Область изобретения
Изобретение относится к новым производным бензотиазолона, а именно к новым производным 7-(2-аминоэтил)-бензотиазолона, и к способу их получения, фармацевтическим композициям, содержащим их, и методам лечения, которые включают их использование. Новые соединения являются агонистами допаминового DА2-рецептора и агонистами β2-адренорецептора.
Производные бензотиазолона известны. Например, в международных патентных заявках, номера публикаций WO 92/08708 и WO 92/23385, описаны биологически активные амины, среди них биологически активные производные аминоэтилбензотиазолона, которые являются агонистами β2-адренорецептора и агонистами допаминового DA2-рецептора, и которые показаны при лечении обструктивных заболеваний дыхательных путей.
В WO 93/24473 описаны 7-(2-аминоэтил)-бензотиазолоновые соединения, которые имеют формулу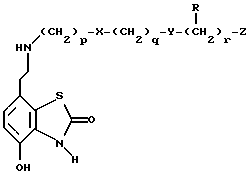
где X и Y независимо являются -S(O)n- или -О-, n равно 0, 1 или 2; p, q и r независимо равны 2 или 3; Z является фенилом, необязательно замещенным галогеном, OR1, NO2 или NR2R3; или Z является пяти- или шестичленным гетероциклом, содержащим N, O или S; и R1, R2 и R3 независимо являются водородом или C1-6 алкилом. Данные соединения являются агонистами β2-адренорецептора и агонистами допаминового DA2-рецептора и показаны при лечении обструктивных заболеваний дыхательных путей.
Настоящее изобретение относится к группе новых производных 7-(2-аминоэтил)-бензотиазолона, которые применимы в качестве агонистов допаминового DA2-рецептора и агонистов β2-адренорецептора.
Описание изобретения
Таким образом, в одном из аспектов данное изобретение относится к соединениям формулы I, включая их оптические изомеры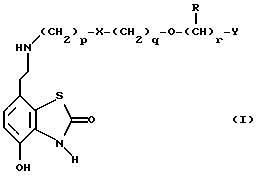
где X представляет -SO2NH- или -NHSO2-,
p, q и r независимо друг от друга равны 2 или 3,
Y представляет тиенил, необязательно замещенный алкилом или галогеном, или фенилтио-, или фенил, необязательно замещенный алкилом или галогеном; и
каждый R независимо представляет H или алкил и его фармацевтически приемлемые соли, сложные эфиры и амиды.
Данные соединения обладают фармакологической активностью. Они демонстрируют агонизм как к допаминовому DA2-pецептору, так и β2-адренорецептору. Они показывают незначительный агонизм или его отсутствие к α1-адренорецептору. Соединения обладают преимущественными длительностью действия и соотношением DA2/B2.
Предпочтительно, в формуле I q равно 2, г предпочтительно равно 2.
Когда Y является фенилом, замещенным алкилом, алкильная группа предпочтительно представляет C1-6, например, C1 или C2-группы, наиболее предпочтительным является метил.
Когда Y представляет фенил, замещенный галогеном, галогеновый заместитель предпочтительно является хлор- или фторзаместителем.
Предпочтительными соединениями данного изобретения являются соединения формулы I, в которых X является SO2NH, p равно 3, а q и r каждый равны 2. Другими предпочтительными соединениями являются соединения формулы I, в которых X представляет NHSO2, a и p, и q, и r - все равны 2.
Подходящие фармацевтически приемлемые соли соединений формулы I включают кислотно-аддитивные соли, полученные с неорганическими и органическими кислотами. Эти соединения могут также образовывать соли с подходящими основаниями. Примеры подходящих солей включают гидрохлорид, цитрат, D,L-лактат, полусульфат, полутатрат, D-глюконат, метансульфонат, p-толуолсульфонат, полуфумарат, бензоат, ксинафоат, полусукцинат, З-гидрокси-2-нафтоат, полуэмбонат, полумалеат, D-камфорсульфонат, 10-ундеканоат, манделат, нафтален-1-сульфонат, нафтален-2-сульфонат, 4-метоксибензоат, 4-хлорбензоат, 5-метилсалицилат, сахаринат, монометилсуберат, полусуберат и дифенилацетат.
Подходящие фармацевтически приемлемые сложные эфиры соединений формулы I включают фенилалкиловые и алкиловые сложные эфиры.
Подходящие амиды включают в себя незамещенные или моно- или дизамещенные алкил- или фениламиды.
Наиболее предпочтительными соединениями данного изобретения являются:
3-[2-(4-гидрокси-2-оксо-3Н-1,3-бензотиазол-7-ил)этиламино] -N- [2-(2-фенилэтокси)этил]пропансульфонамид;
N-[2-[2-(4-гидрокси-2-оксо-3Н-1,3-бензотиазол-7-ил)этиламино] этил]-2-(2-фенилэтокси)этансульфонамид;
3-[2-(4-гидрокси-2-оксо-3Н-1,3-бензотиазол-7-ил)этиламино] -N- [2-[2-(5-метил-2-тиенил)этокси]этил]пропансульфонамид;
N-[2-[2-(4-фторфенил)этокси] этил] -3-[2-(4-гидрокси-2-оксо-3Н- 1,3-бензотиазол-7-ил)этиламино]пропансульфонамид;
N-[2-[2-(4-хлорфенил)этокси] этил] -3-[2-(4-гидрокси-2-оксо- 5Н-1,3-бензотиазол-7-ил)этиламино]пропансульфонамид;
3-[2-(4-гидрокси-2-оксо-3H-1,3-бензотиазол-7-ил) этиламино] - N-[2-[2-(4-метилфенил)этокси]этил]пропансульфонамид;
(R, S)-3-[2-(4-гидрокси-2-оксо-3H-1,3-бензотиазол-7-ил)- этиламино]-N-[2-(2-фенил-1-пропокси)этил]пропансульфонамид;
3-[2-(4-гидрокси-2-оксо-3H-1,3-бензотиазол-7-ил)этиламино] -N-[2-[2-(2-метилфенил)этокси]этил]пропансульфонамид и
3-[2-(4-гидрокси-2-оксо-3H-1,3-бензотиазол-7-ил)этиламино] -N- [2-[2-фенилтиоэтокси)этил]пропансульфонамид;
предпочтительно в виде солей, и, наиболее предпочтительно, в виде гидрохлорида.
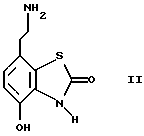
Данное изобретение также относится к способу получения соединений формулы I, который включает избирательное восстановительное алкилирование соединения формулы II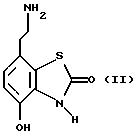
соединением формулы III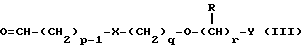
в которой p, q, r, R, X и Y имеют значения, определенные выше, в присутствии восстановителя.
Восстановителем может быть, например, водород в присутствии катализатора, такого как платина, оксид платины, палладий, оксид палладия, никель Ренея или родий, на носителе, например, древесном угле, с использованием спирта, например, этанола или сложного эфира, например, этилацетата или простого эфира, например, тетрагидрофурана или воды в качестве растворителя для реакции или смеси растворителей, при нормальных или повышенных температуре и давлении. Предпочтительной температурой является комнатная температура. Предпочтительное давление 1-3 атмосферы. Альтернативным восстановителем может быть боргидрид натрия или гидрид металла, например, цианоборогидрид натрия. Выбор подходящих растворителей для использования с гидридным восстановителем будет зависеть от конкретного гидрида, используемого в данном случае, что хорошо известно специалистам в данной области. Подходящие растворители включают спирты, например, этанол или метанол.
В процессе этой реакции могут образовываться промежуточные иминовые соединения, которые могут быть восстановлены при описанных выше условиях с получением соединений формулы I.
Соединения формулы II можно получить известными методами, например, методом, описанным в J.Med.Chem., 1987, 30, 1116.
Альдегиды формулы III можно получить многими известными методами, per se. Например, диоксиды изотиазолидина (как в Примере 1в, например) могут быть восстановлены с помощью DIBAL в толуоле; ацетали (как в примере 2б, например) можно гидролизовать 70% водной уксусной кислотой; и сложные эфиры (как в примере 3г, например) можно восстановить DIBAL в толуоле. Конкретный синтез определенных предшественников соединений описан в примерах и может быть адаптирован для многих различных целей.
Альдегиды формулы III также можно получить из соответствующих спиртов частичным окислением, используя ДМСО, DCC и безводную фосфорную кислоту; или используя хлорхромат пиридиния или дихромат пиридиния.
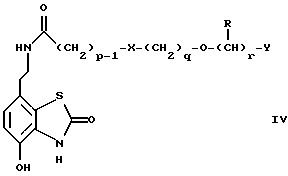
Данное изобретение также относится к дополнительному способу получения соединений формулы I, включающему избирательное восстановление соединения формулы IV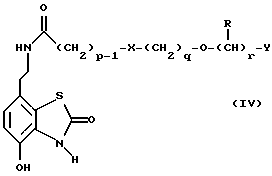
в которой p, q, r, R, X и Y имеют значения, указанные выше.
Подходящие восстановители включают электрофильные восстановители, например, диборан и алан (гидрид алюминия) или нуклеофильные восстановители, например, комплексные гидриды металлов, такие как натрий бис(2-метоксиэтокси)алюминий гидрид. Предпочтительным восстановителем является диборан. Растворитель должен быть инертным в условиях реакции. Апротонные растворители являются предпочтительными, например, тетрагидрофуран, диэтиловый эфир или 1,2-диметоксиэтан. Реакцию можно проводить при температуре от приблизительно 0 до приблизительно 100oC, предпочтительно при температуре кипения.
Соединения формулы IV можно получить реакцией амина формулы II с подходящей кислотой формулы V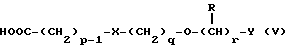
или с соответствующим хлорангидридом кислоты стандартными методами. Например, реакция может быть проведена в присутствии дициклогексилкарбодиимида с использованием метода Sheehan и Hess, J. Am. Chem. Soc., 1955, 77, 1067; или 1,1'- карбонилдиимидазола, как описано в Staab, Angew. Chem. Int. Ed. Engl. , 1962, 1, 351; или бромтрипирролидинфосфония гексафторфосфата в таком растворителе, как ДМФ, как описано в Примере 1д. Кислоты, которые необходимы для этой реакции, можно получить из соответствующих сложных эфиров гидролизом, используя гидроксид лития в водном метаноле по методу Примера 16. В примерах 1а, 2г, 3г, 4е, 5г, 6г, 7г, 8г и 9г описаны конкретные способы получения сложных эфиров, и эти способы можно адаптировать для получения других сложных эфиров для дальнейшего получения кислот, которые реагируют с аминами формулы II. Хлорангидриды кислот можно получить из кислот, например, реакцией с оксалилхлоридом или тионилхлоридом в толуоле при температуре в интервале от комнатной температуры до температуры кипения.
Соединения данного изобретения также можно получить некоторыми другими способами.
Одним из таких способов является алкилирование соединения формулы II или его соли, сложного эфира или амида алкилирующим агентом формулы VI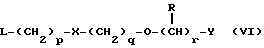
в которой p, q, r, R, X и Y имеют значения, указанные выше, a L обозначает легко уходящую группу, например, галоген, такой как хлор, бром, или йод, или алкил- или арилсульфонилокси группу, например, метансульфонилокси группу.
Реакцию можно проводить, например, в присутствии основания, например, неорганического основания, такого как карбонат натрия или калия, или органического основания, такого как триэтиламин, N,N'- диизопропилэтиламин или пиридин.
Реакцию можно проводить в растворителе, например, в простом эфире, таком как тетрагидрофуран или диоксан, в кетоне, таком как бутанон или метилизобутилкетон, в замещенном амиде, например, диметилформамиде, или в хлорированном углеводороде, например, хлороформе, при температуре в интервале от комнатной температуры до температуры кипения растворителя. Предпочтительно, реакцию проводят при комнатной температуре.
Алкилирующий агент формулы VI можно получить из соответствующего спирта (т. е. соединения, в котором L представляет группу ОН), методами, известными специалистам в данной области. Например, спирт может реагировать с галогенирующим агентом с получением соединения формулы VI, в котором L представляет атом галогена. Подходящие галогенирующие агенты включают, например, трифенилфосфинтетрагалогенметановые аддукты (традиционно получаемые in situ, например, реакцией трифенилфосфина и тетрабромида углерода). Реакция может проходить в присутствии растворителя, такого как ацетонитрил или хлорированный углеводород, такой как дихлорметан, например, при температуре в интервале от 0 до 30oC.
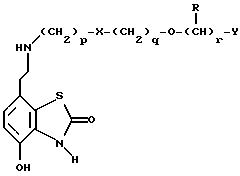
Другим способом является избирательное восстановление соединения формулы VII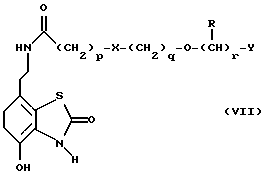
в которой p, q, r, R, X и Y имеют значения, указанные выше.
Подходящие восстановители включают электрофильные восстановители, например, диборан и алан (гидрид алюминия) или нуклеофильные восстановители, например, комплексные гидриды металлов, такие как натрий бис(2-метоксиэтокси)алюминий гидрид. Предпочтительным восстановителем является диборан. Растворитель должен быть инертным в условиях реакции. Апротонные растворители являются предпочтительными, например, тетрагидрофуран, диэтиловый эфир или 1,2-диметоксиэтан. Реакцию можно проводить при температуре от приблизительно 0 до приблизительно 100oC, предпочтительно, при температуре кипения.
Соединения формулы VII можно получить взаимодействием аминов и кислот или хлорангидридов кислот стандартными методами. Например, взаимодействие можно проводить в присутствии дициклогексилкарбодиимида или 1,1'-карбонилдиимидазола или бромтрипирролидинфосфония гексафторфосфата, как это описано выше в отношении соединений формулы IV. Амины, необходимые для реакции, можно получить реакцией соединений формулы VI, в которой L представляет легко уходящую группу, например, галоген, такой как хлор или бром, с фталимидом в присутствии основания. Полученные имиды затем можно обработать гидразингидратом в этаноле с получением соединений формулы VI, в которых уходящая группа заменена на аминогруппу.
В описанных выше процессах может возникнуть необходимость в защите каких-либо функциональных групп, например, гидрокси- или аминогрупп, присутствующих в исходных материалах. Подходящие защитные группы и методы их удаления описаны, например, в "Protective Groups in Organic Synthesis" T.W.Greene и P.G.W. Wuts, John Wiley and Sons Inc., 1991.
Другой способ получения соединений формулы I включает удаление защитных групп у соответствующего защищенного соединения формулы I, в котором одна или более функциональных групп защищены, и где желательно или необходимо превращение полученного соединения формулы I в его фармацевтически приемлемую соль, сложный эфир или амид либо наоборот.
Фармацевтически приемлемые соли можно получить, например, реакцией соединения формулы I с соответствующей кислотой в присутствии подходящего растворителя.
Фармацевтически приемлемые сложные эфиры соединений формулы I можно получить при помощи стандартных методов, например, реакцией этерификации или переэтерификации.
Фармацевтически приемлемые амиды соединений формулы I можно получить при помощи стандартных методов, например, реакцией соединения формулы I с кислотой или хлорангидридом кислоты.
Промежуточные соединения формулы IV являются новыми, и, таким образом, дополнительным аспектом данного изобретения являются соединения формулы IV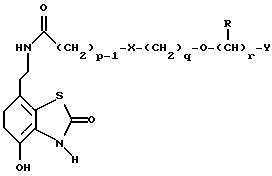
в которой p, q, r, R, X и Y имеют значения, указанные выше.
Промежуточные соединения формулы VII, определенные выше, также являются новыми и, таким образом, также представлены в данном изобретении.
Кроме того, данное изобретение включает альдегиды формулы III, которые определены выше и которые также являются новыми.
Помимо этого данное изобретение включает кислоты формулы V и соответствующие хлорангидриды кислот, которые являются новыми.
Соединения формулы I и их соли, сложные эфиры и амиды являются агонистами допаминового DA2-рецептора. Сродство к связыванию тестируемых соединений к сайтам связывания DA2-рецептора на мембранах гипофиза быка можно определить по замещению [3Н]-N-n-пропилнорапоморфина и [3H]-спиперона, соответственно, в присутствии или отсутствии негидролизуемого аналога ГТФ, D.R. Sibley, A. DeLean and I.Greese, Anterior Pituitary Dopamione Receptors, Demonstration of Interconvertible High and Low Affinity States of Dopamine DA2-receptor, J. Biol. Chem., 1982, 257(11), 6351-6361. Активность DA2-рецептора можно также продемонстрировать в функциональном скрининге на изолированной ушной артерии кролика, как описано Brown и O'Connor, Br.J. Pharmacol. , 1981, 73, 189P. Данные соединения являются также агонистами β2-адренорецептора. Эту активность можно показать на изолированной трахее морской свинки, как описано I.G.Dougall, D.Harper., D.M.Jackson and P.Left, Br.J.Pharmacol., 1991, 104, 1057. Активность в отношении α1- рецептора можно проанализировать, используя скрининг изолированной ушной артерии кролика, как описано ниже в Фармакологическом примере.
Соединения формулы I и их соли, сложные эфиры и амиды показаны для использования в лечении ряда заболеваний дыхательных путей, включая такие состояния, как астма, включая бронхиальную астму, аллергическую астму, наследственную астму (например, поздняя астма и повышенная чувствительность дыхательных путей); и бронхиты и подобное (см. , например, UK Patent N 2022078 и Br.J.Pharmacol., 1987, 24, 4983).
Соединения формулы I и их соли, сложные эфиры и амиды также показаны для использования в лечении различных других болезненных состояний, например, воспалительных и аллергических заболеваний кожи, рака, например, мелкоклеточный рак легких, застойная сердечная недостаточность и глаукома.
Термин "лечение", как он использован здесь, включает как профилактику, так и облегчение симптомов заболевания.
Таким образом, дополнительным аспектом данного изобретения является использование соединений формулы I или их фармацевтически приемлемых солей, сложных эфиров или амидов для лечения.
Далее данное изобретение включает использование соединений формулы I или их фармацевтически приемлемых солей, сложных эфиров или амидов для получения лекарственных средств для лечения обструктивных заболеваний дыхательных путей, особенно для лечения астмы или хронических бронхитов.
К тому же данное изобретение относится к способу лечения заболеваний дыхательных путей, который включает введение пациенту, страдающему таким заболеванием или предрасположенным к нему, терапевтически эффективного количества соединения формулы I или его фармацевтически приемлемых солей, сложных эфиров или амидов.
Обычная суточная единичная доза может составлять, например, 1 мкг - 10 мг для местного введения, предпочтительно 10-500 мкг, например, разделенная на два-три приема, или 10 мкг - 100 мг для перорального введения, предпочтительно 100 мкг - 10 мг, например, разделенная на два-три приема.
Соединения формулы I или их фармацевтически приемлемые соли, сложные эфиры или амиды можно использовать как таковые или в составе соответствующих фармацевтических композиций.
Введение можно осуществить при помощи ингаляции и другими путями, например, пероральным или внутривенным введением.
Назальное или внутрилегочное введение можно осуществить с помощью соответствующих ингаляционных устройств.
Например, дозирующие ингаляционные устройства можно использовать для введения соединения, диспергированного в подходящем пропелленте, и с добавлением или без дополнительных наполнителей, таких как этанол, поверхностно-активные вещества, смазывающие вещества и стабилизаторы.
Подходящие пропелленты включают углеводороды, хлорфторуглероды и гидрофторалканы или смеси любых таких пропеллентов. Особенно предпочтительными пропеллентами являются P134a и P227, каждый из которых можно использовать отдельно или в сочетании с другими пропеллентами и/или поверхностно-активными веществами, и/или другими наполнителями, например, в сочетании друг с другом.
Также можно использовать распыляемые водные суспензии или, предпочтительно, растворы, доведенные до соответствующих значений pH и/или тоничности, или без этого, в аэрозольных устройствах как для одноразового, так и для многократного введения.
Можно использовать порошковые ингаляторы для введения препарата, одного или в сочетании с фармацевтически приемлемым носителем, в последнем случае как в виде тонко измельченного порошка, так и в виде необходимой смеси. Порошковый ингалятор может быть с одной дозой или многодозовым, и в нем может использоваться порошок или капсула, содержащая порошок.
Дозирующий ингалятор, аэрозольный или порошковый ингаляторы хорошо известны, и доступно множество таких приспособлений.
Изобретение иллюстрируется, но не ограничивается каким-либо образом следующими примерами, в которых температура приведена в градусах Цельсия. При необходимости реакции проводят в инертной атмосфере азота или аргона. Когда необходимо, производили разделение с помощью препаративной ВЭЖХ, используя колонки Novapak, Bondapak и Hypersil (зарегистрированные товарные знаки), заполненные BDSC-18, двуокисью кремния с обращенной фазой. Флэш-хроматографию проводили, используя кремнезем Fisher Matrix 60, 35-70 микрон.
Пример 1
3-[2-(4-гидрокси-2-оксо-3H-1.3-бензотиазол-7- ил)этиламино] -N-[2-(2-фенилэтокси)этил]пропансульфонамида гидрохлорида)
а) Метил 3-[2-(2-фенилэтокси)этиламиносульфонил]пропаноат
2-(2-фенилэтокси)этанамин1 (1.95 г) перемешивают в дихлорметане при комнатной температуре. Добавляют триэтиламин (3.36 мл), затем метил-3-(хлорсульфонил)-пропаноат2 и смесь перемешивают в течение ночи при комнатной температуре. Смесь разводят дополнительным объемом дихлорметана, промывают разбавленной соляной кислотой, затем водой, затем сушат (MgSO4). Растворитель удаляют в вакууме, в результате чего получают бледно-желтое масло, которое затем очищают флэш-хроматографией (элюент - 0,1% этанол:дихлорметан) и получают соединение, названное в заголовке, в виде бледно-желтого масла (1.68 г).
Масс-спектр: FAB 316 (М+Н);
1H ЯМР (360 МГц, CDCl3) δ : 2.75 (2H, т), 2.85 (2H, т), 3.18-3.35 (4H, м), 3.46-3.57 (2H, м), 3.60-3.77 (5H, м), 4.44 (1H, шир.т), 7.12-7.35 (5H, м).
1Chem. Вег., 1964, 97, 510-519.
2J.Am. Chem.Soc., 1950, 72, 128-132.
б) 3-[2-(2-фенилэтокси)этиламиносульфонил]пропановая кислота
Продукт стадии а) (1.68 г) растворяют в этаноле (30 мл). Добавляют раствор гидроксида лития (0.45 г) в воде (30 мл) и смесь перемешивают в течение ночи при комнатной температуре. Добавляют воду и смесь промывают эфиром. Водный слой подкисляют разбавленной соляной кислотой и экстрагируют эфиром. Эфирный экстракт промывают водой, затем насыщенным солевым раствором, затем высушивают (MgSO4). Растворитель удаляют в вакууме с получением соединения заголовка в виде твердой белой массы (0.97 г), которую затем используют без дальнейшей очистки.
Т.пл. 80-82oC;
Масс-спектр: ESI 300 (M-H);
1H ЯМР (360 МГц, CDCl3) δ : 2.77-2.95 (4H, м), 3.20-3.47 (4H, м), 3.51-3.58 (2H, М), 3.69 (2H, т), 4.66 (1H, Т), 7.18-7.38 (5H, м).
в) 2-[2-(2-фенилэтокси)этил]-3-изотиазолидинон-1,1'- диоксид
Продукт стадии б) (34 г) растворяют в диметилформамиде (200 мл). К этому раствору, который перемешивают, добавляют 1,1'- карбонилдиимидазол (20.12 г) и смесь перемешивают 2 часа. Добавляют триэтиламин (15.7 мл) и смесь перемешивают при комнатной температуре 60 часов. Смесь выливают в разбавленную соляную кислоту и экстрагируют этилацетатом (3 раза). Объединенные органические экстракты промывают насыщенным раствором бикарбоната натрия, затем насыщенным солевым раствором, высушивают (MgSO4) и растворитель удаляют в вакууме с получением бледно-желтого масла, которое затем очищают флэш-хроматографией (элюент - 50% этилацетат:бензин) и получают соединение заголовка в виде масла (27.4 г).
Масс-спектр: ESI 301 (M+NH4);
1H ЯМР (360 МГц, CDCl3) δ : 2.87 (2H, т), 2.99 (2H, т), 3.51 (2H, т), 3.64-3.68 (4H, м), 3.76 (2H, т), 7.18-7.43 (5H, м).
г) 3-[2-(4-гидрокси-2-оксо-3H-1,3-бензотиазол-7-ил) этиламино] -N-[2-(2-фенилэтокси)этил]пропансульфонамида гидрохлорид
Продукт стадии в) (0.62 г) механически размешивают в толуоле (20 мл), затем охлаждают до -70oC. Добавляют гидрид диизобутилалюминия, в течение 15 мин поддерживая температуру ниже -58oC. Смесь перемешивают в течение 10 мин, после чего TCX показывает отсутствие исходного материала. Осторожно добавляют этилацетат (9 мл), поддерживая температуру ниже -60oC. Смеси дают нагреться до комнатной температуры и добавляют 10% раствор тартрата калия-натрия. После перемешивания в течение 1 часа смесь экстрагируют этилацетатом (3 раза). Объединенные органические экстракты высушивают (MgSO4), затем приблизительно 70% растворителя удаляют в вакууме. К смеси добавляют метанол (20 мл) и снова приблизительно 70% растворителя удаляют в вакууме, эту процедуру повторяют еще два раза. Этот раствор разбавляют метанолом (20 мл) и добавляют 7-(2-аминоэтил)-4-гидрокси-1,3- бензотиазол-2(3H)-она гидробромид (0.64 г). pH доводят до значения 4 при помощи ледяной уксусной кислоты. Добавляют цианоборгидрид натрия (0.14 г), затем сульфат натрия (50 мг) и смесь перемешивают в течение 72 часов. Смесь делают основной добавлением концентрированного водного раствора гидроксида аммония. Летучие продукты удаляют в вакууме и осадок очищают флэш-хроматографией (элюент - 10-25% метанол в хлороформе). Продукты затем очищают ВЭЖХ с обращенной фазой (элюент - 25% метанол в 0.1% водной трифторуксусной кислоте), в результате чего после превращения в соль соляной кислоты получают соединение заголовка в виде твердой белой массы (0.417 г).
Т.пл. 205-206oC;
Масс-спектр: FAB 408 (M+H);
1H ЯМР (360 МГц, d6 ДМСО) δ : 2.00 (2H, т), 2.73-2.92 (4H, м), 2.96-3.19 (8H, м), 3.44 (2H, т), 3.60 (2H, т), 6.75 (1H, д), 6.85 (1H, д), 7.13-7.35 (6H, м), 8.92 (2H, с), 10.41 (1H, с), 11.77 (1H, с).
Элементный анализ
Найдено: C, 51.48; H, 6.25; N, 8.41; S, 12.50%
Вычислено для C22H29N3O5S2•HCl: C, 51.20; H, 5.86; N, 8.14; S, 12.43%.
В альтернативном способе приведенные выше стадии а) и б) повторяют, затем проводят стадии д) и е), которые приведены ниже:
д) N-[2-(4-гидрокси-2-оксо-3H-1,3-бензотиазол-7-ил)-этил] - 3-[2-(2-фенилэтокси)этиламиносульфонил]пропанамид
Раствор продукта стадии б) (3,89 г), гидрохлорид 7- (2-аминоэтил)-4-гидрокси-1,3-бензотиазол-2(3H)-она (3.22 г), бромтрипирролидинфосфония гексафторфосфат, PyBroP, (6.32 г) в диметилформамиде (50 мл) охлаждают до -15oC и добавляют диизопропилэтиламин (9.0 мл) по каплям в течение более 5 мин. Раствор перемешивают при -15oC 5 минут, после чего позволяют нагреваться до 13oC в течение более 4 часов. После этого смесь добавляют по каплям в течение более 40 мин в разбавленную соляную кислоту (2N, 500 мл) и после перемешивания в течение выходных дней твердую массу собирают фильтрованием. Эту твердую массу высушивают в вакууме, в результате чего получают соединение подзаголовка (4.5 г).
Масс-спектр: FAB 492 (M-H).
е) 3-[2-(4-гидрокси-2-оксо-3H-1,3-бензотиазол-7-ил)этиламино] -N-[2-(2-фенилэтокси)этил]пропансульфонамида гидрохлорид
К раствору продукта стадии д) (0.28 г) в тетрагидрофуране (2 мл) добавляют борантетрагидрофуран (2.44 мл 1М раствора) в течение 5 мин. Смесь нагревают с обратным холодильником в течение 3 час и после охлаждения осторожно добавляют метанол (1 мл). Летучие продукты удаляют в вакууме, осадок повторно растворяют в метаноле (5 мл) и добавляют концентрированную соляную кислоту (1 мл). Летучие продукты опять удаляют в вакууме. Остаток разделяют между водой и этилацетатом, водный слой собирают и опять экстрагируют этилацетатом. Затем водный слой делают основным с помощью гидрокарбоната натрия и четыре раза экстрагируют хлороформом. Объединенные хлороформные экстракты сушат и летучие соединения удаляют в вакууме с получением после превращения в гидрохлорид соединения заголовка (0.070 г).
Пример 2
N-[2-[2-(4-гидрокси-2-оксо-3H-1.3-бензотиазол-7- ил)этиламино]этил]-2-(2-фенилэтокси)этансульфонамида гидрохлорид
а) 2-(2-фенилэтокси)этансульфонилхлорид
Перемешиваемую суспензию 2-(2-фенилэтокси)этантиола (1.0 г) в воде (40 мл) насыщают при 5-10oC хлором в течение 20 мин. Смесь промывают сильной струей жидкости под током азота, чтобы удалить избыток хлора. Затем смесь экстрагируют дихлорметаном (2 раза), объединенные органические экстракты промывают водой и высушивают (CaCl2). Растворитель удаляют в вакууме с получением масла, которое затем подвергают азеотропной перегонке с толуолом с получением соединения заголовка в виде желтого масла (1.36 г), которое затем используют без дальнейшей очистки.
Масс-спектр: EI 248/250 (M);
1H ЯМР (360 МГц, CDCl3) δ : 2.90 (2H, т), 3.74 (2H, т), 3.88 (2H, т), 3.99 (2H, т), 7.13-7.36 (5H, м).
б) N-(2.2-диметоксиэтил)-2-(2-фенилэтокси)этансульфонамид
Перемешиваемый раствор продукта стадии а) (1.0 г) в дихлорметане (20 мл) и пиридин (0.358 мл) обрабатывают, добавляя по каплям в течение 5 мин раствор диметилацеталя аминоацетальдегида (0.438 мл) в дихлорметане (5 мл). Смесь перемешивают при комнатной температуре в течение 2 суток. Смесь промывают водой, затем высушивают (CaCl2). Летучие продукты удаляют в вакууме, в результате чего получают оранжевое масло, которое затем очищают флэш-хроматографией (элюент - эфир) с получением соединения заголовка в виде желтого масла (0,42 г).
Масс-спектр: 335 (M+NH4);
1H ЯМР (360 МГц, CDCl3) δ : 2.90 (2H, т), 3.13 (2H, т), 3.26 (2H, т), 3.38 (6H, с), 3.73 (2H, т), 3.85 (2H, т), 4.38 (1H, т), 4.46 (1H, т), 7.14-7.35 (5H, м).
в) N-[2-[2-(4-гидрокси-2-оксо-3H-1,3-бензотиазол-7- ил)этиламино]этил] -2-(2-фенилэтокси)этансульфонамида гидрохлорид
Раствор продукта стадии б) (0.26 г) в 70% водной уксусной кислоте (5 мл) нагревают до 100oC в течение 2 часов, после чего TCX показала отсутствие исходного материала. Растворитель удаляют в вакууме. Остаток собирают в метаноле (10 мл) и к этому перемешиваемому раствору добавляют 7- (2-аминоэтил)- 4-гидрокси-1,3-бензотиазол-2(3H)-она гидробромид (0.238 г) и цианоборгидрид натрия (0.038 г), а затем уксусную кислоту (1 капля). Смесь перемешивают при комнатной температуре 24 часа. Смесь делают щелочной добавлением концентрированного водного раствора гидроксида аммония. Летучие продукты удаляют в вакууме и остаток очищают флэш-хроматографией (элюент - 17% этанол в дихлорметане) с получением светло-желтой смолы. Массу затем очищали ВЭЖХ с обращенной фазой (элюент - 30-45% ацетонитрил в 0,1% водной трифторуксусной кислоте) с получением после превращения в соль соляной кислоты соединения заголовка в виде белого порошка (0.080 г).
Масс-спектр: 466 (M+H);
1H ЯМР (360 МГц, d6 ДМСО) δ : 2.81-2.90 (4H, м), 3.04-3.09 (4H, шир. д), 3.26-3.28 (2H, м), 3.24 (2H+H2O), 3.63 (2H, с), 3.74 (2H, т), 6.78 (1H, д), 6.87 (1H, д), 7.19-7.30 (5H, м), 7.43 (1H, т), 9.10 (2H, с), 10.16 (1H, с), 11.78 (1H, с).
Элементный анализ
Найдено: C, 49.35; H, 5.69; N, 8.30; S, 14,94%
Вычислено для C21H27N3O5S2•HCl: C, 49.31; H, 5.67; N, 8.22; S, 12.52%
В альтернативном способе стадию (а) повторяют с последующим проведением стадий (г) - (ж), представленных ниже.
г) Метил-[2-(2-фенилэтокси)этилсульфониламино]ацетат
Суспензию гидрохлорида метилглицината (2.52 г) в дихлорметане (30 мл) перемешивают при -18oC и добавляют диизопропилэтиламин (8 мл) в течение 10 мин. К этой смеси добавляют по каплям продукт стадии (а) (2.64 г) в дихлорметане (10 мл) в течение 10 мин, поддерживая температуру ниже -5oC. Охлаждающую баню убирают и позволяют перемешиваемой смеси нагреться до комнатной температуры. По прошествии следующих 50 мин смесь промывают 5% водным гидросульфатом калия и высушивают (Na2SO4), летучие продукты удаляют в вакууме с получением соединения заголовка в виде коричневого масла (58.5 г).
Масс-спектр: FAB 302 (M+H);
1H ЯМР (360 МГц, CDCl3) δ : 2.90 (2H, т), 3.36 (2H, т), 3.72-3.85 (7H, м), 3.96 (2H, т), 4.70 (1H, шир. с), 7.20-7.31 (5H, м).
д) 2-[(2-фенилэтокси)этилсульфониламино]уксусная кислота
Раствор продукта стадии (г) (3.2 г) в метаноле (30 мл) охлаждают на ледяной бане и обрабатывают раствором гидрата гидроксида лития (1.06 г) в воде (7 мл) в течение 5 минут. Ледяную баню убирают и раствор перемешивают в течение 16 часов. Смесь подкисляют концентрированной соляной кислотой (3 мл) и концентрируют в вакууме до объема приблизительно 15 мл. Остаток смешивают с бикарбонатом натрия и экстрагируют эфиром. Затем водную фазу снова подкисляют насыщенным водным раствором гидросульфата калия и экстрагируют эфиром. Этот экстракт высушивают (MgSO4) и летучие продукты удаляют в вакууме с получением твердой массы. Далее продукт очищают перекристаллизацией из смеси дихлорэтана и толуола с получением соединения заголовка в виде массы кристаллического твердого вещества (1.57 г).
Масс-спектр: FAB 288 (M+H);
1H ЯМР (360 МГц, CDCl3) δ : 2.90 (2H, т), 3.32 (2H, т), 3.74-3.78 (4H, м), 3.89 (2H, т), 4.66 (1H, т), 7.20-7.33 (5H, м), 8.07 (1H, шир. с).
e) N-[2-(4-гидрокси-2-оксо-3H-1,3-бензотиазол-7-ил)-этил] - 2-[2-(2-фенилэтокси)этилсульфониламино]ацетамид
Раствор продукта стадии (д) (70 г) в диметилформамиде (605 мл) охлаждают до -10oC и добавляют 1,1'-карбомилдиимидазол (39.5 г). Смесь перемешивают 119 минут, затем добавляют 7-(2-аминоэтил)-4-гидрокси-1,3-бензотиазол-2(3H)-она гидрохлорид (68.87 г), а затем по каплям добавляют триэтиламин (34 мл) в течение 3 минут, поддерживая температуру на уровне -10oC. После следующих 10 минут при температуре -10oC смеси дают нагреться до комнатной температуры и перемешивают в течение 24 часов. Эту смесь затем по каплям добавляют в разбавленную соляную кислоту (2N, 1870 мл) и водную смесь экстрагируют этилацетатом. Затем органические продукты промывают разбавленной соляной кислотой, водным бикарбонатом натрия и затем сушат (MgSO4). Летучие продукты удаляют в вакууме с получением соединения заголовка в виде пены (92.21 г).
Масс-спектр: FAB 480 (M+H);
1H ЯМР (360 МГц, d6 ДМСО) δ : 2.61 (2H, т), 2.81 (2H, т), 3.29-3.32 (4H, м, перекрывающийся с водой), 3.56 (2H, с), 3.61 (2H, т), 3.74 (2H, т), 6.70 (1H, д), 6.80 (1H, д), 7.16-7.53 (5H, м), 8.03 (1H, шир. т).
ж) N-[2-[2-(4-гидрокси-2-оксо-3H-1,3-бензотиазол-7- ил)этиламино]этил] -2-(2-фенилэтокси)этансульфонамида гидрохлорид
Раствор продукта стадии (е) (38.7 г) в тетрагидрофуране (200 мл) охлаждают на бане ацетон/лед и добавляют борогидрид лития (727 мл, 2М раствор в тетрагидрофуране) в течение 30 минут. Через 15 минут охлаждающую баню убирают и добавляют триметилсилилхлорид (205 мл) и перемешивают в течение 162 часов при комнатной температуре. Смесь охлаждают до -20oC и осторожно добавляют метанол. После этого добавляют еще 50 мл метанола, насыщенного хлористым водородом, и смеси позволяют нагреться до комнатной температуры. Один час спустя смесь нагревают с обратным холодильником в течение 30 минут, затем охлаждают и летучие продукты удаляют в вакууме. Добавляют воду (400 мл), колбу энергично встряхивают и после охлаждения на бане ацетон/лед в течение 30 минут воду декантируют. Добавляют дополнительное количество воды (100 мл), процесс повторяют. Полутвердую оставшуюся массу затем растворяют в горячем этаноле (40 мл), обрабатывают активированным углем (3 г) и перемешивают в течение 1 часа. Смесь фильтруют и растворитель удаляют в вакууме. Остаток повторно растворяют в горячем этаноле (80 мл) и оставляют стоять при комнатной температуре на 16 часов. Затем выпавшие кристаллы разрыхляют и разбивают шпателем, взбалтывают и оставляют стоять еще на 16 часов. Затем твердое вещество собирают фильтрованием и промывают этанолом, эфиром, затем сушат в вакууме с получением соединения заголовка (23 г).
Пример 3
3-[2-(4-гидрокси-2-оксо-3H-1,3-бензотиазол-7- ил)этиламино] -N-[2-[2-(5-метил-2-тиенил)этокси]этил] пропансульфонамида гидрохлорид
а) 2-[2-(5-метил-2-тиенил)этокси]уксусная кислота
Гидрид натрия (0.622 г) суспендируют в безводном диметилформамиде (5 мл), обрабатывают, добавляя по каплям раствор 2-(5-метил-2-тиенил)этанола (1.0 г) в безводном диметилформамиде (5 мл). Смесь перемешивают при комнатной температуре в атмосфере азота в течение 2 часов, затем добавляют раствор хлоруксусной кислоты (0.664 г) в безводном диметилформамиде (5 мл). Смесь механически перемешивают при комнатной температуре в течение ночи. Летучие продукты удаляют в вакууме, остаток гасят водой и экстрагируют этилацетатом. pH водного слоя доводят до значения 2, используя разбавленную соляную кислоту, и затем экстрагируют этилацетатом. Этот экстракт промывают водой, насыщенным солевым раствором, затем сушат (MgSO4). Растворитель удаляют в вакууме с получением коричневого масла (1.51 г), которое затем используют без дальнейшей очистки.
Масс-спектр: 200 (М);
1H ЯМР (360 МГц, CDCl3) δ : 2.45 (3H, с), 3.05-3.09 (2H, т), 3.75-3.83 (2H, т), 4.17 (2H, с), 6.56 (1H, д), 6.63 (1H, д).
б) 2-[2-(5-метил-2-тиенил)этокси]ацетамид
Продукт стадии а) (6.48 г) растворяют в толуоле (55 мл) и добавляют по каплям оксалилхлорид (2.877 мл) при комнатной температуре в атмосфере азота. Добавляют каплю диметилформамида и смесь перемешивают 3 часа, после чего TCX показала отсутствие исходного материала. Летучие продукты удаляют в вакууме с получением коричневого масла (6.7 г), которое затем по каплям добавляют к перемешиваемому концентрированному раствору гидроксида аммония (50 мл) при 0oC. Смеси позволяют нагреться до комнатной температуры и перемешивают в течение 4 час. В осадок выпадает коричневое твердое вещество. Это твердое вещество собирают фильтрованием и промывают водой, в результате чего получают соединение заголовка (2.02 г).
Масс-спектр: 200 (М+Н);
1H ЯМР (360 МГц, CDCl3) δ : 2.45 (3H, с), 3.04 (2H, т), 3.73 (2H, т), 3.97 (2H, с), 5.61 (2H, шир. с), 6.57 (1H, д), 6.63 (1H, д).
в) 2-[2-(5-метил-2-тиенил)этокси]этанамин
Раствор боран-тетрагидрофурана (1.0 М в ТГФ, 21.7 мл) по каплям добавляют в перемешиваемый раствор продукта стадии б) (1.25 г) в безводном тетрагидрофуране (100 мл). Реакционную смесь нагревают с обратным холодильником в атмосфере инертного газа в течение 5 часов. Реакционную смесь охлаждают и осторожно добавляют метанол (10 мл). Растворители удаляют в вакууме и остаток растворяют в метаноле (100 мл), к которому добавляют концентрированную соляную кислоту (удельный вес 1.18, 0.45 мл). Этот раствор нагревали с обратным холодильником в течение 15 мин, затем растворитель удаляли в вакууме. Остаток очищали флэш-хроматографией (элюент - дихлорметан:5% метанол) с получением названного в подзаголовке соединения в виде твердой белой массы (0.916 г).
Масс-спектр: 186 (M+H);
1H ЯМР (360 МГц, CDCl3) δ : 2.45 (3H, с), 3.04 (2H, т), 3.73 (2H, т), 3.97 (2H, с), 5.61 (2H, шир.с), 6.57 (1H, д), 6.63 (1H, д).
г) Метил 3-[2-[2-(5-метил-2-тиенил)этокси]этиламиносульфонил] пропаноат
Продукт стадии в) в форме гидрохлорида (1.50 г) перемешивают в дихлорметане (25 мл) в атмосфере азота. Добавляют триэтиламин (2.21 мл), затем метил-3-(хлорсульфонил)пропаноат (1.18 г). Смесь механически перемешивают при комнатной температуре в течение ночи. Раствор разбавляют дополнительным объемом дихлорметана и органические продукты промывают разбавленной соляной кислотой, затем водой, затем высушивают (MgSO4). Смесь фильтруют и летучие продукты удаляют в вакууме с получением соединения заголовка в виде масла (1.4 г).
Масс-спектр: 336 (M+H);
1H ЯМР (360 МГц, CDCl3) δ : 2.38 (3H, с), 2.80 (2H, т), 3.00 (2H, т), 3.26-3.36 (4H, м), 3.56 (2H, т), 3.64 (2H, т), 3.72 (3H, с), 4.71 (1H, т), 6.57 (1H, д), 6.60 (1H, д).
д) 3-[2-(4-гидрокси-2-оксо-3H-1,3-бензотиазол-7-ил) этиламино] -N-[2-[2-(5-метил-2-тиенил)этокси]этил]пропансульфонамида гидрохлорид
Продукт стадии г) (0.60 г) растворяют в толуоле (30 мл) в атмосфере азота и охлаждают до -78oC. По каплям добавляют диизобутилалюминийгидрид (1.5 М раствор в толуоле, 1.78 мл) и смесь выдерживают при температуре -78oC 10 мин. Реакцию
останавливают добавлением этилацетата, затем 10% водным раствором тартрата калия натрия. Смесь нагревают до комнатной температуры и после перемешивания в течение 1 часа экстрагируют толуолом (3 раза). Объединенные органические экстракты промывают водой, сушат (MgSO4), затем приблизительно 70% растворителя удаляют в вакууме. К смеси добавляют метанол (20 мл) и опять приблизительно 70% растворителя удаляют в вакууме, эту процедуру повторяют еще два раза. Этот раствор разбавляют метанолом (20 мл) и добавляют 7-(2-амино-этил)-4-гидрокси-1,3-бензотиазол-2 (3H)-она гидрохлорид (0.516 г). pH доводят до 4 при помощи ледяной уксусной кислоты. Добавляют цианоборгидрид натрия (0.114 г) и смесь перемешивают в течение 2 часов в атмосфере азота. Смесь делают основной добавлением концентрированного водного раствора гидроксида аммония. Летучие продукты удаляют в вакууме и остаток очищают флэш-хроматографией (элюент - 15-20% метанол:дихлорметан). Продукт затем дополнительно очищают ВЭЖХ с обращенной фазой (элюент - 25-85% метанол в 0.1% водной трифторуксусной кислоте) с получением после превращения в соль соляной кислоты соединения заголовка в виде твердого белого вещества (0.12 г).
Т.пл. 210-212oC;
Масс-спектр: FAB 500 (М+Н);
1H ЯМР (360 МГц, d6 ДМСО) δ : 2.00 (2H, кв), 2.37 (3H, с), 2.85 (2H, т), 2.93 (2H, т), 3.01-3.17 (8H, м), 3.46 (2H, т), 3.58 (2H, т), 6.60 (1H, д), 6.65 (1H, д), 6.75-6.88 (2H, м), 7.31 (1H, т), 8.93 (2H, с), 10.15 (1H, с), 11.7 (1H, шир. с).
Пример 4
N-[2-[2-(4-фторфенил)этокси] этил] -3-[2-(4- гидрокси-2-оксо-3H-1,3-бензотиазол-7-ил)этиламино] пропансульфонамида гидрохлорид
а) т-бутил-3-[2-(4-фторфенил)этокси]пропаноат
2-(4-фторфенил)этанол (20.5 г) и Тритон-B (2.4 мл, 40% в метаноле) смешивают друг с другом и метанол удаляют в вакууме. Добавляют трет-бутилакрилат (21.38 мл) и раствор нагревают при температуре 50oC в течение 2 часов, затем перемешивают при комнатной температуре в течение ночи. Раствор разбавляют водой и экстрагируют диэтиловым эфиром. Объединенные органические экстракты промывают насыщенным раствором соли, высушивают (MgSO4) и фильтруют. Летучие продукты удаляют в вакууме с получением соединения заголовка (37.91 г), которое используют без дальнейшей очистки.
1H ЯМР (360 МГц, CDCl3) δ : 1.46 (9H, с), 2.47 (2H, т), 2.84 (2H, т), 3.55-3.69 (4H, м), 6.93-6.98 (2H, м), 7.15-7.18 (2H, м).
б) 3-[2-(4-фторфенил)этокси]пропановая кислота
Продукт стадии а) (37.91 г) растворяют в дихлорметане (50 мл) и добавляют трифторуксусную кислоту (50 мл). Раствор перемешивают при комнатной температуре в течение 1 часа, затем разбавляют водой и экстрагируют этилацетатом (4 раза). Объединенные органические экстракты промывают водой (4 раза), затем насыщенным раствором соли, высушивают (MgSO4) и фильтруют. Летучие продукты удаляют в вакууме, в результате чего получают масло, это масло собирают в диэтиловый эфир и экстрагируют водным раствором бикарбоната натрия (3 раза). Объединенные водные слои подкисляют концентрированной соляной кислотой, затем экстрагируют диэтиловым эфиром (х 4). Объединенные органические экстракты промывают насыщенным солевым раствором, высушивают (MgSO4) и фильтруют. Летучие продукты удаляют в вакууме с получением соединения подзаголовка (19.95 г), которое использовали без дальнейшей очистки.
Масс-спектр: 212 (М);
1H ЯМР (360 МГц, CDCl3) δ : 2.62 (2H, т), 2.85 (2H, т), 3.66 (2H, т), 3.73 (2H, т), 6.94-6.99 (2H, м), 7.14-7.18 (2H, м), 7.37 (1H, шир. с).
в) 2-[2-(4-фторфенил)этокси]этилизоцианат
Продукт стадии б) (15.49 г), триэтиламин (11.2 мл) и дифенилфосфорилазид (15.7 мл) нагревают в толуоле (150 мл) при 80oC в течение 5 часов в атмосфере азота. Смеси дают остыть, затем оставляют при комнатной температуре на ночь. Смесь разбавляют водой и экстрагируют этилацетатом (3 раза). Объединенные органические экстракты промывают водным раствором бикарбоната натрия, разбавленной соляной кислотой, насыщенным солевым раствором, затем сушат (MgSO4) и фильтруют. Летучие продукты удаляют в вакууме и остаток очищают флэш-хроматографией (элюент - 50% диэтиловый эфир:изогексан) с получением соединения заголовка (5.54 г).
Масс-спектр: 209 (М);
1H ЯМР (360 МГц, CDCl3) δ : 2.89 (2H, т), 3.38 (2H, т), 3.56 (2H, т), 3.68 (2H, т), 6.95-7.01 (2H, м), 7.17-7.21 (2H, м).
г) Метил N-[2-[2-(4-фторфенил)этокси]этил]карбамат
Продукт стадии в) (9.0 г) растворяют в метаноле (300 мл) и к этому перемешиваемому раствору добавляют метоксид натрия (4.65 г). Смесь механически перемешивают при комнатной температуре в течение 3 часов. Летучие продукты удаляют в вакууме и остаток экстрагируют этилацетатом. Этот этилацетат промывают солевым раствором, сушат (MgSO4) и фильтруют. Летучие продукты удаляют в вакууме и часть остатка далее очищают флэш-хроматографией (элюент - 60% диэтиловый эфир:изогексан), в результате чего получают соединение заголовка (0.375 г).
Масс-спектр: FAB 242 (М+Н);
1H ЯМР (360 МГц, CDCl3) δ : 2.84 (2H, т), 3.34 (2H, т), 3.49 (2H, т), 3.63 (2H, т), 3.67 (3H, с), 6.95-7.00 (2H, м), 7.14-7.18 (2H, м).
Элементный анализ
Найдено: C, 59.1; H, 6.83; N, 5.83%
Вычислено для C12H16FNO3: C, 59.74; H, 6.68; N, 5.81%.
д) 2-[2-(4-фторфенил)этокси]этанамин
Продукт стадии г) (8.0 г) растворяют в этиленгликоле и к этому добавляют гидроксид калия (48 г) и гидразингидрат (8.3 мл). Перемешиваемую смесь нагревают до 140oC в течение 4 часов, затем оставляют остывать до комнатной температуры на ночь. Смесь разбавляют водой и экстрагируют диэтиловым эфиром (3 раза). Объединенные органические экстракты промывают солевым раствором, высушивают (MgSO4) и фильтруют. Летучие продукты удаляют в вакууме, в результате чего получают соединение заголовка (5.36 г).
Масс-спектр: 184 (М+Н);
1H ЯМР (360 МГц, CDCl3) δ : 2.82-2.88 (4H, м), 3.47 (2H, т), 3.64 (2H, т), 6.95-6.99 (2H, м), 7.16-7.20 (2H, м).
е) Метил 3-[2-[2-(4-фторфенил)этокси]этиламиносульфонил] пропаноат
Соединение заголовка (8.32 г) получают в соответствии со способом Примера 3 г) с использованием 2-[2-(4-фторфенил)этокси] этанамина (5.36 г), триэтиламина (4.6 мл) и метил-3- (хлорсульфонил)пропаноата (5.6 г) в диэтиловом эфире (150 мл).
Т.пл. 49oC;
Масс-спектр: 184 (М+Н);
1H ЯМР (360 МГц, CDCl3) δ : 2.78-2.87 (4H, м), 3.24-3.28 (2H, м), 3.34 (2H, т), 3.51-3.56 (2H, т), 3.65 (2H, т), 3.73 (3H, с), 6.97-7.01 (2H, м), 7.15-7.19 (2H, м).
Элементный анализ
Найдено: C, 50.52; H, 6.24; N, 4.16; S, 9.40%
Вычислено для C14H20FNO5S: C, 50.44; H, 6.05; N, 4.20; S, 9.62%.
ж) N-[2-[2-(4-фторфенил)этокси] этил]-3-[2-(4-гидрокси- 2-оксо-3H-1.3-бензотиазол-7-ил)этиламино]пропансульфонамида гидрохлорид
Соединение заголовка (0.092 г) получают в соответствии со способом Примера 3д) с использованием метил 3-[2-[2-(4- фторфенил)этокси]этиламиносульфонил] пропаноата (1.0 г), диизобутилалюминийгидрида (1.5 М раствор в толуоле, 4 мл), 7-(2- аминоэтил)-4-гидрокси-1,3-бензотиазол-2-(3H)-она гидрохлорида (0.875 г) и цианоборгидрида натрия (0.354 г).
Т.пл. 195-197oC;
Масс-спектр: FAB 498 (М+Н);
1H ЯМР (360 МГц, d6 ДМСО) δ : 1.99 (2H, кв), 2.78-2.87 (4H, м), 3.04-3.16 (8H, м), 3.45 (2H, т), 3.59 (2H, т), 6.86 (1H, д), 6.85 (1H, д), 7.01 (2H, т), 7.26-7.31 (3H, м), 8.90 (2H, с), 10.15 (1H, с), 11.77 (1H, шир. с).
Элементный анализ
Найдено: C, 49.01; H, 5.74; N, 7.96; S, 11.50%
Вычислено для C22H29NF3O5S2•HCl• 0,5H2O: C 49.48; H, 5.47; N, 7.87; S, 12.01%.
Пример 5
N-[2-[2-(4-хлорфенил)этокси] этил] -3-[2-(4- гидрокси-2-оксо-3H-1,3-бензотиазол-7-ил)этиламино] пропансульфонамида гидрохлорид
а) 2-[2-(4-хлорфенил)этокси]уксусная кислота
2-(4-хлорфенил)этанол (10 г) перемешивают в 50% водном гидроксиде натрия (70 мл), добавляют тетрабутиламмонийбромид (1.4 г) и смесь перемешивают 1 час. Добавляют трет-бутилбромацетат (28.6 мл) в толуоле (140 мл) и перемешивание продолжают в течение 18 часов. Добавляют воду (50 мл) и через два часа смесь охлаждают на льду и подкисляют до значения pH 1 при помощи концентрированной соляной кислоты. Органический слой отделяют, а водный слой экстрагируют этилацетатом. Объединенные органические экстракты промывают солевым раствором и высушивают (MgSO4). Растворитель удаляют в вакууме, в результате чего получают бледно-желтое масло. Это масло растворяют в дихлорметане (100 мл) и добавляют трифторуксусную кислоту (100 мл), смесь нагревают с обратным холодильником в течение 1 часа. Летучие продукты удаляют в вакууме, а остаток собирают в раствор гидроксида натрия и промывают этилацетатом. Водный слой подкисляют концентрированной соляной кислотой и экстрагируют этилацетатом, этот этилацетат промывают водой, высушивают (MgSO4) и летучие продукты удаляют в вакууме, в результате чего получают соединение заголовка в виде твердого светло-коричневого вещества (16.4 г), которое используют без дальнейшей очистки.
Масс-спектр: EI 214/6 (М).
б) 2-[2-(4-хлорфенил)этокси]ацетамид
Продукт стадии а) (16.4 г) растворяют в толуоле (300 мл) и добавляют по каплям оксалилхлорид (13 мл) при комнатной температуре в атмосфере азота. Смесь перемешивают в течение 1 часа, затем добавляют диметилформамид (0.3 мл). Через 2 часа летучие продукты удаляют в вакууме, в результате чего получают коричневое масло, которое по каплям добавляют в перемешиваемый раствор концентрированного гидроксида аммония (60 мл). Выпавшее в осадок твердое вещество собирают фильтрованием, промывают водой и изогексаном, в результате чего получают соединение заголовка (6.6 г).
Т.пл. 106-109oC;
Масс-спектр: FAB 214/6 (m+H);
1H ЯМР (360 МГц, CDCl3) δ : 2.89 (2H, т), 3.73 (2H, т), 3.93 (2H, с), 5.87 (1H, шир. с), 6.22 (1H, шир. с), 7.16 (2H, д), 7.28 (2H, д).
в) 2-[2-(4-хлорфенил)этокси]этанамин
Продукт стадии б) небольшими порциями добавляют в перемешиваемый раствор борантетрагидрофурана (1.0 М в ТГФ, 85 мл). Реакционную смесь нагревают с обратным холодильником в атмосфере инертного газа в течение 3 часов. Реакционную смесь охлаждают и осторожно добавляют метанол (10 мл). Растворители удаляют в вакууме, а остаток растворяют в метаноле (100 мл), к которому добавляют концентрированную соляную кислоту (удельный вес 1.18, 4 мл). Этот раствор нагревают с обратным холодильником в течение 30 минут, затем растворитель удаляют в вакууме. Остаток собирают в воду и промывают эфиром. Добавляют бикарбонат натрия и водный слой экстрагируют этилацетатом. Этот этилацетат промывают водой, солевым раствором и высушивают (MgSO4). Летучие продукты удаляют в вакууме, в результате чего получают соединение заголовка в виде масла (5.5 г), которое затем используют без дальнейшей очистки.
Масс-спектр EI 200/2 (М+Н).
г) Метил 3-[2-[2-(4-хлорфенил)этокси]этиламиносульфонил] пропаноат
Продукт стадии в) (1.1 г) перемешивают в дихлорметане (30 мл) в атмосфере азота, добавляют триэтиламин (1.52 мл), затем метил-3-(хлорсульфонил)пропаноат (2.04 г). Смесь перемешивают при комнатной температуре в течение 3 часов. Летучие продукты удаляют в вакууме, а остаток очищают флэш-хроматографией на силикагеле (элюент - 60% эфир:бензин), в результате чего получают соединение заголовка (1.1 г) в виде масла.
Масс-спектр: FAB 350/2 (m+H);
1H ЯМР (360 МГц, CDCl3) δ : 2.80 (2H, т), 2.82 (2H, т), 3.26 (2H, кв), 3.33 (2H, т), 3.54 (2H, т), 3.67 (2H, т), 3.72 (2H, с), 4.63 (1H, т), 7.14 (2H, д), 7.27 (2H, д).
д) N-[2-[2-(4-хлорфенил)этокси] этил]-3-[2-(4-гидрокси- 2-оксо-3H-1,3-бензотиазол-7-ил)этиламино]пропансульфонамида гидрохлорид
Продукт стадии г) (1.1 г) растворяют в толуоле (50 мл) в атмосфере азота и охлаждают до -78oC. По каплям добавляют диизобутилалюминийгидрид (1.5 М раствор в толуоле 2.6 мл) и смесь выдерживают при температуре -78oC 10 минут. Реакцию останавливают добавлением 10% соляной кислоты в этаноле и смеси позволяют нагреться до комнатной температуры. Смесь выливают в 10% соляную кислоту и экстрагируют эфиром. Объединенные органические экстракты промывают водой, сушат (MgSO4), затем приблизительно 70% растворителя удаляют в вакууме. К смеси добавляют метанол (20 мл) и опять приблизительно 70% растворителя удаляют в вакууме, эту процедуру повторяют еще два раза. Этот раствор разбавляют метанолом (20 мл) и добавляют 7-(2-аминоэтил)- 4-гидрокси-1,3-бензотиазол-2(3H)-она гидрохлорид (0.92 г). pH доводят до 4 при помощи ледяной уксусной кислоты. Добавляют цианоборгидрид натрия (0.247 г) и смесь перемешивают в течение 2 часов в атмосфере азота. Смесь делают основной добавлением концентрированного водного раствора гидроксида аммония. Летучие продукты удаляют в вакууме и остаток очищают флэш-хроматографией (элюент - 2.5-10% метанол: дихлорметан). Продукт затем очищают с помощью ВЭЖХ с обращенной фазой (элюент - 50-100% метанол в 0.1% водной трифторуксусной кислоте), в результате чего после превращения в соль соляной кислоты получают соединение заголовка в виде твердого белого вещества (0.089 г).
Т. пл. 190-193oC;
Масс-спектр: FAB 514/6 (М+Н);
1H ЯМР (360 МГц, d6 ДМСО) δ : 1.99 (2H, м), 2.82 (4H, м), 3.12 (8H, м), 3.45 (2H, т), 3.60 (2H, т), 6.76 (1H, д), 6.87 (1H, д), 7.30 (5H, м), 8.87 (2H, шир. с), 10,14 (1H, с), 11.77 (1H, шир. с).
Элементный анализ
Найдено: C, 46.68; H, 5.55; N, 7.47; S, 10,86%
Вычислено для C22H28N3O5S2 •HCl•H2O: C, 46.47; H, 5,50; N, 7.39; S, 11.28%.
Пример 6
3-[2-(4-гидрокси-2-оксо-3H-1,3-бензотиазол-7- ил)этиламино] -N-[2-[2-(4-метилфенил)этокси]этил] пропансульфонамида гидрохлорид
а) 2-[2-(4-метилфенил)этокси]уксусная кислота
2-(4-метилфенил)этанол (8.16 г) перемешивают в 50% водном гидроксиде натрия (25 мл), добавляют т-бутилбромацетат (9.05 мл) в толуоле (30 мл) вместе с тетрабутиламмонийбромидом (2.2 г) и смесь перемешивают в течение 18 часов. Добавляют ледяную воду (100 мл), затем эфир, органический слой отделяют и затем водный слой подкисляют при помощи соляной кислоты. Подкисленный водный слой экстрагируют эфиром, этот экстракт промывают солевым раствором и сушат (MgSO4). Растворитель удаляют в вакууме, в результате чего получают соединение заголовка в виде твердого белого вещества (1.1 г), которое используют без дальнейшей очистки.
1H ЯМР (360 МГц, CDCl3) δ : 2.32 (3H, с), 2.91 (2H, т), 3.77 (2H, т), 4.10 (2H, с), 7.11 (4H, м).
б) 2-[2-(4-метилфенил)этокси]ацетамид
Соединение заголовка (0.77 г) получают в соответствии со способом Примера 1, стадия (б) с использованием 2-[2-(4- метилфенил)этокси]уксусной кислоты (1.1 г), оксалилхлорида (1.44 г), концентрированного гидроксида аммония (20 мл) и толуола (40 мл).
Т. пл. 106-107oC;
Масс-спектр: FAB 194 (М+H, 100);
в) 2-[2-(4-метилфенил)этокси]этанамина гидрохлорид
Соединение, названное в заголовке, (6.25 г) получают в соответствии со способом Примера 5, стадия (в) с использованием 2- [2-(4-метилфенил)этокси] ацетамида (5.79 г), боран- тетрагидрофурана (1.0 М в ТГФ, 75 мл) и тетрагидрофурана (40 мл).
Масс-спектр: FAB 180 (М+H, 100);
1H ЯМР (360 МГц, CDCl3) δ : 2.30 (3H, с), 2.87 (2H, т), 3.17 (2H, д), 3.65-3.73 (4H, м), 7.11 (4H, м), 8.30 (2H, шир. с).
г) Метил 3-[2-[2-(4-метилфенил)этокси]этиламиносульфонил] пропаноат
Соединение заголовка (2.49 г) получают в соответствии со способом Примера 5, стадия (г) с использованием 2- [2-(4-метилфенил)этокси]этанамина гидрохлорида (3.5 г), триэтиламина (4.74 мл), метил-3-(хлорсульфонил)пропаноата (3.08 г) и дихлорметана (80 мл).
Масс-спектр: FAB 330 (М+H, 119 (100));
д) 3-[2-(4-гидрокси-2-оксо-3H-1.3-бензотиазол-7-ил) этиламино] -N-[2-[2-(4-метилфенил)этокси]этил]пропансульфонамида гидрохлорид
Названное в заголовке соединение (0.2 г) получают в соответствии со способом Примера 5, стадия (д) с использованием метил-3-[2-[2-(4-метилфенил)этокси]этиламиносульфонил]пропаноата (2.49 г), диизобутилалюминийгидрида (1.5 М раствор в толуоле, 7.5 мл), 7-(2-аминоэтил)-4-гидрокси-1,3-бензотиазол- 2(3H)-она гидрохлорида (2.2 г) и цианоборгидрида натрия (0.48 г).
Т. пл. 213-215oC;
Масс-спектр: FAB 494 (М+H);
1H ЯМР (360 МГц, d6 ДМСО) δ : 1.99 (2H, м), 2.25 (3H, с), 2.74-2.84 (4H, м), 3.06-3.15 (8H, м), 3.45 (2H, т), 3.57 (2H, т), 6.75 (1H, д), 6.87 (1H, д), 7.10 (4H, м), 7.30 (1H, т), 8.61 (2H, с), 10.11 (1H, с), 11.77 (1H, шир. с).
Пример 7
3-[2-(4-гидрокси-2-оксо-3H-1.3-бензотиазол-7- ил)этиламино]-N-[2-(2-фенил-1-пропокси)этил] пропансульфонамида гидрохлорид
а) 2-(2-фенил-1-пропокси)уксусная кислота
Соединение заголовка (14,95 г) получают в соответствии со способом Примера 6, стадия (а) с использованием 2-фенил-1-пропанола (13.6 г), трет-бутилбромацетата (19.5 г), тетрабутиламмонийбромида (3.2 г), толуола (70 мл) и 50% водного гидроксида натрия (25 мл).
1H ЯМР (360 МГц, CDCl3) δ : 1.32 (3H, д), 3.08 (1H, м), 3.67 (2H, т), 4.07 (2H, с), 7.23-7.34 (5H, м).
б) 2-(2-фенил-1-пропокси)ацетамид
Соединение заголовка (4.39 г) получают в соответствии со способом Примера 5, стадия (б) с использованием 2-(2-фенил-1-пропокси)уксусной кислоты (5.0 г), оксалилхлорида (4.5 мл), концентрированного гидроксида аммония (20 мл) и толуола (30 мл).
Масс-спектр: GC 134 (М-59);
1H ЯМР (360 МГц, CDCl3) δ : 1.31 (3H, д), 3.05 (1H, м), 3.61 (2H, м), 3.90 (2H, кв.), 7.22 (3H, м), 7.33 (2H, м).
в) 2-(2-фенил-1-пропокси)этанамина гидрохлорид
Соединение заголовка (3.58 г) получают в соответствии со способом Примера 5, стадия в) с использованием 2-(2-фенил-1-пропокси)ацетамида (3.86 г), боран-тетрагидрофурана (1.0 М в ТГФ, 50 мл) и тетрагидрофурана (40 мл).
Масс-спектр: GC 180 (М+H);
г) Метил 3-[2-(2-фенил-1-пропокси)этиламиносульфонил]- пропаноат
Соединение заголовка (2.0 г) получают в соответствии со способом Примера 5, стадия (г) с использованием 2-(2-фенил-1- пропокси)этанамина гидрохлорида (3.58 г), триэтиламина (5.65 мл), метил-3-(хлорсульфонил)пропаноата (3.73 г) и дихлорметана (70 мл).
Масс-спектр: GC 224 (М-105);
1H ЯМР (360 МГц, CDCl3) δ : 1.27 (3H, д), 2.75 (2H, т), 3.02 (1H, м), 3.20-3.29 (4H, м), 3.45-3.56 (4H, м), 3.72 (3H, с), 4.42 (1H, т), 7.21-7.34 (5H, м).
д) 3-[2-(4-гидрокси-2-оксо-3H-1,3-бензотиазол-7-ил) этиламино] -N-[2-(2-фенил-1-пропокси)этил]пропансульфонамида гидрохлорид
Соединение заголовка (0.164 г) получают в соответствии со способом Примера 5, стадия (д) с использованием метил 3-[2- (2-фенил-1-пропокси)этиламиносульфонил] пропаноата (1.01 г), диизобутилалюминийгидрида (1.5 М раствор в толуоле, 3 мл), 7-(2- аминоэтил)-4-гидрокси-1,3-бензотиазол-2(3H)-она гидрохлорида (0.89 г) и цианоборгидрида натрия (0.19 г).
Т. пл. 183-184oC;
Масс-спектр: FAB 494 (М+Н);
Элементный анализ
Найдено: C, 52.42; H, 6.31; N, 8.28; S, 11.74%
Вычислено для C23H31N3O5S2•HCl: C, 52.11; H, 6.08; N, 7.93; S, 12.10%.
Пример 8
3-[2-(4-гидрокси-2-оксо-3H-1,3-бензотиазол-7-ил)этиламино] -N- [2-[2-(2-метилфенил)этокси]этил]пропансульфонамида гидрохлорид
а) 2-[2-(2-метилфенил)этокси] уксусная кислота
Соединение подзаголовка (8.91 г) получают в соответствии со способом Примера 5, стадия (а) с использованием 2-(2- метилфенил)этанола (5.0 г), трет-бутилбромацетата (5.47 г), тетрабутиламмонийбромида (0.78 г), толуола (80 мл), 50% водного гидроксида натрия (40 мл), трифторуксусной кислоты (20 мл) и дихлорметана (20 мл).
1H ЯМР (360 МГц, CDCl3) δ : 2.34 (3H, с), 2.97 (2H, т), 3.75 (2H, т), 4.13 (2H, с), 7.12-7.17 (4H, м), 8.14 (1H, шир. с).
б) 2-[2-(2-метилфенил)этокси]ацетамид
Соединение подзаголовка (5.02 г) получают в соответствии со способом Примера 5, стадия (б) с использованием 2-[2-(2-метилфенил)этокси]уксусной кислоты (6.726 г), оксалилхлорида (6.22 мл), концентрированного гидроксида аммония (60 мл) и толуола (60 мл).
Масс-спектр: FAB 194 (М+H);
1H ЯМР (360 МГц, CDCl3) δ : 2.22 (3H, с), 2.86 (2H, т), 3.60 (2H, т), 3.80 (2H, с), 7.06-7.19 (4H, м), 7.25 (2H, шир. с).
в) 2-[2-(2-метилфенил)этокси]этанамина гидрохлорид
Соединение подзаголовка (5.4 г) получают в соответствии со способом Примера 5, стадия (в) с использованием 2-[2-(2- метилфенил)этокси]ацетамида (5.13 г), раствора боран-тетрагидрофурана (1.0 М в ТГФ, 53.5 мл) и тетрагидрофурана (100 мл).
Масс-спектр: FAB 180 (М++H);
г) Метил 3-[2-[2-(2-метилфенил)этокси]этиламиносульфонил] пропаноат
Соединение подзаголовка (5.4 г) получают в соответствии со способом Примера 5, стадия (г) с использованием гидрохлорида 2- [2-(2-метилфенил)этокси] этанамина (5.4 г), триэтиламина (8.73 мл), метил 3-(хлорсульфонил)пропаноата (4.66 г) и дихлорметана (100 мл).
Масс-спектр: FAB 330 (М+Н);
1H ЯМР (360 МГц, CDCl3) δ : 2.32 (3H, с), 2.71 (2H, т), 2.91 (2H, т), 3.24 (2H, т), 3.34 (2H, т), 3.53 (2H, м), 3.62 (2H, т), 3.70 (3H, с), 4.64 (1H, т), 7.14 (4H, м).
д) 3-[2-(4-гидрокси-2-оксо-3H-1,3-бензотиазол-7-ил) этиламино] -N-[2-[2-(2-метилфенил)этокси]этил]пропансульфонамида гидрохлорид
Продукт стадии г) (2.0 г) растворяют в толуоле (100 мл) в атмосфере азота и охлаждают до -78oC. По каплям добавляют диизобутилалюминийгидрид (1.5 М раствор в толуоле, 6.22 мл) и смесь выдерживают при температуре -78oC в течение 10 минут. Реакцию останавливают добавлением этилацетата, затем 10% водного раствора калия натрия тартрата. Реакционную смесь нагревают до комнатной температуры и после перемешивания в течение 1 часа экстрагируют толуолом. Объединенные органические экстракты промывают водой, сушат (MgSO4), затем приблизительно 70% растворителя удаляют в вакууме. К смеси прибавляют метанол (50 мл) и опять приблизительно 70% растворителя удаляют в вакууме, эту процедуру повторяют еще два раза. Раствор разбавляют метанолом (50 мл) и добавляют 7-(2- аминоэтил)-4-гидрокси-1,3-бензотиазол-2(3H)-она гидрохлорид (1.8 г). pH доводят до значения 4 при помощи ледяной уксусной кислоты. Добавляют цианоборгидрид натрия (0.114 г) и смесь перемешивают в течение 18 часов в атмосфере азота. Смесь делают основной добавлением концентрированного водного раствора гидроксида аммония. Летучие продукты затем удаляют в вакууме. Осадок очищают флэш-хроматографией на кремнеземе (элюент - 1% метанол: дихлорметан). Затем вещество очищают ВЭЖХ с обращенной фазой (элюент - 35-85% метанол в 0.1% водной трифторуксусной кислоте), в результате чего после превращения в соль соляной кислоты получают соединение заголовка в виде твердого белого вещества (0.38 г).
Т. пл. 184-187oC;
Масс-спектр: FAB 494 (М+Н);
1H ЯМР (360 МГц, d6 ДМСО) δ : 1.74-1.77 (2H, м), 1.99 (2H, т), 2.27 (3H, с), 2.79-2.85 (4H, м), 3.07-3.16 (6H, м), 3.45-3.48 (2H, м), 3.55-3.61 (2H, м), 6.74 (1H, д), 6.88 (1H, д), 7.08-7.17 (4H, м), 7.32 (1H, т), 8.81 (1H, с), 10.17 (1H, с), 11.77 (1H, шир. с).
Пример 9
3-[2-(4-гидрокси-2-оксо-3H-1,3-бензотиазол-7-ил)этиламино] -N- [2-(2-фенилтиоэтокси)этил]пропансульфонамида гидрохлорид
а) трет-бутил 2-(2-фенилтиоэтокси)ацетат
2-фенилтиоэтанол (6.1 г), трет-бутилбромацетат (7.9 мл), тетрабутиламмонийбромид (1.3 г), толуол (80 мл) и 75% водный гидроксид натрия (40 мл) перемешивают вместе в течение 72 часов. Органический слой отделяют, а водный слой экстрагируют дихлорметаном. Объединенные органические экстракты промывают солевым раствором, высушивают (MgSO4) и растворитель удаляют в вакууме, в результате чего получают соединение заголовка (12.07 г).
Масс-спектр: FAB 269 (М+Н).
б) 2-(2-фенилтиоэтокси)ацетамид
Продукт стадии а) (12.07 г) растворяют в дихлорметане (50 мл) и добавляют трифторуксусную кислоту (50 мл), смесь перемешивают в течение 2 часов. Летучие продукты удаляют в вакууме, а остаток собирают в водный раствор бикарбоната натрия и промывают эфиром. Водный слой подкисляют концентрированной соляной кислотой, затем экстрагируют этилацетатом. Этилацетат промывают солевым раствором, высушивают (MgSO4) и летучие продукты удаляют в вакууме. Остаток растворяют в толуоле (50 мл) и добавляют по каплям оксалилхлорид (50 мл) при комнатной температуре в атмосфере азота. По каплям добавляют диметилформамид (0.3 мл), смесь перемешивают в течение 45 минут. Летучие продукты удаляют и неочищенный хлорид кислоты по каплям добавляют в перемешиваемый раствор концентрированного гидроксида аммония (50 мл) при -10oC. Выпавшее в осадок твердое вещество собирают фильтрованием, промывают водой и эфиром, в результате чего получают соединение заголовка (3.53 г).
Масс-спектр: FAB 212 (М+Н);
1H ЯМР (360 МГц, CDCl3) δ : 3.13 (2H, т), 3.72 (2H, т), 3.95 (2H, с), 5.58 (1H, шир. с), 6.56 (1H, шир. с), 7.20-7.39 (5H, м).
в) 2-(2-фенилтиоэтокси)этанамина гидрохлорид
Соединение заголовка в виде соли соляной кислоты (6.0 г) получают в соответствии со способом Примера 5, стадия (в) с использованием 2-(2-фенилтиоэтокси)ацетамида (5.5 г), раствора борантетрагидрофурана (1.0 М в ТГФ, 60 мл) и тетрагидрофурана (60 мл).
г) Метил 3-[2-(2-фенилтиоэтокси)этиламиносульфонил]пропаноат
Соединение заголовка (2.85 г) получают в соответствии со способом Примера 5, стадия (г) с использованием 2-(2- фенилтиоэтокси)этанамина гидрохлорида (6.0 г), триэтиламина (4 мл), метил-3-(хлорсульфонил)пропаноата (5.8 г) и дихлорметана (50 мл).
Масс-спектр: FAB 348 (М+105).
д) 3-[2-(4-гидрокси-2-оксо-3H-1,3-бензотиазол-7-ил) этиламино] -N-[2-(2-фенилтиоэтокси)этил]пропансульфонамида гидрохлорид
Соединение заголовка (0.106 г) получают в соответствии со способом Примера 8, стадия (д) с использованием метил-3-[2- (2-фенилтиоэтокси)этиламиносульфонил] пропаноата (1.4 г), диизобутилалюминийгидрида (1.5 М раствор в толуоле, 3 мл), 7-(2-аминоэтил)-4-гидрокси-1,3-бензотиазол-2(3H)-она гидрохлорида (1.2 г) и цианоборгидрида натрия (0.3 r).
Т. пл. 193-194oC;
Масс-спектр: FAB 512 (М+Н);
1H ЯМР (360 МГц, d6 ДМСО) δ : 2.06 (2H, м), 2.85 (2H, м), 3.08 (6H, м), 3.15 (4H, м), 3.46 (2H, т), 3.59 (2H, т), 6.76 (1H, д), 7.19 (1H, м), 7.31-7.36 (4H, м), 9.06 (2H, с), 10.17 (1H, с), 11.78 (1H, шир. с.).
Фармакологический пример
Сродство связывания соединений примеров, приведенных выше, к сайтам связывания DA2-рецепторов на мембранах гипофиза быка определяют по вытеснению [3H] -N-н-пропилнорапоморфина и [3H]-спиперона, соответственно, в присутствии или в отсутствие негидролизуемого аналога ГТФ, D. R. Sibley, A. DeLean and I. Creese, Anterior Pituitaru Dopamione Receptors, Demonstration of Interconvertible High and Low Affinity States of the D-2 Dopamine Receptor, J. Biol. Chem., 1982, 257(11), 6351-6361.
Активность в отношении DA2-рецепторов демонстрируют в функциональном исследовании на изолированной ушной артерии кролика, как описано Brown и O' Connor, Br. J. Pharmacol., 1981, 73, 189P.
Активность в отношении β2-адренорецепторов была показана на изолированной трахее морской свинки, как описано I. G. Dougall, D. Harper., D.M. Jackson and P. Leff, Br. J. Pharmacol., 1991, 104, 1057.
Активность в отношении α1-рецепторов определяют на изолированной ушной артерии кролика следующим способом.
Изолированная ушная артерия кролика
Кроликов-самцов NZW (2.5-3 кг) забивают внутривенной инъекцией пентобарбитоната натрия (60 мг/кг). Уши удаляют, проксимальную часть средней ушной артерии выделяют и вводят в нее канюлю, используя полипропиленовую канюлю (внешний диаметр 0.75 мм). После удаления артерию очищают от примыкающей соединительной ткани и препарируют, получая 6 колец шириной 5 мм с сохранением слоя круговой гладкой мускулатуры. Ткани помещают на тонкие крючки из вольфрамовой проволоки (диаметр 0.25 мм) в 20 мл ванночку для органов с раствором Кребса следующего состава (мМ): NaCl 117.56; NaHCO3 25.00; KCl 5.36; NaH2PO4 0.89; MgSO4 1.18; глюкоза 11.10 и CaCl2 2.55. В раствор также включают кокаин (30 мкМ) и пропанолол (1 мкМ) для того, чтобы блокировать нейронный захват и β-рецепторы, соответственно. Для предотвращения окисления катехоламина также добавляют аскорбат (100 мкМ). Температуру этого раствора поддерживают на уровне 37oC и через него непрерывно пропускают 95% O2 и 5% CO2. Верхний проволочный крючок соединяют с датчиком силы смещения Ормед, нижний крючок соединяют с неподвижной подложкой в ванночке. Изменения изометрической силы регистрируют при помощи Advance Bryans AB500 горизонтально-плоскостного самописца.
Экспериментальная часть
Общее описание
В начале какого эксперимента к каждому кусочку ткани прикладывают нагрузку 1.0 г. Такую нагрузку прикладывают повторно два или три раза в течение стабилизационного периода продолжительностью приблизительно 60 мин до тех пор, пока она не станет постоянной. В то же время, когда переустанавливают нагрузку, ванночки промывают. Кривые зависимости концентрация-действие (E/[A] ) строят, используя накопительное добавление агониста с приращением 0.5 log10. Реакции (сокращения) регистрируют как процент от максимального ответа при использовании стандартного агониста.
Количественное определение агонистического действия
В качестве стандартного агониста используют фенилэфрин. Сначала строят кривую E/[A] для фенилэфрина. Затем фенилэфрин отмывают и строят кривую E/[A] для тестируемого соединения. Реакции соединения, вызывающие агонизм, выражают в процентах от максимального ответа на фенилэфрин. Значение асимптоты кривой тестируемого соединения относительно фенилэфрина показывает собственную активность соединения. Собственную активность фенилэфрина принимают за 1).
Значение p[A50] является мерой активности (силы) агониста. Это отрицательный логарифм концентрации агониста, вызывающей половину максимального ответа. Для соединений, чья собственная активность значительно меньше 1, например, ≅0.8, можно рассчитать значения эффективности (τ) и значения сродства (pKA), используя сравнительный метод анализа. При таком анализе принимается, что фенилэфрин действует как полный агонист в данной системе, и, следовательно, он используется для определения операционных модельных параметров Em и n (Leff, et al., "Estimation of agonist affinity and efficacy by direct and operational model fitting. , '' J.Pharmacol. Methods., 1989, 23, 225- 237). Эти параметры могут затем использоваться для проведения сравнительного анализа тестируемых соединений. Сродство выражают в виде pKA (отрицательный логарифм концентрации агониста, достаточной для того, чтобы занять половину рецепторов).
Количественное определение антагонистического действия
Соединения, которые не проявили себя в качестве агонистов, исследовали на антагонистическую активность, инкубируя ткани с максимально высокими концентрациями этого соединения, с последующим построением кривых E/[A], фенилэфриновых кривых. Степень сдвига вправо таких фенилэфриновых кривых сравнивают с контрольными кривыми фенилэфрина, что позволяет оценить сродство тестируемого соединения. Такую оценку сродства выражают в значениях pA2 (отрицательный логарифм такой концентрации антагониста, которая дает двукратное смещение вправо контрольной E/[A] кривой).
Подтверждение α1-опосредованного агонизма
Празосин был принят в качестве стандартного α1-антагониста. Если в этих условиях тестируемое соединение демонстрирует агонизм, затем при достижении асимптоты кривой E/[A] тестируемого соединения добавляют празосин (1 мкМ) для того, чтобы проверить, является ли эта реакция обратимой. Если α1-антагонист устраняет ответ на испытуемое соединение, то это предполагает, что агонизм является опосредованным α1.
Данные по активности
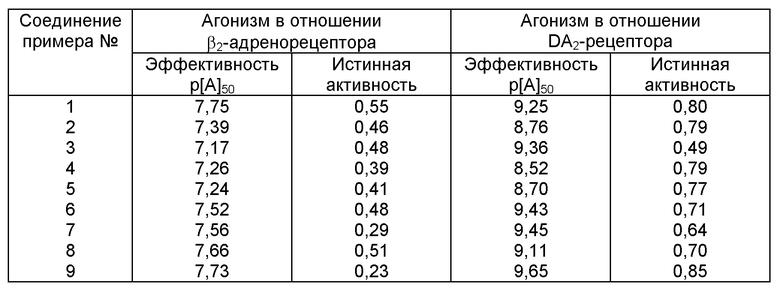
Таблица, приводимая ниже, включает данные по агонистической активности β2-адренорецептора и DA2-рецептора допамина соединений 1-9 заявки.
Активность β2-адренорецептора проиллюстрирована в исследовании, описанном Dougall et al. in Br.J.Pharmacol., 1991, 104, 1057 (см. тест, соответствующий стр. 11, строки 21-23 WO 97/10227). В этом исследовании для получения количественных данных по активности β2-адренорецепторов используют изолированную трахею морской свинки, содержащей β2-адренорецепторы, которые при стимулировании приводят к расслаблению гладкой мускулатуры. В качестве контроля применяют изопреналин, который считается полным агонистом этой системы.
Агонистические кривые концентрация-действие получают в результате кумулятивного добавления исследуемого соединения к препарату из изолированной трахеи морской свинки, а ответы записывают в виде процентной величины ответа изопреналина.
Величину pA50 которая является мерой эффективности агониста, определяют как отрицательный логарифм концентрации агониста, вызывающей ответ, составляющий половину максимального ответа.
Если исследуемое соединение вызывает расслабление гладкой мускулатуры, то может быть определена его истинная активность. Истинная активность - это ответ, вызываемый исследуемым соединением, разделенный на максимальный ответ, вызываемый изопреналином.
Величина pA50 и истинная активность каждого соединения из примеров представлены в нижеследующей таблице.
Активность рецептора допамина (DA2) проиллюстрирована в исследовании, описанном Brown et al. in Br.J.Pharmacol., 1981, 73, 189P (см. текст, соответствующий стр. 11, строки 19-21 WO 97/10227), с применением в качестве контроля соединения, идентифицируемого как "58075AB". Величины pA50 и истинная активность каждого соединения из примеров включены в таблицу наряду с результатами исследования β2-адренорецептора.
Как ясно показывают данные из таблицы, соединения в соответствии с данным изобретением демонстрируют хороший агонизм в отношении β2-адренорецептора и DA2-рецептора допамина, обеспечивая высокую применимость этих соединений при лечении заболеваний дыхательных путей.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ 7-(2-АМИНОЭТИЛ)БЕНЗОТИАЗОЛОНА ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ПРОИЗВОДНЫЕ N-[2-(4-ГИДРОКСИ-2-ОКСО-3H-1,3-БЕНЗОТИАЗОЛ-7-ИЛ)ЭТИЛАМИДА]ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ПРОЯВЛЯЮЩАЯ АГОНИСТИЧЕСКУЮ АКТИВНОСТЬ В ОТНОШЕНИИ β-АДРЕНОРЕЦЕПТОРОВ | 1993 |
|
RU2114108C1 |
| БИОЛОГИЧЕСКИ АКТИВНЫЕ ЭТАНАМИНЫ БЕНЗОТИАЗОЛОНА | 1996 |
|
RU2177476C2 |
| ПРОИЗВОДНЫЕ 3-Н-1,2,3-ТРИАЗОЛО-[4,5-D]ПИРИМИДИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1996 |
|
RU2174518C2 |
| ПРОИЗВОДНЫЕ N-ГЕТЕРОАРИЛ-ПИРИДИНСУЛЬФОНАМИДА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ), ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ПРОТИВОДЕЙСТВИЯ ОДНОМУ ИЛИ БОЛЕЕ ВОЗДЕЙСТВИЯМ ЭНДОТЕЛИНА | 1996 |
|
RU2172738C2 |
| КОНДЕНСИРОВАННОЕ ГЕТЕРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ | 2005 |
|
RU2389731C2 |
| ИНГИБИТОРНЫЕ СОЕДИНЕНИЯ | 2013 |
|
RU2673079C2 |
| ГЕТЕРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ | 2006 |
|
RU2382781C2 |
| ПИРИМИДИНАМИДНЫЕ СОЕДИНЕНИЯ КАК ИНГИБИТОРЫ PGDS | 2006 |
|
RU2420519C2 |
| СОЕДИНЕНИЯ ТИЕНОПИРИМИДИНА И ИХ ПРИМЕНЕНИЕ | 2004 |
|
RU2331648C2 |
| ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛИДИНОНА В КАЧЕСТВЕ АГЕНТОВ МУСКАРИНОВЫХ РЕЦЕПТОРОВ | 2002 |
|
RU2288919C2 |
Изобретение относится к новым производным бензотиазолона общей формулы I, где Х представляет -SО2NН- или -NНSО2-; р, q и r независимо друг от друга представляют 2 или 3; Y представляет тиенил, необязательно замещенный С1-6алкилом или галогеном, либо фенилтио- или фенил, необязательно замещенный С1-6алкилом или галогеном; каждый из R независимо представляет Н или С1-6алкил; его оптические изомеры и фармацевтически приемлемые соли. Способ получения соединения формулы I или его фармацевтически приемлемой соли путем селективного восстановительного алкилирования соединения формулы II соединением формулы III в присутствии восстановителя или путем селективного восстановления соединения формулы IV. Амид формулы VI, кислота формулы V и альдегид формулы III в качестве промежуточных соединений для получения бензотиазолона формулы I. Фармацевтическая композиция, проявляющая агонистическое действие в отношении DА2-рецепторов и β2-андренорецепторов, содержащая соединение формулы I или его фармацевтически приемлемую соль, диспергированная в пропелленте, необязательно включающем наполнители, смазывающие агенты или стабилизаторы, или в виде сухого порошка для ингаляции, необязательно включающего фармацевтически приемлемый носитель. Технический результат - получение новых соединений бензотиазолона. 9 с. и 9 з.п.ф-лы, 1 табл. 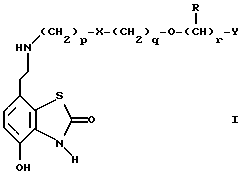
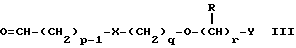
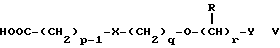
отличающееся тем, что
X представляет -SO2NH- или -NHSO2-;
p, q и r независимо друг от друга представляют 2 или 3;
Y представляет тиенил, необязательно замещенный C1-6 алкилом или галогеном, либо фенилтио- или фенил, необязательно замещенный C1-6 алкилом или галогеном;
каждый из R независимо представляет H или C1-6 алкил, его оптические изомеры и его фармацевтически приемлемые соли.
2-[2-(4-гидрокси-2-оксо-3Н-1,3-бензотиазол-7-ил)этиламино] -N-[2-(2-фенилэтокси)этил]пропансульфонамид;
N-[2-[2-(4-гидрокси-2-оксо-3Н-1,3-бензотиазол-7-ил)-этиламино] этил]-2-(2-фенилэтокси)этансульфонамид;
3-[2-(4-гидрокси-2-оксо-3Н-1,3-бензотиазол-7-ил)этиламино] -N-[2-[2-(5-метил-2-тиенил)этокси]этил]пропансульфонамид;
N-[2-[2-(4-фторфенил)этокси] этил] -3-[2-(4-гидрокси-2-оксо-3Н-1,3-бензотиазол-7-ил)этиламино]пропансульфонамид;
N-[2-[2-(4-хлорфенил)этокси] этил] -3-[2-(4-гидрокси-2-оксо-3Н-1,3-бензотиазол-7-ил)этиламино]пропансульфонамид;
3-[2-(4-гидрокси-2-оксо-3Н-1,3-бензотиазол-7-ил)этиламино]-N-[2-[-(4-метилфенил)этокси]этил]пропансульфонамид;
(R, S)-3-[2-(4-гидрокси-2-оксо-3Н-1,3-бензотиазол-7-ил)этиламино]-N-[2-(2-фенил-1-пропокси)этил]пропансульфонамид;
3-[2-(4-гидрокси-2-оксо-3Н-1,3-бензотиазол-7-ил)этиламино] -N-[2-[2-(2-метилфенил)этокси]этил]пропансульфонамид; и
3-[2-(4-гидрокси-2-оксо-3Н-1,3-бензотиазол-7-ил)этиламино] -N[2-(2-фенилтиоэтокси)этил]пропансульфонамид.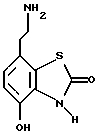
соединением формулы III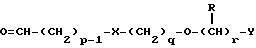
в котором p, q, r, R, X и Y имеют значения, указанные в п.1, в присутствии восстановителя.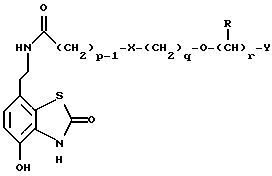
в котором p, q, r, R, X и Y имеют значения, указанные в п.1.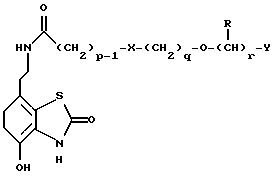
в которой p, q, r, R, X и Y имеют значения, указанные в п.1.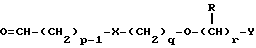
в которой p, q, r, R, X и Y имеют значения, указанные в п.1.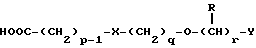
в которой p, q, r, R, X и Y имеют значения, указанные в п.1.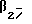 адренорецепторов и включающая соединение бензотиазолона формулы I или его фармацевтически приемлемую соль по любому из пп.1 - 10, в виде распыляемой водной суспензии или раствора.
адренорецепторов и включающая соединение бензотиазолона формулы I или его фармацевтически приемлемую соль по любому из пп.1 - 10, в виде распыляемой водной суспензии или раствора.
Приоритет по пунктам и признакам:
15.09.1995 по п.1, где X представляет -SO2NH- или -NHSO2; p, q, r независимо друг от друга представляют 2 или 3; Y представляет фенил, необязательно замещенный галогеном; R представляет H;
по п.9, где соединения представляют собой: 3-[2-(4-гидрокси-2-оксо-3Н-1,3-бензотиазол-7-ил)этиламино] -N-[2-(2-фенилэтокси]этил]пропансульфонамид; N-[2-[2-(4-гидрокси-2-оксо-3Н-1,3-бензотиазол-7-ил)этиламино] этил]-2-(2-фенилэтокси)этансульфонамид; 3-[2-(4-гидрокси-2-оксо-3Н-1,3-бензотиазол-7-ил)этиламино]-N-[2-[2-(5-метил-2-тиенил)этокси]этил]пропансульфонамид;
по пп.2 - 8, 11 - 18;
10.07.1996 по п.1, где Y представляет фенилтио, необязательно замещенный C1-6 алкилом;
по п.9, где соединения представляют собой: N-[2-[2-(4-фторфенил)этокси] этил]-3-[2-(4-гидрокси-2-оксо-3Н-1,3-бензотиазол-7-ил)этиламино]пропансульфонамид; N-[2-[2-(4-хлорфенил)этокси]этил]-3-[2-(4-гидрокси-2-оксо-3Н-1,3-бензотиазол-7-ил)этиламино] пропансульфонамид; 3-[2-(4-гидрокси-2-оксо-3Н-1,3-бензотиазол-7-ил)этиламино] -N-[2-(2-фенилэтокси)этил]пропансульфонамид; (R,S)-3-[2-(4-гидрокси-2-оксо-3Н-1,3-бензотиазол-7-ил)этиламино]-N-[2-(2-фенил-1-пропокси)этил] пропансульфонамид; 3-[2-(4-гидрокси-2-оксо-3Н-1,3-бензотиазол-7-ил)этиламино]-N[2-[2-(2-метилфенил)этокси]этил]пропансульфонамид; 3-[2-(4-гидрокси-2-оксо-3Н-1,3-бензотиазол-7-ил)этиламино] -N[2-(2-фенилтиоэтокси)этил]пропансульфонамид;
по п.10;
12.09.1996 по п.1, где Y представляет тиенил, необязательно замещенный C1-6 алкилом или галогеном.
| Дорожная спиртовая кухня | 1918 |
|
SU98A1 |
| Домовый номерной фонарь, служащий одновременно для указания названия улицы и номера дома и для освещения прилежащего участка улицы | 1917 |
|
SU93A1 |
| Автоматический огнетушитель | 0 |
|
SU92A1 |
