Изобретение относится к новым соединениям, способам их получения и фармацевтическим препаратам, в которые они входят и способам лечения, основанным на их использовании.
В международной патентной заявке N WO 92/08708 (опубликованной после даты приоритета этой заявки) описан ряд биологически активных аминов и их действие как агонистов β2-адренорецепторов и агонистов допамина DA2.
Открыта группа производных от 7-(2-аминоэтил)-бензотиазолонов, обладающих значительными преимуществами перед уже известными веществами.
В соответствии с предлагаемым изобретением предложены соединения общей формулы I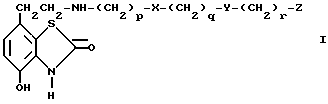
где
X и Y независимо одна от другой означают -S(O) n или -O-,
n означает 0, 1 или 2,
p, q и r независимо одна от других означают 2 или 3,
Z означает фенил с возможным замещением галогенами, -OR1, -NO2 или NR2R3 или представляет собой 5- или 6- членный гетероцикл, содержащий n, O или S, причем R1, R2 и R3 независимо одна от других означают водород или алкил C1-C6, и их фарамацевтически приемлемые производные.
Кроме того, в соответствии с изобретением предложен способ получения соединений общей формулы I и их фармацевтически приемлемых производных предусматривающий:
а) алкилирование соединения формулы II, или его производного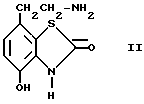 III
III
при помощи алкилирующего соединения общей формулы III,
L-(CH2)p-X-(CH2)q-Y-(CH2)r-Z
где
p, q, r, X, Y и Z имеют вышеуказанные значения, а L означает временную группу,
b) алкилированные соединения формулы II, указанной выше, при помощи соединения общей формулы IV
O=CH-(CH2)p-1-X-(CH2)q-Y-(CH2)-Z IV
где
p, q, r, X, Y и Z имеют вышеуказанные значения, в присутствии восстановителя,
c) избирательное восстановление соединения общей формулы V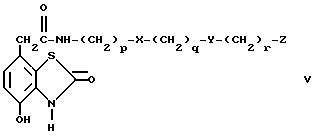
где
p, q, X, Y и Z имеют вышеуказанные значения,
d) избирательное восстановление соединения общей формулы Va,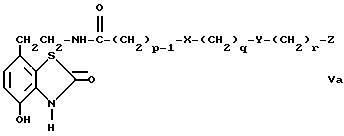
где
p, q, r, X, Y и Z имеют вышеуказанные значения,
e) удаление защитной группы у соответствующего защищенного соединения общей формулы I, у которого защищена одна или несколько функциональных групп, и при желании или необходимости преобразование соединения общей формулы I в фармацевтически приемлемое производное, или наоборот.
При этом на шаге а) временная группа L представляет собой либо галогенидгруппу, например хлорид, бромид или иодид, либо алкил или арилсульфонилоксигруппу, например метансульфонилоксигруппу или p-толуолсульфонилоксигруппу. Реакция в предпочтительном случае проводится в присутствии основания, например неорганического основания, такого как натрия или калия карбонат, или органического основания, такого как триэтиламин, N,N-диизопропилэтиламин или пиридин. Реакцию удобнее проводить в растворителе, таком как эфир, например в тетрагидрофуране или в диоксане; в кетоне, например бутаноне или метил-изобутил-кетоне; в амиде с замещениями, например в диметилформамиде, или в хлорированном углеводороде, например хлороформе, при температуре в пределах от температуры окружающей среды до температуры перегонки растворителя.
Алкилирующее соединение общей формулы III можно получить из соответствующего спирта (т. е. соединения, в котором L означает OH) известными способами. Например, спирт можно подвергнуть реакции с галогенирующим средством, в результате чего получается соединение общей формулы III, где L означает атом галогена к приемлемым галогенирующим средствам относятся, например, трифенилфосфин-тетрагалогенметановые добавки (удобно получать их на месте, например, путем реакции трифенилфосфина с тетрабромидом углерода). Реакцию можно проводить в присутствии растворителя, такого как ацетонитрил или хлорированный углеводород, например дихлорметан, при температуре 0 - 30oC.
На шаге b) восстановителем может служить водород в присутствии катализатора, например платины, оксида платины, палладия, оксида палладия, никеля Ренея или родия, на основе из активированного угля. В качестве растворителя при этом используется спирт, например этанол; сложный эфир, например этилацетат; простой эфир, например тетрагидрофуран; вода или смесь растворителей; температура и давление могут быть нормальными или повышенными. Кроме того, в качестве восстановителя может быть использован гидрид, такой как диборан или гидрид металла, например натрия борогидрид, натрия цианборогидрид или лития-алюминия гидрид. Подбор растворителя для такой реакции зависит от того, какой именно гидрид служит восстановителем; растворитель выбирают из числа спиртов (метанол или этанол) и эфиров (диэтиловый эфир, t-бутил-метиловый эфир или тетрагидрофуран).
Алкилирование при помощи соединения общей формулы IV может привести к образованию промежуточного имина, восстановление которого при описанных условиях дает соединение общей формулы I.
Соединения формулы II и общей формулы IV и спирты общей формулы III либо известны, либо могут быть получены известными способами.
На этапах c) и d) реакцию можно проводить с использованием традиционных приемов восстановления. Восстановитель может быть электрофильным (например, диборан) или нуклеофильным (например, комплексный металлогидрид, такой как лития-алюминия гидрид или натрия бис(2-метоксиэтокси)алюминия гидрид). В предпочтительном случае используют растворитель, который в условиях реакции является инертным. Предпочтительными являются апротические растворители, например тетрагидрофуран, диэтиловый эфир или 1,2-диметоксиэтан. Реакцию можно проводить при температуре 0 - 100oC.
Соединения общей формулы V и общей формулы Va можно получить путем соединения амина с кислотой или с кислым хлоридом по стандартной методике. Например, соединение проводят в присутствии дициклогексилкарбодиимида по способу Шигана и Гесса (Sheehan и Hess, J.Am. Chem. Soc., 1955, 77, 1067) или 1,1-дикарбонилдиимидазола, как описано у Штааба (Staab, Angew. Chem. Int. Ed. Endl. , 1962, 1, 351). Амины, необходимые для проведения этой реакции, либо известны, либо могут быть получены известными способами, например, как описано в J.Med.Chem., 1987, 30, 1166.
Промежуточные соединения общей формулы Va являются новыми, таким образом согласно одному из аспектов предлагаемого изобретения предлагаются соединения общей формулы Va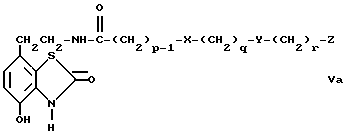
где
p, q, r, X, Y и Z имеют вышеуказанные значения.
Остальные подробности получения соединений общей формулы I перечислены в примерах.
При осуществлении упомянутых выше процессов может понадобиться защитить функциональные группы исходных материалов, например, гидрокси- или аминогруппы, поэтому на шаге e) может понадобиться удалить одну или несколько защитных групп. Приемлемые защитные группы и способы их удаления описаны, например, в "Protective Groups in Organic Synthesis", T.W. Greene and P.G.M Wuts, John Wiley and Sons Inc., 1991. Гидроксильные группы можно защищать, например, арилметиловыми группами, такими как фенилметил, дифенилметил или трифенилметил или производные терагидропиранила.
К приемлемым группам для защиты аминогрупп относятся арилметилы, такие как бензил, (R, S)- α -фенилэтил, дифенилметил, трифенилметил, и ацилы: ацетил, трихлорацетил или трифторацетил. Можно использовать традиционные способы снятия защиты. Арилметиловые группы удаляют, например, посредством гидрогенолиза в присутствии металлического катализатора, например палладия на активированном угле. Тетрагидропираниловые группы расщепляют посредством гидролиза в кислой среде. Ациловые группы снимают путем гидролиза с основанием, таким как натрия гидроксид или калия карбонат, а такие группы, как трихлорацетил, удаляют путем восстановления, например, с цинком или уксусной кислотой.
К фармацевтически приемлемым производным соединений общей формулы I относятся их фармацевтически приемлемые соли, сложные эфиры и амиды.
К подходящим фармацевтически приемлемым солям соединений общей формулы I относятся соли органических и неорганических кислот: гидрохлориды, гидробромиды, сульфаты, фосфаты, малеаты, тартраты, цитраты, бензоаты, 4-метоксибензоаты, 2- или 4-гидроксибензоаты, 4-хлорбензоаты, бензолсульфонаты, p-толуолсульфонаты, нафталинсульфонаты, метансульфонаты, сульфаматы, аскорбаты, салицилаты, ацетаты, дифенилацетаты, трифенилацетаты, адипаты, фумараты, сукцинаты, лактаты, глутараты, глюконаты, гидроксинафталинкарбоксилаты, например 1-гидрокси или 3-гидрокси-2-нафталинкарбоксилаты или олеаты. Упомянутые соединения могут также образовывать соли с некоторыми основаниями. К таким солям относятся соли щелочных металлов, например натрия и калия, и щелочноземельных металлов, например кальция и магния. Соединения общей формулы I могут быть получены в виде солей, в том числе в виде фармацевтически приемлемых солей. При необходимости такие соли можно преобразовать в свободные основания посредством традиционных процедур. Фармацевтически приемлемые соли получают в результате реакции соединений общей формулы I с соответствующей кислотой или основанием в присутствии соответствующего растворителя.
К подходящим фармацевтически приемлемым сложным эфирам соединений общей формулы I относятся алкил C1-C6-эфиры, например этиловый эфир. Эфиры можно получить традиционными способами, например посредством эстерификации или транс-эстерификации.
К подходящим амидам относятся незамещенные, а также одно- или двузамещенные алкил C1-C6 или фениламиды, которые можно получить традиционными способами, например, посредством реакции сложного эфира соответствующей кислоты с аммиаком или соответствующим амином.
Соединениям общей формулы I бывает свойственна таутометрия, также они могут содержать один или более асимметричных углерода, из-за чего возникает оптическая и/или диастереометрия. Диастереомеры разделяют традиционными способами, например, посредством хроматографии или кристаллизации фракций. Различные оптические изомеры выделяют путем разделения рацемической или другой смеси соединений с использованием традиционных приемов, например, кристаллизации фракций или жидкостной хроматографии высокого давления. С другой стороны, нужные оптические изомеры получают в результате реакции между исходными материалами с соответствующей оптической активностью при условиях, не приводящих к рацемизации.
Под термином "алкил" мы подразумеваем прямые, разветвленные или циклические насыщенные и ненасыщенные алкиловые группы.
Если Z означает фенил, содержащий в качестве заместителя галоген, -OR1, -NO2 или -NR2R3, то мы предпочитаем, чтобы такой заместитель был только один. Этот фенил может быть присоединен к группе -(CH2)p-X-(CH2)q-Y-(CH2)r- в орто-, мета- или параположении, но предпочтительными являются орто- и параположениия.
К 5- или 6-членным гетероциклам, обозначаемым буквой Z, относятся фуранил, пиридинил и тиенил. Однако мы предпочитаем соединения общей формулы I, в которых Y означает фенил.
Галогены, которые могут в нем содержаться, - это хлор, бром и фтор. Мы предпочитаем соединения общей формулы I, в которых Z означает фенил.
Предпочтительны соединения общей формулы I, в которых хотя бы одна из букв X и Y означает -O-.
Предпочтительны соединения общей формулы I, в которых r равно 2.
Предпочтительны соединения общей формулы I, в которых p+q равно 5.
К группам, обозначаемым -(CH2)p-X-(CH2)q-Y-(CH2)r, относятся следующие:
-(CH2)3-S-(CH2)2-O-(CH2)2-
-(CH2)3-SO2-(CH2)2-O-(CH2)2-
-(CH2)3-O-(CH2)2-O-(CH2)2-
-(CH2)2-O-(CH2)3-O-(CH2)2-
-(CH2)2-O-(CH2)2-O-(CH2)2-
-(CH2)2-SO2-(CH2)3-O-(CH2)2
-(CH2)2-S-(CH2)3-O-(CH2)2-
Соединения общей формулы I полезны своим фармакологическим воздействием на животных. В частности эти соединения являются агонистами β2-адренорецепторов. Их действие можно продемонстрировать на изолированной трахее морской свинки, как описано в I. G. Dougall, D. Harper, D.M. Jackson, P. Leff, Br. J. Pharmacol, 1991, 104, 1057. Эти соединения также являются агонистами допамина DA2. Афинность испытываемых соединений к площадкам связывания DA2 в мембранах коровьего гипофиза можно определить по вытеснению [3H]-N-n-пропилнорапоморфина и [3H]-спиперона в присутствии или в отсутствии негидролизуемого аналога гуанозина-5'-трифосфата, соответственно как описано в D.R. Sibley, A. DeLean, I. Creese, Anterior Pituitary Dopamine Receptors, Demonstration of Interconvertible High and Low Affinity States of the D-2 Dopamine Receptor, J. Biol. Chem, 1982, 257 (11), 6351-6361. Активность типа DA2 можно также продемонстрировать на функциональном экране, изолированной ушной артерии кролика, как описано в Brown и O'Connor, Br. J. Pharmacol., 1981, 73, 189P. Кроме того, упомянутые соединения дают хорошие соотношения активности DA2 : β2.
Соединения общей формулы I показаны к применению при лечении ряда состояний, известных как обратимое обструктивное заболевание дыхательных путей. Специалистам выражение "обратимое обструктивное заболевание дыхательных путей" должно быть хорошо понятно. Под ним подразумевается астма, в том числе бронхиальная астма, аллергическая астма, наследственная, приобретенная, пылевая астма, особенно хроническая или застарелая астма (например, поздняя астма и повышенная чувствительность дыхательных путей), бронхит и т.п. (патент Великобритании N 2022078 и Br. J. Pharmacol, 1987, 24, 4983). Особый интерес представляет астма. Под термином "лечение" в настоящем документе подразумевается и профилактика, и облегчение симптомов заболевания.
Итак, в соответствии с одним из аспектов изобретения предлагается способ лечения и профилактики обратимого обструктивного заболевания дыхательных путей, предусматривающий введение пациенту, страдающему упомянутым заболеванием или восприимчивому к нему, терапевтически эффективного количества соединения общей формулы I или его фармацевтически приемлемого производного.
Соединения общей формулы I также показаны к применению для лечения различных других состояний, например воспалительных и аллергических заболеваний кожи, застойной сердечной недостаточности и глаукомы.
При использовании в упомянутых выше целях дозы упомянутых соединений зависят от состава используемого соединения, способа введения и ожидаемого результата. Однако, как правило, удовлетворительного результата можно добиться, применяя соединение общей формулы I, в суточной дозе около 1 мкг - около 20 мг на 1 кг живого веса животного, предпочтительно в несколько приемов 1-4 раз в сутки или в форме, обеспечивающей длительную подачу препарата в организм. Для человека суточная доза колеблется в пределах 70 мкг - 1400 мг, а одноразовые формы, удобные для употребления, содержат 20 мкг - 1400 мг соединения в смеси с жидким или твердым фармацевтическим разбавителем или носителем.
Соединения общей формулы I можно применять в чистом виде или в составе соответствующих фармацевтических препаратов местно, внутрь или парентерально.
К составам, удобным для местного применения на легких, относятся аэрозоли, например порошкообразные составы в упаковке под давлением и без давления; к составам, удобным для применения внутрь, относятся таблетки, капсулы и драже; к составам, удобным для нанесения на кожу, относятся кремы, например эмульсии типа масло-в-воде или вода-в-масле; к составам, пригодным для внутривенного введения, относятся инъекции и инфузии; к составам, удобным для применения на глазах, относятся капли и мази.
В соответствии с изобретением также предлагается фармацевтический препарат, содержащий предпочтительно не более 80%, а более предпочтительно не более 50% по массе соединения общей формулы I, или его фармацевтически приемлемое производное в смеси с фармацевтически приемлемым разбавителем или носителем.
Примерами таких разбавителей и носителей являются: для таблеток и драже - лактоза, крахмал, тальк, стеариновая кислота; для капсул - винная кислота или лактоза; для растворов, применяемых парентерально, - вода, спирты, глицерин, растительные масла.
Если соединение общей формулы 1 применяется на легких, его можно вдыхать в виде порошка, подаваемого под давлением и без давления. Подача порошкообразного соединения общей формулы 1 под давлением осуществляется при помощи сжатого или сниженного газа. Если порошок подается без давления, то тонко измельченный активный ингредиент можно смешать с более крупными частицами фармацевтически приемлемого носителя, имеющими диаметр, например, до 100 мкм. К подходящим инертным носителям относится, например, кристаллическая лактоза.
Соединения общей формулы 1 имеют следующие по сравнению с известными веществами подобного состава: они менее токсичны, более эффективны, действуют более продолжительно, имеют более широкий спектр действия, являются более мощными, дают меньше побочных эффектов, легче всасываются и обладают другими полезными фармакологическими свойствами.
Предлагаемое изобретение может быть проиллюстрировано, но отнюдь не ограничено, следующими примерами, в которых температура измеряется градусами шкалы Цельсия. Реакции проводятся в инертной азотной или аргоновой атмосфере. Выделение веществ проводится посредством жидкостной хроматографии высокого давления в обращенно-фазной колонне DYNAMAXTM60A C-18.
Пример 1. 4-Гидрокси-7-[2-[3-[2-[2-фенилэтокси]этилсульфонил] пропиламин]этил]1,3-бензотиазол-2(3H)-она гидрохлорид.
a) 3-[2-[2-Фенилэтокси]этилтио]пропановая кислота.
Раствор 2-[2-фенилэтокси]этантиола(2,13 г) в сухом диметилформамиде (10 мл) добавили по каплям к охлажденной (0oC) перемешиваемой суспензии натрия гидрида (0,60 г, 80% в масле) в диметилформамиде (ДМФ) (50 мл). Смесь перемешивали при 0oC в течение 90 мин. Затем по каплям добавили раствор 3-бромпропановой кислоты (3,15 г) в сухом ДМФ (10 мл) и реакционную смесь перемешивали при комнатной температуре 16 ч. Добавили воду (250 мл) и подкислили смесь до pH 2/3 при помощи концентрированной соляной кислоты. Затем водный раствор несколько раз экстрагировали эфиром и соединили эфирные слои, которые промывали водой и солевым раствором, высушили (MgSo4) и испарили при пониженном давлении. Получили сырую кислоту, которую очистили посредством хроматографии на силикагеле с резким понижением давления с дихлорметаном : эфиром в соотношении 6 : 1 (1 капля уксусной кислоты/100 мл элюента). Получили указанное в подзаголовке соединение (2,15 г).
1H-ЯМР (CDCl3) δ : 2,6 - 2,8 (m, 4H), 2,81 (t, 2H), 2,89 (t, 2H), 3,6 - 3,76 (m, 4H), 7,2 - 7,4 (m, 5H).
b) 3[2-[2-Фенилэтокси]этилсульфонил]пропановая кислота.
Раствор калия пероксимоносульфата (15,6 г, OXONETM) в воде (50 мл) добавили по каплям к охлажденному (0oC) раствору вещества, полученного на шаге a) (2,15 г), в метаноле (50 мл). Когда добавление было закончено, ледяную баню убрали и перемешивали реакционную смесь при комнатной температуре 4 ч. Затем все вылили в воду и три раза экстрагировали хлороформом. Органические экстракты соединили, промыли водой, высушили (MgSO4) и испарили при пониженном давлении, получив указанное в подзаголовке соединение в виде белого твердого вещества (1,91 г, 79%).
Масс-спектр: производное El TMS 343 [(M-15)+];
1H-ЯМР (CDCl3) δ : 2,76 (t, 2H), 2,91 (t, 2H), 3,19 (m, 4H), 3,72 (t, 2H), 3,86 (t, 2H), 7,15 - 7,3 (m, 5H).
c) N-[2-[4-гидрокси-2-оксо-3H-1,3-бензотиазол-7-ил]-3[2-[2- фенилэтокси] этилсульфонил]пропанамид.
К перемешиваемому раствору 7-[2-аминоэтил]-4-гидрокси-1,3-бензотиазол-2(3H)-она гидробромида (1,62 г) и вещества, полученного на этапе b) (1,75 г), в ДМФ (25 мл) добавили триэтиламин (0,70 мл), 1-гидроксибензотриазола гидрат (0,98 г) и, наконец, дициклогексилкарбодиимид (1,49 г). Все это перемешивали при комнатной температуре в течение 16 ч. Добавили ледяную уксусную кислоту (0,1 мл) и перемешивали еще 15 мин. Удалили ДМФ при пониженном давлении, а остаток развели этилацетатом (50 мл). Взвешенную дициклогексилмочевину удалили путем фильтрации. Фильтрат промыли насыщенным водным натрия бикарбонатом и высушили (MgSO4). Растворитель удалили при пониженном давлении, а полученный остаток очистили путем хроматографии в колонке на силикагеле с дихлорметаном : этанолом в соотношении 95 : 5. Получили вещество, указанное в подзаголовке (1,89 г, 71%).
Т. пл. 142 - 144oC.
Масс-спектр: FAB + ve 479 [(M+H)+];
1H-ЯМР (DMCO-d6) δ : 2,50 (m, 2H), 2,61 (t, 2H), 2,81 (t, 2H), 3,2 - 3,4 (brm, 6H+D2O), 3,64 (t, 2H), 3,75 (t, 2H), 6,70 (d, 1H), 6,80 (d, 1H), 7,15 - 7,30 (m, 5H), 8,14 (t, 1H), 10,0 (brs, 1H), 11,5 (s, 1H).
Анализ: найдено, %: C 55,03; H 5,55; N 5,90; S 13,07.
C22H26N2O6S2
Вычислено, %: C 55,21; H 5,48; N 5,85; S 13,39.
d) 4-Гидрокси-7-[2-[3-[2-[2-фенилэтокси] этилсульфонил]пропиламин]этил] -1,3-бензотиазол-2-(3H)-она гидрохлорид.
Раствор борана в тетрагидрофуране (1,0 М в ТГФ, 15 мл) добавили по каплям к перемешиваемому раствору продукта этапа c) (2,06 г) в сухом тетрагидрофуране (100 мл). Реакционную смесь подвергли кипячению в инертной атмосфере в колбе с обратным холодильником пока тонкослойная хроматография не показала, что исходного материала больше не осталось. Реакционную смесь охладили и добавили метанол (3,5 мл, ОСТОРОЖНО!). Затем кипятили в колбе с обратным холодильником в течение 30 мин. Растворители удалили при пониженном давлении, а осадок растворили в метаноле (100 мл). Добавили концентрированную соляную кислоту (отн. пл. 1,18, 0,75 мл). Все это кипятили в колбе с обратным холодильником в течение 30 мин. После охлаждения и удаления при пониженном давлении метанола получили маслянистый осадок, который после тритурирования эфиром дал сырое соединение, указанное в заголовке, в виде бледно-желтого твердого вещества. Порции указанного в заголовке соединения очищали путем подготовительной обращенно-фазной жидкостной хроматографии с метанолом и 0,1%-ной трифторуксусной кислотой в качестве элюента. И, наконец, получение гидрохлорида путем растворения в небольшом количестве этанола и обработки сухой эфирной соляной кислотой с последующим удалением растворителей дало названное в заголовке соединение в виде белого порошка.
Т. пл. 201 - 203oC.
Масс-спектр: FAB + ve 465 [(M+H)+];
1H-ЯМР (DMCO-d6) δ : 2,01 (m, 2H), 2,80 (m, 4H), 2,98 (brs, 2H), 3,10 (t, 4H), 3,36 (t, 2h), 3,66 (t, 2H), 3,77 (t, 2H), 6,77 (d, 1H), 6,88 (d, 1H), 7,2 - 7,35 (m, 5H), 8,98 (brs, 2H), 10,13 (brs, 1H), 11,77 (s, 1H).
Анализ: найдено, %: C 52,31; H 5,85; N 5,54; S 12,54; Cl 7,48.
C22H28N2O5S2.HCl
Вычислено,%: C 52,73; H 5,83; N 5,90; S 12,79; Cl 7,08.
Пример 2. 4-гидрокси-7-[2-[2-[3-[2-фенилэтокси]пропокси]этиламин]этил] -1,3-бензотиазол -2(3H)-она гидрохлорид.
а) 2-[3-[2-Фенилэтокси]пропокси]уксусная кислота.
Указанное в подзаголовке соединение приготовили по способу, описанному в примере 1а), с использованием 3-[2-фенилэтокси]пропанола (приготовленного из 2-фенилметил-1,3-диоксана по способу, описанному в Can. J. Chem., 1974, 52, 888).
1H-ЯМР(CDCl3) δ 1,89 (m, 2H), 2,90 (q, 2H), 3,49-3,60 (m, 6H), 4,05 (s, 2H), 7,21-7,30 (m, 5H).
b) N-[2-[4-Гидрокси-2-оксо-3H-1,3-бензотиазол-7-ил]этил]-2-[3-[2-фенилэтокси] пропокси]ацетамид.
Указанное в подзаголовке соединение приготовили по способу, описанному в примере 1 с).
Т.пл. 150 - 151oC.
Масс-спектр: FAB + ve 431[(M+H)+];
1H-ЯМР (DMCO-d6) δ : 1,73 (m, 2H), 2,63 (t, 2H), 2,79 (t, 2H), 3,2-3,4 (brm, 6H+D2O), 3,54 (t, 2H), 3,76 (brs, 2H), 6,69 (d, 1H), 6,79 (d, 1H), 7,16-7,29 (m, 5H), 8,12 (t, 1H), 9,92 (s, 1H), 11,61 (s, 1H).
Анализ: найдено, %: C 60,90; H 6,02; N 6,40; S 6,91.
C22H26N2O5S
Вычислено,%: C 61,37; H 6,09; N 6,51; S 7,45.
с) 4-Гидрокси-7-[2-[2-[3-[2-фенилэтокси] пропокси] этиламин]этил]-1,3-бензотиазол -2(3H)-она гидрохлорид.
Указанное в заголовке соединение приготовили по способу, описанному в примере 1d).
Т. пл. 159 - 160oC.
Масс-спектр: FAB + ve 417 [(M+H)+;
1H-ЯМР (DMCO-d6) δ : 1,75 (t, 2H), 2,79 (t, 2H), 2,87 (t, 2H), 3,12 (m, 4H), 3,45 (m, 4H+D2O), 3,58 (m, 4H), 6,77 (d, 1H), 6,85 (d, 1H), 7,18-7,27 (m, 5H), 8,99 (brs, 2H), 10,16 (s, 1H), 11,8 (brs, 1H)
Анализ: найдено, %: C 58,33; H 6,54; N 6,37; S 6,79; Cl 7,96,
C22H28N2O4S.HCl
Вычислено,%: C 58,33; H 6,23; N 6,18; S 7,08; Cl 7,83.
Пример 3. 4-Гидрокси-7-[2-[3-[2-[2-фенилэтокси]этокси]пропиламин]этил] -1,3-бензотиазол-2 (3H)-она гидрохлорид.
а) 3-[2-[-2-Фенилэтокси]этокси]пропаннитрил.
Смесь из 3-(2-фенилэтокси) этанола (8,0 г, приготовлена из 2-фенилметил 1,3-диоксолана по способу, описанному в Can. J. Chem., 1974, 52, 888), 3-бромпропаннитрила (5,6 мл), натрия гидроксида (50 г) и тетрабутиламмония хлорида (0,5 г) в дихлорметане (100 мл) и воде (100 мл) перемешивали при комнатной температуре на протяжении 72 ч. Смесь разбавили водой и отделили органический слой. Водный слой экстрагировали новой порцией дихлорметана. Соединенные органические экстракты промыли разбавленной водной соляной кислотой и водой, высушили (MgSO4) и испарили при пониженном давлении. Получили сырой продукт. Вещество очистили путем хроматографии на силикагеле с резким понижением давления с эфиром : петролейным эфиром (т. кип. 60 - 80oC) в соотношении 1 : 1 в качестве элюента. Получили указанное в подзаголовке соединение в виде масла (9,84 г, 90%).
Масс спектр: EI 219 (M+;
1H-ЯМР (CDCl3) δ 2,55 (t, 2H), 2,90 (t, 2H), 3,61-3,74 (m, 8H), 7,18-7,36 (m, 5H).
b) 3-[2-[2-Фенилэтокси]этокси]пропаналь.
Диизобутилалюминия гидрид (3,3 мл, 1,5 M в толуоле) добавили по каплям к охлажденному (0oC) перемешиваемому раствору 3-[2-[2-фенилэтокси] этокси] пропаннитрила (1,0 г) (этап а)) в тетрагидрофуране. Через 30 мин смесь нагрели до комнатной температуры и перемешивали 2 ч. Осторожно добавили воду и 10%-ную водную соляную кислоту и перемешивали еще 5 мин. Реакционную смесь экстрагировали несколько раз эфиром, эфирные экстракты соединили, промыли насыщенным водным раствором натрия бикарбоната и солевым раствором, высушили (MgSO4), испарили при пониженном давлении и получили указанное в подзаголовке соединение в виде желтого масла, которое без очистки передали на следующий этап.
c) 4-Гидрокси-7-[2-[3-[2-[2-фенилэтокси]этокси]пропиламин]этил]-1,3-бензотиазол-2 (3H)-она гидрохлорид.
Натрия цианоборогидрид (0,333 г) добавили к перемешиваемому раствору 3-[2-[2-фенилэтокси]этокси]пропаналя (этап b)) (2,2 г), 6%-ной водной уксусной кислоты (2 мл) и 7-[2- аминоэтил]-4-гидрокси-1,3-бензотиазол-2(3H)-она гидробромида (2,05 г) в метаноле (180 мл). Реакционную смесь перемешивали 2 ч при комнатной температуре и к этому времени хроматографический анализ показал, что весь исходный материал израсходован. Реакционную смесь подщелочили при помощи концентрированного раствора гидроксида аммония в воде, удалили метанол при пониженном давлении и получили сырой продукт. Хроматографическая очистка силикагелем с метанолом в хлороформе в качестве элюента, обращенно-фазная подготовительная жидкостная хроматография высокого давления с 0,1%-ной водной трифторуксусной кислотой в метаноле в качестве элюента и приготовление соли-гидрохлорида дали указанное в заголовке соединение в виде белого твердого вещества.
Т. пл. 186 - 190oC.
Масс-спектр: FAB + ve 417 [(M+H)+];
1H-ЯМР (DMCO-d6) δ 1,80-1,88 (m, 2H) 2,78-2,86 (m, 4H), 2,97 (t, 2H), 3,09 (t, 2H), 3,46 (t, 2H), 3,47-3,58 (m, 4H), 3,60 (t, 2H), 6,76 (d, 1H), 6,88 (d, 1H), 7,16-7,29 (m, 5H), 8,70 (brs, 2H), 10,13 (s, 1H), 11,76 (brs, 1H).
Анализ: найдено,%: C 55,24; H 5,98; N 5,92; S 6,36; C 17,35,
C22H28N2O4S.HCl.1,42 H2O
Вычислено,%: C 55,20; H 6,41; N 5,88; S 6,70; Cl 7,41.
Пример 4. 4-Гидрокси-7-[2-[2-[2-[2-фенилэтокси]этокси]этиламин]этил]-1,3- бензотиазол-2(3H)-она гидрохлорид.
а) 2-[2-[-2-Фенилэтокси]этокси]уксусная кислота.
Натрия гидрид (60%-ная дисперсия в масле, 0,86 г) несколько раз промыли петролейным эфиром и суспендировали в тетрагидрофуране (5 мл). К суспензии по каплям добавили раствор 2-[2-фенилэтокси]этанола (1,5 г приготовлен из 2-фенилметил-1,3-диоксолана по способу, описанному в Can. J. Chem., 1974, 52, 888) в тетрагидрофуране (10 мл) и нагревали смесь до 55 oC в течение 15 мин, а затем 2 ч перемешивали при комнатной температуре. Добавили хлоруксусную кислоту (0,85 г) в тетрагидрофуране (5 мл) и продолжали перемешивание в течение 17 ч при комнатной температуре. Тетрагидрофуран удалили при пониженном давлении, а осадок разделили между насыщенным водным раствором натрия бикарбоната и диэтиловым эфиром (эфирный слой оставили). Выделенный водный слой подкислили разбавленной водной соляной кислотой и экстрагировали диэтиловым эфиром. Органические экстракты промыли насыщенным солевым раствором, высушили (MgSO4), испарил при пониженном давлении и получили светло-коричневое масло (1,48 г). Хроматография на силикагеле с эфиром : петролейным эфиром (т. кип. 60 - 80oC) 1 : 1 в качестве элюента дала указанное в подзаголовке соединение (1,07 г).
1H-ЯМР (CDCl3) δ : 2,94 (t, 2H), 3,49-3,82 (m, 6H), 4,16 (s, 2H), 7,18-7,34 (m, 5H).
b) N-[2-[4-гидрокси-2-оксо-3H-1,3-бензотиазол-7-ил] этил] -2-[2- [2-фенилэтокси]этил]ацетамид.
Указанное в подзаголовке соединение приготовили в соответствии с процедурой, описанной в примере 1c).
Масс-спектр: FAB + ve 417 [(M+H)+];
1H-ЯМР (DMCO-d6) δ : 2,61 (t, 2H), 2,79 (t, 2H), 3,31 (m, 6H), 3,60 (t, 2H), 3,82 (s, 2H), 6,69 (d, 1H), 6,79 (d, 1H), 7,15-7,29 (m, 5H), 7,72 (t, 1H), 9,91 (brs, 1H), 11,61 (brs, 1H).
c) 4-Гидрокси-7-[2-[2-[2-[2-фенилэтокси] этокси] этиламин] этил]- 1,3-бензоатиазол-2(3H)-она гидрохлорид
Указанное в заголовке соединение приготовили по способу, описанному в примере 1d).
Т. пл. 123oC.
Масс-спектр: FAB + ve 403 [(M+H)+];
1H-ЯМР (DMCO-d6) δ : 2,78 (t, 2H), 2,85 (t, 2H), 3,09 (m, 4H), 3,56 (m, 6H), 3,65 (t, 2H), 6,77 (d, 1H), 6,83 (d, 1H), 7,08-7,29 (m, 5H), 9,00 (s, 2H), 10,15 (s, 1H), 11,69 (s, 1H).
Анализ: найдено,%: C 56,62; H 6,15; N 6,43; Cl 9,40,
C21H26N2O4S. HCl с избытком 0,18 моль HCl
Вычислено,%: C 56,62; H 6,14; N 6,29; Cl 9,37.
Пример 5. 4-Гидрокси-7-[2-[2-[3-[2-фенилэтокси]пропилтио] этиламин]этил] -1,3-бензотиазол-2(3H)-она гидрохлорид.
a) 3-Меркаптопропанол.
Раствор тиомочевины (36 г) в воде (100 мл) смешали с 3-бромопропанолом (33 мл) и кипятили в колбе с обратным холодильником в течение 4 ч. Затем смеси позволили немного остыть и добавили 10%-ный водный раствор натрия гидроксида (190 мл). Смесь снова кипятили в колбе с обратным холодильником в течение 3 ч, затем позволили ей остыть и держали 17 ч при комнатной температуре. Смесь подкислили до pH 4 при помощи концентрированной серной кислоты и экстрагировали диэтиловым эфиром. Органические экстракты соединили, высушили (MgSO4), сконцентрировали при пониженном давлении и получили сырой продукт в виде желтой жидкости. Дистилляция дала соединение, указанное в подзаголовке (14,67 г).
Масс спектр: EI 92 (M)+;
1H-ЯМР (CDCl3) δ : 1,40 (m, 1H), 1,91 (m, 3H), 2,63 (q, 2H), 3,75 (t, 2H).
b) 2-Фенилметил-1,3-оксатиан.
К раствору тиола (14,67 г, этап a)) в толуоле (200 мл) добавили р-толуолсульфоновую кислоту (1 г) и фенилацетальдегид (18,3 мл). Реакционную смесь кипятили в колбе с обратным холодильником, используя аппарат Дина-Старка. Когда собрали требуемое количество воды, смесь охладили, промыли насыщенным натрия бикарбонатом, насыщенным солевым раствором и высушили (K2CO3). Сырой продукт дистиллировали (т. кип. 100 - 110oC/0,3 мбар) и получили желтую жидкость (19,65 г).
Масс спектр: EI 194 (M)+;
1H-ЯМР (CDCl3) δ : 1,66 (d, 1H), 1,95 (m, 1H), 2,7 (m, 1H), 2,94 (m, 2H), 3,10 (m, 1H), 3,53 (t, 1H), 4,14 (d, 1H), 4,90 (t, 1H), 6,69-7,32 (m, 5H).
c) 3-[2-Фенилэтокси]пропантиол.
Кальциевые опилки (3,5 г) добавили в несколько приемов к жидкому аммиаку (500 мл) и все это интенсивно перемешивали в течение 10 мин. Добавили по каплям тиоацеталь (шаг b), 10 г) в эфире (7 мл) к темно-синему раствору. Добавление продолжалось 7 мин. Реакционную смесь 2 ч перемешивали, затем гасили хлоридом аммония пока не прекратилось бурление. Избыток аммония испаряли в азоте под вытяжкой в течение ночи. Оставшееся твердое вещество подкислили до pH 1 - 2 при помощи 10%-ной водной соляной кислоты и экстрагировали продукт этилацетатом. Органические слои соединили, промыли водой и солевым раствором, высушили (MgSO4), сконцентрировали при пониженном давлении и получили соединение, указанное в подзаголовке (8,29 г).
Масс спектр: EI 196 (M)+;
1H-ЯМР (CDCl3) δ : 1,29 (d, 1H), 1,86 (m, 2H), 2,56 (q, 2H), 2,87 (t, 2H), 3,49 (t, 2H), 3,64 (t, 2H), 6,97-7,31 (m, 5H).
d) 2-[3-[2-Фенилэтокси]пропилтио]уксусная кислота.
Натрия гидрид (60%, 3,38 г) промыли петролейным эфиром и суспендировали в диметилформамиде (5 мл) при 0oC. Добавили по каплям раствор тиола (шаг c)) в диметилформамиде (8,29 г в 10 мл).
Перемешивание продолжали 2 ч при 0 - 8oC, затем добавили по каплям раствор бромуксусной кислоты (5,88 г) в диметилформамиде (15 мл). Затем, чтобы облегчить перемешивание, добавили еще диметилформамида (20 мл). Через 17 ч при комнатной температуре диметилформамид удалили при пониженном давлении. Остаток разделили между насыщенным водным раствором натрия бикарбоната и диэтиловым эфиром (эфирный слой отставили). Водный слой подкислили соляной кислотой до pH 1 - 2 и экстрагировали диэтиловым эфиром. Эфирные экстракты соединили, промыли водой и солевым раствором, высушили (MgSO4) и выпарили при пониженном давлении. Хроматография сырого продукта на силикагеле с петролейным эфиром (т. кип. 60 - 80oC) : эфиром в соотношении 1 : 1 в качестве элюента дала указанное в подзаголовке соединение (7,10 г).
1H-ЯМР (CDCl3) δ : 1,86 (m, 2H), 2,70 (t, 2H), 2,87 (t, 2H), 3,21 (s, 2H), 3,51 (t, 2H), 3,63 (t, 2H), 7,17-7,30 (m, 5H), 9,74 (s, 1H).
e) N-[2-[4-гидрокси-2-оксо-3H-1,3-бензотиазол-7-ил] этил] -2-[3- [2-фенилэтокси]пропилтио]ацетамид.
Указанное в подзаголовке соединение приготовили по способу, описанному в примере 1c), с использованием 7-[2-аминоэтил]-4-гидрокси-1,3-бензотиазол-2(3H)-она гидробромида. После хроматографии на силикагеле с дихлорметаном : этанолом в соотношении 9 : 1 в качестве элюента получили указанное в подзаголовке соединение (1,08 г).
Масс-спектр: FAB + ve 447 [(M+H)+];
1H-ЯМР (DMCO-d6) δ : 1,70 (m, 2H), 2,65 (t, 2H), 2,67 (t, 2H), 2,78 (t, 2H), 3,05 (s, 2H), 3,28 (q, 2H), 3,41 (t, 2H), 3,53 (t, 2H), 6,71 (d, 1H), 6,83 (d, 1H), 7,15-7,43 (m, 5H), 8,05 (s, 1H), 9,9 (s, 1H), 11,62 (s, 1H).
f) 4-Гидрокси-7-[2-[2-[3-[2-фенилэтокси]пропилтио]этиламин]этил] -1,3-бензотиазол-2(3H)-она гидрохлорид.
Указанное в заголовке вещество приготовили, как описано в примере 1d). Сырой продукт очистили посредством обращенно-фазной жидкостной хроматографии высокого давления с метанолом в 0,1%-ной водной трифторуксусной кислоте в качестве элюента.
Т. пл. 209 - 211oC
Масс-спектр FAB + ve 433 [(M + H)+];
1H-ЯМР (DMCO-d6) δ: 1,82 (m, 2H)2,62 (m, 4H), 2,87 (m, 4H), 2,93 (m, 2H), 3,16 (m 2H), 3,53 (t, 2H), 3,65 (t, 2H), 6,83 (d, 1H), 6,94 (d, 1H) 7,26 - 7,37 (m, 5H), 9,02 (s, 2H), 10,21 (s, 1H), 11,83(s, 1H).
Анализ: найдено, %: C 55,36; H 6,35; N 6,12; S 13,30,
C22H28N2O3S2.HCl с избытком 0,46 моль H2O
Вычислено, %: C 55,36; H 6,32; N 5,87; S 13,41.
Пример 6. 4-Гидрокси-7-[2-[2-[3-[2-фенилэтокси]пропилсульфонил]этиламин] этил] -1,3-бензотиазол-2(3H)-она гидрохлорид.
a) 2-[3-[2-Фенилэтокси]пропилсульфонил]уксусная кислота.
Указанное в подзаголовке соединение приготовили из 2-[3-[2- фенилэтокси] пропантио] уксусной кислоты (пример 5d)) по способу, описанному в примере 1b).
Масс-спектр: FAB + ve 287 [M + H)+];
1H-ЯМР (CDCl3) δ: 2,12 (m, 2H), 2,87 (t, 2H), 3,41 (t, 2H), 3,58 (t, 2H) 3,67 (t, 2H), 3,97 (s, 2H), 7,00 - 7,43 (m, 5H), 8,79 (s, 1H).
b) N-[2-[4-гидрокси-2-оксо-3H-1,3-бензотиазол-7-ил] этил] -2-[3-[2- фенилэтокси]пропилсульфонил]ацетамид.
Указанное в заголовке соединение приготовили по способу, описанному в примере 1c). Сырой продукт очистили посредством хроматографии на силикагеле с резким понижением давления с дихлорметаном метанолом в соотношении 9:1 в качестве элюента.
Масс-спектр: FAB + ve 479 [(M + H)+];
1H-ЯМР (DMCO-d6) δ 1,92 (q, 2H), 2,62 (t, 2H), 2,81 (t,2H), 3,27 (m, 4H), 3,49 (t, 2H), 3,58 (t, 2H), 4,04 (s, 2H), 6,70 (d, 1H), 6,83 (d, 1H), 7,17-7,29 (m, 5H), 8,47 (t, 1H), 9,96 (s, 1H), 11,66 (d, 1H).
c) 4-Гидрокси-7-[2-[2-[3-[2-фенилэтокси]пропилсульфонил]этиламин] этил] -1,3-бензотиазол-2(3H)-она гидрохлорид.
Указанное в заголовке вещество приготовили, как описано в примере 1d). Сырой продукт очистили посредством обращенно-фазной жидкостной хроматографии высокого давления с метанолом в 0,1%-ной водной трифторуксусной кислоте в качестве элюента.
Т. пл. 217 - 220oC.
Масс-спектр: FAB + ve 465 [(M + H)+];
1H-ЯМР (DMCO-d6) δ: 1,91 (quin, 2 H), 2,81 (t, 2H), 2,87 (t, 2H), 3,20 (m, 4H), 3,34 (t, 2H), 3,51 (t, 2H), 3,57 (q, 4H), 6,77 (d, 1H), 6,86 (d, 1H), 7,17 - 7,31 (m, 5H), 9,27 (s, 2H), 10,15 (s, 1H), 11,77 (s, 1H).
Анализ: найдено, %: C 52,57; H 6,05; N 5,73; S 12,61,
C22H28N2O5S2.HCl
Вычислено, %: C 52,73; H 5,83; N 5,59; S 12,79.
Пример 7. 4-Гидрокси-7-[2-[3-[2-[2-фенилэтокси]этилтио] пропиламин]этил] -1,3-бензотиазол-2(3H)-она гидрохлорид.
a) N-[2-[4-гидрокси-2-оксо-3H-1,3-бензотиазол-7-ил] этил] -3-[2-[2-фенилэтокси]этилтио]пропанамид.
Указанное в подзаголовке вещество приготовили по способу, описанному в примере 1c), с использованием вещества из примера 1a).
Масс-спектр: FAB + ve 447 [(M + H)+;
1H-ЯМР (DMCO-d6) δ: 2,26 - 2,33 (t, 2H), 2,54 - 2,72 (m, 6H) 2,75 - 2,83 (t, 2H), 3,19 - 3,28 (q, 2H), 3,50 - 3,63 (2xt, 4H), 6,68 (d, 1H), 6,78 (d, 1H), 7,15 - 7,3 (m, 5H), 9,89 (s, 1H), 11,60 (brs, 1H).
b) 4-Гидрокси-7-[2-[3-[2-[2-фенилэтокси]этилтио]пропиламин] этил]-1,3-бензотиазол-2(3H)-она гидрохлорид.
Указанное в подзаголовке соединение приготовили, как описано в примере 1d).
Т. пл. 211 - 213oC.
Масс-спектр: FAB + ve 433 [(M + H)+];
1H-ЯМР (DMCO-d6) δ: 1,85 (m, 2H), 2,59 (t, 2H), 2,65 (t, 2H), 2,81 (t, 2H), 2,85 (t, 2H) 2,97 (t, 2H), 3,08 (m, 2H), 3,56 (t, 2H), 3,61 (t, 2H), 6,76 (d, 1H), 6,87 (d, 1H), 7,17 - 7,30 (m, 5H), 8,9 (brs, 2H), 10,14 (s, 1H), 11,76 (s, 1H).
Анализ: найдено, %: C 56,49; H 6,40; N 6,12; S 13,78; Cl 7,98,
C22H28N2O3S2.HCl
Вычислено, %: C 56,33; H 6,23; N 5,97; S 13,67; Cl 7,56.
Пример 8. 4-Гидрокси-7-[2-[3-[2-[2-фенилэтокси]этилсульфонил] пропиламин]этил]-1,3-бензотиазол-2(3H)-она 4-метилбензолсульфонат.
a) 4-Гидрокси-7-[2-[3-[2-[2-фенилэтокси]этилсульфонил] пропиламин]этил] -1,3-бензотиазол-2(3H)-она.
Водный раствор (500 мл) указанного в заголовке соединения из примера 1 (4,9 г) смешали с избытком водного натрия гидрокарбоната. Свободное основание экстрагировали хлороформом, экстракты соединили, промыли водой, высушили (MgSO4), профильтровали и удалили хлороформ при пониженном давлении. Получили указанное в подзаголовке соединение в виде белого твердого вещества (4,22 г, 91%).
Т. пл. 69 - 70oC.
b) 4-Гидрокси-7-[2-[3-[2-[2-фенилэтокси]этилсульфонил] пропиламин]этил] -1,3-бензотиазол-2(3H)-она 4-метилбензолсульфонат
Часть свободного основания растворили в метаноле и добавили один молярный эквивалент 4-метилбензолсульфоновой кислоты. Раствор выпарили при пониженном давлении и собранное твердое вещество рекристаллизовали (метанол/вода). Получили указанное в заголовке соединение в виде белых игольчатых кристаллов.
Т. пл. 170 - 171oC.
Пример 9. 4-Гидрокси-7-[2-[3-[2-[2-фенилэтокси]этилсульфонил] пропиламин]этил]-1,3-бензотиазол-2(3H)-она гемисукцинат.
Указанное в заголовке соединение приготовили по способу, описанному в примере 8a) и b), с использованием янтарной кислоты.
Т. пл. 182 - 183oC, блестящие белые бляшки (рекристаллизация из метанола/воды).
Пример 10. 4-Гидрокси-7-[2-[3-[2-[2-фенилэтокси]этилсульфонил] пропиламин]этил]-1,3-бензотиазол-2(3H)-она гексаноат.
Указанное в заголовке соединение приготовили по способу, описанному в примере 8a) и b), с использованием гексановой кислоты.
Т. пл. 131 - 132oC, белые иголки (рекристаллизация из метанола/воды).
Пример 11. 4-Гидрокси-7-[2-[3-[2-[2-фенилэтокси]этилсульфонил] пропиламин]этил]-1,3-бензотиазол-2(3H)-она тартрат.
Указанное в заголовке соединение приготовили по способу, описанному в примере 8a) и b), с использованием винной кислоты.
Т. пл. 158 - 162oC (рекристаллизация из метанола/воды).
Пример 12. 4-Гидрокси-7-[2-[3-[2-[2-фенилэтокси]этилсульфонил] пропиламин]этил]-1,3-бензотиазол-2(3H)-она 1-гидрокси-2-нафтоат (Xinafoate).
Указанное в заголовке соединение приготовили по способу, описанному в примере 8a) и b), с использованием 1-гидрокси-2-нафтовой кислоты.
Т. пл. 176 - 177oC, белые иголки (рекристаллизация из метанола/воды).
Пример 13. 4-Гидрокси-7-[2-[3-[2-[2-[2-аминофенил]этокси] этилсульфонил] пропиламин]этил]-1,3-бензотиазол-2(3H)-она дигидрохлорид.
a) Метил 3-[2-[2-[2-нитрофенил]этокси]этилсульфонил]пропаноат.
Концентрированную азотную кислоту (3,25 мл) добавляли по каплям в течение получаса к перемешиваемому охлажденному (лед/соль) раствору метил 3-[2-[2-фенилэтокси] этилсульфонил] пропаноата (15,12 г) (приготовленного из кислоты, полученной по процедуре из примера 1b)) в трифторуксусной кислоте. Реакционную смесь оставили нагреваться до комнатной температуры, затем в течение ночи перемешивали, разбавили водой и несколько раз экстрагировали этилацетатом. Органические экстракты соединили, промыли водой и солевым раствором, высушили (MgSO4) и сконцентрировали при пониженном давлении. Получили сырую смесь изомерных метил 3-[2-[2-[2-нитрофенил]этокси]этилсульфонил] пропаноатов. Указанное в подзаголовке соединение отделили от других изомеров посредством жидкостной хроматографии высокого давления с нормальными фазами с гексаном : этилацетатом в соотношении 1 : 1 в качестве элюента.
1H-ЯМР (CDCl3) δ : 2,80-2,84 (t, 2H), 3,17-3,24 (m, 4H), 3,32-3,36 (m, 2H), 3,71-3,78 (m, 2H), 3,84-3,87 (t, 2H), 7,36-7,41 (m, 2H), 7,54 (t, 2H), 7,91 (d, 1H).
b) 3-[2-[2-[2-Нитрофенил]этокси]этилсульфонил]пропановая кислота.
Металлический литий (0,59 г) растворили в метаноле (200 мл). Добавили воду (100 мл), затем к охлажденному раствору (лед/соль) по каплям добавили соединение, полученное на этапе a) (6,05 г), в метаноле (50 мл). Реакционную смесь оставили нагреваться до комнатной температуры, затем перемешивали всю ночь. Удалили при пониженном давлении растворитель, а остаток разбавили водой. Основной водный раствор промыли этилацетатом, (который отставили), подкислили до pH 2 (концентрированной соляной кислотой) и экстрагировали этилацетатом. Эти экстракты соединили, промыли водой и солевым раствором, высушили (MgSO4) и выпарили растворители при пониженном давлении. Полученный сырой продукт очистили посредством хроматографии на силикагеле с резким понижением давления и с дихлорметаном : этанолом в соотношении 9 : 1 в качестве элюента. Получили указанное в подзаголовке соединение.
Масс-спектр: TS 349 [(M+NH4)+].
c) N-[2-[4-гидрокси-2-оксо-3H-1,3-бензотиазол-7-ил]этил]-3- [2-[2-[2-нитрофенил]этокси]этилсульфонил]пропанамид.
Указанное в подзаголовке соединение приготовили, как описано в примере 1c), с использованием 7-[2-аминоэтил]-4-гидрокси-1,3 безотиазол-2(3H)-она гидробромида и продукта этапа b). Указанное в подзаголовке соединение получили после хроматографии на силикагеле с 6%-ным этанолом в хлороформе в качестве элюента.
Масс-спектр FAB + ve 524 [(M+H)+];
1H-ЯМР (DMCO-d6) δ: 2,5 (m, 4H), 2,60 (t, 2H), 3,09 (t, 2H), 3,24 (m, 4H), 3,68 (t, 2H), 3,74 (t, 2H), 6,70 (d, 1H), 6,80 (d, 1H), 7,47 (t, 1H), 7,55 (d, 1H), 7,62 (t, 1H), 7,91 (d, 2H), 8,14 (t, 1H), 9,91 (s, 1H), 11,62 (s, 1H).
d) N-[2-[4-гидрокси-2-оксо-3H-1,3-бензотиазол-7-ил] этил]-3-[2- [2-[2-аминофенил]этокси]этилсульфонил]пропанамид.
Гидразина гидрат (10 мл) по каплям добавили к перемешиваемой суспензии свежепромытого никеля Ренея, соединения, полученного на шаге c) (2,21 г), и этанола (50 мл). Когда реакция закончилась никель Ренея отфильтровали (ОСТОРОЖНО: ОГНЕОПАСНО!), а этанол выпарили при пониженном давлении. Остаток разделили между водой и дихлорметаном и водный слой экстрагировали новыми порциями дихлорметана. Экстракты соединили, промыли водой и солевым раствором, высушили (MgSO4), выпарили при пониженном давлении и получили сырой продукт, который использовали без очистки.
Масс-спектр FAB + ve 494 [(M+H)+];
1H-ЯМР (DMCO-d6) δ: 2,4-2,8 (t, 8H), 3,2-3,4 (m, 6H), 3,58 (t, 2H), 3,76 (t, 2H), 6,46 (t, 1H), 6,59 (d, 1H), 6,70 (d, 1H), 6,80 (d, 1H), 6,86-6,93 (m, 2H), 8,16 (t, 1H), 9,93 (brs, 1H), 11,63 (s, 1H).
e) 4-Гидрокси-7-[2-[3-[2-[2-[2-аминофенил] этокси]этилсульфонил] пропиламин]этил]-1,3-бензотиазол-2(3H)-она дигидрохлорид.
Указанное в заголовке соединение приготовили, как описано в примере 1d), с использованием продукта этапа d). Сырой продукт очистили посредством обращенно-фазной хроматографии с 0,1%-ной водной трифторуксусной кислотой в ацетонитриле в качестве элюента.
Т. пл. 65oC (размягчается).
Масс-спектр FAB + ve 480 [(M+H)+];
1H-ЯМР (DMCO-d6) δ: 2,00-2,08 (m, 4H), 2,8-3,3 (m, 10H), 3,39-3,45 (m, 2H), 3,71 (t, 2H), 3,81 (t, 2H), 4,5 (brs, 3H), 6,77 (d, 1H), 6,89 (d, 1H), 7,28-7,36 (m, 4H), 9,04 (brs, 2H), 10,15 (s, 1H), 11,6 (s, 1H).
Пример 14. 4-Гидрокси-7-[2-[3-[2-[2-[4-нитрофенил]этокси]этилсульфонил] пропиламин] этил]-1,3-бензотиазол-2(3H)-она гидрохлорид.
a) N -[2-[4-гидрокси-2-оксо-3H-1,3-бензотиазол-7-ил] этил]-3-[2-[2-[4- нитрофенил]этокси]этилсульфонил]пропанамид.
Процедура, описанная в примере 1с, в ходе которой использовались 7-[2-аминоэтил] -4-гидрокси-1,3-бензотиазол-2-(3H)-она гидробромид и 3-[2-[2-[4-нитрофенил] этокси] -этилсульфонил] пропановая кислота (пример 13 a)), дала указанное в подзаголовке вещество (после хроматографической очистки на силикагеле с 8%-ным этанолом в дихлорметане в качестве элюента).
Масс-спектр: FAB + ve 524 [(M + H)+];
1H-ЯМР (DMCO-d6) δ: 2,45 (t, 2H), 2,60 (t, 2H), 2,97 (t, 2H), 3,2-3,4(m, 6H), 3,69-3,77(m 4H), 6,70 (d, 1H), 6,80 (d, 1H), 7,53 (d,2H), 8,12 (d, 3H), 9,91 (brs, 1H), 11,6 (brs, 1H).
b) 4-Гидрокcи-7-[2-[3-[2-[2-[4-нитрофенил] этокси] этилсульфонил]пропиламин] этил]-1,3-бензотиазол-2(3H)-она гидрохлорид.
Указанное в заголовке соединение приготовили, как описано в примере 1d). Сырой продукт реакции очистили обращенно-фазной хроматографией с ацетононитрилом в 0,1%-ной водной трифторуксусной кислоте в качестве элюента.
Т.пл. 75 - 78oC.
Масс-спектр: FAB + ve 510 [(M + H)+];
1H-ЯМР (DMCO-d6) δ: 2,01 (quin, 2H), 2,83 (t, 2H), 2,98 (t, 4H), 3,12 (t 4H), 3,39 (t, 2H), 3,70-3,79 (m, 4H), 6,76 (d, 1H), 6,88 (d, 1H), 7,55 (d, 2H), 8,16 (d, 2H), 8,83 (brs 2H), 10,13 (s, 1H), 11,76 (s, 1H).
Пример 15. 7-[2-[2-[3-[2-[4-Фторфенил]этокси]пропилсульфонил]этиламин] этил]-4-гидрокси-1,3-бензотиазол-2(3H)-она гидрохлорид.
a) 2-[4-Фторфенил]этил аллиловый эфир.
2-[4-Фторфенил] этанол (7,0 г) медленно добавили к перемешиваемой суспензии натрия гидрида (1,25 г, 60% в виде дисперсии в масле, предварительно промытая петролейным эфиром (т.кип. 60 - 80oC)) в ДМФ (50 мл). Смесь перемешивали 30 мин при комнатной температуре. Затем медленно добавили аллил бромид (6,0 г) и все это перемешивали в течение ночи. Водный и эфирный слои разделили. Водный слой экстрагировали новой порцией эфира, эфирные экстракты соединили, промыли водой и высушили (MgSO4). Эфир удалили при пониженном давлении и получили сырое вещество, указанное в подзаголовке, в виде бесцветного масла (8,7 г, 96%).
Масс-спектр: El 180 (M)+;
1H-ЯМР (CDCl3) δ: 2,9 (t, 2H), 3,6(t, 2H), 4,0 (t, 2H) 5,2 (m, 2H), 5,9 (m, 1H), 6,95 (m, 2H), 7,2 (m, 2H).
Эту реакцию успешно повторили в 5-кратном масштабе.
b) 2-[3-[2-[4-Фторфенил]этокси]пропилтио]уксусная кислота.
Соединение, полученное на шаге a) (15 г), и тиоглеколевую кислоту (6,8 мл) перемешивали при комнатной температуре в конической колбе при доступе воздуха в течение 2 ч. Затем добавили еще порцию тиогликолевой кислоты (3,4 мл). Еще полчаса перемешивали и после этого реакция завершилась. Сырой продукт хроматографировали на силикагеле с дихлорметаном:уксусной кислотой в соотношении 99 : 1 в качестве элюента и получили указанное в подзаголовке вещество в виде бесцветного масла (19,69 г, 87%).
Масс-спектр: FAB + ve 273 [M + H)+];
1H-ЯМР (CDCl3) δ : 1,87 (m, 2H), 2,71 (t, 2H), 2,85 (t, 2H), 3,31 (d, 2H), 3,51 (t, 2H), 3,65 (t, 2H), 6,98 (m, 2H), 7,18 (m, 2H).
c) 2-[3-[2-[4-Фторфенил]этокси]пропилсульфонил]уксусная кислота.
Раствор калия гидрокарбоната (150 г) в воде (500 мл) в течение 20 мин добавляли к перемешиваемой смеси кислоты, полученной на шаге b) (39,5 г), с водой (50 мл). Добавили в несколько приемов водный раствор OXONETM (278 г в 400 мл) и перемешивали реакцию всю ночь. Затем добавили воду (1 л) и все экстрагировали эфиром, (чтобы удалить некислые материалы). Затем водный слой подкислили 20%-ной водной серной кислотой и три раза экстрагировали эфиром. Эфирные слои соединили, промыли водой и солевым раствором, высушили (MgSO4), сконцентрировали при пониженном давлении и получили бледно-желтое масло (41,7 г). Отстоявшись, масло дало белый твердый осадок. Его выделили фильтрацией и промыли небольшими порциями эфира : пентана в соотношении 1 : 1. Получили указанное в подзаголовке соединение.
Т.пл. 47 - 48oC.
Масс-спектр: FAB + ve 305 [(M + H)+];
1H-ЯМР (CDCl3) δ: 2,12 (m, 2H), 2,84 (t, 2H), 3,32 (m, 2H), 3,58 (t, 2H), 3,65 (t, 2H), 4,00 (s, 2H), 6,98 (m, 2H), 7,16 (m, 2H), 8,80 (brs, 1H).
d) N-[2-[4-гидрокси-2-оксо-3H-1,3-бензотиазол-7-ил] этил] -2-[3-[2-[4- фторфенил]этокси]пропилсульфонил]ацетамид.
Карбонил-диимидазол (1,94 г) добавили к перемешиваемому раствору кислоты (шаг c) (3,64 г) в ДМФ (15 мл) и перемешивали еще 40 мин при комнатной температуре. К этому раствору добавили 7-[2-аминоэтил]-4-гидрокси-1,3 бензотиазол-2(3H)-она гидробромид (3,48 г), затем триэтиламин (1,7 мл). Все это оставили на ночь. Затем реакционную смесь медленно добавили к быстро перемешиваемой смеси 10%-ной водной соляной кислоты и эфира (каждого по 100 мл). Постепенно осело бледно-желтое твердое вещество, которое отфильтровали, промыли пентаном и высушили в вакууме. Получили указанное в подзаголовке соединение желто-коричневого цвета, которое использовали без очистки.
Масс-спектр: FAB + ve 497 [(M + H)+];
1H-ЯМР (DMCO-d6) δ: 1,91 (m, 2H), 2,61 (t, 2H), 2,80 (t, 2H), 3,30 (m, 4H), 3,48 (t, 2H), 3,56 (t, 2H), 4,03 (s, 2H), 6,70 (d, 1H), 6,82 (d, 1H), 7,10 (t, 2H), 7,28 (m, 2H), 8,46 (t, 1H), 9,95 (s, 1H), 11,66 (s, 1H).
e) 7-[2-[2-[3-[2-[4-Фторфенил] этокси]пропилсульфомил]этиламин] этил]-4-гидрокси-1,3-бензотиазол-2(3H)-она гидрохлорид.
Указанное в заголовке соединение приготовили, как описано в примере 1d), с использованием соединения, полученного на шаге d). Сырой продукт очистили посредством хроматографии с обращенными фазами с 35%-ным тетрагидрофураном в 0,1%-ной водной трифторуксусной кислоте в качестве элюента.
Т.пл. 240 - 245oC.
Масс-спектр: FAB + ve 483 [(M + H)+];
1Н-ЯМР (DMCO-d6) δ: 1,91 (m, 2H), 2,80 (t, 2H), 2,85 (t, 2H), 3,14-3,21 (m, 4H), 3,24(2H + D2O), 3,49 (t, 2H), 3,54 (q, 4H), 6,76 (d, 1H), 6,87 (d, 1H), 7,10 (t, 2H), 7,27 (t, 2H), 9,14 (s, 2H), 10,15 (s, 1H), 11,78 (s, 1H).
Анализ: найдено, %: C 50,58, H 5,62; N 5,61; S 12,26,
C22H27N2FO5S2.HCl;
Вычислено, %: C 50,91; H 5,44; N 5,40; S 12,36.
Пример 16. 4-Гидрокси-7-[2-[2-[3-[2-[2-тиенил]этокси]пропилтио]этиламин] этил]- 1,3-бензотиазол-2(3H)-она гидрохлорид.
Указанное в заголовке вещество приготовили, как описано в примере 15, с использованием 2-тиенилэтанола, не проводилось лишь окисление по примеру 15c). За исключением этого была соблюдена процедура примера 1b).
Т. пл. 220 - 221oC.
Масс-спектр: FAB + ve 471 [(M + H)+];
1H-ЯМР (DMCO-d6) δ: 1,90-1,98 (m, 2H); 2,53 (m, 2H), 2,85 (t, 2H), 3,03 (t, 2H), 3,16 (m, 2H), 3,25 (m, 2H), 3,37 (m, 2H), 3,53 (m, 2H), 3,60 (t, 2H), 6,76 (d, 1H), 6,88 (m, 2H), 6,94 (m, 1H), 7,33 (dd, 1H), 9,09 (brs, 2H), 10,14 (s, 1H), 11,78 (brs, 1H).
Анализ: найдено, %: C 46,67; H 5,51; N 5,68; S 18,42,
C20H26N2O5S3.HCl
Вычислено, %: C 47,37; H 5,37; N 5,52; S 18,97.
Пример 17. 4-Гидрокси-7-[2-[3-[2-[2-[2-пиридил]этокси]этилтио]пропиламин]этил]- 1,3-бензотиазол-2(3H)-она дигидрохлорид.
а) 2-[2-[2-Бромэтилтио]этил]-1,3-диоксолан.
К перемешиваемому охлажденному (< 0oC) раствору 2-[2-[1,3-диоксолан-2-ил]-этилтио]этанола (13,6 г, приготовлен путем конденсации меркаптоэтанола с 2-[2-бромэтил] -1,3-диоксоланом с использованием натрия гидрида) в сухом ацетонитриле (150 мл) добавили трифенилфосфин (20 г) и углерода тетрабромид (38 г). Раствор перемешивали 4 ч. Растворитель удаляли при пониженном давлении, а остаток адсорбировали силикагелем. Вещество хроматографировали на силикагеле с резким понижением давления с 10%-ным этилацетатом/петролейным эфиром в качестве элюента и получили указанное в подзаголовке соединение в виде прозрачного масла (4,45 г), которое без дальнейшей очистки передали на следующий этап.
b) 2-[2-[2-[2-[2-Пиридил]этокси]этилтио]этил]-1,3-диоксолан.
Вещество, полученное на шаге а) (6,12 г), тетра-n-бутиламмония гидросульфат (1 г) и 2-пиридилэтанол (2,85 мл) перемешивали с дихлорметаном и 20%-ным водным раствором натрия гидроксида (каждого по 20 мл), пока газовая хроматография не показала, что реакция завершилась. Органический слой отделили, промыли водой и высушили (MgSO4). После удаления растворителей при пониженном давлении осталось масло. Его очистили посредством хроматографии на силикагеле с резким понижением давления с этилацетатом : петролейным эфиром, т. кип. 60 - 80oC в соотношении 4 : 1 в качестве элюента. Получили указанное в подзаголовке соединение в виде желтого масла (0,770 г).
1H-ЯМР (CDCl3) δ : 1,92 (m, 2H), 2,65 (m, 4H), 3,07 (t, 2H), 3,62 (t, 2H), 3,85 (m, 4H), 3,95 (m, 2H), 4,94 (t, 1H), 7,13 (m, 1H), 7,22 (d, 1H), 7,60 (td, 1H), 8,53 (d, 1H).
c) 3-[2-[2-[2-Пиридил]этокси]этилтио]пропаналь.
Продукт этапа b) (0,850 г) растворили в 80%-ной муравьиной кислоте (10 мл) и оставили при комнатной температуре на 22 ч. Смесь разделили между эфиром и водой. Слои разделили и водный слой экстрагировали эфиром. Эфирные экстракты соединили, промыли солевым раствором, высушили (MgSO4) и выпарили при пониженном давлении. Получили указанное в заголовке соединение в виде масла (0,70 г).
1H-ЯМР (CDCl3) δ : 2,69 (m, 4H), 2,87 (t, 2H), 3,06 (t, 2H), 3,64 (t, 2H), 3,85 (t, 2H), 7,13 (td, 1H), 7,21 (d, 1H), 7,60 (td, 1H), 8,53 (d, 1H), 9,74 (s, 1H).
d) 4-Гидрокси-7-[2-[3-[2-[2-[2-пиридил]этокси]этилтио]пропиламин]этил]- 1,3-бензотиазол-2(3H)-она дигидрохлорид.
Продукт этапа c) (0,700 г) растворили в метаноле (20 мл). К этому раствору добавили 7-[2-аминоэтил] -4-гидрокси-1,3-бензотиазол-2(3H)-она гидробромид (0,655 г), натрия цианоборогидрид (0,100 г) и водную 6%-ную уксусную кислоту (чтобы довести pH до 6). Раствор перемешивали в течение ночи при комнатной температуре, потом его подщелочили концентрированным раствором аммония гидроксида. Растворитель удаляли при пониженном давлении, а остаток хроматографировали в колонке с силикагелем с метанолом в дихлорметане в качестве элюента. Полученные фракции соединили и очистили посредством обращенно-фазной хроматографии с ацетонитрилом в 0,1%-ной водной трифторуксусной кислоте в качестве элюента, и после приготовления гидрохлорида вышло чистое вещество, указанное в заголовке.
Т. пл. 50 - 60oC (размягчается).
Масс-спектр : FAB + ve 434[(M + H)+];
1H-ЯМР (DMCO-d6) δ : 1,86 (m, 2H), 2,55 (t, 2H), 2,62 (t, 2H), 2,88 (m, 2H), 2,91 (m, 2H), 2,95 (m, 2H), 3,28 (t, 2H), 3,58 (t, 2H), 3,85 (t, 2H), 6,78 (d, 1H), 6,88 (d, 1H), 7,85 (t, 1H), 7,96 (d, 1H), 8,45 (t, 1H), 8,78 (d, 1H), 9,17 (brs, 2H), 10,17 (brs, 1H), 11,78 (brs, 1H).
Анализ: найдено, %: C 47,69; H 6,08; N 7,79; S 10,88; Cl 12,84,
C21H27N3O3S2.2HCl 1,5H2O
Вычилено, %: C 47,27; H 6,05; N 7,88; S 12,02; Cl 13,29.
Пример 18. 4-Гидрокси-7-[2-[2-[3-[2-(4-гидроксифенил)этокси]пропил-сульфонил] этиламино]этил]-1,3-бензотиазол-2(3H)-он, хлоргидрат.
а) 4-(2-Пропенилокси)этил-1-(фенилметокси)бензол.
К суспензии свободного от масла гидрида натрия (1,6 г) в сухом диметилформамиде при перемешивании в атмосфере азота порциями добавляют 2-[4-(фенилметокси)фенил] этанол. По окончании добавления смесь перемешивают в течение 4 ч, после чего по каплям добавляют пропенилбромид (7,98 г). Реакционную смесь перемешивают в течение ночи при комнатной температуре, затем при перемешивании выливают в воду (1,5 л). Смесь экстрагируют эфиром, экстракты промывают рассолом, сушат (MgSO4), фильтруют, фильтраты упаривают досуха, получают масло, выход 15,3 г (86%).
Масс-спектр (электронный удар): 268 (M).
Спектр ПМР (360 МГц, CDCl3) δ , м.д.: 2,85 (2H, т), 3,62 (2H, т), 4,0 (2H, д), 5,2 (2H, м), 5,9 (1H, м), 7,0 (4H, кв), 7,4 (5H, м).
b) 2-[3-[2-[4-(Фенилметокси)фенил]этокси]пропилтио] уксусная кислота.
Смесь продукта стадии а) (15,3 г) объединяют с меркаптоуксусной кислотой (5,82 г) и 2,2'-азобис(2-метилпропионитрила) (AlBN, 0,2 г) нагревают в открытой колбе при 60oC в течение 90 мин. Смесь охлаждают и очищают флэш-хроматографией на силикагеле, элюируя сначала изогексаном, содержащим эфир (20%), затем смесью изогексан - эфир (50:50), получают продукт в виде бесцветного масла, которое медленно кристаллизуется (15,6 г, 76%).
Масс-спектр (электронный удар): 360 (М).
Спектр ПМР (360 МГц, CDCl3) δ , м.д.: 1,85 (2H, м), 2,70 (2H, т), 2,80 (2H, т), 3,20 (2H, т), 3,51 (2H, т), 3,62 (2H, т), 5,03 (2H, с), 7,00 (4H, кв.), 7,40 (5H, м).
c) 3-[2-[4-(Фенилметокси)фенил]этокси]пропилсульфонилуксусная кислота.
К раствору сульфида со стадии b) (15,2 г) в метаноле (120 мл) при перемешивании при температуре 0oC небольшими порциями добавляют раствор OXONER (79 г) в воде (300 мл). Образовавшийся густой осадок перемешивают при комнатной температуре в течение 4 ч, добавляют дополнительное количество воды и смесь экстрагируют хлороформом. Экстракты промывают рассолом, сушат (MgSO4), фильтруют, фильтраты упаривают досуха, получают твердое вещество белого цвета, которое кристаллизуют из тулуола, получают названное соединение в виде белых игольчатых кристаллов (14,0 г, 84%) т. пл. 105 - 107oC.
Масс-спектр (электронный удар): 348 (M-CO2).
Спектр ПМР (360 МГц, CDCl3+DMCO) δ , м.д.: 2,10 (2H, м), 2,80 (2H, т), 3,30 (2H, т), 3,60 (2х2H, т), 3,95 (2H, с), 5,04 (2H, с), 7,00 (4H, кв.), 7,40 (5H, м).
d) 2-[3-[2-(4-Гидроксифенил)этокси]пропилсульфонил]уксусная кислота.
Смесь кислоты со стадии c) (5,0 г), 10% Pd/C (5,0 г) и формиата аммония (10,0 г) в этаноле (250 мл) перемешивают в течение 48 ч, катализатор отделяют фильтрованием, фильтрат упаривают досуха. Твердый остаток кристаллизуют из этанола, получают названное соединение в виде твердого вещества белого цвета (1,3 г, 34%), т.пл. 172 - 2oC.
Масс-спектр (бомбардировка быстрыми атомами): 301 (M-H).
Спектр ПМР (360 Мгц, DMCO-d6) δ , м.д.: 1,88 (2H, м), 2,67 (2H, т), 3,33 (2H, м), 3,45 (4H, м), 3,64 (2H, с), 6,84 (4H, кв.), 7,5 (1H, уш. с.).
e) N-[2-(4-Гидрокси-2-оксо-3H-1,3-бензотиазол-7-ил)этил]-2-[3- [2-(4-гидроксифенил)этокси]пропилсульфонил]ацетамид.
Раствор кислоты со стадии d) (5,4 г) в сухом диметилформамиде (100 мл) в атмосфере азота обрабатывают 1,1'-карбонилдиимидазолом (5,3 г) и смесь перемешивают в течение 90 мин, после чего добавляют бромгидрат 7-(2-аминоэтил)-4-гидрокси-1,3-бензотиазол-2(3H)-она (5,28 г), а затем триэтиламин (1,8 г). Смесь перемешивают при комнатной температуре в течение ночи и медленно при интенсивном перемешивании добавляют ее к смеси разбавленной соляной кислоты (200 мл) и эфира (100 мл). Эфирный слой отделяют, водную фазу фильтруют, фильтрат экстрагируют этилацетатом. Экстракты упаривают досуха, остаток объединяют с отфильтрованным твердым продуктом, растворяют в этаноле и обрабатывают активированным углем. Уголь и растворитель отделяют, остаток очищают быстрой хроматографией на силикагеле (элюент этилацетат - изогексан, 3: 1). После упаривания фракций получают соединение в виде твердого вещества белого цвета (2,5 г, 28%), т.пл. 183 - 4oC.
Масс-спектр (бомбардировка быстрыми атомами): 495 (M+H).
Спектр ПМР (360 Мгц, DMCO-d6) δ , м.д.: 1,91 (2H, м), 2,65 (4H, м), 3,30 (4H, м), 3,47 (4H, м), 4,04 (2H, с), 6,77 (2H, кв.), 6,83 (4H, кв.), 8,45 (1H, т), 9,15 (1H, с), 9,96 (1H, с), 11,61 (1H, с).
Элементный анализ: найдено, %: C 53,44, H 5,59, N 5,74, S 12,92. Брутто-формула C22H26N2O7S2. Вычислено, %: C 53,43, H 5,30, N 5,66, S 12,97.
f) 4-Гидрокси-7-[2-[2-[3-[2-(4-гидроксифенил)этокси-пропилсульфонил] -этиламино] этил]1,3-бензотиазол-2(3H)-он, хлоргидрат.
К амиду со стадии e) (0,49 г) в тетрагидрофуране (20 мл) в атмосфере азота при перемешивании добавляют 1 M раствор борана в тетрагидрофуране (3,5 мл) и кипятят в течение 120 мин. Смесь охлаждают, реакцию гасят добавлением метанола и нескольких капель концентрированной соляной кислоты. Смесь упаривают досуха, остаток очищают флэш-хроматографией на силикагеле (элюент хлороформ - этанол, 9:1), получают фракцию, которую затем очищают жидкостной хроматографией высокого давления (ВДЖХ). Полученный продукт кристаллизуют из разбавленной соляной кислоты, получают названное соединение в виде твердого вещества белого цвета (0,053 г, 10%), т. пл. 167 - 8oC.
Масс-спектр (ESILoop): 481 (M+H).
Спектр ПМР (360 Мгц, DMCO-d6) δ , м.д.: 1,91 (2H, м), 2,68 (2H, м), 2,90 (2H, м), 3,45 (6H, м), 6,82 (2H, кв.), 6,85 (4H, кв.) 9,50 (1H, с), 10,19 (1H, с), 11,80 (1H, с).
Пример 19. 4-Гидрокси-7-[2-[2-[3-[2-(4-метоксифенил)этокси]пропилсульфонил]этиламино] этил]-1,3-бензотиазол-2(3H)-он, хлоргидрат.
а) 2-[3-[2-(4-Метоксифенил)этокси]пропилсульфонил]этанол.
Данное соединение может быть получено, исходя из 2-(4-метоксифенил)этанола, с использованием 2-меркаптоэтанола в соответствии с приведенными выше методиками стадий a) и b) с последующим окислением промежуточного сульфида перекисью водорода в среде водного ацетонитрила.
Масс-спектр (ESI Loop: 303 (M+H).
Спектр ПМР (360 МГц, CDCl3) δ , м.д.: 2,08 (2H, м), 2,59 (1H, т), 2,82 (2H, т), 3,10 (2H, т), 3,14 (2H, т), 3,54 (2H, т), 3,61 (2H, т), 3,79 (3H, т), 4,07 (2H, кв.), 6,97 (4H, кв.).
b) 2-[3-[2-(4-Метоксифенил)этокси] пропилсульфонил] этиловый эфир бензойной кислоты.
Раствор продукта стадии a) (7,5 г) в метиленхлориде (70 мл) обрабатывают триэтиламином (4,2 мл), затем добавляют по каплям бензоилхлорид (3,5 мл). Смесь перемешивали в течение 2 ч, промывают разбавленной соляной кислотой, сушат (MgSO4), фильтруют, фильтрат упаривают досуха, получают названное соединение в виде масла (11,0 г).
Масс-спектр (бомбардировка быстрыми атомами): 407 (M+H).
c) 1-[3-[2-(4-Метоксифенил)этокси]пропилсульфонил]этилен.
Раствор эфира со стадии b) (11,0 г) в этилацетате (100 мл) обрабатывают диазабициклоундеканом (ДБУ, 7,0 мл). Через 2 ч добавляют дополнительно 1 мл ДбУ и смесь перемешивают в течение ночи. Реакционную массу промывают насыщенным водным раствором бикарбоната натрия, водой, рассолом и сушат (MgSO4), фильтруют, фильтрат упаривают досуха, получают масло, которое очищают быстрой хроматографией на силикагеле (элюент метиленхлорид - этилацетат, 10 : 1), получают названное соединение в виде масла (5,24 г).
Масс-спектр: 284 (М).
Спектр ПМР (360 Мгц, CDCl3) δ , м.д: 2,01 (2H, м), 2,79 (2H, т), 3,02 (2H, м), 3,51 (2H, т), 3,58 (2H, т), 3,79 (3H, т), 5,95 (1H, д), 6,14 (1H, д), 6,37 (1H, м), 6,7 (4H, кв).
d) 4-Гидрокси-7-[2-[2-[3-[-2-(4-метоксифенил)этокси] пропилсульфонил] этиламино] этил]-1,3-бензотиазол-2(3H)-он, хлоргидрат.
Продукт со стадии c) (5,24 г) в метаноле (10 мл) обрабатывают хлоргидратом 7-(2-аминоэтил)-4-гидрокси-1,3-бензотиазол-2(3H)-она (5,0 г) и триэтиламином (2,85 мл) и кипятят в течение 2 ч, после чего охлаждают и добавляют соляную кислоту (0,4 мл). Смесь упаривают, остаток очищают флэш-хроматографией на силикагеле (элюент метиленхлорид - метанол, 20 : 1). Продукт последовательно кристаллизуют из смеси метанол - HCl, метанола и воды, получают названное соединение (0,474 г), т.пл. 226oC.
Масс-спектр (ESI ацетат аммония): 481 (M+H), 479 (M-H).
Спектр ПМР (360 Мгц, DMCO-d6) δ , м.д: 1,90 (2H, м), 2,74 (2H, т), 3,84 (2H, т), 3,16 (2H, т), 3,22 (2H, т), 3,36 (2H, т), 3,39 (2H, т), 3,55 (4H, т), 3,70 (3H, с), 6,76 (1H, д), 6,85 (3H, м), 7,14 (2H, д), 9,02 (2H, уш.с. ), 10,14 (1H, с), 11,77 (1H, с).
Элементный анализ: найдено,%: С 49,22, H 5,75, N 5,44, S 11,44, Cl 6,64, брутто-формула: C23H30N2O6S2. HCl с 2,4 моль воды, вычислено,%: C 49,26, H 6,42, N 4,99, S 11,42, Cl 6,54.
Получение фармосоставов.
Сухая порошкообразная готовая препаративная форма, содержащая смесь соединения формулы I и разбавителя, например лактозы, где разбавитель присутствует в количестве до 99% от массы готовой препаративной формы, например 50 - 95% или 75 - 95% от массы готовой препаративной формы.
Водный раствор готовой препаративной формы может содержать просто водный или солевой раствор, в котором концентрация соединения находится в интервале 1,5 мкг/мл - 3 мг/мл, предпочтительно 1,5 мкг/мл - 1,5 мг/мл, например 0,5 мг/мл - 1,0 мг/мл. pH раствора можно установить при помощи HCl, например до pH около 3,5.
Примеры фармацевтических составов.
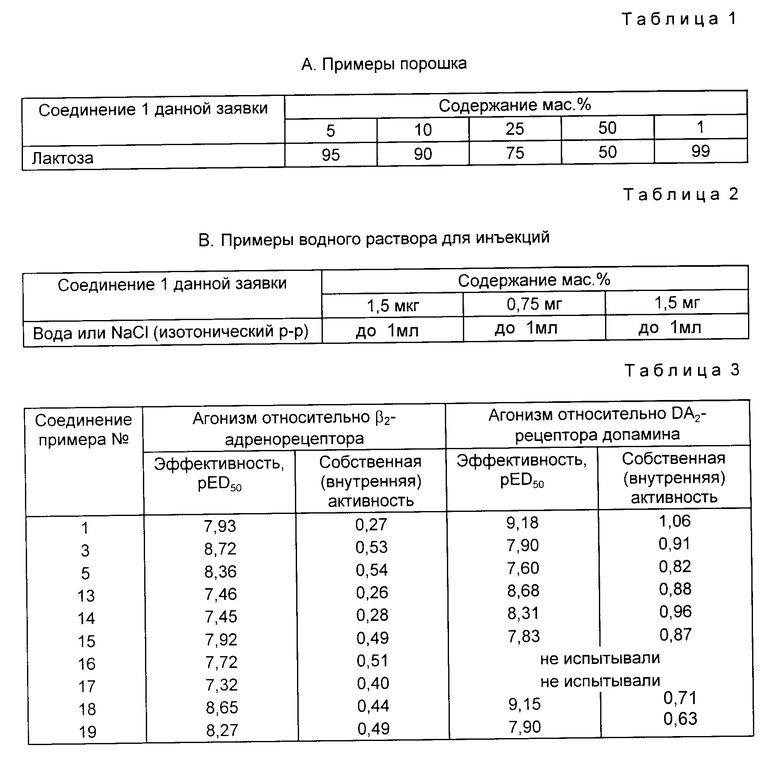
Соединение, полученное по любому из примеров 1 - 13, смешивают с наполнителем - лактозой и тонко измельчают с получением готовой формы окончательного состава, указанного в табл. 1. Альтернативно до смешения активного компонента и носителя, каждый из них также тонко измельчают.
Заранее определенное количество соединения по примерам 1 - 13 (например 150 мг) растворяют в воде для инъекций (100 мл), доводят до pH 3,5 с помощью HCl, фильтруют, ампулируют и стерилизуют (см. табл. 2).
В табл. 3 и далее по тексту приведены данные по биологической активности, касающиеся измерения агонизма к β2 -андренорецептору и агонизма к DA2-рецептору допамина.
Методика: бета2-рецепторный анализ длительно действующих соединений на изолированном препарате трахеи морских свинок.
Введение: трахеи морских свинок содержат бета-адренорецепторы, стимуляция которых вызывает релаксацию гладкой мышцы. Эту изолированную тканевую систему используют для получения количественной информации по новым продолжительно действующим бета2-лигандам.
Изолированные трахеальные кольца морских свинок. Самцов морских свинок-альбиносов Dunkin-Hartley умерщвляли цервикальным смещением и удаляли целые трахеи. После очистки от соседних соединительных тканей трахею разрезали на шесть кольцевых сегментов, каждый шириной трех хрящевых пучков, и затем суспендировали в 20 мл ванной для органов, содержащей раствор Кребса.
Суспензию сохраняли при 37oC и непрерывно газировали кислородом, содержащим 5% CO2. В раствор Кребса добавляли индометацин (2,8 мкМ), кокаин (30 мкМ), кортикостерон (10 мкМ), аскорбат (1 мМ), CGP20712A (1 мкМ) и фентоламин (3 мкМ): индометацин для предотвращения развития тонуса гладких мышц вследствие синтеза продуктов циклооксигеназы, кокаин и кортикостерон для ингибирования процессов поглощения 1 и поглощения 2 соответственно, аскорбат для предотвращения окисления катехоламина и CGP20712A и фентоламин для того, чтобы избежать любых осложняющих влияний активации бета1- и альфа-адренорецепторов соответственно. Трахеальные кольца находились в подвешенном состоянии между двумя проволочными крючкам (из вольфрама или нержавеющей стали), один из них был присоединен к датчику изометрической силы Ormed Beam, другой - к стационарной опоре в ванне для органа. Изменения в изометрической силе регистрировали на 2-канальных планшетных самописцах Advance Bryans.
Протоколы эксперимента. В начале каждого эксперимента к ткани прилагали силу в 1,0 г и эту силу восстанавливали после периода уравновешивания в 60 мин до тех пор, пока она не оставалась постоянной.
Ткани затем сокращали 3 мкМ метахолина, являющегося агонистом мускаринового рецептора. После того, как получали устойчивую ответную реакцию, для каждой ткани строили кумулятивную кривую (0,5 log единичного приращения): концентрация изопреналина - эффект (E/[A]). После промывания ткни оставляли на 60 мин и затем снова сокращали 3 мкМ метахолина. Когда достигали устойчивую ответную реакцию, добавляли 10 мкМ испытуемого соединения или наполнителя (тот факт, что это максимально эффективная концентрация, определяли в отдельных экспериментах). Когда достигали устойчивой релаксации на испытуемое соединение (или после 40 мин, если релаксацию не наблюдали), строили E/[A] -кривую изопреналина. Ответные реакции регистрировали как релаксацию в процентах сокращения, индуцированного метахолином.
Допуская, что соединение вызывает релаксацию и что добавленная концентрация максимально эффективна, можно рассчитать собственную (внутреннюю) активность (α) и эффективность (τ) агониста. Внутренняя активность является просто ответной реакцией, вызванной испытуемым соединением, деленной на максимальную релаксацию, вызванную изопреналином. Эффективность можно оценить, допуская, что изопреналин действует как полный агонист в этой системе, и таким образом используя его для определения параметров оперативных моделей Em и n (Leff et al., 1989). Эти параметры можно затем использовать для проведения сравнительного анализа испытуемого соединения, позволяющего оценить его эффективность.
Установленное сродство (рКА) испытуемого агониста получают, исходя из сдвига вправо контрольной изопреналиновой кривой, который получается в присутствии испытуемого агониста, т.е. уровень концентраций, позволяющий установить сродство агониста как в анализе Schild.
Получив установленное сродство и эффективность испытуемого соединения, можно установить его биологическую активность (Эффективность) (p[A50]).
Пример. Изопреналин (стандартный агонист).
p[A50] = p[ED50] = 7,14
εm= 92,0 %
n = 1,18
Испытуемое соединение (сальметерол).
Отклик на 10-5М = 41,3%.
Следовательно внутренняя активность (α) = 41,3/92,0 = 0,44. Поскольку α /Em = Гn/1 + Гn, то 0,44 = Г1,18/1 + Г1,18, таким образом Г (эффективность) = 0,82.
Изопреналиновый контроль p[A50] = 7,1.
Изопреналин в присутствии 10-5М сальметерола p[A50] = 5,1.
Таким образом соотношение концентраций (CR) = 100 и поскольку CR - 1 = [A]KA, то pKA = 7,0
Получив Г и KA, можно установить активность (p[A50]) испытуемого соединения
[A50] = KA/(2+Гn)1/n-1)).
Таким образом используя вышеуказанные значения, получаем p[A50] = 7,14.
DA2-рецепторный анализ на препарате изолированной ушной артерии кролика.
Введение. Препарат ушной артерии кролика содержит пресинаптические допаминовые рецепторы (DA2), стимулирование которых ингибирирует выделение норадреналина из симпатических нейронов. Эту изолированную тканевую систему используют для получения количественной информации (оценки сродства и эффективности) по новым DA2-лигандам.
Кроличья изолированная стимулированная на участке ушная артерия.
Самцов кроликов NZV (2,5 - 3,0 кг) умерщвляли внутривенной инъекцией пентобарбитон-натрия (60 мг/кг). Уши удаляли и проксимальную часть артерии среднего уха обнажали и канюлировали, используя полипропиленовую канюлю (внешний диаметр 0,75 мм). После удаления артерию очищали от прилипшей соединительной ткани и получали 6 колец (по 3 от каждой артерии) шириной 5 мм, сохраняя поверхность кругового слоя гладких мышц. Ткани устанавливали на тонких крючках из вольфрамовой проволоки (диаметром 0,25 мм) в ваннах для органов на 20 мл, содержащих раствор Кребса следующего состава (мМ : NaCl 117,56; NaHCO3 25,00; KCl 5,36; NaH2PO4 0,89; MgSO1 1,18; глюкоза 11,10 и CaCL2 2,55. В раствор Кребса для блокирования нейронального поглощения и β -рецепторов включали кокаин (30 мкМ) и пропанолол (1 мкМ). Этот раствор выдержали при 37oC и непрерывно газировали смесью 95%-ной O2 и 5%-ной CO2. Верхний проволочный крючок был присоединен к Ormed-датчику силового смещения, нижний крючок был присоединен к стационарной опоре в ванной. Сокращения тканей вызывали стимуляцией участка в течение 10 с каждые 2 мин. Используемые параметры стимуляции были следующие: продолжительно 0,5 мс, 5 Гц, 15 В. Изменения в изометрической силе регистрировали на планшетах-самописцах Advance Bryans AB500.
Протоколы эксперимента. Общий: в начале каждого эксперимента к каждой ткани прилагали силу в 1,0 г. Эту силу восстанавливали в течение периода стабилизации 60 мин до тех пор, пока она не оставалась постоянной. В конце этого периода начинали электрическую стимуляцию. Воспроизводимые ответные реакции на стимуляцию устанавливали приблизительно после 60 мин. Кривые концентрация агониста - эффект (E[a]) были построены кумулятивными добавлениями агониста при 0,5 log10 единичных приращений. Ответные реакции регистрировали как выраженное в процентах ингибирование стимулированного подергивания.
Количественная оценка агониста. В качестве стандартного агониста принимали 58075AB (6,7 ADTN). После того, как ответная реакция на электрическую стимуляцию становилась стабильной, каждую ткань подвергали действию дозы (300 мкМ) 58075AB, которое можно наблюдать визуально. После достижения максимальной ответной реакции агонист вымывали. Затем строили кривые E/[A] для испытуемых соединений. Ответные реакции соединений, которые вызывают агонизм, выражают как процент ответной реакции на дозу, действие которой можно наблюдать визуально. Путем использования высокой частоты (5 Гц) стимуляции возможно идентифицировать соединения с меньшей эффективностью, чем 58075AB.
Величина относительно 58075AB указывает внутреннюю активность соединений при том, что полагают, что 58075AB имеет величину 1. Для соединения с величинами, значительно меньшими чем 1, возможно рассчитать величины эффективности. Величина p[ED50], представляет показатель активности агониста. Он является отрицательным логарифмом концентрации агониста, которая вызывает ответную реакцию, соответствующую половине максимального ответа. Для соединений, которые имеют величины, значительно меньшие чем 1, возможно рассчитать величины аффинности (сродства) из измеренных величин p[ED50] и установленных величин эффективности. Эти величины сродства показаны как pKa (отрицательный логарифм концентрации агониста, которая связывает половину рецепторов).
Соединения, которые не демонстрируют агонизм, исследовали в качестве антагонистов, инкубируя ткани с наиболее высокой возможной концентрацией, и затем строили E/[A]-кривые 58075AB E/[A]. Степень смещения вправо этих кривых 58075AB по сравнению с контрольными кривыми 58075AB позволяет установить аффинность (сродство) испытуемых соединений. Такие оценки аффинности приводятся в виде величин pA2 (отрицательный логарифм концентрации антагониста, которая вызывает 2-кратное смещение вправо контрольной кривой E[A]).
Сравнительное изучение антагонистов. (-)-Сульпирид выбирали в качестве стандартного антагониста DA2-рецептора, чтобы подтвердить, что агонизм, наблюдаемый с испытуемыми соединениями, медиирован DA2 -рецептором. Его величина константы равновесной диссоциации (Кв) составляет приблизительно 10 нМ. В экспериментах, использующих этот антагонист, E/[A] -кривые антагониста строили за 60 мин до инкубирования и через 60 мин после инкубирования с 100 нМ (-)-сульпирида. Для селективных DA2-агонистов эта концентрация должна вызывать 11-кратное параллельное смещение вправо контрольной кривой. Там, где было возможно рассчитать эти смещения, они даются как отношения концентраций (CR). Если полностью определенные E/[A]-кривые не могут быть получены после обработки (-)-сульпиридом, агонизм описывается только как (-)-сульпирид чувствительный или (-)-сульпирид нечувствительный.
Использование: в химии гетероциклических соединений, проявляющих агонистическую активность в отношении β2-адренорецепторов. Сущность изобретения: производные 7-(2-аминоэтил)бензтиазолона формулы I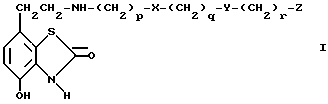
где X и Y независимо означают -S(O)n- или -O-, n означает 0,1 или 2, p, q и r независимо означают 2 или 3, Z означает фенил с возможным замещением галогеном, -OR1, NO2 или NH2 или Z представляет собой 5- или 6-членный ненасыщенный гетероцикл, содержащий N или S, при этом R1 означает водород или алкил C1 - C6 или их фармацевтически приемлемые соли. Описаны способы их получения и содержащие их фармацевтические композиции, а также промежуточные соединения для их получения формулы Va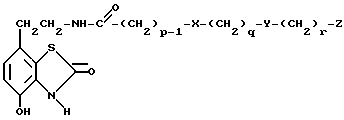
где p, q и r, X, Y имеют указанные значения, а Z - фенил, необязательно замещенный атомом галогена, NO2, NH2 или OR1, где R1 - H или C1 - C6-алкил. 4 с. и 7 з.п.ф-лы, 3 табл.
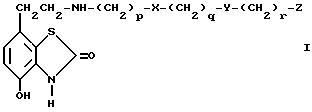
где заместители X и Y независимо друг от друга представляют собой S(O)n- или -O-;
n принимает значения 0, 1 или 2;
p, q и r независимо друг от друга принимают значения 2 или 3;
заместитель Z представляет собой фенил, необязательно замещенный атомом галогена, NO2, NH2 или OR1, где заместитель R1 - H или C1-C6-алкил, или заместитель Z представляет собой 5- или 6-членный ненасыщенный гетероцикл, содержащий N или S,
или их фармацевтически приемлемые соли.
4-гидрокси-7-[2-[3-[2-[2-фенилэтокси] этилсульфонил] пропиламин] этил]-1,3-бензотиазол-2(3H)-он,
4-гидрокси-7-[2-[2-[3-[2-фенилэтокси] пропокси] этиламин] этил]-1,3-бензотиазол-2(3H)-он,
4-гидрокси-7-[2-[3-[2-[2-фенилэтокси] этокси] пропиламин] этил]-1,3-бензотиазол-2(3H)-он,
4-гидрокси-7-[2-[2-[2-[2-фенилэтокси] этокси] этиламин] этил]-1,3-бензотиазол-2(3H)-он,
4-гидрокси-7-[2-[2-[3-[2-фенилэтокси]пропилтио]этиламин]этил]-1,3-бензотиазол-2(3H)-он,
4-гидрокси-7-[2-[2-[3-[2-фенилэтокси] пропилсульфонил] этиламин] этил]-1,3-бензотиазол-2(3H)-он,
4-гидрокси-7-[2-[3-[2-[2-фенилэтокси]этилтио]пропиламин]этил]-1,3-бензотиазол-2(3H)-он,
4-гидрокси-7-[2-[3-[2-[2-[2-аминофенил]этокси]этилсульфонил] пропиламин] этил]-1,3-бензотиазол-2-(3H)-он,
4-гидрокси-7-[2-[3-[2-[2-[4-нитрофенил] этокси]этилсульфонил]пропиламин] этил]-1,3-бензотиазол-2-(3H)-он,
7-[2-[2-[3-[2-[4-фторфенил] этокси]пропилсульфонил]этиламин] этил]-4-гидрокси-1,3-бензотиазол-2(3H)-он,
4-гидрокси-7-[2-[2-[3-[2-[2-тиенил] этокси] пропилтио] этиламин] этил-1,3-бензотиазол-2(3H)-он или
4-гидрокси-7-[2-[3-[2-[2-[2-пиридил] этокси] этилтио]пропиламин] этил]-1,3-бензотиазол-2(3H)-он,
или фармацевтически приемлемую соль любого из них.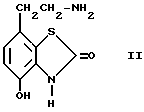
или его производного соединением общей формулы IV
O=CH-(CH2)p-1-X-(CH2)q-Y-(CH2)r-Z,
где p, q, r, X, Y и Z имеют значения, указанные в п.1,
в присутствии восстановителя.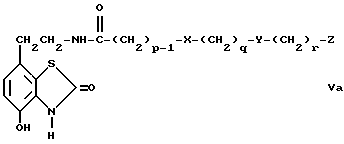
где p, q, r, X, Y и Z имеют значения, указанные в п.1.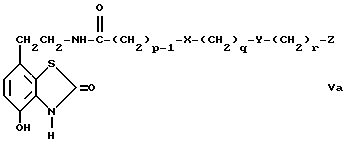
где p, q, r, X, Y имеют значения, указанные в п.1;
Z - фенил, необязательно замещенный атомом галогена, NO2, NH2 или OR1, где R1 - H или C1-C6-алкил.
| EP, 0174811, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| EP, 0478446, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
