Изобретение относится к пространственно затрудненным производным пиперидина, которые могут быть использованы в качестве эффективных стабилизаторов различных органических материалов, в частности полимеров, от действия света и кислорода.
Известны различные N-алкоксизатрудненные аминопроизводные, содержащие одно пиперидиновое кольцо. Например, О-алкиловые производные пиперидина с водородом в 4-положении описаны Курумада и др. J.Polym. Sci., Polym. Chem. Ed. 23, 1477-1491 (1985); Боламэн и др. Rec. Trav. Chim.Pays-Bas, 97, 313-319 (1978) и Шелл и др., Доклады Акад. Наук СССР, раздел Химия, 200, 137-139 (1971). Аналогичные производные с бензоилокси в 4-положении указаны Курумада и др. в J.Polym, Sci., Polym. Chem. Ed. 22, 277-281 (1984). В патенте США N 4547537 предложены N-алкоксипиперидиновые соединения с тетрагидро-1,4-оксазин-2-оновой группой, соединенной с пиперидиновым кольцом. N-аралоксизамещенные на блокированном пиперидиновом кольце соединения описаны также Кеана и др., J.Org. Chem. 36, 209-211 (1971) и Ховардом и др., J. Org. Chem., 43, 4279-4283 (1976). N-альфа-окси-алкоксизамещенные пиперидиноны были указаны Уилсоном Trans. Far. Soc., 3008-19 (1971). Moad et al, Aust. J. Chem., 36, 1573-1588 (1983) описывают различные О-заместители, содержащие ненасыщенные и/или карбоксильные группы в цепи. Наконец, Фуджита и др. J. Polym. Sci. , Polym. Lett. Ed. 16, 515-(1978) описывают дипепиридиноксидиоксоспиросоединения, которые обладают способностью предотвращать разложение некоторых синтетических полимеров.
Известны затрудненные амины, которые могут быть использованы для стабилизации сшиваемых при нагревании алкидных или акрилатных лаков для покрытия металлов (патент США N 4426472) или для стабилизации катализируемых кислотой лаков, высушиваемых в сушильной печи, на основе полиэфиракрилатных или алкидных смол (патенты США NN 4344876 и 4426471).
Известные затрудненные амины, в частности производные 2,2,6,6-тетраалкилпиридины, отличны по структуре от предлагаемых производных пиридина, так как не содержат OR-группу у N-атома затрудненного амина.
Указанные затрудненные амины могут быть использованы самостоятельно или в комбинации с поглотителями ультрафиолетового излучения для улучшения эксплуатационных свойств покрытий на их основе.
Однако по-прежнему имеется необходимость в поиске соединений, позволяющих уменьшить фотоокисление и фоторазложение полимерных покрытий, обеспечивающих при этом более высокую эффективность при поддержании физической целостности покрытия.
Эффективность покрытия можно увеличить за счет повышения способности к предотвращению образования трещин, снижения хрупкости, предотвращения коррозии, эрозии, потери блеска, образования царапин и пожелтения покрытий.
Предлагаемые согласно настоящему изобретению производные 2,2,6,6-тетраалкилпиридина формулы 1 могут быть использованы для стабилизации отверждаемых в условиях окружающей среды и катализируемых кислотой термореактивных покрытий. При этом указанные соединения сохраняют физическую целостность покрытия в более высокой степени, значительно уменьшают потерю блеска и пожелтение.
Таким образом, согласно настоящему изобретению предлагаются ранее не известные производные 2,2,6,6-тетраметилпиперидина формулы А: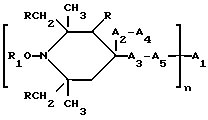 ,
,
в которой:
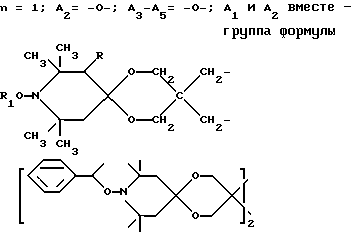
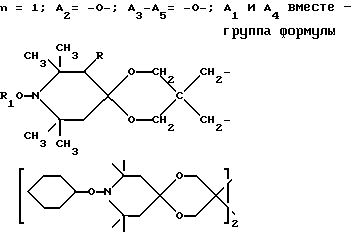
n выбрано из 1-4; R означает водород; R1 означает C1-C18-алкил, C5-C8-циклоалкенил, C5-C12-циклоалкил, возможно замещенный C1-C9-алкилом, C7-C9-аралкил, возможно замещенный C1-C9-алкилом, C6-C10-бициклоалкил; A3-A5 означает -О- или -N(R3), где R3 - водород, C2-C4-алканоил, C1-C8-алкил, при условии: а) когда n=1, и A3-A5 является NR3, то A2-A4 является водородом, а A1 является водородом, C1-C18-алкилом, или b) когда n=1, и A3-A5 является -О-, A1 вместе с A4 образует C2-C8-алкилен, а A2 является кислородом или CH2; или с) когда n=1, A3-A5 является -O- и A2 = O, то A1 вместе с A4 образуют группу формулы I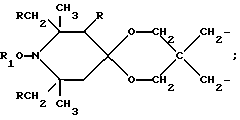
или, при условии: а) когда n=2, A3-A5 означает NR3, A2-A4 - водород, то A1 является группой формулы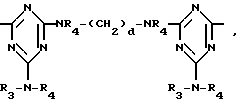
в которой
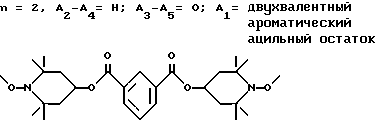
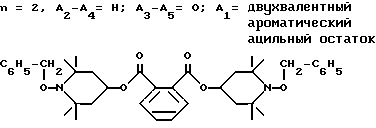
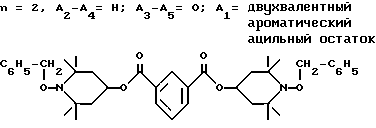
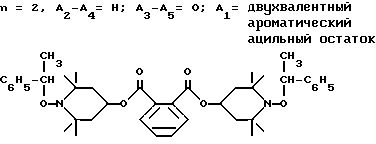
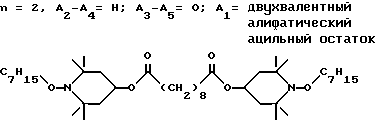
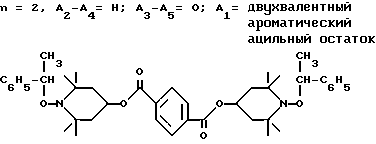
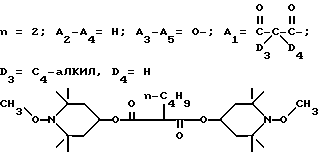
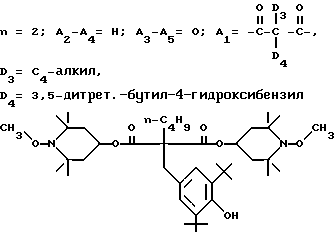
d - число, выбранное из 2-10, а 2 из 4-х значений R4 означает водород, или A1 является ксилиленом, C1-C12 алкиленом, двухвалентным ацильным остатком ароматической, алифатической или аралифатической карбоновой кислоты с числом атомов углерода до 20, или A1 является группой формулы II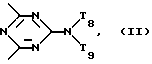
где
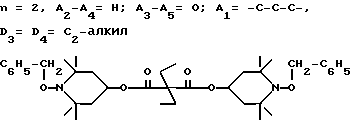
T8 и T9 независимо друг от друга означают H или C1-C12 алкил, или оба T8 и T9 вместе означают 3-оксапентаметилен; или b) когда n=2, A3-A5 является -О-, A2-A4 - водород, то A1 является ксилиленом, C1-C12 алкиленом, двухвалентным ацильным остатком ароматической, алифатической или аралифатической карбоновой кислоты с числом атомов углерода до 20; или A1 является группой формулы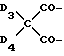 ,
,
в которой
D3 и D4 означают H, C1-C6 алкил или 3,5-ди-трет.-бутил-4-гидроксибензил; или при условии, что а) когда n=3, A3-A5 является NR3, A2-A4 - водород, то A1 является 2,4,6-триазинилом или группой формулы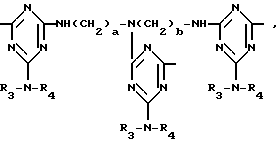
в которой
а и b независимо друг от друга означают 2 или 3; или при условии, когда n=4, A3-A5 является N(R3), A2-A4 - водород, то A1 является остатком формулы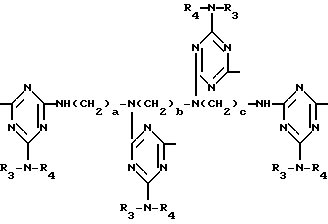 ,
,
в которой
a, b и с независимо друг от друга означают 2 или 3, R3 и R4 в триазиновых остатках означают: R3 - C1-C8алкил, водород, C2-C4алканоил; R4 - группа формулы III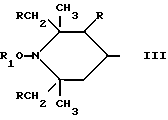 ,
,
а также водород или C4-C12алкил.
Предпочтительны производные 2,2,6,6-тетраметилпиперидина формулы (А), в которой R - водород, R1 - C1-C18алкил, C5-C8циклоалкил или C7-C9аралкил; когда n равно 2, A3-A5 означает -О-, A2-A4 означает H и A1 представляет собой C2-C8алкилен, ксилилен, двухвалентный ацильный радикал алифатической или ароматической карбоновой кислоты, содержащей до 12 атомов углерода или группу формулы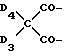 ,
,
где
D3 и D4 являются независимо водородом, C1-C6алкилом или 3,5-ди-трет.-бутил-4-гидроксибензилом; или A3-A5 означают -N(R3)-, A2-A4 означают H и A1 представляет собой C2-C12алкилен, ксилилен или, при условии, что R3 не является алканоилом, A1 может быть также двухвалентным ацильным радикалом алифатической или ароматической дикарбоновой кислоты, содержащей до 12 атомов углерода, или может быть группой формулы II,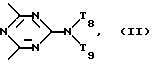
в которой
T8 и T9 независимо представляют собой водород, или C1-C12алкил, или T8 и T9 вместе являются З-оксапентаметиленом; когда n равно 3, а A3-A5 означают -N(R3)-, A2-A4 означают -H и A1 представляет собой 2,4,6-триазинил; или A1 означает группу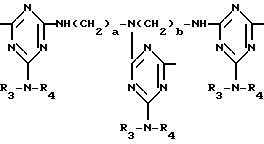 ,
,
где
а и b независимо равны 2 или 3; когда n равно 4, A3-A5 означают -N(R3)-, A2-A4 означают H и A1 группа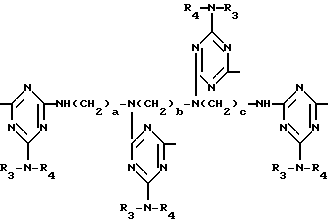 ,
,
где
a, b и с независимо равны 2 или 3; или когда n=1, а A3-A5 является -О- и A2 = О, то A1 вместе с A4 образуют группу формулы I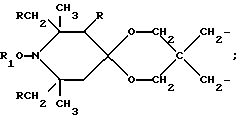
Особенно предпочтительны производные 2,2,6,6-тетраметилпиперидина формулы A, где R - водород, R1 - C1-C18алкил, циклогексил, циклогексенил, метилциклогексил или фенилалкил; n равно 1, 2 или 4; когда n равно 1 и A3-A5означает -О-, A2 означает -О- и A1 и A4 вместе являются C2-C8 алкиленом или A1-A4 месте образуют группу формулы I: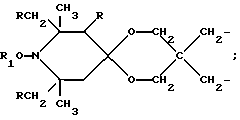
когда
n равно 2, а A3-A5 означают -О-, A2-A4 - водород и A1 означает C2-C8алкилен, ксилилен, группу -CO-R11-CO-, где R11 означает C2-C8алкилен, или группу формулы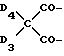 ,
,
где
D3 и D4 независимо означают водород, C1-C4алкил или 3,5-ди-трет.-бутил-4-гидроксибензил; когда n равно 1 и A3-A5 означает -N(R3)-, R3 - водород, C1-C8алкил или C2-C4алканоил, A2-A4- H и A1 - водород или C1-C12алкил; когда n равно 2, A3-A5 означают -N(R3)-, A2-A4 - H и A1 - C2-C8алкилен или -CO-R11-CO-; когда n равно 4 и A3-A5 означают -N(R3)-, A2-A4 - H, а A1 группа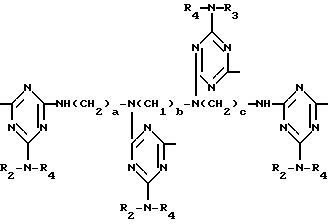 ,
,
где
a, b и c независимо равны 2 или 3;
Из указанных групп соединений наиболее предпочтительны соединения, выбранные из группы: ди(1-метокси-2,2,6,6-тетраметилпиперидин-4-ил)себацинат; ди(1-циклогексилокси-2,2,6,6-тетраметилпиперидин-4-ил)себацинат; ди(1-циклогексилокси-2,2,6,6-тетраметилпиперидин-4-ил)изофталат; ди(1-октилокси-2,2,6,6-тетраметилпиперидин-4-ил)себацинат; 3,15-дициклогексилокси-2,2,4,4,14,14,16,16-октаметил-7,11,18,21- тетраокса3,15-диазатриспиро[5.2.2.5.2.2] хенэйкозан; ди(1-циклогексилокси-2,2,6,6-тетраметилпиперидин-4-ил)сукцинат.
Более конкретно, предлагаемое изобретение относится к соединению, например, соответствующему одной из формул A'- R'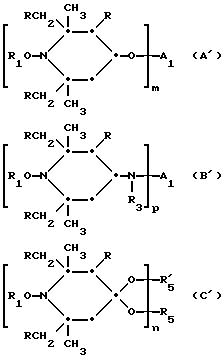
R5 и R'5 вместе являются C2-C8 алкиленом и когда n равно 2, R5 и R'5 вместе являются (-CH2)2C(CH2-)2,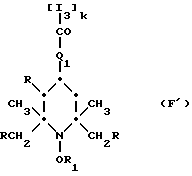
Q означает -O- или -N(R3)-; T3CO означает одновалентный или двухвалентный остаток A1;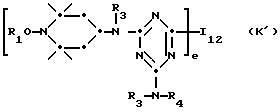
T12 является группой -N(R')-(CH2)d-N(R')- или 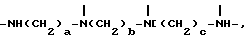 где a, b и c независимо друг от друга являются 2 или 3, d равно 2-10 или c равно 0, где R - R3, A1 имеют вышеуказанные значения, m, n, p означают 1, 2.
где a, b и c независимо друг от друга являются 2 или 3, d равно 2-10 или c равно 0, где R - R3, A1 имеют вышеуказанные значения, m, n, p означают 1, 2.
Предложенные соединения формулы 1 могут быть использованы для стабилизации отверждаемых в условиях окружающей среды или при катализе кислотой, при нагревании полимерных систем в зависимости от типа смол.
К таким смолам, как правило, относятся следующие смолы. Смолами для покрытий, отверждаемыми при окружающих условиях, могут быть алкидные смолы, термопластичные акрилатные смолы, акрилатные алкидные смолы, полиуретановые смолы или полиэфирные смолы; вышеупомянутые смолы могут быть модифицированы силиконами, изоцианатами, эпоксидами, изоциануратами, кетиминами или оксазолидинами. Такие смолы могут быть сложными эфирами целлюлозы, такой как нитроцеллюлоза, или ацетобутират целлюлозы, или такие смолы могут быть эпоксидными смолами, отверждаемыми полиаминами или другими агентами отверждения.
Применяемые алкидные, акрилатные, полиэфирные и эпоксидные смолы описаны в книге С.Паула, Покрытия поверхностей: Наука и технология, (1985) с.70-310. Немодифицированные и модифицированные алкидные смолы, которые могут быть стабилизированы соединениями предлагаемого изобретения, являются известными смолами, которые используют в торговле, для текущего ремонта и для окончательной полировки автомобилей. Например, такие покрытия основаны на алкидных смолах, акрил-алкидных или акрилатных смолах и алкил/силиконовых смолах, возможно сшитых изоцианатами или эпоксидными смолами.
Смолами, катализируемыми кислотой для термореактивных покрытий, могут быть, например, сшиваемые акрилатные, полиэфирные, полиуретановые, полиамидные или алкидные смолы. К ним относятся также смеси этих смол или смеси со сшивающими агентами, такими как меламиновые смолы.
Лаками на основе акрилатной смолы являются известные, высыхающие при нагревании лаки на основе акрилатной смолы или термореактивные смолы, включающие акриловые/меламиновые системы, которые описаны, например, в книге Г.Киттеля, Учебник по лакам и покрытиям, т. I, ч. 2, с. 735-742 (Берлин, 1972), Лаковые смолы (1977) Г.Вагнера и Н.Ф.Саркса на с. 229-238 и в книге С.Паула Покрытия поверхностей: Наука и Технология (1985).
Полиэфирными лаками являются известные, высыхающие при нагревании лаки, описанные, например, в книге Г.Вагнера и Г.Ф.Саркса (см. выше) на с. 86-99.
Лаки на основе алкидной смолы, которые могут быть стабилизированы, являются известными, высыхающими при нагревании лаками, которые используются, в частности, для покрытия автомобилей (автомобильные полирующие лаки), например, лаки на основе алкидных/меламиновых смол (например, Г.Вагнер и Г.Ф.Саркс, см. выше, с. 99-123). К другим сшивающим агентам относятся гликолауриловые смолы, блокированные изоцианатами, или эпоксидные смолы.
При промышленном использовании эмали с высоким содержанием твердых частиц на основе сшиваемых акрилатных, полиэфирных, уретановых или алкидных смол отверждают дополнительным кислотным катализатором. Световые стабилизаторы, содержащие основную азотную группу, в общем случае являются малопригодными для такого применения. Образование соли между кислотным катализатором и световым стабилизатором приводит к несовместимости или нерастворимости и осаждению соли и к более низкому уровню отверждения, к более низкому световому защитному действию и к более слабой стойкости к действию влажности.
Покрытия, отверждаемые при окружающих условиях, а также покрытия, катализируемые кислотой и отверждаемые при нагревании, стабилизированные в соответствии с предлагаемым изобретением, пригодны как для полирующих покрытий металлов, так и для придания оттенков твердым материалам, в частности при подкрашивании. Лаки, стабилизированные в соответствии с предлагаемым изобретением, наносят обычно двумя методами: либо разовым покрытием, либо двойным покрытием. В соответствии с последним методом сначала наносят содержащее пигмент основное покрытие, а уже на него наносят второй слой прозрачным лаком.
Количество используемого соединения I находится в пределах 0,1-10 мас.% в пересчете на не содержащий растворителя связывающий материал, предпочтительно 0,5-5 мас.%. Связывающие материалы могут быть растворены или диспергированы в известных органических растворителях или в воде, или они могут не содержать растворителя.
Когда используют двухслойную полировку, то соединение I может быть включено в прозрачное покрытие или как в прозрачное покрытие, так и в основное покрытие, содержащее пигмент. При производстве модифицированных акрилом алкидных смол или акрилатных смол полимеризуемые производные 2,2,6,6-тетраметилпиридина могут быть полимеризованы в смолу. Включение связывающего агента в лаках может быть осуществлено поликонденсацией при производстве алкидных или полиэфирных смол. В этом случае имеется дополнительное преимущество, заключающееся в том, что световые стабилизаторы не могут быть удалены в результате экстракции или миграции, в результате чего их действие является весьма продолжительным.
Чтобы достичь максимальной световой стабильности, иногда желательно одновременно использовать другие известные светостабилизаторы. Примерами могут служить УФ-абсорберы типа бензофенонов, бензотриазолов, производного акриловой кислоты или оксаланилида, или арил-симм.-триазины, или содержащие металл световые стабилизаторы, например органические соединения никеля. В двухслойных системах эти дополнительные световые стабилизаторы могут быть добавлены в прозрачное покрытие и/или основное покрытие, содержащее пигмент.
Если используют такие составы, сумма всех световых стабилизаторов составляет 0,2-20 мас.%, предпочтительно 0,5-5 мас.%, в пересчете на образующую пленку смолу.
Примеры Уф-абсорберов, которые могут быть использованы в комбинации с вышеупомянутыми (затрудненными) аминосоединениями, приведены ниже:
(а) 2-(2'-оксифенил)-бензотриазолы, например, 5'-метил-, 3', 5'-ди-трет. -бутил-, 5'-трет. -бутил-, 5'-(1,1,3,3-тетраметилбутил)-, 5-хлор-3, 5'-ди-трет. -бутил-, 5-хлор-З'-трет.-бутил-5'-метил-, 3'-втор.-бутил-5'-трет.-бутил-, 4-октилокси-, 3',5'-ди-трет.-амиловое производное;
(b) 2-окси-бензофеноны, например, 4-окси-, 4-метокси-, 4-октилокси-, 4-децилокси-, 4-додецилокси-, 4-бензилокси-, 4,2', 4'-триокси- и 2'-окси- 4,4'-диметоксипроизводное;
(с) акрилаты, например, этиловый сложный эфир или изооктиловый сложный эфир альфа-циано- β,β -диметилакриловой кислоты, метиловый сложный эфир альфа-карбометоксикоричной кислоты, метиловый сложный эфир или бутиловый сложный эфир альфа-циано- β -метил-пара-метоксикоричной кислоты, метиловый сложный эфир альфа-карбометокси-пара-метоксикоричной кислоты, N-( β -карбометокси- β -циановинил)-2-метилиндолин;
(d) соединения никеля, например, никелевые комплексы 2,2'-тиобис[4-(1,1,3,3-тетраметилбутил)-фенола], в соотношении 1 : 1 или 1 : 2, возможно с дополнительными лигандами такими, как н-бутиламин, триэтаноламин или N-циклогексил-диэтаноламин; дибутилдитиокарбамат никеля, соли никеля моноалкилового сложного эфира 4-окси-3,5-ди-трет.-бутилбензилалкилфосфиновой кислоты, такого как метилового, этилового или бутилового сложного эфира, комплексы никеля кетоксимов, таких как 2-окси-4-метилфенилундецилкетоксим, комплексы никеля с 1-фенил-4-лаурил-5-оксипиразолом, возможно с дополнительными лигандами;
(е) диамиды щавелевой кислоты, например, 4,4'-диоктилоксиоксанилид, 2,2'-диоктилокси-5,5'-дитрет.-бутил-оксанилид, 2,2'-ди-додецилокси- 5,5'-дитрет. -бутил-оксанилид, 2-этокси-2'-этил-оксанилид, N,N'-бис-(3-диметиламинопропил)-оксаламид, 2-этокси-5-трет. -бутил-2'-этилоксанилид и их смеси с 2-этокси-2'-этил-5,4'-трет.-бутил-оксанилидом, и смеси орто- и пара-, а также орто- и пара-этокси-двузамещенных оксанилидов;
(f) оксифенил-s-триазины, такие как 2,6-бис-(2,4-диметилфенил)-4-(2-окси-4-октилоксифенил)-s-триазин или соответствующие 4-(2,4-диоксифенил)-производные.
Особую ценность для предлагаемых композиций представляют бензотриазолы с высокой молекулярной массой и низкой летучестью, такие как 2-[2-окси-3,5-ди(альфа,альфа-диметилбензил)-фенил]-бензотриазол, 2-[2-окси-3,5-дитрет.-октилфенил] -бензотриазол, 2-(2-окси-З-альфа,альфа-диметилбензил-5-трет.-октилфенил)- бензотриазол, 2-(2-окси-З-трет. -октил- 5-альфа,альфа-диметилбензилфенил)-бензотриазол, 2-(2-окси-3,5-дитрет.-амилфенил)-бензотриазол, 2-[2-окси-З-трет. -бутил-5-(2-(омега-оксиокта-(этиленокси)-карбонил)- этилфенил]-бензотриазол, 5-хлор-2-[2-окси-3,5-ди(альфа,альфа-диметилбензил)-фенил] - бензотриазол, 5-хлор-2-(2-окси-3,5-дитрет.-бутилфенил)-бензотриазол, 5-хлор-2-[2-окси-3-трет.-бутил-5-(2-октилоксикарбонилэтил)-фенил]- бензотриазол, 2-[2-окси-3-втор.-додецил-5-метилфенил]-бензотриазол и гексаметиленди[ β -(3-трет.-бутил-4-окси-5-(2-бензотриазолил)- фенил)-пропионат].
Наиболее предпочтительными бензотриазолами, используемыми в предлагаемых композициях, являются 2-[2-окси-3,5-ди(альфа,альфа-диметилбензил)-фенил]-бензотриазол и 2-[2-окси-З-трет.-бутил-5-(2-(омега-окси-окта)этиленокси)карбонил) этилфенил]-бензотриазол.
Другими ингредиентами, которые могут содержать эмали или покрытия, являются антиоксиданты, например антиоксиданты пространственно затрудненных производных фенола, соединения фосфора, такие как фосфиты, фосфины или фосфониты, пластификаторы, выравнивающие добавки, катализаторы отверждения, загустители, диспергирующие агенты и промоторы адгезии.
К фосфитам и фосфонитам относятся трифенилфосфит, дифенилалкилфосфиты, фенилдиалкилфосфиты, три-(нонилфенил)фосфиты, трилаурилфосфит, триоктадецилфосфит, дистеарилпентаэритритдифосфит, трис(2,4-дитрет. -бутилфенил)фосфит, дифосфит дизодецил-пентаэритрита, дифосфит ди(2,4-дитрет.-бутилфенил)пентаэритрита, трифосфит тристеарил-сорбита, тетракис(2,4-дитрет.-бутилфенил)-4,4'-дифенилендифосфонит.
Стабилизаторы необходимы для того, чтобы придать большую прочность отвержденным эмалям (измеряемую определением блеска под углом 20o ,искажения отражения, трещин или царапин); стабилизаторы не должны замедлять отверждение (нормальная сушка для автомобиля заканчивается при температуре 121oC, а низкотемпературную сушку осуществляют при температуре 82oC), которое измеряют твердостью, адгезией, стойкостью к растворителям и стойкостью к воздействию влажности; эмаль не должна желтеть при отверждении и, кроме того, изменение цвета при воздействии света должно быть минимальным; стабилизаторы должны быть растворимы в органических растворителях, которые в общем случае используют для нанесения покрытий, таких как метил-амилкетон, ксилол, н-гексилацетат, спирт и т.п.
Предлагаемые в соответствии с изобретением затрудненные световые стабилизаторы, замещенные на N-атоме OR1-группой, удовлетворяют каждому из этих требований и обеспечивают как таковые или в комбинации с УФ-абсорбером прекрасную световую стабилизацию для отвержденных покрытий.
Приводимые ниже примеры показывают использование соединений изобретения в различных отверждаемых при окружающих условиях и катализируемых кислотой термореактивных покрытиях. Части и проценты являются массовыми.
Пример 1. Стабилизация ароматического уретанового лака.
Куски досок из туи с чистым радиальным срезом, имеющие размеры 1,27 х 20,32 х 30,48 см, использовали для испытания производимого промышленностью ароматического уретанового лака (Флекто-Варатан 90). Одну половину каждой доски покрывали двумя слоями нестабилизированного лака. Равное количество лака, содержащего 7 мас.% (в пересчете на твердые частицы смолы) световых стабилизаторов наносили на другую половину доски в два слоя. После выдерживания в течение 2 нед при окружающей температуре деревянные доски выставляли на открытый воздух под углом 45o на 5 мес. Блеск под углом 60o каждой половины доски измеряли в верхней, средней и нижней частях доски и усредняли (ASTM D 523, ASTM - Американское Общество по Испытанию Материалов). Из-за неудовлетворительной однородности древесины сохранение блеска одного и того же лака несколько отличается в зависимости от доски. Применение нестабилизированного контрольного лака к каждой панели позволяет осуществить более корректное измерение улучшения блеска благодаря присутствию светового стабилизатора (см. табл. 1)
Пример 2. Стабилизация акрилатной алкидной полирующей эмали.
Производимую промышленностью акрилатную алкидную эмаль, пигментированную не дающим осадка алюминиевым пигментом и подкрашенную синью, стабилизировали указанным количеством поглотителя ультрафиолетового излучения и соединения формулы I (по массе в пересчете на твердые частицы смолы), а затем наносили распылителем на панели Бондерит 40, образованные алкидной грунтовкой. Затем покрытию давали возможность затвердеть при комнатной температуре в течение 14 дн, далее панели выставляли на открытом воздухе под углом 5o в течение 8 мес. Измеряли блеск под углом 20oC для каждой панели (результаты испытания приведены в табл. 2).
Пример 3. Стабилизация алкидной эмали в масляной среде.
Алкидную эмаль в масляной среде, пигментированную не дающим осадка алюминиевым пигментом и подкрашенную яркой синью, стабилизировали указанными количествами поглотителя ультрафиолетового излучения и блокированного аминового производного формулы I, а затем наносили распылителем на пластины из холоднокатаной стали, обработанные эпоксидной грунтовкой.
Покрытию давали возможность отвердеть при комнатной температуре в течение 2 нед, затем пластины для ускорения испытания атмосферных влияний выставляли под воздействие устройства типа Ксеноно Дуговой Прибор для Моделирования Погодных Явлений на 840 ч. Показатели блеска под углом 20o для пластин определяли перед и после воздействия (в табл. 3 указаны полученные результаты в % сохранения блеска).
Пример 4. Стабилизация термореактивного акрилатного лака.
Производимый промышленностью ярко-синий термопластический акрилатный лак для металлов стабилизировали при помощи 2% каждого из УФ-абсорберов и блокированным амином формулы I (массовые, в пересчете на общую массу твердых частиц смолы), а затем наносили с использованием распылителя на панели Бондерит 40, обработанные алкидной грунтовкой. После выдерживания при окружающей температуре в течение 2 нед панели выставляли под воздействие Ксенонового Дугового Устройства для Моделирования Погодных Явлений на 1250 ч (сохранение блеска под углом 20o для панелей см. табл. 4).
Пример 5. Стабилизация акрилатной алкидной полирующей эмали.
Акрилатную алкидную эмаль из примера 2, пигментированную не дающим осадка алюминиевым пигментом, стабилизировали указанным количеством световых стабилизаторов (по массе в пересчете на твердые частицы смолы), а затем при помощи распылителя наносили на панели Бондерит 40, обработанные алкидной грунтовкой. Затем покрытию давали возможность затвердеть при окружающей температуре в течение 14 дн, после чего панели подвергали обработке с использованием СУФ-установки для моделирования природных явлений (значения блеска под углом 60o для проб в различные моменты времени приведены в табл. 5)
Пример 6. Стабилизация термореактивной акрилатной эмали.
Куски листовой стали, покрытые грунтовкой на основе полиэфирной/эпоксидной смолы, покрывали серебряным слоем для металлов толщиной примерно 0,02 мм и сушили воздухом в течение 3 мин. Затем на него распылением наносили прозрачное покрытие толщиной примерно 0,038 мм. Прозрачное покрытие состояло из термореактивной акрилатной эмали, состоящей на 70% из сополимера оксиэтилакрилата, стирола, акрилонитрила, бутилакрилата и акриловой кислоты и на 30% из меламиновой смолы. Кроме того, она содержала 0,5% пара-толуол(моно)сульфокислоты и стабилизаторы, указанные в табл. 6. Через 15 мин сушки воздухом листы с покрытием сушили в течение 30 мин при температуре 121oC. Отвержденные пробы обрабатывали на СУФ-установке и определяли время, в течение которого теряется 50% блеска под углом 20o.
Пример 7. Стабилизация термореактивной акрилатной эмали.
Прозрачную термореактивную акрилатную эмаль на основе связывающего агента, состоящего на 70% сополимерной формы из оксиэтилакрилата, бутилакрилата, бутилметакрилата, стирола и акриловой кислоты и на 30% из меламиновой смолы, смешивали с 0,5% пара-толуол(моно)сульфокислоты и стабилизатором (см. табл. 7).
Производимые промышленностью стальные листы, обработанные грунтовкой, использовали в качестве субстрата. Эти листы покрывали серебряным основным слоем для металлов, который стабилизировали 1% блокированного амина (ТинувинR 440) и 1% УФ-поглотителя (Соединение В), наносили на панель слоем толщиной примерно 0,015 - 0,020 мм. Через 3 мин на основное покрытие наносили распылением прозрачное покрытие толщиной 0,04 - 0,05 мм. Через 10 мин сушки воздухом пробы сушили в течение 30 мин при температуре 121oC. Высушенные пробы обрабатывали СУФ-устройством, моделирующим явления природы, и через некоторые промежутки времени определяли разницу в отражениях (OO).
Пример 8. Стабилизация акрилат-кетиминовой эмали.
Систему: акрилатное кетиминовое основное покрытие и прозрачное покрытие стабилизировали в прозрачном покрытии указанным количеством поглотителя ультрафиолетового излучения и затрудненным аминовым производным формулы 1. Основное покрытие наносили распылением толщиной 0,02 мм на лист холоднокатаной стали, загрунтованный кетимин-ацетоацетатной грунтовкой. Затем его покрывали прозрачным слоем толщиной 0,06 мм эмали на основе ненасыщенного акрилата-кетимина (влажный на влажный). Листы сушили в течение 45 мин при температуре 60oC, а затем обрабатывали на СУФ-установке. В этой установке пробы подвергали обработке, моделирующей различные явления природы, повторяющимися циклами по 4 ч во влажной атмосфере при температуре 50oC, затем в течение 4 ч под воздействием УФ-света при температуре 60oC (в табл. 8 приведены результаты испытаний на блеск под углом 20o при различных интервалах обработки).
Пример 9. Стабилизация полиэфирной-меламиновой эмали.
Полиэфирное-меламиновое покрытие для витков, катализируемое паратолуол(моно)сульфокислотой, составляли таким образом, чтобы оно включало бензотриазоловый УФ-поглотитель и блокированный аминный световой стабилизатор формулы I, являющийся предметом предлагаемого изобретения. Этот состав наносили с использованием стержня с намотанной проволокой автоматического устройства для нанесения на доски, покрытые витками с толщиной в сухом виде 0,02 мм. Затем доски сушили при температуре 500oF (260oC) в печи в течение 45 с, в течение которых максимальная температура металла достигала 435oF (225oC). Две окрашенные системы, фтало/синь и систему, пигментированную окисью бронзы, подвергали испытанию. Доски обрабатывали в Южной Флориде под углом 45o на солнце в течение 17 мес (в табл. 9 приведены изменения цвета (ΔE) досок).
Пример 10. Стабилизация термореактивной акрилатной эмали.
Составляли композицию термореактивной акрилатной эмали из примера 6 таким образом, чтобы она включала блокированный аминный световой стабилизатор, являющийся предметом предлагаемого изобретения. Покрытые витками алюминиевые пластины грунтовали эпоксидной грунтовкой и затем покрывали слоем толщиной примерно 0,02 мм серебряного основного покрытия для металлов и, наконец, слоем примерно 0,06 мм прозрачной полирующей эмали. После сушки в течение 5 - 10 мин воздухом покрытые пластины сушили в течение 30 мин при температуре 30oC.
Пластины после покрытия обрабатывали в СУФ-установке и в различные моменты времени определяли блеск проб под углом 20o (см. табл. 10).
Пример 10a. Сравнение с прототипом (US, патент 4344876) в акриловом лаке, подвергаемом горячему отверждению.
Получают прозрачный лак из 57,3 ч. акриловой смолы, подвергаемой горячему отверждению (Paral-oid® AT 410, Rohm & Haas Co., 73%-ный раствор в ксилоле); 17,9 ч. меламиновой смолы (Cymel® 301, Amer. Cyanamid Co.); 1,8 ч. ацетобутирата целлюлозы (CAB 551, Eastman Chem.); 12,0 ч. бутилацетата; 10,2 ч. н-бутанола; 0,3 ч. вспомогательного средства, способствующего розливу (Modaflow® Monsanto Со. ); 0,5 ч. п-толуолсульфокислоты в качестве катализатора отверждения.
К прозрачному лаку добавляют приведенные в табл. 11 стабилизаторы, растворенные в ксилоле, в количестве 1 мас.%, в расчете на твердое связующее (без разбавителя). Стабилизованные таким образом лаковые составы наносят на алюминиевые листы, грунтованные основным лаком, пигментированным серебряным слоем для металлов. Образцы выдерживают в течение 30 мин при комнатной температуре, а затем отверждают в течение 30 мин при 130oC. Отвержденный прозрачный лак имеет толщину около 40 мкм.
Отвержденные образцы в течение 12-и месяцев подвергают влиянию погодных условий во Флориде (под углом 5o к югу). Затем измеряют блеск под углом 20o поверхности лака по нормам ASTM D 523.
Использованные стабилизаторы: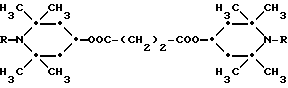 ,
,
S-1: R = -CH3 Соединение LS B примера 5 US, патент 4344876 (колонна 20);
S-2: R = -COCH3 Соединение LS D примера 5 US, патент 4344876;
S-3: R = -OC8H17 Соединение N 19 изобретения;
S-4: R =  Соединение N 9 изобретения.
Соединение N 9 изобретения.
Эффективность новых стабилизаторов значительно выше эффективности стабилизатора по прототипу.
Пример 10b. Сравнение с прототипом (US, патент 4344876) в отверждаемом при комнатной температуре полиуретановом лаке.
Получают прозрачный лак из компонентов A и B.
Компонент A: 75 ч. сложного полиэфира, содержащего гидроксильные группы (Macrynal® SM 510 N, Hoechst AG); 15 ч. ацетата бутилгликоля; 6 ч. смеси ароматических растворителей (Solvesso® 100); 3,7 ч. метил-изобутилкетона; 0,1 ч. цинкоктоата в качестве катализатора отверждения; 0,2 ч. вспомогательного средства, способствующего розливу (BYK® 300, Mallinckrodt Co.).
Компонент B: 30 ч. ароматического полиизоцианата (Desmodur® N 75, Bayer AG).
Перед смешением компонентов A и B к лаку A добавляют нижеуказанные стабилизаторы в виде ксилольного раствора в количестве 1% в расчете на содержание твердых веществ A+B. Готовый прозрачный лак разбавляют ксилолом до тех пор, пока его можно будет наносить распылением, а затем его напыляют на алюминиевые листы, грунтованные основным лаком, пигментированным серебряным слоем для металлов.
Образцы подвергают отверждению в течение 3 дн при комнатной температуре. Толщина отвержденного прозрачного лака составляет около 45 мкм. Отвержденные образцы обрабатывают UVCON-установкой (UVB-313), моделирующей явления природы, циклами в течение 4-х ч под воздействием УФ-света при 60oC и 4 ч конденсации при 50oC. Через равные промежутки времени определяют блеск поверхности образцов по нормам ASTM D 523, (см. табл. 12).
Предлагаемые стабилизаторы S-3 и S-4 сохраняют блеск дольше и тем самым имеют более высокую эффективность.
Пример 10с. Сравнение с прототипом (US, патент 4344876) в присутствии УФ-абсорбера.
Получают прозрачный лак из 58,3 ч. акриловой смолы, подвергаемой горячему отверждению (Viacryl® VC 373, Vianova AG); 27,3 ч. меламиновой смолы (Maprenal® MF 590, Hoechst AG); 5,4 ч. ксилола; 4,0 ч. ацетата бутилгликоля; 4,0 ч. смеси ароматических растворителей (Solvesso® 150); 1,0 ч. вспомогательного средства, способствующего розливу (Bayrilon® Bayer A AG).
Приведенные в табл. 13 стабилизаторы растворяют в ксилоле и добавляют к прозрачному лаку. После разбавления ксилолом прозрачный лак распылением наносят на алюминиевые листы, грунтованные основным лаком, пигментированным серебряным слоем для металлов.
Образцы выдерживают в течение 30 мин, а затем отверждают при 130oC в течение 30 мин. Отвержденный прозрачный лак имеет слой толщиной 40-50 мкм. Отвержденные образцы обрабатывают UVCON-установкой, моделирующей явления природы (UVB-313), циклом в течение 8 ч под воздействием УФ-света при 70oC и 4 ч конденсации при 50oC. Через равные промежутки времени определяют блеск поверхности образцов по нормам ASTM D 523.
В сочетании со стандартным УФ-абсорбером предлагаемый стабилизатор имеет более длительную эффективность, чем стабилизатор согласно прототипу.
Результаты нижеследующих примеров показывают, что благодаря использованию добавок согласно изобретению достигаются очень хорошие результаты по отверждению и стабилизации лаков (эмалей).
Пример 10d. Получают прозрачную термореактивную акрилатную эмаль на основе связующего, состоящего из 70 мас.% мономеров 2-оксиэтилакрилата, стирола, акрилонитрила, бутилакрилата и акриловой кислоты и 30 мас.% меламиновой смолы. В качестве катализатора отверждения добавлена п-толуолсульфокислота, в количестве 0,5 мас%. В полученную эмаль добавлены 1 мас.% 2-[2-окси-3,5- (α,α -диметилбензил)фенил] -2Н-бензотриазол в качестве УФ-поглотителя и 2 мас.% стабилизатора согласно изобретению.
Добавлялись следующие, описываемые в предлагаемом изобретении стабилизаторы:
Соединение B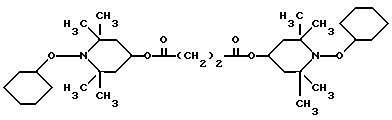 .
.
Соединение C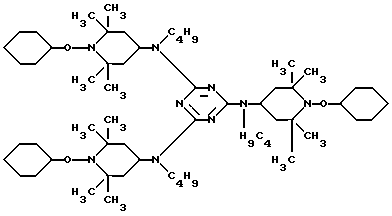 .
.
Соединение E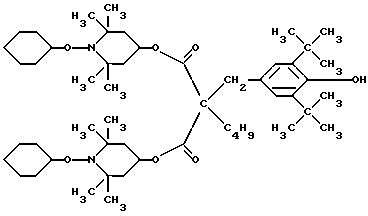 .
.
Соединение P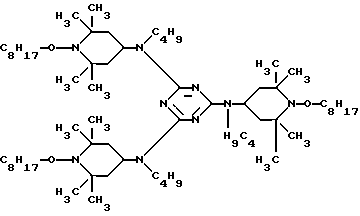 .
.
Стальные листы размером 4'' х 12'' (9,16 х 30,48 см), обработанные грунтующим серебрянометаллическим базисным лаком, затем покрывались испытуемой эмалью, толщиной 0,020 мм и высушивались в течение 3 мин на воздухе. Затем на основное покрытие наносили распылением прозрачное лаковое покрытие толщиной 0,038 мм, сушили в течение 15 мин на воздухе и затем в течение 30 мин при 121oC.
Затем эти листы испытывались на возможность влияния стабилизатора на катализатор отверждения. В соответствии со стандартом ASTM D-1474-68 определялась твердость по Кноппу отвержденной пленки лака. Результаты приведены в табл. 14.
Результаты показывают, что связующее лака затвердевает нормально и что заявляемые согласно изобретению соединения не оказывают воздействия на катализаторы отверждения.
Ниже приведены примеры полиалкилпиперидиновых исходных соединений, которые используют для получения блокированных аминовых производных формулы A'(это относится к выбранной процедуре получения): ди-(2,2,6,6-тетраметилпиперидин-4-ил)адипат; ди-(2,2,6,6-тетраметилпиперидин-4-ил)себацинат; ди-(2,2,6,6-тетраметилпиперидин-4-ил)фталат; альфа, альфа'-(ди-2,2,6,6-тетраметилпиперидин-4-окси)-пара-ксилол; ди-(2,2,6,6-тетраметилпиперидин-4-ил)сукцинат; ди-(2,2,6,6-тетраметилпиперидин-4-ил)малонат; ди-(1-окси-2,2,6,6-тетраметилпиперидин-4-ил)изофталат; 4-окси-1-метокси-2,2,6,6-тетраметилпиперидин; ди-(1-окси-2,2,6,6-тетраметилпиперидин-4-илокси)-пара-ксилол; 1-этокси-4-окси-2,2,6,6-тетраметилпиперидин; (2,2,6,6-тетраметилпиперидин-4-ил)-[4-(2-оксоазепин-1-ил)-2,2,6,6- тетраметилпиперидин-4-ил] ацетат.
Когда R3 - C2-C4алканоил, R3 является, например, пропионилом, бутирилом, предпочтительно ацетилом.
Если какими-либо заместителями являются C2-C12алкилены, они могут быть, например, этиленом, пропиленом, 2,2-диметилпропиленом, тетраметиленом, гексаметиленом, октаметиленом, декаметиленом или додекаметиленом. C5-C8 - циклоалаленил является, в частности, циклогексиленом.
Приводимые ниже соединения являются примерами полиалкилпиперидиновых исходных соединений, которые используют при получении блокированных производных пиперидина формулы В': N,N'-бис-(2,2,6,6-тетраметилпиперидин-4-ил)-гексаметилен-1,6-диамин; N, N'-бис-(2,2,6,6-тетраметилпиперидин-4-ил)-гексаметилен-1,6- диацетамид; N-н-бутил-N-(2,2,6,6-тетраметилпиперидин-4-ил)-4-окси-3,5-дитрет. - бутилбензамид; N,N'-бис-(2,2,6,6-тетраметилпиперидин-4-ил)-N, N'-дибутиладипамид; N,N'-бис-(2,2,6,6-тетраметилпиперидин-4-ил)-пара- фенилендиметилендиамин; 4-(3-метил-4-окси-5-трет.-бутил-бензоилацетамидо)-2,2,6,6- тетраметилпиперидин; 1-оксил-2,2,6,6-тетраметилпиперидин-4-он.
Приводимые ниже соединения являются примерами полиакрилпиперидиновых исходных соединений, которые используют при получении блокированных аминовых производных формулы C': 9-аза-8,8,10,10-тетраметил-1,5-диоксаспиро[5.5]ундекан; 9-аза-8,8,10,10-тетраметил-3-этил-1,5-диоксаспиро[5,5] ундекан; 2,2,6,6-тетраметилпиперидин-4-спиро-2'-(1', 3'-диоксан)-5'-спиро-5'' -(1'', 3''-диоксан)-2''-спиро-4'''-(2"',2''',6''',6'''- тетраметилпиперидин).
Исходные блокированные аминовые производные включают в случае формулы J': поли-{[6-[(1,1,3,3-тетраметилбутил)-имино]-1,3,5-триазин-2,4-диил] [2-(1-оксил-2,2,6,6-тетраметилпиперидил)-амино] -гексаметилен-4 [4-(1-оксил-2,2,6,6-тетраметилпиперидил]-имино]}.
Производные 2,2,6,6-тетраметилпиперидина, являющиеся предметом предлагаемого изобретения, обычно получают окислением соответствующего блокированного амина подходящим пероксисоединением, таким как перекись водорода или трет. -бутилгидроперекись в присутствии в качестве катализатора карбонила металла или окиси металла с последующим восстановлением промежуточного оксида в целевое N-оксипроизводное, как правило, каталитической гидрогенизацией.
Кроме того, О-алкилпроизводные могут быть получены с использованием нескольких способов. Например, N-оксипроизводное может быть алкилировано в присутствии гидрида натрия галоидуглеводородом, таким как бензилбромид или этилиодид. N-метоксипроизводные могут быть получены в результате термической обработки хлорбензольного раствора нитроксильного радикала и дитрет.-бутилперекиси. Продукт получают при реакции между нитроксилрадикалом и метиловым радикалом, образующимся при β -делении трет.-бутоксирадикала.
Другие N-алкоксипроизводные могут быть синтезированы при помощи реакции нитроксилрадикалов с углеводородными радикалами, образованными в результате термического разложения дитрет.-бутилперекиси в присутствии углеводородных растворителей, таких как циклогексан, толуол и этилбензол.
Предпочтительный способ состоит в получении N-алкоксипроизводных непосредственно из затрудненных аминов. Например, смесь 4-бензоилокси-2,2,6,6-тетраметилпиперидина, водного раствора трет.-бутилгидроперекиси, окиси молибдена и этилбензола дает 90%-ный выход N-альфа-метилбензилоксипиперидина. Было установлено, что молибден (VI) увеличивает эффективность как окисления затрудненного амина в нитроксилрадикал, так и реакции нитроксилрадикалов с углеводородами.
Хотя эти способы приведены для получения N-алкоксипроизводных, они в равной степени применимы ко всем OR1-группам.
Исходные блокированные амины широко доступны на рынке или могут быть получены с использованием известных приемов.
По этому поводу можно обратиться к Курумада и др., J. Polym. Sci., Polym. Chem. Ed. 23, 1477-1491 (1985), Моаду и др. Aust. J. Chem., 36, 1573-1588 (1983) и патенту CША N 4547537.
Производные формулы I особенно эффективны для стабилизации органических веществ от фотохимического воздействия. К таким органическим веществам относятся полимерные материалы, которые перечислены ниже.
1. Полимеры моноолефинов и диолефинов, например, полипропилен, полиизобутилен, полибутен-1, полиметилпентен-1, полиизопрен и полибутадиен, а также полимеры циклоолефинов, например, циклопентена или норборнена, полиэтилен (которые могут быть сшитыми), например, полиэтилен высокой плотности (ПЭВП), полиэтилен низкой плотности (ПЭНП) и линейный полиэтилен низкой плотности (ЛПЭНП).
2. Смеси полимеров, упомянутых в пункте 1, например, смеси полипропилена с полиизобутиленом, полипропилена с полиэтиленом (например, ПП/ПЭВП, ПП/ПЭНП) и смеси различных типов полиэтилена (например, ПЭНП/ПЭВП).
3. Сополимеры моноолефинов и диолефинов друг с другом или с другими виниловыми мономерами, такими как, например, этилен/пропилен, линейный полиэтилен низкой плотности (ЛПЭНП) и его смеси с полиэтиленом низкой плотности (ПЭНП), пропилен/бутен-1, этилен/гексен, этилен/этилпентен, этилен/гептен, этилен/октен, пропилен/изобутилен, этилен/бутен-1, пропилен/бутадиен, изобутилен/изопрен, этилен/алкилакрилаты, этилен/алкилметакрилаты, этилен/винилацетат или сополимеры этилен/акриловая кислота и их соли (мономеры) и тримеры этилена с пропиленом и диеном, таким как гексадиен, дициклопентадиен или этилиденнорборнен, а также смеси таких сополимеров и их смеси с полимерами, упомянутыми в пункте 1 выше, например, сополимеры полипропилен/этиленпропилен, ПЭНП/ЭВА, ПЭНП/ЭАА, ЛПЭНП/ЭВА и ЛПЭНП/ЭАА.
За. Углеводородные смолы (например, C5-C9) и их гидрогенизированные модификации (например, придающие липкость агенты).
4. Полистирол, поли-(пара-метилстирол), поли-( α -метилстирол).
5. Сополимеры стирола или α -метилстирола с диенами или акриловыми производными, такие как стирол-бутадиен, стирол/акрилонитрил, стирол/алкилметакрилат, стирол/малеиновый ангидрид, стирол/бутадиен/этилакрилат, стирол/акрилонитрил/метилакрилат, смеси с высокой ударной прочностью из сополимеров стирола и другого полимера, таких как, например, из полиакрилатдиенового полимера или этилен/пропилен/диенового тримера, блок-сополимеры стирола, такие как, например, стирол/бутадиен/стирол, стирол/изопрен/стирол, стирол/этилен/бутадиен/стирол или стирол/этилен/пропилен/стирол.
6. Графт-сополимеры стирола или α -метилстирола, такие как, например, стирол на полибутадиене, стирол на полибутадиен-стироле или полибутадиен-акрилонитриле; стирол и акрилонитрил (или метакрилонитрил) на полибутадиене; стирол и малеиновый ангидрид или малеимид на полибутадиене; стирол, акрилонитрил и малеиновый ангидрид или малеимид на полибутадиене; стирол, акрилонитрил и метилметакрилат на полибутадиене, стирол и алкилакрилаты или метакрилаты на полибутадиене, стирол и акрилонитрил на тримере этилена/пропилена/диена, стирол и акрилонитрил на полиакрилатах или полиметакрилатах, стирол и акрилонитрил на сополимерах акрилата/бутадиена, а также смеси их с сополимерами, перечисленными в пункте 5, например, смеси сополимеров, известные как ABS-, MBS-, ASA- или AES-полимеры.
7. Содержащие галоген полимеры, такие как полихлоропрен, хлорированные каучуки, хлорированный или сульфохлорированный полиэтилен, гомо- и сополимеры эпихлоргидрина, полимеры из содержащих галоген виниловых соединений, например, поливинилхлорид, поливинилиденхлорид, поливинилфторид, поливинилиденхлорид, а также их сополимеры, например, винилхлорид/винилиденхлорид, винилхлорид/винилацетат или винилиденхлорид/винилацетат-сополимеры.
8. Полимеры, которые получают из α,β -ненасыщенных кислот и их производных, такие как полиакрилаты и полиметакрилаты, полиакриламид и полиакрилонитрил.
9. Сополимеры из мономеров, упомянутых в пункте 8, друг с другом или с другими ненасыщенными мономерами, такие как, например, сополимеры акрилонитрил/бутадиен, акрилонитрил/алкилакрилат, акрилонитрил/алкоксиалкилакрилат или акрилонитрил/винилгалогенид или тримеры акрилонитрил/алкилметакрилат/бутадиен.
10. Полимеры, которые получают из ненасыщенных спиртов и аминов или их ацильных производных, или их ацеталей, такие как поливиниловый спирт, поливинилацетат, поливинилстеарат, поливинилбензоат, поливинилмалеат, поливинилбутират, полиаллилфталат или полиаллилмеламин, а также их сополимеры с олефинами, упомянутыми выше в пункте 1.
11. Гомополимеры и сополимеры циклических простых эфиров, такие как полиалкиленгликоли, окись полиэтилена, окись полипропилена или их сополимеры с бис-глициловыми простыми эфирами.
12. Полиацетали, такие как полиоксиметилен, и такие полиоксиметилены, которые содержат окись этилена в качестве сомономера, полиацетали, модифицированные термопластичными полиуретанами, акрилатами или MBS.
13. Окислы и сульфиды полифенилена, и смеси окислов полифенилена с полистиролом или полиамидами.
14. Полиуретаны, которые получают из простых полиэфиров, сложных полиэфиров или полибутадиенов с концевыми гидроксильными группами на одном конце и алифатическими или ароматическими полиизоцианатами на другом конце, а также их предшествующие соединения (полиизоцианаты, полиолы или предполимеры).
15. Полиамиды и сополиамиды, которые получают из диаминов и дикарбоновых кислот и/или из аминокарбоновых кислот, или соответствующих лактамов, таких как полиамид 4, полиамид 6, полиамид 6, полиамид 6/6, 6/10, 6/9, 6/12 и 4/5, полиамид 11, полиамид 12, ароматические полиамиды, полученные в результате конденсации мета-ксилолдиамина и адипиновой кислоты, полиамиды, полученные из гексаметилендиамина и изофталевой и/или терефталевой кислоты, и при необходимости эластомера в качестве модификатора, например, поли-2,4,4-триметилгексаметилентерефталамид или полиметафениленизофталамид. Другие сополимеры вышеупомянутых полиамидов с полиолефинами, сополимерами олефинов, иономерами или химически связанными или графт-эластомерами, или с простыми полиэфирами, такими как, например, полиэтиленгликоли, полипропиленгликоли или политетраметиленгликоли. Полиамиды или сополиамиды, модифицированные при помощи ЭПДМ или ABS. Полиамиды, конденсированные в процессе обработки (RIM-полиамидные системы).
16. Полимочевины, полиимиды и полиамидимиды.
17. Сложные полиэфиры, которые получают из дикарбоновых кислот и диолов и/или из оксикарбоновых кислот, или соответствующих лактонов, такие как полиэтилентерефталат, полибутилентерефталат, поли-1,4-диметилолциклогексантерефталат, поли-[2,2-(4-оксифенил)-пропан]терефталат и полиоксибензоаты, а также блок-сополимеры простых эфиров и сложных эфиров, полученные из простых полиэфиров, содержащих гидроксильные концевые группы.
18. Поликарбонаты и сложные полиэфиркарбонаты.
19. Полисульфоны, простые полиэфирсульфоны и простые полиэфиркетоны.
20. Сшитые полимеры, которые получают из альдегидов с одной стороны и фенолов, мочевин и меламинов - с другой, такие как фенол/формальдегидные смолы, смолы из мочевины/формальдегида и меламин/формальдегидные смолы.
21. Высыхающие и невысыхающие алкидные смолы.
22. Смолы из ненасыщенных сложных полиэфиров, которые получают из сложных сополиэфиров насыщенных и ненасыщенных дикарбоновых кислот с многоатомными спиртами, и виниловыми соединениями в качестве сшивающих агентов, а также их содержащие галогенпроизводные с низкой воспламеняемостью.
23. Термореактивные акриловые смолы, полученные из замещенных акриловых сложных эфиров, такие как эпоксиакрилаты, уретанакрилаты и сложные полиэфиракрилаты.
24. Алкидные смолы, смолы сложных полиэфиров или акрилатные смолы в смеси с меламиновыми смолами, смолами мочевины, полиизоцианатами или эпоксидными смолами в качестве сшивающих агентов.
25. Сшитые эпоксидные смолы, которые получают из полиэпоксидов, например, бис-глицидиловых простых эфиров или из циклоалифатических диэпоксидов.
26. Натуральные полимеры, такие как целлюлоза, каучук, желатин и их производные, которые химически модифицированы в полимеры-гомологи, такие как ацетаты целлюлозы, пропионаты целлюлозы и бутираты целлюлозы, или простые эфиры целлюлозы, такие как метилцеллюлоза: канифоль и ее производные.
27. Смеси полимеров, упомянутых выше, например, ПП/ЭПДМ, полиамид 6/ЭПДМ или ABS, ПВХ/ЭВА, ПВХ/ABS, ПВХ/MBS, ПХ/ABS, ПБТП/ABS, ПХ/ASA, ПХ/ПБТ, ПВХ/акрилаты, ПОМ/термопластический ПУР, ПХ/термопластический ПУР, ПОМ/акрилат, ПОМ/MBS, ППЭ/HIPS* (* - полистирол с высокой прочностью на удар), ППЭ/ПА 6.6 и сополимеры, ПА/ПХНП, ПА/ПП, ПА/ППЭ.
Предлагаемые соединения добавляют в полимеры в концентрации 0,05 - 5 мас. %, в пересчете на материал, подлежащий стабилизации. В предпочтительном варианте 0,1 - 2,5 мас.% стабилизатора в пересчете на стабилизируемый материал добавляют в последний.
Такое добавление может быть осуществлено в процессе полимеризации или после полимеризации, например, при помощи смешения соединения и, если это необходимо, других добавок с расплавом с использованием приемов, известных в этой области техники, перед или в процессе формования или при помощи введения растворенных или диспергированных соединений в полимер.
Другими добавками (присадками), которые используют с предлагаемыми соединениями, могут быть другие стабилизаторы, такие как феноловые антиоксиданты, дезактиваторы металлов, фосфиты, тиодипропионовые сложные диэфиры, соли жирных кислот, УФ-поглотители или комплексные соли никеля. Другими добавками могут быть пигменты, наполнители, пластификаторы, ингибиторы воспламенения или антистатики.
Как указано выше, стабилизаторы, являющиеся предметом настоящего изобретения, могут быть использованы в количестве от примерно 0,05 до примерно 5 мас. % от стабилизируемой композиции. Это количество может варьироваться в зависимости от конкретного субстрата и применения. Предпочтительной областью является область от примерно 0,1 до примерно 2,5%.
Соединения, являющиеся предметом предлагаемого изобретения, могут быть использованы как таковые или вместе с фенолами в фотографических слоях в качестве световых стабилизаторов желтого цвета, в качестве стабилизаторов темных оттенков цианового цвета, в качестве агентов, препятствующих образованию пятен в фуксиновых слоях (в частности, для двух эквивалентных фуксиновых пар) и в качестве термостабилизаторов для фуксиновых пар.
Пример 11. Ди-(1-метокси-2,2,6,6-тетраметилпиперидин-4-ил)изофталат.
 .
.
Раствор 30,0 г (73 ммоль) ди-(1-оксид-2,2,6,6-тетраметилпиперидин-4-ил)изофталата и 27,8 г (190 ммоль) дитрет.-бутилперекиси в 70 мл хлорбензола нагревали в течение 6 ч в атмосфере азота в склянке Фишера-Портера (температура бани 145 - 150oC). Сырую реакционную смесь подвергали хроматографии на силикагеле (98 : 2 гептан : этилацетат), чтобы получить твердое вещество, которое подвергали перекристаллизации из метанола, в результате чего получали соединение, указанное в заголовке: белок - кристаллическое твердое вещество, т.пл. 99 - 101oC.
Результаты анализа для C28H44N2O6:
рассчитано: C 66,6; H, 8,8; N, 5,66.
найдено: C 66,4; H, 8,7; N, 5,5.
Пример 12. Ди-(1-метокси-2,2,6,6-тетраметилпиперидин-4-ил)себацинат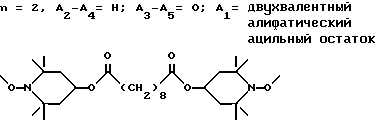 .
.
4-Бензоилокси-1-метокси-2,2,6,6-тетраметилпиперидин (8,0 г, 32 ммоль) перемешивали 2 ч при температуре 60 - 70oC (в атмосфере азота) с 2,2 г (39 ммоль) гидрата окиси калия в 300 мл 1 : 1 (о/о) метанол : вода. Растворитель удаляли при пониженном давлении, чтобы получить белое твердое вещество, которое разделяли между водой (100 мл) и дихлорметаном (150 мл). Водный слой промывали дихлорметаном (2 х 150 мл). Органические слои соединяли и промывали водой (100 мл) и насыщенным раствором хлорида натрия (100 мл), затем сушили над сульфатом магния и концентрировали, чтобы получить 5,7 г сырого 4-окси-1-метокси-2,2,6,6-тетраметилпиперидина, белого твердого вещества с т. пл. 92,5 - 93,5oC. ИК: 3250 см-1.
Раствор 5,4 г (29 ммоль) 4-окси-1-метокси-2,2,6,6-тетраметилпиперидина, 3,2 г (13,9 ммоль) диметилсебацината и 200 мл толуола подвергали дистилляции в течение 45 мин с тем, чтобы азеотропно удалить оставшуюся воду. Раствору давали возможность охладиться и добавляли 150 мг амида лития. Реакционную смесь медленно дистиллировали в течение 5 ч, чтобы удалить метанол вместе с некоторым количеством толуола. Оставшийся толуол затем удаляли при пониженном давлении. Реакционную смесь охлаждали до 5oC и добавляли воду (20 мл). Органический материал растворяли в этилацетате (200 мл). Водный слой экстрагировали этилацетатом (2 х 100 мл). Соединенные органические слои промывали водой (2 х 50 мл) и насыщенным раствором хлорида натрия (50 мл), затем сушили над сульфатом магния и концентрировали при пониженном давлении. Сырую жидкость подвергали хроматографии на силикагеле (95 : 5 гептан : этилацетат), чтобы получить 5,4 г (68% общий выход) соединения, указанного в заголовке, бесцветной жидкости. ИК: 1750 см-1.
Результаты анализа для C30H56N2O6
рассчитано: C 66,6; H 10,4; N 5,2:
найдено: C 66,7; H 10,5; N 5,0.
Пример 13. α,α′ -(Ди-1-этокси-2,2,6,6-тетраметилпиперидин-4-илокси)-пара-ксилол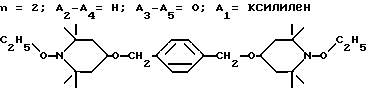 .
.
Смесь 9,0 г (20,1 ммоль) α,α′ -(ди-1-окси-2,2,6,6-тетраметилпиперидин-4-илокси)-пара-ксилола, 1,8 г (44,2 ммоль) гидрида натрия и 100 мл тетрагидрофурана (ТГФ) подвергали дефлегмации в атмосфере азота в течение 2 ч. Реакционную смесь охлаждали до 50oC и добавляли избыточное количество этилиодида (7,5 г, 48,2 ммоль). Реакционную смесь дефлегмировали в течение 2 ч. Затем добавляли дополнительное количество гидрида натрия (1,8 г), этилиодида (7,5 г) и 1,0 мл трет.-бутилового спирта, а реакционную смесь подвергали дефлегмации в течение 16 ч. Реакционную смесь охлаждали и добавляли метанол. Реакционную смесь разделяли между водой (600 мл) и диэтиловым простым эфиром (200 мл). Водный слой экстрагировали простым эфиром (200 мл). Соединенные органические слои промывали водой (200 мл) и насыщенным раствором хлорида натрия (200 мл), затем сушили над сульфатом магния и концентрировали, чтобы получить желтое масло. Это масло подвергали хроматографии на силикагеле (4 : 1 гексан : этилацетат), чтобы получить сырое твердое вещество, которое последовательно подвергали перекристаллизации из холодного метанола и гексана. Выход равен 7,9 г (78%) белого твердого вещества, т. пл. 99 - 110oC.
Результаты анализа для C30H52N2O4:
рассчитано: C 71,4; H 10,4; N 5,5
найдено: C 71,6; H 10,8; N 5,5.
Пример 14. Ди-(1-циклогексилокси-2,2,6,6-тетраметилпиперидин-4-ил) себацинат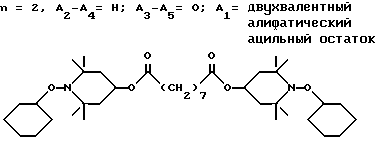 .
.
Смесь 20,0 г (41,6 ммоль) ди-(-2,2,6,6-тетраметилпиперидин-4-ил)себацината, 4,3 г (334 ммоль) 70% водного раствора трет.-бутил гидроперекиси, 1,3 г (9,0 ммоль) трехокси молибдена и 125 мл циклогексана нагревали при температуре дефлегмации 2,3 ч. Воду собирали при помощи ловушки Дина-Старка. Красную реакционную смесь охлаждали и переносили в склянку Фишера-Портера. Свежий циклогексан (25 мл) использовали для того, чтобы тщательно промыть колбу и промывочные жидкости добавляли в толстостенную склянку. Эту склянку погружали в масляную ванну (140oC) на 3 ч, после чего бесцветную реакционную смесь охлаждали до комнатной температуры и фильтровали. Фильтрат перемешивали с 10 г сульфита натрия в 90 мл воды в течение 2 ч, чтобы разложить непрореагировавшую гидроперекись, затем разбавляли этилацетатом (200 мл) и водой (100 мл). Органический слой промывали 10%-ным раствором сульфита натрия (100 мл), водой (100 мл), насыщенным раствором хлорида натрия (100 мл), затем сушили над сульфатом магния и концентрировали при пониженном давлении. Сырой продукт подвергали очистке с использованием испарительной хроматографии (силикагель, 100 : 2, гептан : этилацетат), в результате чего получали 17,8 г (выход 68%) белого твердого вещества, т.пл. 56 - 59oC.
Результаты анализа для C40H72N2O6:
рассчитано: C 71,0, H 10,7, N 4,1,
найдено: C 71,0, H 10,2, N 4,2.
Пример 15. Ди-(1-циклогексилокси-2,2,6,6-тетраметилпиперидин-4-ил) изофталат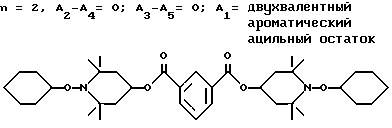 .
.
Раствор 34,2 г ди-(1-оксил-2,2,6,6-тетраметилпиперидин-4-ил)изофталата и 54 мл ди-трет.-бутилперекиси в 250 мл циклогексана нагревали на 22 ч в атмосфере азота в склянке Фишера-Портера (температура бани 140oC). Растворитель выпаривали при пониженном давлении. Продукт подвергали перекристаллизации из пентана, чтобы получить белое твердое вещество, т. пл. 140 - 142oC.
Результаты анализа для C38H56N2O6:
рассчитано: C 71,2; H 9,4; N 4,4
найдено: C 71,4; H 9,1; N 4,2.
Пример 16. 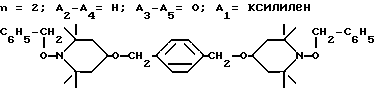 -(Ди-1-бензилокси-2,2,6,6-тетраметилпиперидин-4-илокси)-параксилол
-(Ди-1-бензилокси-2,2,6,6-тетраметилпиперидин-4-илокси)-параксилол
α,α′ .
Смесь 27,7 г (61,7 ммоль) 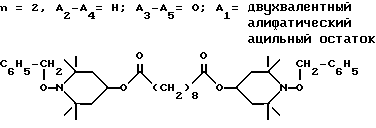 -(ди-1-окси-2,2,6,6-тетраметилпиперидин-4-илокси)-пара-ксилола, 44,4 г (18,5 ммоль) 97%-ного раствора гидрида натрия и 200 мл тетрагидрофурана спокойно дефлегмировали до прекращения выделения водорода. Затем добавляли бензилбромид 31,6 г (185 ммоль) по каплям и реакционную смесь нагревали до температуры дефлегмации на 3 ч, затем перемешивали в течение ночи при комнатной температуре. Избыточное количество гидрида натрия подвергали разложению метанолом. Добавляли толуол (500 мл) и реакционную смесь фильтровали, чтобы удалить соли. Фильтрат промывали водой (3 х 1000 мл) и насыщенным раствором хлорида натрия (500 мл), затем сушили над сульфатом магния и концентрировали, чтобы получить масло. Масло кристаллизовали из метанола, чтобы получить 29,7 г (выход 77%) белого твердого вещества, т. пл. 126 - 129oC.
-(ди-1-окси-2,2,6,6-тетраметилпиперидин-4-илокси)-пара-ксилола, 44,4 г (18,5 ммоль) 97%-ного раствора гидрида натрия и 200 мл тетрагидрофурана спокойно дефлегмировали до прекращения выделения водорода. Затем добавляли бензилбромид 31,6 г (185 ммоль) по каплям и реакционную смесь нагревали до температуры дефлегмации на 3 ч, затем перемешивали в течение ночи при комнатной температуре. Избыточное количество гидрида натрия подвергали разложению метанолом. Добавляли толуол (500 мл) и реакционную смесь фильтровали, чтобы удалить соли. Фильтрат промывали водой (3 х 1000 мл) и насыщенным раствором хлорида натрия (500 мл), затем сушили над сульфатом магния и концентрировали, чтобы получить масло. Масло кристаллизовали из метанола, чтобы получить 29,7 г (выход 77%) белого твердого вещества, т. пл. 126 - 129oC.
Результаты анализа для C40H56N2O4:
рассчитано: C 76,4; H 9,0; N 4,5
найдено: C 76,0; H 9,1; N 4,4.
Пример 17. Ди-(1-бензилокси-2,2,6,6-тетраметилпиперидин-4-ил)себацинат.
Раствор 40,0 г (83 ммоль) ди(2,2,6,6-тетраметилпиперидин-4-ил)себацината в 130 мл толуола нагревали до 80oC. Добавляли гексакарбонилмолибдена (1,0 г) и также добавляли в течение 15 мин 5,0М раствор трет.-бутилгидроперекиси (266 мл, 1,33 ммоль). Реакционную смесь облучали в течение 24 ч УФ-лампой, при этом внутреннюю температуру поддерживали на уровне 85 - 95oC. Реакционную смесь фильтровали и фильтрат выпаривали до объема приблизительно 100 мл. Раствор подвергали хроматографии на силикагеле (9 : 1, гептан : этилацетат), чтобы получить масло, которое кристаллизовали из метанола. После перекристаллизации из этанола получали 16,8 г (выход 29%) белого твердого вещества, т. пл. 64 - 68oC.
Результаты анализа для C42H64N2O6:
рассчитано: C 72,8; H 9,3; N 4,0
найдено: C 72,7; H 9,2; N 4,0.
Пример 18. Ди-(1-бензилокси-2,2,6,6-тетраметилпиперидин-4-ил)фталат.
 .
.
Это соединение получают в соответствии с процедурой, описанной в примере 7, за тем исключением, что гексакарбонил молибдена добавляли перед нагреванием реакционной смеси, т. пл. 141 - 143oC.
Результаты анализа для C40H52N2O6:
рассчитано: C 73,1; H 8,0; N 4,3
найдено: C 73,0; H 7,7; N 4,2.
Пример 19. Ди(1-бензилокси-2,2,6,6-тетраметилпиперидин-4-ил)изофталат.
 .
.
Смесь 35,0 г (78,7 ммоль) ди(2,2,6,6-тетраметилпиперидин-4-ил)изофталата, 1,0 г гексакарбонилмолибдена и 75 мл толуола нагревали до 90oC в атмосфере азота. В течение 5 мин добавляли 4,2М раствор трет.-бутилгидроперекиси в толуоле (225 мл, 945 ммоль). Реакционная смесь становилась красной. После добавления реакционную смесь облучали в течение 6 ч (внутренняя температура 85oC) УФ-лампой. Добавляли еще одну порцию 1,0 г гексакарбониламолибдена и реакционную смесь облучали еще в течение 16 ч. Затем смесь фильтровали и концентрировали. Сырой остаток подвергали хроматографии на силикагеле (9 : 1, гексан : этилацетат). Менее полярный из двух основных продуктов подвергали перекристаллизации из смеси 9 : 1 этанол : дихлорметан, чтобы получить 14,8 г (выход 29%) белого твердого вещества, т. пл. 135 - 141oC, которое представляет собой соединение, указанное в заголовке.
Результаты анализа для C40H52N2O6:
рассчитано: C 73,1; H 8,0; N 4,3
найдено: C 72,9; H 7,7; N 4,6.
Пример 20. Ди(1-бензилокси-2,2,6,6-тетраметилпиперидин-4-ил)диэтилмалонат.
 .
.
Это соединение получали из ди(2,2,6,6-тетраметилпиперидин-4-ил)диэтилмалоната, трет. -бутилгидроперекиси, толуола и гексакарбониламолибдена в соответствии с процедурой, описанной в примере 19, т. пл. 122 - 123oC.
Результаты анализа C39H58N2O6:
рассчитано: C 72,0; H 9,0; N 4,3
найдено: C 72,0; H 9,3; N 4,7.
Пример 21. Ди-[1-( α -метилбензилокси)-2,2,6,6-тетраметилпиперидин- 4-ил]фталат.
 .
.
Это соединение получали из 40,0 г ди(1-оксил-2,2,6,6-тетраметилпиперидин-4-ил)фталата, 200 мл этилбензола и 2,0 г окиси молибдена, которые нагревали до 110oC (в атмосфере азота). Затем по каплям в течение 1 ч добавляли 65 г 70%-ной трет.-бутилперекиси в воде. Реакционную смесь подвергали дефлегмации в течение 3 ч после того, как добавление гидроперекиси закончено. Сырой продукт подвергали хроматографии на силикагеле (9 : 1, гексан : этилацетат), чтобы получить 51,0 г (выход 88%) мягкого стекловидного продукта.
Результаты анализа для C42H56N2O6:
рассчитано: C 73,7; H 8,2; N 4,1
найдено: C 74,1; H 8,4; N 4,1.
Пример 22. Ди-(1-бензилокси-2,2,6,6-тетраметилпиперидин-4-ил) изофталат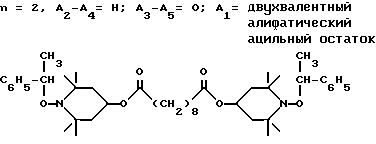 .
.
Смесь 40,0 г (83 ммоль) ди(2,2,6,6-тетраметилпиперидин-4-ил)себацината, 2,0 г окиси молибдена и 250 мл этилбензола нагревали до 110oC (в атмосфере азота). По каплям в течение 30 мин добавляли производимый промышленностью раствор 70%-ной трет. -бутилгидроперекиси в воде (64,3 г, 499 ммоль). Воду собирали в ловушку Дина-Старка. Нагревание продолжали в течение 90 мин после добавления. Реакционную смесь фильтровали и выпаривали. Полученное в результате сырое масло растворяли в гептане (300 мл) и этот раствор пропускали через короткую колонну из силикагеля. Первые 350 мл фильтрата, которые являются почти чистыми после ТСХ, выпаривали, чтобы получить 41,7 г (выход 70%) соединения, указанного в заголовке, в виде вязкого масла.
Результаты анализа для C44H67N2O6:
рассчитано: C 72,8; H 9,6; N 3,9
найдено: C 72,9; H 9,7; N 3,8.
Пример 23. Ди-(1-гептилокси-2,2,6,6-тетраметилпиперидин-4-ил) себацинат.
 .
.
Смесь 35,0 г (72,8 ммоль) ди-(2,2,6,6-тетраметилпиперидин-4-ил)себацината, 58,3 г (582 ммоль) 90%-ного водного раствора трет.-бутилгидроперекиси, 2,0 г трехокиси молибдена и 250 мл гептана нагревали до температуры 140oC в склянке Фишера-Портера. Давление поддерживали на уровне 40-50 фунтов/дюйм2 (2,8-3,5 кг/см2) при помощи периодической вентиляции. Нагревание прекращали через 7 ч. Добавляли дополнительную порцию (20,0 г) 90%-ного раствора трет.-бутилгидроперекиси и реакционную смесь нагревали на 1 ч до 140oC. Реакция становилась почти бесцветной к этому моменту. Реакционную смесь охлаждали и фильтровали, чтобы удалить катализатор. Органическую фазу отделяли, сушили над сульфатом магния и концентрировали до общего объема в 100 мл. Этот раствор пропускали через силикагель с гептаном в качестве элюента. Фильтрат выпаривали с выходом 36,9 г (72%) соединения, указанного в заголовке, которое имело вид почти бесцветного масла.
Результаты анализа для C42H80N2O6:
рассчитано: C 71,1; H 11,4; N 3,95
найдено: C 71,3; H 11,8; N 3,9.
Пример 24. Ди-(1- α -метилбензилокси-2,2,6,6-тетраметилпиперидин- 4-ил)терефталат.
 .
.
Суспензию 40,0 г (90,0 ммоль) ди-(2,2,6,6-тетраметилпиперидин-4-ил)терефталата, 2,0 г трехокиси молибдена и 250 мл этилбензола нагревали до температуры 110oC, быстро добавляли трет.-бутилгидроперекись (70%, 69,5 г, 540 ммоль). Никакой реакции не происходит до тех пор, пока не будет удалена вода при помощи азеотропной дистилляции и внутренняя температура не достигнет 115oC. Нагревание продолжали в течение 6 ч. Почти бесцветной реакционной смеси давали возможность охладиться, затем ее фильтровали и выпаривали, в результате чего получали розовое твердое вещество. Это твердое вещество подвергали перекристаллизации (9 : 1, 2 - пропанол : метиленхлорид), чтобы получить 48,4 г соединения, указанного в заголовке, белого твердого вещества, т. пл. 150 - 152oC. Вторую порцию в 5,3 г получали из маточного раствора. Общий выход 53,7 г (87%).
Результаты анализа для C42H56N2O6:
рассчитано: C 73,7; H 8,2; N 4,1
найдено: C 74,0; H 8,2; N 4,0.
Пример 25. Ди-(1- α -метилбензилокси-2,2,6,6-тетраметилпиперидин- 4-ил)изофталат.
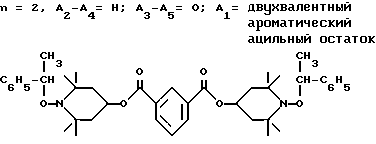 .
.
Смесь 40,0 г (90,0 ммоль) ди-(2,2,6,6-тетраметилпиперидин-4-ил)изофталата, 2,0 г трехокиси молибдена и 250 мл этилбензола нагревали до 110oC, по каплям в течение 45 мин добавляли трет.-бутилгидроперекись (70%, 69,5 г, 540 ммоль). Реакционная смесь становилась красной при добавлении. Воду удаляли при помощи азеотропной дистилляции. Смесь подвергали дефлегмации в течение 4 ч после того, как добавление закончено. Катализатор отделяли фильтрацией, а фильтрат выпаривали, чтобы получить желтое масло. Разгонку по Кугельрору (110oC, 0,1 мм рт.ст.) осуществляли с целью удаления летучих побочных продуктов. Остаток, вязкое масло, растворяли в гексане и пропускали через силикагель. После выпаривания получали сырое твердое вещество, которое подвергали перекристаллизации из этанола, чтобы получить 39,8 г (выход 65%) соединения, указанного в заголовке, белого порошка, т. пл. 118 - 134oC.
Результаты анализа для C42H56N2O6:
рассчитано: C 73,7; H 8,2; N 4,1
найдено: C 73,4; H 8,3; N 4,1.
Пример 26. Ди-(1-этокси-2,2,6,6-тетраметилпиперидин-4-ил)себацинат.
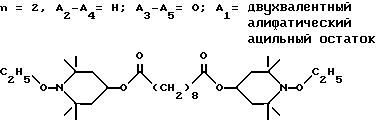 .
.
Раствор 7,8 г (32,5 ммоль) себацилхлорида в 20 мл дихлорметана по каплям добавляли в течение 15 мин в раствор 13,1 г (65,1 ммоль) 1-этокси-4-окси-2,2,6,6-тетраметилпиперидина, 7,0 г триэтиламина и 100 мл дихлорметана. Реакционная смесь начинала кипеть в процессе добавления. Смесь спокойно дефлегмировали еще в течение часа. Добавляли простой эфир (800 мл), осадок отделяли фильтрацией, а фильтрат промывали 1н раствором HCl (2 х 100 мл), водой (200 мл) и насыщенным раствором бикарбоната натрия (300 мл). Органический раствор сушили над сульфатом магния и выпаривали, чтобы получить масло. После очистки с использованием хроматографии (силикагель, 19 : 1, гексан : этилацетат) получали 12,7 г (выход 69%) соединения в виде бесцветного масла, указанного в заголовке.
Результаты анализа для C32H50N2O6:
рассчитано: C 67,6; H 10,6; N 4,9
найдено: C 67,3; H 10,8; N 4,8.
Пример 27. Ди(1-куменилокси-2,2,6,6-тетраметилпиперидин-3-ил) себацинат.
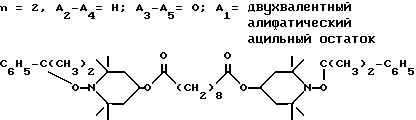 .
.
Соединение из заголовка примера получали в соответствии с процедурой из примера 26 за тем исключением, что использовали 1-куменилоксисоединение. Реакционная температура достигала 33oC в процессе добавления, а затем реакционную смесь перемешивали в течение 1 ч при окружающей температуре. Продукт имел вид белого твердого вещества, т.пл. 94 - 96oC.
Результаты анализа для C46H72N2O6:
рассчитано: C 73,76; H 9,69; N 3,74
найдено: C 74,0; H 9,8; N 3,8.
Пример 28. 8- α -метилбензилокси-7,7,9,9-тетраметил-8-аза-1,4- диоксаспиро[4.5]декан.
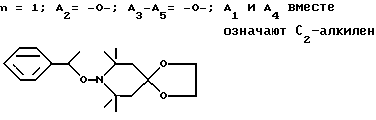 .
.
Смесь 38,1 г (191 ммоль) 7,7,9,9-тетраметил-8-аза-1,4-диоксаспиро[4.5] декана, 73,8 г (574 ммоль) 70%-ного водного раствора трет.-бутилгидроперекиси, 2,0 г трехокиси молибдена и 130 мл этилбензола подвергали дефлегмации в течение 6 ч. Воду собирали в ловушку Дина-Старка. Катализатор отделяли фильтрацией, а фильтрат концентрировали при пониженном давлении. Остаток растворяли в гептане и пропускали через силикагель. Использовали разгонку по Кугельрору (120oC, 0,1 мм рт.ст.), чтобы удалить летучие побочные продукты. Соединение из заголовка примера кристаллизовали при отстаивании.
Результаты анализа для C19H29NO3:
рассчитано: C 71,4; H 9,1; N 4,4
найдено: C 70,3; H 9,2; N 4,4.
Пример 29. 3,15-Ди- α -метилбензилокси-2,2,4,4,14,14,16,16- октаметил-7,11,18,21,-тетраокса-3,15-диазатриспиро[5.2.2.5.2.2]-генэйкозан.
 .
.
Соединение из заголовка примера получали из 2,2,4,4,14,14,16,16-октаметил-7,11,18,21-тетраокса-3,15- диазатриспиро[5.2.2.5.2.2] -генэйкозана в соответствии с процедурой, описанной в примере 29. Катализатор отделяли фильтрацией, а фильтрат концентрировали, чтобы получить масло, которое кристаллизовали из этанола, чтобы получить 19,6 г (выход 65%) белого порошка, т.пл. 150-153oC.
Пример 30. 3,15-Дициклогексилокси-2,2,4,4,14,14,16,16-октаметил- 7,11,18,21-тетраокса-3,15-диазатриспиро[5.2.2.5.2.2]-генэйкозан.
 .
.
Смесь 16,7 г (37,9 ммоль) 3,15-диоксил-2,2,4,4,14,14,16,16-октаметил-7,11,18,21-тетраокса- 3,15-диазатриспиро[5,2,2,5,2,2, ]-генэйкозана, 22,8 г (227 ммоль) 90%-ного водного раствора трет.-бутилгидроперекиси, 2,0 трехокиси молибдена и 125 мл циклогексана нагревали в склянке Фишера-Портера при 155 - 160oC (температуры бани) в течение 6 ч. Давление поддерживали на уровне 40 - 50 фунтов/дюйм2 (2,8 - 3,5 кг/см2) при помощи периодической вентиляции. Катализатор отделяли фильтрацией, а фильтрат концентрировали. Остаток растворяли в гексане и пропускали через силикагель. В результате кристаллизации из 2-пропанола получали 8,0 (35%) белого твердого вещества, т.пл. 163 - 175oC.
Результаты анализа для C35H62N2O6:
рассчитано: C 69,3; H 10,3; N 4,6
найдено: C 68,7; H 10,3; N 4,7.
Пример 31. Ди-(1-метокси-2,2,6,6-тетраметилпиперидин-4-ил)-н-бутилмалонат.
 .
.
Смесь диэтил-н-бутилмалоната (11,5 г, 53,4 ммоль), 4-окси-1-метокси-2,2,6,6-тетраметилпиперидина (2,0, г, 107 ммоль), амида лития (120 г) и ксилола(100 мл) подвергали дистилляции до тех пор, пока дистиллят не достигнет постоянной температуры 137oC. Ксилол выпаривали и остаток растворяли в гептане. Добавляли уксусную кислоту и полученный в результате осадок отделяли фильтрацией. Фильтрат концентрировали, чтобы получить 26,2 г (выход 98%) ярко-желтого масла.
Результаты анализа для C27H50N2O6:
рассчитано: C 65,0; H 10,1; N 5,6
найдено: C 65,0; H 10,4; N 5,5.
Пример 32. Ди-(1-метокси-2,2,6,6-тетраметилпиперидин-4-ил) (3,5-ди-трет. -бутил-4-оксибензил)-н-бутилмалонат.
 .
.
Смесь 11,9 г (45,2 ммоль) N,N-диметил-3,5-дитрет.-бутил-4-оксибензиламина, 18,8 г (37,7 ммоль) ди-(1-метокси-2,2,6,6-тетраметилпиперидин-4-ил)-н-бутилмалоната, 173 мг амида лития и 100 мл тетрагидрофурана дефлегмировали в течение 90 мин. Реакционную смесь разбавляли этилацетатом (350 мл). Органический раствор промывали 1н раствором HCl (2 х 100 мл), водой (2 х 250 мл) и насыщенным раствором NaHCO3 (250 мл), затем сушили над сульфатом магния и выпаривали, чтобы получить коричневое масло. После кристаллизации из смеси 9 : 1 метанола : воды получали 14,9 г (выход 55%) белого твердого вещества с т.пл. 111 - 113oC.
Результаты анализа для C42H72N2O7:
рассчитано: C 70,3; H 10,1; N 3,9
найдено: C 70,2; H 10,2; N 3,9.
Пример 33. Ди(1-циклогексилокси-2,2,6,6-тетраметилпиперидин-4-ил)-н- бутилмалонат.
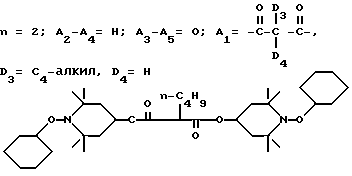 .
.
Соединение из заголовка примера, вязкое масло, получали с 89%-ным выходом в соответствии с процедурой, описанной в примере 31, используя 1-циклогексилоксисоединение в качестве исходного вещества.
Результаты анализа для C37H66N2O6:
рассчитано: C 70,0; H 10,5; N 4,4
найдено: C 69,8; H 10,7; N 4,4.
Пример 34. Ди(1-циклогексилокси-2,2,6,6-тетраметилпиперидин-4-ил) (3,5-ди-трет.-бутил-4-оксибензил)-н-бутилмалонат.
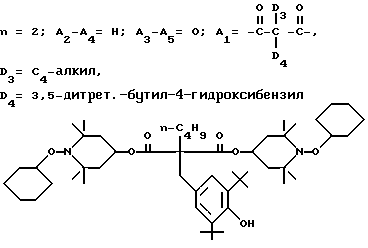 .
.
Соединение из заголовка примера, белок - кристаллическое твердое вещество, получали с 80%-ным выходом в соответствии с процедурой, описанной в примере 32, используя 1-циклогексилоксисоединение, т. пл. 184 - 185oC (этилацетат).
Результаты анализа для C52H88N2O7:
рассчитано: C 73,2; H 10,4, N 3,3
найдено: C 73,7; H 10,8, N 3,3.
Пример 35. Поли-{ [6-(1,1,3,3-тетраметилбутил)амино] -1,3,5-триазин- 2,4-диил] [2-(1-циклогексилокси-2,2,6,6-тетраметилпиперидин)-имино] -гексаметилен-[4-(1-циклогексилокси-2,2,6,6-тетраметилпиперидил)- имино]}.
Смесь 46,9 г N-оксилового предшествующего соединения, 40,2 г (402 ммоль) 90% трет. -бутилгидроперекиси, 6,0 г окиси молибдена и 200 мл циклогексана нагревали на 3 ч до 155oC под давлением. Бесцветную реакционную смесь разбавляли смесью простого эфира, метиленхлорида и толуола, и фильтровали. Фильтрат перемешивали с 5%-ным водным раствором сульфита натрия (400 мл) в течение 45 мин. Органическую фазу промывали водой, сушили над сульфатом магния и фильтровали. Затем фильтрат концентрировали, добавляли метанол, чтобы осадить продукт. Выход составил 19,2 г (36%) белого твердого порошка с т.пл. 187 - 200oC.
Результаты анализа для (C47H86N8O2)n:
рассчитано: C 71,0; H 10,9; N 14,1
найдено: C 68,2; H 10,6; N 14,0.
Пример 36. Ди-(1-циклогексилокси-2,2,6,6-тетраметилпиперидин-4- ил)сукцинат.
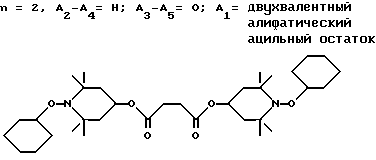 .
.
Двухфазную смесь 70%-ного водного раствора трет.-бутилгидроперекиси (103,9 г, 807 ммоль), циклогексана (200 мл) и хлорида натрия (15 г) встряхивали в делительной воронке. Органическую фазу сушили над сульфатом магния, фильтровали и добавляли к 40,0 г ди-(2,2,6,6-тетраметилпиперидин-4-ил)сукцината. Добавляли окись молибдена (2,0 г) и смесь дефлегмировали в течение 1 ч. Воду собирали в ловушку Дина-Старка. Всю реакционную смесь затем переносили в склянку Фишера-Портера и нагревали 6 ч до 140oC. Добавляли дополнительное количество трет.-бутилгидроперекиси (90%, 10,1 г, 101 ммоль) и нагревание осуществляли еще 4 ч. Бесцветную реакционную смесь фильтровали, концентрировали и растворяли в гептане (20,0 мл). Раствор в гептане пропускали через короткую колонну из силикагеля с гептаном. В результате последующего выпаривания получали масло, которое перекристаллизовывали из этанола, чтобы получить 41,2 г (69%) белого порошка, т.пл. 122 - 126oC.
Результаты анализа для C34H60N2O6:
рассчитано: C 68,9; H 10,2; N 4,7
найдено: C 68,4; H 10,5; N 4,5.
Пример 37. Ди-(1- α -метилбензилокси-2,2,6,6-тетраметилпиперидин-4-ил) сукцинат.
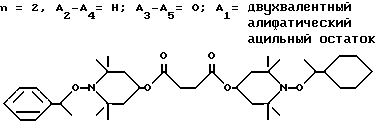 .
.
Соединение из заголовка примера получали в соответствии с процедурой, описанной в примере 25, используя в качестве исходного соединения соответствующий сукцинат. После кристаллизации из этанола получали с 78%-ным выходом белое твердое вещество, т.пл. 85 - 88oC.
Результаты анализа для C38H56N2O6:
рассчитано: C 71,7; H 8,9; N 4,4
найдено: C 71,5; H 8,6; N 4,3.
Пример 38. Ди-(1-нонилокси-2,2,6,6-тетраметилпиперидин-4-ил)себацинат.
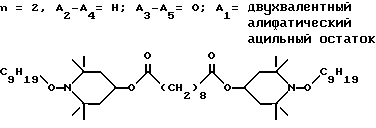 .
.
Соединение из заголовка примера получали в соответствии с процедурой, описанной в примере 36, используя соответствующий себацинат в качестве исходного соединения и нонан за тем исключением, что реакционную смесь дефлегмировали в течение 22 ч при атмосферном давлении. Сырой продукт пропускали через короткую колонну из силикагеля с гептаном в качестве элюента, в результате чего получали с 73%-ным выходом бесцветное масло.
Результаты анализа для C46H88N2O6:
рассчитано: C 72,2; H 11,6; N 3,7
найдено: C 71,6; H 11,5; N 4,7.
Пример 39. Ди-(1-октадецилокси-2,2,6,6-тетраметилпиперидин-4-ил) себацинат.
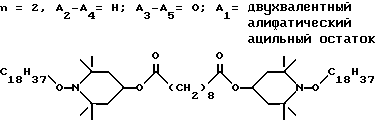 .
.
Реакцию осуществляли в склянке Фишера-Портера в атмосфере азота. В реакционный сосуд загружали 15,0 г (31,2 ммоль) ди(2,2,6,6-тетраметилпиперидин-4-ил)себацината, 101 г октадекана, 25,3 г (253 ммоль) 90%-ной трет.-бутилгидроперекиси и 1,25 г трехокиси молибдена. Склянку Фишера-Портера помещали в масляную ванну и температуру ванны поддерживали на уровне 143oC в течение 1,3 ч. Нагревание продолжали еще в течение 3,2 ч при температуре 145±3oC. Бесцветную реакционную смесь охлаждали до комнатной температуры, разбавляли гексаном (100 мл) и фильтровали, чтобы удалить твердые частицы. Твердые частицы промывали гексаном (2 х 50 мл). Органический раствор перемешивали в течение 90 мин с 16,1 г сульфата натрия в 200 мл воды, чтобы разложить непрореагировавшую гидроперекись. Добавляли этилацетат (200 мл) и органический раствор промывали водой (4 х 250 мл), сушили над сульфатом магния и концентрировали, чтобы получить 121 г бесцветного масла. Сырой продукт подвергали очистке при помощи испарительной хроматографии (силикагель, гептан, затем 20 : 1 смесь гептан : этилацетат), чтобы получить 20,8 г (выход 66%) соединения из заголовка примера в виде бесцветного масла.
Результаты анализа для C64H124NO6:
рассчитано: C 75,5; H 12,3; N 2,75
найдено: C 75,1; H 12,6; N 3,2.
Пример 40. Ди(1-циклогексилокси-2,2,6,6-тетраметилпиперидин-4-ил) фталат.
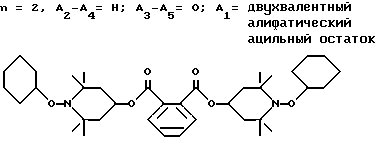 .
.
Смесь 30,0 г (67,5 ммоль) ди-(2,2,6,6-тетраметилпиперидин-4-ил)фталата, 27,5 г (214 ммоль) 70%-ного водного раствора трет.-бутил-гидроперекиси, 2,0 г трехокиси молибдена и 200 мл циклогексана нагревали до температуры дефлегмации. Воду собирали в ловушку Дина-Старка. Через 75 мин реакционная смесь становилась красной. В течение 30 мин добавляли еще одну порцию трет.-бутилгидроперекиси (42,5 г, 70%, 330 ммоль). После того, как собирали дополнительную воду, реакционную смесь переносили в склянку Фишера-Портера и нагревали до 140oC на 4,5 ч. Почти бесцветную реакционную смесь обрабатывали с 6,9 г (90%, 69 ммоль) трет.-бутилгидроперекиси и нагревали до 140oC на 90 мин, чтобы удалить последние следы розового цвета. Реакционную смесь охлаждали, фильтровали и перемешивали с раствором 43 г сульфита натрия в 530 мл воды в течение 2 ч. Добавляли дихлорметан (600 мл) и органический слой отделяли, сушили над сульфатом магния и концентрировали, чтобы получить сырое твердое вещество. После очистки (разделение Уотерза 500А ВЭЖХ, 25 : 1, гептан : этилацетат) получали 31,0 г (выход 72%) белого твердого вещества, т.пл. 149-151oC.
Результаты анализа для C38H60N2O6:
рассчитано: C 71,2; H 9,2; N 4,4
найдено: C 71,1; H 9,3; N 4,3.
Пример 41. (1-Циклогексилокси-2,2,6,6-тетраметилпиперидин-4-ил) (1-метокси-2,2,6,6-тетраметилпиперидин-4-ил)фталат.
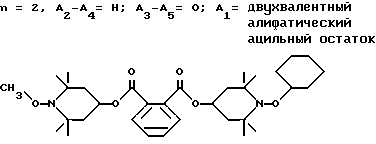 .
.
Соединение из заголовка примера в виде стеклообразного вещества получали в качестве побочного продукта в примере 40.
Масс-спектр: М+: 572.
Пример 42. 1-Циклогексилокси-4-(н-додециламино)-2,2,6,6- тетраметилпиперидин.
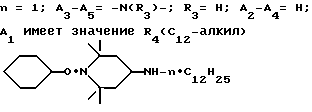 .
.
Уксусную кислоту (22,0 г, 367 ммоль) добавляли по каплям в раствор 15,0 г (59,2 ммоль) 1-циклогексилокси-2,2,6,6-тетраметилпиперидин-4-она и 54,9 г (296 ммоль) додециламина в 200 мл сухого тетрагидрофурана, содержащего молекулярные сита 5А (25 г). Реакционную смесь нагревали при добавлении. Затем смесь разбавляли тетрагидрофураном (150 мл) и охлаждали до 21oC. Одной порцией добавляли цианборгидрид натрия (4,46 г, 7,1 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 4 ч, затем фильтровали. Фильтрат концентрировали и остаток разделяли между простым эфиром (400 мл) и 5%-ным раствором гидрата окиси натрия (250 мл). Органический слой сушили над сульфатом магния, фильтровали и концентрировали. Остаточный додециламин удаляли разгонкой по Кугельрору (110oC, 0,3 мм). Сырой продукт подвергали очистке при помощи хроматографии (силикагель), чтобы получить 20,4 г (выход 82%) соединения из заголовка примера в виде почти бесцветного масла.
Результаты анализа для C27H54N2O:
рассчитано: C 76,7; H 12,9; N 6,6
найдено: C 76,7; H 13,2; N 6,7.
Пример 43. 1- α -метилбензилокси-4-(н-додециламино)-2,2,6,6- тетраметилпиперидин.
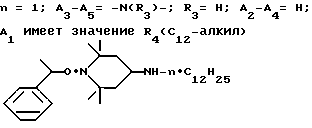 .
.
Соединение из заголовка примера, бесцветное масло, получали из 1- α -метилбензилокси-2,2,6,6-тетраметилпиперидин-4-она и додециламина в соответствии с процедурой, описанной в примере 42.
Результаты анализа для C29H52N2O:
рассчитано: C 78,3; H 11,8; N 6,3
найдено: C 77,7; H 11,6; N 6,6.
Пример 44. 1- α -метилбензилокси-4-(н-бутиламино)-2,2,6,6-тетраметилпиперидин.
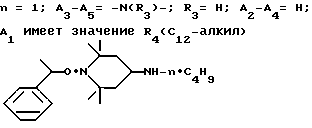 .
.
Соединение из заголовка примера, бесцветное масло, получали из 1- α -метилбензилокси-2,2,6,6-тетраметилпиперидин-4-она и бутиламина в соответствии с процедурой, описанной в примере 42, за тем исключением, что тетрагидрофуран заменяют ацетонитрилом, а молекулярные сита не используют.
Результаты анализа для C21H36N2O:
рассчитано: C 75,8; H 10,9; N 8,4
найдено: C 74,7; H 11,0; N 8,6.
Пример 45. N,N'-ди-(1-циклогексилокси-2,2,6,6-тетраметилпиперидин- 4-ил)-1,6-гександиамин.
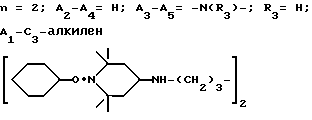 .
.
Смесь 15,0 г (59,1 ммоль) 1-циклогексилокси-2,2,6,6-тетраметилпиперидин-4-она, 3,4 г гександиамина, 500 мг окиси платины и 150 г этанола подвергали гидрогенизации в аппарате Парра в течение 4 ч. Катализатор удаляли фильтрацией. Фильтрат концентрировали в полученное масло, подвергали очистке при помощи испарительной хроматографии (силикагель : этилацетат, затем этилацетат : метанол), чтобы получить 9,7 г (выход 55%) соединения из заголовка примера в виде желтого масла.
Результаты анализа для C36H70N4O2:
рассчитано: C 73,2; H 11,9; N 9,5
найдено: C 72,7; H 12,1; N 9,3.
Пример 46. 1-Циклогексилокси-4-(н-бутиламино)-2,2,6,6- тетраметилпиперидин.
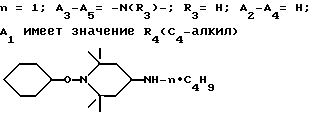 .
.
Соединение из заголовка примера, бесцветное масло, получали из 1-циклогексилокси-2,2,6,6-тетраметилпиперидин-4-она и бутиламина в соответствии с процедурой, описанной в примере 45.
Результаты анализа для C19H38N2O:
рассчитано: C 73,5; H 12,3; N 9,0
найдено: C 73,0; H 12,6; N 8,6.
Пример 47. Ди-(1-нонилокси-2,2,6,6-тетраметилпиперидин-4-ил)сукцинат.
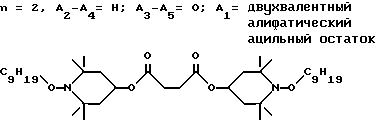 .
.
Соединение из заголовка примера, ярко-желтое масло, получали из ди-(2,2,6,6-тетраметилпиперидин-4-ил)сукцината и нонана в соответствии с процедурой из примера 36.
Результаты анализа для C46H76N2O6:
рассчитано: C 70,5; H 11,2; N 4,1
найдено: C 70,6; H 11,3; N 4,0.
Пример 48. Ди-(1-децилокси-2,2,6,6-тетраметилпиперидин-4-ил)себацинат.
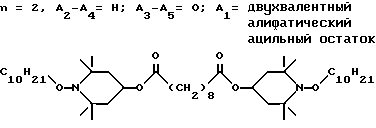 .
.
Трет. -бутилгидроперекись (55,0 г 70%-ного водного раствора, 427 ммоль) по каплям добавляли в течение 15 мин в смесь 25,0 г (52,0 ммоль ди-(2,2,6,6-тетраметилпиперидин-4-ил)себацината, 1,5 г (10,4 ммоль) трехокиси молибдена и 225 мл н-декана, которую нагревали до 90oC. Реакционную смесь дефлегмировали в течение 7,5 ч, а воду собирали в ловушку Дина-Старка. Реакционную смесь охлаждали до комнатной температуры, затем перемешивали в течение 2 ч с раствором 26 г сульфита натрия в 500 мл воды. Реакционную смесь разбавляли этилацетатом (200 мл). Органический слой сушили над сульфатом магния и концентрировали в масло. Неочищенный продукт очищали при помощи испарительной хроматографии (силикагель, 97 : 3, гептан : этилацетат), чтобы получить 29,2 г (выход 71%) соединения из заголовка примера в виде бесцветного масла.
Результаты анализа для C48H92N2O6:
рассчитано: C 72,7; H 11,7; N 3,5
найдено: C 73,1; H 12,2; N 3,5.
Пример 49. Ди-(1-додецилокси-2,2,6,6-тетраметилпиперидин-4-ил) себацинат.
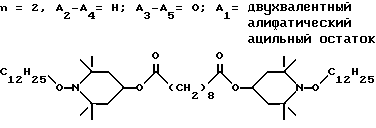 .
.
Соединение из заголовка примера, бесцветное масло, получали из ди-(2,2,6,6-тетраметилпиперидин-4-ил)себацината и н-додекана в соответствии с процедурой, приведенной в примере 48.
Результаты анализа для C52H100N2O6:
рассчитано: C 73,5; H 11,9; N 3,3
найдено: C 73,2; H 12,2; N 3,2.
Пример 50. Ди-[1-1-метилциклогексилокси)-2,2,6,6-тетраметилпиперидин- 4-ил]себацинат.
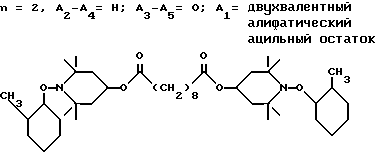 .
.
Трет. -бутилгидроперекись (70% 133,9 г, 1,04 ммоль), метилциклогексан (250 мл) и хлорид натрия (20 г) перемешивали в делительной воронке. Органический слой сушили над сульфатом магния. Раствор трет.-бутилгидроперекиси-метилциклогексана смешивали с 50,0 г (104 ммоль) ди-(2,2,6,6-тетраметилпиперидин-4-ил)себацината, 3,0 г трехокиси молибдена и 100 мл метилциклогексана. Реакционную смесь нагревали до температуры дефлегмации 4,5 ч и воду собирали в ловушку Дина-Старка. Реакцию охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали при пониженном давлении, чтобы получить масло, которое подвергали очистке с использованием испарительной хроматографии (силикагель, 19 : 1, гептан : этилацетат), чтобы получить 51,6 г (выход 72%) бесцветного масла.
Результаты анализа для C42H76N2O6:
рассчитано: C 71,6; H 10,9; N 4,0
найдено: C 71,4; H 11,0; N 3,9.
Пример 51.
Стадия А: 4-Бензоилокси-1-(2-циклогексен-1-илокси)-2,2,6,6- тетраметилпиперидин.
Раствор 33,6 г (122 ммоль) 4-бензоилокси-н-оксил-1-2,2,6,6- тетраметилпиперидина, 23,0 г (157 ммоль) ди-трет.-бутилперекиси и 70 мл циклогексана нагревали в склянке Фишера-Портера до температуры 138oC на 6,5 ч. Реакционную смесь подвергали хроматографии на силикагеле (200 : 1, гептан : этилацетат), чтобы получить 34,1 г (81% выход) бесцветного масла.
Результаты анализа для C22H31NO3:
рассчитано: C 73,9; H 8,7; N 3,9
найдено: C 73,7; H 8,8; N 3,9.
Стадия B: 1-(2-циклогексен-1-илокси)-4-окси-2,2,6,6- тетраметилпиперидин.
Соединение из заголовка стадии, белое твердое вещество (температура плавления 66 - 69oC) получали при помощи гидролиза (КОН-вода-метанол) 4-бензоилокси-1-(2-циклогексен-1-илокси)-2,2,6,6- тетраметилпиперидина.
Результаты анализа для C15H27NO2:
рассчитано: C 71,1; H 10,7; N 5,5
найдено: C 71,7; H 11,4; N 5,5.
Стадия C: Ди-[1-(2-циклогексен-1-илокси)-2,2,6,6-тетраметилпиперидин- 4-ил]себацинат.
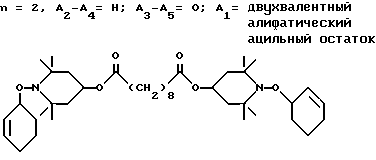 .
.
Соединение из заголовка стадии, бесцветное масло, получали реакций 1-(2-циклогексен-1-илокси)-4-окси-2,2,6,6-тетраметилпиперидина и себацилхлорида в соответствии с процедурой, описанной в примере 26.
Результаты анализа для C40H68N2O6:
рассчитано: C 71,4; H 10,2; N 4,2
найдено: C 71,6; H 10,7; N 4,0.
Пример 52.
Стадия A: 4-Бензоилокси-1-трет.-бутокси-2,2,6,6-тетраметилпиперидин.
Раствор 56,0 г (203 ммоль) 4-бензоилокси-1-окси-2,2,6,6- тетраметилпиперидина, 125 мл хлорбензола и 58,9 г трет.-бутилиодида охлаждали до 5oC. По каплям в течение 110 мин добавляли гидрид три-н-бутилолова (29 г, 100 ммоль), при этом температуру поддерживали ниже 20oC. Реакционную смесь концентрировали при пониженном давлении, а затем подвергали очистке при помощи хроматографии на силикагеле (200 : 1, затем 100 : 3, гептан : этилацетат), чтобы получить 30,1 г белого твердого вещества (т. пл. 80 - 82,5oC).
Результаты анализа для C20H31NO3:
рассчитано: C 72,0: H 9,4; N 4,2
найдено: C 71,9; H 9,6; 4,1.
Стадия B: 1-Трет.-бутокси-4-окси-2,2,6,6-тетраметилпиперидин.
Соединение из заголовка стадии, белое твердое вещество (т. пл. 115 - 116oC) получали при помощи гидролиза (КОН-вода-метанол) 4-бензилокси-1-трет. -бутокси-2,2,6,6-тетраметилпиперидина.
Результаты анализа для C13H27NO2:
рассчитано: C 68,1; H 11,9; N 6,1
найдено: C 68,5; H 12,4; N 6,1.
Стадия C: Ди-(1-трет.-бутокси-2,2,6,6-тетраметилпиперидин-4-ил) -себацинат.
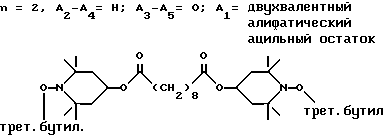 .
.
Соединение заголовка стадии, белок твердое вещество (т.пл. 62 63oC) получали в результате реакции 1-трет.-бутокси-4-окси-2,2,6,6-тетраметилпиперидина и себацилхлорида в соответствии с процедурой, описанной в примере 26.
Результаты анализа для C36H68N2O6:
рассчитано: C 69,2; H 11,0; N 4,4
найдено: C 69,5; H 11,3; N 4,4.
Пример 53. 4-Ацетамидо-1-циклогексилокси-2,2,6,6-тетраметилпиперидин.
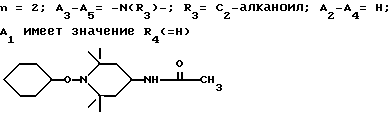 .
.
Перемешиваемую смесь 10,0 г 4-ацетамидо-2,2,6,6-тетраметилпиперидин-1-оксила, 16 мл 70%-ного водного раствора трет.-бутилгидроперекиси и 0,67 г трехокиси молибдена в 75 мл циклогексана нагревали при дефлегмации в колбе, снабженной устройством Дина-Старка. После того, как собрано 6 мл воды, реакционную смесь переносили в аппарат Фишера-Портера и нагревали до 140oC в течение 4 ч при давлении 30 фунтов на дюйм2 (2,11 кг/см2). Обесцвеченную реакционную смесь фильтровали и фильтрат промывали водой, водным раствором сульфита натрия, соляным раствором, сушили над сульфатом магния и концентрировали, в результате чего получали 13,61 г белого твердого вещества. После перекристаллизации из гептана получали 10,39 г соединения из заголовка примера в виде белого кристаллического твердого вещества, т. пл. 140 - 144oC.
Результаты анализа для C17H32N2O2:
рассчитано: C 68,9; H 10,9; N 9,5
найдено: C 68,6; H 11,3; N 9,3.
Пример 54. 4-Амино-1-циклогексилокси-2,2,6,6-тетраметилпиперидин.
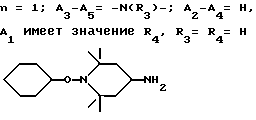 .
.
Раствор 5,0 г ацетамида из примера 53 в 7,5 мл воды и 7,5 мл концентрированной хлористоводородной кислоты нагревали до температуры дефлегмации и выдерживали 10 ч. Затем реакционную смесь быстро охлаждали насыщенным раствором карбоната натрия и экстрагировали этилацетатом. Соединенные органические экстракты промывали водой, соляным раствором, сушили над сульфатом магния и выпаривали, чтобы в результате получить светло-коричневое масло. В результате перегонки (Кугельрор, 140oC @1,5 мм) получали соединение из заголовка примера в виде прозрачного бесцветного масла.
Пример 55. N,N'-бис(1-циклогексилокси-2,2,6,6-тетраметилпиперидин- 4-ил)сукцинамид.
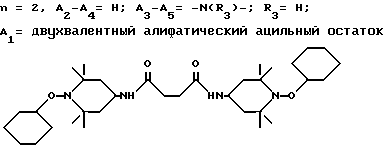 .
.
Смесь 1-циклогексилокси-4-амино-2,2,6,6-тетраметилпиперидина (2,65 г), диметилсукцината (1,46 г) и метилата натрия (50 мг) нагревали до 180 - 190oC в течение 4 ч. Сырой продукт подвергали перекристаллизации из этанола/воды, чтобы получить 1,38 г соединения из заголовка примера в виде белого твердого вещества, т. пл. 230 - 234oC.
Результаты анализа для C34H62N4O4;H2O:
рассчитано: C 67,1; H 10,6; N 9,2
найдено: C 66,8; H 10,7; N 9,1.
Пример 56. Бис(1-метокси-2,2,6,6-тетраметилпиперидин-4-ил)сукцинат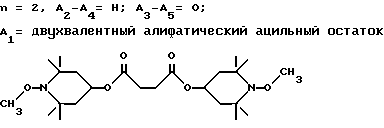 .
.
Смесь 20,0 г (107 ммоль) 4-окси-1-метокси-2,2,6,6-тетраметилпиперидина, 7,41 г (50,7 моль) диметилсукцината, 0,08 г амида лития и 100 мл толуола нагревали до температуры дефлегмации 7 ч. Метанол отгоняли из реакционной смеси вместе с некоторым количеством толуола. Реакционную смесь охлаждали и добавляли 0,21 г ледяной уксусной кислоты в толуоле. Осадок удаляли фильтрацией. Фильтрат концентрировали, чтобы получить масло, которое подвергали очистке при помощи ВЭЖХ (Преп. Уотерза 500 А, смесь 29 : 1, гептан : этилацетат), чтобы получить 14,5 г (выход 63%) соединения из заголовка примера.
Результаты анализа для C24H44N2O6:
рассчитано: C 63,1; H 9,7; N 6,1
найдено: C 63,1; H 9,9; N 6,0.
Пример 57. Бис(1-октилокси-2,2,6,6-тетраметилпиперидин-4-ил)сукцинат.
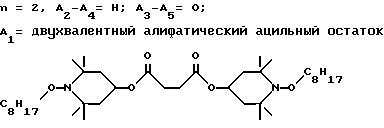 .
.
Раствор 143,1 г (1,11 моль) 70%-ного водного раствора трет.-бутилгидроперекиси добавляли в течение 4 ч в смесь при дефлегмации 55,0 г (139 ммоль) бис(2,2,6,6-тетраметилпиперидин-4-ил)сукцината, 1,0 г трехокиси молибдена и 350 мл н-октана. Реакционную смесь нагревали при температуре дефлегмации 16 ч после завершения добавления до удаления красной окраски. Смесь фильтровали, чтобы удалить твердые частицы. Фильтрат концентрировали, чтобы получить остаток, который подвергали очистке при помощи испарительной хроматографии, чтобы получить 53,6 г (выход 59%) соединения из заголовка примера в виде желтого масла.
Результаты анализа для C38H72N2O6:
рассчитано: C 69,9; H 11,1; N 4,3
найдено: C 70,0; H 11,7; N 4,4.
Пример 58. Бис(1-метокси-2,2,6,6-тетраметилпиперидин-4-ил)терефталат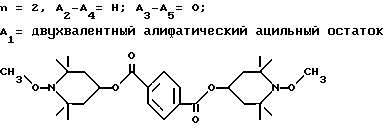 .
.
Смесь 7,36 г (16,6 ммоль) бис(2,2,6,6-тетраметилпиперидин-4-ил) терефталата, 0,1 г трехокиси молибдена и 25 мл хлорбензола нагревали до 130oC в атмосфере азота. В реакционную смесь в течение 1 ч добавляли раствор 40,2 г (132 ммоль) гидроперекиси кумола в хлолрбензоле. Ловушку Дина-Старка использовали для удаления воды из реакции. Смесь нагревали при дефлегмации 4 ч после того, как добавление закончено, затем охлаждали и фильтровали. Фильтрат концентрировали, а остаток подвергали очистке фильтрацией через силикагель с использованием 19 : 1 смеси гептана : этилацетата в качестве элюента. После кристаллизации из метанола получали 5,0 г (выход 60%) соединения из заголовка примера, белый порошок, т. пл. 179 - 181oC.
Результаты анализа для C28H44N2O6:
рассчитано: C 66,6; H 8,8; N 5,5
найдено: C 66,7; H 8,9; N 5,4.
Пример 59. 1-Метокси-4-н-бутиламино-2,2,6,6-тетраметилпиперидин.
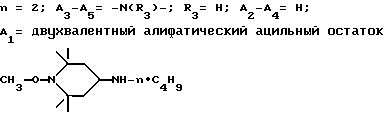 .
.
Смесь 10,1 г (54,5 ммоль) 1-метокси-2,2,6,6-тетраметилпиперидин-4-она, 27,9 г (382 ммоль) н-бутиламина, 100 мл метанола и 1,0 г 5% палладия на углероде подвергали гидрогенизации [50 фунтов/дюйм2 (3,5 кг/см2, 25oC] в течение 5 ч. Катализатор удаляли фильтрацией. После выпаривания фильтрата получали масло, которое подвергали очистке при помощи фракционной перегонки, чтобы получить 10,6 г (выход 80%) соединения из заголовка примера, бесцветное масло, т. кип. 93 - 100oC (0,25 мм).
Результаты анализа для C14H30N2O:
рассчитано: C 69,4; H 12,5; N 11,6
найдено: C 68,4; H 12,2; N 11,2.
Пример 60. Бис[N-1-циклогексилокси-2,2,6,6-тетраметилпиперидин-4-ил)-N-бутил] себацинамид.
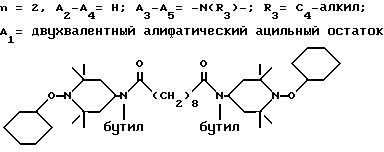 .
.
70%-ный Водный раствор трет. -бутилгидроперекиси (41,4 г, 321 ммоль) разделяли между 200 мл циклогексана и 50 мл насыщенного водного раствора хлорида натрия. Раствор смеси трет.-бутилгидроперекись/циклогексан, 19,0 г (32 ммоль) бис[N-(2,2,6,6-тетраметилпиперидин-4-ил)-N-бутил]себацинамида и 0,4 г трехокиси молибдена нагревали в склянке Фишера-Портера при температуре 150 - 160oC 2 ч. Реакционную смесь фильтровали и фильтрат выпаривали. Остаточное масло подвергали очистке при помощи испарительной хроматографии на силикагеле (3 : 1, гептан : этилацетат). После кристаллизации из метанола получали 13,1 г (выход 52%) соединения из заголовка примера, белое твердое вещество, т. пл. 124 - 128oC.
Результаты анализа для C48H90N4O4:
рассчитано: C 73,2; H 11,5; N 7,1
найдено: C 72,7; H 11,7; N 7,0.
Пример 61. Бис[N-(1-октилокси-2,2,6,6-тетраметилпиперидин-4-ил)- N-бутил]-себацинамид.
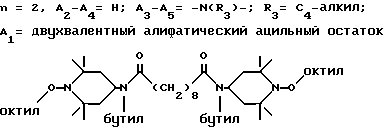 .
.
Раствор 70%-ного водного раствора трет.-бутилгидроперекиси (33,0 г, 256 ммоль) насыщали хлоридом натрия и экстрагировали 200 мл н-октана. Смесь 19,0 г (32 ммоль) бис[N-(2,2,6,6-тетраметилпиперидин-4-ил)-N-бутил] себацинамида, 0,2 г трехокиси молибдена и половине раствора трет.-бутилгидроперекиси/октана нагревали при температуре дефлегмации 30 мин. Воду собирали при помощи ловушки Дина-Старка. Затем в кипящую реакционную смесь в течение 2 ч добавляли остаток раствора трет.-бутилгидроперекиси/октана. Реакционную смесь нагревали до температуры дефлегмации еще на 4,5 ч, затем обрабатывали 20,0 г (155 ммоль) 70%-ного водного раствора трет.-бутилгидроперекиси и нагревали до дефлегмации еще 2 ч до удаления красной окраски. Твердые частицы удаляли фильтрацией, а фильтрат выпаривали. В результате очистки сырого продукта испарительной хроматографией на силикагеле (4 : 1, гептан : этилацетат) получали 16,0 г (выход 59%) соединения из заголовка примера, бледно-желтое масло.
Результаты анализа для C52H102N4O4:
рассчитано: C 73,7; H 12,1; N 6,6
найдено: C 74,0; H 12,0; N 6,5.
Пример 62. 1,6-Бис[N-(1-циклогексилокси-2,2,6,6-тетраметилпиперидин- 4-ил)-ацетиламино]гексан.
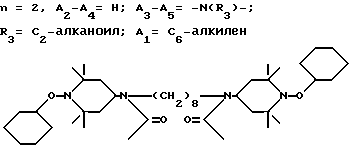 .
.
Смесь 8,0 г (16,7 ммоль) 1,6-бис[N-(2,2,6,6-тетраметилпиперидин- 4-ил)-ацетиламино] гексана, 21,5 г (167 ммоль) 70%-ного водного раствора трет. -бутилгидроперекиси, 0,1 г трехокиси молибдена и 100 мл циклогексана нагревали до температуры дефлегмации 2 ч. Воду собирали в ловушку Дина-Старка. Красную реакционную смесь переносили в склянку Фишера-Портера и нагревали до 150 - 160oC 1 ч до удаления красного цвета. Твердые частицы удаляли фильтрацией, а фильтрат выпаривали, чтобы получить масло. После растирания масла в метаноле получали 8,4 г (выход 74%) соединения из заголовка примера, белое твердое вещество, т. пл. 65 - 72oC.
Результаты анализа для C40H74N4O4:
рассчитано: C 71,2; H 11,1; N 8,3
найдено: C 70,5; H 11,0; N 8,2.
Пример 63. Бис(1-циклооктилокси-2,2,6,6-тетраметилпиперидин- 4-ил)себацинат.
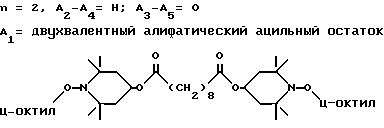 .
.
Смесь 75 г (156 ммоль) бис(2,2,6,6-тетраметилпиперидин-4-ил)себацината, 1,3 г трехокиси молибдена и 475 мл циклооктана нагревали до 118oC. В эту реакционную смесь добавляли 130 г (1,01 моль) 70%-ного водного раствора трет. -бутилгидроперекиси в течение 5 ч. Реакционную смесь выдерживали при дефлегмации в процессе добавления, а воду собирали в ловушку Дина-Старка. Красную реакционную смесь нагревали 7 ч после завершения добавления для удаления красной окраски. Твердые частицы удаляли фильтрацией, а фильтрат выпаривали, чтобы получить желтое масло. После очистки с использованием испарительной хроматографии (20 : 1, гептан : этилацетат) получали 104 г (91%) соединения из заголовка примера, ярко-желтое масло.
Пример 64. Бис(1-октилокси-2,2,6,6-тетраметилпиперидин-4-ил)себацинат.
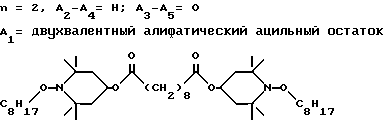 .
.
70%-ный Водный раствор трет.-бутилгидроперекиси (140 г, 1,09 моль) добавляли в течение 6 ч в смесь 75,4 г (0,157 моль) бис(2,2,6,6-тетраметилпиперидин-4-ил)себацината, 1,25 г (8,7 ммоль) трехокиси молибдена и 570 мл н-октана, которую предварительно нагревали до 115oC в атмосфере азота. В процессе добавления реакционную смесь поддерживали при дефлегмации. Воду собирали в ловушку Дина-Старка. После завершения добавления красную реакционную смесь нагревали при дефлегмации (95 - 97oC) 7 ч до удаления красной окраски. Трехокись молибдена удаляли фильтрацией. Желтый фильтрат перемешивали при окружающей температуре в течение 30 мин с 15 г активированного древесного угля (ДАРКО), чтобы удалить часть желтой окраски, а затем концентрировали при пониженном давлении. Сырой продукт подвергали очистке при помощи испарительной хроматографии на силикагеле (100 : 3, гептан : этилацетат), чтобы получить 92,9 г (выход 80%) соединения из заголовка примера, бесцветное масло.
Результаты анализа для C44H84N2O6:
рассчитано: C 71,7; H 11,5; N 3,8
найдено: C 71,6; H 11,5; N 3,6.
Пример 65. Бис(1-гептилокси-2,2,6,6-тетраметилпиперидин-4-ил)сукцинат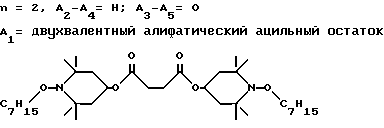 .
.
70%-ный Водный раствор трет. -бутилгидроперекиси (78,9 г, 0,613 моль) добавляли в течение 30 мин в смесь 55 г (0,139 ммоль) бис(2,2,6,6-тетраметилпиперидин-4-ил)сукцината, 1,0 г трехокиси молибдена и 400 мл н-гептана, которую поддерживали при температуре 110oC. Воду собирали в ловушку Дина-Старка. После того, как добавление завершено, реакционную смесь нагревали при дефлегмации на 30 мин. Еще одну порцию 70%-ного водного раствора трет. -бутилгидроперекиси (100 г, 0,777 моль) добавляли в красную реакционную смесь в течение 90 мин. Реакционную смесь нагревали при дефлегмации 16 ч до удаления красной окраски. Трехокись молибдена удаляли фильтрацией, а фильтрат выпаривали при пониженном давлении. Остаток подвергали очистке с использованием испарительной хроматографии (19 : 1, затем 9 : 1 смесь гептана : этилацетата) на силикагеле, чтобы получить 63,3 г (выход 73%) соединения из заголовка примера в виде прозрачной желтой жидкости.
Результаты анализа для C36H68N2O6:
рассчитано: C 69,2; H 11,0; N 4,5
найдено: C 68,8; H 11,1; N 4,5.
Пример 66. Бис(1-декагидронафтилокси-2,2,6,6-тетраметилпиперидин- 4-ил)себацинат.
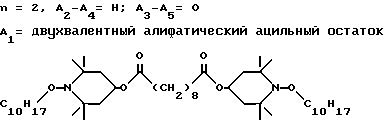 .
.
Смесь 25,0 г (0,052 моль) бис(2,2,6,6-тетраметилпиперидин-4-ил)себацината, 55,0 г (0,427 моль) 70%-ного водного раствора трет.-бутилгидроперекиси, 1,5 г (0,010 моль) трехокиси молибдена и 180 мл декагидронафталина нагревали при дефлегмации 4,5 ч до тех пор, пока не исчезнет красная окраска. Воду собирали ловушкой Дина-Старка. Трехокись молибдена удаляли фильтрацией, а фильтрат перемешивали с раствором 26 г сульфита натрия в 500 мл воды, чтобы разложить непрореагировавшую трет.-бутилгидроперекись. Двухфазную смесь разбавляли этилацетатом (200 мл) и органический слой промывали насыщенным раствором хлорида натрия, сушили над сульфатом магния и концентрировали при пониженном давлении, чтобы получить масло. После очистки с использованием испарительной хроматографии (силикагель, 19 : 1, гептан : этилацетат) получали 28,8 г (выход 71%) соединения из заголовка примера в виде бледно-желтого масла.
Результаты анализа для C48H84N2O6:
рассчитано: C 73,4; H 10,8; N 3,6
найдено: C 74,9; H 11,5; N 3,4.
Пример 67. Бис(1-циклододецилокси-2,2,6,6-тетраметилпиперидин- 4-ил)себацинат.
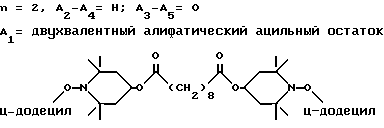 .
.
Смесь 30,1 г (62,6 ммоль) бис(2,2,6,6-тетраметилпиперидин-4-ил)себацината, 44 г (439 ммоль) 90%-ного раствора трет.-бутилгидроперекиси, 0,5 г трехокиси молибдена и 207 г циклододекана нагревали в склянке Фишера-Портера до 135-145oC 7,5 ч. Реакционную смесь разбавляли гептаном (350 мл) и подвергали очистке с использованием испарительной хроматографии на силикагеле (гептан, затем 20 : 1 смесь гептана : этилацетата), чтобы получить 49,4 г (выход 93%) соединения из заголовка примера, бесцветного вязкого масла.
Пример 68. N,N',N'',N'''-тетракис[2,4-бис[N-(1-циклогексилокси-2,2,6,6- тетраметилпиперидин-4-ил)-бутиламино] -1,3,5-триазин-6-ил] -1,5,8,12- тетразадодекан.
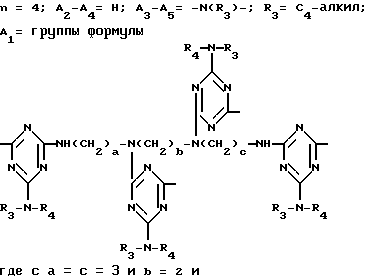 .
.
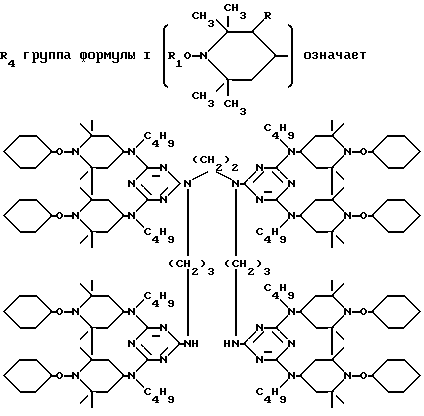 .
.
В кипящую смесь 30,0 г (13,0 моль) N,N',N'',N'''-тетракис-[2,4-бис[N-(2,2,6,6-тетраметилпиперидин-4-ил)- бутиламино] -1,3,5-триазин-6-ил] -1,5,8,12-тетразадодекана, 1,0 г трехокиси молибдена и 300 мл циклогексана добавляли 88,7 г (689 ммоль) 70%-ного водного раствора трет.-бутилгидроперекиси в течение 1 ч. Реакционную смесь нагревали до температуры дефлегмации еще в течение часа после того, как добавление завершено. Воду и некоторое количество органического дистиллята (100 мл) собирали ловушкой Дина-Старка и удаляли из реакционной смеси. Затем реакционную смесь переносили в склянку Фишера-Портера и нагревали 5 ч до температуры 130 - 135oC. В смесь добавляли еще одну порцию 70%-ного раствора трет.-бутилгидроперекиси (14,0 г, 107 ммоль) и нагревание продолжали еще в течение 3 ч. Реакционную смесь охлаждали и твердый осадок удаляли фильтрацией. Фильтрат концентрировали, разбавляли 19 : 1 смесью гептана : этилацетата и подвергали очистке с использованием испарительной хроматографии на силикагеле (19 : 1, гептан : этилацетат), чтобы получить 24,0 г (59%) соединения из заголовка примера в виде бледно-желтого стеклообразного вещества с температурой размягчения 108 - 123oC.
Результаты анализа для C172H314N33O8:
рассчитано: C 69,8; H 10,7; N 15,1
найдено: C 66,6; H 10,6; N 14,7.
Пример 69. 2,4,6-Трис-[N-(1-циклогексилокси-2,2,6,6-тетраметилпиперидин- 4-ил)-н-бутиламино]-1,3,5-триазин.
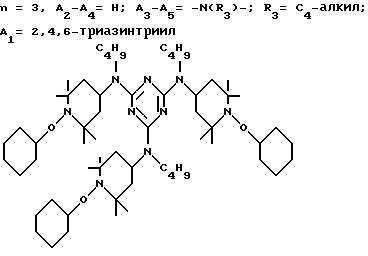 .
.
В кипящую смесь 13,5 г (19,0 ммоль) 2,4,6-трис-[N-(2,2,6,6-тетраметилпиперидин-4-ил)-н-бутиламино] -1,3,5- триазина, 0,2 г трехокиси молибдена и 175 мл циклогексана добавляли 36,6 г (284 ммоль) 70%-ного водного раствора трет. -бутилгидроперекиси в течение 10 мин. Красную реакционную смесь нагревали при температуре дефлегмации в течение часа и затем переносили в склянку Фишера-Портера, используя 25 мл свежего циклогексана. Затем реакционную смесь нагревали 3 ч до 150oC, чтобы удалить красную окраску. Твердый осадок удаляли фильтрацией, а фильтрат концентрировали, остаток подвергали очистке с использованием испарительной хроматографии на силикагеле (19 : 1, гептан : этилацетат). Продукт кристаллизовали из изопропилового спирта, чтобы получить 7,6 г (выход 40%) белого порошка, т. пл. 189 - 194oC.
Результаты анализа для C60H111N9O39:
рассчитано: C 71,6; H 11,1; N 12,5
найдено: C 71,8; H 11,1; N 12,6.
Пример 70. 2,4-Бис[N-(1-циклогексилокси-2,2,6,6-тетраметилпиперидин- 4-ил)-н-бутиламино]-6-трет.-октиламино-1,3,5-триазин.
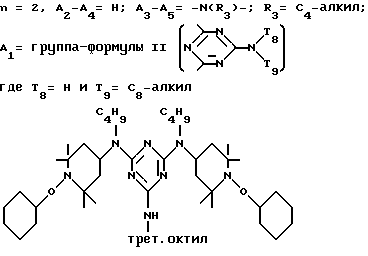 .
.
Смесь 22,2 г (35,3 ммоль) 2,4-бис[N-(2,2,6,6-тетраметилпиперидин-4-ил)-н-бутиламино] -6- трет.-октиламино-1,3,5-триазина, 0,3 г трехокиси молибдена, 35,3 г (353 ммоль) 90%-ного водного раствора трет.-бутилгидроперекиси и 250 мл циклогексана нагревали 5 ч до температуры 150 - 155oC в склянке Фишера-Портера. Давление поддерживали ниже 45 фунтов/дюйм2 (3,164 кг/см2) при помощи периодической вентиляции. Затем смесь охлаждали и твердые материалы удаляли фильтрацией. Фильтрат содержит два основных продукта, которые разделяли комбинированием испарительной хроматографии (39 : 1, гептан : этилацетат) и жидкостной хроматографии среднего давления (Преп. Уотерза 500 A, 99 : 1, гептан - этилацетат, а затем этилацетат). Соединение из заголовка примера является более полярным продуктом, бледно-желтым стеклом, т. пл. 85 -103oC. Выход 11 г (38%).
Результаты анализа для C49H92N8O2:
рассчитано: C 71,3; H 11,2; N 13,6
найдено: C 71,2; H 12,0; N 13,6.
Пример 71. 2,4-Бис[N-(1-циклогексилокси-2,2,6,6-тетраметилпиперидин- 4-ил)-н-бутиламино]-6-морфолино-1,3,5-триазин.
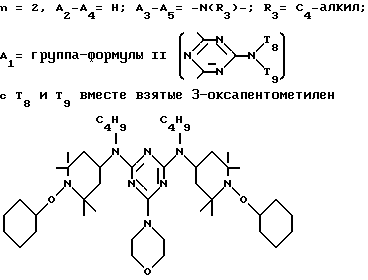 .
.
Смесь 10,0 г (13,7 ммоль) 2-хлор-4,6-бис[N-(1-циклогексилокси-2,2,6,6- тетраметилпиперидин-4-ил)-н-бутиламино] -1,3,5-триазина, 1,43 г (16,5 ммоль) морфолина, 0,8 г гидрата окиси натрия и 30 г толуола нагревали при температуре дефлегмации 4 ч. Воду собирали ловушкой Дина-Старка. Жидкую фазу сливали, а остаточный твердый остаток промывали толуолом. Соединенные органические растворы сушили над сульфатом магния и концентрировали, чтобы получить вязкое масло. В результате очистки при помощи ЖХСД (Преп. Уотерза 500 A, 29 : 1, гептан - этилацетат) получали 5,9 г (выход 55%) соединения из заголовка примера, белый порошок, т. пл. 159 - 163oC.
Результаты анализа для C45H82N8O3:
рассчитано: C 69,0; H 10,6; N 14,3
найдено: C 69,5; H 10,6; N 14,3.
Пример 72. 2,4-Бис[N-(1-циклогексилокси-2,2,6,6-тетраметилпиперидин- 4-ил)-н-бутиламино]-6-диэтиламино-1,3,5-триазин.
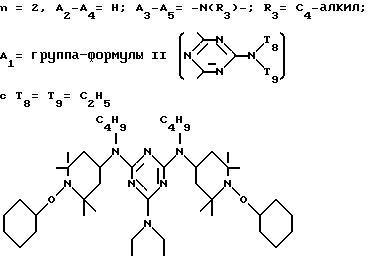 .
.
Смесь 50,0 г (271 ммоль) цианурхлорида, 22,0 г 50%-ного водного раствора гидрата окиси натрия, 22 мл воды и 150 мл толуола, охлажденную до 0oC, смешивали с раствором 19,8 г (271 ммоль) диэтиламина в 25 мл толуола в течение 45 мин. Реакционную температуру поддерживали в пределах О - 5oC в течение добавления. После того, как добавление закончилось, реакционную смесь перемешивали в течение 2 ч при окружающей температуре. В смесь добавляли воду, чтобы растворить хлорид натрия и фазы разделяли. Органическую фазу сушили над сульфатом магния и концентрировали, чтобы получить масло, которое кристаллизовали из гептана, чтобы получить 42,0 г (выход 70%) 2,4-дихлор-6-диэтиламино-1,3,5-триазина, белого кристаллического вещества, т. пл. 77 - 79oC. 4-н-Бутиламино-2,2,6,6-тетраметилпиперидин (31,7 г, 149 ммоль) добавляли в течение 15 мин в суспензию 15,0 г (67,8 ммоль) 2,4-дихлор-6-диэтиламино-1,3,5-триазина, 150 мл ксилола и 7,0 г порошкообразного гидрата окиси натрия, которую предварительно нагревали до 70oC. Реакционную смесь нагревали при дефлегмации 23 ч. Хлорид натрия удаляли фильтрацией, а фильтрат концентрировали в вязкое масло. После дальнейшей концентрации масла при помощи разгонки по Кугельрору (140 - 150oC, 0,05 мм) получали остаток, который кристаллизовали из метанола-воды, чтобы получить 25,8 г (выход 66%) 2,4-бис-[N-(2,2,6,6-тетраметилпиперидин-4-ил)-н-бутиламино] -6-диэтиламино-1,3,5-триазина с т. пл. 74 - 76oC.
Смесь 14,0 г (24,4 моль) 2,4-бис[N-(2,2,6,6-тетраметилпиперидин-4-ил)- н-бутиламино]-6-диэтиламино-1,3,5-триазина, 31,4 г (244 ммоль) 70%-ного водного раствора трет.-бутилгидроперекиси, 0,15 г трехокиси молибдена и 140 мл циклогексана нагревали при температуре дефлегмации 3 ч. Красную смесь затем переносили в склянку Фишера-Портера и нагревали до температуры 145 - 155oC 3 ч до исчезновения красной окраски. Твердые продукты удаляли фильтрацией, а фильтрат концентрировали, чтобы получить в результате остаток, который подвергали очистке с использованием испарительной хроматографии (39 : 1, гептан : этилацетат), чтобы получить 4,9 г (26%) соединения из заголовка примера, белое твердое вещество, т. пл. 91 - 99oC.
Результаты анализа для C45H84N8O2:
рассчитано: C 70,3; H 11,0; N 14,6
найдено: C 70,2; H 10,9; N 14,4.
Пример 73. 2,4,6-Трис[N-(1-октилокси-2,2,6,6-тетраметилпиперидин- 4-ил)-н-бутиламино] -1,3,5-триазин.
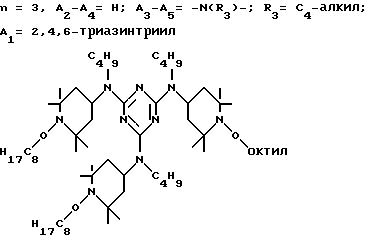 .
.
2,4,6-Трис[(2,2,6,6-тетраметилпиперидин-4-ил)-н-бутиламино] -1,3,5-триазин (50,0 г, 70,2 ммоль) добавляли в смесь 108,4 г (842 ммоль) 70%-ного водного раствора трет.-бутилгидроперекиси, 1,0 г трехокиси молибдена и 350 мл н-октана, которую предварительно нагревали до 50oC. Реакционную смесь тщательно доводили до дефлегмации. Ловушку Дина-Старка использовали, чтобы удалить воду из реакции. Реакционную смесь нагревали при дефлегмации 16 ч. Твердые частицы удаляли фильтрацией, а фильтрат концентрировали, чтобы получить вязкий маслянистый остаток. После очистки с использованием испарительной хроматографии (З9 : 1, гептан : этилацетат) получали 15,0 г (выход 19%) соединения из заголовка примера.
Результаты анализа для C66H129N9O3:
рассчитано: C 72,3; H 11,9; N 11,5
найдено: C 72,2; H 12,1; N 11,4.
Пример 74. N, N', N'',N'''-тетракис-[2,4-бис [N-(1-октилокси-2,2,6,6-тетраметил-пиперидин-4-ил)-н-бутиламино] -1,3,5-триазин-6-ил] -1,5,8,12-тетразододекан.
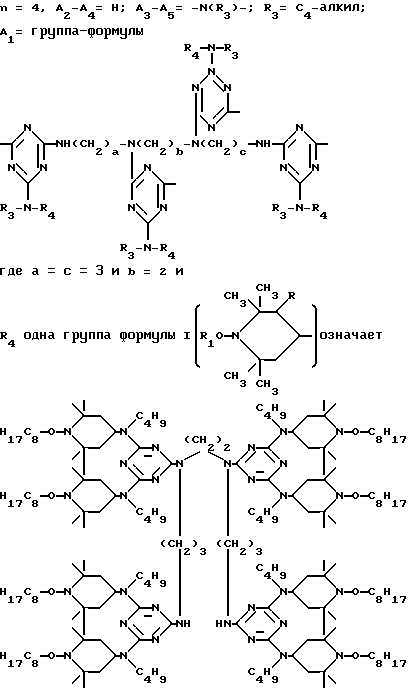 .
.
94,6 г (736 ммоль) 70%-ного Водного раствора трет.-бутилгидроперекиси экстрагировали 300 мл н-октана. Смесь 100 мл трет.-бутилгидроперекиси /октана, 50,0 г (23,0 ммоль) N,N',N'',N'''-тетракис-[2,4-бис[N-(2,2,6,6- тетраметилпиперидин-4-ил)-бутиламино] -1,3,5-триазин-6-ил] -1,5,8,12- тетразадодекана, 0,5 г трехокиси молибдена и 100 мл н-октана нагревали до дефлегмации. Воду собирали ловушкой Дина-Старка. Оставшийся раствор трет.-бутилгидроперекиси/октана, затем добавляли в течение 2,5 ч в красную реакционную смесь. Реакционную смесь нагревали при дефлегмации 4 ч после добавления, а затем обрабатывали при помощи 60 г (470 моль) 70%-ного трет.-бутилгидроперекиси и нагревали при дефлегмации еще 4 ч., чтобы удалить красную окраску. Реакционную смесь охлаждали, а твердый материал удаляли фильтрацией. Фильтрат перемешивали с 300 мл 5%-ной водного раствора сульфита натрия, сушили над сульфатом магния и концентрировали. Остаток подвергали очистке при помощи испарительной хроматографии (19 : 1, гептан - этилацетат) на силикагеле, чтобы получить 20,2 г (выход 27%) соединения из заголовка примера, желтого стекла, т. пл. 81 - 91oC.
Результаты анализа для C108H326N32O8:
рассчитано: C 70,6; H 11,4; N 14,0
найдено: C 68,7; H 11,9; N 14,0.
Пример 75. N,N'-бис(1-циклогексилокси-2,2,6,6-тетраметилпиперидин- 4-ил)-N, N'-бис{ 2,4-бис[N-(1-циклогексилокси-2,2,6,6-тетраметилпиперидин- 4-ил)-н-бутиламино]-1,3,5-триазин-6-ил}-гексаметилендиамин.
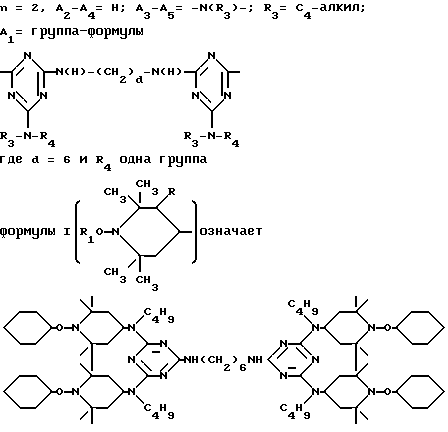 .
.
Раствор 19,0 г (32,1 ммоль) 1,6-бис(N,N'-1-циклогексилокси-2,2,6,6- тетраметилпиперидин-4-ил)аминогексана в 50 мл толуола добавляли в течение 20 мин в смесь 11,9 г (64,3 ммоль) цианурхлорида, 100 мл толуола, 6,8 г карбоната натрия и 30 мл воды, которую предварительно охлаждали до 0oC. Реакционную температуру поддерживали в интервале 2 -5oC в течение добавления. Реакционную смесь затем перемешивали при окружающей температуре в течение 2 ч. Осадок отделяли фильтрацией и промывали последовательно водой и толуолом, затем растворяли в дихлорметане. Толуольный раствор сушили над сульфатом магния и выпаривали, чтобы получить остаток, который добавляли в дихлорметановый раствор. После выпаривания этого раствора получали 12,1 г (выход 42%) N, N'-бис(1-циклогексилокси- 2,2,6,6-тетраметилпиперидин-4-ил)-N, N'-бис(2,4-дихлор-1,3,5-триазин-6- ил) гексаметилендиамина.
Смесь 7,0 г (7,9 ммоль) N,N'-бис(1-циклогексилокси-2,2,6,6- тетраметилпиперидин-4-ил)-N,N'-бис(2,4-дихлор-1,3,5-триазин-6- ил)гексаметилендиамина, 13,0 г (41,9 ммоль) 1-циклогексилокси-4-н- бутиламино-2,2,6,6-тетраметилпиперидина, 2,5 г гидрата окиси натрия и 100 мл ксилола нагревали при температуре дефлегмации в атмосфере азота 30 ч. Твердые частицы удаляли фильтрацией. Фильтрат концентрировали, остаток частично растворяли в кипящем дихлорметане. Горячий раствор фильтровали, чтобы удалить нерастворимые примеси, затем частично выпаривали и разбавляли этанолом, чтобы получить 12,2 г (78%) соединения из заголовка примера, белый порошок, т. пл. 254 - 257oC (разл.).
Результаты анализа для C18H216N18O6:
рассчитано: C 71,5; H 11,0; N 12,7
найдено: C 70,4; H 10,7; N 12,4.
Пример 76. 2,4-Бис[N-(1-метокси-2,2,6,6-тетраметилпиперидин- 4-ил)-н-бутиламино]-6-диэтиламино-1,3,5-триазин.
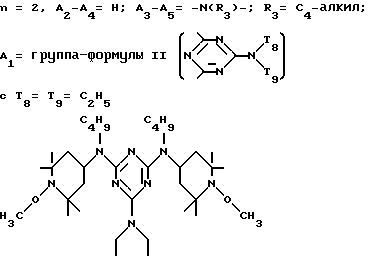 .
.
Смесь 5,0 г (22,6 ммоль) 2,4-дихлор-6-диэтиламино-1,3,5-триазина, 14,0 г (57,8 ммоль) 1-метокси-4-н-бутиламино-2,2,6,6-тетраметилпиперидина, 50 мл ксилола и 1,8 г гидрата окиси натрия нагревали до температуры дефлегмации, которую поддерживали в течение 18 ч. Соли удаляли фильтрацией, а фильтрат концентрировали с получением масла. После очистки с использованием колонны для хроматографии (гептан) получали 9,7 г (выход 68%) соединения из заголовка примера, белое твердое вещество, т. пл. 109 - 112oC.
Результаты анализа для C35H68N8O2:
рассчитано: C 66,4; H 10,8; N 17,7
найдено: C 66,2; H 10,9; N 17,4.
Пример 77. 2,4-Бис[N-(1-циклогексилокси-2,2,6,6-тетраметилпиперидин- 4-ил)-н-бутиламино]-6-н-бутиламино-1,3,5-триазин.
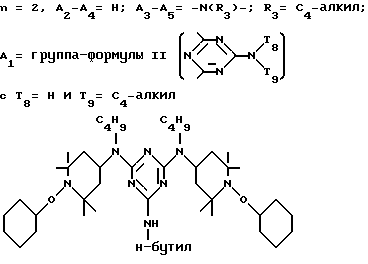 .
.
Смесь 5,0 г (22,5 ммоль) 2,4-дихлор-6-н-бутиламино-1,3,5-триазина, 21,1 г (67,8 ммоль) 1-циклогексилокси-4-н-бутиламино-2,2,6,6-тетраметилпиперидина, 100 мл ксилола и 5,4 г 50%-ного водного раствора гидрата окиси натрия нагревали до температуры дефлегмации, которую поддерживали 16 ч. Реакционную смесь разделяли между простым эфиром и водой. Слой простого эфира промывали 1 н раствором HCl (2 х 100 мл), насыщенным раствором бикарбоната натрия (100 мл) и насыщенным раствором хлорида натрия (100 мл), затем сушили над сульфатом магния и концентрировали. Сырой продукт подвергали очистке с использованием испарительной хроматографии (гептан) и кристаллизовали из этанола, чтобы получить 7,6 г (выход 44%) стеклообразного соединения из заголовка примера, т. пл. 80 - 86oC.
Результаты анализа для C45H84N8O2:
рассчитано: C 70,3; H 11,0; N 14,6
найдено: C 70,2; H 11,1; N 14,6.
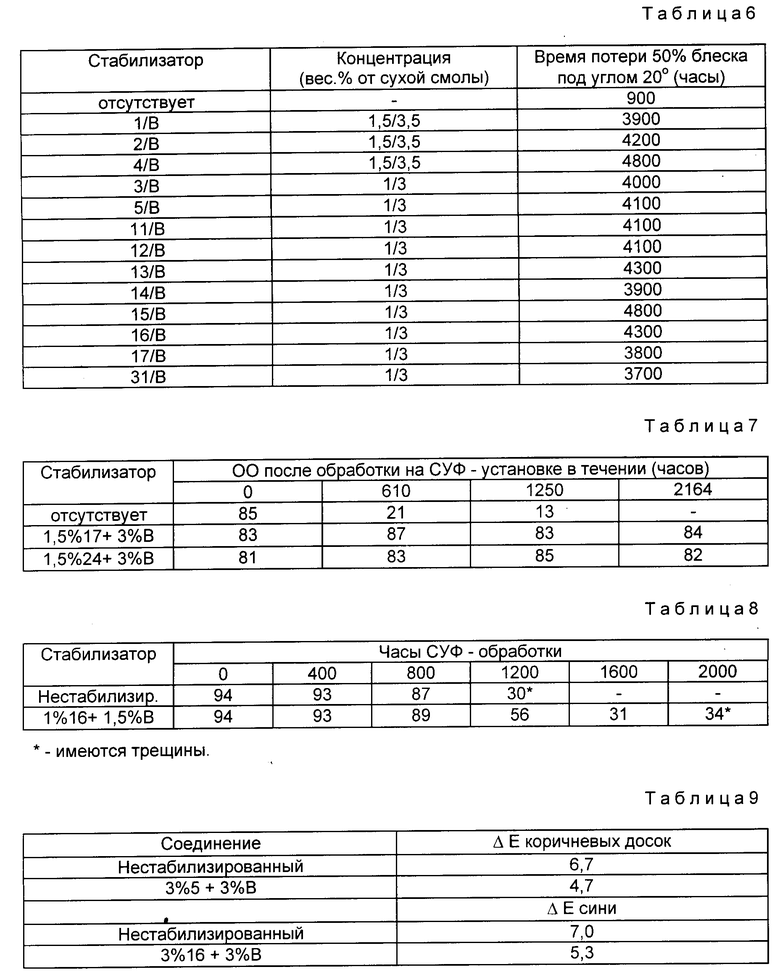
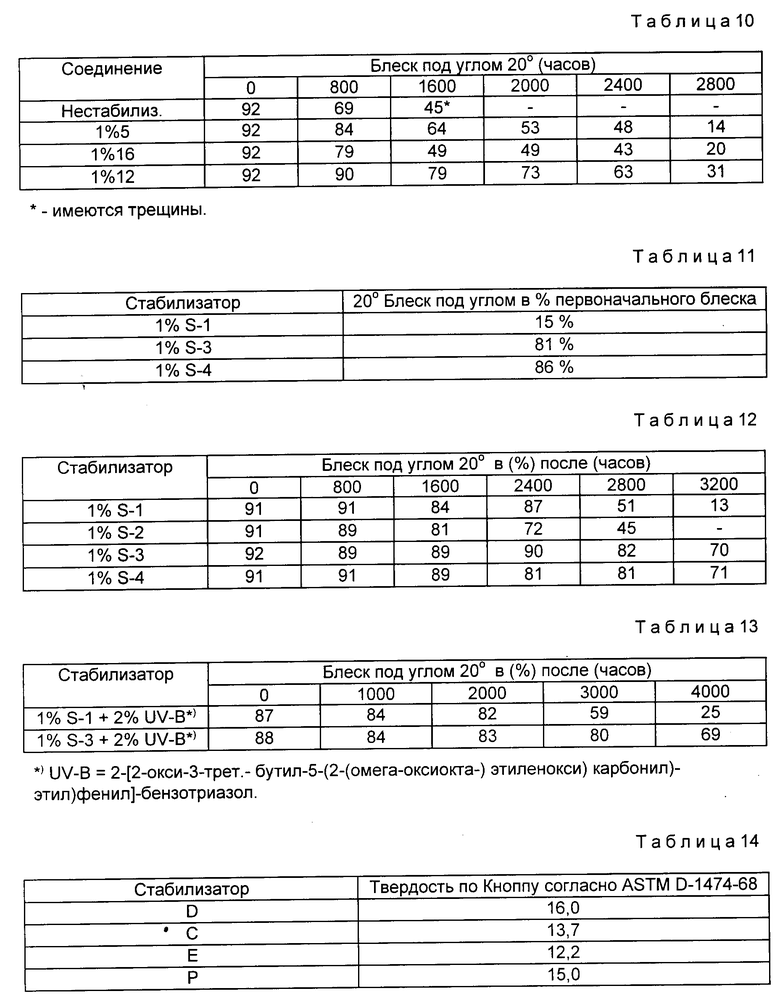
Изобретение относится к производным 2,2,6,6-тетраметилпиперидина формулы I: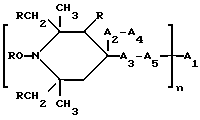
где R1 -C1-C18-алкил, R - водород, C5-C8-циклоалкенил и др.; A3-A5 представляет собой, -O- или -(NR3), где R3 = водород, C2-C4-алканоил и др. а) когда п = 1, A3-A5 = NR3, то A1 означает одновалентный остаток и водород, C1-C18 - алкил, в) п = 1, когда A2-A4 - водород либо A1 вместе с A4 образует C2-C8 алкилен, A2 означает кислород или CH2, A3-A5= -O- или с) п = 1, A3-A5= -O- и A2 = O, то A1 вместе с A4 образуют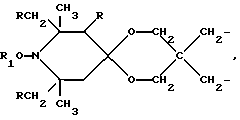
когда п = 2, A5-A5 является NR3, A2-A4 - водород, то A1 группа формулы: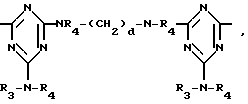
d = 2-10 или A1 группа формулы 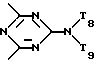 , T8 и T9 - водород, C1-C12-алкил или T8 и T9 вместе - триоксапентаметилен и т.д. Соединения формулы I могут быть использованы в качестве эффективных стабилизаторов различных органических материалов, в частности полимеров, от действия света и кислорода. 6 з.п. ф-лы, 14 табл.
, T8 и T9 - водород, C1-C12-алкил или T8 и T9 вместе - триоксапентаметилен и т.д. Соединения формулы I могут быть использованы в качестве эффективных стабилизаторов различных органических материалов, в частности полимеров, от действия света и кислорода. 6 з.п. ф-лы, 14 табл.
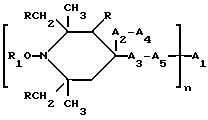
где n = 1 - 4;
R - водород;
R1 - C1 - C18-алкил, C5 - C8-циклоалкенил, C5 - C12-циклоалкил, возможно замещенный C1 - C9-алкилом, C6 - C10-бициклоалкил, C7 - C9-аралкил, возможно замещенный C1 - C9-алкилом;
A3 - A5 представляет собой -O- или - N(R3)-, где R3 - водород, C2 - C4-алканоил, C1 - C8-алкил, при условии, что a) когда n = 1 и A3 - A5 являются NR3, то A2 - A4 - водород, а A1 - водород или C1 - C18-алкил, или b) когда n = 1 и A3 - A5 - -O-, то A1 вместе с A4 образуют C2 - C8-алкилен, а A2 - кислород или CH2, или c) когда n = 1, а А3 - A5 является -O- и A2 = 0, то A1 вместе с A4 образуют группу формулы I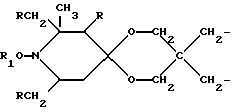
или при условии, что a) когда n = 2, A3 - A5 является NR3, A2 - A4 - водород, то A1 - группа формулы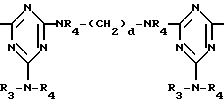
в которой d = 2 - 10;
2 из 4-х значений означают водород при том, что R4 также обозначает тетраметилпиперидинильный остаток формулы III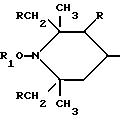
или A1 является ксилиленом, C1 - C12-алкиленом, двухвалентным ацильным остатком ароматической, алифатической или аралифатической карбоновой кислоты с числом атомов углерода до 20 или A1 является группой формулы II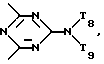
где T8 и T9 независимо друг от друга - водород или C1 - C12-алкил или T8 и T9 вместе означает 3-оксапентаметилен;
b) когда n = 2, A3 - A5 является -O-, A2 - A4 - водород, то A1 является ксилиленом, C1 - C12-алкиленом, двухвалентным ацильным остатком ароматической, алифатической или аралифатической карбоновой кислоты с числом атомов углерода до 20 или A1 является группой формулы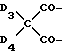
в которой D3 и D4 - водород, C1 - C6-алкил или 3,5-ди-трет-бутил-4-гидроксибензил;
или при условии, что a) когда n = 3, A3 - A5 является NR3, A2 - A4 - водород, то A1 является 2,4,6-триазинилом или группой формулы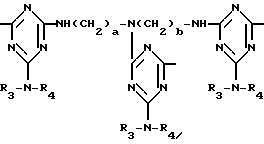
в которой a и b = 2 или 3 независимо друг от друга;
или при условии, когда n = 4, A3 - A5 является N(R3), A2 - A4 - водород, то A1 является остатком формулы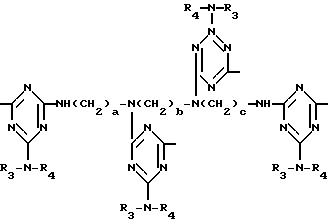
в которой a, b и c = 2 или 3 независимо друг от друга;
R3 и R4 в триазиновых остатках означают R3 - водород, C1 - C8-алкил, C2 - C4-алканоил;
R4 обозначает группу формулы III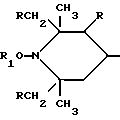
а также водород или C4 - C12-алкил.
| Шелл и др | |||
| Доклады Академии Наук СССР | |||
| Раздел: Химия | |||
| Мяльно-трепальный станок для обработки тресты лубовых растений | 1922 |
|
SU200A1 |
| J.Org.Chem., 43, 1976, p | |||
| АППАРАТ ДЛЯ ТЕМПИРОВАНИЯ АРТИЛЛЕРИЙСКИХ СНАРЯДОВ, СНАБЖЕННЫХ ТРУБКАМИ С ЧАСОВЫМ МЕХАНИЗМОМ | 1925 |
|
SU4279A1 |
| US, патент, 4344876, кл | |||
| Разборный с внутренней печью кипятильник | 1922 |
|
SU9A1 |
