na - 4,0
Кукурузный крахмал, паста (5 мас.%/об.) - 0,75
Стеарат магния - 1,0
d) Капсула - мг/капсула
Соединение X - 10,0
Лактоза Ph.Eur. - 488,5
Стеарат магния - 1,5
e) Инъекция I - 50 мг/мл
Соединение X - 5,0 мас.%/об.
Раствор гидроксида натрия 1M - 15,0 мас.%/об.
Соляная кислота 0,1 M для доведения pH до 7,6
Полиэтиленгликоль 400 - 4,5 мас.%/об.
Вода для инъекций - До 100%
f) Инъекция II - 10 мг/мл
Соединение X - 1,0 мас.%/об.
Фосфат натрия BP - 3,6 мас.%/об.
Раствор гидроксида натрия 1,0 M - 15,0 мас.%/об.
Вода для инъекций - До 100%
g) Инъекция III - I мг/мл, забуференный до pH 6
Соединение X - 0,1 мас.%/об.
Фосфат натрия BP - 2,26 мас.%/об.
Лимонная кислота - 0,38 мас.%/об.
Полиэтиленгликоль 400 - 3,5 мас.%/об.
Вода для инъекций - До 100%
Вышеуказанные композиции могут быть получены традиционными методами, обычно применяемыми в фармацевтике. Таблетки (a)-(c) могут быть покрыты энтеросолюбильным покрытием в соответствии со стандартной техникой, например покрытием из ацетатфталата целлюлозы.
Схемы формул I - V даны в конце текста.
Хиназолиновые производные формулы (I), его фармацевтически приемлемая соль или его сложный эфир, где R1 - C1-C4-алкил; R2 - водород, C1-C4 - алкил; R3 - C1-C4 - алкил; C3-C4 - алкенил, C3-C4 - алкинил; Ar - фенилен, который может иметь необязательно один или два заместителя, выбранных из галогена, C1-C4 - алкила, C1-C4 - алкокси, или пиридиндиил, обладают противоопухолевой активностью. Хиназолиновое кольцо может необязательно иметь в одном из 5-, 7- и 8-положениях заместитель, выбранный из галогена и C1-C4-алкила. Соединение (I) получают взаимодействием кислоты (II) с соединением (III), где R1, R2, R3 имеют указанные значения, а R4 - водород или защитная группа, R5 - защитная группа, защитные группы удаляют традиционным способом. Соединение (I) используется в качестве активного ингредиента в фармацевтической композиции. 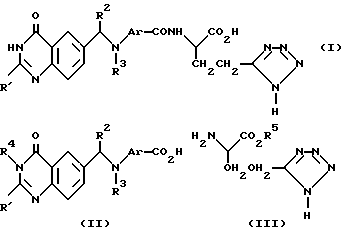 3 с. и 9 з.п. ф-лы, 2 табл.
3 с. и 9 з.п. ф-лы, 2 табл.
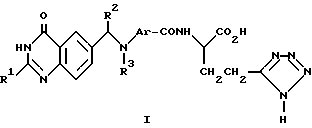
где R1 - С1 - С4-алкил, хиназолиновое кольцо может необязательно иметь (в одном из 5-, 7- и 8-положений) один заместитель, выбранный из галогена и С1 - С4-алкила;
R2 - водород или С1 - С4-алкил,
R3 - С1 - С4-алкил, С3 - С4-алкенил или С3 - С4-алкинил;
Ar - фенилен, который может иметь необязательно один или два заместителя, выбранных из галогена, С1 - С4-алкила и С1 - С4-алкокси, или Ar - пиридиндиил,
или его фармацевтически приемлемая соль, или его сложный эфир.
R2 - водород;
R3 - метил, этил, пропил, проп-2-енил или проп-2-инил;
Ar - 1,4 фенилен, который может иметь, но необязательно, один фторзаместитель, либо Ar - пиридин-2,5-диил (с CONH-группой во 2-положении),
или его фармацевтически приемлемая соль.
2-{ п-[N-(2-метил -4-оксо-3,4-дигидрохиназолин-6- ил-метил)-N-(проп-2-инил)амино] бензамидо}-4-( тетразол-5-ил)масляную кислоту,
2-{ o-фторо-п-[N-( 2-метил-4-оксо-3,4-дигидрохиназолин -6-илметил)-N-(проп-2-инил) амино]бензамидо}-4- (тетразол-5-ил)масляную кислоту,
2-{ п-[N-(2,7-диметил-4-оксо-3,4- дигидрохиназолин -6-илметил)-N-метиламино]бензамидо} -4-(тетразол-5-ил)масляную кислоту,
2-{ o-фторо-п-[N-(2,7-диметил -4-оксо-3,4-дигидрохиназолин -6-илметил)-N-метиламино]бензамидо} -4-(тетразол-5-ил)масляную кислоту,
2-{ п-[N-(2,7-диметил -4-оксо-3,4-дигидрохиназолин -6-илметил)-N-(проп -2-инил)амино]бензамидо}-4- (тетразол-5-ил)масляную кислоту,
2-{ o-фторо-п-[N- (2,7-диметил-4-оксо-3,4- дигидрохиназолин-6- илметил)-N-(проп-2-инил)амино/бензамидо} -4-(тетразол-5-ил)масляную кислоту;
2-{ п-[N-(7-хлоро -2-метил-4-оксо-3,4-дигидрохиназолин -6-илметил)-N-(проп-2-инил)амино]бензамидо }-4-(тетразол -5-ил)масляную кислоту,
2-{ o-фторо-п-[N-(7-хлоро-2-метил-4-оксо- 3,4-дигидрохиназолин-6-илметил)-N (проп-2-инил)амино] бензамидо}-4-(тетразол-5- ил)масляную кислоту и
2-{ п-[N-(7-бромо-2-метил-4-оксо -3,4-дигидрохиназолин-6- илметил)-N-(проп-2-инил) амино]бензамидо}-4-(тетразол-5- ил)масляную кислоту.
(2S)-2-п-[N-(2-метил-4-оксо-3,4 -дигидрохиназолин-6- илметил)-N-(проп-2-инил)амино]бензамидо} -4-(тетразол-5-ил)масляную кислоту,
(2S)-2-{ o-фторо-п-[N -(2-метил-4-оксо-3,4- дигидрохиназолин-6-илметил)-N-(проп-2-инил)амино]бензамидо} -4-(тетразол-5-ил)масляную кислоту,
(2S)-2-{ п-[N-(2,7-диметил-4- оксо-3,4-дигидрохиназолин -6-илметил)-N-метиламино] бензамидо}-4-( тетразол-5-ил)масляную кислоту,
(2S)-2-{ o-фторо-п-[N-(2,7 -диметил-4-оксо-3,4-дигидрохиназолин -6-илметил)-N-метиламино/бензамидо} -4-(тетразол-5-ил)масляную кислоту,
(2S)-2-{ п-[N-(2,7-диметил-4 -оксо-3,4-дигидрохиназолин- 6-илметил)-N-(проп-2-инил)амино] бензамидо}-4-(тетразол -5-ил)масляную кислоту,
(2S)-2-{ o-фторо-п- [N-(2,7- диметил-4-оксо -3,4-дигидрохиназолин-6- илметил)-N-(проп-2-инил) амино]бензамидо}-4-(тетразол-5-ил)масляную кислоту,
(2S)-2-{ п-[N-(7-хлоро-2-метил-4-оксо-3,4-дигидрохиназолин-6-илметил)-N- (проп-2-инил)амино]бензамидо} -4-(тетразол-5-ил)масляную кислоту,
(2S)-2-{ o-фторо-п- [N-(7-хлоро-2-метил-4-оксо -3,4-дигидрохиназолин-6-илметил)-N-( проп-2-инил)амино]бензамидо}-4- (тетразол-5-ил)масляную кислоту и
(2S)-2-{ п- [N-(7- бромо-2-метил-4-оксо-3,4- дигидрохиназолин-6-илметил)-N-(проп-2-инил)амино] бензамидо}-4- (тетразол-5-ил)масляную кислоту.
(2S)-2-{ o-фторо-п-[N-(2,7 -диметил-4-оксо-3,4-дигидрохиназолин -6-илметил)-N-(проп-2-инил) амино]бензамидо}-4-(тетразол-5-ил)масляная кислота.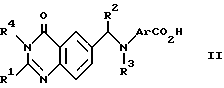
где R1, R2 и R3 имеют значения, указанные в п.1;
R4 - водород или защитная группа,
с соединением формулы (III)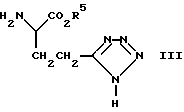
где R5 - защитная группа,
после чего защитные группы удаляют традиционными способами.
| ЕР, патент, 373891, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |

