Данное изобретение относится к пиримидиновым производным, сконденсированным с гетероциклическим кольцом, и к способам использования их в лечении гиперпролиферативных заболеваний, таких как раковые опухоли и псориаз; растеноз; заболевание почек и панкреатит, и для предотвращения биастоцильных имплантаций, например, контрацепции у млекопитающих.
Многие из теперешних режимов лечения рака используют соединения, которые ингибируют синтез ДНК. Такие соединения являются обычно токсичными для клеток, но их токсическое действие на быстро делящиеся опухолевые клетки может быть благоприятным. Для усиления селективности действия в отношении раковых клеток были разработаны альтернативные подходы к поиску противораковых агентов, которые действуют по иным механизмам, чем ингибирование синтеза ДНК.
Известно, что клетки могут становиться злокачественными или раковыми в силу трансформации части их ДНК в онкоген (т.е. ген, который при активации ведет к образованию злокачественных опухолевых клеток). Многие онкогены кодируют белки, которые представляют аберрантные тирозинкиназы, способные вызывать трансформацию клеток. Альтернативно, сверхэкспрессия нормальной прото-онкогенной тирозинкиназы также может приводить в результате к пролиферативным нарушениям, иногда приводящим к злокачественному фенотипу.
Рецепторные тирозинкиназы являются крупными ферментами, которые спанируют клеточную мембрану и обладают внеклеточным связывающим доменом факторов роста, таких как фактор эпидермального роста, трансмембранным доменом и внутриклеточной частью, которая функционирует как киназа, фосфорилируя специфические тирозиновые остатки в белках, и следовательно оказывая влияние на пролиферацию клеток. Известно, что такие киназы часто аберрантно экспрессируются в обычных раковых опухолях людей, таких как рак грудной железы, рак желудочно-кишечного тракта, такой как рак толстой кишки, прямой кишки или рак желудка, лейкемия и рак яичников, бронхиальный или панкреатический рак. Показано было также, что рецептор фактора эпидермального роста (EGFR), который обладает активностью тирозинкиназы, мутируется (подвергается мутации) и/или сверхэкспрессируется во многих раковых опухолях человека, таких как опухоли мозга, легких, сквамозных клеток, мочевого пузыря, желудка, груди, головы и шеи, эзофагиальные, гинекологические и тироидные (щитовидной железы) опухоли.
Соответственно, признано было, что ингибиторы рецепторных тирозинкиназ являются полезными в качестве селективных ингибиторов роста злокачественных клеток млекопитающих. Например, эрбстатин, ингибитор тирозинкиназы, селективно аттенуирует у атимических голых мышей рост трансплантированной карциномы молочной железы человека, которая экспрессирует рецепторную тирозинкиназу эпидермального фактора роста (EGFR), но не оказывая влияния на рост других карцином, которые не экспрессируют EGF рецептор (рецептор фактора эпидермального роста).
Было также показано, что разнообразные другие соединения, такие как производные стирола, обладают свойствами ингибирования тирозинкиназы. Совсем недавно, в трех европейских патентных публикациях, а именно EP 0556226 A1, EP 0602851 A1 и EP 0520722 A1, было отмечено, что некоторые сконденсированные с гетероарилом производные пиримидина обладают противо-раковыми свойствами, что является результатом их свойств ингибировать тирозинкиназу. В PCT публикации WO 92/20642 также раскрываются бис- моно и бициклические арильные и гетероарильные соединения как ингибиторы тирозинкиназы. В европейской патентной публикации EP 0496617 A1 раскрываются некоторые пиразоло[3,4-d] пиримидины и пирроло[2,3-d]пиримидины, которые обладают свойством ингибировать аденозинкиназу.
В европейской патентной публикации EP 0475413 A2 раскрываются некоторые карбоциклические нуклеозидные аналоги в качестве полезных иммуноподавляющих веществ. В европейской патентной публикации EP 0414386 A1 раскрываются некоторые пиридо[2,3-d]пиримидины в качестве фунгицидов, инсектицидов и митицидов. В публикации II Farmco-Ed. Sc, том 35, fasc. 4, стр. 308-323 (1980) описывается синтез и противоаллергичекая активность производных 9-арил-8-азааденина.
В находящихся на рассмотрении патентных заявках США (NN 08/200359 и 08/413300) и заявке PCT N PC8836A, принадлежащих заявителю данной заявки, описываются необязательно замещенные индолил- и фениламино хиназолины, соответственно, которые полезны в лечении гиперпролиферативных заболеваний, вовлекающих рецепторные тирозинкиназы. В дополнение к сказанному патент США 4012513 раскрывает некоторые производные 1-(гетероциклической)-индол-3-ил-уксусной кислоты, которые проявляют противовоспалительную, анальгетическую и антипиретическую (жаропонижающую) активность.
Хотя описанные выше противораковые соединения вносят значительный вклад в данную область техники, в данной области все еще продолжается поиск улучшенных противораковых фармацевтических веществ.
Краткое раскрытие изобретения.
Данное изобретение направлено на соединения формулы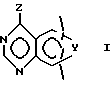
и его стереоизомеры, фармацевтически приемлемые соли, где Y представляет -NR4-CR3= CR3-, -CH= CR3-N= CH-, -CR3=N-CR3=CR3-, CR3=CR3-NR4-, -NR4-CR3= N- или -N=N-NR4-;
Z представляет NR1R2, где R1 представляет H и R2 представляет фенил, замещенный группой (R5)m или Q, или R1R2N представляет группу формулы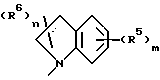
в которой пунктирная линия представляет необязательную двойную связь;
каждый R3 присоединен к атому углерода в Y и независимо выбран из: гидрокси, (C1-C4)алкокси, гидрокси(C1-C4)алкокси, амино(C2-C4)алкил, амино(C2-C4)алкокси, (C1-C4)алкокси(C2- C4)алкокси, гидрокси(C1-C4)алкил(C1- C4)алкилендиокси, (C1-C4)алкокси(C1- C4)алкил(C1-C4)алкилендиокси, моно-N- или ди-N, N-(C1-C4)aлкиламино(C2-C4)алкокси, 3- или 4-(C1-C4)алкокси-(2-гидрокси)-(C3- C4)алкокси, карбокси(C1-C4)алкокси, морфолино(C2-C4)алкокси,
имидазол-1-ил(C2- C4)алкокси, 4(C1-C4)алкилпиперазин-1- ил-(C2-C4)алкокси,
(C1-C4)алкокси(C1- C4)алканоилокси, нитро, гидроксиламино, амино, фенил, пиридил, пирроло, имидазоло, тиазоло, бензимидазоло, пиридонил, моно-N- или ди-N, N-(C1-C4)алкиламино, (C1-C4)алканоиламино, гидрокси(C2-C4)алкиламино,
(C1-C4)алкокси (C2-C4)алкиламино, (C1-C4)алкилсульфонамидо, морфолино, (C1-C4)алкил-пиперазин-1-ил, бис(C1-C4)алкан-сульфонамидо, ди-N, N-(C1- C4)алкиламино(C2-C4)алкиламино, (C1-C4)алкиламино(C2-C4)алкиламино,
пиперидин-1-ил, имидазол-1-ил, пирролидин-1-ил, (C1-C4)алкокси(C1-C4)алкилкарбониламино, карбокси, (C1-C4)алкоксикарбонил,
(C1-C4)алкоксикарбонил(C1-C4)алкокси, амидо, моно-N- или ди-N,N-(C1-C4)алкиламинокарбонил, моно-N- или ди-N,N-(гидpoкси(C2-C4)aлкиламинокарбонил, (C1-C4)алкил, гидрокси(C1-C4)алкил, моно-N- или ди-N, N-((C1-C4)алкокси(C1- C4)алкиламино(C1-C4)алкил, моно-N- или ди-N,N-(C1-C4)алкиламино(C1-C4)алкил, (C1-C4)алканоиламино(C1-C4)алкил,
(C1-C4)алкилтио, (C1-C4)алкокси(C2- C4)алкилтио или гидрокси(C2-C4)алкилтио;
каждый R4 присоединен к N-атому в Y и независимо выбран из: водорода, бензила, фенила, (C2-C4)алкила, гидрокси(C2-C4)алкила или каждой из таких групп, как гидрокси(C2-C4)алкил, амино(C2-C6)алкил, (C2-C4)алкоксикарбонил, замещенной амино, галогеном, гидрокси, (C2-C4)алканоилокси, (C1-C4)алкокси, моно-N- или ди-N,N-(C1-C4)алкиламино, моно-N- или ди-N,N-(гидрокси(C2-C4)алкиламино, моно-N- или ди-N,N-(C1-C4)алкокси(C2-C4)алкил)амино, сульфониларил(C1-C4)алкиламин, (C1-C4)алканоиламино, имидазол-1-ил, пиперидино, морфолино, пиперазин-1- ил, 4-(C1-C4)алкилпиперазин-1-ил, пиридил, пирроло, имидазоло, тиазоло, пиридонил, карбокси, (C1-C4)алкоксикарбонил, карбамоил, моно-N- или ди-N,N-(C1-C4)aлкилкарбамоил, карбоксамидо, моно-N- или ди-N,N-(C1-C4)алкилкарбоксамидо, или моно-N- или ди-N,N-(C1-C4)алкил)карбоксамидо;
каждый R5 независимо выбран из моно-, ди- или трифторметила, галогена, нитро, гидрокси, амино, азидо, изотиоциано, (C1-C4)алкила, фенила, тиенила, (C1-C4)алкокси, бензилокси, фенокси, (C2-C6)алкенила, (C2-C6)алкинила, (C1-C4)алкилендиокси, циано, бензоиламино, трифторметилкарбониламино, (C1-C4)алканоиламино, (C1-C4)алканоил, моно-N- или ди-N,N-(C1-C4)алкиламино,
(C1-C4)алкилсульфониламино, трифторметилсульфониламино, (C1-C4)алкилтио, (C1-C4)алкилсульфинила или (C1-C4)алкилсульфонила, пиррол-1-ила, пиперидин-1-ила или пирролидин-1-ила; указанные фенил, бензилокси, фенокси и бензоиламино являются необязательно монозамещенными галогеном, нитро, трифторметилом, гидрокси или (C1-C4)алкилом; указанный (C1-C4)алкилендиокси связан с обоих концов с соседними атомами углерода бензольного фрагмента или две группы R5 с атомами углерода, к которым они присоединены, образуют группу, выбранную из имидазолила, пирроло и пиразолила;
каждый R6 независимо выбран из гидрокси, амино, моно-N- или ди-N,N-(C1-C4)алкиламино, сульфо или (C1-C4)алкокси (при условии, что такие группы не присоединены к атому углерода кольца, который непосредственно примыкает к атому азота кольца) или R6 в каждом случае независимо представляет карбокси, гидрокси(C1-C4)алкил, (C1-C4)алкокси(C1-C4)алкил, амино(C1-C4)алкил, моно-N- или ди-N, N-(C1-C4)алкиламино(C1-C4)алкил, морфолино(C1-C4)алкил, 4-(C1-C4)алкил- пиперазин-1-ил(C1-C4)алкил, карбокси(C1-C4)алкил, (C1-C4)алкоксикарбонил, сульфо(C1-C4)алкил, пиридил(C1-C4)алкил или (C1-C4)алкил;
m представляет целое число от 1 до 3;
n представляет 0, 1 или 2;
p представляет 0 или целое число от 1 до 3;
при условии, что, когда Y в направлении, показанном стрелкой в формуле 1, представляет -CR3=N-CR3=CR3-, p=0, m=1 и Z - замещенный фенил, тогда R5 не является 4-этокси, 4-метокси, 4- трифторметокси, 4-трет-бутилом или 4-изопропилом;
Q представляет 9- или 10-членный бициклический гетероарильный циклический фрагмент или его гидрированное производное, содержащие один или два гетероатома азота и необязательно содержащие дополнительный гетероатом, выбранный из азота, кислорода и серы, и может необязательно нести один или два заместителя, выбранных из галогена, гидрокси, оксо, амино, нитро, карбамоила, (C1-C4)алкила, (C1-C4)алкокси, (C1-C4)алкиламино, ди-[(C1-C4)алкил] амино, (C2-C4)алканоиламино, (C2-C4)алкенила и (C2-C4)алкинила при условии, что, когда Y в направлении, показанном стрелкой в формуле 1, представляет -NR4-CR3=CR3-, R3 представляет CH3 и R4 представляет H, тогда R5 не является группой 4-CH3, 3,5-(CH3)2, 2,6-(CH3)2, 2-C2H5, 4-н-C4H9, 2-Cl, 4-Cl, 3,4-Cl2, 2-F или 3-CF3.
Предпочтительны соединения формулы 1, в которых Y в направлении, показанном стрелкой в формуле 1, выбран из -NR4-CR3=CR3-, -CH=CR3-N=CH-, -CR3= CR3-NR4-;
Особенно соединения 1, в которых Y в направлении, показанном стрелкой в формуле 1, представляет CR3=CR3-NR4-, a R4 представляет водород;
Среди соединений формулы 1 следует выделить соединение, в котором NR1R2 представляет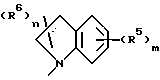
и R5, R6, m и n имеют значения, определенные выше;
в частности, в котором каждый R5 независимо выбран из 4-гидрокси, 4-амино, 5-фтора, 5-гидрокси, 5-амино, 6-галогена, 6-метила, 6-этенила, 6-этинила, 6-нитро и 7-метила и каждый R6 независимо выбран из гидрокси, амино, моно-N- или ди-N,N-(C1-C4)алкиламино, сульфо или (C1-C4)алкокси (при условии, что такие группы не присоединены к атому углерода кольца, который непосредственно примыкает к атому азота кольца) или R6 в каждом случае независимо представляет карбокси, гидрокси(C1-C4)алкил, (C1-C4)алкокси(C1-C4)алкил, амино(C1-C4)алкил, моно-N- или ди-N, N-(C1-C4)алкиламино(C1-C4)алкил, морфолино(C1-C4)алкил, 4-(C1-C4)алкил- пиперазин-1-ил(C1-C4)алкил, карбокси(C1-C4)алкил, (C1-C4)алкоксикарбонил, сульфо(C1-C4)алкил, пиридил(C1-C4)алкил и (C1-C4)алкил;
Также предпочтительны соединения формулы 1, в которых R1представляет H, и R2 представляет (R5)m-замещенный фенил, в котором R5 и m имеют значения, определенные выше;
Особенно соединение, в котором каждый R5 независимо выбран из 4-фтор-3-хлора, 3-трифторметила, 4-фтор-3-трифторметила, 3-нитро-4-хлора, 3-нитро-4-фтора, 4-фтор-3-брома, 3-иод-5-амино, 3-метил-4-фтора, 4-амино, 3-фтора, 3-гидрокси, 3-амино, 3-галогена, 3-метила, 3-этенила, 3-этинила, 3-нитро и 4-метила;
Среди указанных соединений следует выделить соединение - в котором R1 представляет H, и R2 представляет Q;
в частности, соединение, в котором Q выбран из 4-, 5-, 6-индолила, 1H-бензимидазол-4-ила, 1H-бензимидазол-5-ила, 1H-индазол-4-ила, 1H-индазол-5-ила, 1H-индазол-6-ила, 1H-индазол-7-ила, 1H-бензотриазол-4-ила, 1H-бензотриазол-5-ила, 1H-бензотриазол-6-ила, 5- или 6-бензоксазолила, 5- или 6-бензотиазолила, бензо[с] [2,1,3]тиадиазол-4-ила, 2-, 3-, 4-, 5-, 6-, 7- или 8-хинолила, 1-, 3-, 4-, 5-, 6-, 7- или 8-изохинолила, 4-, 5-, 6-, 7- или 8-циннолинила, 5-, 6-, 7- или 8-хиназолинила или 2-, 5- или 6-хиноксалинила, который может необязательно нести один или два заместителя, выбранных из фтора, брома, хлора, метила, этила, этенила, этинила и метокси;
Особенно соединение, в котором Q выбран из 5-индолила, 1H-индазол-5-ила, 1H-бензотриазол-5-ила, 6-бензотиазолила, бензо[с] [2,1,3] тиадиазол-4-ила, 5-хинолила, 6-хинолила, 8-хинолила, 5-изохинолила или 5-хиноксалинила, который может необязательно нести один или два заместителя, выбранных из фтора, брома, хлора, метила, этила, этенила, этинила и метокси;
Среди указанных соединений можно назвать соединения, выбранные из группы, состоящей из следующих соединений: (3-этинилфенил)-(7H-пирроло[2,3-d] пиримидин-4-ил)-амина гидрохлорид; (3-хлорофенил)-(7H-пирроло[2,3-d] пиримидин-4-ил)-амина гидрохлорид; 4-(6-хлор-2,3-дигидро-индол-1-ил)7H-пирроло[2,3-d] пиpимидинa гидрохлорид; (7H-пирроло[2,3-d]пиримидин-4-ил)m-толиламина гидрохлорид; (1H-индол-5-ил)-(7H-пирроло[2,3-d]пиримидин-4-ил)-амина гидрохлорид; (6-метилиндолин-1-ил)-(7H-пирроло[2,3-d] пиримидин-4-ил)-амина гидрохлорид; (бензо[b]тиен-5-ил)-(7H-пирроло[2,3-d]пиримидин-4-ил)-амин; (6-хлор-5-фториндолин-1-ил)-(7H-пирроло[2,3-d]пиримидин-4-ил)-амин; (1H-индазол-5-ил)-(7H-пирроло[2,3-d]пиримидин-4-ил)-амин; 1-(4-м-толиламино-пирроло[2,3-d] пиримидин-7-ил)-этанона гидрохлорид; (5-иодо-7H-пирроло[2,3-d] пиримидин-4-ил)-m-толиламин; (3-хлорфенил)-(1H-[1,2,3] триазол[4,5-d]пиpимидин-7-ил)-амина гидрохлорид; (3-хлорфенил)-пиридо[4,3-d] пиримидин-4-ил-амина гидрохлорид; (1H-индол-5-ил)-пиридо[4,3-d]пиримидин-4-ил-амина гидрохлорид; (3-этинилфенил)-(7-метилпиридо[4,3-d]пиримидин-4-ил)-амина гидрохлорид; (3-хлорфенил)(7-метилпиридо[4,3-d]пиримидин-4-ил)-амина гидрохлорид; (3-этинилфенил)-(пиридо[4,3-d]пиримидин-4-ил)-амина гидрохлорид; (6-бром-5-фториндолин-1-ил)-(пиридо[4,3-d] пиримидин-4-ил)-амин; (6-хлор-5-фториндолин-1-ил)-(пиридо[4,3-d] пиpимидин-4-ил)-aмин; (1H-индазол-5-ил)-(пиридо[4,3-d] пиримидин-4-ил)-амин; (бензо[b] тиен-5-ил)-(пиридо[4,3-d] пиpимидин-4-ил)-амин; (3-метил-4-гидроксифенил)(6-метилпиридо[4,3-d] пиримидин-4-ил)-амин; (6-йодиндолин-1-ил)-(пиридо[4,3-d] пиримидин-4-ил)-амин; (бензо[b] тиен-5-ил)-(пиридо[4,3-d] пиримидин-4-ил)-амин; (3-этинил-фенил)-(9H-пурин-6-ил)-амин; (1H-индол-5-ил)-(9H-пурин-6-ил)-амина гидрохлорид; (3-хлорфенил)-(9H-пурин-6-ил)-амина гидрохлорид; 4-(6-хлор-2,3-дигидроиндол-1-ил)-пиридо[3,4-d]пиримидин; (пиридо[3,4-d]пиримидин-4-ил)-(m-толил)амин; (1H-индaзoл-5-ил)-(пиpидo[3,4-d] пиpимидин-4-ил)-aмин; (1H-индол-5-ил)-(пиридо[3,4-d] пиримидин-4-ил)-амин; (фенил)-(пиридо[2,3-d] пиримидин-4-ил)-амин; (3-хлорфенил)-(пиридо[2,3-d] пиримидин-4-ил)-амин; (3-хлорфенил)-(пиридо[3,4-d]пиримидин-4-ил)-амин; (3-бромфенил)-(пиридо[3,4-d] пиpимидин-4-ил)-aмин: (фенил)-(пиридо[3,4-d]пиримидин-4-ил)-амин; 4-(6-хлор-2,3-дигидроиндол-1-ил)-пиpидo[3,4-d]пиpимидин; (пиридо[3,4-d] пиримидин-4-ил)-(m-толил)-амин; (1H-индазол-5-ил)-пиридо[3,4-d] пиримидин-4-ил-амин; (1H-индол-5-ил)-пиридо[3,4-d]пиримидин-4-ил-амин; фенил-пиридо[2,3-d]пиримидин-4-ил-амин; 3-хлорфенил-пиридо[2,3-d]пиpимидин-4-ил-aмин; 3-хлорфенил-пиридо[3,4-d] пиримидин-4-ил-амин; 3-бромфенил-пиридо[3,4-d] пиримидин-4-ил-амин; фенил-(пиридо[3,4-d]пиpимидин-4-ил)-aмин; (7-6ензолсульфонил-7H-пирроло[2,3-d]пиpимидин-4-ил)-(3-этинилфенил)-амин; 4-(6-хлор-2,3-дигидроиндол-1-ил)-5H-пирроло[3,2-d] пиpимидин-6-ол; (3-этинилфенил)-[7-(2-морфолин-4-ил-этил)-7H-пирроло[2,3-d]пиримидин- 4-ил)] -амин; (3-этинилфенил)-[7-(2-метоксиэтил)-7H-пирроло[2,3-d]пиримидин- 4-ил)] -амин; (3-этинилфенил)-{7-[2-(2-метоксиэтокси)-этил]-7H- пирроло[2,3-d]пиримидин-4-ил} -амин; (7-аллилпирроло[2,3-d]пиримидин- 4-ил)-(3-этинилфенил)-амина гидрохлорид; (3-этинилфенил)-(7-метилпирроло[2,3-d]nиpимидин-4-ил)-амина гидрохлорид; (5-бром-7H-пирроло[2,3-d] пиримидин-4-ил)-(3-этинилфенил)-амин; (3-этинилфенил)-(5-иод-7H-пирроло[2,3-d] пиримидин-4-ил)-амин; 4-(3-этинилфениламино)-7H-пирроло[2,3-d] пиримидин-5-карбоновая кислота; (3-этинилфенил)-(5-метил-7H-пирроло[2,3-d]пиримидин-4-ил)-амина гидрохлорид; N-(5-иод-7H-пирроло[2,3-d] пиримидин-4-ил)-N-m-толилацетамид; 4-(3-этинилфениламино)-7H-пирроло[2,3-d]пиримидин-5-карбоновой кислоты метилового эфира гидрохлорид; (1H-индазол-5-ил)-(6-метил-пиридо[3,4-d] пиримидин-4-ил)-амина гидрохлорид; бензо[b]тиофен-5-ил-(6-метилпиридо[3,4-d] пиpимидин-4-ил)-амина гидрохлорид; (3-этинил-4-фторфенил)-(6- метилпиридо[3,4-d]пиpимидин-4-ил)-амин; 2-метил-4-(6-метилпиридо[3,4-d] пиримидин-4-иламино)-фенола дигидрохлорид; 4-(4-бром-7-метил-2,3- дигидроиндол-1-ил)-6-метилпиридо[3,4-d]пиpимидинa гидрохлорид; 4-(6-бpoм-7-мeтил-2,3-дигидpоиндoл-1-ил)-6-мeтилпиpидo[3,4-d] пиpимидинa гидрохлорид; 4-(6-бром-5-фтор-2,3-дигидроиндол-1-ил)-6-метилпиридо[3,4-d] пиримидина гидрохлорид; (3-хлор-4-фторфенил)-(6-метилпиридо[3,4-d] пиримидина-4-ил)-амина гидрохлорид; (6-метилпиридо[3,4-d] пиримидин- 4-ил)-(3-трифторметил-фенил)-амина гидрохлорид; (4-фтор-3-метилфенил)-(6- метилпиридо[3,4-d] пиримидин-4-ил)-амина гидрохлорид; 2-иод-4-(6-метилпиридо[3,4-d] пиримидин-4-иламино)-фенола гидрохлорид; (4-бром-3-фторфенил)-(6-метилпиридо[3,4-d] пиримидин-4-ил)-амина гидрохлорид; 4-(6,7-диметил-2,3-дигидроиндол-1-ил)-пиридо[3,4-d] пиримидина гидрохлорид; (3-этинилфенил)-пиридо[3,4-d] пиримидин-4-ил-амина гидрохлорид; бензо[b]тиофен-5-ил-пиридо[3,4-d] пиримидин-4-ил)-амина гидрохлорид; (3-этинилфенил)-(6-метилпиридо[3,4-d] пиримидин-4-ил)-амина гидрохлорид; 4-(6-хлор-2,3-дигидроиндол-1-ил)-6-метилпиридо[3,4-d] пиримидин; (3-этинилфенил)-(5-метилсульфанил-7H-пирроло[2,3-d] пиримидин-4-ил)-амин; (3-этинилфениламино)-(7H-пирроло[2,3-d] пиpимидин-5-ил-карбонитрил; (1H-индол-5-ил)-(6-метилпиридо[3,4-d]пиpимидин-4-ил-амина метансульфонат.
Особенно соединение выбранное из группы, состоящей из следующих соединений:
(1H-индол-5-ил)-(6-метилпиридо[3,4-d] пиримидин-4-ил)-амин; (3-этинилфенил)-(6-метилпиридо[3,4-d] пиримидин-4-ил)-амин; (3-этинилфенил)-(7H-пирроло[2,3-d] пиримидин-4-ил)-амин; (3-хлорфенил)-(7H-пирроло[2,3-d]пиpимидин-4-ил)-амин; (3-этинилфенил)-(5-метил-7H-пирроло[2,3-d]пиримидин-4-ил)-амин; 4-(3-этинилфениламино)-7H-пирроло[2,3-d]пиримидин-5-карбоновой кислоты метиловый эфир; 4-(3-этинилфениламино)-7H-пирроло[2,3-d]пиpимидин- 5-карбонитрил; (1H-индол-5-ил)-пиридо[3,4-d] пиpимидин-4-ил-aмин; (3-хлор-4-фторфенил)-(6-метилпиридо[3,4-d] пиримидин-4-ил)-амин; бензо[b]тиофен-5-ил(6-метилпиридо[3,4-d] пиримидин-4-ил)-амин; (3-этинилфенил)-пиридо[3,4-d]пиримидин-4-ил-амин; (4-фтор-3-метилфенил)-(6-метилпиридо[3,4-d]пиримидин-4-ил)-амина гидрохлорид; 4-(6-хлор-2,3-дигидроиндол-1-ил)-пиридо[3,4-d]пиримидин (пиридо[3,4-d]пиримидин-4-ил)-(m-толил)амин; (6-метилпиридо[3,4-d]пиримидин- 4-ил)-(3-трифторметилфенил)-амина гидрохлорид; (1H-индазол-5-ил)-(6- метилпиридо[3,4-d]пиpимидин-4-ил)-амин; (3-этинилфенил)-(5-метилсульфанил- 7H-пирроло[2,3-d] пиpимидин-4-ил)-амин; (1H-индол-5-ил)-(7H-пирроло[2,3-d] пиpимидин-4-ил)-амин; (5-бром-7H-пирроло[2,3-d]пиримидин-4-ил)-(3- этинилфенил)-амин; (3-этинилфениламино)-(7H-пирроло[2,3-d]пиримидин- 5-ил-карбонитрил.
Изобретение предоставляет также соединения формулы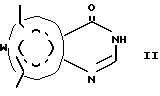
в которой W в направлении, указанном стрелками, выбран из =CHC(CH3)-NCH= , -CH= N-C(CH3)=CH- и =C(CH3)-NH-CH=, а пунктирный круг указывает на то, что кольцо является ароматическим. Согласно еще одному аспекту изобретения предоставляется соединение, описанное выше, в котором W представляет -CH= C(CH3)-N=CH-.
Еще один аспект изобретения предоставляет соединение, описанное выше, в котором W представляет -CH2=N-C(CH3)=CH-.
Еще один аспект изобретения предоставляет соединение, описанное выше, в котором W представляет =C(CH3)-NH-CH=.
Данное изобретение предоставляет также способ лечения гиперпролиферативных расстройств, который включает назначение для приема млекопитающему, нуждающемуся в таком лечении гиперпролиферативных нарушений, количества соединения формулы 1 эффективного для лечения.
Согласно еще одному аспекту изобретения предоставляется способ, описанный выше, при котором гиперпролиферативным заболеванием является рак.
Еще один аспект изобретения предоставляет способ, описанный выше, при котором заболеванием является рак мозга, легких, сквамозных клеток, мочевого пузыря, желудка, панкреатический рак, рак печени, почек, толстой кишки, груди, головы, шеи, эзофагеальный, гинекологический рак или рак щитовидной железы.
Следующий аспект изобретения предоставляет способ, описанный выше, при котором гиперпролиферативным нарушением или расстройством является заболевание неракового характера.
Еще один аспект изобретения предоставляет способ, описанный выше, при котором нераковым пролиферативным нарушением является псориаз или доброкачественная гиперплазия простаты.
Изобретение далее предоставляет фармацевтическую композицию для лечения гиперпролиферативного расстройства у млекопитающих, которая включает соединение формулы 1 в количестве эффективном для лечения гиперпролиферативного заболевания и фармацевтически приемлемый носитель.
В настоящей заявке некоторые термины, которые используются, определяются следующим образом:
Под термином галоид имеется в виду хлор, бром, иод или фтор.
Алкил означает углеводородную группу с прямой цепью, или, когда количество атомов углерода составляет 3 или выше, циклическую или разветвленную углеводородную группу, необязательно ненасыщенную.
Используемое здесь выражение "реакционно инертный растворитель" относится к растворителю, который не взаимодействует с исходными материалами, реагентами, промежуточными соединениями или продуктами каким-либо образом, который пагубно влияет на выход желаемого продукта.
Другие признаки и преимущества изобретения очевидны из описания и формулы изобретения, которые отражает изобретение.
Подробное описание изобретения.
Соединения формулы 1 или их фармацевтически приемлемые соли и пролекарства могут быть получены с помощью любого известного способа, который может быть применим для получения химически родственных соединений.
Как показано на схеме (см. в конце текста), соединения формулы 1 могут обычно получаться из 4-хлор или гидрокси производных соответствующим образом замещенных сконденсированных с гетероарилом пиримидинов 1 с использованием соответствующего замещенного амина ZH2.
В типичном случае пиримидин 1 сконденсированный с соответствующим образом замещенным 4-галоидгетероарилом (или пиримидин, сконденсированный с гетероарилом, несущий подходящую замещаемую уходящую группу в 4-положении, такую как арилокси, алкил-сульфинилокси, такую как трифторметансульфонилокси, арилсульфинилокси, силокси, циано, пиразоло, триазоло или тетразоло), предпочтительно галоидгетероарильной, таким как 4-хлоргетероарильное производное, подвергается реакции с соответствующим амином 2 в растворителе, таком как (1-6)C-спирт, диметилформамид (ДМФ), N-метилпирролидин-2-он, хлороформ, ацетонитрил, тетрагидрофуран (ТГФ), диметилсульфоксид (ДМСО) 1,4-диоксан, пиридин или другой апротонный растворитель. Реакция сочетания или присоединения может проводиться в присутствии основания, предпочтительно карбоната или гидроокиси щелочного или щелочно-земельного металла, или третичного аминового основания, такого как пиридин, 2,6-лютидин, коллидин, N-метил-морфолин, триэтиламин, диэтилизопропиламин, 4-диметиламино-пиридин или N,N-диметиланилин. Эти основания называются здесь далее "подходящими основаниями". Смесь поддерживается при температуре от температуры окружающей среды до температуры дефлегмации, предпочтительно примерно от 35oC до температуры дефлегмации, до тех пор, пока по существу уже не сможет обнаруживаться остающийся сконденсированный с 4-галоидгетероарилом пиримидин, в типичном случае примерно от 2 часов до 72 часов. Реакция предпочтительно проводится в инертной атмосфере, такой как азотный газ. В случае пирролопиримидинов реакцию предпочтительно осуществлять в метаноле при температуре примерно 90-140oC в герметизированной трубке в отсутствии дополнительного основания.
Обычно реагенты сочетаются в стехиометрическом количестве, когда используется подходящее аминовое основание, хотя, для тех соединений, для которых используется соль (обычно HCl соль) амина, предпочтительно использовать избыток амина 2, обычно дополнительный эквивалент амина 2. Альтернативно, если аминовое основание не используется, может использоваться избыток аминового реагента.
Для тех соединений, в случае которых используется стерически или пространственно затрудненный амин (такой как 2-алкил-индолин) или очень реакционноспособный сконденсированный с 4-галоидгетероарилом пиримидин, предпочтительно использовать трет-бутиловый спирт или полярный апротонный растворитель, такой как диметилформамид или N-метилпирролидин-2-он, в качестве растворителя.
Другие соединения формул 1 могут получаться с помощью следующих реакций после указанного выше сочетания.
Соединения формулы 1, в которой R3 или R5 представляет первичный амино или гидроксиамино, могут быть получены с помощью восстановления соединений формулы 1, в котором R3 или R5 представляет нитро группу.
Восстановление может удобно осуществляться с помощью любой из многих процедур, известных для таких преобразований. Восстановление может, например, осуществляться с помощью гидрирования раствора нитро соединения в реакционно инертном растворителе в присутствии подходящего металлического катализатора, такого как палладий или платина. Дополнительным подходящим восстанавливающим агентом является, например, активированный металл, такой как активированное железо (получаемое с помощью порошка железа разбавленным раствором кислоты, такой как соляная кислота). Так, например, восстановление может осуществляться с помощью нагревания смеси нитро соединения и активированного металла в растворителе, таком как смесь воды и спирта, например, метанола или этанола, до температуры в интервале, например, 50-150oC, обычно при равной или близкой к 70oC температуре.
Для получения соединений формулы 1, в которой R5 или R6 включает в себе первичный или вторичный амино фрагмент (иной, чем амино группа, предназначенная для реакции с хиназолином), такой свободный амин предпочтительно защищается перед описанной выше реакцией с последующим снятием защиты после описанной выше реакции с 4-галоидхиназолином.
Что касается описания защитных групп и их использования, смотри работу T. W. Greene и P.G.M. Wuts, "Protective Groups in Organic Synthesis" второе издание, Джон Вили энд Санз, Hью-Йорк, 1991.
Защитные группы азота являются хорошо известными в технике и включают (1-6) C-алкоксикарбонил, необязательно замещенный бензилоксикарбонил, арилоксикарбонил, тритил, винилоксикарбонил, O-нитрофенилсульфонил, дифенилфосфинил, п-толуолсульфонил и бензил. Добавление защитной группы азота может осуществляться в хлорированном углеводородном растворителе, таком как метиленхлорид или 1,2-дихло-хлорэтан, или в простом эфирном растворителе, таком как диглим, глим или ТГФ, в присутствии или в отсутствии третичного аминового основания, такого как триэтиламин, диизопропилэтиламин или пиридин, предпочтительно, триэтиламина, при температуре примерно от 0oC до 50oC, предпочтительно примерно при температуре окружающей среды. Альтернативно, защитные группы удобным образом присоединяются с использованием условий Шоттен-Бауманна.
После описанной выше реакции присоединения амина защитная группа может удаляться с помощью методов деблокирования известных специалистам в данной области техники, таких как с использованием трифторуксусной кислоты в метиленхлориде, для защищенных трет-бутоксикарбонилом продуктов.
Соединения формулы 1, в которой R3 представляет гидрокси, могут предпочтительно получаться с помощью расщепления соединения формулы 1, в которой R3 представляет (1-4)C-алкокси.
Реакция расщепления может удобно осуществляться с помощью любой из многих процедур, известных для такого преобразования. Для реакций O-дезалкилирования может применяться обработка сконденсированного с гетероарилом пиримидинового производного формулы 1 расплавленным хлоргидратом пиридина (20-30 экв. ). Альтернативно, реакция может осуществляться, например, с помощью обработки сконденсированного с гетероарилом пиримидинового производного (1-4)C-алкилсульфидом щелочного металла, таким как этантиолат натрия, или, например, с помощью обработки диарилфосфидом щелочного металла, таким как дифенилфосфид лития. Альтернативно реакция расщепления может удобно осуществляться, например, с помощью обработки сконденсированного с гетероарилом пиримидинового производного тригалогенидом бора или алюминия, таким как трехбромистый бор. Такие реакции предпочтительно осуществляются в присутствии реакционного инертного растворителя и при подходящей температуре.
Для получения соединений формулы 1, в которой R3 представляет (1-4)C-алкилсульфинил или (1-4)C-алкилсульфонильную группу, предпочитается окисление соединения формулы 1, в которой R3 представляет (1-4)C-алкилтио группу.
Подходящим окисляющим агентом является, например, агент, известный в технике для окисления тио в сульфинил и/или сульфонил, например, перекись водорода, надкислота (такая как 3-хлорнадоксибензойная кислота или пероксиуксусная кислота), пероксисульфат щелочного металла (такой как пероксимоносульфат калия), трехокись хрома или газообразный кислород в присутствии платины. Окисление обычно осуществляется в как можно мягких условиях и с требуемым стехиометрическим количеством окисляющего агента для того, чтобы снизить риск сверх окисления и вреда для других функциональных групп. Обычно реакция осуществляется в подходящем растворителе, таком как метиленхлорид, хлороформ, ацетон, тетрагидрофуран или трет-бутилметиловый эфир, и при температуре, например, -25 oC 50oC, удобным образом при равной или близкой к температуре окружающей среды, то есть в интервале 15-35oC. Когда требуется соединение, несущее сульфинильную группу, может использоваться более мягкий окисляющий агент, например, метапериодат натрия или калия, удобным образом в полярном растворителе, таком как уксусная кислота или этанол. Очевидно понятно, что, когда требуется соединение формулы 1, содержащее (1-4)C-алкилсульфонильную) группу, оно может получаться с помощью окисления соответствующего (1-4)C-алкилсульфинильного соединения, а также соответствующего (1-4)C-алкилтио соединения.
Для получения тех соединений формулы 1, в которой R3 - (2-4)C-алканоиламино, или замещенный (2-4)C-алканоиламино, уреидо, 3-фенилуреидо, бензамидо или сульфонамидо, подходящим является ацилирование или сульфонилирование соединения формулы 1, в которой R3 представляет амино.
Подходящим ацилирующим агентом является, например, любой агент известный в технике для ацилирования амино в ациламино, например, ацилгалогенид (например, (2-4)C-алканоилхлорид или бромид, или бензоил-хлорид или -бромид), ангидрид алкановой кислоты или смешанный ангидрид (например, (2-4)C-алкановокислотный ангидрид, такой как уксусный ангидрид или смешанный ангидрид, образуемый по реакции алкановой кислоты и (1-4)C-алкоксикарбонилгалогенида, например, (1-4)C-алкоксикарбонилхлорида, в присутствии подходящего основания). Для получения соединений формулы 1, в которой R1 представляет уреидо или 3-фенилуреидо, подходящим ацилирующим агентом является, например, цианат, например, цианат щелочного металла, такой как цианат натрия, или например, изоцианат, такой как фенилизоцианат. Реакции N-сульфонилирования могут осуществляться с использованием подходящих сульфонилгалогенидов или сульфонилангидридов, в присутствии третичного аминового основания. Обычно ацилирование или сульфонилирование осуществляется в реакционно инертном растворителе при температуре в интервале, например, от -30 до 120oC, удобно при температуре близкой или равной температуре окружающей среды.
Для получения соединений формулы 1, в которой R3 представляет (1-4)C-алкокси или замещенный (1-4)C-алкокси; или R1 представляет (1-4)C-алкиламино или замещенный моно-N- или ди-N,N-(1-4)C-алкиламино, предпочитается алкилирование, предпочтительно, в присутствии подходящего основания, соединения формулы 1, в которой R1 представляет гидрокси или амино, как это соответствует.
Подходящим алкилирующим агентом является, например, любой агент известный в технике для алкилирования гидрокси в алкокси или замещенный алкокси, или для алкилирования амино в алкиламино, например, алкил- или замещенный алкил-галогенид, например, (1-4)C-алкилхлорид, бромид или иодид, или замещенный (1-4)C-алкилхлорид, бромид или иодид, в присутствии подходящего основания в реакционно инертном растворителе, и при температуре в интервале, например, от 10 до 140oC, удобным образом при температуре близкой или равной температуре окружающей среды.
Для получения тех соединений формулы 1, в которой R3 представляет амино-, окси- или циано-замещенный (1-4)C-алкильный заместитель соответствующей является реакция соединения формулы 1, в которой R3 представляет (1-4)C-алкильный заместитель, несущий замещаемую группу, с соответствующим амином, спиртом или цианидом, как это соответствует, предпочтительно, в присутствии подходящего основания.
Реакция предпочтительно осуществляется в реакционно инертном растворителе или разбавителе и при температуре в интервале, например 10-100oC, удобным образом при близкой или равной температуре окружающей среды.
Для получения соединений формулы 1, в которой R3, R5, или R6 представляет карбокси заместитель или заместитель, который включает карбокси группу, желателен гидролиз соединения формулы 1, в которой R3, R5, R6 представляет (1-4)C-алкоксикарбонильный заместитель или заместитель, который включает (1-4)C-алкоксикарбонильную группу.
Гидролиз может удобно проводиться, например, в основных условиях, таких как гидролиз, осуществляемый с помощью гидроокиси щелочного металла, как иллюстрируется в сопровождающих примерах.
Для получения соединений формулы 1, в которой R3 представляет амино, (1-4)C-алкиламино, ди-((1-4)C-алкил) амино, пирролидин-1-ил, пиперидино, морфолино, пиперазин-1-ил, 4-(1-4)C-алкилпиперазин-1-ил или (1-4)C-алкилтио, предпочитается реакция соединения формулы 1, в которой R3 замещаемая группа, с соответствующим амином или тиолом, удобным образом, в присутствии подходящего основания.
Реакция предпочтительно осуществляется в реакционно инертном растворителе или разбавителе и при температуре в интервале, например, 10-180oC, удобным образом в интервале 100-150oC.
Для получения соединений формулы 1, в которых R3 представляет 2-оксопирролидин-1-ил или 2-оксопиперидин-1-ил, удобной является циклизация в присутствии подходящего основания, соединения формулы 1, в которой R3 - галоид-(2-4)C-алканоиламино группа.
Реакция предпочтительно осуществляется в реакционно инертном растворителе или разбавителе, при температуре в интервале, например, от 10 до 100oC, удобным образом при равной или близкой к температуре окружающей среды.
Для получения соединений формулы 1, в которой R3 - карбамоил, замещенный карбамоил, алканоилокси или замещенный алканоилокси, удобным является карбамоилирование или ацилирование соединений формулы 1, в которой R3 представляет гидрокси.
Подходящими ацилирующими агентами являются, например, любые агенты известные в технике для ацилирования гидроксиарильных фрагментов в алканоилоксиарил. Например, могут применяться (2-4)C-алканоилгалогениды, (2-4)C-алканоилангидриды или смешанные ангидриды, и их подходящие замещенные производные, типичным образом в присутствии подходящего основания. Альтернативно, (2-4)C-алкановые кислоты или замещенные подходящим образом производные их могут сочетаться с соединениями формулы 1, в которой R3 представляет гидрокси, с помощью конденсирующего агента, такого как карбодиимид. Для получения соединений формулы 1, в которой R3-карбамоил или замещенный карбамоил, подходящими карбамоилирующими агентами являются, например, цианат или алкил- или арил-изоцианат, типичным образом в присутствии подходящего основания. Альтернативно, подходящее промежуточное соединение, такое как хлорформиат или имидазолилкарбонильное производное сконденсированного с гетероарилом пиримидина формулы 1, в которой R3 представляет гидрокси может получаться, например, с помощью обработки указанного производного фосгеном (или эквивалентом фосгена) или карбонилдиимидазолом. Получающееся в результате промежуточное соединение может затем подвергаться реакции с соответствующим амином или замещенным амином с получением желаемых карбамоильных производных.
Для получения сконденсированных с гетероарилом производных пиримидина формулы 1, в которой R3 - аминокарбонил или замещенный аминокарбонил, предпочтительным является аминолиз подходящего промежуточного соединения, производимого из сконденсированного с гетероарил-сконденсированного пиримидина формулы 1, в которой R3 представляет карбокси.
Активирование или сочетание соединения формулы 1, в которой R3 представляет карбокси, может проводиться с помощью разнообразия способов известных специалистам в данной области. Подходящие способы включают активирование карбоксила, такого как галоидангидрид кислоты, азид кислоты или симметричный или смешанный ангидрид, или активный сложный эфир соответствующей реакционноспособности, для сочетания с желаемым амином. Примеры таких типов промежуточных соединений и их получение и использование в реакциях присоединения с аминами, можно найти широко в литературе; например, в работе М.Бодански и А. Бодански, "The Practice of Peptide Synthesis" Спринглер-Верлаг, Нью-Йорк, 1984.
Получающиеся в результате соединения формулы 1, могут выделяться и очищаться с помощью стандартных методов, таких как удаление растворителя и перекристаллизация или хроматография, если необходимо.
Необязательно замещенные индолы и индолины, полезные при осуществлении изобретения, и способы их получения, описываются в находящейся совместно на рассмотрении заявке США N 08/200359, приведенной здесь для сведения. В дополнение к способам, описанным там, получение различных индолинов, индолов, оксиндолов и изатинов, полезных в качестве промежуточных соединений дополнительно описывается в публикации "Heterocyclic Compounds with Indole and Carbazole Systems". W.C. Sumpter и F.M Miller, в томе 8 серии "The Chemistry of Heterocyclic Compounds", Interscience Publishers Inc., Нью-Йорк, 1954, и ссылках, приведенных там.
Замещенные анилины, полезные в практическом воплощении изобретения и способы их получения описываются в находящейся совместно на рассмотрении заявке США 08413300 и международной заявке N PCT/IB95/00436, приведенных здесь для сведения.
Соединения формулы ZH, в которой ZH представляет QNH2, используемые в практическом воплощении изобретения, и способы их получения описываются в европейской патентной заявке N EP 0496617 A1, приведенной здесь для сведения.
Некоторые соединения формулы 1 могут существовать в сольватированной также как и в несольватированной формах, таких как, например, гидратированные формы. Следует понимать, что данное изобретение охватывает все такие сольватированные, а также несольватированные формы, которые обладают активностью в отношении гиперпролиферативных заболеваний.
Подходящими фармацевтически приемлемыми солями сконденсированных с гетероарилом пиримидиновых производных данного изобретения являются, например, кислотно-аддитивные соли сконденсированных с гетероарилом пиримидиновых производных изобретения, которые являются достаточно основными (щелочными), например, кислотно-аддитивные соли, например, с неорганическими или органическими кислотами, например, соляной, бромистоводородной, серной, фосфорной, метансульфоновой, бензолсульфоновой, трифторуксусной, лимонной, молочной или малеиновой кислотой. В дополнение к сказанному подходящими фармацевтически приемлемыми аддитивными солями основания пиримидиновых производных изобретения, сконденсированных с гетероарилом, которые являются достаточно кислыми, являются соли щелочных металлов, например, литиевая, натриевая или калиевая соль; соли щелочно-земельных металлов, например, кальциевая или магниевая соль; аммониевые соли; или соли с органическими основаниями, которые дают фармацевтически приемлемый катион, например, соль с метиламином, диметиламином, триметиламином, пиперидином, морфолином или трис-(2-гидроксиэтил) амином. Все такие соли охватываются объемом данного изобретения, и они могут получаться с помощью общепринятых способов. Например, они могут получаться просто путем введения в контакт кислотного и основного фрагментов, обычно в стехиометрическом соотношении, или в водной, неводной, или частично водной среде, как это подходит. Соли выделяются или с помощью фильтрования, или осаждением нерастворителем, предпочтительно эфирным или углеводородным растворителем, с последующим фильтрованием, или путем выпаривания растворителя, или в случае водных растворов, путем лиофилизации.
Некоторые из соединений формулы 1 имеют асимметрические атомы углерода. Такие диастереомерные смеси могут разделяться на индивидуальные диастереомеры на основе их физико-химических различий с помощью способов, известных самих по себе, например, с помощью хроматографии и/или фракционной кристаллизации. Энантиомеры могут разделяться с помощью превращения энантиомерной смеси в диастереомерную смесь с помощью реакции с подходящим оптически активным соединением (например, спиртом), разделения диастереомеров и превращения (например, гидролизом) индивидуальных диастереомеров в соответствующие чистые анатиомеры. Все такие изомеры, включая диастереомеры и энантиомеры, считаются частью данного изобретения.
Соединения данного изобретения являются сильными ингибиторами онкогенных и протоонкогенных белковых тирозинкиназ семейства erb, таких, как рецептор фактора эпидермального роста (EGFR), erbB2, HER3, или HER4, и таким образом все могут найти терапевтическое использование в качестве противопролиферативных агентов (например, противораковых) для млекопитающих, в частности людей. В частности, соединения данного изобретения являются терапевтическими или профилактическими агентами для лечения разнообразных опухолей человека (ренальных, печени, почек, мочевого пузыря, груди, желудка, яичников, толстой кишки, простаты, панкреатической опухоли (поджелудочной железы), легких, вульвы, щитовидной железы, гепатической карциномы, саркомы, глиобластомы, различных опухолей головы и шеи), и других гиперпластических состояний, таких как доброкачественная гиперплазия кожи (например, псориаз) или простаты (например, BPH). Кроме того, ожидается, что гетероарилсконденсированные пиримидины настоящего изобретения могут обладать активностью против ряда лейкемий и лимфоидных злокачественных образований.
Можно также ожидать, что соединения формулы 1, могут быть полезными при лечении дополнительных нарушений, при которых вовлечены аберрантная экспрессия лиганд-рецепторных взаимодействий, активирование или сигнальные события, связанные с различными белковыми тирозинкиназами, (например, IGF-рецепторами), активность которых ингибируется соединениями формулы 1.
Такие нарушения могут включать нарушения нейронального, глиального, астроцитарного, гипоталамического характера и другой гландулярной, макрофаговой, эпителиальной, стромальной и бластоколической природы, при которых могут быть вовлечены аберрантная функция, экспрессия, активация или сигнализирование тирозинкиназы. В дополнение к сказанному соединения формулы 1 могут иметь терапевтическую полезность при воспалительных, ангиогенных и иммунологических расстройствах, при которых вовлечены, как идентифицированные, так и еще неидентифицированные тирозинкиназы, которые ингибируются соединениями формулы 1.
Активность ин витро данных соединений в ингибировании рецепторных тирозинкиназ (и таким образом последующего пролиферативного ответа, например рака) может определяться с помощью процедуры, описанной подробно ниже. Активность соединений формулы 1 ин витро может определяться по степени ингибирования фосфорилирования экзогенного субстрата (например, Lys3 - Гастрин или поли GluTyr (4:1) рэндом сополимер (1. Posner и др., J. Biol. Chem. 267 (29), 20638-47 (1992)), по тирозину под действием рецепторной киназы фактора эпидермального роста с помощью испытываемого соединения относительно контроля. Очищенный от аффинности растворимый рецептор человека EGF (96 нг) получается в соответствии с процедурой, описанной в работе G.N. Gill, W. Weber, Methods in Enzymology 146, 82-88 (1987) из клеток A431 (Американской коллекции культур, Роксилл, МД) и предварительно инкубировался в мицкроцентрифуге с EGF (2 мкг/мл) в смеси буфер фосфорилирования + ванадат (PBV : 50 мМ HEPES, pH 7.4; 125 мМ NaCl; 24 мМ MgCl2; 100 мкМ ортованадат натрия), в общем объеме 10 мкл, в течение 20-30 минут при комнатной температуре. Испытываемое соединение, растворенное в диметилсульфоксиде (ДМСО), разбавляется в PBV, и 10 мкл смешивается со смесью EGF рецептор/EGF, и инкубируется в течение 10-30 минут при 30oC. Реакция фосфорилирования инициируется добавлением 20 мкл смеси 33P-ATP/субстрат (120 мкМ Lys3-Гастрин (последовательность аминокислот обозначенная буквенным кодом, KKKGPWLEEEEEAYGWLDF), а 50 мМ Hepes pH 7.5, 40 мкМ ATP, 2 мк Ci V-[33P]-ATP) к смеси EGFr/EGF и инкубируется в течение 20 минут при комнатной температуре. Реакция останавливается добавлением 10 мкл стоп-раствора (0.5 М EDTA, pH 8; 2 мМ ATP) и 6 мкл 2 норм. HCl. Трубки центрифугируются при 1400 оборот. в мин. 4oC, в течение 10 минут. 35 мкл супернатанта на каждой пробирке отбирается пипеткой на 2.5 см кружок бумаги Ватман Р81, промывается четыре раза 5% уксусной кислотой, 1 литр на промывку, и затем сушится на воздухе. Это приводит в результате к связыванию субстрата с бумагой с потерей свободного ATP при промывке. Введенный [33P] измеряется с помощью жидкостного сцинтилляционного подсчета. Введение в отсутствии субстрата (например, lyys3-гастрин) вычитается из всех величин в качестве основы, и процент ингибирования вычисляется по сравнению с контролем, в случае которого испытываемое соединение не присутствует.
Такие анализы, осуществляемые в интервале доз испытываемых соединений, позволяют определить приблизительные IC50 величины ингибирования ин витро активности EGFR киназы. Хотя ингибирующие свойства соединений формулы 1 изменяются в зависимости от структурных изменений, как это и ожидается, в общем, активность, проявляемая этими агентами, определенная описанным выше образом, находится в интервале IC50=0.0001-30 мкМ.
Активность соединений формулы 1 ин витро может быть определена по степени ингибирования роста опухоли с помощью испытываемого соединения относительно контроля. Ингибирующее действие различных соединений на рост опухоли измеряется в соответствии с методиками авторов Corbett T.H. и др. "Tumor Induction Relationship in Development of Transplantable Cancers of the Colon in Mice for Chemotherapy Assays, with a Note on Carcinogen Structure, Cancer Res. , 35, 2434-2439 (1975) и Corbett T.H. и др., "A Mouse Colon-tumor Model for Experimental Therapy", Cancer Chemother. Pep. (Part 2)" 5, 169-186 (1975), с незначительными видоизменениями. Опухоли индуцируются на левом боку с помощью подкожных инъекций 1•106 выращенных опухолевых клеток в log фазе (клетки карциономы груди человека MOA-MB-468 или головы и шеи человека HN5), суспендированных в 0.10 мл RPM1 1640. После прохождения достаточного периода времени, для того, чтобы опухоли стали ощутимыми (2-3 мм в диаметре), испытываемых животных (атимические мыши) подвергали лечению соединением (подготовленным в виде препаративной формы путем растворения в ДМСО типичным образом в концентрации 50-100 мг/мл с последующим 1:9 разбавлением в 0.1% Рлуроник P105 в 0.9% солевом растворе) интраперитональным (iр) или оральным (po) способами путем введения дважды ежедневно (т.е., каждые 12 часов) в течение 5-20 последовательных дней. Для того, чтобы определить противо-опухолевый эффект, опухоль измеряется в миллиметрах с использованием циркулей Вернье по двум диаметрам, и размер опухоли (мг) (т.е. вес опухоли TuW) вычисляется с использованием формулы: Вес опухоли = (длина • [ширина]2)/2, в соответствии с методами Geran R.I., и др. Protocols for Screening Chemical Agents and Natural Products Against Animal Tumors and other Biological Systems". Третье издание Cancer Chemother. Результаты выражаются в виде процента ингибирования по формуле: Ингибирование (%) = (TuWконтроля - TuWиспытыв.)/TuWконтроля • 100%. Участок имплантации опухоли в боку дает воспроизводимые эффекты доза/ответ для различных хемотерапевтических агентов, а способ измерения (по диаметру опухоли) является надежным методом оценки степени роста опухоли.
Назначение для приема соединений данного изобретения может производиться для приема любым способом, который обеспечивает возможность доставки соединений к участку действия (например, к раковым клеткам). Эти способы включают оральные пути, интрадуоденальные пути введения, парэнтеральные инъекции (включая внутривенную, подкожную, внутримышечную, внутрисосудистую или вливание), топическое или местное назначение и проч.
Назначаемое количество гетроарил-сконденсированного пиримидинового производного будет, конечно зависеть от субъекта, подвергаемого лечению, от тяжести заболевания, от способа назначения и от опыта и знаний предписывающего лечащего врача. Однако, эффективная дозировка составляет в интервале приблизительно 0.1-100 мг/кг, предпочтительно от 1 до 35 мг/кг в виде одной или раздельных доз. Для человека среднего веса 70 кг данное количество будет составлять до 0.05-7 г/день, предпочтительно от 0.2 до 2.5 г/день.
Композиция может быть, например, в форме подходящей для орального назначения в виде таблеток, капсул, пилюль, порошков, препаративных форм с замедленным высвобождением активного агента, в виде растворов, суспензий, для перэнтеральных инъекций - в виде стерильного раствора, суспензии или эмульсии, для топического назначения в виде мази или крема, или для ректального назначения в виде суппозиторий или медицинских свечей. Фармацевтическая композиция может быть в форме единичных доз, подходящих для разового приема в предписанных дозировках. Фармацевтические композиции включают обычный фармацевтический носитель или эксципиент и соединение согласно изобретению в качестве активного ингредиента. В дополнение они могут включать другие медицинские или фармацевтические агенты, носители, адъюванты и др.
Фармацевтическая композиция согласно изобретению может содержать 0.1-95% соединения, предпочтительно 1-70%. В любом случае композиция или препаративная форма, предназначенная для введения, содержит соединение согласно изобретению в количестве эффективном для облегчения или уменьшения признаков заболевания у субъекта, подвергаемого лечению, т.е. пролиферативных заболеваний, в ходе лечения.
Примеры парентеральных форм назначения включают растворы или суспензии соединения согласно изобретению формулы 1 в стерильных водных растворах, например, применяются водные растворы пропиленгликоля или декстрозы. Такие дозированные формы могут быть подходящим образом забуферены, если необходимо.
Подходящие фармацевтические носители включают инертные разбавители или наполнители, воду и различные органические растворители. Эти фармацевтические композиции могут, если необходимо, содержат дополнительные ингредиенты, такие как вкусовые или ароматизирующие добавки, связующие, эксципиенты и аналогичные. Таким образом для орального приема таблетки, содержащие различные эксципиенты, такие как лимонная кислота, могут применяться вместе с различными дезинтегрирующими агентами, такими как крахмал, альгиновая кислота и некоторые комплексные силикаты, и со связующими агентами, такими как сахароза, желатин и камедь акации. Дополнительно для целей таблетирования часто полезными являются смазочные агенты, такие как стеарат магния, лаурилсульфат натрия и тальк. Твердые композиции аналогичного типа могут также применяться в виде заполненных мягких или твердых желатиновых капсул. Предпочтительные материалы следовательно включают лактозу или молочный сахар и полиэтиленгликоли с высоким молекулярным весом. Когда для орального приема желательны водные суспензии или эликсиры, существенный активный ингредиент в них может комбинироваться с различными подслащивающими или вкусовыми агентами, красящими веществами или красителями и, если необходимо, эмульгирующими или суспендирующими агентами, вместе с разбавителями, такими как вода, этанол, пропиленгликоль, глицерин или их сочетания.
Способы получения различных фармацевтических композиций с определенным количеством активного ингредиента известны или очевидны для специалистов в данной области. Например, смотрите публикацию Remington's Pharmaceutical Sciences, Mack Publishing Company, Easter, PA., 15-е издание (1975).
Противораковое лечение, описанное выше, может применяться в виде единственного метода терапии или может включать в дополнение к гетероарил-сконденсированному пиримидиновому производному данного изобретения одно или более других противораковых веществ. Такое совместное лечение может достигаться путем одновременного, последовательного, циклического или раздельного дозирования индивидуальных компонентов лечения.
Должно быть понятно, что изобретение не ограничивается конкретными воплощениями, показанными и описанными здесь, и могут производиться различные изменения и модификации без отклонения от сути и объема данной новой концепции изобретения, определенного приведенными ниже пунктами формулы изобретения.
Пример 1. Хлоргидрат (3-этинил-фенил)-(7H-пирроло[2,3-d]пиримидин- 4-ил)-амина.
К 4-хлор-7H-пирроло[2,3-d]пиримидину (10,0 г, 0,065 моля) в сухом пиридине (90 мл) добавлялся м-аминофенилацетилен (9.2 г, 0,078 моля), и смесь нагревалась на масляной бане с температурой 85oC в течение 2 дней. Реакционная смесь охлаждалась до температуры окружающей среды и концентрировалась в вакууме. Получающийся остаток очищался с помощью флеш хроматографии на силикагеле (375 г, 40 мкм меш) с использованием 5% метанол/метиленхлорид, давая целевое соединение в виде розово-оранжевого твердого вещества (1.9 г, 12%). HRMS (масс-спектр высокого разрешения). Вычислено 235.0984, найдено 235.1000; анал. RP18-ВЭЖХ время удерживания: (RT) 3.48 мин.
Указанное выше соединение растворялось в минимальном количестве метанола, и по каплям добавлялся раствор HCl в этиловом эфире (HCl, барботированный в 2 мл этилового эфира) до тех пор, пока смесь не становилась мутной. Выпавшая в осадок HCl соль сушилась в вакууме, промывалась один раз этиловым эфиром, и сушилась в вакууме до постоянной массы. Т.пл.: 196-198oC.
Пример 2. (3-Хлор-фенил)-(7-пиppoлo[2,3-d]пиримидин-4-ил)-амин.
Следуя процедуре, описанной в примере 1, получали целевое соединение из 4-хлор-7H-пирроло[2,3-d] пиримидина и 3-хлоранилина (3.4 %). LS-MS : 245 (MH+); анализ. RP-18 ВЭЖХ RT: 3.74 мин.; HCl соль т. пл.: 227-228oC.
Пример 3. Хлоргидрат 4-(6-хлор-2,3-дигидро-индол-1-ил)-7H- пирроло[2,3-d]пиримидина.
В соответствии с процедурой, описанной в примере 1, целевое соединение получалось из 4-хлор-7H-пирроло[2,3-d] пиримидина и 6-хлор-2,3-дигидроиндол-1-ила (4.3%). HRMS: Вычислено 271.0750, найдено: 271.0729; анил. RP18-ВЭЖХ RT: 4.88 мин., HCl соль т. пл. 266oC (разл.).
Пример 4. Хлоргидрат (7H-пирроло[2,3-d]пиримидин-4-ил)(M-толил)-амина.
В соответствии с процедурой, описанной в примере 1, целевое соединение получалось из 4-хлор-7H-пирроло[2,3-d]пиримидина и т-толуидина (34%). HRMS: Вычислено: 225.1140, Найдено: 225.1131; анал. RP18-ВЭЖХ RT: 3,45 мин.; HCl соль т. пл.: 219oC.
Пример 5. Хлоргидрат (1H-индол-5-ил)-(7H-пирроло[2,3-d]пиримидин- 4-ил)-амина.
Целевое соединение получалось из 4-хлор-7H-пирроло[2,3-d]пиримидина и 5-аминоиндола (7%) в соответствии с процедурой, описанной в примере 1. HRMC: Вычислено: 250.1093, Найдено: 250.1081; анал. RP18-ВЭЖХ RT: 2,58 мин.; HCl соль т. пл.: 218-221oC.
Пример 6. (Фенил)-(7-пирроло[2,3-d]пиримидин-4-ил)-амин.
С использованием процедуры, аналогичной процедуре, описанной в примере 1, данный продукт получался с выходом 16% из 4-хлор-7H-пирроло[2,3-d]пиримидина (1.0 экв. ) и анилина (5.0 экв.) в пиримидине. (Т. пл. 234-236oC; GC-MS 211 (MH+); анал. RP18-ВЭЖХ RT: 3.11 мин.).
Соединения примеров 7-10 получались в соответствии с методом примера 1 из соответствующих исходных материалов (см. в конце текста).
Пример 11. Хлоргидрат 1-(4-м-Толиламино)-1-(пирроло[2,3-d] пиримидин- 7-ил)-этанона.
К (3-метил-фенил)-(7H-пирроло[2,3-d] пиримидин-4-ил)-амину (пример 4) (0.168 г, 0,75 ммоля), растворенному в горячем ацетонитриле (7 мл), добавлялся гидрид натрия (36 мг, 0,90 ммоля, 60% дисперсия в минеральном масле). После перемешивания при температуре окружающей среды в течение 0.75 часа добавлялся ацетилхлорид (0.11 мл, 1,5 ммоля), и перемешивание продолжалось в течение 48 часов. Смесь концентрировалась в вакууме, растиралась в горячем этилацетате и фильтровалась. Фильтрат концентрировался в вакууме, давая оранжевый твердый остаток. Твердое вещество растиралось в метиленхлориде и фильтровалось, давая целевое соединение в виде светло-желтого твердого вещества (0.11 г, 55%). LC-MS: 267 (MH+); анал. RP18-ВЭЖХ RT: 3.53 мин.
Пример 12. 5- Иoд-7H-пиppoлo[2,3-d]пиримидин-4-ил)-(м-толил)-амина.
К 1-(4-толиламино-пирроло[2,3-d] пиримидин-7-ил)-этанону (113 мг, 0.42 ммоля) в сухом метаноле (4 мл) и метиленхлориде (1 мл) добавлялся карбонат натрия (45 мг, 0.42 ммоля). После перемешивания при температуре окружающей среды в течение 0.75 часа добавлялся N-иодсукцинимид (190 мг, 0.85 ммоля), и перемешивание продолжалось в течение 48 часов. Смесь концентрировалась в вакууме и распределялась между метиленхлоридом и водой. Органическая фаза промывалась дважды водой, сушилась над сульфатом натрия и концентрировалась в вакууме. Получающийся остаток очищался с помощью флэш хроматографии на силикагеле (7 г, 40 мкм меш) с использованием 2% метанол/метиленхлорид, давая целевое соединение в виде оливковых игл (6 мг, 4%. LC-MS: 351 (MH+); анал. RP18-ВЭЖХ RT: мин).
Пример 13. Хлоргидрат (3-хлор-фенил)-(1H-[1,2,3]триазоло[4,5-d] пиримидин-7-ил)-амина.
К 3-хлоранилину (0,39 мл, 3,6 ммоля) в сухом диметилциклогексиламине (0,55 мл, 3,6 ммоля) добавлялась пятиокись фосфора (0,52 г, 3,6 ммоля). После нагревания на масляной бане с температурой 170oC в течение 0.5 часа добавлялся 8-азагипоксиксантин (0.50 г, 3,6 ммоля), и перемешивание продолжалось при 170oC в течение 23 часов. Смесь охлаждалась до температуры окружающей среды, и добавлялась 2М гидроокись натрия, до тех пор, пока среда не становилась щелочной. Твердые вещества отфильтровывались и промывались последовательно водой, метиленхлоридом и метанолом. Получающееся в результате твердое вещество сушилось в вакууме, давая целевое соединение в виде рыжевато-коричневого порошка (0.26 г, 29%). LC-MS 247 (MH+); анал. RP18-ВЭЖХ RT: 3.76 мин.
Пример 14. Хлоргидрат (3-хлор-фенил)-(пиридо[4,3-d]пиримидин-4-ил)-амина.
К 4-гидрокси-пиридо[4,3-d] пиримидину (0.13 г, 0.90 ммоля) в хлорокиси фосфора (2 мл) добавлялся сухой пиридин (0.15 мл, 1.8 ммоля). Подсоединялись конденсор и CaCl2 осушающая трубка, и суспензия нагревалась с обратным холодильником (в условиях дефлегмации) в течение 3 часов. Конечный прозрачный раствор концентрировался в вакууме (CaCl2 осушающая трубка) с последующим пропусканием толуола. Получающийся 4-xлop-пиpидo[4,3-d]пиpимидин растворялся в сухом пиридине (1.5 мл). Добавлялся 3-хлоранилин (0,096 мл, 0.90 ммоля), и смесь нагревалась на масляной бане при 85oC в течение 23 часов. Реакционная смесь охлаждалась до температуры окружающей среды и концентрировалась в вакууме. Масляный остаток распределялся между метиленхлоридом и водой, фильтровался, и водная фаза экстрагировалась метиленхлоридом. Объединенные органические фазы сушились над сульфатом натрия и концентрировались в вакууме. Получающийся остаток очищался с помощью флеш-хроматографии на силикагеле (5 г, 40 мкм меш) с использованием смеси 5% метанол/метиленхлорид, давая целевое соединение в виде не совсем белого твердого вещества (3 мг, 1.3%). LC-MS 257 (MH+); анал. RP18-ВЭЖХ RT: 3.85 мин.
Пример 15. Хлоргидрат (1H-индол-5-ил)-(пиридо[4,3-d]пиримидин- 4-ил)-амина.
К суспензии 4-гидрокси-пиридо[4,3-d]пиримидина (0.103 г, 0,70 ммоля) в сухом пиридине (2 мл), охлаждаемой в водно-ледяной ванне добавлялся по каплям трифторуксусный ангидрид (0.20 мл, 1.4 ммоля). После перемешивания в течение 0,5 часа добавлялся по каплям раствор 5-аминоиндола (0,204 г, 1,5 ммоля) в сухом диметилформамиде (ДМФ) (1.5 мл). Холодная ванна оставлялась подогреваться до температуры окружающей среды, и перемешивание продолжалось в течение 24 часов. Смесь концентрировалась в вакууме и распределялась между метиленхлоридом и водой. Водная фаза экстрагировалась метиленхлоридом, и объединенные органические фазы промывались водой, сушились над сульфатом натрия и концентрировались в вакууме. Получающийся остаток очищался с помощью флэш хроматографии на силикагеле (11 г, 40 мкм меш) с использованием смеси 5% метанол/метиленхлорид давая целевое соединение в виде оранжевого твердого вещества (37 мг, 20%) LC-MS: 262 (MH+); анал. RP18-ВЭЖХ RT: 2.02 мин.
Пример 16. Хлоргидрат (3-Этинилфенил)-(7-метил-пиридо[4,3-d]пиримидин- 4-ил)-амина.
С использованием процедуры, аналогичной процедуре, описанной в примере 15, данный продукт получался с выходом 28% из 4-гидрокси-7-метил-пиридо[4,3-d]пиримидина (1.0 экв.) и м-аминофенилацетилена (40.0 экв.) в пиридине. HCl соль получалась из очищенного свободного основания в соответствии с процедурой, описанной в примере 1. (Т. пл. 240-241oC; GC-MS: 261 (MH+); анал. RP18-ВЭЖХ RT: 3.73 мин.).
Пример 17. Хлоргидрат (3-хлор-фенил)-(7-метил-пиридо[4,3-d]пиримидин- 4-ил)-амина.
С использованием процедуры, аналогичной процедуре, описанной в примере 16, данный продукт получался с выходом 34% из 4-гидрокси-7-метил-пиридо[4,3-d] пиримидина (1,0 экв.) и м-хлоранилина (40,0 экв.) в пиридине. HCl соль получалась из очищенного свободного основания в соответствии с процедурой, данной в примере 1. (Т. пл. 255-256oC; GC-MS: 270 (MH+); анал. RP18-ВЭЖХ RT: 4.05 мин.).
Пример 18. Хлоргидрат (3-этинил-фенил)-(пиридо[4,3-d]пиримидин- 4-ил)-амина.
В соответствии с процедурой, описанной в примере 16 целевое соединение получалось из 4-гидрокси-пиридо[4,3-d] пиримидина и м-аминофенилацетилена (5%) LC-MS: 247 (MH+); анал RP18-ВЭЖХ RT: 3.41 мин.
Соединения примеров 19-24 получались согласно методике примера 15 из соответствующих исходных материалов (см. в конце текста).
Пример 25. (3-Этинил-фенил)-(9H-пурин-6-ил)-амин.
К 6-хлорпурину (1.0 г, 6,5 ммолей) в сухом пиридине (10 мл) добавлялся м-аминофенилацетилен (0.91 г, 7.8 ммоля). Смесь нагревалась на масляной бане с температурой 85oC в течение 23 часов. Смесь охлаждалась до температуры окружающей среды и концентрировалась в вакууме. Масляный остаток распределялся между метиленхлоридом и водой, затем фильтровался, давая целевое соединение в виде светло-оранжевого твердого вещества (50 мг, 3.3%). LC-MS: 236 (MH1); анал. RP18-ВЭЖХ RT: 3.25 мин.
Пример 26. Хлоргидрат (1H-индол-5-ил)(9H-пурин-6-ил)-амина.
В соответствии с процедурой, описанной в примере 9, целевое соединение получалось из 6-хлорпурина и 5-аминоиндола (70%). TS-MS: 251 (MH+); анал. RP18-ВЭЖХ RT: 2.44 мин.
Пример 27. Хлоргидрат (3-хлор-фенил)-(9H-пурин-6-ил)-амина.
Следуя процедуре, описанной в примере 1, целевое соединение получалось из 4-хлор-7H-пирроло[2,3-d]пиримидина и 3-хлоранилина (3.4%). LC-MS:BP: 245 (MH+); анал. RP18-ВЭЖХ RT: 3.74 мин.; HCl соль т. пл.: 227-228oC.
Пример 28. 4-(6-хлор-2,3-дигидро-индол-1-ил)-пиридо[3,4-d]пиримидин.
4-Xлopпиpидo[3,4-d] пиримидин (0.10 г, 0,60 ммоля), 6-хлориндолин (0.10 г, 0.66 ммоля) и пиридин (0.14 г, 1,81 ммоля) объединялись в ДМФ (1 мл) и нагревались при 70oC в течение 3 часов. Реакционная смесь охлаждалась до комнатной температуры, а затем добавлялась к метиленхлориду (150 мл). Органический слой промывался насыщенным карбонатом натрия и водой, а затем сушился над сульфатом натрия. Растворитель удалялся с помощью роторного испарения, и остаток очищался с помощью хроматографии на колонке (силикагель, 9:2:1-метиленхлорид: гексаны: метанол), давая бледно-желтый остаток (0.048 г, 28%). Т. пл. 194-6oC; LCMS 283 (MH+).
Продукты примеров 29-31 получались в соответствии со способом примера 1 из 4-хлорпиридо[3,4-d]пиримидина (1 экв.) и указанного амина.
Пример 29. (Пиридо[3,4-d]пиримидин-4-ил)-(м-толил)-амин.
Данный продукт получался с выходом 44% из м-анизидина (1.1 экв.). Т. пл. 172oC; LC-MS 237 (MH+).
Пример 30. (1H-Индазол-5-ил)-(пиридо[3,4-d]пиримидин-4-ил)-амин.
Данный продукт получался с выходом 96% из 5-аминоиндазола (1.1 экв.). Т. пл. 258oC; LC-MS: 263 (MH+).
Пример 31. (1H-Индол-5-ил)-(пиридо[3,4-d]пиримидин-4-ил)-амин.
Данный продукт получался с выходом 15% из 5-аминоиндола (1.1 экв.) и 4-хлорпиридо[3,4-d]пиримидина (1 экв.). Т. пл. 265oC; LC-МS: 262 (MH+).
Пример 32. (Фенил)-(пиридо[2,3-d]пиримидин-4-ил)-амин.
4-Хлорпиридо[2,3-d]пиримидин (0.15 г, 0.91 ммоля) добавлялся осторожно к раствору анилина (0,15 г, 1,61 ммоля) в воде (1,5 мл). Данный раствор нагревался в течение 0.5 часа на паровой бане, охлаждался, а затем подщелачивался концентрированной гидроокисью аммония. Сырой осадок собирался фильтрованием и перекристаллизовывался из 95% этанола, давая целевой продукт в виде желтых кристаллов (0,054 г, 27%). Т. пл. 258oC; LC-MS: 223 (MH+).
Продукты примеров 33-36 получались в соответствии с процедурой примера 5 из 4-хлорпиридо[2,3-d] пиримидина (1 экв.) и соответствующего замещенного анилина.
Пример 33. (3-Хлор-фенил)-(пиридо[2,3-d]пиримидин-4-ил)-амин.
Данный продукт получался с выходом 61% из м-хлоранилина (1,8 экв.). Т. пл. 228oC; LC-MS: 257 (MH+).
Пример 34. (3-Хлор-фенил)-(пиридо[3,4-d]пиримидин-4-ил)-амин.
Данный продукт получался с выходом 37% из м-хлоранилина (1,8 экв.). Т. пл. 228oC; MCBP: 257 (MH+).
Пример 35. (3-Бром-фенил)-(пиридо[3,4-d]пиримидин-4-ил)амин.
Данный продукт получался с выходом 26% из м-броманилина (1.8 экв.). Т. пл. 206oC; LC-MS: 301 (MH+).
Пример 36. (Фенил)(пиридо[3,4-d]пиримидин-4-ил)амин.
Данный продукт получался с выходом 22% из анилина (1.8 экв.). Т. пл. 161oC; LC-МS: 223 (MH+).
Пример 37. (4-(6-Хлор-2,3-дигидро-индол-1-ил)-пиридо[3,4-d]пиримидин.
4-Хлорпиридо[3,4-d] пиримидин (0.10 г, 0,60 ммоля), 6-хлориндолин (0.10 г, 0,66 ммоля) и пиридин (0,14 г, 1,81 ммоля) объединялись в ДМФ (1 мл) и нагревались при 70oC в течение 3 часов. Реакционная смесь охлаждалась до комнатной температуры, а затем добавлялась к метиленхлориду (150 мл). Органический слой промывался насыщенным карбонатом натрия и водой, а затем сушился над сульфатом натрия. Растворитель удалялся с помощью роторного испарения, и остаток очищался с помощью хроматографии на колонке (силикагель, 9:2: 1 - метиленхлорид: гексаны: метанол), давая целевой продукт в виде бледно- желтого остатка (0,048 г, 28%). Т. пл. 194-196oC; LCMS: 283 (MH+).
Пример 38. (Пиридо[3,4-d]пиримидин-4-ил)(м-толил)амин.
Данный продукт получался с выходом 44% из м-анизидина (1.1 экв.). Т. пл. 172oC; LC-MS: 237 (MH+).
Продукты примеров 39-40 получались в соответствии с процедурой примера 1 из 4-хлорпиридо[3,4-d]пиримидина (1 экв.) и соответствующего амина.
Пример 39. (1H-Индазол-5-ил)(пиридо[3,4-d]пиримидин-4-ил)-амин.
Данный продукт получался с выходом 96% из 5-аминоиндазола (1.1 экв.) и 4-хлорпиридо[3,4-d]пиримидина (1 экв.). Т. пл. 258oC. LC-MS: 263 (MH+).
Пример 40. (1H-Индол-5-ил)(пиридо[3,4-d]пиримидин-4-ил)-амин.
Данный продукт получался с выходом 15% из 5-аминоиндола (1.1 экв.) и 4-хлорпиридо[3,4-d]пиримидина (1 экв.). Т. пл. 265oC; LC-MS: 262 (MH+).
Пример 41. (Фенил)-пиридо[2,3-d]пиримидин-4-ил-амин.
4-Xлopпиpидo[2,3-d]пиримидин (0,15 г, 0.91 ммоля) осторожно добавлялся к раствору анилина (0,15 г, 1,61 ммоля) в воде (1,5 мл). Раствор нагревался в течение 0.5 часа на паровой бане, охлаждался, а затем подщелачивался концентрированной гидроокисью аммония. Сырой осадок собирался фильтрованием и перекристаллизовывался из 95% этанола, давая желтые кристаллы (0,054 г, 27%). Т. пл. 258oC; LC-MS: 223 (MH+).
Пример 42. (3-Хлор-фенил)-(пиридо[2,3-d]пиримидин-4-ил)-амин.
С использованием процедуры, аналогичной процедуре, описанной в примере 5, данный продукт получался с выходом 61% из м-хлоранилина (1.8 экв.) и 4-хлорпиридо[2,3-d]пиримидина (1 экв.). Т. пл. 228oC; LC-МS: 257 (MH+).
Пример 43. (3-Хлор-фенил)-(пиридо[3,4-d]пиримидин-4-ил)-амин.
C использованием процедуры, аналогичной процедуре, описанной в примере 5, данный продукт получался с выходом 37% из м-хлоранилина (1,8 экв.) и 4-хлорпиридо[3,4-d]пиримидина (1 экв.). Т. пл. 228oC LC-MS: 257 (MH+).
Пример 44. (3-Бром-фенил)-(пиридо[3,4-d]пиримидин-4-ил)-амин.
Данный продукт получался с выходом 26% из м-броманилина (1,8 экв.) и 4-хлорпиридо[3,4-d] пиримидина (1 экв. ) с использованием процедуры аналогичной описанной в примере 5. Т. пл. 206oC; LC-MS: 301 (MH+).
Пример 45. (Фенил)-(пиридо[3,4-d]пиримидин-4-ил)-амин.
Данный продукт получался с выходом 22% из анилина (1.8 экв.) и 4-xлopпиpидo[3,4-d]пиримидина (1 экв.) с использованием процедуры аналогичной процедуре, описанной в примере 5. Т. пл. 161oC; LCMS: 223 (MH+).
Пример 46. (7-Бензолсульфонил-7H-пирроло[2,3-d]пиримидин-4-ил)-(3- этинил-фенил)-амин.
К 4-хлор-7H-пирроло[2,3-d] пиримидину (1,0 г, 0.0065 моля) в сухом ТГФ (10 мл) в атмосфере азота при -78oC добавлялся по каплям с помощью шприца на протяжении 15 минут н-бутиллитий (2.5 М в гексане; 2.88 мл, 0.0072 моля). Охлаждающая баня удалялась, и раствор перемешивался в течение 1 часа. Получающаяся в результате соль пирроло аниона выпадала в осадок в виде тонко дисперсного твердого вещества в мутном бесцветном растворе. После того, как суспензия повторно охлаждалась до -78oC, добавлялся бензолсульфонилхлорид (1.26 г, 0.0072 моля) с помощью шприца. Получающаяся в результате желтая реакционная смесь оставлялась медленно подогреваться до комнатной температуры на протяжении ночи. Серо-белая суспензия выливалась в 2% водный бикарбонат натрия (50 мл) и экстрагировалась два раза диэтиловым эфиром (20 мл). Объединенные экстракты промывались водой и сушились (карбонатом калия) и выпаривались, давая светло янтарное масло, которое кристаллизовалось из эфира, продукт собирался фильтрованием, давая 1.4 г (74%) белого твердого вещества. LC-MS = 294 (MH+) RP18-ВЭЖХ RT: 4:40 мин.
Указанное выше соединение растворялось в метаноле и добавлялся м-аминофенилацетилен (0,159 г, 0.0013 моля), и реакционная смесь нагревалась в масляной ванне с температурой 85oC в течение 2 дней. Реакционная смесь охлаждалась до температуры окружающей среды и концентрировалась в вакууме. Остаток растирался с диэтиловым эфиром, давая целевой продукт в виде белого твердого вещества (0,234 г, 92%). LC-MS = 375 (MH+), RP18-ВЭЖХ RT: 3.48 мин.
Пример 47. 4-(6-Хлор-2,3-дигидро-индол-1-ил)-5H-пирроло[3,2-d] пиримидин-6-ол.
К раствору 4-(6-хлор-2,3-дигидро-индол-1-ил)-5-амино-6- метилацетилпиримидина (541 мг, 1.55 ммоля) в 40 мл этанола добавлялось 25 мол.% 10%-ного палладия на угле (125 мг) и 0.11 мл 1 норм. HCl (1.55 ммоля). Реакционная смесь гидрировалась в течение 3 часов при 50 фунт./кв.дюйм (3.515 кг./кв. см). Реакционная смесь фильтровалась через целит и концентрировалась в вакууме. Коричневый остаток суспендировался в метаноле, и белый твердый целевой продукт отфильтровывался (279 мг, 63%) LC-MS: 287 (М+), RP18-ВЭЖХ RT: 5.61 мин. Т. пл.: 250oC (разл.).
Пример 48. (3-Этинил-фенил)-[7-(2-морфолин-4-ил-этил)-7H- пирроло[2,3-d] пиримидин-4-ил]-амин.
К раствору 184 мг (1.4 ммоля) 4-(2-гидроксиэтил)морфолина в 10 мл толуола добавлялось 276 мг (2.0 ммоля) безводного карбоната калия, а затем 32 мг (1.3 ммоля) 97% гидрида натрия. Спустя 30 минут, добавлялось 343 мг (1.0 ммоль) сульфонилированного 4-хлор-7H-пирроло[2,3-d]пиримидина, и реакционная смесь нагревалась при 100oC в течение 2 часов. Реакционная смесь затем распределялась между этилацетатом и водой, и водный слой экстрагировался двумя дополнительными порциями этилацетата. Объединенные органические фазы промывались водой, сушились над сульфатом магния и концентрировались в вакууме. Остаток хроматографировался на силикагеле с использованием 10% метанол/метиленхлорид; давая янтарное масло (140 мг, 55%). LC-MS: 267 (М+).
Указанный выше продукт растворялся в метаноле и добавлялся м-аминофенилацетилен (0.123 г, 0.001 моля), и реакционная смесь нагревалась в запаянной трубке в масляной ванне с температурой 120oC в течение 12 часов. Реакционная смесь охлаждалась до температуры окружающей среды и концентрировалась в вакууме. Остаток растирался с диэтиловым эфиром, давая целевой продукт в виде белого твердого вещества (0.135 г, 74%). LC-MS = 348 (MH+), RP18-ВЭЖХ RT: 3.33 мин.
Пример 49. (3-Этинил-фенил)-[7-(2-метокси-этил)-7H-пирроло[2,3-d] пиримидин-4-ил]-амин.
Данный продукт получался с выходом 81% из 4-хлор-7-(2-мeтoкcиэтил)-7H-пиppoлo[2,3-d] пиpимидинa (1.0 экв. ) и м-аминофенилацетилена (1.2 экв.) в метаноле с использованием процедуры, аналогичной описанной в примере 47. Т. пл. 240-241oC; LC-MS: 292 (MH+); RP18-ВЭЖХ RT: 4.16 мин.
Пример 50. (3-Этинил-фенил)-{ 7-[2-(2-мeтoкcи-этoкcи)-этил]-7H- пирроло[2,3-d]пиримидин-4-ил}-амин.
С использованием процедуры, аналогичной процедуре, описанной в примере 47, данный продукт получался с выходом 81% из 4-хлор-7-[2-(2-метокси-этокси)-этил] -7H-пирроло[2,3-d] пиримидина (1.0 экв.) и м-аминофенилацетилена (1.2 экв.) в метаноле. Т.пл. 240-241oC; LC-MS: 336 (М+); RP18-ВЭЖХ RT: 4.29 мин.
Пример 51. Хлоргидрат (7-аллил-пирроло[2,3-d]пиримидин-4-ил)-(3- этинил-фенил)-амина.
К 4-хлор-7H-пирроло[2,3-d]пиримидину (1.3 г, 8.5 ммолей) в сухом ТГФ (30 мл) добавлялся гидрид натрия (1.0 г, 0.25 ммоля, 60% дисперсия в минеральном масле). После перемешивания при температуре окружающей среды в течение 1 часа, добавлялся аллилиодид (0.93 мл, 10 ммолей), и перемешивание продолжалось в течение 48 часов. Реакционная смесь концентрировалась в вакууме, растиралась в горячем этилацетате и фильтровалась. Фильтрат концентрировался в вакууме, давая оранжевый твердый остаток. Твердое вещество растиралось в метиленхлориде и фильтровалось, давая 4-хлор-7-аллил-пирролопиримидин в виде светло-желтого порошка (0.58 г, 36%). TS-MC: 194 (MH+). К 4-хлор-7-аллил-пирроло[2,3-d] пиримидину (0.5 г, 2.6 ммоля) в сухом метаноле (5 мл) добавлялся м-аминофенилацетилен (0.36 г, 3.1 ммоля). Суспензия нагревалась в запаянной трубке под давлением при 125oC в течение 20 часов. Реакционная смесь охлаждалась до температуры окружающей среды и концентрировалась в вакууме. Получающееся в результате масло очищалось с помощью флэш хроматографии на силикагеле (50 г, 40 мм меш) с использованием 3% метанол/метиленхлорид, давая целевой продукт в виде желтого порошка (0.29 г, 41%). Т. пл. 150-150oC; TS-MS: 275 (MH+).
Пример 52. Хлоргидрат (3-этинил-фенил)-(7-метил-пирроло[2,3-d] пиримидин-4-ил)-амина.
В соответствии с процедурой, описанной в примере 51, целевое соединение получалось из 4-хлор-7H-пирроло[2,3-d]пиримидина и метилиодида, и м-аминофенилацетилена (75%), TS-MS: 249 (MH+); т. пл. 204-205oC.
Пример 53. (5-Бpoм-7H-пиppoлo[2,3-d]пиримидин-4-ил)-(3-этинил- фенил)-амин.
К 4-хлор-7H-пирроло[2,3-d] пиримидину (0.21 г, 1.4 ммоля) в сухом метиленхлориде (10 мл) добавлялся N-бромсукцинимид (0.26 г, 1.5 ммоля) при температуре окружающей среды. Реакционная смесь перемешивалась в течение 18 часов, и получающееся в результате твердое вещество фильтровалось с метиленхлоридными промывными водами и сушилось в вакууме, давая 5-бром-4-хлор-7H-пирроло[2,3-d] пиримидин в виде рыжевато-коричневого порошка (0.28 г, 88%). GC-MS: 233 (MH+), RT: 4.42 мин.
К 5-бpoм-4-xлop-7H-пиppoлo[2,3-d]пиpимидину (0.13 г, 0,57 ммоля) в сухом метаноле (2 мл) добавлялся м-аминофенилацетилен (0.08 г, 0.68 ммоля). Суспензия нагревалась в запаянной трубке под давлением при 125oC в течение 18 часов. Реакционная смесь охлаждалась до температуры окружающей среды и концентрировалась в вакууме. Получающееся в результате масло очищалось с помощью флэш хроматографии на силикагеле (10 г, 40 мм меш) с использованием смеси 3% метанол/метиленхлорид, давая целевое соединение в виде желтого порошка (71 мг, 39%). TS-MS: 314 (MH+); Т. пл. 208oC (разл.).
Пример 54. (3-Этинил-фенил)-(5-иод-7H-пирроло[2,3-d]пиримидин-4-ил)-амин.
К (3-мeтил-фeнил)-(7H-пиppoлo[2,3-d]пиpимидин-4-ил)-амину (17 мг, 0,076 ммоля) в сухом метиленхлориде (1 мл) добавлялся N-иодсукцинимид (19 мг, 0,083 ммоля). Реакционная смесь перемешивалась при температуре окружающей среды в течение 2 часов, затем фильтровалась с метиленхлоридными промывными водами и сушилась в вакууме, давая целевое соединение в виде серо-рыжевато-коричневого порошка (12 мг, 46%). TS-MS: 351 (MH+).
Пример 55. 4-(3-Этинил-фениламино)-7H-пирроло[2,3-d]пиримидин- 5-карбоновая кислота.
К 5-бpoм-4-xлop-7H-пиppoлo[2,3-d]пиpимидину (0.87 г, 3.7 ммоля) в сухом ТГФ (29 мл), охлажденному в ванне из сухого льда и ацетона, добавлялся по каплям н-бутиллитий (3.4 мл, 8.4 ммоля, 2.5 М в гексанах). Реакционная смесь перемешивалась в течение 1 часа, затем резко охлаждалась борботированием CO2. К получающейся в результате оливковой суспензии добавлялась вода (1 мл), и смесь перемешивалась в течение 5 минут при температуре окружающей среды. Реакционная смесь концентрировалась в вакууме, растиралась с этилацетатом и сушилась в вакууме, давая 4-хлор-7H-пирроло[2,3-d]пиримидин-5-карбоновую кислоту в виде порошка цвета авокадо (0.80 г, 74%). TS-МS: 198 (MH+).
К 4-хлор-7H-пирроло[2,3-d] пиримидин-5-карбоновой кислоте (0.38 г, 1.9 ммоля) в сухом метаноле (4 мл) добавлялся м-аминофенилацетилен (0.47 г, 4,0 ммоля). Суспензия нагревалась в запаянной трубке под давлением при 125oC в течение 18 часов. Реакционная смесь охлаждалась до температуры окружающей среды, фильтровалась с метиленхлоридными смывками и сушилась в вакууме, давая целевое соединение в виде рыжевато-коричневого порошка (0.30 г, 54%). TS-MS: 278 (MH+). Т. пл. 190oC (разл.).
Пример 56. Хлоргидрат (3-этинил-фенил)-(5-метил-7H-пирроло[2,3-d] пиримидин-4-ил)-амин.
К 5-бром-4-хлор-7H-пирролопиримидину (0.28 г, 1.2 ммоля) в сухом ТГФ (9 мл), охлажденному в ванне из сухого льда и ацетона, добавлялся по каплям н-бутиллитий (1.1 мл, 2.7 ммоля, 2.М в гексанах). Реакционная смесь перемешивалась в течение 1 часа, затем добавлялся метилиодид (0.12 мл, 1.9 ммоля). Раствор перемешивался в течение 1 часа при температуре окружающей среды, и добавлялась вода (1 мл). Реакционная смесь концентрировалась в вакууме и разбавлялась этилацетатом и водой. Органическая фаза промывалась дважды водой, сушилась над сульфатом натрия и концентрировалась в вакууме, давая 4-xлop-5-мeтил-7H-пиppoлo[2,3-d] пиpимидин (0.17 г, 85%). GC-МS: 167 (M+), RT: 3.15 мин.
К 4-хлор-5-метил-7H-пирролопиримидину (0.17 г, 1.0 ммоль) в сухом метаноле (3 мл) добавлялся м-аминофенилацетилен (0.14 г, 1.2 ммоля). Суспензия нагревалась в запаянной трубке под давлением при 125oC в течение 18 часов. Реакционная смесь охлаждалась до температуры окружающей среды и концентрировалась в вакууме. Получающийся в результате остаток очищался с помощью флэш хроматографии на силикагеле (15 г, 40 мм меш) с использованием 5% метанол метиленхлорид, давая целевое соединение в виде желтого твердого вещества (0.11 г, 43%). TS-MS: 249 (М+).
Пример 57. N-(5-Иод-7H-пирроло[2,3-d]пиримидин-4-ил)-N-м-толил-ацетамид.
К (3-метил-фенил)-(7H-пирроло[2,3-d] пиримидин-4-ил)-амину (0,75 г, 3.4 ммоля), растворенному в горячем ацетонитриле (30 мл), добавлялся гидрид натрия (0,16 г, 4,0 ммоля, 60% дисперсия в минеральном масле). После перемешивания при температуре окружающей среды в течение 0.75 часа, добавлялся ацетилхлорид (0.48 мл, 6.7 ммоля), и перемешивание продолжалось в течение 48 часов. Реакционная смесь концентрировалась в вакууме, растиралась в горячем этилацетате и фильтровалась. Фильтрат концентрировался в вакууме, давая оранжевый твердый остаток. Данное твердое вещество очищалось с помощью флэш хроматографии на силикагеле (13 г, 40 мм меш) с использованием 1:3 смеси этилацетат/гексаны, давая 1-(4-м-толиламино-пирроло[2,3-d]пиримидин-7-ил)-этанон в виде желтого твердого вещества (0.21г). TS-MS: 309 (MH+).
К 1-(4-м-толиламино-пирроло[2,3-d]пиримидин-7-ил)-этанону (0,21 г, 0.79 ммоля) в сухом метиленхлориде (5 мл) и сухом метаноле (2 мл) добавлялся карбонат натрия (0.17 г, 1.6 ммоля). После перемешивания при температуре окружающей среды в течение 0.75 часа добавлялся N-иодсукцинимид (0.35 г, 1.6 ммоля). Реакционная смесь перемешивалась при температуре окружающей среды в течение 48 часов, а затем концентрировалась в вакууме. Остаток разбавлялся метиленхлоридом и водой. Водная фаза экстрагировалась один раз метиленхлоридом. Органическая фаза промывалась дважды водой, сушилась над сульфатом натрия и концентрировалась в вакууме. Получающийся остаток очищался с помощью флэш хроматографии на силикагеле (11 г, 40 мм меш) с использованием смеси 2% метанол/метиленхлорид, давая целевое соединение в виде желтого твердого вещества (30 мг). TS-MS: 393 (MH+). Т. пл. 178-179oC.
Пример 58. Хлоргидрат метилового эфира 4-(3-этинил-фениламино)-7H- пирроло[2,3-d]пиримидин-5-карбоновой кислоты.
К 4-(3-этинил-фениламино)-7H-пирроло[2,3-d]пиримидин-5-карбоновой кислоте (0.108 г, 0.39 ммоля) в сухом метиленхлориде (2 мл) добавлялся раствор оксалилхлорида (0.17 мл, 1.9 ммоля) в сухом метиленхлориде (4 мл) с последующим добавлением 1 капли сухого ДМФ. Суспензия перемешивалась при температуре окружающей среды в течение 1 часа, затем концентрировалась в вакууме. К получающемуся твердому веществу добавлялся сухой ацетон (2 мл) и сухой метанол (1 мл). Раствор перемешивался при температуре окружающей среды в течение 15 часов, затем концентрировался в вакууме. Остаток разбавлялся этилацетатом и водой, и твердое вещество отфильтровывалось и сушилось в вакууме, давая целевое соединение в виде рыжевато-коричневого порошка (40 мг, 35%). TS-MS: 293 (MH+). Т. пл. 256oC (разл.).
Пример 59. (3-Этил-фениламино)-7H-пирроло[2,3-d]пиримидин-5-ил- карбонитрил.
К 5-Бром-4-хлор-7H-пирроло[2,3-d]пиримидину (0.31 г, 1.3 ммоля) в сухом ТГФ (4 мл), охлажденному в ванне из сухого льда и ацетона, добавлялся по каплям н-бутиллитий (1.3 мл, 3.3 ммоля, 2.5 М в гексанах). Реакционная смесь перемешивалась в течение 1 часа, затем добавлялся п-толуолсульфонилцианид (0.44 г, 2.5 ммоля), суспендированный в сухом ТГФ (7 мл). Раствор перемешивался в течение 18 часов при температуре окружающей среды, затем разбавлялся водным хлоридом аммония. Фазы разделялись, и органическая фаза промывалась водой и водным хлоридом натрия. Органическая фаза сушилась над сульфатом натрия и концентрировалась в вакууме. Получающийся в результате остаток очищался с помощью флэш хроматографии на силикагеле (15 г, 40 мм меш) с использованием смеси 3% метанол/метиленхлорид, давая 4-хлор-5-циано-7H-пирроло[2,3-d]пиримидин в виде желтого твердого вещества (52 мг).
К 4-Хлор-5-циано-7H-пирроло[2,3-d]пиримидину (52 мг, 0.29 ммоля) в сухом метаноле (3 мл) добавлялся м-аминофенилацетилен (41 мг, 0.35 ммоля). Суспензия нагревалась в запаянной трубке под давлением при 125oC в течение 18 часов. Реакционная смесь охлаждалась до температуры окружающей среды, фильтровалась небольшим количеством метанола, и сушилась в вакууме, давая целевое соединение в виде белого твердого вещества (27 мг, 36%). TS-MS: 260 (MH+); анал. RP18-ВЭЖХ RT: 3.70 мин.
Пример 60. Хлоргидрат (1H-индазол-5-ил)-(6-метил-пиридо[3,4-d] пиримидин-4-ил)амина.
6-Meтил-пиpидo[3,4-d] пиpимид-4-oн (200 мг, 1.24 ммоля), осажденный на полимере трифенилфосфин (2.06 г, примерно 3,0 ммол. Р/г смолы, 6,20 ммоля) и безводный четыреххлористый углерод (1.20 мл, 12.40 ммолей) объединялись в 1,2-дихлорэтане (6 мл). Реакционная смесь нагревалась до 60oC в атмосфере сухого азота в течение 18 часов. Добавлялся 5-Аминоиндазол (221 мг, 1.66 ммоля), и нагревание продолжалось при 60oC еще в течение 18 часов. Полимер с нанесенным на него трифенилфосфином отфильтровывался и промывался несколько раз хлороформом. Фильтрат и промывные воды концентрировались в вакууме и подвергались флэш хроматографии на силикагеле в смеси 10% метанол/0.1% триэтиламин/89.9% метиленхлорид, давая 207 мг свободного основания целевого продукта (LC-МC: 277 (MH+). Данный материал растворялся в минимальном объеме хлороформа, и по каплям при перемешивании добавлялся 1 моль эквивалент HCl в эфире. Реакционная смесь разбавлялась эфиром (4 объема), и выпавший в осадок HCl соль целевого продукта отфильтровывалась и сушилась в вакууме (188 мг; т. пл. 208oC; LC-MS: 277 (MH+); анал. RP18-ВЭЖХ: 2.71 мин.).
Пример 61. (3-Этинил-4-фтор-фенил)-(6-метил-пиридо[3,4-d] пиримидин-4-ил)-амин.
Данное вещество получалось из 6-метил-пиридо[3,4-d]пиримид-4-она (1.0 экв. ) и 5-амино-бензо[b]тиофена (1.5 экв.), как описано в примере 60. Полимер, отфильтровывался и промывался несколько раз смесью 30 % метанола/ 70% хлороформа. К фильтрату и промывным водам добавлялся триэтиламин (3.0 экв.) перед тем, как они концентрировались в вакууме. Остаток подвергался флэш хроматографии на силикагеле с использованием смеси 10% метанол/метиленхлорид, давая 135 мг продукта в виде свободного основания (LC-MC: 293 (MH+)). Данный материал растворялся в минимальном объеме хлороформа, и по каплям при перемешивании добавлялся 1 эквивалент HCl в эфире. Реакционная смесь разбавлялась эфиром (4 объема), и выпавший в осадок целевой продукт отфильтровывался и сушился в вакууме. (152 мг, т. пл. 273-276oC, LC-MS: 293 (MH+), анал, RP-ВЭЖХ: 4.10 мин).
Пример 62. (3-Этинил-4-фтор-фенил)-(6-метил-пиридо[3,4-d]пиримидин- 4-ил)-амин.
6-Meтил-пиpидo[3,4-d] пиpимид-4-oн (44 мг, 0.27 ммоля), полимер с нанесенным на него трифенилсфосфином (0.452 г примерно 3.0 ммол. Р/г смолы, 1,55 ммоля) и безводный четыреххлористый углерод (0.261 мл, 2.71 ммоля) объединялись в 1,2-дихлорэтане (1,25 мл). Реакционная смесь нагревалась до 60oC в атмосфере сухого азота в течение 2 часов. Добавлялся 3-этинил-4-фтор-анилин (55 мг, 0.407 ммоля), и нагревание продолжалось при 60oC в течение 6 часов. Полимер отфильтровывался и промывался несколько раз смесью 50% метанол/хлороформ. Фильтрат и промывные воды концентрировались в вакууме и подвергались флэш хроматографии на силикагеле с использованием градиента от 0 до 10% метанол/метиленхлорид, давая целевой продукт (10 мг, т. пл. 225oC, LC-MS: 279 (MH+), анал. RP-ВЭЖХ: 3.94 мин.).
Пример 63. Дихлоргидрат 2-метил-4-(6-метил-пиридо[3,4-d]пиримидин- 4-иламино)-фенола.
Данное вещество получалось из 6-метил-пиридо[3,4-d]пиримид-4-она (1.0 экв. ) и 4-амино-о-крезола (1.5 экв.), как описано в примере 61. Полимер с нанесенным на него трифенилфосфином отфильтровывался и промывался несколько раз 50% метанол/хлороформ. Триэтиламин (3.0 жкв.) добавлялся к фильтрату перед концентрированием в вакууме. Остаток подвергался флеш хроматографии на силикагеле с использованием градиента от 0 до 15 метанола и метиленхлорида, давая 314 мг целевого продукта в виде свободного основания (LC-MS: 267 (MH+)). Данное вещество превращалось в дихлоргидратную соль путем растворения в хлороформе и титрования с 2 эквивалентами 1М HCl в эфире. Выпавший в осадок целевой продукт отфильтровывался и сушился в вакууме, (т. пл. 298-305oC, LC-MS: 267 (MH+); анал. RP-ВЭЖХ: 2.88 мин.).
Пример 64. Хлоргидрат 4-(4-Бром-7-метил-2,3-дигидро-индол-1-ил)-6- метилпиридо[3,4-d]пиримидина.
Данный продукт получался из 6-метил-пиридо[3,4-d]пиримид-4-она (1.0 экв. ) и 4-бром-7-метил-индолина (1,5 экв.) в соответствии с процедурой, описанной для примера 61. Сырой или неочищенный продукт из фильтрата подвергался флэш хроматографии на силикагеле с использованием смеси этилацетат:гексаны: метанол (9: 2: 1), давая свободное основание целевого продукта, которое превращалось в целевой продукт, как описано для примера 60 (33%, т. пл. 232-244oC, LC-MS: 355 (MH+), анал. RP-ВЭЖХ: 5.20 мин.).
Пример 65. Хлоргидрат 4-(6-бром-7-метил-2,3-дигидро-индол-1-ил)-6- метилпиридо[3,4-d]пиримидина.
Данный продукт получался из 6-метил-пиридо[3,4-d]пиримид-4-она (1.0 экв. ) и 6-бром-7-метил-индолина (1.5 экв.) в соответствии с процедурой, описанной для примера 61. Сырой продукт из фильтрата подвергался флэш хроматографии на силикагеле с использованием смеси этилацетат:гексаны:метанол (9:2: 1), давая свободное основание целевого продукта, которое превращалось в целевой продукт в виде соли, как описано для примера 60, (34%, т. пл. 212-229oC, LC-MS: 355 (MH+) анал. RP-ВЭЖХ: 4.90 мин.).
Пример 66. Хлоргидрат 4-(6-бром-5-фтор-2,3-дигидро-индол-1-ил)-6- метилпиридо[3,4-d]пиримидина.
Данный продукт получался из 6-метил-пиридо[3,4-d]пиримид-4-она (1.0 экв. ) и 6-бром-5-фтор-индолина (1,5 экв.) согласно процедуре, описанной для примера 61. Неочищенный продукт из фильтрата подвергался флэш хроматографии на силикагеле с использованием смеси 2% метанол/98% метиленхлорид, давая свободное основание целевого продукта, которое превращалось в целевой продукт, как описано для примера 60 (36%, т. пл. 262-264oC; LC-MS: 361 (MH+), анал. RP-ВЭЖХ: 4,83 мин.).
Пример 67. Хлоргидрат (3-хлор-4-фтор-фенил)-(6-метил-пиридо[3,4-d] пиримидин-4-ил)-амина.
Данный продукт получался из 6-метил-пиридо-[3,4-d]пиримид-4-она (1.0 экв. ) и 3-хлор-4-фтор-анилина (1,5 экв.) в соответствии с процедурой, описанной для примера 61. Сырой продукт из фильтрата подвергался хроматографии на силикагеле с использованием градиента 0% -10% метанол/метиленхлорид, давая свободное основание целевого продукта, которое превращалось в целевой продукт, как описано для примера 60 (47%, т. пл. 251-258oC; LC-MS: 289,291 (MH+), анал. RP-ВЭЖХ 4.18 мин.).
Пример 68. Хлоргидрат (6-метил-пиридо[3,4-d] пиримидин-4-ил-(3- трифторметил-фенил)-амина.
Данный продукт получался из 6-метил-пиридо[3,4-d]пиримид-4-она (1,0 экв. ) и 3-трифторметиланилина (1.5 экв.) в соответствии с процедурой, описанной для примера 61. Неочищенный продукт из фильтрата хроматографировался на силикагеле с использованием градиента 0%-5% метанол/метиленхлорид, давая свободное основание целевого продукта, которое превращалось в целевой продукт, как описано для примера 60 (33%, т. пл. 269-270oC; LC-MS: 305 (MH+), анал. RP-ВЭЖХ: 4.30 мин.).
Пример 69. Хлоргидрат (4-фтор-3-метил-фенил)-(6-метил-пиридо[3,4-d] пиримидин-4-ил)-амина.
Данный продукт получался из 6-метил-пиридо[3,4-d]пиримид-4-она (1.0 экв. ) и 4-фтор-3-метил-анилина (1,5 экв.) в соответствии с процедурой, описанной для примера 61. Сырой продукт из фильтрата хроматографировался на двуокиси кремния с использованием смеси 4% метанол/96% метиленхлорид, давая свободное основание целевого продукта, которое превращалось в целевой продукт, как описано для примера 60 (41%, т. пл. 246-250oC, LC-MS: 269 (MH+), анал, RP-ВЭЖХ: 3.79 мин.).
Пример 70. Хлоргидрат 2-иод-4-(6-метил-пиридо[3,4-d]пиримидин-4- иламино)-фенола.
6-Метил-пиридо[3,4-d]пиримид-4-он (161 мг, 1,00 моль) добавлялся к полимеру с нанесенным на него трифенилфосфином (1,66 г, примерно 3 ммоля Р/г полимера; 5 ммолей) наряду с CCl4 (1,54 г, 10,0 ммолей) в 1,2-дихлорэтане (6 мл). Реакционная смесь нагревалась до 60oC в течение 2 часов, а затем смола отфильтровывалась и промывалась 1,2-дихлорэтаном. Фильтрат собирался в колбе, содержащей 4-гидрокси-3-иод-анилин (0,235 г, 1,00 ммоль) и концентрировался к до 5 мл с помощью выпаривания. После 12 часов нагревания с обратным холодильником в атмосфере азота с последующим охлаждением до 20oC целевой продукт собирался с. помощью фильтрования (347 мг, 83%; т. пл. 261-265oC; LC-MS: 379 (MH+); анал. RP-ВЭЖХ: 3.20 мин.).
Пример 71. Хлоргидрат (4-бром-3-фтор-фенил)-(6-метил-пиридо[3,4-d] пиримидин-4-ил)-амина.
Данный продукт получался из 6-метил-пиридо[3,4-d]пиримид-4-она (1 экв.) и 3-бром-4-фтор-анилина (1.0 экв.) и выделялся в соответствии со способом, применяемым для примера 70 (53%, т. пл. 251-254oC LC-MS: 333, 335 (MH+); анал. RP-ВЭЖХ: 4.07 мин.).
Пример 72. Хлоргидрат 4-(6,7-диметил-2,3-дигидро-индол-1-ил)- пиридо[3,4-d]пиримидина.
К 4-хлор-пиридо[3,4-d]пиримидину (200 мг, 1.21 ммоля) в изопропаноле (3 мл) добавлялся 6,7-диметилиндолин (211 мг, 1.44 ммоля) и пиридин (190 мг, 2,41 ммоля). Реакционная смесь нагревалась до температуры дефлегмации в атмосфере сухого азота в течение 6 часов. Растворитель удалялся в вакууме, и остаток растворялся в хлороформе и промывался насыщенным водным карбонатом натрия. Органическая фаза сушилась над сульфатом натрия, концентрировалась в вакууме и подвергалась флэш хроматографии на двуокиси кремния в смеси 45% ацетон/гексаны, давая 60 мг свободного основания целевого продукта (LC-MC: 278 (MH+)). Данный материал растворялся в минимальном объеме 10% метанола в метиленхлориде, и по каплям при перемешивании добавлялся 1 моль эквивалент HCl в эфире. Реакционная смесь разбавлялась четырьмя объемами эфира, и выпавший в осадок целевой продукт отфильтровывался и сушился в вакууме (58 мг; т. пл. 248oC; GC-MS: 277 (М+); анал. RP-ВЭЖХ: 4.06 мин.).
Пример 73. Хлоргидрат (3-этинил-фенил)-(пиридо[3,4-d]пиримидин- 4-ил)-амина.
К 4-хлор-пиридо[3,4-d] пиримидину (250 мг, 1.50 ммоля) в N-метилпирролидин-2-оне (0.5 мл) добавлялся 3-этиниланилин (212 мг, 1.81 ммоля) и пиридин (237 мг, 3,0 ммоля). Реакционная смесь нагревалась до 80oC в атмосфере сухого азота в течение 3 часов. Реакционная смесь растворялась в хлороформе и промывалась насыщенным водным карбонатом натрия и солевым раствором. Органическая фаза сушилась над сульфатом натрия, концентрировалась в вакууме и подвергалась флэш хроматографии на двуокиси кремния с использованием градиента 40% - 70% ацетон/гексаны, давая 120 мг продукта. Данный продукт растворялся в минимальном количестве хлороформа, титровался 1 экв. HCl в эфире и разбавлялся эфиром. Полученный в результате осадок отфильтровывался и сушился в вакууме, давая желтый целевой продукт (133 мг, т. пл. 233-235oC; LC-MS: 247 (MH+); анал. RP-ВЭЖХ: 3.45 мин.).
Пример 74. Хлоргидрат (бензо[b] тиофен-5-ил)-(пиридо[3,4-d]пиримидин- 4-ил)-амина.
К 4-хлор-пиридо[3,4-d] пиримидину (250 мг, 1.50 ммоля) в N-метилпирролидин-2-оне (0.5 мл) добавлялся бензо[b] тиофен-5-ил-амин (270 мг, 1.81 ммоля) и пиридин (237 мг, 3.0 ммоля). Реакционная смесь нагревалась до 80oC в атмосфере сухого азота в течение 3 часов. Реакционная смесь растворялась в хлороформе и промывалась насыщенным водным карбонатом натрия и солевым раствором. Органическая фаза сушилась над сульфатом натрия, концентрировалась в вакууме и подвергалась флэш хроматографии на двуокиси кремния в смеси 40% ацетон-гексан, давая 180 мг продукта. Данный продукт растворялся в минимальном количестве хлороформа, титровался с 1 экв. HCl в эфире и разбавлялся эфиром. Полученный в результате осадок отфильтровывался и сушился в вакууме, давая желтый целевой продукт (188 мг; т. пл. 280-282oC; LC-MS: 279 (MH+); анал. RP-ВЭЖХ: 3,63 мин.).
Пример 75. Хлоргидрат (3-этинил-фенил)-(6-метил-пиридо[3,4-d] пиримидин-4-ил)-амина.
Данное вещество получалось из 4-хлор-6-метил-пиридо[3,4-d]пиримидина (1.0 экв. ) и 3-этиниланилина (1.1 экв.), как описано для примера 74. После экстрагирования, хроматография остатка на двуокиси кремния в смеси от 20% до 80% ацетон-гексаны давала 166 мг целевого продукта в виде его свободного основания, которое превращалось в целевой продукт (т. пл. 250-252oC; LC-MS: 261 (MH+); анал. RP-ВЭЖХ: 3.69 мин.).
Пример 76. (4-(6-Хлор-2,3-дигидро-индол-1-ил)-6-метил- пиридо[3,4-d]пиримидин.
Данный материал получался из 4-хлор-6-метил-пиридо[3,4-d]пиримидина (1.0 экв. ) и 6-хлориндолина (1.1 экв.) как описано в примере 74. Препаративная хроматография с обращенной фазой (C18) с использованием градиента 15%-70% ацетонитрил/pH 4,5, 50 мМ ацетат аммония с последующей лиофилизацией соответствующих фракций давала целевой продукт (30%) (т. пл. 232-234oC; LC-MS: 297 (MH+), анал. RP-ВЭЖХ: 4.33 мин.).
Пример 77. Метансульфонат (1H-индол-5-ил)-(6-метил-пиридо[3,4-d] пиримидин-4-ил)-амина.
Данное вещество получалось из 4-хлор-6-метил-пиридо[3,4-d]пиримидина (1.0 экв. ) и 5-аминоиндола (1.1 экв.), как описано для примера 74. Препаративная хроматография с обращенной фазой (C18) с использованием градиента 15%-70% ацетонитрил/pH 4.5, 50 мМ ацетат аммония с последующей лиофилизацией соответствующих фракций давала свободное основание (30%) целевого продукта (т. пл. 262-263oC; LC-МS: 276 (MH+); анал. RP-ВЭЖХ: 2.98 мин.). Данное вещество превращалось в целевой продукт с помощью растворения в минимальном количестве хлороформа с последующим добавлением 1 экв. метансульфоновой кислоты. Разбавление эфиром осаждало целевой продукт, который отфильтровывался и сушился в вакууме (т. пл. 317-318oC).
Пример 78. (3-Этинил-фенил)-(5-метилсульфанил-7H-пирроло[2,3-d] пиримидин-4-ил)-амин.
К 5-бpoм-4-xлop-7H-пиppoлo[2,3-d]пиримидину (0,18 г, 0.77 ммоля) в сухом ТГФ (2 мл), охлажденному в ванне из сухого льда и ацетона добавлялся по каплям н-бутиллитий (0.77 мл, 1.9 ммоля, 2.5 М в гексанах). Смесь перемешивалась в течение 1 часа, затем добавлялся диметилдисульфид (0.077 мл, 0.77 ммоля), суспендированный в сухом ТГФ (1 мл). Раствор перемешивался в течение 2,5 часов при - 78oC, а затем разбавлялся водным хлоридом аммония. Фазы разделялись, и водная фаза экстрагировалась 2 раза этилацетатом. Объединенные органические фазы сушились над сульфатом натрия и концентрировались в вакууме, давая 4-хлор-5-метилсульфанил-7H-пирроло[2,3-d]пиримидин в виде оранжевого твердого вещества (150 мг).
К 4-хлор-5-метилсульфанил-7H-пирроло[2,3-d]пиримидину (150 мг, 0.75 ммоля) в сухом метаноле (2 мл) добавлялся м-аминофенилацетилен (110 мг, 0.90 ммоля). Раствор нагревался в запаянной трубке под давлением при 125oC в течение 5.5 часов. Реакционная смесь охлаждалась до температуры окружающей среды, фильтровалась с небольшим количеством метанола и сушилась в вакууме, давая целевое соединение в виде рыжевато-коричневого порошка (57 мг, 27%), TS-MS: 281 (MH+); анал. RP18-ВЭЖХ RT: 4.74 мин.
Получение 1. Пиридо[4,3-d]пиримидон из 4-аминоникотиновых кислот.
6-Метил-4-аминоникотиновая кислота (420 мг, 2,74 ммоля) и сухой формамид нагревались до 165oC в течение 6 часов в атмосфере азота. Реакционная смесь охлаждалась до комнатной температуры, и формамид удалялся в вакууме. Остальной остаток очищался с помощью ВЭЖХ с обращенной фазой (линейный градиент 5-100% ацетонитрил при pH 4.50, 50 мМ ацетат аммония на протяжении 1 часа со скоростью потока 23.0 мл/мин.), давая целевой продукт (50%, GCMS RT = 1.48 мин., M+ = 195).
Получение 2. 6-Метил-пиридо[3,4-d]пиримид-4-он.
5-Амино-2-метил-4-пиридинкарбоновая кислота получалась по методике Palt. K. ; Celadnik, M; Dvorackova, D.; Kubalda, B., Cesk. Farm., 32(8). 275-278 (1983). Данная карбоновая кислота превращалась в целевой продукт с помощью нагревания в формамиде при 165oC в соответствии с методикой Robins, R.; Hitchings, G.; J. Am. Chem. Soc. 77, 2256 (1955).

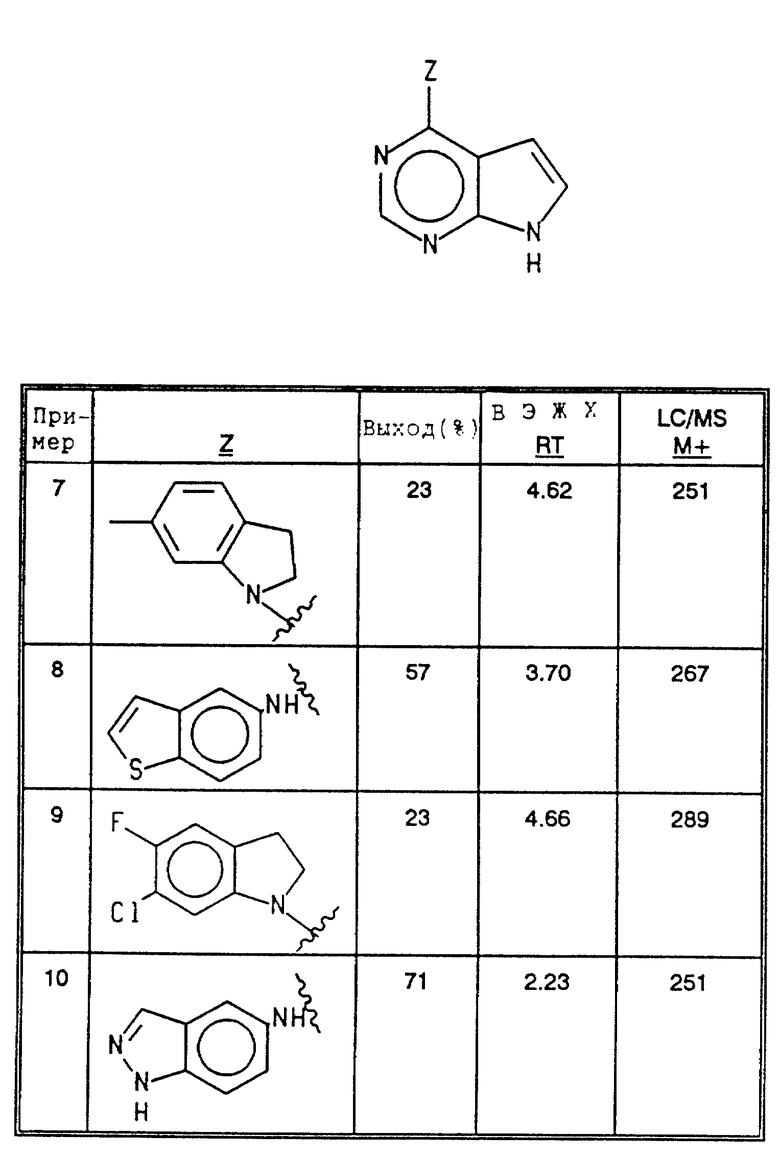
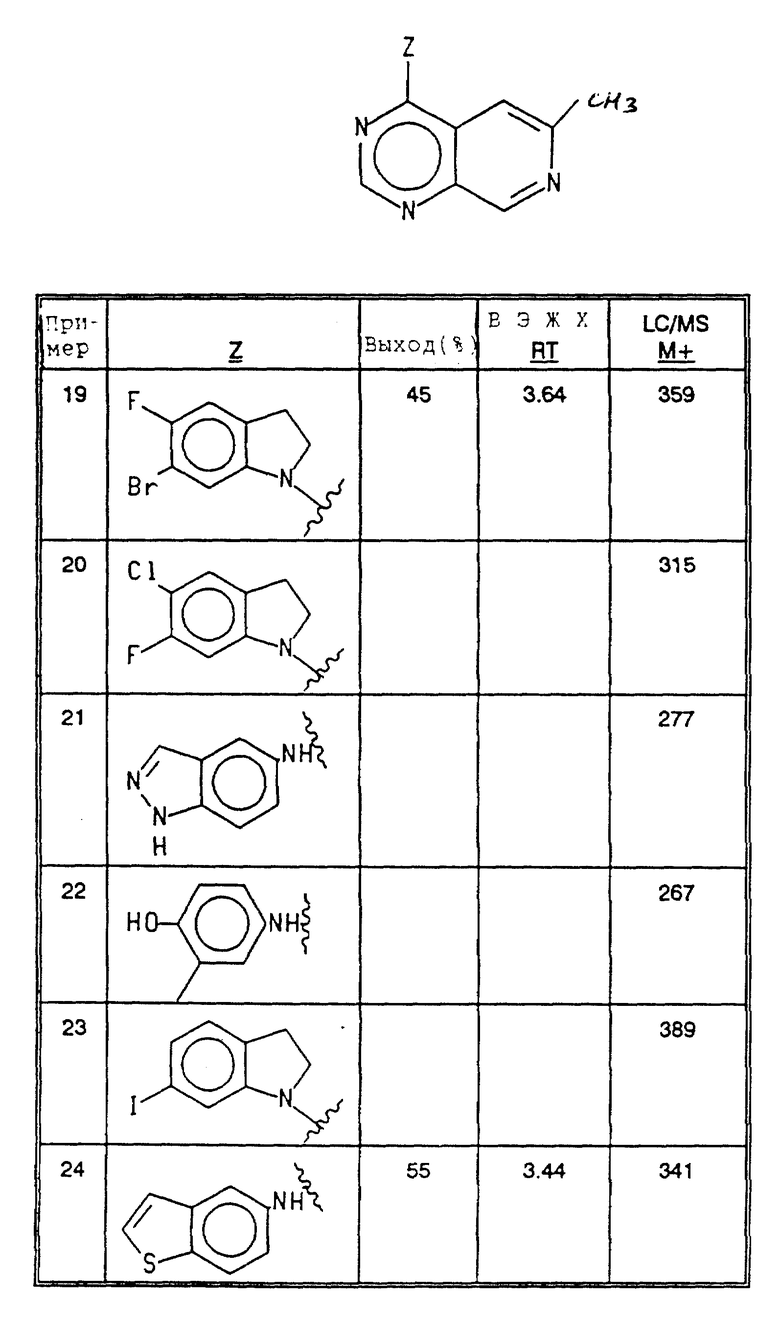
Соединения формулы I
и его стереоизомеры, фармацевтически приемлемые соли, где Y представляет -NR4-CR3= CR3-, CH= CR3-N=CH-, CR3=N-CR3=CR3-, -CR3=CR3-NR4-,
-NR4-CR3= N-, N=N-NR4-; Z - NR1R2, R1 - H, R2 - фенил, замещенный группой (R5)m или Q, или R1R2N представляет группу формулы A
(другие обозначения см. в п.1 ф-лы), могут быть использованы при лечении гиперпролиферативных заболеваний, таких как раковые опухоли и псориаз, астеноз, заболевание почек и панкреатит. 11 з.п. ф-лы.
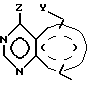
и его стереоизомеpы, фармацевтически приемлемые соли, где Y представляет -NR4-CR3= CR3-, -CH= CR3-N= CH-, -CR3=N-CR3=CR3-, -CR3=CR3-NR4-, -NR4-CR3= N- или -N=N-NR4-;
Z представляет NR1R2, где R1 представляет Н и R2 представляет фенил, замещенный группой (R5)m или Q, или R1R2N представляет группу формулы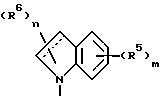 в которой пунктирная линия представляет необязательную двойную связь; каждый R3 присоединен к атому углерода в Y и независимо выбран из: гидрокси, (С1-С4)алкокси, гидрокси(С2-С4)алкокси, амино(С2-С4)алкил, амино(С2-С4)алкокси, (С1-С4)алкокси(С2-С4)алкокси,
в которой пунктирная линия представляет необязательную двойную связь; каждый R3 присоединен к атому углерода в Y и независимо выбран из: гидрокси, (С1-С4)алкокси, гидрокси(С2-С4)алкокси, амино(С2-С4)алкил, амино(С2-С4)алкокси, (С1-С4)алкокси(С2-С4)алкокси,
гидрокси(С1-С4)алкил(С1-С4)алкилендиокси, (С1-С4)алкокси(С1-С4) алкил(С1-С4)алкилендиокси, моно-N- или ди-N,N-(C1-C4)aлкиламино(С2-С4)алкокси, 3- или 4-(С1-С4)алкокси-(2-гидрокси)-(С3-С4)алкокси, карбокси(С1-С4)алкокси, морфолино (С2-С4)алкокси, имидазол-1-ил(С2-С4)алкокси, 4(С1-С4)алкилпиперазин-1-ил-(С2-С4)алкокси, (С1-С4)алкокси(С1-С4)алканоилокси, нитро, гидроксиламино, амино, фенил, пиридил, пирроло, имидазоло, тиазоло, бензимидазоло, пиридонил, моно-N- или ди-N, N-(C1-С4)алкиламино, (С1-С4)алканоиламино, гидрокси(С2-С4)алкиламино,
(С1-С4)алкокси(С2-С4)алкиламино, (С1-С4)алкилсульфонамидо, морфолино, (С1-С4)алкил-пиперазин-1-ил, бис(С1-С4)алкан-сульфонамидо, ди-N, N-(C1-C4)aлкилaминo(C2-C4)aлкилaминo, (С1-С4)алкиламино(С2-С4)алкиламино,
пиперидин-1-ил, имидазол-1-ил, пирролидин-1-ил, (С1-С4)алкокси(С1-С4)алкилкарбониламино, карбокси, (С1-С4)алкоксикарбонил, (С1-С4)алкоксикарбонил (С1-С4)алкокси, амидо, моно-N- или ди-N,N-(C1-C4)алкиламинокарбонил, моно-N- или ди-N, N-(гидpoкcи(C2-C4)aлкил)-аминокарбонил, (С1-С4)алкил, гидрокси(С1-С4)алкил, моно-N- или ди-N,N-((С1-С4)алкокси(С1-С4)алкил)амино(С1-С4)алкил, моно-N- или ди-N, N-(C1-C4)aлкилaминo(C1-C4)aлкил, (С1-С4)алканоиламино (С1-С4)алкил,
(С1-С4)алкилтио, (С1-С4)алкокси(С2-С4)алкилтио или гидрокси(С2-С4)алкилтио; каждый R4 присоединен к N-атому в Y и независимо выбран из: водорода, бензила, фенила, (С2-С4)алкила, гидрокси(С2-С4)алкила или каждой из таких групп, как гидрокси(С2-С4)алкил, амино (С2-С6)алкил, (С2-С4)алкоксикарбонил, замещенной амино, галогеном, гидрокси, (С2-С4)алканоилокси, (С1-С4)алкокси, моно-N- или ди-N,N-(С1-С4)алкиламинo, моно-N- или ди-N,N-(гидрокси(С2-С4)алкил)амино, моно-N- или ди-N,N-(С1-С4)aлкoкcи(С2-С4)aлкил)aминo, сульфониларил(С1-С4), (С1-С4)алканоиламино, имидазол-1-ил, пиперидино, морфолино, пиперазин-1-ил, 4-(С1-С4)алкилпиперазин-1-ил, пиридил, пирроло, имидазоло, тиазоло, пиридонил, карбокси, (С1-С4)алкоксикарбонил, карбамоил, моно-N- или ди-N,N-(С1-С4)aлкилкарбамоил, карбоксамидо, моно-N- или ди-N,N-(С1-С4)алкилкарбоксамидо, или моно-N- или ди-N,N-(С1-С4)алкил)карбоксамидо; каждый R5 независимо выбран из моно-, ди- или трифторметила, галогена, нитро, гидрокси, амино, азидо, изотиоциано, (С1-С4)алкила, фенила, тиенила, (С1-С4)алкокси, бензилокси, фенокси, (С2-С6)алкенила, (С2-С6)алкинила, (С1-С4)алкилендиокси, циано, бензоиламино, трифторметилкарбониламино, (С1-С4)алканоиламино, (С1-С4)алканоил, моно-N- или ди-N,N-(С1-С4)алкиламино, (С1-С4)алкилсульфониламино, трифторметилсульфониламино, (С1-С4)алкилтио, (С1-С4)алкилсульфинила или (С1-С4)алкилсульфонила, пиррол-1-ила, пиперидин-1-ила или пирролидин-1-ила; указанные фенил, бензилокси, фенокси и бензоиламино являются необязательно монозамещенными галогеном, нитро, трифторметилом, гидрокси или (С1-С4)алкилом; указанный (С1-С4)алкилендиокси связан с обоих концов с соседними атомами углерода бензольного фрагмента или две группы R5 с атомами углерода, к которым они присоединены, образуют группу, выбранную из имидазолила, пирроло и пиразолила; каждый R6 независимо выбран из гидрокси, амино, моно-N- или ди-N, N-(С1-С4)алкиламино, сульфо или (С1-С4)алкокси (при условии, что такие группы не присоединены к атому углерода кольца, который непосредственно примыкает к атому азота кольца) или R6 в каждом случае независимо представляет карбокси, гпдрокси (С1-С4)алкил, (С1-С4)алкокси(С1-С4)алкил, амино(С1-С4)алкил, моно-N- или ди-N, N-(С1-С4)aлкилaминo(С1-С4)aлкил, морфолино(С1-С4)алкил, 4-(С1-С4)алкил-пиперазин-1-ил(С1-С4)алкил, карбокси(С1-С4)алкил, (С1-С4)алкоксикарбонил,
сульфо(С1-С4)алкил, пиридил(С1-С4)алкил или (С1-С4)алкил; m представляет целое число от 1 до 3; n представляет 0, 1 или 2; р представляет 0 или целое число от 1 до 3; при условии, что, когда Y в направлении, показанном стрелкой в формуле 1, представляет -CR3=N-CR3=CR3-, р=0, m=1 и Z-замещенный фенил, тогда R5 не является 4-этокси, 4-метокси, 4-трифторметокси, 4-трет-бутилом или 4-изопропилом; Q представляет 9- или 10-членный бициклический гетероарильный циклический фрагмент или его гидрированное производное, содержащие один или два гетероатома азота и необязательно содержащие дополнительный гетероатом, выбранный из азота, кислорода и серы, и может необязательно нести один или два заместителя, выбранных из галогена, гидрокси, оксо, амино, нитро, карбамоила, (С1-С4)алкила, (С1-С4)алкокси, (С1-С4)алкиламино, ди-[(С1-С4)алкил]амино, (С2-С4)алканоиламино, (С2-С4)алкенила и (С2-С4)алкинила при условии, что, когда Y в направлении, показанном стрелкой в формуле 1, представляет -NR4-CR3=CR3-, R3 представляет CH3 и R4 представляет Н, тогда R5 не является группой 4-СН3, 3,5-(СН3)2, 2,6-(CH3)2, 2-С2Н5, 4-н-С4H9, 2-Сl, 4-Сl, 3,4-Cl2, 2-F или 3-СF3.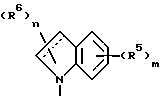
и R5, R6, m и n имеют значения, определенные выше.
сульфо(С1-С4)алкил, пиридил(С1-С4)алкил и (С1-С4)алкил.
| Способ получения производных хиназолина или их солей | 1976 |
|
SU858563A3 |
| 1967 |
|
SU414386A1 | |
| Устройство для передачи сигналов времени и кодовой информации о текущем времени в составе телевизионных сигналов | 1975 |
|
SU520722A1 |
| Способ определения величины нечувствительности системы регулирования турбины | 1975 |
|
SU556226A1 |
| Ультразвуковой искатель | 1975 |
|
SU602851A1 |
| Ди -ацилокси(ароилокси)ацильные перекиси-инициаторы полимеризации винилхлорида | 1976 |
|
SU602498A1 |
| Сплав на основе железа | 1974 |
|
SU475413A1 |
| US 4012513 A, 15.03.77 | |||
| Автоматический огнетушитель | 0 |
|
SU92A1 |
