Изобретение относится к соединениям, которые можно использовать в пролекарственной терапии под действием ферментов, направляемых антителами, так называемой ADEPT - терапии, способам их получения, содержащим их фармацевтическим композициям и способам их применения, а также к двухкомпонентным системам, содержащим 1) конъюгат энзима и антитела или фрагмента антитела и 2) соединение настоящего изобретения. Эти соединения представляют особый интерес как пролекарства для использования вместе с энзимами карбоксипептидазы G, особенно карбоксипептидазы G2 (CPG2).
Были раскрыты многие цитотоксические соединения, которые находят эффективное применение в химиотерапии раковых заболеваний.
Азотистые иприты представляют одно важное семейство таких цитотоксических соединений. Клинические применение цитотоксических соединений, в целом, и азотистых ипритов, в частности, было ограничено из-за малой селективности цитотоксического действия на опухолевые и нормальные клетки.
Один из подходов к решению этой проблемы состоит в разработке так называемых пролекарств, которые представляют собой производные этих цитотоксических лекарств, и часто весьма простые производные, цитотоксические свойства которых значительно ниже, чем у исходных лекарств. Было предложено введение таких пролекарств пациентам в определенных режимах за счет чего пролекарство превращается в цитотоксическое лекарство только в участке, где оно должно действовать.
Один из подходов включает связывание исходного цитотоксического азотистого иприта с аминокислотой с образованием пролекарства, которое можно превратить в исходный азотистый иприт в участке, где он должен действовать, под действием энзима. Этот подход можно внедрить в практику за счет использования конъюгата антитело/энзим в ассоциации с пролекарством. Конъюгат-антитело/ энзим образуется из антител, селективных к опухолям, и энзима, который впоследствии превращает пролекарство в цитотоксическое лекарство. В клинической практике конъюгат антитело/энзим вначале вводят пациенту и предоставляют ему связываться с опухолью. После соответствующего промежутка времени, в течение которого конъюгат антитело/энзим выводится из остальных частей организма, пролекарство вводят пациенту. Превращение пролекарства под влиянием локализованного энзима в цитотоксическое лекарство происходит, главным образом, в области опухоли. Такая система описана в международной заявке PCT/GB88/00181, опубликованной как WO88/07378 и как патент США N 4975278.
Известные пролекарства для ADEPT-терапии, расщепленные под действием CPG приводят к образованию ароматических производных азотистого иприта в виде их активных лекарственных форм. Однако существует необходимость в более активных лекарствах, получаемых при CPG расщеплении для повышения терапевтической эффективности против опухолевых клеток. Необходимо также повысить селективность ADEPT терапии с CPG, то есть, степень токсичности по отношению к опузолевым клеткам по сравнению со здоровыми клетками.
Настоящее изобретение основано на обнаружении новых пролекарств для применения в ADEPT терапии, которые расщепляются под действием CPG, и которые приводят к получению гораздо более активных цитотоксических лекарств, нежели известные продукты реакций, катализируемых CPG. CPG в природе действует как фолат-разлагающий энзим, который специфически гидролизует глутаминовую и аспарагиновую кислоты в производные фолата /Sherwood, P.F. et al., Eur. J. Biochem. /1085/, 148, 447-453/. Энзимы карбоксипептидазы G не распознают неклассические аналоги фолатов /Kalghatgi, K.K.et.al., Cancer Research /1979/, 39, 3441-3445/, и поэтому считаются консервативными в специфичности субстратов. Неожиданно оказалось, что пролекарства настоящего изобретения представляют собой субстраты для CPG энзимов, таких как CPG1, но особенно для CPG2 энзимов. CPG2 представляет собой экзопептидазу, которая специфична для L - глутамата. Известно, что она гидролизует фрагмент глутаминовой кислоты из фолиевой кислоты и ее аналогов, и глутамил-р-аминобензойной кислоты за счет расщепления по -CO-NH- части структуры - ароматическое кольцо -CO-NH-Glu. В настоящем изобретении этой частью структуры для сравнительных целей является -ароматическое кольцо -X-CO-NH-Glu, где X представляет -NH-, -O- или -CH2-; изменяя, тем самым, расстояние между точкой расщепления и ароматическим кольцом, а также изменяя распределение электронной плотности по связи -CO-NH-, особенно, если X представляет NH или O. При этом нельзя предсказать, как CPG2 распределит эти пространственные и электронные различия. Соединения, в которых X представляет -CH2-, раскрыты в Chemical Abstracts 75/5/ 2 августа 1971, абстр.N 36603 м, и у Карпавикус с сотр. Изв. Академии Наук СССР, сер. хим.N 91970, 2150-2153 /см.соединения VI, VIII, IX и XI в таблице 1/.
В соответствии с одной из особенностей настоящего изобретения предложены соединения формулы I, которые являются пролекарственными субстратами для CPG энзимов.
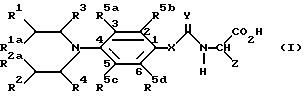
где R1 и R2 каждый независимо представляет хлор, бром, иод, OSO2Me, или OSO2 фенил, где фенил необязательно замещен 1, 2, 3, 4 или 5 заместителями, независимо выбранными из C1-4- алкила галоида, -CN или -NO2/;
R1a и R2a каждый независимо представляет водород, C1-4-алкил или C1-4-галоидалкил;
R3 и R4 каждый независимо представляет водород, C1-4-алкил или C1-4- галоидалкил;
R5a, R5b и R5d каждый независимо представляет водород, C1-4-алкил, необязательно содержащий одну двойную или одну тройную связь, C1-4-алкокси, галоид, циано, -NH2, -CONR7R8/ где R7 и R8 имеют указанные ранее значения/, -NH/C1-4- алкил/, -N/C1-4-алкил/2 и C2-5- алканоил; или R5a и R5b вместе представляют a/ C4-алкилен, необязательно содержащий одну двойную связь;
b/ C3 алкилен; или
c/ -CH= CH-CH=CH-, -CH=CH-CH2- или -CH2-CH=CH- каждый необязательно замещенный 1,2,3 или 4 заместителями, причем каждый из указанных заместителей выбирают из группы, состоящей из C1-4-алкила, C1-4-алкокси, галоида, циано, нитро, C2-5-алканоила и -CONR7R8/ где R7 и R8 имеют указанные ранее значения/;
X представляет O, NH или -CH2-;
Y представляет O;
Z представляет -V-W, где V представляет -CH2-T-, где
T представляет -CH2-, -O-, -S-, -/SO/- или -/SO2/- при условии, что если V содержит серу или кислород в качестве второго атома, W отличается от -COOH/, а указанная группа необязательно содержит, кроме того один или два заместителя Q1 и/или Q2 у атома углерода, где Q1 и Q2 каждый независимо представляет C1-4-алкил или галоид; или, если Q1 и Q2 связаны с соседними атомами углерода, Q1 и Q2 вместе могут дополнительно представлять C3-C4-алкиленовый радикал, необязательно замещенный 1,2,3 или 4 заместителями, независимо выбранными из группы, состоящей из C1-4 алкила и галоида; а W представляет
/1/ COOH,
/2/-/C=O/-O-R6, где R6 представляет C1-6-алкил, C3-6-циклоалкил или арил /как указано в пункте 3 далее/;
/3/ -/C=O/-NR7R8, где R7 и R8 каждый независимо представляет водород или C1-6-алкил, C3-6-циклоалкил, арил, гетероарил, связанные с атомом азота через углерод, или C7-9-аралкильную группу, где арил представляет фенил; гетероарил является 5 или 6 членным кольцом, содержащим 1-3 гетероатома, выбранные из группы, состоящей из атома и серы; арильный фрагмент per se, гетероарильный фрагмент и арильный фрагмент арильной группы могут быть замещены по углероду 1-4 заместителями, выбранными из группы, состоящей из -COOH, OH, -NH2, -CH2-NH2, -/CH2/1-4-COOH, тетразол-5-ил и -SO3H, и алкильный фрагмент может необязательно содержать метильную группу;
/4/ - SO2NHR9, где R9 имеет значения, указанные для R7, но может дополнительно представлять -CF3, -CH2CF3 или арил, как указано ранее;
/5/ SO3R10, где R10 представляет H, C1-6-алкил или C3-6-циклоалкил,
/6/ PO3R10, R10, где радикалы R10, которые могут быть одинаковы или различны, имеют указанные ранее значения,
/7/ группу тетразолил-5-ила;
/8/ -CONH-SO2R11, где R11представляет /a/ C3-7 циклоалкил; /b/ C1-6-алкил, необязательно замещенный заместителями, выбранными из группы, состоящей из арила в соответствии с приведенными ранее определениями, C1-4-алкила, CF3 или галоида; и /c/ перфтор-C1-6-алкил; где арил представляет фенил, или фенил с 1-5 заместителями, где эти заместители выбирают из группы, состоящей из галоида, -NO2, -CF3, C1-4-алкила, C1-4-алкокси, -NH2, -NHCOCH3, -CONH2, -OCH2COOH, -NH/C1-4-алкила/, -N/C1-4-алкила/2, -NHCOOC1-4-алкила, -OH, -COOH, -CN и -COOC1-4-алкила; и
/9/ -M-Неt, где M представляет S, SO или SO2, а Нет представляет 5-6-членное гетероциклическое ароматическое кольцо, связанное с M через атом углерода ароматического кольца, причем указанное ароматическое кольцо содержит 1,2,3 или 4 гетероатама, выбранные из группы, состоящей из O, N и S, причем указанное ароматическое кольцо необязательно замещено по атомам углерода кольца 1, 2, 3 или 4 заместителями, выбранными из группы, состоящей из -OH, -SH, -N, -CF3, NH2 и галоида;
и соли указанных соединений формулы I.
Соединения формулы I содержат по крайней мере один асимметричный атом углерода, то есть атом углерода с заместителем -COOH в формуле I. Более того, в зависимости от значений R1, R2, R3, R4, Q1 и Q2, соединения формулы I могут иметь дополнительные асимметричные атомы углерода. Следует учитывать, что настоящее изобретение включает все такие формы соединений формулы I, включая рацемические формы, а также отдельные оптические изомеры, которые обладают полезными физиологическими свойствами указанных ранее композиций настоящего изобретения; причем специалистам известно, как можно разделить такие изомеры и как можно определить их физиологические свойства. Соединения настоящего изобретения, предпочтительно, имеют L-конфигурацию у атома углерода, который замещен -COOH в формуле I.
Настоящее изобретение включает соли соединений формулы I. Следует, однако, учитывать, что для фармацевтического использования указанные соли должны быть фармацевтически приемлемыми, но и другие соли могут найти применение, например, при получении соединений формулы I и их фармацевтически приемлемых солей. Можно получить полиморфные формы соединений настоящего изобретения, и эти формы также включены в объем изобретения.
Если относительно какого-либо заместителя указано, что он представляет или содержит алкильную группу, то такая группа может быть разветвленной или неразветвленной. Если относительно какого-либо заместителя указано, что он представляет или содержит C1-6-алкильную группу, то желательно, чтобы такая группа содержала 1 - 4 атома углерода, и была, например, метилом, этилом, н-пропилом или изопропилом, предпочтительно метилом или этилом, но наиболее предпочтительно метилом. Если относительно какого-либо заместителя указано, что он представляет или содержит C1-4-алкильную группу, то такая группа может быть, например, метилом, этилом, н-пропилом или изопропилом, предпочтительно метилом или этилом, но наиболее предпочтительно метилом.
Предпочтительными значениями для R1 и R2 являются I, Br, Cl, OSO2Me и OSO2фенил, где фенил замещен 1 или 2 /особенно 1/ заместителями /определенными ранее/ в 2- или 4-положениях. Наиболее предпочтительными значениями для R1 и R2 являются I, Br, Cl и -OSO2Me. Предпочтительными значениями для R1a и R2a являются -CH3 или водород, но предпочтительно водород.
Предпочтительными значениями для R3 и R4 являются водород, метил и CF3, но особенно водород.
Предпочтительными значениями для
R5a-d являются водород, фтор, хлор, метил, -CONH2 и CN. Если фенильное кольцо замещено так, что, по крайней мере один из R5a, R5b, R5c и R5d отличен от водорода, тогда предпочтительно, чтобы только один или два из R5a, R5b, R5c и R5d были отличны от водорода. В таких случаях предпочтительно далее, чтобы R5b и R5d были водородами, и, чтобы R5a и/или R5c отличались от водорода. Однако наиболее предпочтительно, чтобы R5a-b были водородами.
Предпочтительно чтобы X представлял O или N, особенно O. В другом варианте изобретения наиболее предпочтительно, чтобы X представлял N.
Предпочтительное значение для V составляет -CH2-CH2-. В другом варианте изобретения предпочтительное значение для V, если W представляет тетразол-5-ил, составляет -CH2-S-.
Если W представляет группу, определенную в пункте 3, арил, предпочтительно, представляет замещенный фенил, и гетероарил, предпочтительно, представляет 5 или 6-членное кольцо, содержащее 1 или 2 гетероатома, причем предпочтительно чтобы этими гетероатомами был азот, а гетероарил представлял бы пиридил или пиримидил. Предпочтительными заместителями на арильном фрагменте per se, гетероарильном фрагменте или аралкильном фрагменте аралкильной группы являются -COOH, -CH2-COOH, или тетразол-5-ил.
Если W представляет группу, определенную в пункте 8/b/, где W представляет C1-6-алкильную группу, необязательно замещенную арилом, необязательно замещенная арильная группа, предпочтительно, представляет фенил, замещенный -CONH2, -CH2-COOH и/или -COOH, но наиболее предпочтителен незамещенный фенил.
Если W представляет группу, определенную в пункте /9/, предпочтительно, чтобы Het представлял 5- или 6-членное ароматическое кольцо, содержащее 1, 2, 3 или 4 атома азота. Предпочтительно, чтобы азот был единственным гетероатомом в кольце. Таким образом конкретные группы включают пиридил, пирролил, 1,2,3-триазинил и 1,2,4-триазинил.
Конкретные значения для W представлены в определениях 1, 2, 3, 7, 5, 6 и 9 и предпочтительные в пунктах 1, 2, 3, 7 и 9. Более конкретные значения для W представлены: -COOH, -CONH2, -CONHR8 /где R8 имеет указанные ранее значения, и особенно, если R8 представляет фенил/, тетразол-5-ил, -CONH-SO2R11 /где R11 представляет определения /b/ и /c/, как указано ранее/ и группой -M-Het /где M представляет S, а Het представляет 5-членное гетероциклическое ароматическое кольцо с 3 или 4 гетероатомами, необязательно замещенными по углероду, где гетероциклическое кольцо содержит 3 гетероатома, атомом галоида или цианогруппой/.
Если W представляет -CONH-SO2R11, где R11 представляет перфтор-C1-6-алкильную группу, перфторалкильная группа, предпочтительно, содержит 1 - 4 атома углерода, особенно 1 или 2 атома углерода.
Более конкретные значения для W представляют -COOH, тетразол-5-ил или -CONH/арил/ /при этом арил определен в п. 3 ранее/. В рассматриваемом контексте арил представляет, предпочтительно, замещенный фенил, а предпочтительными заместителями являются -COOH, -CH2-COOH или тетразол-5-ил.
Предпочтительные конкретные соединения настоящего изобретения с точки зрения их применимости в АДЕРТ, включают:
/S/-2-/4-бис/-2-хлорэтил/амино/феноксикарбониламино/-4-/1Н- 1,2,3,4-тетразол-5-ил/масляную кислоту и ее соли;
N-/4-бис/2-хлорэтил/амино/-3-фторфенилкарбамоил/-L-глутаминовую кислоту и ее соли;
N-/4-/бис/2-хлорэтил/амино/фенилкарбамоил/-L-глутаминовую кислоту и ее соли;
но наиболее предпочтительным соединением изобретения является N-/4-/бис/2-хлорэтил/амино/феноксикарбонил/-L-глутаминовая кислота и ее соли.
Другим предпочтительным соединением, которое, как было показано, обладает высокой активностью в тестах, является N-4-/бис/-2-иодоэтил/амино/феноксикарбонил/-L-глутаминовая кислота и ее соли.
Конкретные подгруппы соединений настоящего изобретения, представляющих интерес, можно получить, выбирая любое из вышеуказанных конкретных или общих определений для R1-R4, R5a-d, X, Y, W, Q1 или Q2, либо отдельно, либо в сочетании с любым другим конкретным или общим определением для R1-R4, R5a-d, X, Y, W, Q1 или Q2.
Кроме того, лекарства, полученные в результате CPG расщепления тестовых соединений настоящего изобретения для осуществления АДЕРТ, менее стабильны в физиологических условиях, нежели известные лекарственные продукты CPG2 катализируемых реакций. Уменьшение стабильности приводит к снижению токсичности по отношению к здоровым клеткам, нежели в том случае, когда лекарство стабилизируется аналогично известным продуктам CPG катализируемых реакций для АДЕРТ, если часть активного лекарства, полученного в области опухоли, перемещается в общую циркуляцию. Так проведенные тесты продемонстрировали, что даже после внутривенного введения лекарства /то есть, активного лекарства, не пролекарства/, через 15 мин лекарство не обнаруживается в плазме.
Соединения настоящего изобретения образуют соли с различными органическими и неорганическими кислотами и основаниями, и такие соли входят в объем настоящего изобретения. Эти соли включают аммонийные соли, такие соли щелочных металлов, как соли натрия и калия, такие соли щелочноземельных металлов, как соли кальция и магния, соли органических оснований; например, соли дициклогексиламина, N-метил-D-глюкамина, соли таких аминокислот как аргинин, лизин, и т.п. Можно также получить соли органических и неорганических кислот, например, HCl, HBr, H2SO4, H3PO4, метансульфоновой, толуолсульфоновой и камфорсульфоновой кислот. Предпочтительными кислотами являются сильные кислоты со значениями pKa, менее или равными 2, и, особенно, со значениями pKa, менее или равными 1. Предпочтительны физиологически приемлемые соли, хотя могут быть полезны и другие соли; например при выделении или очистке продукта.
Соли можно получать обычными способами, например, осуществляя взаимодействие форм свободной кислоты или свободного основания продукта с одним или более эквивалентами соответствующего основания или кислоты в растворителе, или в такой среде, в которой эта соль не растворяется, или в таком растворителе, как вода, которую затем удаляют в вакууме или сушкой вымораживанием, или заменяя катионы имеющейся соли на другой катион на подходящей ионообменной смоле.
В соответствии с еще одной отличительной особенностью изобретения предложен способ получения соединений формулы I и их солей, который включает удаление защиты у соединения формулы Ia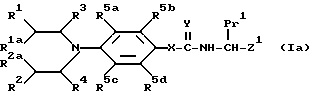
/где R1, R2, R1a, R3, R4, R5a, R5b, R5c, R5d; X, Y, Q1 и Q2 имеют указанные ранее значения, а Z1 представляет указанные ранее для Z значения, при условии, что если W представляет карбоксильную группу, она представлена в защищенной форме /обозначается Pr2/, и Pr1 также представляет карбоксильную группу в защищенной форме /которая может быть одинакова или может отличаться от Pr2/,
и при желании, превращение с полученного таким образом соединения формулы I в его соль.
Pr1 и Pr2 могут быть представлены, например, бензилоксикарбонильными группами, трет-бутоксикарбонильными группами, 2-/триметилсилил/этиловым сложным эфиром, диметил-трет-бутилсилиловым сложным эфиром, тетрагидропирановым сложным эфиром, тетрагидрофурановым сложным эфиром, метоксиметиловым сложным эфиром или бензилоксиметиловым сложным эфиром или другими общеизвестными карбоксизащитными группами, например, группами, образующими сложные эфиры, для удаления защиты галогенолизом или в результате кислотного катализа (Greene, T.W. и Wuts. P.G.M. в Protective Groups in Organic Synthesis, 2nd. Edition, Wiley-Ynterscience, 1990).
Если Pr1 и/или Pr2 представляют бензилоксикарбонильную группу, удаление защиты предпочтительно осуществлять гидрированием. Такое гидрирование можно осуществить любым удобным способом, например, в присутствии платины или никеля Рэнея, но предпочтительно использовать палладий на угле. Гидрирование удобно вести в присутствии инертного растворителя, предпочтительно, непротонного растворителя, особенно этилацетата, тетрагидрофурана, или такого полярного апротонного растворителя, как диметилформамид, предпочтительно, при температуре 0-100oC, более предпочтительно при температуре 15 - 50oC, и наиболее предпочтительно при комнатной температуре; и предпочтительно в течение 1 - 24 ч.
Если Pr1 и/или Pr2 представляет трет-бутоксикарбонильную группу, реакцию удаления защиты удобно вести в присутствии кислоты, особенно такой сильной кислоты, как трифторуксусная кислота, HCl, HBr, HJ или муравьиная кислота. Если нужно использовать растворитель, предпочтительны такие инертные непротонные растворители, как CH2Cl2 или диэтиловый эфир. Обычно реакцию ведут при температуре 0 - 100oC, обычно при 0 - 30oC, и обычно при комнатной температуре.
Соединения формулы Ia /как было указано ранее/ и их соли являются новыми, и, таким образом, составляют следующую отличительную черту изобретения.
Следующей отличительной чертой изобретения является создание способа получения соединений формулы Ia, где X = 0, и Y = 0, и их солей, который включает осуществление взаимодействия соединения формулы II: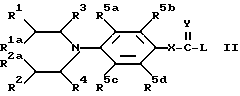
(где R1, R2, R1a, R2a, R3, R4, R5a, R5b, R5c и R5d имеют указанные ранее значения, X = 0, Y = 0, а Z представляет отщепляемый атом или группу)
с соединением формулы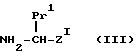
(где Pr1 и Z1 имеют указанные ранее значения),
в результате чего получают соединение формулы Ia, и, при желании, превращение соединения формулы Ia в его соль. Желательно, чтобы L представлял Cl, Br, J, 4-нитрофенокси или пентафторфенокси. Реакцию удобно вести в присутствии растворителя при температуре от -10 до 100oC, и, предпочтительно, при температуре 20 - 50oC.
Предпочтительно вести реакцию в органическом растворителе /особенно хлороформе, этилацетате, толуоле, диметилформамиде и CH2Cl2/, предпочтительно при 5 - 50oC, предпочтительно в течение 1 - 24 ч. Соединения формулы /II/ составляют следующий аспект изобретения, причем эти соединения можно получить, например, осуществляя взаимодействие соответствующего фенола, содержащего группу хлорэтиланилина, либо с арилхлорформатом, например нитрофенилхлорформатом /особенно, 4-нитрофенилхлорформатом/ или с фосгеном, в результате чего получают соединение формулы /II/.
Предпочтительно вести реакцию в присутствии органического растворителя /особенно этилацетата или хлороформа/, предпочтительно при 15 - 50oC /особенно при комнатной температуре/, предпочтительно в течение 1 - 10 ч.
Соединения формулы /III/, где W представляет группу /3/, как было указано ранее, можно получить из соединений формулы /III/, где W представляет группу /2/, как указано ранее, используя стандартные условия. Предпочтительные стандартные условия включают реакцию с азотсодержащими нуклеофильными соединениями /особенно с аммиаком или первичным или вторичным амином/, предпочтительно, при 25 - 30oC, предпочтительно в течение 24 ч.
Соединения формулы /III/, где W представляет тетразол-5-ильную группу, можно получить из соответствующего нитрила известными способами, например, по способу Finnegan, W.G. et al., JAC5 80, 1978, 3909 с последующим удалением защиты у амина /Pr3/, в результате чего получают соединения формулы /III/, где W представляет тетразол-5-ил.
Соединения формулы /III/, где W представляет группу /9/, как было указано ранее, можно получить, восстанавливая соединения формулы /XV/, как указано ранее, до соответствующего первичного спирта в стандартных условиях /особенно с использованием диборана, или реакции смешанного ангидрида с последующим восстановлением боргидридом натрия/. Соответствующий первичный спирт превращают стандартными способами, например, используя метансульфонилхлорид и триметиламин при 0oC в присутствии CH2Cl2 в такую отщепляемую группу, как Br, J или мезилат; причем эту отщепляемую группу замещают группой формулы /XVI/:
HS - Het, (XVI)
(где Het имеет указанные ранее значения для W = /9/), в результате чего после обработки в окислительных условиях получают соединения формулы: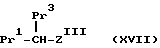
где ZIII представляет Z, как определено ранее, при условии, что W представляет -S-Het, -/S = O/-Het или -SO2-Het, а Pr1 и Pr3 представляют защищенные карбоксильные и -NH2 группы соответственно, как было определено ранее). Подходящие окислительные условия включают обработку окислительным агентом /особенно 3-хлорпербензойной кислоты/. Удаление защиты у -NH2 группы соединений формулы /XVII/ в стандартных условиях, как указано ранее, осуществляют с образованием соединения формулы /III/, где W представляет группу /9/, как определено ранее.
Следующей отличительной чертой настоящего изобретения является предложенный способ получения соединений формулы Ia, где X представляет N, а Y = 0, и их солей, который включает осуществление взаимодействия соединения формулы /IV/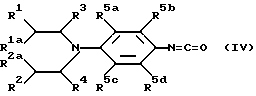
(где R1, R2, R1a, R2a, R3, R4 и R5a-d имеют указанные ранее значения) или их солей с соединением формулы /III/, как определено ранее, в результате чего получают соединение формулы Ia, и, при необходимости, превращение соединения формулы Ia в его соль. Реакцию можно вести, например, в органическом растворителе/предпочтительно, полярном апротонном, особенно CH2Cl2 или этилацетате/, предпочтительно, при комнатной температуре, предпочтительно, в течение 1 - 5 ч.
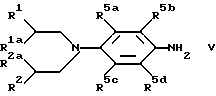
В соответствии со следующей отличительной чертой изобретения предложен способ получения соединений формулы /IV/ и их солей, который включает осуществление взаимодействия соединения формулы /V/, где R1, R2, R1a, R2a, R3, R4 и R5a-d имеют указанные ранее значения/ с соединением формулы L1 - /C=O/ - L2/ где L1 и L2 представляют отщепляемые группы/, в результате чего получают соединение формулы /IV/. Значения для L1 и L2 включают Cl, CCl3, имидазолил и арилокси /особенно фенокси/. Предпочтительные условия реакции включают проведение реакции в органическом растворителе /предпочтительно, полярном апротонном растворителе, особенно в этилацетате/ при 5 - 25oC в течение 15 мин.
В особенности со следующей отличительной особенностью настоящего изобретения предложен способ получения соединений формулы /Ia/, как определено ранее, где X представляет -CH2-, а Y = O, и их солей, который включает: осуществление взаимодействия соединения формулы /II/ или его соли, как указано ранее, при условии, что X представляет -CH2-, Y = O, а L желательно представляет пентафторфенокси, Cl, -O-/CO/-C1-6-алкил, предпочтительно, разветвленный алкил, особенно C1-4-алкил, а продукт соответствующего фенилуксусную кислоту содержащего анилинового производного иприта, прореагировашего с карбодиимидом /особенно с дициклогексилкарбодиимидом/; с соединением формулы /III/ как определено ранее, используя стандартные условия реакции /например, осуществляя реакцию в присутствии полярного апротонного растворителя, такого как диметилформамид, этилацетат или тетрагидрофуран, в течение 1 - 24 ч при 20 - 50oC/ в результате чего получают соединение формулы /Ia/, и при необходимости превращения соединения формулы /Ia/ в его соль. Соединения формулы /II/ можно получить, используя стандартные способы, из соответствующих фенилуксусных кислот, содержащих анилино-ипритные группы.
В соответствии со следующим аспектом изобретения предложен способ соединения формулы /Ia/, как определено ранее, где W представляет группу /8//, то есть, -CONH-SO2R11/, или его соли, который включает: осуществление взаимодействия соединения формулы /III/, как определено ранее, при условии, что W представляет только группу /8/; с соединением формулы /II/, как определено ранее, в условиях реакции известных per se, за счет чего получают соединение формулы /Ia/, где W представляет группу /8/, и, при желании, превращение соединения формулы /Ia/ в его соль. Предпочтительные условия реакции включают проведение реакции в присутствии органического растворителя /предпочтительно, полярного апротонного растворителя, особенно этилацетата или дихлорметана/, предпочтительно, при 5 - 50oC /особенно при комнатной температуре/, предпочтительно, в течение 1 - 5 ч.
Соединения формулы /III/ составляют следующий аспект настоящего изобретения, и эти соединения можно получить, удаляя защитную группу амина соединения формулы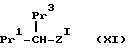
/где Pr1 представляет карбоксильную группу в защищенной форме, как определено ранее, Pr3 представляет -NH2 группу в защищенной форме, особенно бензилоксикарбонильное произвольное или фталимидо производное, а Z1 представляет Z, как определено ранее, при условии, что W представляет группу /8/ в условиях реакции, известных per se, в результате чего получают соединение формулы /III/, где W представляет группу /8/, и, при необходимости, превращение соединения формулы /III/ в его соль. Предпочтительные условия включают гидрирование в присутствии палладия на угле в органическом растворителе /предпочтительно, полярном апротонном, особенно в этилацетате или тетрагидрофуране/, предпочтительно, при комнатной температуре, и предпочтительно в течение 1 - 24 ч. Остальные предпочтительные условия реакции включают удаление защиты в присутствии HBr в уксусной кислоте, предпочтительно при комнатной температуре, предпочтительно, в течение 1 - 24 ч.
Соединения формулы /XI/ можно получить, осуществляя взаимодействие соединения формулы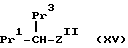
(где Pr1 и Pr3 имеют указанные ранее значения, а Z11 представляет Z, как указано ранее, при условии, что W представляет только COOH/; с соединением формулы R11SO2NH2, где R11 имеет указанные ранее значения в W = /8/, в результате чего получают соединение формулы /XI/ в стандартных реакционных условиях. Стандартные условия включают проведение реакции в инертном растворителе /особенно в дихлорметане/ в присутствии карбодиимида /особенно дициклогексилкарбодиимида/, и основания /особенно 4//N,N-диметиламино/пиперидина.
В соответствии со следующим аспектом настоящего изобретения предложен способ получения соединений формулы /I/, где W представляет следующие группы /определенные ранее/: /4/ /то есть, -SO2NHR9/, /5/, /то есть, -SO3R10/ и /6/ /то есть -PO3R10R10/ причем способ этот включает осуществление взаимодействия соединения формулы: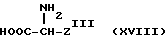
(где ZIII представляет Z, как определено ранее, при условии, что W представляет следующие группы /определенные ранее/:
/4/ /то есть = SO2NHR9, /5/ /то есть - SO3R10/ и /6/ /то есть -PO3R10R10//; с соединениями формулы /II/ /как определено ранее, в стандартных условиях; или с соединениями формулы /Y/ /как определено ранее/ в стандартных условиях; в результате чего получают соединение формулы /I/, где W представляет группу /4/, /5/ и /6/, и, при необходимости, превращение соединения формулы I в его соль. Стандартные условия включают проведение реакции с основанием /особенно с триэтиламином/ в присутствии органического растворителя /предпочтительно, апротонного, полярного, особенно дихлорметана/, предпочтительно при комнатной температуре. Соединения формулы /XVIII/ известны /доступны от Sigma Chemical Co/ или их можно получить из известных соединений стандартными способами.
В соответствии с еще одной отличительной особенностью настоящего изобретения предложен способ получения соединения формулы /Ia/ /определенной ранее/, где X представляет N или O, Y представляет O, R1 и R2 представляют Cl, Br, J или OSO2Me /особенно Cl/, R1a представляет H, R2a представляет H, R3= H, а R4 = H, и их солей, который включает осуществление взаимодействия соединений соединения формулы /XIX/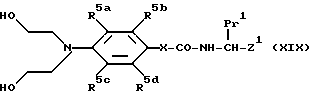
(где R5a-d, Pr1 и Z1 имеют указанные ранее значения, а X = O или NH),
либо с фосфорсодержащим галоидирующим агентом /особенно пентахлоридом фосфора/ или тионилхлоридом в присутствии органического растворителя/ предпочтительно неполярного апротонного, особенно CH2Cl предпочтительно, с нагреванием при кипении с обратным холодильником в течение 1 - 2 ч/ и особенно 90 мин/; или метилсульфонилхлоридом в присутствии органического растворителя /предпочтительно, полярного апротонного, особенно пиридина/, в результате чего получают соединение формулы /Ia/, где X = O или NH, Y представляет O, R1 и R2 представляет Br, I, OSO2Me или Cl, R1a = H, R2a = H, R3 = H и R4 = H, и, при необходимости превращение соединений формулы /Ia/ в их соли. Если используют метилсульфонилхлорид, гидроксильные группы в соединении формулы /XIX/ можно превратить либо в Cl либо / и OSO2Me в зависимости от используемой температуры реакции. Дихлорсоединения /то есть, R1 = R2 = Cl/ можно получить, проводя реакцию при 70oC в течение 15 мин; и соответствующее соединение, в котором R1 = Cl, R2 = OSO2Me можно получить, проводя реакцию при 50oC в течение 10 мин.
Если R1 и R2 представляют Br, а и R1 и R2 представляют J, метансульфонилангидрид предпочтительно, заменяют метансульфонил хлоридом, так как это снимает все проблемы конкуренции галоида в реакции. Соединение формулы XIX /0,002 М/ растворяют в CHCl3 /30 мл/. Триэтиламин /1,12 мл/ и метилсульфонилангидрид /0,008 М/ добавляют при комнатной температуре, полученную смесь перемешивают в течение 2 ч, а затем промывают водой. Полученный таким образом продукт сушат над сульфатом магния, фильтруют и выпаривают до масла. Это масло растворяют в сухом ДМФ и добавляют литийиодид /или бромид; 0,005/, перемешивают при 80oC в течение 2 ч, охлаждают, выливают в воду и экстрагируют эфиром. Полученный при этом продукт сушат над сульфатом магния, фильтруют, выпаривают досуха и очищают на хроматографической колонке с мгновенным испарением.
Соединения формулы /XIX/ можно получить при взаимодействии соединения формулы /XX/: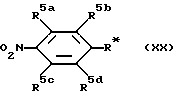
(где R5a-d имеет определенные ранее значения, а R* поедставляет -N = C = O или -O-CO-Z, где Z представляет отщепляемую группу, как определено ранее)
с соединением формулы /III/ /как определено ранее) до получения соединения формулы XXI: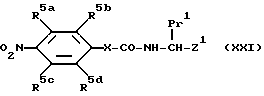
(где R5a-d, Pr1 и Z1 имеют указанные ранее значения, а X = O или NH/. Полученные таким образом соединения гидрируют /предпочтительно в присутствии палладия на угле/ до окончания соединения, соответствующего формуле /XXI/, но с -NH2 вместо -NO2.
Полученное таким образом соединение подвергают взаимодействию с этиленоксидом в водной кислоте /предпочтительно в смеси уксусная кислота /вода 1: 1/, предпочтительно в течение 1 - 2 дней, предпочтительно, при комнатной температуре, в результате чего получают соединение формулы /XIX/. Пример такого способа представлен на схеме 5.
Способ получения соединений формулы /I/, где W представляет группу тетразол-5-ила, включает осуществление взаимодействия соединения формулы XVIII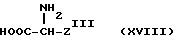
(где ZIII представляет Z как определено ранее, при условии, что W представляет группу тетразол-5-ила; с соединениями формулы /II/, как определено ранее, в стандартных условиях; и для получения соединения формулы I, где W представляет группу тетразол-5-ила, и, при необходимости, превращения соединения формулы I в их соли. Стандартные условия включают реакцию в полярном апротонном растворителе /особенно ДМФ/ в присутствии основания /предпочтительно, диметиламинопиридина, особенно триэтиламина/ в течение по крайней мере 2 ч/ предпочтительно 20 ч/ при температуре 20 - 50oC /особенно 25oC/.
Ин витро цитотоксическая эффективность пролекарств и лекарств
Ин витро цитотоксическую эффективность пролекарств, пролекарств плюс энзим и лекарств определяют в анализе на цитотоксичность, аналогичном описанному Shehan et al. /J. Natl. Cancer Just 82, 1107 - 1112, 1990/. Lovo клетки /ЕСАСС N 87060101/ разбавляют в ДМЕМ среде /содержащей 10% FCS, 1% глютамина и 0,2% гентамицина/, помещенной в микротитровальные пластины с 96 ячейками при плотности 2500 клеток/ячейку, и инкубируют в течение ночи при 37oC в 5% CO2. Различные концентрации пролекарств, соответствующие лекарства в качестве контроля или пролекарства плюс энзим /IU и CPG2 активность/ячейку - одна единица энзима это количество, необходимое для гидролиза 1 мкмоля метотрексата/мин/мл при 37oC/ добавляют в эти ячейки, и после 1 ч или 24 ч инкубирования клетки промывают, добавляют свежую среду и клетки инкубируют при 37oC в 5% CO2. Через 3 дня после добавления соединения в ячейки добавляют ТСА /16% конечная концентрация/, и оценивают количество клеточного протеина, прилипшего к пластинам, добавляя SRB краситель /Skehan et al./. Изменяют оптическую плотность на 540 нм, и выражают как процент ОД540 /оптическая плотность на 540 нм/ в контрольной ячейке, в которой соединение отсутствует. Эффективность выражают как концентрацию, необходимую для ингибирования роста клеток на 50% /IC50/. Само по себе пролекарство должно обычно обладать низкой активностью в тесте по сравнению с пролекарством в присутствии CPG2 /то есть, энзим CPG2 необходим для активации превращения пролекарства в лекарство/. Непосредственное добавление химически синтезированного лекарства/ не нуждающегося в CPG2 активации/ действует в анализе как контроль.
Используя описанную методику, проводят оценку представительных соединений, тестированных в изобретении, и оказывается, что они дают по крайней мере 10-кратное увеличение активности в присутствии CPG2 по сравнению с активностью в отсутствие CPG2 спустя 1 ч; тем самым демонстрируя и подтверждая эффективность использования соединений настоящего изобретения.
В соответствии с еще одним аспектом настоящего изобретения предложены фармацевтические композиции, содержащие в качестве пролекарственного ингредиента, по крайней мере одно соединение формулы I или физиологически приемлемую соль указанного соединения в смеси с фармацевтически приемлемым носителем или разбавителем. Желательно, чтобы композиция содержала эффективное количество пролекарства, которое нужно использовать с CPG /предпочтительно, CPG2/-опухоле-селективным конъюгатом антител, уже локализованным на опухоли.
В соответствии с еще одним аспектом изобретения предложены стерильные фармацевтические композиции для инъекций, содержащие в качестве пролекарственного ингредиента по крайней мере одно соединение формулы I или физиологически приемлемую соль указанного соединения в сочетании с фармацевтически приемлемым носителем или разбавителем. Композиция должна содержать эффективное количество этого пролекарства.
В соответствии с еще одним аспектом настоящего изобретения предложена двухкомпонентная система (причем каждый компонент должен использоваться в сочетании с другим), которая включает:
i/ первый компонент, который представляет собой антитело или его фрагмент, способный связывать данный антиген, причем это антитело или его фрагмент конъюгированы с CPG /предпочтительно, карбоксипептидазой G2/ энзимом, который способен превращать соединение формулы I или его физиологически приемлемую соль в цитотоксическое лекарство; и
ii/ второй компонент, то есть соединение формулы I или его фармацевтически приемлемую соль, превращаемую под действием CPG/ предпочтительно, карбоксипептидазы G2/ энзима в цитотоксическое лекарство.
Антитело или его фрагмент, предпочтительно, способны связываться со связанным с опухолью антигеном.
Конкретным антителом, способным связываться со связанным с опухолью антигеном, является мышиное моноклональное антитело A5B7. Антитело A5B7 связывается с человеческим карциноэмбрионным антигеном /CEA/ и наиболее подходит для обеспечения доставки лекарства к карциноме прямой кишки. A5B7 доступно от ДАКО Ltd. 16 Manor Courtyard, Hughenden Avenue, High Wycombe, Bucks HP13 5RE, England, United Kingdom. Фрагменты антител можно получить из целевого IgG антитела, такими обычными способами, как, например, F/ab'/2 фрагменты, как предложено Mariani M. et. al. /1991/, Molecular Immunology, 28, 69 - 77. Вообще, конъюгат антитело /или фрагмент антитела/ - энзим должен быть по крайней мере двухвалентным, то есть, иначе говоря, должен быть способен связывать по крайней мере 2 опухолевых антигена /которые могут быть одинаковым или различны/. Молекулы антител можно гуманизировать обычными способами, например "CDR прививкой" как раскрыто в EP 239400, или за счет прививки полностью вариабельных участков на постоянные участки организма человека, как раскрыто в патенте США 4816567. Гуманизированные антитела могут пригодиться для уменьшения иммуногенности антител /или фрагментов антител/. Гуманизированный вариант антитела A5B7 раскрыт в PCT WO92/1059.
Гибридома, которая продуцирует моноклональное антитело A5B7, депонирована в Европейской Коллекции Культур Клеток Животных, в разделе Биологическом, PHLS Центра Прикладной микробиологии и Исследований, Porton Down, Salisbury, Wiltshire SP4 OJG, United Kinhdom. Дата депонирования 14 июля 1993 г., регистрационный номер 93071411. Антитело A5B7 можно получить из депонированной гибридомы стандартными способами известными из Fenge C, Fraune E & Schuegerl K b "Production of Biologicals from Animal Cells in Culture" (Spier RE, Griffiths JR & Meignier B, eds) Butterworth - Heinemann, 1991, 262 - 265 and Anderson BL & Gruenberg ML b "Commercial Production of Monoclonal Antibodies" (Seaner S. , ed), Marcel Dekker, 1987, 175 - 195. Эти клетки могут потребовать время от времени реклонирования за счет ограниченного разбавления для поддержания высоких уровней продуцирования антител.
Другие антитела, применимые в АДЕРТ, описаны в следующих источниках. Антитела BW 431/26 описаны в Haisma, H.J. et al., Cancer Immunal. Immunother. , 34: 343 - 348 /1992/. антитела L6, 96, 5, и IF5 описаны в Европейском патенте 302473. Антитело 18, 88 описано в международной патентной заявке WO90/07929. Антитело B72,3 описано в европейском патенте N 292745. Антитело СЕМ231 описано в европейском патенте N 382411. Антитела HMFG-1 и HMFG-11 /Unipath Ltd. Basingstoke, Hants, United Kingdom/ реагируют с муцино-подобными гликопротеиновыми молекулами на мембранах глобул молочного жира, и могут быть использованы для того чтобы обеспечить доставку к раковым образованиям груди и яичников. Антитело SM3 /Chemicon Internstional Ltd. London, United Kindom/ реагирует с ядерным протеином муцина, и может быть использовано для того чтобы обеспечить доставку к раковым образованиям груди и яичников. Антитела 85A12 /Unipath Ltd.Basingstoke, Hants, United Kingdom/ реагируют с опухолевым антигеном CEA. Антитело PR4D1 /Serotec, Oxford, United Kingdom/ реагирует с антигеном, связанным с раком толстой кишки. Антитело E29 /Дако Ltd, High Wycomne, United Kingdom/ реагирует с антигеном эпителиальных мембран. Антитело C242 можно получить из CANAG Diagnostics Gothenberg, Sweden.
Обычно антитела, пригодные для использования в АДЕРТ, плохо интернализуются опухолевыми клетками, которые они распознают. Это позволяет доставленному на место пролекарство-активирующему энзиму оставаться на поверхности клеток и за счет этого создавать активное лекарство на опухоли за счет циркулирующего пролекарства. Интернализацию антител можно оценить известными способами, например, по способу Jafrezou et al., Cancer Research 52: 1352 /1992/ и Press et al., Cabcer Research, 48:2249 /1988/.
Крупномасштабное выделение CPG2 из Pseudomonas RS-16 описано Sherwood et al. /1985/, Eur, J. Biochem 148, 447 - 453. Получение F/ab1/2 и IgG антител, соединенных с CPG энзимом, можно осуществлять известными способами, и это описано, например, в PCT WO 89/10140. CPG можно получить из центра прикладной микробиологии и исследований Porton Down, Salisbury, Wiltshire SP4 OJG, United Kingdom. CPG можно получить рекомбинантными методиками. Нуклеотидная кодирующая последовательность для CPG2 была опубликована Minton, N.P. et al. , Gene 31 /1984/, 31-38. Сообщалось об экспрессии этой кодирующей последовательности в E. coli /Chamber. S. P. et al., Appl. Microbiol. Biotechnol. /1988/, 29, 572 - 578/ и в Saccharomyces cerevisiae (Clareke, L.E. et al., J. Gen Miceoniol, /1985/ 131, 897 - 904/. Синтез полного гена описан M.Edwards в Am. Biotech. Lab. /1987/, 5, 38 - 44. Об экспрессии гетерологичных протеинов в E.Coli сообщается F.A.O. Marston в ДНК Cloning Vol. 111, Practical Approach Series. JRL Press (Editor Д.М. Glover). 1987, 69 - 88. Об экспрессии протеинов в дрожжи сообщается в Methods in Enzymology, Volume 194, Academic Press 1991, Edited by C.Guthrie and G.R.Flink.
CPG энзим доступен от Sigma Chemical Company, Fancy, Poole, Dorset, U.K. , CPG энзим описан в: Coldman, P. and Levy, C.C., PNAS США, 58:1299 - 1306 /1967/ и в Levy, C. C. and Colman P., J. Biol. Chem., 242: 2933 - 2938 /1967/. Энзим карбоксипептидаза G3 описан Jasuda, N. et al., Biosci. Biotech. Biochem, 56: 1536 - 1540 /1992/. Энзим карбоксипептидаза G2 описан в европейском патенте 121 352.
В еще одном аспекте настоящего изобретения предложен способ доставки цитотоксического лекарства в определенное место, который включает введение пациенту первого компонента, причем этот первый компонент содержит антитело или его фрагмент, способные связываться с определенным антигеном, причем это антитело или его фрагмент конъюгированы с CPG энзимом /предпочтительно, карбоксипептидазой G2/, способным превращать соединение формулы I или его физиологически приемлемую соль в цитотоксическое лекарство; с последующим введением пациенту второго компонента, который содержит соединение формулы I или его физиологически приемлемую соль, превращаемую под действием CPG энзима /предпочтительно, карбоксипептидазы G2/ в цитотоксическое лекарство.
Сайтом, в который должно быть доставлено цитотоксическое лекарство, предпочтительно, являются опухолевые клетки, обычно присутствующие у млекопитающих, имеющих опухоли, например у человека.
Когда указанный первый компонент вводят пациенту, у которого имеется опухоль, антитело или фрагмент антитела направляет конъюгат к участку опухоли и связывает конъюгат с опухолевыми клетками.
После того, как несвязанный конъюгат практически удаляется из организма пациента, например, по истечении соответствующего промежутка времени после ускорения выведения, например, по способу Br. J. Canaer /1990/, 61, 659 - 662, пациенту можно вводить второй компонент. Весьма желательно, чтобы практически весь несвязанный конъюгат был выведен из организма хозяина до введения второго компонента, так как в противном случае цитотоксическое лекарство может образоваться не только в опухолевых участках, что может привести к общей токсичности для пациента, а не сайт-специфической токсичности.
Соединения настоящего изобретения можно использовать в таких композициях, как таблетки, капсулы или эликсиры для орального приема, суппозитории для ректального введения, стерильные растворы или суспензии для парэнтерального или внутримышечного введения и т.п.
Соединения настоящего изобретения можно вводить пациентам /животным или человеку/ при необходимости такого лечения в дозах, которые обеспечат оптимальную терапевтическую эффективность. Хотя доза обычно меняется в зависимости от пациента и характера и тяжести заболевания, специального питания, сопутствующих медикаментов и других факторов, которые следует учитывать специалистам, доза обычно составляет от около 1 до 150 мг на кг веса пациента в день, причем ее можно вводить в виде единичной дозы или множества доз. Предпочтительный интервал доз составляет от около 10 до 75 мг/кг веса пациента в день; более предпочтительно от около 10 до 40 мг/кг веса пациента в день.
Естественно, эти интервалы доз можно установить на основании единичной дозы, необходимой для того, чтобы обеспечить разделенные дневные дозы, и как указано ранее, эта доза будет меняться в зависимости от характера и тяжести заболевания, веса пациента, специальной диеты и других факторов.
Обычно эти дозы приготавливают в виде фармацевтических композиций, что будет описано далее.
Около 50 - 500 мг соединения или смеси соединений формулы I или его фармацевтически приемлемой соли, смешивают с физиологически приемлемым носителем, наполнителем, эксципиентом, связующим, консервантом, стабилизатором, вкусовой добавкой и т.д. в единичной дозовой форме, как это принято в фармацевтической практике. Количество активного вещества в этих композициях или препаратах таково, что достигается подходящая доза в указанном интервале значений.
Иллюстративными адъювантами, которые можно включать в таблетки, капсулы и т. п. являются следующие: такие связующие, как смола трагаканта, акации, кукурузный крахмал или желатин; такие эксципиенты, как микрокристаллическая целлюлоза; такие разрыхляющие агенты, как кукурузный крахмал, прежелатинизированный крахмал, альгининовая кислота и т. п.; такие скользящие, как стеарат магния; подслащивающие агенты, такие, как сахароза, лактоза или сахарин; такие вкусовые агенты, как мята перечная, винтергреновое /гаультериевое/ или вишневое масло. Если единичную дозу приготавливают в виде капсулы, она может содержать, помимо перечисленных материалов, такой жидкий носитель, как жирное масло. Могут присутствовать другие материалы в качестве покрытий, или агентов для другого типа модификации физических форм единичной дозы. Так, например, таблетки могут быть покрыты щеллаком, сахаром или тем и другим вместе. Сироп или эликсир может содержать активное соединение, сахарозу в качестве подслащивающего агента, металл и пропил-парабены в качестве консервантов, краситель и вкусовой агент, например вишневый или апельсиновый.
Как было указано ранее, соединения формулы I и их физиологически приемлемые соли, а также коньюганты, указанные ранее, можно вводить обычными способами, включая внутривенный, внутрибрюшинный, оральный, внутрилимфатический или непосредственно в опухоль. Внутривенное введение предпочтительно, например внутривенное вливание.
Стерильные композиции для инъекций или инфузий можно приготовить в соответствии с обычной фармацевтической практикой, растворяя или суспендируя активное вещество в таком носителе, как вода для инъекций, такое природное масло, как кунжутное, кокосовое, арахисовое, хлопковое и т.д., или в таких синтетических жировых носителях, как этилолеат или т.п. При необходимости можно включать буферные агенты, консерванты, антиоксиданты и т.п. Предпочтительные стерильные композиции для инъекций или инфузий, полученные в соответствии с общепринятой фармацевтической практикой, включают растворение пролекарства в таком носителе, как вода, необязательно содержащая соли, сахар /особенно декстран/, буферирующие агенты и/или сорастворители /особенно полиэтиленгликоль, пропиленгликоль или диметилизосорбид/.
В одном варианте настоящего изобретения предложено соединение формулы I в форме свободной кислоты, причем эту свободную кислоту можно приготовить в форме для нагревательного введения непосредственно перед введением пациенту. Так, например, соединение формулы I в форме свободной кислоты можно смешать с буфером, за счет чего его превращают в физиологически приемлемую соль непосредственно перед введением.
Предпочтительная единичная дозовая форма соединения настоящего изобретения содержит соединение формулы I или его фармацевтически приемлемую соль, в стерильной, высушенной замораживанием форме для восстановления, в инъектируемый /инфузируемый раствор в ампуле. Ампулы, предпочтительно, содержат от 500 мг до 2 г /особенно 1 г/ указанного соединения.
Нижеследующие примеры иллюстрируют получение соединений формулы I и их введение в фармацевтические композиции, причем эти примеры не следует рассматривать как лимитирующие изобретение. В этих примерах, если нет других указаний, осуществляют следующие стандартные процедуры.
i/ Все процессы выпаривания проводят в роторном испарителе в вакууме, а процедуры обработки осуществляют после удаления твердых остатков за счет фильтрования;
ii/ все операции проводят при комнатной температуре, то есть в интервале 18 - 25oC в атмосфере инертного газа, например, в аргоне;
iii/ хроматографическую обработку/ с мгновенным испарением/ осуществляют на Merck Kieselgel двуокиси кремния /Агт. 9385/, полученной от E.Merck, Darmstadt, W.Germfny;
iv/ выходы приведены только в иллюстративных целях, и они необязательно представляют максимально достижимые;
v/ конечные продукты формулы I имеют остаточную чистоту по данным микроанализов, а их структура подтверждена с помощью ЯМР и масс-спектрометрии;
vi/промежуточные соединения охарактеризованы не полностью, а их чистоту оценивают по данным тонкослойной хроматографии, ИК и или ЯМР;
vii/ температуры плавления даны без поправок, и их определяют, используя прибор с масляной баней; температуры плавления конечных продуктов формулы I определяют после кристаллизации из таких обычных органических растворителей, как этанол, метанол, ацетон, эфир или гексан, или в их смесях.
Пример 1
N-/4-/N,N-бис/2-хлорэтил/амино/-феноксикарбонил/-L -глутаминовая кислота
Раствор дибензил N-/4-//N, N-бис/2-хлорэтил/амино/феноксикарбонил/-L-глутамата /см./2/в схеме 1//6 г/ в 100 мл этилацетата гидрируют над 30% палладием на угле /0.6 г/ в течение 2 ч. Когда поглощается теоретическое количество водорода, катализатор удаляют фильтрованием, а полученный фильтрат выпаривают досуха. Остаток в горячий эфир /25 мл/, и добавляют гексан до помутнения. После охлаждения получают N-/4-/N,N-бис/2-хлорэтил/амино/феноксикарбонил/- L-глутаминовой кислоты /см./3/ в схеме 1/ в виде твердого кристаллического вещества 3,4 г /79% выход/. Т.пл. 87 - 89oC.
ЯМР 7,0 /д, 2H/, 6,6 /д, 2H/, 6,2 /д, 1H/, 4,4 /м, 1H/, 3,5- 3,7 /м, 8H/, 2,0-2,6 /м, 4H/.
Элементный анализ:
Рассчитано: C 47,2; H 4,95; N 6,88
Найдено: C 47,5; H 5,1; N 6,7
Указанное в заглавии соединение получают также в другой полиморфной форме, не растворяющейся в эфире, с т. плавления 128 - 130oC.
Исходный материал дибензил-N-4-/N,N-бис/2-хлорэтил/амино/феноксикарбонил/-L-глутамат получают следующим образом.
Раствор 1,43 г 4-нитрофенил-хлорформата в 15 мл хлороформа добавляют к смеси 4-/бис/2-хлорэтил/амино/фенолгидрохлорида /Biochem. Pharmacol. 17 893 /1968// 1,93 г/, триэтиламина /2 мл/ и хлороформа /20 мл/. После 2 ч при комнатной температуре смесь выпаривают досуха, а остаток обрабатывают на силикагеле Merck Агт 9385. После элюирования смесью гексан/ этилацетат и перекристаллизации из смеси бензол: петролейный эфир /3:1/ получают O-/4-/N, N-бис/2-хлорэтил/амино/фенил/-O'-/4-нитрофенил/-карбонил /см./1/ на схеме 1/ в виде желтоватого твердого продукта /1,4 г/ /50%/ Т. пл. = 66 - 67oC.
3.8 мл триэтиламина добавляют к 5,5 г полученного продукта в 40 мл хлороформа, с последующим добавлением тозилата дибензилового сложного эфира, L -глутаминовой кислоты /13,75 г/. Полученную смесь перемешивают и нагревают при 60oC в течение 4 ч, и выпаривают досуха. Остаток обрабатывают хроматографически на силикагеле /Merck Агт 9385/ и элюируют 3% этилацетатов в хлороформе до получения требуемого исходного материала дибензил-2-//бис-/2-хлорэтил/амино/феноксикарбонил/глутамата /см. /2/на схеме 1/ /6,2 г/ /выход 77%/ в виде белого твердого вещества, Т. пл. 85-87oC.
Пример 2
N-/4-/N,N-бис/2-хлорэтил/-амино/-3-метилфеноксикарбонил/-L- глутаминовая кислота
Повторяют способ примера 1, используя дибензил-N-/4-/N,N-бис- /2-хлорэтил/амино/-3-метил-феноксикарбонил/-L-глутамат вместо дибензил-N-/4-/N, N-бис/-2-хлорэтил/-амино/феноксикарбонил/-L -глутамата до получения N-/4-/N, N-бис/2-хлорэтил/амино/-3- метилфеноксикарбонил/-L-глутаминовой кислоты в виде твердого вещества белого цвета, T пл. 160 - 162oC.
ЯМР: 7,3 /д, 2H/; 6,7 /д, 2H/, 6,0 /д, 1H/, 4,2 /м, 1H/, 3,7 /м, 8H/, 2,3 - 2,0 /м, 4H/.
Элементный анализ:
Рассчитано: C 47,3; H 5,2; N 10,3; Cl 17,5
Найдено: C 46,8; H 5,2; N 10,3; Cl 18,0
Дибензил N-/4-/N, N-/2-хлорэтил/амино/-3-метилфеноксикарбонил/- L-глутамат, используемый в качестве исходного материала, получают способом, аналогичным описываемому в примере 1, используя 4-/N,N-бис/2-хлорэтил/амино/-3-метилфенол вместо 4-/N,N-бис(2-хлорэтил)аминофенола.
4-/N, N-бис/2-хлорэтил/амино/-3-метилфенол получают следующим образом /см. схему 2/.
/a/ 40 г этиленоксида барботируют в раствор 4-амино-м-крезола /12,3 г/ в смеси уксусная кислота/вода /1:1/ /500 мл/. Полученной смеси дают остыть до комнатной температуры и оставляют на 48 ч, а затем выпаривают досуха. Остаток обрабатывают хроматографически на силикагеле, элюируя этилацетатом до получения 4-/N, N-бис/2-гидроксиэтил/амино/-3-метилфенола в виде масла /6,2 г/. ЯМР: см. таблицу 2.
/b/ 4,2 г бензилбромида добавляют к смеси полученного таким образом продукта /6 г/, гидроксида калия /1,6 г/ и этанола /40 мл/. Полученную смесь перемешивают и кипятят с обратным холодильником в течение 2 ч, охлаждают и концентрируют выпариванием. Остаток выливают в 100 мл воды, дважды экстрагируют этилацетатом, сушат и выпаривают до получения 2,2'-/4-бензилокси-2-толуидино/диэтанола в виде твердого продукта /7,2 г/, T. пл. 70 - 72oC. ЯМР, см. таблицу 3.
/c/ Пентахлорид фосфора /11,4 г/ добавляют порциями к полученному продукту /7 г/ в 50 мл хлороформа при 10 - 20oC. Затем полученную смесь нагревают при кипячении с обратным холодильником в течение 90 мин, охлаждают и выливают в воду. Органическую фазу выделяют и промывают водным бикарбонатом натрия, водой и выпаривают досуха. Остаток обрабатывают хроматографически на силикагеле. После элюирования смесью гексан/этилацетат /2:1/ получают в виде масла 4-бензилокси-3-метил-N,N-бис/2-хлорэтил/-анилин /2,2 г/. ЯМР: см. таблицу 4.
Вместо описанной реакции можно использовать альтернативную реакцию. 2,5 мл метансульфонилхлорида добавляют при 0 - 5oC к раствору полученного на стадии /b/ продукта /2,6 г/ на 8 мл пиридина. Затем полученную смесь нагревают при 70oC в течение 15 мин, охлаждают и выливают в разбавленный раствор лимонной кислоты /100 мл/. Полученную смесь экстрагируют дважды этилацетатом, сушат и выпаривают до получения 4-бензилокси-3-метил-N,N-бис/2-хлорэтил/- анилина в виде масла /2,6 г, 84%/ ЯМР: см. таблицу 4.
d/ Эфирный HCl /насыщенный/ добавляют к полученному продукту /2 г/ в 25 мл этанола до полного растворения. Добавляют 300 мг 30% палладия на угле в качестве катализатора, и полученную смесь перемешивают в атмосфере водорода до тех пор, пока не поглощается соответствующее количество водорода. Катализатор удаляют фильтрованием, и полученный фильтрат выпаривают до получения 4-/N, N-бис/2-хлорэтил/амино-3-метилфенолгидрохлорида в виде твердого вещества /950 мг/ T. пл. = 164 - 167oC.
Пример 3
N-/4-/N, N-бис/2-хлорэтил/амино/фенилкарбамоил/-L-глутаминовой кислоты гидрохлорид
Насыщенный раствор хлористого водорода в эфире /120 мл/ добавляют к раствору ди-трет-бутил-N-/4-/N, N-бис/2-хлорэтил/-амино/- фенилкарбамоил/-L-глутамата /4,4 г/ в 20 мл этилацетата. После часа при комнатной температуре полученную смесь выпаривают до твердого вещества. Это вещество тщательно растирают с эфиром до получения N-/4-/N,N-бис/2-хлорэтил/амино/фенилкарбамоил/-L-глутамовой кислоты гидрохлорида /3,5 г/ в виде серого твердого вещества. T/ пл. = 148 - 150oC /см. схему 3/.
ЯМР 7,2 /д, 2H/, 6,7 /д, 2H/, 4,2 /н, 1H/, 3,7 /м, 8H/, 2,4 - 1,8 /м, 4H/,
Элементный анализ:
Рассчитано: C 46,7; H 6,8; N 7,0
Найдено: C 47,1; H 6,5; N 7,5
Исходный материал, ди-трет-бутил-N-/4-/N, N-бис//2-хлорэтил/- амино/фенилкарбамоил/-L-глутамат получают следующим образом.
Смесь p-фторнитробензола /14,1 г/ и диэтаноламина /30 мл/ перемешивают и нагревают при 130oC в течение 2 ч. Полученную смесь охлаждают до около 60oC и выливают в 1 л воды, содержащей 10 мл 48% раствора каустика. После охлаждения до 15oC осадок отфильтровывают и сушат до получения 2,2'-/4-нитро- анилинодиэтанола /20,7 г/ /92%/. T. пл. 102 - 104oC.
Добавляют 30 мл тионилхлорида при охлаждении, к смеси полученного продукта /20 г/, 200 мл дихлорметана и 7 мл пиридина. После добавления эту смесь нагревают и кипятят с обратным холодильником в течение 1 ч. После охлаждения смесь разбавляют равным объемом дихлорметана, и осторожно дважды промывают водой, сушат и выпаривают до получения /N,N-бис-/2-хлорэтил//- 4-нитроанилина в виде твердого вещества /21 г/ T. пл. 81,3oC.
К раствору полученного продукта /0,53 г/ в свежеперегнанном тетрагидрофуране /20 мл/, добавляют 30% палладий на угле в качестве катализатора /100 мг/. Полученную смесь перемешивают в атмосфере водорода в течение 2 ч, затем катализатор удаляют фильтрованием. Полученный фильтрат выпаривают досуха, а остаток снова растворяют в 20 мл эфира, и в некотором избытке добавляют раствор газообразного хлористого водорода в эфире. В результате получают 4-/N, N-бис/2- хлорэтил/амино/анилинийхлорид в виде твердого продукта, который сушат. Выход 0,5 г, T. пл. 238 - 240oC /с разложением/.
К раствору трифосгена /Aldrich /200 мг/ в 10 мл хлороформа при 0 - 5oC добавляют полученный таким образом продукт /539 мг/, а затем 0,83 мл триэтиламина. Спустя 15 мин при комнатной температуре добавляют раствор ди-трет-бутилового сложного эфира L-глутаминовой кислоты /0,31 г/ в 5 мл хлороформа. Полученную смесь оставляют при комнатной температуре на 18 ч, промывают водой, сушат и выпаривают досуха. Остаток обрабатывают хроматографически на силикагеле Merck и элюируют смесью гексан-этилацетат /3:1/ до получения целевого исходного материала ди-трет-бутил-4/бис-2-/хлорэтиламино// фенилкарбамоил-L-глутамата /0,44 г/ в виде масла /см. схему 3/.
ЯМР: δ 7,2/д/ и 6,65 /дд, 4H/, ароматика, 4,1 /м, 1H/, 3,66 /с, 8H/ 1,7 - 2,2 /м, 4H/, 1,38 /с, 9H/ и 1,42 /с, 9H/.
Пример 4
N-/4-/N,N-бис/2-хлорэтил/амино/фенилкарбамоил/-L-глутаминовая кислота
Далее описывается способ получения N-/4-/N, N-бис/2-хлорэтил/ -амино/фенилкарбамоил/-L-глутаминовой кислоты альтернативный способу примера 3. Исходный материал дибензил-N /4-/N,N-бис-/2-хлорэтил/амино /фенилкарбамоил/-L-глутамат получают аналогично способу соответствующей стадии примера 5.
Раствор дибензил-N-/4-/N, N-бис-/2-хлорэтил/амино/фенил/ карбамоил/-L-глутамат /1,138 г/ в 15 мл ДМФ гидрируют над 10% Pd/C в течение 16 ч. После фильтрования и выпаривания в вакууме остаток растворяют в CHCl3 /20 мл//. Через 18 ч кристаллический остаток отфильтровывают и сушат в вакууме до получения N-/4-/N,N-бис- /2-хлорэтил/амино/фенилкарбамоил/-L-глутаминовой кислоты. Выход 93% /730 мг/. После перекристаллизации из ацетона /CHCl3 образуются микроскопические палочки, T. пл. 116 - 118oC.
ЯМР /CD3COCD3/: δ : 8,0 /с, 1H/, 7,2 /д, 2H/, 6,6 /д, 2H/, 6,2 /д, 2H/, NH/; 4,4 /м, 1H/, 3,6 /м, 8H/, 2,5 - 1,9 /м, 4H/.
Пример 5
N-/4-/N, N-бис/2-хлорэтил/-амино/-3-фторфенилкарбамоил/-L- глутаминовая кислота
К раствору дибензил-N-/4-/N, N-бис/2-хлорэтил/амино/-3-фторфенилкарбамоил/ -L-глутамата /0.4 г/ в 10 мл этилацетата добавляют 30% палладий на угле /50% влажности/ /160 мг/, и полученную смесь перемешивают в атмосфере водорода в течение 1 ч. После отфильтровывания катализатора фильтрат выпаривают досуха. После тщательного растирания маслянистого остатка со смесью этилацетата /гексан получают N-/4/-N,N-бис/2-хлорэтил/амино/-3-фторфенилкарбамоил-L-глутаминую кислоту в виде белого порошка /210 мг/, т.пл. 111 - 114oC. Исходный материал, дибензил-N-4-/N,N-бис/2-хлорэтил/амино/-3-фторфенилкарамоил/- L-глутаминовую кислоту получают следующим образом.
Суспензию 4-/N,N-бис-/2-хлорэтил/амино/-3-фторанилинийоксалата /3,5 г/ в безводном этилацетате /200 мл/ и карбоната калия /5,5 г/ охлаждают в атмосфере аргона до 5oC. К этой смеси добавляют раствор фосгена в толуоле /1,9 М, 5,5 мл/. После добавления смесь перемешивают при комнатной температуре в течение 10 мин: фильтруют, и полученный фильтрат сушат над сульфатом магния. Полученный сухой фильтрат, добавляют сразу к смеси дибензилглутамата р-толуолсульфоната /5 г/, карбоната калия /2 г/ и этилацетата /100 мл/. Добавляют 2 мл триэтиламина, и полученную смесь перемешивают в течение 20 мин при комнатной температуре. Полученную смесь фильтруют, а полученный фильтрат выпаривают досуха. Остаток обрабатывают хроматографически на силикагеле, элюируя смесью этилацетат/гексан /1:2/ до получения целевого исходного материала в виде масла, которое кристаллизуется. Выход 5,5 г. Т. пл. 81 - 84oC. 4-/N, N-бис/2-хорметил/амино/-3-фторанилинийоксалат получают по способу примера 3, за исключением того, что в качестве исходного материала используют 3,4-дифторнитробензол вместо р-фторнитробензола до получения 4-N/N,N-бис-/2-гидроксиэтил/амино/-фторнитробензола, Т.пл. 99 - 101oC.
Полученный таким образом продукт обрабатывают тионилхлорилом по способу примера 3 до получения 4-/N,N-бис/2-хлорэтил/-3-фторнитробензоата. Т.пл. 66-68oC.
Полученный таким образом продукт гидрируют по способу примера 3, используя в качестве растворителя этилацетат. Полученную смесь фильтруют, а полученный фильтрат выпаривают до малого объема и снова растворяют в эфире. В избытке добавляют насыщенный раствор щавелевой кислоты в эфире, и собирают целевой продукт 4-/N,N-бис/2-хлорметил/амино/-3-фторанилинийоксалат. Т.пл. 146 - 148oC.
Пример 6
N-/4-/N,N-бис/2-хлорэтил/амино/-3-хлорфенилкарбамоил/-L-глутаминовая кислота
Раствор дибензил-N-//4-/N, N-бис/2-хлорэтил/амино/-3-хлорфенилкарбамоил/-L-глутамата /350 мг/ в 30 мл этилацетата, содержащий 70 мг 30% палладия на угле /50% влажности/, перемешивают в атмосфере водорода в течение 1 ч. После отфильтровывания катализатора полученный фильтрат выпаривают досуха, а остаток тщательно растирают со смесью эфир/этилацетат до получения N-/4-/N, N-бис-/2-хлорэтил/амино/-3-хлорфенилкарбамоил/-L- глутаминовой кислоты в виде масла.
ЯМР: δ 8,7 /с, 1H/, 7,6 /с, 1H/, 7,1 - 7,4 /м, 2H/, 6,5 /д, 1H/, 4,2 / м, 1H/, 3,3-3,6 /м, 8H/, 1,7 - 2,4 /м, 4H/.
Исходный материал, дибензил-4-/N,N-бис-/2-хлорэтил/амино-3-хлоркарбамоил/-L-глутамат получают по способу примера 3, за исключением того, что в качестве исходного материала используют 4-фтор-3-хлорбензол вместо 4-фторнитробензола для получения 4-/N, N- бис/2-гидроксиэтиламино//3-хлорнитробензола в виде масла оранжевого цвета.
ЯМР: δ 8,0 - 8,2 /м, 2H/, 7,3 /1H/, 4,7 /т, 2H/, 3,5 /м, 8H/.
Полученный таким образом продукт обрабатывают тионилхлоридом по способу примера 3 до получения 4-/N,N-бис-3-хлорэтиламино/-3-хлорнитробензола в виде масла.
ЯМР, δ : CDCl3/8,3 /д, 1H/, 8,1 /кв,1H/, 7,25 /д, 1H/, 3,8 /т, 4H/, 3,6 /т, 4H/.
Полученный продукт гидрируют, используя в качестве растворителя этилацетат, по способу примера 3. Катализатор отфильтровывают, а полученный фильтрат выпаривают до малого объема, снова растворяют в эфире и в избытке добавляют насыщенный эфирный раствор щавелевой кислоты, полученный оксалат отфильтровывают. Т.пл. 118 - 121oC.
Полученный продукт превращают в дибензол-N-/4-/N, N-бис/2-хлорэтил/амино/-2-хлорфенилкарбамоил/- L-глутаминовую кислоту по способу примера 5. Получают масло:
ЯМР, δ : 7,1 - 7,4 /м, 13H/, 5,1 /с, 2H/, 5,0 /с, 2H/, 4,6 /м, 1H/, 3,5 /с, 8H/, 2,0-2,6 /м, 4H/.
Пример 7
Типичные фармацевтические композиции, содержащие соединение настоящего изобретения
А.Капсулы с сухим наполнителем, содержащие по 100 мг пролекарства на капсулу
Ингредиент - Кол-во мг на капсулу
Соединение - 100
Лактоза - 149
Стеарат магния - 1
Капсула /размер N 1/ - 250
Соединение можно измельчить в порошок до N 60, а затем стеарат магния и лактозу просеять через фильтровальную ткань N 60 на этот порошок. Объединенные ингредиенты можно затем перемешивать в течение около 10 мин, а затем заполнить ими сухие желатиновые капсулы N 1.
B. Таблетки
Обычно таблетки содержит 100 мг соединения, 82 мг прежелатинированного крахмала /82 мг/, 82 мг микрокристаллической целлюлозы и 1 мг стеарата магния.
C. Суппозитории
Обычные композиции суппозиториев для ректального введения могут содержать 50 мг соединения, 0,25 - 0,5 мг динатрийкальций эдетата и 775 - 160 мг полиэтиленгликоля. Другие композиции суппозиториев можно получить, заменяя, например, динатрийкальций эдетат бутилированным гидрокситолуолом /0,04 - 0,08 мг/, а полиэтиленгликоль - гидрированным растительным маслом /675 - 1400 мг/, например Suppocire L, Wocobee FS, Wecobee M, Witepsols и т.п.
Д. Инъекции
Типичные компенсации для инъекций должны содержать 500 мг соединения, 0,05 мл бензилового спирта и 0,15 М бикарбоната натрия для инъекций /0,5 мл/.
Пример 8 (см. схему 4)
N-/4-/N-/2-хлорэтил/-N-/2-мезилоксиэтил/амино/феноксикарбонил/-L- глутаминовая кислота
Ди-трет-бутиловый сложный эфир N-/4-//N-/2-хлорэтил/, N-/2-мезилоксиэтил/амино/фенилоксикарбонил/-L-глутаминовой кислоты /6, где R1 = Cl, а R2 = OSO2Me/ /100 мг/ суспендируют в трифторуксусуной кислоте /TFA/ /2,2 мл/, и перемешивают в течение 40 мин при комнатной температуре. TFA удаляют при пониженном давлении; оставшееся масло разбавляют 1 мл этилацетата, и выпаривают до получения N-/4-/N-/2-хлорэтил/-N-/2-мезилоксиэтил/-амино/феноксикарбонил/- L-глутаминовой кислоты -1,02 TFA -0,16 EtOAc /9, где R1 = Cl, а R2 = OSO2Me/, 90 мг выход 95%.
ЯМР, δ : -CH2-CH2-OSO2Me/-CH2-CH2Cl
3,15 /с, 3H/, 3,70 /м, 6H/
остальное 4,30 /т, 1H/, 4,49 /т, 1H/,
Ароматика 6,75 /д, 2H/, 6,92 /д, 2H/, 1,8 - 2,3 /м, 4H/, 4,02 /м, 1H/, 7,90 /д, 1H/.
Исходный материал, 4-//N-/2-хлорэтил/, N-/2-мезилоксиэтил//-амино/фенилкарбонил-L-глутаминовой кислоты ди-трет-бутиловый сложный эфир получают следующим образом.
Раствор L-глутаминовой кислоты ди-трет-бутилового сложного эфира, гидрохлорида /4,26 г/ и 4 мл триэтиламина в 30 мл сухого хлороформа перемешивают с охлаждением раствором 4-нитрофенилхлороформа /2,92 г// доступен от Aldrich/ в течение 5 мин. Спустя 5 ч при комнатной температуре растворитель выпаривают, а остаток снова растворяют в этилацетате /70 мл/, фильтруют и выпаривают досуха. Остаток обрабатывают хроматографически на силикагеле, элюируя хлороформом до получения ди-трет-бутилового сложного эфира 4-нитрофенилоксикарбонил-L-глутаминовой кислоты /2/ в виде масла, 5,02 г /82%/
ЯМР: 7,33 /д, 2H/, 8,24 /д, 2H/, 1,46 /с, 9H/, 1,50 /с, 9H/, 2,0-2,4 /м, 4H/, 4,32 /м, 1H/, 5,90 /д, 1H/.
Раствор полученного таким образом продукта /2/ /5,01 г/ в уксусной кислоте /30 мл/ гидрируют над 10% палладием на угле в течение 3 дней. После фильтрования раствор охлаждают, и добавляют 5 мл этиленоксида, и оставляют при комнатной температуре на 22 ч. Растворитель выпаривают, а остаток разделяют между этилацетатом и водой. Органическую фазу выделяют, промывают водой, сушат над сульфатом натрия и выпаривают досуха. Остаток хроматографически обрабатывают на силикагеле; элюируют этилацетатом в хлороформе /2:1/ до получения ди-трет-бутилового сложного эфира 4-/бис/2-гидроксиэтил/амино/фенилоксикарбонил-L-глутаминовой кислоты /4/ /3,93 г/, 69%. Т.пл. 91 - 93oC.
Раствор полученного продукта /4/ /0,86/ в 3 мл пиридина перемешивают с 0,6 мл метансульфонилхлорида при 2oC в течение 20 мин, а затем при 50oC в течение 10 мин. Реакционную смесь разделяют между этилацетатом и водой. Органическую фазу выделяют, промывают водой, сушат над сульфатом натрия и выпаривают досуха. Остаток обрабатывают хроматографически на силикагеле, и элюируют смесью этилацетат : дихлорметан /1 : 9/ до получения ди-трет-бутилового сложного эфира 4-//2-хлорэтил/ /2-мезилокси/-этил/аминофенилоксикарбонил-L-глутаминовой кислоты /6/ в виде масла /0,44 г/. Выход 43%.
ЯМР: δ : 3,15 /с, 3H/, 3,70 /м, 6H/, 4,29 /т, 2H/, 6,75 /д, 2H/, 6,92 /д, 2H/, 1,41 /с, 9H/, 1,42 /с, 9H/, 1,8-2,3 /м, 2H/, 3,97 /м, 1H/, 7,92 /д, 1H/.
Примеры 9 - 15
Соединения, перечисленные далее в таблице 1, получены по способу примера 2, с использованием исходных материалов, и промежуточных соединений, перечисленных в таблицах 2 - 8 далее (все таблицы приведены в конце описания).
Так, соединения примеров 9 - 15 получают по способу примера 2. Соединение примера 10 получают, заменяя 4-амино-м-крезол, использованный на стадии /a/, на 4-амино-3-изопропилфенол /G:l- man, H. et al., J. Org. Chem. 19/1954/, 1067 - 78/. Соединение примера 11 получают, заменяя 4-амино-м-крезол на 4-амино-2-метилфенол /доступный от Aldrich/, а соединение примера 12 получают, заменяя 4-амино-м-крезол- на 4-амино-3-фторфенол/, полученный по способу Journal; of the Chemical Society /1964/, p.473/. Соединение примера 13 получают, заменяя 4-амино-м-крезол на 4-аминонафт-1-ол/ Aldrich Chemical Co Ltd/ и соединение примера 14 получают, заменяя 4-амино-м-крезол на 4-амино-2-хлорфенол /полученный по способу Journal of the American Chemical Society, 45, 2192 /1923/. Соединение примера 15 получают, заменяя 4-амино-м-крезол на 4-амино-3-хлорфенол /Berichte, p.2065 /1938/ и Organic Synthesis; Collected, vol. 4, p. 148/
Исходные материалы и промежуточные соединения для получения соединений примеров 9 - 15 и их свойства перечисленны далее в таблицах 2 - 8.
Пример 16
γ - анилид N/4-/N,N-бис/2-хлорэтил/амино/феноксикарбонил/-L- глутаминовой кислоты
Супензию α -бензил-4-/N,N-бис/2-хлорэтил/амино/феноксикарбонил -L-глутаминовой кислоты γ -анилид//2,0 г/ в 50 мл этилацетата гидрируют над 10% палладием на угле /0,15 г/ в течение 4 ч. Катализатор удаляют фильтрованием, а полученный фильтрат выпаривают досуха при пониженном давлении при 35oC. Продукт, 4-/N,N-бис/2- хлорэтил/амино/феноксикарбонил-L-глутаминовой кислоты γ -анилид/ 1 в схеме 6/ получают в виде кристаллов белого цвета, 1,4 г /83%/. Т.пл. 110oC.
Элементный анализ:
Рассчитано: C 54,8; H 5,22; N 8,71
Найдено: C 54,5; H 5,62; N 8,31
Исходный материал, γ -анилид α -бензил-4-/N,N-бис/ 2-хлорэтил/-амино/феноксикарбонил-L-глутаминовой кислоты, получают описанным далее способом.
2 мл триэтиламина добавляют к смеси γ -анилида альфа-бензил-р-тозил-L-глутаминовой кислоты /2,8 г/ и 2,0 г O-/4-/N, N-бис/2-хлорэтил/амино/фенил/-O'-/4-нитрофенил/карбоната в 30 мл дихлорметана. Полученную смесь перемешивают при комнатной температуре в течение 16 ч, и растворители удаляют в вакууме. Остаток хроматографически обрабатывают на силикагеле /Merck Агт 9385/ и элюируют 10% этилацетатом в дихлорметане до получения γ -анилида α -бензил-4-/N, N-бис/2-хлорэтил/амино/феноксикарбонил-L-глутаминовой кислоты в виде белого твердого вещества, 1,9 г /64%/.
Исходный материал, O-/4-/N, N-бис/2-хлорэтил/амино-O'-/ 4-нитрофенил/карбонат, получают описанным далее способом.
Раствор 10 мл триэтиламина в 10 мл дихлорметана добавляют к смеси 7,25 г 4-нитрофенил-хлорформата и 10 г 4-/ N,N-бис/2- хлорэтил/амино/фенолгидрохлорида в 100 мл дихлорметана в течение 2 ч. После перемешивания в течение 16 ч при комнатной температуре растворители удаляют при пониженном давлении, а остаток обрабатывают хроматографически на силикагеле /Merck Агт. 9385/. Элюирование дихлорметаном и выпаривание элюатов приводит к получению продукта в виде масла красного цвета. После тщательного растирания с гексаном получают твердое вещество желтого цвета, которое перекристаллизовывают из смеси бензол/гексан до получения O-/4-/N,N-бис/2-хлорэтил/амино/фенол-O'-/4-нитрофенил/карбоната в виде оранжевых кристаллов, 10,4 г /71%/. Т.пл. 68oC.
Исходный материал γ -анилид р-тозил- α -бензил-L-глутаминовой кислоты получают следующим образом.
α -бензиловый сложный эфир N-трет -ВОС-L-глутаминовой кислоты /10 г/ и 6,1 г дициклогексилкарбодиимида растворяют в 120 мл дихлорметана и перемешивают при комнатной температуре в течение 10 мин. 2,8 мл анилина добавляют, и полученную смесь перемешивают при комнатной температуре в течение 16 ч. Эту смесь фильтруют, промывают последовательно насыщенным раствором бикарбоната натрия /2х100 мл/ и водой /100 мл/, а затем выпаривают. Полученный твердый продукт перекристаллизовывают из смеси EtOAc/гексана до получения бесцветных пластин, 8,2 г /67%/. γ -анилид /12,0 г/ и р-толуолсульфоновую кислоту /5,4 г/ в 300 мл бензола кипятят с обратным холодильником в течение 40 мин, и оставляют охлаждаться в течение ночи. Осадок фильтруют, сушат с подсосом и перекристаллизовывают из EtOAc/ /MeOH до получения бесцветных пластинок γ -анилида тозил- α -бензил-L-глутаминовой кислоты, 8,2 г / 58%/
Пример 17
N-/4-/N,N-бис/2-хлорэтил/амино/феноксикарбонил/-L-глутаминовой кислоты γ -трет-бутиламид
По способу примера 16, используя γ трет-бутиламид γ -бензил-4-/N,N-бис/2-хлорэтил/амино/феноксикарбонил-L-глутаминовой кислоты вместо γ -анилида 4-/N,N-бис/2-хлорэтил/амино/феноксикарбонил- L-глутаминовой кислоты получают γ -трет-бутиламид 4-/N,N-бис/2-хлорэтил/амино/ феноксикарбонил-L-глутаминовой кислоты /2 схеме 6/, который кристаллизуют из смеси этилацетат/гексан до получения бесцветных кристаллов, Т.пл. 129oC.
Элементный анализ:
Рассчитано: C 51,9; H 6,32; N 9,09
Найдено: C 52,1; H 6,33; N 8,96
γ -трет-бутиламид α -бензил-4-/N,N-битс/2-хлорэтил/амино/ феноксикарбонил-L-глутаминовой кислоты получают по способу примера 16 для производного γ -анилида.
Пример 18
N-/4-/N, N-бис/2-хлорэтил/амино/-3-метилфенилкарбамоил/-L- глутаминовая кислота.
Раствор ди-трет-бутил-4-/N, N-бис/2-хлорэтил/амино/-3- метилфенилкарбамоил-L-глутамата /0,6 г/ в 6 мл дихлорметана охлаждают до 0oC и добавляют 15 мл трифторуксусной кислоты. Затем этот раствор оставляют при 0oC на три дня. Затем этот раствор выпаривают досуха до получения /4-/N,N-бис/2-хлорэтил/амино/-3- метилфенилкарбамоил-L-глутаминовой кислоты в виде масда. Выход 0,49 г.
ЯМР, δ : 8,46 /с, 1H/, 7,15 /м, 3H/, 6,4 /д, 1H/, 4,18 /м, 1H/, 3,55 /м, 4H/, 3,35 /м, 4H/, 2,3 /м, 2H/, 2,23 /с, 3H/, 2,0 /м, 2H/.
Исходный материал, ди-трет-бутил-4-/N,N-бис/2-хлорэтил/амино/ -3-метилфенилкарбамоил-L-глутамат, получают описанным далее способом.
27,5 г карбоната калия добавляют к раствору 3-метил-4-нитроанилина/ J.of Org. Chem. 33, 3498 /1968/ /7,6 г/ в 150 мл этилацетата, с последующим прикапыванием 1,9 М раствора фосгена в 27,5 мл толуола, поддерживая температуру ниже 30oC. Затем полученную смесь перемешивают при комнатной температуре в течение 1 ч. Ди-трет-бутиловый сложный эфир L-глутаминовой кислоты /13 г/ добавляют к смеси, которую затем перемешивают при комнатной температуре в течение ночи. Затем раствор фильтруют, промывают водой, и органический слой сушат над сульфатом магния и выпаривают до масла. Его затем хроматографически обрабатывают на силикагеле, элюируют смесью гексан-этилацетат 3:1 до получения ди-трет-бутил/3-метил-4- нитрофенилкарбамоил-L-глутамата в виде масла. Выход 11,19 г, 51%.
ЯМР: δ : 9,16 /с, 1H/, 8,0 /д, 1H/, 7,43 /м, 2H/, 6,70 /д, 1H/, 4,1 /м, 1H/, 2,51 /с, 3H/, 2,25 /м, 2H/, 1,8 /м, 2H/, 1,41 /д, 8H/.
Раствор ди-трет-бутил-3-метил-4-нитрофенил-карбамоилглутамата 4,7 г/ в 125 мл этилацетата гидрируют над 30% Po-C /0,5 г/. Полученную смесь фильтруют через целит /очищенные и кальцинированные частицы диатомовой земли размером 20 - 45 мкм, которые, вместе с тем, можно получить от Fluka Chemical L+d/, и выпаривают до твердого вещества темного цвета. Его хроматографически обрабатывают на силикагеле, элюируя смесью этилацетат; гексан /1:1/ до получения ди-трет-бутил-3-метил-4-аминофенилкарбамоил-L-глутамата в виде масла, выход 3,71 г, 83%.
ЯМР, δ : 7,98 /с, 1H/, 6,9 /м, 2H/, 6,52 /д, 1H/, 6,15 /д, 1H/, 4,53 /с, 2H/, 4,15 /м, 1H/, 2,25 /м, 2H/, 2,04 /с, 3H/, 1,8 /м, 2H/, 1,45 /д, 18H/
4,8 г этиленоксида барботируют через раствор ди-трет-бутил-3-метил-4-аминофенилкарбамоил-L-глутамата /5 г/ в 25 мл ледяной уксусной кислоте и 25 мл воды. Этот раствор перемешивают при комнатной температуре в течение 24 ч. После выпаривания досуха остаток снова растворяют в этилацетате, промывают водой, органический слой сушат над сульфатом магния и выпаривают до получения ди-трет-бутил-4-/N,N-бис/2-гидроксиэтил/амино/-3-метилфенилкарбамоил- L-глутамата, который используют далее без дополнительной очистки.
ЯМР δ : 8,35 /с, 1H/, 7,1 /м, 3H/, 6,35 /д, 1H/, 4,15 /м, 1H/, 3,35 /м, 4H/, 3,05 /м, 4H/, 2,25 /м, 2H/, 2,20 /с, 3H/, 1,8 /м, 2h/, 1,45 /д, 18H/
3,8 мл метансульфонилхлорида прикапывают к раствору ди- трет-бутил-4-/N, N-бис/2-гидроксиэтил/амино/-3-метилфенилкарбомаил-L- глутамата /4 г/ в 60 мл пиридина в атмосфере азота, поддерживая температуру ниже 30oC. После добавления раствор перемешивают в течение 2 ч при 80oC. Раствор охлаждают и выливают на 10% лимонную кислоту /500 мл/, экстрагируют этилацетатом, промывают водой, органический слой сушат над сульфатом магния, а затем выпаривают до получения масла коричневого цвета. Это масло хроматографируют на силикагеле, элюируя смесью гексан-этилацетат 5:1 до получения ди-трет-бутил-4-/N, N-бис/2-хлорэтиламино/-3-метилфенилкарбамоил-L- глутамат в виде масла. Выход 7,22 г, 28% ЯМР, δ : 8,42 /с, 1H/, 7,1 /м, 3H/, 6,36 /д, 1H/, 4,13 /м, 1H/, 3,50 /м, 4H/, 3,31 /м, 4H/, 2,3 /м, 2H/, 2,23 /с, 3H/, 1,9 /м, 2H/, 1,4 /с, 9H/, 1,35 /с, 9H/.
Последовательность реакций для этого примера приведена на схеме 7.
Пример 19
N-/4-/N,N-бис/2-хлорэтил/амино/бензилкарбонил/-L-глутаминовая кислота
Ди-трет-бутил-4-/N, N-бис/2-хлорэтил/амино/бензилкарбонил-L- глутамат /0,5 г/ растворяют в 1,5 мл CH2Cl2 и добавляют 1,5 мл трифторуксусной кислоты. Полученный раствор перемешивают при комнатной температуре в течение 2 ч. Затем раствор выпаривают до получения указанного в заглавии соединения в виде масла /0,8 г/.
ЯМР: δ : 8,22 /д, 1H/, 7,10 /д, 2H/, 6,66 /д, 2H/, 4,19 /м, 1H/, 3,69 /с, 8H/, 2,24 /м, 2H/, 1,9 /м, 2H/, 3,32 /с, 2H/.
Исходный материал, ди-трет-бутил-4-/N,N-бис/2-хлорэтил/-амино/ бензилкарбонил-L-глутамат, получают следующим образом.
4,05 г 1-гидроксибензотриазола добавляют к раствору 4-нитрофенилуксусной кислоты /5,4 г/ в 75 мл диметилформамида. К этой смеси добавляют ди-трет-бутиловый сложный эфир L-глутаминовой кислоты /7,77 г/, а затем дициклогексилкарбодиимид /6,2 г/. Затем полученную смесь перемешивают в течение 18 ч при комнатной температуре. Полученную смесь фильтруют, и фильтрат промывают насыщенным раствором бикарбоната натрия, водой и 0,5 М соляной кислотой, а затем сушат и выпаривают досуха. Остаток обрабатывают хроматографически на силикагеле, элюируя смесью гексан/этилацетат /2:1/ до получения ди-трет-бутил-4-нитробензилкарбонил-L-глутамата в виде масла, 9,7 г /77%/.
ЯМР, δ : 8,2 /д, 2H /, 7,45 /д, 2H/, 6,45 /д, 1H/, 4,45 /м, 1H/, 3,15 /с, 2H/, 2,1 /м, 4H/, 1,4 /д, 9H/, 1,35 /с, 9H/.
Раствор ди-трет-бутил-4-нитробензилкарбонил-L-глутамата /9,7 г/ в 200 мг этилацетата гидрируют над 30% Pd/C/ 900 г/. Полученную смесь фильтруют через целит, и выпаривают до получения желтого масла /ди-трет-бутил-4-аминобензилкарбонил-L-глутамата/, которое используют без дополнительной очистки.
ЯМР, δ : 8,23 /д, 1H/, 6,92 /д, 2H/, 6,50 /д, 2H/, 4,86 /с, 2H/, 4,15 /м, 1H/, 3,24 /с, 2H/, 2,22 /м, 2H/, 1,8 /м, 2H/, 1,4 /д, 18H/. Выход 8,2 г /90%/.
К раствору ди-трет-бутил-4-аминобензилоксикарбонил-L-глутамата /8,2 г/ в 40 мл ледяной уксусной кислоты и 40 мл воды добавляют 7,1 г этиленоксида. Полученную смесь перемешивают при комнатной температуре в течение 24 ч. Этот раствор выпаривают досуха, снова растворяют в эфире, промывают водой, органический слой сушат над сульфатом магния и выпаривают до масла /ди-трет-бутил-4-/N, N-бис/2-гидроксиэтил/-амино/-бензилкарбонил-L- глутамата/ /6,3 г/, которое используют без дополнительной очистки /6,3 г/.
ЯМР, δ : 8,16 /д, 1H/, 7,02 /д, 2H/, 6,58 /д, 2H/, 4,1 /м, 1H/, 3,4 /м, 8H/, 3,25 /с, 2H/, 2,25 /м, 2H/, 1,8 /м, 2H/, 1,4 /с, 18H/.
2,91 мл метансульфонилхлорида прикапывают к раствору ди-трет-бутил-4-/N, N-бис-/2-гидроксиэтил/амино/бензилкарбонил-L- глутамата /2,88 г/ в 45 мл пиридина в атмосфере аргона, поддерживая температуру ниже 25oC. После добавления раствор перемешивают при 80oC в течение 1 ч. Затем раствор охлаждают и выливают в 10% лимонную кислоту /500 мл/, экстрагируют эфиром, промывают водой, органический слой сушат над сульфатом магния, а затем выпаривают до масла коричневого цвета. Это масло обрабатывают хроматографически на силикагеле, элюируя смесью гексан-этилацетат 2:1 до получения ди-трет-бутил-4-/N, N-бис/ 2-хлорэтил/амино/бензилкарбонил-L-глутамата в виде масла /1,3 г/ /42%/.
ЯМР, δ : 8,19 /д, 1H/, 7,10 /д, 2H/, 6,66 /д, 2H/, 4,10 /м, 1H/, 3,69 /с, 8H/, 3,32 /с, 2H/, 2,21 /м, 2H/, 1,9 /м, 2H/, 1,37 /д, 18H/. Выход 1,32 г /42%/.
Последовательность реакций для этого примера приведена на схеме 8.
Пример 20
N-/4-/N,N-бис-/2-хлорэтил/амино/фенилкарбамоил/-L-глутаминовая кислота
Раствор ди-трет-бутил-4-/N, N-бис-/2-хлорэтил/амино/фенилкарбамоил-L-глутамата/, полученного по способу примера 3/ /500 мг/ в 10 мл 98% муравьиной кислоты, оставляют при комнатной температуре на 24 часа. Раствор выпаривают досуха, а остаток хроматографируют на силикагеле Merck Aгт 9385 в смеси дихлорметан/этилацетат/муравьиная кислота /7:2:1/ до получения указанного в заглавии соединения в виде масла, которое кристаллизуется. Т. пл. 117-119oC.
Пример 21
N-/4-N,N-бис/2-бромэтил/амино/-3-фторфенилкарбамоил/-L-глутаминовая кислота
Раствор дибензил-4-/N, N-бис/2-бромэтил/амино-3-фторфенилкарбамоил-L-глутамата /0,5 г/ в 10 мл этилацетата и 30% Pd/C /100 мл/ перемешивают в атмосфере водорода в течение 6 ч. Катализатор отфильтровывают, а полученный фильтрат выпаривают до масла, которое и представляет указанное в заглавии соединение
ЯМР, δ : 8,6 /с, 1H/, 7,35 /дд, 1H/, 7,1-6,8 /м, 2H/, 6,5 /д, 1H/, 4,2 /м, 1H/, 3,7-3,2 /м, 8H/, 2,4-1,6 /м, 4H/.
Исходный материал для этой реакции получают способом, аналогичным способу примера 5, но вместо тионилхлорида используют тионилбромид.
ЯМР для соединения примера 23: δ : 8,6 /шир., 1H/, 7,6 /м, 1H/, 7,2 /м, 2H/, 4,25 /м, 1H/, 3,6-3,3 /м, 8H/, 2,4-1,7 /м, 4H/. Исходные материалы и промежуточные соединения, использованные для получения соединений примеров 21 - 24, а также их свойства указаны далее в таблицах 10 - 13 (см. в конце описания).
Соединения 25 - 32 получают по способу примера 1, за исключением описываемых далее.
Данные ЯМР для каждого из соединений примеров 25 - 32 представлены в табл. 15 (см. в конце описания).
Соединение примера 25 получают по способу примера 1, но используя α -бензил-4-/N, N-бис/2-хлорэтил/-амино/феноксикарбонил-L-глутамин в реакции гидрирования/ его получение описано далее/. Тетрагидрофуран добавляют к этилацетату в реакции гидрирования для облегчения солюбилизации вышеуказанного альфа-бензильного соединения.
Соединения примеров 26 и 27 получают по способу примера 1 за счет гидрогенолиза промежуточных α -бензил-4-/N,N-бис/2-хлорэтил/амино/феноксикарбонил-L-мезилглутамина и α -бензил-4-/N,N-бис/2-хлорэтил/амино/феноксикарбонил-L-/3-метилфенил/- глутамина, соответственно.
Соединение примера 28 получают по способу примера 1 за счет гидрирования бензил-4-/N,N-бис/2-хлорэтил/амино/феноксикарбонил- γ -/5-тетразолил/- α -амино-L-бутирата. Соединение примера 28 получают также вторым способом, который приводится далее. Следуют способу примера 1, но используют /S/-2-амино-4/1H-1,2,3,4-тетразол-5-ил/масляную кислоту /Z. Grzonka et al., Tetrahedron 33, 2299 - 2302, 1077/ вместо дибензилового эфира Z-глутаминовой кислоты в реакции с O-/4-/N,N-бис/2-хлорэтил/амино/фенил/O'-/4-нитрофенил/карбонатом. Реакцию ведут в сухом ДМФ с двумя эквивалентами триэтиламина в течение 20 ч при 25oC. После выпаривания досуха остаток растворяют в этилацетате, промывают разбавленной лимонной кислотой, сушат и выпаривают досуха. Полученный продукт медленно кристаллизуется из этилацетата. Остаток перекристаллизовывают из этилацетата до получения соединения примера 28. Т. пл. 173 - 175oC.
Соединение примера 29 получают способом, аналогичным описанному для первого получения примера 28, но 1-/4-N,N-бис/-2-хлорэтиламино/фенокси/карбониламино/-1-бензилоксикарбонил-2- /5-тиотетразол/-этан используют вместо бензил-4/N,N-бис/2-хлорэтил/амино/феноксикарбонил- γ -/5-тетразолил/- α -амино-L-бутирата. После аналогичной обработки реакционной смеси получают смолу, которую очищают на хроматографической колонке с силикагелем, элюируя 4% муравьиной кислотой/этилацетатом/ по объему/.
Соединение примера 29 также получают вторым способом, описываемым далее.
1-Амино-1-карбокси-2-/5-тиотетразол/этан/600 мг, 2,89 мМ/ суспендируют в сухом ДМФ /48 мл/, триэтиламин /0,806 мл, 578 мМ/ добавляют, и суспензию перемешивают.
O-/4-/N, N-бис/2-хлорэтил/амино/фенил/-O'-/4-нитрофенил/карбонат /1,0 г, 2,89 мМ/ добавляют сразу в твердом виде, и полученный раствор перемешивают в течение 20 ч при комнатной температуре. ДМФ удаляют в вакууме. Остаток растворяют в этилацетате и разбавленной лимонной кислоте. Этилацетатный слой промывают водой, сушат над сульфатом натрия, фильтруют, и выпаривают. Полученный неочищенный продукт обрабатывают хроматографически на силикагеле и элюируют 4% муравьиной кислотой в этилацетате до получения 4-/N,N-бис/2-хлорэтил/амино/феноксикарбониламино-1-карбокси-2-/5-тиотетразол/ этана в виде стеклообразного твердого вещества /0,963 г/.
ЯМР /ДМСО-d6/ δ : 3,69 /с, 8H/, 3,8 /дд, 1H/, 4,38 /м, 1H/, 6,72 /д, 2H/, 6,93 /д, 2H/, 8,12 /д, 1H/.
Соединения примеров 30 и 31 получают по способу примера 1 за счет гидрирования α -бензил-4/N,N-бис/2-хлорэтил/амино/-
феноксикарбонил- γ -/3-/бензилоксикарбонилметил/-фенил/-L-глутамина и α -бензил-4-/N, N-бис/2-хлорэтил/амино/феноксикарбонил- γ -3-/5-тетразолил/-фенил/-L-глутамина соответственно.
Соединение примера 32 получают по способу примера 1 за счет гидрирования α -бензил-4-/N,N-бис/2-хлорэтил/амино/феноксикарбонил- γ -/3-/бензилоксикарбониламино/фенил/-L-глутамина.
Промежуточные соединения, используемые для получения соединений примеров 25 - 32
α -бензил-4-/N,N-бис/2-хлорэтил/амино/феноксикарбонил-L-глутамин, который используют для получения соединения примера 25, получают по способу примера 1 из O-/4-/N,N-бис/2-хлорэтил/амино/-фенил-O'-/4-нитрофенил/карбоната, но заменяя тозилат дибензилового сложного эфира L-глутаминовой кислоты на α -бензил-L-глутамин /L. Zervas et al., J. Am. Chem. Soc. 87 /1/, 99-104, 1965/. Полученный продукт очищают с помощью флеш-хроматографии на силикагеле, элюируя 80% EtOAc/20% гексаном. Продукт получают в виде твердого вещества белого цвета после тщательного растирания с эфиром.
α -бензил-4/N, N-бис/2-хлорэтил/амино/бензилоксикарбонил- γ -мезил-L-глутамин, который используют для получения соединения примера 26, получают следующим образом.
N-BOC- α -бензил-L-глутамат /E. Klieger et al., Ann., 673, 196 - 207, 1964/ /10 г/ в 50 мл сухого дихлорметана обрабатывают диметиламинопиридином /ДМАР/ /3,9 г/ и дициклогексилкарбодиимидом /6,73 г/. Затем в реактор вводят метансульфонамид /3,04 г/, и реакцию ведут при 25oC в течение 20 ч. Дихлорметан выпаривают, а остаток снова растворяют в этилацетате. Этилацетатный раствор промывают затем 0,25 М лимонной кислотой, затем водой, а затем сушат над безводным сульфатом натрия. После выпаривания этилацетатного экстракта получают остаток, который очищают на силикагеле, используя флеш-хроматографию, используя в качестве элюента метиленхлорид, затем 5% метанол/метиленхлорид, а затем 10% метанол/метиленхлорид до получения Boc-защищенного ацилсульфонамида: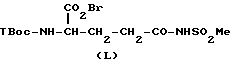
ЯМР δ : 1,37 /с, 9H/, 1,98 /м, 2H/, 2,34 /т, 2H/, 5,11 /д, 2H/, 7,26 /д, 1H/, 7,36 /с, 5H/, 11,64 /с, 1H/
3,6 г Boc-защищенного ацилсульфонамида суспендируют в 50 мл этилацетата с последующим добавлением 8 эквивалентов HCl насыщенного этилацетата /22,4 мл, 3,1 М раствор/. Исходный материал, сульфонамид, помещают в раствор, и реакцию ведут в течение 20 ч в атмосфере аргона при 25oC. α -бензил- γ -мезил-L-глутамин, гидрохлорид формулы:
получают в виде белого твердого вещества, которое отфильтровывают и промывают безводным эфиром перед тем, как сушат в эксикаторе, ЯМР, δ : 2,08 /м, 2H/, 2,52 /м, 2H/, 3,20 /с, 3H/, 4,09 /т, 1H/, 5,15 - 5,35 /дд, 2H/, 7,39 - 7,50 /м, 5H/, около 9 /шир. 3H/.
α -бензил-4/N,N-бис/2-хлорэтил/амино/феноксикарбонил-L-мезилглутамин получают по способу примера 1 из O-/4-N,N-бис/2-хлорэтил/амино/фенил-O'-/4-нитрофенил/карбоната, но заменяя тозилат сложного дибензилового эфира L-глутаминовой кислоты на α -бензил- γ -мезил-L-глутамингидрохлорид. Продукт этой реакции очищают на хроматографической колонке с силикагелем элюируя 10% муравьиной кислотой в этилацетате /по объему/.
α -бензил-4/N, N-бис/2-хлорэтил/амино/феноксикарбонил- γ -/3-метилфенил/-L-глутамин, который используют при получении соединения примера 27, получают следующим образом:
α -бензил-4/N,N-бис/2-хлорэтил/амино/феноксикарбонил-L-глутамат получают по способу примера 1 из O-/4-/N,N-бис/2-хлорэтил/амино/фенил/-O'-/4-нитрофенил/карбоната, но заменяя дибензиловый сложный эфир L-глуматиновой кислоты на α -бензил-L-глутамат /Ref. C. Coutsogeorgopou los et al, J. Am. Chem. Soc. 83, 1885, 1961/. Реакцию, кроме того, ведут в сухом ДМФ при 25oC в течение 2 ч. Полученный продукт очищают на хроматографической колонке с силикагелем Merck Art 9385, используя ацетат/гексановые смеси в интервале от 70/30 соответственно, до 100% этилацетата. Данные ЯМР приведены в таблице 17.
α -бензил-4/N,N-бис/2-хлорэтил/амино/феноксикарбонил-L-глутамат /300 мг/ в 5 мл сухого тетрагидрофурана обрабатывают 1,1 эквивалентами триэтиламина. К реакционной смеси медленно добавляют изобутилхлорформат /0,08 мл, 1,1 экв. / в 10 мл сухого ТГФ при -25oC. Спустя 15 мин добавляют 1,1 эквивалента /0,08 мл/ м-толуидина в 5 мл сухого тетрагидрофурана. Полученной смеси дают нагреться до 25oC, а затем непрерывно перемешивают в течение 18 ч. Материал реакции затем фильтруют, и полученный фильтрат выпаривают до получения остатка, который очищают с помощью флэш-хроматографии, используя в качестве элюента смеси этилацетат/гексан. После выпаривания соответствующих фракций получают α -бензил-4- /N,N-бис/2-хлорэтил/амино/феноксикарбонил γ -/3-метилфенил/- L-глутамин в виде белого кристаллического твердого вещества. ЯМР данные для этого продукта представлены в таблице 17.
1-/4-/N, N-бис/2-хлорэтил/амино/феноксикарбониламино/-1- бензилоксикарбонил-2-/5-тиотетразол/-этан, который используется для получения соединения примера 29, получают следующим образом.
1-Амино-1-карбокси-2/5-тиотетразол/этан получают, добавляя β -хлоранилин /18,50 г, 115,6 ммоля/ (Sigma Chemicals Co) и 5-тио- тетразол /11,79 г, 115,6 ммоля// полученный по способу европейской патентной публикации N 33965 /к 230 мл 2M раствора гидроксида натрия при перемешивании. Затем реакционную смесь нагревают от комнатной температуры до 90oC в течение полутора ч. Реакционной смеси дают остыть до комнатной температуры, а затем охлаждают далее в бане лед/метанол, полученную смесь подкисляют до pH 4,0 концентрированной соляной кислотой и перемешивание продолжают в течение получаса. Полученный осадок отфильтровывают с подсосом до получения 6,9 г. Снова проверяют pH маточного раствора и обнаруживают, что он повысился до около 5,5. Вышеописанную процедуру повторяют, и получают еще 2,3 г продукта. Его сушат в высоком вакууме.
ЯМР, δ : 3,30 - 3,50 мд/ м, 2H/, 4,16 - 4,22 мд /кв, 1H/, 7,47 /мд /шир, H2O в обмене с NH2.
Элементный анализ: Рассчитано: C 23,2; H 4,4N 33,8 /+1 моль H2O/
Найдено: C 23,1; H 4,4; N 33,7
/9,6% H2O/
1-/4-/N, N-бис/2-хлорэтил/амино/феноксикарбониламино/-1-бен- зилоксикарбонил-2-/5-тиотетразол/-этан получают по способу примера 1 из 0-/4-/N,N-бис/2-хлорэтил/амино/-фенил/-0'-/4-нитрофенил/ карбоната, но заменяя дибензиловый сложный эфира L-глутаминовой кислоты на γ -/5-тиотетразолил/- α -бензилокси-карбонил-амино- L-масляную кислоту /Ref. Z. Grzonka et al. Tetrahedron Letters 33, 2399 - 2302, 1977/. В качестве растворителя используют сухой диметилформамид, и реакцию ведут при 25oC. Реакционную смесь обрабатывают спустя 2 часа, и полученный продукт очищают с помощью флеш-хроматографической колонки с силикагелем, элюируя смесью 2% муравьиновй кислоты в этилацетат/метиленхлориде /3: 1 по объему/. Данные ЯМР для этого продукта приведены в таблице 16 (см. в конце описания).
α -бензил-4-/N,N-бис/2-хлорэтил/амино/феноксикарбонил- γ -/3-/бензилоксикарбонилметил/фенил/-L-глутамин, который используют в примере 27, получают следующим образом.
Соль присоединения p-толуолсульфоновой кислоты к бензиловому эфиру 3-аминофенилуксусной кислоты получают, добавляя 3-аминофенилуксусную кислоту /10 г/ и моногидрат p-толуолсульфоновой кислоты /13,2 г/ к бензиловому спирту /27,2 мл/ в 30 мл толуола. Полученную смесь нагревают при кипячении с обратным холодильником, образующуюся воду собирали в приемник Дина-Старка. Когда всю воду отгоняют, смесь оставляют остывать до 25oC, перед тем, как разбавить диэтиловым эфиром, и помещают на 1 час в баню со льдом. Кристаллический p-толуолсульфонат отфильтровывают и сушат в эксикаторе /23,5 г/. ЯМР, δ : 2,31 /с, 3H/, 3,82 /с, 2H/, 5,11 /с, 2H/, 7,11 /д, 2H/, 7,25 - 7,45 /м, 8H/, 7,52 /д, 2H/.
α -бензил-4-/N,N-бис/2-хлорэтил/амино/феноксикарбонил- γ -/3-/бензилоксикарбонилметил/фенил/-L-глутамин получают по способу примера 27, но заменяя м-толуидин на 3-аминофенилуксусной кислоты бензиловый сложный эфир.
α -бензил-4-/N, N-бис/2-хлорэтил/амино/феноксикарбонил- γ -/3-/5-тетразолил/фенил/-L-глутамин, который используют в примере 31, получают по способу примера 27, но заменяя м-толуидин на 3-/тетразол-5-ил/-анилин. 3-/тетразол-5-ил/анилин, который используют в качестве исходного материала, получают следующим способом. К раствору 5-/3-нитрофенил/тетразола /Finnegan, W.G; Henry R.A., и Lolquist R. J. A.C.S.
80: 3908 /1958// /52 г/ в этаноле /2,5 л/ добавляют 10% палладий на угле /5 г/, и полученную смесь перемешивают в атмосфере водорода в течение 16 ч. Катализатор отфильтровывают, и полученный фильтрат выпаривают до получения целевого исходного материала /40,5 г, T. пл. 188 - 189oC/.
α -бензил-4-/N, N-бис/2-хлорэтил/амино/феноксикарбонил- γ - /3-/бензилоксикарбониламино/фенил/-L-глутамин, который используют в примере 32, получают по способу примера 27, но заменяя м-толуидин на 3-/бензилоксикарбониламино/анилин.
ЯМР δ : ClCH2CH2N: 3,68 /м, 8H/
ароматика: 6,68 - 7,74 /м, 1H/
ArCH2O: 5,12 /с, 2H/, 5,14 /с, 2H/
α CH : 4,18 /м, 1H/
остальные: 1,9 - 2,15 /м, 2H/, 2,45 /м, 2H/, 8,12 /д, 1H/, 9,7 /с, 1H/, 9,9 /c, 1H/.
3-/бензилоксикарбониламино/анилин, используемый в качестве промежуточного соединения, в примере получения 32, получают следующим образом. К холодному раствору /0o/ м-фенилендиамина /10 г/ в 200 мл этилацетата добавляют водный KHCO3 /9 г в 300 мл воды/ при перемешивании. Прикапывают бензилхлорформат /13,2 мл/ в 100 мл этилацетата в течение 10 мин, перемешивают при 0oC в течение одного часа, а затем подкисляют /pH 2/, добавляя M.HCl /водный/. Полученный продукт экстрагируют этилацетатом /250 мл/, промывают рассолом, сушат над сульфатом магния и выпаривают в "вакууме" до получения масла. Это масло обрабатывают хроматографически на силикагеле Merck Aгт 9385, элюируя смесью гексан/этилацетат /7:3/ до получения 8 г промежуточного соединения в виде твердого вещества с низкой температурой плавления /выход 36%/.
ЯМР, δ : 9,32 /с, 1H/, 7,30 /м, 5H/, 6,80 /т, 1H/, 6,70 /м, 1H/, 6,50 /м, 1H/, 6,12 /м, 1H/, 5,0 /с, 2H/, 4,88 /с, 2H/.
Пример 33
N-/4-/N,N-бис/2-иодоэтил/амино/феноксикарбонил/-L- глутаминовая кислота
4-/N, N-бис/2-иодоэтил/амино/феноксикарбонил-L-глутаминовой кислоты ди-трет-бутиловый сложный эфир /188 мг//см. схема 9-соединение 14/ суспендируют в трифторуксусной кислоте /TFA/ /4 мл/ и перемешивают в течение 30 мин при комнатной температуре. TFA удаляют при пониженном давлении, оставшееся масло разбавляют этилацетатом /3 мл/ и выпаривают до получения 4-/N,N-бис/2-иодоэтил/ амино/феноксикарбонил-L-глутаминовая кислота-1,4-TFA-0,8EtOAc /162 мг//выход 82%/; /Соединение 15 на схеме 9/.
ЯМР; δ : 1,84 - 2,01 /м, 1H/, 2,36 /м, 2H/, 3,31 /т, 4H/, 3,72 /т, 4H/, 4,02 /м, 1H/, 6,66 /д, 2H/, 6,94 /д, 2H/, 7,92 /д, 1H/.
4-/N, N-бис/2-иодоэтил/амино/феноксикарбонил-L-глутаминовой кислоты ди-трет-бутиловый сложный эфир, который используют в качестве промежуточного соединения, получают следующим образом:
a/ Раствор ди-трет-бутилового сложного эфира бис/2-мезилоксиэтил/амино/феноксикарбонил-Z-глутаминовой кислоты /1,0 г/ /см. схема 9, соединение 11/ в 50 мл ацетонитрила перемешивают с иодидом натрия /1,0 г/ при 70oC в течение 20 ч. Реакционную смесь фильтруют, и полученный фильтрат концентрируют в вакууме. Остаток обрабатывают хроматографически на силикагеле, элюируя этилацетатом в циклогексане /1:5/ до получения ди-трет-бутилового сложного эфира 4-/N,N-бис/2-иодоэтил/амино/феноксикарбонил-L- глутаминовой кислоты /см. схему 9, соединение 14/ в виде масла /0,75 г/ /68% выход/. ЯМР, δ : 1,41 /c, 9H/, 1,43 /c, 9H/, 1,81 - 1,95 /м, 1H/, 2,34 /м, 2H/, 3,31 /т, 4H/, 3,72 /т, 4H/, 4,00 /м, 1H/, 6,67 /д, 2H/, 6,93 /д, 2H/, 7,91 /д, 1H/.
b/ ди-трет-бутиловый сложный эфир бис/2-мезилоксиэтил/амино- феноксикарбонил-L-глутаминовой кислоты
Ди-трет-бутиловый сложный эфир 4-/N,N-бис/2-гидроксиэтил/амино/ феноксикарбонил-L-глутаминовой кислоты /2,53 г/ /полученный по способу примера 8 - см. схему 4/ в 9 мл пиридина перемешивают с метансульфонилхлоридом /1,8 мл/ при 2oC в течение 20 мин, а затем при 80oC в течение 11 мин. Реакционную смесь разделяют между этилацетатом и лимонной кислотой /в воде /10%/. Выделяют органическую фазу, промывают водой, сушат над сульфатом натрия и выпаривают досуха. Остаток хроматографически обрабатывают на силикагеле, элюируя этилацетатом в дихлорметане /1:9/ до получения ди-трет-бутилового сложного эфира 4-/N,N-бис/2-мезилоксиэтил/амино/ феноксикарбонид-L-глутаминовой кислоты /соединение 11 на схеме 9/ в виде масла /0,95 г/, /выход 28%/
ЯМР, δ : 1,41 /c, 9H/, 1,43 /c, 9H/, 1,8 - 1,99 /м, 1H/, 2,34 /м, 2H/, 3,16 /с, 6H/, 3,72 /т, 4H/, 3,9 /м, 1H/, 4,31 /т, 4H/, 6,78 /д, 2H/, 6,91 /д, 1H/, 7,9 /д, 1H/.
Пример 34
N-/4-/N,N-бис/2-бромэтил/амино/феноксикарбонил/-L-глуминовая кислота
Ди-трет-бутиловый сложный эфир 4-/N,N-бис/2-бромэтил/амино/феноксикарбонил/-L-глутаминовой кислоты /133 мг/ /см. схему 9-соединение /12/ /суспендируют в ТГФ /4 мл/ и перемешивают в течение 30 мин при комнатной температуре. ТГФ удаляют при пониженном давлении, оставшееся масло разбавляют этилацетатом /3 мл/ и выпаривают до получения 4-/N,N-бис/2-бромэтил/амино/феноксикарбонил-L-глутаминовая кислота - 1,3 TFA-0,9 EtOAc /126 мг/, /выход 80%/ /см. схему 9 соединения 13/.
ЯМР, δ : 1,83 - 2,01 /м, 1H/, 2,36 /м, 2H/, 3,58 /т, 4H/, 3,76 /т, 4H/, 4,03 /т, 1H/, 6,71 /д, 2H/, 6,94 /д, 2H/, 7,92 /д, 1H/.
Ди-трет-бутиловый сложный эфир 4-/N,N-бис/2-бромэтил/амино/феноксикарбонил-L-глутаминовую кислоту, которую используют в качестве промежуточного соединения, получают следующим образом.
Раствор ди-трет-бутилового сложного эфира бис/2-мезилоксиэтил/амино/феноксикарбонил-L-глутминовой кислоты /0,48 г/ /соединение 11 на схеме 9, полученное по способу примера 8, см. также схему 4, в 30 мл ацетонитрила перемешивают с литийбромидом /0,26 г/ при 70oC в течение 22 ч. Реакционную смесь фильтруют, а полученный фильтрат концентрируют в вакууме. Остаток обрабатывают хроматографически на силикагеле, элюируя смесью этилацетат в дихлорметане /1: 5/ до получения ди-трет-бутилового сложного эфира 4-/N,N-бис/2-бромэтил/амино/феноксикарбонил-L-глутаминовой кислоты /соединение 12 на схеме 9/ в виде масла /0,37 г/ /выход 83%/.
ЯМР, δ : /с, 9H/, 1,43 /с, 9H/, 1,8 - 1,98 /м, 1H/, 2,34 /м, 2H/, 3,58 /т, 4H/, 3,76 /т, 4H/, 3,97 /м, 1H/, 6,72 /д, 2H/, 6,93 /д, 2H/, 7,93 /д, 1H/.
Пример 35
N-/4-/N,N-бис/2-хлорэтил/амино/2-фтор-фенилкарбамоил/-L- глутамат
Указанное в заглавии соединение //ЯМР /ДМСО/: δ : 8,0 /с, 1H/, 7,65 /т, 1H/, 6,6 /м, 3H/, 4,2 /м, 3H/, 3,7 /с, 8H/, 2,28 /м, 2H/, 1,8 /м, 2H// получают из промежуточного дибезнол-4-/N, N-бис/2-хлорэтил/амино/2-фторфенилкарбамоил-L-глутамата /ЯМР/ДМСО/: 7,62 /т, 1H/, 7,35 /с, 10H/, 6,8 /д, 1H/, 6,57 /м, 2H/, 5,1 /д, 4H/, 4,33 /м, 1H/, 3,7 /с, 8H/, 2,43 /м, 2H/, 2,0 /м, 2H/ способом, аналогичным соответствующей стадии примера 5.
Промежуточное соединение получают следующим образом.
Этиленоксид /6,6 г/ добавляют к 3-фтор-4-нитроанилину /1,3 г/ в ледяной уксусной кислоте /30 мл/, и реакционную смесь оставляют в закрытой колбе при комнатной температуре на 72 часа. Раствор выпаривают при пониженном давлении до половины исходного объема, разбавляют насыщенным водным раствором хлористого натрия, и трижды экстрагируют этилацетатом. Этилацетатные экстракты объединяют, промывают насыщенным водным бикарбонатом натрия, выпаривают, и остаток очищают с помощью флеш-хроматографии на колонке с силикагелем. После элюирования гексаном, содержащим 50% /объем/объем/ этилацетата для удаления непрореагировавшего исходного материала и монозамещенного продукта. После элюирования этилацетатом получают 2', 2'-/3-фтор-4-нитроанилино/диэтанол /Т. пл. 99 - 101oC/.
Полученный продукт /280 мг/ растворяют в 7,5 мл дихлорметана, 0,1 мл пиридина добавляют, и полученный раствор охлаждают на бане лед/вода. 0,025 мл тионилхлорида добавляют по каплям при перемешивании. После завершения добавления реакционную смесь нагревают при кипячении с обратным холодильником в течение 1 часа, а затем оставляют при комнатной температуре на 20 ч. Полученный раствор разбавляют 10 мл дихлорметана и трижды промывают водой, сушат над сульфатом натрия, выпаривают, а остаток перекристаллизовывают из метанола до получения продукта - /N,N-бис/2-хлорэтил/-3-фтор-4-нитроанилина. Т.пл. 97 - 98oC.
Полученный таким образом продукт /200 мг/ в 7,5 мл тетрагидрофурана перемешивают в течение 16 ч в атмосфере водорода в присутствии палладия на угле /20 мг 5% вес/вес/. Катализатор удаляют фильтрованием, и полученный раствор выпаривают. Полученный остаток растворяют в минимальном объеме метанола, а неочищенный продукт осаждают, добавляя избыток диэтилового эфира, насыщенного хлористым водородом. После перекристаллизации из метанол/диэтилового эфира получают продукт -4-/N,N-бис/2-хлорэтил/амино-2-фторанилинийхлорид /т. пл. 195 - 200oC, с разложением.
Полученный продукт превращают в целевое промежуточное соединение способом, аналогичным способу соответствующей стадии примера 5.
Примеры 36 - 43
Структуры и данные элементного анализа для соединений примеров 36 - 43 представлены в табл. 17 (см. в конце описания).
Соединения, перечисленные в таблице 17, получают по способу примера 16. Так соединение примера 36 получают, заменяя анилин в примере 16 на бензил-34-аминобензоат /Aldrich Chemical Co Ltd/. Аналогично, соединения примеров 37 - 43 получают, заменяя анилин примера 16 на бензил-4-аминобензоат, втор-бутиламин, н-пропиламин, изо-пропиламин, циклогексиламин, бензиламин или р-бензилоксианилин.
Пример 44
N-4-/N,N-бис/2-хлорэтил/амино/фенилкарбамоил/-L-глутаминовая кислота
Раствор промежуточного дибензил-4-/N,N-бис-/2-хлорэтил/амино/ фенилкарбамоил-L-глутамата /1,138 г/ в ДМФ /15 мл/ гидрируют над 10% Pd/C в течение 16 ч. После фильтрования и выпаривания в вакууме, остаток растворяют в CHCl3 /20 мл/. Спустя 18 ч кристаллический осадок отфильтровывают и сушат в вакууме до получения N-4-/N,N-бис/2-хлорэтил/амино/фенилкарбамоил/-L-глутаминовой кислоты. Выход 730 мг /93%/. После перекристаллизации из смеси ацетон /CHCl3/ образуются микроскопические палочки. Т.пл. 116 - 118oC.
ЯМР /CD3COCD3/ δ : 8,0 /с, 1H/, 7,2 /д, 2H/, 6,6 /д, 2H/, 6,2 /д, 2H/NH, 4,4 /м, 1H/, 3,6 /м, 8H/, 2,5 - 1,9 /м, 4H/.
Дибензильное призводное получают следующим образом /схема 10/ p-толуолсульфонат дибензилглутамата /доступен от Bachem U.K 0,25 г/ растворяют в сухом метиленхлориде /10 мл/ в атмосфере аргона и при охлаждении до 0oC. Добавляют 0,162 мл пиридина с последующим быстрым добавлением фосгена в толуоле /1,93М, 0,311 мл/. Раствор перемешивают при 0oC в течение 2 ч, добавляют 0,05 мл пиридина, а затем сразу весь N-4-/N,N-бис/2-хлорэтил/амино/анилинийхлорид. Полученную смесь перемешивают в течение 10 мин при 0oC, а затем в течение 18 ч при комнатной температуре. Затем реакционную смесь разбавляют этилацетатом и водой. Органический слой промывают по очереди разбавленной лимонной кислотой /2х/, водой и насыщенным рассолом; сушат и выпаривают до получения целевого дибензильного промежуточного соединения в виде твердого продукта.
Возможен альтернативный способ получения дибензильного производного.
К раствору трифосгена /1 г/ в 80 мл хлорформа добавляют при 10oC 4-/N, N-бис/2-хлорэтил/амино/анилинийхлорид /2,7 г/. Поддерживая температуру при 10oC, добавляют 4,15 мл триэтиламина, и полученную смесь перемешивают и оставляют нагреваться до комнатной температуры в течение 15 мин. К этой смеси добавляют сразу весь тозилат дибензилглутамата и триэтиламин /1,7 мл/. Спустя 1,5 часа при комнатной температуре полученную смесь разбавляют 100 мл хлороформа, дважды промывают водой, сушат и выпаривают досуха. Остаток обрабатывают хроматографически на силикагеле Merck Aгт 9385, элюируя этилацетатом и гексаном до получения дибензил-4-/N,N-бис/2-хлорэтил/амино/-фенилкарбамоил-L-глутамата /2,5 г/. Т. пл. 119 - 122oC.
Пример 45
4-/N, N-бис/2-хлорэтил/амино/фенил/карбамоил- γ -/N/3-карбоксиметил/анилино/-L-глутамат /соединение 7 на схеме 11/.
Указанное в заглавии соединение получают следующим образом /см. схему 11/. Раствор промежуточного соединения /соединение 6 на схеме 11, 1 г/ в 20 мл ТГФ гидрируют над 30% Pd/C /100 мг/ в течение 4 ч. Полученную смесь фильтруют через целит и выпаривают до получения масла коричневого цвета, которое затем очищают с помощью флеш-хроматографии, используя 1 и 3% смеси муравьиной кислоты /этилацетата в качестве элюента, выпаривают соответствующие фракции, и после взаимодействия с эфиром получают 300 мг указанного в заглавии соединения.
ЯМР /ClCH2CH2N/ δ : 3,66 /с, 8H/; ароматика, 6,66 - 7,46 /м, 8H/, α CH: 4,22 /м, 1H/, остальные 1,66 - 2,06 /м, 2H/, 2,39 /м, 2H/, 3,49 /с, 2H/, 6,29 /д, 1H/, 8,33 /с, 1H/, 9,93 /с, 1H/, 12,5 /шир.с, 2H/.
Промежуточное соединение получают следующим образом:
/a/ соль присоединения p-толуолсульфоновой кислоты и бензолового эфира 3-амино-фенилукуксусной кислоты получают, добавляя 3-аминофенилуксусную кислоту /10 г/ моногидрат p-толуолсульфоновой кислоты /13,2 г/ к бензиловому спирту /27,2 мл/ в 30 мл толуола. Полученную смесь кипятят с обратным холодильником, а образующуюся воду собирают ловушкой Дина-Старка. После того, как отгоняют всю воду, смесь оставляют охлаждаться до 25oC перед разбавлением диэтиловым эфиром, и затем помещают на 1 час на баню со льдом. Кристаллический p-толуолсульфонат отфильтровывают, а полученный продукт сушат в эксикаторе /23,5 г/.
ЯМР, δ : 2,31 /c, 3H/, 3,82 /c, 2H/, 5,11 /c, 2H/, 7,11 /д, 2H/, 7,25 - 7,45 /м, 8H/, 7,52 /д, 2H/.
/b/ N-Boc-бензил-L-глутамат /5 г/ в 20 мл сухого ДМФ при 5oC нагревают с /1,1 экв. 2,45 г/ гидроксибензотриазола /HOBT/, и реакционную смесь перемешивают при этой температуре в атмосфере аргона в течение 10 мин. /DCCl/ Дициклогексилкарбодиимид /1,1 экв. , 3,37 г/ добавляют затем, и реакционную смесь перемешивают в течение еще мин при 5oC перед тем, как дать ей нагреться до 25oC, и перемешивают еще 45 мин. Соль присоединения p-толуолсульфоновой кислоты и бензилового эфира аминофенилуксусной кислоты /продукт, полученный в /a// /1,1 экв., 6 г/, вместе с 1,1 экв. триэтиламина /2,23 мл/ в 10 мл сухого ДМФ добавляют, и реакционную смесь перемешивают при 25oC в течение еще 20 ч. Осадок дициклогексилмочевины отфильтровывают, и ДМФ фильтраты выпаривают досуха. Затем остаток снова растворяют в EtOAc. Этилацетатный раствор промывают NaHCO3 /водным/, затем рассолом, сушат над безводным сульфатом натрия.
После выпаривания этилацетатных экстрактов получают остаток, который затем очищают с помощью флеш-хроматографии, используя 30, 40 и 50% EtOAc /гексановые смеси в качестве элюента. После выпаривания соответствующих фракций получают 5 г продукта /соединение 4 на схеме 11/.
ЯМР, δ 1,39 /с, 9H/, 1,81-2,10 /м, 2H/, 2,38 /м, 2H/, 3,69 /с, 8H/, 4,03 /м, 1H/, 5,12 /м, 4H/, 6,92 /д, 1H/, 7,21 /м, 1H/, 7,35 /м, 11H/, 7,42 /д, 1H/, 7,52 /с, 1H/, 9,89 /с, 1H/.
/c/ Продукт, полученный в /b/ /5 г/ суспендируют в 10 мл эфира и добавляют 5 мл дихлорметана /для повышения растворимости/, а затем 8 экв. насыщенного эфирного HCl. Затем реакционную смесь оставляют при перемешивании на 20 ч при 25oC. Полученный на этой стадии продукт представляет несмешивающееся масло. Затем эфир выпаривают, а остаток дважды азеотропно перегоняют с толуолом перед добавлением эфира и упаривают до получения 5 г продукта /соединение 5 на схеме 11/ в виде желтой пены.
ЯМР /CDCl3/ δ : 2,35 /м, 1H/, 2,65 /м, 2H/, 3,50 /с, 2H/, 4,22 /шир. с , 1H/, 5,01 /с, 2H/, 6,8-7,6 /м, 14H/, 8,65 /шир. с., 3H/, 9,35 /с, 1H/.
/d/ 1,1 экв. /0,66 г/ 1,1-карбонилдиимидазола в 20 мл сухого ТГФ при 5oC обрабатывают раствором, содержащим 1 г 4-/N,N-бис/2-хлорэтил/амино/анилинийхлорида. Реакционную смесь перемешивают затем при 5oC в течение 15 мин перед добавлением 1 экв. /1,84 г/ продукта, полученного в /c/, с 1,1 экв. /0,56 мл/ триэтиламина в 10 мл сухого ТГФ, и перемешивают в течение еще 2 ч при 25oC. Остаток триэтиламингидрохлорида отфильтровывают, и ТГФ фильтрат выпаривают, а остаток снова растворяют в EtOAc. Затем этилацетатный раствор промывают водой, 0,25 M лимонной кислотой, рассолом, и сушат над безводным сульфатом натрия. После выпаривания этилацетата получают остаток, который очищают с помощью флеш-хроматографии, используя 30, 40 и 50% этилацетат/гексановые смеси в качестве элюента. После выпаривания соответствующих фракций получают целевое промежуточное соединение /соединение 6 на схеме 11/,
ЯМР /CDCH2CH2N/ δ : 3,65 /с, 8H/; ароматика; 6,63-7,5 /м, 18H/, ArCH2O: 5,10 /с, 2H/, 5,12 /с, 2H/, α CH; 4,32 /м, 1H/, остальные: 1,83-2,13 /м, 2H/, 2,41 /м, 2H/, 6,42 /д, 1H/, 8,25 /с, 1H/, 9,91 /с, 1H/.
Пример 46
4-/N, N-бис/2-хлорэтил/амино/феноксикарбонил-L-глутаминовой кислоты γ -/3,5-дикарбокси/анилид
По способу примера 16, используя α -бензил-4-/N, N-бис/2- хлорэтил/амино/феноксикарбонил-L-глутаминовой кислоты γ -/3,5-дикарбоксибензил/анилид вместо α -бензил-4-/N,N-бис/2- хлорэтил/амино/феноксикарбонил-L-глутаминовой кислоты γ -анилида получают γ -/3,5-дикарбокси/анилид-4-/N, N-бис/2-хлорэтил/- амино/феноксикарбонил-L-глутаминовой кислоты в виде бесцветных кристаллов /Т. плавления 167-170oC/.
Элементный анализ:
Рассчитано: C 49,6; H 4,87; N 6,68
Найдено: C 49,7; H 4,9; N 6,7
α -бензил-4-бензил-4-/N, N-бис/2-хлорэтил/амино/- феноксикарбонил-L-глутаминовой кислоты- γ -/3,5-дикарбоксибензил/- анилид получаю способом, аналогичным способу примера 16 для γ анилино производного.
Данные In Vivo
Антиопухолевая активность Lo Vo опухолевых ксенотрансплантантов
Направляемая антителами ферментов пролекарственная терапия (Antibody-directed enzyme prodrug therapy, ADEPT) была разработана как двухкомпонентная нацеливающая система для лечения рака.
"ZD2767"
Первый компонент представляет собой конъюгат фрагмента F(ab')2 моноклонального антитела A5B7 и фермента карбоксипептидазы G2.
Второй компонент представляет пролекарственное соединение N-{4-[N,N-бис(2-иодэтил)амино]феноксикарбонил}-L-глютаминовую кислоту.
"ZD9063"
Первый компонент представляет собой конъюгат фрагмента F(ab')2 моноклонального антитела A5B7 и фермента карбоксипептидазы G2.
Второй компонент представляет пролекарственное соединение N-{4-[N,N-бис(2-хлорэтил)амино] феноксикарбонил} -L-глютамино-гамма- (3,5-дикарбокси)анилид.
Антиопухолевые испытания
В опытах были использованы ксенотрансплантанты опухоли Lo Vo прямой кишки человека, выращенные в голых мышах без зобной железы (in athymic nube mice). По данным иммуногистологии примерно 60% опухолевых клеток экспрессировали CEA. В пальпируемые опухоли (5-6 мм в диаметре) вводили первый компонент и спустя 3 дня, когда опухоли достигали примерно 8-9 мм в диаметре, вводили внутрибрюшинно 0,2-0,3 г пролекарственного соединения в виде трех доз с часовым интервалом за 2 ч.
Антиопухолевый эффект на ксенотрансплантаты Lo Vo показан в таблице 18 (см. в конце описания) и на чертеже. Данные показывают, что ZD2767 и ZD9063 обладают значительной антиопухолевой активностью In vivo. Тн
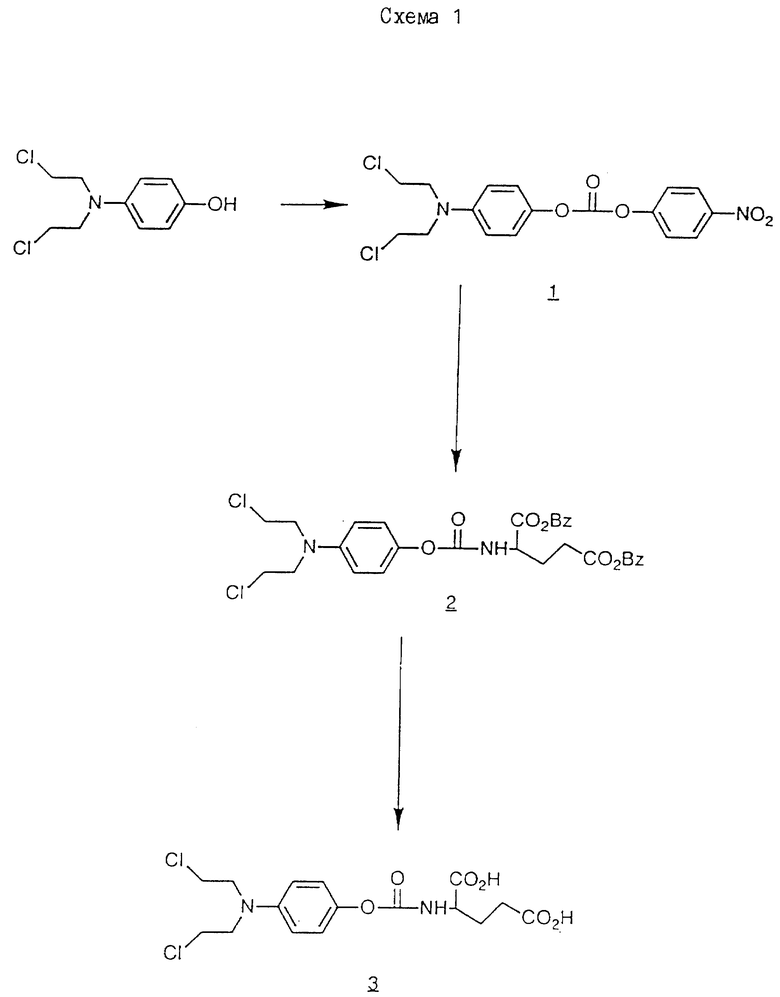
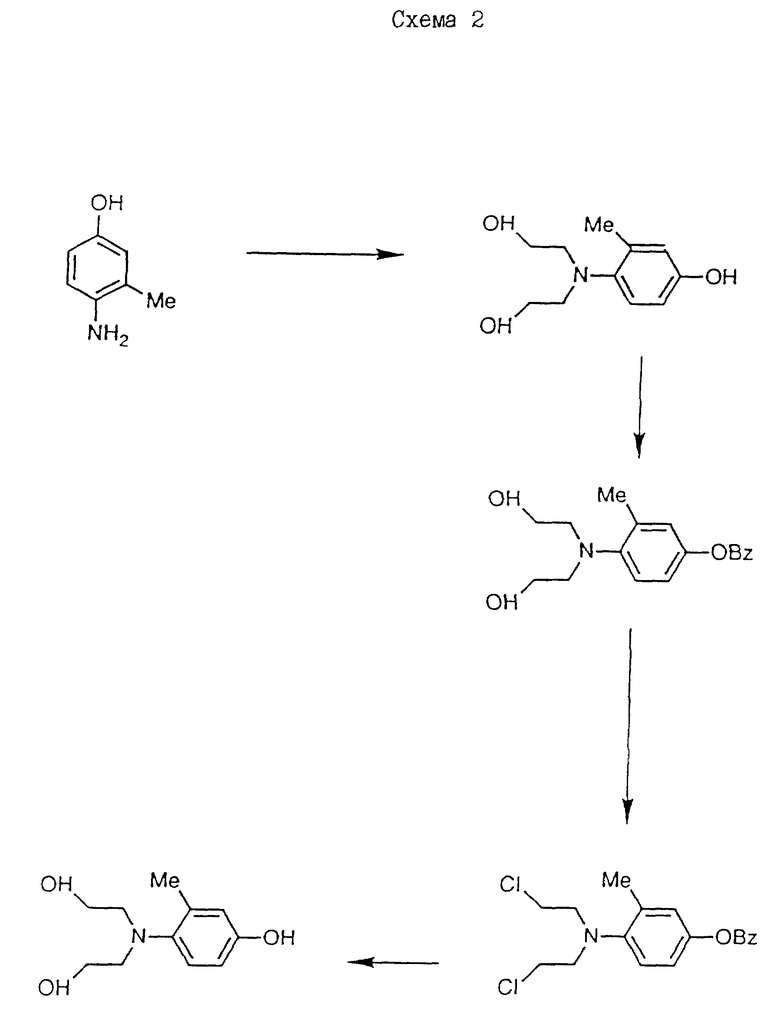

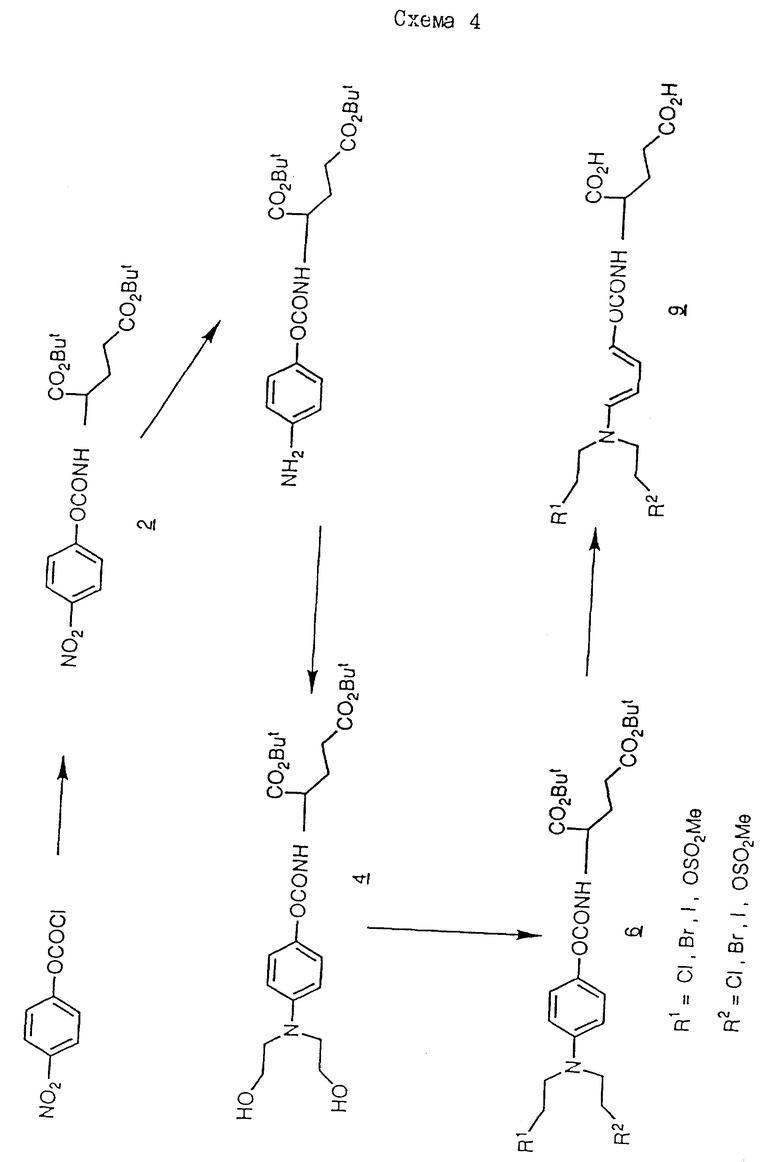
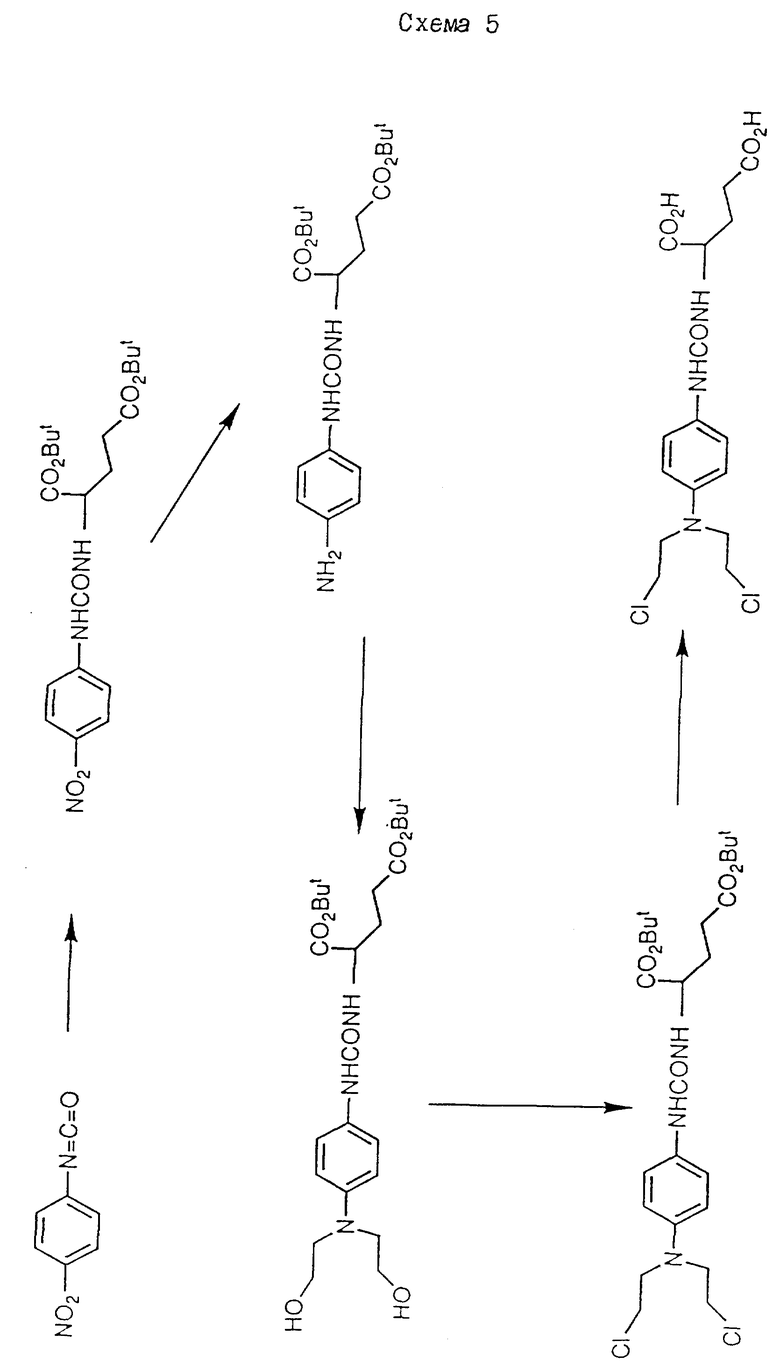
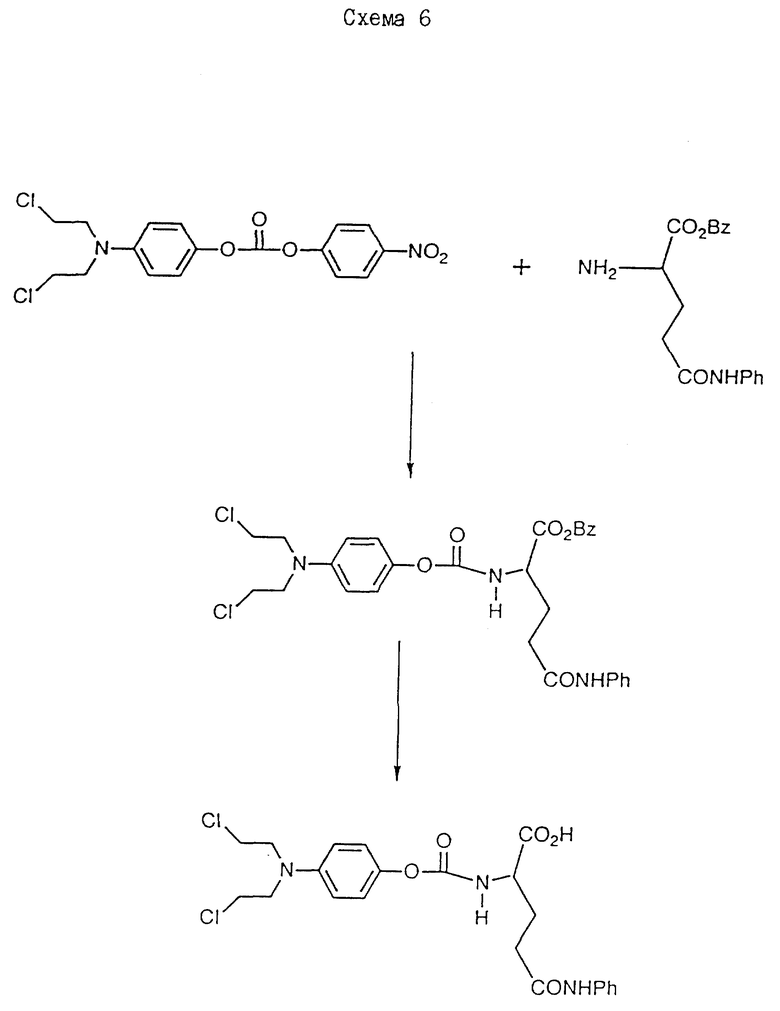
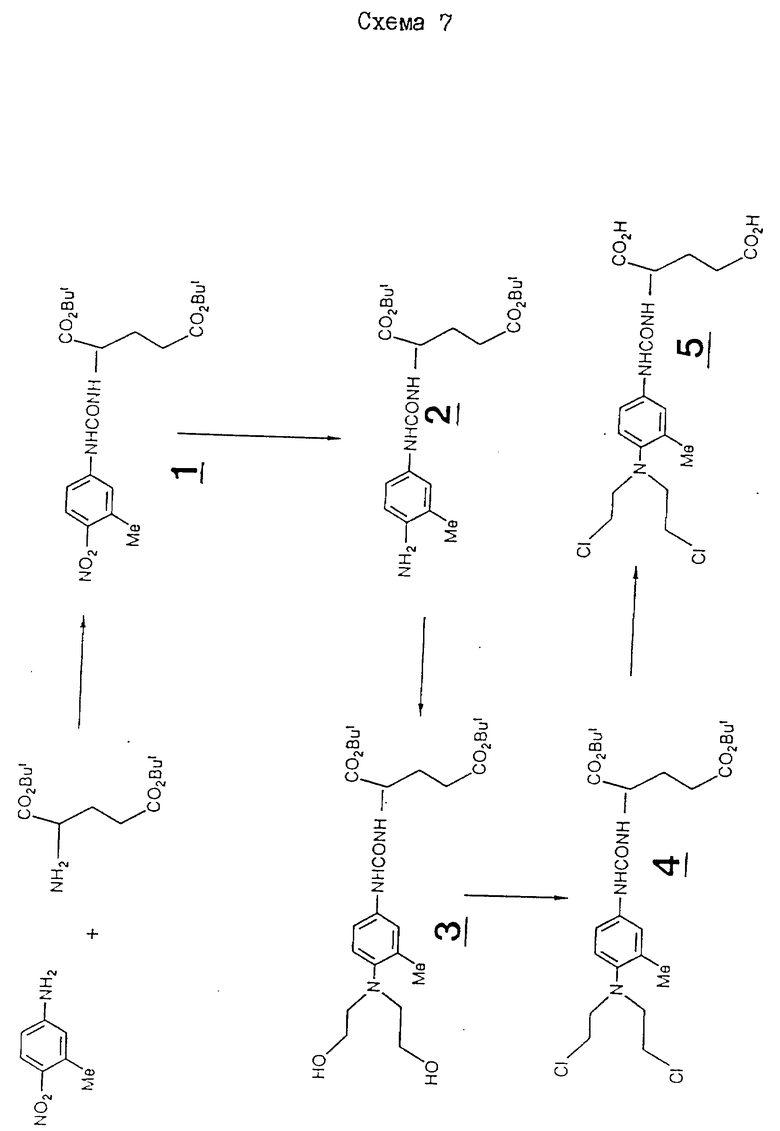
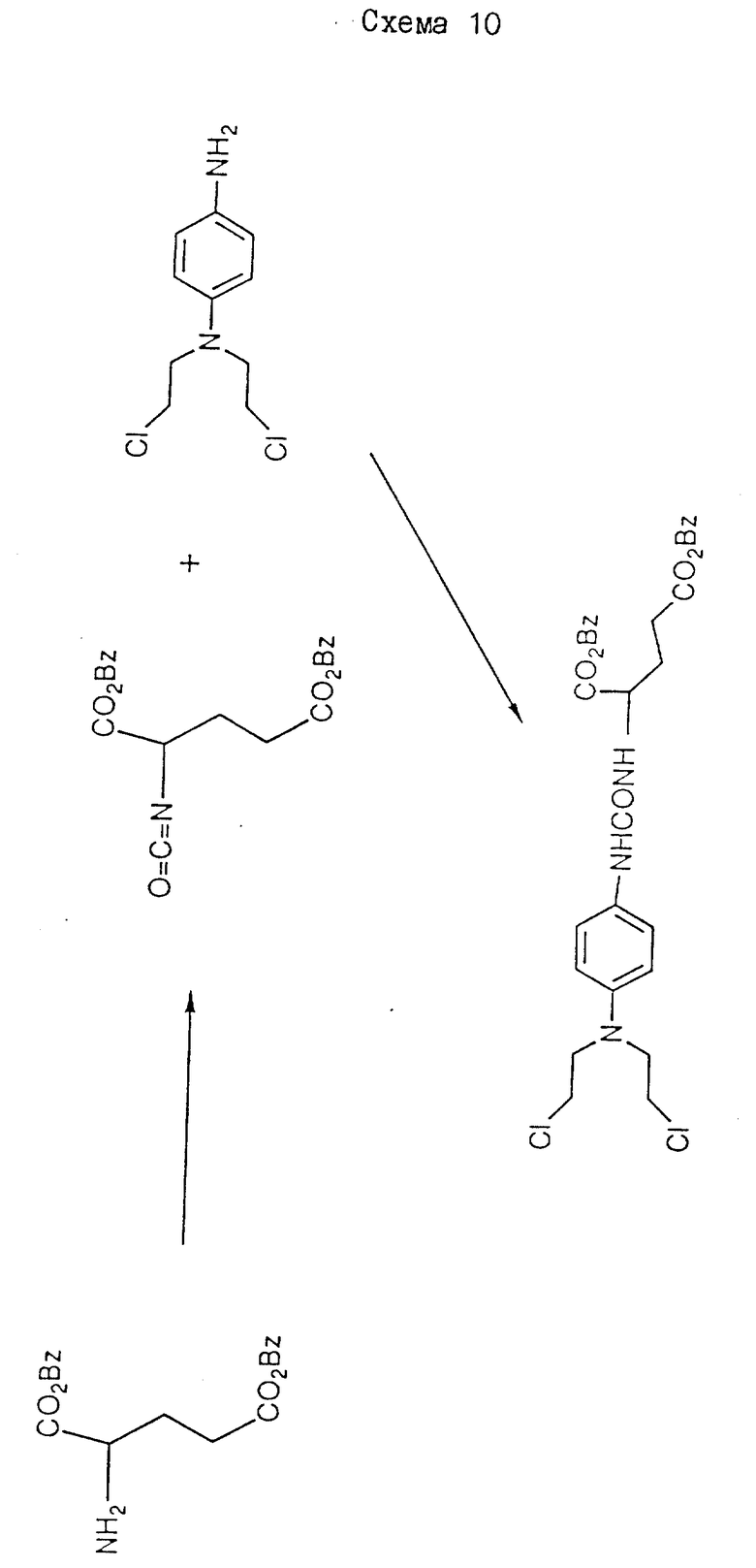
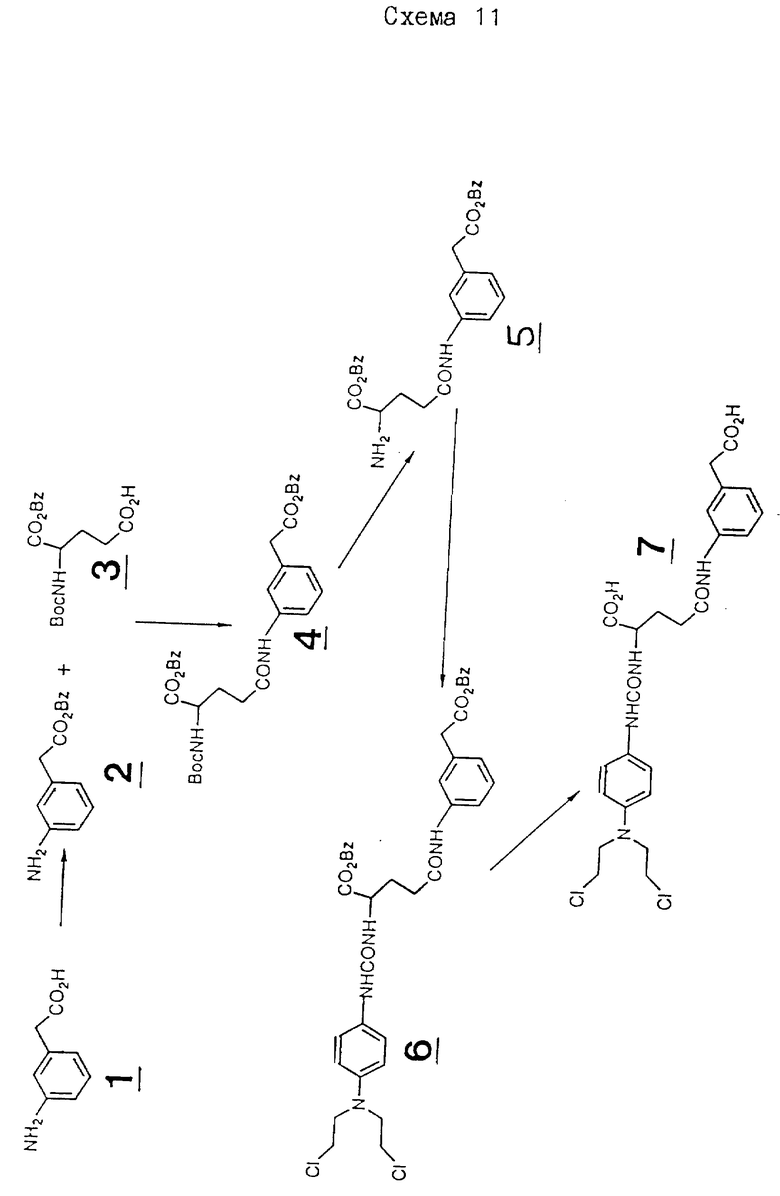
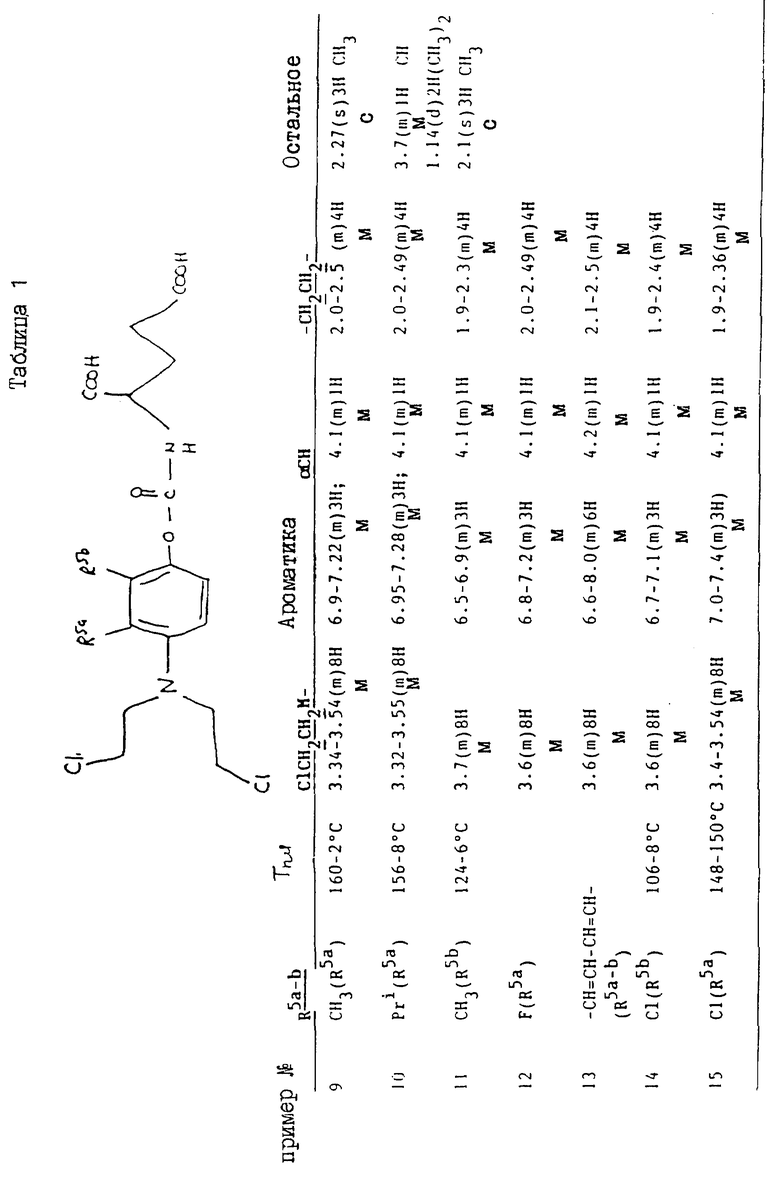

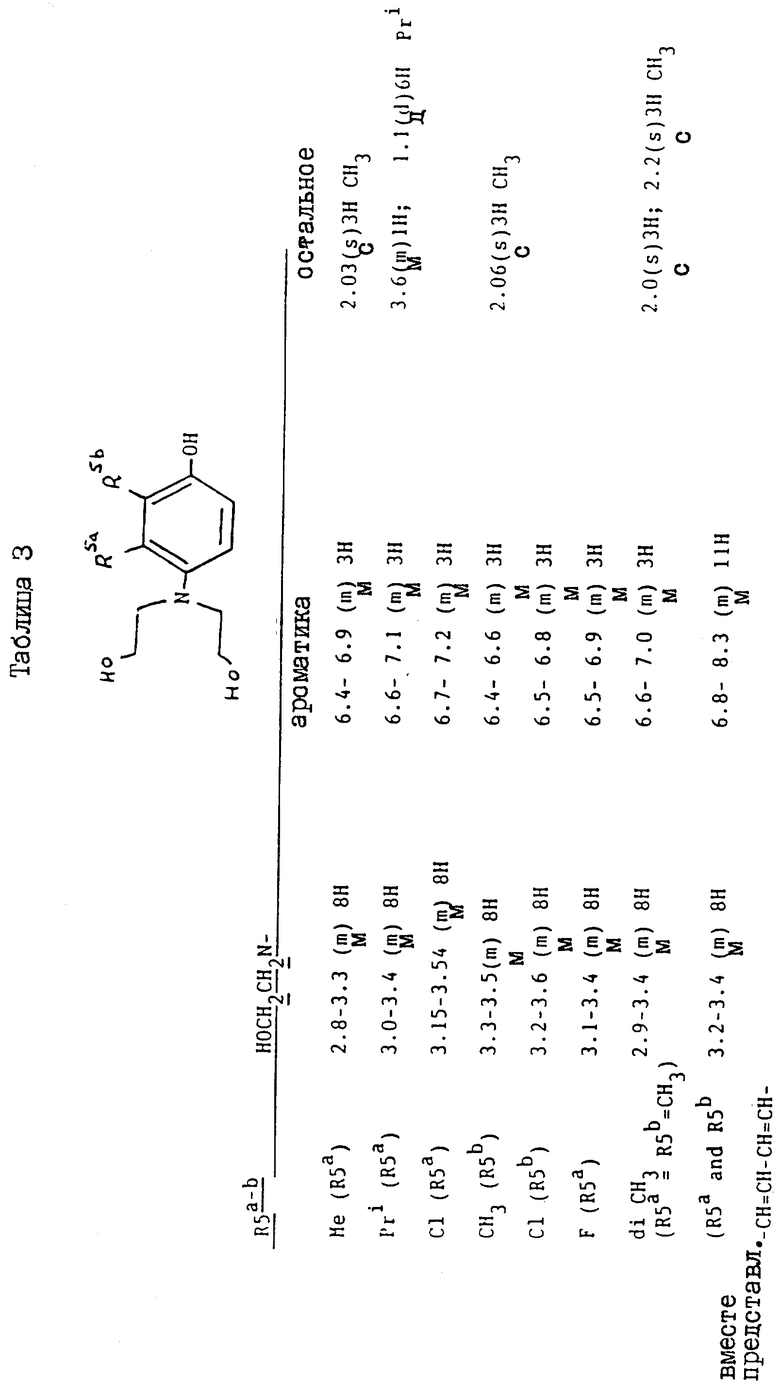
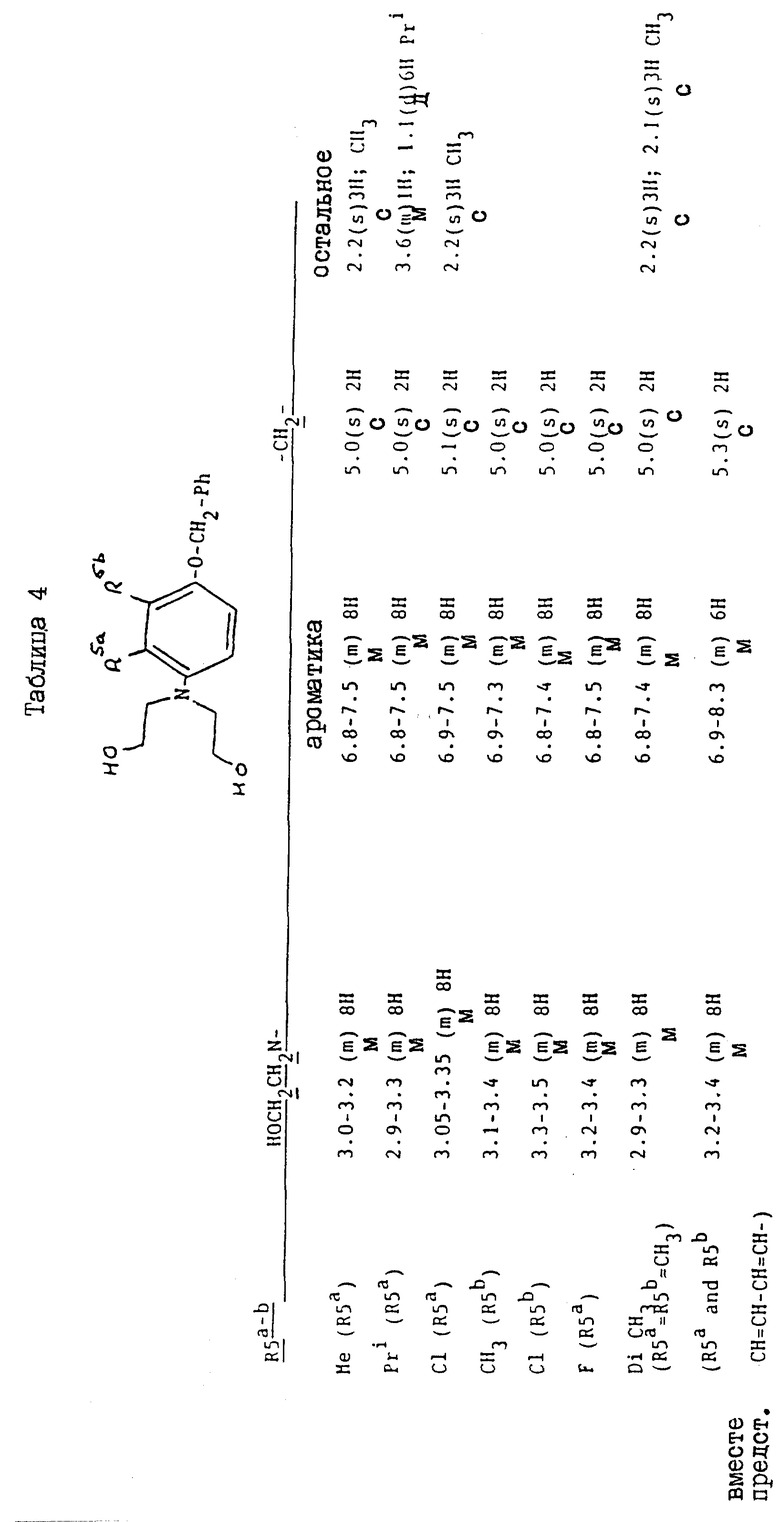
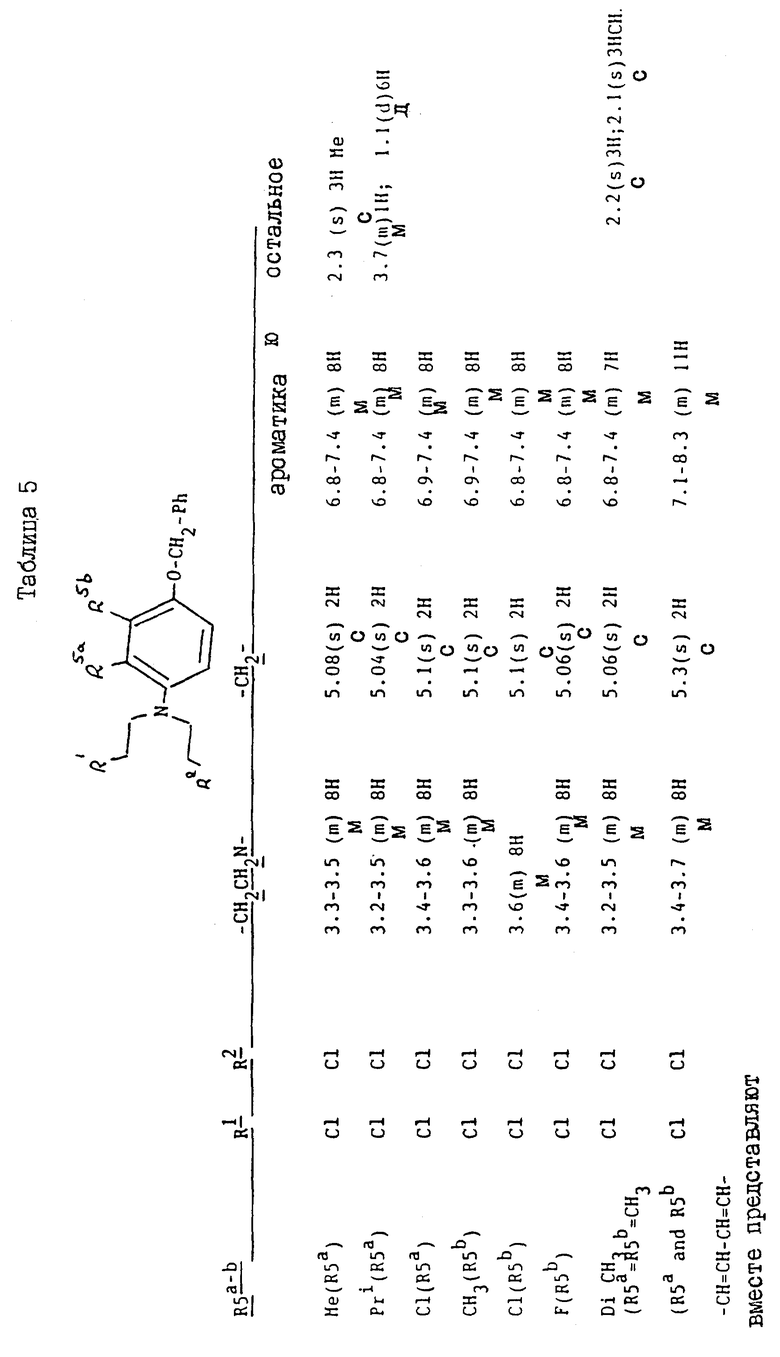
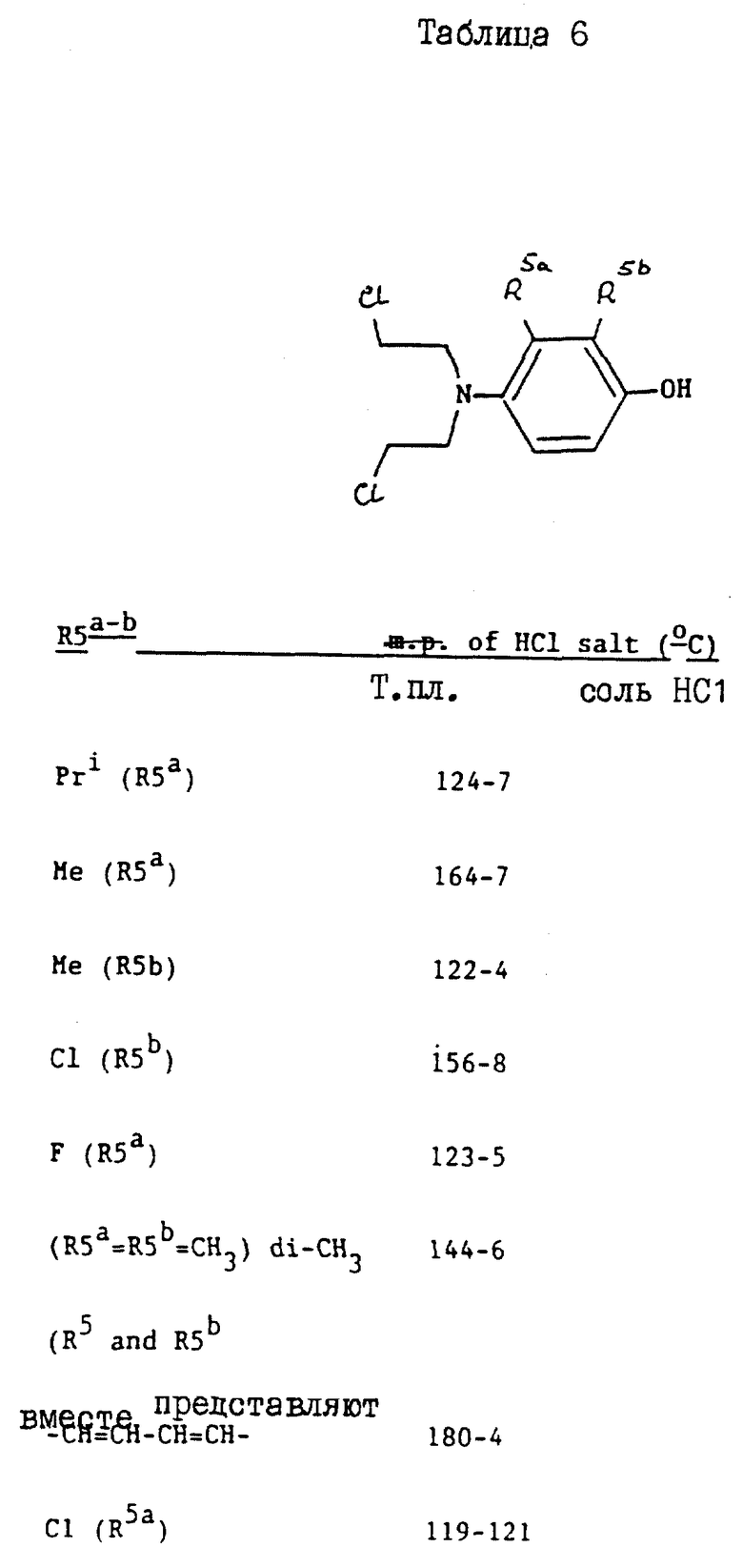
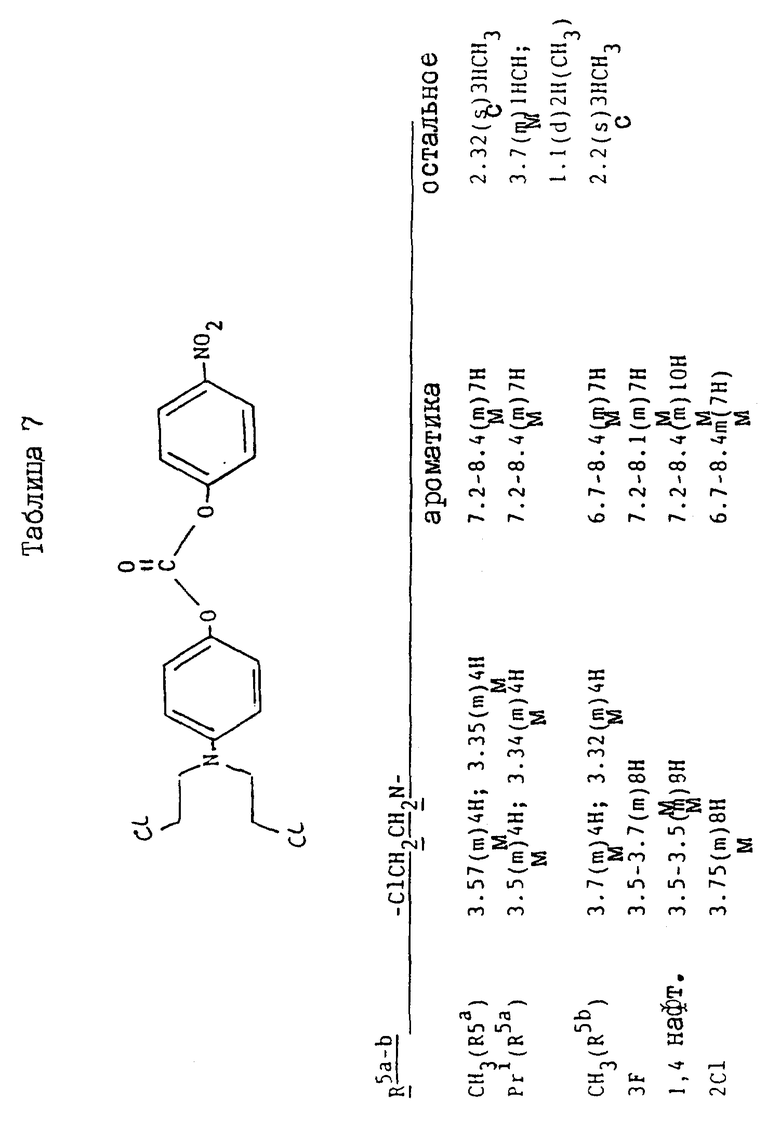
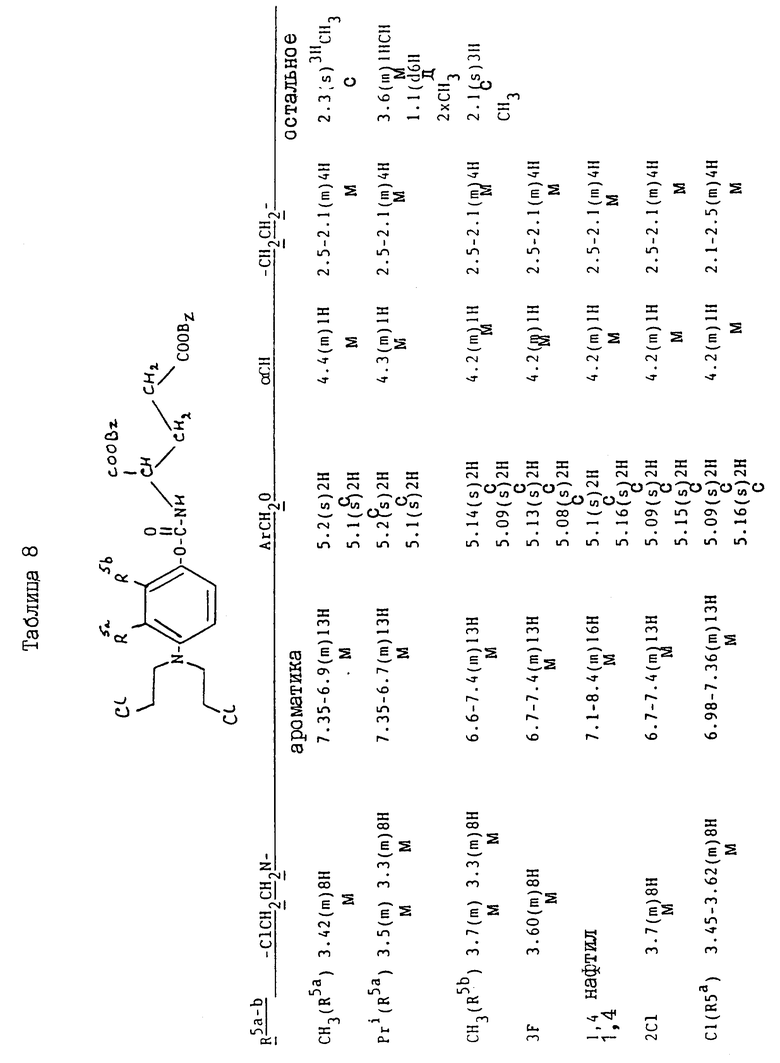
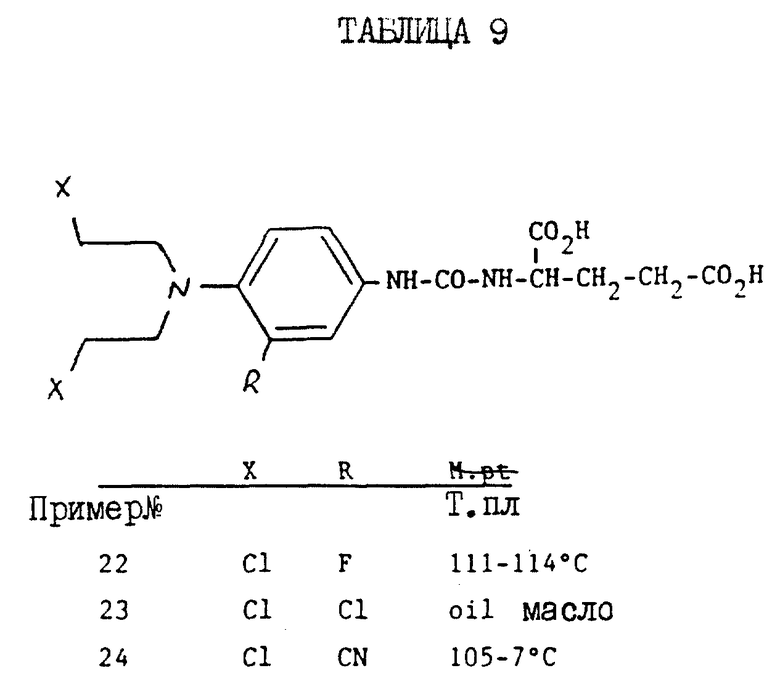
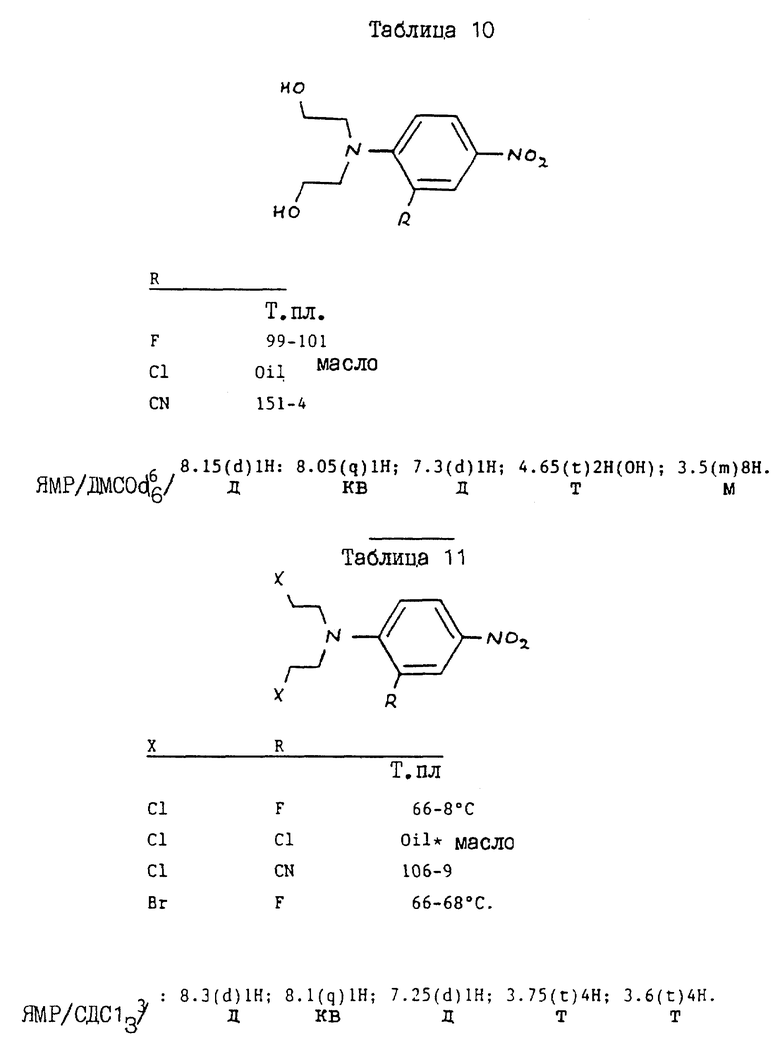
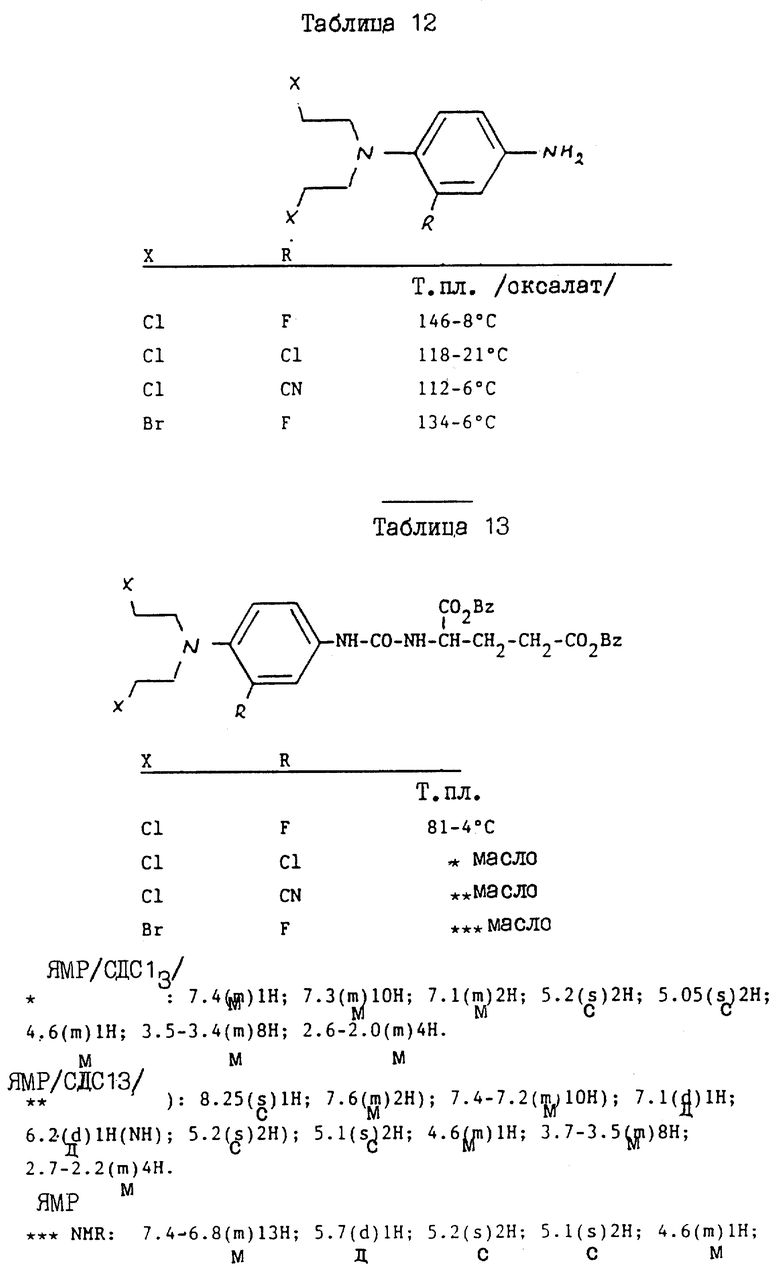
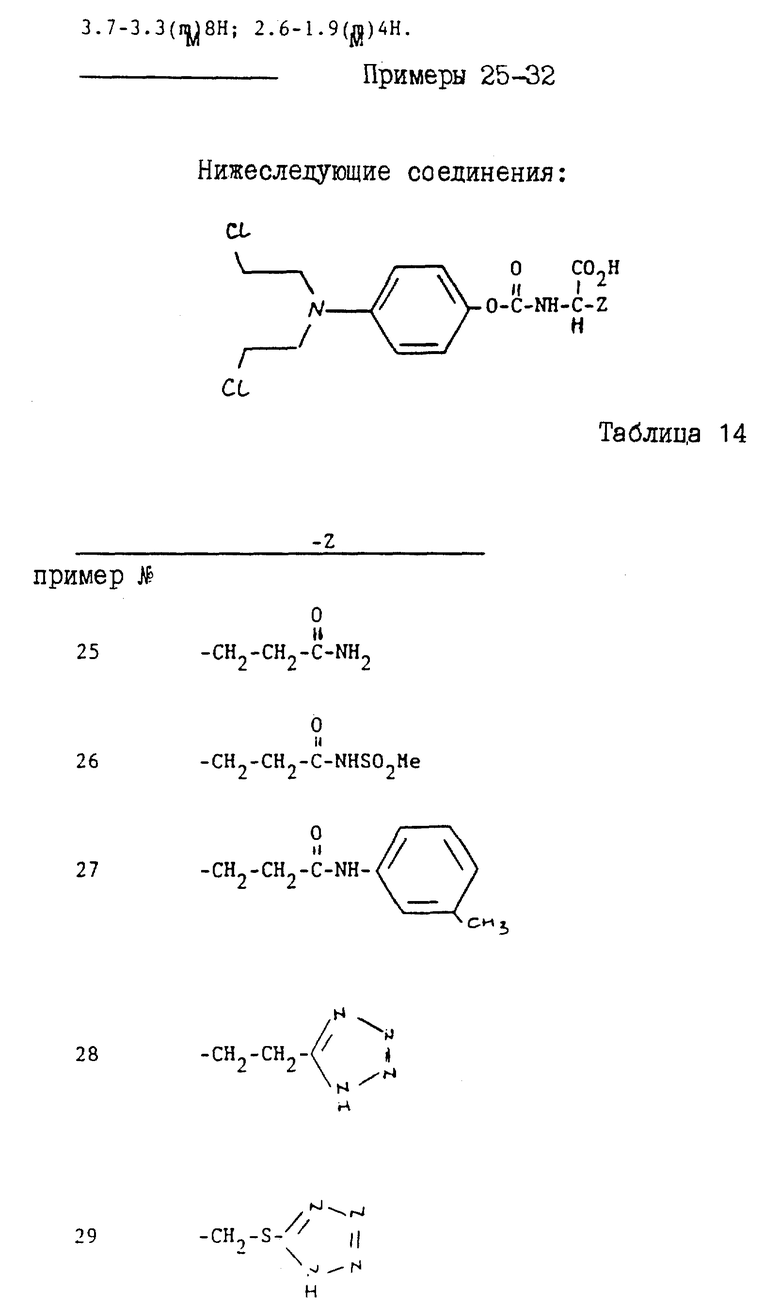
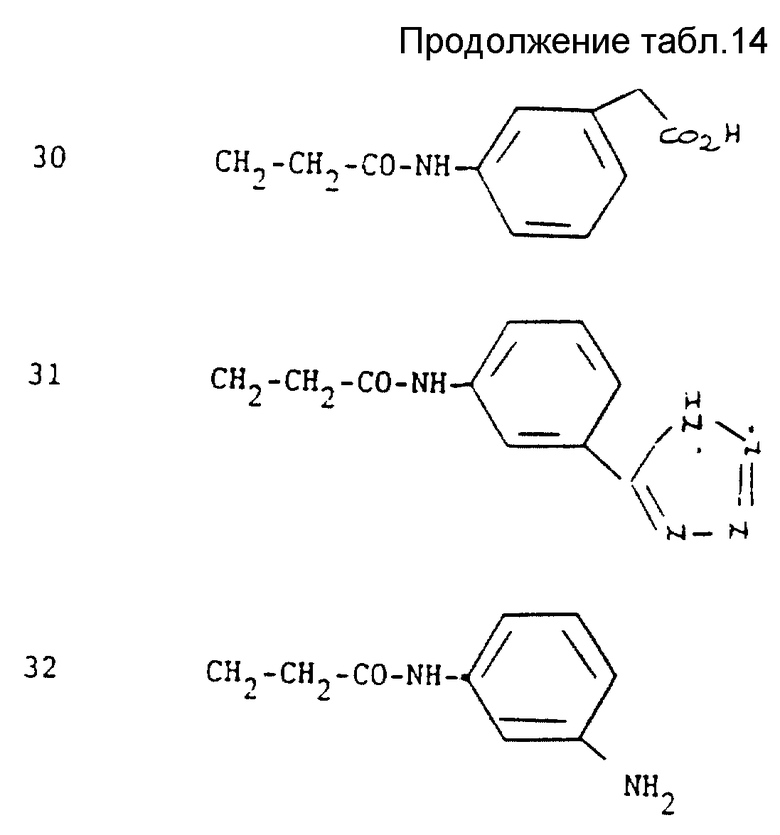




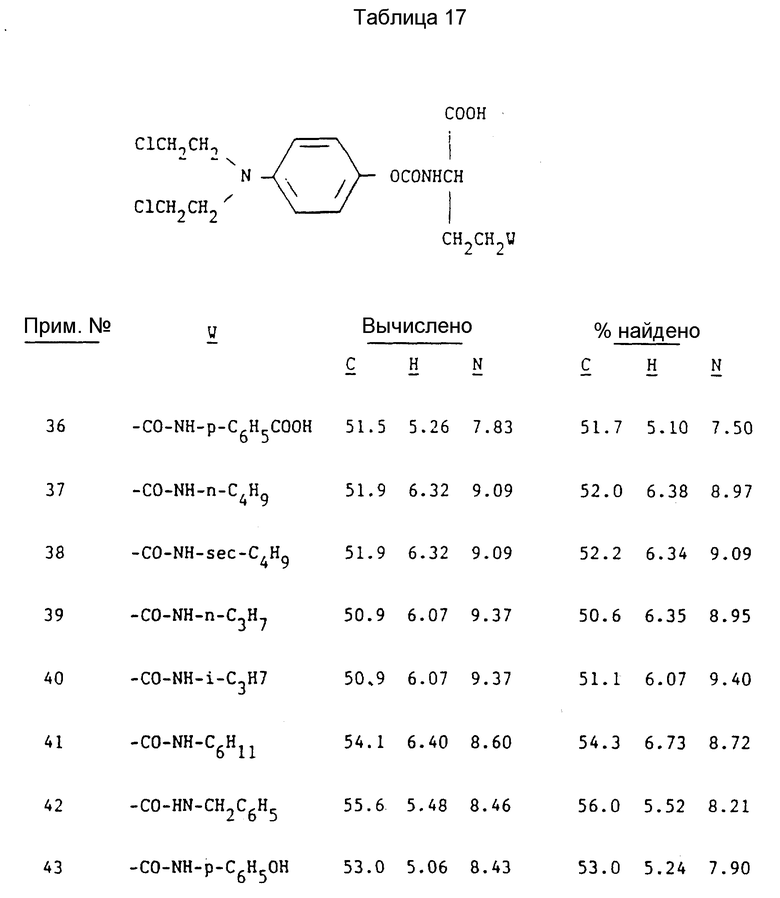

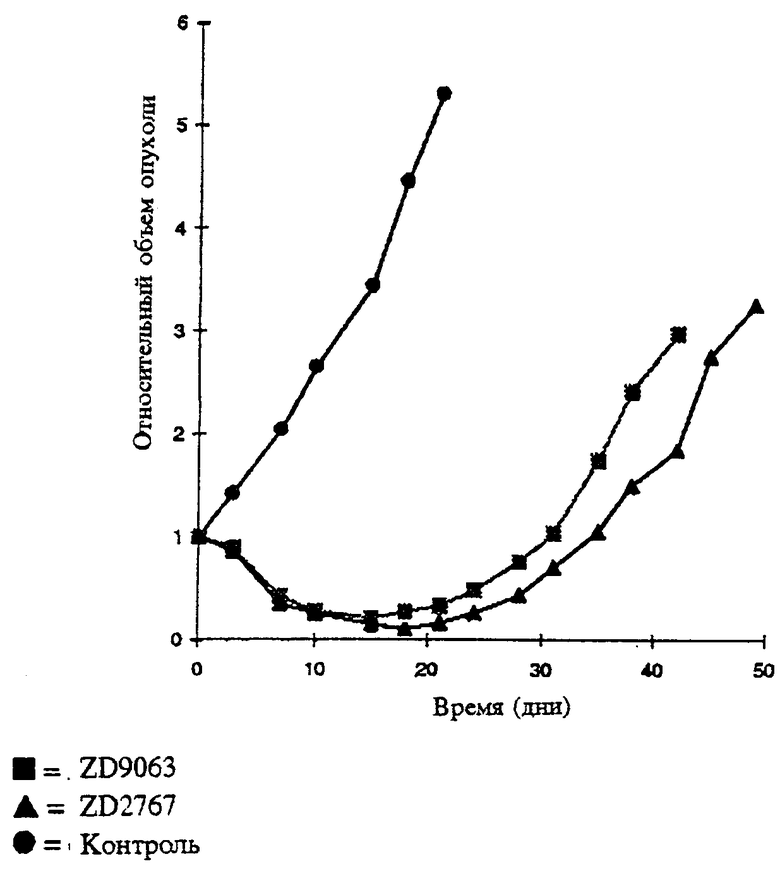
Изобретение касается новых соединений формулы I, где R1 и R2 каждый независимо представляет хлор, бром, иод или OSO2Me; R1a и R2b - водород; R3 и R4 - водород; R5a и R5b -водород, C1-4-алкил, галоид; R5a и R5b вместе -СН= СН-СН= СН-; R5c и R5d - водород, циано, галоид; Х представляет O, NH, -СН2-; Y представляет O; Z представляет -V-W, где V представляет -СН2Т-, в которой Т представляет -CH2- или -S-; W представляет СООН, -(С=O)NR7R8, где R7 - водород, C1-6-алкил и др.; R8- водород, и их соли. Способ получения соединений формулы I заключается в удалении защиты у соединения формулы Iа. Предложены фармацевтическая композиция, содержащая эффективное количество соединения формулы I, а также двухкомпонентная система доставки цитотоксического лекарства, содержащая антитело или его фрагмент, способные связывать определенный антиген, причем антитело или его фрагмент конъюгированы с энзимом СРG, способным превращать соединение формулы I или его соль в цитотоксическое лекарство, и содержит дополнительно соединение формулы I или его соль, превращаемые под действием энзима СРG в цитотоксическое лекарство. 4 c. и 12 з.п ф-лы, 18 табл., 1 ил.
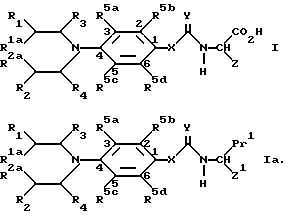
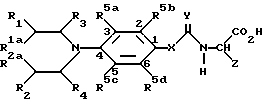
где R1 и R2 каждый независимо представляет хлор, бром, иод или OSO2Me;
R1a и R2a каждый независимо представляет водород;
R3 и R4 каждый независимо представляет водород;
R5a и R5b каждый независимо представляет водород С1-4-алкил, галоид или R5a и R5b вместе представляют -CH=CH-CH=CH;
R5с и R5d представляют водород, циано, галоид;
Х представляет 0, NH или -CH2-;
Y представляет 0;
Z представляет -V-W, где V представляет -CH2-Т-, в которой Т представляет -CH2- или -S- (при условии, что когда вторым атомом в Y является сера, группа W отлична от -COOH), и W представляет собой: i) -COOH; ii) -(C=O)-NR7R8, где R7 представляет водород или С1-6-алкил, С3-6-циклоалкил; фенил- или С7-9-фенилалкил, и где фенильная группа сама по себе или фенильная группа в С7-9-фенилалкильной группе могут иметь от одного до четырех заместителей, выбираемых из группы, включающей -COOH, -ОН, NH2, -(CH2)1-4-COOH и тетразол-5-ил; R8 представляет водород; iii) тетра-ол-5-ильную группу; iv) -CONH-SO2R11, где R11 представляет С1-6-алкил,
и соли указанного соединения формулы I, при условии, что исключаются следующие соединения:
2-(2-{4-[бис-(2-хлорпропил)-амино]-фенил}-ацетиламино)-пентандиоевая кислота;
2-(2-{ 4-[бис-(2-хлорэтил)-амино] -фенил} -ацетиламино)-пентандиоевой кислоты 5-метиловый эфир;
2-(2-{ 4-[бис-(2-хлорэтил)-амино] -фенил} -ацетиламино)-пентандиоевая кислота;
2-(2-{ 4-[бис-(2-хлорэтил)-амино] -фенил} -ацетиламино)-4-карбамоил-масляная кислота.
a) (S)-2{ 4-[N, N-бис(2-хлорэтил)амино] феноксикарбониламино} -4-(1H-1,2,3,4-тетразол-5-ил)масляная кислота и ее соли;
b) N-{4-[N,N-бис(2-хлорэтил)амино]-3-фторфенилкарбамоил}-L-глютаминовая кислота и ее соли;
с) N-{4-[N,N-бис(2-хлорэтил)амино]фенилкарбамоил}-L-глютаминовая кислота и ее соли.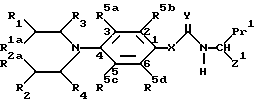
где R1, R2, R1а, R2а, R3, R4, R5а, R5b, R5c, R5d, X и Y имеют значения, определенные в п.1;
Z1 представляет Z, как определено в п.1, при условии, что, когда W представляет карбонильную группу, она присутствует в защищенной форме (обозначено Pr2) и Pr1 также представляет карбоксильную группу в защищенной форме (которая может быть такой же, или может отличаться от Pr2),
и, при желании, превращают полученное таким образом соединение формулы I (как определено в п.1) в его соль, при условии, что исключаются следующие соединения и их соли:
2-(2-{4-[бис-(2-хлорпропил)-амино]-фенил}-ацетиламино)-пентандиоевая кислота;
2-(2-{ 4-[бис-(2-хлорэтил)-амино] -фенил} -ацетиламино)-пентандиоевой кислоты-5-метиловый эфир;
2-(2-{ 4-[бис-(2-хлорпропил)-амино] -фенил} -ацетиламино)-пентандиоевая кислота;
2-(2-{ 4-[бис-(2-хлорпропил)-амино] -фенил} -ацетиламино)-пентандиоевой кислоты-5-этиловый эфир;
2-(2-{ 4-[бис-(2-хлорпропил)-амино] -фенил}-ацетиламино-4-карбамоил-масляная кислота.
2-(2-{4-[бис-(2-хлорпропил)-амино]-фенил}-ацетиламино)-пентандиоевая кислота;
2-(2-{ 4-[бис-(2-хлорэтил)-амино]-фенил}-ацетиламино)-пентандиоевой кислоты-5-метиловый эфир;
2-(2-{ 4-[бис-(2-хлорэтил)-амино]-фенил}-ацетиламино)-пентандиоевая кислота;
2-(2-{ 4-[бис-(2-хлорэтил)-амино]-фенил}-ацетиламино)-пентандиоевой кислоты-5-этиловый эфир;
2-(2-{ 4-[бис-(2-хлорэтил)-амино] -фенил} -ацетиламино)-4-карбамоил-масляная кислота.
Приоритеты по пунктам:
23.07.92 по п.5.
| US 4975278 A, 1991 | |||
| Шланговое соединение | 0 |
|
SU88A1 |
| СПОСОБ ПОЛУЧЕНИЯ ФОСФОРИЛИРОВАННЫХ S-АЛКИЛИЗОТИОМОЧЕВИН | 0 |
|
SU222373A1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ N-КАРБАМОИЛ-о- ФЕНИЛЕНДИАМИНА | 0 |
|
SU292965A1 |
| RU 94038047 A1, 10.06.96. | |||

